26
Data Visualization in Cheminformatics Simon Xi Computational Sciences CoE Pfizer Cambridge
| Date post: | 09-Dec-2015 |
| Category: |
Documents |
| Upload: | tommy-poernomo |
| View: | 212 times |
| Download: | 0 times |

Data Visualization in Cheminformatics
Simon XiComputational Sciences CoE
Pfizer Cambridge

My BackgroundProfessional ExperienceSenior Principal Scientist, Computational Sciences CoE, Pfizer
Cambridge9-year experience in pharmaceutical research with a focused on
developing cheminformatics and bioinformatics applications for research scientists
EducationMSc in Molecular Cell Biology in UTDallasMSc in Software Engineering in SMUFinishing Ph.D in Bioinformatics in Boston University

What we will cover today
• Introduction to drug discovery• Cheminformatics basics• Encoding of the chemical structures• Visualizing data and structures• Design and optimization of compound library• A case study

The Billion Dollar Molecules
Drug Name2006 World-Wide Sales Primary Use
Lipitor $14,385M cholesterolNexium $5,182M heartburnAdvair $6,129M asthmaPrevacid $3,425M heartburnPlavix $6,057M anticoagulantSingulair $3,579M asthmaSeroquel $3,560M depressionEffexor $3,722M depressionNorvasc $4,866M hypertension
Lipitor – 14 billion annual sales

