164
Copyright page ©2014 Saman Mazahreh ALL RIGHTS RESERVED

Copyright page
©2014
Saman Mazahreh
ALL RIGHTS RESERVED

THE DISARRAY IN THE REGULATORY APPROVAL PROCESS FOR
INTRODUCING NEW MEDICINES TO PATIENTS AROUND THE WORLD
By
SAMAN MAZAHREH
A Dissertation submitted to the
Graduate School-Newark
Rutgers, The State University of New Jersey
in partial fulfillment of the requirements
for the degree of
Doctor of Philosophy
Global Affairs
written under the direction of
Dr. Carlos Seiglie
and approved by
____________________
Dr. Carlos Seiglie
____________________
Dr. Douglas Coate
____________________
Dr. Mariana Spatareanu
____________________
Dr. Susan Sultzbaugh
Newark, New Jersey
May, 2014

ii
ABSTRACT
THE DISARRAY IN THE REGULATORY APPROVAL PROCESS FOR
INTRODUCING NEW MEDICINES TO PATIENTS AROUND THE WORLD
By: Saman Mazahreh
Dissertation Director:
Dr. Carlos Seiglie
The pharmaceutical regulatory framework is a complex system requiring great diligence
when requesting approval for a new medicine in different countries around the world.
The hypothesis was that cost of an application for a new drug approval would play a
major role in the country’s ability to review and approve a new drug. A study of the
history of regulation, current gaps in the system, and regional harmonization efforts was
conducted. Research and data collection on 129 countries was completed. Data included
economic, development, political, and health indicators. Statistical analysis was
conducted and some results confirmed part of the hypothesis while other results indicated
that further study is needed. Stability of a government was confirmed to be a key factor
in the drug approval process. The more stable the government, the more educated the
population, the higher life expectancy because of increased access to medicine.
As part of the research that led to the results above, it’s important to understand which
governance structure is in place or needs to be installed? What are the next steps moving
forward in gathering additional data from other companies, governments, and conducting
a bigger research project that can confirm the results of this study? The World Health
Organization, World Bank, International Monetary Fund, and other technical cooperative
groups serve as forums that are collaborating to achieve better faster results. The term
Global Affairs suggests that there are multiple subjects being discussed. The results of

iii
this paper imply and confirm that a multidisciplinary approach must be taken in order to
enhance the current global pharmaceutical regulatory process.

iv
Preface
The idea to venture into this work came about when I was working in Clinical Trial
Management for a major pharmaceutical company. The problem at the time was patients
in underdeveloped and developing countries did not have access to medicine that was
readily available in developed countries. At the time, it was the case of a drug I worked
on that is a potent chemical entity that fights infections. The drug received approval in
the United States and Europe and other developed countries were soon to follow. In my
role at the time, I was the project manager responsible for supporting all of the clinical
trials for that drug. Once approval was achieved, only a few clinical trials continued as
part of commitments to the health authorities. However, since not all countries had
approval or access to the drug, some patients in developing countries did not have the
option to receive that drug. We would receive letters from doctors in developing
countries indicating that their patients tried all of the current marketed medicines in their
country but were not cured. The doctors heard about our drug and wanted to get samples
for the patients. Unfortunately, it wasn’t as simple as sending a letter. In order to ship the
medicine, there were multiple regulatory hurdles, importation documentation, and proof
that the drug would not introduce an increased risk to the patient. In the end, we were
able to ship the medicine. In some cases, the patient’s health improved and we received
thank you letters; in other cases, the patient had ‘expired’ by the time the medicine
reached their hospital. So, today, we have this dissertation reviewing the current
regulatory process and some of the political and economic reasons of the current situation.
Access to medicine is getting better and global regulatory harmonization seems to be a

v
realistic goal. Below are some quotes that summarize the importance of access to
medicine.
From: FDA Global Engagement Report, April 2012
“As our world transforms and becomes increasingly globalized, we must come together
in new, unprecedented, even unexpected, ways to build a public health safety net for
consumers around the world.”
Margaret Hamburg, FDA Commissioner
“Today we recognize that to successfully protect U.S. public health, we must think, act,
and engage globally. Our interests must be broader than simply those within our own
borders.”
Margaret Hamburg, FDA Commissioner
“Globalization creates real opportunities to collaborate and leverage our collective
expertise and resources. Investments globally are critical to FDA’s success domestically.
Mary Lou Valdez, FDA’s Associate Commissioner for International Programs
“By helping countries build their regulatory capacities, we strengthen their power to
improve the safety and value of goods their own people consume, while also building
confidence in the imports they send to the United States.”
Margaret Hamburg, FDA Commissioner
From: George W. Merck’s famous “Medicine is for the people” speech made in 1950
https://www.merck.com/about/code_of_conduct.pdf
“We try never to forget that medicine is for the people. It is not for the profits. The
profits follow, and if we have remembered that, they have never failed to appear. How
can we bring the best of medicine to each and every person? We cannot rest until the
way has been found with our help to bring our finest achievements to everyone.”
Acknowledgement and Dedication:
I would like to acknowledge the unwavering support of Dr. Seiglie and my dissertation
committee members. Thank you! Without the committee members, I would not have
reached this point! Special thanks to Dr. Seiglie for his patience, leadership, and
continued encouragement that helped guide me through the challenges. Thank you for
caring. Special thanks to Dr. Coate for the inquisitive questions that made me think
differently about my approach to the problem. Special thanks to Dr. Spatareanu for

vi
actively supporting me, making me smile and having an open dialogue about industry and
economic theory. Last but not least, I cannot thank Dr. Sultzbaugh enough for her time
and dedication to this project while working full-time in industry and being a full-time
mom. Her great insight on industry, pharmacy background, and overall professionalism
has taken me to a new level.
I would also like to acknowledge Ann Martin and the Division of Global Affairs staff for
continuously raising the bar, and holding us to a higher standard. I would not be here
today without the continued support of all the staff. Thank you! I must mention and
thank my fellow PhD students for their continued support and creating a motivational
atmosphere to continuing working through all of the challenges, whether it is in the DGA
lounge, 8th
floor of Hill Hall, Library, or McGovern’s! I would like to thank all of my
friends and Kappa Xi Kappa Fraternity brothers for their support and encouraging me to
go over and above! I would like to thank my colleagues at work for their continued
support in guiding me through tough times or simply listening to my challenges.
Lastly, I want to acknowledge my family for their words of encouragement and believing
in me. I would like to thank my father for his words of wisdom in making the most
difficult issues so simple and for his words of empowerment and dedication. For setting
a great example of hard work paying off and to never give up. I would like to thank my
mother for her unending love, work ethic, and faith. I would not be the person I am today
if it were not for your teachings and installation of the belief to work for the greater good.
I would like to thank my siblings for their support, encouragement and always believing
in me. I would like to especially thank my nephews and nieces for always putting a
smile on my face and encouraging me to go over and above.

vii
Dedication:
I dedicate this to:
- My Grandmother, who has been calling me Dr. Saman since the day I was born.
While she was probably expecting and MD, I know she is watching me from
above with a smile on her face.
- My Parents for their unconditional love, teachings, inspirational conversations,
and helping me believe that I can do anything.
Saman Mazahreh
April 22, 2014

viii
Table of Contents
Abstract ii
Preface, Acknowledgement and Dedication iv
Table of Contents viii
List of Abbreviations and Acronyms ix
List of Tables xi
List of Illustrations and Figures xii
Introduction xiii
Chapter 1 1
Chapter 2 21
Chapter 3 49
Chapter 4 68
Chapter 5 86
Bibliography 92
Appendix I 96
Appendix II 101
Appendix III 126
Appendix IV 129
Appendix V 140
Appendix VI 142
Curriculum Vitae 148

ix
List of Abbreviations and Acronyms
AHC APEC Harmonization Center
AMRH The African Medicines Regulatory Harmonization
AP Asia Pacific
APEC Asia-Pacific Economic Cooperation
API Active Pharmaceutical Ingredient
ASEAN Association of South-East Asian Nations
CARICOM The Caribbean Community
CFR Code of Federal Register
CHMP Committee for Medicinal Products for Human Use
CIRS Center for Innovation in Regulatory Science
CMC Chemistry, Manufacturing, and Controls
CPMP Committee for Proprietary Medicinal Products
CPP Certificate of Pharmaceutical Product
CTA Clinical Trial Application
CTD Common Technical Document
DCP Decentralized Procedure
EAC East African Community
EC European Commission
EEMEA Eastern Europe, Middle East and Africa
EFI’A European Free Trade Association
EFPIA European Federation of Pharmaceutical Industries Associations
EFTA European Free Trade Agreement
EMA / EMEA* European Medicines Agency
EU European Union
FDA Food and Drug Administration
FD&C Food, Drug and Cosmetic Act
FIH First In Human
GCC Gulf Cooperation Council
GCC-DR Gulf Central Committee for Drug Registration
GCG Global Cooperation Groups
GCP Good Clinical Practice
GDP Gross Domestic Product
GMP Good Manufacturing Practice
HA Health Authority
ICDRA International Conference of Drug Regulatory Authorities
ICH International Conference on Harmonization
IFPMA International Federation of Pharmaceutical Manufacturers
Associations
IMF International Monetary Fund
IND Investigation New Drug
IOM Institute of Medicine
IRB Investigational Review Board
JP Japan; Japan Pharmacopeia

x
JPMA Japan Pharmaceutical Manufacturers Association
LSIF Life Sciences Innovation Forum
MERCOSUR Mercado Comun del Sur – Argentina, Brazil, Uruguay, Paraguay,
Venezuela, Bolivia
MoH Ministry of Health
MRA Mutual Recognition Agreement
MRP Mutual Recognition Procedure
NAFTA North American Free Trade Agreement
NCE New Chemical Entity
NDA New Drug Application
NGO Non-Governmental Organization
NME New Molecular Entity
OLS Ordinary Least Squares
PAHO Pan-American Health Organization
PANDRH Pan-American Network for Drug Regulatory Harmonization
PhRMA Pharmaceutical Research and Manufacturers of America
PMDA Pharmaceuticals and Medical Devices Agency, Japan
RHIs Regional Harmonization Initiatives
RMS Reference Member States
SADC South African Development Community
SC Steering Committee
SICA Central American Integration System
SwissMedic Switzerland Agency for Therapeutic Products
USA United States of America
USAID United States Agency for International Development
USP United States Pharmacopeia
WHO World Health Organization
*EMEA was the original name for the European Medicines Agency, but was then
amended to EMA.

xi
List of Tables
Table 1: Summary of laws and amendments introduced in the US regulations 43
Table 2: Variable Definitions 78
Table 3: Means and standard deviations of important variables 79
Table 4: Statistical analysis results using Ordinary Least Squares (OLS) model 80-82
Table 5: Statistical analysis results using Cox Hazard model 83
Table 6: Statistical analysis results using OLS model (using time instead of log.time) 143

xii
List of illustrations and figures
Figure1 – NDA review time per country (days) 5
Figure 2: NDA fees per country (USD) 6
Figure 3: NDA fee per country without the USA (USD) 7
Figure 4: Drug Development cycle for a new molecular entity (NME) 16
Figure 5: Standard NDA Regulatory Review Process and Milestones 20
Figure 6: Evolution of the ICH 55

xiii
Introduction
Theoretical modeling can help proactively identify faults or benefits of any system.
Theoretical statistical analysis can be very predictive and can help inform a system’s
potential output. Using empirical data that has been generated from actual outcomes in a
statistical analysis can yield powerful results that can confirm or dispute the theoretical
results. The approach taken in writing this dissertation combines all three pathways in
scoping the issue, leveraging industry data, and conducting statistical analysis to draw
conclusions.
The author has extensive experience in the pharmaceutical industry and therefore, a
practical approach was taken in the methodology and writing style. This approach and
writing style is slightly different than other dissertations that use the standard approach
that uses theoretical modeling in attempting to address an issue or answer a question.
The author used experience from the field, researched academic publications, collected
regulatory, economic, and political stability data in framing the situation. There are two
sides to the story (government and industry), and both have been represented in an
objective manner in this paper.
The diversity of pharmaceutical regulatory requirements makes marketing new drugs a
very complex and costly process that could delay public access to innovative and
essential drugs. In recent years, the pharmaceutical industry has become increasingly
global, taking advantage of the growing opportunities in the rapidly expanding new
markets in Asia, Latin America, the Middle East, and Africa. This globalization trend
created the need for a new strategic approach to pharmaceutical regulations, leading to
more international cooperation and harmonization. Globalization of pharmaceutical

xiv
regulatory standards has become a necessity and a goal for many groups of neighboring
countries in several regions of the world to reduce unnecessary and duplicative
requirement, rationalize time and costs, and create a transparent regulatory process that
improves access to medicines. Attempts to address this known gap have been fragmented.
In this paper, the following will be discussed:
1. Literature review of published work
2. History and current state of pharmaceutical regulation
3. Regional attempts to address the regulatory gaps
4. The role of politics and economics as it relates to the gaps
5. Draw conclusions, identify a potential path forward, and future research
Data has been collected form 129 countries1
on the lead-time for reviewing and
approving a New Drug Application (NDA) as well as the fees required by the respective
health authority. Economic data on the same countries was collected from the World
Bank; fragility score as well as democracy/stability index data were collected from the
Polity project for the same countries2.
The hypothesis is that countries with a higher GDP per capita, higher democracy score,
and lower fragility index will have a higher fee and shorter, predictive, lead-time for
review of new pharmaceutical drug applications. In other words, if the state is stable,
infrastructure will be in place to review the NDA in a timely manner for a set cost that the
applicant will be able to afford because the return on investment for the application will
yield substantial gains from sales.
1 See appendix I for a full list of countries surveyed and data collected
2 Data from the World Bank’s World Development Indicators were used in this study, see references
section.
In addition, data from the Polity project was used to better understand the political stability of each country.
See references section.

xv
The hypothesis will be addressed by first studying the evolution of the regulatory system
for the pharmaceutical industry. Then look at the regional harmonization efforts and how
countries have acknowledged that having separate systems for each country or even
region is not sustainable. Next, a statistical analysis will be completed of all the data that
has been gathered. The data set includes the results of the survey conducted, the data
researched from the World Health Organization, The World Bank, The Polity Index, The
Penn World Table, and other sources. The specific variables studied are:
- NDA review time
- NDA fees
- Life expectancy in a given country
- Literacy rate
- GDP per capita
- Health expenditure
- Physicians per 1000 people
- Democracy score
- Fragility index
- Openness to trade
A statistical regression will be run on the variables and results should show that a country
with high GDP, high literacy rate, and high health expenditure will have a high cost for a
NDA and short time for review of a NDA relative to the other countries.

Chapter 1: Literature Review
This section is broken into two parts. Part A focuses on the pharmaceutical regulation
sources and references while Part B identifies the references for the economic data and
regulation.
A. Pharmaceutical Regulation
Several sources3 have been used to generate the data and information contained in this
document. There have been several key websites, journals, an industry survey, and
textbooks researched to gain the amount of information needed for this dissertation.
Evidence from the available literature indicates there has been limited focus on the
discussion of global regulation and the economic factors contributing to the disarray in
global regulatory frameworks.
In looking at the overall spectrum of areas of research, a broad approach was taken to
retrieve information to support this dissertation. This approach is three fold: governance
structure, regional cooperation and local regulation. The first step is a review of the
current global governance structure that exists for all medicinal issues. The World Health
Organization (WHO), under the authority of the United Nations (UN) serves as the main
body that ties all of the public health issues globally4. Drug Regulation is one of the
topics covered by one of the committees within the WHO – International Conference of
Drug Regulatory Authorities (ICDRA). Previous conferences were researched and cited
for the content relevant to this research5. The conferences covered include: Hong Kong
3 Sources used include an industry survey, data from the World Bank, World Health Organization, and
ministry of health websites, among others. Refer to bibliography for a full list. 4 WHO is the directing and coordinating authority for health within the UN www.who.int
5 The ICDRA meetings occur every two years in different cities attended by Ministry of Health
representatives from most of the WHO’s member countries. The conferences cover multiple topics, one of
which is regulatory harmonization.

2
2002, Madrid 2004, Seoul 2006, Bern 2008, Singapore 2010, and Tallinn 2012. The
conferences occur every two years and representatives from the different ministries of
health discuss and develop plans to harmonize elements of the regulatory process. For
example, outcomes include decisions on the various governments adopting specific
protocols and specifications for approval of manufacturers of Active Pharmaceutical
Ingredients (API). However, no specific global legislation is proposed at these
conferences. Another organization that provides regulatory harmonization across all
continents is the International Conference on Harmonization (ICH). While the ICH does
not have authority to enforce guidelines or penalize organizations, the industry and
regulatory authorities have frequently adopted their published guidelines6.
Second, there are currently regional cooperation groups that discuss public health issues –
one of the main issues is regulatory harmonization. In this particular case, an example of
‘regional cooperation’ is the Gulf Cooperation Council (GCC) and Pan-American Health
Organization (PAHO). These organizations have developed milestones against certain
harmonization goals and review the plans during their regional meetings. Publications
from the conferences that were held were also reviewed and used for discussion in
Chapter 3 of this dissertation7.
Last, we have local (country level) regulatory bodies. These are usually under the
authority of the Ministry of Health (MoH). Where it exists, the country specific website
was researched for each country to confirm requirements for a new NDA. During the
research, it was observed that all of the websites focused on information for immediate
6 ICH has published guidelines in an effort to harmonize requirements. This will be discussed in more
detail in chapter 3. http://www.ich.org/ 7 Asia-Pacific Economic Cooperation. www.apec.org, APEC Harmonization Center. www.apec-ahc.org,
European Medicines Agency www.emea.eu , Gulf Cooperation Council. www.gcc-sg.org , Southern
African Development Community. www.sadc.int , Pan American Health Organization. www.paho.org

3
consumer need. It was very difficult to navigate through the websites and find the link to
the regulatory process. In many cases, no such link existed. The websites for the
countries are listed in Appendix II8.
In addition to reviewing the global governance structure for the health regulation, there
are also independent centers of excellence on regulatory requirements that have been
published on this topic. Information from their publications has been used for this work.
The Center for Innovation in Regulatory Science (CIRS) has evaluated specific country
requirements within specific regions to influence policy in the region9. Papers were
reviewed and elements of the study methodology were used to support the arguments
made in this paper. In a 201210
publication focusing on ICH countries (USA, EU, JP),
CIRS summarizes the NDA review timelines across the 3 major agencies (FDA, EMA,
PMDA)11
covering years from 2002 – 2011. The article highlights that there is a wide
variation in review times across agencies as well as within agencies primarily due to the
application category (priority vs. standard). Not all 3 agencies agree on which products
are priority versus standard. In the period between 2002 and 2011, 45% of the 249
approvals by the FDA and 17% of the 241 approvals by the PMDA were designated as
priority. This compares to just 5% of EMA for the same period of time (McAuslane and
Wang). McAuslane and Wang12
identified 69 common products that were approved by
all three health authorities from 2002 – 2011. Of the 69 products, 41 of the approvals in
8 Appendix II contains all of the websites found for each country that has a website. Some of the websites
are in the local language only. 9 Neil McAuslane and Tina Wang wrote the publication in March 2012 for CIRS titled: “New Drug
Approvals in ICH Countries: 2002 – 2011” http://www.cirsci.org/ 10
Ibid 11
The Food and Drug Administration (FDA) is the USA’s health authority. The European Medicines
Agency (EMA) is the European health authority. The Pharmaceuticals and Medical Devices Agency is the
health authority for Japan. 12
Ibid

4
the USA were priority reviews. McAuslane and Wang also highlight that the majority
(46) of the 69 applications were submitted to the FDA first, 26 of the 69 were submitted
to the FDA and EMA at the same time, while 53 of the 69 applications were submitted to
Japan’s PMDA more than a year after the initial submissions to the FDA and EMA13
.
McAuslane and Wang do not identify reasons for products being submitted
simultaneously to the US and EU regulatory authorities or why the Japan health authority
received most of the applications more than a year later. Japan requires local ethnic
clinical studies in addition to the basic applications submitted to the US and EU. These
studies are not usually conducted early in the drug development cycle because
manufacturers prefer to wait for the Phase III clinical study results to ensure a high
probability of success for the approval of the product, in any country. Since the
companies do not conduct the specific studies required for Japan only, the results of the
ethnic sensitivity studies come after the initial submission to the US and EU. As a result,
the new drug application to the Japanese health authority comes approximately a year
after the US and EU submission.
Another source of valuable data was industry survey that was conducted within a
pharmaceutical company. Industry data on approval time and fees were collected from
129 countries (plots of the data collected are included below). This survey was initiated
in 2011 and data collection was completed in 2012. Figure 1 shows the amount of review
time per country. The time is indicated in days and represents review of standard
applications, not priority or orphan drugs. The figure shows variability in many of the
countries where the most advanced countries are the troughs at approximately 365 days
and underdeveloped countries at the 3-year mark (1095 days). Due to the high number of
13
Ibid

5
countries, it was not possible to display the names of all 129 countries in Figure 1.
However, the data points for all the 129 countries are represented.
Figure 1 – NDA review time per country (days)

6
Figure 2: NDA fees per country (USD)
Figure 2 includes the entire list of countries. The US FDA price tag of $2MM skews the
table and most developing countries’ fees appear to be irrelevant in comparison. The
smaller peaks are the developed EU countries, Japan, Australia, and Canada. The
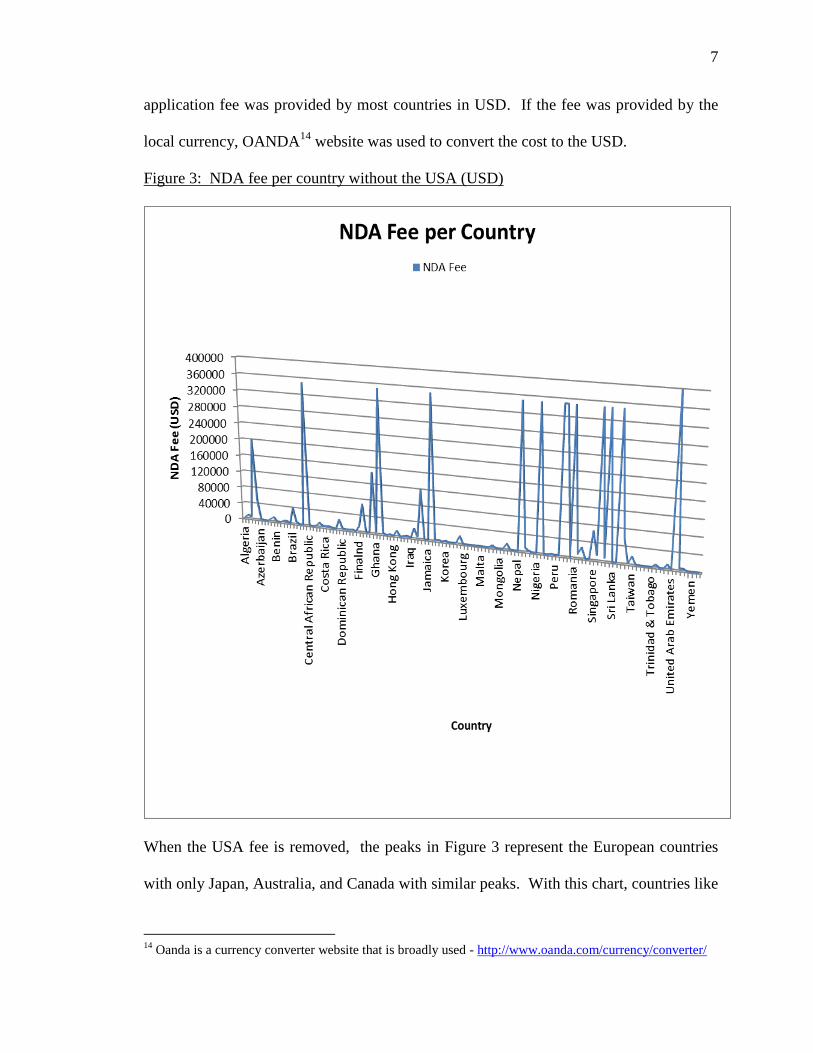
7
application fee was provided by most countries in USD. If the fee was provided by the
local currency, OANDA14
website was used to convert the cost to the USD.
Figure 3: NDA fee per country without the USA (USD)
When the USA fee is removed, the peaks in Figure 3 represent the European countries
with only Japan, Australia, and Canada with similar peaks. With this chart, countries like
14
Oanda is a currency converter website that is broadly used - http://www.oanda.com/currency/converter/

8
Singapore, Brazil, and Switzerland are the smaller peaks ranging from $50,000 to
$100,000 USD in application cost.
The Thomson Reuters IDRAC15
files have summaries of country specific requirements,
processes, and fees. However, the database is limited in the number of countries covered
and does not compare countries to identify gaps and opportunities. Information from this
source was used to compare data collected from a survey conducted with many of the
countries. See Appendix I for the regulatory processes from selected countries and an in-
depth description of the Singapore regulatory review process.
Additional academic publications on regulation were reviewed to better understand the
landscape of current published work. Braithwaite and Drahos16
discuss the regulatory
framework in multiple industries and focus on business regulation. A chapter of the book
dedicated to drug regulation focuses on the business as well as the current regulatory
regimes that govern the industry. The chapter begins by looking at the history of the
pharmaceutical regulatory process from its very inception and also discusses regulated
and unregulated drugs. Braithwaite and Drahos discuss key themes such as the EU
leadership in the harmonization of the technical requirements, the global regulatory
structure, regional, industry, individual, business, and professional actors in framing the
current landscape of the regulatory framework. Information from this text was used for
Chapters 2 and 3 as well as to build on the opportunities and challenges that currently
exist in the global regulatory process.
15
IDRAC files are available by subscription. A description of the available data can be found at the
following location: http://thomsonreuters.com/business-unit/science/pdf/IDRAC-cfs-en.pdf 16
Braithwaite, J., Drahos, P., Global Business Regulation, Cambridge 2000

9
Abraham and Smith17
compiled a collection of articles that discuss different points of
view in the pharmaceutical regulatory process. Relevant chapters included: The
regulatory laws and political culture in the United States and Germany, Europeanization
of medicines regulation, globalization of medicines control, New molecules, Markets and
changing drug regulatory practices, The limitations of current ethical regulations.
Abraham and Smith identify the gaps in the different regulatory authorities and stop short
of identifying a path forward.
Berry and Martin18
also contains a collection of articles covering a broad range of
regulatory questions mainly within the United States. The book also included topics on
generic drugs, biological, non-prescription (over-the counter medicine) and the impact on
the industry. These include: pharmaceutical regulation before and after the Food, Drug,
and Cosmetic Act, Modernizing the food and drug administration, The new drug
approval- process - before and after, Ways, means, and evolving trends in the U.S.
registration of drug products from foreign countries. This book focused on the US
regulatory process, and is supportive of the write up of the details outlined in Chapter 2 in
this paper.
In addition to published works, information from the ministry of health or drug regulatory
health authority websites for many of the countries was researched and used for
supportive information. See Appendix II for a listing of the countries with their matching
website. This is not an all-inclusive list and information extracted from these websites
gave additional details about the specific regulatory requirements in each country.
17
Abraham, J., and Helen Lawton Smith, Regulation of the Pharmaceutical Industry, New York : Palgrave
Macmillan, 2003 18
Berry, Ira, Martin, Robert P., The Pharmaceutical Regulatory Process, New York: Informa Healthcare,
2008.

