89
Comparative Genomics The Finale Angela Pena, Ambily Sivadas, Amit Rupani Shimantika Sharma, Juliette Zerick Keerti Surapaneni, Artika Nath, Hema Nagrajan
| Date post: | 28-Dec-2015 |
| Category: |
Documents |
| Upload: | leslie-oneal |
| View: | 215 times |
| Download: | 0 times |

Comparative Genomics The FinaleAngela Pena, Ambily Sivadas, Amit RupaniShimantika Sharma, Juliette Zerick Keerti Surapaneni, Artika Nath, Hema Nagrajan

Outline
Results
• Goal 1 – PCR Assay
• Goal 2 – Comparative genome analysis
• Goal 3 – Haemolysis study
• Goal 4 – Virulent factors
• Discussion

Goal 1Identification and characterization of target genes for PCR Assay

Identification of target genes
A CB
A C
Hhae
NTHi
PCR products of different size
Fw primer Rv primer
One copy of B or multiple copies? Is A-C organized the same way in both organisms?Identify candidate clusters/genes for assay development and conserved regions for primer/probe design

Cluster Analysis – Genome Set

Cluster statistics
Total clusters: 8402 Total common to all genomes : 361 Total unique to Hhae: 82 Total unique to Hinf: 38 Total unique to Pathogenic strains: 0
Protein sequences were clustered using Blastclust

Metabolism protein syntheis /post transtransportDNA repair replication chaperone/protein foldingproteolysis tRNA processing DNA repair/ replication secretioncompetencecell division cell cycle regulationHypothetical ATP/DNA bindingunknown transcriptionstress response secretionantiobiotic resistence
Common clusters – Functional breakdown

Target identification - Protocol 1
1. Take all proteins common to all 25 genomes
• Common = Most conserved proteins

1. Cluster Analysis: Take all proteins common to all 25 genomes
• Common = Most conserved proteins
2. Compute and compare inter-cluster distances for Hhae vs Hinf
• Look for species specific patterns
• Look for including unique genes
Target identification - Protocol 1

Protocol II:BLAST Everything
Our method is this: for every unique Hhae gene, we will locate its corresponding contig
We checked the flanking regions (on the contig) for conserved genes.
We will then locate the conserved genes in the Hinf genome and see if they are adjacent.
Since a wide net can be cast with BLASTn searches, this includes homologs.
If a pistol just isn't working for you . . .
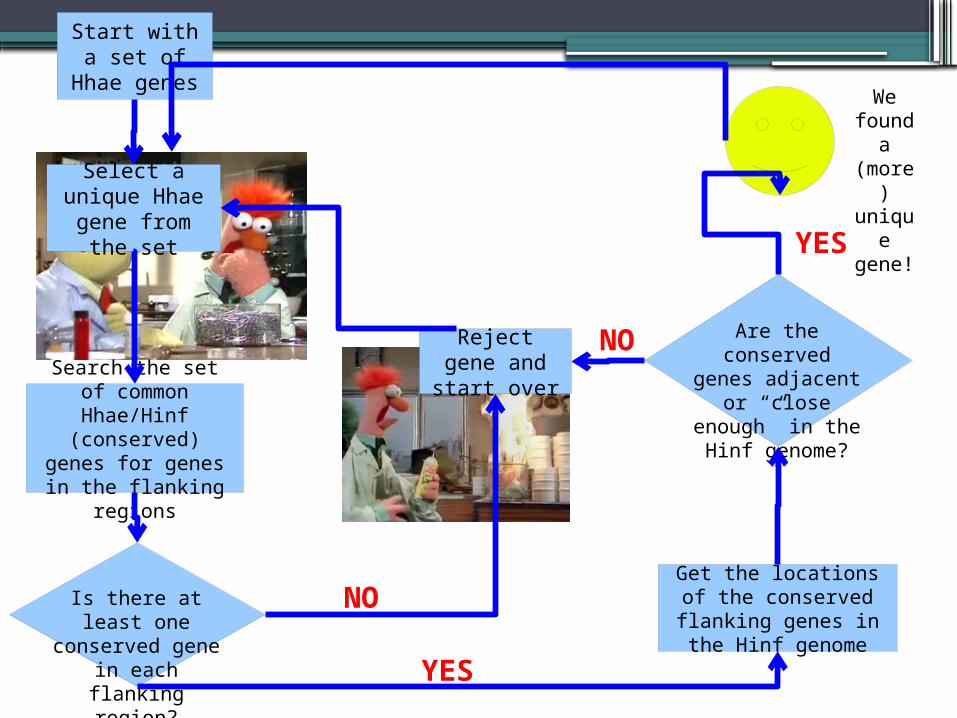
Start with a set of Hhae
genes
Select a unique Hhae gene from
the set
Get the locations of the conserved flanking genes in the Hinf
genome
Search the set of common Hhae/Hinf
(conserved) genes for genes in the flanking
regions
Is there at least one conserved gene in each
flanking region?
Are the conserved genes adjacent or “close enough” in the Hinf genome?
Reject gene and start over
We found a (more) unique gene!
NO
NO
YES
YES