$0
$5
$10
$15
$20
$25
1970
1972
1974
1976
1978
1980
1982
1984
1986
1988
1990
1992
1994
1996
1998
2000
# NM
Es
604020200
Source: Source: PhRMAPhRMA annual survey, 2000annual survey, 2000
Total R&D Investment ($ Billions)
Industry Productivity vs. InvestmentThe Challenge
Nature Reviews Drug Discovery 3, 451-456 (2004)
NME/$
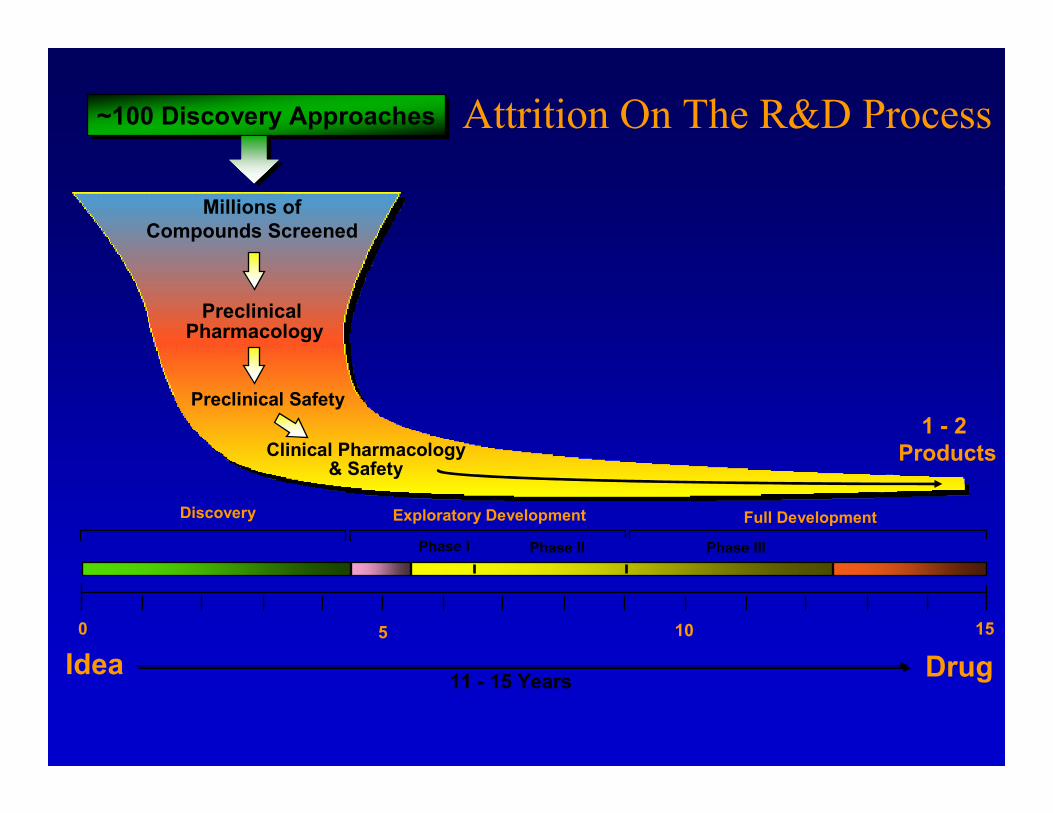
Preclinical Pharmacology
Preclinical Safety
Millions ofCompounds Screened
Idea Drug11 - 15 Years
~100 Discovery Approaches~100 Discovery Approaches
1 - 2 Products
Discovery Exploratory Development Full Development
Phase I Phase II Phase III
0 155 10
Clinical Pharmacology& Safety
Attrition On The R&D Process
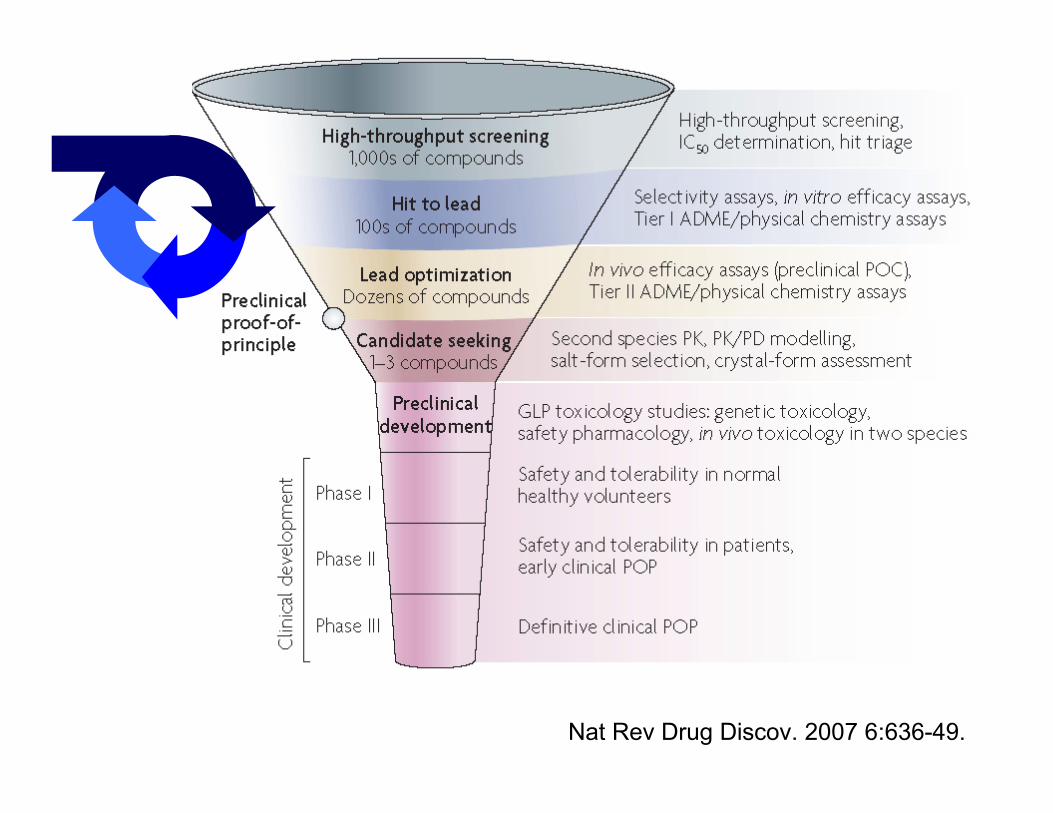
Nat Rev Drug Discov. 2007 6:636-49.

