COST Chemistry D36 3rd Workshop and 5th Management Committee Meeting Structure-performance relationships at the surface of functional materials Benahavís (Málaga, Spain), 21 st to 23 rd of October, 2009
Transcript
COST Chemistry D36 3rd Workshop
and
5th Management Committee Meeting
Structure-performance relationships
at the surface of functional materials
Benahavís (Málaga, Spain), 21 st to 23 rd of October, 2009
The main objective of the COST D36 Action is to increase the fundamental
knowledge and understanding of the chemistry occurring at surfaces and
interfaces and the factors that tune it. An interdisciplinary, combined effort is the
approach. A fundamental approach is advocated, even for industrially oriented
research projects. This requires precisely defined problems at all levels and an
interdisciplinary approach i.e. synthesis and activation of the materials;
measurement of the surface properties; understanding surface properties at the
atomic, molecular or cluster level and theoretical understanding of these
properties in relation to chemical composition and the structure of the surface.
As a consequence, the secondary objective is to gain advanced knowledge for
modelling/predicting of the structure/composition reactivity/surface properties
relationships of the materials, by means of characterisation of the bulk and
surface properties under real operation conditions and for preparing materials
with tuneable properties.
COST Chemistry D36 3rd Workshop and 5th Management Committee Meeting
4 Benahavís (Málaga, Spain), 21st to 23rd of October, 2009
COST Chemistry D36 3rd Workshop and 5th Management Committee Meeting
Benahavís (Málaga, Spain), 21st to 23rd of October, 2009 5
Sponsors and collaborating institutions:
COST (European cooperation in science and technolog y) http://www.cost.esf.org/
Universidad de Málaga
http://www.uma.es/
Ayuntamiento de Benahavís http://www.benahavis.es/inicio.asp
Junta de Andalucía
http://www.juntadeandalucia.es/index.html
Ayuntamiento de Ronda http://www.turismoderonda.es/
PID Eng&Tech
http://www.pidengtech.com/
COST Chemistry D36 3rd Workshop and 5th Management Committee Meeting
6 Benahavís (Málaga, Spain), 21st to 23rd of October, 2009
Organizers
Dr. M. Olga Guerrero Pérez Universidad de Málaga
Prof. Dr. José Rodríguez Mirasol
Universidad de Málaga
Local Committee
Dr. Jorge Bedia Universidad de Málaga
Dr. Juana M. Rosas
Universidad de Málaga
Mr. Ricardo López Medina Instituto de Catálisis y Petroleoquímica
Ms. Elizabeth Rojas García
Instituto de Catálisis y Petroleoquímica
Mr. Ramiro Ruiz Rosas Universidad de Málaga
Ms. M. José Valero Romero
Universidad de Málaga
BOOK OF ABSTRACTS
Section I: Scientific Program
COST Chemistry D36 3rd Workshop and 5th Management Committee Meeting
Benahavís (Málaga, Spain), 21st to 23rd of October, 2009 9
Wednesday 21 st 17:00 – 17:15 Welcome
Miguel A. Bañares, Action Chair M. Olga Guerrero-Pérez, Local Organizer
17:15 – 18:00 Keynote 1 Chair: Miguel A. Bañares, Instituto de Catálisis y Petroleoquímica (CSIC) (Spain)
K1 José Manuel López-Nieto, Instituto de Tecnología Química (CSIC) (Spain) “Synthesis, Characterization and Catalytic behaviour in partial alkane oxidation of Multicomponent mixed oxidic”
18:00 – 20:00 Oral Session 1 Chair: Sanna Airaksinen, Helsinki University of Technology (Finland)
O1 Maria Ziolek, Adam Mickiewicz University (Poland) “The effect of porosity of niobosilicate supports and VSbOx loading on the ammoxidation of propane” O2 James Sullivan, University College Dublin (Ireland) “Towards 4-way catalysis” O3 Gerhard Mestl, SÜD-CHEMIE AG (Germany) “Towards an optimization of MoVNbTe-catalysts for C3-oxidation” O4 Maricarmen Capel, Instituto de Catálisis y Petroleoquímica (CSIC) (Spain) “Silylation of titanium-containing amorphous silica catalyst: Effect on the alkenes epoxidation with H2O2” O5 Lyuba Ilieva-Gencheva, Institute of Catalysis (BAS) (Bulgary) “Preferential oxidation of CO in H2 rich stream over gold catalysts supported on doped ceria: effect of preparation method and dopants nature” O6 Stanislaw Dzwigaj, Université Pierre et Marie Curie (France) “The Design of Metal-Single site Catalysts for their Application in Catalytic and Photocatalytic Processes“
20:30 Welcome Reception
COST Chemistry D36 3rd Workshop and 5th Management Committee Meeting
10 Benahavís (Málaga, Spain), 21st to 23rd of October, 2009
Thursday 22 nd
9:00 – 9:45 Keynote 2 Chair: Robert Schoonheydt, Catholic University of Leuven (Belgium)
K2 Venceslav Kaucic, National Institute of Chemistry (Slovenia) “Microporous and Mesoporous Materials”
9:45 – 11:05 Oral Session 2 Chair: Sven Jaras, Chemical Technology, KTH (Sweden)
O7 Monica Calatayud, Université Pierre et Marie Curie (France) “Glycerol etherification over alkaline earth metal oxides” O8 Izabela Sobczak, Adam Mickiewicz University (Poland) “Glycerol oxidation on gold catalysts supported on group five metal oxides –a comparative study with other metal oxide and carbon based catalysts” O9 Andrei Parvulescu, Utrecht University (Netherlands) “Etherification of Glycerol and Other Biomass-Derived Polyols: New Routes to Valuable Bulk Chemicals” O10 Rafael Mariscal, Instituto de Catálisis y Petroleoquímica (Spain) “Relevance of the physicochemical properties of CaO catalyst for the methanolysis of triglycerides to obtain biodiesel”
11:05 – 11:35 Cofee Break 11:35 – 13:15 Oral Session 3 Chair: Tomás Cordero, Universidad de Málaga (Spain)
O11 J. Ángel Menéndez, Instituto Nacional del Carbón (Spain) “Influence of porosity and surface groups on catalytic activity of carbon materials for the microwave-assisted CO2 reforming of CH4” O12 Frederik Tielens, Université Pierre et Marie Curie (France) “Niobium Oxide Species in and on Silica Materials; a Molecular Picture” O13 Enrique Rodriguez-Castellón, Universidad de Málaga (Spain) “Study of Nanoporous Catalysts in the Selective Catalytic Reduction of NOx” O14 Anna M. Venezia, ISMN CNR (Italy) “New HDS catalysts supported on thiol functionalized mesoporous silica” O15 Ángel Landa-Cánovas, Instituto de Ciencia de Materiales de Madrid (Spain) “Structural Flexibility in ~SbVO4”
13:15 – 14:15 Lunch 14:45 Visit to Ronda
COST Chemistry D36 3rd Workshop and 5th Management Committee Meeting
Benahavís (Málaga, Spain), 21st to 23rd of October, 2009 11
Friday 23 rd
9:00 – 9:45 Keynote 3 Chair: José Rodríguez-Mirasol, Universidad de Málaga (Spain)
K3 Jean Michel Léger, Université de Poitiers (France) “Carbon powder as conducting supports for electrocatalysts in low temperature fuel cells”
9:45 – 11:05 Oral Session 4 Chair: Guido Mul, Delft University of Technology (Netherlands)
O16 Álvaro Colina, Universidad de Burgos (Spain) “Synthesis of Pt nanoparticles on poly(3,4-ethylenedioxythiophene) modified electrodes for the electrocatalysis of Methanol” O17 David Fermin, University of Bristol (UK) “Electrochemical Hydrogen Loading in Ultrathin Assemblies of Au-Pd Nanostructures” O18 Atilla Cihaner, Atillim University (Turkey) “One More Step Closer to Realizing the Dream of the Polymeric RGB Electrochromics” O19 László Guczi, Chemical Research Center (Hungary) “Modelling of Au/FeOx interface by in situ Sum Frequency Generation Technique”
11:05 – 11:35 Cofee Break 11:35 – 13:15 Oral Session 5 Chair: Jean Michel Léger, Université de Poitiers (France)
