77 Dialogues in Cardiovascular Medicine - Vol 15 . No. 2 . 2010 Editorial R. Ferrari, D. J. Hearse 79 Lead Article End points in chronic heart failure clinical trials - I. S. Anand, V. G. Florea 81 Expert Answers to Three Key Questions Can BNP or NT-pro-BNP be considered surrogate end points for heart failure? T. A. McDonagh 105 Could endothelial dysfunction be a surrogate end point for coronary artery disease? M. Wolfrum, I. Sudano, J. Steffel, T. F. Lüscher 114 Can there be any surrogate for safety? - M. A. Pfeffer, H. Skali 130 Fascinoma Cardiologica Trails of Discovery: Physiology, histology, and biochemistry: the long path leading to the discovery of the atrial natriuretic peptides - N. Fitzgerald, J. D. Fitzgerald 141 Summaries of Ten Seminal Papers - S. H. Kubo 151 Bibliography of One Hundred Key Papers 163 Surrogate endpoints in clinical trials: definition and operational criteria – R. L. Prentice Role of surrogate end points in the evaluation of drugs for heart failure – R. J. Lipicky and M. Packer Mode of death in chronic heart failure. A request and proposition for more accurate classification – R. Narang and others Surrogate end points in clinical trials: are we being misled? T. R. Fleming and D. L. DeMets Are surrogate markers adequate to assess cardiovascular disease drugs? – R. Temple Surrogate end points in heart failure – I. S. Anand and others Reliability of ventricular remodeling as a surrogate for use in conjunction with clinical outcomes in heart failure – M. A. Konstam Key issues in end point selection for heart failure trials: composite end points – J. D. Neaton and others Influence of nonfatal hospitalization for heart failure on subsequent mortality in patients with chronic heart failure S. D. Solomon and others Heart failure as an endpoint in heart failure and non–heart failure cardiovascular clinical trials: the need for a consensus definition – F. Zannad and others Surrogate End Points in Heart Failure Trials: Potentials and Limitations
Transcript
77
Dialogues in Cardiovascular Medicine - Vol 15 . No. 2 . 2010
EditorialR. Ferrari, D. J. Hearse 79
Lead ArticleEnd points in chronic heart failure clinical trials - I. S. Anand, V. G. Florea 81
Expert Answers to Three Key QuestionsCan BNP or NT-pro-BNP be considered surrogate end points for heart failure?T. A. McDonagh 105Could endothelial dysfunction be a surrogate end point for coronary artery disease?M. Wolfrum, I. Sudano, J. Steffel, T. F. Lüscher 114Can there be any surrogate for safety? - M. A. Pfeffer, H. Skali 130
Fascinoma CardiologicaTrails of Discovery: Physiology, histology, and biochemistry: the long pathleading to the discovery of the atrial natriuretic peptides - N. Fitzgerald, J. D. Fitzgerald 141
Summaries of Ten Seminal Papers - S. H. Kubo 151
Bibliography of One Hundred Key Papers 163
Surrogate endpoints in clinical trials: definition and operationalcriteria – R. L. Prentice
Role of surrogate end points in the evaluation of drugs for heartfailure – R. J. Lipicky and M. Packer
Mode of death in chronic heart failure. A request and propositionfor more accurate classification – R. Narang and others
Surrogate end points in clinical trials: are we being misled?T. R. Fleming and D. L. DeMets
Are surrogate markers adequate to assess cardiovascular diseasedrugs? – R. Temple
Surrogate end points in heart failure – I. S. Anand and others
Reliability of ventricular remodeling as a surrogate for use inconjunction with clinical outcomes in heart failure – M. A. Konstam
Key issues in end point selection for heart failure trials:composite end points – J. D. Neaton and others
Influence of nonfatal hospitalization for heart failure onsubsequent mortality in patients with chronic heart failureS. D. Solomon and others
Heart failure as an endpoint in heart failure and non–heartfailure cardiovascular clinical trials: the need for a consensusdefinition – F. Zannad and others
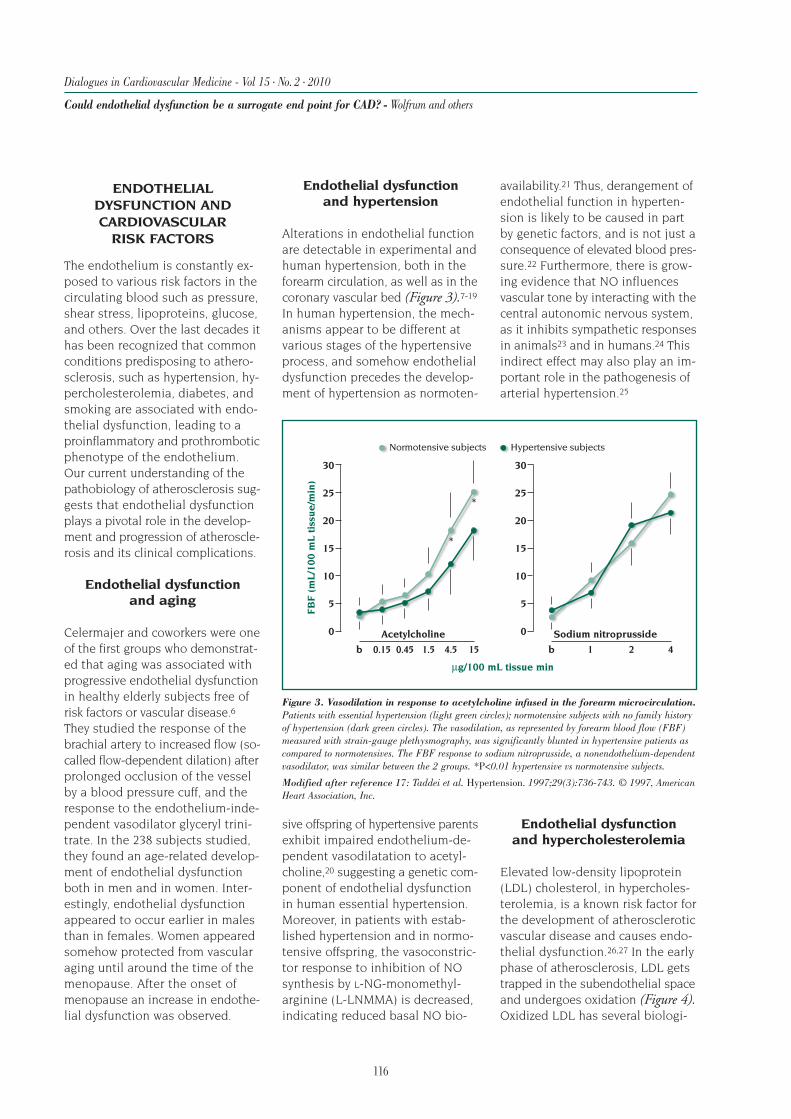
Surrogate End Points in Heart Failure Trials:Potentials and Limitations
81
Dialogues in Cardiovascular Medicine - Vol 15 . No. 2 . 2010
End points in chronic heart failure clinical trialsInder S. Anand, MD, FRCP, DPhil (Oxon), FACC*†, Professor of Medicine andViorel G. Florea, MD, PhD, ScD, FACC*†, Assistant Professor of Medicine*Division of Cardiology - University of Minnesota Medical School; and†Veterans Administration Medical Center - Minneapolis, Minn - USA
Selection of end points for outcomes is an importantstep in a randomized clinical trial. The primary endpoint defines the research question and should ideal-ly be clinically relevant, easily ascertainable in allpatients, capable of unbiased assessment, sensitive tothe hypothesized effects of the treatment, and inex-pensive to measure. Mortality is currently regardedas the most important true end point for evaluationof new heart failure drugs. However, the diminishingrates of these events in sequential trials means thatprogressively larger sample sizes are needed to dis-play benefit from the next therapeutic agent. This ex-plains the increasing use of composite end points andsurrogate end points. The latter are substitutes fortrue end points for the purpose of comparing specificinterventions in a clinical trial; they have no directimportance to the patient, but are biologically rele-vant and are supposed to show a strong and consis-tent relationship with clinical benefit. Another, per-haps more important, aspect is that surrogate endpoints increase our understanding of the diseaseprocess and mechanisms of action of drugs and thusmay help take a more enlightened approach to man-aging patients. We review the potentials and limita-tions of the true and surrogate end points in clinicalstudies of patients with chronic heart failure.
The primary objectives in the treatment of patientswith heart failure (HF) are to improve quality oflife (QoL), delay the progression of the diseaseand increase survival. Randomized clinical trials
represent the standard scientific method for assess-ing the efficacy of any treatment, and the basis for theapproval of new drugs by governmental regulatoryagencies.
The selection of the best response variables for theassessment of the efficacy of a treatment in HF pa-tients is thus still under debate.1,2 Clinical trials con-ducted in thousands of HF patients with such agentsas angiotensin-converting enzyme (ACE) inhibitors,β-receptor blockers, and aldosterone receptor blockershave succeeded in demonstrating incremental benefitson clinically relevant end points, particularly survivaland freedom from hospitalization for HF. Althoughmorbidity and mortality rates remain substantial inpatients with HF, in the setting of clinical trials, a re-markable reduction in all-cause mortality is beingobserved. The diminishing rates of these events in
Keywords: heart failure; clinical trial; end point, drug evaluationAddress for correspondence: Inder S. Anand, MD, FRCP, DPhil(Oxon), FACC, Professor of Medicine, University of Minnesota MedicalSchool. Director, Heart Failure Program, VA Medical Center, Cardiology111-C, One Veterans Drive, Minneapolis, MN 55417(e-mail: [email protected])Dialogues Cardiovasc Med. 2010;15:81-101
MLHFQ Minnesota Living with Heart FailureQuestionnaire
NE norepinephrine
NYHA New York Heart Association (heart failureclass)
QoL quality of life
sequential trials have therefore mandated progres-sively larger sample sizes to display benefit from thenext therapeutic agent. Therefore, the choice of anend point to show the benefit of an agent becomesvery important. In this review, we briefly discuss theadvantages and disadvantages of the common endpoints used in HF trials
The primary end point of a trial should be clinicallyrelevant, easily ascertainable in all patients, capableof unbiased assessment, sensitive to the hypothesizedeffects of the treatment, and inexpensive to measure.2
End points may be categorized as: (i) measures of clin-ical outcomes (eg, death or morbid events as hospi-talization for worsening HF); (ii) measures of symptoms
or clinical status (eg, quality of life, New York HeartAssociation [NYHA] class); or (iii) surrogates (eg, hemo-dynamic measurements, neurohormones, ventricularvolumes and function).
MEASURES OFCLINICAL OUTCOMES
Mortality
Survival in HF clinical trials can be assessed by all-cause mortality, adjusted all-cause mortality, andcause-specific mortality. All-cause mortality is the mostunbiased end point and has routinely been used innumerous HF clinical trials. A reduction in all-cause
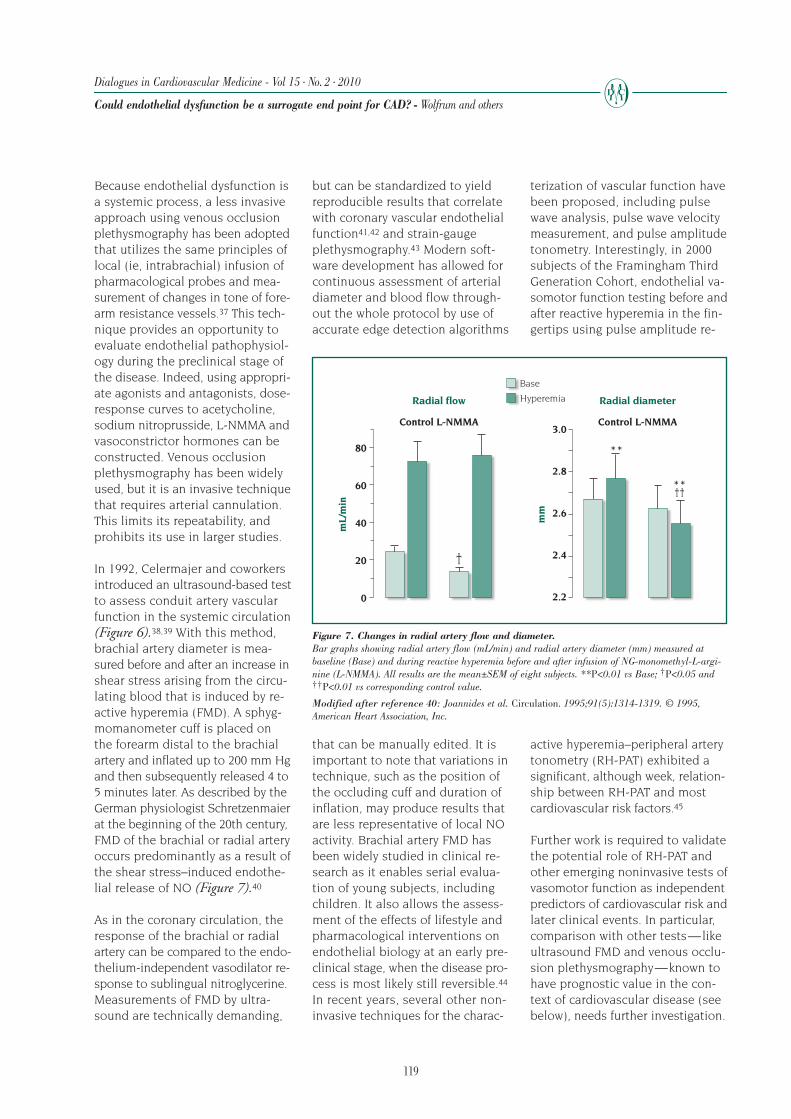
82
Dialogues in Cardiovascular Medicine - Vol 15 . No. 2 . 2010
End points in chronic heart failure clinical trials - Anand and Florea
TRIAL ACRONYMS
A-HeFT African-American–Heart Failure Trial
CAPRICORN Carvedilol Post Infarction Survival Control in Left Ventricular Dysfunction
CHARM Candesartan in Heart failure Assessment of Reduction in Mortality andmorbidity
COMET Carvedilol Or Metoprolol European Trial
COMPANION Comparison of Medical Therapy, Pacing, and Defibrillation in Chronic HeartFailure
CONSENSUS COoperative North Scandinavian ENalapril SUrvival Study
ELITE (I and II) Evaluation of Losartan In The Elderly (first and second trials)
EVEREST Efficacy of Vasopressin antagonism in hEart failuRE: outcome Study withTolvaptan
INSIGHT International Nifedipine once-daily Study Intervention as a Goal inHypertension Treatment
MADIT II Second Multicenter Automatic Defibrillator Implant Trial
MERIT-HF MEtoprolol Randomized Interventional Trial in Heart Failure
MOXCON Effect of Sustained Release Moxonidine on Mortality and Morbidity in Patientswith Congestive Heart Failure
OVERTURE Omapatrilat Versus Enalapril Randomized Trial of Utility in Reducing Events
PRIME II Second Prospective Randomized study of Ibopamine on Mortality and Efficacy
REFLECT Randomized Evaluation of FLosequinan on ExerCise Tolerance
REMATCH Randomized Evaluation of Mechanical Assistance for the Treatment ofCongestive Heart failure
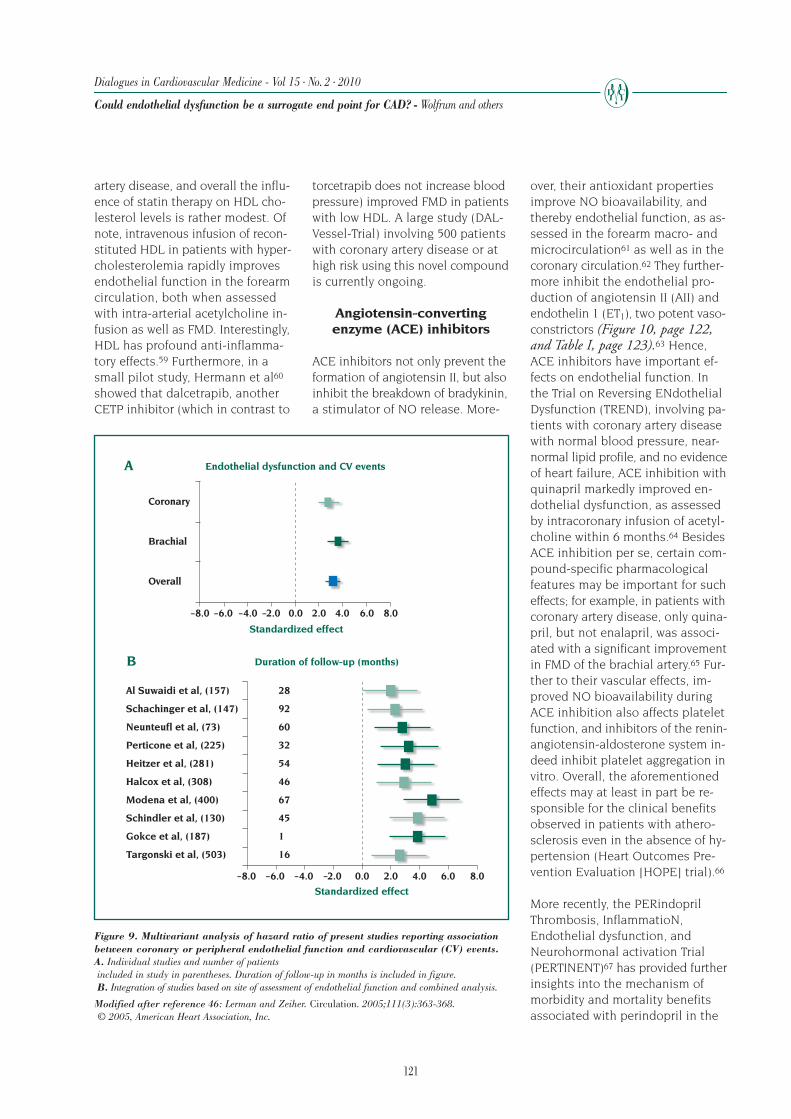
RENAISSANCE Randomized Etanercept North AmerIcan Strategy to Study ANtagonism ofCytokinEs
SAVE Survival And Ventricular Enlargement
SOLVD Studies Of Left Ventricular Dysfunction
Val-HeFT Valsartan–Heart Failure Trial
V-HeFT (I and II) Vasodilator–Heart Failure Trial (first and second trials)
VMAC Vasodilation in the Management of Acute CHF
mortality, or alternatively another beneficial effect onsymptoms or QoL, with assurance of no important in-crease in mortality, is important for regulatory approvalof a drug. Although all-cause mortality has the advan-tage of being a “hard” end point, that is easy to mea-sure, not readily subject to observer bias, and clearlyrepresents an important event for the patients them-selves, it has several limitations. The main concern ofusing only mortality as an end point is that it refers tothe extreme manifestation of HF and occurs in only asmall percentage of patients. Thus, most of the patientsin the study do not contribute to a mortality end point,yet may have important QoL issues. Because the cur-rent management of HF has reduced the event rateconsiderably, if mortality is the primary end point, pa-tients with advanced diseases have to be studied toget enough events for reasonable statistical power ina reasonable period of time. Consequently, patientsin early stages of HF, in whom the disease process ismost likely to be halted or possibly reversed, are notevaluated. Preventive strategy is therefore not assessed.Finally, trials using all-cause mortality as the primaryend point require a large sample size to show a sur-vival advantage of a new drug.
Adjusted all-cause mortality
Several trials have used adjusted all-cause mortality,to control for clinically relevant prognostic variables inCox regression analyses. In the first Vasodilator–HeartFailure Trial (V-HeFT-I),3 mortality at the end of thestudy was lower at a borderline significance in the hy-dralazine/isosorbide (H/I) group compared with placebousing log-rank statistics. However, when a number ofimportant baseline prognostic covariates, such as ejec-tion fraction (EF), history of coronary artery disease,heart rate, and peak oxygen consumption were includedin a Cox regression model, the reduction in mortality inthe H/I group did reach statistical significance. In theCandesartan in Heart failure Assessment of Reductionin Mortality and morbidity (CHARM) overall program,4
the unadjusted hazard ratio (HR) and 95% confidenceinterval (CI) for all-cause mortality was of borderlinesignificance (HR, 0.91; 95% CI, 0.83 to 1.00; P=0.055),but improved significantly (HR, 0.90; 95% CI, 0.82 to0.99; P=0.032) when adjusted for 33 predefined covari-ates. Whether the main treatment comparison shouldbe adjusted remains a subject of debate.5,6 Adjustmentmay help to correct for unexpected baseline imbalance,and may increase statistical power. The covariates cho-sen should predict mortality and be prespecified in theprimary analysis. Often, it is not possible to predictan imbalance at the beginning of the study. However,
even a nonsignificant imbalance in a baseline covari-ate can matter if it is strongly related to mortality. Incontrast, if the correlation with mortality is weak, evena statistically significant imbalance is unimportant.6
Cause-specific mortality
Total mortality is classified into cardiovascular (CV) andnon-CV mortality. CV mortality is further classified intocardiac and vascular. Cardiac death may be suddenand arrhythmic in nature, or result from pump failureand progressive HF. Although cause-specific mortalityappears attractive, there are no “gold standard” defini-tions of different modes of death. The definition ofsudden death, for instance, differs dramatically fromone study protocol to another. Some trials have useda time-dependent definition, such as one hour sincethe onset of new symptoms, as was used in the COoper-ative North Scandinavian ENalapril SUrvival Study(CONSENSUS)7 and Evaluation of Losartan In TheElderly (ELITE)8 trials. The V-HeFT-I trial3 defined sud-den death as either “observed to be instantaneous” or“unwitnessed, but assumed to be instantaneous onthe basis of the clinical setting.” This heterogeneity ofdefinition is also shared by death due to progressiveHF,9 and further complicated by the inclusion of “inter-mediate” classifications, such as death due to “HF orarrhythmias with HF”10 or “sudden death with worsen-ing HF.11” Thus, variations in definition often makecomparison between trials difficult.
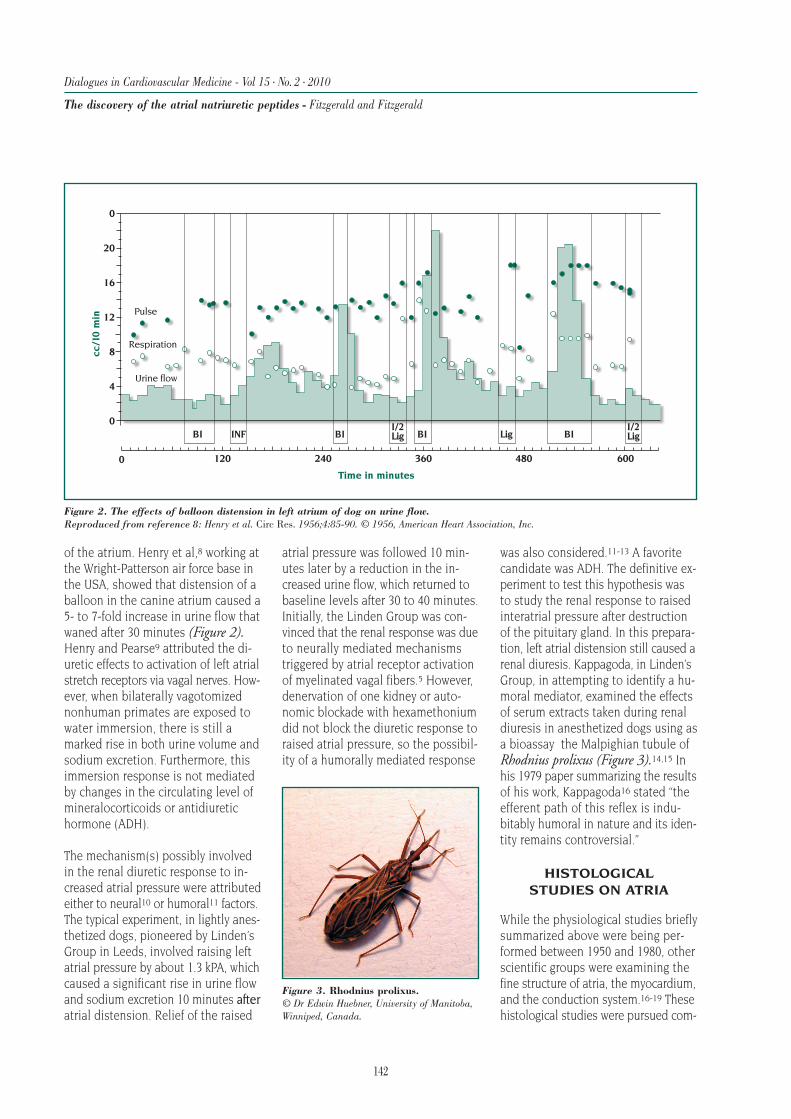
Table I (page 84)3,4,7,10-22 shows the mode of death inseveral landmark HF trials in patients with moderate-to-severe HF and low left ventricular (LV) EF. Despitedifferences in the definitions of mode of death,9 about85% to 90% of all deaths were classified as CV deaths.The remaining 10% to 15% were non-CV deaths. Of theCV deaths, about 80% were classified as cardiac deaths(not shown in the Table). Generally, sudden deathswere more common in patients with less severe HF (eg,V-HeFT I and II) compared with more severe HF (eg,RALES [Randomized ALdactone Evaluation Study] andCOMPANION [Comparison of Medical Therapy, Pacing,and Defibrillation in Chronic Heart Failure]).
The opposite was the case for pump failure deaths thatwere more common in severe HF patients. Therefore,if an intervention, like an implantable cardioverterdefibrillator (ICD), that is expected to reduce suddendeath is being tested, the entire benefit is likely to bein that group. The major disadvantage of cause-spe-cific mortality as an end point is that the mode ofdeath has to be adjudicated. If sudden death can be
83
Dialogues in Cardiovascular Medicine - Vol 15 . No. 2 . 2010
End points in chronic heart failure clinical trials - Anand and Florea
84
assessed accurately, then the use of sudden death as acause-specific mortality end point would be the mostsensitive outcome. Similarly, if a drug were expectedto reduce cardiac events in a HF trial, then the use ofcardiac or CV deaths as the primary outcome wouldbe more appropriate and sensitive than the use of to-tal mortality. Clearly, the added sensitivity conferredby using the CV cause-specific mortality as opposed tototal mortality varies, depending on the proportion ofall deaths that are expected to be CV. For example, inpatients with a low EF and NYHA class IV end-stage HFsuch as those seen in the REMATCH trial (RandomizedEvaluation of Mechanical Assistance for the Treatment
of Congestive Heart failure),23 where over 95% (53 outof 54) of deaths were CV, the difference between usingCV and total deaths is small, and use of all-cause mor-tality becomes a reasonable primary end point. On theother hand, in patients with HF and preserved EF whereonly approximately 60% of the deaths are CV,4,24-27
there could be an important increment in sensitivityby using CV deaths as opposed to total deaths as theprimary end point. This is even more applicable in pri-mary prevention trials where only about 50% of theevents are classified as CV. In such trials, use of cause-specific mortality is likely to significantly improve thesensitivity of end point measurement.
Dialogues in Cardiovascular Medicine - Vol 15 . No. 2 . 2010
End points in chronic heart failure clinical trials - Anand and Florea
Number Total Annual Cardiovascular Sudden Pump Myocardialof mortality mortality death death failure death infarction
Trials patients n (%) y rate (%) n (%) n (%*) n (%*) death, n (%*)
Worsening HF is an important end point in HF trials.However, identifying worsening HF clinically is often achallenge and most clinical trials have used hospital-ization for HF to identify such events. Hospitalizationsare generally adjudicated and classified into eithercardiac or noncardiac. Cardiac hospitalizations are fur-ther categorized into those due to worsening HF, my-ocardial infarction, unstable angina pectoris, syncope,cardiac procedures, arrhythmia-based, heart transplan-tation, complications of cardiac medication or proce-dure, and other causes. Noncardiac causes includepulmonary, vascular, gastrointestinal, and renal causes,as well as noncardiac chest pain, cancer, hypovolemia,complications from noncardiac medication, and othernonspecific reasons.28 Whereas hospitalization as anend point represents a “hard” objective event, it is as-sociated with its own limitations. The threshold for ad-mission and duration of hospitalization differs amonginstitutions and countries, depending in part on reim-bursement or other governmental policies. Moreover,many patients who develop signs and symptoms ofHF may not be hospitalized for these acute episodes,especially in institutions that use multidisciplinarychronic disease management programs. Therefore,these nonhospitalized episodes of HF may not be cap-tured if hospitalization is a requirement to meet theHF end points. Furthermore, the definitions of HF hos-pitalization vary from study to study.29 Most clinicaltrials define hospitalization for HF as a hospital admis-sion, or 24-hour observational stay with at least twosigns and/or symptoms of HF and treatment with loopdiuretics or intravenous vasoactive agents. Some trialprotocols permit the use of IV therapy in the emergencyroom for at least a 4-hour period to be counted as aHF hospitalization.17 The SOLVD trial (Studies Of LeftVentricular Dysfunction) protocol also allowed signifi-cant increase in oral diuretic therapy in the hospitalto be counted toward a HF event.10 Whereas use of IVdiuretics identifies the high-risk patient, it may missthe low-risk patient, with important consequences tothe outcome of a trial. For example, in the OVERTUREtrial (Omapatrilat Versus Enalapril Randomized Trialof Utility in Reducing Events),30 the result of the pri-mary end point of CV death or hospitalization for HFrequiring IV diuretics changed from neutral (HR, 0.94;95% CI, 0.86-1.03; P=0.19) to positive when the posthoc analysis used the SOLVD trial definition and in-cluded significant increase in oral diuretic therapy tobe counted toward HF hospitalization (HR, 0.89; 95%CI, 0.82-0.98; P=0.012).
There are other challenges in the adjudication of wors-ening HF. Comorbidities often associated with HF likechronic obstructive pulmonary disease, atrial fibrilla-tion, and renal dysfunction often make it difficult toassess whether the signs and symptoms of an acuteevent are due to worsening HF or worsening of thecomorbidity.
CAUSE-SPECIFIC HOSPITALIZATIONS
In patients with moderate-to-severe HF and low EF,approximately 30% to 40 % of all-cause hospitaliza-tions are for worsening HF.28,31,32 This proportion de-creases to around 20% to 25% in patients with lesssevere HF.32 Therefore, if a therapy is expected to re-duce only HF hospitalizations, use of all-cause hospi-talizations as the end point is likely to miss even alarge decrease in HF hospitalization. For example, inthe Val-HeFT trial (Valsartan–Heart Failure Trial), theuse of valsartan was associated with a significant 28%reduction in HF hospitalizations, whereas all-causehospitalizations were reduced by a nonsignificant 8%(P=0.15). In this case, the selection of cause-specifichospitalizations for HF helped to increase the sensi-tivity and statistical power. However, the drug beingtested might reduce HF hospitalizations, but increasehospitalizations for another cause such as cancer.Therefore, recording of all-cause hospitalization servesas an important safety end point. Another problem inusing all-cause hospitalizations arises when the pro-tocol only counts the first hospitalization in a time-to-event analysis. Consider a patient whose first hospi-talization is for gallbladder surgery and 6 months lateris readmitted for worsening HF. If the protocol is fo-cused only on time to first hospitalization, then thehospitalization for HF would be missed. In SOLVD,32
approximately 38% of hospitalizations for HF occurredafter a hospitalization for another cause. Therefore,considering time to first all-cause hospitalizationcould lead to a loss in statistical power by inclusionof events that are insensitive, and to loss in eventsthat are truly sensitive to the treatment effect.
When a drug or device reduces mortality, the unbiasedevaluation of recurrent hospitalizations becomes achallenge due to differential follow-up time betweencomparison groups and the competing risk of mortal-ity. Therefore, statistical comparisons of hospitaliza-tion burden that are not adjusted for follow-up time,mortality, and/or multiple hospitalizations can be bi-ased and misleading. It is well known that the likeli-hood of hospitalization and death are related. For ex-ample, death removes the sickest patients who are
Dialogues in Cardiovascular Medicine - Vol 15 . No. 2 . 2010
End points in chronic heart failure clinical trials - Anand and Florea
86
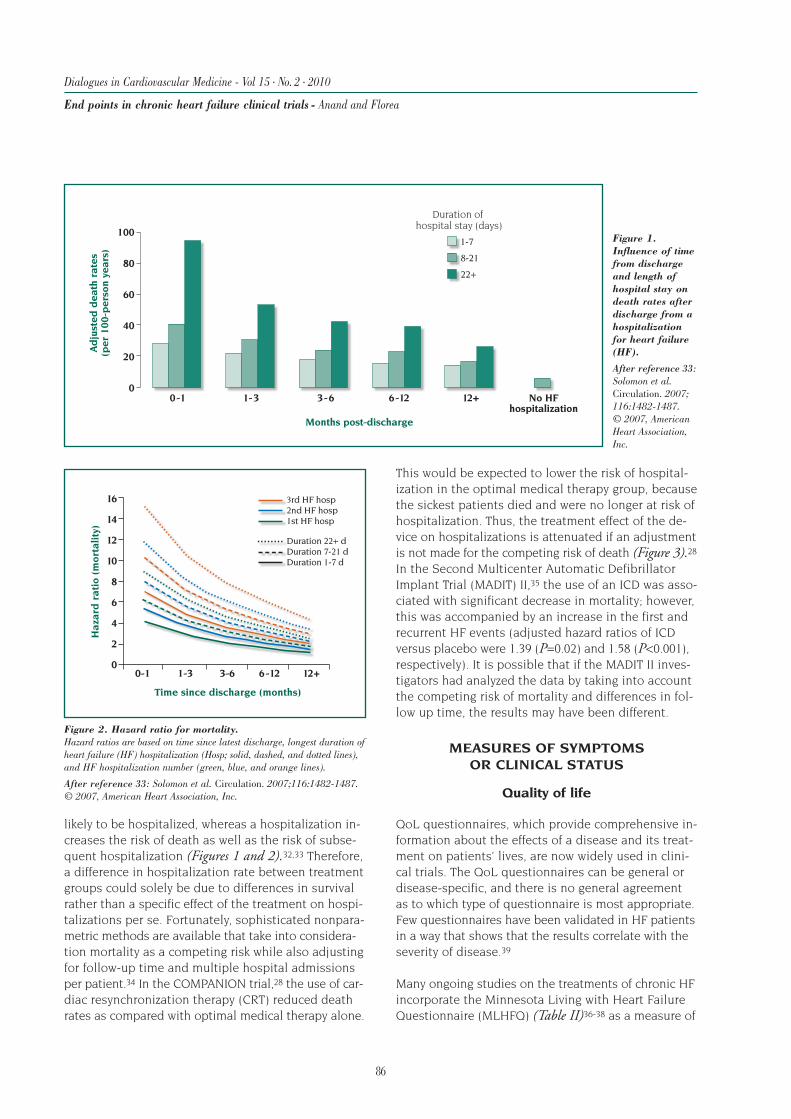
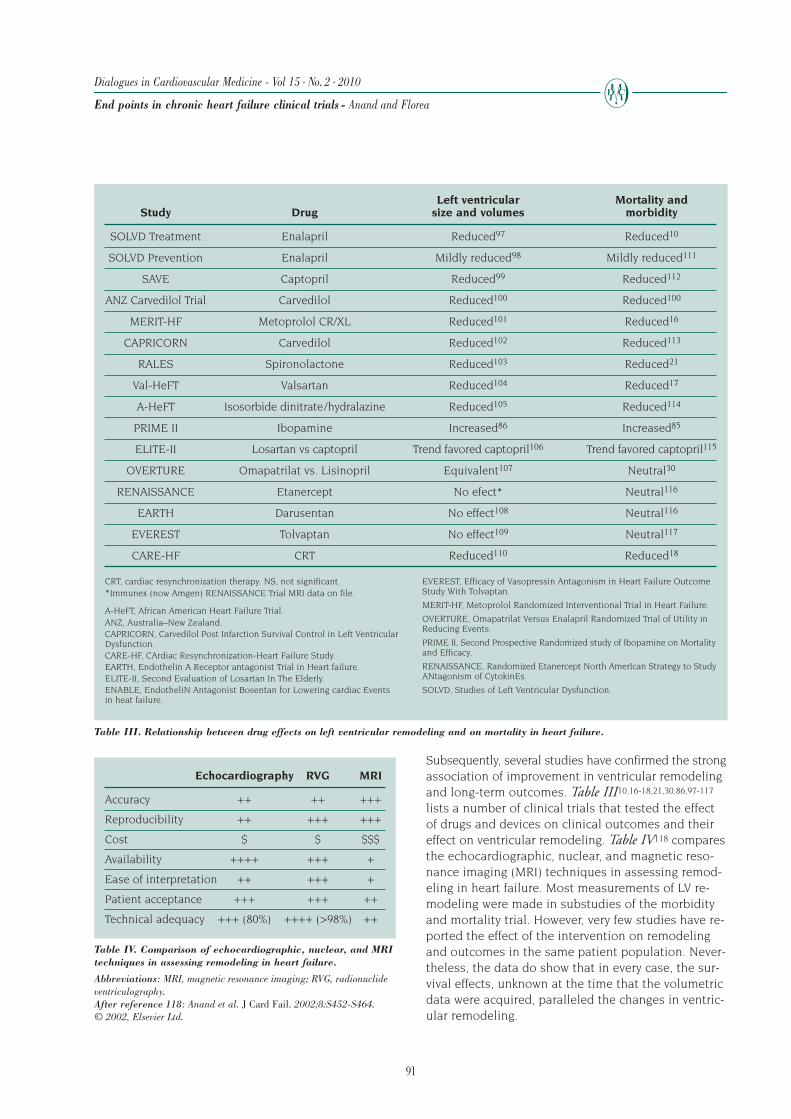
likely to be hospitalized, whereas a hospitalization in-creases the risk of death as well as the risk of subse-quent hospitalization (Figures 1 and 2).32,33 Therefore,a difference in hospitalization rate between treatmentgroups could solely be due to differences in survivalrather than a specific effect of the treatment on hospi-talizations per se. Fortunately, sophisticated nonpara-metric methods are available that take into considera-tion mortality as a competing risk while also adjustingfor follow-up time and multiple hospital admissionsper patient.34 In the COMPANION trial,28 the use of car-diac resynchronization therapy (CRT) reduced deathrates as compared with optimal medical therapy alone.
This would be expected to lower the risk of hospital-ization in the optimal medical therapy group, becausethe sickest patients died and were no longer at risk ofhospitalization. Thus, the treatment effect of the de-vice on hospitalizations is attenuated if an adjustmentis not made for the competing risk of death (Figure 3).28In the Second Multicenter Automatic DefibrillatorImplant Trial (MADIT) II,35 the use of an ICD was asso-ciated with significant decrease in mortality; however,this was accompanied by an increase in the first andrecurrent HF events (adjusted hazard ratios of ICDversus placebo were 1.39 (P=0.02) and 1.58 (P<0.001),respectively). It is possible that if the MADIT II inves-tigators had analyzed the data by taking into accountthe competing risk of mortality and differences in fol-low up time, the results may have been different.
MEASURES OF SYMPTOMSOR CLINICAL STATUS
Quality of life
QoL questionnaires, which provide comprehensive in-formation about the effects of a disease and its treat-ment on patients’ lives, are now widely used in clini-cal trials. The QoL questionnaires can be general ordisease-specific, and there is no general agreementas to which type of questionnaire is most appropriate.Few questionnaires have been validated in HF patientsin a way that shows that the results correlate with theseverity of disease.39
Many ongoing studies on the treatments of chronic HFincorporate the Minnesota Living with Heart FailureQuestionnaire (MLHFQ) (Table II)36-38 as a measure of
Dialogues in Cardiovascular Medicine - Vol 15 . No. 2 . 2010
End points in chronic heart failure clinical trials - Anand and Florea
8-21
22+
1-7
0-1
Ad
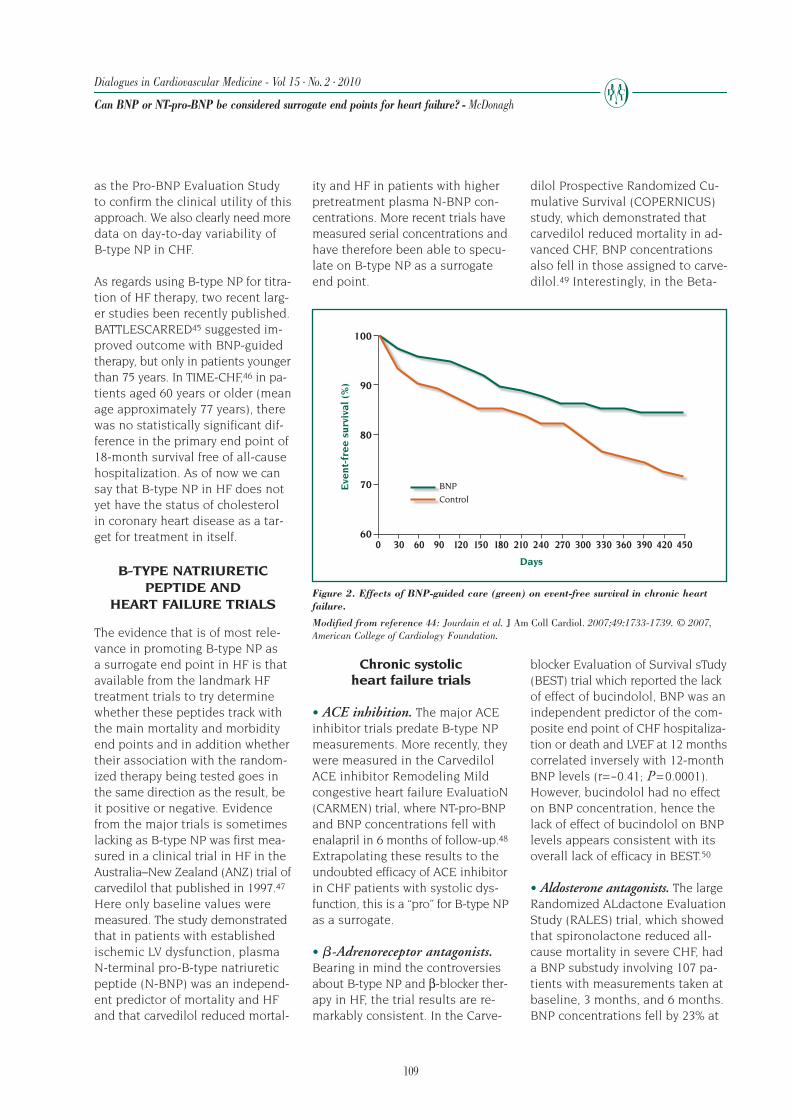
just
edd
eath
rate
s(p
er10
0-p
erso
nye
ars)
Duration ofhospital stay (days)
1-3 3-6
Months post-discharge
6-12 12+ No HFhospitalization
100
80
60
40
20
0
16
14
12
10
8
6
4
2
00-1 1-3 3-6 6-12 12+
Time since discharge (months)
Haza
rdra
tio
(mo
rta
lity
)
3rd HF hosp2nd HF hosp1st HF hosp
Duration 22+ dDuration 7-21 dDuration 1-7 d
Figure 1.Influence of timefrom dischargeand length ofhospital stay ondeath rates afterdischarge from ahospitalizationfor heart failure(HF).
Figure 2. Hazard ratio for mortality.Hazard ratios are based on time since latest discharge, longest duration ofheart failure (HF) hospitalization (Hosp; solid, dashed, and dotted lines),and HF hospitalization number (green, blue, and orange lines).
QoL.36 Statistically significant improvements in the QoLscore have been observed in placebo-controlled studiesof enalapril, flosequinan, pimobendan, vesnarinone, andvalsartan.17,20,40-42 However, flosequinan, pimobendan,and vesnarinone have also been shown to have an
adverse effect on survival,20,43,44 raising the issue of atrade-off between improved QoL and the risk of drug-induced death. Increased mortality with these and oth-er agents clearly indicates that symptomatic benefitin HF does not necessarily predict improved survival.45
Dialogues in Cardiovascular Medicine - Vol 15 . No. 2 . 2010
End points in chronic heart failure clinical trials - Anand and Florea
DID YOUR HEART FAILURE PREVENT YOUFROM LIVING AS YOU WANTED DURING THE PAST MONTH?
