Page 1
Common Otolaryngological
Congenital Abnormalities
Viet Pham, M.D.
Lewis Hutchinson, M.D.
Harold Pine, M.D.
Shraddha Mukerji, M.D.
The University of Texas Medical Branch
Department of Otolaryngology
November 22, 2010
Visual Synopsis of Classic Syndromes and Features
htt
p:/
/ww
w.e
xp
losm
.ne
t/co
mic
s
Page 2
Foreword and
Acknowledgements
Special appreciation to Dr. Hutchinson for his
assistance and contribution
Additional gratitude to Drs. Pine and Mukerji
All clinical photos are presented solely for educational
purposes
All other photos were obtained via a Google search unless
otherwise specified and are used without permission
Page 3
Objective
Highlight typical features of congenital abnormalities
evaluated in the otolaryngology practice
Visual emphasis on classical presentation of commonly
encountered syndromes
Page 4
Down Syndrome (Trisomy 21)
Extra chromosome 21
Meiotic nondisjunction in gamete formation
Mosaicism (1-2%)
Robertsonian translocation (2-3%)
Duplication (rare)
Increased risk with advanced maternal age
Most common cause of intellectual disability
Page 5
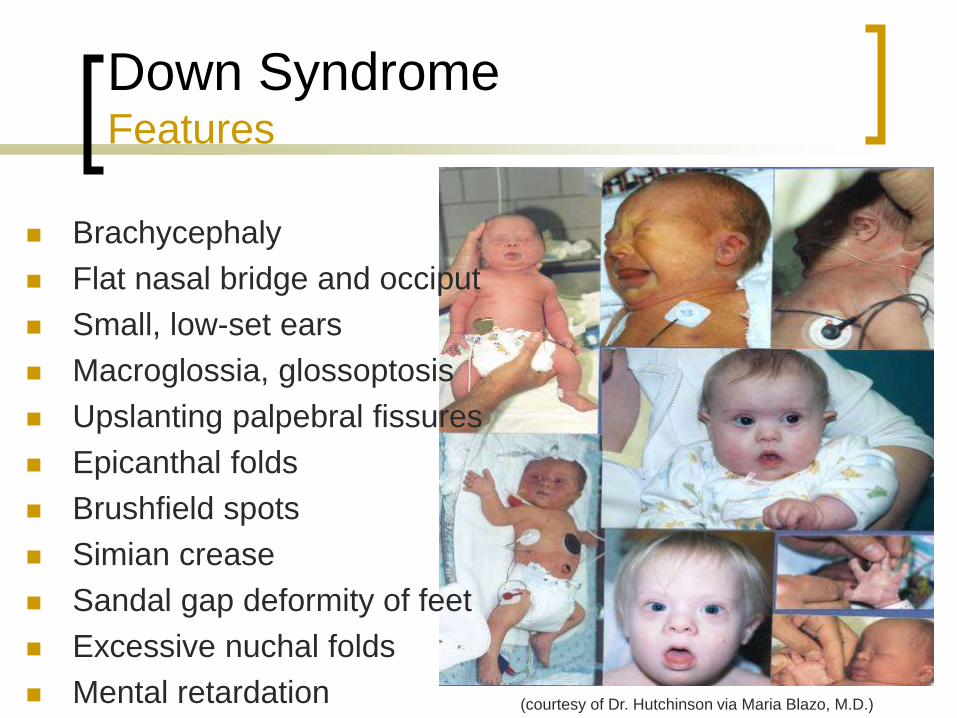
Down Syndrome Features
Brachycephaly
Flat nasal bridge and occiput
Small, low-set ears
Macroglossia, glossoptosis
Upslanting palpebral fissures
Epicanthal folds
Brushfield spots
Simian crease
Sandal gap deformity of feet
Excessive nuchal folds
Mental retardation (courtesy of Dr. Hutchinson via Maria Blazo, M.D.)
Page 6
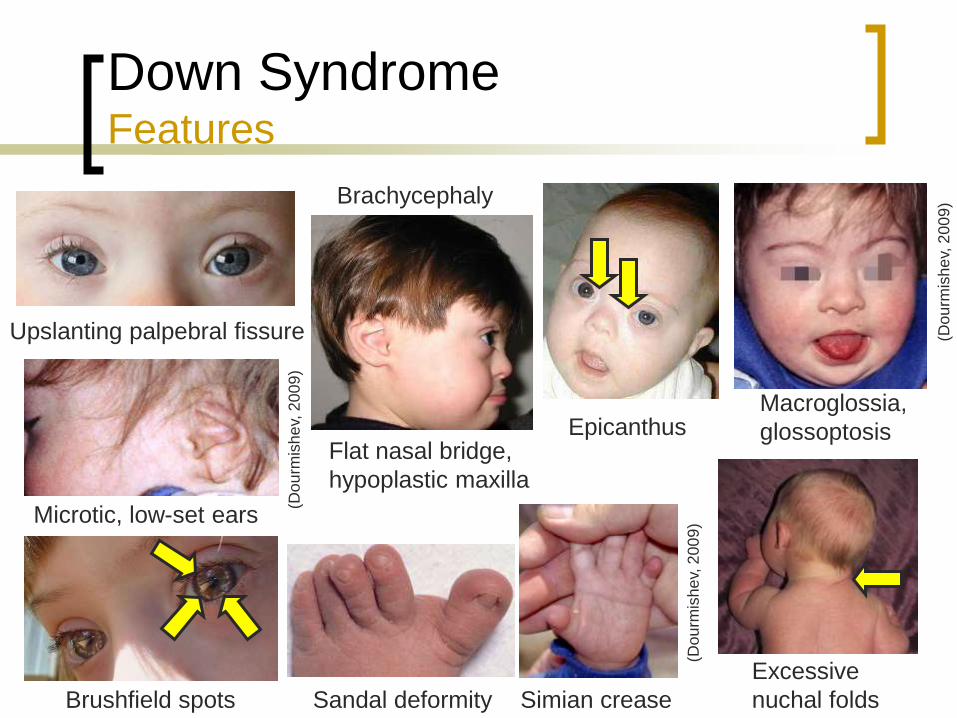
Down Syndrome Features
(Do
urm
ishe
v, 2
00
9)
Simian crease Brushfield spots Sandal deformity
Excessive
nuchal folds
Upslanting palpebral fissure
Macroglossia,
glossoptosis
(Do
urm
ishe
v, 2
00
9)
(Do
urm
ishe
v, 2
00
9)
Microtic, low-set ears
Epicanthus
Brachycephaly
Flat nasal bridge,
hypoplastic maxilla
Page 7
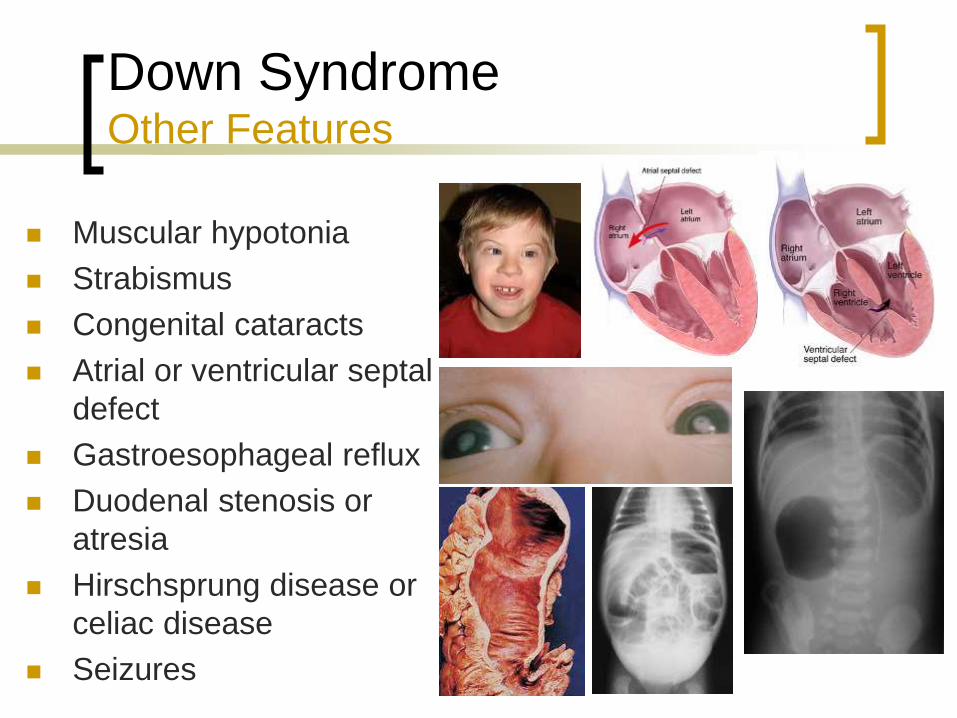
Down Syndrome Other Features
Muscular hypotonia
Strabismus
Congenital cataracts
Atrial or ventricular septal
defect
Gastroesophageal reflux
Duodenal stenosis or
atresia
Hirschsprung disease or
celiac disease
Seizures
Page 8
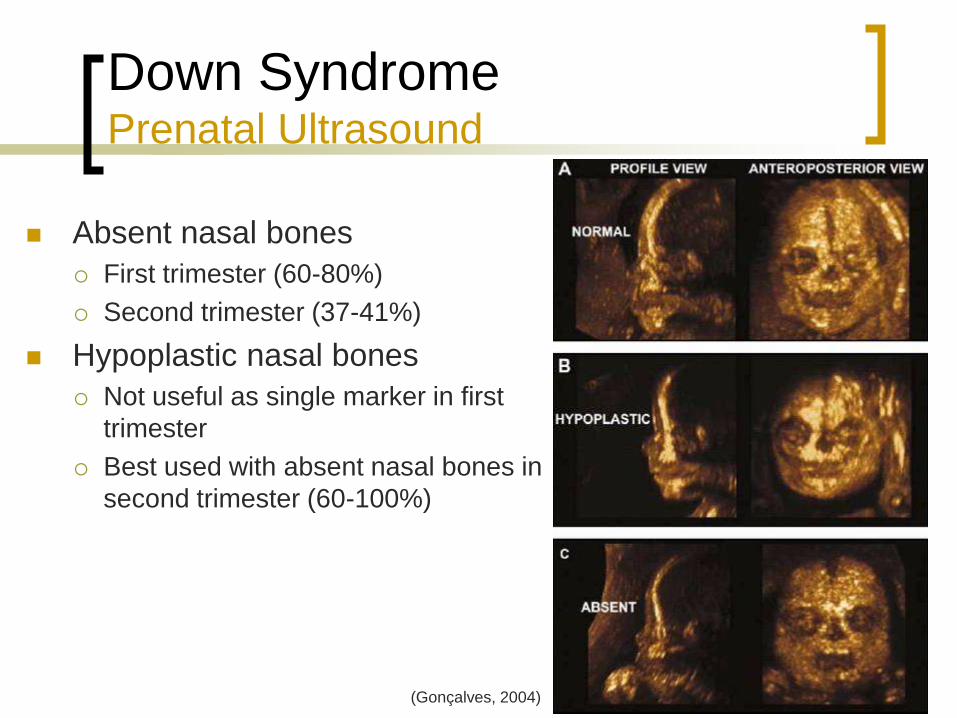
Down Syndrome Prenatal Ultrasound
Absent nasal bones
First trimester (60-80%)
Second trimester (37-41%)
Hypoplastic nasal bones
Not useful as single marker in first
trimester
Best used with absent nasal bones in
second trimester (60-100%)
(Gonçalves, 2004)
Page 9
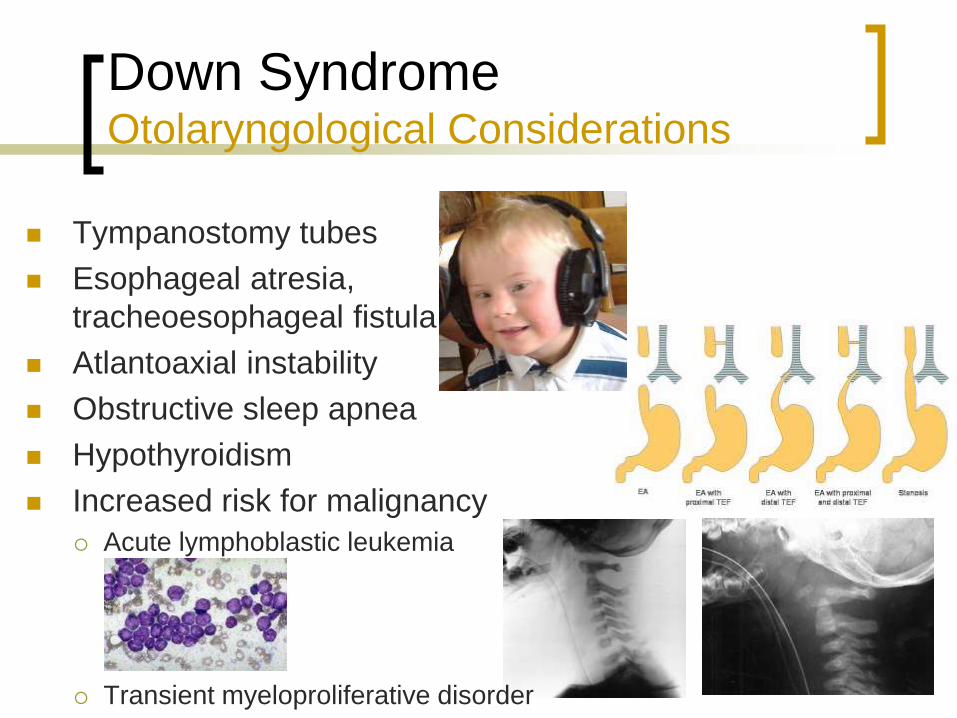
Down Syndrome Otolaryngological Considerations
Tympanostomy tubes
Esophageal atresia,
tracheoesophageal fistula
Atlantoaxial instability
Obstructive sleep apnea
Hypothyroidism
Increased risk for malignancy
Acute lymphoblastic leukemia