10
Economic Regulation
The theory of economic regulation was studied as discussed by Peltzman and Stigler19
.
They argued that regulation is acquired by the industry and is designed and operated
primarily for its benefit. Alternative views of the regulation of industry include (1) the
notion that regulation is in place for the protection and benefit of the general public and
(2) the irrational political machine controls what it is directed to control but the few who
are in a position of influence. Stigler and Peltzman argue that the industry influences the
regulation to benefit itself only and this model is applicable across industries, including
the pharmaceutical industry. This theory may be applicable in the United States for most
industries, but this paper will show reasons why the economic theory of regulation does
not apply to pharmaceuticals, especially outside the US.
Ron Vogel, a Professor of Economics in the Eller College of Management and Research
Professor in the College of Pharmacy at the University of Arizona, covers a wide range of
topics on the pharmaceutical industry with focus on the economics and policy making
within the industry. Vogel argues that drugs were never regulated in the US until 1906,
when the Pure Food and Drugs Act was passed marking the beginning of the federal
regulation of drugs in the USA20
. Emphasis of the 1906 act was made on adulteration
and labeling of food, drinks and drugs, not testing and control of contents. In 1938,
Congress enacted the Food, Drug, and Cosmetic (FD&C) Act which required the drugs
19
George Stigler and Sam Peltzman published many articles on this top and a full listing is included in the
bibliography of this paper. Below are two of the articles referenced in this discussion.
Stigler, George “The Theory of Economic Regulation” The Bell Journal of Economics and Management
Science, Vol. 2, No.1 (Spring 1971), pp.3-21
Peltzman, Sam “George Stigler’s contribution to the Economic Analysis of Regulation” Journal of Political
Economy, 1993, vol. 101, no.5 pp.818 – 832 20
Vogel, Ronald, Pharmaceutical Economics and Public Policy, New York: Pharmaceutical Products Press,
2007

11
be proven to be safe for the use suggested on the label. FDA was given authority to
judge safety by requiring manufacturers to prove new drugs to be safe prior to marketing.
In addition, the law granted the FDA authority to regulate drugs and cosmetics in
addition to food. Therefore, companies started testing the drugs before marketing it to
comply with the laws. Amendments were introduced in 1962 to include more stringent
testing that increased the monetary burden on the pharmaceutical companies. Greater
safety thresholds were introduced mandating 3 phases of drug testing. For the first time,
companies were required to prove the drug was effective against the condition for which
it was being marketed for prior to marketing the product. Lastly, phase 3 studies, which
encompasses a larger patient population was added as a requirement to show both safety
and effectiveness of the drug. This regulation was a win-win situation for the regulators
and the patients21
.
Another valuable resource on the history of regulation and health economics is the World
Health Organization (WHO). In 2011 WHO published a report22
that provides details on
expenditures per country and per region. This information will be used in the
development of the argument for this paper in explaining rationale for identifying a
country with a firm infrastructure vs. those that do not. The 2011 report concludes that
per capita pharmaceutical expenditures in 2005/2006 ranged from $ 7.61 USD in low-
income countries to $ 431.6 USD in high-income countries, with considerable variation
between income groups in each country. The reports also highlights that the total
pharmaceutical expenditure is closely related with both total health expenditures, and
with gross domestic product (GDP).
21
Ibid 22
WHO Report WHO/EMP/MIE/2011.2.6
http://apps.who.int/medicinedocs/documents/s18767en/s18767en.pdf

12
The World Bank website is a valuable resource for providing details about the GDP and
GDP per capita for all the countries in this study. This information will be used to
support the argument that countries with stronger GDPs have a more developed
regulatory infrastructure to support regulation of medicines. The World Bank was also a
valuable resource in attaining information such as literacy rate, physicians per 1000
people, foreign direct investment (FDI), and life expectancy. While not all of the
information was directly used in the model, the information obtained provided supportive
background data in addressing the questions of this paper.
The Penn World Table23
was also a valuable resource for information on the countries’
financial status including real GDP from 1950 – 2011. This information will also be used
to support the argument that countries with stronger GDPSs have a more developed
regulatory infrastructure to support regulation of medicines. The historical trends from
this resource will also help show the progress being made in the emerging markets. The
primary data set used for this paper was the 2010 and 2011 GDP, the economy openness
factor (Open K) to show how much trade a country is involved in, and the size of the
government consumption (KG factor) relative to the GDP. This data set will be
discussed in more detail in Chapter 4. The Polity Project24
was a valuable resource in
attaining information on the democracy score and fragility index for the countries studied.
This information was used to confirm and support the argument of this thesis that the
more stable countries had a higher probability of having a sound pharmaceutical
regulatory process.
23
The Penn World Table can be found at : http://www.rug.nl/research/ggdc/data/penn-world-table 24
The Polity Index: http://www.systemicpeace.org/polity/polity4.htm

13
Overall, research was conducted in different disciplines using data collected from an
industry survey as well as leveraging published data. Combining these sets of data into
one paper and conducting analysis using statistical methods is a first. No other published
work has the combination of these data sets nor has any published work set out to test the
hypothesis set forth in this paper. Previously published work on regulation focused on a
single country, single region, or limited number of countries. In this paper, 129 countries
are included in the study. Other published work that focused on health related topics
discussed health expenditure and/or GDP, political stability, or regulatory process. No
published work combined these factors in a single publication. The following chapters
will discuss the history of regulation, the regional attempts at harmonization, the
economic factors as they impact the regulatory framework, and conclude with results,
potential path forward for the regulatory process, and future work.
25Prior to the submission of an NDA to a regulatory health authority, the manufacturer
must go through the drug development process. For the purpose of this paper, only a
brief description will be given to give background information on the content of the NDA.
A drug manufacturer must go through the initial toxicology stage to prove the new drug
being investigated is safe to test on humans. In order for this step to be complete, animal
studies are conducted where the animals (mice, dog, and/or monkey) are injected with a
strain of the disease being investigated and different dose levels of the new drug. Once
the company identifies the limits that would be appropriate to test on humans, an
Investigation New Drug (IND) Application is filed with the FDA (Clinical Trial
25
This section is written based on knowledge and experience the author gained from working in the
industry

14
Application – CTA in Europe26
). The application includes the toxicology data and a
proposed First-In-Human (FIH) study. The IND application is reviewed and, in some
cases, a special meeting is held with the regulatory authority in case of any questions.
The approval of the IND application marks the start of a Phase I study. Once the Phase I
study is complete, a Phase II study is planned, a clinical study protocol is drafted based
on the results of the Phase I study, and another application is submitted to the health
authority for approval prior to the initiation of the study. Once approval is granted, the
study can begin. If the study is to be conducted in more than one country, a separate
application must be filed to each individual country prior to the start of the study. Once
the Phase II study is complete and all results are analyzed, meeting is typically scheduled
with the regulatory authority to discuss results and plan for Phase III. A Phase III study
protocol is finalized; it is submitted to the health authority in the countries that will be
involved. Once approvals are received, the clinically labeled product is shipped out to
each country for study initiation. Phase III studies are typically global large-scale studies
that can take 2 -3 years to complete. Once the study is complete and results are reviewed
within the company and an expert panel, a decision is made to submit the NDA to the
health authority. The NDA contains information on all of the studies conducted starting
with initial discovery studies. The NDA also contains details on the chemical synthesis
of the new molecular entity and the drug product manufacturing process. The review
time shown earlier in Figure 1 reflects the review time for the NDA only, not the
previous phases.
26
A Clinical Trial Application is a form that a research organization submits to a health authority to request
permission to conduct a clinical study. More details can be found here:
http://ec.europa.eu/health/files/pharmacos/docs/doc2005/10_05/ca_14-2005_en.pdf

15
The other piece of important information is that, as the drug development cycle
progresses, the number of molecules being studied decreases. In the early phases of
discovery, thousands of compounds are investigated. Out of the thousands of compounds
studied, only a few hundred progress to the pre-clinical / toxicology / animal studies. Out
of the few hundred compounds, less than 10 are progressed to the Phase I FIH study. The
number of subjects tested in the Phase I study range from 20 to 100. Compounds
entering Phase II are between 5 to 10 and the number of subjects range from 100 to 500.
Fewer than 3 compounds from the original thousands reach the Pivotal Phase III stage of
development where massive global studies are conducted with thousands of patients from
around the world. The information from all of the studies conducted are included in the
NDA. Additional Phase IV (post approval) studies are conducted to monitor the market
activity. Some countries (other than the most developed) require 12-month post-approval
pharmacovigilance27
data. This is another reason some of the spikes are seen in Figure 1
shown earlier in this chapter. Figure 4 below summarizes the drug development process
for all of the stages.
27
Pharmacovigilance studies focus on the detection, assessment and prevention of adverse reactions to
drugs. In the context of this paper, these are studies that are conducted for drugs on the market.
http://apps.who.int/medicinedocs/en/d/Jh2934e/3.html

16
Figure 4: Drug Development cycle for a new molecular entity (NME)28
To simplify the description of the NDA review process, Figure 5 has been created to
show the generic drug approval process for many of the developing countries that require
a Certificate of Pharmaceutical Product (CPP)29
. A more detailed description of the
regulatory review procedure and process maps for selected countries can be found in
Appendix I. In most countries, a Registration Committee would be in charge of the
overall application and would be the primary contact with the applicant company. The
Registration Committee would have sub-committees that focus on the various sections of
the application. The first step in Figure 5 is for the health authority to validate or confirm
that the application is complete and contains all of the relevant sections. In some cases,
28
Graphic of the drug development cycle representing all clinical studies, number of patients in each phase,
as well as number of compounds studied: http://www.immunetrics.com/applications/drug-discovery.php 29
Certificate of Pharmaceutical Product (CPP) must be issued by one of the major countries (US, EU, JP)
manufacturing or marketing the product confirming the product meets all legal and quality requirements
and is being marketed in that country.
http://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/ImportsandExportsCompliance/uc
m348825.htm

17
the health authority may request additional information from the company that is filing
the submission. Once all of the documents are received and confirmed, the review
timeline starts. Some health authorities have a structured approach and a system in place
tracking all of the applications while others do not have a structured approach.
The second step in the review process is to assign the different sections of the dossier to
different sub-committees. The application is usually broken up into two major sections,
Clinical, and Chemistry Manufacturing, and Controls (CMC). These two major sections
can be divided into several different sub-sections. Depending on the country and the
available capability and capacity, it could be one person reviewing each sub-section or
the entire section. If it is one person reviewing the entire section, then there will be a
great deal of dependency on the approvals of the more advanced countries. During this
stage of the review, it is normal for each reviewer to compile a list of questions and send
them to the applicant for additional clarification. In most cases, the health authority
representative would send the questions to the company representative in their country.
The company representative would then translate and send the questions to the global
regulatory person in the company who would then call on the technical experts to respond
to the questions. There are multiple communication channels and languages that the
information must travel through which adds complexity to the process. Once all of the
questions are answered and the health authority is satisfied, the reviewer(s) would then
provide a recommendation of continue / reject to the Registration Committee within the
health authority who then will issue the decision.
If the decision from Step 2 is to proceed, the health authority may ask the companies for
samples of the investigational product and reference (testing) materials. This step could

18
occur in parallel to Step 2. However, many countries prefer to wait to make sure they are
satisfied with Step 2 before investing resources on testing of a product that may not be
acceptable for other reasons. Since the application contains methods for testing the
product, the health authority will then either test the product samples or use an
independent analytical laboratory to test the product. If the results are within the
specifications set in the application, then a supportive statement is given to the
Registration Committee for approval. If the results are different or new peaks or
interference are seen in the chromatograms30
, questions are sent to the applicant for
clarification. The same steps would then follow as in Step 2 when sending
communication back to the technical experts in the company submitting the application.
The fourth and last step in the approval process is the pricing and labeling. The applicant
company can be requested to provide pricing data from countries where the product is
marketed to compare and generate a baseline. Other administrative information on
registration, marketing materials, and labeling in other countries may also be requested.
The Pricing Unit within the health authority proposes a price based on the information
provided by the applicant company and the country’s local guidelines. The Registration
Committee makes a decision based on the report from the sub-committees (e.g. scientific
assessment, lab analysis, and the cost analysis). While typically outside of the
registration process, the name of the product maybe different from country to country.
This is a negotiation the manufacturer will have with the health authority. The name
could be different from one country to another because the manufacturer may request it
30
Chromatograms are print-outs of chromatography results. Chromatography is a technique used to
separate and analyze mixtures of chemicals and results would show a peak for each chemical within the
mixture.

19
for promotional purposes or the health authority could request it because it may have a
certain connotation that may not be true once translated in the local language31
.
31
This information is based on the authors experience when working on products marketed in different
parts of the world. A tiered approach in this case would mean, as a hypothetical example, that a company
would set a price of $10 for the rich countries, $5 for medium income countries, and $2.50 for low income
countries with each tier having a different brand name. So the same product can be sold different countries
with different brand names.

20
Figure 5: Standard NDA Regulatory Review Process and Milestones32
32
Neil McAuslane and Tina Wang wrote the publication in March 2012 for CIRS titled: “New Drug
Approvals in ICH Countries: 2002 – 2011” http://www.cirsci.org/
Milestones
Official
submissi
on date
Process
Scientific
assessment
start time
Referred
to
Analytical
Laborator
y
Pricing
informatio
n
requested
Registrati
on
committee
decision Start of
queue
time
Submitted to
scientific sub-
committee
Validation
and Queue
Scientific
Assessment
Analysis of
samples
Collection of
pricing data
The authority
checks the
dossier to
ensure that
all
documents
are present
Company is
asked to
provide
additional
documentatio
n, if required
The validated
file waits in a
queue to be
picked up for
scientific
assessment
The dossier is assigned
to a member(s)of
the scientific sub-
committee for pre-
review and
completion of the
scientific report
form
Where necessary, the
company is asked
to provide further
information or
clarification
The report is referred
to the scientific
sub-committee
which makes a
recommendation
on whether the
product should be
registered
If the scientific
sub-committee
recommendation
is positive, the
product is sent to
the Lab for
analysis
Company is
notified and
provides samples
and reference
material for
analysis
Sample is
analyzed and Lab
issues a report on
whether it is
accepted
On acceptance of
the analysis the
company is
notified and asked
to provide pricing
information
Company
provides pricing
data from
countries where
the product is marketed
Other
administrative
information on registration in
other countries
may also be required
The pricing sub-committee
proposes a price based on the
information
provided and
guidelines
The Registration Committee makes
a decision based on the report from
the scientific sub-
committee, Lab and the pricing
sub-committee
Validation
& Queue
Scientific
assessment
Analysis of
samples
Collection of
pricing data

21
Chapter 2: Pharmaceutical Regulation
Medicines are probably as old as mankind and the concept about how their quality has to
be ensured has evolved gradually over time. For example, King of Pontus, Mithridates VI
(~120 BC), formulated a compound preparation which he called “Mithridatium” included
41 different ingredients and was used as a cure for many illnesses until as late as the
1780s33
. It wasn’t until 1540, when the Apothecaries Wares, Drugs and Stuff Act was
implemented in England, which required supervision of the manufacture of all medicines,
including Mithridatium34
. The Act was one of the first British laws to regulate medicines
and it instituted the appointment of four inspectors of “Apothecary Wares, Drugs and
Stuffs.” This could be viewed as the start of pharmaceutical regulations in England.
History of Pharmacopoeias, the official books of drug quality standards, probably dates
back to one of the proclamations of the Salerno Medical Edict issued by Fredrick II of
Sicily (1240), and ordered apothecaries to formulate medicines always in the same way –
forma curiae35
. The first Pharmacopoeias as we know them today were first seen in
Europe from the 16th century (e.g. the first Spanish Pharmacopoeia was issued in 1581).
The standards for the method of manufacture of Mithridatum were established in England
in The London Pharmacopoeia only in 161836
.
The modern medicines regulation started only after breakthrough progress in the 19th
century life sciences, especially in chemistry, physiology and pharmacology, which laid a
solid foundation for the modern drug research and development and started to flourish
33
Griffin JP, Shah RR. History of drug regulation in the UK. In: O’Grady J, Griffin JP, editors. The
Regulation of Medical Products. London: Blackwell BMJ Books; 2003 34
Ibid 35
Ibid 36
Rago, L., Santoso, B. Drug Regulation: History, Present and Future In: van Boxtel, C.J., B. Santoso and
I.R. Edwards, editors. Drug Benefits and Risks: International Textbook of Clinical Pharmacology, IOS
Press and Uppsala Monitoring Center, 2008

22
after World War II. Unfortunate events have catalyzed the development of medicines
regulation more than the evolution of a knowledge base. In 1937 over 100 people in the
United States died of diethylene glycol poisoning following the use of a sulfanilamide
elixir, which used the chemical as a solvent without any safety testing. This enabled the
establishment of The Federal Food, Drug and Cosmetic Act with the premarket
notification requirement for new drugs in 1938. However, in countries with poor
regulatory environment, even recently, medicines contaminated with diethylene glycol
have killed patients37
.
Over one hundred years ago, medicine in the United States was prescribed and dispensed
to patients by a pharmacist. The pharmacist was the one that determined which mix of
herbs or chemicals would treat an illness. In the United States, the first Pharmacopeia
was established in 1820 and the Philadelphia College of Pharmacy was established in
1821 producing trained pharmacists. However, in most of the world, there was little
governance back then of who would certify a person to be a pharmacist or approve the
'medicine' that was prescribed. It was a skill that was passed on from generation to
generation until the medical doctors developed an infrastructure to be in a position to
have the authority to prescribe the 'right' medicine to the patients. In the early 1940s,
chemical and pharmaceutical companies emerged as a strong industry that was able to
influence policy in the US and Europe38
.
37
In 1995-1996, 86 children in Haiti died after using Acetaminophen syrup that used Glycerin
contaminated with Diethylene Glycol. www.cdc.gov/mmwr/preview/mmwrhtml/00043194.html
In 2008 – 2009, 54 children under the age of 3 died from exposure to Diethylene Glycol that was used in
Acetaminophen based teething syrup in Nigeria.
www.cdc.gov/mmwr/preview/mmwrhtml/mm5848a2.html 38
Braithwaite, J., Drahos, P., Global Business Regulation, Cambridge 2000

23
In the nineteenth and early twentieth century in the West, pharmacy was a family
business, progressively subject to regulation by Pharmaceutical Societies. Pharmacists
would mix their own medicines. Patent-medicine peddlers travelled the countryside with
their wares or used local grocers as retail outlets. Some of these family businesses saw
the opportunities in internationalizing. They created new products by learning from
indigenous medicine in exotic places. Many large companies found the tropics
particularly rich sources of new biological materials in much the same way as they now
find them sources of genetic materials.
Regulation favored these international companies, crushing their small business
competitors39
. Pharmacopeias started more as recipe-books than as instruments of
regulation as long ago as the fifth century BC in Greece40
. Pharmacists, like cooks, could
experiment with the basic recipes and market their innovations. It was the medical
profession which slowed down pharmacist control over advice to consumers on drugs41
.
In most Western countries, the medical profession had successfully lobbied for laws to
require potent drugs to be sold only on the prescription of a licensed medical practitioner.
At the end of the twentieth century, developing countries are on the same trajectory;
though for most of the world’s poor population the local pharmacist remains a more
important source of therapeutic power and advice than the doctor or the global
pharmaceutical company.
After the thalidomide disaster of 1961, the medical profession lobbied states to require
more stringent safety testing and then to provide evidence of efficacy before a product
39
Ibid 40
McCoy, Alfred W. ‘Heroin as a Global Commodity: A History of Southeast Asia’s Opium Trade’, in
A.McCoy & A. Block (eds) War on Drugs: Studies in the Failure of US Narcotic Policy. Boulder,
Colorado: Westview Press, 1992. 41
Braithwaite, J., Drahos, P., Global Business Regulation, Cambridge 2000

24
was allowed to be marketed. In the 1970s, manufactures were also required in Western
nations, then progressively in developing counties, to comply with Good Manufacturing
Practices regulations, written by the US Food and Drug Administration (FDA) then
propagated by the World Health Organization (WHO).
By 1990 most pharmacists or ‘bathtub’ manufacturers could not afford the average of
$231 million for the research health regulators in advanced countries required before
allowing doctors to prescribe a new drug (D’Arcy & Harron 1999). Thus, regulation has
favored the strong – the doctors and the big corporations – over the pharmacists and
patent- medicine peddlers. In the poorest countries, where regulation is harder to enforce
and where consumers do not have enough money for the pharmaceutical industry or the
medical profession to be overly worried about chasing business, the global shift of power
to them from the local pharmacists and sellers of traditional medicines is yet to occur.
Current State of the Pharmaceutical Regulatory Framework:
The current system for making sure the medicine gets to the shelf on a pharmacy near
you is as follows: a pharmaceutical company must prove that the new medicine is safe
and efficacious and the company and produce millions of tablets that are within a set
specification – essentially make them over and over and over without any changes
(repeatability). The companies prove this by doing multiple clinical trials in local
markets and globally over the course of the drug development life cycle. All of the work
that needs to be done from discovering the drug to submitting an application for
marketing authorization is in the range of 12 – 15 years.
Here's where the fun begins! When a company is ready to submit their NDA to the
regulatory body, an application fee must be paid (for the United States, it is a little under

25
$2 million dollars). Once the NDA is submitted and the fee is paid, the FDA has 12
months to review and approve the application and issues a PDUFA date42
. There are
different timelines for special cases where there is an unmet medical need and the FDA
will grant a fast-track approval. For the purposes of this paper, we will only look at the
'regular' process for a new medicine across all of the countries. At the same time as the
NDA is submitted to the FDA, the pharmaceutical company can submit NDAs to other
countries and pay the associated fees. There is a small problem, the process isn't that
simple. There are only certain countries that can accept an NDA before any approvals
from major countries (i.e. US, EU, JP). The US, EU, and Japan are known to be leaders
in the pharmaceutical market. Once a major approval is granted by one of the major
markets, the pharmaceutical company will need to acquire a Certificate of Pharmaceutical
Product (CPP) from that government (this is the approval). The CPP can come from the
US, EU, Japan, or any developed country with a strong regulatory system. 75% of the
countries surveyed require a CPP at the time an NDA is submitted (some of the countries
will accept an application ahead of the CPP being available, but will not approve the
NDA until a CPP is submitted).
This means that 75% of the countries will have to wait at least 12-months, in the case of
the US NDA review time, before their government receives the NDA from a
pharmaceutical company. After the country receives the NDA, there are additional
requirements that each country must fulfill prior to approval. For example, China has
specific requirements that must be fulfilled prior to accepting an application (i.e. a
company must complete a local clinical study of 100 patients within China and have
42
PDUFA (Prescription Drug User Fee Act) was adopted in 1992 requiring drug and biologics
manufacturers to pay fees for product applications. The act also requires the FDA to use to issue a decision
date to the manufacturer.