1. Causing swelling in your ankles or legs?2. Making you sit or lie down to rest during the day?3. Making your walking about or climbing stairs difficult?4. Making your working around the house or yard difficult?5. Making your going places away from home difficult?6. Making your sleeping well at night difficult?7. Making your relating to or doing things with your friends or
family difficult?8. Making your working to earn a living difficult?9. Making your recreational pastimes, sports, or hobbies difficult?10. Making your sexual activities difficult?11. Making you eat less of the foods you like?12. Making you short of breath?13. Making you tired, fatigued, or low on energy?14. Making you stay in a hospital?15. Costing you money for medical care?16. Giving you side effects from treatments?17. Making you feel you are a burden to your family or friends?18. Making you fell a loss of self-control in your life?19. Making you worry?20. Making it difficult for you to concentrate or remember things?21. Making you feel depressed?
Modified fromreference 36:Rector et al.Heart Fail.1987;3:198-209.With permission.
88
New York Heart Associationfunctional class
The New York Heart Association (NYHA) classification46
is a 4-point semiquantitative index of functional sta-tus of patients with HF. NYHA class is widely acceptedand useful clinically because it correlates with qualityof life36,37,39,47 and survival.48 When measured seriallyover time, it provides a means of tracking diseaseprogression and response to therapeutic interventions.Although it is “subjective,” NYHA class has also beenused in many clinical trials as a demonstration of effi-cacy for both pharmacologic and device interventions.
To overcome the “subjective” nature of NYHA classifi-cation, various quantitative and objective measure-ments of functional capacity have been developed inrecent years. Although most physicians are experiencedin assigning a NYHA class, the method of assignmentis not standardized and the reproducibility of determin-ing NYHA class has never been established.49 Concernshave also been raised about unblinding. Kubo et al49
developed and validated a patient questionnaire todetermine NYHA classification within the context ofclinical trials where blinded conditions are not possi-ble. Goldman et al50 developed a specific activity scalein which the patient’s functional class was based on theestimated metabolic cost of different activities. How-ever, the output of the specific activity scale was notexactly analogous to NYHA and many of the queriesdid not appear to be relevant to a contemporary pop-ulation.50
Exercise capacity
6-minute walk testThe 6-minute walk test was found to predict long-termmortality and HF hospitalization rates in patients withLV dysfunction of varying cause and severity.51,52 Thetest can be administered safely in an outpatient set-ting without specialized equipment and is well accept-ed by patients. Although the 6-minute walk test hasbeen used as an outcome measure in more than 60randomized clinical trials since 1988, its ability to dis-tinguish between effective or ineffective interventionsin patients with HF has not been fully explored. Somedata have not confirmed the predictive value of the dis-tance walked on survival,53 especially in patients withmild HF and preserved exercise tolerance.54 Olson etal55 performed a systematic literature review investi-gating the utility of the 6-minute walk test as a mea-sure of the effectiveness of treatment in randomizedcontrolled trials of HF and found that the test has not
yet been proven to be robust enough for the identifica-tion of effective pharmacological interventions. Like-wise, it has proved useful in some,56,57 but not in otherstudies58 that assessed CRT. The 6-minute walk dis-tance is therefore considered helpful in clinical descrip-tions of HF patients, but cannot be used as a surrogatemarker for assessing survival in HF trials.
Treadmill or cycle exercise testingTreadmill or cycle exercise testing has generally shownthat therapeutic interventions that lessen symptoms inHF patients also improve exercise tolerance and, con-versely, that symptomatically ineffective drugs producelittle change in exercise capacity. Exercise tolerance,expressed as exercise time or workload achieved on anergometer, has been recognized for several decades asan important prognostic marker in patients with heartdisease.59
Peak VO2In recent years, there has been increased interest indirectly measured maximal oxygen uptake (peak VO2)during exercise. Peak VO2 has been considered by someinvestigators as the best criterion of exercise capacityin patients with chronic HF.60-62 As an objective mea-sure of maximal exercise capacity, peak VO2 has beenfound to be an independent prognostic indicator inHF.63,64 In some HF trials, change in peak VO2 has beenused to assess the effectiveness of the intervention.
Some therapeutic interventions in HF that increaseexercise capacity also improve survival.65 However, animprovement in survival has not been demonstratedwith every therapeutic agent that improves effort tol-erance. Results from the Prospective RandOmizedMIlrinone Survival Evaluation (PROMISE)66 and Ran-domized Evaluation of FLosequinan on ExerCise Tol-erance (REFLECT)67 trials have shown that early treat-ment-induced improvements in exercise tolerancewere unreliable predictors of actual treatment effectson survival.
SURROGATE END POINTS INHEART FAILURE TRIALS
Surrogate end points are those that are not direct mea-sures of clinical outcome, symptoms, or clinical status,but correlate with clinically relevant findings, eitherbecause they signal worsening of the underlying dis-ease or contribute to its pathophysiology.2,68 A validsurrogate should have a strong consistent and biolog-ically relevant relationship with survival and shouldunequivocally reflect the true end points (ie, survival
Dialogues in Cardiovascular Medicine - Vol 15 . No. 2 . 2010
End points in chronic heart failure clinical trials - Anand and Florea
89
and QoL). Importantly, there must be no effects of theintervention on the outcome that are not mediatedthrough or captured by the surrogate.69 A change inthe surrogate measure over time should be associat-ed with a proportional and corresponding change inmortality and should fully capture the net effect oftreatment on the clinical outcome.68,70 Surrogate endpoints have several potential advantages. Clinical trialsevaluating surrogate end points can provide an answerabout the effectiveness of a drug or device with a small-er sample size, in a shorter duration, and are thereforeless expensive to run. Unlike mortality, which onlyprovides an average response in a population, surro-gate end points assess efficacy in every individual andcan assess early stages of the disease. The principaldisadvantage of using surrogates to assess therapiesis the possibility of an incomplete, inadequate, or mis-leading evaluation and the fact that they do not assesslong-term safety of the drug or device.68,71-73 Drugsusually have multiple effects, and resorting to a singlesurrogate end point, focused exclusively on one inter-mediate effect, often precludes the evaluation of otherintended or unintended health effects. Currently, reg-ulatory agencies do not accept surrogate end pointsfor approval of drugs for the treatment of HF. Never-theless, surrogate outcomes are important and indis-pensable in the early development of drugs and de-vices and in establishing a “proof of concept.”
Chronic heart failure (CHF) is the final common path-way of several processes involved in the cardiovascularcontinuum that is initiated by risk factors for cardiovas-cular diseases. Once initiated, cardiovascular diseaseprogresses through structural remodeling of the heartand blood vessels. Factors that contribute to this in-clude activation of various neurohormones, growth fac-tors, and cytokines. Markers of this biological process(eg, LV hypertrophy and enlargement) and factors thatcontribute to it (eg, neurohormones) may be viewedas surrogates of the progression of the disease.
More than 150 clinical, hemodynamic, or exercise vari-ables correlate with survival in patients with HF. How-ever, “a correlate does not a surrogate make.”71 Onlysome of these variables have been tested in clinicaltrials as surrogates, and none have been completelyvalidated. We will only briefly focus on hemodynamicmeasurements, neurohormones, and variables of LVstructure and function (remodeling) as potential surro-gate end points for patients with HF. The role of B-typenatriuretic peptide level as a surrogate end point forHF, endothelial dysfunction as a surrogate end pointfor coronary artery disease, and serum creatinine and
microalbuminuria as surrogate end points for renaldysfunction will be detailed in the following ExpertAnswers section of this Journal.
Hemodynamic measurements
During the 1980s, HF was considered primarily a he-modynamic disorder, and physicians believed thattherapeutic interventions that improved pump functionwould predictably benefit patients. Invasive hemody-namic studies to assess cardiac output and right andLV filling pressures were viewed as crucial in develop-ment programs for new drugs. Later studies, however,have raised important concerns about the validity ofacute hemodynamic changes as surrogate end points.A number of controlled clinical trials conducted sincethe 1990s have shown that drugs like milrinone,66,74
and levosimendan,76 which produce striking hemody-namic benefits, do not necessarily produce long-termclinical benefits and may be associated with increasedmortality. More recently, in the Vasodilation in the Man-agement of Acute CHF (VMAC) study,77 when the recom-binant human brain natriuretic peptide nesiritide wasadded to standard care in patients hospitalized withacutely decompensated CHF, the hemodynamics andsome self-reported symptoms improved more with ne-siritide than intravenous nitroglycerin or placebo. In an-other report by the Nesiritide Study Group,78 nesiritidesignificantly reduced the pulmonary capillary wedgepressure and clinical status. However, follow-up dataon these subjects, which were not part of the study de-sign, suggested that nesiritide may have had an adverseeffect on 30-day mortality79 and a greater deteriorationin renal function as compared to those given placebo(P=0.04).80 These findings have discouraged the use ofhemodynamic variables as surrogate markers for drugefficacy. However, the converse is not true. All the drugsapproved for treatment of HF have long-term beneficialhemodynamic effects, and there are no drugs that wors-en hemodynamics and improve long-term outcomes.
Neurohormones
Several neurohormones play an important role in thepathogenesis and progression of HF.81 Two sets of neu-rohormones with opposing effects are activated in thesyndrome of HF. The vasoconstrictor hormones areantinatriuretic, antidiuretic, and generally have growth-promoting properties, whereas the vasodilator hor-mones are natriuretic, diuretic and have antimitogeniceffects. Norepinephrine (NE) and the natriuretic pep-tides are the most studied neurohormones in HF, and
Dialogues in Cardiovascular Medicine - Vol 15 . No. 2 . 2010
End points in chronic heart failure clinical trials - Anand and Florea
the strongest evidence for their pathogenetic rolecomes from studies showing that modulation of theseneurohormones is associated with changes in clinicalcourse and survival.
Measurements of plasma NE were performed in theSecond Vasodilator–Heart Failure Trial (V-HeFT-II) toexamine the effects of therapy on neuroendocrine ac-tivation and the responses to therapy among patientswith different degrees of activation. The baseline plas-ma NE data were grouped into three relatively homo-geneous strata: plasma NE <600 pg/mL, 600 to 900pg/mL, and >900 pg/mL.82 Cumulative mortality wasfound to differ significantly between strata: NE values<600 pg/mL were associated with the lowest risk, val-ues between 600 and 900 pg/mL were associated withan intermediate risk, and values >900 pg/mL identifieda group at exceedingly high risk. The group treatedwith enalapril had a significantly lower mortality thanthe group treated with hydralazine-isosorbide dinitrate,and this benefit was most evident in patients with NEvalues >900 pg/mL.82 Similarly, in the CONSENSUS tri-al, significant reduction in mortality seen with enalaprilwas confined to patients with baseline NE levels abovethe median.83 Other studies have raised important con-cerns about the validity of plasma NE as a surrogatemarker in HF treatment trials. In the Australia–New
Zealand Carvedilol Heart Failure Trial, highbaseline NE levels did not predict addition-al survival benefit with carvedilol, whichsignificantly reduced HF admissions onlyin those patients with NE levels below themedian.84 The most worrisome examplesof disagreement between survival data andplasma NE values come from studies withibopamine85,86 and moxonidine.87 ThePRIME II (Second Prospective RandomizedStudy of Ibopamine on Mortality and Effi-cacy)85 and MOXCON (Effect of SustainedRelease Moxonidine on Mortality and Mor-bidity in Patients with Congestive Heart
Failure)87 trials were terminated prematurely becauseof the adverse effects of ibopamine and moxonidineon mortality despite significant reductions in plasmaNE. These results limit the use of plasma NE as asurrogate marker for HF trials.
Ventricular remodeling
It is now generally recognized that heart failure pro-gresses through a process of structural remodeling ofthe heart (Figures 4 and 5) to which neurohormonaland cytokine activation make an important contribu-tion.88,89 A number of studies have demonstrated astrong and independent correlation between ventriculardilation and subsequent mortality, particularly amongpatients who have suffered a myocardial infarction.90-93
More importantly, agents that have beneficial effectsin HF also generally attenuate or reverse ventricularremodeling, whereas agents that have failed to improveclinical outcomes either had no effect on remodelingor have been associated with adverse remodeling.94,95
V-HeFT-I and V-HeFT-II were the earliest studies toshow that drugs like the hydralazine-isosorbide dini-trate combination and enalapril, which improved sur-vival, also slowed remodeling, whereas prazosin hadno effect on remodeling or outcomes.3,11,96
90
Dialogues in Cardiovascular Medicine - Vol 15 . No. 2 . 2010
End points in chronic heart failure clinical trials - Anand and Florea
ECHO LV mass 206 gMass/vol 1.1; no MR
ECHO LV mass 288 gMass/vol 0.9; 3+ MR
March 1996 April 1998
EDVESVSVEF
188 mL107 mL81 mL38%
EDVESVSVEF
312 mL255 mL57 mL18%
Figure 4. Left ventricular (LV) remodeling over time.Left ventricular angiogram in the right anterior oblique projection ofa patient 1 month after acute myocardial infarction (March 1996) and2 years later (April 1998). Note that 2 years after infarction, the end-diastolic volume (EDV) was 3 times normal, end-systolic volume (ESV)was 5 times normal, stroke volume (SV) was decreased, and there wasa further decrease in ejection fraction (EF). There was a decrease inthe ventricular mass-to-volume ratio over time, suggesting furtherincrease in wall stress. The globular shape contributed to severemitral regurgitation.
Dialogues in Cardiovascular Medicine - Vol 15 . No. 2 . 2010
End points in chronic heart failure clinical trials - Anand and Florea
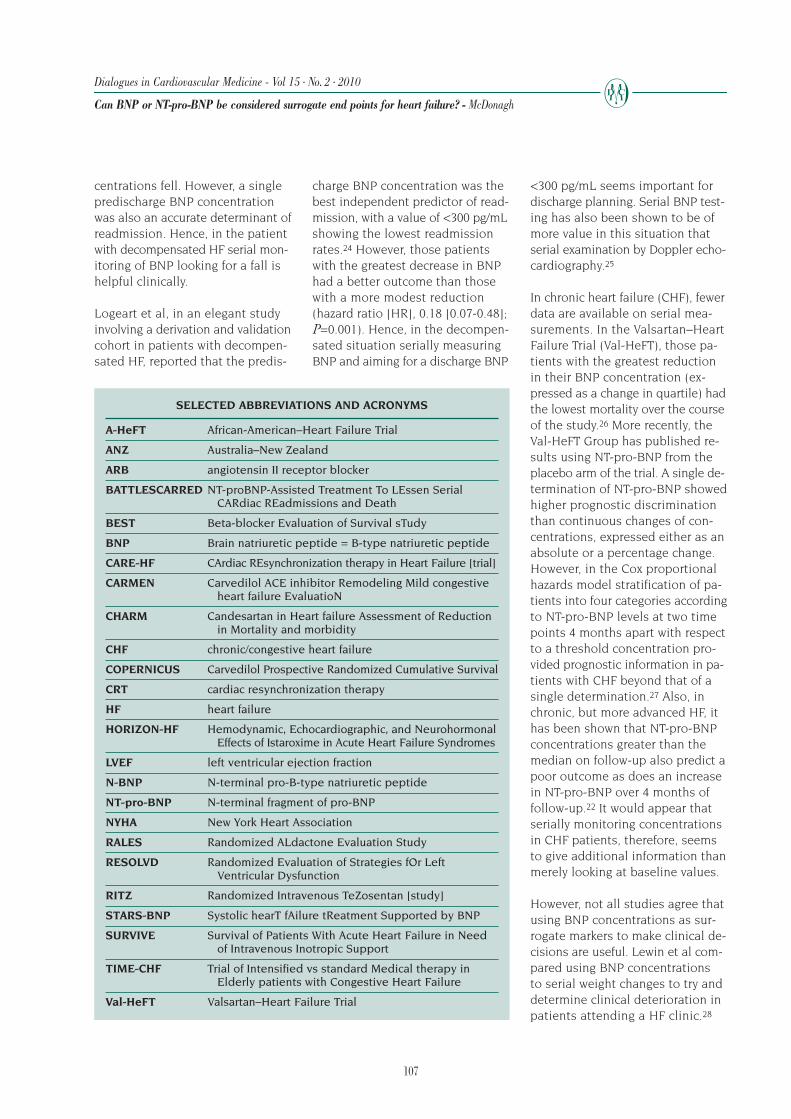
Subsequently, several studies have confirmed the strongassociation of improvement in ventricular remodelingand long-term outcomes. Table III10,16-18,21,30,86,97-117lists a number of clinical trials that tested the effectof drugs and devices on clinical outcomes and theireffect on ventricular remodeling. Table IV118 comparesthe echocardiographic, nuclear, and magnetic reso-nance imaging (MRI) techniques in assessing remod-eling in heart failure. Most measurements of LV re-modeling were made in substudies of the morbidityand mortality trial. However, very few studies have re-ported the effect of the intervention on remodelingand outcomes in the same patient population. Never-theless, the data do show that in every case, the sur-vival effects, unknown at the time that the volumetricdata were acquired, paralleled the changes in ventric-ular remodeling.
Table IV. Comparison of echocardiographic, nuclear, and MRItechniques in assessing remodeling in heart failure.
ELITE-II Losartan vs captopril Trend favored captopril106 Trend favored captopril115
OVERTURE Omapatrilat vs. Lisinopril Equivalent107 Neutral30
RENAISSANCE Etanercept No efect* Neutral116
EARTH Darusentan No effect108 Neutral116
EVEREST Tolvaptan No effect109 Neutral117
CARE-HF CRT Reduced110 Reduced18
CRT, cardiac resynchronization therapy. NS, not significant.*Immunex (now Amgen) RENAISSANCE Trial MRI data on file.
A-HeFT, African American Heart Failure Trial.ANZ, Australia–New Zealand.CAPRICORN, Carvedilol Post Infarction Survival Control in Left VentricularDysfunction.CARE-HF, CArdiac Resynchronization-Heart Failure Study.EARTH, Endothelin A Receptor antagonist Trial in Heart failure.ELITE-II, Second Evaluation of Losartan In The Elderly.ENABLE, EndotheliN Antagonist Bosentan for Lowering cardiac Eventsin heat failure.
EVEREST, Efficacy of Vasopressin Antagonism in Heart Failure OutcomeStudy With Tolvaptan.
MERIT-HF, Metoprolol Randomized Interventional Trial in Heart Failure.
OVERTURE, Omapatrilat Versus Enalapril Randomized Trial of Utility inReducing Events.
PRIME II, Second Prospective Randomized study of Ibopamine on Mortalityand Efficacy.
RENAISSANCE, Randomized Etanercept North AmerIcan Strategy to StudyANtagonism of CytokinEs.
SOLVD, Studies of Left Ventricular Dysfunction.
Echocardiography RVG MRI
Accuracy ++ ++ +++
Reproducibility ++ +++ +++
Cost $ $ $$$
Availability ++++ +++ +
Ease of interpretation ++ +++ +
Patient acceptance +++ +++ ++
Technical adequacy +++ (80%) ++++ (>98%) ++
In SOLVD, the relative benefit of enalapril versus place-bo on ventricular remodeling approximated the rela-tive benefit on outcomes, although this was not testedin the same population.10,97,98,111,119 Similar correla-tions were also seen with the use of captopril betweenmortality and morbidity end points and LV remodel-ing in the Survival And Ventricular Enlargement (SAVE)trial.92,99,112 In the MEtoprolol Randomized Interven-tional Trial in Heart Failure (MERIT-HF), the antiremod-eling effects of metoprolol CR/XL on the left ventricleseen in the MRI substudy101 paralleled the decreasein mortality from worsening HF.16 In the Australia-NewZealand trial of carvedilol in patients with ischemiccardiomyopathy,100 and in the Carvedilol Post Infarc-tion Survival Control in Left Ventricular Dys-function (CAPRICORN) trial, carvedilol hada beneficial effect on ventricular remodel-ing102 and reduced CV mortality.113
In a meta-analysis, carvedilol showed greaterbenefits on LV remodeling compared withimmediate-release metoprolol,120 a findingthat anticipated the subsequent results ofthe Carvedilol Or Metoprolol European Trial(COMET) showing improved survival for pa-tients randomized to carvedilol, comparedwith those randomized to immediate-re-lease metoprolol.121 RALES showed a 30%reduction in mortality with spironolactonein patients with advanced HF.21 A later studyshowed improvement in LV volume andmass with spironolactone.103 In the Val-HeFT trial, beneficial effects of valsartan onthe first morbid event in the overall popu-lation were associated with reduction in LVsize and improvement in left ventricularejection fraction (LVEF).17,104 In the African AmericanHeart Failure Trial (A-HeFT), therapy with the isosor-bide dinitrate / hydralazine combination resulted inregression in LV remodeling105 and increased survivalamong black patients with advanced HF.114
Ibopamine was initially observed to increase ventricu-lar volumes,86 and later found to be associated withexcess mortality.85 The angiotensin receptor blockerlosartan was initially found to have a smaller effecton LV volumes compared with the ACE inhibitor cap-topril,106 and later the ELITE-II study found that mor-tality also tended to be lower with captopril.115 A com-parison of lisinopril122 and the dual vasopeptidaseinhibitor omapatrilat found an equivalent effect onboth these drugs on mortality30 and LV remodeling107
(OVERTURE trial). In the RENAISSANCE trial (Random-
ized Etanercept North AmerIcan Strategy to StudyANtagonism of CytokinEs), the use of the soluble tu-mor necrosis factor antagonist etanercept had no effecton mortality or LV mass and volumes measured withMRI.116 Despite favorable early clinical findings123,124
endothelin receptor antagonist use has been associatedwith neutral to adverse effects on clinical outcomesand no benefits on LV volume or mass.108
We have shown earlier that chronic arginine vasopressinreceptor blockade does not attenuate post–myocardialinfarction ventricular remodeling in the rat model.125
In a well-treated population of stable HF patients,there was no significant effect of tolvaptan therapy on
LV volumes observed during 1 year of therapy.109 Inthe Efficacy of Vasopressin antagonism in hEart failuRE:outcome Study with Tolvaptan (EVEREST) trial,117
tolvaptan initiated for acute treatment of patients hos-pitalized with HF had no effect on long-term mortalityor HF-related morbidity. CRT is associated with im-provement in mortality and morbidity18 and reversesventricular remodeling.18,110
Changes in LV remodeling over time have also beenshown to correspond generally to subsequent changesin mortality, independent of drug effect.92,96,126 Re-cently, Kramer et al127 reported the effects of a drug ordevice on remodeling and mortality in 68 841 patientswith LV dysfunction included in 30 large-outcome ran-domized clinical trials of 24 distinct drug/device ther-apies and in 14 808 patients included in 89 remodel-
92
Dialogues in Cardiovascular Medicine - Vol 15 . No. 2 . 2010
End points in chronic heart failure clinical trials - Anand and Florea
0.5
1.5
1
0.8
Difference in change from baseline, EDV (mL)
Ris
kra
tio
for
de
ath
inla
rge
RC
Ts
–100 –50 0 50
r=0.42; P=0.003
Figure 6. Placebo-corrected change in end-diastolic volume (EDV) from ran-domized clinical trials (RTC) plotted against the mortality odds ratio for thespecific therapy.
After reference 127: Abstract presented at the American College of Cardiology 2008Annual Scientific Session. With permission.
93
ing trials. The odds ratios for death in the outcomerandomized clinical trials correlated significantly withdrug/device effects on LV volumes and EF (Figure 6).127However, there are no studies that have shown that aparticular relative magnitude of change in ventricularremodeling is associated with similar relative magni-tude of benefit on outcomes in the same pop-ulation. Thus, further studies are requiredto reinforce the role of remodeling as acredible surrogate marker in HF trials.95
COMPOSITE END POINTS
With the diminishing rates of mortalityevents, the use of composite end points hasbecome common in HF trials. A compositeor combined outcome is defined as “an eventthat is considered to have occurred if anyone of several different events or outcomesis observed.”128 Each component of a com-posite end point should be clinically rele-vant and sensitive to the hypothesized effectof the treatment, and must be easy to bedetermined.2 Composite end points may compriseany combination of clinical or surrogate end points.Table V shows the advantages and disadvantages ofdifferent primary end point choices.2 If the treatmenteffect is similar for each component of the compositeoutcome, the event rate in a trial will increase. Thatwould help to reduce the sample size, increase thepower and decrease the duration of the study. If, how-ever, the treatment does not have a similar effect onall the composites, the power can actually decrease.
The composite end points become difficult to interpretif the treatment effects go in the opposite direction forsome components or if the effect of treatment is pri-marily on a more common, less serious component ofthe composite. Composite outcomes are typically an-alyzed as time to first event. As discussed in the cause-specific hospitalization section above, this could leadto a substantial loss of information because the eventsafter the first are not counted. Therefore, when compos-ite end points are used, data on all subsequent eventsthat are part of the composite should be collected andevaluated separately as secondary end points. In theCHARM-Added, Alternative, and Preserved trials, theprimary outcome was the composite of cardiovasculardeath or hospitalization for HF analyzed as time to thefirst event. However, all the components of the compos-ite primary end point were separately analyzed as sec-ondary end points.4 In the COMPANION trial, the pri-mary end point was the composite of death from any
cause or hospitalization from any cause.28 The A-HeFTtrial used a unique composite score of weighted val-ues for all-cause mortality, first hospitalization for HF,and change in QoL after 6 months. The time-to-eventanalysis was not used.114,129 Each component of theend point was given a score, with death getting the
worse score, followed by hospitalization and changein QoL. There are several advantages of such a com-posite: each patient contributes to the end point andit integrates the QoL with clinical outcomes. The maindisadvantages of this scoring system are that the timeto death or hospitalization is not taken into account,the weight assigned to each component is arbitrary andnot dependent on any objective criteria, and the rela-tive importance of each component is subjective. More-over, the score has not been validated in other trials.
The role of the clinical event committeein a cardiovascular outcome study
Identification of clinical events is critical to the integri-ty of the clinical research process, but accurately classi-fying clinical events can be problematic. Debate aboutclassification of events has changed the interpretationof some study results and undermined the validityof a study.130,131 Many studies have relied on medicalrecords, death certificates and interpretation of the siteprincipal investigator to establish a clinical event, butthe utility of these source documents may be limiteddue to inconsistencies and omissions in the recordingof important details.132 In prospective studies, disagree-ment between independent reviewers can occur de-spite predefined criteria, casting uncertainty on endpoint assessment.133 Many prospective clinical trialshave used clinical event committees (CECs) to blindlyadjudicate the occurrence of an end point using crite-
Dialogues in Cardiovascular Medicine - Vol 15 . No. 2 . 2010
End points in chronic heart failure clinical trials - Anand and Florea
Advantage Disadvantage
Single outcome Simple Sample size; multipleend points are a reality
Single combined Sample size Interpretation not easyend point if components show
different patterns
Co-primary Eggs not all in Sample size and poweroutcomes one basket
ria defined at the outset of the trial. Heagerty et al134
recently examined the role and usefulness of CECs inexamining all events in the International Nifedipineonce-daily Study Intervention as a Goal in Hyperten-sion Treatment (INSIGHT). More than 28% of investiga-tor-coded primary events and more than 41% of sec-ondary events were reclassified by the CEC. Thesefindings support the use of CECs for end point adju-dication in any large outcome clinical trial.
CONCLUSIONS
In the design of clinical trials, choice of the most appro-priate primary outcome measures is crucial. Althoughall-cause mortality is a simple and most unbiased endpoint that is easy to measure, not subject to observerbias, and clearly represents an important event for thepatient himself, it has several limitations. Mortality isthe extreme manifestation of HF and because it occursin only a small percent of subjects, most patients donot contribute to a mortality end point. Trials usingmortality as the primary end point require a large sam-ple size to show a survival advantage of a new drug.Moreover, patients in early stages of HF, in whom thedisease process is most likely to be halted or possiblyreversed, are not evaluated. Preventive strategy is there-fore not assessed.
To understand whether a treatment makes patients feelbetter and live longer and out of hospital, incorpora-tion of clinical (QoL, mortality, hospitalization), func-tional, structural, and laboratory outcomes may pro-vide a powerful and meaningful composite end pointin some cases. Use of a composite end point also re-sults in a smaller sample size and reduces the dura-tion of the trial if the treatment effect is similar on allthe components of the composite. However, there islittle agreement on the most appropriate compositeend point and the criteria to define a meaningful effecton the composite. There is, therefore, urgent need fora consensus among the trialists and the regulatory au-thorities on some of these issues.
Surrogate end points are physiologic variables that areknown to be statistically associated and are believedto be pathophysiologically related to the clinical out-come. Use of surrogate end points in clinical trials of-fers many potential advantages: fewer patients, shorterfollow-up, and lower cost. However, use of surrogatesrequires a clear understanding of the relationship—both physiologic and statistical—between the surro-gate and the clinical results that are presumed to fol-low. Demonstration of “efficacy” based on surrogate
results must be further subjected to analysis of risk-ben-efit, because serious adverse events may negate anintervention’s clinical utility. An appropriate approachmay be to integrate surrogate end points with clinicalmeasures through use of composites, allowing the sur-rogate finding to augment the clinical outcome, whichmight otherwise not be definitive on its own. Currently,regulatory agencies do not accept surrogate end pointsfor approval of drugs for the treatment of HF and re-quire that new therapies address clinically relevant out-comes before approval. The recent concerns about well-established surrogate end points such as reduction incholesterol and glucose support such policies.135-137
The putative role of B-type natriuretic peptides as sur-rogate end points for HF, as well as endothelial dys-function as a surrogate end point for coronary arterydisease and serum creatinine and microalbuminuriaas surrogate end points for renal dysfunction will bedetailed in the following Expert Answers section ofthis Journal.
REFERENCES
1. Parker M.
How should we judge the efficacy of drug therapy in patients withchronic congestive heart failure? The insights of six blind men.
J Am Coll Cardiol. 1987;9:433-438.
2. Neaton JD, Gray G, Zuckerman BD, Konstam MA.
Key issues in end point selection for heart failure trials: compositeend points.
J Card Fail. 2005;11:567-575.
3. Cohn JN, Archibald DG, Ziesche S, et al.
Effect of vasodilator therapy on mortality in chronic congestive heartfailure. Results of a Veterans Administration Cooperative Study.
N Engl J Med. 1986;314:1547-1552.
4. Pfeffer MA, Swedberg K, Granger CB, et al.
Effects of candesartan on mortality and morbidity in patients withchronic heart failure: the CHARM-Overall programme.
Lancet. 2003;362:759-766.
5. Ford I, Norrie J, Ahmadi S.
Model inconsistency, illustrated by the Cox proportional hazardsmodel.
Stat Med. 1995;14:735-746.
6. Senn SJ.
Covariate imbalance and random allocation in clinical trials.
Stat Med. 1989;8:467-475.
Dialogues in Cardiovascular Medicine - Vol 15 . No. 2 . 2010
End points in chronic heart failure clinical trials - Anand and Florea
95
7. CONSENSUS Trial Study Group.
Effects of enalapril on mortality in severe congestive heart failure.Results of the Cooperative North Scandinavian Enalapril SurvivalStudy (CONSENSUS).
N Engl J Med. 1987;316:1429-1435.
8. Pitt B, Segal R, Martinez FA, et al.
Randomised trial of losartan versus captopril in patients over 65 withheart failure (Evaluation of Losartan in the Elderly Study, ELITE).
Lancet. 1997;349:747-752.
9. Narang R, Swedberg K, Cleland JG.
What is the ideal study design for evaluation of treatment for heartfailure? Insights from trials assessing the effect of ACE inhibitorson exercise capacity.
Eur Heart J. 1996;17:120-134.
10. SOLVD Investigators.
Effect of enalapril on survival in patients with reduced left ventric-ular ejection fractions and congestive heart failure.
N Engl J Med. 1991;325:293-302.
11. Cohn JN, Johnson G, Ziesche S, et al.
A comparison of enalapril with hydralazine-isosorbide dinitrate inthe treatment of chronic congestive heart failure.
N Engl J Med. 1991;325:303-310.
12. Carson P, Anand I, O'Connor C, et al.
Mode of death in advanced heart failure: the Comparison ofMedical, Pacing, and Defibrillation Therapies in Heart Failure(COMPANION) trial.
J Am Coll Cardiol. 2005;46:2329-2334.
13. Singh SN, Fletcher RD, Fisher SG, et al.
Amiodarone in patients with congestive heart failure and asympto-matic ventricular arrhythmia. Survival Trial of AntiarrhythmicTherapy in Congestive Heart Failure.
N Engl J Med. 1995;333:77-82.
14. Digitalis Investigation Group.
The effect of digoxin on mortality and morbidity in patients withheart failure.
N Engl J Med. 1997;336:525-533.
15. Packer M, O'Connor CM, Ghali JK, et al.
Effect of amlodipine on morbidity and mortality in severe chronicheart failure. Prospective Randomized Amlodipine SurvivalEvaluation Study Group.
N Engl J Med. 1996;335:1107-1114.
16. MERIT-HF Study Group.
Effect of metoprolol CR/XL in chronic heart failure: MetoprololCR/XL Randomised Intervention Trial in Congestive Heart Failure(MERIT-HF).
Lancet. 1999;353:2001-2007.
17. Cohn JN, Tognoni G.
A randomized trial of the angiotensin-receptor blocker valsartan inchronic heart failure.
N Engl J Med. 2001;345:1667-1675.
18. Cleland JG, Daubert JC, Erdmann E, et al.
The effect of cardiac resynchronization on morbidity and mortalityin heart failure.
N Engl J Med. 2005;352:1539-1549.
19. Beta-Blocker Evaluation of Survival TrialInvestigators.
A trial of the beta-blocker bucindolol in patients with advancedchronic heart failure.
N Engl J Med. 2001;344:1659-1667.
20. Cohn JN, Goldstein SO, Greenberg BH, et al.
A dose-dependent increase in mortality with vesnarinone amongpatients with severe heart failure.
N Engl J Med. 1998;339:1810-1816.
21. Pitt B, Zannad F, Remme WJ, Cody R, et al.
The effect of spironolactone on morbidity and mortality in patientswith severe heart failure. Randomized Aldactone Evaluation StudyInvestigators.
N Engl J Med. 1999;341:709-717.
22. Bristow MR, Saxon LA, Boehmer J, et al.
Cardiac-resynchronization therapy with or without an implantabledefibrillator in advanced chronic heart failure.
N Engl J Med. 2004;350:2140-2150.
23. Rose EA, Gelijns AC, Moskowitz AJ, et al.
Long-term mechanical left ventricular assistance for end-stageheart failure.
N Engl J Med. 2001;345:1435-1443.
24. Solomon SD, Wang D, Finn P, et al.
Effect of candesartan on cause-specific mortality in heart failurepatients: the Candesartan in Heart failure Assessment of Reductionin Mortality and morbidity (CHARM) program.
Circulation. 2004;110:2180-2183.
25. Ahmed A, Rich MW, Fleg JL, et al.
Effects of digoxin on morbidity and mortality in diastolic heartfailure: the ancillary digitalis investigation group trial.
Circulation. 2006;114:397-403.
26. Cleland JG, Tendera M, Adamus J, Freemantle N,Polonski L, Taylor J.
The perindopril in elderly people with chronic heart failure(PEP-CHF) study.
Eur Heart J. 2006;27:2338-2345.
Dialogues in Cardiovascular Medicine - Vol 15 . No. 2 . 2010
End points in chronic heart failure clinical trials - Anand and Florea
96
27. Massie BM, Carson PE, McMurray JJ, et al.
Irbesartan in patients with heart failure and preserved ejection fraction.
N Engl J Med. 2008;359:2456-2467.
28. Anand IS, Carson P, Galle E, et al.
Cardiac resynchronization therapy reduces the risk of hospitaliza-tions in patients with advanced heart failure: results from theComparison of Medical Therapy, Pacing and Defibrillation inHeart Failure (COMPANION) trial.
Circulation. 2009;119:969-977.
29. Zannad F, Stough WG, Pitt B, et al.
Heart failure as an endpoint in heart failure and non-heart failurecardiovascular clinical trials: the need for a consensus definition.
Eur Heart J. 2008;29:413-421.
30. Packer M, Califf RM, Konstam MA, et al.
Comparison of omapatrilat and enalapril in patients with chronicheart failure: the Omapatrilat Versus Enalapril Randomized Trialof Utility in Reducing Events (OVERTURE).
Circulation. 2002;106:920-926.
31. Carson P, Tognoni G, Cohn JN.
Effect of Valsartan on hospitalization: results from Val-HeFT.
J Card Fail. 2003;9:164-171.
32. Yusuf S, Negassa A.
Choice of clinical outcomes in randomized trials of heart failuretherapies: disease-specific or overall outcomes?
Am Heart J. 2002;143:22-28.
33. Solomon SD, Dobson J, Pocock S, et al.
Influence of nonfatal hospitalization for heart failure on subsequentmortality in patients with chronic heart failure.
Circulation. 2007;116:1482-1487.
34. Ghosh D, Lin DY.
Nonparametric analysis of recurrent events and death.
Biometrics. 2000;56:554-562.
35. Moss AJ, Zareba W, Hall WJ, et al.
Prophylactic implantation of a defibrillator in patients with my-ocardial infarction and reduced ejection fraction.
N Engl J Med. 2002;346:877-883.
36. Rector TS, Kubo SH, Cohn JN.
Patients' self-assessment of their congestive heart failure: content,reliability and validity of a new measure, the Minnesota Livingwith Heart Failure Questionnaire.
Heart Fail. 1987;3:198-209.
37. Green CP, Porter CB, Bresnahan DR, Spertus JA.
Development and evaluation of the Kansas City CardiomyopathyQuestionnaire: a new health status measure for heart failure.
J Am Coll Cardiol. 2000;35:1245-1255.
38. Ware J, Jr., Kosinski M, Keller SD.
A 12-Item Short-Form Health Survey: construction of scales andpreliminary tests of reliability and validity.
Med Care. 1996;34:220-233.
39. Rector TS, Anand IS, Cohn JN.
Relationships between clinical assessments and patients’ perceptionsof the effects of heart failure on their quality of life.
J Card Fail. 2006;12:87-92.
40. Kubo SH, Gollub S, Bourge R, et al.
Beneficial effects of pimobendan on exercise tolerance and qualityof life in patients with heart failure. Results of a multicenter trial.The Pimobendan Multicenter Research Group.
Circulation. 1992;85:942-949.
41. Massie BM, Berk MR, Brozena SC, et al.
Can further benefit be achieved by adding flosequinan to patientswith congestive heart failure who remain symptomatic on diuretic,digoxin, and an angiotensin converting enzyme inhibitor? Resultsof the flosequinan-ACE inhibitor trial (FACET).
Circulation. 1993;88:492-501.
42. Rector TS, Kubo SH, Cohn JN.
Validity of the Minnesota Living with Heart Failure questionnaireas a measure of therapeutic response to enalapril or placebo.
Am J Cardiol. 1993;71:1106-1107.
43. Packer M, Rouleau J, Swedberg K.
Effect of flosequinan on survival in chronic heart failure: preliminaryresults of the PROFILE study [abstract].
Circulation. 1993;88(suppl 1):1-301.
44. Rector TS, Cohn JN.
Assessment of patient outcome with the Minnesota Living with HeartFailure questionnaire: reliability and validity during a randomized,double-blind, placebo-controlled trial of pimobendan. PimobendanMulticenter Research Group.
Am Heart J. 1992;124:1017-1025.
45. Rector TS, Tschumperlin LK, Kubo SH, et al.
Use of the Living With Heart Failure questionnaire to ascertainpatients’ perspectives on improvement in quality of life versus riskof drug-induced death.
J Card Fail. 1995;1:201-206.
46. Fleg JL, Pina IL, Balady GJ, et al.
Assessment of functional capacity in clinical and research applica-tions: An advisory from the Committee on Exercise, Rehabilitation,and Prevention, Council on Clinical Cardiology, American HeartAssociation.
Circulation. 2000;102:1591-1597.
Dialogues in Cardiovascular Medicine - Vol 15 . No. 2 . 2010
End points in chronic heart failure clinical trials - Anand and Florea
97
47. Al-Kaade S, Hauptman PJ.
Health-related quality of life measurement in heart failure: challengesfor the new millennium.
J Card Fail. 2001;7:194-201.
48. Francis GS, Kubo SH.
Prognostic factors affecting diagnosis and treatment of congestiveheart failure.
Development and validation of a patient questionnaire to deter-mine New York Heart Association classification.
J Card Fail. 2004;10:228-235.
50. Goldman L, Hashimoto B, Cook EF, Loscalzo A.
Comparative reproducibility and validity of systems for assessingcardiovascular functional class: advantages of a new specific ac-tivity scale.
Circulation. 1981;64:1227-1234.
51. Bittner V, Weiner DH, Yusuf S, et al.
Prediction of mortality and morbidity with a 6-minute walk test inpatients with left ventricular dysfunction. SOLVD Investigators.
JAMA. 1993;270:1702-1707.
52. Cahalin LP, Mathier MA, Semigran MJ, Dec GW,DiSalvo TG.
The six-minute walk test predicts peak oxygen uptake and survivalin patients with advanced heart failure.
Chest. 1996;110:325-332.
53. Lucas C, Stevenson LW, Johnson W, et al.
The 6-min walk and peak oxygen consumption in advanced heartfailure: aerobic capacity and survival.
Am Heart J. 1999;138:618-624.
54. Roul G, Germain P, Bareiss P.
Does the 6-minute walk test predict the prognosis in patients withNYHA class II or III chronic heart failure?
Am Heart J. 1998;136:449-457.
55. Olsson LG, Swedberg K, Clark AL, Witte KK,Cleland JG.
Six minute corridor walk test as an outcome measure for the as-sessment of treatment in randomized, blinded intervention trials ofchronic heart failure: a systematic review.
Eur Heart J. 2005;26:778-793.
56. Abraham WT, Fisher WG, Smith AL, et al.
Cardiac resynchronization in chronic heart failure.
N Engl J Med. 2002;346:1845-1853.
57. Linde C, Leclercq C, Rex S, et al.
Long-term benefits of biventricular pacing in congestive heartfailure: results from the MUltisite STimulation in cardiomyopathy(MUSTIC) study.
J Am Coll Cardiol. 2002;40:111-118.
58. Young JB, Abraham WT, Smith AL, et al.
Combined cardiac resynchronization and implantable cardioversiondefibrillation in advanced chronic heart failure: the MIRACLEICD Trial.
JAMA. 2003;289:2685-2694.
59. Morris CK, Ueshima K, Kawaguchi T, Hideg A,Froelicher VF.
The prognostic value of exercise capacity: a review of the literature.
Am Heart J. 1991;122:1423-1431.
60. Franciosa JA, Ziesche S, Wilen M.
Functional capacity of patients with chronic left ventricular failure.Relationship of bicycle exercise performance to clinical and hemo-dynamic characterization.
Am J Med. 1979;67:460-466.
61. Lipkin DP, Perrins J, Poole-Wilson PA.
Respiratory gas exchange in the assessment of patients with impairedventricular function.
Oxygen utilization and ventilation during exercise in patients withchronic cardiac failure.
Circulation. 1982;65:1213-1223.
63. Cohn JN, Johnson GR, Shabetai R, et al.
Ejection fraction, peak exercise oxygen consumption, cardiothoracicratio, ventricular arrhythmias, and plasma norepinephrine as deter-minants of prognosis in heart failure.
Circulation. 1993;87:VI5-V16.
64. Florea VG, Henein MY, Anker SD, et al.
Prognostic value of changes over time in exercise capacity andechocardiographic measurements in patients with chronic heartfailure.
Eur Heart J. 2000;21:146-153.