Transient myeloproliferative disorder
Page 10
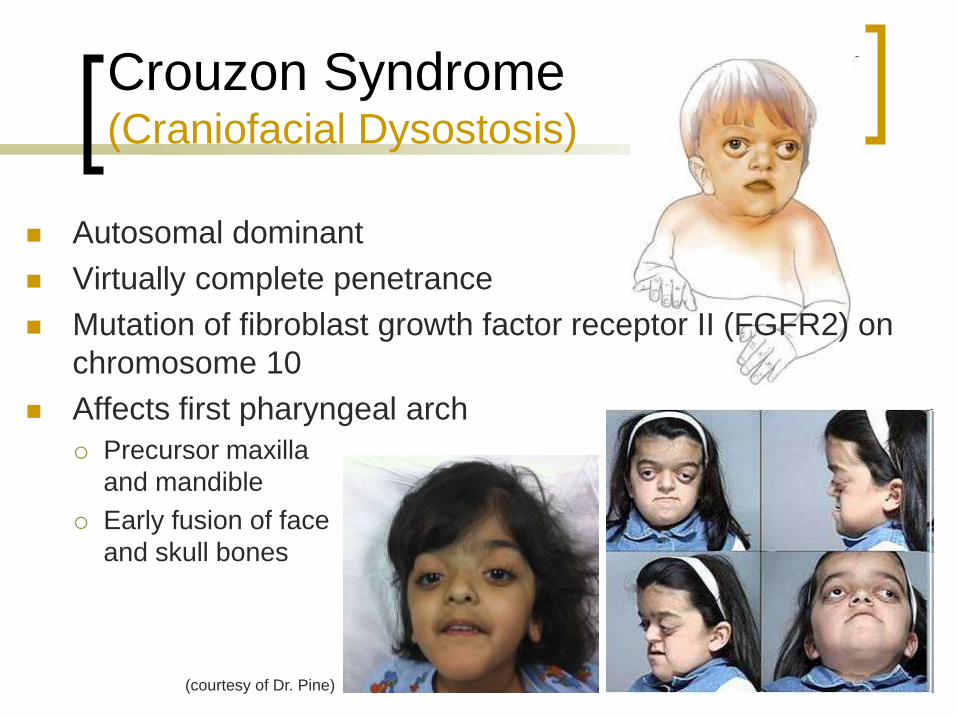
Crouzon Syndrome (Craniofacial Dysostosis)
Autosomal dominant
Virtually complete penetrance
Mutation of fibroblast growth factor receptor II (FGFR2) on
chromosome 10
Affects first pharyngeal arch
Precursor maxilla
and mandible
Early fusion of face
and skull bones
(courtesy of Dr. Pine)
Page 11
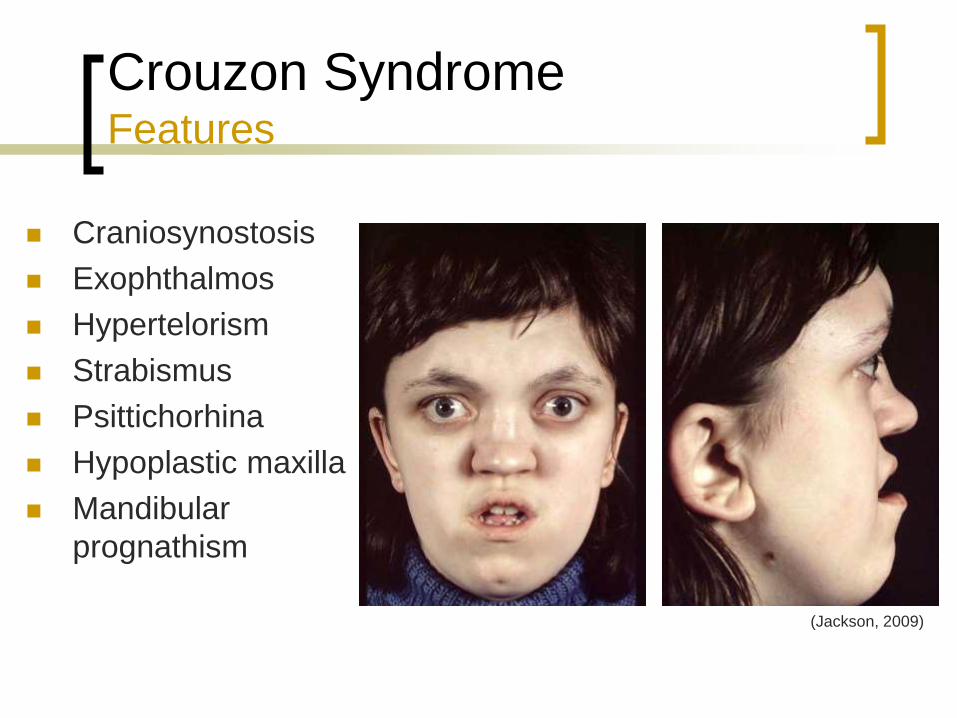
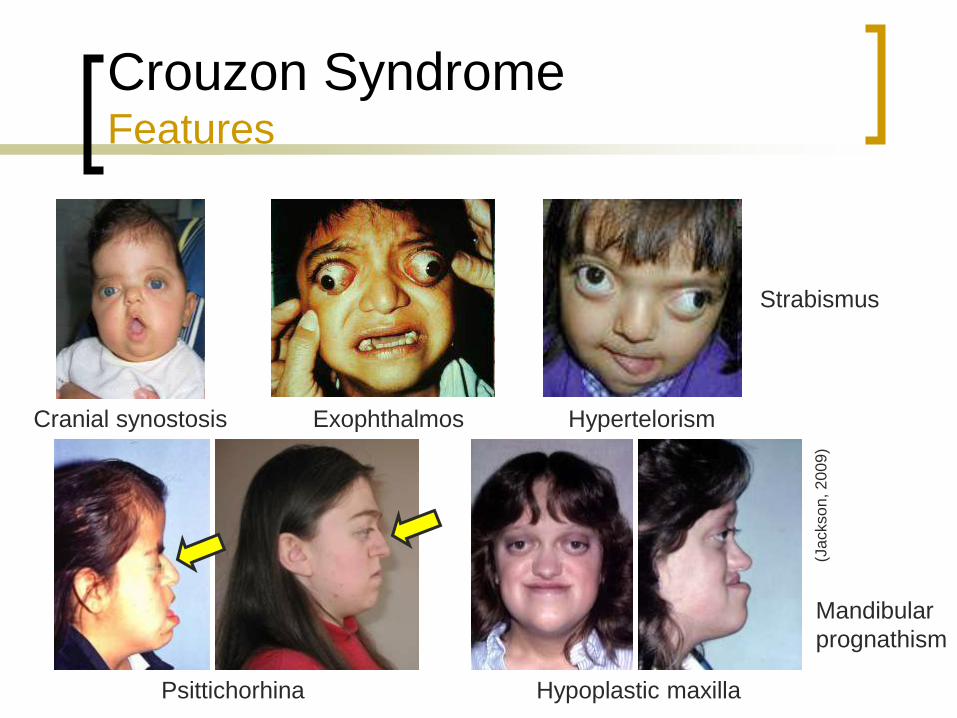
Crouzon Syndrome Features
Craniosynostosis
Exophthalmos
Hypertelorism
Strabismus
Psittichorhina
Hypoplastic maxilla
Mandibular
prognathism
(Jackson, 2009)
Page 12
Crouzon Syndrome Features
Cranial synostosis Exophthalmos Hypertelorism
Strabismus
Hypoplastic maxilla
Mandibular
prognathism
Psittichorhina
(Jackso
n, 2
00
9)
Page 13
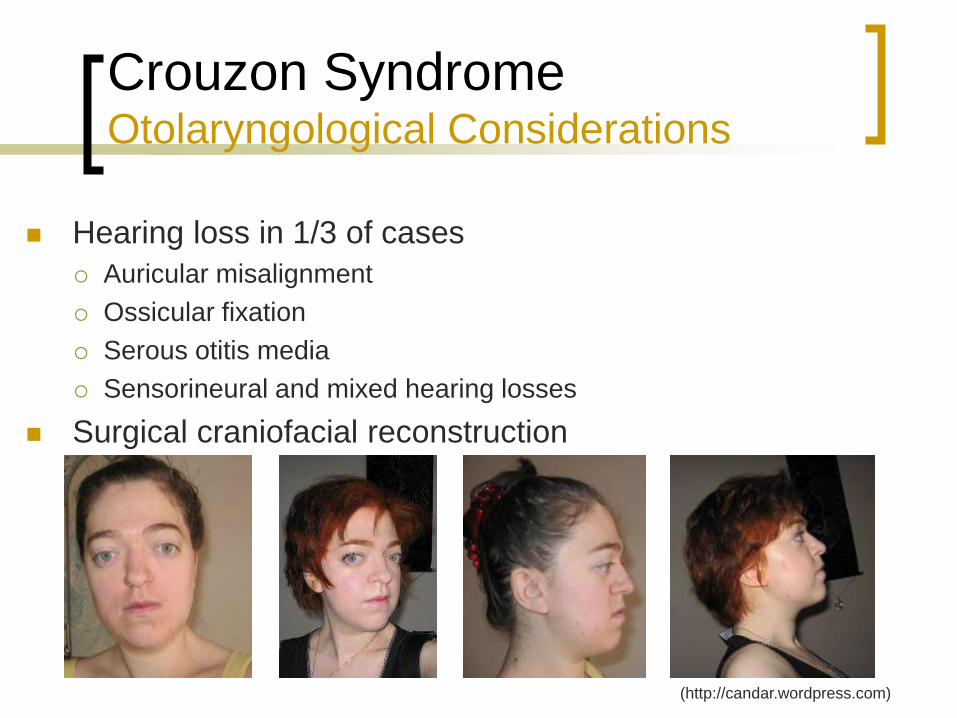
Crouzon Syndrome Otolaryngological Considerations
Hearing loss in 1/3 of cases
Auricular misalignment
Ossicular fixation
Serous otitis media
Sensorineural and mixed hearing losses
Surgical craniofacial reconstruction
(http://candar.wordpress.com)
Page 14
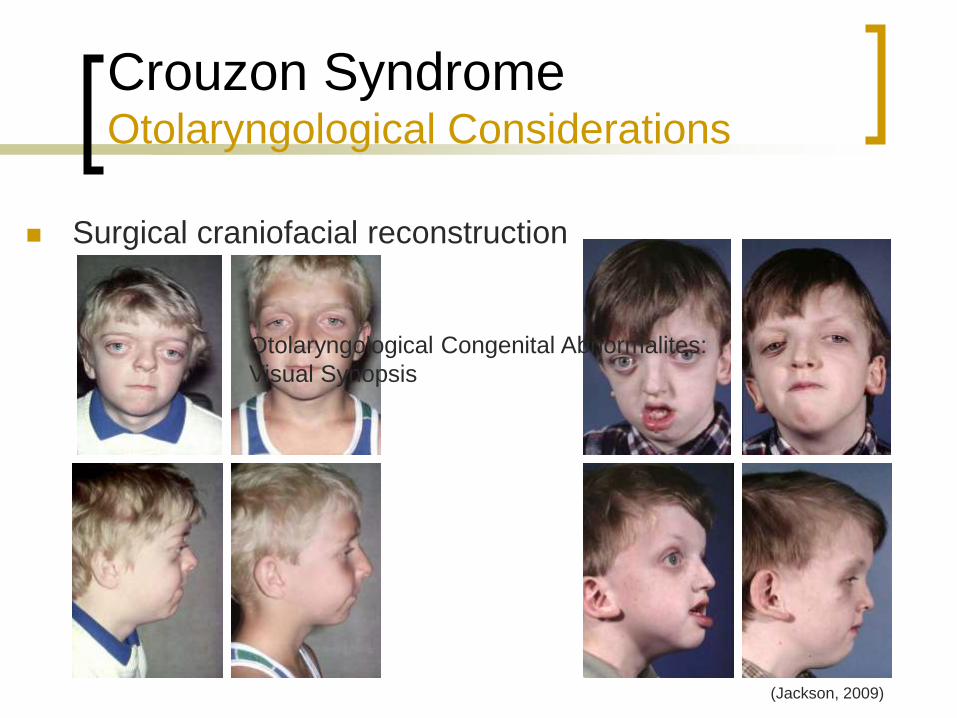
Crouzon Syndrome Otolaryngological Considerations
Surgical craniofacial reconstruction
(Jackson, 2009)
Otolaryngological Congenital Abnormalites:
Visual Synopsis
Page 15
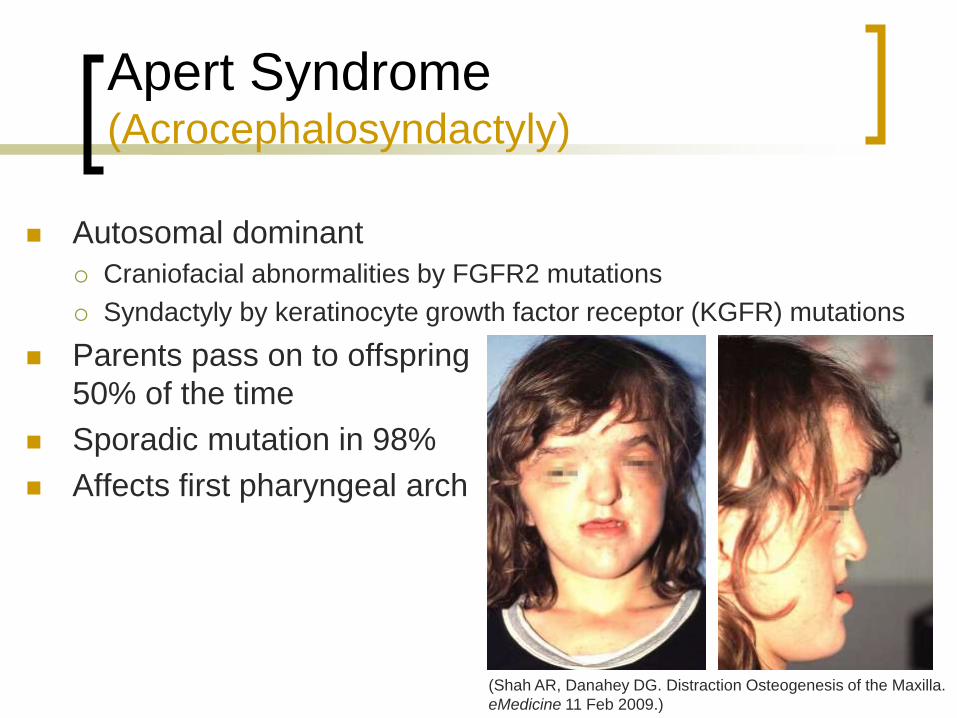
Apert Syndrome (Acrocephalosyndactyly)
Autosomal dominant
Craniofacial abnormalities by FGFR2 mutations
Syndactyly by keratinocyte growth factor receptor (KGFR) mutations
Parents pass on to offspring
50% of the time
Sporadic mutation in 98%
Affects first pharyngeal arch
(Shah AR, Danahey DG. Distraction Osteogenesis of the Maxilla.
eMedicine 11 Feb 2009.)
Page 16
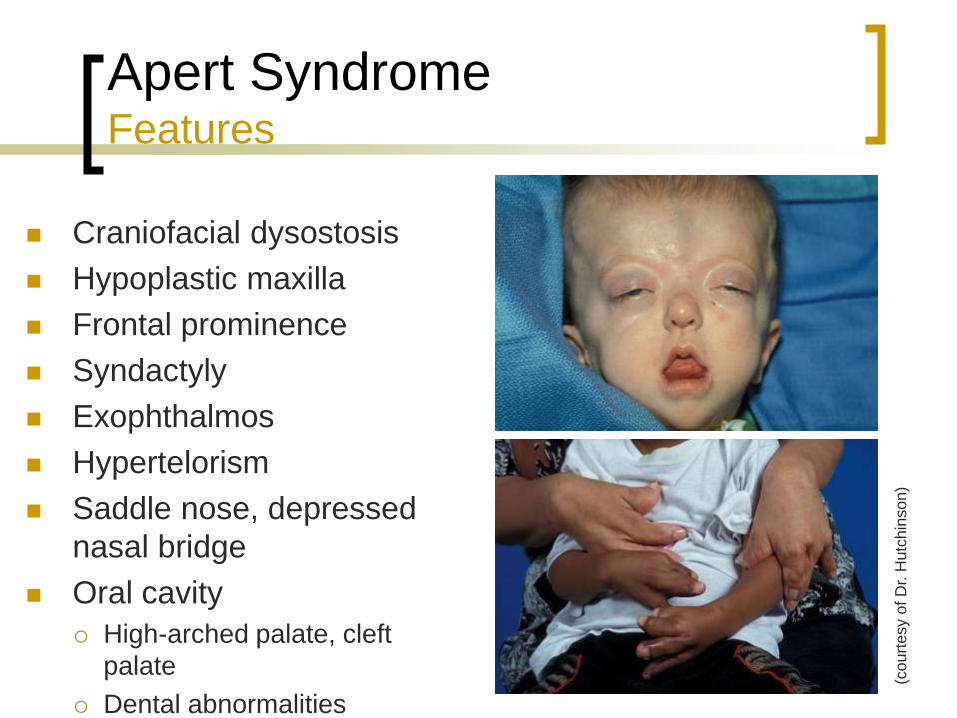
Apert Syndrome Features
Craniofacial dysostosis
Hypoplastic maxilla
Frontal prominence
Syndactyly
Exophthalmos
Hypertelorism
Saddle nose, depressed
nasal bridge
Oral cavity
High-arched palate, cleft
palate
Dental abnormalities
(co
urt
esy o
f D
r. H
utc
hin
so
n)
Page 17
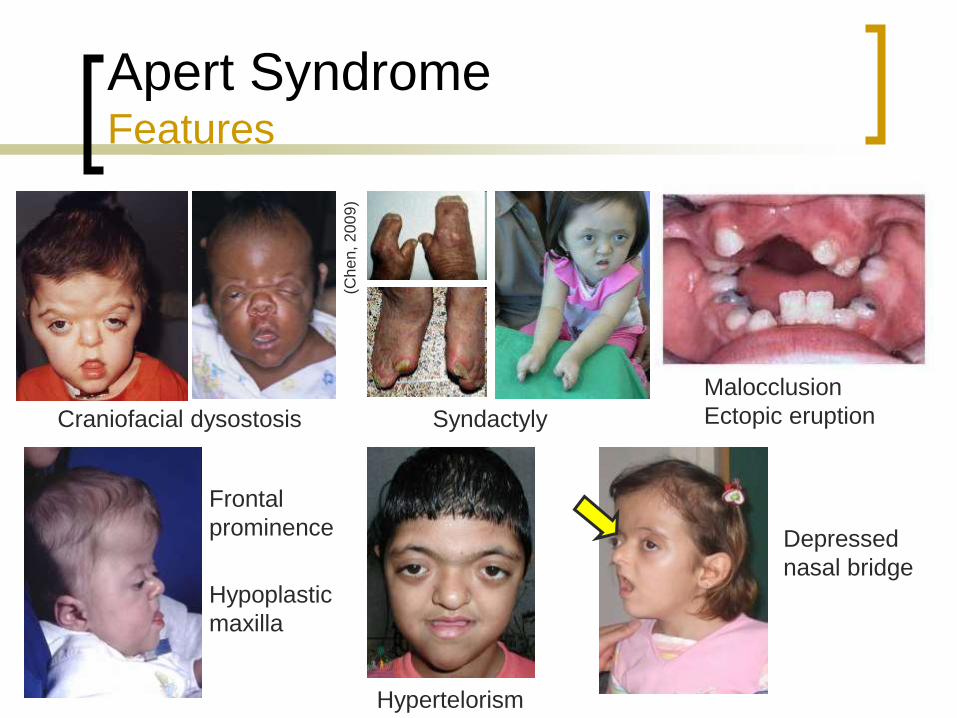
Apert Syndrome Features
Craniofacial dysostosis
Hypoplastic
maxilla
Frontal
prominence
Syndactyly
Depressed
nasal bridge
Malocclusion
Ectopic eruption
Hypertelorism
(Ch
en
, 2
00
9)
Page 18
Apert Syndrome Dr. Hutchinson’s mnemonic
Apert = Crouzon + Syndactyly
Page 19
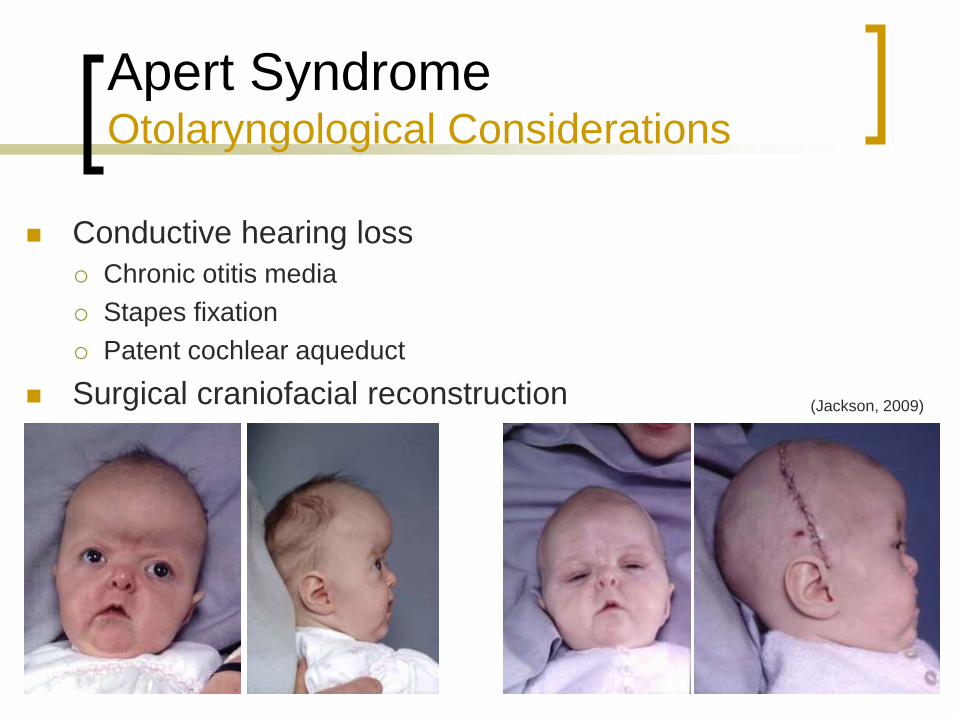
Apert Syndrome Otolaryngological Considerations
Conductive hearing loss
Chronic otitis media
Stapes fixation
Patent cochlear aqueduct
Surgical craniofacial reconstruction (Jackson, 2009)
Page 20
Treacher Collins Syndrome (Mandibulofacial Dysostosis)
Also known as Franceschetti-Zwahlen-Klein syndrome