26
favorable results). In addition to the requirements, many countries do not have the
capacity or capability to review the NDA and therefore delay the approval of the new
medicine, which in turn delays access of a new medicine to their market. Some countries
may never receive the application for not having a regulatory infrastructure or attractive
commercial market and therefore their patients may never have a choice to that medicine.
A significant amount of data has been gathered to show the difference in review time for
an NDA for each country as well as the amount of fees required per country (see chapter
4).
Because some pharmaceutical drug applications may not reach certain countries, patients
that require that particular novel drug may not have access to it because it is not approved
in their country. The alternative approach to receiving the drug would be through a
"compassionate use" or “single patient use” programs43
that are sponsored by the
manufacturing company or the patient would have to go to another country where the
drug is approved in order to obtain that drug (if they can afford it). The drug is not
approved in that country mainly because it is a low GDP country and therefore could not
afford the product or the sponsor company did not find the local market attractive. This
process is not available for every medicine – only to life-threatening diseases where no
alternative medicines exists or does not show efficacy in the named patient. There have
been examples of the sponsor company providing medicine to patients under the
‘compassionate use banner for several compounds. As an example, Schering Plough
43
There is no requirement for any manufacturer to conduct a compassionate use or single patient use
programs in all regions of the world. The studies are typically conducted in the regions where there is an
urgent need for the medicine and the study stops once the medicine is commercially available in that
particular country or region.

27
instituted a compassionate use program for its life saving drugs, Noxafil and Temodar,
for several years after gaining approval in the US and EU.
The current application process is specific for each country or region. In North America,
an application must be submitted to the US FDA. Another application is submitted to the
Canadian Health Authority (HA), and a third is filed with the Mexican HA. For Europe,
there are two different ways of submitting a new drug application: One is to submit a
single application to the EU HA (European Medicines Agency). The second is to submit
an application to each country individually and receive approval from each one of those
countries. For the most part, all of the applications have more or less the same
information. Multiple applications are prepared (extra work and cost for the
pharmaceutical companies), all the applications are reviewed by the various HAs
(overlapping use of government resources – time wasted), and most importantly delaying
access to patients around the world. The first EU option is better and is a potential model
that can be utilized globally. The focus on harmonization within the global technical
community has been on the CMC section as well as the clinical requirements section. In
the next chapter, we will discuss regional harmonization efforts and global technical
harmonization.
The process for Eastern Europe, Middle East & Africa (EEMEA), Asia Pacific (AP), and
South America has not yet been discussed but yield the same issues with duplicate
reviews. All of the countries and regions have some varying requirements (which
essentially are extensions of the first requirements already approved in the US, EU & JP)
which further delays access of the pharmaceutical product to the patients.

28
For example, countries like Egypt, Turkey, and the Philippines require that some type of
local manufacturing occur in their country. The regulations on this issue have been
evolving to include any part of the supply-chain (meaning manufacturing or packaging)
to the entire manufacturing and packaging process occurring locally.
Another example is Southeast Asian countries such as China, Japan, Taiwan, and South
Korea which require local clinical trial testing. This regulation requires companies who
wish to market their product in those countries to conduct clinical trials on a specific
number of local patients instead of counting on the global trials. In the past, a global trial
would be conducted and there may be patients from China, Japan, Taiwan, and South
Korea enrolled in the global clinical study. If enrollment is low at the clinical sites in the
aforementioned countries, and clinical sites in countries in Europe and the Americas are
enrolling at a high rate. This is a safety concern because those countries claim that their
patient population is different than that of Europe or the Americas (i.e. different digestive
system) and therefore the trial is not representative of their people. For example, if a
patient in Europe or the Americas is able to digest a certain amount of active
pharmaceutical ingredient (API) over a specified period of time, the concern is that a
patient in China or Japan may not be able to digest that same quantity. This leads to
overexposure of an active drug to the patient, which in some cases can cause renal failure
or impact other organs. If a manufacturer seeks to market the product locally, they are
required to conduct a separate local study that meets the countries’ requirements and
include the satisfactory results in the application. As a result, added cost, time, and
resources are needed to support a market launch in those countries. The outcome of the
local study could be that the dosage for countries in Southeast Asia differs (higher or

29
lower dose/strength) from other countries. If that were the case, this would mean that
additional drug development is required and the product for those countries would be
treated as a different product than that marketed in the rest of the countries.
Middle Eastern countries such as Jordan require additional post-market surveillance data
from manufacturers. For example, if a drug is approved in the US or Europe, the Jordan
health authority requires that the company submitting an application provide additional
data that shows that the commercially marketed product is safe after it has been marketed
to the general public for at least 12 months (CIRS 2011)44
. This requirement is in
addition to the clinical trials conducted by the manufacturer to prove the drug is safe and
efficacious to the first health authority that approved it.
The Institute of Medicine of the National Academies held a workshop in February 2013
to discuss regulatory harmonization and invited key stakeholders from governmental
agencies, non-governmental organizations, and Industry. The workshop held discussions
mainly on patient access to safe medicine and regulatory processes. The linkage between
safe medicine and regulatory systems is clear. Any product safety failure in any one
country can have ramifications around the world, and therefore the regulatory system is a
key factor in public health safety. The regulatory authorities in low and middle-income
countries often cannot perform all of the necessary responsibilities “…the FDA cannot do
its job well without substantive improvements in the capacity of its counterpart agencies
in the emerging economies” (IOM 2013). Specifically, the committee of regulatory
experts called for the sharing of inspection reports as an important first step in mutual
recognition and international regulatory harmonization.
44
CIRS 2011 Jordan Comparative Report.

30
Peter Honig, Global Vice President of Regulatory Affairs, AstraZeneca Pharmaceuticals,
summarized that harmonized standards in the pharmaceutical industry would45
:
Reduce costly duplication of effort;
Encourage sharing of experience and knowledge among regulators and scientists;
Require fewer clinical trials; and
Optimize use of limited resources
From the industry perspective, Honig highlights that harmonization would increase the
likelihood that a particular molecule will become a successful drug. Reduced
development time, less cumbersome approval processes across countries, and increased
speed to market are all important to companies. In addition, Honig believes that
harmonization would give patients faster access to new medicines and might lower the
costs of drug development, which could lower the price, making new drugs more
affordable in many more markets. An ongoing challenge for the industry and regulators is
to develop shared expectations regarding the use of adaptive trials, conduct of clinical
trials, acceptability of endpoints, and data transparency46
.
45
IOM 2013 46
Ibid

31
Persistent Barriers
Before clinicians can use a pharmaceutical product in a particular country, it needs to be
registered, approved, and marketed there. Honig cites numerous barriers to registration
that currently exist include47
:
China, Korea, and Taiwan require that a new drug be tested in subsets of their
population or in separate studies before it can be approved.
India, Mexico, and Vietnam require that specific numbers of their nationals
participate in clinical trials of the proposed drug.
Egypt, Turkey, and the Philippines require local manufacturing
Such requirements can create logistical difficulties in multiregional trials, when
researchers are required to allocate a certain number or percentage of trial slots to specific
groups of patients. In addition to the logistical issues, forcing a randomization code48
to
allocate certain percentages of patient groups to different centers may skew the study
results or hold up the completion of the study if patients do not enroll. Retaining
Investigational Review Board (IRB) approvals in each of the regions of the countries is
also a barrier as IRBs meet at a set schedule and have specific requirements about
submission of the protocols to be reviewed.
Challenges for existing harmonization initiatives
A large gap in regulatory capacity and expertise between emerging and developed
countries remains (IOM 2013). Programs developed by WHO helped governments in
less developed nations to be sure the drugs and medical devices imported into their
47
Ibid 48
Clinical trials use a randomization scheme where patients or drugs are randomized (or both). Different
treatment groups would receive active or placebo depending on the study design and randomization code.
Forcing a code means that the design of the code makes sure xx number of patients or drugs are used at a
defined time point.

32
countries are safe and effective, without requiring them to use limited resources to
replicate more developed nations’ regulatory infrastructures. This allows them to focus
on the issues of greatest local concern, such as the integrity of the supply chain.
Good Manufacturing Practice (GMP) inspections and re-inspections of manufacturing
and clinical trial sites can become burdensome. A global pharmaceutical company will
often have multiple inspectors coming in from different countries and from different
regulatory authorities in different regions. The incremental value of some of these
duplicate inspections becomes increasingly high and can be better utilized (IOM 2013).
Regulatory Perspective
Different countries take different approaches to medical products regulation, depending
on a number of factors. This is true even when they are geographically proximate,
operate under the same legal framework and rely on the same scientific processes and the
same data to make their decisions (IOM 2013). Some regulatory regimes may be more
risk-averse, while others may prioritize potential benefits. Whether they emphasize risks
or benefits may vary from one instance to another. As a result of these discordant
outcomes from regulatory decision making, patients in one country may have access to
medications that others do not have, which regulators may be hard pressed by patients,
providers, politicians, and the media to explain.
At a conference on regulatory harmonization in 2013, the Chair of the Dutch Medicines
Evaluation Board discussed the example of Avastin in a regulatory body taking this risk
benefit approach. As an example, the FDA revoked approval of Avastin for metastatic
breast cancer. Although the FDA originally approved the drug for this indication,
evidence that it did not extend life or improve the quality of life, while increasing the risk

33
of serious side effects, prompted the FDA’s decision. Yet, Avastin remains approved for
metastatic breast cancer in other countries! Such contradictory situations, some of them
widely publicized, can erode public trust in the system. However, the FDA’s public
report on the reasoning behind its decision is a model of balance and perspective. How
quickly do we forget? Fifty years ago, the FDA held up approval of thalidomide which
was proven to prevent thousands of birth defects that were found in EU after approval.
Schellekns and colleagues49
stated that regulatory systems should be assessed “in terms
of their ability to ensure patient safety, enhance public health, and stimulate innovation.”
Their effectives at achieving this latter aim are much in doubt, as the introduction of new
and innovative drugs has decreased sharply, despite rapid advances in biomedical
research. Schellekns and colleagues50
further stated, “Although the reasons for this
innovation deficit are not fully understood, many observers see the increasing demands of
the regulatory systems as one of the main drivers.” The regulators must take a step back
and ensure the right questions are being answered in assessing a new chemical entity.
Regulators use dossiers prepared by manufacturers in determining where to approve a
new drug. Problems associated with these dossiers are not infrequent. Typical problems
that can contribute to different regulatory decisions include the following51
:
Poor presentation: For example, the dossier presents data in a confusing way or
presents too much data, in which case the drug itself often receives a poor
assessment. Conversely, some dossiers may mask the data shortcomings by
strength of their presentations.
49
Schellekens, H., E. Moors, and H.G. Leufkens. 2011. Drug regulatory systems must foster innovation.
Science 332(6026):174-175 50
Ibid 51
Ibid

34
Coping with innovation: it may be difficult for regulators to assess a new concept,
so the default is to request more information, but whether such requests actually
produce an improved product is debatable. Some advanced therapies, including
gene therapies, may appear to regulators as too risky.
In the end, some of the variance in approval decisions across nations arises through the
dynamics of their individual review committees and their decision-making styles and
processes. Some nations base their processes on precise rules, whereas others base them
on principles. The latter approach gives greater flexibility to regulators but also reduces
the system’s predictability.
The labeling of a drug, which includes the indications for which its use is approved, can
vary among countries and change over time as new information is compiled. Sometimes
the number of indications is expanded and sometimes reduced; particularly if
complications arise that need to be more tightly controlled. A study of approaches used
by the FDA and EMA in the evaluation and approval of new cancer treatment indications
found real difference in the regulatory agencies’ wording nearly half (47%) of the
indications. However, the differences were clinically meaningful in only 10 of these
instances (Trotta et al., 2011).
Similarly, a study of differences in regulatory actions by the FDA and the European
Union related to biologicals appeared at first to suggest these differences were quite
large, but further analysis indicated that clinically relevant difference were much smaller
(Giezen et al., 2008). The more important feature was the timing in the two entities’
actions. The FDA was more likely to advise clinicians about potential problems sooner
than was the EU, and in some cases even to require a “black box warning” sooner. There

35
may always be differences in the ways people look at the data, how they weigh the
potential benefit or harm of specific products, and how they try to respond to their
populations’ unmet medical needs.
Evolution of harmonized regulations in the EU
The efforts to reduce differences in national drug regulation within Europe began in 1963,
when the European Commission (EC) hosted a conference of industry representatives,
doctors, and costumers to discuss harmonizing pharmaceutical laws52
. However, there
was disagreement over whether a drug should have proven “therapeutic potency” before
it could be approved. While doctors, pharmacists, consumers, and trade union
representatives insisted that this requirement was necessary, industry representatives at
the time refused to accept it53
. The result was stalemate.
In 1965, the EU issued its first pharmaceutical directive. It established baseline “criteria
for safety, quality and efficacy as preconditions for marketing authorization for new
medicine”54
. The directive also identified what constitutes medicinal products and
required member states to ensure that submissions of medicinal products to national
authorities were prepared and signed by experts with “the necessary technical or
professional qualifications”55
. These qualifications, however, remained undefined. In
addition, the efficacy test itself was limited: “Therapeutic efficacy would only be
considered lacking in a medicine which failed to produce pharmacodynamics results”56
.
52
Oraz, Luis, Kenneth Kaitin and Louis Lasagna. 1992. Pharmaceutical Regulation in the European
Community: Barriers to Single Market Integration. Journal of Health Politics, Policy and Law 17:859–861. 53
Vogel, David. The Globalization of Pharmaceutical Regulation. Governance: An International Journal of
Policy and Administration, January 1998 (11(1):1-22) 54
See Oraz et al. 55
Ibid 56
Hancher, Leigh. 1990. Regulating for Competition: Government, Law, and the pharmaceutical Industry
in the United Kingdom and France. Oxford: Clarendon Press.

36
Yet even this minimal requirement was implemented by only seven of the twelve
member states57
.
In 1974 the European Commission drafted a directive that permitted only “qualified
persons” to supervise drug production. But the Commission ultimately adopted such a
wide definition of qualified person (a medical professional, a veterinarian, a chemist, a
pharmaceutical technologist, or a biologist) that the directive became meaningless58
. Thus,
through the mid-1970s, virtually no progress had been made in creating a single market
for pharmaceutical products.
In 1975, the EC established the Committee for Proprietary Medicinal Products (CPMP)
with members requested from the regulatory authorities of the member states59
. The
CPMP was given the authority to review all drug applications by EU member states.
Applications would be examined for conformity to European Union safety, quality, and
efficacy standards, and the Committee would then issue an opinion on marketing
approval60
. However the CPMP’s role was only advisory. Member states maintained the
right to deny approval of an application. The multi-state procedure was intended not
only to generate confidence among member states about each other’s scientific
competence, but also to speed the approval of safe and effective drugs. Results were
mixed for this goal as well. Since the multi-state procedure had strict time limits (a state
had four months to evaluate a dossier that another state had already approved) member
states did give priority to multistate submissions. However, because member states still
57
Oraz et al. 1992, 854. 58
Ibid 59
Ibid 60
Buono, Teresa. Biotechnology-Derived Pharmaceuticals: Harmonizing Regional Regulation. Suffolk
Transnational Law Review 18:133, 1995.

37
carefully reviewed each submission rather than leveraging the initial state’s opinion, the
multi-state time limits were frequently violated (van de Gevel 1992). Only in France were
the time limits ever close to the intended plan. In Germany and the United Kingdom it
took an average of two years to review submissions, while in Italy, Spain, the
Netherlands, or Belgium it took three years or more (Howells 1992).
Although all member states agreed to accept test evidence gathered elsewhere in the
Community, there was no real progress towards mutual recognition of drug approval. As
the Committee’s chair noted in 1988, “Each concerned state seemed to conduct its own
assessment and raised its own objections . . . In practice there have been objections with
regard to each and every case dealt with under the Multi-State procedure.” He concluded
that “on the whole, the member states do not yet accept each other’s assessments”61
. The
implicated financial costs were significant. The Cecchini Report on “The Benefits of the
Single Market” estimated that the lost revenues of companies forced to wait over the
agreed 120 day limit was in a range between 100–175 million Euros, while multiple
registration requirements forced the applicants to employ extra staff at a cost of between
40 and 55 million Euros62
.
In 1987, unsatisfied by the outcomes of the multi-state procedure and committed to the
Community’s newly established goal of creating a single European market by 1992, the
EC took a radical new path. It approved a directive establishing a CPMP-administered
“Centralized Procedure.” This procedure was designed especially for biotech and high
technology products, since Brussels reasoned that it would be easier to harmonize
standards that had not yet been created than to require states to change the current
61
See Oraz et al. 62
Cecchini, Paolo. The European Challenge, 1992. Aldershot, England: Wildwood House, 1988.

38
procedures. The CPMP pooled scientific expertise in this new area of medical research in
order to create a Europe-wide consensus as to what represented good manufacturing
practices, adequate laboratory procedures and adequate evaluation criteria (Sauer 1991).
Once a manufacturer submitted its application, the CPMP would have seven months to
complete its initial review and evaluation; applicants were required to respond to both
CPMP and member states’ questions and comments within three months. The member
states then had up to one month to make their final recommendations, after which the
CPMP would send its report to all additional parties63
. While the CPMP’s report was
nonbinding, this new process was intended as the first move towards genuine regional
review and evaluation: “While fundamentally national, it [was] seen by the Commission
as a significant step in the direction of a single evaluation procedure applicable
throughout the Community”64
.
Following approval of the Maastricht Treaty on European Union (1992), which gave the
EU binding authority on some health care issues, the European Commission undertook
another major new initiative to harmonize national drug approval policies. It installed a
new regulatory institution, the European Medicines Evaluation Agency (EMEA) and two
new regulatory procedures65
. The EMEA consists of the CPMP, the corresponding body
for veterinary medicine, “a secretariat, an executive director, and a management board
composed of representatives of the member states, the Commission, and the European
Parliament.” The EU’s goal was to convert the relationship between national regulatory
63
Kanusky, Rosemary. 1994. Pharmaceutical Harmonization in the United States, the European Economic
Community and Japan. Houston Journal of International Law 16:665-707. 64
See Oraz et al. 65
Kingham, Richard, Peter Bogaert, and Pamela Eddy. 1994. The New European Medicines Agency. The
Food and Drug Law Journal 49:301–321.

39
authorities and those of the Union, to finally create a common regional review process for
pharmaceutical products.
An essential objective of the creation of a single European drug approval procedure was
to grow European-wide drug research and development, thus helping the industry to
“confidently continue to hold its place on the world stage in the foreseeable future66
. A
1995 communication from European Commissioner Martin Bangermann noted that over
the last two decades the share of all new medicines developed in the EU had declined
from half to one-third, while in the critical biotechnology sector, 65% of all patents were
American while only 15% were European67
. Increasing the efficiency of European drug
approval was regarded as a way of strengthening the effectiveness of this critical
industry.
The centralized procedure positioned the final regulatory approval at the Union level for
the first time. It permits manufacturers to submit applications directly to the European
Agency, which then refers them to the CPMP for review and evaluation. The latter is
required to issue its opinion within 210 days. If approval is denied, the drug’s sponsor
may file an appeal, which in turn must be reviewed within sixty days. Final approval rests
with the European Commission, which has ninety days to draft its own opinion. If the
Commission grants marketing authorization, it automatically becomes valid throughout
the EU for renewable periods of five years. The EMEA centralized approval process was
intended to be relatively fast, with application to final approval to take a maximum of ten
months68
. This is more than twice as fast as that of many member state drug regulatory
66
Pharmaceuticals: Carving Up Europe’s Drugs Industry. 1995. Economist August 26:57 67
Ibid 68
See Kingham et al.

40
agencies. To help the EMEA meet its own goals, applications can be accepted on CD-
ROM, and the agency’s questions and comments are to be provided by email69
.
As indicated earlier, the centralized procedure specifically targeted biotechnology drugs,
which must be approved by EMEA, as there is no national alternative. Biotechnology
was targeted because of its potential for economic growth and, because since it is such a
new field, individual states had not yet created their own testing infrastructures70
. As
one European official explained, “Global participation means we don’t have to keep on
reinventing the wheel. A new technique such as stereoisomerism, for instance, offers the
chance to put together a unified international approach before separate guidelines are
issued . . . The most obvious candidate here is biotechnology”71
. The centralized
application procedure was intended in part to benefit small firms with limited resources,
which were more likely to be found in the biotechnology sector. However, manufacturers
of any other pharmaceutical products may also choose to use the centralized procedure72
.
The Union also established a decentralized procedure that would be available to all
pharmaceutical products, except those produced through biotechnology73
. This
procedure, based on the principle of mutual recognition, was approved in 1993 and went
into effect on January 1, 1995. If a product has been approved for use in any member
state, its manufacturer could submit an identical scientific and technical application to
any or all other member states. It also must alert both the CPMP and the member state to
which its application was first submitted; the latter then is required to provide its
69
See Green 1995a 70
See Buono 71
Global harmonization on Pharmaceutical Regulations a Step Nearer. 1991. Pharmaceutical Business
News; Financial Times November 15. 72
See Kanusky 73
See Kingham et al.