PCR Assay: Results
Target 1
PCR product
No duplication was found for these genes
A B DC
A B D
Hh
NTHi
3-oxoacyl-(acyl carrier protein) synthase III
Nucleic acid binding protein (hypothetical)
fatty acid/phospholipid
synthesis protein
50S ribosomal
protein1020 bp
170 bp
Hh
NTHi
1250 bp
380 bp

PCR Assay: Results
Target 2
PCR product
A B E
A 1 E
Hh
NTHi
predicted membrane
protein
fructose-biphosphate
aldolase
1451 bp
1934 bp
Hh
NTHi
1934 bp
1451 bp
C D
D2 3 4 5
purine nucleoside
phosphorylase
6

Target validation by Insilico PCR
H. haemolyticus5 strains
Non TypableH. influenzae19 strains + 1 Typeable
1 1775870 2749
Step 1: Multiple Sequence Alignment by ClustalW2 - Overview
905 ntsTarget 1

Step 2: Phylogenetic analysis Neighbor Joining Tree Percentage of Identity usingJalview

H. haemolyticus5 strains
Non TypableH. influenzae20 strains
1 1775870 2749
Forward
Reverse
5’-CTCACTTACGCCACCACGTA-3’
3’-TGCAACAATAATCAGTTCAATATCT-5’
Step 3: Finding primers

Product length: 1354
M21621
Product length: 487
AAZD00000000
Non Typable H. influenzae
H. haemolyticus
In silico PCR Analysis
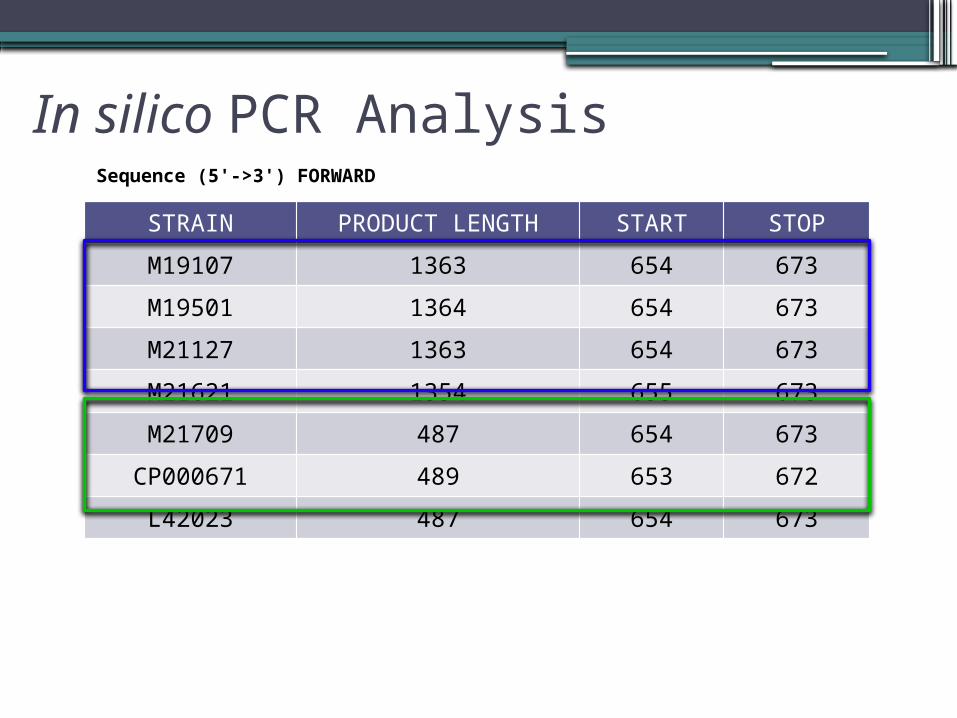
In silico PCR Analysis
STRAIN PRODUCT LENGTH START STOP
M19107 1363 654 673
M19501 1364 654 673
M21127 1363 654 673
M21621 1354 655 673
M21709 487 654 673
CP000671 489 653 672
L42023 487 654 673
Sequence (5'->3') FORWARD

Non TypableH. influenzae20 strains
H. haemolyticus5 strains
1 5372
MSA – Target 2

Goal 2Comparative genomic analysis

Horizontal Gene Transfer• Horizontal gene transfer (HGT), also lateral gene transfer (LGT) refers to the transfer of
genetic material between organisms
Alien Hunter• Predicts putative horizontally transferred regions.• Standalone software• Available at http://www.sanger.ac.uk/Software/analysis/alien_hunter
Usage:./alien-hunter <input_file> <output_file>
INPUT: raw genomic sequence
PREDICTION: HGT regions based on Interpolated Variable Order Motifs (IVOMs)

.sco file

Last time, we got many hits with varied scores that covered almost 90% of the genes in each genome. Hence, we decided to place a threshold on the scores.
• We studied the distribution of scores for each genome by plotting histograms for each genome based on the scores.
• We decided to place a threshold of >70 after studying all the histograms.
Screenshot of M21621

HGT gene count
1 2 3 4 5 60
500
1000
1500
2000
2500
3000
3500
4000
4500
5000
5500
6000
before filteringafter filtering
before filtering after filtering
1699 360
2709 145
3065 225
5601 253
1140 108
2717 185

Insertion elements
• An Insertion element is a short DNA sequence that acts as a simple transposable element.
• A transposable element (TE) is a DNA sequence that can change its relative position (self-transpose) within the genome of a single cell. The mechanism of transposition can be either "copy and paste" or "cut and paste".