O20 Nikolaos Tsiouvaras, Instituto de Catálisis y Petroleoquímica (Spain) “The effect of the Mo precursor on the nanostructure and activity of PtRuMo electrocatalysts for Proton Exchange Membrane Fuel Cells” O21 Luisa Maria Abrantes, Universidade de Lisboa (Portugal) “Electrocatalytic activity of polypyrrole films incorporating palladium particles“ O22 Hubert Girault, Ecole Polytechnique Federale de Lausanne (Switzerland) “Bio-inspired electrochemistry: From oxygen reduction to hydrogen evolution at soft interfaces.” O23 Stanislas Zalis, Heyrovski Institute Prague (Czech Republic) “Density functional and electrochemical studies of the catalytic ethylene oxidation on nanostructured Au and Pt electrodes.” O24 Sergio García, Instituto de Catálisis y Petroleoquímica (Spain) “An FTIR study of ternary PtSn-Rh/C for ethanol electrooxidation: effect of surface composition “
13:15 – 14:30 Lunch
COST Chemistry D36 3rd Workshop and 5th Management Committee Meeting
12 Benahavís (Málaga, Spain), 21st to 23rd of October, 2009
14:45 – 15:30 Keynote 4 Chair: Viorica Parvulescu, Institute of Physical Chemistry I.G. Murgulescue (Romania)
K4 Jaques Fraissard, Laboratoire de Physique Quantique – ESPCI (France) “NMR of physisorbed 129Xe used as a probe to investigate porous solids”
15:30 – 17:10 Oral Session 6 Chair: Maria Ziolek, Adam Mickiewicz University (Poland)
O25 Volker Ribitsch, Universitat Graz (Austria) “Adsorption of proteins on DLC surfaces“ O26 M. Rosa Infante, Instituto de Química avanzada de Cataluña (Spain) “Amino Acid-Based Biocompatible Surfactants” O27 Bjšörn Lindman, University of Lund (Sweden) “Interactions of DNA with cationic surfactants and proteins: Gels, gel nano-particles, microstructure and phase separation” O28 Eduardo Marques, University of Porto (Portugal) “Symmetry-asymmetry effects on the self-assembly of ion-paired surfactant systems” O29 Julian Ross, University of Limerick (Ireland) “Formic Acid as a Hydrogen Source for Vapor Phase Catalytic Reactions”
COST Chemistry D36 3rd Workshop and 5th Management Committee Meeting
Benahavís (Málaga, Spain), 21st to 23rd of October, 2009 13
Poster Contributions
1. Blanco, Gema Instituto de Catálisis y Petroleoquímica (Spain) “Silylation of functionalized commercial silica for the direct synthesis of hydrogen peroxide solution” 2. Zgrablich, Jorge Instituto de Física Aplicada (INFAP) (Argentina) “Attempts to Understand the Enantioselectivity of Chiral Propylene Oxide Adsorption on NEA-Modified Pt Surfaces” 3. Pinazo, Aurora Instituto de Química avanzada de Cataluña (CSIC) (Spain) “Argine-based surfactants: mixtures with 1,2 dipalmitoyl-sn-glycerol-3-phosphate monosodium salt” 4. Pinazo, Aurora Instituto de Química avanzada de Cataluña (CSIC) (Spain) “Lysine-based cationic surfactants: synthesis and study of the effect of the polar group on their biological properties” 5. Pons, Ramon Instituto de Química avanzada de Cataluña (CSIC) (Spain) “Mono acyl lysine based surfactants: self-aggregation” 6. Ivanov, Ivan Institute of Catalysis (BAS) (Bulgary) “Gold supported on ceria doped by Me3+ (Me=Al and Sm) for water gas shift: influence of dopant and preparation method” 7. Andreeva, Donka. Institute of Catalysis (BAS) (Bulgary) “Redox activity of gold-molybdena catalysts: influence of the preparation method” 8. Iliopoulou, Eleni F. CPERI/CERTH (Greece) “FTIR accessibility studies of 2,6 DTBPy adsorption on FCC catalysts” 9. La Mesa, Camilo Sapienza University (Italy) “Supramolecular Assemblies in Association Colloids: from dilute to concentrated regimes” 10. Trejda, Maciej Adam Mickiewicz University (Poland) “New V, Nb, Ta – FAU zeolites – texture and surface properties” 11. Ruíz-Rosas, Ramiro Universidad de Málaga (Spain) “Lignin-based electrospun carbon microforms” 12. Bedia, Jorge Universidad de Málaga (Spain) “2-propanol decomposition on carbon based acid and basic catalysts” 13. Valero-Romero, M. José Universidad de Málaga (Spain) “Catalytic and non-catalytic hydrothermal carbonization of hemp biomass: the carbonaceous product“ 14. Rosas, Juana M. Universidad de Málaga (Spain) “Surface chemistry modification of carbon supported chromium catalysts alter no reduction by XPS analyses“ 15. Pantaleo, Giuseppe ISMN-CNR (Italy) “CH4 combustion activity of Pd catalysts supported on TiO2 incorporated mesoporous SiO2 (SBA-15 and HMS)” 16. Edolfa, Kristine Latvian Institute of Organic Synthesis (Latvia) “Ketonization of aliphatic acids over zinc chromite catalyst” 17. Liotta, Leonarda ISMN-CNR (Italy)
COST Chemistry D36 3rd Workshop and 5th Management Committee Meeting
14 Benahavís (Málaga, Spain), 21st to 23rd of October, 2009
“Supported gold catalysts for Preferential oxidation (PROX) of CO in the presence of excess H2” 18. Pospisil, Lubomir J. Heyrovsky Institute of Physical Chemistry (Czech Republic) “Structure-Reactivity Relationships in ElectronTransfers of Helical Polyaromatic Dications” 19. Mores, Davide Utrecht University (Netherlands) “Coke formation during the Methanol-to-Olefin Conversion: Space- and Time-resolved In-Situ Spectroscopy on H-SAPO-34 and H-ZSM-5” 20. Tirkes, Seha Atilim University (Turkey) “A Neutral State Green Polymeric Electrochromic Based on Acenaphtho[1,2-b]quinoxaline and EDOT” 21. Boghosian, Soghomon University of Patras (Greece) “Molecular structure and reactivity of MoO3/TiO2 catalysts for ethane oxidative dehydrogenation studied by operando Raman spectroscopy“ 22. López-Medina, Ricardo Instituto de Catálisis y Petroleoquímica (Spain) “Nanostructured MoVNbTeO Oxide Catalysts for Selective Oxidation Reactions” 23. Mikolajska, Ewelina Joanna Instituto de Catálisis y Petroleoquímica (Spain) “Operando Studies of VPO catalysts in n-butane selective oxidation reaction. Activity, selectivity and structure transformations” 24. Tielens, Frederik Université Pierre et Marie Curie (France) “Theoretical Study of Thiol Self Assembled Monolayer Formation on Au(111) surfaces” 25. Rojas, Elizabeth Instituto de Catálisis y Petroleoquímica (Spain) “Theoretical Investigation of the Ammonia Adsorption Process on (110)-VSbO4 Surface” 26. Wolfgang, Grünert Ruhr-Universität Bochum (Germany) “Peculiar response of V2O5-WO3/TiO2 DeNOx catalysts to thermal stress an investigation with catalytic and spectroscopic tools“ 27. Zhang, Yongmin Université Pierre et Marie Curie (France) “Synthesis of novel 2:1 permethylated b-cyclodextrin-fullerene conjugates “ 28. Nervi, Carlo Dipartimento di Chimica IFM (Italy) “Electrochemical Functionalization of Glassy Carbon Electrode Surfaces by Organometallic Moieties” 29. Hromadová, Magdaléna J. Heyrovský Institute of Physical Chemistry of ASCR (Czech Republic) “Self–assembled monolayers of atrazine–based thiolates and their interaction with anti–atrazine antibody” 30. Morán, Carmen University of Coimbra (Portugal) “DNA gel particles from single and double-tail surfactants” 31. Dias, Rita University of Coimbra (Portugal) “Adsorption of macromolecules to responsive surfaces” 32. Mendez, Manuel Ecole Polytechnique Fédérale de Lausanne (Switzerland) “Proton Coupled Oxygen Reduction at Liquid-Liquid Interfaces Catalyzed by Cobalt Porphine” 33. Boutonnet, Magali KTH Chemical Science and Engineering (Sweden) “Synthesis of crystalline CeO2 nanoparticles by a novel oil-in-water microemulsion reaction method and its use as catalyst support”
COST Chemistry D36 3rd Workshop and 5th Management Committee Meeting
Benahavís (Málaga, Spain), 21st to 23rd of October, 2009 15