41
assessment report to each country where recognition is sought. Each member state then
has ninety days to decide whether to recognize the first nation’s approval. It must do so
unless “there are grounds for supposing that the authorization of the medicinal product
concerned may present a risk to public health”74
. In cases of disagreement, member
states are urged to request the opinion of the CPMP.
While firms may still apply to individual member states for marketing approval, as of
January 1, 1998, any Member State which receives an application for a product which has
been approved by another member of the Union, must either accept that approval, or refer
the application to the CPMP for binding arbitration. Thus, pharmaceutical products will,
for the first time, be subject to mutual recognition, under the sponsorship of the EMEA.
Unlike the FDA, the EMEA’s role is a coordinating one; the actual processing of
applications is delegated to national regulatory agencies. This in turn means that the
agency must rely on member state authorities not only to administer applications in a
timely fashion, but also to apply similar criteria. The harmonization of national
requirements is even more important for the viability of the EU’s decentralized
procedure, since under this procedure a single national authority will be able to approve a
product for the entire EU. The inspection of manufacturing facilities and the certification
of the reviewers of applications could also vary between national authorities. However,
according to Fernand Sauer, the agency’s executive director, “after fifteen years of
harmonization we now have everything in place so that EMEA and all of the national
authorities practice exactly the same requirements” (Koberstein 1996).
Both new procedures promise to provide important benefits to manufacturers.
Streamlined approval allows cash flow to start sooner. “Successful new drugs earn $1
74
Ibid

42
million per day in global sales revenues. The European Union accounts for about 40% of
global sales” (Green 1995b). Also, faster drug approval will increase the actual life of
drug patents, increasing the potential value of future research75
. Since companies can
submit one application rather than fifteen76
, large firms may save up to $5 million
annually in national clinical staff and testing equipment77
. While the application fee for
submitting a drug to the EMEA is high a typical filing costs approximately 200,000
Euros—this is about half of what it would cost to pay all fifteen national fees. Centralized
approval also allows firms to use the same package inserts and make similar promotional
claims throughout the EU with the required languages.
Evolution of regulations in the US
The US NDA has a total of 19 sections that cover the lifecycle of a molecule from
discovery to commercialization. The bulk of the applications comprised of 2 major
sections – the Clinical section and the Chemistry, Manufacturing and Controls (CMC)
section. Within these 2 sections are clinical trial reports, drug stability reports,
toxicology, and drug-drug interaction reports, among many others. See appendix III for a
listing of all the sections within the NDA.
Reaching an NDA with 19 sections that generates tens of thousands of pages of data and
analysis did not occur over night. The regulations in the US started in 1820 when eleven
physicians met in Washington, D.C., to establish the U.S. Pharmacopeia, the first
compendium of standard drugs for the United States78
. Since then, many appointments
were made by Presidents within the administrations that followed through the 19th
75
Green, Daniel. 1995a. EU Body to Speed Up Approval of New Drugs. Financial Times January 26, 1995. 76
At the time Green wrote the article, there were only 15 member states in the EU. In 2014, there are 28
members states and all have adopted this process. 77
Ibid 78
See www.fda.gov

43
century until the actual implementation of the Food and Drug Act in 1906 that gave
legitimacy to the Food and Drug Administration. Regulations continued to evolve over
time throughout the 20th
century before reaching the 19 sections that make up the NDA.
The 20th
century brought many new requirements that manufacturers had to follow before
introducing a new drug to the market in the US and around the world (Table 1
summarizes some of the regulations introduced). Many of the changes generated a great
deal of controversy from manufacturers and from economists.
Table 1: Summary of laws and amendments introduced in the US regulations in the 20th
century
Year Law Purpose
1906 Food and Drugs Act Prohibited misbranded and adulterated food and
drugs in interstate commerce
1937 The Elixir Sulfanilamide
Incident
Use of diethylene glycol to dissolve Sulfanilamide
was toxic and resulted in over 100 deaths in more
than 15 states (234 of 240 gallons were recalled).
1938 Federal Food, Drug, and
Cosmetic Act
Tightened controls over drugs and food, included
new consumer protection against unlawful cosmetics
and medical devices, and enhanced the
government’s ability to enforce the law
1962 Kefauver-Harris
amendments
Thalidomide incidents – shift from economic to
safety concerns (proof of efficacy & FDA control
over clinical testing)
1983 Tamper-resistant
packaging regulations
Prevent poisonings such as deaths from cyanide
placed in Tylenol capsules
1992 Prescription Drug User
Fee Act
Requires drug and biologics manufacturers to pay
fees for product applications and supplements, and
other services. The act also requires FDA to use
these funds to hire more reviewers to assess
applications.
1997 Food and Drug
Administration
Modernization Act
Reauthorizes the Prescription Drug User Fee Act of
1992 and mandates the most wide-ranging reforms
in agency practices since 1938
2004 FDA Publishes
“Innovation or
Stagnation?”
Examines the path needed to bring therapeutic
products to fruition, and how FDA can collaborate in
the process

44
The Economics of Regulation
The theory of economic regulation was introduced by George Stigler in 1971 and further
built upon by Richard Posner and Sam Peltzman. The main argument is that regulation is
acquired by the industry and is designed and operated primarily for its benefit. Stigler
uses a simple model of regulation: A regulator (Congress or Parliament) faces special
interest pressure from producers and electoral pressure from consumers. The special
interest pressure is always more persuasive than others so producers always win.
Regulations are passed only for the benefit of large firms, not for the benefit or protection
of consumers.
This doesn't mean that regulators will be blatant about this. There are two ways to help a
producer: Via a direct subsidy or via protectionism. Subsidies aren't good--they
encourage new entrants into the market, so producers gain only a short-term benefit.
Protectionism, on the other hand, limits entry into the market--regulators favor this
method. So we see protective regulations like tariffs and occupational licensing fees
installed to help the large producers. Stigler's reasoning draws heavily on the point that
producers simply organize better.
Stigler79
proceeds from two primary premises:
1. The fundamental asset controlled by the state is the power to coerce. Any group
that can control how this power is used can profit.
2. Since we are self-interested actors, we will seek to get the state's coercive power
to support our interests. Efforts to do so, however, are costly.
79
Stigler, George. 1971. “The Theory of Economic Regulation” The Bell Journal of Economics and
Management Science. 2(1): 3-21.

45
Invariably, large firms win. Again, because they organize much better and are better
structured80
:
Large firms have high benefits from mobilizing. Since they are a small group, and
since they are fairly homogeneous, they have no difficulty with collective action
problems.
Small firms don't organize for political reasons because of collective action
problems: low potential benefits.
Consumers don't organize because the costs of doing so are high compared to the
benefits. Basically, consumers remain rationally ignorant.
This theory maybe applicable in the United States for most industries, but this may not be
the case for all of the regulations. Within the US, producers are able to influence policy
on pricing of pharmaceuticals, but not necessarily the regulatory review process and
timing. Outside the US, there are industrial groups in Europe, Japan, Canada and a few
other developed countries that serve a role in influencing policy. In the developing world,
there is an absence of up-to-date regulatory processes as well as industry groups. The
absence of both leaves room for corrupt actions or limitation on the ability to influence
the regulatory process.
In the next section, a review of the impact of the 1962 amendments to the Food, Drug,
and Cosmetic Act will show that the additional regulation limited the number of new
medicines introduced to the market thus limiting revenues for the producers and not
allowing patients access to potential new medicines. In this case, both parties lose and
the economic theory of regulation does not apply. Several economists studied the impact
80
Stigler, George. 1980. “An introduction to privacy in Economics and Politics” The Journal of Legal
Studies. 623-648.

46
of the new amendment on innovation, patient safety, and economic impact. Sam
Peltzman is one of the economists that sought to understand the impact of the added
regulation, specifically from the 1962 amendments requiring manufacturers to complete
efficacy trials. His initial study was to see what the impact of the regulations was on
innovation. The next section will give a summary of his findings.
The Peltzman review of the 1962 amendments to the Food, Drug, and Cosmetic Act:
Sam Peltzman has a serious issue with the regulations introduced in 1962 that required
pharmaceutical companies to conduct efficacy trials shifting the focus from economics to
safety. So much so, he wrote a couple of articles and a book with mathematical models
with results showing that the introduction of additional regulation has significantly
slowed down innovation in the pharmaceutical industry. Peltzman contends that the 1962
amendments decreased the amount of ‘safe’ drugs from reaching the market by more than
50% (Peltzman 1974). Although Peltzman did not set out to criticize the 1962
amendments prior to completing his study, and had given the benefit of the doubt to the
new regulations with favorable assumptions, he summarizes his final outcome to be that
the regulation cost the patients money and lives.
The 1962 amendments were introduced because of the Thalidomide incidents where a
number of people (17) died in the United States and much larger number of people in the
thousands died in Germany, the UK and other European countries. Thalidomide was an
approved drug in Europe, but not the United States. Patients from the United States were
able to get the drug as free samples from the doctor’s office (at that time, samples were
allowed to be provided to patients in anticipation of the drug approval). How can any
patient disagree with requiring a manufacturer to prove that the medicine said

47
manufacturer will market is indeed efficacious and safe? Peltzman suggests that
regulations were in place already to make sure manufacturers produced safe drugs
(proving efficacy came after the 1962 amendments). With that logic, one can concluded
that a manufacturer is able to produce a sugar pill and claim that it treats a certain kind of
illness without having to prove its efficacy. Playing the scenario out to an extreme,
patients would purchase the sugar pill, spend money on it, more importantly spend time
thinking this would help, only to realize that it was indeed not efficacious and did not
treat the intended illness. While it was safe to take (did not cause additional harm), the
patients may have missed the window to get treated with the right medicine that was
efficacious for that particular illness. Peltzman does credit the 1962 amendments with
attaining the goal of reducing the introduction (consumer waste) of ineffective drugs to
the market. However, based on his analysis in 1974, the costs in the process to have
clearly outweighed the benefits (Peltzman 1974).
In another 1974 publication, Peltzman cites an example of a drug that was approved in
the UK but was delayed approval in the US by 5 years (nitrazepam). The suggestion is
that 3700 deaths were seen in the US could have been averted had the drug been available
(without conducting the efficacy trials) because patients had to use other, less safe
sedatives and hypnotics81
. Peltzman later says “…greater risk taking is likely to yield net
benefits” (Petlzman 1974). Meaning that if we have several deaths for not conducting the
additional clinical efficacy trials and had more lives saved, that alone should be sufficient
to dismiss the additional requirements or lay blame on the requirements for the drop in
innovation!
81
Peltzman used pharmacologist William Wardell's estimate that because the relatively safe hypnotic drug
nitrazepam was not cleared for use in the United States until 1971, five years after it was available in
Britain, more than 3,700 Americans may have died from less safe sedatives and hypnotics.

48
From true numbers and an economics perspective, Peltzman may be correct. However,
the reality is that as technology evolves and scientists are able to learn more about new
chemicals because of improved equipment or testing methodology – the numbers don’t
tell the whole story. The science simply can’t be translated into numbers. As Vogel re-
iterates, regulatory harmonization is a key driver in promoting a higher number of new
medicines. The more regulatory authorities that have different requirements, the bigger
the hurdle to promote a new drug that must adhere to the different rules. As a result,
fewer innovative medicines are brought forward to the market. In the 1970s and 1980s,
new medicines introduced by European companies declined from half to a one-third and
only 15% of the biotechnology drugs (Vogel 2000).

49
Chapter 3: Regional Attempts to Close the Regulatory Gaps
Regional drug regulatory harmonization can be described by a number of initiatives
driven mainly by common socioeconomic needs and supported by global organizations
such as WHO and ICH. These initiatives are at different stages of development and
maturity, those including the more established countries being at a more advanced stage
than the less-resourced ones.
Marketing of pharmaceutical products is highly regulated because it involves several
ethical and human health and safety implications. As highlighted in earlier chapters, one
of the main obstacles to international approval of pharmaceutical products is that
different models for regulation of medicines exist in countries across the world. The
diversity of the regulatory requirements in different countries makes pharmaceutical
drugs’ applications and marketing very complex and costly. The added cost and
complexity often delays access of the public to critical and potentially life-saving drugs.
In addition, lack of respect for the Trade-Related Intellectual Property Rights (TRIPs)
agreement, strong price controls, and generic competition in the emerging and developing
countries remain a challenge for companies and could hinder marketing of innovative
drugs in these markets (Lakkis 2010)82
.
Historically, drug regulations in different countries evolved independent of each other,
affected mainly by local politics and economics, as well as the availability of resources
and the public health needs of the specific country. The substantial diversity in the
regulations, laws, and procedures of registering new pharmaceutical products, especially
between developed and developing countries, resulted in delayed patient access to
82
Maha Lakkis is an experienced regulatory affairs professional that has worked in industry for over 30
years and has published several articles on regulation in the pharmaceutical industry.

50
innovative medicines in the developing countries. Even with the acceptance of approval
by major regulatory authorities as evidenced by issuing a CPP (certificate of
pharmaceutical product), new drugs are still delayed by 1 to 2 or more years between the
time products are approved in developed and emerging countries (Lakkis 2010).
Recently, there has been an increased trend toward harmonization through increased
inter-country cooperation initiatives at regional and international levels, making borders
more open for trading among groups of countries with different regulatory, technological,
or financial backgrounds. In 2012, the FDA published a Global Engagement Report
where the FDA Commissioner, Margaret Hamburg, states “…Today we recognize that to
successfully protect U.S. public health, we must think, act, and engage globally. Our
interests must be broader than simply those within our own borders”83
.
In addition, the recent global expansion of the pharmaceutical industry by establishing
multinational manufacturing sites and moving towards marketing of new products in
international markets has made the need to standardize quality, efficacy, and safety
regulations stronger than ever. Therefore, global pharmaceutical regulatory
harmonization has become critically important for companies, international consumers,
and agencies; as a result, the different national regulatory agencies and industry
organizations started to cooperate more closely to harmonize and bring their regulations
closer to international standards (Lakkis 2010). The harmonization activities have been
often supported by intergovernmental initiatives at regional and interregional levels, as
well as by international agencies such as the World Health Organization (WHO) and the
International Conference on Harmonization (ICH) of Technical Requirements for the
83
The 2012 Global Engagement report is the first ever published report that stresses the need for global
cooperation on all fronts – regulation, imports/export, food supply chain, drug supply chain, and adding
FDA international posts to act like “embassies”.

51
Registration of Pharmaceuticals for Human Use (Vogel 2007). Harmonization of
pharmaceutical regulatory standards is important to reduce unnecessary duplication of
requirements, which delays access to new medicines. Practically, the regulatory standards
of safety and efficacy related to the review and availability of new medicines should be
essentially similar unless there are apparent regional and ethnic differences (Lakkis
2010).
International harmonization is characterized by a number of initiatives undertaken in
different regions of the world, driven mainly by common local or regional economic and
social needs. This chapter discusses some of the regional and global drug regulatory
harmonization initiatives. These initiatives are at different stages of development and
maturity, those including the more established countries being at more advanced stages
than the less-resourced ones.
European Harmonization
Europe was the first to lead the most advanced initiative for regional harmonization of
drug regulation. In the 1980s the European Commission (EC), now known as the
European Union (EU), led the first attempt to harmonize pharmaceutical regulatory
requirements, which was very successful in developing and implementing a structure for
harmonizing the drug regulatory laws and regulations across the region, leading to the
creation of a single market and promoting free circulation of pharmaceuticals within the
European member states. This was achieved through the establishment of the European
Council Regulation in July 1993, which resulted in the creation of the European Agency
for the Evaluation of Medical Products (EMEA) in 1995 (EMEA changed its name and
logo in 2010 to European Medicines Agency, EMA) to coordinate and facilitate the

52
European harmonization of pharmaceutical requirements84
. The creation of the EMEA
was a result of recognizing the increasing regulatory complexity, as well as the cost and
time required for development of new medicines; therefore, pharmaceutical companies
needed an effective and efficient regulatory environment within the EU to be more
effective in developing and marketing medicinal products. The most significant
achievement of the EMEA is the implementation of the Centralized Procedure in 1995,
which allows applicants to file one marketing authorization application that is assessed by
a centralized committee, the Committee for Medicinal Products for Human Use (CHMP),
which allows the approved products to be marketed in all EU member states85
.
At the same time, the option to use a specific national registration path in a particular
country only via national procedure, or within a certain number of countries via mutual
recognition procedure (MRP) or decentralized procedure (DCP), remains valid. The MRP
allows any national marketing authorization granted by any national authority of an EU
member state to support an application for mutual recognition by other member states.
On the other hand, in the DCP, the applicant selects a Reference Member State (RMS) to
lead the review procedure while being informed about other included states (Concerned
Member States) selected for marketing the product. Identical dossiers are sent to all
Concerned Member States as well as the RMS. After completing the review, the RMS
communicates its assessment results to the Concerned Member States, who can grant
marketing authorizations separately by the individual health authorities. However, any
Concerned Member State can disagree with the RMS assessment, which allows the
84
Council Regulation (EEC) No. 2309/93, July 22, 1993, laying down community procedure for the
authorization and supervision of medicinal products fro human and veterinary use and establishing a
European Agency for the Evaluation of Medicinal Products (OJ No L 214 of 24. 8.1993). 85
See Centralized Procedure: Directive (EC) No. 726/2004

53
applicant to pursue a dispute resolution phase. These procedures provide a more efficient
and cooperative approach than applying to several countries independently, especially
because these procedures include filing the same dossier, using identical specifications in
all involved countries8687
. These procedures include a scheme for involved states to
cooperate on post marketing surveillance of MRP and DCP products and to establish a
voluntary risk-based model of post market surveillance at the EU level, by sharing the
results of product testing with other participants, thus reducing duplication of procedures.
International Conference on Harmonization (ICH)
While the EC was working on harmonization in the EU, discussions started among
Europe, Japan, and the United States on possible harmonization among the three parties.
As a result, a unique tripartite harmonization project was initiated in 1989 through the
cooperative effort of the regulators and the industry associations in the three regions,
which led to the creation of the ICH in April 199088
. The ICH provided a multiregional
discussion forum for harmonization that led to significant progress in harmonizing
regulatory standards and technical guidelines for the registration of pharmaceutical
products (including new chemical entities and biotechnological products) and improved
the efficiency of global drug development (Lakkis 2010). The established ICH Steering
Committee (SC) consists of six voting members representing the regulatory bodies and
pharmaceutical industries of the three involved regions: it included members of the US
Food and Drug Administration (FDA) and the Pharmaceutical Research and
Manufacturers of America (PhRMA); Japanese Ministry of Health, Labor, and Welfare
(JMHLW), and Japan Pharmaceutical Manufacturers Association (JPMA), as well as the
86
See Mutual Recognition Procedure: Directive 2001/83/EC 87
See Decentralized Procedure: Directive 2004/27/EC 88
See International Conference on Harmonization www.ich.org

54
EU89
. The EU was initially represented by the EC and later transferred to the EMEA, in
addition to the European Federation of Pharmaceutical Industries Associations (EFPIA),
which represented pharmaceutical companies from 16 countries in Western Europe. The
ICH SC also includes nonvoting, non-ICH observer members representing WHO, the
European Free Trade Association (EFI'A) currently represented by SwissMedic (Swiss
Agency for Therapeutic Products), and Health Canada (Canadian Health Authority). The
Secretariat is provided by the International Federation of Pharmaceutical Manufacturers
Associations (IFPMA) (IOM 2013).
ICH’s mission is “to make recommendations towards achieving greater harmonization in
the interpretation and application of technical guidelines and requirements for
pharmaceutical product registration, thereby reducing or obviating duplication of testing
carried out during the research and development of new human medicines” (ICH, 2013).
The ICH was very successful in improving the development and licensing of new drugs
in the three regions in an efficient and cost-effective manner, by harmonizing regulatory
requirements and eliminating unnecessary duplication of clinical trials and animal testing
while maintaining the high standards of quality, safety, and effectiveness of the products.
The success of the ICH process has become a very good model of harmonization, with
over 50 guidelines for the technical requirements in four major categories of drug
development (quality, safety, efficacy, and multidisciplinary) and a dictionary of medical
terms. In addition, it has developed electronic standards, a common technical document
for electronic submission for registration of data on NDAs. The common technical
document includes guidance on formatting trial datasets and data elements, which
89
Ibid

55
facilitates review and enables industry to submit its data to different regulatory authorities
in a single format (Lakkis 2010).
Even though the ICH initially focused on developing guidelines and standards for the
ICH region, from the beginning, it maintained openness and transparency by keeping all
ICH guidelines and documents open to all authorities from all other countries at its
website. It also allowed representatives from non-ICH member states to attend ICH
plenary sessions. As a result, the effect of the successful ICH harmonization spread
beyond the three main ICH regions and its guidelines became increasingly perceived as
the gold standard for regulations in other countries, either by implementing them directly
or by following them when establishing their local regulations. Figure 6 shows an
illustration of how the ICH has evolved over the past 10 years from being a small player
connecting a small group of organizations to a major player driving harmonization across
many regions and implementing more than 50 guidelines.
Figure 6: Evolution of the ICH

56
Global Cooperation Group and Non- ICH Regional Harmonization Initiatives
The ICH successful model of harmonization triggered an international interest in the
process of harmonization, leading to several regional harmonization initiatives (RHIs)
that aimed to develop unified standards and guidelines on drug quality, safety, and
efficacy, across a defined regional group of non-ICH countries, with the aim to improve
access to essential medicines within their regions, using ICH procedures and guidelines.
Therefore, following the completion of the majority of its objectives in 1997, ICH SC
recognized the need to take a further step by expanding its efforts to support these
initiatives (Lakkis 2010). In 1999, it started to establish communication with interested
nonmember states and created a subcommittee designated as the Global Cooperation
Group (GCG) to serve as an information liaison between ICH and non-ICH countries and
to "make information on ICH activities and guidelines available to any country or
company that may be interested"90
. It also invited WHO to join, acting as a link between
ICH and non-ICH countries and regions. The GCG worked with WHO and other
international organizations to achieve acceptance and adoption of ICH guidelines in non-
ICH countries. Currently, WHO attends ICH SC meetings as an observer and has also
become an observer to the work of the GCG as a nonvoting member without decision
making capability. Currently, the most active regional harmonization initiatives include
the following:
- Asia-Pacific Economic Cooperation (APEC)
- Association of Southeast Asian Nations Pharmaceutical Product Working Group
- GCC’s Gulf Central Committee for Drug Registration
- PAHO’s PANDRH
90
See ICH Global Cooperation Group. http://www.ich.org/meetings/gcg-reports.html

57
- Southern African Development Community (SADC) and East African
Community (EAC)
- Individual countries, including Australia, Brazil, China, Chinese Taipei, India,
Republic of Korea, Russia, and Singapore
Asia Pacific Economic Cooperation (APEC)
APEC was established in 1989 among 21 countries of the Pacific Rim (Australia, Brunei
Darussalam, Canada, Chile, China, Hong Kong, Indonesia, Japan, Republic of Korea,
Malaysia, Mexico, New Zealand, Papua New Guinea, Peru, Philippines, Russia,
Singapore, Chinese Taipei, Thailand, United States, and Vietnam) with the objective of
enhancing economic growth and prosperity in the Pacific region91
. Some APEC member
states are also members of other RHIs such as ASEAN and the Pan American Health
Organization (PAHO). APEC, in partnership with ICH, set up a pharmaceutical
harmonization initiative in 2002 through the APEC Life Science Innovation Forum
(LSIF). LSIF established a Regulatory Harmonization Steering Committee and APEC
Network of Pharmaceutical Science to support cooperation on regulation of
pharmaceutical drugs and make medicines accessible to a larger population in the region.
The APEC network of pharmaceutical professionals and policymakers worked toward
harmonizing regulations among member states with the ultimate goal of developing a
single pharmaceutical market. APEC has collaborated with GCG to develop several
harmonization topics including92
:
1. Bridging studies
2. Stability in climatic zones of the APEC region
91
See Asia-Pacific Economic Cooperation www.apec.org
See: APEC harmonization center www.apec-ahc.org
See: APEC Health working group http://www.apechwg.org/portal/PortalHome.asp
See: APE Life Sciences Innovation Forum http://lsif.apec.org/ 92
Ibid