What is Chemoinformatics?
• Use of computer and informational techniques, applied to a range of problems in the field of chemistry.
• These in silico techniques are commonly used in pharmaceutical companies in the process of drug discovery.• Chemistry is a visual science. Data visualization is a key component of cheminformatics.

What is Chemoinformatics?
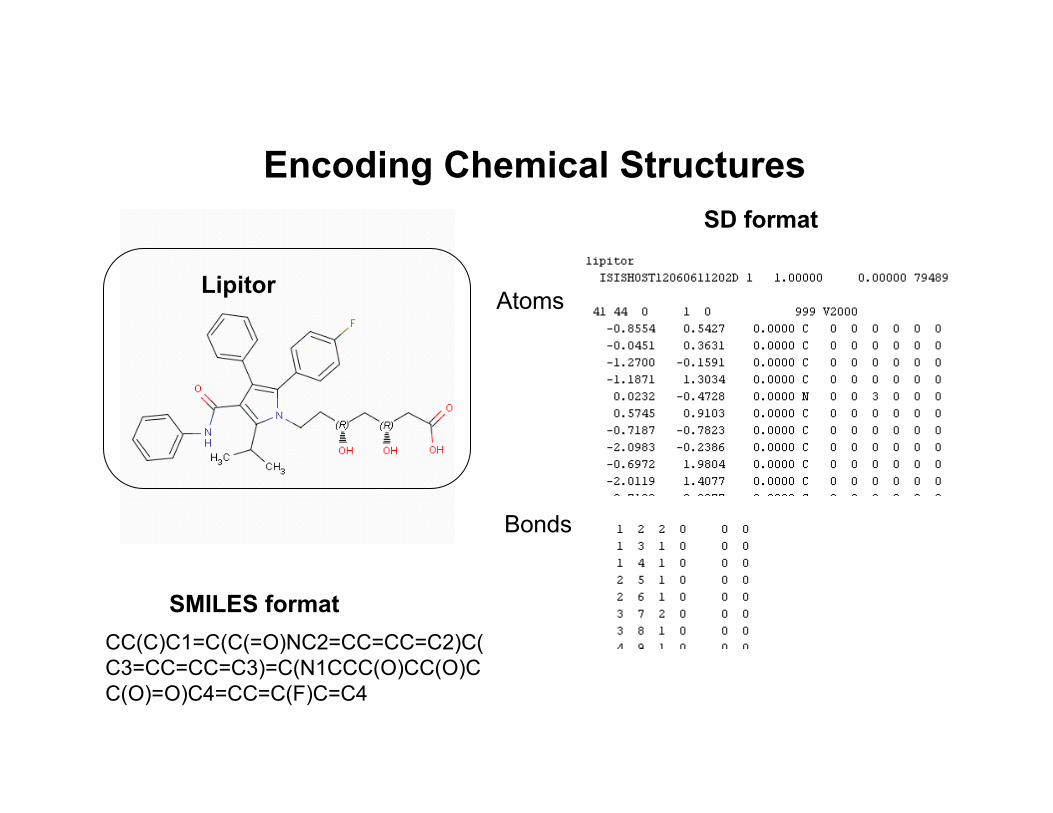
Encoding Chemical Structures
LipitorAtoms
Bonds
SD format
CC(C)C1=C(C(=O)NC2=CC=CC=C2)C(C3=CC=CC=C3)=C(N1CCC(O)CC(O)CC(O)=O)C4=CC=C(F)C=C4
SMILES format