IS Finder

FASTA sequencesWe retrieved FASTA sequences by submitting the accession IDs in NCBI

BLASTWe blasted these insertion sequences against each of the strains and got the location of the insertion sequences in the strain.
A PERL script was written to extract the insertion sequences from their respective contigs in each strain.

Comparative Analysis TableFeature /Strain
Tools M19107 M19501 M21127 M21621 M21639 M21709 Average
Genome size - 1774129 1809865 2029793 1959123 2397857 1808157 1963154
GC content % IGIPT 39.39 38.23 39.11 39.11 38.30 38.73 38.81
Total Number of genes
- 1973 1785 2086 1923 2669 1840 2046
Operons - 76 (27) 69 (14) 71 (25) 79 (21) 87 (36) 70 (14) 73
Virulence factors 124 115 116 115 124 144 115
HGT gene count Alien Hunter
360 145 225 253 108 185 213
Pathogenic - No No Yes Yes Yes No -
Insertion elements IS Finder 6 6 - - 15 - 4.5
Hemolytic activity - Y N Y Y N N -

M19107 – Circular alignment using BRIG

M19501 – Circular alignment using BRIG

M21127– Circular alignment using BRIG

M21621– Circular alignment using BRIG

M21639 – Circular alignment using BRIG

M21709 – Circular alignment using BRIG

Goal 3Identification and Characterization of Haemolysin in Hhae

AIM #1
Look for the hemolysin BA operon present in the H.haemolyticus strains and characterize it as present/absent in the hemolytic and non hemolytic strains

HEMOLYSIN • Hemophilus ducreyi, requires two adajecent genes, hhdB
and hhdA for hemolysis .
• hhdB is an outer membrane protein, which is required for secretion and activation of the hemolysin structural protein, hhdA.
• Once secreted, hhdA interacts with target cell membranes, oligomerizes, and forms pores 2.5 to 3.0 nm in diameter, which lyse the target cell
TWO PROTEIN SECRETION SYESTEM

OUR STRATEGY
•Downloaded the Fasta files of all hemolysin protein sequence of the Pasteurellaceae family from NCBI protein database.
•Blasted the predicted protein sequences of the six strains against these.
Cut off threshold: Identity 70% Coverage 80%

RESULTSStrain Hemolysis Gene A /contig Gene B /contif
Haemophilus haemolyticus M19107
Y 51_11|7343|115961417 amino acidsZP_09185204.1| hemolysin [Haemophilus [parainfluenzae]
51_1216|11855|13366503 amino acidsZP_09185203.1| hemolysin activation/secretion protein [Haemophilus [parainfluenzae]
Haemophilus haemolyticus M21127
Y 20_113|106934|111307|1457 amino acids ZP_09185204.1| hemolysin [Haemophilus [parainfluenzae]
20_112|105150|106760536 amino acids ZP_09185203.1| hemolysin activation/secretion protein Haemophilus [parainfluenzae
Haemophilus haemolyticus M21621
Y 1_361|369207|3735801457 amino acidsZP_09185204.1| hemolysin [Haemophilus [parainfluenzae]
1_362|373754|375349|531 amino acids |ZP_09185203.1| hemolysin activation/secretion protein [Haemophilus [parainfluenzae]
Haemophilus haemolyticus M19501
N None None
Haemophilus haemolyticus M21639
N None None
Haemophilus influenzaM21709
N None None
All hits had 70% and more identity and 95-100 coverage

AIM# 2
•Characterize the domains/motifs/residue in hemolysin.
•Depict the secondary structures in hemolysin.
•Predict the 3D structure of hemolysin.

SIGNAL PEPTIDE & HAEMAGGLUTINATION ACTIVITY DOMAIN
• A signal peptide (25 aa) to transport the hemolysin to outer membrane or periplasm. LipoP cleavage site Spase I at 25-26. NOT LIPOPROTEIN
• Haemagglutination activity domain -suggested that the haemagglutination activity domain is a carbohydrate-dependent haemagglutination activity site which is found in a range of haemagglutinins and haemolysins
N’ terminal
Signal Peptide
Haemagglutination activity domain