58
3. Clinical trials
4. GMP (good manufacturing practice) and GCP (good clinical practice)
implementations
5. Collaborative opportunity for common local clinical data that meet the regulatory
requirements for Korea, Japan, and China.
An APEC Harmonization Center (AHC) website is established under the authority of the
APEC-LSIF for supporting regulatory harmonization efforts and to provide a platform to
address and solve priority concerns of APEC member economies on regulatory
harmonization93
.
Association of South-East Asian Nations (ASEAN)
ASEAN was established among 10 participating Southeast Asian countries (Brunei,
Cambodia, Indonesia, Laos, Malaysia, Myanmar, Philippines, Singapore, Thailand, and
Vietnam) to promote regional economic unity and cooperation aiming to have a single
ASEAN market by 201594
. The initiative included a public health and pharmaceutical
harmonization scheme that includes developing harmonized guidelines for the regulation
of pharmaceuticals and a unified format of drug registration application, the ASEAN
CTD. It also includes a Mutual Recognition Agreement (MRA) on a post marketing alert
system to enhance pharmacovigilance capabilities by sharing information on marketed
medicinal products via one common reporting form and glossary, and supporting the
WHO Counterfeiting Taskforce Program as well as an MRA on bioavailability and
93
Ibid 94
See Association of Southeast Asian Nation http://www.asean.org/

59
bioequivalence by sharing relevant data and establishing one common list of comparators
for generics, similar to that of the WHO95
(Lakkis 2008).
The Gulf Cooperation Council (GCC)
The GCC, also known as the Cooperation Council for the Arab States of the Gulf, was
established in 1981 as a trade bloc among seven Arab states of the Persian Gulf (Bahrain,
Kuwait, Oman, Qatar, Saudi Arabia, United Arab Emirates, and Yemen as member in
Health Council) that share common economic and social interests. The GCC established
its patent office in 1992 and launched the GCC common market on January 1, 2008. In
1999, a pharmaceutical harmonization initiative was launched by a committee called Gulf
Central Committee for Drug Registration (GCC-DR), with the executive office being
located in Riyadh, Saudi Arabia. The GCCDR is composed of two members from each
participating country with the possibility of appointing two ad hoc nonvoting advisors
from its affiliates. The main objective of the GCC-DR is to coordinate health policies and
programs among the participating members via exchange of information, knowledge,
techniques, and expertise96
.
It is responsible for registration of pharmaceutical products, GMP inspection and
compliance, approval of quality control laboratories, and review of technical and post-
market surveillance reports. It also includes a Program of Bioequivalence Studies
according to a consolidated Registration Gulf Act. The GCC-DR adopted the ICH
guidelines as primary source for developing its own. It follows a centralized procedure
for drug registration with harmonized drug registration requirements and drug pricing for
95
See ASEAN Consultative Committee on Standards and Quality Pharmaceutical Product Working Group
http://www.asean.org/communities/asean-economic-community/item/accsq-pharmaceutical-product-
working-group 96
See Gulf Cooperation Council www.gcc-sg.org http://www.gcc-sg.org/eng/

60
all the member states. After centralized approval of a product, authentication and fee
payment should be followed in each of the member states as per their local policies97
.
The Southern African Development Community (SADC)
The SADC, headquartered in Botswana, is an intergovernmental organization created to
improve socioeconomic and political cooperation and integration among 15 southern
African member states (Angola, Botswana, the Democratic Republic of Congo, Lesotho,
Madagascar, Malawi, Mauritius, Mozambique, Namibia, Seychelles, South Africa,
Swaziland, United Republic of Tanzania, Zambia, and Zimbabwe). The ultimate
objective of SADC is to build a region with a high degree of harmonization and
rationalization to enable the pooling of resources and achieving collective self-reliance to
improve the living standards of the people in the region98
. The SADC Health Protocol for
Regional Cooperation and Integration was adopted in August 1999 to harmonize health
standards of quality, safety, and efficacy in the region, to ensure the efficient use of
resources and facilitate faster access to safe and effective medicines99
. Significant
progress has been made by SADC member states in the harmonization of guidelines and
there have been notable achievements on enhanced medicine access in a number of
member states. This was mainly achieved by strengthening regulatory capacities of
member states via "centers of excellence" that provide regional training on evaluation of
drug application dossiers, GMP compliance, and development of local GMP certification
to improve the quality of local productions, as well as training for qualification of testing
laboratories, development of a quality management program, and a market surveillance
97
See ICH GCC-DR working group http://www.ich.org/about/organisation-of-ich/coopgroup/gcc.html 98
See Southern African Development Community www.sadc.int and
http://www.sadc.int/themes/health/pharmaceuticals/ 99
See SADC protocol http://www.sadc.int/documents-publications/show/804

61
and monitoring program that helps fight counterfeit products. The major challenge to this
group is the disparity in the capabilities of the member states, as some members have
very limited resources.
The efforts of the SADC initiatives are part of a larger plan in Africa. There are several
other regional communities in Africa that are doing similar activities as that of SADC.
The East African Community (EAC) is a very active community that also has a working
group on drug regulatory harmonization for its five nation member states (Burundi,
Kenya, Rwanda, the United Republic of Tanzania, and the Republic of Uganda). There
have been setbacks in the initiatives due to limited funding, resources, technical
implementation of website, and lack of having a clear owner to the initiative. The SADC
and EAC working groups now fall under The African Medicines Regulatory
Harmonization (AMRH) Initiative was launched in 2010 to support African countries’
ability to review and approve medicines100
. This initiative provides holistic oversight for
the continent and better aligns resource needs across the continent as well as with
supporting organizations such as the WHO, World Bank, USAID, NGOs, and health
authorities such as FDA, EMA, etc. (ICDRA 2010).
Pan American Health Organization
PAHO is an international public health agency more than 100 years old that aims to
improve health and living standards of the countries of the Americas101
. It serves as the
WHO Regional Office for the Americas, being part of the United Nations system. The
PAHO/WHO Regional Office for the Americas established the Pan-American Network
for Drug Regulatory Harmonization (PANDRH) and its working groups to support drug
100
See East African Community http://www.eac.int/index.php 101
See Pan American Health Organization www.paho.org

62
regulatory harmonization processes102
. Participants in PANDRH include the national
authorities of countries in the Americas, various pharmaceutical interest groups, industry,
and academia. Current members include drug regulatory authorities of all PAHO member
states as well as representatives of the regional pharmaceutical industry associations such
as Latin American Association of Pharmaceutical Industries and Latin American
Federation of the Pharmaceutical Industry, members from academia, consumer groups,
professional associations, and representatives from the five sub-regional economic and
trade integration groups such as the Andean Community (Bolivia, Colombia, Ecuador,
Peru, and Venezuela), CARICOM (the Caribbean community), SICA (Central American
region with Costa Rica, El Salvador, Guatemala, Honduras, and Nicaragua),
MERCOSUR (Mercado Comun del Sur, including Argentina, Brazil, Uruguay, and
Paraguay), and NAFTA (North American Free Trade Agreement, including the United
States, Canada, and Mexico)103
. The harmonization activities of PANDRH include
establishing technical guidelines for harmonization of processes and standards to improve
drug quality, and programs for the strengthening of national regulatory agencies via
technical training programs. Twelve technical working groups (Good Manufacturing
Practices, Bioequivalence and Bioavailability, Good Clinical Practice, Drug
Classification, Counterfeit Drugs, Good Laboratory Practices. Pharmacopoeias, Medical
Plants, Drug Registration, Pharmacovigilance, Vaccines, and Promotion and Marketing)
were established and so far they have adopted eight final technical guidelines. In response
to these RHIs, the GCG endorsed new Terms of Reference in November 2003, to
establish collaborative partnership with RHIs and help them understand ICH guidelines.
102
See ICH PANDRH working group http://www.ich.org/about/organisation-of-ich/coopgroup/pandrh.html 103
Ibid

63
Consequently, GCG invited each of the RHls to designate permanent representatives to
attend the GCG meetings, and a GCG section was established on the ICH website as a
repository for documents and harmonization activities (Lakkis 2012, ICH).
The GCG membership currently includes one representative from each of the six ICH
parties on the Steering Committee, with observers (WHO, Health Canada, and IFPMA,
EFTA, and the ICH secretariat at IFPMA). It also includes a maximum of two permanent
representatives per regional harmonization initiative (APEC, ASEAN, GCC, PANDRH,
and SADC) to solicit and present the views of their regional initiative. In a 2005 meeting
in Brussels, GCG issued a mission statement: "to promote a mutual understanding of
regional harmonization initiatives in order to facilitate the harmonization process related
to ICH guidelines regionally and globally and to facilitate the capacity of drug regulatory
authorities and industry to utilize them." Since then, representatives from RHls have been
invited to listen to technical topics at the level of the Expert Working Groups,
Implementation Working Groups, Discussion Groups, and the SC (Lakkis 2010). In
October 2007, the ICH SC identified its goals:
1. To reduce country and regional differences in technical requirements that impact
the availability and cost of new medicines
2. To promote international movement of pharmaceuticals that are safe, effective,
and of high quality
3. To promote the conduct of clinical trials and data collection that meet
international standards
It also expanded the GCG membership and invited representatives from eight distinct
drug regulatory authorities (Australia, Brazil, China, Chinese Taipei, India, Russia,

64
Singapore, and South Korea) to attend the GCG meetings104
. These representatives are
granted the same access as RHIs at the biannual ICH meetings and invited to listen to
technical topics. In 2008, GCG's role expanded further, shifting from information sharing
to implementation issues and training.
Even though ICH has no legal mandate from the international community, recent
developments within GCG suggest that ICH activities are gaining wider international
interest and participating countries are following the ICH guidelines as the international
norm. It is noteworthy that GCG follows a set of principles when handling requests from
non-ICH parties: (a) ICH will not seek to impose its views on any country, region, or
company, but will serve as a source of information; and (b) ICH will provide non-ICH
member countries or companies with any document related to GCG initiative without
charge105
.
Role of the WHO in Global Regulatory Harmonization
WHO is an intergovernmental health international agency of the United Nations, with
193 member states. It is the only organization that has a legal international mandate from
member states to set global standards for the promotion and protection of public health.
WHO'S constitution specifically states that one of its functions is to "develop, establish,
and promote international standards with respect to food, biologics and pharmaceuticals
and similar products" to improve access to quality essential drugs, especially for poor and
vulnerable populations106
. WHO has been very active since the early 1970s in providing
guidance and assistance to regulatory agencies in the developing countries by
strengthening their national regulatory capacity and helping them to set up infrastructures.
104
See ICH Global Cooperation Groups http://www.ich.org/about/organisation-of-ich/coopgroup.html 105
Ibid 106
See World Health Organization http://www.who.int/about/en/

65
In addition, WHO played a critical role in driving harmonization of quality, safety,
efficacy, and nomenclature requirements on a global level. In addition, in collaboration
with a number of government agencies and individual experts, it developed several
practical guidelines on the quality and safety of drugs and established a mechanism to
review, modify, or adopt ICH guidelines as appropriate (Rago and Santoso 2008). WHO
provided a supportive mechanism for approval in countries with less regulatory capability
by implementing the WHO Certification Scheme and encouraging the use of CPPs issued
by developed regulatory authorities as a valid document to support approval in
developing or underdeveloped countries. WHO also supported global harmonization by
convening the International Conference of Drug Regulatory Authorities (ICDRA) every 2
years since 1980, which provides an important forum for communication, coordination,
and collaboration among drug regulatory authorities of WHO member states107
. The
objectives of the ICDRA are to (a) promote collaboration among drug regulatory
authorities, (b) reach a consensus on matters of common interest, (c) facilitate timely and
adequate exchange of information, and (d) discuss issues of international relevance. Over
the years, ICDRA became a very effective network and a forum where senior drug
regulators can discuss issues, exchange ideas, and set direction for future development108
.
It also played an instrumental role in guiding regulatory authorities from all WHO
regions by determining priorities for action in national and international regulatory issues.
During the 1989 conference, plans began to develop global pharmaceutical regulatory
standards by addressing critical regulatory issues, and at the thirteenth ICDRA
107
See WHO ICDRA http://www.who.int/medicines/areas/quality_safety/regulation_legislation/icdra/en/ 108
Ibid

66
preconference109
, it addressed an important issue, better medicines for children, and
recommended that WHO convene and collaborate with the global pediatric group of
regulators to identify priorities and treatment guidelines needed for neonates, and
establish mechanisms to support drug development for children. ICDRA also
recommended that WHO should do the following:
1. Promote adoption and implementation of the WHO Model Registration Package
as minimum information requirements for product registration
2. Produce guidance and draft regulation for managing confidentiality issues among
regulatory authorities
3. Undertake joint assessment of selected applications using the WHO Model
Registration Package
4. Foster the development of regional, collaborative post-market surveillance and
pharmacovigilance systems to monitor the quality, safety, and efficacy of health
products
5. Establish formal mechanisms for the exchange and use of regulatory information
among all authorities to strengthen capacity and maximize efficiencies
Summary
Drug regulations have evolved over time in response to increasing scientific knowledge
and the complexity of the pharmaceutical industry. The pharmaceutical industry is
becoming increasingly multinational, and pharmaceutical companies are now focusing on
global drug development to take advantage of the opportunities in the rapidly expanding
new markets in Asia, Latin America, the Middle East, and Africa. This globalization
109
See 13th
ICDRA conference
http://www.who.int/medicines/areas/quality_safety/regulation_legislation/icdra/presentations13thICDRA/e
n/

67
trend highlights the need for a new strategic approach of pharmaceutical harmonization.
Globalization of standards has become a necessity and a goal for many groups of
neighboring countries in several regions to reduce unnecessary and duplicative
requirements, rationalize time and costs, and create a transparent regulatory process,
resulting in improved access to medicines both domestically and globally. Global
harmonization became possible thanks to the success of ICH in providing very advanced
and comprehensive guidelines and global standards, and in supporting the development
of RHls leading to harmonized regulatory standards. Drug regulatory harmonization is
beneficial to the pharmaceutical industry as well as consumers: therefore, harmonization
efforts are supported by governments as well as the pharmaceutical industry: PhRMA
groups as well as EFPlA are playing a constructive role in supporting global
harmonization via international regulatory affairs groups that act as a liaison with local
and multinational regulatory and trade organizations to advocate pharmaceutical industry
views (IOM 2013). Substantial progress has been achieved over recent years throughout
the world, but much remains to be done. However, a complete international
harmonization of drug regulations and establishing one set of international standards is
still far from reality, and no single regional initiative can be considered an ideal model for
international application and implementation, but at least a new trend is moving in the
right direction toward global harmonization.

68
Chapter 4 – Methodology and Data Analysis
Introduction
The pharmaceutical industry is often viewed as a money-making machine with market
cap of $1.2 trillion in 2011! It’s also a very expensive business to be in costing major
pharmaceutical companies $4 – 7 Billion USD in R&D investment every year (IMS
2012)110
. These figures are specific to innovative companies manufacturing new
molecular entities, not generics. Still, if the industry’s revenues are so high, why aren’t
all the medicines accessible to everyone around the world? That’s a simple question with
a very complicated answer. This chapter will attempt to address this question in
reviewing economic, social, political, sociopolitical, and education data from over 100
countries around the world. The data will be analyzed to identify the relationship of the
NDA review period (time) with the cost of application (cost), life expectancy (LE), health
expenditure per person, and literacy rate. Said differently, time data is linked to assess
the impact on quality of life, which is linked to the ability of access to medicine. The
quality of life is determined by the life expectancy in a given country, GDP per capita,
literacy rate, and health expenditure per person.
The research strategy is to relate NDA review cycle time to economic, political, and
quality of life characteristics. The hypothesis being that a country with a stable
democracy will have a structured regulatory process which leads to timely access of
newly developed medicine. Additionally, in order to have a structured regulatory process
with defined timelines, a country must have a high literacy rate. The more educated the
people, the stronger the regulatory structure, the longer life expectancy – which is linked
to access to medicine based on a set review time and cost.
110
IMS Health publication in 2012 on the pharmaceutical industry

69
Data, Methodology, and Regression results
Detailed description of the variables
The research strategy for this paper is to relate review time of a new drug application
(time) to:
1. Quality of life indicators (life expectancy and physicians per 1000 people),
2. Economic status (GDP per capita, Health Expenditure per person, openness of
economy, and cost of the applications), and
3. Political status (Democracy score, State fragility index, size of government)
The NDA review time and fee were collected by conducting a survey with regulatory
professionals working for a global pharmaceutical company and therefore represent real-
time data. The data was supplemented and confirmed from a review conducted of the
IDRAC reports published by Thompson Reuters Regulatory Intelligence unit. The
IDRAC reports provided data on 69 countries based on their research. Ministries of
health generally do not publish timelines on their websites or in any other outlet. As a
result, the data collected from the survey will be the only published data available for all
the countries included in this study. The data submitted by the regulatory professionals
was provided in units of either days or months. All of the time data was converted to
days for consistency in the analysis. The cost of the application fee (cost) was provided
in either local currency or USD. All of the cost data was converted into USD by using
the Oanda currency converter website111
.
111
Oanda is a currency converter website that is broadly used - http://www.oanda.com/currency/converter/.
Monetary conversions made in March – May 2012.

70
GDP per capita and Life Expectancy, Physicians per 1000 people, and Health
Expenditure per person were collected from the World Bank world development
indicators report of 2012112
. The variables are defined as:
- GDP per capita: basic economic indicator and measures the level of total
economic output relative the population of a country.
- Life expectancy at a specific age: average number of additional years a person
of that age could expect to live if current mortality levels observed for ages above
that age were to continue for the rest of that person’s life. In particular, life
expectancy at birth is the average number of years a newborn would live if current
age-specific mortality rates were to continue.
- Physicians per 1000 people: number of physicians per 1000 people in a given
country. The number includes general practitioners as well as specialists.
- Total health expenditure: sum of public and private health expenditures as a
ratio of total population. It covers the provision of health services (preventive and
curative), family planning activities, nutrition activities, and emergency aid
designated for health but does not include provision of water and sanitation. Data
are in current U.S. dollars.
The two political stability indicators, Democracy Score and State Fragility Index, were
collected from the Polity project113
. The Polity conceptual scheme is unique in that it
examines concomitant qualities of democratic and autocratic authority in governing
institutions, rather than discreet and mutually exclusive forms of governance. This
perspective envisions a spectrum of governing authority that spans from fully
112
The World Bank publishes “World Development Indicators” for all the countries around the world 113
The polity project is supported by the Political Instability Task Force, Societal-Systems Research Inc.,
and Center for Systemic Peace http://www.systemicpeace.org/polity/polity4.htm

71
institutionalized autocracies through mixed, or incoherent, authority regimes (termed
"anocracies") to fully institutionalized democracies. The democracy score captures this
regime authority spectrum on a 21-point scale ranging from -10 (hereditary monarchy) to
+10 (consolidated democracy). The Polity scores can also be converted to regime
categories: a three-part categorization of "autocracies" (-10 to -6), "anocracies" (-5 to +5
and the three special values: -66, -77, and -88), and "democracies" (+6 to +10). The
Polity scheme consists of six component measures that record key qualities of executive
recruitment, constraints on executive authority, and political competition. It also records
changes in the institutionalized qualities of governing authority. The Polity data include
information only on the institutions of the central government and on political groups
acting, or reacting, within the scope of that authority.
The State Fragility Index and Matrix list all independent countries in the world in which
the total country population is greater than 500,000 in 2010 (164 countries). The Fragility
Matrix scores each country on both Effectiveness and Legitimacy in four performance
dimensions: Security, Political, Economic, and Social, at the end of the year 2010. Each
of the Matrix indicators is rated on a four-point fragility scale: 0 “no fragility,” 1 “low
fragility,” 2 “medium fragility,” and 3 “high fragility” with the exception of the
Economic Effectiveness indicator, which is rated on a five-point fragility scale (including
4 “extreme fragility”). The State Fragility Index, then, combines scores on the eight
indicators and ranges from 0 “no fragility” to 25 “extreme fragility.” A country’s fragility
is closely associated with its state capacity to manage conflict, make and implement
public policy, and deliver essential services while progressing development114
.
114
ibid

72
Open K and KG were extracted from the Penn World Table – Center for International
Comparisons. The Penn World Table provides purchasing power parity and national
income accounts converted to international prices for 189 countries/territories for some or
all of the years 1950-2010115
. Open K is the economy openness at 2005 constant prices
(%). It is a calculated number scoring the openness of economy with regard to trade.
Trade is the sum of exports and imports of goods and services measured as a share of
gross domestic product. KG is the Government Consumption Share of PPP Converted
GDP Per Capita at 2005 constant prices. It is a calculated number identifying the size of
the government consumption. General government final consumption expenditure
(formerly general government consumption) includes all government current
expenditures for purchases of goods and services (including compensation of employees).
It also includes most expenditures on national defense and security, but excludes
government military expenditures that are part of government capital formation.
The data sets described in the section above was collected and uploaded to a spreadsheet.
Linear regression analysis was conducted using Ordinary Least Squares method (OLS)116
.
Definitions and summary statistics for important variables used in this study are
presented in Tables 3 and 4. The summary statistics show extremes between nations
across all of the variables. Health expenditure per capita, for example, ranges from the
low number of just under $15 in the Democratic Republic of Congo and the highest
number being over $8200 in the United States. Physicians per 1000 people ranged from
115
Alan Heston, Robert Summers and Bettina Aten, Penn World Table Version 7.1, Center for
International Comparisons of Production, Income and Prices at the University of Pennsylvania, Nov 2012.
https://pwt.sas.upenn.edu/php_site/pwt_index.php 116
OLS is a method for estimating the unknown parameters in a linear regression model. This method
minimizes the sum of squared vertical distances between the observed responses in the dataset and the
responses predicted by the linear approximation.

73
the low of 0.008 doctors/1000 people in Tanzania to 6.167 doctors/1000 people in Greece.
GDP per capita ranged from $105 in the Democratic Republic of Congo to Luxembourg
at $52,000.
The database collected for this research is unique in that it compares variables that had
not been looked at in the past and therefore the analysis is unavailable. The list of review
time for NDA approval for all of the countries was not found in any one publication. All
of the regression in Table 4 shows the regression results of the log time variable on the
rest of the 9 variables. Regression analysis was not conducted on all variables at the
same time. The first regression results show that the Log of GDP per capita yields a
statistically significant result when regressed alone. The second regression shows that
Log of GDP still yields a statistically significant result when regressed with Cost. In fact,
both variables, Log GDP per capita and Cost, yield statistically significant results. When
regression analysis is conducted with more than one other variable, Log GDP does not
yield a statistically significant result. Health Expenditure, Physicians per 1000 people,
Fragility, and Life expectancy consistently yielded a statistically significant result
indicating that faster drug approval means higher health expenditures and longer life
expectancy values. The results also suggest that faster drug approvals are occurring
because of the higher literacy rate. The results for Literacy rate indicate that for every 4 –
5 days the review time is reduced, there is a 1% increase in literacy rate. Being
conservative and taking the 5 days per 1%, the results indicate that Literacy Rate in a
country would increase by 20% if the review time was 100 days less.
Life expectancy seems to be the most important variable because it implies that:

74
- Countries with lower review time and high NDA fee will have higher life
expectancy
- Population would live 10 more years more if the approval review time is
reduced by 128 days
One of the surprises of the results is the significance level of the NDA fee in regression
results with multiple variables. It was expected that the NDA Fee (cost) would be
significant when correlated with any other variable. The results in Table 4 indicate that
cost is significant when regression conducted with time and one other variable only – this
was expected. However, when 2 or more variables were introduced, cost was significant
at the 10% level – this is the unexpected result. As an example, if NDA fee is increased
by $100k, the NDA review time would only improve by 27 days. The expectation was
the cost would play a bigger role in the review time and other variables. The second
surprise was the insignificance of the democracy score. In all the regression analysis, the
democracy score was consistently insignificant. Fragility, on the other hand, was
consistently significant. This can be interpreted to mean that a country must have
stability, but not necessarily an open democracy to have access to medicine in a timely
manner. This was a very important result when looking at broken states versus an
autocratic regime. The broken state will surely not have access to medicine, but the
autocratic regime seems to have better access as long as there is stability in the country.
Summary of results
Log GDP per capita was statistically significant when regression analysis on log time was
conducted alone or with cost only. When regression analysis conducted with more than
one variable, it was no longer significant. This result was also unexpected as one of the

75
points in the hypothesis was that richer countries would have more stability and less
review time. The results indicate that GDP per capita plays a critical role when it is a
standalone variable, not when combined with other variables. Cost, as discussed earlier,
was also a surprise. While statistically significant in many of the regressions, the actual
value of the improvement on review time was minimal. For example, we would expect
to see $100K in increased cost that for 27 – 40 days improvement in approval time.
Health Expenditure yielded statistically significant results across all data sets. However,
the actual financial value of the Health Expenditure was not very high, similar to the
results of cost. Open K was not statistically significant in any of the regressions. This
result implies that having an open economy does not necessarily yield a faster drug
approval. Physicians per 1000 people had a clearly significant result in the regression
analysis with time alone. Statistically significant results were also seen when regression
analysis was conducted for Physicians per 1000 people with cost, Health Expenditure,
Open K, and LogGDPPC. However, regression results of Physicians per 1000 people
with Fragility or Life Expectancy does not yield a statistically significant result.
Regression analysis of Literacy rate with cost yielded statistically significant results.
However, regression analysis did not yield statistically significant results of Literacy rate
with life expectancy. An example of the result between Literacy and cost is that for
$100k increase in NDA fee, the review time is reduced by 93 days. This result is three
times better than the result seen with cost alone where $100k increase in NDA fee only
yielded 27 – 40 days in a shorter review cycle. Therefore, higher Literacy rate
contributes to reducing the review time when the same increase in cost is applied.