65. Ziesche S, Cobb FR, Cohn JN, Johnson G, Tristani F.
Hydralazine and isosorbide dinitrate combination improves exercisetolerance in heart failure. Results from V-HeFT I and V-HeFT II.The V-HeFT VA Cooperative Studies Group.
Circulation. 1993;87:VI56-VI64.
66. Packer M, Carver JR, Rodeheffer RJ, et al.
Effect of oral milrinone on mortality in severe chronic heart failure.The PROMISE Study.
N Engl J Med. 1991;325:1468-1475.
Dialogues in Cardiovascular Medicine - Vol 15 . No. 2 . 2010
End points in chronic heart failure clinical trials - Anand and Florea
98
67. Packer M, Narahara KA, Elkayam U, et al.
Double-blind, placebo-controlled study of the efficacy of flosequinanin patients with chronic heart failure. REFLECT Study.
Users’ guides to the medical literature: XIX. Applying clinical trialresults. A. How to use an article measuring the effect of an interven-tion on surrogate end points. Evidence-Based Medicine WorkingGroup.
JAMA. 1999;282:771-778.
70. Prentice RL.
Surrogate endpoints in clinical trials: definition and operationalcriteria.
Stat Med. 1989;8:431-440.
71. Fleming TR, DeMets DL.
Surrogate end points in clinical trials: are we being misled?
Ann Intern Med. 1996;125:605-613.
72. Lipicky RJ, Packer M.
Role of surrogate end points in the evaluation of drugs for heartfailure.
J Am Coll Cardiol. 1993;22:179A-184A.
73. Sobel BE, Furberg CD.
Surrogates, semantics, and sensible public policy.
Circulation. 1997;95:1661-1663.
74. Cuffe MS, Califf RM, Adams KF Jr, et al.
Short-term intravenous milrinone for acute exacerbation of chronicheart failure: a randomized controlled trial.
JAMA. 2002;287:1541-1547.
75. Califf RM, Adams KF, McKenna WJ, et al.
A randomized controlled trial of epoprostenol therapy for severecongestive heart failure: the Flolan International RandomizedSurvival Trial (FIRST).
Am Heart J. 1997;134:44-54.
76. Mebazaa A, Nieminen MS, Packer M, et al.
Levosimendan vs dobutamine for patients with acute decompensatedheart failure: the SURVIVE Randomized Trial.
JAMA. 2007;297:1883-1891.
77. Publication Committee for the VMAC Investigators(Vasodilatation in the Management of Acute CHF).
Intravenous nesiritide vs nitroglycerin for treatment of decompen-sated congestive heart failure: a randomized controlled trial.
JAMA. 2002;287:1531-1540.
78. Colucci WS, Elkayam U, Horton DP, et al.
Intravenous nesiritide, a natriuretic peptide, in the treatment ofdecompensated congestive heart failure. Nesiritide Study Group.
N Engl J Med. 2000;343:246-253.
79. Sackner-Bernstein JD, Kowalski M, Fox M, Aaronson K.
Short-term risk of death after treatment with nesiritide for decompen-sated heart failure: a pooled analysis of randomized controlled trials.
JAMA. 2005;293:1900-1905.
80. Sackner-Bernstein JD, Skopicki HA, Aaronson KD.
Risk of worsening renal function with nesiritide in patients withacutely decompensated heart failure.
Circulation. 2005;111:1487-1491.
81. Packer M.
The neurohormonal hypothesis: a theory to explain the mechanismof disease progression in heart failure.
J Am Coll Cardiol. 1992;20:248-254.
82. Francis GS, Cohn JN, Johnson G, Rector TS,Goldman S, Simon A.
Plasma norepinephrine, plasma renin activity, and congestiveheart failure. Relations to survival and the effects of therapy inV-HeFT II. The V-HeFT VA Cooperative Studies Group.
Circulation. 1993;87:VI40-VI48.
83. Swedberg K, Eneroth P, Kjekshus J, Wilhelmsen L.
Hormones regulating cardiovascular function in patients withsevere congestive heart failure and their relation to mortality.CONSENSUS Trial Study Group.
Circulation. 1990;82:1730-1736.
84. Richards AM, Doughty R, Nicholls MG, et al.
Neurohumoral prediction of benefit from carvedilol in ischemic leftventricular dysfunction. Australia-New Zealand Heart Failure Group.
Circulation. 1999;99:786-792.
85. Hampton JR, van Veldhuisen DJ, Kleber FX, et al.
Randomised study of effect of ibopamine on survival in patients withadvanced severe heart failure. Second Prospective Randomised Studyof Ibopamine on Mortality and Efficacy (PRIME II) Investigators.
Lancet. 1997;349:971-977.
86. Rousseau MF, Konstam MA, Benedict CR, et al.
Progression of left ventricular dysfunction secondary to coronaryartery disease, sustained neurohormonal activation and effects ofibopamine therapy during long-term therapy with angiotensin-converting enzyme inhibitor.
Am J Cardiol. 1994;73:488-493.
87. Cohn JN, Pfeffer MA, Rouleau J, et al.
Adverse mortality effect of central sympathetic inhibition with sus-tained-release moxonidine in patients with heart failure (MOXCON).
Eur J Heart Fail. 2003;5:659-667.
Dialogues in Cardiovascular Medicine - Vol 15 . No. 2 . 2010
End points in chronic heart failure clinical trials - Anand and Florea
99
88. Anand IS, Florea VG.
Alterations in ventricular structure: role of left ventricular remodeling.In: Mann DL, ed.
Heart Failure: Companion to Braunwald’s Heart Disease:Elsevier, W. B. Saunders. 2003:229-246.
89. Chatterjee K, Massie B.
Systolic and diastolic heart failure: differences and similarities.
J Card Fail. 2007;13:569-576.
90. Hammermeister KE, DeRouen TA, Dodge HT.
Variables predictive of survival in patients with coronary disease.Selection by univariate and multivariate analyses from the clinical,electrocardiographic, exercise, arteriographic, and quantitativeangiographic evaluations.
Circulation. 1979;59:421-430.
91. Pfeffer MA, Pfeffer JM.
Ventricular enlargement and reduced survival after myocardialinfarction.
Circulation. 1987;75:IV93-IV97.
92. St John Sutton M, Pfeffer MA, Plappert T, et al.
Quantitative two-dimensional echocardiographic measurementsare major predictors of adverse cardiovascular events after acutemyocardial infarction. The protective effects of captopril.
Circulation. 1994;89:68-75.
93. White HD, Norris RM, Brown MA, Brandt PW,Whitlock RM, Wild CJ.
Left ventricular end-systolic volume as the major determinant ofsurvival after recovery from myocardial infarction.
Circulation. 1987;76:44-51.
94. Anand IS, Florea VG.
Traditional and novel approaches to management of heart failure:successes and failures.
Cardiol Clin. 2008;26:59-72.
95. Konstam MA, Udelson JE, Anand IS, Cohn JN.
Ventricular remodeling in heart failure: a credible surrogateendpoint.
J Card Fail. 2003;9:350-353.
96. Cintron G, Johnson G, Francis G, Cobb F, Cohn JN.
Prognostic significance of serial changes in left ventricular ejectionfraction in patients with congestive heart failure.
Circulation. 1993;87:VI17-VI23.
97. Konstam MA, Rousseau MF, Kronenberg MW, et al.
Effects of the angiotensin converting enzyme inhibitor enalapril onthe long-term progression of left ventricular dysfunction in patientswith heart failure.
Circulation. 1992;86:431-438.
98. Konstam MA, Kronenberg MW, Rousseau MF, et al.
Effects of the angiotensin converting enzyme inhibitor enalapril onthe long-term progression of left ventricular dilatation in patientswith asymptomatic systolic dysfunction. SOLVD (Studies of LeftVentricular Dysfunction) Investigators.
Circulation. 1993;88:2277-2283.
99. St John Sutton M, Pfeffer MA, Moye L, et al.
Cardiovascular death and left ventricular remodeling two years aftermyocardial infarction: baseline predictors and impact of long-termuse of captopril: information from the Survival and VentricularEnlargement (SAVE) trial.
Circulation. 1997;96:3294-3299.
100. Doughty RN, Whalley GA, Gamble G, MacMahon S,Sharpe N.
Left ventricular remodeling with carvedilol in patients with conges-tive heart failure due to ischemic heart disease. Australia-NewZealand Heart Failure Research Collaborative Group.
Antiremodeling effects on the left ventricle during beta-blockadewith metoprolol in the treatment of chronic heart failure.
J Am Coll Cardiol. 2000;36:2072-2080.
102. Doughty RN, Whalley GA, Walsh HA, Gamble GD,Lopez-Sendon J, Sharpe N.
Effects of carvedilol on left ventricular remodeling after acute my-ocardial infarction: the CAPRICORN Echo Substudy.
Circulation. 2004;109:201-206.
103. Tsutamoto T, Wada A, Maeda K, et al.
Effect of spironolactone on plasma brain natriuretic peptide andleft ventricular remodeling in patients with congestive heart failure.
J Am Coll Cardiol. 2001;37:1228-1233.
104. Wong M, Staszewsky L, Latini R, et al.Valsartan benefits left ventricular structure and function in heartfailure: Val-HeFT echocardiographic study.
J Am Coll Cardiol. 2002;40:970-975.
105. Cohn JN, Tam SW, Anand IS, Taylor AL,Sabolinski ML, Worcel M.
Isosorbide dinitrate and hydralazine in a fixed-dose combinationproduces further regression of left ventricular remodeling in a well-treated black population with heart failure: results from A-HeFT.
J Card Fail. 2007;13:331-339.
106. Konstam MA, Patten RD, Thomas I, et al.
Effects of losartan and captopril on left ventricular volumes inelderly patients with heart failure: results of the ELITE ventricularfunction substudy.
Am Heart J. 2000;139:1081-1087.
Dialogues in Cardiovascular Medicine - Vol 15 . No. 2 . 2010
End points in chronic heart failure clinical trials - Anand and Florea
100
107. Solomon SD, Skali H, Bourgoun M, et al.
Effect of angiotensin-converting enzyme or vasopeptidase inhibi-tion on ventricular size and function in patients with heart failure:the Omapatrilat Versus Enalapril Randomized Trial of Utility inReducing Events (OVERTURE) echocardiographic study.
Am Heart J. 2005;150:257-262.
108. Anand I, McMurray J, Cohn JN, et al.
Long-term effects of darusentan on left-ventricular remodellingand clinical outcomes in the EndothelinA Receptor Antagonist Trialin Heart Failure (EARTH): randomised, double-blind, placebo-controlled trial.
Lancet. 2004;364:347-354.
109. Udelson JE, McGrew FA, Flores E, et al.
Multicenter, randomized, double-blind, placebo-controlled study onthe effect of oral tolvaptan on left ventricular dilation and functionin patients with heart failure and systolic dysfunction.
J Am Coll Cardiol. 2007;49:2151-2159.
110. Ghio S, Freemantle N, Scelsi L, et al.
Long-term left ventricular reverse remodelling with cardiac resyn-chronization therapy: results from the CARE-HF trial.
Eur J Heart Fail. 2009;11:480-488.
111. SOLVD Investigators.
Effect of enalapril on mortality and the development of heart failurein asymptomatic patients with reduced left ventricular ejectionfractions.
N Engl J Med. 1992;327:685-691.
112. Pfeffer MA, Braunwald E, Moye LA; SAVEInvestigators.
Effect of captopril on mortality and morbidity in patients with leftventricular dysfunction after myocardial infarction. Results of thesurvival and ventricular enlargement trial.
N Engl J Med. 1992;327:669-677.
113. Dargie HJ.
Effect of carvedilol on outcome after myocardial infarction inpatients with left-ventricular dysfunction: the CAPRICORNrandomised trial.
Lancet. 2001;357:1385-1390.
114. Taylor AL, Ziesche S, Yancy C, et al.
Combination of isosorbide dinitrate and hydralazine in blacks withheart failure.
N Engl J Med. 2004;351:2049-2057.
115. Pitt B, Poole-Wilson PA, Segal R, et al.
Effect of losartan compared with captopril on mortality in patientswith symptomatic heart failure: randomised trial—the LosartanHeart Failure Survival Study ELITE II.
Lancet. 2000;355:1582-1587.
116. Mann DL, McMurray JJ, Packer M, et al.
Targeted anticytokine therapy in patients with chronic heart fail-ure: results of the Randomized Etanercept Worldwide Evaluation(RENEWAL).
Circulation. 2004;109:1594-1602.
117. Konstam MA, Gheorghiade M, Burnett JC Jr, et al.
Effects of oral tolvaptan in patients hospitalized for worseningheart failure: the EVEREST Outcome Trial.
JAMA. 2007;297:1319-1331.
118. Anand IS, Florea VG, Solomon SD, Konstam MA,Udelson JE.
Noninvasive assessment of left ventricular remodeling: concepts,techniques, and implications for clinical trials.
J Card Fail. 2002;8:S452-S464.
119. Greenberg B, Quinones MA, Koilpillai C, et al.
Effects of long-term enalapril therapy on cardiac structure andfunction in patients with left ventricular dysfunction. Results of theSOLVD echocardiography substudy.
Circulation. 1995;91:2573-2581.
120. Packer M, Antonopoulos GV, Berlin JA, Chittams J,Konstam MA, Udelson JE.
Comparative effects of carvedilol and metoprolol on left ventricularejection fraction in heart failure: results of a meta-analysis.
Am Heart J. 2001;141:899-907.
121. Poole-Wilson PA, Swedberg K, Cleland JG, et al.
Comparison of carvedilol and metoprolol on clinical outcomes inpatients with chronic heart failure in the Carvedilol Or MetoprololEuropean Trial (COMET): randomised controlled trial.
Lancet. 2003;362:7-13.
122. Udelson JE, Antonopoulos GV, Proulx G, et al.
Comparison of long-term dual vasopeptidase inhibition with oma-patrilat to ACE inhibition with lisinopril on ventricular volumes inpatients with heart failure.
Circulation. 2000;102:II536.
123. Lüscher TF, Enseleit F, Pacher R, et al.
Hemodynamic and neurohumoral effects of selective endothelin A(ETA) receptor blockade in chronic heart failure: the Heart FailureETA Receptor Blockade Trial (HEAT).
Circulation. 2002;106:2666-2672.
124. Spieker LE, Mitrovic V, Noll G, et al.
Acute hemodynamic and neurohumoral effects of selective ETAreceptor blockade in patients with congestive heart failure. ET 003Investigators.
J Am Coll Cardiol. 2000;35:1745-1752.
Dialogues in Cardiovascular Medicine - Vol 15 . No. 2 . 2010
End points in chronic heart failure clinical trials - Anand and Florea
101
125. Chandrashekhar Y, Prahash AJ, Sen S, Gupta S,Roy S, Anand IS.
The role of arginine vasopressin and its receptors in the normaland failing rat heart.
J Mol Cell Cardiol. 2003;35:495-504.
126. Anand IS, Wong M, Latini R, Fisher LD, Chiang YT,Cohn JN.
Relation between changes in ejection fraction over time and subse-quent mortality and morbidity in Val-HeFT.
J Am Coll Cardiol. 2003;41:221A.
127. Kramer DG, Trikalinos TA, Kent DM,Antonopoulos GV, Konstam M, Udelson JE.
Quantitative analysis of ventricular remodeling as a surrogate fordrug/device effects on mortality in patients with heart failure andreduced ejection fraction: an updated analysis.
Abstract presented at the American College of Cardiology 2008Annual Scientific Session.
128. Meinert CL.
Clinical trial dictionary.
Baltimore (Md): The Johns Hopkins Center for Clinical Trials; 1996.
Sulfinpyrazone in the prevention of sudden death after myocardialinfarction.
N Engl J Med. 1980;302:250-256.
131. Temple R, Pledger GW.
The FDA’s critique of the anturane reinfarction trial.
N Engl J Med. 1980;303:1488-1492.
132. Every NR, Parsons L, Hlatky MA, et al.
Use and accuracy of state death certificates for classification ofsudden cardiac deaths in high-risk populations.
Am Heart J. 1997;134:1129-1132.
133. Early Treatment Diabetic Retinopathy StudyResearch Group.
Grading diabetic retinopathy from stereoscopic color fundus photo-graphs—an extension of the modified Airlie House classification.ETDRS report number 10.
Ophthalmology. 1991;98:786-806.
134. Heagerty A, Deverly A, Palmer C, et al.
The role of the critical event committee in a major cardiovascularoutcome study.
Blood Press. 2002;11:339-344.
135. Nissen SE, Wolski K.
Effect of rosiglitazone on the risk of myocardial infarction anddeath from cardiovascular causes.
N Engl J Med. 2007;356:2457-2471.
136. Gerstein HC, Miller ME, Byington RP, et al.
Effects of intensive glucose lowering in type 2 diabetes.
N Engl J Med. 2008;358:2545-2559.
137. Kastelein JJ, Akdim F, Stroes ES, et al.
Simvastatin with or without ezetimibe in familial hypercholes-terolemia.
N Engl J Med. 2008;358:1431-1443.
Dialogues in Cardiovascular Medicine - Vol 15 . No. 2 . 2010
End points in chronic heart failure clinical trials - Anand and Florea
78
Avkiran M, PhDCardiovascular ResearchThe Rayne InstituteSt Thomas’ HospitalLondon, UK
Bassand JP, MDDept of CardiologyUniversity Hospital Jean MinjozBesançon, France
Bertrand ME, MDHôpital CardiologiqueLille, France
Bolli R, MDDivision of CardiologyUniversity of LouisvilleLouisville, KY, USA
Camm JA, MDDept of Cardiac and VascularSciencesSt George’s University of LondonLondon, UK
Coats A, MDFaculty of MedicineUniversity of SydneySydney, Australia
Cobbe SM, MDDept of Medical CardiologyGlasgow Royal InfirmaryGlasgow, UK
Cohn JN, MDRasmussen Center forCardiovascular DiseasePreventionMinneapolis, MN, USA
Cokkinos DV, MD1st Cardiology DeptOnassis Cardiac Surgery CenterAthens, Greece
Cowie M, MD, PhDDept of Clinical CardiologyNational Heart & Lung InstituteLondon, UK
Danchin N, MDDept of CardiologyHôpital EuropéenGeorges PompidouParis, France
Dargie HJ, MDCardiac ResearchWestern InfirmaryGlasgow, UK
Di Pasquale G, MDDpt of CardiologyMaggiore HospitalBologna, Italy
Dzau VJ, MDDuke University MedicalCenter & Health System DUMCDurham, NC, USA
Consulting Editors
Editors in ChiefFerrari R, MD, PhDDept of Cardiology, Arcispedale S. AnnaUniversity of Ferrara, Ferrara, Italy
Hearse DJ, BSc, PhDThe Cardiothoracic Centre, The Rayne InstituteSt Thomas’ Hospital, London, UK
Fernandez-Aviles F, MDInstitute of Hematology andOncology, IDIBAPSHospital University Clinicof BarcelonaBarcelona, Spain
Fox KM, MDDept of CardiologyRoyal Brompton HospitalLondon, UK
Fox KA, MDDept of Cardiological ResearchUniversity of EdinburghEdinburgh, UK
Fuster V, MD, PhDCardiovascular InstituteMount Sinai Medical CenterNew York, NY, USA
Hasenfuss G, MDDept of CardiologyGeorg-August UniversitätGöttingen, Germany
Hori M, MD, PhDDept of Internal Medicineand TherapeuticsOsaka University GraduateSchool of MedicineOsaka, Japan
Katz AM, MDUniversity of ConnecticutSchool of MedicineFarmington, CT, USA
Komajda M, MDDept of CadiologyCHU Pitié-SalpêtrièreParis, France
Komuro I, MD, PhDDept of CardiovascularSciences & MedicineChiba University GraduateSchool of MedicineChiba, Japan
Lakatta EG, MDNational Institute on AgingGerontology Research CenterBaltimore, MD, USA
Libby P, MDCardiovascular MedicineBrigham & Women’s HospitalBoston, MA, USA
Lonn E, MDHamilton Health SciencesGeneral SiteHamilton, Ontario, Canada
Lopez-Sendon JL, MDCCU Dept of CardiologyHospital UniversityGregorio MaranonMadrid, Spain
Maggioni AP, MDANMC Research CenterFirenze, Italy
Marber MS, MD, PhDCardiovascular ResearchThe Rayne InstituteSt Thomas’ HospitalLondon, UK
Oto A, MDMedical Office, HacettepeUniversity School of MedicineAnkara, Turkey
Patrono C, MDDept of PharmacologyUniversity La SapienzaRome, Italy
Pepine CJ, MDDept of MedicineUniversity of FloridaGainesville, FL, USA
Rapezzi C, MDInstitute of CardiologyUniversity of BolognaBologna, Italy
Remme WJ, MD, PhDSticares FoundationRotterdam, The Netherlands
Rosen MR, MDDept of Pharmacology &PediatricsColumbia University Collegeof Physicians & SurgeonsNew York, NY, USA
Ruzyllo W, MDNational Institute of CardiologyWarsaw, Poland
Ryden L, MD, PhDDept of CardiologyKarolinska UniversityHospital SolnaStockholm, Sweden
Schneider MD, MDBaylor College of MedicineHouston, TX, USA
Seabra-Gomes RJ, MDInstituto do CoracaoHospital Santa CruzCarnaxide, Portugal
Sechtem U, MDDept of Internal Medicine& CardiologyRobert Bosch KrankenhausStuttgart, Germany
Simoons ML, MDThoraxcenterErasmus UniversityMedical CenterRotterdam, The Netherlands
Sleight P, MDDept of Cardiovascular MedicineJohn Radcliffe HospitalOxford, UK
Soler-Soler J, MDDept of CardiologyHospital General Vall d’HebronBarcelona, Spain
Steg PG, MDDept of CardiologyHôpital Bichat–Claude BernardParis, France
Swedberg K, MD, PhDDept of MedicineSahlgrenska UniversityHospital OstraGöteborg, Sweden
Tavazzi L, MDDivision of CardiologyPoliclinico San Matteo IRCCSPavia, Italy
Tendera M, MD3rd Division of CardiologySilesian School of MedicineKatowice, Poland
Vanhoutte PM, MDDept of PharmacologyUniversity of Hong KongFaculty of MedicineHong Kong, China
Widimsky P, MD, PhDVinohrady CardiocenterCharles University HospitalPrague, Czech Republic
Wijns WC, MDCardiovascular Center AalstOLV Hospital,Aalst, Belgium
Dialogues in Cardiovascular Medicine - Vol 15 . No. 2 . 2010
Dialogues in Cardiovascular Medicine - Vol 15 . No. 2 . 2010
79
Roberto Ferrari, MD, PhD
David J. Hearse, BSc, PhD, DSc
urrogate” is a word that has many meanings—or, rather, the meaning is sim-
ple and straightforward: all it is really is just another word for “substitute” (in
other words, it’s a substitute for substitute…). But this simple meaning applies
to many different situations that are often quite arcane for the layperson: a
surrogate model (used in engineering), a Surrogate Court (a type of law court), a sur-
rogate proxy (referring to a variety of server in computer sciences), and many others.
However, there is one situation where the word is much in the public eye these days,
and that is “surrogate motherhood” and the ongoing debate around the ethical issues
it raises. It does not fall within the purview of this journal to fuel that particular debate,
but one parallel can be drawn: there is just as heated a debate among cardiologists
around the concept and use of “surrogate end points” in clinical trials, and particularly
so in the context of heart failure trials.
So how do “surrogate end points” (which the National Institutes of Health [NIH] define
as “a biomarker intended to substitute for a [real] clinical end point”) enter the picture
in heart failure? A recent editorial in Dialogues (2009;14:No. 2) on cardiovascular dis-
ease prevention, proudly recalled that 6 out of the 8 years of extended life expectancy
enjoyed by affluent societies over the past century were directly attributable to ad-
vances in cardiology.
However, this sunny outlook must be tempered by two caveats. First of all, the problem
of cardiovascular disease and cardiovascular mortality has by no means been solved.
Cardiovascular mortality is still the number one killer worldwide and will continue to
be so until the year 2030 at least. In actual fact, cardiological success has often meant
merely delaying cardiovascular mortality, for example by transforming an acute disease
into a chronic one. On other occasions, it has meant replacing one evil with another:
thus the success of reperfusion means that today one is far less likely to die from a
myocardial infarction than from its sequelae, chief among which is heart failure. So
the fight must go on to find ever better treatments and procedures. And this is where
the second caveat comes in. In a sense, cardiology is suffering from it’s own success.
Editorial
“S
•••
SURROGATE END POINTS IN HEART FAILURETRIALS: POTENTIALS AND LIMITATIONS
Better drugs and better procedures are increasingly more difficult to develop because we
are trying to improve on an improvement. This requires randomized controlled trials,
in which the trial drug has to prove effectiveness against already effective background
therapies. Thus, because the frontiers of cardiovascular mortality have been so success-
fully pushed back, clinical trials with cardiovascular end points are becoming increasing-
ly difficult because they need more patients or a longer follow-up for the results to be
statistically meaningful. This means, too, that they are becoming ever more expensive.
In turn, this has implications for the pharmaceutical industry: because of the increased
length of studies, a huge share of the patent life of cardiovascular drugs is taken up by
the drug development process so that its “market life” has shrunk to the current average
of 6 to 8 years. As a result, pharmaceutical companies are finding it increasingly hard
to make their investment in research financially sustainable, and several of them are
considering reducing financial investments in cardiovascular research and allocating
their funds instead to other areas where the length of studies and numbers of patients
are more manageable, such as cancer research, for example. Clinical trials for cardio-
vascular drugs thus are at risk of simply pricing themselves out of existence. Thus, while
all agree that cardiovascular research should continue, the real question is whether it
will be able to continue, in terms of economic sustainability.
There is therefore a crying need to find alternative and reliable ways to test cardiovascu-
lar drugs, and particularly so where the needs are among the greatest: in heart failure,
which is the price cardiology is paying for its own success. One of these ways is to use
surrogate end points, and this is what this issue of Dialogues is all about. Many surrogate
end points have been proposed, but here more than anywhere else in medicine the say-
ing applies: “Many are called but few are chosen.” As stated by Desai and Temple (AAPS J.2006;8:E146-E152) “Only a small minority of biomarkers are established surrogate endpoints; blood pressure is an example of a surrogate end point accepted by both clinicians andregulators. It was a plausible surrogate because of the large epidemiologic databases demon-strating a correlation between elevated blood pressure and adverse cardiovascular measures.”This issue of Dialogues will focus on two of the most promising surrogate end points
in heart failure: brain natriuretic peptides and endothelial dysfunction. But alongside
potentials there are also limitations, and surrogate end points have their downside:
each surrogate has its cutoff point below which, while still reflecting an “abnormality,”
it no longer is reliably predictive. Also, overreliance on surrogate end points in the
end risks promoting a sort of treatment based on “numbers” rather than on clinical
reasoning. This issue of Dialogues also looks into this delicate issue.
Obviously, it is too early to substitute surrogate end points for hard end points.
However, the scientific community and clinical trial regulators cannot ignore the prob-
lem and Dialogues in Cardiovascular Medicine is proud to contribute to the debate.
Dialogues in Cardiovascular Medicine - Vol 15 . No. 2 . 2010
Editorial - Ferrari and Hearse
80
•••
103
Dialogues in Cardiovascular Medicine - Vol 15 . No. 2 . 2010
Can there be any surrogate for safety?M. A. Pfeffer, H. Skali
Could endothelial dysfunction be a surrogate end pointfor coronary artery disease?
M. Wolfrum, I. Sudano, J. Steffel, T. F. Lüscher
Expert Answers to Three Key Questions
Can BNP or NT-pro-BNPbe considered surrogate end points for heart failure?
T. A. McDonagh
1
2
3
Surrogate End Points in Heart Failure Trials:Potentials and Limitations
Keywords: B-type natriuretic peptide; heartfailure; end point; BNP; NT-pro-BNPAddress for correspondence:Theresa A. McDonagh, ConsultantCardiologist, Royal Brompton Hospital,London SW3 6NP, UK
iven the recent history ofclinical trials of novel in-terventions for heart fail-ure (HF), in particular new
drug treatments, it would be im-mensely appealing to be able to usea robust surrogate end point thatmight reduce the size, time, andexpense of conducting randomizedclinical trials. Managing to moveaway from conventional mortalityand morbidity end points would beparticularly useful in chronic systolicHF studies where new drugs arebeing tested on top of extremelyefficacious baseline therapy.
So what is a surrogate end point?In clinical trial terminology it is de-fined as a “measure of effect of acertain treatment that may correlatewith a real end point, but doesn'tnecessarily have a guaranteed rela-tionship.” The US National Insti-tutes of Health have also stated thata surrogate end point is “a biomark-er intended to substitute for a clini-cal end point.”1,2
There are a number of potential sur-rogate end points for the complexsyndrome of HF. As HF is not a dis-ease entity in itself, but a clinicalsyndrome arising as a result of nu-merous cardiac pathologies, it is atough task to find a good surrogatefor the morbidity and mortality thatare constant hallmarks of the pres-ence of HF. Numerous neurohor-monal markers, cytokines, hemo-dynamic parameters, and imagingmeasures of remodeling have been
suggested. However, the B-type na-triuretic peptides (B-type NPs, for-merly known as brain natriureticpeptides [BNP]) have emerged asthe strongest contenders for suchsurrogate status.
This article reviews the credentialsof B-type NPs as a surrogate endpoint for HF.
B-TYPE NATRIURETICPEPTIDES ANDHEART FAILURE
The family of B-type NPs consistsof two circulating forms, the inac-tive N-terminal fragment (NT-pro-BNP) and the active peptide (BNP)(Figure 1, page 106). They are pro-duced in the heart predominantlyin response to increased left ven-tricular (LV) wall stress. As such, themajority of focus to date has beenon their potential role in HF.
The circulating concentrations ofboth measurable forms of the pep-tide (BNP and NT-pro-BNP) havelong been known to be elevatedin patients with HF, be it acute orchronic, due to systolic dysfunction,or in the presence of preservedsystolic function,3-6 and in its pre-cursor form—asymptomatic LVdysfunction.7 They are elevated inproportion to the severity of thedisease. Their concentrations risewith worsening New York Heart As-sociation (NYHA) class and declin-ing left ventricular ejection fraction(LVEF).6
Can BNP or NT-pro-BNP be considered surrogateend points for heart failure?Theresa A. McDonagh, MD, FRCP
Royal Brompton Hospital - London - UK
Dialogues in Cardiovascular Medicine - Vol 15 . No. 2 . 2010
The B-type natriuretic peptides(B-type NPs), BNP (brain natri-uretic peptide) and the N-terminalfragment NT-pro-BNP, have beenresearched extensively in heartfailure (HF) over the last 20 years.To date, we know that their circu-lating concentrations are elevatedin patients with HF, and they arenow used diagnostically to excludeHF in patients presenting withdyspnea in primary care or in theemergency department. B-type NPsare arguably the best prognosticmarkers we have in HF. In addition,there is emerging evidence thatmonitoring patients with chronicHF due to systolic dysfunctionaccording to their B-type NP con-centrations may confer improvedclinical outcomes. This article looksat whether B-type NP is robustenough to be used as a surrogateend point in HF in future clinicaltrials.
The evidence base is now such thatB-type NPs are used in clinical prac-tice as diagnostic tools for HF—inparticular as rule-out tests in pa-tients suspected on clinical groundsof having HF.8-10
All these properties would seem topoint to B-type NPs being excellentsurrogate markers for the presenceof HF. However, the picture is a bitmore complex. B-type NPs are notexclusive biomarkers of HF. Whilelow concentrations rule HF outwith a negative predictive value ofaround 98% to 99%, high concen-trations do not necessarily diag-nose HF. Concentrations are alsoraised in other forms of structuraland functional heart disease, eg,acute coronary syndromes, myo-cardial infarction without systolicdysfunction, LV hypertrophy, andvalve disease.11 They are also raisedin patients with renal dysfunctionwhere there is reduced clearance ofthe peptides.12 In addition, in obeseHF patients, values can be lowerthan expected.13,14
B-TYPE NATRIURETICPEPTIDES AND PROGNOSIS
IN HEART FAILURE
Perhaps of greater interest to theirputative role as a surrogate is therelationship between B-type NPsand outcome in HF.
There is now a wealth of data thatB-type NPs are excellent prognosticmarkers in HF. Numerous studiesconfirm that they are independentarbiters of a poor prognosis in allgrades of HF ranging from asymp-tomatic LV dysfunction through toNYHA class IV.15-20 Indeed they ap-pear to be the best single prognosticmarkers we have to date, when weexamine studies that have used mul-tivariable models including estab-lished and novel markers of pooroutcome including, NYHA class,
In addition to their role in predict-ing all-cause and cardiovascularmortality in HF, they also seem tobe effective in determining suddencardiac death: in a study by Bergeret al, an increased BNP concentra-tion greater than the median wasthe only independent predictor ofsudden death in 452 patients withLV systolic dysfunction.21 It would betantalizing to think that we mightbe able to use B-type NPs in the fu-ture as surrogates to select patientsfor expensive device therapy in HF,eg, implantable cardioverter-defi-brillators (ICDs) and cardiac resyn-chronization therapy (CRT andCRT-D) pacemakers.
Similarly, selection of patients forthe scarce resource of cardiac trans-plantation in advanced HF is noto-riously difficult. It usually relies onamalgamating a number of clinicalvariables to try to ascertain whetherthe patient’s prognosis merits theconsiderable risk of a 15% to 20%1-year mortality that is associated
with a transplant. It has beendemonstrated that an NT-pro-BNPconcentration greater than the me-dian value at baseline was the sin-gle best predictor of mortality inpatients referred for considerationof transplantation.22 How we usethe information gained from thissurrogate clinically is as yet unclear,ie, whether we take a single base-line value or a concentration thatfails to fall on follow-up or whetherwe need to incorporate BNP/NT-pro-BNP concentrations into our clini-cal scoring models for assessingprognosis in HF remains to be de-termined.
Some information from studies inacute HF is unraveling the picturefurther about the usefulness of sin-gle or serial B-type NP measure-ments.
Cheng et al demonstrated in a studyof 72 patients admitted to hospitalwith decompensated HF that theclinical end points of death orreadmission to hospital with HFoccurred in those whose BNP con-centrations increased during theadmission.23 There were no clinicalend points in those whose BNP con-
106
Dialogues in Cardiovascular Medicine - Vol 15 . No. 2 . 2010
Can BNP or NT-pro-BNP be considered surrogate end points for heart failure? - McDonagh
proBNP
furin
BNPNT-proBNP
H P L G S P G S A SY T L R A KP PR S
QVM
GC
SG
FCR
KM
D I S SSS
GLC
C KV L
R RH
R
COOH
COOH
108
H P L G S P G S A S Y T L R A P R
KPS
QVM
GC
SG
FCR
KM
D I S SSS
GLC
C KV L
R RH
R
COOH108
H2N77
1 10 70 76
1
H2N
H2N
Figure 1. Cleavage of pro-BNP into NT-pro-BNP and BNP.
Abbreviation: BNP, brain natriuretic peptide; NT-pro-BNP, N-terminal fragment of pro-BNP.
centrations fell. However, a singlepredischarge BNP concentrationwas also an accurate determinant ofreadmission. Hence, in the patientwith decompensated HF serial mon-itoring of BNP looking for a fall ishelpful clinically.
Logeart et al, in an elegant studyinvolving a derivation and validationcohort in patients with decompen-sated HF, reported that the predis-
charge BNP concentration was thebest independent predictor of read-mission, with a value of <300 pg/mLshowing the lowest readmissionrates.24 However, those patientswith the greatest decrease in BNPhad a better outcome than thosewith a more modest reduction(hazard ratio [HR], 0.18 [0.07-0.48];P=0.001). Hence, in the decompen-sated situation serially measuringBNP and aiming for a discharge BNP
<300 pg/mL seems important fordischarge planning. Serial BNP test-ing has also been shown to be ofmore value in this situation thatserial examination by Doppler echo-cardiography.25
In chronic heart failure (CHF), fewerdata are available on serial mea-surements. In the Valsartan–HeartFailure Trial (Val-HeFT), those pa-tients with the greatest reductionin their BNP concentration (ex-pressed as a change in quartile) hadthe lowest mortality over the courseof the study.26 More recently, theVal-HeFT Group has published re-sults using NT-pro-BNP from theplacebo arm of the trial. A single de-termination of NT-pro-BNP showedhigher prognostic discriminationthan continuous changes of con-centrations, expressed either as anabsolute or a percentage change.However, in the Cox proportionalhazards model stratification of pa-tients into four categories accordingto NT-pro-BNP levels at two timepoints 4 months apart with respectto a threshold concentration pro-vided prognostic information in pa-tients with CHF beyond that of asingle determination.27 Also, inchronic, but more advanced HF, ithas been shown that NT-pro-BNPconcentrations greater than themedian on follow-up also predict apoor outcome as does an increasein NT-pro-BNP over 4 months offollow-up.22 It would appear thatserially monitoring concentrationsin CHF patients, therefore, seemsto give additional information thanmerely looking at baseline values.
However, not all studies agree thatusing BNP concentrations as sur-rogate markers to make clinical de-cisions are useful. Lewin et al com-pared using BNP concentrationsto serial weight changes to try anddetermine clinical deterioration inpatients attending a HF clinic.28
107
Dialogues in Cardiovascular Medicine - Vol 15 . No. 2 . 2010
Can BNP or NT-pro-BNP be considered surrogate end points for heart failure? - McDonagh
SELECTED ABBREVIATIONS AND ACRONYMS
A-HeFT African-American–Heart Failure Trial
ANZ Australia–New Zealand
ARB angiotensin II receptor blocker
BATTLESCARRED NT-proBNP-Assisted Treatment To LEssen SerialCARdiac REadmissions and Death
HORIZON-HF Hemodynamic, Echocardiographic, and NeurohormonalEffects of Istaroxime in Acute Heart Failure Syndromes
LVEF left ventricular ejection fraction
N-BNP N-terminal pro-B-type natriuretic peptide
NT-pro-BNP N-terminal fragment of pro-BNP
NYHA New York Heart Association
RALES Randomized ALdactone Evaluation Study
RESOLVD Randomized Evaluation of Strategies fOr LeftVentricular Dysfunction
RITZ Randomized Intravenous TeZosentan [study]
STARS-BNP Systolic hearT fAilure tReatment Supported by BNP
SURVIVE Survival of Patients With Acute Heart Failure in Needof Intravenous Inotropic Support
TIME-CHF Trial of Intensified vs standard Medical therapy inElderly patients with Congestive Heart Failure
Val-HeFT Valsartan–Heart Failure Trial
They found that neither was accurateenough to predict clinical deterio-ration. They pointed out that as yetwe know little about the day-to-day,month-to-month variability of BNPin CHF patients to predict whetherthe changes we are looking for canbe used. Wu et al recently showedthat many short-term therapeuticstudies of inpatients have largelyresulted in statistically significantdeclines in BNP and NT-pro-BNPwith clinical evidence of patient im-provements.29 In contrast, however,many therapeutic studies involvinglong-term outpatient monitoringhave produced changes in BNP/NT-pro-BNP that do not exceed the bi-ological variance. More work clearlyneeds to be done here.
B-TYPE NATRIURETICPEPTIDE AND
TITRATION OF THERAPYIN HEART FAILURE
Another area of interest in B-typeNPs as surrogates in HF concernstheir use as a potential HbA1 orBiochemical Swan Ganz Cathetercapable of monitoring progressionof the disease and therefore prompt-ing decisions of changes in therapy.To some extent the usefulness ofthis approach depends on what hap-pens to B-type NP concentrationswith the drug and device therapieswe give for HF. Diuretics are knownto reduce NP concentrations,30,31
whereas there are reports suggest-ing that digoxin increases their lev-els.32,33 However, it is the actions ofthe disease-modifying drugs that areperhaps the most interesting. Thereis good evidence that both angio-tensin-converting enzyme (ACE) in-hibitors and angiotensin II receptorblockers (ARBs) decrease B-typeNP.34,35 Tsutamoto et al have reportedin 37 patients with CHF that treat-ment with spironolactone for fourmonths significantly reduces BNPconcentration compared to place-
bo.36 The information regardingβ-adrenoreceptor antagonists andB-type NP is a little more confus-ing to date. Data from the Random-ized Evaluation of Strategies fOr LeftVentricular Dysfunction (RESOLVD)study with metoprolol versus place-bo treatment for 24 weeks reporteda rise in BNP despite the expectedimprovement in LV function, reduc-tion in mortality, and fall in angio-tensin II and renin concentrationswith metoprolol.37 However, in anonrandomized Japanese study look-ing at 52 patients with CHF, againcomparing metoprolol with placebo,both atrial natriuretic peptide (ANP)and BNP concentrations fell withthe β-blocker.38 More recent workshows that they may increase BNPconcentrations initially, but chroni-cally they seem to reduce them—this fits with these agents’ effectson long-term reverse remodeling.39
Hence, at the moment, two specu-lative schools of thought exist.The first presume that initially theβ-adrenoreceptor antagonists in-crease B-type NP due to their neg-atively inotropic and chronotropicproperties and that as the beneficialeffect of these drugs on LV functionemerge the peptide concentrationsfall. The second group proposesthat the improvement seen withthese drugs could be explained, atleast in part, by their ability to in-crease NP levels. Irrespective of theeffect of β-blockade, evidence is nowemerging suggesting that when weoptimize therapy in patients withHF, be that by increasing ACE in-hibitor, adding spironolactone, oruptitrating β-adrenoreceptor an-tagonists—which is, after all whatwe do when dealing clinically withHF patients—that B-type NP con-centrations fall.40
CRT also reduces BNP concentra-tions.41 The real question arisingfrom these observations is, howev-
er, do these reductions in B-type NPconcentrations matter? The answeris probably yes as we know, as stat-ed above, that patients whose BNPconcentrations fall during an admis-sion with HF and chronically havea better outlook that those patientswhere the levels fail to fall.
However, randomized studies ofB-type NP–driven care versus usualcare are scarce. Murdoch et al ran-domized small group of 20 patientsattending a HF clinic to usual careor optimization of HF drugs accord-ing to BNP concentrations wherethe BNP target was to be within thenormal range. The study showed agreater suppression of markers ofthe renin-angiotensin-aldosteronesystem in those receiving BNP-driv-en care.42
Richard’s group published a smallstudy of 69 patients attending a HFclinic.43 They were randomized tocare according to their aminotermi-nal portion of BNP (N-BNP) concen-tration/usual care. Those allocatedto N-BNP driven care had a signifi-cantly lower incidence of death orreadmission to hospital at 6 months,(P=0.034), suggesting that this ap-proach may give superior patientoutcomes. Subsequently, the SystolichearT fAilure tReatment Supportedby BNP (STARS-BNP) study report-ed beneficial effects in those ran-domized to BNP driven care in 220outpatients with CHF due to systolicdysfunction.44 There was a statisti-cally significant absolute reductionof 28% in the combined primary endpoint of HF-related death or hospi-talization (P<0.001) (Figure 2).
In summary, to date, observationalevidence shows that monitoringwith B-type NP as a surrogate mark-er can help with discharge planningand risk stratification for intensifi-cation of HF therapy. However, weawait more randomized studies such
108
Dialogues in Cardiovascular Medicine - Vol 15 . No. 2 . 2010
Can BNP or NT-pro-BNP be considered surrogate end points for heart failure? - McDonagh
as the Pro-BNP Evaluation Studyto confirm the clinical utility of thisapproach. We also clearly need moredata on day-to-day variability ofB-type NP in CHF.
As regards using B-type NP for titra-tion of HF therapy, two recent larg-er studies been recently published.BATTLESCARRED45 suggested im-proved outcome with BNP-guidedtherapy, but only in patients youngerthan 75 years. In TIME-CHF,46 in pa-tients aged 60 years or older (meanage approximately 77 years), therewas no statistically significant dif-ference in the primary end point of18-month survival free of all-causehospitalization. As of now we cansay that B-type NP in HF does notyet have the status of cholesterolin coronary heart disease as a tar-get for treatment in itself.