34. Beck, Andrea Chemical Research Center (Hungary) “The effect of preparation method on the formation of highly active Au-promoter oxide perimeter in promoted Au/SiO2 catalysts” 35. Benko, Timea Chemical Research Center (Hungary) “TiO2 and CeO2 promoted Au/SBA-15 in propene total oxidation” 36. Hernandez-Alonso, M. Dolores CIEMAT-PSA (Spain) “Selective photo-oxidation of cyclohezane on TiO2: the role of surface characteristics“ 37. Alekseev, Sergyi Kiev University (Ucrania) “Porous silicon with gold nanoparticles as laser desorption/ionization mass spectrometry platform” 38. Gerda, Vasilyi Kiev University (Ucrania) “Matrix synthesis and functionalisation of the ordered mesoporous carbon by palladium nanoparticles as potential sorbent for hydrogen storage” 39. Syzgantseva, Olga Université Pierre et Marie Curie (France) “Theoretical studies of hydrogen adsorption mechanism on ZrO2”
Abstract The selective oxidative functionalization of short chain paraffins is a formidable challenge for the sustainable use of alkanes as feedstock. The incorporation of several functions in an adequate structure seems to be the way to the development of active and selective catalysts for partial alkane oxidations. Nevertheless, despite there are some achievements, several aspects are still not solved for an industrial applicability, as the selectivity to partial oxidation products. This paper will present an overview on the synthesis, characterization and catalytic behaviour of multicomponent metal oxides, with special attention to metal oxidic bronzes and molecular sieves, as active and selective catalysts for the gas phase partial oxidation of hydrocarbons. Recent examples on the new synthetic procedures and new structures will be also discussed. Introduction The selective oxidative functionalization of short chain paraffins is a formidable challenge for the sustainable use of alkanes as feedstock and has attracted special attention during the last two decades [1]. Two strategies have been mainly developed in alkane oxidation: i) the oxidative dehydrogenation to achieve olefins and ii) the direct oxidation of alkanes to O- or N-containing products. However, only the oxidation of n-butane to maleic anhydride is industrially applied. The oxidative dehydrogenation of short chain alkanes has been extensively studied because it is a very attractive way for alkane functionalization. Mixed metal oxides and metal containing molecular sieves have been proposed as active and relatively selective catalysts in the activation of alkanes. Although the use of N2O rather than oxygen could improve the selectivity to olefins, these catalysts cannot be considered as competitor of steam cracking technologies. Only a few catalytic systems for ethane oxydehydrogenation could have some interest from an industrial point of view. The second way for the functionalization of alkanes could be to replace olefins by alkane due to its low cost. Although a first approach could be to integrate a first dehydrogenation reactor to the conventional olefin oxidation process, the research effort in the last years is being carried out towards the direct oxidation (in one stage) of propane since this could permit the reduction of the reaction steps. Multicomponent mixed metal oxides, MoVTe(Sb)NbO catalysts, reported by Mitsubishi in the early 1990s, seem to be promising in the (amm)oxidation of propane [2] and in the oxidative dehydrogenation of ethane to ethylene [3]. Typically, the most efficient MoVTe(Sb)NbO catalysts present at least two crystalline phases [4, 5]: (i) an orthorhombic (AO)2−2x(A2O)nM20O56 (A = Te or Sb and M =Mo, V, Nb), the so-called M1 (isostructural with Csx(Nb,W)5O14) and (ii) an orthorhombically distorted Te0.33MO3.33 or (Sb2O)M6O19 phase (M = Mo, V, Nb), the so-called M2. In addition, TeMo5O16 (or Sb4Mo10Ox), (V,Nb)-containing Mo5O14, and/or tetragonal bronzes may be present, depending on the catalyst composition and the catalyst preparation procedure. However, the M1 phase itself seems to be active and selective on the partial oxidation of propane and ethane, while M2 is only active and selective in the oxidation of propene to acrolein and/or acrylic acid. A certain composition range seems to be necessary to achieve the best catalytic performance, since the formation of Te2M20O57-type phase strongly depends on both the catalyst composition and the catalyst preparation method, post-synthesis treatment should be also considered in order to prepare effective catalysts [5]. The reported results suggest a molecular structure-performance relationship at the surface of functional materials [4].
20 Benahavís (Málaga, Spain), 21st to 23rd of October, 2009
On the other hand, the different catalytic behaviour of these phases can be explained on the basis of crystal structure, MO6 octahedra form pentagonal, hexagonal and heptagonal channels in M1-phase and only hexagonal channels in M2-phase. More recently, new oxidic bronzes and synthesis strategies have been proposed. A0.5[Mo5-a-bVa
4+XbO14] (A = Rb, Cs, X = no element, Nb, Ta,W, Sb, Bi, Se, Te) with a M1-type structure [6], PMo(W)VNbO mixed oxides with a tetragonal tungsten bronze structure (TTB) [7], or new synthesis procedures in the preparation of other Mo-bronzes [8], or Nb,Mo-containing mesoporous materials [9] have been reported, in which a clear structure-behaviour relationship can also be proposed. Recently it has been proposed that some of these structures should be considered as microporous materials [10] and their catalytic performance is discussed in terms not only of the chemical composition (bulk and surface) but also in terms of catalyst structure (including the nature and size of channels in this type of materials). Acknowledgments Financial support was provided by the DGICYT of Spain (project CTQ2006-09358-BQU). References [1] J.M. López Nieto, Top. Catal. 41 (2006) 3. [2] a) M. Hatano, A. Kayo. EP 318285B1 (1988); b) T. Ushikubo, K. Oshima, A. Kayou, A, T. Umezawa, K. Kiyono, I. Sawaki, EP529853 A2 (1993). [3] a) J.M. López Nieto, P. Botella, M.I. Vázquez, A. Dejoz, WO Pat 0346035 (2003); b) J.M. López Nieto, P. Botella, M.I. Vázquez, A. Dejoz, Chem. Commun. (2002) 1906. [4] a) J.M.M. Millet, H. Roussel, A. Pigamo, J.L. Dubois, J.C. Jumas, Appl. Catal. A: Gen. 232 (2002) 77; b) H. Tsuji, K. Oshima, Y. Koyasu, Chem Mater. 15 (2003) 2112; c) P. DeSanto, D.J. Buttrey, R.K. Grasselli, C.G. Lugmair, A.F. Volpe, B.H.Toby, Topics Catal. 23 (2003) 23. [5] a) P. Botella, E. García-González, J.M. López Nieto, J.M. González-Calbet, Solid State Sciences 7 (2005) 507; b) A.C Sanfiz, T.W. Hansen, A. Sakthivel, A. Trunschke , R. Schlogl, A. Knoester, H.H. Brongersma, M.H. Looi, S.B.A. Hamid, J. Catal. 258 (2008) 35. [5] F. Ivars, B. Solsona, E. Rodríguez-Castellón, J.M. López Nieto J. Catal. 262 (2009) 35. [6] H. Hibst, F. Rosowski, G. Cox, Catal. Today 117 (2006) 234. [7] P. Botella, B. Solsona, E. García-González, J:M. M. González-Calbet, J.M. López Nieto, Chem Comm. (2007) 5040. [8] a) M. Sadakane,N. Watanabe, T. Katou, Y. Nodasaka, W. Ueda, Angew. Chem. Int. Ed. 46 (2007) 1493; b) N. R. Shiju, V.V. Guliants, ChemPhysChem 8 (2007) 1615. [9] L. Yuan, S. Bhatt, G. Beaucage, V.V. Guliants, S. Mamedov, R.S. Soman J. Phys. Chem. B, 109 (2005) 23250. [10] M. Sadakane, K. Kodato, T. Kuranishi, Y. Nodasaka,K. Sugawara, N. Sakaguchi, T. Nagai, Y. Matsui, W. Ueda, Angew. Chem. Int. Ed. 47 (2008) 2493.