HAEMAGLUTININ REPEAT• Haemaglutinin repeat is a highly divergent repeat that
occurs in number of proteins implicated in cell aggregation

hemolysins/cytolysins ShlA of Serratia marcescens, HpmA of Proteus mirabilis, EthA of Edwardsiella tarda, HhdA of Haemophilus ducreyi, the large supernatant proteins LspA1 and LspA2 of H. ducreyi, and the HecAadhesin of E. chrysanthemi .Clantin et al., 2004. The crystal structure of filamentous hemagglutinin secretion domain and its implications for the two-partner secretion pathway.PNAS.
TPS DOMAIN All TPS-secreted proteins contain a distinctive N-proximal module essential for secretion, the TPS domain. TpsA proteins display two conserved regions, C1 and C2, and two less-conserved regions, LC region. ANPNL and NPNGIS is found in this region
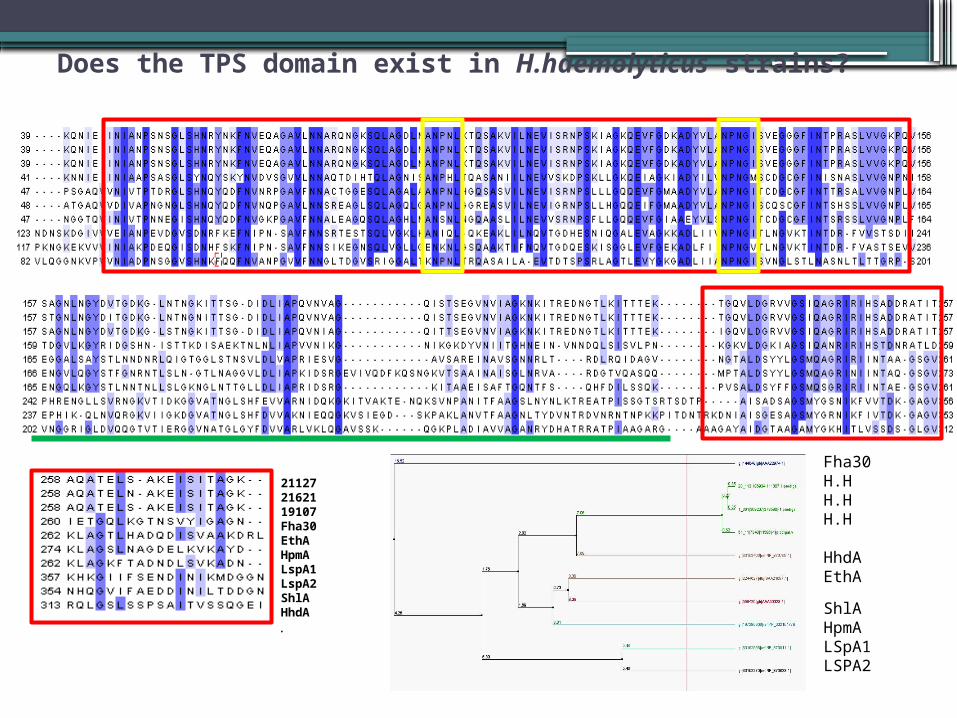
Does the TPS domain exist in H.haemolyticus strains?
211272162119107Fha30EthA HpmALspA1LspA2 ShlAHhdA.
Fha30H.HH.HH.H
HhdAEthA
ShlAHpmALSpA1LSPA2

CONSERVED RESIDUES IN TPS DOMAIN
NPNL & NPNGI These motifs form type I beta -turns, which might play important stabilizing roles. The conserved residues of the TPS domain serve to drive the folding of the TPS domain into a beta -helix and to stabilize the helix
HAD 39-159-PfamOr
TPS 39-270
TPS
ANPNL
NPNLGI

STRATEGY SECONDARY STRUCTURE

AIM #3
•Identify the domains in the hemolysin activator gene •Determine the secondary and 3D
structure of hemolysin activator gene

HEMOLYSIN ACTIVATOR PROTEIN TRANSMEMBRANE PROTEIN
MEMBRANE PROTEINS
α-helical β-barrel
β-barrel membrane protein class are located in the outer membrane of Gram-negative bacteria.
These proteins have membrane spanning segments formed by antiparallel β-strands, creating a channel in the form of a barrel that spans the outer membrane.

DOMAIN IS HEMOLYSIN ACTIVATOR
SP (LipoP) – SPI cleavage site between pos. 19 and 20. NOT LIPOPROTEIN
POTRA_2- polypeptide-transport-associated domain. In ShlB this domain has a chaperone-like function over ShlA.
Activator domain in ShlB is shown to interacts with ShlA during secretion and imposes a conformational change in ShlA to form the active hemolysin.ShlA/B: Serratia marcescens
POTRA_2
Activator Domain SP

Strategy
• Prediction of TransMembrame Beta Barrels (PRED TMBB) Method is powerful when used for discrimination purposes, as it can discriminate with a high accuracy the outer membrane proteins from water soluble in large datasets
• The 'TransMembrane protein Re-Presentation in 2 Dimensions' tool, automates the creation of uniform, two-dimensional, high analysis graphical images/models of alpha-helical or beta-barrel transmembrane proteins.