Representing Structure as Fingerprints
010 0 100 0 1001 00000 1 00……

Compound Similarity Search
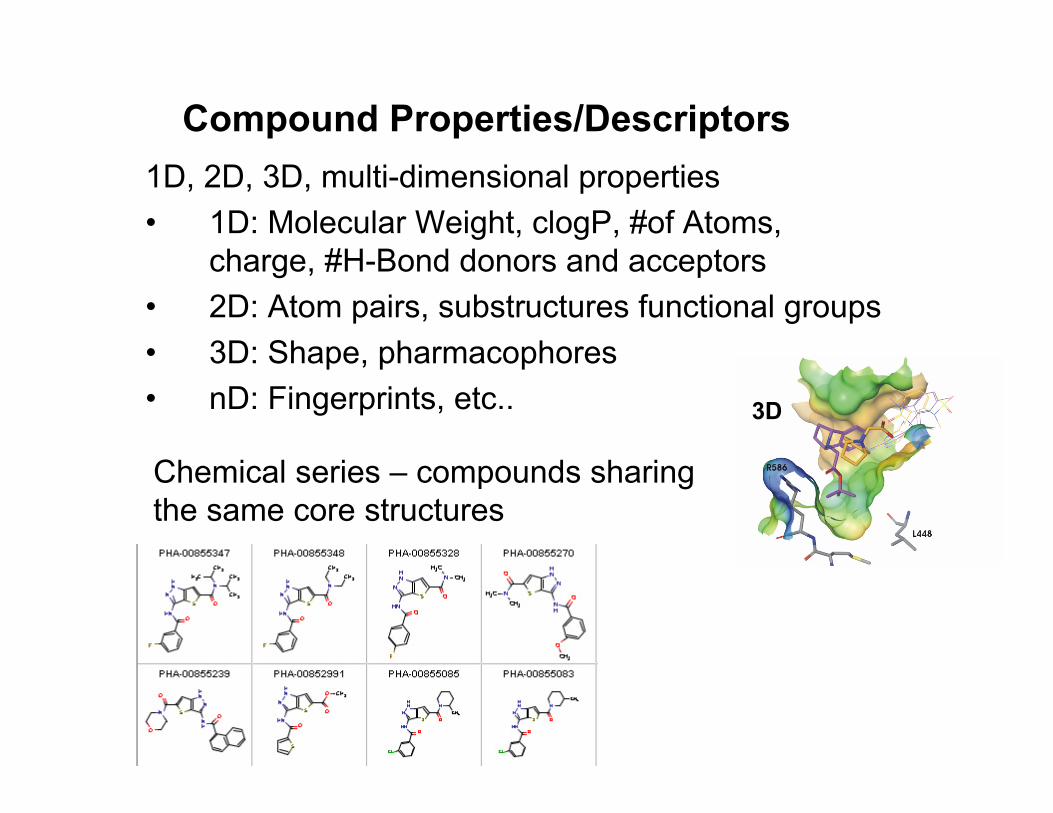
Compound Properties/Descriptors1D, 2D, 3D, multi-dimensional properties• 1D: Molecular Weight, clogP, #of Atoms,
charge, #H-Bond donors and acceptors• 2D: Atom pairs, substructures functional groups• 3D: Shape, pharmacophores• nD: Fingerprints, etc..
Chemical series – compounds sharing the same core structures
3D

Series Classifications –Wards Clustering
Iteratively merging a pair of nodes until all nodes are merged.
At each merging step, two nodes that give minimal variance are chosen and merged into one new node.
Once the tree hierarchy is generated, clusters can be defined by cutting the tree at certain dissimilarity threshold

Toxicity Properties• Inhibition of CYP450 isozymes• PXR transactivation• Human hepatocyte toxicity• Mutagenicity• Mitochondria toxicity• Covalent protein binding• Inhibition of HERG
ADME/Physicochemical Properties• Solubility• Chemical stability• Hydrophobicity/hydrogen bonding
potential• Intestinal mucosal cell permeation• Liver and kidney clearance• Metabolism• Transporters• Charge• Size• Protein binding• Blood-brain barrier permeation• Target cell permeation
Primary pharmacology• In vitro potency• Cell based potency• Functional assays• Selectivity against other targets
What makes a drug?