Benahavís (Málaga, Spain), 21st to 23rd of October, 2009 21
Microporous and Mesoporous Materials
Venčeslav Kau čič
National Institute of Chemistry and University of Ljubljana, Hajdrihova 19, 1000 Ljubljana, Slovenia
Summary Transition metal-modified microporous zeolitic materials (silicate- and phosphate-based) are attractive catalysts due to their hydrothermal stability and high catalytic activity and selectivity. Metal-modified mesoporous materials with larger pore openings have been developed for catalytic processes where larger molecules are involved. The inclusion of nanosized particles of zeolitic microporous materials with larger external surface areas and high surface activity into mesoporous matrices, i.e. the preparation of microporous/mesoporous composites, substantially enhances the catalytic activity of mesoporous materials. The important feature of nanoporous solids based on various metal oxides is also their ability to form thin films with nanometer-scale thickness. Examples of successful preparation and/or functionalisation of new nanoporous solids encompass microporous and mesoporous silicates (MnS-1, MnMCM-41, MnTUD-1), microporous and mesoporous aluminophosphates (FeAPO-36, FeHMA), microporous/mesoporous silicate composites ((Ti,Al)-Beta/MCM-41, (Ti,Al)-Beta/MCM-48, Ti-Beta/SBA-15) as well as cubic mesoporous aluminophosphate thin films. Studies of structure-property relations of new solids have included X-ray diffraction, spectroscopic (XAS, NMR) and electron microscopy characterisation techniques. Porous materials are classified into three categories, microporous with pore openings from 0.3 to 2 nm, mesoporous having pores between 2 and 50 nm, and macroporous with pores greater than 50 nm. Microporous materials are exemplified by crystalline framework solids such as zeolites (aluminosilicates), whose crystal structure defines channels and cages, i.e. micropores, of strictly regular dimensions. Mesoporous materials, exemplified by the silicate MS41 materials family, are amorphous solids exhibiting highly-ordered pore structures and large internal surface areas. Microporous materials are generally prepared hydrothermally from aqueous gels containing a source of the framework building elements (Si, Al, P, etc.), a mineraliser (OH-, F-) regulating the dissolution/condensation processes during the crystallization, and a structure-directing agent, usually an organic amine or ammonium salt. Transition metals can be incorporated into microporous or mesoporous materials by a post-synthetic ion-exchange treatment or by direct framework substitution by the addition of transition metal cations into the synthesis gel. An alternative to a classical hydrothermal synthesis is a microwave oven. The microwave heating is regarded as a novel synthesis tool for microporous and mesoporous materials because it offers several benefits, such as homogeneous nucleation, the promotion of faster crystallisation, rapid synthesis, the formation of uniform crystals, and small crystallites, facile morphology control, the avoidance of undesirable phases by shortening the synthesis time and so on. Recently, it was found that it provides an effective way to control the particle size distribution, crystal morphology, orientation, and even the crystalline phase. Microporous materials are mostly used as heterogeneous acid- and redox catalysts in petroleum industry and in the production of chemicals for various types of shape-selective conversion and separation reactions. The most common reactions, where microporous acid-catalysts are involved, are fluid catalytic cracking, hydrocracking, aliphate alkylation, isomerisation, transformation of aromatics and the conversion of methanol to hydrocarbons. Redox microporous catalysts are also increasingly used for a variety of selective oxidations of various substrates of synthetic hydrocarbons, alcohols, and amines since these reactions can be performed under mild conditions in the liquid phase. An illustrative example is the clean production of adipic acid that is used in the production of nylon with the direct oxidation of cyclohexene with aqueous H2O2 using Ti- or Fe-substituted microporous catalysts. The discovery of mesoporous silicates attracted worldwide attention since they can incorporate relatively large-sized species inside the pores. The extensive research to expand their
22 Benahavís (Málaga, Spain), 21st to 23rd of October, 2009
functionality and improve their hydrothermal and chemical stability by modified and optimised synthetic or post-synthetic routes in recent years has already enabled their application in the field of catalysis. Intensive research efforts have also been driven by the emerging applications such as biosensors, drug delivery, gas separation, energy storage and fuel cell technologies. Investigations in the filed of mesoporous thin films are uprising fast due to their potential applications as chemical and optical sensors, shape-selective membranes and energy-storage devices. The incorporation of transition metals into silicate, aluminophosphate and similar inorganic frameworks generates or moderates catalytic activity of the materials. Here we report on synthesis and structural studies of new micro- and mesoporous materials with the emphasis on the preparation of metal-modified nanosized zeolitic particles, microporous/mesoporous composites and zeolitic thin films.The elucidation of structures of ordered porous materials is essential for the understanding and prediction of their macroscopic physical and chemical properties. In particular, the size and connectivity of the pores determine their molecular sieving capability. The coordination, location, oxidation state and strength of bonding of the divalent and other transition metal ions in materials are directly related to their activity/selectivity in catalytic and other reactions. The conventional single-crystal and powder diffraction methods have been successfully used for structure determinations of crystalline microporous structures. Problems that can arise are mainly due to the small size of the crystallites that often require ab initio powder structure solutions and the low concentration and/or random distribution of metal active sites over the framework or extra-framework positions. The rapid development of synchrotron radiation sources has brought around a tremendous progress in XRD techniques and methods, e.g. anomalous dispersion methods for metal site determination. With the availability of synchrotron radiation sources, X-ray absorption spectroscopy (XAS) techniques have also developed into a widely used tool for structural research of ordered porous materials. XAS analytical methods XANES and EXAFS provide structural information about local symmetry and the average oxidation number of selected atom. Since XAS is selective towards a particular element and sensitive only towards a short-range order, it is one of the most appropriate spectroscopic tools for microporous and mesoporous catalysts characterization. Combining in situ XRD and XAS is an excellent approach to obtain information on reaction-dependent changes of both long-range crystallographic order (XRD) as well as oxidation state and local coordination environment of particular elements (XAS) in a solid catalyst. Nuclear magnetic resonance spectroscopy also offers a wealth of information on structural and dynamical properties of crystalline- as well as amorphous porous materials. The positions and local environments of framework and extra-framework atoms of porous solids can be determined by either studying NMR spectra of 29Si, 27Al, 31P, 69Ga or 71Ga nuclei, or nuclei of charge-compensating ions like 1H, 23Na, 7Li or 133Cs.
Benahavís (Málaga, Spain), 21st to 23rd of October, 2009 23
Carbon powder as conducting supports for electrocat alysts in low temperature fuel cells
Jean-Michel LEGER
Laboratory of Organic Chemistry (LACCO), CNRS-University of Poitiers, 40 Avenue du Recteur Pineau, 86000 Poitiers France
[email protected] Proton Exchange Fuel Cells (PEMFC) are now considered as the most convenient fuel cells for application in a large range of power densities. Applications from micro fuel cells (electronic devices) mid-sized fuel cells to automotive applications are scheduled in the future. However, PEMFC, which work at low temperatures (from room temperature to 100 °C) and in acidic environment (protonic electrolytic membrane) need the development of convenient electrocatalysts. This means catalysts leading to acceptable performances (kinetically speaking) and with a good stability with time. Suitable catalysts are generally noble metals, mainly platinum, possibly modified by other metals or oxides. Due to the costs of platinum, it is obvious that the total amount of noble metal need to be limited for large scale applications. An electrocatalytic reaction is a reaction taking place at the catalyst (electrode) surface. Then the only way to increase the overall rate of the reaction is to increase the active surface of the catalyst. This can be obtained by decreasing the size of the metallic particles of the catalysts. The key problem is then to have an optimized utilization of these particles and it is one of the key roles of the supporting material used in the construction of electrode for fuel cells. Carbon is actually the unique conducting material used for this application, even if some other alternative are explored, for example with oxides. The main key property of the carbon powder for fuel cell is the electrical conductivity. Different preparation procedures are proposed to increase it before the preparation of the catalytic layer itself. The second key point concerns the ability of the catalytic particle to be fixed on the carbon powder surface. This is important to have the highest possible utilization of the catalyst (agglomeration of particles should be limited for example), but also if we considered the stability with time. If the mobility of particle is too high at the carbon surface, fritting and agglomeration of metallic particles occur leading to a decrease of the active area and of the performances of the fuel cell. Another problem concerns the chemical degradation of the carbon materials under the working conditions. The presence of a catalyst such as platinum and of oxygen (cathodic side) can lead to the chemical oxidation of carbon (to produce CO2). This is observed during long term experiment with significant degradation of the carbon layer and consequently migration of platinum particles though the electrolytic membrane. The purpose of this keynote lecture is to discuss of these different points in relation with the use of carbon powder as supporting materials for catalysts in fuel cells. Several examples of the preparation of electrocatalysts and how to put and maintain them at the carbon powder surface will be given. It is important to understand that the preparation procedures are critical. These techniques can be purely chemical (colloidal precursors; micro-emulsion…), electrochemical (electrodeposition…), physical (plasma…). The pretreatment of the carbon powder is also a key point, mainly for the optimization of the utilization of the catalyst. It consist mainly developing procedures to increase the concentration of oxidized sites at the carbon powder surface. These sites allow then a strong interaction with the metallic particles and limit their mobility. Even if some people from fuel cell development still consider that carbon supporting materials are only a secondary problem, less essential than catalyst or membrane for example, it is obvious that the interactions between catalysts and carbon are extremely important. The best catalyst not stabilized at the carbon surface leads always to low performances.
24 Benahavís (Málaga, Spain), 21st to 23rd of October, 2009
References [1] C. Lamy, J-M. Léger , S. Srinivasan, Direct Methanol Fuel Cells: From a 20th Century Electrochemist's Dream to a 21st Century Emerging Technology, in Modern Aspects of Electrochemistry, J'O.M. Bockris, B. E. Conway and R. White (Eds), Kluwer Academic/Plenum Publishers (New-York), vol. 34, (2001) p 53-118. [2] J-M. Léger, C. Coutanceau, C. Lamy, Electrocatalysis for Direct Alcohol Fuel Cell, in “Fuel Cell Catalysis: a surface science approach”, M.T.M. Koper (Ed), J. Wiley & Sons, New Jersey, chap 11 (2009) 343-373. [3] C. Coutanceau, S. Brimaud, C. Lamy, J.-M. Léger, L. Dubau, S. Rousseau, F. Vigier, Review of different methods for developing Nanoelelectrocatalysts for the oxidation of organic compounds, Electrochim. Acta, 53 (2008) 6865. [4] C. Grolleau, C. Coutanceau, F. Pierre, J.M. Léger, Effect of potential cycling on structure and activity of Pt nanoparticles dispersed on different carbon supports, Electrochim. Acta, 53 (2008) 7157. [5] P. Brault, S. Roualdes, A. Caillard, A.-L. Thomann, J. Mathias, J. Durand, C. Coutanceau, J.-M.Leger,C. Charles R. Boswell,Solid polymer fuel cell synthesis by low pressure plasmas: a short review, Eur. Phys. J. Appl. Phys. 34 (2006) 151.