Autosomal dominant
TCOF1 gene on chromosome 5q
New mutation in up to 60%
Complete penetrance, variable expression
First and second pharyngeal arches,
grooves, and pouches
Page 21
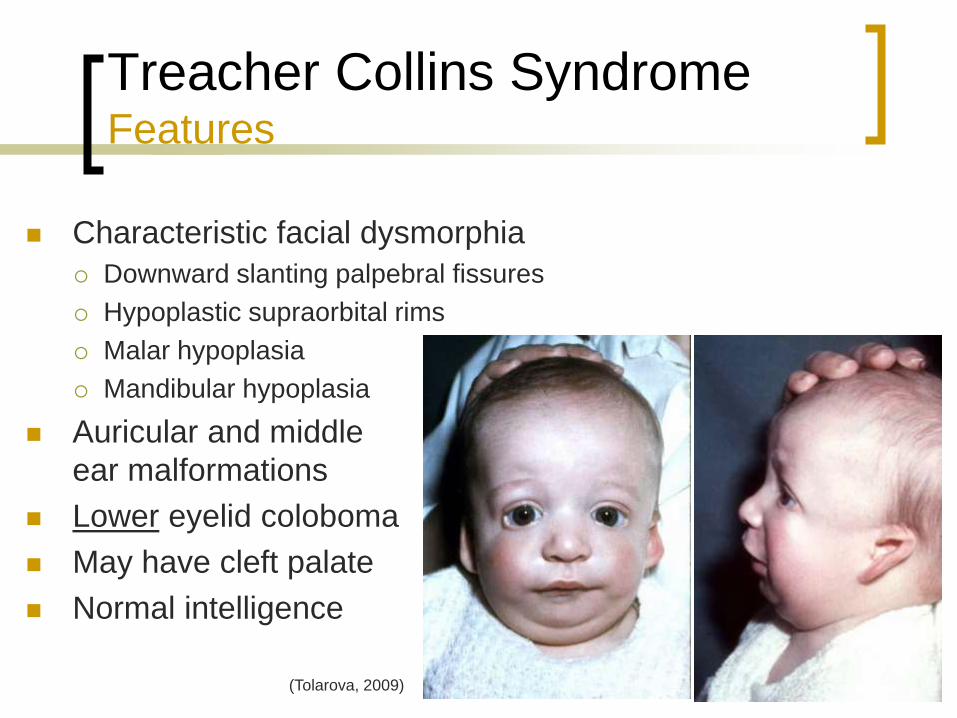
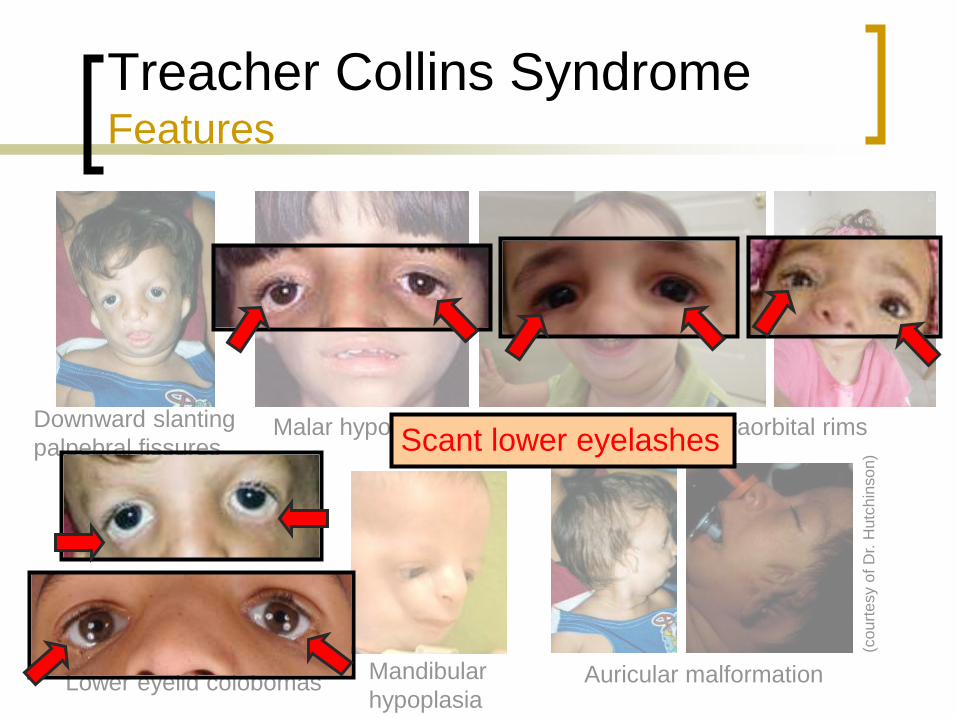
Treacher Collins Syndrome Features
Characteristic facial dysmorphia
Downward slanting palpebral fissures
Hypoplastic supraorbital rims
Malar hypoplasia
Mandibular hypoplasia
Auricular and middle
ear malformations
Lower eyelid coloboma
May have cleft palate
Normal intelligence
(Tolarova, 2009)
Page 22
Treacher Collins Syndrome Features
Downward slanting
palpebral fissures
Lower eyelid colobomas
Hypoplastic supraorbital rims
Mandibular
hypoplasia Auricular malformation
Malar hypoplasia
(cou
rtesy o
f D
r. H
utc
hin
so
n)
Page 23
(cou
rtesy o
f D
r. H
utc
hin
so
n)
Treacher Collins Syndrome Features
Downward slanting
palpebral fissures
Lower eyelid colobomas
Hypoplastic supraorbital rims
Mandibular
hypoplasia Auricular malformation
Malar hypoplasia Scant lower eyelashes
Page 24
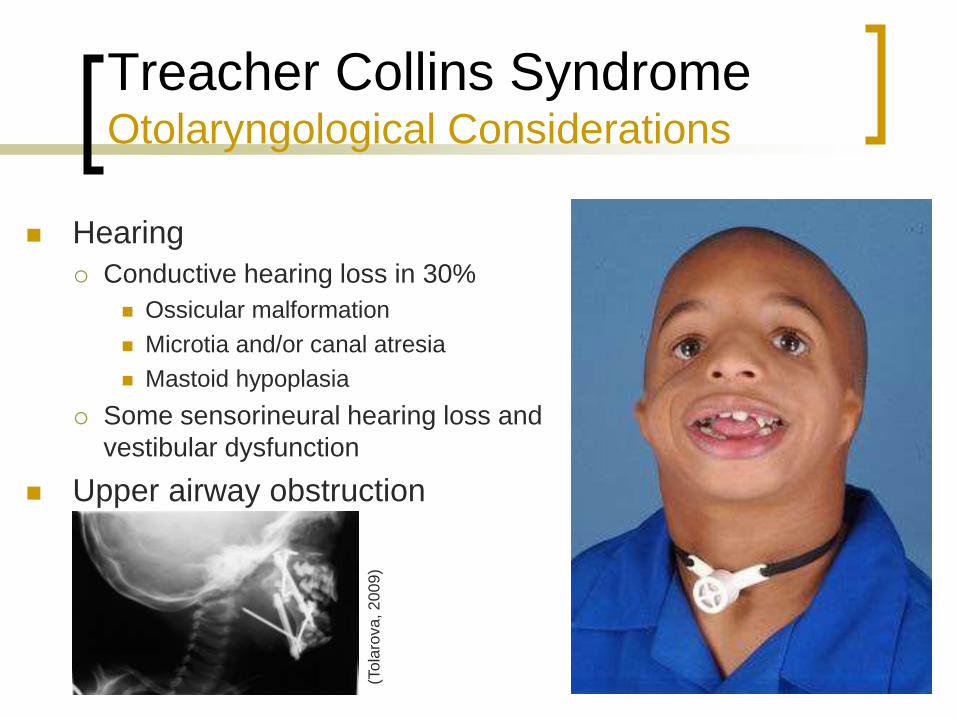
Treacher Collins Syndrome Otolaryngological Considerations
Hearing
Conductive hearing loss in 30%
Ossicular malformation
Microtia and/or canal atresia
Mastoid hypoplasia
Some sensorineural hearing loss and
vestibular dysfunction
Upper airway obstruction
(To
laro
va
, 2
00
9)
Page 25
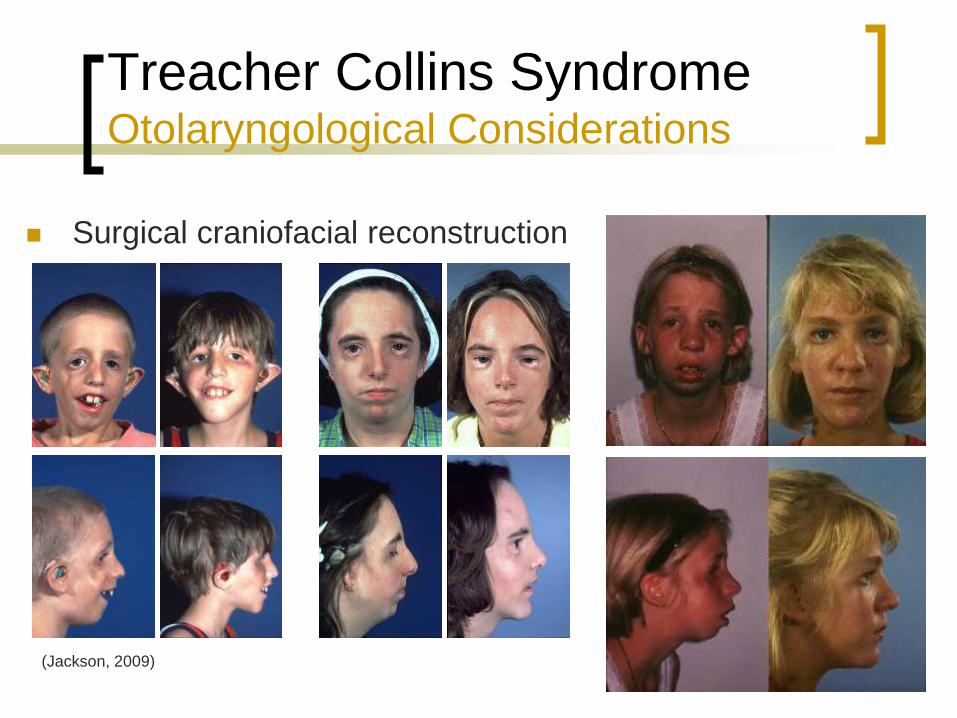
Treacher Collins Syndrome Otolaryngological Considerations
Surgical craniofacial reconstruction
(Jackson, 2009)
Page 26
Goldenhar Syndrome Oculoauriculovertebral Dysplasia
Diverse etiologies
In utero vascular disruption with hematoma
Disturbed neural crest cells at 30-45 days gestation
No single genetic locus
First and second branchial arch
Hemifacial microsomia
when no internal organ or
vertebral disruption
Page 27
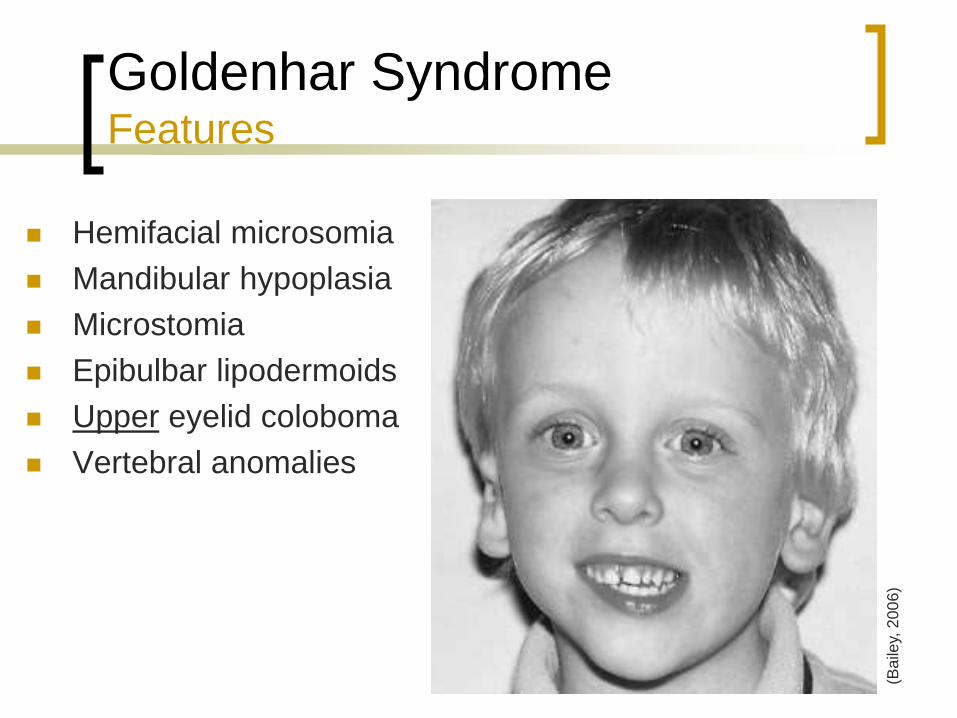
Goldenhar Syndrome Features
Hemifacial microsomia
Mandibular hypoplasia
Microstomia
Epibulbar lipodermoids
Upper eyelid coloboma
Vertebral anomalies
(Ba
iley,
20
06
)
Page 28
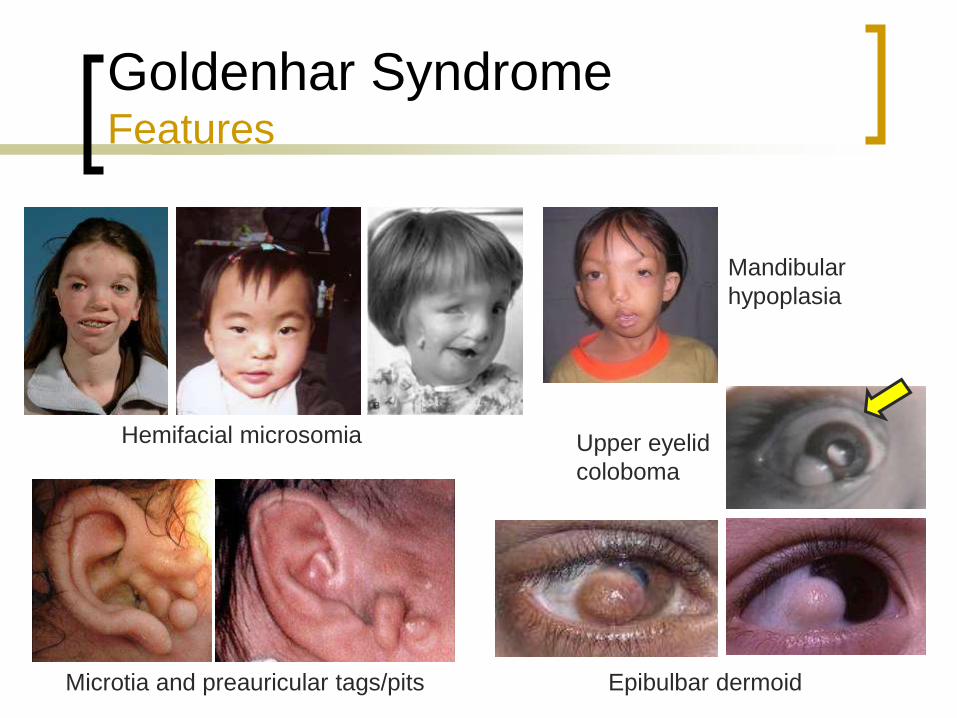
Goldenhar Syndrome Features
Hemifacial microsomia
Epibulbar dermoid
Mandibular
hypoplasia
Microtia and preauricular tags/pits
Upper eyelid
coloboma
Page 29
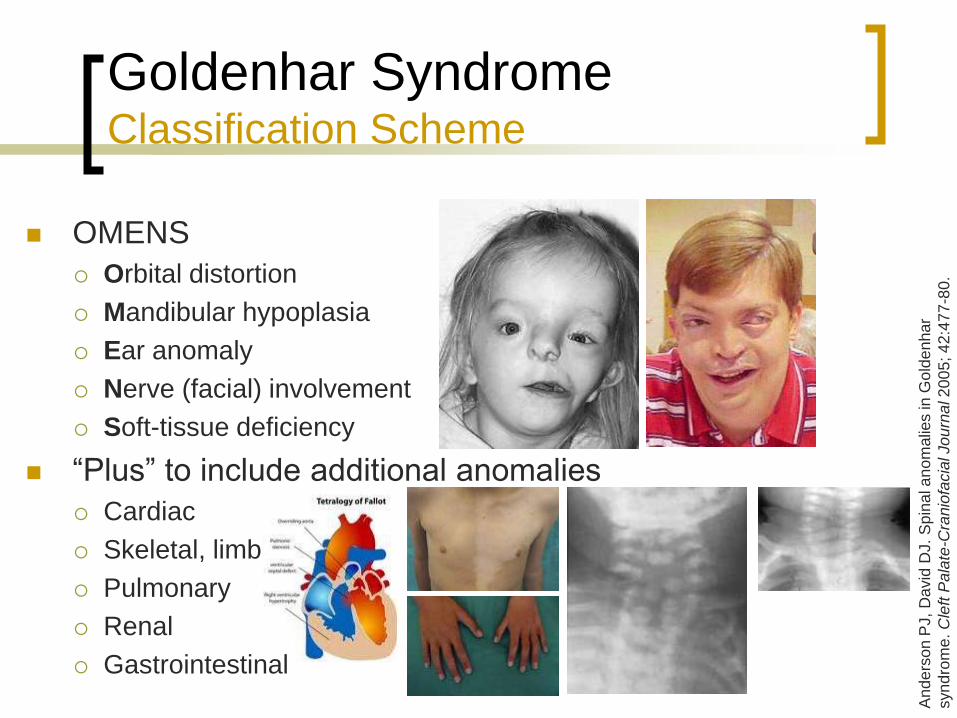
OMENS
Orbital distortion
Mandibular hypoplasia
Ear anomaly
Nerve (facial) involvement
Soft-tissue deficiency
“Plus” to include additional anomalies
Cardiac
Skeletal, limb
Pulmonary
Renal
Gastrointestinal
Goldenhar Syndrome Classification Scheme
An
de
rso
n P
J,
David
DJ.
Sp
ina
l a
no
malie
s in G
old
en
har
syn
dro
me
. C
left
Pa
late
-Cra
nio
facia
l Jo
urn
al 2
00
5; 4
2:4
77
-80.