Work Flow
Sequence
Discrimination scoreSequence

Step 1: Find Discrimination Score
Haemophilus haemolyticus M19107 –hemolystic Contig : 51_1216 Start/End: |11855|13366 Length: 503 amino acids

STEP2Predicted structure 2D of hemolysin activator Haemophilus haemolyticus M19107 –hemolystic
CYTOPLASMIC
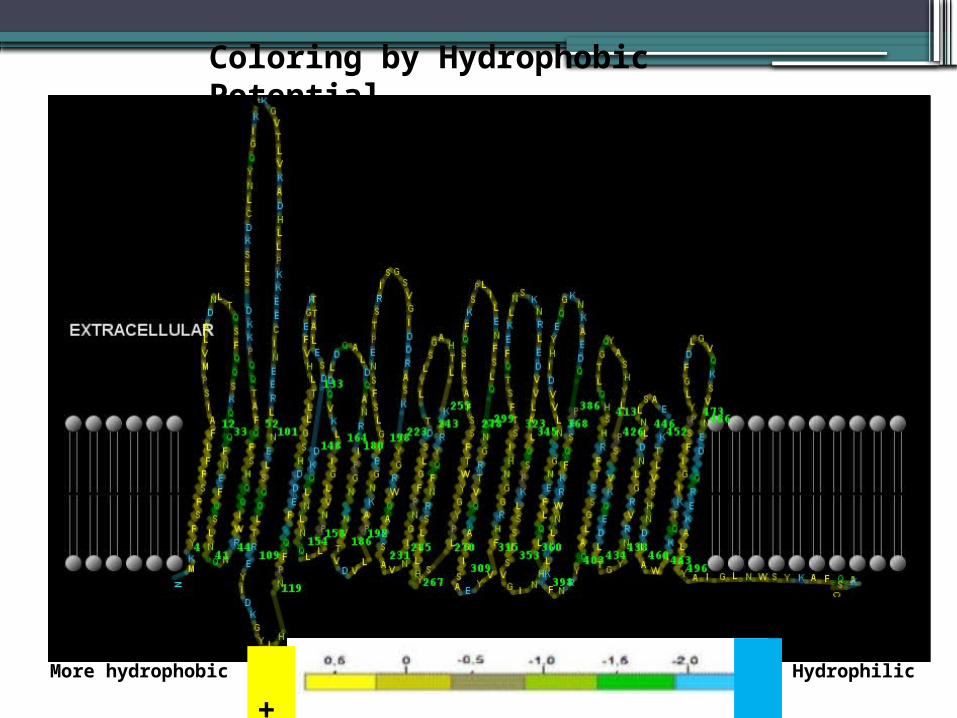
Coloring by Hydrophobic Potential
+ -More hydrophobic Hydrophilic


3D STRUCTRES

STRUCTURE PREDICTION METHODS
Homology
Modelling
De novo protein
structure prediction
Protein Threadin
g
Requires a template with a high
percentage identity
Models the structure based on general
principles that govern protein
folding energetics
Works when homology modelling
fails

PROTEIN THREADING APPROACH • It follows Protein Threading approach to predict the
structure of the target protein sequence.
• In practice, when the sequence identity in a sequence alignment is low (i.e. <25%), homology modeling may not produce a significant prediction. In this case, if there is distant homology found for the target, protein threading can generate a good prediction.
• Protein threading, also known as fold recognition, is a method of protein modeling (i.e. computational protein structure prediction) which is used to model those proteins which have the same fold as proteins of known structures, but do not have homologous proteins with known structure.
• We used RaptorX server which predicts the structure based on protein threading method.

3D STRUCTURES OF HEMOLYSIN FROM RaptorX
Segment 1 (37-158) Segment 2 (159-1457)

3D STRUCTURES OF ACTIVATOR FROM RaptorX
POTRA_2
Activator Domain SP
Activator Domain Segment 1 (149-500)
POTRA_2 Domain Segment 2 (67-136)
Segment 3 (1-66)
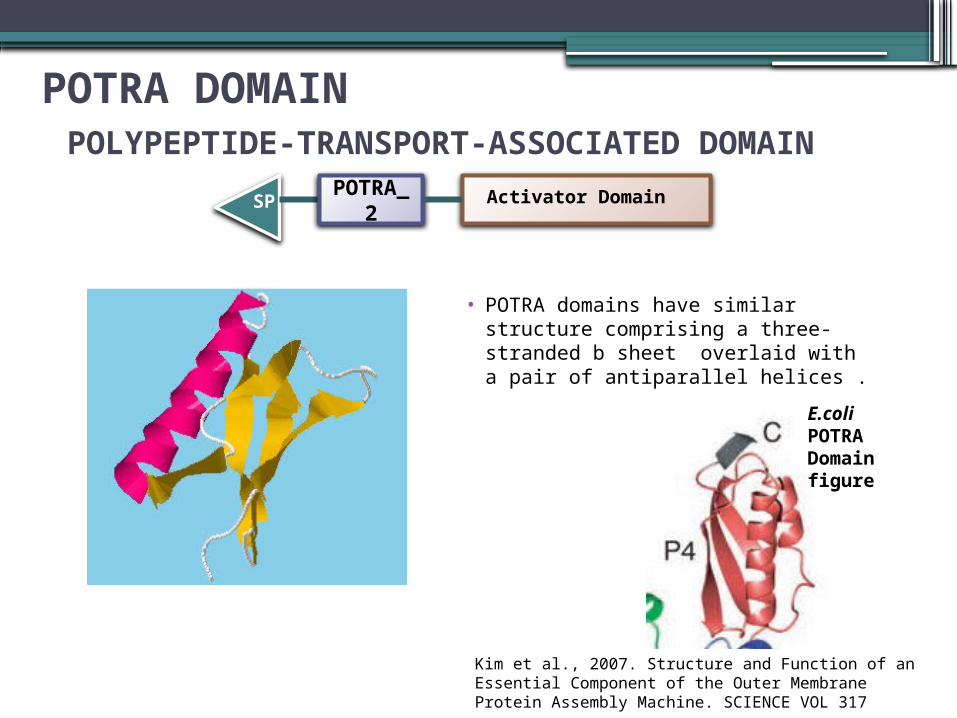
POTRA DOMAIN POLYPEPTIDE-TRANSPORT-ASSOCIATED DOMAIN
• POTRA domains have similar structure comprising a three-stranded b sheet overlaid with a pair of antiparallel helices .
POTRA_2
Activator Domain SP
Kim et al., 2007. Structure and Function of an Essential Component of the Outer Membrane Protein Assembly Machine. SCIENCE VOL 317
E.coli POTRA Domain figure