B-TYPE NATRIURETICPEPTIDE AND
HEART FAILURE TRIALS
The evidence that is of most rele-vance in promoting B-type NP asa surrogate end point in HF is thatavailable from the landmark HFtreatment trials to try determinewhether these peptides track withthe main mortality and morbidityend points and in addition whethertheir association with the random-ized therapy being tested goes inthe same direction as the result, beit positive or negative. Evidencefrom the major trials is sometimeslacking as B-type NP was first mea-sured in a clinical trial in HF in theAustralia–New Zealand (ANZ) trial ofcarvedilol that published in 1997.47
Here only baseline values weremeasured. The study demonstratedthat in patients with establishedischemic LV dysfunction, plasmaN-terminal pro-B-type natriureticpeptide (N-BNP) was an independ-ent predictor of mortality and HFand that carvedilol reduced mortal-
ity and HF in patients with higherpretreatment plasma N-BNP con-centrations. More recent trials havemeasured serial concentrations andhave therefore been able to specu-late on B-type NP as a surrogateend point.
Chronic systolicheart failure trials
• ACE inhibition. The major ACEinhibitor trials predate B-type NPmeasurements. More recently, theywere measured in the CarvedilolACE inhibitor Remodeling Mildcongestive heart failure EvaluatioN(CARMEN) trial, where NT-pro-BNPand BNP concentrations fell withenalapril in 6 months of follow-up.48
Extrapolating these results to theundoubted efficacy of ACE inhibitorin CHF patients with systolic dys-function, this is a “pro” for B-type NPas a surrogate.
• �-Adrenoreceptor antagonists.Bearing in mind the controversiesabout B-type NP and β-blocker ther-apy in HF, the trial results are re-markably consistent. In the Carve-
dilol Prospective Randomized Cu-mulative Survival (COPERNICUS)study, which demonstrated thatcarvedilol reduced mortality in ad-vanced CHF, BNP concentrationsalso fell in those assigned to carve-dilol.49 Interestingly, in the Beta-
blocker Evaluation of Survival sTudy(BEST) trial which reported the lackof effect of bucindolol, BNP was anindependent predictor of the com-posite end point of CHF hospitaliza-tion or death and LVEF at 12 monthscorrelated inversely with 12-monthBNP levels (r=–0.41; P=0.0001).However, bucindolol had no effecton BNP concentration, hence thelack of effect of bucindolol on BNPlevels appears consistent with itsoverall lack of efficacy in BEST.50
• Aldosterone antagonists. The largeRandomized ALdactone EvaluationStudy (RALES) trial, which showedthat spironolactone reduced all-cause mortality in severe CHF, hada BNP substudy involving 107 pa-tients with measurements taken atbaseline, 3 months, and 6 months.BNP concentrations fell by 23% at
109
Dialogues in Cardiovascular Medicine - Vol 15 . No. 2 . 2010
Can BNP or NT-pro-BNP be considered surrogate end points for heart failure? - McDonagh
3 months and 6 months (P=0.004and P=0.05, respectively) in thegroup assigned to spironolactone;another result in the correct direc-tion for BNP as a surrogate.51
• Angiotensin receptor blockers. Inthe Val-HeFT trial, BNP rose overtime in the placebo group. Valsar-tan caused a sustained reductionin BNP. This effect of valsartan isconsistent with the clinical bene-fits reported, ie, a reduction in the
combined composite primary endpoint of morbidity and mortality by13.3%.26 Data from other ARB stud-ies, particularly the Candesartan inHeart failure Assessment of Reduc-tion in Mortality and morbidity(CHARM) program, are awaited asthey have still to report their neu-rohormonal substudies.
• Hydralazine plus nitrate. TheAfrican-American–Heart FailureTrial (A-HeFT) examined the effectsof combination therapy with hy-dralazine and isosorbide dinitratecompared with placebo in African-American patents with CHF due to
systolic dysfunction. Patients wereon standard optimal medication forHF. BNP concentrations fell signifi-cantly in treatment group (39 pg/mLversus 8 pg/mL in the placebo arm;P=0.05).52 Again this is consistentwith the improvement in the primaryend point, which was a compositescore including all-cause mortality,first HF hospitalization, and changein quality of life (P=0.01). There wasalso a reduction in all-cause mor-tality (P=0.02).
• CRT. Device therapy with CRTpacemakers has become a standardtherapy for patients with more symp-tomatic CHF (NYHA class >III) dueto systolic dysfunction who alsohave a wide QRS complex on theECG. The CArdiac REsynchroniza-tion therapy in Heart Failure (CARE-HF) trial demonstrated a strikingmortality reduction in those as-signed to CRT. NT-pro-BNP concen-trations were measured at baseline,3 months, and 18 months. There wasa sustained reduction in its concen-tration compared with the placebogroup at both 3 and 18 months offollow-up (P<0.001) (Figure 3).53,54
In these large randomized trials inCHF there does seem to be remark-able consistency between B-type NPmeasurements and the main endpoint.
Studies in acute HF
Investigators have only really em-barked upon large randomized trialsin acute HF relatively recently. How-ever, some data on B-type NP areemerging from these and producingdifferent though not necessarily in-consistent results.
In the Survival of Patients WithAcute Heart Failure in Need ofIntravenous Inotropic Support(SURVIVE) trial, which comparedthe effects of levosimendan versusdobutamine in 1327 patients withacute decompensated HF and lowejection fraction requiring inotrop-ic therapy, there was no differencein all-cause mortality at 180 days.However, BNP concentrations fellsignificantly in the levosimendan-treated group.55 BNP was only mea-sured during the in-hospital infusionphase of the trial, but we can saythat the transient reduction did notgo in the same direction as the 180-day outcome measure. In contrast,in the recent Hemodynamic, Echo-cardiographic, and NeurohormonalEffects of Istaroxime in Acute HeartFailure Syndromes (HORIZON-HF)study looking at the effects of thenew inotrope istaroxime, in pa-tients with decompensated HF,there were significant changes inthe hemodynamic end points in fa-vor of istaroxime, but no change inBNP concentrations.56
In the Randomized IntravenousTeZosentan (RITZ) trial of a lowdose of the endothelin antagonisttezosentan in patients admitted withacute HF, tezosentan doses of 1 to25 mg/h were efficacious in improv-ing the hemodynamics and reduc-
110
Dialogues in Cardiovascular Medicine - Vol 15 . No. 2 . 2010
Can BNP or NT-pro-BNP be considered surrogate end points for heart failure? - McDonagh
Medical therapy
CRT
n=362 n=3700
n=341 n=3233
n=308 n=28218
NT-
pro
-BN
P(p
g/m
L)
Months of follow up
4500
4000
3500
3000
2500
2000
1500
1000
500
0
P<0.0001
P<0.0001
Figure 3. The effects of CRT on NT-pro-BNP concentrations in the CARE-HF trial.
ing BNP, showing a move of bothend points in the same direction.55
Overall, to date, we could concludethat for short-time inotropic studiesor studies of other agents in acuteHF, B-type NP does not appear tobe a reliable surrogate marker.
Small proof-of-conceptand pilot studies
Several smaller trials of newer ther-apeutic strategies are already usingchanges in B-type NPs as explorato-ry end points to build a more robustbody of evidence prior to embark-ing on larger definitive outcomestudies. Such trials, eg, those usingerythropoietins, seem to show re-ductions in B-type NPs in HF pa-tients with concomitant improve-ments in clinical status.58,59
CONCLUSIONS
The evidence to date points to B-type NP having the potential to bea useful surrogate end point in HFtrials. B-type NPs are raised in HFsyndromes; elevated concentrationsand levels that fail to fall duringtherapy portend a poor prognosis;and titration of known efficaciousdrugs according to their concentra-tions seems to be beneficial. Inlarge randomized trials of interven-tions in chronic systolic HF, B-typeNP changes seem to be in the samedirection as the harder end points.
However, before we can recommendusing B-type NP as surrogate pri-mary end point for clinical trialsacross the HF spectrum, we willneed to see much more data emerg-ing from the more recent large ran-domized HF trials. This should pro-vide us with more information onhow B-type NP fares as an endpoint in trials of HF with preservedLVEF and in acute HF syndromesin addition to those already seen inCHF due to systolic dysfunction.
REFERENCES
1. Cohn JN.
Improving outcomes in heart failure.
Eur Heart J. 1998;19:1124-1125.
2. De Gruttola VG, Clax P, DeMets DL,et al.
Considerations in the evaluation of surro-gate endpoints in clinical trials. Summaryof a National Institutes for Health workshop.
Controlled Clin Trials. 2001;22:485-502.
3. Lang CC, Motwani JG, Rahman AR,Coutie WJ, Struthers AD.
Effect of angiotensin-converting enzyme in-hibition on plasma brain natriuretic peptidelevels in patients with heart failure.
Clin Sci (Lond). 1992;83:143-147.
4. Davis M, Espiner E, Richards G,et al.
Plasma brain natriuretic peptide in assess-ment of acute dyspnoea.
Lancet. 1994;343:440-444.
5. Lubien E, DeMaria A,Krishnaswamy P, et al.
Utility of B-natriuretic peptide in detectingdiastolic dysfunction: comparison withDoppler velocity recordings.
Circulation. 2002;105:595-601.
6. Valli N, Georges A, Corcuff JB,Barat JL, Bordenave L.
Assessment of brain natriuretic peptide inpatients with suspected heart failure: compar-ison with radionuclide ventriculography data.
Clin Chim Acta. 2001;306:19-26.
7. McDonagh TA, Robb SD,Murdoch DR, et al.
Biochemical detection of left-ventricularsystolic dysfunction.
Lancet. 1998;351:9-13.
8. Cowie MR, Struthers AD, Wood DA,et al.
Value of natriuretic peptides in assessmentof patients with possible new heart failurein primary care.
Lancet. 1997;350:1349-1353.
9. Cowie MR, Jourdain P, Maisel A,et al.
Clinical applications of B-type natriureticpeptide (BNP) testing.
Eur Heart J. 2003;24:1710-1718.
10. Maisel AS, Krishnaswamy P,Nowak RM, et al.
Rapid measurement of B-type natriureticpeptide in the emergency diagnosis of heartfailure.
N Engl J Med. 2002;347:161-167.
11. McDonagh TA, Holmer S,Raymond I, Luchner A, Hildebrant P,Dargie HJ.
NT-proBNP and the diagnosis of heart fail-ure: a pooled analysis of three Europeanepidemiological studies.
Eur J Heart Fail. 2004;6:269-273.
12. Luchner A, Hengstenberg C,Lowel H, Riegger GA, Schunkert H,Holmer S.
Effect of compensated renal dysfunction onapproved heart failure markers: direct com-parison of brain natriuretic peptide (BNP)and N-terminal pro-BNP.
Hypertension. 2005;46:118-123.
13. Rivera M, Cortes R, Salvador A,et al.
Obese subjects with heart failure have lowerN-terminal pro-brain natriuretic peptideplasma levels irrespective of aetiology.
Eur J Heart Fail. 2005;7:1168-1170.
14. Khush KK, Gerber IL, McKeown B,et al.
Obese patients have lower B-type and atrialnatriuretic peptide levels compared withnonobese.
Congest Heart Fail. 2006;12:85-90.
15. Richards AM, Nicholls MG,Yandle TG, et al.
Plasma N-terminal pro-brain natriureticpeptide and adrenomedullin: new neurohor-monal predictors of left ventricular functionand prognosis after myocardial infarction.
Circulation. 1998;97:1921-1929.
16. Tsutamoto T, Wada A, Maeda K,et al.
Attenuation of compensation of endogenouscardiac natriuretic peptide system in chronicheart failure—Prognostic role of brain na-triuretic peptide concentration in patientswith chronic symptomatic left ventriculardysfunction.
Circulation. 1997;96:509-516.
111
Dialogues in Cardiovascular Medicine - Vol 15 . No. 2 . 2010
Can BNP or NT-pro-BNP be considered surrogate end points for heart failure? - McDonagh
17. McDonagh TA, Cunningham AD,Morrison CE, et al.
Left ventricular dysfunction, natriuretic pep-tides, and mortality in an urban population.
Heart. 2001;86:21-26.
18. Gardner RS, Chong V, Morton I,McDonagh TA.
N-terminal brain natriuretic peptide is amore powerful predictor of mortality thanendothelin-1, adrenomedullin and tumournecrosis factor-alpha in patients referred forconsideration of cardiac transplantation.
N-terminal pro-brain natriuretic peptide.A new gold standard in predicting mortali-ty in patients with advanced heart failure.
Eur Heart J. 2003;24:1735-1743.
20. Omland T, Aakvaag A, Bonarjee VV,et al.
Plasma brain natriuretic peptide as an in-dicator of left ventricular systolic functionand long-term survival after acute myocar-dial infarction. Comparison with plasmaatrial natriuretic peptide and N-terminalproatrial natriuretic peptide.
Circulation. 1996;93:1963-1969.
21. Berger R, Huelsman M,Strecker K, et al.
B-type natriuretic peptide predicts suddendeath in patients with chronic heart failure.
Circulation. 2002;105:2392-2397.
22. Gardner RS, Chong KS, Morton JJ,McDonagh TA.
A change in N-terminal pro-brain natriureticpeptide is predictive of outcome in patientswith advanced heart failure.
Eur J Heart Fail. 2007;9:266-271.
23. Cheng V, Kazanagra R, Garcia A,et al.
A rapid bedside test for B-type peptide predictstreatment outcomes in patients admitted fordecompensated heart failure: a pilot study.
J Am Coll Cardiol. 2001;37:386-391.
24. Logeart D, Thabut G, Jourdain P,et al.
Predischarge B-type natriuretic peptide assayfor identifying patients at high risk of re-admission after decompensated heart failure.
J Am Coll Cardiol. 2004;43:635-641.
25. Gackowski A, Isnard R,Golmard JL, et al.
Comparison of echocardiography and plasmaB-type natriuretic peptide for monitoring theresponse to treatment in acute heart failure.
Eur Heart J. 2004;25:1788-1796.
26. Anand IS, Fisher LD, Chiang YT,et al.
Changes in brain natriuretic peptide andnorepinephrine over time and mortality andmorbidity in the Valsartan Heart FailureTrial (Val-HeFT).
Circulation. 2003;107:1278-1283.
27.Masson S, Latini R, Anand IS, et al.
Prognostic value of changes in N-terminalpro-brain natriuretic peptide in Val-HeFT(Valsartan Heart Failure Trial).
J Am Coll Cardiol. 2008;52:997-1003.
28. Lewin J, Ledwidge M, O’LoughlinC, McNally C, McDonald K.
Clinical deterioration in established heartfailure: what is the value of BNP andweight gain in aiding diagnosis?
Eur J Heart Fail. 2005;7:953-957.
29. Wu AH.
Serial testing of B-type natriuretic peptideand NTpro-BNP for monitoring therapy ofheart failure: the role of biologic variationin the interpretation of results.
Am Heart J. 2006;152:828-834.
30. Anderson JV, Woodruff PW,Bloom SR.
The effect of treatment of congestive heartfailure on plasma atrial natriuretic peptideconcentration: a longitudinal study.
Br Heart J. 1988;59:207-211.
31. Tsutsui T, Tsutamoto T, Maeda K,Kinoshita M.
Comparison of neurohumoral effects of short-acting and long-acting loop diuretics inpatients with chronic congestive heart failure.
J Cardiovasc Pharmacol. 2001;38(suppl 1):S81-S85.
32. Tsutamoto T, Wada A, Maeda K,et al.
Digitalis increases brain natriuretic peptide inpatients with severe congestive heart failure.
Am Heart J. 1997;134(5 pt 1):910-916.
33. Kobusiak-Prokopowicz M,Swidnicka-Szuszkowska B, Mysiak A.
[Effect of digoxin on atrial natriuretic pep-tide (ANP), brain natriuretic peptide (BNP)and cyclic 3', 5'-guanosine monophosphate(cGMP) in patients with chronic congestiveheart failure]. Article in Polish.
Pol Arch Med Wewn. 2001;105:475-482.
34. Yoshimura M, Yasue H, Tanaka H,et al.
Responses of plasma concentrations of Atype natriuretic peptide and B type natriuret-ic peptide to alacepril, an angiotensin-con-verting enzyme inhibitor, in patients withcongestive heart failure.
Br Heart J. 1994;72:528-533.
35. Tsutamoto T, Wada A, Maeda K,et al.
Relationship between plasma levels of cardiacnatriuretic peptides and soluble Fas: plas-ma soluble Fas as a prognostic predictor inpatients with congestive heart failure.
J Card Fail. 2001;7:322-328.
36. Tsutamoto T, Wada A, Maeda K,et al.
Effect of spironolactone on plasma brainnatriuretic peptide and left ventricular re-modeling in patients with congestive heartfailure.
J Am Coll Cardiol. 2001;37:1228-1233.
37. The RESOLVD Investigators.
Effect of metoprolol CR in patients with is-chaemic and dilated cardiomyopathy.
Circulation. 2002;101:378-384.
38. Hara Y, Hamada M, Shigematsu Y,et al.
Effect of beta-blocker on left ventricularfunction and natriuretic peptides in patientswith chronic heart failure treated with an-giotensin-converting enzyme inhibitor.
Jpn Circ J. 2000;64:365-369.
39. Fung JW, Yu CM, Yip G, et al.
Effect of beta blockade (carvedilol or meto-prolol) on activation of the renin-angiotensin-aldosterone system and natriuretic peptidesin chronic heart failure.
Am J Cardiol. 2003;92:406-410.
112
Dialogues in Cardiovascular Medicine - Vol 15 . No. 2 . 2010
Can BNP or NT-pro-BNP be considered surrogate end points for heart failure? - McDonagh
40. Maeda K, Tsutamoto T, Wada A,et al.
High levels of plasma brain natriuretic pep-tide and interleukin-6 after optimized treat-ment for heart failure are independent riskfactors for morbidity and mortality in patientswith congestive heart failure.
J Am Coll Cardiol. 2000;36:1587-1593.
41. Yu CM, Fung JW, Zhang Q, et al.
Improvement of serum NT-ProBNP predictsimprovement in cardiac function and favor-able prognosis after cardiac resynchroniza-tion therapy for heart failure.
J Card Fail. 2005;11(5 suppl):S42-S46.
42. Murdoch DR, McDonagh TA,Byrne J, et al.
Titration of vasodilator therapy in chronicheart failure according to plasma brain na-triuretic peptide concentration: randomizedcomparison of the hemodynamic and neuro-endocrine effects of tailored versus empiricaltherapy.
Treatment of heart failure guided by plasmaaminoterminal brain natriuretic peptide(N-BNP) concentrations.
Lancet. 2000;355:1126-1130.
44. Jourdain P, Jondeau G, Funck F,et al.
Plasma brain natriuretic peptide-guidedtherapy to improve outcome in heart failure:the STARS-BNP Multicenter Study.
J Am Coll Cardiol. 2007;49:1733-1739.
45. Lainchbury JG, Richards AM,Troughton RW, et al.
NT-proBNP guided treatment for chronicheart failure: results from the Battlescarredtrial.
J Am Coll Cardiol. 2010;55:53-60.
46. Pfisterer M, Buser P, Rickli H,et al.
BNP-guided heart failure therapy: the Trialof Intensified vs Standard Medical Therapyin Elderly Patients With Congestive HeartFailure (TIME-CHF) randomized trial.
JAMA. 2009;301:383-392.
47. Richards AM, Doughty R,Nicholls MG, et al.
Neurohumoral prediction of benefit fromcarvedilol in ischemic left ventricular dys-function. Australia-New Zealand HeartFailure Group.
Circulation. 1999;99:786-792.
48. Rosenberg J, Gustafsson F,Remme WJ, Riegger GA,Hildebrandt PR.
Effect of beta-blockade and ACE inhibitionon B-type natriuretic peptides in stable pa-tients with systolic heart failure.
Cardiovasc Drugs Ther. 2008;22:305-311.
49. Hartmann F, Packer M, Coats AJ,et al.
Prognostic impact of plasma N-terminalpro-brain natriuretic peptide in severe chroniccongestive heart failure: a substudy of theCarvedilol Prospective RandomizedCumulative Survival (COPERNICUS) trial.
Circulation. 2004;110:1780-1786.
50. Frantz RP, Lowes BD, Grayburn PA,et al.
Baseline and serial neurohormones in pa-tients with congestive heart failure treatedwith and without bucindolol: results of theneurohumoral substudy of the Beta-BlockerEvaluation of Survival Study (BEST).
J Card Fail. 2007;13:437-444.
51. Rousseau MF, Gurne O, Duprez D,et al.
Beneficial neurohormonal profile of spirono-lactone in severe congestive heart failure: re-sults from the RALES neurohormonal substudy.
J Am Coll Cardiol. 2002;40:1596-1601.
52. Cohn JN, Tam SW, Anand IS,Taylor AL, Sabolinski ML, Worcel M.
Isosorbide dinitrate and hydralazine in afixed-dose combination produces further re-gression of left ventricular remodeling in awell-treated black population with heartfailure: results from A-HeFT.
J Card Fail. 2007;13:331-339.
53. Fruhwald FM, Fahrleitner-Pammer A, Berger R, et al.
Early and sustained effects of cardiac resyn-chronization therapy on N-terminal pro-B-type natriuretic peptide in patients withmoderate to severe heart failure and cardiacdyssynchrony.
Eur Heart J. 2007;28:1592-1597.
54. Richardson M, Freemantle N,Calvert MJ, Cleland JG, Tavazzi L.
Predictors and treatment response with car-diac resynchronization therapy in patientswith heart failure characterized by dyssyn-chrony: a pre-defined analysis from theCARE-HF trial.
Eur Heart J. 2007;28:1827-1834.
55. Mebazaa A, Nieminen MS,Packer M, et al.
Levosimendan vs dobutamine for patientswith acute decompensated heart failure: theSURVIVE Randomized Trial.
JAMA. 2007;297:1883-1891.
56. Gheorghiade M, Blair JE,Filippatos GS, et al.
Hemodynamic, echocardiographic, andneurohormonal effects of istaroxime, a novelintravenous inotropic and lusitropic agent:a randomized controlled trial in patientshospitalized with heart failure.
J Am Coll Cardiol. 2008;51:2276-2285.
57. Cotter G, Kaluski E, Stangl K,et al.
The hemodynamic and neurohormonal ef-fects of low doses of tezosentan (an endothelinA/B receptor antagonist) in patients withacute heart failure.
Eur J Heart Fail. 2004;6:601-609.
58. Parissis JT, Kourea K, Panou F,et al.
Effects of darbepoetin alpha on right andleft ventricular systolic and diastolic func-tion in anemic patients with chronic heartfailure secondary to ischemic or idiopathicdilated cardiomyopathy.
Am Heart J. 2008;155:751-757.
59. Palazzuoli A, Silverberg DS,Iovine F, et al.
Effects of beta-erythropoietin treatment onleft ventricular remodeling, systolic function,and B-type natriuretic peptide levels in pa-tients with the cardiorenal anemia syndrome.
Am Heart J. 2007;154:645-715.
113
Dialogues in Cardiovascular Medicine - Vol 15 . No. 2 . 2010
Can BNP or NT-pro-BNP be considered surrogate end points for heart failure? - McDonagh
ver the past two decadesit has become obviousthat the endothelium isnot an inert, single-cell
monolayer of the vessels. Rather, theendothelium constitutes the largestand most extensive tissue in thebody. It forms a highly selective per-meability barrier constituting a con-tinuous, uninterrupted, smooth, andnonthrombogenic surface.
The importance of the endotheliumwas first recognized by its effect onvascular tone. The pioneering exper-iments of Furchgott and Zawadzki—
the former was later awarded theNobel Prize in 1998 together withLouis Ignarro and Ferid Murad—proposed an endothelium-derivedrelaxing factor that was subsequent-ly shown to be nitric oxide (NO).1
NO is a free radical gas with an invivo half-life of a few seconds, whichis readily able to cross biologicalmembranes. It is synthesized by NOsynthase (NOS) from L-arginine inpresence of the cofactor tetrahydro-biopterin (BH4) and is released fromendothelial cells mainly in responseto shear stress produced by bloodflow or pharmacological stimulants
Could endothelial dysfunction be a surrogateend point for coronary artery disease?Mathias Wolfrum, MD; Isabella Sudano, MD, PhD; Jan Steffel, MD;Thomas F. Lüscher, MD, FESC, FRCP
Cardiology - Cardiovascular Center - University Hospital Zürich - and Cardiovascular Research - Institute of PhysiologyUniversity of Zürich-Irchel - SWITZERLAND
Dialogues in Cardiovascular Medicine - Vol 15 . No. 2 . 2010
Keywords: heart failure; atherosclerosis;cardiovascular disease; coronary arterydisease; surrogate end point; endothelialdysfunctionAddress for correspondence:Thomas F. Lüscher, MD, FESC, FRCP,Professor and Chairman of Cardiology,CardioVascular Center, University Hospital,Rämistr. 100, CH-8091 Zürich, Switzerland(e-mail: [email protected])Dialogues Cardiovasc Med. 2010;15:114-129
Cardiovascular disease still ac-counts for most of the morbidity andmortality in Western countries. Theunderlying cause of most forms ofcardiovascular disease, specificallymyocardial infarction and stroke,is atherosclerosis. Atherosclerosisdevelops over decades and may leadto vascular occlusion with devas-tating clinical consequences. Theprocess is initiated by endothelialdysfunction, followed by intimalthickening with deposition of lipo-proteins, and invasion of macro-phages and other white blood cells.Atherosclerosis leads to angina pec-toris; plaque rupture or endothelialerosion leads to platelet activation,initiation of the coagulation cas-cade, thrombus formation, andeventually vascular occlusion. Thelatter events account for most ofthe morbidity in stroke and acutecoronary syndromes.
SELECTED ABBREVIATIONS AND ACRONYMS
ACTION A Coronary disease Trial Investigating Outcome withNifedipine GITS
ARB angiotensin receptor blocker
CARATS Coronary Artery Reactivity After Treatment withSimvastatin [trial]
CCB calcium channel blocker
CETP cholesterol ester transport protein
COMET Carvedilol Or Metoprolol European Trial
ENCORE I and II Evaluation of Nifedipine and Cerivastatin On Recovery ofcoronary Endothelial function [First and Second trials]
eNOS endothelial nitric oxide synthase
FMD flow-mediated [arterial] dilation
HOPE Heart Outcomes Prevention Evaluation
INTACT International Nifedipine Trial on AntiatherosclerotiCTherapy
such as acetylcholine (Figure 1).2,3The healthy endothelium is placedin an anatomically strategic posi-tion between the circulating bloodand vascular smooth muscle cellsof the media, and is able to respondappropriately to physical and chem-ical signals by the production of awide range of factors that regulatevascular tone, cellular adhesion,thromboresistance, smooth musclecell proliferation, and vessel wallinflammation (Figure 1).4 Impor-tantly, healthy endothelium, via therelease of NO, inhibits platelet andleukocyte adhesion to the vascularsurface, while, through the releaseof tissue plasminogen activator aswell as plasminogen activator in-hibitor-1 and tissue factor, it main-tains a balance of profibrinolytic andprothrombotic activity (Figure 2).5
Dialogues in Cardiovascular Medicine - Vol 15 . No. 2 . 2010
Could endothelial dysfunction be a surrogate end point for CAD? - Wolfrum and others
Anti-inflammatory(inhibition of leukocyte adhesion
and migration)
Antithrombotic(inhibition of platelet adhesion
and aggregation)
Anticoagulant andprofibrinolytic
Endothelium-dependentvasodilation
Antihypertrophic(inhibition of vascular smooth muscle
cell proliferation and migration)
HEALTHYENDOTHELIUM
Figure 2. The healthy endothelium not only mediates endothelium-dependent vasodila-tion, but also actively suppresses thrombosis, vascular inflammation, and hypertrophy.Nitric oxide is a particularly important mediator of both endothelium-dependent vasodilation andanti-inflammatory and antithrombotic effects of the endothelium, and endothelium-dependent vaso-motion is therefore thought to represent a “read-out” of other important functions of the endothelium.
Figure 1: Endothelium-derived vasoactive substances.NO is released from endothelial cells in response to shear stress and to activation of a variety of receptors. NO inhibits thrombocyte ag-gregation and leukocyte adhesion, and exerts vasodilating and antiproliferative effects on smooth muscle cells. Through activation ofthe ETA-receptor, ET1 leads to vasoconstriction and cell proliferation; in contrast, activation of the ETB-receptor results in vasodilation(via release of NO and prostacyclin).
The endothelium is constantly ex-posed to various risk factors in thecirculating blood such as pressure,shear stress, lipoproteins, glucose,and others. Over the last decades ithas been recognized that commonconditions predisposing to athero-sclerosis, such as hypertension, hy-percholesterolemia, diabetes, andsmoking are associated with endo-thelial dysfunction, leading to aproinflammatory and prothromboticphenotype of the endothelium.Our current understanding of thepathobiology of atherosclerosis sug-gests that endothelial dysfunctionplays a pivotal role in the develop-ment and progression of atheroscle-rosis and its clinical complications.
Endothelial dysfunctionand aging
Celermajer and coworkers were oneof the first groups who demonstrat-ed that aging was associated withprogressive endothelial dysfunctionin healthy elderly subjects free ofrisk factors or vascular disease.6
They studied the response of thebrachial artery to increased flow (so-called flow-dependent dilation) afterprolonged occlusion of the vesselby a blood pressure cuff, and theresponse to the endothelium-inde-pendent vasodilator glyceryl trini-trate. In the 238 subjects studied,they found an age-related develop-ment of endothelial dysfunctionboth in men and in women. Inter-estingly, endothelial dysfunctionappeared to occur earlier in malesthan in females. Women appearedsomehow protected from vascularaging until around the time of themenopause. After the onset ofmenopause an increase in endothe-lial dysfunction was observed.
Endothelial dysfunctionand hypertension
Alterations in endothelial functionare detectable in experimental andhuman hypertension, both in theforearm circulation, as well as in thecoronary vascular bed (Figure 3).7-19In human hypertension, the mech-anisms appear to be different atvarious stages of the hypertensiveprocess, and somehow endothelialdysfunction precedes the develop-ment of hypertension as normoten-
sive offspring of hypertensive parentsexhibit impaired endothelium-de-pendent vasodilatation to acetyl-choline,20 suggesting a genetic com-ponent of endothelial dysfunctionin human essential hypertension.Moreover, in patients with estab-lished hypertension and in normo-tensive offspring, the vasoconstric-tor response to inhibition of NOsynthesis by L-NG-monomethyl-arginine (L-LNMMA) is decreased,indicating reduced basal NO bio-
availability.21 Thus, derangement ofendothelial function in hyperten-sion is likely to be caused in partby genetic factors, and is not just aconsequence of elevated blood pres-sure.22 Furthermore, there is grow-ing evidence that NO influencesvascular tone by interacting with thecentral autonomic nervous system,as it inhibits sympathetic responsesin animals23 and in humans.24 Thisindirect effect may also play an im-portant role in the pathogenesis ofarterial hypertension.25
Endothelial dysfunctionand hypercholesterolemia
Elevated low-density lipoprotein(LDL) cholesterol, in hypercholes-terolemia, is a known risk factor forthe development of atheroscleroticvascular disease and causes endo-thelial dysfunction.26,27 In the earlyphase of atherosclerosis, LDL getstrapped in the subendothelial spaceand undergoes oxidation (Figure 4).Oxidized LDL has several biologi-
Dialogues in Cardiovascular Medicine - Vol 15 . No. 2 . 2010
Could endothelial dysfunction be a surrogate end point for CAD? - Wolfrum and others
30
25
20
15
10
5
0
µg/100 mL tissue min
Acetylcholine
Normotensive subjects Hypertensive subjects
FB
F(m
L/1
00
mL
tiss
ue
/min
)
b 0.45 4.5 150.15 1.5
30
25
20
15
10
5
0 Sodium nitroprusside
b 1 42
*
*
Figure 3. Vasodilation in response to acetylcholine infused in the forearm microcirculation.Patients with essential hypertension (light green circles); normotensive subjects with no family historyof hypertension (dark green circles). The vasodilation, as represented by forearm blood flow (FBF)measured with strain-gauge plethysmography, was significantly blunted in hypertensive patients ascompared to normotensives. The FBF response to sodium nitroprusside, a nonendothelium-dependentvasodilator, was similar between the 2 groups. *P<0.01 hypertensive vs normotensive subjects.
cal effects; it is proinflammatory,it inhibits endothelial nitric oxidesynthase (eNOS), it promotes vaso-constriction and adhesion, stimu-lates cytokines such as interleukin-1(IL-1) and increases platelet aggre-gation. The release of cytokinesattracts monocytes to the vascularendothelium. After their migrationinto subendothelial compartments,monocytes differentiate under theinfluence of monocyte colony stim-ulating factor (M-CSF) into macro-phages.28 These macrophages takeup modified LDL by their scavengerreceptor and in turn differentiateinto foam cells (Figure 4). Accumu-lation of these LDL-laden macro-phages leads to the formation of“fatty streaks,” the earliest manifes-tation of atherosclerosis, which laterturn into fibrous plaques. In con-trast to LDL cholesterol, high-den-
sity lipoprotein (HDL) cholesterolexerts a protective effect and in-versely correlates with morbidityand mortality. HDL is an importantantioxidant. Consequently, intrave-nous infusion of HDL restores theimpaired flow-mediated dilation(FMD) by improving NO bioavail-ability in the brachial artery of pa-tients with endothelial dysfunctiondue to hypercholesterolemia(Figure 5, page 118).29
Endothelial dysfunctionand diabetes
Cardiovascular disease is the majorcomplication of diabetes. Experi-mental and clinical studies haveshown that diabetes is associatedwith endothelial dysfunction. Bothnon–insulin-dependent and insulin-dependent diabetes mellitus are
associated with impaired endothe-lium–dependent vasodilation evenin patients without other risk factorslike hypertension or hypercholes-terolemia.30-32 Although abnormal-ities leading to endothelial dysfunc-tion may differ between type 1 andtype 2 diabetes, decreased NObioactivity, reflecting a defectiveL-arginine/nitric oxide pathway, maybe, in part, responsible for the in-creased cardiovascular risk associat-ed with type 1 and 2 diabetes.
ASSESSMENTOF ENDOTHELIALDYSFUNCTION
Several years after Furchgott andZawadzki’s seminal demonstrationof endothelium-dependent relax-ations to acetylcholine in the isolat-ed rabbit aorta,1 similar responses
117
Dialogues in Cardiovascular Medicine - Vol 15 . No. 2 . 2010
Could endothelial dysfunction be a surrogate end point for CAD? - Wolfrum and others
Figure 4. Endothelium and atherosclerosis.Interplay of lipids, inflammatory cells, and mediators, and the vessel wall in atherogenesis. See text for details.
could be demonstrated in the hu-man forearm.7,33 In certain endothe-lium-denuded vessels such as thehuman coronary artery, acetylcholinecauses vasoconstriction due to adirect effect on vascular smoothmuscle cells.34 In line with this ob-servation, intracoronary infusion ofacetylcholine in the catheter labo-ratory induces small increases inepicardial coronary artery diameterin patients without coronary arterydisease and no risk factors, but pro-found paradoxical vasoconstrictionin patients with coronary disease.35
In later studies, Quyyumi et al con-firmed that the impaired responseto acetylcholine in patients withcoronary disease or cardiovascular(CV) risk factors was largely due toreduced coronary availability of en-dothelium-derived NO.36 The directassessment of coronary endothelialfunction—due to its invasive na-ture—is restricted, limited to pa-tients with advanced disease, andrepeated testing during serial fol-low-up is difficult and costly.
118
Dialogues in Cardiovascular Medicine - Vol 15 . No. 2 . 2010
Could endothelial dysfunction be a surrogate end point for CAD? - Wolfrum and others
Figure 6. Flow-mediated dilation(FMD) of thebrachial artery.A. Ultrasound probeheld in stereotacticclamp with microme-ter adjustment.B. Continuous mea-surement of brachialartery diameter be-fore, during, andafter inflation andrelease of sphygmo-manometer cuff onforearm. The analogvideo signal is ac-quired with a videoprocessing systemthat computes theartery diameter inreal-time. FMDStudio, A system forReal-Time Measure-ment, Institute ofClinical Physiology,Pisa, Italy, see refs38 and 39.
A
B
Figure 5. Forearm arterial vasodilator response.A. Forearm blood flow during intra-arterial infusion of acetylcholine. Endothelium-dependent vasodilation to acetylcholine is impaired in hypercholes-terolemic compared with normocholesterolemic controls (P<0.0001), whereas there is no difference in the vasodilator response to sodium nitroprusside, anendothelium-independent vasodilator. In hypercholesterolemic subjects, there is a significant improvement in endothelium-dependent vasodilation to acetyl-choline after administration of reconstituted HDL. B. Improvement in endothelium-dependent vasodilation to acetylcholine induced by reconstituted HDLis prevented by intra-arterial coinfusion of L-NMMA, an inhibitor of NO synthesis, identifying improved NO bioavailability as the responsible mechanismsfor the improvement in endothelial function. C. Flow-mediated dilation of the brachial artery in hypercholesterolemic subjects before and after intravenousinfusion of reconstituted HDL. There is a significant improvement in endothelium-dependent vasodilation of the brachial artery, which does not depend onacetylcholine or its receptors.
Because endothelial dysfunction isa systemic process, a less invasiveapproach using venous occlusionplethysmography has been adoptedthat utilizes the same principles oflocal (ie, intrabrachial) infusion ofpharmacological probes and mea-surement of changes in tone of fore-arm resistance vessels.37 This tech-nique provides an opportunity toevaluate endothelial pathophysiol-ogy during the preclinical stage ofthe disease. Indeed, using appropri-ate agonists and antagonists, dose-response curves to acetycholine,sodium nitroprusside, L-NMMA andvasoconstrictor hormones can beconstructed. Venous occlusionplethysmography has been widelyused, but it is an invasive techniquethat requires arterial cannulation.This limits its repeatability, andprohibits its use in larger studies.
In 1992, Celermajer and coworkersintroduced an ultrasound-based testto assess conduit artery vascularfunction in the systemic circulation(Figure 6).38,39 With this method,brachial artery diameter is mea-sured before and after an increase inshear stress arising from the circu-lating blood that is induced by re-active hyperemia (FMD). A sphyg-momanometer cuff is placed onthe forearm distal to the brachialartery and inflated up to 200 mm Hgand then subsequently released 4 to5 minutes later. As described by theGerman physiologist Schretzenmaierat the beginning of the 20th century,FMD of the brachial or radial arteryoccurs predominantly as a result ofthe shear stress–induced endothe-lial release of NO (Figure 7).40
As in the coronary circulation, theresponse of the brachial or radialartery can be compared to the endo-thelium-independent vasodilator re-sponse to sublingual nitroglycerine.Measurements of FMD by ultra-sound are technically demanding,
but can be standardized to yieldreproducible results that correlatewith coronary vascular endothelialfunction41,42 and strain-gaugeplethysmography.43 Modern soft-ware development has allowed forcontinuous assessment of arterialdiameter and blood flow through-out the whole protocol by use ofaccurate edge detection algorithms
that can be manually edited. It isimportant to note that variations intechnique, such as the position ofthe occluding cuff and duration ofinflation, may produce results thatare less representative of local NOactivity. Brachial artery FMD hasbeen widely studied in clinical re-search as it enables serial evalua-tion of young subjects, includingchildren. It also allows the assess-ment of the effects of lifestyle andpharmacological interventions onendothelial biology at an early pre-clinical stage, when the disease pro-cess is most likely still reversible.44
In recent years, several other non-invasive techniques for the charac-
terization of vascular function havebeen proposed, including pulsewave analysis, pulse wave velocitymeasurement, and pulse amplitudetonometry. Interestingly, in 2000subjects of the Framingham ThirdGeneration Cohort, endothelial va-somotor function testing before andafter reactive hyperemia in the fin-gertips using pulse amplitude re-
active hyperemia–peripheral arterytonometry (RH-PAT) exhibited asignificant, although week, relation-ship between RH-PAT and mostcardiovascular risk factors.45
Further work is required to validatethe potential role of RH-PAT andother emerging noninvasive tests ofvasomotor function as independentpredictors of cardiovascular risk andlater clinical events. In particular,comparison with other tests—likeultrasound FMD and venous occlu-sion plethysmography—known tohave prognostic value in the con-text of cardiovascular disease (seebelow), needs further investigation.
119
Dialogues in Cardiovascular Medicine - Vol 15 . No. 2 . 2010
Could endothelial dysfunction be a surrogate end point for CAD? - Wolfrum and others
Hyperemia
Base
†
mL/
min
Radial flow
Control L-NMMA
80
60
40
20
0
††**
**
mm
Radial diameter
Control L-NMMA3.0
2.8
2.6
2.4
2.2
Figure 7. Changes in radial artery flow and diameter.Bar graphs showing radial artery flow (mL/min) and radial artery diameter (mm) measured atbaseline (Base) and during reactive hyperemia before and after infusion of NG-monomethyl-L-argi-nine (L-NMMA). All results are the mean±SEM of eight subjects. **P<0.01 vs Base; †P<0.05 and††P<0.01 vs corresponding control value.
ENDOTHELIALDYSFUNCTION AND MAJORCARDIOVASCULAR EVENTS
Over the last years, several studies,utilizing different tests of vasomo-tor function, have established en-dothelium-dependent vasomotorfunction as an independent predic-tor of the long-term risk of majorcardiovascular events, includingsudden cardiac death, myocardialinfarction, and stroke. Of note, in2005, Lerman and Zeiher publisheda multivariant meta-analysis of 10studies involving a total of 2500 sub-jects, which analyzed the relation-ship between coronary or peripheralendothelial dysfunction and car-diovascular events.46 Their findingsstrongly supported the notion thatendothelial dysfunction is inde-pendently associated with the riskof major cardiovascular events,even after adjustment for presenceof coronary artery disease and/orcardiovascular risk factors (Figures 8and 9).46,47
The prognostic value of endothelialdysfunction was not only investigat-ed in patients with known athero-sclerosis, but also in those with riskfactors. In postmenopausal womenwith newly diagnosed hypertension,Modena and coworkers examinedFMD noninvasively in the brachialartery.48 Patients with persistent en-dothelial dysfunction after 6 monthsdespite appropriate blood pressure–lowering therapy had an increasedrisk of nonfatal cardiovascular eventsover the next 5 years. Althoughtreatment was not standardized, thetype of antihypertensive therapy orthe degree of blood pressure low-ering did not explain the differencein outcome. This study strongly sug-gests a possible value of endothe-lial function as a screening test forthe primary prevention of cardio-vascular disease and for assessingtherapy.
EFFECT OFCARDIOVASCULAR DRUGS
ON ENDOTHELIALDYSFUNCTION
Statins
Statins are well established in thesecondary prevention of cardiovas-cular disease; indeed, these drugsimprove the prognosis of patientswith atherosclerotic vascular diseaseeven in the presence of so-callednormal cholesterol plasma levels.49,50
Of note, statins upregulate eNOSexpression, leading to improved NObioavailability, improved endothe-lial function, and reduced transientmyocardial ischemia, suggesting thatthese vascular biological effects ofHMG-coenzyme A reductase inhib-itors may importantly contribute tothe clinical benefits.51-54 Indeed, inthe forearm circulation of patientswith hypercholesterolemia, statinsimprove FMD within weeks of treat-ment, an effect that is lost afterstopping the treatment.55 In thecoronary circulation of patients withcoronary artery disease, treatment
with either simvastatin (CoronaryArtery Reactivity After Treatmentwith Simvastatin [CARATS] trial)56
or cerivastatin (Evaluation of Nife-dipine and Cerivastatin On Recoveryof coronary Endothelial function[ENCORE I] trial)57 for 6 monthsfailed to improve the paradoxicalvasoconstriction of epicardial coro-nary arteries to acetylcholine. Al-though smaller studies suggesteda rapid benefit of statin treatmenton coronary vasomotion,58 largertrials showed that it probably takesmore than 6 months to improvecoronary vasomotion, an interpre-tation in line with the observationthat in clinical trials Kaplan-Meiersurvival curves of placebo- andstatin-treated patients diverge onlyafter 1 to 2 years.