Page 30
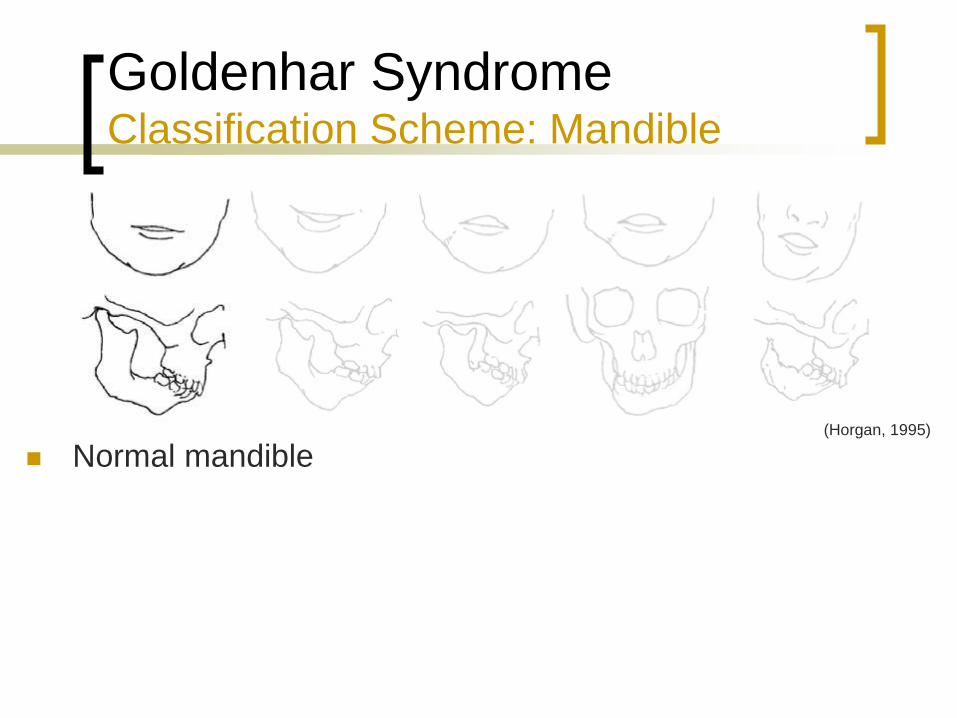
Goldenhar Syndrome Classification Scheme: Mandible
Normal mandible (Horgan, 1995)
Page 31
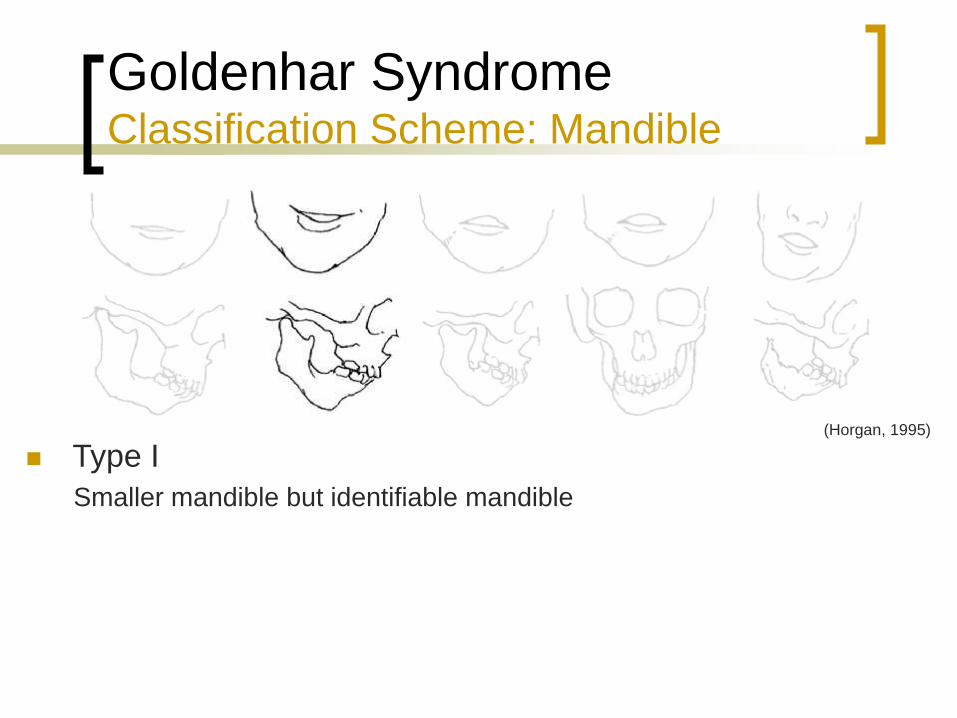
Goldenhar Syndrome Classification Scheme: Mandible
Type I
Smaller mandible but identifiable mandible
(Horgan, 1995)
Page 32
Goldenhar Syndrome Classification Scheme: Mandible
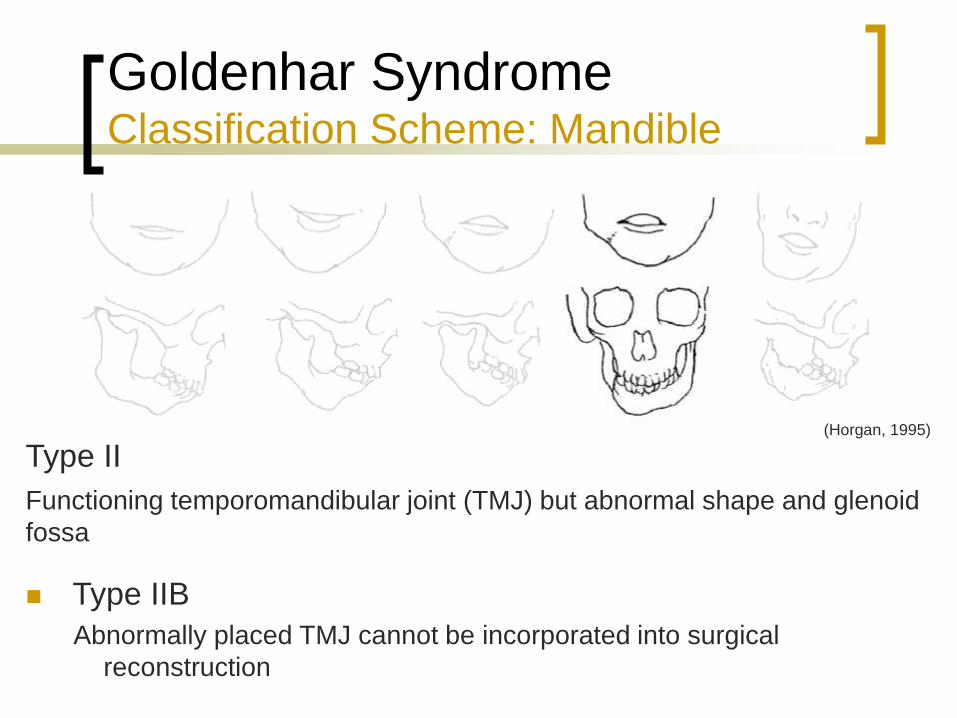
Type II
Type IIA
Glenoid fossa is in an acceptable position
Functioning temporomandibular joint (TMJ) but abnormal shape and glenoid
fossa
(Horgan, 1995)
Page 33
Goldenhar Syndrome Classification Scheme: Mandible
Type II
Type IIB
Abnormally placed TMJ cannot be incorporated into surgical
reconstruction
Functioning temporomandibular joint (TMJ) but abnormal shape and glenoid
fossa
(Horgan, 1995)
Page 34
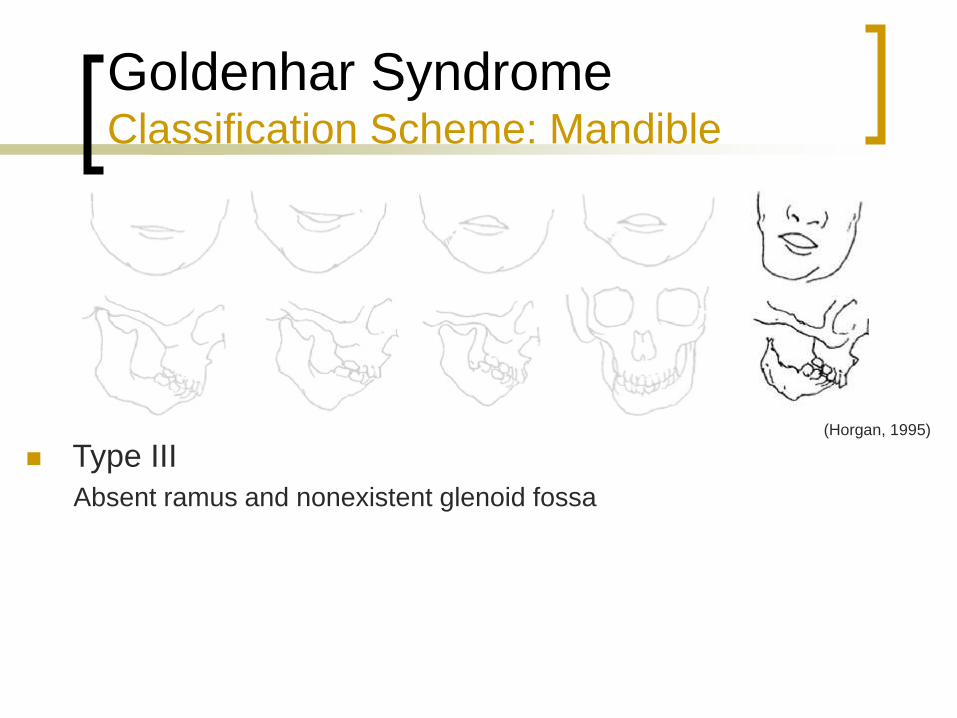
Goldenhar Syndrome Classification Scheme: Mandible
Type III
Absent ramus and nonexistent glenoid fossa
(Horgan, 1995)
Page 35
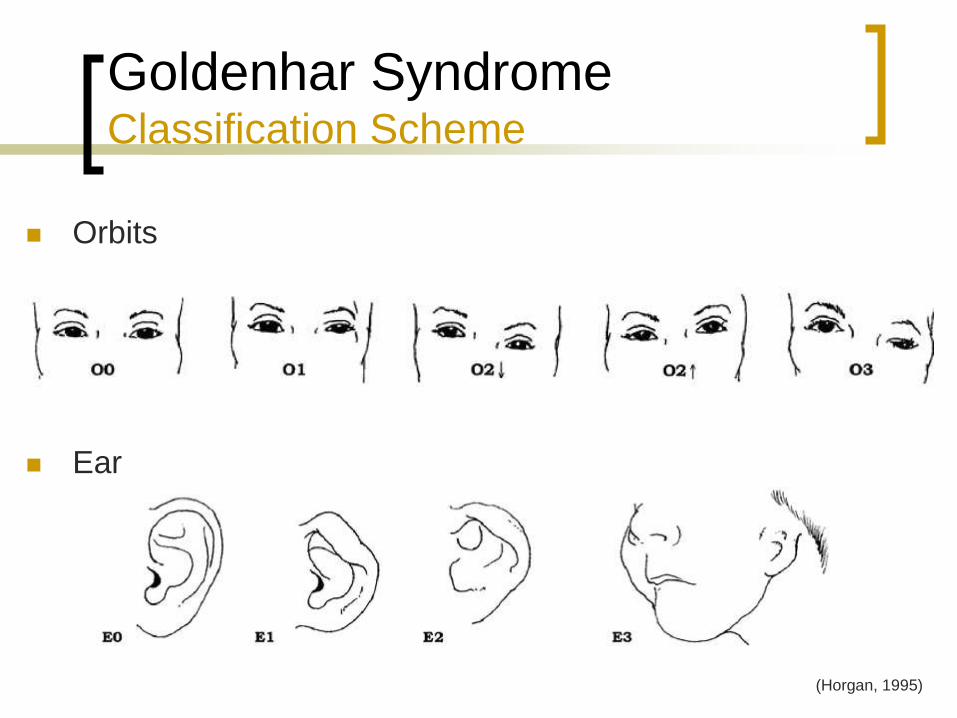
Goldenhar Syndrome Classification Scheme
Orbits
Ear
(Horgan, 1995)
Page 36
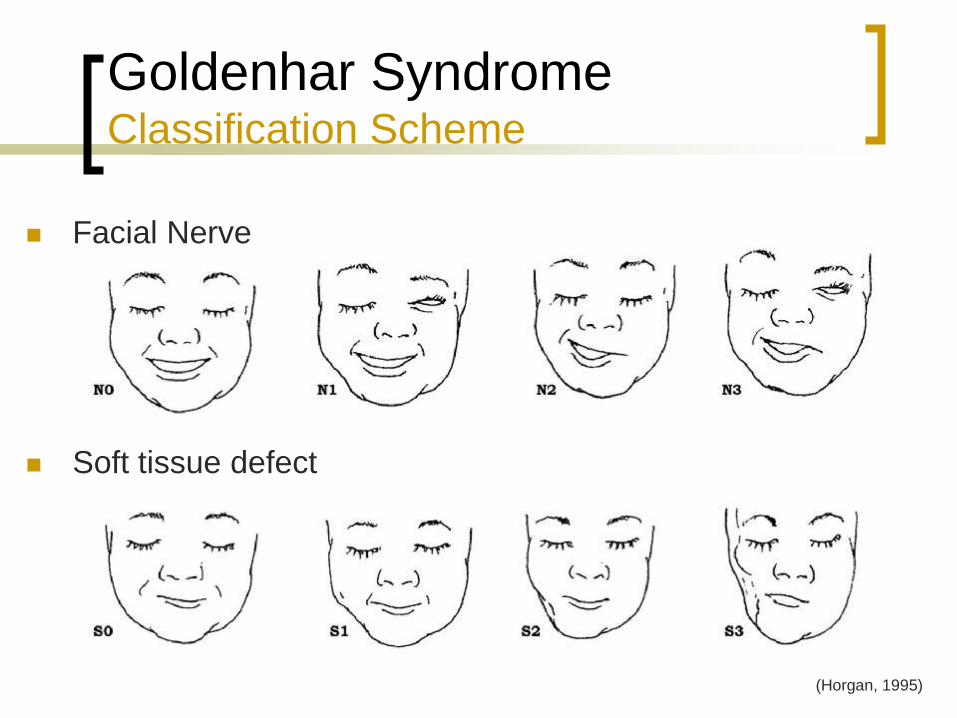
Goldenhar Syndrome Classification Scheme
Facial Nerve
Soft tissue defect
(Horgan, 1995)
Page 37
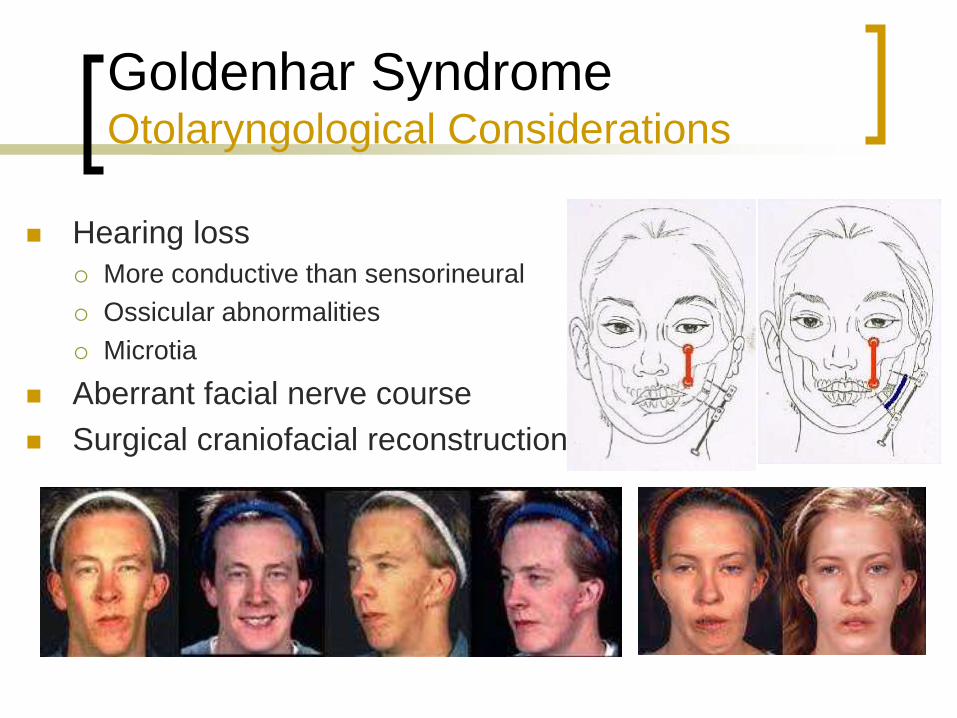
Goldenhar Syndrome Otolaryngological Considerations
Hearing loss
More conductive than sensorineural
Ossicular abnormalities
Microtia
Aberrant facial nerve course
Surgical craniofacial reconstruction
Page 38
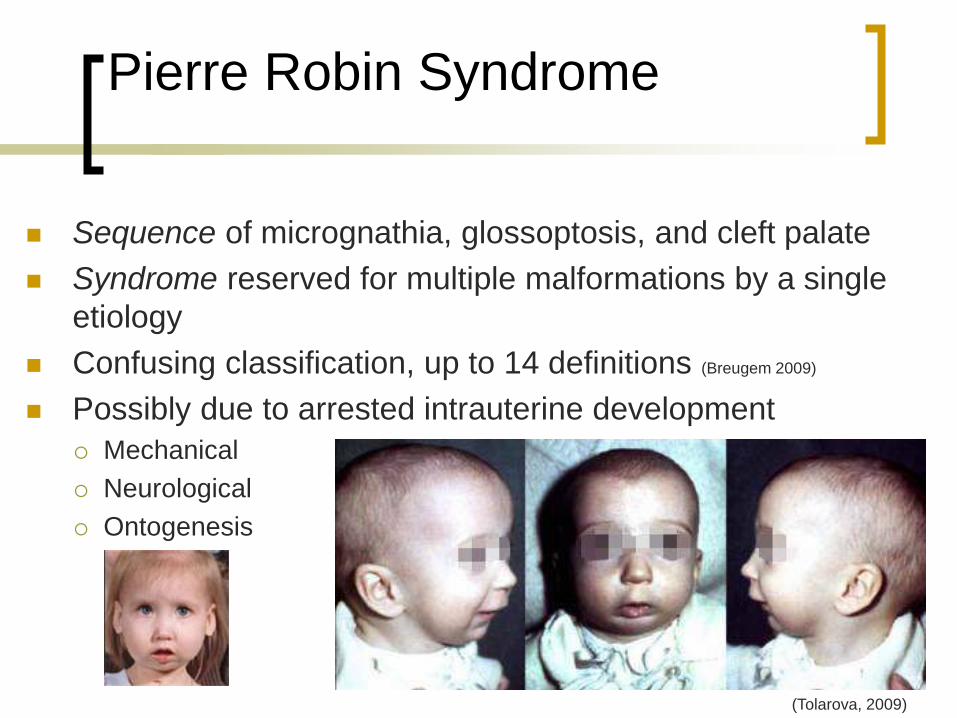
Pierre Robin Syndrome
Sequence of micrognathia, glossoptosis, and cleft palate
Syndrome reserved for multiple malformations by a single
etiology
Confusing classification, up to 14 definitions (Breugem 2009)
Possibly due to arrested intrauterine development
Mechanical
Neurological
Ontogenesis
(Tolarova, 2009)
Page 39
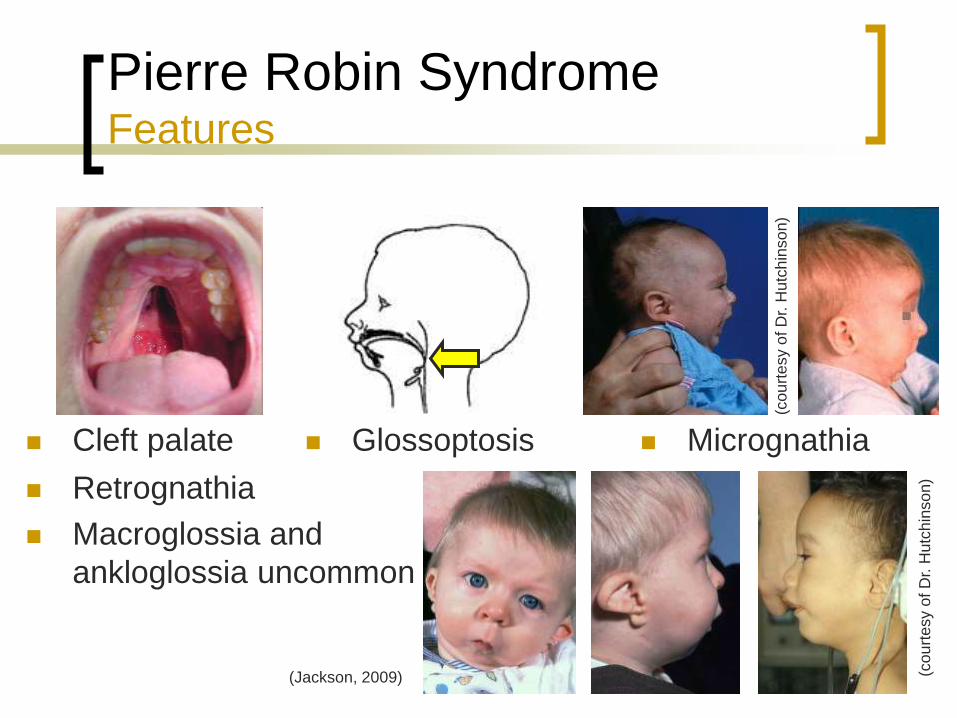
Pierre Robin Syndrome Features
Cleft palate Glossoptosis
Retrognathia
Macroglossia and
ankloglossia uncommon
Micrognathia
(Jackson, 2009) (co
urt
esy o
f D
r. H
utc
hin
so
n)
(co
urt
esy o
f D
r. H
utc
hin
so
n)
Page 40
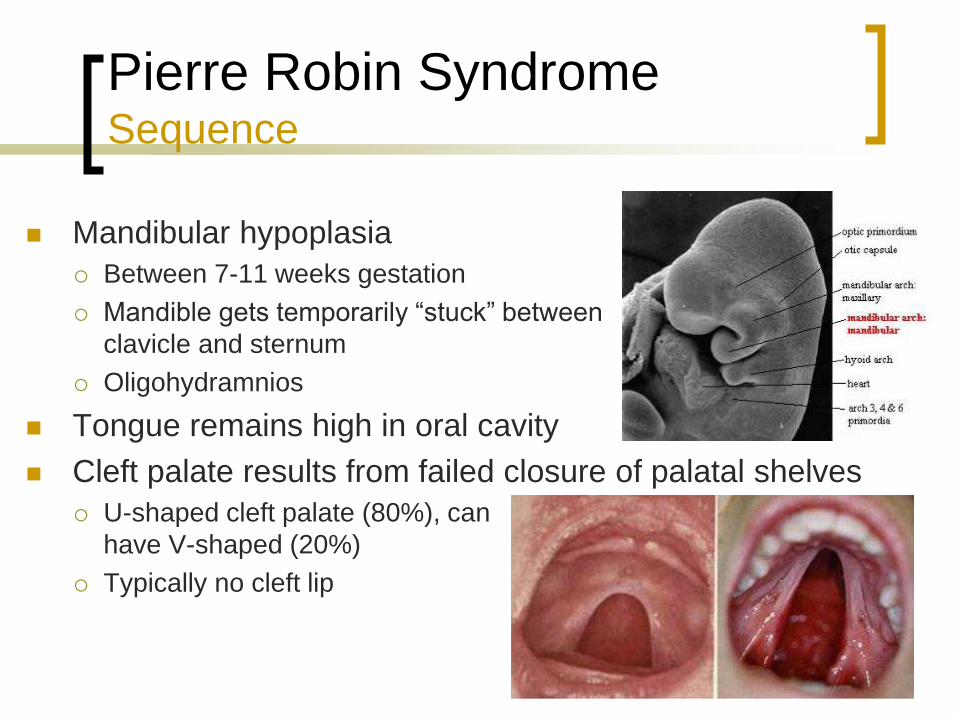
Pierre Robin Syndrome Sequence
Mandibular hypoplasia
Between 7-11 weeks gestation
Mandible gets temporarily “stuck” between