Goal 4Identify and characterize the potential virulence factors in Haemophilus haemolyticus

Human Immune System
An Immune System (IS) is a system of biological structures and processes within an organism that protects again disease.
In order to function properly, IS must detect a wide variety of agents, from viruses to parasitic worm, and distinguish them from the organism’s own healthy tissue.
Pathogens can rapidly evolve and adapt to new environments to avoid detection and destruction by the immune system:
Phase variation

Phase variation
Phase variation is defined as the random switching of phenotype at frequencies that are much higher (sometimes >1%) than classical mutation rates.
Is a widespread source of intraespecific genotypic and phenotypic variation
Several different mechanisms are exploited by bacteria to switch gene and/or protein expression “on” or “off”
Combinatorial math:
A bacterium with just 20-phase variable loci can exist in 220 different states (more than a million)

Multiple defense mechanisms have evolved to recognize and neutralize pathogens:
Immune System
AdaptiveInnate Creates immunological memory after an initial response to a specific pathogen
Non-specific response to microorganism or toxins found in the cell
Microbes are identified by pattern recognition receptors, eg. LPS
Generic response
Does not confer long-lasting immunity against the pathogen.
1. Inflammatory response2. Activation of Complement System3. Antimicrobial peptides
Immune-Evasion target
Major component of innate IS

Complement System
Biochemical cascade that attacks the surfaces of foreign cells It can contain over 20 different proteins
Complement the killing of pathogens by antibodies:
- produces peptides that attract immune cells- opsonize (coat) the surface of a pathogen, marking it for the destruction
- increase vascular permeability

Host Immune Evasion
Microorganisms have developed many ways to evade complement actions:
• Trapping endogenous C1 inhibitor• Inactivating antibodies through capture of their FC regions• Mimicking structural regulators• Degradation crucial components of Complement System

How we did the analysis ?Virulence Factor Data Base Search of VF in Haemophilus genus
132 VF were retrieved
Upload matrix to MeV
Blastp against all the 25 genomes of Haemophilus
constraints:At least 40% identity
At least 70% of query coverage
Search in NCBI for all VF in Haemophilus genus
RefSeq protein sequences Data Base
Build a matrix with presence/absence +3 Presence-3 Absence
Heat Map was build HCL was generated

Results….
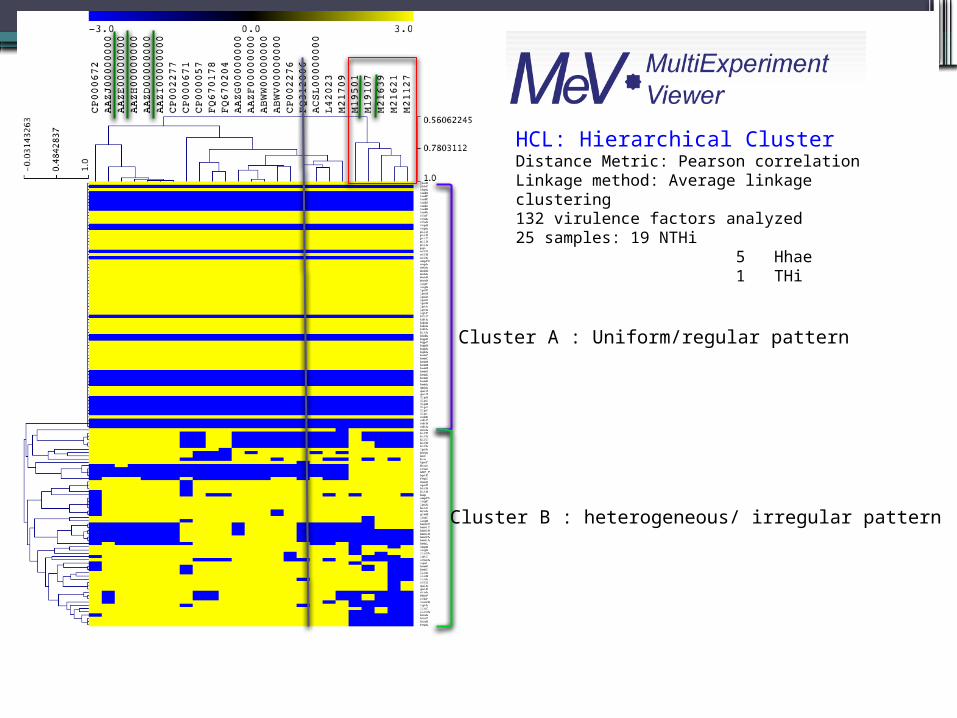
HCL: Hierarchical ClusterDistance Metric: Pearson correlationLinkage method: Average linkage clustering132 virulence factors analyzed25 samples: 19 NTHi 5 Hhae 1 THi
Cluster A : Uniform/regular pattern
Cluster B : heterogeneous/ irregular pattern