Drug-Likeness: Rule of Five
Proposed by C. Lipinski to describe ‘drug-like’ molecules. Molecules displaying good oral absorption and /or distribution properties are likely to possess the following characteristics:
– Molecular Weight < 500– logP < 5.0– H-donors < 5– H-acceptors (number of N and O atoms) < 10
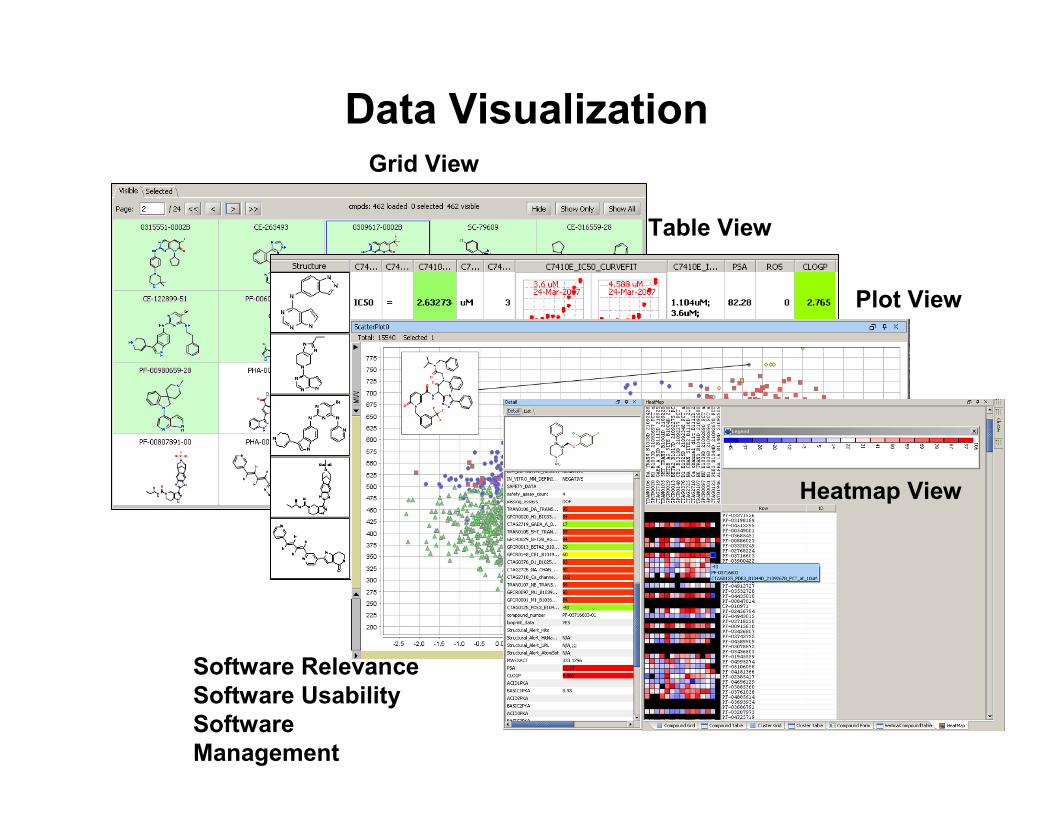
Data VisualizationGrid View
Table View
Plot View
Heatmap View
Software RelevanceSoftware UsabilitySoftware Management

Building Predictive Models using Machine Learning Techniques
• Use computational models to understand Structure-ActivitiveRelationship (SAR)
• Use computational models to run virtual screen to guide compound selection for synthesis
Interpretability of Predictive Models
The good part The not so good part
Can we derive this for non-linear models?