clavicle and sternum
Oligohydramnios
Tongue remains high in oral cavity
Cleft palate results from failed closure of palatal shelves
U-shaped cleft palate (80%), can
have V-shaped (20%)
Typically no cleft lip
Page 41
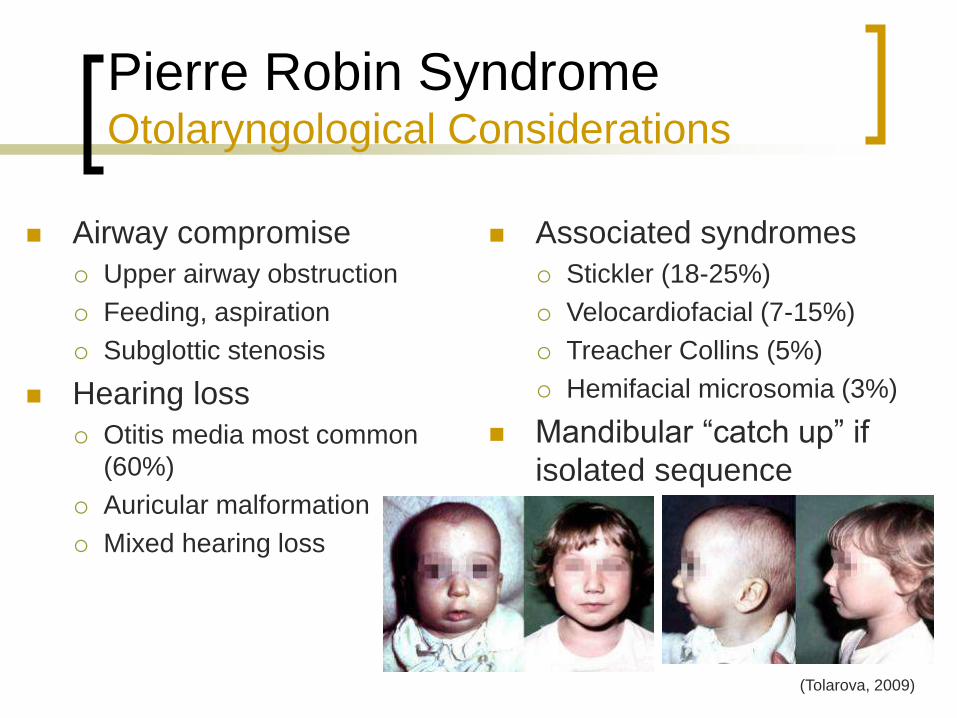
Pierre Robin Syndrome Otolaryngological Considerations
Airway compromise
Upper airway obstruction
Feeding, aspiration
Subglottic stenosis
Hearing loss
Otitis media most common
(60%)
Auricular malformation
Mixed hearing loss
Associated syndromes
Stickler (18-25%)
Velocardiofacial (7-15%)
Treacher Collins (5%)
Hemifacial microsomia (3%)
Mandibular “catch up” if
isolated sequence
(Tolarova, 2009)
Page 42
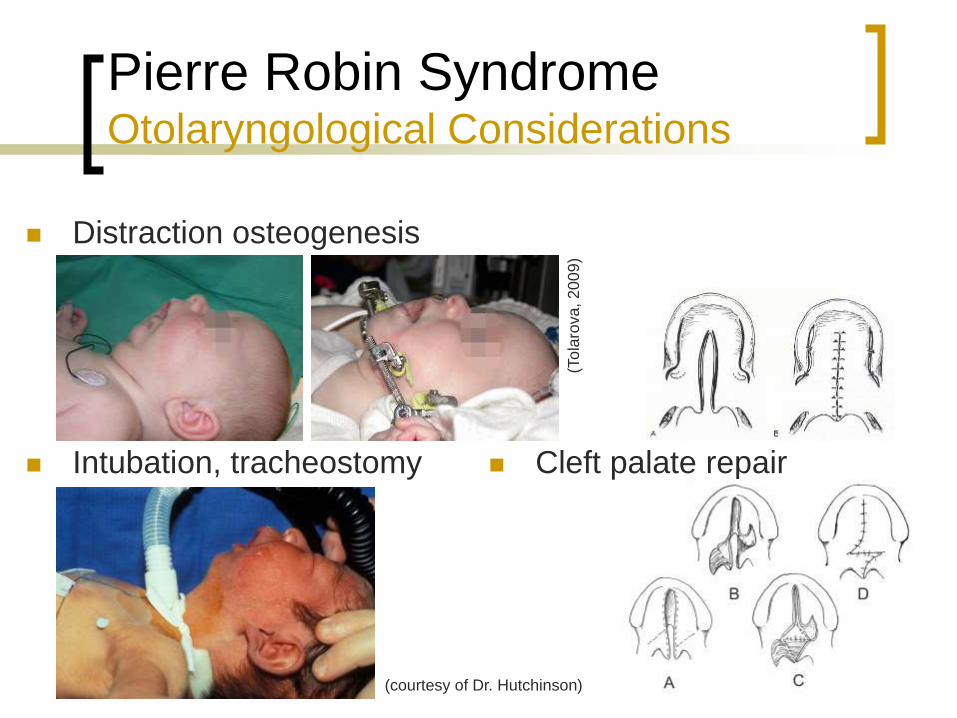
Pierre Robin Syndrome Otolaryngological Considerations
Distraction osteogenesis
Intubation, tracheostomy
(To
laro
va
, 2
00
9)
Cleft palate repair
(courtesy of Dr. Hutchinson)
Page 43
Stickler Syndrome
Autosomal dominant
Mutations of type II and XI collagen
COL2A1 gene on chromosome 12
COL11A1 and COL11A2 genes on chromosome 6
COL9A1 is rare recessive variant
Craniofacial, ocular, and
arthopathic features
Page 44
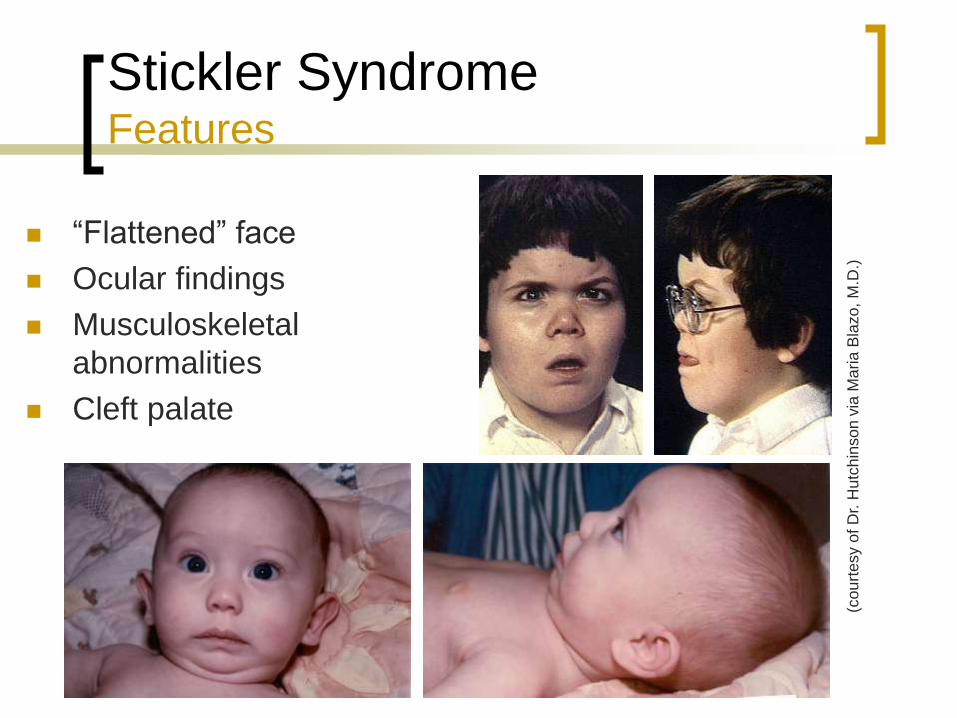
Stickler Syndrome Features
“Flattened” face
Ocular findings
Musculoskeletal
abnormalities
Cleft palate
(cou
rtesy o
f D
r. H
utc
hin
so
n v
ia M
aria
Bla
zo
, M
.D.)
Page 45
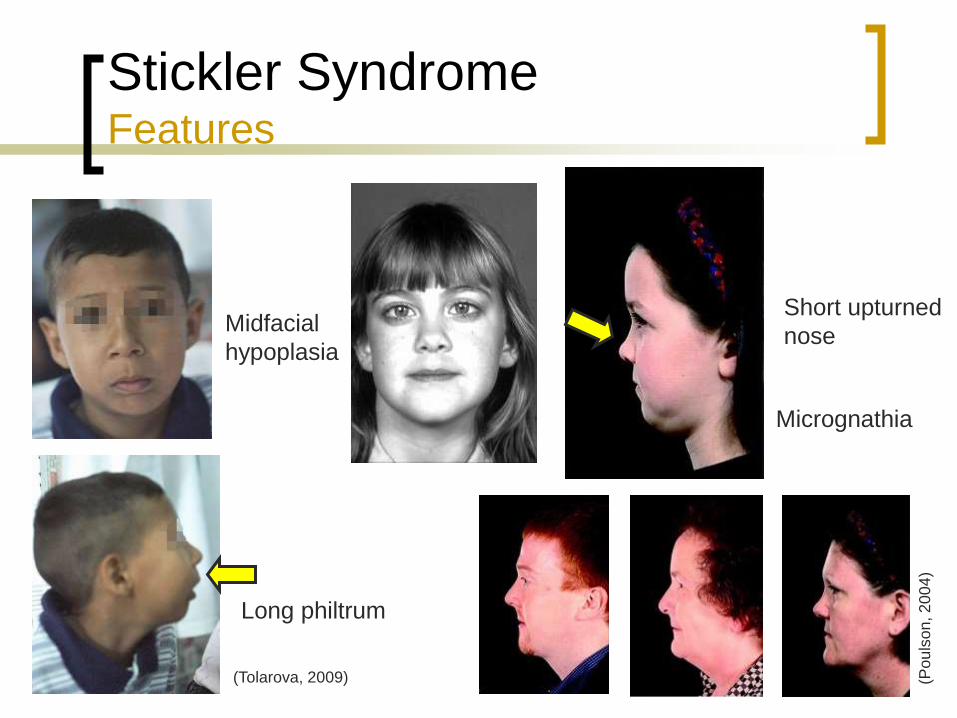
Stickler Syndrome Features
(Tolarova, 2009) (Po
uls
on
, 2
00
4)
Midfacial
hypoplasia
Long philtrum
Short upturned
nose
Micrognathia
Page 46
Stickler Syndrome Features
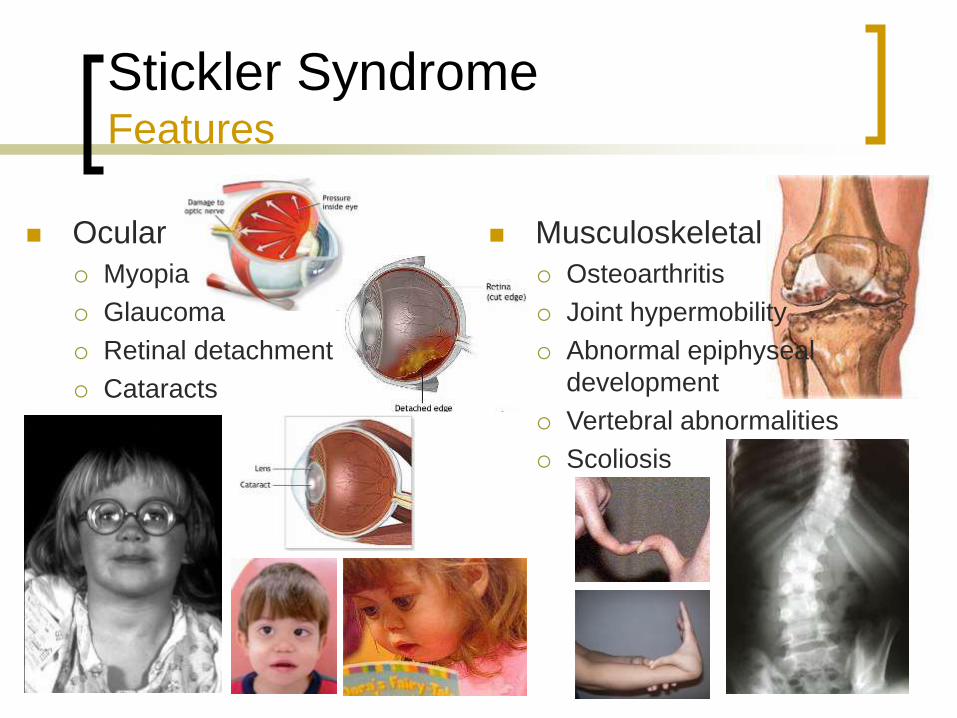
Ocular
Myopia
Glaucoma
Retinal detachment
Cataracts
Musculoskeletal
Osteoarthritis
Joint hypermobility
Abnormal epiphyseal
development
Vertebral abnormalities
Scoliosis
Page 47
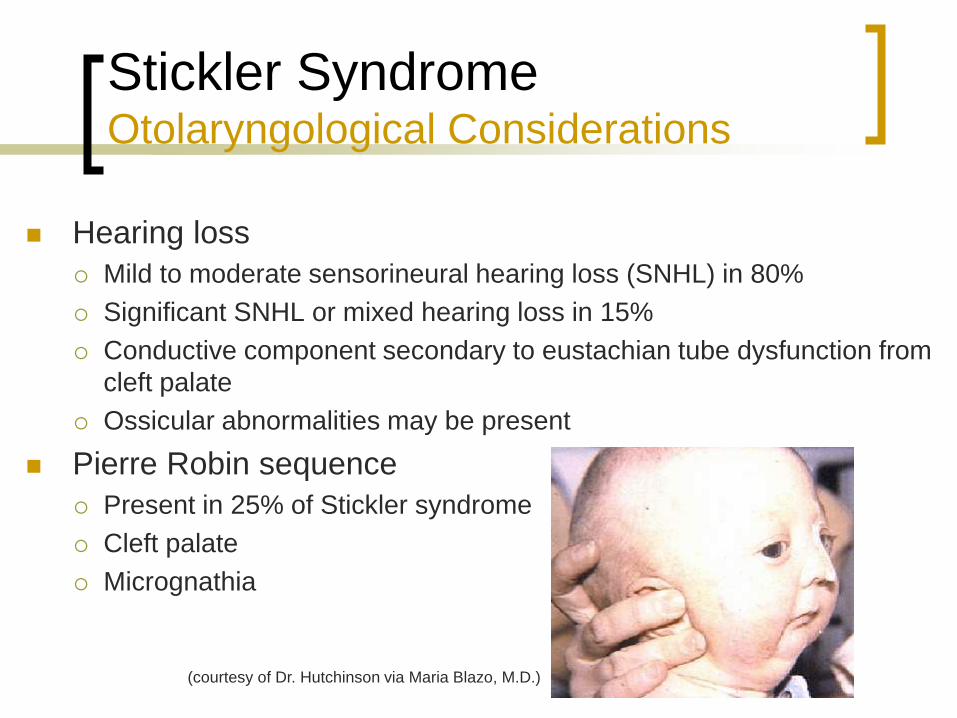
Stickler Syndrome Otolaryngological Considerations
Hearing loss
Mild to moderate sensorineural hearing loss (SNHL) in 80%
Significant SNHL or mixed hearing loss in 15%
Conductive component secondary to eustachian tube dysfunction from
cleft palate
Ossicular abnormalities may be present
Pierre Robin sequence
Present in 25% of Stickler syndrome
Cleft palate
Micrognathia
(courtesy of Dr. Hutchinson via Maria Blazo, M.D.)