76
Fragility alone gives statistically significant results. Fragility yields statistically
significant results in regression analysis with all other variables except when Life
Expectancy is included in the analysis. Fragility yields statistically significant results in
regression analysis with Democracy score and Open K. This result implies and confirms
that a stable and open democracy that trades with other governments will have a lower
review cycle and therefore access to medicine. Life Expectancy yields a very high
statistically significant result in regression analysis alone with time or with GDP per
capita. Life Expectancy yields statistically significant results in regression analysis with
cost and physicians per 1000 people as well as Open K and Health Expenditure. Life
Expectancy does not yield statistically significant results in regression analysis with
Fragility and GDP per capita. These results indicate that Fragility and Life Expectancy
are equally important to access to medicine. This result makes sense as stability is
required in order to have access to medicine which would support an increase in Life
Expectancy. The results indicate that lack of education in a given country leads to a
limited number of educated people (low literacy). Infrastructure in a country cannot be
built without stability and an educated population. Therefore, capabilities cannot be
established to review the complex applications for new medicines and economic growth
is limited.
A summary of regression results using the Cox Hazard model can be found in Table 5.
The results of the Cox Hazard117
model yielded that Life Expectancy is a very important
117
Cox Hazard is a survival model that relates the time that passes before some event occurs to one or more
covariates that may be associated with that quantity of time. In a proportional hazards model, the unique
effect of a unit increase in a covariate is multiplicative with respect to the hazard rate. For example, taking
a drug may halve one's hazard rate for a stroke occurring, or, changing the material from which a
manufactured component is constructed may double its hazard rate for failure. Other types of survival
models such as accelerated failure time models do not exhibit proportional hazards. The accelerated failure
time model describes a situation where the biological or mechanical life history of an event is accelerated.

77
variable giving a result of nearly 1.04. This means that life increases by 10 years with 128
days less of approval time. This result is in line with the OLS outcomes discussed earlier.
Using the Cox Hazard survival model was an experiment to see what the results would
yield and were added to this section.
In addition, the same analysis for the results in Table 4 was conducted using time, instead
of log time, as the dependent variable. The data are summarized in Table 6 in Appendix
VI – regression analysis data with dependent variable time. Cost is not statistically
significant with time but is statistically significant with log time as shown in Table 4.
Adjusted R2 is not as good with time as the dependent variable as seen in table 6.
Adjusted R2 is higher in all the regression analysis results when log time is the dependent
variable (Table 4). This suggests that, because countries’ review time can be divided into
groups, applying the log function to time better represents the industry trends.

78
TABLE 2
VARIABLE DEFINITIONS
Health Expend*
Health expenditure per capita in a given country (USD)
Open K**
Percentage of economy openness at 2005 constant prices
Cost+
Fee charged by a health authority in a country to review a new drug
application (USD)
LG GDP PC** Log of GDP per capita in a given country
Dem Score***
Classification of a country based on regime; Full Democracy (10),
Democracy (6 to 9), Open Anocracy (1 to 5), Closed Anocracy (0
to -5), Autocracy (-6 to -10), and Failed (-66 to -77)
Phys / 1000* Number of physicians per 1000 people in a country
Frag. Index***
Fragility Index scores each country on both effectiveness and
legitimacy in four performance dimensions: Security, Political,
Economic, and Social, at the end of the year 2010. The higher the
number (0 – 25), the more fragile the country
Life Expectancy*
Average life expectancy in a given country (combined
male/female) (years)
KG** Government consumption share of PPP converted GDP per capita
at 2005 constant prices
Time+
Amount of time needed to review a new drug application (days)
118
Sources:
* World Bank
** Penn World Table
*** Polity Project – Center for System Peace
+ Survey results and research
118
Accelerated review of life saving medicines had a shorter approval time.

79
TABLE 3
MEANS AND STANDARD DEVIATIONS OF IMPORTANT VARIABLES FOR
COUNTRIES STUDIED FOR THE YEARS 2010 – 2011
VARIABLE
MEAN STANDARD
DEVIATION OBSERVATIONS (# OF COUNTRIES)
Health Expend
1205.14 1928.70 125
Open K
91.34 56.92 129
Cost (USD)
57335.3 198029.5 129
LG GDP PC
7.94 1.62 124
Dem Score
4.91 10.35 128
Phys / 1000
1.97 1.48 106
Frag. Index
7.71 6.03 124
Life Expectancy 70.21 9.60 128
KG 9.62 5.59 129
Time (days) 744 265.39 129

80
TABLE 4
RESULTS OF REGRESSION ANALYSIS OF LOG APPROVAL TIME ON COUNTRY ECONOMIC AND POLITICAL
CHARACTERISTICS ACROSS 129 COUNTRIES, 2010 - 2011
Variables OLS
(t-Stat) (1) (2) (3) (4) (5) (6) (7)
LG GDP PC -0.077 -0.056 -0.017 -0.018
(-3.73)* (-2.85)* (-0.88) (-0.93)
Cost -7.52E-07 -2.82E-07 -2.81E-07 -3.08E-07 -3.05E-07
(-4.75)* (-1.70) (-1.71) (-1.80) (-1.80)
Health Expend -0.00010 -0.00013 -0.00011 -9.98E-05 -0.00011
(-5.50)* (-8.65)* (-6.37)* (-5.11)* (-5.98)*
Open K -0.00033 -0.00031
(-0.66) (-0.63)
Constant 7.15 7.03 6.81 6.69 6.68 6.85 6.71
(42.38) (44.52) (45.08) (203.15) (204.40) (42.44) (125.01)
Adjusted R2
0.095 0.23 0.39 0.37 0.38 0.38 0.38
# of observations 124 124 121 125 125 121 125
*statistically significant

81
TABLE 4 (continued)
Variables OLS
(t-Stat) (8) (9) (10) (11) (12) (13) (14)
LG GDP PC 0.0020 0.013
(0.092) (0.63)
Cost -3.04E-07 -3.21E-07 -3.16E-07 -2.64E-07 -2.55E-07 -2.45E-07 -2.67E-07
(-1.79) (-1.94) (-1.88) (-1.62) (-1.59) (-1.51) (-1.69)
Health Expend -0.00010 -7.40E-05 -7.36E-05 -6.10E-05 -7.48E-05 -7.68E-05 -6.35E-05
(-5.77)* (-3.71)* (-3.50)* (-2.70)* (-3.55)* (-1.51) (-2.99)*
Open K -0.00032 2.03E-06 6.25E-05 0.00029 0.00017 0.00028 0.00030
(-0.63) (0.0040) (0.12) (0.55) (0.33) (0.53) (0.59)
Dem Score -0.0019
(-0.70)
Phys / 1000 -0.078 -0.080 -0.034
(-3.30)* (-3.22)* (-1.29)
Frag. Index 0.021 0.022 0.024 0.013
(2.86)* (3.75)* (3.62)* (1.77)
Life Expectancy -0.010
(-2.26)*
Constant 6.72 6.78 6.75 6.49 6.45 6.32 7.21
(122.94) (107.47) (39.34) (54.46) (71.72) (29.87) (20.83)
Adjusted R2
0.38 0.44 0.43 0.48 0.46 0.46 0.48
# of observations 125 106 103 103 121 117 121

82
TABLE 4 (continued)
Variables OLS
(t-Stat) (15) (16) (17) (18)
LG GDPPC 0.013
(0.58)
Cost -2.66E-07 -4.62E-07 -4.82E-07 -2.58E-07
(-1.64) (-3.14)* (-3.30)* (-1.57)
Health Expend -5.59E-05 -6.23E-05
(-2.43)* (-2.72)*
Open K 0.00034 0.00060 0.00042 0.00029
(0.64) (1.07) (0.77) (0.54)
KG 0.0015 0.00054 0.0027
(0.23) (0.084) (0.43)
Phys / 1000 -0.023 -0.051 -0.051 -0.032
(-0.82) (-1.90) (-1.91) (-1.20)
Frag. Index 0.017 0.031 0.029 0.020
(2.05)* (4.01)* (4.03)* (2.75)*
Life Expectancy -0.0064
(-1.18)
Constant 6.94 6.22 6.38 6.46
(17.24) (24.53) (48.26) (49.14)
Adjusted R2
0.48 0.45 0.44 0.48
# of observations 103 100 103 103
*statistically significant

83
TABLE 5 (Cox Hazard)
Variables (Hazard Ratio)
(z value)
(1) (2) (3)
Dummy<60 0.58 0.60
(-4.19)* (-3.91)*
Cost 1.000001 1.000001
(3.38)* (3.04)*
Life Expectancy 1.04
(5.56)*
# of observations 129 129 128
Dummy: countries with life expectancy under 60 is 24 of 129
*statistically significant

84
Discussion:
Life expectancy was the most important variable yielding statistically
significant results in both the OLS and Cox Hazard models. While the
focus of this study has been using OLS regression, Cox Hazard analysis
was another attempt at generating results to confirm outcomes. According
to the data and results, people in a given country would live 10 years
longer if the approval time is reduced by 128 days. While that sounds
simple, it is not. In order for a country to reduce the cycle time for
reviewing all of the drug applications, it will need medical experts
(literacy rate) who are capable of reviewing the new drug applications. In
order to have experts review the applications, a country must be stable
(fragility index) in order for the political system to develop and thrive
(democracy score). Once a stable political system is in place, economy
will grow because a strong political system attracts investments. If a
country sustains political stability and investments are made in the
country, the people living in the country can thrive in education (literacy
rates) and obtain high-income jobs (GDP per capita).
Based on the above, a conclusion was made that all of the indicators were
linked and cannot be separated. While the OLS results did show that one
variable alone could yield statistically significant results, the true result
was when there are several variables were analyzed at the same time. A
surprise variable in this analysis was cost. It was expected that cost would
play a big role, maybe even bigger than life expectancy or literacy rate. In

85
reviewing the data, cost ranged from zero to $2M. Qatar, for example, is
one of the wealthiest countries in the world (high GDP per capita) does
not have a fee for a new drug application. This input could potentially
skew the result. The United States, on the other hand, has a cost of $2M
for an application – nearly 4 times as much as the next highest application
fee. Removing Qatar alone or the United States alone out of the dataset
would probably not change anything. Removing both extremes (groups of
nations) could potentially change the output. However, removing the
United States risks other variables such as Literacy rate and life
expectancy to be impacted negatively which could change the previously
seen results.
The other story to be told here is that the US, EU, Japan, Australia,
Canada with firm regulatory infrastructure and high cost deliver approvals
on time, every time. This group of nations is in the upper tier for all of the
variables. They have a high GDP per capita, high literacy rate, excellent
democracy and fragility scores, and have a similar timeline for review and
approval of new drug applications.

86
Chapter 5
Conclusion
This work began with a basic question. Why does it take a long time
before a new drug is available to patients in all of the countries? The
initial thought was to look at the general regulations, determine the gaps,
leverage or develop a governance structure that would allow for a global
approval, and the question would be answered. We now know it is not
that simple. There was a complex history of the evolution of regulation
globally and how the regulation is impacted by the economics and
political landscape in a country or a region. While many questions were
addressed in this paper, there is still more work to be done.
Studying the different variables in each country and reviewing the results
of the regression analysis made things abundantly clear. The issue is not
that governments do not want to have medicines available to their
populations. The issue is not necessarily that people cannot afford it,
although that is concern. The issue is that in order for access to medicine
to reach the top of the agenda, more pressing issues need to be addressed.
For example, having a stable government with an educational system in
place is a must before reestablishing or enhancing a drug regulatory
process. Cost of the application fee was thought to be a clear reason why
some countries were unable to have access to the medicine. After the
regression analysis, it’s very clear that a stable government with an
educational system is a must and how much money is paid for the NDA is

87
not as critical, because without a stable government and an educated
people, the NDA fee becomes insignificant. By taking the unique
approach of evaluating multiple variables across the political and
economic systems more insight to the key factors is gleaned as discussed
in Chapter 4 which can be further studied and built upon.
The term Global Affairs suggests that there are multiple subjects being
discussed. The results of this paper imply and confirm that a
multidisciplinary approach must be taken in order to address current
global issues. Economics are dependent on the politics and politics are
dependent on the economic policies. History and international law are
closely connected with the financial and political systems. Development
indicators are an outcome and a direct result of the interdependencies of
these subject fields. The results of this study confirm this statement in that
the regulatory system cannot be studied independently of the political and
economic development indicators. If a single system is studied
independently, the results will not be sufficient. This is confirmed in the
results shown in Chapter 4. Cost is statistically significant on its own, but
not when analyzed with other factors.
Furthermore, economists and policy makers must become active
participants with the healthcare and regulatory professionals. In the
research conducted, it was evident that industry leaders and regulatory
bodies (ministries of health) are heavily involved in the current
discussions on regulatory issues, harmonization, and access to medicine.

88
What is recommended is having additional government representatives
from other departments sit at the table to make it a more fruitful
discussion. The current process has Global Cooperation Groups (GCG)
focused on pharmaceutical regulations work in conjunction with bigger
regional organizations. A GCG is under the umbrella of APEC, ASEAN,
SADC, EAC, PAHO, GCC and so forth. The GCG sits in a silo, discusses
the issues, and reports them to the bigger groups. This is a problem.
Instead of having regulators and industry professionals recommending
policy, economists and politicians need to inform these policies, not be
informed. A holistic approach is needed to support a viable approach that
can yield a realistic path forward.
At the National Academies Institute of Medicine (IOM) conferences as
well as WHO’s International Conference on Drug Regulatory Authorities
(ICDRA), for example, executives from all the major industry players as
well as high level officials from all of the regulatory bodies were present
discussing key issues in pharmaceutical regulation. However, there were
no representatives from the ministries of commerce, foreign affairs,
interior, or education. This is a key learning from this study – that is
having the right people at the table to inform policy making. Based on the
research conducted, there is a lack of ‘other’ voices on the table when it
comes to global regulatory harmonization and policy making.
Regulatory / policy implications

89
The hypothesis was that countries with a higher GDP per capita, higher
democracy score, and lower fragility index will have a higher fee and
shorter time for review. In other words, if the state is stable, infrastructure
will be in place to review the NDA in a timely manner for a set cost that
the applicant will be able to afford because the return on investment for
the application will yield substantial gains. The results of the analysis
from Chapter 4 confirm part of the hypothesis. The cost of the NDA is
statistically significant on its own. But it is not statistically significant
when analyzed with other variables. Fragility of a country and life
expectancy results were the strongest among all of the variables and
statistically significant.
Based on the regional harmonization efforts, a recommendation for a
potential regulatory model that will help alleviate the resource constraints
and enable faster approval of medicines globally is possible. The model
would involve an agreement where shared resources from neighboring
countries and regions work together to approve a new drug. As a result,
the approval would be instantly applied to all of the countries involved.
The current harmonization efforts in leveraging resources are only on a
regional level. In order for a model to be successful, a broader effort must
be made in utilizing expertise from more developed regulatory systems to
support the underdeveloped systems. The support cannot be only
consulting, but rather more hands on of coaching and training. As
indicated earlier, the cooperation and collaboration has to be across

90
multiple disciplines for this to be a success. This means that ministries of
education need to be involved in the educational system of a developing
country. This means that other ministries (commerce, interior, foreign)
must be involved to understand the impact of economic and development
indicators on a successful model. The model used to develop a
harmonized process in the European Union can be a significant starting
point. The EU had multiple players around the table discussing how the
harmonization model could play out. They were successful not only
because of the right players, but also because they had a head start in a
developed regulatory model and strong economies. The developing world
is lacking behind on education, economic and political status, and
therefore, the effort to bring about a globally harmonized process will be
an extremely challenging project.
Future Work:
This study focused on the regulation of new innovative pharmaceutical
products. There are different classifications for all of the medicines and
therefore this study did not cover all of them. There are regulations for
generic medicine, regulation for Over the Counter (OTC) products,
regulation for Biologics, and regulation for conducting clinical trials. The
work conducted for this paper could be expanded into these other fields
and confirm if the same results would be reached. Generics and OTC
products follow a different model in terms of review time and cost, and
therefore, may not have similar outcomes. The regulations of these

91
different medicines can be the same as pharmaceutical products studies
and it can be different. In some cases, such as the OTC products, there
may be limited or no regulation versus a regulatory process that is exactly
equivalent to the pharmaceutical products studied in this paper.
Deciphering which regulatory system applies in the different countries and
applying the statistical analysis will be challenging and results would be
very interesting to see. Generics also follow a different process that the
innovative pharmaceutical products studied in this paper. The correlation
between the variables for generics could be clearer and more
straightforward. However, because generics are also prescription
products, the CPP requirement would still apply and therefore a similar
trend in some of the variables should be expected. Once policy makers,
regulators, and industry leaders can clearly understand where the biggest
opportunity (based on data) lies, there will be more traction on a
multidisciplinary approach to global regulatory harmonization.

92
Bibliography
1. Abraham, J., Helen Lawton Smith. 2003. Regulation of the Pharmaceutical
Industry. New York : Palgrave Macmillan.
2. APEC Harmonization Center. www.apec-ahc.org
3. Asia-Pacific Economic Cooperation. www.apec.org
4. Berry, Ira, Martin, Robert P. 2008. The Pharmaceutical Regulatory Process.
New York: Informa Healthcare.
5. Braithwaite, John. 1994. Prospects for Win-Win International Rapprochement
of Regulation. In Regulatory Co-operation for an Interdependent World.
Paris: OECD.
6. Braithwaite, J., Drahos, P. 2000. Global Business Regulation. Cambridge.
7. Buono, Teresa. 1995. Biotechnology-Derived Pharmaceuticals: Harmonizing
Regional Regulation. Suffolk Transnational Law Review 18:133.
8. Cecchini, Paolo. 1988. The European Challenge, 1992. Aldershot, England:
Wildwood House.
9. Center for Innovation in Regulatory Science: http://www.cirsci.org/
10. Council Regulation (EEC) No. 2309/93. July 22. 1993. Laying down
community procedures for the authorization and supervision of medicinal
products for human and veterinary use and establishing a European Agency
for the Evaluation of Medicinal Products (OJ No L 214 of 24. 8. 1993).
11. Davies, Norman. 1997. Europe: A History. London: Pimlico.
12. D’Arcy, P.F. & D.W.G Harron (eds). 1991. Proceedings of the First
International Conference on Harmonization. Belfast: Queen's University of
Belfast.
13. Drug Regulation. Economist November 16:84. 1991.
14. European Medicines Agency www.emea.eu
a. Centralized Procedure: Directive (EC) No.726/2004.
b. Decentralized Procedure: Directive 2004/83/EC.27/EC
c. Mutual Recognition Procedure: Directive 2001/
15. Food and Drug Administration www.fda.gov
16. Global harmonization on Pharmaceutical Regulations a Step Nearer.
Pharmaceutical Business News; Financial Times November 15, 1991.
17. Giezen, T.J., A. K. Mantel-Teeuwisse, S.M. Straus, H. Schellekns, H. G.
Leufkens, and A.C. Egberts. 2008. Safety-related regulatory actions for
biologicals approved in the United States and the Euripoean Union. JAMA
300(16):1887-1896.
18. Grabowski, H., J. Vernon, and L. Thomas. 1988. Estimating the Effects of
Regulations on Innovation: An International Comparative Analysis of the
Pharmaceutical Industry. Journal of Law and Economics 21 (1): 133-163.
19. Grabowski, Henry and John Vernon. The Regulation of Pharmaceuticals;
Balancing the Benefits and Risks. American Enterprise Institute Studies in
Government Regulation

93
20. Green, Daniel. Fast-track to Approval. Financial Times April 24, 1997.
_____. 1995a. EU Body to Speed Up Approval of New Drugs. Financial Times
January 26.
_____. 1995b. European Medicines Evaluation Agency: Fast-track Approvals
Service. Financial Times April 25.
21. Griffin JP, Shah RR. 2003. History of drug regulation in the UK. In: O’Grady
J, Griffin JP, editors. The Regulation of Medical Products. London: Blackwell
BMJ Books.
22. Gulf Cooperation Council. www.gcc-sg.org
23. Hancher, Leigh. 1990. Regulating for Competition: Government, Law, and the
pharmaceutical Industry in the United Kingdom and France. Oxford:
Clarendon Press.
24. Howells, J. Pharmaceuticals and Europe 1992: The Dynamics of Industrial
Change. Environment and Planning 29:33–49, 1992.
25. Institute of Medicine (IOM). 2013. International Regulatory Harmonization
Amid Globalization of Drug Development: Workshop Summary. Washington,
DC: The National Academies Press.
26. International Conference on Harmonization. www.ich.org
27. ICH Global Cooperation Group. www.ich.org/cache/compo/276-254- 1 .html
28. ICH Global Cooperation Group. Terms of Reference, November 2003.
www.ich.org/LOB/media/MEDIA341Zpdf
29. The International Conference of Drug Regulatory Authorities, Bern,
Switzerland, September 16-19, 2008.
30. Jordan, David. 1992. International Regulatory Harmonization: A New Era in
Prescription Drug Approval. Vanderbilt Journal of Transnational Law
25:471–505.
31. Kahn, Alfred, 1988. The Economics of Regulation: Principles and Institutions.
Cambridge, Mass.: MIT Press.
32. Kanusky, Rosemary. 1994. Pharmaceutical Harmonization in the United
States, the European Economic Community and Japan. Houston Journal of
International Law 16:665-707.
33. Kingham, Richard, Peter Bogaert, and Pamela Eddy. 1994. The New
European Medicines Agency. The Food and Drug Law Journal 49:301–321.
34. Knight, Kenneth, George Kozmetsky and Helen R. Baca. 1976. Industry
Views of the Role of the Federal Government in Industrial Innovation,
University of Texas at Austin.
35. Koberstein, Wayne. 1996. Fernand Sauer: Tending the Garden.
Pharmaceutical Executive 16:36–7.
36. Lakkis, M. 2010. Global and Regional Drug Regulatory Harmonization
Initiatives. Drug Information Journal, 44:289-297.
37. Lakkis M. 2008. The ASEAN harmonization scheme: another CTD-another
challenge to the industry. Regul Focus. November: 39-43.
38. Lakkis M. 2008. The WHO certification scheme and international
pharmaceutical product marketing. Regu1 Focus. June: 34-37
39. Life Sciences Innovation Forum. www.apec.org/apec/documen ts-
reports/life-sciences-innovation-forum/

94
40. McAuslane, Neil and Wang, Tina. 2012. “New Drug Approvals in ICH
Countries: 2002 – 2011”. Center for Innovation in Regulatory Sciences
(CIRS).
41. McCoy, Alfred W. 1980. Drug Traffic: Narcotics and Organized Crime in
Australia. Sydney: Harper & Row.
42. McCoy, Alfred W. 1992. ‘Heroin as a Global Commodity: A History of
Southeast Asia’s Opium Trade’, in A.McCoy & A. Block (eds) War on Drugs:
Studies in the Failure of US Narcotic Policy. Boulder, Colorado: Westview
Press.
43. Oraz, Luis, Kenneth Kaitin and Louis Lasagna. 1992. Pharmaceutical
Regulation in the European Community: Barriers to Single Market Integration.
Journal of Health Politics, Policy and Law 17:859–861.
44. Pan American Health Organization. www.paho.org
45. Peltzman, S. 1974. Regulation of Pharmaceutical Innovation. Washington,
DC: American Enterprise Institute.
46. Peltzman, S.1973. An Evaluation of Consumer Protection Legislation: The
1962 Drug Amendments, 81 Journal of Political Economy 81 (5) 1049-1091.
47. Peltzman, Sam. 2010. “Regulation and the Natural Progress of Opulence”
Institute of Economic Affairs 30 (2) 33-39.
48. Peltzman, Sam. 1993. “George Stigler’s contribution to the Economic
Analysis of Regulation” Journal of Political Economy. 101 (5): 818-832.
49. Peltzman, Sam. 1974. Regulation of Pharmaceutical Innovation; The 1962
Amendments. American Enterprise Institute for Public Policy Research,
Washington, D.C.
50. Peltzman, Sam. 1976. “Toward a more general theory of regulation” The
Journal of Law and Economics. 211-240.
51. The Penn World Table : http://www.rug.nl/research/ggdc/data/penn-world-
table
52. Pharmaceuticals: Carving Up Europe’s Drugs Industry. 1995. Economist
August 26:57.
53. The Polity Index: http://www.systemicpeace.org/polity/polity4.htm
54. Rago, L., Santoso, B. 2008. Drug Regulation: History, Present and Future In:
van Boxtel, C.J., B. Santoso and I.R. Edwards, editors. Drug Benefits and
Risks: International Textbook of Clinical Pharmacology. IOS Press and
Uppsala Monitoring Center.
55. Sauer, Fernand. 1991. Harmonization of Biotechnological Regulations in the
European Community. In Drug Biotechnology Regulation: Scientific Basis
and Practices, eds., Yuan-yuan H. Chiu and John L. Gueriguian. New York:
M. Dekker.
56. Schellekens, H., E. Moors, and H.G. Leufkens. 2011. Drug regulatory systems
must foster innovation. Science 332(6026):174-175
57. Southern African Development Community. www.sadc.int
58. Southern African Development Community strategy
http://www.sadc.int/themes/health/pharmaceuticals/
59. Stigler, George. 1971. “The Theory of Economic Regulation” The Bell
Journal of Economics and Management Science. 2(1): 3-21.