Multiple Parameter Optimization in Combinatorial Library Design
Given a 100x100x100 virtual library space and a set of predictive models for various properties (e.g. potency, ADME, selectivity), select the best 300 compounds for synthesis with the highest probability of being potent and drug-like and with diverse sampling of the chemical space
N
NN N
R3
R1 R2
For example diaminopyrimidine library
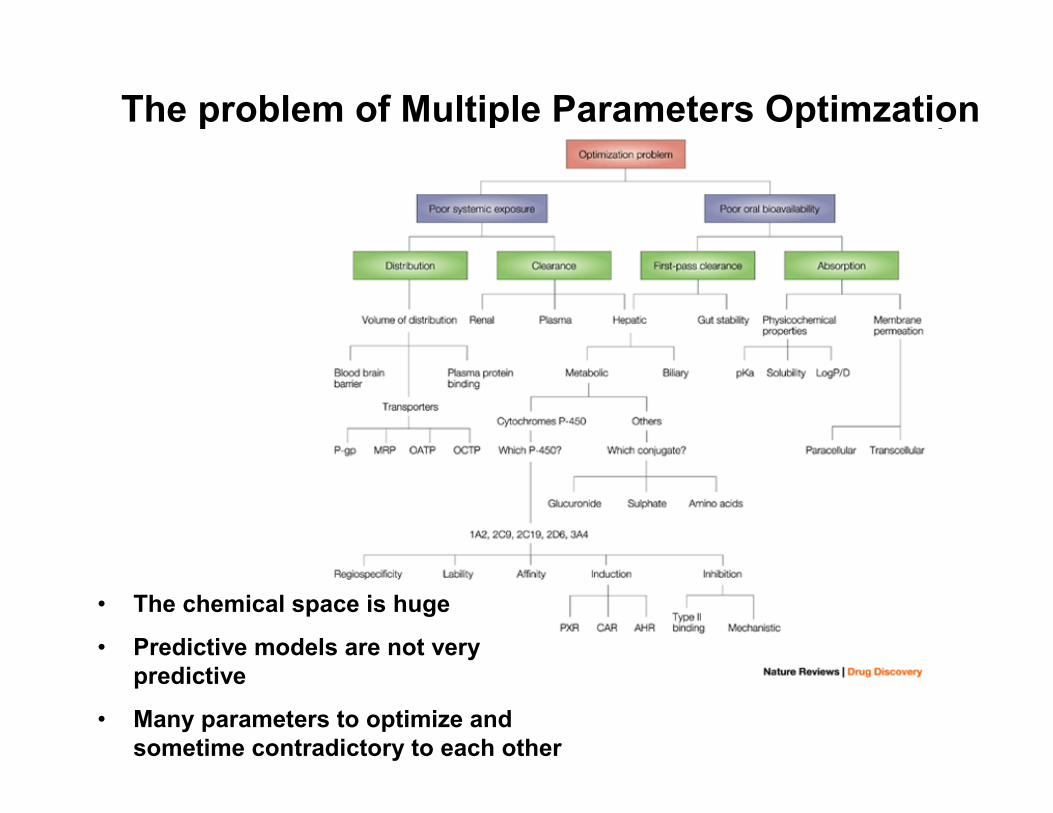
The problem of Multiple Parameters Optimzation
• The chemical space is huge
• Predictive models are not very predictive
• Many parameters to optimize and sometime contradictory to each other
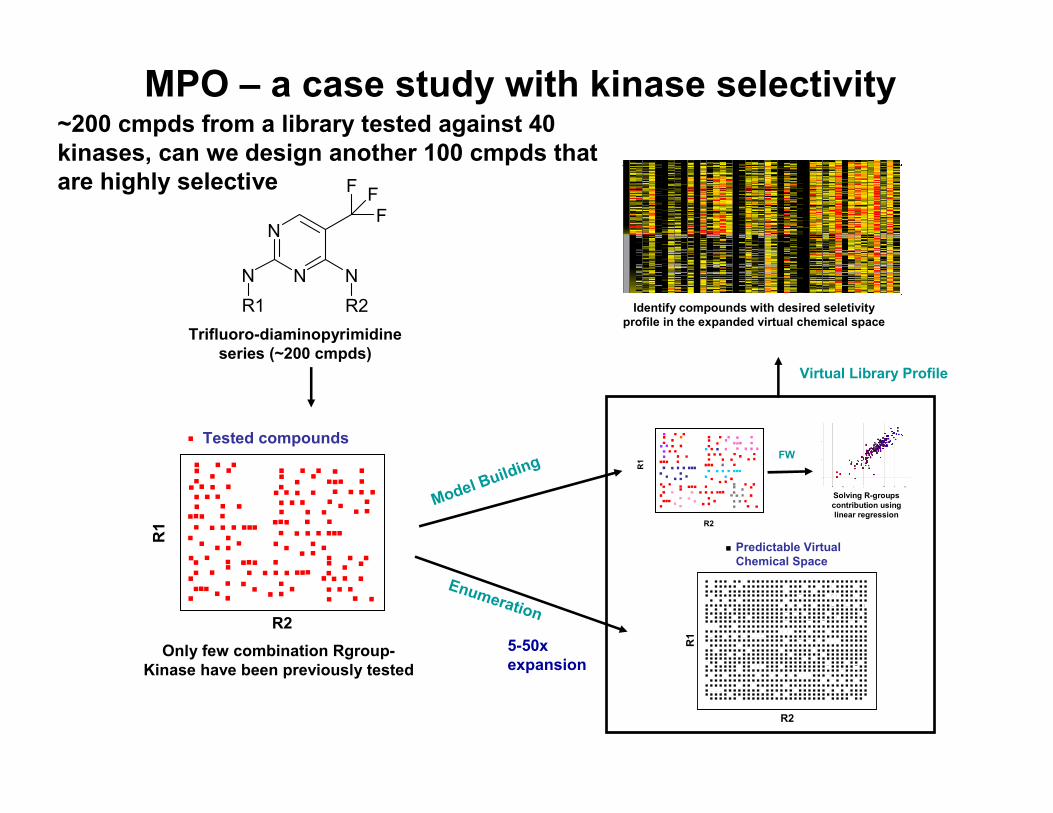
MPO – a case study with kinase selectivity
N
NN N
F FF
R1 R2Trifluoro-diaminopyrimidine
series (~200 cmpds)
R1
R2
Tested compounds
Only few combination Rgroup-Kinase have been previously tested
R2
Model Building R1 FW
Solving R-groups contribution using linear regression
Identify compounds with desired seletivityprofile in the expanded virtual chemical space
Virtual Library Profile
Enumeration
Predictable Virtual Chemical Space
R2
R1
5-50x expansion
~200 cmpds from a library tested against 40 kinases, can we design another 100 cmpds that are highly selective