Benahavís (Málaga, Spain), 21st to 23rd of October, 2009 25
NMR of physisorbed 129Xe used as a probe to investigate porous solids
Jacques Fraissard
University P. and M. Curie, ESPCI, Laboratory ˝ Physique Quantique˝, 10 rue Vauquelin, 75231 Paris, France
The fundamental idea was to find a chemically inert molecule, detectable by NMR and particularly sensitive to physical interactions with other species, which could be used as a probe to determine the properties of its environment [1]. The 129 xenon isotope is this ideal probe. Chemical shifts and relaxation times of xenon are solely affected by intermolecular interactions and are exquisitely sensitive to the atom’s surrounding. This sensitivity to its environment means that the Xe nucleus can report on a wide variety of attributes of the physical systems in which it finds itself: gas, liquids, cages in a zeolite, nanochannels in a molecular solid, clathrates, proteins in solution, amorphous polymers, etc. It can be used also for imaging and gas diffusion measurements. Several reviews have been published on these applications [2-3]. By using optical polarization techniques [4] the sensitivity of detection can be increased by several orders of magnitude and is particularly useful for several studies (porous materials, microimaging, polymers and elastomers, etc.). We will present some examples of the applications of the Xe-NMR technique to the characterization of microporous and mesoporous solids, including carbon nanotubes. We will add also few words about the characterization of solid polymers and proteins interactions.
References [1] T. Ito and J. Fraissard, Proceedings of the 5th International Zeolite Conference, Naples, 1980 L.V.C. Rees (ed.), Heyden, London, 1980, p. 510. [2] D. Raftery, B.F. Chmelka, NMR Basic Principles and Progress. B. Blümich, Ed ; Springer-verlag, Berlin, Heidelberg, 30 (1994) 111. [3] J.L. Bonardet, J. Fraissard, A. Gedeon, M.A. Springuel-Huet, Catal.Rev.-Sci.Eng., 41(2) (1999) 115. [4] D. Raftery, H. Long, T. Meersmann, P.J. Grandinetti, L. Revey and A. Pines, Phys. Rev.Lett.,66 (1991) 584.
Benahavís (Málaga, Spain), 21st to 23rd of October, 2009 29
The effect of porosity of niobosilicate supports an d VSbOx loading on the ammoxidation of propane
H. Golinska a,b, E. Rojas a, R. Lopez-Medina a, M. Ziolek b,*, Miguel A. Bañares a,
M.O. Guerrero-Pérez c,*
a Catalytic Spectroscopy Laboratory, Instituto de Catálisis y Petroleoquímica; CSIC; Marie Curie 2; E-29049-Madrid (Spain); b Adam Mickiewicz University, Faculty of Chemistry, Grunwaldzka 6,
60-780 Poznan, Poland; cDepartamento de Ingeniería Química. Universidad de Málaga; E-29071-Málaga (Spain)
Introduction Vanadium-antimony oxides are well known as catalysts for selective oxidation and ammoxidation reactions [1,2]. The activity and selectivity of catalysts in these processes are greatly dependent on the loading (Sb:V atomic ratio), method of preparation, gas phase composition during the thermal treatment, and the nature of the support [3]. Moreover, the role of the nature of antimony complex used during the preparation of the catalysts was stressed [4,5]. Sb-V-Ox catalysts with an excess of V are highly active and selective for propane oxidative dehydrogenation while an excess of Sb affords Sb-V-Ox catalyst more efficient for propane ammoxidation [6,7]. The idea of this work was to use niobosilicate supports exhibiting different porosity (mesopores or macropores) as supports for VsbOx binary oxides introduced with step by step impregnation. The effect of porosity, vanadium-antimony oxides loading, and the sequence of the impregnation (first vanadium next antimony or reverse) on the effectivness in ammoxidation of propane has been studied. Experimental Two niobosilicate supports were synthesized: mesoporous NbMCM-41 (denoted NbM; Si/Nb=64) and macroporous SiNbOx. They were impregnated stepwise with antimony and vanadium precursors (NH4VO3 – BDH Chemicals Ltd. And (CH3COO)3Sb – Aldrich) using V/Sb atomic ratios of 1 or 0.5. and ~25 wt % of Sb. The other group of materials were prepared by the sequenced impregnation starting from vanadium and next antimony sources, and with the atomic excess of vanadium (3 wt % of Sb and 1.5 wt % of V). The samples used in the ammoxidation reactions and their texture parameters estimated by XRD and nitrogen adsorption, are shown in Table 1. The gas phase ammoxidation of propane in the temperature range of 623 – 773 K was studied on the prepared catalysts, which were characterized before and after reactions with Raman spectroscopy. Results and discussion The pristine NbMCM-41 material exhibits very well ordered hexagonal arrangement of mesopores of 2.2 nm diameter and high pore volume and surface area of ~1000 m2/g estimated from XRD and nitrogen adsorption measurements. The data in Table 1 show that the use of high loading of binary SbV oxides almost totally block mesopores in NbMCM-41 ordered mesoporous material and causes the dramatically decrease of the surface area and pore volume. The use of macroporous niobiosilica allow to leave 0.3 cm3/g free pore volume in the catalyst after VsbOx loading. These texture parameters determine the catalytic activity and selectivity in ammoxidation of propane (Table 2). The catalytic tests for acidity and basicity indicated that 0.5VSb/SiNbOx reveals lower acidity than 0.5VSb/NbM with the same oxides loading. This feature together with texture parameters cause a very high selectivity in the formation of acrylonitrile on 0.5VSb/SiNbOx catalyst.
30 Benahavís (Málaga, Spain), 21st to 23rd of October, 2009
Table 1 . Composition of the catalysts and texture parameters The order of V and Sb in the symbol of catalysts indicates the sequence of impregnation (e.g. SbV/M means the first vanadium was loaded and next antimony)
Table 2 . The results of ammoxidation af propene at 773 K
Taking into account the results of catalytic activity and TEM, SEM, XRD, Raman spectroscopy study one could define the following conclusions from this work.
• NbMCM-41 as support for Sb-V-Ox phase interacts strongly with vanadium when a low loading of V (1.5 wt.%) is used and vanadium is the first component introduced during the stepwise impregnation.
• The higher loading of antimony and vanadium (25 and 5 wt.% respectively) results in the formation of needle/stake Sb0.95V0.95O4 rutile crystals; the increase of vanadium content to 10 wt.% gives rise to the domination of plate shaped SbxVyO5 phase.
• The mesopores in NbMCM-41 modified by the high loading of Sb-V-Ox phases are almost completely blocked by the bimetallic oxides.
• The use of macroporous SiNbOx as the support for VsbOx phase leads to the higher selectivity in the formation of acrylonitrile.
Acknowledgements COST action D36, WG No D36/0006/06, the Polish Ministry of Science (Grant No. 118/COS/2007/03) and . Spanish Ministry of Science and Innovation (CTQ2008/02461/PPQ) are acknowledged for the financial support References [1] R. K. Grasselli, Catal. Today 49 (1999) 141. [2] S. Larrondo, B. Irigoyen, G. Baronetti, N. Amadeo, Appl. Catal. A, 250 (2003) 279. [3] G. Centi, S. Perathoner, F. Trifiro, Appl. Catal. A, 157 (1997). [4] M.O. Guerrero-Perez, M.A. Banares, Catal. Today 96 (2004) 265. [5] M.O. Guerrero-Perez, J.L. G. Fierro, M.A. Banares, Top. Catal. 41 (2006) 43. [6] M.O. Guerrero-Perez, J.L. G. Fierro, M.A. Banares, Catal. Today 78 (2003) 387.