95
60. Stigler, George. 1980. “An introduction to privacy in Economics and
Politics” The Journal of Legal Studies. 623-648.
61. Trotta, F., H.G. Leufkens, J. H. Schellens, R. Laing, and G. Tafuri. 2011.
Evaluation of oncology drugs at the European Medicines Agency and U.S.
Food and Drug Administration: When differences have an impact on clinical
practice. Journal of Clinical Oncology 29(16):2266-2272
62. van Boxtel, C.J., B. Santoso and I.R. Edwards. 2008. Drug Benefits and
Risks: International Textbook of Clinical Pharmacology, IOS Press and
Uppsala Monitoring Center.
63. Van de Gevel, A.J.W. 1992. The Pharmaceutical Industry. CEPSWorking
Paper No. 42
64. Viscusi, W. Kip, Joseph E. Harrington, Jr., John M. Vernon. 2005. Economics
of regulation and antitrust. Cambridge, Mass.: MIT Press.
65. Vogel, David. 1995. Trading Up: Consumer and Environmental Regulation in
a Global Economy. Cambridge: Harvard University Press.
66. Vogel, David. 1998. The Globalization of Pharmaceutical Regulation.
Governance: An International Journal of Policy and Administration. 11(1): 1-
22.
67. Vogel, Ronald. 2007. Pharmaceutical Economics and Public Policy, New
York: Pharmaceutical Products Press.
68. World Health Organization. www.who.int
69. World Health Organization. International Conference of Drug Regulatory
Authorities.
www.who.int/medicines/areas/quality_safety/regulation_legislation/icdra/en/i
ndex.html
70. WHO ICDRA Meetings
http://www.who.int/medicines/areas/quality_safety/regulation_legislation/icdr
a/en/
71. WHO Medicine Expenditure Report –WHO/EMP/MIE/2011.2.6
http://apps.who.int/medicinedocs/documents/s18767en/s18767en.pdf

96
Appendix I: List of countries included in the survey with data collected

97
Country / Region Cost
(USD)
CPP Time for approval
(days)
Algeria 3300 Y 995
Argentina 10000 Y 795
Armenia 5600 Y 730
Australia 200,000 N 300
Austria 50000 N 365
Azerbaijan 2500 Y 730
Bahrain 148 Y 815
Bangladesh 100 Y 485
Belarus 6000 Y 545
Belgium 11470 N 540
Benin 960 Y 1095
Bhutan 100 Y 485
Bolivia 5000 Y 665
Bosnia and Herzegovina 5500 Y 635
Botswana 104.8 Y 1095
Brazil 40000 Y 1095
Bulgaria 5500 N 365
Burkina Faso 1055 Y 1095
Cameroon 960 Y 1095
Canada 350000 N 365
Central African Republic 870 Y 1095
Chad 680 Y 1095
Chile 2500 Y 445
China 11000 Y 730
Colombia 4500 Y 395
Costa Rica 3600 Y 730
Croatia 5000 Y 595
Cyprus 2000 N 365
Democratic Republic of
Congo
950 Y 1095
Denmark 25000 N 365
Dominican Republic 3600 Y 730
Ecuador 2500 Y 545
Egypt 2500 Y 730
El Salvador 3600 Y 730
Ethiopia 800 Y 730
Finland 15000 N 365
France 70000 N 400

98
Country / Region Cost
(USD)
CPP Time for approval
(days)
Gabon 1055 Y 1095
Georgia 2500 Y 485
Germany 150000 N 365
Ghana 4500 Y 730
Greece 350000 N 365
Guatemala 3600 Y 730
Guinea 655 Y 1095
Honduras 3600 Y 730
Hong Kong 351 Y 815
Hungary 14,384 Y 619
India 1000 Y 905
Indonesia 3000 Y 575
Iran 5000 Y 1095
Iraq 165 Y 1095
Ireland 25000 N 730
Israel 5528 Y 730
Italy 120,000 N 600
Ivory Coast 680 Y 1095
Jamaica 75 Y 545
Japan 350000 N 365
Jordan 2500 Y 730
Kazakhstan 4000 Y 730
Kenya 1000 Y 730
Korea 3696 Y 1095
Kuwait 356 Y 1095
Kyrgyzstan 500 Y 730
Lebanon 2567 Y 1095
Libya 20000 Y 730
Luxembourg 200 N 540
Madagascar 750 Y 1095
Malawi 420 Y 730
Malaysia 1350 Y 730
Mali 1055 Y 1095
Malta 150 N 365
Mauritania 861 Y 1095
Mauritius 677 Y 1095
Mexico 6300 Y 730
Moldova 1100 Y 730
Mongolia 0 Y 730

99
Country / Region Cost
(USD)
CPP Time for approval
(days)
Montenegro 2300 Y 1095
Morocco 15000 Y 1085
Mozambique 610 Y 730
Namibia 555.1 Y 1095
Nepal 100 Y 485
Netherlands 350000 N 365
New Zealand 10000 N 460
Nicaragua 3600 Y 730
Niger 1055 Y 1095
Nigeria 1590 Y 730
Norway 350000 N 365
Oman 195 Y 730
Pakistan 1100 Y 995
Panama 3600 Y 730
Peru 1500 Y 1095
Philippines 2910 Y 905
Poland 350000 N 365
Portugal 350000 N 365
Qatar 0 Y 1095
Romania 350000 N 365
Russian Federation 10000 Y 1095
Saudi Arabia 27000 Y 1095
Senegal 960 Y 1095
Serbia 2500 Y 815
Singapore 69100 Y 730
Slovakia 12000 N 480
Slovenia 350000 N 365
South Africa 7715 Y 1095
Spain 350000 N 365
Sri Lanka 100 Y 605
Sudan 750 Y 1095
Sweden 350000 N 365
Switzerland 60000 N 480
Syria 2000 Y 1095
Taiwan 20000 Y 905
Tajikistan 2050 Y 730
Tanzania 1000 Y 730
Thailand 70 Y 765
Togo 861 Y 1095

100
Country / Region Cost
(USD)
CPP Time for approval
(days)
Trinidad & Tobago 125 Y 545
Turkey 8000 Y 605
Turkmenistan 1400 Y 730
Uganda 1000 Y 730
Ukraine 11000 Y 1095
United Arab Emirates 300 Y 605
United Kingdom 400000 N 365
United States 1958800 N 365
Uzbekistan 5500 Y 905
Venezuela 5232 Y 1095
Vietnam 1000 Y 545
Yemen 700 Y 1095
Zambia 1000 Y 730
Zimbabwe 1000 Y 730

101
Appendix II: Examples of the regulatory process for selected countries119
119
The diagrams are from the Thomson Reuters IDRAC files. The intent of showing the examples in this
section is to illustrate the similarities and differences between the countries. For example, some countries
identify how much time is needed from stage to stage, while others do not. Some countries identify the
different segments or milestones, others do not. Some countries have an appeal process, others do not.

102
Australia
There are eight major milestones or phases in the approval process that is designed to
take up to 13 – 14 months.
Phase I Phase II Phase III Phase IV
Phase V Phase VI Phase VII Phase VIII

103
Chile
There are two major sections of the review process – The pharmaceutical and the
analytical evaluation sections.

104
China

105
Colombia

106
Croatia
Czech Republic

107
Egypt
Estonia

108
EU – Centralized Procedure

109
EU – Mutual Recognition Procedure / Decentralized Process
110
France

111
Indonesia
Israel

112
Italy

113
Japan
Lebanon

114
Netherlands

115
Norway
116
Peru

117
Phillipines
118
Saudi Arabia

119
Singapore
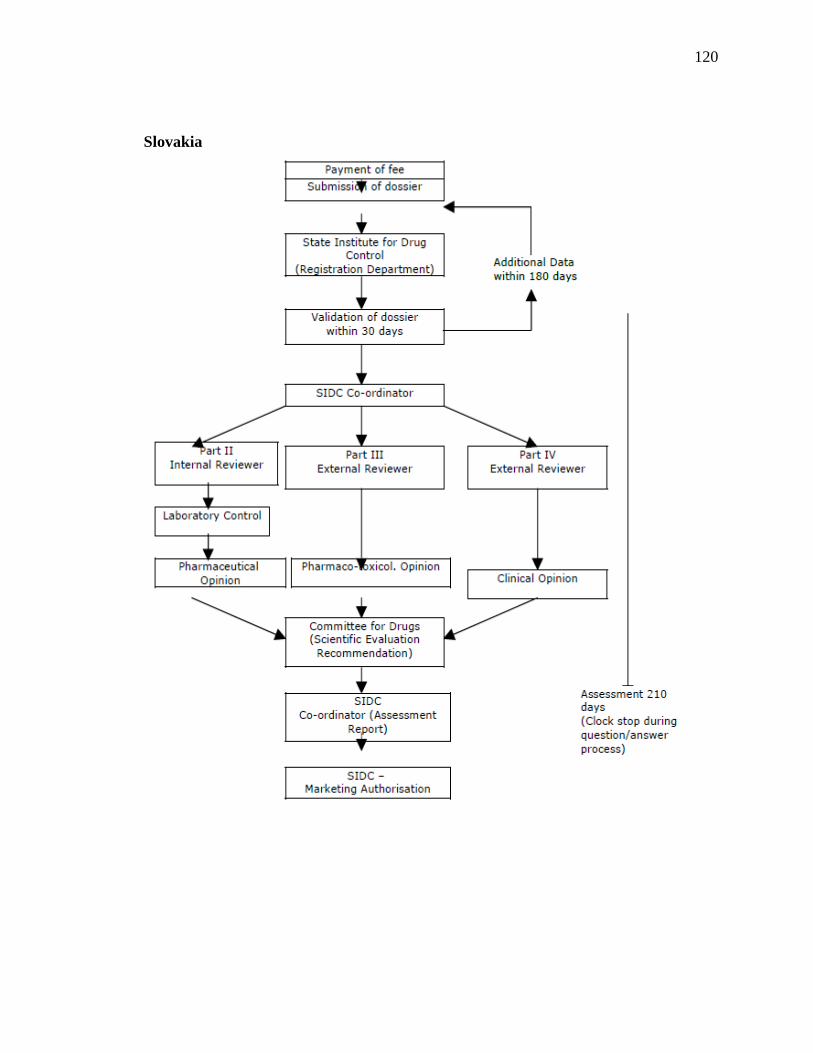
120
Slovakia

121
South Korea
Taiwan

122
United Kingdom
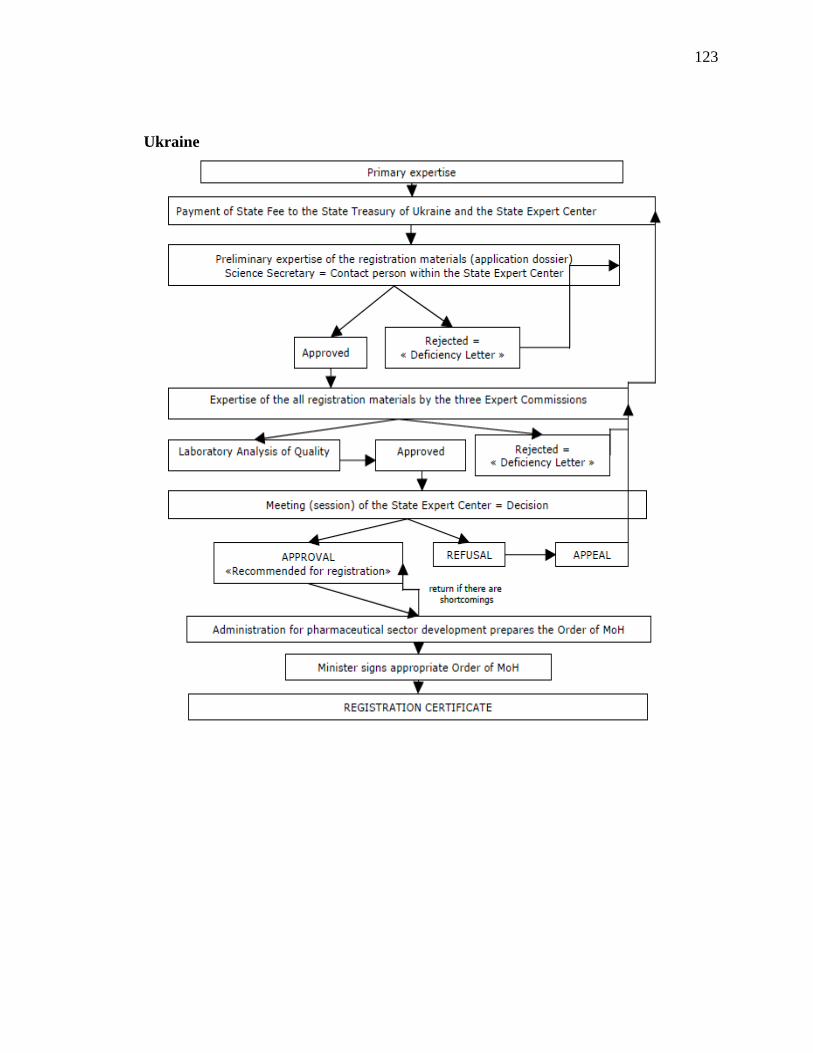
123
Ukraine

124
USA
Serbia

125
Venezuela

126
Appendix III: Listing of countries with matching website for the Drug Health
Authority

127
No Country Link to website of the health authority
1 Algeria http://www.sante.gov.dz/
2 Armenia
http://www.moh.am/?lang=en
http://www.pharm.am/index.php?langid=3
3 Australia www.tga.gov.au
4 Austria http://www.basg.gv.at
5 Azerbaijan http://www.health.gov.az http://www.pharma.az
6 Belarus http://minzdrav.gov.by/ru
7 Bosnia and
Herzegovina http://www.almbih.gov.ba/
8 Botswana http://www.moh.gov.bw/
9 Brazil www.anvisa.gov.br
10 Costa
Rica www.ministeriodesalud.go.cr
11 Croatia http://www.almp.hr/
12 Cyprus www.moh.gov.cy/phs
13 Denmark http://laegemiddelstyrelsen.dk/
14 Dominican
Republic www.drogasyfarmacias.gov.do
15 Ecuador www.inh.gob.ec
16 El Salvador www.salud.gob.sv
17 Ethiopia www.fmhaca.gov.et
18 Finland http://www.fimea.fi/frontpage
19 France http://ansm.sante.fr/
20 Georgia http://www.moh.gov.ge
21 Germany http://www.bfarm.de/DE/Home/home_node.html
22 Ghana www.fdbghana.gov.gh/
23 Guatemala www.mspas.gob.gt
24 Honduras www.salud.gob.hn
25 Hong
Kong http://www.drugoffice.gov.hk
26 Hungary http://www.ogyi.hu/uj_beadvanyok/
27 Iceland http://www.lyfjastofnun.is/
28 Indonesia http://www.pom.go.id/
29 Ireland http://www.imb.ie/
30 Israel http://www.health.gov.il/UnitsOffice/HD/MTI/Drugs/Pages/default.aspx
31 Italy www.agenziafarmaco.it
32 Jamaica www.moh.gov.jm
33 Japan http://www.pmda.go.jp/english/index.html
34 Jordan http://www.jfda.jo/
35 Kazakhstan http://www.dari.kz
36 Kenya www.pharmacyboardkenya.org/
37 Kyrgyzstan
http://www.med.kg
http://www.pharm.kg
38 Lebanon http://www.moph.gov.lb/Drugs/Pages/Drugs.aspx

128
No Country Link to website of the health authority
39 Malawi www.pmpb.mw
40 Malaysia http://www.bpfk.gov.my
41 Malta www.medicinesauthority.gov.mt
42 Mexico www.cofepris.gob.mx
43 Moldova http://www.ms.gov.md/
44 Mongolia http://english.moh.mn/
45 Montenegro http://calims.me
46 Namibia www.nmrc.com.na
47 Nicaragua www.minsa.gob.ni
48 Nigeria www.nafdac.gov.ng
49 Panama www.minsa.gob.pa
50 Philippines http://www.fda.gov.ph/
51 Russian
Federation http://rosminzdrav.ru/docs/mzsr/regulation/1
52 Saudi
Arabia www.sfda.gov.sa
53 Serbia http://www.alims.gov.rs/index_eng.php
54 Singapore
http://www.hsa.gov.sg/publish/hsaportal/en/health_products_regulation/
western_medicines/licences/Fees1.html
55 Slovakia http://www.sukl.sk/en
56 South
Africa www.mccza.com
57 Switzerland www.swissmedic.ch
58 Syria
http://www.moh.gov.sy/ar/%D8%A7%D9%84%D8%B1%D8%A6%D9
%8A%D8%B3%D9%8A%D8%A9/tabid/56/Default.aspx
59 Tajikistan www.health.tj and http://www.pharmnadzor.tj
60 Tanzania www.tfda.or.tz/
61 Thailand http://drug.fda.moph.go.th/drug/
62 Trinidad
&Tobago www.health.gov.tt
63 Uganda www.nda.org.ug
64 Ukraine http://www.pharma-center.kiev.ua/view/
65 United
Kingdom http://www.mhra.gov.uk/Aboutus/Whoweare/index.htm
66 United
States
http://www.fda.gov/ForIndustry/UserFees/PrescriptionDrugUserFee/defa
ult.htm
67 Uzbekistan http://www.minzdrav.uz
68 Venezuela http://www.inhrr.gob.ve/
69 Zambia www.moh.gov.zm
70 Zimbabwe www.mcaz.co.za

129
Appendix IV: Changes in the US drug regulation over time120
120
Source: www.fda.gov

130
Changes in the US drug regulation over time
Date Event
1820 Eleven physicians meet in Washington, D.C., to establish the U.S.
Pharmacopeia, the first compendium of standard drugs for the United States.
1883 Dr. Harvey W. Wiley becomes chief chemist, expanding the Bureau of
Chemistry's food adulteration studies. Campaigning for a federal law, Dr.
Wiley is called the "Crusading Chemist" and "Father of the Pure Food and
Drugs Act." He retired from government service in 1912 and died in 1930.
1898 Association of Official Agricultural Chemists (now AOAC International)
establishes a Committee on Food Standards headed by Dr. Wiley. States
begin incorporating these standards into their food statutes.
1902 The Biologics Control Act is passed to ensure purity and safety of serums,
vaccines, and similar products used to prevent or treat diseases in humans.
Congress appropriates $5,000 to the Bureau of Chemistry to study chemical
preservatives and colors and their effects on digestion and health. Dr. Wiley's
studies draw widespread attention to the problem of food adulteration. Public
support for passage of a federal food and drug law grows.
1906 The original Food and Drugs Act is passed by Congress on June 30 and
signed by President Theodore Roosevelt. It prohibits interstate commerce in
misbranded and adulterated foods, drinks and drugs.
The Meat Inspection Act is passed the same day.
Shocking disclosures of insanitary conditions in meat-packing plants, the use
of poisonous preservatives and dyes in foods, and cure-all claims for
worthless and dangerous patent medicines were the major problems leading
to the enactment of these laws.
1911 In U.S. v. Johnson, the Supreme Court rules that the 1906 Food and Drugs
Act does not prohibit false therapeutic claims but only false and misleading
statements about the ingredients or identity of a drug.
1912 Congress enacts the Sherley Amendment to overcome the ruling in U.S. v.
Johnson. It prohibits labeling medicines with false therapeutic claims
intended to defraud the purchaser, a standard difficult to prove.