Page 48
Waardenburg Syndrome
Autosomal dominant
Multiple genes
PAX3 (Types 1 and 3)
MITF, SNAI2 (Type 2)
EDN3, EDNRB, SOX10 (Type 4)
Autosomal recessive for Type 4
Variable penetrance
Hearing loss
Dystopia canthorum
Pigmentary abnormalities
(co
urt
esy o
f D
r. H
utc
hin
so
n)
Page 49
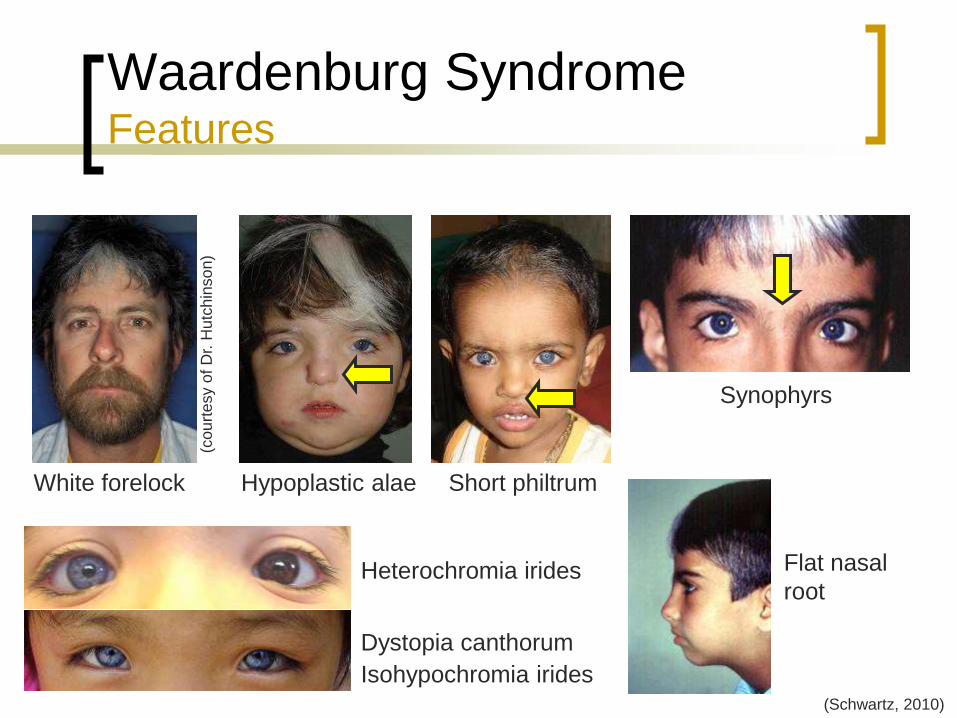
Waardenburg Syndrome Features
Dystopia canthorum
Flat nasal root
Hypoplastic nasal alae
Synophyrs
Heterochromia irides
Isohypochromia irides
White forelock
Vitiligo
Cleft lip and palate (10%)
(Schwartz, 2010)
Page 50
Waardenburg Syndrome Features
Flat nasal
root Heterochromia irides
Dystopia canthorum
Synophyrs
(Schwartz, 2010)
White forelock Hypoplastic alae Short philtrum
(cou
rtesy o
f D
r. H
utc
hin
so
n)
Isohypochromia irides
Page 51
Waardenburg Syndrome Features
Major
Heterochromia irides
White forelock
Dystopia canthorum
Congenital sensorineural
hearing loss
Affected first-degree relative
Minor
Congenital leucoderma
Synophyrs
Broad high nasal root
Hypoplastic nasal alae
Premature graying hair
Page 52
Waardenburg Syndrome Diagnosis
Major
Heterochromia irides
White forelock
Dystopia canthorum
Congenital sensorineural
hearing loss
Affected first-degree relative
Minor
Congenital leucoderma
Synophyrs
Broad high nasal root
Hypoplastic nasal alae
Premature graying hair
Diagnosis
2 major features
1 major features + 2 minor features
Page 53
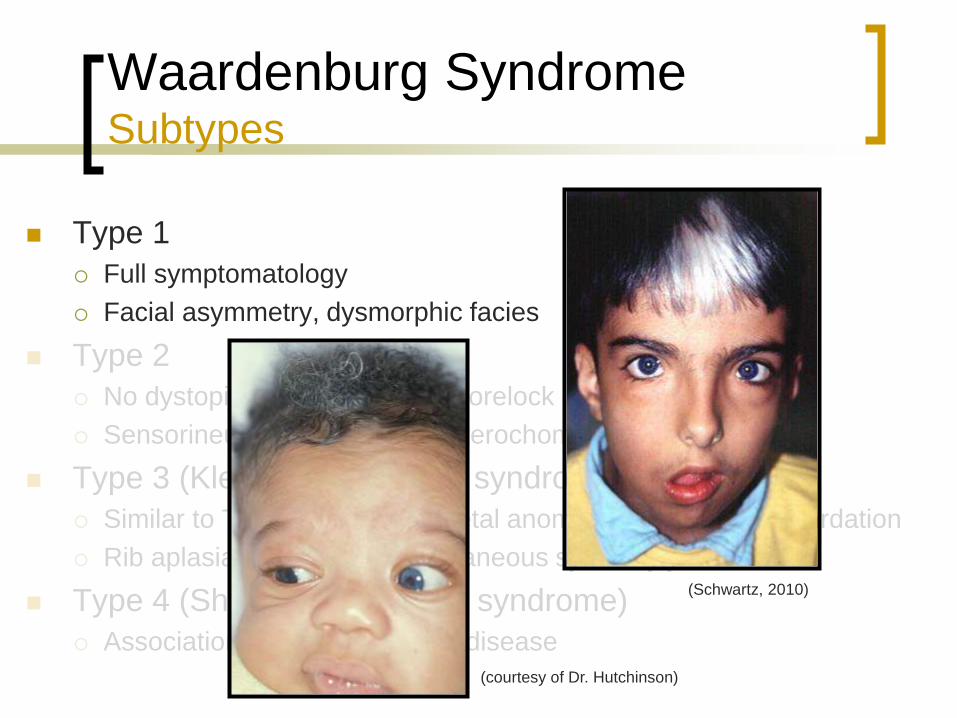
Waardenburg Syndrome Subtypes
Type 1
Full symptomatology
Facial asymmetry, dysmorphic facies
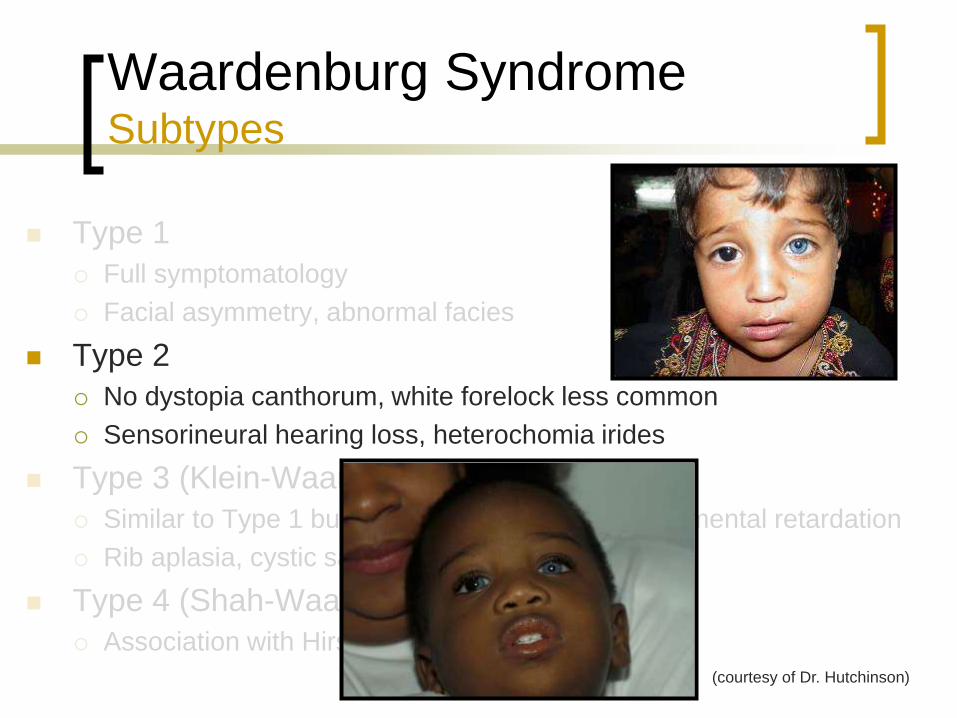
Type 2
No dystopia canthorum, white forelock less common
Sensorineural hearing loss, heterochomia irides
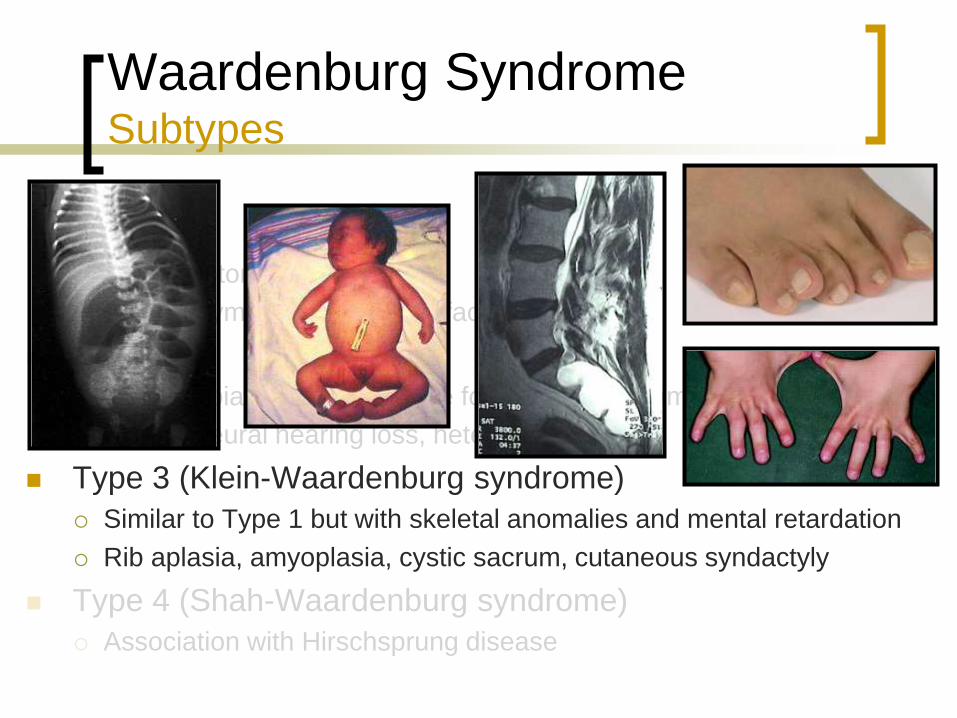
Type 3 (Klein-Waardenburg syndrome)
Similar to Type 1 but with skeletal anomalies and mental retardation
Rib aplasia, cystic sacrum, cutaneous syndactyly
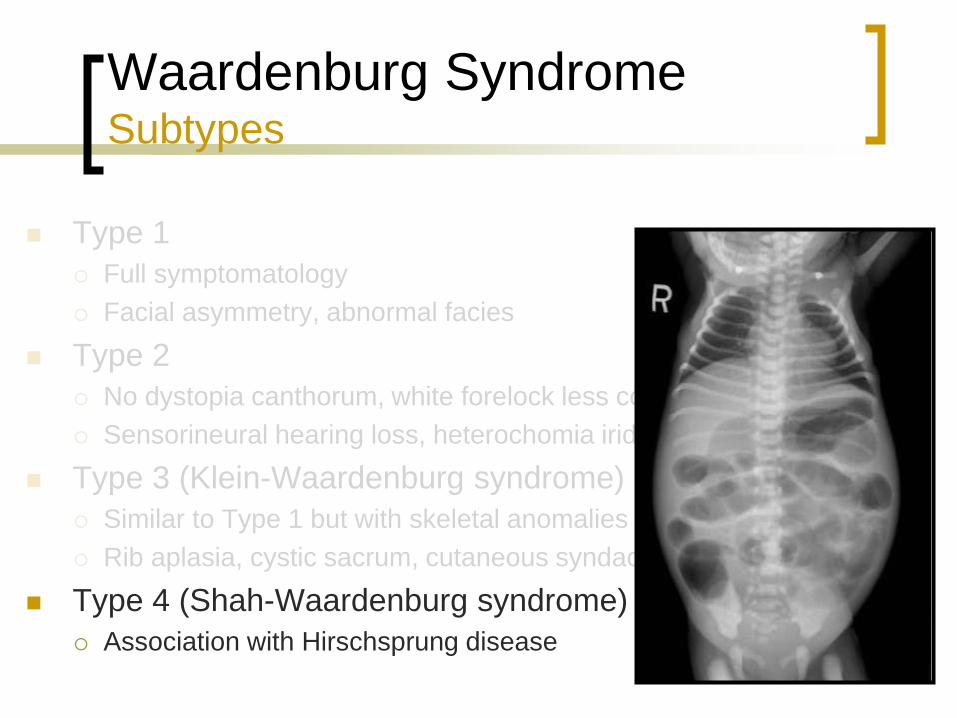
Type 4 (Shah-Waardenburg syndrome)
Association with Hirschsprung disease
(Schwartz, 2010)
(courtesy of Dr. Hutchinson)
Page 54
Waardenburg Syndrome Subtypes
Type 1
Full symptomatology
Facial asymmetry, abnormal facies
Type 2
No dystopia canthorum, white forelock less common
Sensorineural hearing loss, heterochomia irides
Type 3 (Klein-Waardenburg syndrome)
Similar to Type 1 but with skeletal anomalies and mental retardation
Rib aplasia, cystic sacrum, cutaneous syndactyly
Type 4 (Shah-Waardenburg syndrome)
Association with Hirschsprung disease
(courtesy of Dr. Hutchinson)
Page 55
Waardenburg Syndrome Subtypes
Type 1
Full symptomatology
Facial asymmetry, abnormal facies
Type 2
No dystopia canthorum, white forelock less common
Sensorineural hearing loss, heterochomia irides
Type 3 (Klein-Waardenburg syndrome)
Similar to Type 1 but with skeletal anomalies and mental retardation
Rib aplasia, amyoplasia, cystic sacrum, cutaneous syndactyly
Type 4 (Shah-Waardenburg syndrome)
Association with Hirschsprung disease
Page 56
Waardenburg Syndrome Subtypes
Type 1
Full symptomatology
Facial asymmetry, abnormal facies
Type 2
No dystopia canthorum, white forelock less common
Sensorineural hearing loss, heterochomia irides
Type 3 (Klein-Waardenburg syndrome)
Similar to Type 1 but with skeletal anomalies and mental retardation
Rib aplasia, cystic sacrum, cutaneous syndactyly
Type 4 (Shah-Waardenburg syndrome)
Association with Hirschsprung disease
Page 57
Waardenburg Syndrome Otolaryngological Considerations
Congenital sensorineural deafness
Typically not progressive
Hearing amplification
Cochlear implantation
Cleft lip or palate repair
Cosmetic considerations
Page 58
Beckwith-Wiedemann
Syndrome
Imprinting defect at chromosome 11p15
Most cases are sporadic
Autosomal dominant familial inheritance in 15%
Most common overgrowth syndrome in infancy
Five common features
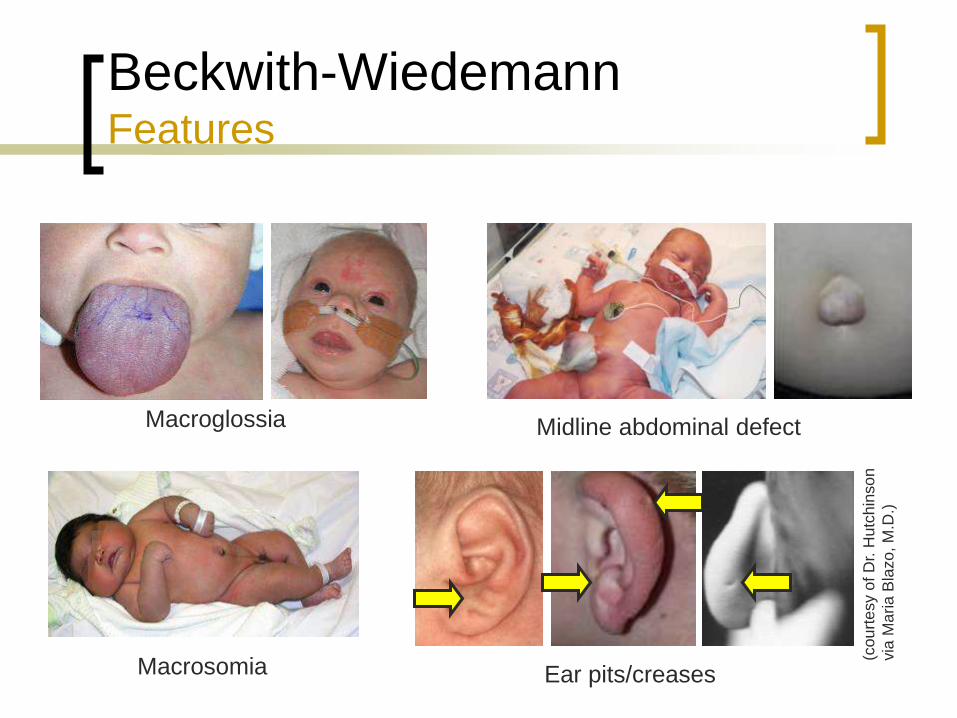
Macroglossia
Macrosomia
Midline abdominal wall defect
Ear pits/creases
Neonatal hypoglycemia
(courtesy of Dr. Hutchinson)
Page 59
Beckwith-Wiedemann
Syndrome
Imprinting defect at chromosome 11p15
Most cases are sporadic
Autosomal dominant familial inheritance in 15%
Most common overgrowth syndrome in infancy
Five common features
Macroglossia
Macrosomia
Midline abdominal wall defect
Ear pits/creases
Neonatal hypoglycemia
Page 60
Beckwith-Wiedemann Features
Macroglossia
Macrosomia Ear pits/creases
Midline abdominal defect
(co
urt
esy o
f D
r. H
utc
hin
so
n
via
Ma
ria
Bla
zo
, M
.D.)