Benahavís (Málaga, Spain), 21st to 23rd of October, 2009 31
Towards 4-way catalysis
James A Sullivan
UCD School of Chemistry and Chemical Biology, Belfield, Dublin 4, Ireland. [email protected]
Nox and Particulate Matter (PM) remain the two most intractable emissions from diesel engines and a goal of the automotive industry is to combine strategies for their removal into a single catalytic bed. Nox is a primary and secondary pollutant contributing directly to acid rain and causing respiratory problems while contributing indirectly to photochemical smog [1]. PM defaces urban environments, carry possible carcinogens that can lodge in the alveoli of the lung and contribute to global warming (through the reduction of the albedo of arctic ice) [2]. For the past 20 years emissions from gasoline powered vehicles have been deNOxed through reduction of Nox to N2 through reduction with CO and unburned hydrocarbons (HC) present in the exhaust mixture [3]. On diesel engines the net concentration of oxidants (NO / O2) is significantly greater than that of reductants (CO/ HC) and standard three way catalysts are unable to reduce Nox so other control strategies are required [4]. The most common and effective is the Nox Storage and Reduction (NSR) system in which NO is oxidised to NO2 over a Pt catalyst and subsequently this is stored on a Nox storage material (BaO) as a nitrate (Ba(NO3)2). Once the Nox trap is saturated, a pulse of hydrocarbons regenerates it, releasing and reducing NO2 and restarts the cycle [5]. Regarding PM control technologies, the current after-treatment system relies on a particulate filter which strains larger particles from the stream followed by oxidation either with O2 through a brief high temperature excursion or with NO2 (through a C(s) + NO2 CO + NO reaction) [6]. In systems where NO2 is used to combust the particulates a Pt catalyst is added to the formulation in order to catalyse NO (which is present in the exhaust mixture) oxidation to NO2. Note that this is the same first step that operates in the NSR system described above and therefore the combination of these two systems into one catalytic bed is a possibility. Such a combination would reduce the overall volume and mass of any after-treatment systems that a diesel exhaust would require and this would have knock on effects on the fuel efficiency (and therefore the CO2 emissions per km travelled) of the vehicle. Recently promotions in soot combustion have been reported in the presence of a Nox trap [7]. In the current work we have studied combinations of Nox trapping materials and Model PM in order to determine the mechanism of this reported promotional effect and we have also studied the effects of PM on the efficiency of a Nox trap. In the former case we have determined that the localised transient increase in NO2(g) upon periodic regeneration of the trap causes the promotional effect upon soot combustion [8] while in the latter case we have, using temperature programmed techniques, transient kinetic analysis and in-situ FTIR, demonstrated that the presence of PM decreases the efficiency of a Nox trap. The reason for the latter finding is a competition between the NO2 generated over Pt sites. In an NSR system this should adsorb on (and react with) the NOx trapping component to generate a surface nitrate. However, in the presence of PM the NO2 is reduced to NO (in the process of combusting PM) which cannot be trapped by the Nox storage component. This confirms that in an NSR system there is significant mobility in the NO2 generated through NO oxidation which in turn suggests that the contact between the NO oxidation component and the Nox trapping component of such systems is not crucial. References [1] W. Kenneth, C.F. Warner, “Air Pollution, its origin and control” Harper and Row Publishers Inc. 1976 [2] J. Hansen, L. Nazarenko, Proc. Natl. Acad. Sci. 101 (2004) 423–428. [3] K.C. Taylor, Catal. Rev. Sci. Eng., 35, (4), 457, 1993.TWC [4] K.C. Taylor, Cat. Sci. and Tech., Anderson, J.R., Boudart, M., Ed., Springer-Verlag, 5, (1984). [5] S. Poulston, R.R. Rajaram, Catal. Today 81 (2003) 603. [6] A.P. Walker, Top. Catal. 28 (1–4) (2004) 165–170. [7] F. Jacquot, J.-F. Brilhac, P. Phillips, at the 4th International Conference on Environmental Catalysis, Heidleberg, June, 2005 [8] JA Sullivan, O Keane, and A Cassidy, Applied Catalysis B: Environmental 75 (2007) 102–106.
Benahavís (Málaga, Spain), 21st to 23rd of October, 2009 33
Towards an optimization of MoVNbTe-catalysts for C3 -oxidation
G. Mestl
Süd-Chemie AG Propylene is one of the key building block petrochemicals used as feedstock for a variety of polymers and intermediates. Mayjor propylene derivatives include polypropylene, acrylonitrile, propylene oxide, cumene/phenol, oxo alcohols, acrylic acid, oligomers, and other miscellaneous intermediates used, in turn in a wide range of end-use applications including automotive, construction, consumer durables, packaging, and electronics. The global propylene demand grew form 16,4 million tons in 1980 to around 30 million tons in 1990, corresponding to an average annual growth of 6,2 percent. In the decade ending in 2000, the demand grew at an average rate of 5,7 percent per year, reaching 52 million tons. Now at the end of this decade, the propylene demand has reached about 81 million tons at a growth rate of about 5,3 percent per annum. Driven by high polypropylene and other propylene derivative demand, propylene growth rate will exceed ethylene growth rate (see Fig.1 [1]).
Based on announced cracker projects olefin expansions will fall short of increased propylene demand for next few years. Future additions of predominately gas based crackers in Middle East, motivated by low NGL prices will reduce worldwide average of propylene yield from steam cracking even further. Hence, propylene demand will remain strong. There will be an imbalance between ethylene and propylene growth rate creating a propylene supply gap. The Asian gap between supply and demand is substantial and is causing very strong propylene pricing. The acrylic acid, currently produced from propylene in a two step process, demand showed an annual growth of 4% through 2005 and the demand for acrylic acid is forecast to increase four percent per annum. Acrylate esters such as butyl, ethyl, ethylhexyl and methyl acrylates account for the majority of acrylic acid demand. These products are utilized as the acrylic monomer component in a variety of coatings, adhesives, paper and leather finishes and as co-monomers and property modifiers in plastics production. Gains will thus be stimulated by growth in demand for these end-use products, in particular industrial and specialty coatings, paper finishes and plastics additives. The producers are expected to focus their attention on the production of higher growth specialty acrylates (such as ethylene methyl acrylate); and acrylic acid polymers. The latter include superabsorbent polymers (SAPs) used in baby diapers, adult incontinence products and feminine hygiene products; water treatment polymers and detergent additives. Growth in acrylic acid demand necessitate plant expansions throughout the coming decade, strong demand for derivatives has led to very tight acrylic acid supplies on several occasions.
34 Benahavís (Málaga, Spain), 21st to 23rd of October, 2009
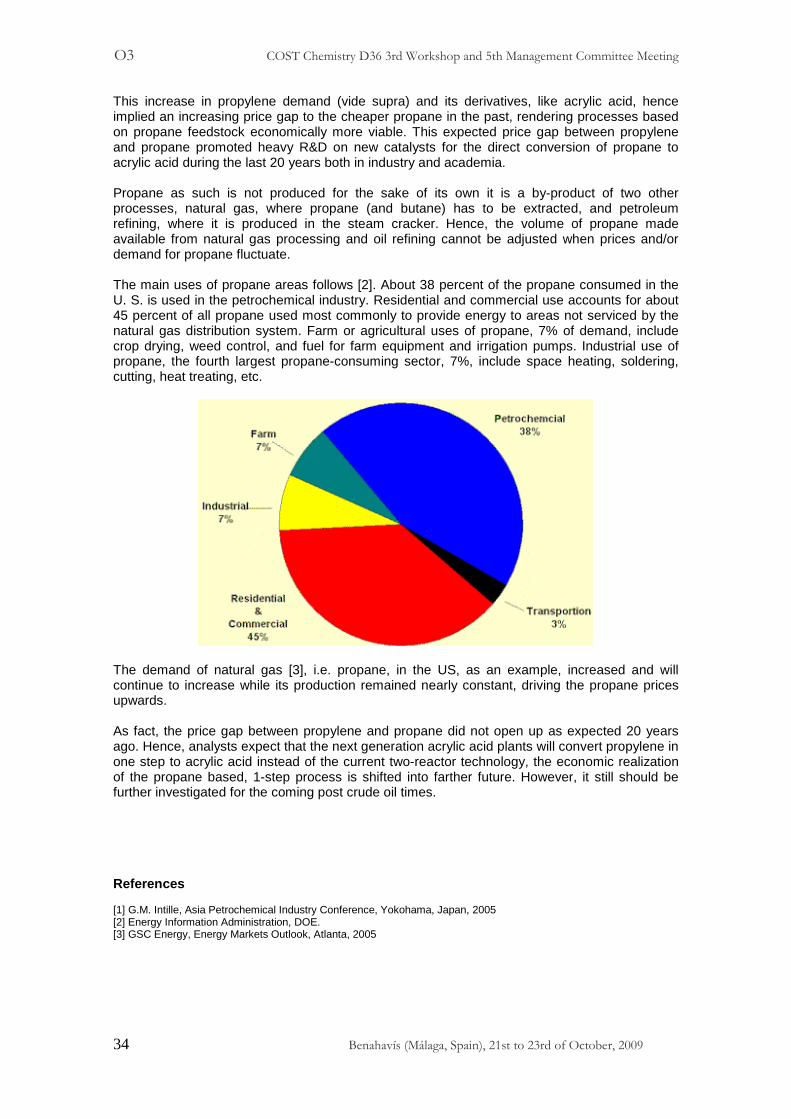
This increase in propylene demand (vide supra) and its derivatives, like acrylic acid, hence implied an increasing price gap to the cheaper propane in the past, rendering processes based on propane feedstock economically more viable. This expected price gap between propylene and propane promoted heavy R&D on new catalysts for the direct conversion of propane to acrylic acid during the last 20 years both in industry and academia. Propane as such is not produced for the sake of its own it is a by-product of two other processes, natural gas, where propane (and butane) has to be extracted, and petroleum refining, where it is produced in the steam cracker. Hence, the volume of propane made available from natural gas processing and oil refining cannot be adjusted when prices and/or demand for propane fluctuate. The main uses of propane areas follows [2]. About 38 percent of the propane consumed in the U. S. is used in the petrochemical industry. Residential and commercial use accounts for about 45 percent of all propane used most commonly to provide energy to areas not serviced by the natural gas distribution system. Farm or agricultural uses of propane, 7% of demand, include crop drying, weed control, and fuel for farm equipment and irrigation pumps. Industrial use of propane, the fourth largest propane-consuming sector, 7%, include space heating, soldering, cutting, heat treating, etc.