Cluster A : Uniform/regular pattern
• tad locus
• Type IV pili
• LPS biosynthesis
• tad locus
• tad locus• LPS biosynthesis
• Adherence
• Transferring-binding protein 1
• Exopolysaccharide
• Haemophilus iron transport locus
• Hemoglobing and hemoglobin-haptoglobin binding proteins
• Heme biosynthesis
• Cytolethal distending toxin

Cluster B : heterogeneous/ irregular pattern
Heme/hemopexin-binding complex
hxuAhxuBhxuCFepA Ferric enterobactin receptor, E. coli
BtucrfaCABC_ThptE
vitamin B12 receptor protein, E. coli1,5 Heptosyltransferase IATP binding cassette transporter familyLipopolysaccharidae heptosyltransferase I
Immunoglobulin A proteaseIgA1

Prevalence Ratio Analysis Patho vs Asymptomatic - Adherence
HAEMOAGGLUTINATING PILI (hifABCDE operon)
Function• Promote adherence to respiratory mucus
and human oropharyngeal epithelial cells• Facilitates colonization
Mechanism: Binding to the Anton antigen (An-Wj) common to buccal epithelial cells and erythrocytes

Prevalence Ratio analysis Patho vs Asymptomatic - AdherenceHAEMOAGGLUTINATING PILI (hifABCDE operon)
Role in virulence
• Expression of pili is a phase-variable phenomenon
• Variation in (TA) repeat units within the overlapping promoter region of hifA and hifB regulates the transcription of the gene
• 11 repeat units – reduced expression• 10 repeat units – maximal expression• 9 repeat units – transcriptional silencing
Found in M19501(5,4),AAZD00000000 (9),AAZE00000000(9),AAZJ00000000(5)
Pathogenic – Hi F3047 strain (9)
In M19107 – only hifA and hifB – split into two contigs –hence no info on TA repeats

Prevalence Ratio Analysis Patho vs Asymptomatic - Adherence
High Molecular Weight Protein 1/2
Function• Adhesins that mediate attachment to
human epithelial cells
Structure features• Autotransport protein• Secretion of these adhesins
HMW1A/HMW1B requires accessory proteins called HMW1B/HMW2B and HMW1C/HMW2C

Prevalence Ratio Analysis Patho vs Asymptomatic-LOS
lic3A
rfaC
Characteristics: Biosynthesis pathway of LOS is producing a branched oligosaccharide attached to a lipid A via two 3-deoxy-D-manno-2-octulosonic acid (KDO) molecules.
rfaC gene product adds the first heptose (Hep I) to KDO
rfaC mutants are shown to produce truncated LPS
rfaC is absent in all H.influenzae, but present in all H. haemolyticus
rfaC mutants also shown to decrease haemolytic activity and expression in E Coli
So, may be, retaining rfaC helps H. hae retain its haemolytic activity
siaA
lgtA
lex2B

Prevalence Ratio Analysis Patho vs Asymptomatic-LOS
lic3A
rfaC
Sialic Acid TransporterLic3A,SiaA or LsgB
Characteristics: Sialic acid is added as terminal nonreducing sugar to LOS – important for bacterial virulence.
These genes code for sialyltransferases which incorporates sialic acid into LOS
In the absence of this transporter, Hinf cannot survive when exposed to serum
Found to be absent in all H.haemolyticus
siaA
lgtA
lex2B

Prevalence Ratio Analysis Patho vs Asymptomatic-LOS
lic3A
rfaC
Phase variable glycosyl transferasesLex2B, LgtB
Characteristics:
Contributes to the significant intrastrain heterogeneity of lipopolysaccharide (LPS) composition in H. influenzae
And phase variable expression
Found to be absent in all H. haemolyticus
siaA
lgtA
lex2B

Prevalence Ratio Analysis Patho vs Asymptomatic- Immuno-evasion
•No difference in pattern observed in this case!

Prevalence Ratio Analysis Patho vs Asymptomatic- Iron acquisition
HxuABC
HxuA binds to heme-hemopexinHxuB releases HxuA from the cell surface into the mediumHxuC is involved in the transport of heme within the cell
Function• Using host heme-
hemopexin as the source of heme iron for growth
Absent in all H.haemolyticus

IgA1
IgA1
YadA is a potent serum resistance factor as it inhibits the classical pathway of complement, Yersinia adhesin A
Fba - Fibronectin-binding protein, streptococcus spp
Protein EmrsA, glmM, galU, galE, manA, manB
IgA1 – serine protease, cleaves IgA1
Protein E – adhesine protein, captures vitonecting (Vn), which prevents the formation of MAC
MAC (Membrane Attack Complex) it forms transmembrane channels, disrupting the phospholipid bilayer of target cells, leading
to cell lysis and death.