Although a first attempt to raise LDLwith the cholesterol ester transportprotein (CETP) inhibitor torcetrapibwas disappointing, HDL remains apotential target for the preventionof vascular disease. Indeed, low HDLis a principal risk factor for the de-velopment of premature coronary
120
Dialogues in Cardiovascular Medicine - Vol 15 . No. 2 . 2010
Could endothelial dysfunction be a surrogate end point for CAD? - Wolfrum and others
EventNo event
Ep
icar
dia
llum
inal
area
chan
ge(%
)
Acetylcholine-induced
vasoreactivity
Coldpressor
test
Flow-dependent
dilation
Nitroglycerin-induced
vasodilation
Cardiovascular event:
40
30
20
10
0
–10
–20
–30
P<0.001
P=0.001
P=0.002P=0.009
Figure 8. Changes in epicardial luminal area.Epicardial luminal area changes in response to various vasoreactivity tests in patients with (darkcolumns) and without (light columns) cardiovascular events during longterm follow-up. Data areshown as mean±SEM.
artery disease, and overall the influ-ence of statin therapy on HDL cho-lesterol levels is rather modest. Ofnote, intravenous infusion of recon-stituted HDL in patients with hyper-cholesterolemia rapidly improvesendothelial function in the forearmcirculation, both when assessedwith intra-arterial acetylcholine in-fusion as well as FMD. Interestingly,HDL has profound anti-inflamma-tory effects.59 Furthermore, in asmall pilot study, Hermann et al60
showed that dalcetrapib, anotherCETP inhibitor (which in contrast to
torcetrapib does not increase bloodpressure) improved FMD in patientswith low HDL. A large study (DAL-Vessel-Trial) involving 500 patientswith coronary artery disease or athigh risk using this novel compoundis currently ongoing.
Angiotensin-convertingenzyme (ACE) inhibitors
ACE inhibitors not only prevent theformation of angiotensin II, but alsoinhibit the breakdown of bradykinin,a stimulator of NO release. More-
over, their antioxidant propertiesimprove NO bioavailability, andthereby endothelial function, as as-sessed in the forearm macro- andmicrocirculation61 as well as in thecoronary circulation.62 They further-more inhibit the endothelial pro-duction of angiotensin II (AII) andendothelin 1 (ET1), two potent vaso-constrictors (Figure 10, page 122,and Table I, page 123).63 Hence,ACE inhibitors have important ef-fects on endothelial function. Inthe Trial on Reversing ENdothelialDysfunction (TREND), involving pa-tients with coronary artery diseasewith normal blood pressure, near-normal lipid profile, and no evidenceof heart failure, ACE inhibition withquinapril markedly improved en-dothelial dysfunction, as assessedby intracoronary infusion of acetyl-choline within 6 months.64 BesidesACE inhibition per se, certain com-pound-specific pharmacologicalfeatures may be important for sucheffects; for example, in patients withcoronary artery disease, only quina-pril, but not enalapril, was associ-ated with a significant improvementin FMD of the brachial artery.65 Fur-ther to their vascular effects, im-proved NO bioavailability duringACE inhibition also affects plateletfunction, and inhibitors of the renin-angiotensin-aldosterone system in-deed inhibit platelet aggregation invitro. Overall, the aforementionedeffects may at least in part be re-sponsible for the clinical benefitsobserved in patients with athero-sclerosis even in the absence of hy-pertension (Heart Outcomes Pre-vention Evaluation [HOPE] trial).66
More recently, the PERindoprilThrombosis, InflammatioN,Endothelial dysfunction, andNeurohormonal activation Trial(PERTINENT)67 has provided furtherinsights into the mechanism ofmorbidity and mortality benefitsassociated with perindopril in the
121
Dialogues in Cardiovascular Medicine - Vol 15 . No. 2 . 2010
Could endothelial dysfunction be a surrogate end point for CAD? - Wolfrum and others
Endothelial dysfunction and CV events
Duration of follow-up (months)
Standardized effect
Standardized effect
Al Suwaidi et al, (157)
Schachinger et al, (147)
Neunteufl et al, (73)
Perticone et al, (225)
Heitzer et al, (281)
Halcox et al, (308)
Modena et al, (400)
Schindler et al, (130)
Gokce et al, (187)
Targonski et al, (503)
28
92
60
32
54
46
67
45
1
16
Coronary
Brachial
Overall
0.0 2.0 4.0 6.0–8.0 –6.0 –4.0 –2.0 8.0
0.0 2.0 4.0 6.0–6.0–8.0 –4.0 –2.0 8.0
B
A
Figure 9. Multivariant analysis of hazard ratio of present studies reporting associationbetween coronary or peripheral endothelial function and cardiovascular (CV) events.A. Individual studies and number of patientsincluded in study in parentheses. Duration of follow-up in months is included in figure.B. Integration of studies based on site of assessment of endothelial function and combined analysis.
EUROPA trial (European trial onreduction Of cardiac events withPerindopril in stable coronary Arterydisease).68 It was found that perin-dopril restored the balance betweenangiotensin II and bradykinin in fa-vor of bradykinin, improved endo-thelial function, and decreased theendothelial cell apoptosis rate.67
Angiotensinreceptor blockers (ARBs)
ARBs also have pronounced effectson vascular function. Candesartanreduces vasoconstriction to endo-genous ET-1 and improves tonic NOrelease in the forearm of hyperten-sive patients (Figure 10, Table I).63,69Treatment of hypertensive patients
with irbesartan enhanced both en-dothelium-dependent and -inde-pendent vascular vasodilation re-sponses.70 Furthermore, someARBs may reduce thromboxaneA2–dependent platelet activation,possibly independent of the angio-tensin II receptor.71 In head-to-headcomparison, angiotensin-II–receptorblockade appears to have strongeranti-inflammatory and antiaggre-gatory effects compared with ACEinhibition; indeed, both ACE inhi-bition (with enalapril) and angio-tensin-II–receptor blockade (withirbesartan) reduce serum metallo-protease-9 protein levels as well asenzyme activity to a similar extent,while in patients with coronary ar-tery disease only irbesartan also
reduced high-sensitivity C-reactiveprotein, interleukin-6, and plateletaggregation.72 Interestingly, thecombination of an ACE-inhibitor(ramipril) and an ARB (candesartan)has a synergistic effect in improvingendothelial function (as measuredby flow-mediated vasodilation).73
Calciumchannel blockers (CCBs)
Several CCBs have been success-fully used to improve endothelialfunction in patients with hyperten-sion.74 Indeed, long-term treatmentwith nifedipine has been shown toimprove endothelium-dependentvasodilation to acetylcholine in theforearm circulation of hypertensive
122
Dialogues in Cardiovascular Medicine - Vol 15 . No. 2 . 2010
Could endothelial dysfunction be a surrogate end point for CAD? - Wolfrum and others
Figure 10. Possible mechanisms underlying the effect of angiotensin-converting enzyme inhibitors (blue) and angio-tensin II receptor antagonists (green) on endothelial function.
Dialogues in Cardiovascular Medicine - Vol 15 . No. 2 . 2010
Could endothelial dysfunction be a surrogate end point for CAD? - Wolfrum and others
Improvement ofTherapy endothelial function
ACE-InhibitorsHirooka et al, Hypertension 1992 Captopril Acute Yes (ACh)Creager et al, Hypertension 1992 Captopril 7-8 weeks No (MCh)
Enalapril 7-8 weeks No (MCh)Taddei et al, J Hypertens 1998 Lisinopril Acute No (ACh); Yes (BK);
1 and 2 months No (ACh); Yes (BK);Lyons et al, J Hypertens 1994 Enalapril 6 weeks Yes (LNMMA)Millgard et al, J Hum Hypertens 1998 Captopril Acute Yes (MCh)
3 months Yes (MCh)Mancini et al, Circulation 1996 Quinapril 6 months Yes (ACh)Schiffrin et al, Am J Hypertens 1995 Cilazapril 1 and 2 years Yes (ACh)Yavuz et al, J Renin Ang Aldost Syst 2003 Enalapril 6 months Yes (FMD)Ghiadoni et al, Hypertension 2003 Perindopril 6 months Yes (FMD)Souza-Barbosa et al, J Clin Hypertens 2006 Quinapril 12 weeks Yes (FMD)Pasini et al, Am J Hypertension 2007 Zofenapril 8 weeks Yes (FMD)Koh et al, Eur Heart J 2007 Ramipril 2 months Yes (FMD)
ARBGhiadoni et al, Hypertension 2000 Candesartan 2 months No (ACh)
12 months Yes* (ACh)Bragulat et al, Br J Biomed Sci 2003 Irbesartan 6 months Yes* (ACh)Yavuz et al, J Renin Ang Aldost Syst 2003 Losartan 6 months No (FMD)Ghiadoni et al, Hypertension 2003 Telmisartan 6 months No (FMD)Souza-Barbosa et al, J Clin Hypertens 2006 Irbesartan 12 Weeks Yes (FMD)Flammer et al, J Hypertens 2007 Losartan 4 weeks Yes (FMD)Hirooka et al, Clin Exp Hypertens 2008 Valsartan 1 year Yes (FMD)Koh et al, Eur Heart J 2007 Candesartan 2 months Yes (FMD)Benndorf et al, J Cardiovasc Pharmacol 2008 Telmisartan 6 weeks Yes (FMD)
ββ-BlockerSchiffrin et al, J Hypertens 1996 Atenolol 2 years No (ACh)Dawes et al, Br J Clin Pharmacol 1999 Nebivolol acute Yes (LNMMA)Ghiadoni et al, Hypertension 2003 Nebivolol 6 months No (FMD)Ghiadoni et al, Hypertension 2003 Atenolol 6 months No (FMD)Bank et al, Am J Hypertens 2007 Metoprolol 5 months No (FMD)Bank et al, Am J Hypertens 2007 Carvedilol 5 months Yes (FMD)
Ca-AntagonistsHirooka et al, Hypertension1992 Nifedipine acute No (ACh)Millgard et al, J Hum Hypertens 1998 Nifedipine acute No (MCh)Schiffrin et al, J Hypertens 1996 Nifedipine 1 year Yes (ACh)Ghiadoni et al, Hypertension 2003 Nifedipine 6 months No (FMD)Taddei et al, Hypertension 1997 Lacidipine 2 and 8 months Yes (ACh)Lyons et al, J Hypertens 1994 Amlodipine 6 weeks Yes (LNMMA)ENCORE Investigators, Circulation 2003 Nifedipine 6 months Yes (ACh)Perticone et al, Cardiovasc Res 1999 Isradipine 2 and 6 months Yes (ACh)Ghiadoni et al, Hypertension 2003 Amlodipine 6 months No (FMD)Oshuma et al, Hyp Res 2005 Efonidine 4 weeks Yes (FMD)Oshuma et al, Hyp Res 2005 Nifedipine 4 weeks No (FMD)Sudano et al, Hypertension 2007 Nifedipine 6 months Yes (ACh)Hirooka et al, Clin Exp Hypertens 2008 Amlodipine 1 year No (FMD)
OthersPanza et al, J Am Coll Cardiol 1993 Diuretic, verapamil, Chronic vs No (ACh)
Taddei et al, Hypertension 1994 Potassium acute Yes (ACh)Souza-Barbosa et al, J Clin Hypertens 2006 Hydrochlorothiazide 12 weeks Yes (FMD)
Table I. Changesin epicardial luminal area.
Abbreviations:ACE: angiotensinconverting enzyme;ARB: angiotensinReceptor Blocker;Ca: calcium; ACh: acetylcholine;MCh: metacholine;LNMMA: L-N-mono-methylarginine;FMD: flow-mediateddilation. *This effect wasparalleled by anenhanced endothe-lium-independentvasodilatation tosodium nitroprus-side. Modified afterreference 63:Sudano et al. HotTopics in Cardiolo-gy. 2009 (No. 15).
patients, while the response to sodi-um nitroprusside remained unaf-fected. In contrast, ACE inhibitionimproved the response to brady-kinin, but not acetylcholine in thispatient population. Furthermore,chronic treatment with nifedipine(even though the drug was stoppedbefore testing) blunted the vaso-constriction to endothelin-1.75 Mostinterestingly, in hypercholesterol-emia, the CCB nifedipine improvesendothelial function independentof its effect on blood-pressure or plasma lipids, most likely by areduction in NO degradation.76
Indeed, oral treatment with dihy-dropyridine calcium antagonistsreduces oxidative stress, therebyimproving bioavailability of NO inthe forearm circulation.77 Manymolecules belonging to this phar-macological class improve endothe-lial function in humans (Table I).63This effect is likely to be specific todihydropyridine calcium antago-nists, while nondihydropyridinecalcium antagonists, eg, verapamil,have no effect on endothelial func-tion (Table I).63
In patients with coronary arterydisease, CCBs were extensively in-vestigated as to their effects on endothelial function and plaque sizeand progression. Initially, the Inter-national Nifedipine Trial on Anti-atherosclerotiC Therapy (INTACT)suggested that nifedipine suppress-es disease progression (as demon-strated by the appearance of newlesions by quantitative coronary ar-teriography).77 In ENCORE I, nife-dipine markedly suppressed theparadoxical vasoconstriction toacetylcholine in epicardial coronaryarteries of patients undergoing per-cutaneous coronary intervention inanother than the target artery.57 Asthis effect was noted even after thedrug had been stopped for severaldays and persisted up to 18 monthas demonstrated in ENCORE II,78
the calcium antagonist appears toconsistently improve coronary en-dothelial function. The effect of di-hydropyridine calcium antagonistson endothelial function are sum-marized in (Figure 11).63 These ef-fects may explain the anti-ischemicproperties of this class of drugs as
demonstrated in the A Coronary dis-ease Trial Investigating Outcomewith Nifedipine GITS (ACTION).79
β-Blockers
In contrast to the previously dis-cussed drugs, most β-blockers donot improve endothelial function.Indeed, hypertensive patients treat-ed with atenolol for 1 and 3 years,as well as 5 months with metopro-lol had no improvement in endo-thelial function (Table I).63 In con-trast, new-generation β-blockers,such as nebivolol and carvedilol,seem to improve this response(Table I and Figure 12).63 As far asnebivolol is concerned, its NO re-leasing properties80 may explainthis difference as it was demonstrat-ed in patients with hypertension(Table I).63 Interestingly, even inhealthy subjects, infusion of nebi-volol improves endothelial func-tion.80 Similar effects were shownwith carvedilol, a selective β1- andα1-antagonist with marked antioxi-dant properties, not only in hyper-tensives, but also in patients with
124
Dialogues in Cardiovascular Medicine - Vol 15 . No. 2 . 2010
Could endothelial dysfunction be a surrogate end point for CAD? - Wolfrum and others
Figure 11.Possible mecha-nisms underlying
the effect of calci-um antagonists on
endothelialfunction.
Abbreviations: seelegend to Figure 10.
Modified afterreference 63:
Sudano et al. HotTopics in Cardiology.
2009 (No. 15).
Growthfactors
Foam cell
diabetes or coronary artery disease(Table I).63,81,82 This is of particularinterest since carvedilol was supe-rior to metoprolol in the CarvedilolOr Metoprolol European Trial(COMET) in preventing death.83
To conclude, many drugs with prov-en efficacy in the prevention of ma-jor cardiovascular events have alsobeen shown to improve endothelialfunction in the peripheral and/orcoronary circulation. However, theresults of some studies have to beinterpreted with caution. Indeed,in some cases promising data frominitial studies could not be subse-quently confirmed.48 Several reasonsmay explain these discrepancies.
Besides type 1 errors, patient num-bers and consequently statisticalpower of many studies were notsufficient. Furthermore, the sophis-ticated tests to assess endothelialfunction require extensive trainingand certification of the operatorsinvolved. Better standardization tominimize the effect of environmen-tal or physiological influences,
such as exercise, eating, caffeineingestion, and variations in tem-perature is also needed.
Thus there is a need of further largeprospective studies with highlytrained investigators to ensure ap-propriate data quality to evaluatethe effects of earlier and newer drugson the vessel wall, and the endo-thelium in particular. Such trialsshould be part of a comprehensiveprogram that would also includelarge morbidity and mortality trialsto assess whether the beneficial effect on endothelial function of agiven drug is truly followed by im-proved prognosis in patients withcardiovascular disease.
SUMMARY
Numerous clinical studies havedocumented the presence of endo-thelial dysfunction at all clinicalstages of atherosclerotic vasculardisease and shown that the pres-ence and the degree of endothelialdysfunction are independently asso-ciated with the risk of major cardio-
vascular events, even after adjust-ment for the presence of coronaryartery disease and/or cardiovascularrisk factors.46 Assessment of en-dothelial function as an integrativemarker reflecting the effects of riskfactors on the vessel wall has be-come a highly valuable clinical re-search tool in cardiovascular dis-ease. However, whether it can beused routinely in daily clinical prac-tice on top of careful evaluation ofclassic risk factors to improve car-diovascular risk prediction, stratifyindividual cardiovascular risk, anddocument the effect of therapeuticinterventions on vascular functionrequires further investigationthrough large prospective studies.The evaluation of emerging simplerand more easily applicable nonin-vasive technologies also needs fur-ther investigation and validation.
Original research of the investigators has been supported by the Swiss NationalResearch Foundation, the MERCATORFoundation Switzerland, the Foundation for Cardiovascular Research, Zurich,
Switzerland, and educational grants of Bayer,Leverkusen, Germany, and a strategic Alliance with Pfizer, New York, USA.
125
Dialogues in Cardiovascular Medicine - Vol 15 . No. 2 . 2010
Could endothelial dysfunction be a surrogate end point for CAD? - Wolfrum and others
Figure 12. Possible mecha-nisms underlyingthe effect ofcarvedilol andnebivolol on en-dothelial function.
Abbreviations: seelegend to Figure 10. Modified after reference 63: Sudano et al. HotTopics in Cardiology.2009 (No. 15).
Growthfactors
Foam cell
REFERENCES
1. Furchgott RF, Zawadzki JV.
The obligatory role of endothelial cells inthe relaxation of arterial smooth muscle byacetylcholine.
Nature. 1980;288(5789):373-376.
2. Palmer RM, Ashton DS, Moncada S.
Vascular endothelial cells synthesize nitricoxide from L-arginine.
Nature. 1988;333(6174):664-666.
3. Stamler JS, Singel DJ, Loscalzo J.
Biochemistry of nitric oxide and its redox-activated forms.
Science. 1992;258(5090):1898-1902.
4. Lüscher TF, Noll G.
Endothelial function as an end-point in interventional trials: concepts, methods andcurrent data.
J Hypertens Suppl. 1996;14(2):S111-S119.
5. Landmesser U, Hornig B, Drexler H.
Endothelial function: a critical determinantin atherosclerosis?
Aging is associated with endothelial dysfunc-tion in healthy men years before the age-related decline in women.
J Am Coll Cardiol. 1994;24(2):471-476.
7. Linder L, Kiowski W, Buhler FR,Lüscher TF.
Indirect evidence for release of endothelium-derived relaxing factor in human forearmcirculation in vivo. Blunted response in es-sential hypertension.
Circulation. 1990;81(6):1762-1767.
8. Panza JA, Quyyumi AA, Brush JE Jr,Epstein SE.
Abnormal endothelium-dependent vascularrelaxation in patients with essential hyper-tension.
N Engl J Med. 1990;323(1):22-27.
9. Hirooka Y, Imaizumi T, Masaki H,et al.
Captopril improves impaired endothelium-dependent vasodilation in hypertensive patients.
Hypertension. 1992;20(2):175-180.
10. Panza JA, Casino PR, Kilcoyne CM,Quyyumi AA.
Role of endothelium-derived nitric oxide inthe abnormal endothelium-dependent vas-cular relaxation of patients with essentialhypertension.
Circulation. 1993;87(5):1468-1474.
11. Panza JA, Quyyumi AA, CallahanTS, Epstein SE.
Effect of antihypertensive treatment on en-dothelium-dependent vascular relaxation inpatients with essential hypertension.
J Am Coll Cardiol. 1993;21(5):1145-1151.
12. Panza JA, Casino PR, Badar DM,Quyyumi AA.
Effect of increased availability of endotheli-um-derived nitric oxide precursor on endothe-lium-dependent vascular relaxation in nor-mal subjects and in patients with essentialhypertension.
Circulation. 1993;87(5):1475-1481.
13. Taddei S, Mattei P, Virdis A,Sudano I, Ghiadoni L, Salvetti A.
Effect of potassium on vasodilation toacetylcholine in essential hypertension.
Hypertension. 1994;23(4):485-490.
14. Creager MA, Roddy MA.
Effect of captopril and enalapril on endothe-lial function in hypertensive patients.
Hypertension. 1994;24(4):499-505.
15. Panza JA, Casino PR, KilcoyneCM, Quyyumi AA.
Impaired endothelium-dependent vasodila-tion in patients with essential hypertension:evidence that the abnormality is not at themuscarinic receptor level.
J Am Coll Cardiol. 1994;23(7):1610-1616.
16. Taddei S, Virdis A, Mattei P, et al.
Aging and endothelial function in normoten-sive subjects and patients with essential hypertension.
Circulation. 1995;91(7):1981-1987.
17. Taddei S, Virdis A, Mattei P, et al.
Hypertension causes premature aging of en-dothelial function in humans.
Hypertension. 1997;29(3):736-743.
18. Treasure CB, Klein JL, Vita JA, et al.
Hypertension and left ventricular hypertrophyare associated with impaired endothelium-mediated relaxation in human coronary resistance vessels.
Circulation. 1993;87(1):86-93.
19. Egashira K, Suzuki S, Hirooka Y,et al.
Impaired endothelium-dependent vasodila-tion of large epicardial and resistance coronary arteries in patients with essentialhypertension. Different responses to acetyl-choline and substance P.
Hypertension. 1995;25(2):201-206.
20. Taddei S, Virdis A, Mattei P,Arzilli F, Salvetti A.
Endothelium-dependent forearm vasodila-tion is reduced in normotensive subjects withfamilial history of hypertension.
L-NG-nitro arginine produces an exaggeratedhypertension in anesthetized SHR.
Eur J Pharmacol. 1991;197(2-3):239-240.
24. Lepori M, Sartori C, Trueb L,Owlya R, Nicod P, Scherrer U.
Haemodynamic and sympathetic effects ofinhibition of nitric oxide synthase by sys-temic infusion of N(G)-monomethyl-L-argi-nine into humans are dose dependent.
J Hypertens. 1998;16(4):519-523.
126
Dialogues in Cardiovascular Medicine - Vol 15 . No. 2 . 2010
Could endothelial dysfunction be a surrogate end point for CAD? - Wolfrum and others
25. Sartori C, Lepori M, Scherrer U.
Interaction between nitric oxide and thecholinergic and sympathetic nervous systemin cardiovascular control in humans.
Pharmacol Ther. 2005;106(2):209-220.
26. Multiple Risk Factor InterventionTrial Research Group.
Multiple risk factor intervention trial. Riskfactor changes and mortality results.
Impaired endothelium-dependent vasodilationin patients with insulin-dependent diabetesmellitus.
Circulation. 1993;88(6):2510-2516.
33. Joannides R, Richard V, HaefeliWE, Linder L, Lüscher TF, Thuillez C.
Role of basal and stimulated release of nitricoxide in the regulation of radial artery cal-iber in humans.
Hypertension. 1995;26(2):327-331.
34. Bossaller C, Habib GB, YamamotoH, Williams C, Wells S, Henry PD.
Impaired muscarinic endothelium-dependentrelaxation and cyclic guanosine 5'-mono-phosphate formation in atherosclerotic humancoronary artery and rabbit aorta.
J Clin Invest. 1987;79(1):170-174.
35. Ludmer PL, Selwyn AP, Shook TL,et al.
Paradoxical vasoconstriction induced byacetylcholine in atherosclerotic coronaryarteries.
N Engl J Med. 1986;315(17):1046-1051.
36. Quyyumi AA, Dakak N, AndrewsNP, et al.
Nitric oxide activity in the human coronarycirculation. Impact of risk factors for coro-nary atherosclerosis.
J Clin Invest. 1995;95(4):1747-1755.
37. Benjamin N, Calver A, Collier J,Robinson B, Vallance P, Webb D.
Measuring forearm blood flow and interpret-ing the responses to drugs and mediators.
Hypertension. 1995;25(5):918-923.
38. Gemignani V, Bianchini E, Faita F,et al.
Ultrasound measurement of the brachialartery flow-mediated dilation without ECGgating.
Ultrasound Med Biol. 2008;34(3):385-391.
39. Gemignani V, Faita F, Ghiadoni L,Poggianti E, Demi M.
A system for real-time measurement of thebrachial artery diameter in B-mode ultra-sound images.
IEEE Trans Med Imaging. 2007;26(3):393-404.
40. Joannides R, Haefeli WE, Linder L,et al.
Nitric oxide is responsible for flow-dependentdilatation of human peripheral conduit arteries in vivo.
Circulation. 1995;91(5):1314-1319.
41. Corretti MC, Anderson TJ,Benjamin EJ, et al.
Guidelines for the ultrasound assessment ofendothelial-dependent flow-mediated vaso-dilation of the brachial artery: a report ofthe International Brachial Artery ReactivityTask Force.
J Am Coll Cardiol. 2002;39(2):257-265.
42. Anderson TJ, Uehata A, GerhardMD, et al.
Close relation of endothelial function in thehuman coronary and peripheral circulations.
J Am Coll Cardiol. 1995;26(5):1235-1241.
43. Irace C, Ceravolo R, NotarangeloL, et al.
Comparison of endothelial function evalu-ated by strain gauge plethysmography andbrachial artery ultrasound.
Atherosclerosis. 2001;158(1):53-59.
44. Woo KS, Chook P, Yu CW, et al.
Effects of diet and exercise on obesity-relatedvascular dysfunction in children.
Circulation. 2004;109(16):1981-1986.
45. Hamburg NM, Keyes MJ, LarsonMG, et al.
Cross-sectional relations of digital vascularfunction to cardiovascular risk factors inthe Framingham Heart Study.
Circulation. 2008;117(19):2467-2474.
46. Lerman A, Zeiher AM.
Endothelial function: cardiac events.
Circulation. 2005;111(3):363-368.
47. Schachinger V, Britten MB,Zeiher AM.
Prognostic impact of coronary vasodilatordysfunction on adverse long-term outcomeof coronary heart disease.
Circulation. 2000;101(16):1899-1906.
48. Modena MG, Bonetti L, Coppi F,Bursi F, Rossi R.
Prognostic role of reversible endothelialdysfunction in hypertensive postmenopausalwomen.
J Am Coll Cardiol. 2002;40(3):505-510.
49. Shepherd J, Cobbe SM, Ford I,et al.
Prevention of coronary heart disease withpravastatin in men with hypercholesterolemia.West of Scotland Coronary PreventionStudy Group.
N Engl J Med. 1995;333(20):1301-1307.
50. Long-Term Intervention withPravastatin in Ischemic Disease(LIPID) Study Group.
Prevention of cardiovascular events anddeath with pravastatin in patients withcoronary heart disease and a broad rangeof initial cholesterol levels.
N Engl J Med. 1998;339(19):1349-1357.
127
Dialogues in Cardiovascular Medicine - Vol 15 . No. 2 . 2010
Could endothelial dysfunction be a surrogate end point for CAD? - Wolfrum and others
51. Dupuis J, Tardif JC, Cernacek P,Theroux P.
Cholesterol reduction rapidly improves endothelial function after acute coronarysyndromes. The RECIFE (reduction of cholesterol in ischemia and function of theendothelium) trial.
Circulation. 1999;99(25):3227-3233.
52. John S, Schlaich M, Langenfeld M,et al.
Increased bioavailability of nitric oxide afterlipid-lowering therapy in hypercholesterol-emic patients: a randomized, placebo-controlled, double-blind study.
Circulation. 1998;98(3):211-216.
53. Laufs U, La Fata V, Plutzky J,Liao JK.
Upregulation of endothelial nitric oxidesynthase by HMG CoA reductase inhibitors.
Circulation. 1998;97(12):1129-1135.
54. van Boven AJ, Jukema JW,Zwinderman AH, Crijns HJ, Lie KI,Bruschke AV.
Reduction of transient myocardial ischemiawith pravastatin in addition to the conven-tional treatment in patients with anginapectoris. REGRESS Study Group.
Circulation. 1996;94(7):1503-1505.
55. Stroes ES, Koomans HA, de Bruin TW, Rabelink TJ.
Vascular function in the forearm of hyper-cholesterolaemic patients off and on lipid-lowering medication.
Lancet. 1995;346(8973):467-471.
56. Vita JA, Yeung AC, Winniford M,et al.
Effect of cholesterol-lowering therapy oncoronary endothelial vasomotor function inpatients with coronary artery disease.
Circulation. 2000;102(8):846-851.
57. ENCORE Investigators.
Effect of nifedipine and cerivastatin on coro-nary endothelial function in patients withcoronary artery disease: the ENCORE IStudy (Evaluation of Nifedipine and Ceri-vastatin On Recovery of coronary Endothe-lial function).
Circulation. 2003;107(3):422-428.
58. Anderson TJ, Meredith IT, YeungAC, Frei B, Selwyn AP, Ganz P.
The effect of cholesterol-lowering and anti-oxidant therapy on endothelium-dependentcoronary vasomotion.
N Engl J Med. 1995;332(8):488-493.
59. Pajkrt D, Doran JE, Koster F, et al.
Antiinflammatory effects of reconstitutedhigh-density lipoprotein during human endotoxemia.
J Exp Med. 1996;184(5):1601-1608.
60. Hermann F, Enseleit F, Spielker LE,et al.
Cholesterylestertransfer protein inhibitionand endothelial function in type II hyper-lipidemia.
Thromb Res. 2009;123(3):460-465.
61. Ghiadoni L, Magagna A, Versari D,et al.
Different effect of antihypertensive drugs onconduit artery endothelial function.
Hypertension. 2003;41(6):1281-1286.
62. Mancini GB, Henry GC, Macaya C,et al.
Angiotensin-converting enzyme inhibitionwith quinapril improves endothelial vaso-motor dysfunction in patients with coronaryartery disease. The TREND (Trial onReversing ENdothelial Dysfunction) Study[published erratum appears in Circulation1996;94:1490].
Circulation. 1996;94(3):258-265.
63. Sudano I, Flammer AJ, Steffel J,Noll G, Lüscher TF.
The vascular endothelium in hypertension:target and promoter?
Hot Topics in Cardiology. 2009 (No. 15).
64. Mancini GB, Henry GC, Macaya C,et al.
Angiotensin-converting enzyme inhibitionwith quinapril improves endothelial vaso-motor dysfunction in patients with coronaryartery disease. The TREND (Trial onReversing ENdothelial Dysfunction) Study.
Circulation. 1996;94(3):258-265.
65. Anderson TJ, Elstein E, Haber H,Charbonneau F.
Comparative study of ACE-inhibition, angiotensin II antagonism, and calciumchannel blockade on flow-mediated vasodi-lation in patients with coronary disease(BANFF study).
J Am Coll Cardiol. 2000;35(1):60-66.
66. Yusuf S, Sleight P, Pogue J,Bosch J, Davies R, Dagenais G.
Effects of an angiotensin-converting-enzymeinhibitor, ramipril, on cardiovascular eventsin high-risk patients. The Heart OutcomesPrevention Evaluation Study Investigators.
N Engl J Med. 2000;342(3):145-153.
67. Ceconi C, Fox KM, Remme WJ, et al.
ACE inhibition with perindopril and endo-thelial dysfunction. Results of a substudy ofthe EUROPA study: PERTINENT.
Cardiovasc Res. 2007;73(1):237-246.
68. Fox KM.
Efficacy of perindopril in reduction of cardio-vascular events among patients with stablecoronary artery disease: randomised, double-blind, placebo-controlled, multicentre trial(the EUROPA study).
Lancet. 2003;362(9399):782-788.
69. Ghiadoni L, Virdis A, Magagna A,Taddei S, Salvetti A.
Effect of the angiotensin II type 1 receptorblocker candesartan on endothelial func-tion in patients with essential hypertension.
Hypertension. 2000;35(1 pt 2):501-506.
70. Bragulat E, Larrousse M, Coca A,de la Sierra A.
Effect of long-term irbesartan treatment onendothelium-dependent vasodilation in es-sential hypertensive patients.
Br J Biomed Sci. 2003;60(4):191-196.
71. Monton M, Jimenez A, Nunez A,et al.
Comparative effects of angiotensin II AT-1-type receptor antagonists in vitro on humanplatelet activation.
J Cardiovasc Pharmacol. 2000;35(6):906-913.
128
Dialogues in Cardiovascular Medicine - Vol 15 . No. 2 . 2010
Could endothelial dysfunction be a surrogate end point for CAD? - Wolfrum and others
72. Schieffer B, Bunte C, Witte J, et al.
Comparative effects of AT1-antagonism andangiotensin-converting enzyme inhibition onmarkers of inflammation and platelet aggre-gation in patients with coronary artery disease.
J Am Coll Cardiol. 2004;44(2):362-368.
73. Koh KK, Quon MJ, Lee Y, et al.
Additive beneficial cardiovascular and meta-bolic effects of combination therapy with rami-pril and candesartan in hypertensive patients.
Eur Heart J. 2007;28(12):1440-1447.
74. Taddei S, Virdis A, Ghiadoni L,Uleri S, Magagna A, Salvetti A.
Lacidipine restores endothelium-dependentvasodilation in essential hypertensive patients.
Hypertension. 1997;30(6):1606-1612.
75. Sudano I, Virdis A, Taddei S, et al.
Chronic treatment with long-acting nifedip-ine reduces vasoconstriction to endothelin-1in essential hypertension.
Hypertension. 2007;49(2):285-290.
76. Verhaar MC, Honing ML, van Dam T, et al.
Nifedipine improves endothelial function inhypercholesterolemia, independently of aneffect on blood pressure or plasma lipids.
Retardation of angiographic progression ofcoronary artery disease by nifedipine. Resultsof the International Nifedipine Trial onAntiatherosclerotic Therapy (INTACT). INTACT Group Investigators.
Lancet. 1990;335(8698):1109-1113.
78. Lüscher TF, Pieper M, Tendera M,et al.
A randomized placebo-controlled study onthe effect of nifedipine on coronary endo-thelial function and plaque formation inpatients with coronary artery disease: theENCORE II study.
Eur Heart J. 2009;30(13):1590-1597.
79. Poole-Wilson PA, Lubsen J,Kirwan BA, et al.
Effect of long-acting nifedipine on mortali-ty and cardiovascular morbidity in patientswith stable angina requiring treatment(ACTION trial): randomised controlled trial.
Lancet. 2004;364(9437):849-857.
80. Cockcroft JR, Chowienczyk PJ,Brett SE, et al.
Nebivolol vasodilates human forearm vas-culature: evidence for an L-arginine/NO-dependent mechanism.
J Pharmacol Exp Ther. 1995;274(3):1067-1071.
81. Bank AJ, Kelly AS, Thelen AM,Kaiser DR, Gonzalez-Campoy JM.
Effects of carvedilol versus metoprolol onendothelial function and oxidative stress inpatients with type 2 diabetes mellitus.
Am J Hypertens. 2007;20(7):777-783.
82. Matsuda Y, Akita H, Terashima M,Shiga N, Kanazawa K, Yokoyama M.
Carvedilol improves endothelium-dependentdilatation in patients with coronary arterydisease.
Am Heart J. 2000;140(5):753-759.
83. Poole-Wilson PA, Swedberg K,Cleland JG, et al.
Comparison of carvedilol and metoprolol onclinical outcomes in patients with chronicheart failure in the Carvedilol Or MetoprololEuropean Trial (COMET): randomisedcontrolled trial.
Lancet. 2003;362(9377):7-13.
129
Dialogues in Cardiovascular Medicine - Vol 15 . No. 2 . 2010
Could endothelial dysfunction be a surrogate end point for CAD? - Wolfrum and others
130
Dialogues in Cardiovascular Medicine - Vol 15 . No. 2 . 2010
he randomized controlledclinical trial (RCT) is the cur-rent foundation for obtainingvital information upon which
to base clinical decisions. The RCTis, however, a relatively new tool inour armamentarium of clinical in-vestigation. The processes of ran-domization, blinding, and when ap-propriate, use of placebo, reducebiases and provide a framework forvalid statistical comparisons. Mod-ern RCTs assessing risks for clinicalevents were first used in infectiousdiseases. The 1948 trial which pro-vided proof that in patients withtuberculosis those randomly as-signed to therapy with streptomycinhad a lower mortality at 6 months(7% of streptomycin patients and27% of the bed rest alone group) ishistorically the first RCT demonstrat-ing a survival benefit of a therapy.1
In our view, the roots of clinical out-comes RCTs in cardiovascular med-icine can be traced to the originalVeterans Association (VA) Coopera-tive Study Group on Antihyperten-sive Agents.2 Under the leadershipof the late Dr Edward Freis, thisstill active clinical trials organiza-tion tested the hypothesis that theadverse association between ele-vated blood pressure and cardiovas-cular outcomes highlighted by thepioneering epidemiologic work fromthe Framingham study3 could befavorably modified by reducingblood pressure with pharmacologictherapies. The Veterans investiga-tors established in patients withsevere diastolic hypertension that,compared with placebo, those ran-
domized to receive the combinationof three antihypertensive agents(hydralazine, hydrochlorothiazide,and reserpine) had a lower incidenceof the composite of death and non-fatal cardiovascular outcomes.2 Thishistoric paradigm shifting trial pro-vided the rationale for pharmaco-logic use of antihypertensive thera-pies in hypertension to reduceadverse cardiovascular outcomes,not just blood pressure. By today’sstandards, the first VA trial, withrelatively few events and a compos-ite of predominantly nonfatal out-comes, would not be considered asdefinitive. However, this demon-strated the importance of conduct-ing RCTs to impact cardiovascularprognosis and serves as the founda-tion for RCTs testing clinical out-comes.
In the ensuing 50 years, the totalityof the evidence for the importanceof pharmacologic blood pressurelowering in hypertension has beenbased on the compilation of RCTs
Can there be any surrogate for safety? Marc A. Pfeffer, MD, PhD; Hicham Skali, MD, MSc
Department of Medicine - Division of Cardiology - Brigham and Women’s Hospital - Harvard Medical School - Boston, Mass - USA
T
Keywords: clinical trial; evidence-basedmedicine; end point; heart failure; safety;therapy; prognosisAddress for correspondence:Marc A. Pfeffer, MD, PhD, Brigham andWomen’s Hospital, 75 Francis Street,Boston, MA 02115, USA(e-mail: [email protected])Dialogues Cardiovasc Med. 2010;15:130-139
Clinical outcomes–focused random-ized clinical trials are the currentgold standard for evidence-basedmedicine. However, it is impracticaland not feasible to attempt to an-swer all questions with large clinicaloutcomes trials. Surrogate markersare often utilized as alternativemeasures in smaller and shorter-duration trials. However, they arenot always a reliable barometer ofintervention-induced alterations inprognosis. This manuscript high-lights several prominent instanceswhere the clinical effects producedby a therapy were not adequatelypredicted by changes in an alleged-ly reliable and important surrogate.In addition, trials targeting onlysurrogate markers do not have suf-ficient exposure to unmask potentialsafety issues of the intervention. In highlighting the limitations ofsurrogate-marker–based trials, weemphasize the need for more defini-tive clinical outcomes trials to betterguide clinical practice. SELECTED ABBREVIATIONS
AND ACRONYMS
ARB angiotensin-II receptor blocker
CETP cholesteryl-ester transfer protein
CKD chronic kidney disease
ESA erythropoiesis-stimulating agent
MACE major adverse cardiac events
PRO patient-reported outcomes
RCT randomized controlled trial
that demonstrate major reductionsin risk for cardiovascular events.4
In general, the greater the extent ofblood pressure lowering achieved,the lower the risk for subsequentcardiovascular events. It is impor-tant to note that these large placebocontrolled clinical outcome RCTsalso generate quantitative data re-garding the adverse experiences thatcan be attributed to these pharma-cologic therapies. This aspect of thelarge clinical outcome assessingRCTs should not be undervalued, astherapeutic decisions are based onthe best available estimate of bothpotential benefits as well as thesafety and tolerability of the inter-vention.
END POINTS IN RCTs
The medical community is familiarwith the components of well-de-signed RCTs. Although many empha-size the inclusion and exclusioncriteria, sample size, event rates, andduration, the fundamental reasonto conduct a RCT is to determinewhether the primary objective is al-tered by the intervention. By defi-nition, the primary objective is theend point upon which the statisticalframework for accepting or rejectingthe hypothesis is based. As is cov-ered by Anand and Florea5 in thisissue of Dialogues in CardiovascularMedicine, few would argue that tri-als comparing rates of death or all-cause mortality are testing the mostdefinitive, unbiased, and irrefutableof all end points. In cardiovascularmedicine, we have been fortunateto have had multiple definitive mor-tality trials that have shaped ourevidence-based practice. The use ofβ-blockers in patients with sympto-matic heart failure and reducedejection fraction is firmly based onseveral RCTs that had all-cause mor-tality as their primary objective.6
These major RCTs generated con-vincing data of the survival-prolong-
ing actions of these agents. Thestrength of survival-improving dataoffers the strongest impetus to im-plement a new therapy into clinicalpractice to improve public health. Asimportant and convincing as RCTswith the primary end point of all-cause mortality can be, it is imprac-tical to consider that our new ther-apies will continue to be testedagainst this highest bar (survival).Indeed, RCTs designed to testwhether rates of death are altered bya therapy will, by definition, be con-fined to only the severest patientswith end stages of disease or beprohibitively large in both samplesize and duration.7
COMPOSITE CLINICALOUTCOMES: DEATH ANDNONFATAL CLINICAL
EVENTS
Most modern clinical outcomesRCTs are designed to determinewhether the intervention alters ratesof death and a prespecified combi-nation of important nonfatal events.Incorporating nonfatal events re-duces the required sample size andextends the populations that can beexamined. The concept is that pre-venting clinically well-defined andimportant nonfatal events such asstroke, myocardial infarction, or hos-pitalization for heart failure wouldbe an important objective.8 Al-though not as definitive as death,those who survive one of these clin-ically impactful, though initially non-fatal events, would, with longer fol-low-up, have a higher risk of deaththan those with a more benigncourse. In general, this assumptionis usually correct in that subsequentmortality is severalfold higher in thepatients who have had a nonfatalcardiovascular event compared withsurvivors with a more benign clini-cal course.9 Time to the first occur-rence of any of the prespecifiedcomposites is probably the most
commonly used analysis of modernmajor outcome RCTs. An intrinsicproblem with the time-to-first-eventanalysis of RCTs that utilizes a clini-cal composite outcome is that theanalysis gives equal statisticalweight to all of the components ofthe composite. As such, a more se-rious event such as death that oc-curs after any of the nonfatal com-ponents is ignored in the primaryoutcome analysis.
In cardiovascular RCTs, the benefitof using a composite of fatal andnonfatal clinical outcomes oftentermed MACE (major adverse car-diac events) to increase event ratesand reduce sample size can be off-set when the influence of the thera-peutic modality being tested is notconsistent across the prespecifiedcomponents of the primary out-come. Cogent arguments have beenmade to narrow the focus of the pri-mary composite to the pattern ofevents that the therapeutic agentbeing examined is mechanisticallymost likely to alter: the so-calledcause-specific or targeted events.10
However, inconsistencies of the ef-fect size and even direction are notuncommon results with compositeoutcomes.7 Moreover, when nonfa-tal end points are dichotomized(yes/no), the broad range of severityof the nonfatal event is not consid-ered. In most RCTs, a myocardialinfarction that is detected by a mi-nor troponin elevation has the sameweight as one that results in car-diogenic shock. Similarly, strokefrom which the patient makes anexcellent functional recovery wouldbe statistically considered the sameas a disabling stroke.