The demand of natural gas [3], i.e. propane, in the US, as an example, increased and will continue to increase while its production remained nearly constant, driving the propane prices upwards. As fact, the price gap between propylene and propane did not open up as expected 20 years ago. Hence, analysts expect that the next generation acrylic acid plants will convert propylene in one step to acrylic acid instead of the current two-reactor technology, the economic realization of the propane based, 1-step process is shifted into farther future. However, it still should be further investigated for the coming post crude oil times. References [1] G.M. Intille, Asia Petrochemical Industry Conference, Yokohama, Japan, 2005 [2] Energy Information Administration, DOE. [3] GSC Energy, Energy Markets Outlook, Atlanta, 2005
Introduction Despite numerous reports in the literature, the epoxidation of terminal alkenes remains a challenge in petrochemistry. Many different methods have been developed for the preparation of epoxides. Among the non-zeolitic substrates, Ti–SiO2-supported catalysts remain prominent for their effectiveness in the epoxidation of alkenes with organic hydroperoxides, though it is generally believed that they do not effectively epoxidize alkenes with hydrogen peroxide. Nevertheless, we have reported a very simple route for the preparation titanium-silica-supported catalysts which are very active and selective in the epoxidation of alkenes with hydrogen peroxide [1,2]. However, it has been reported that Ti-SiO2 samples show a lower intrinsic activity and lower selectivity toward the use of H2O2 for alkene oxidation than either TS-1 or Ti-β owing to their high hydrophilicity. It has been proposed that the hydrophilic/hydrophobic property of Ti zeolites plays an important role in their activity for liquid phase oxidations [1]. We have conducted silylation of Ti-SiO2 in order to enhance their activity in epoxidation with dilute H2O2 by increasing their hydrophobicity. Here, our objective is to improve the catalytic activity in the epoxidation of alkenes with H2O2 by silylation of Ti-containing amorphous silica. Experimental Methods Catalysts were prepared as follows: titanium isopropoxide (Aldrich, reagent grade) (0.65 g) was dispersed in 2-propanol (25 ml), the solution was heated to 353 K under stirring and then 5 g of silica (Grace Davison, XPO 2407) were added and the suspension was stirred for 2 h. The solid was filtered out and washed twice with 25 ml of 2-propanol, dried at 383 K, and finally calcined at 773 K for 5 h. Two silylant reagents: 1,1,1,3,3,3-hexamethyldisilanaze (HMDS) and tetramethyldisilazane (TMDS) were used for the silylation of the samples. The procedure was as follows: the silylant reagent fed continuously by a syringe pump to a continuous flow of N2 on the sample bed with a temperature of 473 K for 2 h, then a nitrogen flow was fed for 2 h. The silylation reagent/catalyst ratio was of 0,23. These solids were characterized by elemental analysis, DRS UV-Vis and X-ray photoelectron spectroscopy (XPS) techniques. The catalysts were used in the epoxidation of 1-octene and cyclohexene. In a typical run, a suspension of alkene (0.2 mol), tert-butanol (11 g) and 1 g of catalyst was heated at 333 K, and then 4 g of an organic solution of 5 wt % of H2O2 (in 1 t-butanol) were added to the reaction vessel. The organic compounds were analysed by GC-FID (Hewlett Packard 6890-plus, equipped with a HP-WAX capillary column). The hydrogen peroxide was measured by standard iodometric titration. Results and Conclusion The elemental analysis (Table 1) shows a higher amount of carbon deposited on the catalyst when TMDS is used. This observation indicates that silylation with TMDS is more effective than HMDS. This observation can be due to the higher volume of trimethylsilane groups than dimethylsilane. DRS UV-Vis spectra (Figure 1) of the samples are similar with only slight differences. The silylated samples showed an absorption peak centered at 220 nm, typical of isolated titanium in tetrahedral coordination [4]. The slight shift in band position and the increase in bandwidth in the spectrum of reference sample point to distorted tetrahedral environment of the titanium. Solid-state 29Si MAS-NMR (Figure 2) confirmed the presence of –SiCH3 groups bound to the surface of the samples. Distinct resonances can be clearly distinguished for the siloxane units [Q3 at ≈−104 ppm for Si(Osi)3(OH) units; Q4 at ≈−114 ppm for Si(Osi)4 units]. Only two signals are observed (Q4 and Q3) on the nonsilylated catalyst. After silylation, a new signal was detected at 12,5 ppm, which can be assigned to (CH3)3Si– (Osi) in the Cat-Sil-HMDS
36 Benahavís (Málaga, Spain), 21st to 23rd of October, 2009
sample and at 20,5 assigned to (CH3)2SiH– (Osi) in the Cat-Sil-TMDS sample. The Q3/Q4 ratio (Table 1) in the reference Catalyst was estimated as 0.081. After silylation this ratio decreases. The Cat-sil-TMDS sample exhibited the lowest ratio which indicates that the silylation coverage in this sample is higher than in the Cat-Sil-HMDS one.
Figure 1 : DRS UV–vis spectra of samples Figure 2 : 29Si CP-MAS NMR spectra of samples under ambient conditions.
Table 1 : Carbon and hydrogen composition and Q3/Q4 ratio of samples
The silylated samples showed higher conversion of hydrogen peroxide and selectivity to epoxide than the original counterpart. This effect was more evident when higher concentration of H2O2 was employed. This effect could be attributed to the higher hydrophobicity of silylated sample. Silylation treatment of Ti/SiO2 catalysts enhances significantly the activity in the expoxidation of alkenes with H2O2. The use of TMDS in the extend of the silylation is higher with TMDS than with HMDS as a consequence of the smaller size of the former sylilating agent. References [1] M. C. Capel-Sanchez J. M. Campos-Martin, J. L. G. Fierro, M. P. de Frutos, A. Padilla Polo, Chem. iuse., (2000) 855-856 [2] M. C. Capel-Sanchez, J. M. Campos-Martin, and J. L. G. Fierro, J. Catal., 217 (2003) 195–202 [3] T. Tatsumi, K. A. Koyano and N. Igarashi, Chem. iuse., (1998) 325-326 [4] V. A. de la Peña O’Shea, M. C. Capel-Sanchez, G. Blanco-Brieva, J. M. Campos-Martin, J. L. G. Fierro, Angew. Chem. Int. Ed. 42 (2003) 5851-5854
Benahavís (Málaga, Spain), 21st to 23rd of October, 2009 37
Preferential oxidation of CO in H 2 rich stream over gold catalysts supported on doped ceria: effect of preparation method and dopants nat ure
L. Ilieva 1, G. Pantaleo 2, I. Ivanov 1, A. M. Venezia 2, D. Andreeva 1
1Institute of Catalysis, BAS, “Acad. G. Bonchev” St., bl.11, 1113 Sofia, Bulgaria 2Istituto per lo Studio di Materiali Nanostrutturati, CNR, I- 90146 Palermo, Italy
Introduction The preferential oxidation of CO in H2-rich stream (PROX) is one of the most promising approaches for the purification of hydrogen. The low temperature polymer electrolyte membrane (PEM) fuel cells are extremely sensitive to trace of CO contamination. At the operating temperature (80-100oC) the PROX catalysts have to be highly active as well as they need to be highly selective, minimizing the loss of hydrogen by unwanted oxidation. Gold-based catalysts are potentially capable of being effectively employed in fuel cells [1]. Schubert et al. [2] have studied the effect of metal oxide support by comparing different Au catalysts. They have established that Au/CeO2 represented the best compromise regarding the PROX activity, selectivity and long term stability. Recently, a detailed study of PROX over Au on CeO2 doped by Sm, La and Zn is given in Ref. [3]. The present investigation is focused on the comparison between the properties and the catalytic performance in PROX over nanosized gold catalysts supported on doped ceria with nano-dimensions. The ceria supports were modified by the addition of rare earth metals (RE=La, Sm, Gd or Y), applying two different preparation methods: mechanochemical activation (MA) or co-precipitation (CP). The influence of the preparation techniques and the nature of the dopant on the structure and catalytic activity are discussed. Experimental Two series of doped ceria supports were synthesized: (i) the supports were prepared by CP from a solution of the corresponding metal nitrates in appropriate ratio with a solution of K2CO3; (ii) a mixture of cerium hydroxide and the corresponding oxide of the dopant was subjected to MA. Prior to gold deposition the mixed support was activated in a UV disintegrator. The amount of Re2O3 modifier was 10 wt%. Gold (2 wt%) was introduced by deposition-precipitation method. The catalysts were denoted as AuCeSm, AuCeGd, AuCeLa and AuCeY, CP or MA. AuCe sample was used as a reference. The catalysts were characterized by XRD, HRTEM, HAADF, TPR and Raman spectroscopy. The catalytic test was performed with feed gas: 1% CO, 70% H2 and 1% O2, WHSV=60 000 ml g-1 h-1. Results The XRD results showed that MA catalysts are double phases, in addition to ceria, lines of the oxides of dopants were also registered; the calculated values of lattice parameter of ceria differed insignificantly. CP samples were single phases, the changes in ceria lattice parameters more clearly depends on the ionic radius of the modifier. For both series of preparation the ceria particles were nanosized with average particle size < 10 nm. A relatively higher number of smaller gold particles were registered in MA samples compared to CP ones. However, there were no big differences in the average size of gold depending on the dopant and the method of preparation. The main line of CeO2 dominates in the Raman spectra. A weak line at 548 cm-1, assigned to the oxygen vacancies created by the presence of the Me3+ modifiers was observed only in the case of CP preparation method. It shows that a deeper modification of ceria structure occurs. The values of the full width at the half of maximum (FWHM) of the main ceria line were calculated. For both preparation methods the differences between the FWHM of AuCe and gold catalysts on doped ceria were very significant. Since the average size of undoped and doped ceria are in the same order, the observed widening can be connected to the formation of oxygen vacancies in ceria structure. Different reasons could be responsible for the formation of the oxygen vacancies in ceria. In the presence of nanogold particles, a strong modification of the ceria surface leading to Ce3+ and neighbour oxygen vacancies has been already observed [4]. Supplementary oxygen vacancies are generated by modification of ceria on adding Me3+
38 Benahavís (Málaga, Spain), 21st to 23rd of October, 2009
ions [5]. The latter are strongly dependent on the preparation method of the supports. In agreement with these results for all gold catalysts supported on doped ceria the calculated H2 consumption for ceria surface layers reduction, estimated by TPR, was higher than that of AuCe sample. However the H2 consumption for CP samples was lower compared to the corresponding MA ones. These experimental results were unexpected. The eventual explanation could be that the amount of oxygen vacancies in doped ceria, prepared by CP is higher than that for the MA samples, however in the first case they are located preferentially around the Me3+ dopant. The PROX activities, expressed as degree of CO conversion and selectivity to CO2, are compared in Fig. 1. It is seen that only AuCeSmMA catalysts exhibited higher activity and selectivity than the AuCe sample. The modification of ceria with rare earths using CP leads to higher activity and selectivity of the gold catalysts in respect to Au on undoped ceria. The maxima in activity were observed in the interval of 80-100°C. Generally the CP catalysts are more active and selective than the corresponding MA ones. Both the degree of CO conver- sion and the selectivity to CO2 are the highest for AuCeYCP sample. This catalyst shows also very good long term stability during the catalytic test at 100oC for 20 hs.