131
Changes in the US drug regulation over time
Date Event
1913 Gould Amendment requires that food package contents be "plainly and
conspicuously marked on the outside of the package in terms of weight,
measure, or numerical count."
1914 In U.S. v. Lexington Mill and Elevator Company, the Supreme Court issues
its first ruling on food additives. It ruled that in order for bleached flour with
nitrite residues to be banned from foods, the government must show a
relationship between the chemical additive and the harm it allegedly caused
in humans. The court also noted that the mere presence of such an ingredient
was not sufficient to render the food illegal. The Harrison Narcotic Act
requires prescriptions for products exceeding the allowable limit of narcotics
and mandates increased record-keeping for physicians and pharmacists who
dispense narcotics.
1927 The Bureau of Chemistry is reorganized into two separate entities.
Regulatory functions are located in the Food, Drug, and Insecticide
Administration, and non-regulatory research is located in the Bureau of
Chemistry and Soils.
1930 McNary-Mapes Amendment authorizes FDA standards of quality and fill-of-
container for canned food, excluding meat and milk products.
The name of the Food, Drug, and Insecticide Administration is shortened to
Food and Drug Administration (FDA) under an agricultural appropriations
act.
1933 FDA recommends a complete revision of the obsolete 1906 Food and Drugs
Act. The first bill is introduced into the Senate, launching a five-year
legislative battle.
1938 Elixir of Sulfanilamide, containing the poisonous solvent diethylene glycol,
kills 107 persons, many of whom are children, dramatizing the need to
establish drug safety before marketing and to enact the pending food and
drug law.
1937 The Federal Food, Drug, and Cosmetic (FDC) Act of 1938 is passed by
Congress, containing new provisions:
Extending control to cosmetics and therapeutic devices.
Requiring new drugs to be shown safe before marketing-starting a

132
Changes in the US drug regulation over time
Date Event
new system of drug regulation.
Eliminating the Sherley Amendment requirement to prove intent to
defraud in drug misbranding cases.
Providing that safe tolerances be set for unavoidable poisonous
substances.
Authorizing standards of identity, quality, and fill-of-container for
foods.
Authorizing factory inspections.
Adding the remedy of court injunctions to the previous penalties of
seizures and prosecutions.
Under the Wheeler-Lea Act, the Federal Trade Commission is charged with
overseeing advertising associated with products otherwise regulated by FDA,
with the exception of prescription drugs.
1939 First Food Standards issued (canned tomatoes, tomato purée, and tomato
paste).
1940 FDA transferred from the Department of Agriculture to the Federal Security
Agency, with Walter G. Campbell appointed as the first Commissioner of
Food and Drugs.
1941- Insulin Amendment requires FDA to test and certify purity and
potency of this lifesaving drug for diabetes.
1943 In U.S. v. Dotterweich, the Supreme Court rules that the responsible officials
of a corporation, as well as the corporation itself, may be prosecuted for
violations. It need not be proven that the officials intended, or even knew of,
the violations.
1945 Penicillin Amendment requires FDA testing and certification of safety and
effectiveness of all penicillin products. Later amendments extended this
requirement to all antibiotics. In 1983 such control was found no longer
needed and was abolished.
1949 FDA publishes guidance to industry for the first time. This guidance,
"Procedures for the Appraisal of the Toxicity of Chemicals in Food," came to
be known as the "black book."

133
Changes in the US drug regulation over time
Date Event
1950 In Alberty Food Products Co. v. U.S., a court of appeals rules that the
directions for use on a drug label must include the purpose for which the drug
is offered. Therefore, a worthless remedy cannot escape the law by not
stating the condition it is supposed to treat.
Oleomargarine Act requires prominent labeling of colored oleomargarine, to
distinguish it from butter.
Delaney Committee starts congressional investigation of the safety of
chemicals in foods and cosmetics, laying the foundation for the 1954 Miller
Pesticide Amendment, the 1958 Food Additives Amendment, and the 1960
Color Additive Amendment.
1951 Durham-Humphrey Amendment defines the kinds of drugs that cannot be
safely used without medical supervision and restricts their sale to prescription
by a licensed practitioner.
1952 In U.S. v. Cardiff, the Supreme Court rules that the factory inspection
provision of the 1938 FDC Act is too vague to be enforced as criminal law.
FDA consumer consultants are appointed in each field district to maintain
communications with consumers and ensure that FDA considers their needs
and problems.
1954 Miller Pesticide Amendment spells out procedures for setting safety limits for
pesticide residues on raw agricultural commodities.
First large-scale radiological examination of food carried out by FDA when it
received reports that tuna suspected of being radioactive was being imported
from Japan following atomic blasts in the Pacific. FDA begins monitoring
around the clock to meet the emergency.
1955 HEW Secretary Oveta Culp Hobby appoints a committee of 14 citizens to
study the adequacy of FDA's facilities and programs. The committee
recommends a substantial expansion of FDA staff and facilities, a new
headquarters building, and more use of educational and informational
programs.
1962 Thalidomide, a new sleeping pill, is found to have caused birth defects in
thousands of babies born in Western Europe. News reports on the role of Dr.
Frances Kelsey, FDA medical officer, in keeping the drug off the U.S.

134
Changes in the US drug regulation over time
Date Event
market, arouse public support for stronger drug regulation.
Kefauver-Harris Drug Amendments passed to ensure drug efficacy and
greater drug safety. For the first time, drug manufacturers are required to
prove to FDA the effectiveness of their products before marketing them.
Consumer Bill of Rights is proclaimed by President John F. Kennedy in a
message to Congress. Included are the right to safety, the right to be
informed, the right to choose, and the right to be heard.
1965 Drug Abuse Control Amendments are enacted to deal with problems caused
by abuse of depressants, stimulants and hallucinogens.
1966 FDA contracts with the National Academy of Sciences/National Research
Council to evaluate the effectiveness of 4,000 drugs approved on the basis of
safety alone between 1938 and 1962.
Child Protection Act enlarges the scope of the Federal Hazardous Substances
Labeling Act to ban hazardous toys and other articles so hazardous that
adequate label warnings could not be written.
Fair Packaging and Labeling Act requires all consumer products in interstate
commerce to be honestly and informatively labeled, with FDA enforcing
provisions on foods, drugs, cosmetics, and medical devices.
1968 FDA Bureau of Drug Abuse Control and Treasury Department Bureau of
Narcotics are transferred to the Department of Justice to form the Bureau of
Narcotics and Dangerous Drugs (BNDD), consolidating efforts to police
traffic in abused drugs.
Reorganization of federal health programs places FDA in the Public Health
Service.
FDA forms the Drug Efficacy Study Implementation (DESI) to implement
recommendations of the National Academy of Sciences investigation of
effectiveness of drugs first marketed between 1938 and 1962.
1970 In Upjohn v. Finch the Court of Appeals upholds enforcement of the 1962
drug effectiveness amendments by ruling that commercial success alone does
not constitute substantial evidence of drug safety and efficacy.
FDA requires the first patient package insert: oral contraceptives must
contain information for the patient about specific risks and benefits.
The Comprehensive Drug Abuse Prevention and Control Act replaces

135
Changes in the US drug regulation over time
Date Event
previous laws and categorizes drugs based on abuse and addiction potential
compared to their therapeutic value.
Environmental Protection Agency established; takes over FDA program for
setting pesticide tolerances.
1972 Over-the-Counter Drug Review begun to enhance the safety, effectiveness
and appropriate labeling of drugs sold without prescription.
1973 The U.S. Supreme Court upholds the 1962 drug effectiveness law and
endorses FDA action to control entire classes of products by regulations
rather than to rely only on time-consuming litigation.
Consumer Product Safety Commission created by Congress; takes over
programs pioneered by FDA under 1927 Caustic Poison Act, 1960 Federal
Hazardous Substances Labeling Act, 1966 Child Protection Act, and PHS
accident prevention activities for safety of toys, home appliances, etc.
1976 Medical Device Amendments passed to ensure safety and effectiveness of
medical devices, including diagnostic products. The amendments require
manufacturers to register with FDA and follow quality control procedures.
Some products must have pre-market approval by FDA; others must meet
performance standards before marketing.
1977 Saccharin Study and Labeling Act passed by Congress to stop FDA from
banning the chemical sweetener but requiring a label warning that it has been
found to cause cancer in laboratory animals.
Introduction of the Bioresearch Monitoring Program as an agency-wide
initiative ensures the quality and integrity of data submitted to FDA and
provides for the protection of human subjects in clinical trials by focusing on
preclinical studies on animals, clinical investigations, and the work of
institutional review boards.
1979 In the hours following the Three Mile Island nuclear emergency of March 28,
1979, FDA contracted with firms in Missouri, Michigan, and New Jersey to
prepare and package enough doses of potassium iodide to protect those
threatened with thyroid cancer if exposed to radiation. Nearly one quarter of a
million bottles-enough for every household in the area-were delivered to
Harrisburg, Pennsylvania within 72 hours.

136
Changes in the US drug regulation over time
Date Event
1980 Infant Formula Act establishes special FDA controls to ensure necessary
nutritional content and safety.
1982 Tamper-resistant Packing Regulations issued by FDA to prevent poisonings
such as deaths from cyanide placed in Tylenol capsules. The Federal Anti-
Tampering Act passed in 1983 makes it a crime to tamper with packaged
consumer products.
FDA publishes first Red Book (successor to 1949 "black book"), officially
known as Toxicological Principles for the Safety Assessment of Direct Food
Additives and Color Additives Used in Food.
1983 Orphan Drug Act passed, enabling FDA to promote research and marketing
of drugs needed for treating rare diseases.
1984 Fines Enhancement Laws of 1984 and 1987 amend the U.S. Code to greatly
increase penalties for all federal offenses. The maximum fine for individuals
is now $100,000 for each offense and $250,000 if the violation is a felony or
causes death. For corporations, the amounts are doubled.
1985 AIDS test for blood approved by FDA in its first major action to protect
patients from infected donors.
1986 Childhood Vaccine Act requires patient information on vaccines, gives FDA
authority to recall biologics, and authorizes civil penalties.
1987 Investigational drug regulations revised to expand access to experimental
drugs for patients with serious diseases with no alternative therapies.
1988 Food and Drug Administration Act of 1988 officially establishes FDA as an
agency of the Department of Health and Human Services with a
Commissioner of Food and Drugs appointed by the President with the advice
and consent of the Senate, and broadly spells out the responsibilities of the
Secretary and the Commissioner for research, enforcement, education, and
information.
1989 FDA issues a nationwide recall of all over-the-counter dietary supplements
containing 100 milligrams or more of L-Tryptophan, due to a clear link

137
Changes in the US drug regulation over time
Date Event
between the consumption of L-tryptophan tablets and its association with a
U.S. outbreak of Eosinophilia Myalgia Syndrome (EMS), characterized by
fatigue, shortness of breath, and other symptoms. By 1990 the Centers for
Disease Control and Prevention confirm over 1,500 cases of EMS, including
38 deaths, and FDA prohibits the importation of l-tryptophan.
1991 Regulations published to Accelerate the Review of Drugs for life-threatening
diseases.
1992 Generic Drug Enforcement Act imposes debarment and other penalties for
illegal acts involving abbreviated drug applications.
Prescription Drug User Fee Act (PDUFA) requires drug and biologics
manufacturers to pay fees for product applications and supplements, and
other services. The act also requires FDA to use these funds to hire more
reviewers to assess applications.
1993 A consolidation of several adverse reaction reporting systems is launched as
MedWatch, designed for voluntary reporting of problems associated with
medical products to be filed with FDA by health professionals.
1995 FDA declares cigarettes to be "drug delivery devices." Restrictions are
proposed on marketing and sales to reduce smoking by young people.
1996 Federal Tea Tasters Repeal Act repeals the Tea Importation Act of 1897 to
eliminate the Board of Tea Experts and user fees for FDA's testing of all
imported tea. Tea itself is still regulated by FDA.
1997 Food and Drug Administration Modernization Act reauthorizes the
Prescription Drug User Fee Act of 1992 and mandates the most wide-ranging
reforms in agency practices since 1938. Provisions include measures to
accelerate review of devices, regulate advertising of unapproved uses of
approved drugs and devices, and regulate health claims for foods.
1999 ClinicalTrials.gov is founded to provide the public with updated information
on enrollment in federally and privately supported clinical research, thereby
expanding patient access to studies of promising therapies.
A final rule mandates that all over-the-counter drug labels must contain data
in a standardized format. These drug facts are designed to provide the patient

138
Changes in the US drug regulation over time
Date Event
with easy-to-find information, analogous to the nutrition facts label for foods.
2000 The U. S. Supreme Court, upholding an earlier decision in Food and Drug
Administration v. Brown & Williamson Tobacco Corp. et al., ruled 5-4 that
FDA does not have authority to regulate tobacco as a drug. Within weeks of
this ruling, FDA revokes its final rule, issued in 1996, that restricted the sale
and distribution of cigarettes and smokeless tobacco products to children and
adolescents, and that determined that cigarettes and smokeless tobacco
products are combination products consisting of a drug (nicotine) and device
components intended to deliver nicotine to the body.
Federal agencies are required to issue guidelines to maximize the quality,
objectivity, utility, and integrity of the information they generate, and to
provide a mechanism whereby those affected can secure correction of
information that does not meet these guidelines, under the Data Quality Act.
2003 The Medicare Prescription Drug Improvement and Modernization Act
requires, among other elements, that a study be made of how current and
emerging technologies can be utilized to make essential information about
prescription drugs available to the blind and visually impaired.
To help consumers choose heart-healthy foods, the Department of Health and
Human Services announces that FDA will require food labels to include
trans-fat content, the first substantive change to the nutrition facts panel on
foods since the label was changed in 1993. An obesity working group is
established by the Commissioner of Food and Drugs, charged to develop an
action plan to deal with the nation's obesity epidemic from the perspective of
FDA. In March 2004 the group releases "Calories Count: Report of the
Obesity Working Group," which addresses issues connected to the food label,
obesity therapeutics, research needs, the role of education, and other topics.
2004 Project BioShield Act of 2004 authorizes FDA to expedite its review
procedures to enable rapid distribution of treatments as countermeasures to
chemical, biological, and nuclear agents that may be used in a terrorist attack
against the U. S., among other provisions.
Passage of the Food Allergy Labeling and Consumer Protection Act requires
the labeling of any food that contains a protein derived from any one of the
following foods that, as a group, account for the vast majority of food
allergies: peanuts, soybeans, cow's milk, eggs, fish, crustacean shellfish, tree
nuts, and wheat.

139
Changes in the US drug regulation over time
Date Event
A ban on over-the-counter steroid precursors, increased penalties for making,
selling, or possessing illegal steroids precursors, and funds for preventive
education to children are features of the Anabolic Steroid Control Act of
2004.
FDA publishes "Innovation or Stagnation? -- Challenge and Opportunity on
the Critical Path to New Medical Products," which examines the critical path
needed to bring therapeutic products to fruition, and how FDA can
collaborate in the process, from laboratory to production to end use, to make
medical breakthroughs available to those in need as quickly as possible.
2005 Formation of the Drug Safety Board is announced, consisting of FDA staff
and representatives from the National Institutes of Health and the Veterans
Administration. The Board will advise the Director, Center for Drug
Evaluation and Research, FDA, on drug safety issues and work with the
agency in communicating safety information to health professionals and
patients

140
Appendix V: NDA required sections in the United States

141
The New Drug Application (NDA)
In the US, pharmaceuticals fall under the Food, Drug, and Cosmetic Act (FD&C Act)
which contains 10 chapters and significant amendments totaling hundreds of pages. The
FD&C is part of the Code of Federal Register (CFR). There are 50 titles in the CFR and
title 21 refers to Food and Drugs totaling 4436 pages of regulation!
Below is the listing of NDA sections121
:
1. Index
2. Labeling
3. Summary (21 CFR 314.50 (c) )
4. Chemistry Section
a. Chemistry, Manufacturing and Controls (CMC)
b. Samples (21 CFR 314.50 (e)(1); 21 CFR 601.2 (a)) (Submit only upon FDA’s
request)
c. Methods validation package (e.g., 21 CFR 314.50(e)(2)(i); 21 CFR 601.2)
5. Nonclinical pharmacology and toxicology section (e.g., 21 CFR 314.50(d)(2); 21 CFR
601.2)
6. Human pharmacokinetics and bioavailability section (e.g., 21 CFR 314.50(d)(3); 21
CFR 601.2)
7. Clinical microbiology section (e.g., 21 CFR 314.50(d)(4))
8. Clinical data section (e.g., 21 CFR 314.50(d)(5); 21 CFR 601.2)
9. Safety update report (e.g., 21 CFR 314.50(d)(5)(vi)(b);21 CFR 601.2)
10. Statistical section (e.g., 21 CFR 314.50(d)(6); 21 CFR 601.2)
11. Case report tabulations (e.g., 21 CFR 314.50(f)(1);21 CFR 601.2)
12. Case report forms (e.g., 21 CFR 314.50 (f)(2); 21 CFR 601.2)
13. Patent information on any patent that claims the drug/biologic (21 U.S.C. 355(b) or
(c))
14. A patent certification with respect to any patent that claims the drug/biologic (21
U.S.C. 355 (b)(2) or (j)(2)(A))
15. Establishment description (21 CFR Part 600, if applicable)
16. Debarment certification (FD&C Act 306 (k)(1))
17. Field copy certification (21 CFR 314.50 (l)(3))
18. User Fee Cover Sheet (PDUFA Form FDA 3397, GDUFA Form FDA 3794, BsUFA
Form FDA 3792, or MDUFMA Form FDA 3601)
19. Financial Disclosure Information (21 CFR Part 54)
121
Information retrieved from www.fda.gov
http://www.fda.gov/drugs/developmentapprovalprocess/formssubmissionrequirements/drugmasterfilesdmfs
/ucm073080.htm

142
Appendix VI: Regression analysis data with Time as the dependent variable

143
TABLE 6
RESULTS OF REGRESSION ANALYSIS OF APPROVAL TIME ON COUNTRY ECONOMIC AND POLITICAL
CHARACTERISTICS ACROSS 129 COUNTRIES, 2010 - 2011
Variables OLS
(t-Stat) (1) (2) (3) (4) (5) (6) (7)
LG GDP PC -47.50 -34.88 -4.01 -6.33
(-3.04)* (-2.28)* (-0.26) (-0.42)
Cost -0.00044 -7.42E-05 -7.40E-05 -0.00013 -0.00013
(-3.59)* (0.00013) (-0.58) (-0.99) (-0.99)
Health Expend -0.081 -0.087 -0.082 -0.074 -0.076
(-5.57)* (-7.80)* (-6.20)* (-4.94)* (-5.58)*
Open K -0.72 -0.72
(-1.86) (-1.88)
Constant 1118 1044 875 845 844 954 905
(8.84) (8.52) (7.47) (33.55) (33.39) (7.73) (22.08)
Adjusted R2
0.063 0.15 0.32 0.33 0.32 0.33 0.34
# of observations 124 124 121 125 125 121 125
*statistically significant

144
TABLE 6 (continued)
Variables OLS
(t-Stat) (8) (9) (10) (11) (12) (13) (14)
LG GDP PC 3.38 12.64
(0.23) (0.76)
Cost -0.00013 -0.00013 -0.00012 -0.00013 -0.00013 -0.00012 -0.00015
(-0.97) (-1.10) (-1.05) (-1.07) (-0.97) (-0.90) (-1.35)
Health Expend -0.075 -0.056 -0.056 -0.040 -0.052 -0.055 -0.035
(-5.44)* (-3.97)* (3.80)* (-2.46)* (-3.06)* (-3.13)* (-2.26)*
Open K -0.72 -0.43 -0.39 -0.13 -0.33 -0.25 -0.044
(-1.87) (-1.22) (-1.07) (-0.33) (-0.79) (-0.59) (-0.12)
Dem Score -0.44
(-0.21)
Phys / 1000 -49.84 -50.52 -28.45
(-2.99)* (-2.90)* (-1.50)
Frag. Index 12.15 13.01 12.64 7.16
(2.29)* (2.78) (2.71)* (1.40)
Life Expectancy -7.93
(-2.47)*
Constant 906 934 902 755 738 623 1296
(21.69) (21.08) (7.48) (8.77) (10.24) (3.67) (5.19)
Adjusted R2
0.33 0.42 0.41 0.42 0.35 0.35 0.41
# of observations 125 106 103 103 121 117 121

145
TABLE 6 (continued)
Variables OLS
(t-Stat) (15) (16) (17) (18)
LG GDPPC 11.56
(0.73)
Cost -0.00015 -0.00027 -0.00028 -0.00015
(-1.33) (-2.56) (-2.68)* (-1.27)
Health Expend -0.031 -0.036
(-1.89) (-2.17)*
Open K -0.020 0.144 0.018 -0.057
(-0.052) (0.36) (0.048) (-0.15)
KG 0.96 0.27 1.50
(0.21) (0.060) (0.34)
Phys / 1000 -19.21 -37.08 -37.15 -26.36
(-0.97) (-1.96) (-1.98) (-1.38)
Frag. Index 9.60 19.26 17.07 12.48
(1.64) (3.48)* (3.41)* (2.34)*
Life Expectancy -4.75
(-1.23)
Constant 1077 549 681 727
(3.74) (3.06) (7.29) (7.73)
Adjusted R2
0.41 0.38 0.38 0.40
# of observations 103 100 103 103
*statistically significant

146
TABLE 6 (continued)
Variables OLS
(t-Stat) (19) (20) (21) (22) (23) (24) (25)
GDP PC -1.55e-11 -1.60e-11 1.97e-11
(-1.08) (-1.12) (0.89)
Cost -0.00038
(-2.09)*
Open K 0.077
(0.17)
Frag. Index 21.16 20.90 13.30 12.97
(5.07)* (5.43)* (2.29)* (2.23)*
Life Expectancy -7.061 6.55 -13.67 -12.92
(-1.89) (-1.74) (-5.68)* (-5.38)*
Democracy 0.162 -2.67
(0.07) (-1.09)
Constant 512 522 699 1074 1049 1650 1600
(7.78) (13.88) (24.86) (3.59) (3.50) (9.77) (9.51)
Adjusted R2
0.17 0.19 0.0014 0.22 0.22 0.22 0.24
# of observations 124 124 128 123 123 128 128
*statistically significant

147
TABLE 6 (continued)
Variables OLS
(t-Stat) (26) (27) (28) (29) (30) (31) (32) (33)
LG GDP PC -3.5e-11
(-2.27)*
Cost -0.00029 -0.00024 -0.00020 0.00093
(-2.46)* (-2.03)* (-1.74) (-3.06)*
Literacy rate -5.37 -2.033 -4.032
(-3.16)* (-0.85) (-2.42)*
Phys / 1000 -74.85 -65.29 -31.23 -15.72
(-4.32)* (-3.76)* (-1.37) (-0.73)
Frag. Index 18.80
(3.35)*
Life Expectancy -8.62 -8.31
(-2.25)* (-1.93)*
Constant 799 800 1343 557 702 1175 1459 1094
(18.74) (19.22) (5.49) (6.86) (26.74) (8.01) (7.09) (7.75)
Adjusted R2
0.144 0.18 0.21 0.25 0.031 0.11 0.14 0.20
# of observations 106 106 106 103 129 73 73 73
*statistically significant

148
Curriculum Vitae
SUMMARY
A technically oriented, experienced, dedicated team leader with extensive knowledge & experience in the drug development process [from discovery through launch] and exceptional expertise in clinical trial supply management. Proven track [12 years] of successful strategy development and project execution through all stages of drug development, across continents and cultures building lasting bridges along the way. Hands on experience in sterile and solid dosage manufacturing, demand & supply forecasting, and project management, execution & leadership.
EXPERIENCE
Novartis Consumer Health May’13 – Present
Associate Director, Project Management (05/13 – Present)
Product Sustainment and Strategy PMO
Merck & Co. Nov '09 – May’13
Senior Manager, Business Consulting (11/09 – 05/13)
Organizational Strategy Management and Operations, Merck Manufacturing Division
Schering-Plough Corp Nov '01 – Nov'09
Senior Manager, Global Development and Commercialization PMO, Global Supply Chain (1/09 – 11/09)
Project Manager, Global Clinical Supplies Planning Kenilworth, NJ (6/05 – 1/09)
Investigational Material Specialist, Global Clinical Supplies Kenilworth, NJ (11/01 – 6/05) EDUCATION Rutgers University, Newark, NJ 05/14 Doctor of Philosophy, Global Affairs Rutgers University, Newark, NJ 05/10 Master of Science, Global Affairs New Jersey Institute of Technology, Newark, NJ 05/05 Master of Science, Pharmaceutical Engineering New Jersey Institute of Technology, Newark, NJ 05/01 Bachelor of Science, Chemical Engineering INTERNATIONAL EXPERIENCE Oss, Netherlands; Lucerne, Switzerland; Antwerp, Belgium; Cork, Ireland; Mexico City, Mexico Middle East (Jordan, Syria, Lebanon, Egypt), France, Spain, Italy, Germany, England LANGUAGE Fluent in English and Arabic Familiar in French, Dutch & German LinkedIn Profile: http://www.linkedin.com/profile/view?id=28066458&trk=tab_pro

