These issues plague most moderntrials and clinicians need to be ableto evaluate the totality of evidencegenerated in the RCT by probingbeneath the surface of the primaryend point. Clinical judgment must
131
Dialogues in Cardiovascular Medicine - Vol 15 . No. 2 . 2010
Can there be any surrogate for safety? - Pfeffer and Skali
132
be used in conjunction with statis-tical testing to more fully under-stand the impact of a therapy. Withor without formal assignments ofweight, clinicians make their own as-sessments of the contributions ofthe components of the compositeclinical outcome to the primary ob-jective. Indeed, clinical trial expertscontinue to develop new metrics toconsider some of these deficienciesimposed by time to first event of acomposite outcome analysis. Majorefforts are now under way to alsoincorporate patient-reported out-comes (PRO)11 along with clinicalevent reporting to attempt to achievea better barometer of potential al-terations in the disease progressionin clinical trials.12 These novel ap-proaches to clinical assessments aremade to provide a more global as-sessment of the potential therapeu-tic impact of the tested intervention.
USE OF SURROGATES
As discussed by Anand and Florea5
and many others,13,14 in an effortto conduct RCTs that provide anindirect measure of morbidity andmortality without requiring extremesample sizes and duration of follow-up, surrogates or substitutes forclinical outcome end points havebeen evaluated. In general, theseare readily quantifiable laboratorymeasures, which, based on epi-demiologic data, are correlated witheither a favorable or unfavorableclinical outcome.15 The underlyingassumption is that therapeuticallyproduced alterations in the surro-gate measure of disease severitywould predict in a quantifiable fash-ion the clinical progression of thedisease. There are many attractiveadvantages to using biologicallyplausible and, indeed, even epi-demiologically important markersof disease severity as end points inclinical trials. In some respects, thisapproach mirrors individual prac-
tice of medicine whereby physiciansfollow a patient, not by events, butby indirect markers we associatewith disease severity or progression.Much needed—indeed essential fordrug development—smaller RCTsusing surrogate end points providethe basis for dose ranging, hypoth-esis-generating RCTs necessary forproviding a rationale and the pre-liminary information needed to plana much larger major clinical out-comes trial. This use of surrogateend points is appropriate and es-sential for early studies evaluatingnew therapeutic approaches. How-
ever, it must be clearly acknowl-edged that these surrogate markerplanning studies cannot providesufficient data regarding either theeffectiveness of the intervention inimproving clinical outcomes or itspotential for adverse events, whichare both needed for informed ther-apeutic decisions.13
CLEAR DISSOCIATION BETWEEN THE INFLUENCEON THE SURROGATE ANDTHE CLINICAL OUTCOMES
Unfortunately, there have been nu-merous discordant findings betweentherapies that clearly altered an es-tablished surrogate marker of dis-ease progression in a favorable di-rection only to be later found in a
more definitive clinical outcome RCTto have either a neutral or evenharmful action (Table I). This man-uscript will highlight some of thesediscordant instances in the field ofcardiovascular, diabetes, and renaldiseases, emphasizing the impor-tance of conducting RCTs with clin-ical outcomes as the primary ob-jective to obtain the best estimatesof both clinical effectiveness as wellas safety (Table II, page 135).
Ventricular premature beats
The Cardiac Arrhythmia SuppressionTrial (CAST) provided one of theearly and most profound examplesof a therapy that favorably altereda highly respected surrogate and yethad a greatly unanticipated detri-mental effect on survival.16 Theepidemiology linking frequency ofventricular premature depolariza-tions to heightened risk of death inhigh-risk patients was firmly estab-lished.17 In the 1970s and 1980s, amajor effort in caring for patientswith acute and chronic myocardialinfarction and heart failure was todetect and suppress ventricular ar-rhythmias. CAST was designed tocompare several antiarrhythmicagents for effectiveness in improv-ing survival. Patients with recentmyocardial infarction, left ventriculardysfunction, and evidence of fre-quent ventricular arrhythmias werescreened to confirm that their ex-trasystoles could be suppressed byan antiarrhythmic drug prior to ran-domization. The primary objectiveof the RCT was survival, not arrhyth-mia control. CAST was stopped byits safety committee when it hadbecome clear that those randomizedto either antiarrhythmic (encainideor flecainide) were more likely to diethan those on placebo.16 A nega-tive impact on survival was alsodemonstrated with moricizine, thethird antiarrhythmic examined.18
These counterintuitive and unex-
Dialogues in Cardiovascular Medicine - Vol 15 . No. 2 . 2010
Can there be any surrogate for safety? - Pfeffer and Skali
SURROGATE-BASED TRIALS ≠OUTCOME
EFFICACY TRIALS
SURROGATES CANNOT PROVIDESAFETY DATA
Table I. Two warnings concerning the useof surrogate markers in clinical trials.
133
pected findings resulted in a majorshift in patient management. It isimportant to emphasize how en-trenched the concept of ventriculararrhythmia suppression was priorto the CAST results and to considerthe magnitude of patients that werebeing unwittingly treated with harm-ful agents.
Ventricular contractility
At approximately the same time,another entrenched icon of cardio-vascular perceptions that improvingcontractility in those with systolicdysfunction heart failure would im-prove clinical outcomes was similar-ly abolished by mortality data fromRCTs that were sufficiently sized toaddress clinical outcomes ratherthan surrogates. With impaired con-tractility or depressed ejection frac-tion as the pathophysiologic rootfor the signs, symptoms, and im-paired prognosis of these patients,it was understandable, logical, andhighly anticipated that positive in-
otropic agents that had been shownin small RCTs to improve multipleimportant surrogates of impairedcardiac performance such as wedgepressure, cardiac output, and leftventricular ejection fraction wouldresult in improved patient out-comes.19-21 The finding of dose-de-pendent increases in mortality withthese agents despite the favorablehemodynamic changes "cast" furtherdistrust on the use of surrogatemarker RCTs to direct clinical care.
Blood pressure
Since the historic VA Cooperativetrial, blood pressure lowering bypharmacologic therapies has con-sistently and quantitatively beenassociated with improved prognosis.However, the limits of this associa-tion are being uncovered. In the recently reported Action to ControlCardiOvascular Risk in Diabetes–Blood Pressure (ACCORD) BloodPressure trial, patients with type 2diabetes mellitus and an average
baseline systolic blood pressure of139 mm Hg, were randomized to in-tensive vs standard blood pressurecontrol strategies.22 Despite achiev-ing a large blood pressure differ-ence (between-group difference 14 mm Hg), patients randomized tothe intensive strategy had no sig-nificant reduction in the risk of ex-periencing the composite outcomeof cardiovascular death, nonfatalstroke, or nonfatal myocardial in-farction (Figure 1).22 Moreover, thisgroup was more likely to have hypo-tension, hyperkalemia, or hypoten-sion, hyperkalemia, or serum crea-tinine elevations. These findings illustrate the hazards of extrapolat-ing results from a higher to a lowerblood pressure range.
Kidney function
The African American Study ofKidney disease and hypertension(AASK) Study Group trial offers an-other example of a discrepant effectof an intervention on a surrogateand a meaningful clinical end point.The primary end point of the trialwas the change in glomerular filtra-tion rate, another established sur-rogate marker for progression ofrenal disease and increased risk ofdeath; the prespecified secondaryoutcomes included mortality andprogression to end-stage renal dis-ease. While the patients randomizedto amlodipine displayed, at leastinitially, an increase in glomerularfiltration rate (GFR) and better bloodpressure control, they demonstrat-ed a higher propensity to progressto a worse outcome of death or end-stage renal disease when comparedwith ramipril.23 In fact, because ofthis deleterious effect, the amlodi-pine arm was stopped early by theData Safety Monitoring Board.24
Another widely held belief is thatreductions in urinary protein excre-tion, or microalbuminuria, could be
Dialogues in Cardiovascular Medicine - Vol 15 . No. 2 . 2010
Can there be any surrogate for safety? - Pfeffer and Skali
ACCORD BP trial: SBP & Outcomes140
130
120
1100 1 2 3 4 5 6 7 8
Post-randomization (years)
Average after 1st year: 133.5;Standard vs 119.3; Intensive, delta = 14.2
SB
P (
mm
Hg
)
CV death, MI, strokeTotal mortality
208 (1.87)150 (1.28)
237 (2.09)144 (1.19)
0.88 (0.73-1.06)1.07 (0.85-1.35)
0.200.55
Intensiven=2363
Standardn=2371
Events (%/year) Events (%/year) HR (95% CI) P
Figure 1. Surrogate markers and findings from the Action to Control Cardiovascular Riskin Diabetes (ACCORD) Blood Pressure Trial.
translated into reduction in risk ofsubsequent clinical events. Thereis evidence that an angiotensin-con-verting enzyme (ACE) inhibitor oran angiotensin-II receptor blocker(ARB) reduces the surrogate markerof urinary protein excretion and im-proves clinical outcomes (increasein serum creatinine, need for dialy-sis or death).25,26 Therapeutic regi-mens combining both inhibitors ofthe renin-angiotensin system havebeen shown in several small studiesto reduce the degree of proteinuria,possibly to a greater extent thaneither agent alone.27 However, clin-ical outcomes RCTs have not shownsuperiority of this combination.The VALsartan In Acute myocardialiNfarcTion (VALIANT) investigatorsshowed that post–myocardial in-farction patients with signs andsymptoms of heart failure and/orleft ventricular systolic dysfunctionrandomized to a combination ofcaptopril and valsartan experiencedmore adverse events without anyfurther improvement in survivalcompared with patients randomizedto either of the two agents.28
More recently, in a different patientpopulation, high-risk vascular pa-tients, the ONgoing TelmisartanAlone and in combination withRamipril Global Endpoint Trial(ONTARGET) tested whether thecombination of the angiotensin-con-verting-enzyme inhibitor ramipriland the ARB telmisartan would besuperior to either one alone in reducing the risk of the primarycomposite outcome (death fromcardiovascular causes, myocardialinfarction, stroke, or hospitalizationfor heart failure) and demonstratedan increase in adverse events with-out any associated benefit in thecombination arm.29 More specifical-ly, to address the mechanistic inter-action between the surrogate endpoint of urinary albumin excretionand a prespecified renal composite
outcome (need for dialysis, doublingof serum creatinine or death), theauthors showed that despite an ex-pected finding of less increase inurinary albumin excretion with thecombination therapy than withramipril, the primary renal outcomewas statistically significantly in-creased with the combination ther-apy than with either ramipril ortelmisartan.30 Had a trial been con-ducted solely with the intent of de-termining the effect of combinationtherapy on proteinuria, the effecton the renal outcomes would havebeen missed. These results, as when-ever there are contrasting effects ona surrogate and a clinical end point,can be explained by unintended ef-fects on the clinical end point (in-creased renal outcomes) independ-ently from, and not through, theintermediary surrogate marker.15
This, again, emphasizes the cautionneeded whenever surrogate mark-ers are utilized for expanding orproving the clinical utility of an in-tervention, and that a clear benefi-cial effect on hard clinical outcomesneeds to be demonstrated.31
Low-density lipoprotein (LDL) cho-lesterol lowering with statins may beconsidered one of the most reliableassociations between a laboratorysurrogate and clinical outcomes.32
Unquestionably, cardiovascular mor-bidity has been reduced and mortal-ity improved with the use of statinsacross a broad range of populations.In the high-risk populations withboth manifest cardiovascular dis-ease and elevated LDL, the firstclinical outcomes trial in the field—
the Scandinavian Simvastatin Sur-vival Study (4S)33 provided the mostdefinitive data on both a relative andabsolute scale about the benefitsof this therapy. Within a very shorttime, complementary clinical out-come data became available froma series of other clinical outcomeRCTs with other statins in patientswith prevalent cardiovascular dis-ease and lower cholesterol valuesas well as those without manifestatherosclerosis (primary prevention).Meta-analyses of these placebocontrolled clinical outcome RCTshave shown very consistent mor-bidity mortality benefits.34
Extrapolation of these clinical ben-efits through LDL lowering of theother populations requires caution.In patients with symptomatic heartfailure, both the Effects of n-3 PUFAand Rosuvastatin on Mortality-Morbidity of Patients With Symp-tomatic CHF (GISSI HF) study 35and the COntrolled ROsuvastatinMultinational Trial in Heart Failure(CORONA)36 were well-conductedRCTs designed with sufficient sta-tistical power to detect differencesin an important cardiovascular out-come composite. In each trial, thestatin (rosuvastatin) was quite ef-fective in reducing LDL, with thestatin-treated group manifestinglarge cholesterol and C-reactiveprotein (CRP) reductions comparedwith placebo. However, both trialswere neutral on their primary clini-cal outcomes (GISSI HF: time todeath, and time to death or admis-sion to hospital for cardiovascularreasons; and CORONA: death fromcardiovascular causes, nonfatalmyocardial infarction, or nonfatalstroke). This same drug was, how-ever, effective in reducing cardiovas-cular events in the Justification forthe Use of statins in primary Pre-vention: an Intervention Trial Eval-uating Rosuvastatin trial (JUPITER),a primary prevention trial.37
134
Dialogues in Cardiovascular Medicine - Vol 15 . No. 2 . 2010
Can there be any surrogate for safety? - Pfeffer and Skali
This issue of effectiveness of a ther-apy on a surrogate without ensur-ing similar clinical outcomes wasalso apparent in the two majorstatin clinical outcome trials con-ducted in patients on dialysis. TheGerman Diabetes and Dialysis Study(GDDS)38 compared atorvastatinand placebo, while A Study to Eval-uate the Use of Rosuvastatin inSubjects on Regular Hemodialysis:An Assessment of Survival and Car-diovascular Events (AURORA)39
compared rosuvastatin and placebo.In both cases, the statin was effec-tive in lowering LDL without result-ing in a statistically different clini-cal event rate between the placeboand the statin-treated groups. Thesefour well-done trials introduce theconcept of competing risk as wellas the difficulties of extrapolatingclinical outcome benefits from onepopulation to another even thoughthe direction and magnitude of theapparent favorable change in thesurrogate was comparable to otherRCTs that showed important reduc-tions in clinical events.
SAFETY IS MOLECULE- SPECIFIC: SURROGATEMARKER STUDIES ARE GENERALLY
INSUFFICIENT TO DETECTSAFETY ISSUES
The strong association between LDLand atherosclerotic risk and thepervasive assumption couplingLDL lowering with better prognosisplaced this particular surrogate inthe unique company with bloodpressure lowering for internationalregulatory agencies to consider ap-proval of agents that favorably mod-ify LDL without conducting preap-proval pivotal clinical outcome RCTs.As such, a member of the statinclass could gain approval predomi-nately on the basis of LDL lower-ing with less patient drug exposureinformation than would generally be
required for compounds claimingclinical rather than surrogate bene-fits. Cerivastatin gained regulatoryapproval on the basis of RCTs tar-geting LDL lowering and apparentsafety within the framework of themore limited total patient-time experience relative to the exposurethat would have been obtainedwhen effectiveness in clinical out-comes is required. The approximate-ly tenfold increase in the rare, butdreaded, risk of statin-induced rhab-domyolysis was only uncovered inpostmarketing surveillance in a non-research setting.40 This example un-derscores that the sample size gen-erally needed for the demonstrationof an apparent favorable effect ona surrogate can be woefully insuffi-cient for an assessment of the safe-
ty of a specific molecule. The corol-lary is that RCTs large enough totest for modifications of clinical out-comes rather than a surrogate lab-oratory measurement are requiredto provide a fuller assessment ofsafety.
SURROGATE STUDIES LACK OF
SAFETY/HDL RAISING
The Investigation of Lipid LevelManagement to Understand itsImpact in Atherosclerotic Events(ILLUMINATE) trial provides themost vivid recent example of a phar-macologic therapy favorably influ-encing a strongly accepted surrogate,yet having adverse consequenceson clinical outcomes. In epidemio-
135
Dialogues in Cardiovascular Medicine - Vol 15 . No. 2 . 2010
Can there be any surrogate for safety? - Pfeffer and Skali
Table II. Prominent examples of discrepancies between surrogate end markers andclinical end points.
Abbreviations: BP, blood pressure; CCB, calcium channel blocker; CV, cardiovascular; ESRD, endstage renal disease; GFR, glomerular filtration rate; HDL, high-density lipoprotein; LDL, low-densitylipoprotein. Trial acronyms: CAST, Cardiac Arrhythmia Suppression Trial; VEST, VESnarinone Trial; AASK,African-American Study of Kidney disease and hypertension; CORONA, COntrolled ROsuvastatinMulti-national Trial in Heart Failure; GISSI-HF, Effects of n-3 PUFA and Rosuvastatin on Mortality-Morbidity of Patients With Symptomatic CHF; GDDS, German Diabetes and Dialysis Study; AURORA,A Study to Evaluate the Use of Rosuvastatin in Subjects on Regular Hemodialysis: An Assessment ofSurvival and Cardiovascular Events; ILLUMINATE, Investigation of Lipid Level Management toUnderstand its Impact in Atherosclerotic Events; ONTARGET, ONgoing Telmisartan Alone and incombination with Ramipril Global Endpoint Trial; TREAT, Trial to Reduce cardiovascular Events withAranesp Therapy; ACCORD-BP, Action to Control CardiOvascular Risk in Diabetes–Blood Pressure.
logic studies, lower HDL cholesterolis strongly associated with higherrisk for atherosclerotic events evenwith adjustment for other conven-tional risks factors.41 Cholesteryl-es-ter transfer protein (CETP) inhibitorswere developed to block the reversecholesterol transport pathway andraise high-density lipoprotein (HDL)levels while decreasing LDL levels.Indeed, the magnitude of the HDLincrease of approximately 80% wasunprecedented and, in addition,there was a further 20% lowering of LDL in those treated with torce-trapib compared with placebo.42
These apparently favorable alter-ations in plasma cholesterol were soprofound that on average the grouptreated with torcetrapib had higherHDL than LDL. However, when ex-amined under the rigors of an RCTof 15 000 patients evaluating clini-cal outcomes rather than the surro-gate of plasma cholesterol, the trialwas terminated for safety reasons asthe mortality of the active treatmentgroup exceeded that with placebo.42
ACCEPTANCE OF A SURROGATE CAN STIFLE RESEARCH
Practicing medicine requires inte-grating the best available, thoughincomplete, data to make clinicaldecisions. Often, results of RCTsfrom a well-specified populationare extrapolated to represent thebest available information for theindividual patient not representedin the trial. Favorable directionalchanges in a laboratory measurethat have an association with betterprognosis offers both the patientand physician feedback of a per-ceived improvement. In the absenceof more definitive benefit/risk data,as is often the case, treatment thenbecomes directed toward improvingthe surrogate. In some instances,the perception of benefit becomesso pervasive and opinions so solid-
ified that few are willing to put asidetheir preconceived notions to par-ticipate in clinical research. Indeed,clinical guidelines are of necessitydeveloped on the best availablecurrent data, which are admittedlynondefinitive. Most guidelines pro-vide a level of evidence for each ofthe recommendations with the high-est being multiple clinical outcomeRCTs.43 Unfortunately, this level isrelatively infrequent and most rec-ommendations are based on lessfirm data. However, it is importantto note that guidelines are not in-tended to stifle research, but ratherto identify areas where data neededfor rational clinical decision mak-ing are lacking or inadequate. Theseguidelines could be used to indi-cate areas of uncertainty where re-search should be encouraged. Whenasking an individual whether or notthey wish to voluntarily participatein clinical research, the informedconsent process delineates the gapsin our knowledge, the precise ques-tion being addressed, and the po-tential risks of the intervention be-ing examined. Patients consentingto participate in RCTs understandthe uncertainty and choose to bepart of a process that generates in-formation for future clinical deci-sion-making.
Anemia treatment with erythropoie-sis-stimulating agents (ESAs) forpatients with chronic kidney disease(CKD) who do not require dialysisrepresents an area where extrapola-tions of data from other popula-tions (dialysis and more severe ane-mia) and assumptions based onepidemiology were for many yearsthe best available data. Small RCTsshowing that ESAs were effectivein raising hemoglobin provided thebasis for their use in moderatelyanemic CKD patients not requiringdialysis. Practice patterns and guide-lines made recommendations forESA use,44 which for some signified
that the question had a robust an-swer. Many felt that placebo-con-trolled trials with ESAs in thesepatients were unnecessary and forsome even unethical. In the absenceof RCT with clinical end points,physicians were basing therapeuticdecisions predominantly on con-sensus opinions.
When clinical outcome RCTs wereeventually undertaken in this field,the assumption of benefit was soprofound that the trials did not testagainst placebo. Instead, strategiescomparing different hemoglobintargets were the objectives of twomajor RCTs that assessed clinicaloutcomes. Correction of Hemoglo-bin and Outcomes In Renal insuffi-ciency (CHOIR) found that a strate-gy to use epoetin alfa to a target of13.5 g/dL resulted in more patientsexperiencing the composite endpoint of death, myocardial infarc-tion, heart failure, or stroke relativeto those randomized to a target of11.3 g/dL.45 They reported moredeaths and heart failure events inthose randomized to the higher he-moglobin target. If true, this wouldindicate a very narrow therapeuticrange for epoetin alfa with a pre-sumed benefit of therapy in thelow arm (achieved hemoglobin of11.3 g/dL), while causing deathsand heart failure in the higher arm(achieved hemoglobin of 12.6 g/dL).The Cardiovascular Risk reductionby Early Anemia Treatment withEpoetin beta (CREATE) also pre-sumed benefits and used two activearms to different targets. This small-er trial also showed numericallymore first cardiovascular events inthose randomized to higher hemo-globin levels.46
Trial to Reduce cardiovascular Eventswith Aranesp Therapy (TREAT) wasbeing conducted before the resultsof CHOIR and CREATE. TREAT wasdesigned as a placebo-controlled
136
Dialogues in Cardiovascular Medicine - Vol 15 . No. 2 . 2010
Can there be any surrogate for safety? - Pfeffer and Skali
137
Dialogues in Cardiovascular Medicine - Vol 15 . No. 2 . 2010
Can there be any surrogate for safety? - Pfeffer and Skali
blinded study to ascertain whetherthe composite cardiovascular mor-bidity and mortality outcome aswell as a composite of death anddialysis outcome would be alteredby treating diabetics with anemia(hemoglobin �11 g/dL).47 At thestart of TREAT, many questionedthe ethics of having a placebo groupeven with ESA rescue should he-moglobin falls below 9 g/dL. Withthe results of CHOIR and CREATE,the concern shifted to the activetherapy arm.48 However, as thelargest of the RCTs, TREAT had ac-cumulated more clinical outcomesdata than all other trials combinedand provided the most reliablerisk-benefit assessment. Despiteachieving hemoglobin separation,no differences in the primary car-diovascular or renal composite out-comes were observed. The largerplacebo-controlled experience didnot show a hazard for death or heartfailure, but did show a doubling inthe rates of stroke compared withplacebo. The prespecified test forassessment of fatigue showed rathermodest benefits compared withplacebo, providing the physicianand patients with critical risk-ben-efit data needed to make informeddecisions. For most patients withmoderate anemia not undergoingdialysis, the increased risk for strokeuncovered in this placebo-con-trolled trial will outweigh the po-tential benefits. In the absence ofplacebo-controlled trials, the fieldwould have been led by the falsesense of security of comparing twodoses of the active therapy, whichby design can only lead to a recom-mendation for using the therapy.49
This is an example of where use ofa surrogate to drive clinical prac-tice was so entrenched as to stifleappropriate placebo-controlled clin-ical outcomes testing RCTs. TheTREAT experience also makes thepoint that definitive trials shouldbe encouraged rather than second-
guessed. Once trials are under way,their Data Safety Monitoring Com-mittee is entrusted with decisionsaffecting patients in the trial as wellas future patients by striving forthe robust answer to the tested hy-pothesis.
CONCLUSION
The practice of medicine remains acombination of both art and science.Clinical decisions regarding an in-dividual patient always require ex-trapolations of best available data.It is unreasonable to expect thatmajor RCTs with clinical outcomesand safety data will be available forall clinical decisions.50 Favorablemodifications in a surrogate markerof disease progression need to beviewed in the context of uncertaintyfor association with clinical out-comes and for certainty in the inad-equacy of safety information. Whenthe surrogate is the best availableinformation to guide clinical deci-sions, its limitations should be un-derstood and further research en-couraged.51
REFERENCES
1. Medical Research Council.
Treatment of pulmonary tuberculosis withstreptomycin and para-aminosalicylic acid;a Medical Research Council investigation.
Br Med J. 1950;2:1073-1085.
2. VA Cooperative Study Group.
Effects of treatment on morbidity of hyper-tension.
JAMA. 1967; 202:1028-1033.
3. Kannel WB, Dawber TR, Kagan A,Revotskie N, Stokes JI.
Factors of risk in the development of coronaryheart disease--six year follow-up experience.The Framingham Study.
Effects of ACE inhibitors, calcium antagonistsand other blood pressure lowering drugs:results of prospectively designed overviewsof randomised trials.
Lancet. 2000;355:1955-1964.
5. Anand IS, Florea VG.
End points in chronic heart failure clinicaltrials.
Dialogues Cardiovasc Med. 2010;15:81-101.
6. McMurray JJ.
Clinical practice. Systolic heart failure.
N Engl J Med. 2010;362:228-238.
7. Skali H, Pfeffer MA, Lubsen J,Solomon SD.
Variable impact of combining fatal andnonfatal end points in heart failure trials.
Circulation. 2006;114:2298-3303.
8. Braunwald E, Cannon CP, McCabe CH.
Use of composite end- points in thromboly-sis trials of acute myocardial infarction.
Am J Cardiol. 1993;72:3G-12G.
9. Lewis EF, Velazquez EJ, SolomonSD, et al.
Predictors of the first heart failure hospital-ization in patients who are stable survivorsof myocardial infarction complicated bypulmonary congestion and/or left ventricu-lar dysfunction: a VALIANT study.
Eur Heart J. 2008;29:748-756.
10. Yusuf S, Negassa A.
Choice of clinical outcomes in randomizedtrials of heart failure therapies: disease-spe-cific or overall outcomes?
Am Heart J. 2002;143:22-28.
11. Packer M.
Proposal for a new clinical end point toevaluate the efficacy of drugs and devicesin the treatment of chronic heart failure.
J Card Fail. 2001;2:176-182.
12. Cleland JG.
How to assess new treatments for the man-agement of heart failure: composite scoringsystems to assess the patients’ clinical journey.
Eur J Heart Fail. 2002;4:243-247.
13. Fleming TR, DeMets DL.
Surrogate end points in clinical trials: arewe being misled?
Ann Intern Med. 1996;125:605-613.
14. Gheorghiade M, Adams KF Jr,Gattis WA, Teerlink JR, Orlandi C,O'Connor CM.
Surrogate end points in heart failure trials.
Am Heart J. 2003;145(2 suppl):S67-S70.
15. Levey AS, Cattran D, Friedman A,et al.
Proteinuria as a surrogate outcome in CKD:report of a scientific workshop sponsored bythe National Kidney Foundation and theUS Food and Drug Administration.
Effect of antiarrhythmic agent moricizineon survival after myocardial infarction: theCardiac Arrhythmia Suppression Trial–II.
N Engl J Med. 1992;327:227-233.
19. Packer M, Carver JR, RodehefferRJ, et al; PROMISE Study ResearchGroup.
Effect of oral milrinone on mortality in severe chronic heart failure.
N Engl J Med. 1991;325:1468-1475.
20. Packer M, Narahara KA,Elkayam U, et al.
Double-blind, placebo-controlled study ofthe efficacy of flosequinan in patients withchronic heart failure. Principal Investigatorsof the REFLECT Study.
J Am Coll Cardiol. 1993;22:65-72.
21. Cohn JN, Goldstein SO,Greenberg BH, et al; VesnarinoneTrial Investigators.
A dose-dependent increase in mortality withvesnarinone among patients with severeheart failure.
N Engl J Med. 1998;339:1810-1816.
22. Cushman WC, Evans GV, ByingtonR, et al; ACCORD Study Group.
Effects of intensive blood-pressure control intype 2 diabetes mellitus.
N Engl J Med. 2010 March 14 [Epubahead of print].
23. Wright JT Jr, Bakris G, Greene T,et al.
Effect of blood pressure lowering and anti-hypertensive drug class on progression ofhypertensive kidney disease: results from theAASK trial.
JAMA. 2002;288:2421-2431.
24. Alderman MH.
Hypertension control and kidney disease:some questions answered, many remain.
JAMA. 2002;288:2466-2467.
25. Brenner BM, Cooper ME, de Zeeuw D, et al.
Effects of losartan on renal and cardiovascu-lar outcomes in patients with type 2 diabetesand nephropathy.
N Engl J Med. 2001;345:861-869.
26. Lewis EJ, Hunsicker LG, Clarke WR, et al.
Renoprotective effect of the angiotensin-receptor antagonist irbesartan in patientswith nephropathy due to type 2 diabetes.
N Engl J Med. 2001;345:851–860.
27. Kunz R, Friedrich C, Wolbers M,Mann JF.
Meta-analysis: effect of monotherapy andcombination therapy with inhibitors of therenin angiotensin system on proteinuria inrenal disease.
Ann Intern Med. 2008;148:30-48.
28. Pfeffer MA, McMurray JJ,Velazquez EJ, et al.
Valsartan, captopril, or both in myocardialinfarction complicated by heart failure, leftventricular dysfunction, or both.
N Engl J Med. 2003;349:1893-1906.
29. Yusuf S, Teo KK, Pogue J, et al.
Telmisartan, ramipril, or both in patients athigh risk for vascular events.
N Engl J Med. 2008;358:1547-1559.
30. Mann JF, Schmieder RE,McQueen M, et al.
Renal outcomes with telmisartan, ramipril,or both, in people at high vascular risk (theONTARGET study): a multicentre, ran-domised, double-blind, controlled trial.
Lancet. 2008;372:547-553.
31. Parving HH, Brenner BM,McMurray JJ, et al.
Dual renin-angiotensin system blockadeand kidney disease.
J Am Coll Cardiol. 2009;54:278-279.
32. Wilson PW, D'Agostino RB, Levy D, Belanger AM, Silbershatz H,Kannel WB.
Prediction of coronary heart disease usingrisk factor categories.
Circulation. 1998;97:1837-1847.
33. Scandinavian SimvastatinSurvival Study (4S) Investigators.
Randomised trial of cholesterol lowering in4444 patients with coronary heart disease:the Scandinavian Simvastatin SurvivalStudy (4S).
Efficacy and safety of cholesterol-loweringtreatment: prospective meta-analysis ofdata from 90,056 participants in 14 ran-domised trials of statins.
Lancet. 2005;366:1267-1278.
35. Tavazzi L, Maggioni AP,Marchioli R, et al.
Effect of rosuvastatin in patients withchronic heart failure (the GISSI-HF trial):a randomised, double-blind, placebo-con-trolled trial.
Lancet. 2008;372:1231-1239.
36. Kjekshus J, Apetrei E, Barrios V,et al.
Rosuvastatin in older patients with systolicheart failure.
N Engl J Med. 2007;357:2248-2261.
138
Dialogues in Cardiovascular Medicine - Vol 15 . No. 2 . 2010
Can there be any surrogate for safety? - Pfeffer and Skali
37. Ridker PM, Danielson E,Fonseca FA, et al.
Rosuvastatin to prevent vascular events in menand women with elevated C-reactive protein.
N Engl J Med. 2008;359:2195-2207.
38. Wanner C, Krane V, März W, et al.
Atorvastatin in patients with type 2 diabetesmellitus undergoing hemodialysis.
N Engl J Med. 2005;353:238-248. Erratumin: N Engl J Med. 2005;353:1640.
39. Fellström BC, Jardine AG,Schmieder RE, et al.
Rosuvastatin and cardiovascular events inpatients undergoing hemodialysis.
N Engl J Med. 2009;360:1395-1407.
40. Hilts PJ.
Drug’s Problems Raise Questions on Warnings.
New York Times. August 21, 2001. Page F2,accessed March 4, 2010.
41. Gordon DJ, Probstfield JL,Garrison RJ, et al.
High-density lipoprotein cholesterol andcardiovascular disease: four prospectiveAmerican studies.
Circulation. 1989;79:8-15.
42. Barter PJ, Caulfield M, Eriksson M,et al.
Effects of torcetrapib in patients at highrisk for coronary events.
N Engl J Med. 2007;357:2109-2122.
43. Kushner FG, Hand M, Smith SC Jr,et al.
2009 Focused updates: ACC/AHA guidelinesfor the management of patients with ST-ele-vation myocardial infarction (updating the2004 guideline and 2007 focused update)and ACC/AHA/SCAI guidelines on percuta-neous coronary intervention (updating the2005 guideline and 2007 focused update) areport of the American College of CardiologyFoundation/American Heart AssociationTask Force on Practice Guidelines.
J Am Coll Cardiol. 2009;54:2205-2241.
44. Locatelli F, Covic A, Eckardt KU,Wiecek A, Vanholder R.
Anaemia management in patients withchronic kidney disease: a position statementby the Anaemia Working Group of EuropeanRenal Best Practice (ERBP).
Nephrol Dial Transplant. 2009;24:348-354.
45. Singh AK, Szczech L, Tang KL, et al.
Correction of anemia with epoetin alfa inchronic kidney disease.
N Engl J Med. 2006;355:2085-2098.
46. Drueke TB, Locatelli F, Clyne N,et al.
Normalization of hemoglobin level in patientswith chronic kidney disease and anemia.
N Engl J Med. 2006;355:2071-2084.
47. Pfeffer MA, Burdmann EA, Chen CY, et al.
A trial of darbepoetin alfa in type 2 diabetesand chronic kidney disease.
N Engl J Med. 2009;361:2019-2032.
48. Pfeffer MA.
An ongoing study of anemia correction inchronic kidney disease.
N Engl J Med. 2007;356:959-961.
49. Pfeffer MA.
Critical missing data on erythropoiesis-stimulating agents in CKD: first beat placebo.
Am J Kidney Dis. 2008;51:366-369.
50. Smith GC, Pell JP.
Parachute use to prevent death and majortrauma related to gravitational challenge:systematic review of randomised controlledtrials.
BMJ. 2003;327:1459-1461.
51. Shaughnessy AF, Slawson DC.
POEMs: patient-oriented evidence thatmatters.
Ann Intern Med. 1997;126:667.
139
Dialogues in Cardiovascular Medicine - Vol 15 . No. 2 . 2010
Can there be any surrogate for safety? - Pfeffer and Skali
Dialogues in Cardiovascular Medicine - Vol 15 . No. 2 . 2010
141
EARLY ATTEMPTS TO UN-DERSTAND HOW ATRIALDISTENSION INDUCES
CHANGES IN HEART RATEAND RENAL DIURESIS
nderstanding the physiologi-cal control of cardiovascularhomeostasis has engagedthe curiosity of scientists for
centuries. The story of the discoveryof atrial natriuretic peptide is a casein point (Figure 1). An aspect of par-ticular interest has been the possiblemechanisms involved in the modula-tion of cardiac function in relation tochanges in pressure and volume inthe cardiac atria.
Bainbridge1 was specifically interestedin the reflex control of the changes inheart rate induced by large changesin venous inflow, which he termed“venous plethora,” or similar changesin heart rate induced by relativelysmall changes in atrial pressure, asopposed to changes in volume. Basedon extensive canine experiments heconcluded that:
the reflex acceleration of the rate of the
heart which takes place when the venous
inflow is increased in the normal animal
is caused by impulses arising within the
heart and the effect of the stimulus is an
adequate rise in venous pressure.
The neural basis for these reflexchanges in heart rate was identified byNonindez in 19372 and described ingreater detail by Paintal (19533; 19794).He identified four types of sensory re-ceptors present in both left and rightatria classifying them as: (i) Type A;(ii) Type B with medullated fibers;(iii) endings with vagal nonmedullatedfibers; and (iv) endings with fibersrunning along sympathetic nerves.These neural receptors are found atthe junction between the veins withthe atrium. The Type B nerve endingsrespond preferentially to stretch.Numerous experiments in the 1950sand 60s were based on intravenous
infusions and perfusions, attemptingto determine the reflex pathways ofboth the right and left atria. There wasa wide range of often conflicting obser-vations made using this experimentaldesign. In order to overcome theseconfounding factors, Linden’s Groupin the Department of Physiology atthe University of Leeds developed thenovel technique of inflating small(2- to 3-mm long) balloons carefullyplaced at the junction of the pulmonaryvein and left atrium. A more complexballoon catheter was devised by Kappa-goda et al5 in order to distend the su-perior vena-caval right atrial junction.A series of studies performed between1958 and 1979 using the anesthetizedcanine preparation clarified the roleof the autonomic sensory and efferentnerves in mediating cardiac rate andcontractile responses to changes inatrial pressure.6,7
In addition to changes in cardiac func-tion induced by increases in atrial pres-sure (or, more specifically, induced byincreases in atrial stretch), other ob-servations were made, which suggestedthat a rise in atrial pressure could actas “volume receptors,” based on theobservation that such a rise couldalso regulate extracellular fluid vol-ume by increasing urine flow (Henryet al, 19568). Several techniques wereused to study the mechanisms thatmight be involved in regulating extra-cellular fluid volume, including nega-tive pressure breathing, immersionin water, as well as distension of theatrium and stretching localized areas
U
Trails of DiscoveryPhysiology, histology, and biochemistry: the long path leading
to the discovery of the atrial natriuretic peptidesNicola Fitzgerald; J. Desmond Fitzgerald, BSc, FRCP, FFPM
Materia Medica - Mere, Nr Knutsford - UK
Figure 1. Structure of humannatriuretic peptide receptor-c complexed
with atrial natriuretic peptide.
Keywords: atrial natriuretic peptide; physiology;histology; heart failure; cardiac function;nesiritide; drug discoveryAddress for correspondence:Dr J. Desmond Fitzgerald, Materia Medica,Mere Croft, Chester Road, Mere, Nr Knutsford,Cheshire WA16 6LG, UK(e-mail: [email protected])
of the atrium. Henry et al,8 working atthe Wright-Patterson air force base inthe USA, showed that distension of aballoon in the canine atrium caused a5- to 7-fold increase in urine flow thatwaned after 30 minutes (Figure 2).Henry and Pearse9 attributed the di-uretic effects to activation of left atrialstretch receptors via vagal nerves. How-ever, when bilaterally vagotomizednonhuman primates are exposed towater immersion, there is still amarked rise in both urine volume andsodium excretion. Furthermore, thisimmersion response is not mediatedby changes in the circulating level ofmineralocorticoids or antidiuretichormone (ADH).
The mechanism(s) possibly involvedin the renal diuretic response to in-creased atrial pressure were attributedeither to neural10 or humoral11 factors.The typical experiment, in lightly anes-thetized dogs, pioneered by Linden’sGroup in Leeds, involved raising leftatrial pressure by about 1.3 kPA, whichcaused a significant rise in urine flowand sodium excretion 10 minutes afteratrial distension. Relief of the raised
atrial pressure was followed 10 min-utes later by a reduction in the in-creased urine flow, which returned tobaseline levels after 30 to 40 minutes.Initially, the Linden Group was con-vinced that the renal response was dueto neurally mediated mechanismstriggered by atrial receptor activationof myelinated vagal fibers.5 However,denervation of one kidney or auto-nomic blockade with hexamethoniumdid not block the diuretic response toraised atrial pressure, so the possibil-ity of a humorally mediated response
was also considered.11-13 A favoritecandidate was ADH. The definitive ex-periment to test this hypothesis wasto study the renal response to raisedinteratrial pressure after destructionof the pituitary gland. In this prepara-tion, left atrial distension still caused arenal diuresis. Kappagoda, in Linden’sGroup, in attempting to identify a hu-moral mediator, examined the effectsof serum extracts taken during renaldiuresis in anesthetized dogs using asa bioassay the Malpighian tubule ofRhodnius prolixus (Figure 3).14,15 Inhis 1979 paper summarizing the resultsof his work, Kappagoda16 stated “theefferent path of this reflex is indu-bitably humoral in nature and its iden-tity remains controversial.”
HISTOLOGICALSTUDIES ON ATRIA
While the physiological studies brieflysummarized above were being per-formed between 1950 and 1980, otherscientific groups were examining thefine structure of atria, the myocardium,and the conduction system.16-19 Thesehistological studies were pursued com-
Dialogues in Cardiovascular Medicine - Vol 15 . No. 2 . 2010
The discovery of the atrial natriuretic peptides - Fitzgerald and Fitzgerald
pletely independently of the physio-logical studies. The atrial endocardi-um has a subendocardial plexus com-posed of varying sizes of nerve trunkscomprising possibly end-nets andunencapsulated nerve endings, whichvary according to species and stainingtechnique. The plexus comprises fibersoriginating from both sympathetic andvagal nerves. Ultrastructural studies inpig atria reveal a range of organellesassociated with these nerve fibers in-cluding glycogen granules, Golgi-likeend-organs, and mitochondria-filledvesicles.18 Somewhat surprisingly, theauthors of related histological studiesquoted in these papers to which ref-erence is made failed to mention theexistence of large numbers of mem-brane-bound specific granules whichwere initially thought to contain lipo-fuscin and lysosomal granules or cat-echolamines. Their characterization,based solely on histological studies,is entirely dependent on the stainingtechniques utilized, which may explainwhy they were overlooked.
In 1964, Jamieson and Palade19 pub-lished a careful analysis of “specificgranules in atrial muscle cells” in whichfirstly they emphasized that up untilthat period the atrial mammalian my-ocardium had been much less exten-sively studied than the ventricularmyocardium. They described large pop-ulations of spherical electron-opaquegranules (0.3-0.4 µ) found in musclefibers of mammalian atria (Figure 4).
These granules are absent from theventricle. They noticed that the gran-ules were intimately connected withthe Golgi complex, and carefully ana-lyzed the tissue so as to determinewhether the granules contained cate-cholamine or other material. Severalprevious publications had claimedthat the granules contained catechol-amines, but Jamieson and Palade sum-marized the evidence against this as:(i) ventricular muscle contains cate-cholamines, but its cells have no gran-
ules; (ii) chromaffin reaction fails todemonstrate catecholamines in atrialgranules; (iii) autoradiographic stud-ies show no uptake of H3-dopamine;and (iv) do not contain 5-hydroxytrip-tamine. They emphasized that isola-tion of the content of the granuleswould be difficult because they are
only a small fraction of the total atrialtissue and concluded their paper stat-ing “in this case, a highly differentiatedcontractile cell appears to possess asecond specialized function in its abil-ity to form and store a population ofgranules, presumably secretory, innature.”
In 1971, Berger and Bencosme20 pub-lished a study demonstrating that theatrial granules do not respond to spe-cific staining for epinephrine and nor-epinephrine that was simultaneously
demonstrable in the rat adrenal medul-la. They also published a lengthy re-view in the same year describing indetail all the studies that had beendone previously to identify specificgranules in both mammalian and non-mammalian vertebrate cardiocytes.21
They concluded their critical reviewby emphasizing that despite intensivestudy the “secretory nature” of specif-ic granules had not yet been demon-strated. They concluded somewhatprophetically that:
should the actual secretory function of these
granules become established, not only
would the present concepts of myocardial
function need revision, but the regulation
of function in more distant organs may be
involved.