Is H. haemolyticus an opportunistic or pathogenic bacterium ?
Ambiental Opportunistic PathogenicInnate IS Adaptive IS
HuxABC
FepA
IgA1
Exopolysaccharides
ompP2
Protein E
YadA
fba
Btuc
rfaC/hptE

DISCUSSION

Hydrogenase-4 10-gene operon
• First identified in E. Coli in 1997• hyfABCDEFGHIJ - hyfR (transcriptional activator)• The proteins encoded by the hyf operon are proposed to
constitute a proton-translocating formate hydrogenlyase• Hyf catalyzes dihydrogen production and ion transport when
the cells are grown at a starting pH of 7.5• This operon is silent in E. Coli – Hyd-3 is the active H2
evolving operon.

Hydrogenase and Virulence
• As per recent studies, Hyd-4 in Yersinia enterocolitica helps in gut colonization▫Using H2 produced during fermentation by intestinal
microflora• Also, hydrogenases facilitated respiratory hydrogen
use by Salmonella enterica and is considered essential for virulence
• So, understanding the expression and role of this hydrogenase operon could provide critical information for▫Characterization of Hhae▫Understanding a new mode of virulence in Hhae (May
be!)

ABC transporter system
•ATP-binding cassette (ABC) transporter system
•One of the largest protein families. •Found in all species and are evolutionarily
related. •Functionally diverse and have roles in a
wide range of important cellular functions.
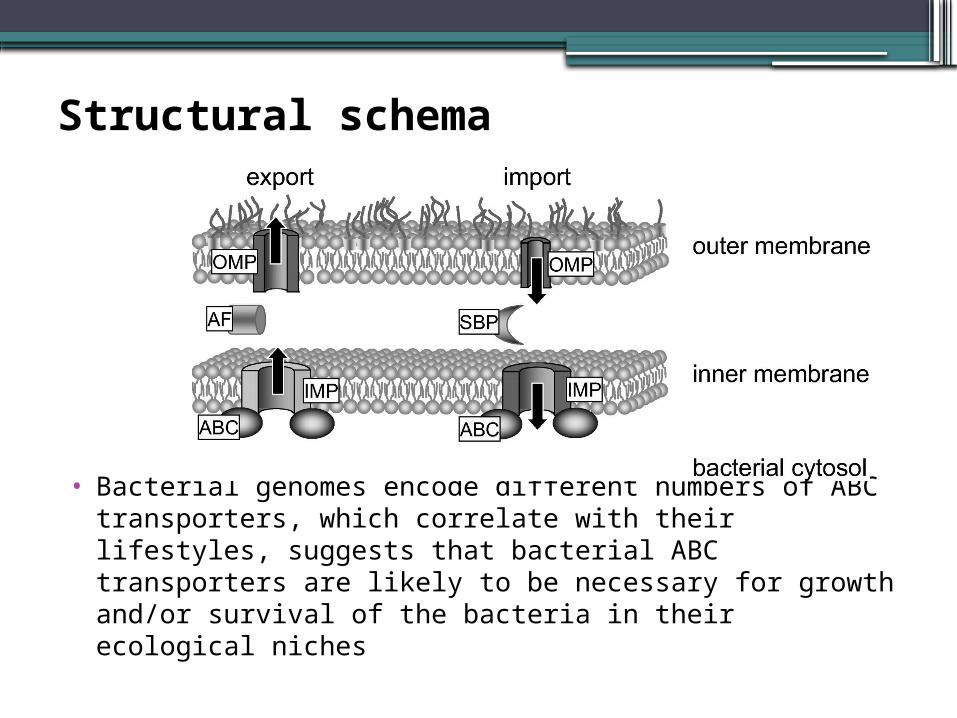
Structural schema
• Bacterial genomes encode different numbers of ABC transporters, which correlate with their lifestyles, suggests that bacterial ABC transporters are likely to be necessary for growth and/or survival of the bacteria in their ecological niches

ABC Transporters and Virulence
• Virulence associated with uptake of nutrients▫ Polyamine, glutamine, sugar
• Virulence associated with uptake of metal ions▫ such as iron, zinc, and manganese
• Virulence associated with cell attachment• ABC transporter (outer membrane) proteins are sometimes
immunogenic too.• Based on the role of the ABC system in virulence, certain
components could be potential targets for developing vaccines too
• In our case, one of the closest homologs is the SalX gene from Pasteurella multocida– which is an ABC-type antimicrobial peptide transport system, ATPase component.
• Hence, characterizing this ABC transporter system could be insightful.




![Untitled-1 [] · 2016-08-23 · Deepu Sivadas, Hemant Deshmukh, Murali Mohan and other team members were all present at the Control Room to cheer this feat, which signals Nigeria's](https://static.documents.pub/doc/80x56/5f7ce1aad3aaab767e045b3f/untitled-1-2016-08-23-deepu-sivadas-hemant-deshmukh-murali-mohan-and-other.jpg)