Predictive models - Leave-One-Out Validations

Experimental Validation of PredictionsKSS pIC50 vs. FW pIC50
r2=0.45 r2=0.59 r2=0.92 r2=0.86
r2=0.74 r2=0.83 r2=0.63 r2=0.88
r2=0.85 r2=0.81 r2=0.81 r2=0.85
~40 cmpds in two series were selected for KSS testing
More promiscuous
More selective

Cheminformatics Challenges for Drug Discovery
• Information retrieval and knowledge managment - rapidly and efficiently present all relevant data/knowledge to scientists atthe right time and right place
• Predictive models - drastically improve the accuracy and interpretability of in silico models for potency and ADME endpoints
• Computer-aided design – provide easy to use software applications to help scientists analyze/visualize their data andmake efficient use of prior knowledge during compound designs

References
1. Agrafiotis, D. K., Lobanov, V. S. and Salemme, F. R. (2002) Combinatorial informatics in the post-genomics ERA. Nat Rev Drug Discov. 1, 337-3462. Lipinski, C. and Hopkins, A. (2004) Navigating chemical space for biology and medicine. Nature. 432, 855-8613. Paolini, G. V., Shapland, R. H., van Hoorn, W. P., Mason, J. S. and Hopkins, A. L. (2006) Global mapping of pharmacological space. Nat Biotechnol. 24, 805-815

