Thus, by 1979, the physiologists werespeculating that pressure changesin the atria lead to changes in renalfunction, either by neural or humoralmechanisms. Attempts to identify acirculating mediator modulating renalfunction were unsuccessful. The can-didate humoral factors triggered byraised atrial pressure included: (i) de-creased secretion of AVP22; (ii) inhibi-tion of the RAS; (iii) increased secretionof catecholamines and or dopamine;(iv) an ouabain-like factor secreted bythe pituitary; and (v) ADH.23,24
On the other hand, histological studiesperformed in Departments of Pathol-ogy measuring the granularity of theatria showed that their content changedfollowing differing experimental inter-ventions in rats, such as bilateraladrenalectomy or infusion of sodiumchloride.25 De Bold and Bencosme,having developed a markedly improvedmorphometric histochemical tech-nique,22,26-28 permitted for the firsttime the accurate quantification ofchanges in atrial granularity. In 1979,using this new morphometric tech-nique, de Bold (Figure 5, page 144)published a sole author paper25 show-ing that sodium and water deprivationsignificantly increased atrial granulari-
Dialogues in Cardiovascular Medicine - Vol 15 . No. 2 . 2010
The discovery of the atrial natriuretic peptides - Fitzgerald and Fitzgerald
143
Figure 4. Specific granules in atrialmuscle cells.Longitudinally sectioned atrial muscle cell fromthe anterior wall of rat right atrium. The sarco-plasmic core contains a population of sphericalgranules (ag), profiles of the Golgi complex (G),numerous mitochondria, an occasional residualbody (rb), and a few lipid droplets (l). Sarco-plasmic layers between myofibrils contain anoccasional granule.
ty. This change correlated with increas-es in blood hematocrit values and hispaper concluded:
the findings point to a relationship between
atrial-specific granules and the regulation of
water-electrolyte balance in these situations.
DISCOVERY OF ANP (ATRIAL NATRIURETIC
POLYPEPTIDE)
It is clear that by 1979/80 several sep-arate scientific investigators were pursuing the identity of a circulatingfactor, which mediated changes inurinary sodium and volume, triggeredby changes in atrial tension or volume.Given the key role of sodium balancein blood pressure control, the identi-fication of a new mediator of bloodvolume control in addition to the es-tablished importance of angiotensinand aldosterone assumed considerableimportance.29
In contrast to the complex experimen-tal physiological studies on cardiorenaldiuretic mechanisms, several groupsused increasingly sophisticated histo-chemical methods to determine thenature of the atrial granules and quan-tified changes in different rat models.It was shown by Cantin’s group at theClinical Research Institute, Montreal,
that the atrial granules contained pro-teins.30 At the nearby Queen’s Univer-sity Department of Pathology, Ben-cosme, who had been studying theseatrial granules for the past 10 years,recruited Dr de Bold from Argentina to
work on the histology of atrial gran-ules for his PhD thesis in 1968. Simi-larly, Hatt’s group working in Parisshowed in 1976 that changes in theoral intake of sodium and water in ratswere associated with an increase inatrial granularity after 5 weeks of re-stricted sodium intake, while treatmentwith DOCA and increased sodium inthe diet caused a significant reductionin atrial granules.31
Assessing quantitative changes repro-ducibly in atrial granule content byhistological measurements was diffi-cult. Results depended on the age ofthe rats, the specificity of the histo-chemical stains, as well as the tech-nique for embedding and sectioningthe tissue. These variables were over-come by de Bold, who utilised a com-bination of embedding, microtome,and highly specific lead-hematoxylin-tartrazine staining methods. In addi-tion, sophisticated statistical softwarewas used in order to ensure precisionof the measurements by a light micro-
scopic technique. Having improvedthe precision of measurement, de Boldthen performed a complex series ofexperiments applying the improvedmorphological methodology to deter-mine the effects on atrial granularityof water deprivation, DOCA injection(1-2.4 mg/kg), and bilateral adrenalec-tomy. Serial measurements of atrialgranularity combined with simultane-ous measurement of blood hematocritmeasurements showed a significantcorrelation between changes in hema-tocrit and the degree of atrial granu-larity in water-deprived rat experiments,ie, hypergranularity is accompaniedby high hematocrit values.28
It would seem that by 1979 the scenewas set for the characterization of thenature of atrial granules and the rela-tionship between granule changes andcardiorenal function. As with so manypivotal scientific discoveries, the an-swer depended on a seemingly obviousexperiment. In this case, de Bold, incollaboration with Sonnenberg’s groupin Toronto, studied the effects of atrialand ventricular homogenates on renalfunction in anesthetized rats.32,33 Theextract was prepared by homogenizingrat atria in phosphate buffer salineand storing the supernatant at –70ºC.Aliquots (2 mL) of either atrial or ven-tricular supernatant were administeredintravenously to anesthetized rats.The atrial extract induced a 30-foldincrease in urinary sodium and chlo-ride excretion, accompanied by a 10-fold rise in urine volume (Table I).33
Extracts of the rat ventricle did not af-fect urine function. The simplicity andclarity of the result is reminiscent ofBayliss and Starling’s single experimentin which they scraped mucosal mem-brane from the duodenum, ground itup with sand and 0.4% HCl, and in-jected the filtrate, which resulted in an increase in pancreatic secretion.34
The atrial extract experiment was first published as an abstract in 1980followed by a more detailed paper in 1981.33
144
Dialogues in Cardiovascular Medicine - Vol 15 . No. 2 . 2010
The discovery of the atrial natriuretic peptides - Fitzgerald and Fitzgerald
Predictably, the race was now on toidentify the nature of the active prin-ciple in the atrial supernatant. A re-view of the literature on ANP followingthese two publications on the renaleffects of atrial extracts reveals a veri-table explosion of scientific interest.This is exemplified in Figure 6, whichdepicts the number of papers pub-lished on ANP between 1980 and 1990.These data may not be strictly accu-rate since different investigators useddifferent terminologies in order to de-scribe the structure and function ofthe peptides isolated from the atria.De Bold, who identified a sequence ofANP in 1983, called it cardionatrin I,35
while Cantin’s group in the nearbyMontreal Institute describe their peptides as ANF-H1(73aa) and car-diodilatin (LEU94-ARG109:106aa).36
Laragh’s group in New York calledatrial natriuretic factor auriculin,37
while the Japanese group of Inagamiat Vanderbilt University, having isolat-ed peptides from the human material,called it atrial natriuretic factor.38 Incontrast, Needleman’s group at theDepartment of Pharmacology of theWashington University in St Louis,Mo, refer to it as atriopeptin.39 Thisapparent confusion in terminology
has been described primarily to illus-trate the nature of the intense, andprobably competitive, scientific inter-est triggered by de Bold’s initial ex-periments.
In concert with the literature expan-sion, there was also an increase inpatent activity relating to structurefunction aspects of ANP. A brief surveyof the worldwide database for patents,entitled “atrial peptide,” identified atleast 30 patents up to 1990. de Bold’sgroup was the first to apply for a pa-tent for the circulating form of ANF, in1983. Patents were granted to PhilipNeedleman in 1985, while a patentsubmitted by the University of Kingstonon de Bold’s behalf was dated 1987.
In the period up to 1990, eight com-mercial companies issued patents onanalogs of atrial peptides, includingMerck. Presumably, the basis for thiscommercial interest was the emergingviews on the possible role of ANP invascular homeostasis, especially es-sential hypertension. The basis for thisview was that ANP had been shown, by1986, to inhibit the renin-angiotensinsystem, reduce plasma aldosteronelevels, as well as having arterial andvenodilator actions.40
An early study of ANP in human volun-teers from Espiner’s group in Auck-land, New Zealand, showed that abolus injection (100 µg) of syntheticANP increased urinary sodium and
145
Dialogues in Cardiovascular Medicine - Vol 15 . No. 2 . 2010
The discovery of the atrial natriuretic peptides - Fitzgerald and Fitzgerald
1200
1000
800
600
400
200
0
Year
Articles
Nu
mb
er
of
art
icle
s
1980 1981(1)
1982(3)
1983(10)
1984 1985 1986 1987 1988 1989 1990
◆
Figure 6.Number of arti-cles on atrialnatriuretic pep-tides per yearbetween 1980and 1990.Data kindly sup-plied by Ms T Hore,Library, RoyalSociety of Medicine,London (2009).
Table I. Urinary re-sponse to intravenousinjection of atrial myocardial extract inrats (compared withventricular myocardialextracts and vehicle).Average urinary data before, during and afterinjection as well as at theend of the experiment.Group A: atrial extract(2 mL); Group B: ven-tricular extract; Group C:vehicle.
µµL/min nEq/min nEq/min nEq/ming kidney wt g kidney wt g kidney wt g kidney wt
Before A 6.52±1.45 334±97 908±213 912±139
(40-60 min) B 5.02±1.07 379±105 1011±190 902±138
C 5.02±1.07 182±57 444±93 531±103
During A 47.04±5.64**†† 7235±689**†† 8769±798**†† 2145±177**†
(60-80 min) B 12.17±2.80 1191±301 2032±355 1208±138
C 15.08±4.03 573±199 1285±310 1170±227
End A 7.25±1.51 1135±190 1581±289 713±74
(160-180 min) B 6.55±1.57 1038±196 1340±330 693±86
C 11.19±1.51 1338±188 2068±286 859±124
*,** Statistically significant differences from group B (P<0.01, P<0.001)†,†† Statistically significant differences from group C (P<0.01, P<0.001)
V, urine volume; UCl V, total chloride excretion; UK V, total potassium excretion; UNa V, total sodium excretion
. . . .
. . . .
volume within 30 minutes of adminis-tration. Curiously, the peptide was ob-tained from BACHEM, a US chemicalcompany (αANP28aa).41 As such, thecompound would not have been sub-ject to routine toxicological evaluation.Subsequently, the same investigatorsreported on the effects in congestiveheart failure of a synthetic analog(1LEU-ANP:L364,343) supplied byMerck. The compound reduced arteri-al pressure and increased cardiac out-put, but there was no significant in-crease in urine volume or sodium.42
Subsequently, synthetic compoundswere developed that either inhibitedthe breakdown of ANP by endogenous
endopeptidases, or selectively blockedANP receptors.43 These approaches to modulating endogenous ANP didnot lead to therapeutic products. Thesubsequent sophistication of the na-triuretic system is summarized inTable II. It shows the impressive andrapid expansion of scientific knowl-edge arising from de Bold’s atrial ex-tract experiment over the past 30 years.It is yet a further example of the piv-otal role of translational medicine inseeking improved therapy. In this in-stance, the main health-related con-tribution appears to be in providingvalidated biomarkers using plasmaBNP levels for the assessment of car-
diovascular risk,44 including outcomesin pulmonary embolism,45 coronaryartery events46 and the managementof acute heart failure.47
The main therapeutic use of ANP hasbeen in the application of recombinantBNP in the management of acute decompensated heart failure. The re-combinant BNP compound (nesiritide)has dual actions (Figure 7). Firstly, itcauses an increase in glomerular fil-tration rate by a direct effect in relax-ing afferent renal arterial vessels andconstriction of efferent arterials. Atthe same time it has potent vasodila-tor actions, reducing systemic bloodpressure by arteriovenous vasodila-tion, and as a consequence reducesglomerular filtration rates. Thus, thenet effect of nesiritide in acute decom-pensated heart failure is critically dose-dependent. Use of high bolus dosesof nesiritide, which can acutely reducesymptoms associated with heart fail-ure, results in an increase in the inci-dence of renal failure and death.48
When nesiritide is used by careful in-fusion, it has a net beneficial effect.49
In order to preserve the desirable renaleffects and eliminate the systemic hy-potension, a novel ANP agonist (CD-NP) is being evaluated in decompen-sated heart failure for its natriureticefficacy and improved safety profile.50
In Japan, ANP is used extensively (ie,carperitide).
REFLECTIONS
This account of the discovery of ANPillustrates how scientific observation,ie, atrial distension-induced renal di-uresis, can focus a single-minded ap-proach to elucidating the physiologicalmechanisms. The physiological ap-proach is epitomized by Linden’steam, who spent more than 10 yearsexamining the possible neural reflexesinvolved in the cardiorenal response.This is epitomized in the Proceedingsof the Leeds symposium held in 1976,which the author attended, on atrial
146
Dialogues in Cardiovascular Medicine - Vol 15 . No. 2 . 2010
The discovery of the atrial natriuretic peptides - Fitzgerald and Fitzgerald
reflexes, in which the emphasis of theparticipants was almost exclusively onneural mechanisms. For some inexpli-cable reason, the literature describingthe presence of noncatecholamineatrial granules was not mentioned atthe meeting, nor in the subsequentlypublished monograph (1979) on atrialreceptors.10 The histological approachinitiated by Palade and maintained by Bencosme21 focused again single-mindedly on understanding atrial gran-ule function, making no reference inthe histological papers to the role ofatrial neural networks.
Another unexplained aspect is thefailure of the Queen’s University re-searchers to collaborate with Cantinand Genest, working in the much larg-er Institute of Clinical Science basedin the University of Montreal. It wouldseem from the sequence of literaturepublications that the initial interest ofBencosme in atrial histology startedbefore he joined the Department ofPathology in Queen’s University, King-ston, Ontario. He recruited de Bold in1968 from the National University ofCordoba, in Argentina, where he hadgraduated in clinical biochemistry, sub-sequently gaining a PhD in pathologyat Queen’s University. Currently he isDirector of the Cardiovascular Endo-crinology Laboratory in the Universityof Ottawa Heart Institute. The key ex-periment showing the natriuretic effectsof rat atrial extracts was performed incollaboration with Sonnenberg, at theDepartment of Physiology, Universityof Toronto. Sonnenberg’s expertise wasin studying renal tubule and medullaryduct mechanisms.51-53 He studied theeffects of blood volume expansion onrenal function in 1971.54 Following thejoint paper with de Bold in 1981, hepublished several further studies onthe renal effects of ANF. The first inter-national symposium on ANF, held inApril, 1985, was co-chaired by Sonnen-berg and not de Bold. Interestingly,Cantin also published on the natureof atrial granules in 197430 showing
their peptidic nature and the effect ofsalt loading in rats on atrial granulari-ty. Thus, Linden’s group and de Boldall suggested that the effect of atrialdistension on renal function was hu-morally mediated, but only de Bolddid the critical proof-of-concept study.Most investigators assumed that thecardiorenal diuretic response, if hu-morally mediated, involved known fac-tors such as renin (Cantin) or ADH(Linden). The possibility of a peptidehormone of atrial origin was clearlyde Bold’s idea. Somewhat inexplicably,Linden published a subsequent paperin 199555 on atrial receptor function,but makes no reference in either paperto de Bold and ANF.
As this article was going to print, thelatest paper on ANP published by deBold was dated March 2010.56
POSTSCRIPT
By coincidence, during a visit of theauthor to Queen’s University in 1976,he met Dr de Bold. De Bold mentionedhis work on atrial granules and gavethe author a paper describing theirnoncatecholamine characteristics.Returning to work in ICI Pharmaceuti-cals Division that year as Biology Di-rector, the author discussed the paperwith Dr Harry Gregory, a notable pep-tide chemist. However, other researchpriorities prevented following thislead further.
The author was also privileged to betutored by Professor Ron Linden in theDepartment of Physiology in Leedsbetween 1970 and 1972, endeavoringto improve his own cardiovascular experimental skills. Professor Lindenwas extremely kind and ran an out-standing cardiovascular physiologygroup. Sadly Professor Linden diedon April 11th, 2010.
Many thanks to Miss Inge Bristow for her expert proofreading and correction
of the manuscript.
REFERENCES
1. Bainbridge FA.
The Influence of venous filling upon therate of the heart.
J Physiol. 1915;50:65-84.
2. Nonindez JF.
Identification of the receptor areas in thevenae cavae and pulmonary veins whichinitiate reflex cardiac acceleration(Bainbridge’s reflex).
Cardiac Receptors. Cambridge, UK:Cambridge University Press; 1979:193-211.
17. Sano T, Mizuhiria V, Matzuda A,eds.
Electrophysiology and ultrastructure of theheart.
New York, NY: Grune and Stratton Inc. 1967.
18. Tranum-Jensen J.
The ultrastructure of the sensory end-organs(baroreceptors) in the atrial endocardium ofyoung mini-pigs.
J Anat. 1975;119: 255-275.
19. Jamieson JD, Palade GE.
Specific granules in atrial muscle cells.
J Cell Biol. 1964;23:151-172.
20. Berger JM, Bencosme SA.
Fine structural cytochemistry of granules inatrial cardiocytes.
J Mol Cell Cardiol. 1971;3:111-120.
21. Bencosme SA, Berger JM.
Specific granules in mammalian and non-mammalian vertebrate cardiocytes. In:Bajusz E, Jasmin G, eds.
Meth Achievm Exp Pathol. 1971;5:173-213.
22. de Bold AJ, Bencosme SA.
Studies on the relationship between the cate-colamine distribution at the atrium and thespecific granules found in atrial muscle cells.II. Studies on the sedimentation pattern ofthe atrial noradrenaline and adrenaline.
Cardiovasc Res. 1973;7:364-369.
23. Ledsome JR, Linden RJ,O’Connor WJ.
The mechanisms by which distension of theleft atrium produces diuresis in anaesthetiseddogs.
J Physiol (Lond). 1961;159:87-92.
24. Clarkson EM, Raw SM, de Wardener HE.
Further observations on a low molecularweight natriuretic substance in the urine ofnormal man.
Kidney Int. 1979;16;710-718.
25. de Bold AJ.
Heart atria granularity effects of changesin water-electrolyte balance.
Proc Soc Exp Biol Med. 1979;161:508-511.
26. de Bold AJ, Bencosme SA.
Selective light microscopic demonstration of the specific granulation of the rat atrialmyocardium by lead-hematoxylin-tartrazine.
Stain Technol. 1975;50:203-205.
27. Solcia E, Capella C, Vassalo G.
Lead-hematoxylin as a stain for endocrinecells.
Histochemie. 1969;20:116-126.
28. de Bold AJ.
Morphometric assessment of granulation inrat atrial cardiocytes: effect of age.
J Mol Cell Cardiol. 1978;10:717-724.
29. Blaustein M.
Sodium ions, calcium ions, blood pressureregulation: a reassessment and a hypothesis.
Am J Physiol. 1977;232:C165-C181.
30. Huet M, Cantin M.
Ultrastructural cytochemistry of atrialmuscle cells. II. Characterisation of theprotein content of specific granules.
Lab Invest. 1974;30:154-163.
31. Marie JP, Guillemot H, Hatt PY.
Le degré de granulation des cardiocytes auri-culaires: étude planimétrique au cours de dif-férents apports d’eau et de sodium chez le rat.
Pathol Biol. (Paris). 1976;24:549-561.
32. Sonnenberg H, Veress AT,Borenstein HB, de Bold AJ.
Rapid and potent natriuretic response tointravenous injection of atrial myocardialextract in rats.
Physiologist. 1980;23:13.
33. de Bold AJ, Borenstein HB,Veress AT, Sonnenberg H.
A rapid and potent natriuretic response tointravenous injection of atrial myocardialextract in rats.
Life Sci. 1981;28:89-94.
34. Bayliss WM, Starling EH.
The mechanism of pancreatic secretion.
J Physiol (Lond). 1902;28:325-353.
35. de Bold AJ, Flynn TG.
Cardionatrin I—A novel heart peptide withpotent diuretic and natriuretic properties.
Life Sci. 1983;33:297-304.
36. Lazure C, Seidah M, Chrétien M,et al.
Atrial pronatriodilatin: a precursor fornatriuretic factor and cardiodilatin.
FEBS Lett. 1984;172:80-86.
37. Maack T, Marion DM, CamargoMJ, et al.
Effects of auriculin (atrial natriureticfactor) on blood pressure, renal function,and the renin aldosterone system in dogs.
Am J Med. 1984;77:1069-1075.
38. Misono KS, Grammer RT,Fukumi H, Inagami T.
Rat atrial natriuretic factor: isolation,structure and biological activities of fourmajor peptides.
Biochem Biophys Res Commun.1984;123:444-459.
39. Needleman P.
Atriopeptin biochemical pharmacology.
Fed Proc. 1986;45:2096-2100.
40. Laragh JH.
Atrial natriuretic hormone, the reninaldosterone axis and blood pressure-electrolyte homeostasis.
N Engl J Med. 1986;313:1330-1340.
148
Dialogues in Cardiovascular Medicine - Vol 15 . No. 2 . 2010
The discovery of the atrial natriuretic peptides - Fitzgerald and Fitzgerald
41. Richards AM, Ikram H, Yandle TG,et al.
Renal, haemodynamic and hormonaleffects of human a atrial natriuretic peptidein healthy volunteers.
Haemodynamic effects of atrial peptideinfusion in heart failure.
Lancet. 1986;2:1242-1245.
43. Jardine AG, Northridge DB,Connell JMC.
Harnessing the therapeutic potential ofatrial natriuretic peptide.
Klin Wochenschr. 1989;67:902-906.
44. Bolliger D, Seeberger MD,Filipovic M.
Pre-operative cardiac risk assessment innon-cardiac surgery: are natriureticpeptides the magic bullet?
J Am Coll Cardiol. 2009;54:1607-1608.
45. Fisher AJ, Corris PA.
Identification of those at risk after acutepulmonary embolism.
Thorax. 2009;64:869-875.
46. Shaw LJ, Polk DM, Kahute TA, et al.
Prognostic accuracy of �-natriuretic peptidemeasurements and coronary artery calciumin asymptomatic subjects (from the EarlyIdentification of Subclinical Atherosclerosisby Noninvasive Imaging Research[EISNER] study).
Am J Cardiol. 2009;104:1245-1250.
47. Stanton E, Hansem M,Wijeysundera HC, et al; PRAISE-IIInvestigators.
A direct comparison of the natriureticpeptides and their relationship to survivalin chronic heart failure of a presumed non-ischaemic origin.
Eur J Heart Fail. 2005;7;557-565.
48. Yancy CW, Crum H, Massie BM,et al.
Safety and efficacy of outpatient nesiritidein patients with advanced heart failure:results of the Second Follow Up SerialInfusions of Nesiritide (FUSION II) Trial.
Circ Heart Fail. 2008;1:9-16.
49. Hernandez AF, O’Connor CM,Starling RC, et al.
Rationale and design of the Acute Study of Clinical Effectiveness of Nesiritide inDecompensated Heart Failure Trial(ASCEND-HF).
Am Heart J. 2009;157:271-277.
50. Lee CY, Chen HH, Lisy O, et al.
Pharmacodynamics of a novel designernatriuretic peptide, CD-NP, in a first-in-human clinical trial in healthy subjects.
J Clin Pharmacol. 2009;49:668-673.
51. Pearse GW, Sonnenberg H.
Effects of spinal section and renaldenervation on the renal response to bloodvolume expansion.
Can J Physiol Pharmacol. 1965;43:211-224.
52. Sonnenberg H Cupples WA, de Bold AJ, Veres AT.
Intrarenal localization of the natriureticeffect of cardiac atrial extract.
Can J Physiol Pharmacol. 1982;60:1149-1152.
53. Sonnenberg H.
Mechanisms of release and renal tubularaction of atrial natriuretic factor.
Fed Proc. 1986;45;2106-2110.
54. Sonnenberg H.
The renal response to blood volumeexpansion in the rat: proximal tubularfunction and urinary excretion.
Can J Physiol Pharmacol. 1971;49:525-535.
55. Linden RJ.
Atrial receptors and heart volumes. In:Kappagoda CT, Kaufman MP, eds.
Control of the Cardiovascular andRespiratory Systems in Health andDisease. New York, NY: Plenum Press;1995:125-134.
56. de Bold ML, Etchepare A,Martinuk A, de Bold AJ.
Cardiac hormones ANF and BNP modulateproliferation in the unidirectional mixedlymphocyte reaction.
J Heart Lung Transplant. 2010;29:323-326.
149
Dialogues in Cardiovascular Medicine - Vol 15 . No. 2 . 2010
The discovery of the atrial natriuretic peptides - Fitzgerald and Fitzgerald
Dialogues in Cardiovascular Medicine - Vol 15 . No. 2 . 2010
Surrogate endpoints in clinical trials: definitionand operational criteria
R. L. Prentice. Stat Med. 1989
1Surrogate end points in heart failureI. S. Anand et al. J Am Coll Cardiol. 2002
6
Role of surrogate end points in the evaluationof drugs for heart failure
R. J. Lipicky and M. Packer. J Am Coll Cardiol. 1993
2
Key issues in end point selection forheart failure trials: composite end points
J. D. Neaton et al. J Card Fail. 2005
8
Summaries of Ten Seminal PapersSpencer H. Kubo, MD
Cardiovascular Division - University of Minnesota - Phillips-Waagenstein Bldg - 516 Delaware Street SEMinneapolis - MN 55455 - USA (e-mail: [email protected])
Dialogues Cardiovasc Med. 2010;15:151-161
4Surrogate end points in clinical trials:
are we being misled?T. R. Fleming and D. L. DeMets. Ann Intern Med. 1996
9Influence of nonfatal hospitalization
for heart failure on subsequent mortalityin patients with chronic heart failure
S. D. Solomon et al. Circulation. 2007
Heart failure as an endpoint in heart failureand non–heart failure cardiovascular clinical trials:
the need for a consensus definitionF. Zannad et al. Eur Heart J. 2008
10
Reliability of ventricular remodelingas a surrogate for use in conjunction with
clinical outcomes in heart failureM. A. Konstam. Am J Cardiol. 2005
7
5
Mode of death in chronic heart failure. A requestand proposition for more accurate classification
R. Narang et al. Eur Heart J. 1996
3
Selection of seminal papers by Inder S. Anand, MD, FRCP,D. Phil (Oxon.), FACC & Viorel G. Florea, MD, PhD, ScD, FACC
University of Minnesota Medical School - VA Medical Center - Cardiology111-C - One Veterans Drive - Minneapolis, MN 55417 - USA
Highlights of the years by Ian Mudway, MDLung Biology - Division of Life Sciences Franklin Williams Building
150 Stamford Street - London SE1 9NN - UK
www.dialogues-cvm.org
Are surrogate markers adequate to assesscardiovascular disease drugs?
R. Temple. JAMA. 1999
Surrogate End Points in Heart Failure Trials:Potentials and Limitations
Soviet nuclear-powered submarine K-278“Komsomolets” sinks in the Barents Sea,
with the loss of 41 lives;legendary Italian film director Sergio Leone dies;
and NATO celebrates its 40th anniversary
1989
152
Dialogues in Cardiovascular Medicine - Vol 15 . No. 2 . 2010
Summaries of Ten Seminal Papers - Kubo
tatistics reign supreme in clinical trials, and thispaper discussing the concept of surrogate endpoints written from the perspective of a statisticianhas particular seminal value. Prentice starts outwith an acknowledgement that there are multiple
levels that motivate interest in surrogates, including thepossible reduction in sample size or trial duration when arare and distal end point is replaced by a more frequentand proximate end point. Further, in some cases, the trueend point measurement may be invasive, uncomfortable,or expensive, so that the surrogate is often preferable froma patient safety viewpoint. Finally, end points that are closein time to the treatment may be more readily interpretedthan more distal end points such as death. Thus, cliniciansoften find it easier to discuss and track the efficacy of treat-ments based on surrogate end points, such as low-densitylipoprotein reduction. In contrast, discussions based ona true end point such as mortality can be very difficultbecause mortality can be confounded by secondary treat-ments or comorbidities.
Prentice then details what should be required for a surro-gate to be a valid and unambiguous end point. He main-tains that it is important to restrict the use of a surrogateto response variables that can substitute for a true responsevariable for certain purposes, and proposes a restrictivecriterion: “Hence, I define a surrogate end point to be aresponse variable for which a test of the null hypothesisof no relationship to the treatment groups under compar-ison is also a valid test of the corresponding null hypoth-esis based on the true end point.”
Prentice notes that this criterion requires the surrogatevariable to capture any relationship between the treatmentand the true end point. He goes on to develop an opera-tional criterion that involves complicated and sophisticatedstatistical techniques that are well beyond the general graspof a clinical cardiologist, but are important “tests” for thevalidity of a proposed surrogate. He cites several examplesthat highlight the intent and the practicality of his opera-tional definition, including a response to the NationalCancer Institute request for proposals to identify surrogate
end points that would allow cancer screening programs tobe evaluated in a more timely fashion than would be pos-sible based on mortality rates. He analyzes the examplesof tumor response and disease recurrence, which are pop-ular surrogates in cancer treatment trials. In the cardio-vascular arena, he cites the possible surrogates for totalcardiovascular mortality, such as ejection fraction in trialsof thrombolytic agents, blood pressure reduction in trialsof antihypertensive drugs, and blood cholesterol levels intrials of cholesterol-lowering drugs. These surrogates satis-fy his operational criterion, as long as the treatments donot differentially affect mortality via pathways that bypassthe proposed surrogate and, secondly, that the mortalityrates do not depend on other treatments.
Prentice concludes with a pessimistic prediction concerningthe potential of the surrogate end point concept becausethe surrogate must have “precisely the same relationshipto the true end point under each of the treatment strategiesbeing compared.” He cites the example of the MultipleRisk Factor Intervention Trial (MRFIT) in which importantdifferences in blood pressure, smoking habits, and bloodcholesterol between the treatment and control patientsdid not lead to the anticipated improvements in coronarymortality.
S
Surrogate endpoints in clinical trials: definition andoperational criteria
R. L. Prentice
Stat Med. 1989;8:431-440
153
Dialogues in Cardiovascular Medicine - Vol 15 . No. 2 . 2010
Summaries of Ten Seminal Papers - Kubo
his paper is a collaborative effort between aleader in the Division of Cardio-Renal DrugProducts in the Center for Drug Evaluation andResearch of the Food and Drug Administrationand a leader in the development and conduct
of many clinical trials. They start with a summary state-ment about drug approval: “If a drug does not lessen symp-toms or prolong life, there would be little support for itsapproval by a regulatory agency and little reason for itsclinical use by physicians.” They focus on two importantquestions, including whether surrogate end points can beused to distinguish effective from ineffective drugs andsecondly, can the use of a surrogate end point provide re-liable information about the effect of a drug on symptomsor survival?
The authors point to the well-known results of the CardiacArrhythmia Suppression Trial (CAST) and of the ProspectiveRandomized Milrinone Survival Evaluation (PROMISE) trial,which “considerably weakened our faith in the reliabilityand validity of surrogate end points in the evaluation ofdrugs for heart failure.” They next examine the relative mer-its of various end points that might be considered clinical-ly relevant in the treatment of heart failure (HF), includingan assessment of symptoms and functional capacity withthe New York Heart Association (NYHA) classification,quality-of-life instruments, and exercise tolerance tests.The discussion on the assessment of survival supports theview also discussed by Temple (see above) that mortalitystudies are seen as necessary for the assessment of safety,rather than evidence of efficacy.
The final section of this paper discusses the basis for theapproval of new drugs in heart failure and looks at an inter-esting hypothetical situation. A developmental programfor a new drug that has characterized the effects on symp-toms and survival could have one of four outcomes. Ap-proval would be likely if the drug relieved symptoms andprolonged life. Conversely, there would be no debate aboutnonapproval if a drug worsened symptoms and reducedlife expectancy. But what would happen in the other twoscenarios in which the effects on symptoms and survival
were discordant? Would a drug be approved if it improvedsymptoms, but also reduced survival? Many researchers inthe field have specifically asked this question of patientsand have been able to quantify this tradeoff. Interestingly,because of the disabling nature of HF symptoms, many pa-tients with advanced HF were indeed very willing to accepta reduced survival if their symptoms could be improved.What about the final scenario in which a drug worsenedsymptoms but improved survival? This type of agent mightbe considered valuable in patients with very mild symptomsand a longer life expectancy, much like the discussionsregarding the use of lipid-lowering drugs and antihyper-tensive agents.
The authors conclude “From these comments, it shouldbe apparent that drugs are approved on the basis of anassessment of their benefits viewed in the context of theirrisks.” In other words, there is no prescribed formula andthe “totality” of the data must be considered. While thiscase-by-case approach seems reasonable and appropriate,it also exposes the opportunity for variability in the inter-pretation and final recommendation by different reviewersand in different time periods.
T
A large-scale battle breaks out between US forcesand local militias in Mogadishu, Somalia;
the United Nations Mission in Haiti is preventedfrom entering the country by the government,triggering resumption of economic sanctions;and American actor River Phoenix dies
of a drug overdose at the age of 23
1993
Role of surrogate end points in the evaluation of drugsfor heart failure
R. J. Lipicky, M. Packer
J Am Coll Cardiol. 1993;22(4 suppl A):179A-184A
nformation on the cause and mode of death is veryimportant to understand the effects of differenttreatments and could have a major bearing on newstrategies to reduce mortality in heart failure. Fur-ther, as the authors point out, the prevention of
sudden death may require a different strategy to the pre-vention of death due to circulatory failure. This statementwas certainly prophetic since, in 2009, we now have thewidespread use and acceptance of implantable cardioverter-defibrillators, which have been shown conclusively to re-duce death due to life-threatening ventricular arrhythmias.
However, data on the cause and mode of death have beenreported sporadically and variably. These investigators per-formed a valuable meta-analysis of 593 studies that re-ported more than 50 deaths due to chronic heart failure.They kept only 27 studies that included patients with treat-ed symptomatic heart failure and reported results usingcategories of the cause of death that could be equated withcommon definitions (the fact that only 27 of 593 studiescould be analyzed emphasizes the sporadic and variablenature of death reporting). The principal finding from thisanalysis was that the proportion of patients dying of pro-gressive heart failure was 43% in studies that only includedpatients with New York Heart Association (NYHA) ClassIII-IV heart failure. In contrast, studies that enrolled fewersick patients (NYHA Class I-II or higher mean left ventric-ular ejection fraction) showed that deaths due to suddendeath and noncardiovascular deaths were more common.
An interesting concept discussed in this paper is the dif-ferentiation between mode of death and cause of death.For example, the mode of death can be classified as suddendeath, which is defined in most studies as either occurringinstantly, or within 15 minutes (or 1 hour) of the onset ofnew symptoms. Within the mode of sudden death, thecause of death could be an arrhythmia, myocardial infarc-tion (MI), electromechanical dissociation, myocardial rup-ture, stroke, pulmonary embolism etc.
The authors also point out that a surprising number ofstudies did not record where the patient died. For exam-
ple, it is implicit that, for patients who die in the hospital,some event anticipated their death and that there shouldbe some objective information to sort out worsening heartfailure, arrhythmias, etc.
Finally, the authors propose a novel classification schemefor death in heart failure, which they term the ACME Sys-tem. Their simple chart asks for information in 4 domains,including: A) Activity and Place of death (hospital, out ofhospital, witnessed); C) Cause of death; M) Mode of death(sudden, circulatory failure, stroke, other cardiovascular,noncardiovascular); and E) Events associated with death(worsening heart failure, preceding chest pain, MI, arrhyth-mia, syncope, or other vascular event. While interesting,it does not appear that this classification scheme enjoyedwidespread adoption among clinical trials. Nonetheless,the authors should be commended for their attempt to en-hance consistency and regularity in the reporting of causeand mode of death.
154
Dialogues in Cardiovascular Medicine - Vol 15 . No. 2 . 2010
Summaries of Ten Seminal Papers - Kubo
Mode of death in chronic heart failure. A request andproposition for more accurate classification
R. Narang, J. G. Cleland, L. Erhardt, S. G. Ball, A. J. Coats, A. J. Cowley, H. J. Dargie,A. S. Hall, J. R. Hampton, P. A. Poole-Wilson
Eur Heart J. 1996;17:1390-1403
I
The Taliban capture Kabul in Afghanistan,driving out President Burhanuddin Rabbani;
Alija Izetbegovic is elected presidentof Bosnia-Herzegovina; and
American musician Bill Monroe,“The Father of Bluegrass,” dies
1996
155
Dialogues in Cardiovascular Medicine - Vol 15 . No. 2 . 2010
Summaries of Ten Seminal Papers - Kubo
ne of the strongest “rebuttals” to the use ofsurrogate end points comes from two bio-statisticians with vast experience in the de-sign and execution of clinical trials. In thisreview, these authors clearly make the point
that pivotal phase 3 trials should only include clinical endpoints and, “…except for rare circumstances in which thevalidity of the surrogate end point has already been rigor-ously established, the primary end point should be the trueclinical outcome.”
This paper makes a strong argument to stay with the “tradi-tional” clinical end points, which the authors define as “aclinical event relevant to the patient, that is, the event ofwhich the patient is aware and wants to avoid. Examplesare death, loss of vision, symptomatic events….and otherevents causing a reduction in quality of life.”
The authors concede that trials with these outcomes oftenhave a long duration and are expensive, and so they aresympathetic to the consideration of surrogate end points.However, for a surrogate end point to be an effective sub-stitute for the clinical outcome, the effects of the interven-tion must reliably predict the overall effect on the clinicaloutcome. It is this requirement that is usually difficult tofulfill and the authors provide examples to illustrate howsurrogate end points have been misleading about the trueclinical effects. In the cardiovascular arena, the authors citethree well-known examples where reliance on a surrogatewas inappropriate. The Cardiac Arrhythmia SuppressionTrial (CAST) showed that antiarrhythmic agents couldeffectively suppress ventricular arrhythmias but lead toan increase in mortality. In heart failure, the ProspectiveRandomized Milrinone Survival Evaluation (PROMISE)and PROspective randomized FlosequInone LongevityEvaluation (PROFILE) trials showed that milrinone andflosequinan both increased exercise tolerance, but werealso associated with an increase in mortality. In the lipid-lowering arena, the Coronary Drug Project showed thatclofibrate and niacin could reduce cholesterol levels, butneither drug reduced total mortality. Further, a meta-analy-sis of 50 randomized controlled trials showed an average
reduction in cholesterol level of 10% and an average reduc-tion in death from coronary heart disease of 9%, but anunintentional increase in noncardiovascular death of 24%.
This review also includes a discussion on the possiblemechanisms by which surrogate end points could be mis-leading. They include the possibility that the surrogate isnot the causal pathway of the disease, that there are manycausal pathways of the disease (and the intervention affectsonly one pathway), that the surrogate is not in the path-way of the intervention’s effect, and that the interventionmight also affect the true clinical outcome by unintendedmechanisms that are independent of the disease process.The authors provide an informative table that speculateson the reasons for failures of surrogate end points usingthese 4 possible mechanisms.
Some of the authors’ concluding statements effectivelystate their case:• “Effects on surrogate end points often do not predict thetrue clinical effects of interventions.”• “The validity of a surrogate end point has rarely beenrigorously established.”• “Surrogate end points should be used where they performbest—in screening for promising new therapies throughevaluation of biologic activity in preliminary phase 2 trials.”
O
Surrogate end points in clinical trials: are we being misled?
T. R. Fleming, D. L. DeMets
Ann Intern Med. 1996;125:605-613
René Lacoste, French tennis playerand multiple grand slam winner, dies;the civil trial of O. J. Simpson beginsin Santa Monica, California; and
former Bulgarian president Andrei Lukanovis assassinated
1996
156
Dialogues in Cardiovascular Medicine - Vol 15 . No. 2 . 2010
Summaries of Ten Seminal Papers - Kubo
egulating the approval of new drugs is theprimary goal of the Center for Drug Evalua-tion and Research of the Food and Drug Ad-ministration (FDA), so it was interesting tohave this institution’s views about surrogate
end points. Temple notes the use of surrogate end pointshas been met with rising enthusiasm as well as rising con-cern. This mixed reception reflects both the potential ofsurrogates to bring needed treatments to patients manyyears before trials that depend on mortality and the poten-tial of surrogates to mislead clinicians into adopting ther-apies that are not effective or even harmful.
The FDA defines a surrogate end point as a “…laboratorymeasurement or physical sign that is used in therapeutictrials as a substitute for a clinically meaningful end pointthat is a direct measure of how a patient feels, functions,or survives, and is expected to predict the effect of thetherapy.” It is important to note that the Food, Drug, andCosmetic Act does not specifically define what end pointscan provide evidence of effectiveness. Indeed, it statesthat the FDA should approve a drug unless it finds a “lackof substantial evidence (defined as adequate and well-con-trolled clinical investigations) that the drug will have theeffect it is represented to have under the conditions of useprescribed, recommended, or suggested in the proposedlabeling.” In 1992, however, an FDA regulation on “acceler-ated approval” provided some indirect guidance allowingapproval based on “an effect on a surrogate end point thatis reasonably likely, based on epidemiologic, therapeutic,pathophysiologic, or other evidence, to predict clinicalbenefit.”
Temple’s main argument against the use of surrogates, how-ever, is based on a safety issue. He maintains that a drugcould have an unexpected unfavorable outcome that canonly be assessed with a study that has a sufficient samplesize and followed for a sufficient period of time to detectany significant safety issues. He notes that reliance on asurrogate is usually an alternative to a large outcome trial.He argues that the lack of a robust safety database, whichusually comes with a large outcome trial, makes the ap-
proval process hazardous, which is sometimes expressedby the phrase “there is no surrogate for safety.”
But there is hope for surrogates after all! If the validity ofa surrogate is accepted, there are several ways to gathersufficient data on safety that do not require a large random-ized trial. Under certain circumstances, it is appropriate torefer to safety data from other closely related populationsand data from other related agents in the same population.For example, angiotensin-converting enzyme (ACE) inhib-itors had been studied in 7000 patients with symptomaticheart failure and in 95 000 patients with acute myocardialinfarction, and these data made it reasonable to concludethat ACE inhibitors would not cause harm in hypertensivepatients.
Temple concludes with specific case examples that illus-trate FDA practice. The only surrogates used for approvalof cardiovascular drugs are blood pressure and serumcholesterol level. For a drug to be approved for heart failure,“…evidence of a symptomatic benefit needs to be support-ed by showing that there is at least no adverse effect onsurvival.” Finally, he notes that “surrogate end points arethus neither consistent successes nor consistent failures.”
Are surrogate markers adequate to assess cardiovasculardisease drugs?
R. Temple
JAMA. 1999;282:790-795
R
M. Night Shyamalan’s film “The Sixth Sense”is released into theaters;
Istanbul and northwest Turkey are hitby a 7.4 magnitude earthquake killingmore than 17 000; and Apple releases
the Power Macintosh G4
1999
157
Dialogues in Cardiovascular Medicine - Vol 15 . No. 2 . 2010
Summaries of Ten Seminal Papers - Kubo
nand et al provide a beautiful summary ofthe many issues regarding the use of surro-gate end points in heart failure trials. Surro-gate end points are attractive because theycan lead to smaller sample size and shorter
trial durations. Nonetheless, there are several risks, includ-ing the possibility of an incomplete, inadequate or mis-leading evaluation. This review comprehensively guides thereader through all of the important issues and also ana-lyzes the potential of suggested surrogate end points forclinical studies of patients with heart failure.
The authors outline the levels of evidence that must be pro-vided for a surrogate end point to be considered an ade-quate substitute for a real end point. First, there must bea strong and consistent relationship between the surrogateand outcomes. Second, changes in the surrogate shouldpredict a change in the morbid event. Third, there shouldbe a consistent proportionality between the change in thesurrogate and the true end point. Finally, these associationsneed to be replicated in a variety of different populationsusing a spectrum of therapeutic interventions. To this setof “Koch postulates” the authors also make a strong state-ment that there must be a sound pathophysiologic basisthat connects the surrogate to the primary outcome.
The next part of this paper is a review of several proposedsurrogates that have proven not to be reliable, includinghemodynamic measurements, ventricular arrhythmias, andautonomic nervous system markers (eg, heart rate vari-ability). There was great interest in peak exercise oxygenuptake as an “objective” assessment of functional capacity.However, the PROspective randomized MIlrinone SurvivalEvaluation (PROMISE) and Randomized Evaluation ofFLosequinan on ExerCise Tolerance (REFLECT) trials, whichevaluated efficacy showed that milrinone and flosequinancould increase peak oxygen uptake, but also reduce sur-vival. We now know that several trials completed in 2009failed on this end point so that peak VO2 has not been con-sidered a viable surrogate. Other trials focused on neuro-hormones, including norepinephrine, but the SecondProspective Randomized Study of Ibopamine on Mortality
(PRIME II) and Beta-blocker Evaluation of Survival sTudy(BEST) trials showed that changes in norepinephrine couldgo in opposite directions to mortality. The available datain 2002 were highly promising for B-type natriuretic pep-tide (BNP) as a surrogate. However, more recent data hasshown great variability in BNP levels, which make it anunreliable marker for pivotal trials.