Page 61
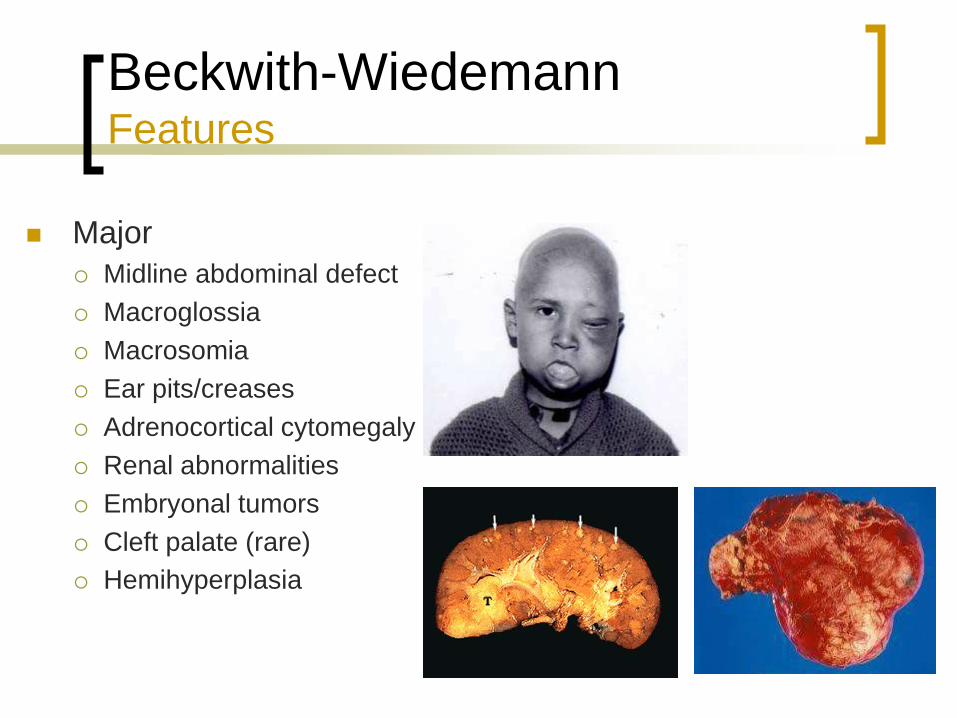
Beckwith-Wiedemann Features
Major
Midline abdominal defect
Macroglossia
Macrosomia
Ear pits/creases
Adrenocortical cytomegaly
Renal abnormalities
Embryonal tumors
Cleft palate (rare)
Hemihyperplasia
Page 62
Beckwith-Wiedemann Features
Major
Midline abdominal defect
Macroglossia
Macrosomia
Ear pits/creases
Adrenocortical cytomegaly
Renal abnormalities
Embryonal tumors
Cleft palate (rare)
Hemihyperplasia
Minor
Neonatal hypoglycemia
Polyhydramnios
Prematurity
Facial nevus flammeus
Hemangioma
Characteristic facies (i.e.
midface hypoplasia)
Cardiac anomalies
Diastasis recti
Advanced bone age
Page 63
Beckwith-Wiedemann Diagnosis
Major
Midline abdominal defect
Macroglossia
Macrosomia
Ear pits/creases
Adrenocortical cytomegaly
Renal abnormalities
Embryonal tumors
Cleft palate (rare)
Hemihyperplasia
Minor
Neonatal hypoglycemia
Polyhydramnios
Prematurity
Facial nevus flammeus
Hemangioma
Characteristic facies (i.e.
midface hypoplasia)
Cardiac anomalies
Diastasis recti
Advanced bone age
Diagnosis
At least 2 common features
3 major features
2 major features + 3 minor features
Page 64
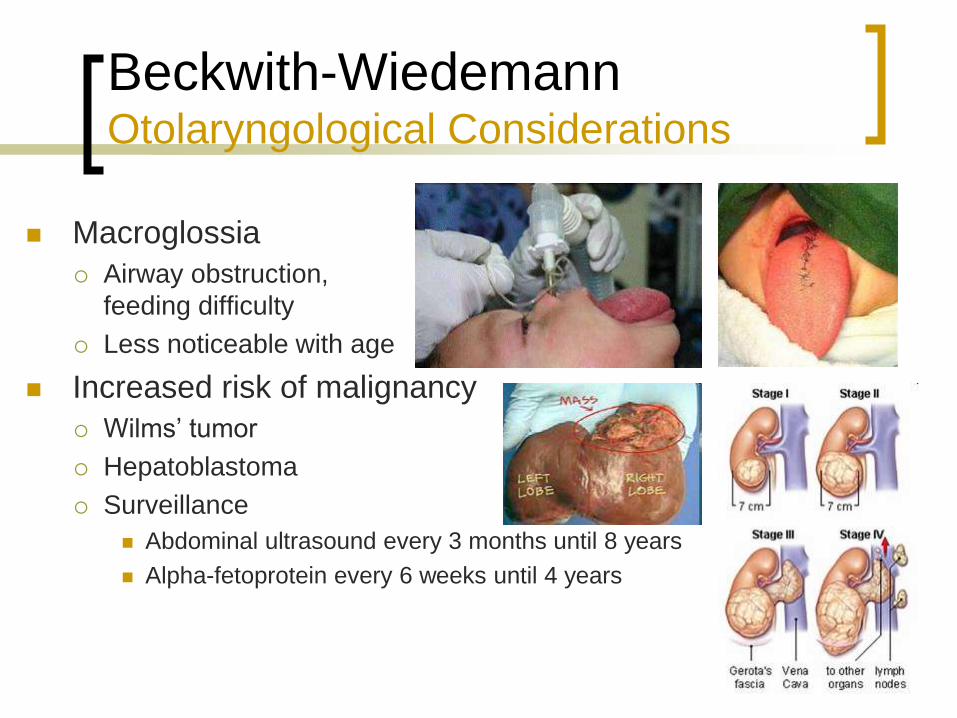
Beckwith-Wiedemann Otolaryngological Considerations
Macroglossia
Airway obstruction,
feeding difficulty
Less noticeable with age
Increased risk of malignancy
Wilms’ tumor
Hepatoblastoma
Surveillance
Abdominal ultrasound every 3 months until 8 years
Alpha-fetoprotein every 6 weeks until 4 years
Page 65
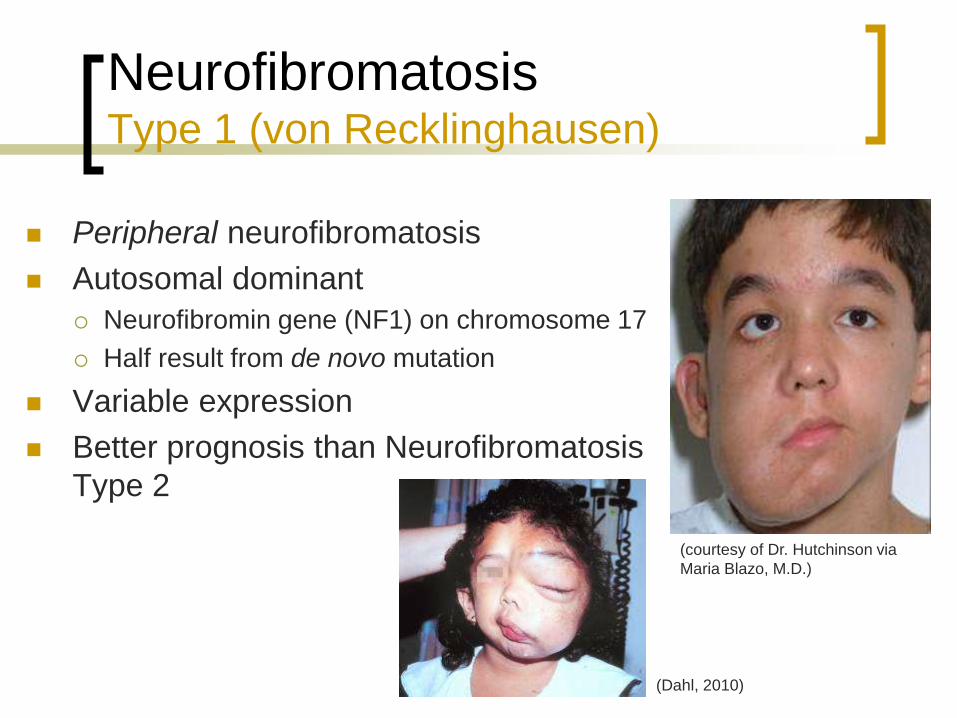
Neurofibromatosis Type 1 (von Recklinghausen)
Peripheral neurofibromatosis
Autosomal dominant
Neurofibromin gene (NF1) on chromosome 17
Half result from de novo mutation
Variable expression
Better prognosis than Neurofibromatosis
Type 2
(Dahl, 2010)
(courtesy of Dr. Hutchinson via
Maria Blazo, M.D.)
Page 66
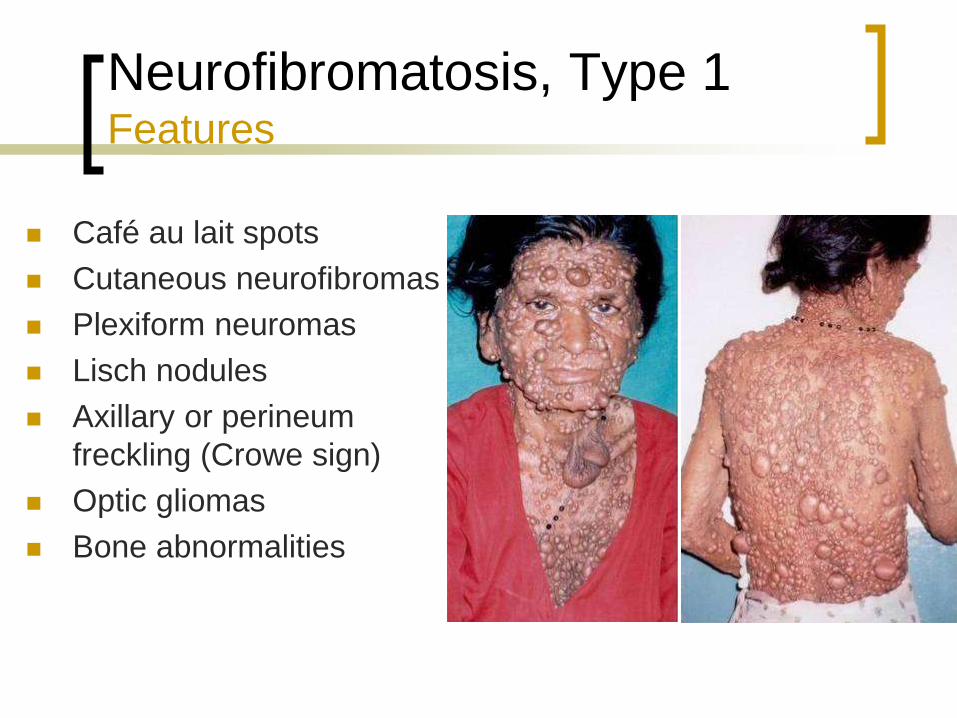
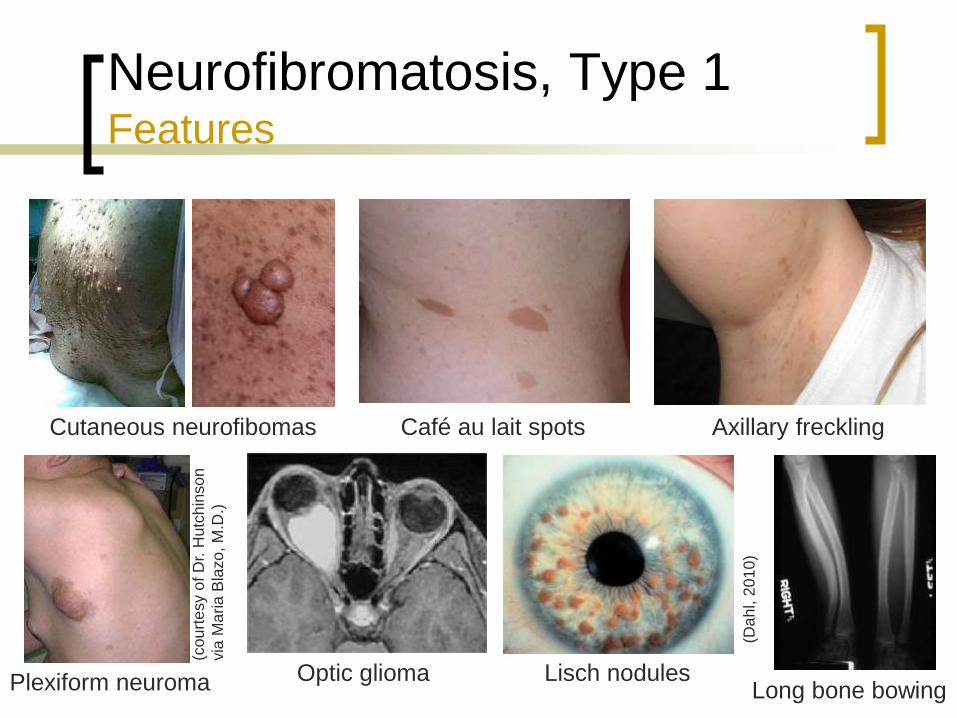
Neurofibromatosis, Type 1 Features
Café au lait spots
Cutaneous neurofibromas
Plexiform neuromas
Lisch nodules
Axillary or perineum
freckling (Crowe sign)
Optic gliomas
Bone abnormalities
Page 67
Neurofibromatosis, Type 1 Features
Cutaneous neurofibomas
Plexiform neuroma Optic glioma Lisch nodules Long bone bowing
Café au lait spots Axillary freckling
(Da
hl, 2
01
0)
(co
urt
esy o
f D
r. H
utc
hin
so
n
via
Ma
ria
Bla
zo
, M
.D.)