Anand et al then summarize the literature addressing theutility of left ventricular (LV) dimensions and ejection frac-tion (EF). One of the most important studies at that timewas a report by Cintron that found a significant and pro-portionate relationship between the direction and magni-tude of change in EF over time and 1-year mortality. Theauthors state, “Thus, LV dimensions and their derivative EFseem to fulfill most of the criteria for surrogate end points:baseline LV dimensions and EF are significantly related toprognosis; changes in these measurements reflect changesin mortality; and both the direction and the magnitude ofchange in these variables cause a proportional change insurvival.” The reader is referred to the Lead article in thisissue, which updates more recent data. Particularly strik-ing is Table III, which shows a significant relationship be-tween the change in left ventricular end-diastolic volumeand mortality across many different trials.
The authors conclude “At present, the one perfect surrogatemarker for mortality and quality of life in assessing patientswith HF remains elusive.” However, there does appear to bea solid foundation for considering LV dimensions and EF.
A
Controversial Dutch politician Pim Fortuyn is shotdead 9 days before the general election;
a remote-control bomb explodes during a holidayparade in Kaspiysk, Russia, killing 43 people;
and floods ravage Central Europe
2002
Surrogate end points in heart failure
I. S. Anand, V. G. Florea, L. Fisher
J Am Coll Cardiol. 2002;39:1414-1421
onstam’s editorial makes a compelling argu-ment that ventricular remodeling deservesstrong consideration as a surrogate end pointin clinical trials of heart failure patients. Thisproposal corroborates one of the important
recommendations that are described in detail in the sum-mary article by Anand and Florea (see section titled “Ven-tricular remodeling”).
Ventricular remodeling is characterized by an enlargementof the ventricular chamber, LV hypertrophy, a change froma normal conical shape to an abnormal spherical shape,and a reduction in ejection fraction. It is the characteristicpathophysiologic mechanism underlying the natural courseof heart failure. Since it is a direct manifestation of the dis-ease process, it is much more than a simple surrogate. Mostimportantly, therapeutic interventions that attenuate orreverse remodeling are uniformly associated with improvedclinical outcomes (morbidity and mortality). Thus, “…thegreatest beneficial impact on clinical outcomes…appearsto be caused by those therapies that affect the underlyingpathophysiologic process of ventricular remodeling.”
Furthermore, recent updates to heart failure guidelineshave focused attention of the need to intervene early tohalt the development or progression of clinical heart fail-ure. In these early treatment scenarios, it is not possibleto focus on symptoms (there may not be any!) or survival(because it can be so far removed). Instead, benefits arebest achieved (and tracked) through interventions thatprevent or regress ventricular remodeling.
Konstam provides a summary of many trials of differenttherapeutic agents that support the above statements. Hediscusses results from the Vasodilator Heart Failure Trial(V-HeFT), several trials with angiotensin-converting enzymeinhibitors, the accumulating evidence with angiotensinreceptor blockers, β-blockers, and recent experience withaldosterone receptor blockers. He also refers to a meta-analysis of 72 trials that demonstrated that changes inejection fractions and/or left ventricular volumes correlatedwith drug effects on clinical outcomes, similar to the analy-
sis summarized by Anand. The key here is the consistencyof the effect—all the therapies that have beneficial actionson the clinical end points of symptoms and survival alsohave a beneficial effect on left ventricular remodeling.
Konstam’s closing suggestions are particularly relevant forthose involved in the development of clinical trials. Theseinclude: “On the basis of the accumulated evidence, thismagnitude of reduction in left ventricular volumes….cantherefore serve as a surrogate marker….it is feasible toconstruct trial designs in which composites are constructedbetween indexes of ventricular structure and/or functionand those of clinical outcomes….A drug’s effect in inhibit-ing or reversing the remodeling process should, at the least,be taken as supportive of the clinical outcome effects….In this way, recognition of the validity of markers of remod-eling as surrogates for clinical outcome can translate intothe improved efficiency of drug development and approval.”
It is recognized that the role of ventricular volumes, or in-deed any surrogate, will continue to be challenged, espe-cially by the purists who insist on true clinical outcomes.But the accumulated data on the viability of ventricularvolumes is strong and compelling for many experts in thefield.
K
Pope John Paul II makes 2005 the InternationalYear of the Eucharist in Catholicism;
Four Royal Canadian Mounted Police officersare gunned down in Mayerthorpe, Alberta, Canadain the deadliest day in Canadian law enforcementin over 120 years; and several hundred Iraqis diein a stampede on Al-Aaimmah bridge in Baghdad
2005
Reliability of ventricular remodeling as a surrogate for usein conjunction with clinical outcomes in heart failure
M. A. Konstam
Am J Cardiol. 2005;96:867-871
158
Dialogues in Cardiovascular Medicine - Vol 15 . No. 2 . 2010
Summaries of Ten Seminal Papers - Kubo
159
Dialogues in Cardiovascular Medicine - Vol 15 . No. 2 . 2010
Summaries of Ten Seminal Papers - Kubo
n 2004, the Heart Failure Society of America spon-sored a 2-day workshop that brought together abroad representation of academic cardiologists, stat-isticians, the Food and Drug Administration (FDA),and industry sponsors to discuss many of the com-
mon issues and problems that were challenging the designof pivotal trials that were necessary for regulatory approvalin the United States. Devices for heart failure were the“hot topic” of the decade, but it became very apparent thatthere were key issues for device trials that were not relevantin the traditional drug studies. Sponsors had brought for-ward many innovative devices and trials, but, because theywere conducted individually, there was little consistencyor coherency.
The main objective of this workshop, then, was to bringall the main groups together to discuss common issues andsee if a consensus could be reached on some of the moreimportant trial design issues.
In this paper, Neaton and his colleagues provide a com-prehensive review on the use of composite end points inheart failure trials. They draw on their expertise and vastexperience serving on steering committees, Data and SafetyMonitoring Boards, Clinical Events committees, and alsoFDA Advisory Panels. They provide an outstanding reviewon the use of composite end points, and discuss the keyadvantages and disadvantages of this approach.
The primary rationale for using a composite primary out-come is sample size. In both time-to-event trials and suc-cess/failure trials, the use of a composite can lead to asmaller sample size or trial duration. A second reason touse composites is to avoid the problem of competing risks,which is why the end point of hospitalization is typicallycombined with mortality. It is conceivable that an interven-tion could reduce the number of hospitalizations becauseit increased mortality! Thus, combining the outcomes ofdeath and hospitalization into a composite reduces thechance for bias and makes the results more interpretable.Finally, a composite can help to make sure that the primaryend point can be assessed in all patients. For example, if
one is assessing quality of life as an end point, one wayto account for missing data from patients who die priorto the end of the study is to include death as part of thecomposite.
This paper also summarizes many of the cautions and po-tential problems of composites. There can be loss of powerif the treatment effect is not similar for all of the compo-nents and, even worse, when components of the compositego in opposite directions. A second problem with compos-ites relates to the differential weighting of the individualcomponents. For example, should death be given the sameweight as a hospitalization or a change in quality of life?If the composite does assign different relative weight, it isimportant that the weights be validated against some cred-ible outcome (eg, mortality).
The authors also provide 6 excellent examples from trialsthat used composite end points to highlight the utility andpotential problems from “real life” trials.
It is unfortunate that only one paper came out of this 2-daymeeting. But, for all those who are involved in trial designand contemplating the use of a composite end point, thisreference is a must read!
I
Key issues in end point selection for heart failure trials:composite end points
J. D. Neaton, G. Gray, B. D. Zuckerman, M. A. Konstam
J Card Fail. 2005;11:567-575
Chinese novelist Li Yaotang (李尧棠), who wroteunder the pen name of Ba Jin (巴金), dies;
the world's largest bank, Mitsubishi UFJ FinancialGroup, is formed by the merger of theMitsubishi Tokyo Financial Group and
Osaka-based UFJ Holdings; and the Qinghai-TibetRailway, the world highest, is completed
2005
160
Dialogues in Cardiovascular Medicine - Vol 15 . No. 2 . 2010
Summaries of Ten Seminal Papers - Kubo
ospitalization rates (both all-cause andheart failure [HF]-specific) are one of themost commonly used end points in HFtrials, and the Lead article by Anand andFlorea in this Journal provides an excellent
summary on how this end point is used in trials and someof the important features to consider in data collectionand interpretation of the results. A very common trial de-sign feature is to combine hospitalization with death in acomposite end point, in order to avoid misinterpretationof an intervention that reduced hospitalization due to thefact that more patients died.
This paper by Solomon et al is a classic description of ad-ditional levels of relation between death and hospitaliza-tion. Death removes the sickest patients who are likely tobe hospitalized. In addition, as shown in this paper, hospi-talization increases the risk of death as well as the risk ofsubsequent hospitalization. Adding to the complexity, if adevice increases survival, it increases the period duringwhich a hospitalization could occur. Thus, statisticians em-ploy nonparametric methods to consider mortality as acompeting risk as well as adjusting for follow up time.
This study analyzed data from the Candesartan in HeartFailure: Assessment of Reduction in Mortality and Morbid-ity (CHARM) study, which evaluated the effect of cande-sartan in three independent, concurrently performed trialsof 7599 patients representing a broad spectrum of symp-tomatic heart failure and either reduced or preserved leftventricular ejection fraction. Data from the CHARM trialswere examined to assess the influence of a nonfatal hos-pitalization for HF on subsequent death. Of the 7572 pa-tients included in this analysis, 1455 had at least one hos-pitalization. Of the 1891 deaths, 586 occurred after hospitaldischarge for a first HF hospitalization. The estimated crudehazard for all-cause mortality after HF hospitalization was4.55 times that of patients never hospitalized. The risk ofdying was also related to the length of the HF hospitaliza-tion, with long hospitalizations (>22 days) carrying morethan double the mortality risk of short HF hospitalizations(<7 days).
The authors discuss important clinical implications of theirfindings. The high risk of death following hospitalizationindicates that patients are particularly vulnerable in theearly discharge period and might be candidates for height-ened surveillance. Indeed, many programs have been im-plemented by hospitals across the country that employearly intervention with early clinic visits (within 3 days ofdischarge) and home visits to detect and treat HF problems.In particular, we have found countless cases where med-ication reconciliation appeared to have aborted a post-dis-charge complication.
Further, the observation that hospitalization increases therisk of death and subsequent rehospitalization has beenused in the design of many different clinical trials. Thus, ifone is designing an intervention that should reduce hos-pitalizations (eg, hemodynamic monitors; home scales), oneimportant inclusion criterion would be to enroll patientswho have been hospitalized in the previous 6 months. Thisinclusion criterion would have the effect of enriching thenumber of anticipated events (hospitalizations) and there-fore increase the power of the study.
Influence of nonfatal hospitalization for heart failure onsubsequent mortality in patients with chronic heart failure
S. D Solomon, J. Dobson, S. Pocock, H. Skali, J. J. McMurray, C. B. Granger, S. Yusuf, K. Swedberg,J. B. Young, E. L. Michelson, M. A. Pfeffer; Candesartan in Heart failure: Assessment of Reductionin Mortality and morbidity (CHARM) Investigators
Circulation. 2007;116:1482-1487
H
Israeli airplanes strike a target suspected of beinga nuclear site in the Deir ez-Zor region of Syria;
Italian tenor Luciano Pavarotti dies frompancreatic cancer, aged 71; and Ang Lee’s film
“Lust, Caution” wins the Golden Lionat the Venice Film Festival
2007
161
Dialogues in Cardiovascular Medicine - Vol 15 . No. 2 . 2010
Summaries of Ten Seminal Papers - Kubo
ardiovascular clinical trialists, biostatisticians,National Institutes of Health scientists, regu-lators, and pharmaceutical industry scientistsmet to discuss issues that related to the de-sign of clinical trials at a Cardiovascular Clin-
ical Trialists Workshop, in December 2005. These meetingscan be very important since they bring all the major stake-holders in a single place to debate strengths and weak-nesses and new data. This manuscript summarizes thediscussion that focused on the definition of heart failureas an end point.
Several clinical trials have established common criteria todefine myocardial infarction and stroke, but a consistentdefinition for heart failure was lacking. Since different trialsused different definitions, it has been difficult to interpretdata across trials, determine the true incidence of heartfailure, and whether or not there are important trends overtime. Some of the challenges, of course, are related to thefact that heart failure is not a single event, like myocardialinfarction and stroke, but rather is a clinical syndrome re-lated to multiple causes and with many different clinicalmanifestations. This review also discusses another levelof complexity by highlighting the difference between heartfailure and non–heart failure clinical trials. In heart failuretrials, the diagnosis of heart failure is already establishedand so one could use hospitalization or administration ofintravenous medications as an indicator of a heart failureevent. In contrast, in non–heart failure trials, the diagnosisof heart failure is not established and so hospitalizationmight not be a very sensitive marker. Moreover, in non–heartfailure trials, some documentation of heart dysfunction(eg, echocardiography or B-type natriuretic peptide [BNP]level) would be necessary to be sure that the symptom(eg, edema) is due to heart failure and not some othercomorbidity.
This review also contains some useful discussion of sever-al important topics including the utility of adjudicationcommittees who are blinded to treatment assignment toreduce bias and maximize consistency in counting heartfailure events. Difficulties with counting heart failure hos-
pitalizations include the fact the thresholds for hospitaladmission and the administration of intravenous medica-tions differ across institutions and regions of the world.Some trials have attempted to reduce this variability byimplementing decision rules for what constitutes a hospi-talization and treatment algorithms for different medica-tions, but it is always difficult to implement this level ofconsistency in a multicenter and multinational clinical trial.
Although the authors state that “it is impractical to createa heart failure definition that is clinically relevant and sat-isfies all types of trials across multiple disciplines,” theydo provide a standard uniform set of criteria that can beused as a framework to define heart failure across trials,that include: (i) objective evidence of cardiac dysfunction(eg, imaging or BNP) and that the patient is receiving treat-ment; (ii) that the event is clinically meaningful; and (iii)that the event captures the course of the disease. Thesecriteria may not be perfect, but it is clearly easier to movethe field forward if all are working from a common platform!
Heart failure as an endpoint in heart failure andnon–heart failure cardiovascular clinical trials:the need for a consensus definition
F. Zannad, W. G. Stough, B. Pitt, J. G. Cleland, K. F. Adams, N. L. Geller, C. Torp-Pedersen,B. A. Kirwan, F. Follath
Eur Heart J. 2008;29:413-421
C
Fidel Castro resigns as President of Cubadue to ill health and is replaced by his
younger brother Raúl Castro;Australian Prime Minister Kevin Rudd delivers
a formal apology to the children of native aboriginesforcibly removed from their families between
1869 and 1969; and the European Organizationfor Nuclear Research circulates a beamthrough along the entire length of theLarge Hadron Collider for the first time
Dialogues in Cardiovascular Medicine - Vol 15 . No. 2 . 2010
Abraham WT, Fisher WG, Smith AL, et al. Cardiac resynchronization in chronic heart failure.
N Engl J Med. 2002;346:1845-1853.
Ahmed A, Rich MW, Fleg JL, et al. Effects of digoxin on morbidity and mortality in diastolic heartfailure: the ancillary digitalis investigation group trial.
Circulation. 2006;114:397-403.
Al-Kaade S, Hauptman PJ. Health-related quality of life measurement in heart failure:challenges for the new millennium.
J Card Fail. 2001;7:194-201.
Anand I, McMurray J, Cohn JN, et al. Long-term effects of darusentan on left-ventricular remodellingand clinical outcomes in the EndothelinA Receptor Antagonist Trialin Heart Failure (EARTH): randomised, double-blind, placebo-controlled trial.
Lancet. 2004;364:347-354.
Anand IS, Carson P, Galle E, et al. Cardiac resynchronization therapy reduces the risk of hospitalizationsin patients with advanced heart failure: results from the Compari-son of Medical Therapy, Pacing and Defibrillation in Heart Failure(COMPANION) trial.
Circulation. 2009;119:969-977.
Anand IS, Florea VG, Fisher L. Surrogate end points in heart failure.
J Am Coll Cardiol. 2002;39:1414-1421.
Anand IS, Florea VG, Solomon SD, Konstam MA, Noninvasive assessment of left ventricular remodeling: concepts,Udelson JE. techniques, and implications for clinical trials.
J Card Fail. 2002;8:S452-S464.
Anand IS, Florea VG. Alterations in ventricular structure: role of left ventricular remod-eling. In: Mann DL, ed. Heart Failure: Companion to Braunwald’sHeart Disease.
Elsevier, W. B. Saunders. 2003:229-246.
Anand IS, Florea VG. Traditional and novel approaches to management of heart failure:successes and failures.
Cardiol Clin. 2008;26:59-72.
Bibliography of One Hundred Key Papersselected by Inder S. Anand, MD, FRCP, DPhil(Oxon) and Viorel G. Florea
Division of Cardiology - University of Minnesota Medical School - and Veterans Administration Medical CenterMinneapolis, Minn - USA (e-mail: [email protected])
Surrogate End Points in Heart Failure Trials:Potentials and Limitations
Anand IS, Wong M, Latini R, Fisher LD, Chiang YT, Relation between changes in ejection fraction over time and subse-Cohn JN. quent mortality and morbidity in Val-HeFT.
J Am Coll Cardiol. 2003;41:221A.
Beta-Blocker Evaluation of Survival Trial Investigators. A trial of the beta-blocker bucindolol in patients with advancedchronic heart failure.
N Engl J Med. 2001;344:1659-1667.
Bristow MR, Saxon LA, Boehmer J, et al. Cardiac-resynchronization therapy with or without an implantabledefibrillator in advanced chronic heart failure.
N Engl J Med. 2004;350:2140-2150.
Bucher HC, Guyatt GH, Cook DJ, Holbrook A, Users’ guides to the medical literature: XIX. Applying clinicaltrial results. A. How to use an article measuring the effect of anintervention on surrogate end points. Evidence-Based MedicineWorking Group.
JAMA. 1999;282:771-778.
Califf RM, Adams KF, McKenna WJ, et al. A randomized controlled trial of epoprostenol therapy for severe con-gestive heart failure: the Flolan International Randomized SurvivalTrial (FIRST).
Am Heart J. 1997;134:44-54.
Carson P, Anand I, O'Connor C, et al. Mode of death in advanced heart failure: the Comparison ofMedical, Pacing, and Defibrillation Therapies in Heart Failure(COMPANION) trial.
J Am Coll Cardiol. 2005;46:2329-2334.
Carson P, Tognoni G, Cohn JN. Effect of Valsartan on hospitalization: results from Val-HeFT.
J Card Fail. 2003;9:164-171.
Chatterjee K, Massie B. Systolic and diastolic heart failure: differences and similarities.
J Card Fail. 2007;13:569-576.
Cintron G, Johnson G, Francis G, Cobb F, Cohn JN. Prognostic significance of serial changes in left ventricular ejectionfraction in patients with congestive heart failure.
Circulation. 1993;87:VI17-VI23.
Cleland JG, Tendera M, Adamus J, Freemantle N, The perindopril in elderly people with chronic heart failurePolonski L, Taylor J. (PEP-CHF) study.
Eur Heart J. 2006;27:2338-2345.
Cohn JN, Archibald DG, Ziesche S, et al. Effect of vasodilator therapy on mortality in chronic congestive heartfailure. Results of a Veterans Administration Cooperative Study.
N Engl J Med. 1986;314:1547-1552.
Cohn JN, Goldstein SO, Greenberg BH, et al. A dose-dependent increase in mortality with vesnarinone amongpatients with severe heart failure.
N Engl J Med. 1998;339:1810-1816.
Cohn JN, Johnson G, Ziesche S, et al. A comparison of enalapril with hydralazine-isosorbide dinitrate inthe treatment of chronic congestive heart failure
N Engl J Med. 1991;325:303-310.
Cohn JN, Pfeffer MA, Rouleau J, et al. Adverse mortality effect of central sympathetic inhibition with sus-tained-release moxonidine in patients with heart failure (MOXCON).
Eur J Heart Fail. 2003;5:659-667.
164
Dialogues in Cardiovascular Medicine - Vol 15 . No. 2 . 2010
Bibliography of One Hundred Key References
Cohn JN, Tam SW, Anand IS, Taylor AL, Sabolinski ML, Isosorbide dinitrate and hydralazine in a fixed-dose combinationWorcel M. produces further regression of left ventricular remodeling in a well-
treated black population with heart failure: results from A-HeFT.
J Card Fail. 2007;13:331-339.
Cohn JN, Tognoni G. A randomized trial of the angiotensin-receptor blocker valsartan inchronic heart failure.
N Engl J Med. 2001;345:1667-1675.
Colucci WS, Elkayam U, Horton DP, et al. Intravenous nesiritide, a natriuretic peptide, in the treatment ofdecompensated congestive heart failure. Nesiritide Study Group.
N Engl J Med. 2000;343:246-253.
CONSENSUS Trial Study Group. Effects of enalapril on mortality in severe congestive heart failure.Results of the Cooperative North Scandinavian Enalapril SurvivalStudy (CONSENSUS).
N Engl J Med. 1987;316:1429-1435.
Cuffe MS, Califf RM, Adams KF Jr, et al. Short-term intravenous milrinone for acute exacerbation of chronicheart failure: a randomized controlled trial.
JAMA. 2002;287:1541-1547.
Dargie HJ. Effect of carvedilol on outcome after myocardial infarction inpatients with left-ventricular dysfunction: the CAPRICORN ran-domised trial.
N Engl J Med. 1997;336:525-533.
Doughty RN, Whalley GA, Gamble G, MacMahon S, Left ventricular remodeling with carvedilol in patients with con-Sharpe N. gestive heart failure due to ischemic heart disease. Australia-New
Zealand Heart Failure Research Collaborative Group.
J Am Coll Cardiol. 1997;29:1060-1066.
Doughty RN, Whalley GA, Walsh HA, Gamble GD, Effects of carvedilol on left ventricular remodeling after acuteLopez-Sendon J, Sharpe N. myocardial infarction: the CAPRICORN Echo Substudy.
Circulation. 2004;109:201-206.
Every NR, Parsons L, Hlatky MA, et al. Use and accuracy of state death certificates for classification ofsudden cardiac deaths in high-risk populations.
Am Heart J. 1997;134:1129-1132.
Fleg JL, Pina IL, Balady GJ, et al. Assessment of functional capacity in clinical and research applica-tions: an advisory from the Committee on Exercise, Rehabilitation,and Prevention, Council on Clinical Cardiology, American HeartAssociation.
Circulation. 2000;102:1591-1597.
Fleming TR, DeMets DL. Surrogate end points in clinical trials: are we being misled?
Ann Intern Med. 1996;125:605-613.
Florea VG, Henein MY, Anker SD, et al. Prognostic value of changes over time in exercise capacity and echo-cardiographic measurements in patients with chronic heart failure.
Eur Heart J. 2000;21:146-153.
Ford I, Norrie J, Ahmadi S. Model inconsistency, illustrated by the Cox proportional hazardsmodel.
Stat Med. 1995;14:735-746.
165
Dialogues in Cardiovascular Medicine - Vol 15 . No. 2 . 2010
Bibliography of One Hundred Key References
Franciosa JA, Taylor AL, Cohn JN, et al. African-American Heart Failure Trial (A-HeFT): rationale, design,and methodology.
J Card Fail. 2002;8:128-135.
Francis GS, Cohn JN, Johnson G, Rector TS, Goldman S, Plasma norepinephrine, plasma renin activity, and congestive heartSimon A. failure. Relations to survival and the effects of therapy in V-HeFT
II. The V-HeFT VA Cooperative Studies Group.
Circulation. 1993;87:VI40-VI48.
Francis GS, Kubo SH. Prognostic factors affecting diagnosis and treatment of congestiveheart failure.
Curr Probl Cardiol. 1989;14:625-671.
Gerstein HC, Miller ME, Byington RP, et al. Effects of intensive glucose lowering in type 2 diabetes.
N Engl J Med. 2008;358:2545-2559.
Ghosh D, Lin DY. Nonparametric analysis of recurrent events and death.
Biometrics. 2000;56:554-562.
Goldman L, Hashimoto B, Cook EF, Loscalzo A. Comparative reproducibility and validity of systems for assessingcardiovascular functional class: advantages of a new specific activityscale.
Circulation. 1981;64:1227-1234.
Greenberg B, Quinones MA, Koilpillai C, et al. Effects of long-term enalapril therapy on cardiac structure and func-tion in patients with left ventricular dysfunction. Results of theSOLVD echocardiography substudy.
Circulation. 1995;91:2573-2581.
Groenning BA, Nilsson JC, Sondergaard L, Fritz-Hansen T, Antiremodeling effects on the left ventricle during beta-blockadeLarsson HB, Hildebrandt PR. with metoprolol in the treatment of chronic heart failure.
J Am Coll Cardiol. 2000;36:2072-2080.
Hammermeister KE, DeRouen TA, Dodge HT. Variables predictive of survival in patients with coronary disease.Selection by univariate and multivariate analyses from the clinical,electrocardiographic, exercise, arteriographic, and quantitativeangiographic evaluations.
Circulation. 1979;59:421-430.
Hampton JR, van Veldhuisen DJ, Kleber FX, et al. Randomised study of effect of ibopamine on survival in patients withadvanced severe heart failure. Second Prospective Randomised Studyof Ibopamine on Mortality and Efficacy (PRIME II) Investigators.
Lancet. 1997;349:971-977.
Heagerty A, Deverly A, Palmer C, et al. Simvastatin with or without ezetimibe in familial hypercholesterolemia.
N Engl J Med. 2008;358:1431-1443.
Konstam MA, Kronenberg MW, Rousseau MF, et al. Effects of the angiotensin converting enzyme inhibitor enalapril onthe long-term progression of left ventricular dilatation in patientswith asymptomatic systolic dysfunction. SOLVD (Studies of LeftVentricular Dysfunction) Investigators.
Circulation. 1993;88:2277-2283.
Konstam MA, Patten RD, Thomas I, et al. Effects of losartan and captopril on left ventricular volumes inelderly patients with heart failure: results of the ELITE ventricularfunction substudy.
Am Heart J. 2000;139:1081-1087.
166
Dialogues in Cardiovascular Medicine - Vol 15 . No. 2 . 2010
Bibliography of One Hundred Key References
Konstam MA, Rousseau MF, Kronenberg MW, et al. Effects of the angiotensin converting enzyme inhibitor enalapril onthe long-term progression of left ventricular dysfunction in patientswith heart failure.
Circulation. 1992;86:431-438.
Konstam MA, Udelson JE, Anand IS, Cohn JN. Ventricular remodeling in heart failure: a credible surrogate endpoint.
J Card Fail. 2003;9:350-353.
Kubo SH, Gollub S, Bourge R, et al. Beneficial effects of pimobendan on exercise tolerance and quality oflife in patients with heart failure. Results of a multicenter trial.The Pimobendan Multicenter Research Group.
Circulation. 1992;85:942-949.
Lipicky RJ, Packer M. Role of surrogate end points in the evaluation of drugs for heartfailure.
J Am Coll Cardiol. 1993;22:179A-184A.
Lüscher TF, Enseleit F, Pacher R, et al. Hemodynamic and neurohumoral effects of selective endothelin A(ETA) receptor blockade in chronic heart failure: the Heart FailureETA Receptor Blockade Trial (HEAT).
Circulation. 2002;106:2666-2672.
Mann DL, McMurray JJ, Packer M, et al. Targeted anticytokine therapy in patients with chronic heart failure:results of the Randomized Etanercept Worldwide Evaluation(RENEWAL).
Circulation. 2004;109:1594-1602.
Massie BM, Berk MR, Brozena SC, et al. Can further benefit be achieved by adding flosequinan to patientswith congestive heart failure who remain symptomatic on diuretic,digoxin, and an angiotensin converting enzyme inhibitor?Results of the flosequinan-ACE inhibitor trial (FACET).
Circulation. 1993;88:492-501.
Massie BM, Carson PE, McMurray JJ, et al. Irbesartan in patients with heart failure and preserved ejectionfraction.
N Engl J Med. 2008;359:2456-2467.
MERIT-HF Study Group. Effect of metoprolol CR/XL in chronic heart failure: MetoprololCR/XL Randomised Intervention Trial in Congestive Heart Failure(MERIT-HF).
Lancet. 1999;353:2001-2007.
Naito Y, Tsujino T, Fujioka Y, Ohyanagi M, Iwasaki T. Augmented diurnal variations of the cardiac renin-angiotensinsystem in hypertensive rats.
Hypertension. 2002;40:827-833.
Morris CK, Ueshima K, Kawaguchi T, Hideg A, The prognostic value of exercise capacity: a review of the literature.Froelicher VF. Am Heart J. 1991;122:1423-1431.
Narang R, Swedberg K, Cleland JG. What is the ideal study design for evaluation of treatment for heartfailure? Insights from trials assessing the effect of ACE inhibitorson exercise capacity.
Eur Heart J. 1996;17:120-134.
Neaton JD, Gray G, Zuckerman BD, Konstam MA. Key issues in end point selection for heart failure trials: compositeend points.
J Card Fail. 2005;11:567-575.
167
Dialogues in Cardiovascular Medicine - Vol 15 . No. 2 . 2010
Bibliography of One Hundred Key References
Nissen SE, Wolski K. Effect of rosiglitazone on the risk of myocardial infarction anddeath from cardiovascular causes.
N Engl J Med. 2007;356:2457-2471.
Packer M. The neurohormonal hypothesis: a theory to explain the mechanismof disease progression in heart failure.
J Am Coll Cardiol. 1992;20:248-254.
Packer M, Antonopoulos GV, Berlin JA, Chittams J, Comparative effects of carvedilol and metoprolol on left ventricularUdelson JE. ejection fraction in heart failure: results of a meta-analysis.
Am Heart J. 2001;141:899-907.
Packer M, Califf RM, Konstam MA, et al. Comparison of omapatrilat and enalapril in patients with chronicheart failure: the Omapatrilat Versus Enalapril Randomized Trialof Utility in Reducing Events (OVERTURE).
Circulation. 2002;106:920-926.
Packer M, Carver JR, Rodeheffer RJ, et al. Effect of oral milrinone on mortality in severe chronic heart failure.The PROMISE Study.
N Engl J Med. 1991;325:1468-1475.
Packer M, Narahara KA, Elkayam U, et al. Double-blind, placebo-controlled study of the efficacy of flosequinanin patients with chronic heart failure. REFLECT Study.
J Am Coll Cardiol. 1993;22:65-72.
Packer M, O'Connor CM, Ghali JK, et al. Effect of amlodipine on morbidity and mortality in severe chronicheart failure. Prospective Randomized Amlodipine SurvivalEvaluation Study Group.
N Engl J Med. 1996;335:1107-1114.
Packer M, Rouleau J, Swedberg K. Effect of flosequinan on survival in chronic heart failure: preliminaryresults of the PROFILE study [abstract].
Circulation. 1993;88(suppl 1):1-301.
Parker M. How should we judge the efficacy of drug therapy in patients withchronic congestive heart failure? The insights of six blind men.
J Am Coll Cardiol. 1987;9:433-438.
Pfeffer MA, Braunwald E, Moye LA; SAVE Investigators. Effect of captopril on mortality and morbidity in patients with leftventricular dysfunction after myocardial infarction. Results of thesurvival and ventricular enlargement trial.
N Engl J Med. 1992;327:669-677.
Pfeffer MA, Pfeffer JM. Ventricular enlargement and reduced survival after myocardialinfarction.
Circulation. 1987;75:IV93-IV97.
Pfeffer MA, Swedberg K, Granger CB, et al. Effects of candesartan on mortality and morbidity in patients withchronic heart failure: the CHARM-Overall programme.
Lancet. 2003;362:759-766.
Pitt B, Poole-Wilson PA, Segal R, et al. Effect of losartan compared with captopril on mortality in patientswith symptomatic heart failure: randomised trial—the LosartanHeart Failure Survival Study ELITE II.
Lancet. 2000;355:1582-1587.
168
Dialogues in Cardiovascular Medicine - Vol 15 . No. 2 . 2010
Bibliography of One Hundred Key References
Pitt B, Segal R, Martinez FA, et al. Randomised trial of losartan versus captopril in patients over 65 withheart failure (Evaluation of Losartan in the Elderly Study, ELITE).
Lancet. 1997;349:747-752.
Pitt B, Zannad F, Remme WJ, Cody R, et al. The effect of spironolactone on morbidity and mortality in patientswith severe heart failure. Randomized Aldactone Evaluation StudyInvestigators.
N Engl J Med. 1999;341:709-717.
Poole-Wilson PA, Swedberg K, Cleland JG, et al. Comparison of carvedilol and metoprolol on clinical outcomes inpatients with chronic heart failure in the Carvedilol Or MetoprololEuropean Trial (COMET): randomised controlled trial.
Lancet. 2003;362:7-13.
Prentice RL. Surrogate endpoints in clinical trials: definition and operationalcriteria.
Stat Med. 1989;8:431-440.
Publication Committee for the VMAC Investigators Intravenous nesiritide vs nitroglycerin for treatment of decompen-(Vasodilatation in the Management of Acute CHF). sated congestive heart failure: a randomized controlled trial.
JAMA. 2002;287:1531-1540.
Rector TS, Anand IS, Cohn JN. Relationships between clinical assessments and patients’ perceptionsof the effects of heart failure on their quality of life.
J Card Fail. 2006;12:87-92.
Richards AM, Doughty R, Nicholls MG, et al. Neurohumoral prediction of benefit from carvedilol in ischemic leftventricular dysfunction. Australia-New Zealand Heart Failure Group.
Circulation. 1999;99:786-792.
Rousseau MF, Konstam MA, Benedict CR, et al. Progression of left ventricular dysfunction secondary to coronaryartery disease, sustained neurohormonal activation and effects ofibopamine therapy during long-term therapy with angiotensin-converting enzyme inhibitor.
Am J Cardiol. 1994;73:488-493.
Sackner-Bernstein JD, Kowalski M, Fox M, Aaronson K. Short-term risk of death after treatment with nesiritide for decompen-sated heart failure: a pooled analysis of randomized controlled trials.
JAMA. 2005;293:1900-1905.
Senn SJ. Covariate imbalance and random allocation in clinical trials.
Stat Med. 1989;8:467-475.
Singh SN, Fletcher RD, Fisher SG, et al. Amiodarone in patients with congestive heart failure and asympto-matic ventricular arrhythmia. Survival Trial of AntiarrhythmicTherapy in Congestive Heart Failure.
N Engl J Med. 1995;333:77-82.
Sobel BE, Furberg CD. Surrogates, semantics, and sensible public policy.
Circulation. 1997;95:1661-1663.
Solomon SD, Skali H, Bourgoun M, et al. Effect of angiotensin-converting enzyme or vasopeptidase inhibitionon ventricular size and function in patients with heart failure: theOmapatrilat Versus Enalapril Randomized Trial of Utility inReducing Events (OVERTURE) echocardiographic study.
Am Heart J. 2005;150:257-262.
169
Dialogues in Cardiovascular Medicine - Vol 15 . No. 2 . 2010
Bibliography of One Hundred Key References
Solomon SD, Wang D, Finn P, et al. Effect of candesartan on cause-specific mortality in heart failurepatients: the Candesartan in Heart failure Assessment of Reductionin Mortality and morbidity (CHARM) program.
Circulation. 2004;110:2180-2183.
SOLVD Investigators. Effect of enalapril on mortality and the development of heart failurein asymptomatic patients with reduced left ventricular ejectionfractions.
N Engl J Med. 1992;327:685-691.
Spieker LE, Mitrovic V, Noll G, et al. Acute hemodynamic and neurohumoral effects of selective ET(A)receptor blockade in patients with congestive heart failure.ET 003 Investigators.
J Am Coll Cardiol. 2000;35:1745-1752.
St John Sutton M, Pfeffer MA, Moye L, et al. Cardiovascular death and left ventricular remodeling two yearsafter myocardial infarction: baseline predictors and impact of long-term use of captopril: information from the Survival and VentricularEnlargement (SAVE) trial.
Circulation. 1997;96:3294-3299.
St John Sutton M, Pfeffer MA, Plappert T, et al. Quantitative two-dimensional echocardiographic measurements aremajor predictors of adverse cardiovascular events after acute myo-cardial infarction. The protective effects of captopril.
Circulation. 1994;89:68-75.
Swedberg K, Eneroth P, Kjekshus J, Wilhelmsen L. Hormones regulating cardiovascular function in patients with severecongestive heart failure and their relation to mortality. CONSENSUSTrial Study Group.
Circulation. 1990;82:1730-1736.
Taylor AL, Ziesche S, Yancy C, et al. Combination of isosorbide dinitrate and hydralazine in blacks withheart failure.
N Engl J Med. 2004;351:2049-2057.
Temple R, Pledger GW. The FDA’s critique of the anturane reinfarction trial.
N Engl J Med. 1980;303:1488-1492.
Tsutamoto T, Wada A, Maeda K, et al. Effect of spironolactone on plasma brain natriuretic peptide and leftventricular remodeling in patients with congestive heart failure.
J Am Coll Cardiol. 2001;37:1228-1233.
Udelson JE, Antonopoulos GV, Proulx G, et al. Comparison of long-term dual vasopeptidase inhibition withomapatrilat to ACE inhibition with lisinopril on ventricular volumesin patients with heart failure.
Circulation. 2000;102:II536.
Udelson JE, McGrew FA, Flores E, et al. Multicenter, randomized, double-blind, placebo-controlled study onthe effect of oral tolvaptan on left ventricular dilation and functionin patients with heart failure and systolic dysfunction.
J Am Coll Cardiol. 2007;49:2151-2159.
Ware J Jr, Kosinski M, Keller SD. A 12-Item Short-Form Health Survey: construction of scales andpreliminary tests of reliability and validity.
Med Care. 1996;34:220-233.
170
Dialogues in Cardiovascular Medicine - Vol 15 . No. 2 . 2010
Bibliography of One Hundred Key References
White HD, Norris RM, Brown MA, Brandt PW, Left ventricular end-systolic volume as the major determinant ofWhitlock RM, Wild CJ. survival after recovery from myocardial infarction.
Circulation. 1987;76:44-51.
Wong M, Staszewsky L, Latini R, et al. Valsartan benefits left ventricular structure and function in heartfailure: Val-HeFT echocardiographic study.
J Am Coll Cardiol. 2002;40:970-975.
Yusuf S, Negassa A. Choice of clinical outcomes in randomized trials of heart failuretherapies: disease-specific or overall outcomes?
Am Heart J. 2002;143:22-28.
Zannad F, Stough WG, Pitt B, et al. Heart failure as an endpoint in heart failure and non-heart failurecardiovascular clinical trials: the need for a consensus definition.
Eur Heart J. 2008;29:413-421.
Ziesche S, Cobb FR, Cohn JN, Johnson G, Tristani F. Hydralazine and isosorbide dinitrate combination improves exercisetolerance in heart failure. Results from V-HeFT I and V-HeFT II.The V-HeFT VA Cooperative Studies Group.
Circulation. 1993;87:VI56-VI64.
171
Dialogues in Cardiovascular Medicine - Vol 15 . No. 2 . 2010
Bibliography of One Hundred Key References
GENERALINSTRUCTIONS
• Manuscripts should be provided onword-processor disks (3.5-in, for IBM,IBM-compatible, or Apple computers)with three hard copies (text and fig-ures) printed on one side of standard-sized white bond paper, double-spaced, with 2.5-cm margins. Pagesmust be numbered. Standard typedpage = 25 lines of 75 characters(including spaces) double-spaced,2.5-cm margins = a total of 275words per page.
• All texts should be submitted inEnglish. In the case of translations,the text in the original language shouldbe included.
• On the title page, provide title ofmanuscript (title should be concise,not exceeding 120 characters, includingspaces), short running title, keywords,and acknowledgments, as well as fullnames (first name, middle name(s),and last name) with highest academicdegrees (in country-of-origin language),affiliations/address, telephone No.,fax No., and E-mail address.
• Illustrations (photographs, tables, graphs,figures–high-quality printouts, glossyprints, and/or high-quality scans asjpg files) should be of good quality orprofessionally prepared, numberedaccording to their order, with properorientation indicated (eg, “top,” or“left”), and SHORT legends provided,not repetitive of text.As figures and graphs may need to bereduced or enlarged, all absolute valuesand statistics should be provided.All illustrations should be cited in thetext, with distinct numbering for fig-ures and tables. Illustrations will bereproduced in full color only whenclearly necessary, eg, images from nu-clear medicine or histology.
• Include HEADINGS using a consistentstyle for the various levels of headings,to highlight key points and facilitatecomprehension of the text.The Publisher reserves the right to addor delete headings when necessary.
• Abbreviations should be used sparinglyand expanded at first mention.
• Use Système International (SI) units.
• Use generic names of drugs.
• All references should be cited in thetext and numbered consecutively us-ing superscript arabic numerals. Theauthor-date system of citation is NOTacceptable. “In press” references are tobe avoided. In the bibliography, titlesof journals should be abbreviated ac-cording to the Index Medicus. All au-thors should be listed up to six; if thereare more, only the first three shouldbe listed, followed by “et al” (Uniformrequirements for manuscripts submitted tobiomedical journals: see www.icmje.org ).Where necessary, references will bestyled to Dialogues in CardiovascularMedicine copyediting requirements.Authors bear total responsibility forthe accuracy and completeness of allreferences and for correct text citation.Example of style for references:
2. Darling RC, Brewster DC, Ottinger LW.Autopsy study of unoperated abdominal aorticaneurysms: the case for early resection.Circulation. 1977;56(suppl II):II161-II164.
3. Schulman JL. Immunology of influenza. In:Kilbourne ED, Alfade RT, eds. The InfluenzaViruses and Influenza. Orlando, Fla: AcademicPress Inc; 1975:373-393.
• Copyediting: all contributions toDialogues in Cardiovascular Medicine willbe styled by the Publisher’s editorialdepartment according to the specifica-tions of the current edition of theAmerican Medical Association Manual ofStyle, Williams & Wilkins. Page proofswill be sent to authors for approval andshould be returned within 5 days. If thistime is exceeded, changes made by theeditorial department will be assumedto be accepted by the author. Authorsare responsible for all statements madein their work, including changes madeby the editorial department and au-thorized by the author. The Publisherwill edit Editorials, Abstracts, andSeminal Paper Summaries to requiredsize if their length does not complywith specific requirements.
• Copyright of articles will be transferredto the Publisher of Dialogues inCardiovascular Medicine. For reproduction
of existing work, it is the author’s re-sponsibility to obtain copyright fromthe author(s) (including self) and thepublisher(s) and provide copies of theseauthorizations with the manuscript.
LEAD ARTICLE
The lead article should not exceed30 standard typed pages (7000 to8000 words), including an abstract ofno more than 200 words, no more than50 references, and a minimum of 5 -maximum of 10 illustrations (figures and/or tables). A maximum of 5-10 keywordsshould be included. The 3 questions forthe respondents should be introducedin or after the conclusion. A separate listof “10 references of seminal papers”as well as a separate list of “100 KeyReferences” should be provided.
RESPONDENT ARTICLES
Respondent articles should not exceed15 standard typed pages (3000 to4000 words), including an abstract ofno more than 125 words, no more than10 references, and a mimimun of 3 -maximum of 5 illustrations (figures andtables). A maximum of 5-10 keywordsshould be included.
SEMINAL PAPERSUMMARIES
Seminal paper summaries take up onepage of Dialogues in Cardiovascular Medicine:the length of each summary should IM-PERATIVELY be comprised between500 and 600 words, ie, not exceed3000 characters. Summaries that aretoo short or too long will be returned tothe author or edited by the Publisher.No figures, tables or references shouldbe included in seminal paper summaries.
FASCINOMA CARDIOLOGICAARTICLES
Fascinoma Cardiologica articles (A Lexiconof the Heart; Icons of Cardiology; Plants andthe Heart; Trails of Discovery, etc) should notexceed 2000 words (8 standard typedpages), should include 3 to 5 illustra-tions (figures and tables), and cite nomore than 15 references. A maximum of5-10 keywords should be included.No abstract.