Page 68
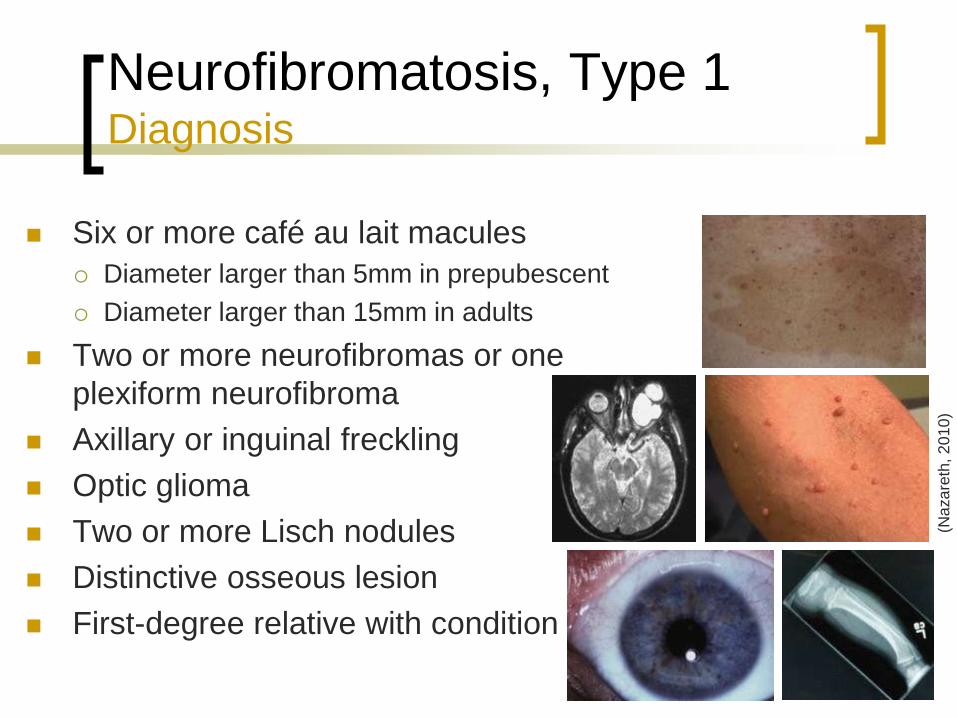
Neurofibromatosis, Type 1 Diagnosis
Six or more café au lait macules
Diameter larger than 5mm in prepubescent
Diameter larger than 15mm in adults
Two or more neurofibromas or one
plexiform neurofibroma
Axillary or inguinal freckling
Optic glioma
Two or more Lisch nodules
Distinctive osseous lesion
First-degree relative with condition
(Na
za
reth
, 2
01
0)
Page 69
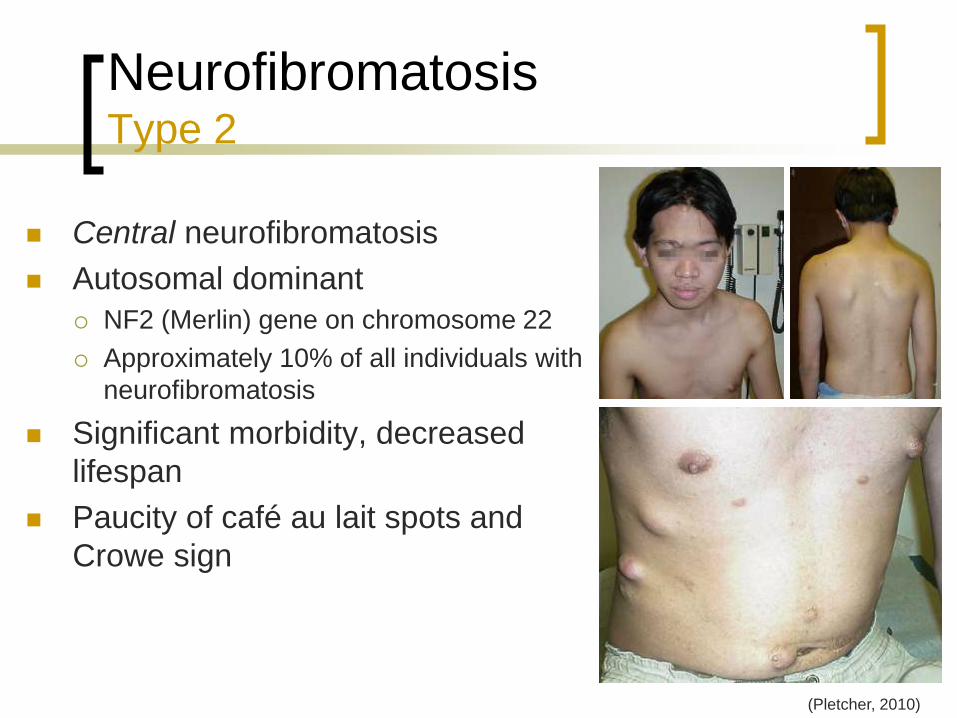
Neurofibromatosis Type 2
Central neurofibromatosis
Autosomal dominant
NF2 (Merlin) gene on chromosome 22
Approximately 10% of all individuals with
neurofibromatosis
Significant morbidity, decreased
lifespan
Paucity of café au lait spots and
Crowe sign
(Pletcher, 2010)
Page 70
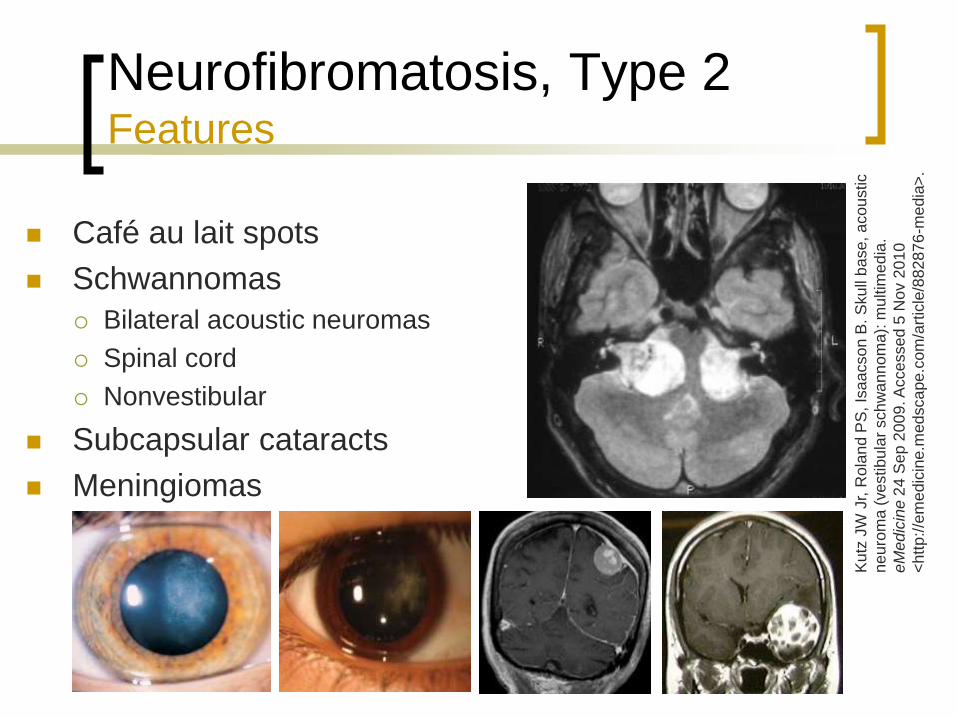
Neurofibromatosis, Type 2 Features
Café au lait spots
Schwannomas
Bilateral acoustic neuromas
Spinal cord
Nonvestibular
Subcapsular cataracts
Meningiomas
Ku
tz J
W J
r, R
ola
nd
PS
, Is
aa
cso
n B
. S
ku
ll b
ase
, a
co
ustic
ne
uro
ma (
ve
stib
ula
r sch
wa
nn
om
a):
mu
ltim
ed
ia.
eM
ed
icin
e 2
4 S
ep
20
09
. A
cce
sse
d 5
Nov 2
01
0
<h
ttp
://e
me
dic
ine.m
edsca
pe.c
om
/art
icle
/88287
6-m
edia
>.
Page 71
Neurofibromatosis, Type 2 Diagnosis
Bilateral vestibular schwannomas
Presumptive
Affected first-degree relative
Unilateral vestibular
schwannoma
Or two of the following:
Meningioma
Glioma
Schwannoma
Juvenile posterior subcapsular
or cortical cataract
Suggestive
Unilateral vestibular
schwannoma
Two of the following:
Meningioma
Glioma
Schwannoma
Juvenile posterior subcapsular
or cortical cataract
Or multiple meningiomas
Page 72
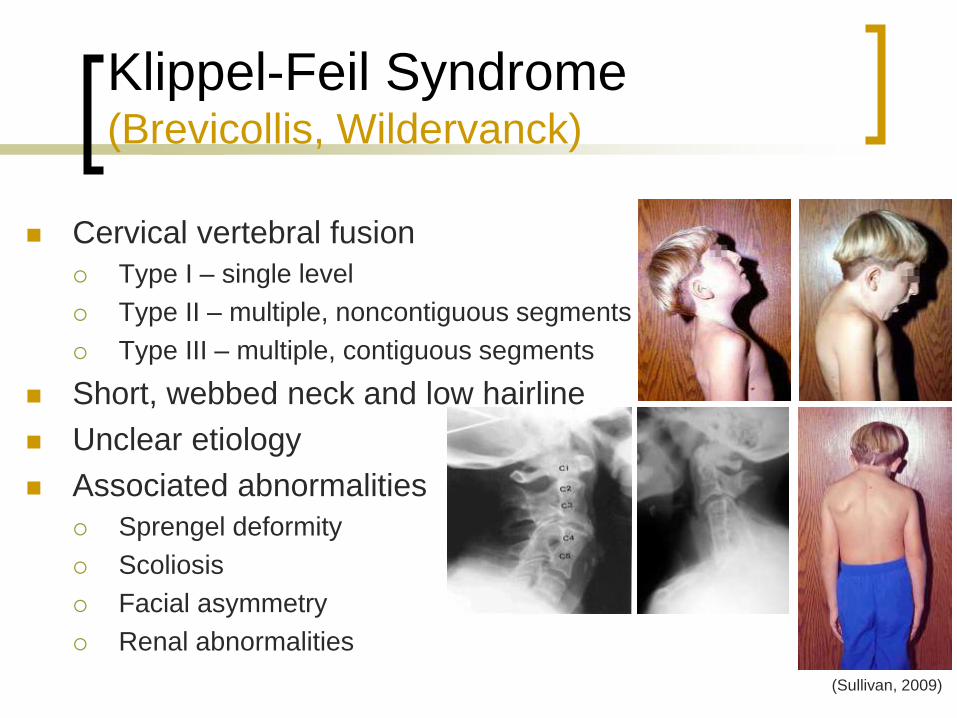
Klippel-Feil Syndrome (Brevicollis, Wildervanck)
Cervical vertebral fusion
Type I – single level
Type II – multiple, noncontiguous segments
Type III – multiple, contiguous segments
Short, webbed neck and low hairline
Unclear etiology
Associated abnormalities
Sprengel deformity
Scoliosis
Facial asymmetry
Renal abnormalities
(Sullivan, 2009)
Page 73
Other Syndromes Without Craniofacial Features
Usher
Hearing loss with defective inner ear
Type I – deafness and vestibular dysfunction
Type II – nonprogressive hearing loss and normal vestibular function
Type III – progressive hearing loss and half vestibular function
Progressive vision loss from retinitis pigmentosa
Pendred
Sensorineural hearing loss
Thyroid goiter
Jervell and Lange-Neilsen
Defective potassium channel from KCNQ1 and KCNE1
mutations
Sensorineural hearing loss and palpitations (long QT syndrome)
Yeah Yeah
Yeah
Page 74
Conclusion
Many syndromes will
present to the
otolaryngologist
Warrant otolaryngological
intervention
Attention to coexisting
conditions
Many affected individuals
are aware of the social
stigma related to their
condition http://www.explosm.net/comics
Page 75
References
Admiraal RJ, et al. Hearing impairment in Stickler syndrome. Adv Otorhinolaryngol 2002; 61:216-23.
Bailey BJ, Johnson JT, Newlands SD, eds. Head and Neck Surgery – Otolaryngology, 4th Ed. Philadelphia:
Lippincott, 2006. pp 1311-2,1380-6,2794.
Breugem CC, Courtemanche DJ. Robin sequence: clearing nosologic confusion. Cleft Palate Craniofac J
2010; 47:197-200.
Chen H. Apert syndrome. eMedicine 2 Sep 2009. Accessed 12 Oct 2010
<http://emedicine.medscape.com/article/941723-overview>.
Chen H. Crouzon syndrome. eMedicine 10 Sep 2009. Accessed 12 Oct 2010
<http://emedicine.medscape.com/article/942989-overview>.
Chen H. Down syndrome. eMedicine 22 Mar 2010. Accessed 31 Oct 2010
<http://emedicine.medscape.com/article/943216-overview>.
Crawford AH, Schorry EK. Neurofibromatosis update. J Pediatr Orthop 2006; 26:413-23.
Dahl AA, Grostern RJ. Neurofibromatosis-1. eMedicine 21 May 2010. Accessed 5 Nov 2010
<http://emedicine.medscape.com/article/1219222-overview>.
DeBaun MR, et al. Epigenetic alterations of H19 and LIT1 distinguish patients with Beckwith-Wiedemann
syndrome with cancer and birth defects. Am J Hum Genet 2002; 70:604-11.
DeBaun MR, Tucker MA. Risk of cancer during the first four years of life in children from The Beckwith
Wiedemann Syndrome Registry. J Pediatr 1998; 132:398-400.
DeBella K, Szudek J, Friedman JM. Use of the national institutes of health criteria for diagnosis of
neurofibromatosis 1 in children. Pediatrics 2000; 105(3 Pt 1):608-14.
Page 76
References
Dourmishev AL, Janniger CK. Down syndrome. eMedicine 1 Jul 2009. Accessed 4 Nov 2010
<http://emedicine.medscape.com/article/1113071-overview>.
Dourmishev LA, Janniger CK. Waardenburg syndrome. eMedicine 2 Jun 2009. Accessed 30 Oct 2010
<http://emedicine.medscape.com/article/1113314-overview>.
Elliott M, et al. Clinical features and natural history of Beckwith-Wiedemann syndrome: presentation of 74 new
cases. Clinical Genetics 1994; 46:168-74.
Farrer LA, et al. Waardenburg syndrome (WS) type I is caused by defects at multiple loci, one of which is near
ALPP on chromosome 2: first report of the WS consortium. Am J Hum Genet 1992; 50:902-13.
Ferry RJ. Beckwith-Wiedemann syndrome. eMedicine 15 Apr 2010. Accessed 23 Oct 2010
<http://emedicine.medscape.com/article/919477-overview>.
Flint PW, et al, eds. Cummings Otolaryngology: Head and Neck Surgery, 5th Ed. Philadelphia: Mosby
Elsevier, 2010. ch 147, 184.
Gould HJ, Caldarelli DD. Hearing and otopathology in Apert syndrome. Archives of Otolaryngology 1982;
108:347-9.
Gonçalves LF, Espinoza J, Lee W, et al. Phenotypic characteristics of absent and hypoplastic nasal bones in
fetuses with Down syndrome: description by 3-dimensional ultrasonography and clinical significance. J
Ultrasound Med 2004; 23:1619-27.
Handzic J, et al. Hearing levels in Pierre Robin syndrome. Cleft Palate Craniofac J 1995; 32:30-6.
Hata T, Todd MM. Cervical spine considerationswhen anesthesizing patients with Down syndrome.
Anesthesiology 2005; 102:680-5.
Page 77
References
Heike CL, Hing AV. Craniofacial microsomia review. Gene Review. Eds. Pagon RA, et al. 19 Mar 2009.
Accessed 23 Oct 2010 <http://www.ncbi.nlm.nih.gov/bookshelf/br.fcgi?book=gene&part=m-hfm-ov>.
Horgan JE, et al. OMENS-plus: analysis of craniofacial and extracraniofacial anomalies in hemifacial
microsomia. Cleft Palate Craniofac J 1995; 32:405-12.
Jackson IT, Malhotra G. Congenital syndromes. eMedicine 2 Jul 2009. Accessed 12 Oct 2010
<http://emedicine.medscape.com/article/1280034-overview>.
Jakobsen LP, et al. The genetic basis of the Pierre Robin Sequence. Cleft Palate Craniofac J 2006; 43:155-9.
Lee KJ, ed. Essential Otolaryngology – Head and Neck Surgery, 9th Ed. New York: McGraw Hill, 2008. pp
139-59.
Liu XZ, Newton VE, Read AP. Waardenburg syndrome type II: phenotypic findings and diagnostic criteria. Am
J Med Genet 1995; 55:95-100.
Nazareth MR, Helm TN. Neurofibromatosis. eMedicine 16 Jun 2010. Accessed 5 Nov 2010
<http://emedicine.medscape.com/article/1112001-overview>.
Nowak CB. Genetics and hearing loss: a review of Stickler syndrome. J Commun Disord 1998; 31:437-54.
Orvidas LJ, et al. Hearing and otopathology in Crouzon syndrome. Layrngoscope 1999; 109:1372-5.
Peterson-Falzone SJ, Hardin-Jones MA, Karnell MP. Cleft Palate Speech, 4th Ed. St. Louis: Mosby, 2010.
Pletcher BA. Neurofibromatosis, type 2. eMedicine 3 Mar 2010. Accessed 5 Nov 2010
<http://emedicine.medscape.com/article/1178283-overview>.
Poulson AV, et al. Clinical features of type 2 Stickler syndrome. J Med Genet 2004; 41:e107.
Page 78
References
Pron G, et al. Ear malformation and hearing loss in patients with Treacher Collins syndrome. Cleft Palate
Craniofac J 1993; 30:97-103.
Robin NH, Falk MJ, Haldeman-Englert CR. FGFR-related craniosynostosis syndromes. Gene Review. Eds.
Pagon RA, et al. 27 Sept 2007. Accessed 19 Oct 2010
<http://www.ncbi.nlm.nih.gov/bookshelf/br.fcgi?book=gene&part=craniosynostosis>.
Saenz RB. Primary care of infants and young children with Down syndrome. Am Fam Physician 1999; 59:381-
90,392,395-6.
Samartzis DD, Herman J, Lubicky JP. Classification of congenitally fused cervical patterns in Klippel-Feil
patients: epidemiology and role in the development of cervical spine-related symptoms. Spine 2006;
31:F798-804.
Samartzis D, Lubicky JP, Herman J. Symptomatic cervical disc herniation in a pediatric Klippel-Feil patient:
the risk of neural injury associated with extensive congenitally fused vertebrae and a hypermobile
segment. Spine 2006; 31:F335-8.
Sangkhathat S, et al. Novel mutation of Endothelin-B receptor gene in Waardenburg-Hirschsprung disease.
Pediatr Surg Int 2005; 21:960-3.
Schwartz RA, Jozwiak S, Krantz I. Waardenburg syndrome. eMedicine 4 Jun 2010. Accessed 24 Oct 2010
<http://emedicine.medscape.com/article/950277-overview>.
Spivey PS, Bradshaw WT. Recognition and management of the infant with Beckwith-Wiedemann syndrome.
Adv Neonatal Care 2009; 9:279-84.
Sullivan AJ. Klippel-Feil syndrome. eMedicine 23 Jun 2009. Accessed 5 Nov 2010
<http://emedicine.medscape.net/article/1264848-diagnosis>.
Page 79
References
Tewfik TL, Trinh N, Teebi AS. Pierre Robin Syndrome. eMedicine 4 Mar 2010. Accessed 21 Oct 2010
<http://emedicine.medscape.com/article/844143-overview>.
Tolarova MM. Pierre Robin Malformation. eMedicine 25 Mar 2009. Accessed 21 Oct 2010
<http://emedicine.medscape.com/article/995706-overview>.
Tolarova MM, Wong GB, Varma S. Mandibulofacial dysostosis. eMedicine 24 Nov 2009. Accessed 17 Oct
2010 <http://emedicine.medscape.com/article/946143-overview>.
Verheij JB, et al. Shah-Waardenburg syndrome and PCWH associated with SOX10 mutations: a case report
and review of the literature. Eur J Paediatr Neurol 2006; 10:11-7.
Watanabe A, et al. Epistatic relationship between Waardenburg syndrome genes MITF and PAX3. Nat Genet
1998; 18:283-6.