Figure 1 . Catalytic activity and selectivity in PROX over studied gold catalysts: (A) – gold on undoped ceria and doped ceria supports, prepared by MA; (B) – gold on undoped ceria and doped ceria supports, prepared by CP.
Conclusions Gold catalysts supported on ceria doped by rare earth metals were synthesized by different methods and studied in the PROX reaction. It was established that catalysts prepared by co-precipitation were more active and selective than samples made by mechanochemical activation. A CP gold catalyst on yttrium-modified ceria exhibited the highest catalytic activity and selectivity, and high stability. In the studied catalysts, the average sizes of gold and ceria nanoparticles were of the same order. The most possible explanation should be associated with the influence of the preparation method and the nature of dopants applied. Acknowledgements This study was performed in the frame of the D36/003/06 COST program. L. I. and D. A. acknowledge the support by National Science Fund, MES of Bulgaria (project ТК-Х-1709). RZ acknowledges PUNTA (IMPULSA 01), PAPIIT IN106507 and CONACYT 55154 project for the financial support. References [1] D. Cameron, R. Holliday, D. Thompson, J. Power Sources 118 (2003) 298 and ref. therein. [2] M.M. Schubert, V. Pizak, J. Garche, R.J. Behm, Catal. Lett. 76 (2001) 143. [3] G. Avgouropoulos, M. Manzoli, F. Boccuzzi, T. Tabakova et al, J. Catal. 256 (2008) 237. [4] T. Tabakova, F. Boccuzzi, M. Manzoli, D. Andreeva, Appl. Catal. A: Gen. 252 (2003) 385. [5] A. Trovarelli, Catal. Rev. Sci. Eng. 38 (1996) 439.
aUPMC Univ. Paris 6, CNRS, UMR 7197, Laboratoire de Réactivité de Surface, 4 Place Jussieu, 75252 Paris Cedex 05, France,
bUPMC Univ. Paris 6, CNRS, UMR 7142, Laboratoire des Systèmes Interfaciaux à l’Echelle Nanométrique, 4 Place Jussieu, 75252 Paris Cedex 05, France
cKyoto University, Department of Molecular Engineering, Kyoto, 615-8510, Japan *[email protected]
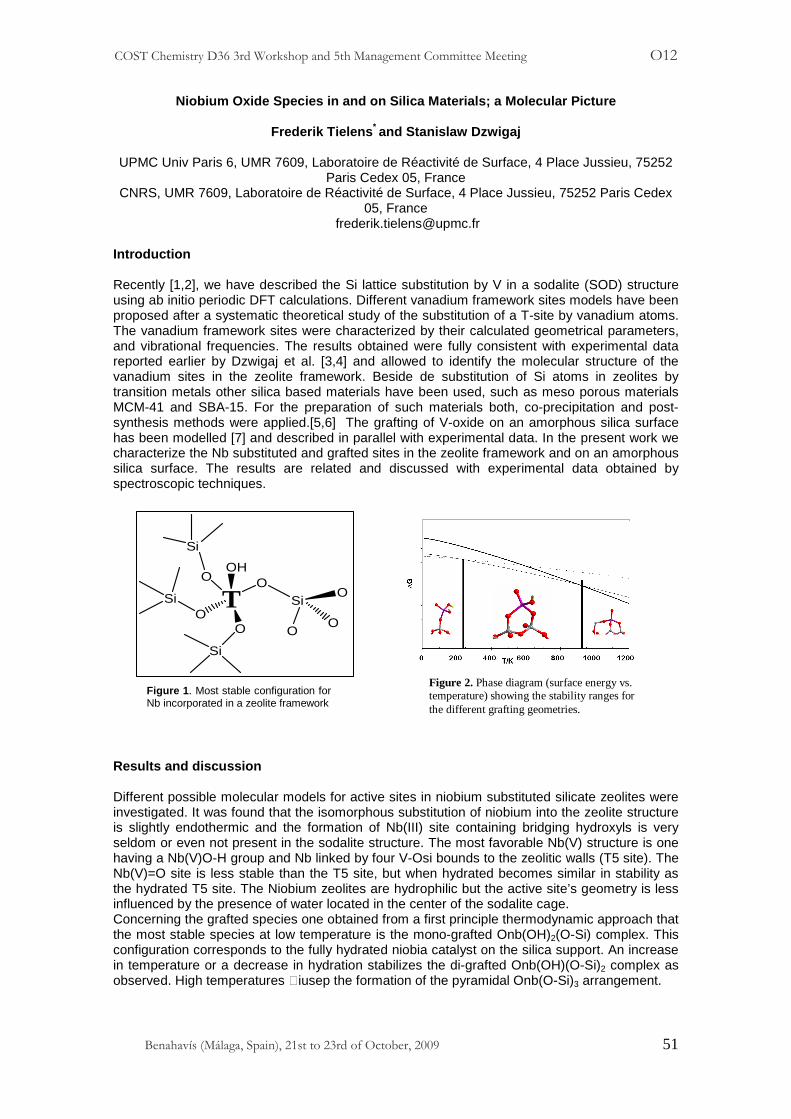
Isolated tetrahedral Ti atoms at zeolite framework sites are considered to be active sites of both catalytic and photocatalytic processes [1,2]. Therefore, the incorporation of transition metal ions into the zeolite framework appears to be the important task. We were shown [3,4] that the incorporation of transition metal ions into the lattice T-atom sites of BEA zeolite is strongly favored when, in the first step, BEA is dealuminated by treatment by nitric acid solution and then, in the second step, the incorporation of transition metal ions results from the reaction between the cationic metal species of the precursor solution and the SiO-H groups of vacant T-atom sites created by dealumination of BEA zeolite. The objective of the present work is to extend the method proposed earlier for vanadium and cobalt and the solid-liquid interface [2-4] to titanium and the solid-gas interface, with TiCl4 vapor as the precursor. The use of TiCl4 vapor has the advantage to obtain a single isolated tetrahedral Ti(IV) in framework sites. The series of TixSiBEA zeolites were prepared, characterized by different spectroscopic techniques and their catalytic and photocatalytic properties investigated in selective oxidation of propene and photocatalytic decomposition of N2O in the presence of CO. The samples prepared by two-step postsynthesis method, hereafter referred to as TixSiBEA (x = 0.3, 0.8, 1.5, 3.2, 5.8 and 9.0 Ti wt %) are white. The incorporation of Ti at tetrahedral Ti(IV) framework sites is evidenced by XRD and the consumption of SiO-H groups by FTIR, 29Si MAS NMR, 1H – 29Si CP MAS NMR and 1H MAS NMR. The progressive increase of the d302 spacing with Ti content is taken as evidence for the incorporation of Ti into the framework because the Ti-O bond distance (1.79 Å, for tetracoordinated Ti) is longer than that of Si-O (typically 1.60-1.65 Å in zeolites). After incorporation of Ti ions in SiBEA, the intensity of a broad IR band at 3520 cm-1 due to H-bonded SiO-H groups and a peak at ~ -101 ppm in 29Si MAS NMR spectra are significantly reduced, confirming the reaction between TiCl4 vapor and silanol groups. The lowest intensity of this peak is observed for Ti9.0SiBEA with the highest Ti content.. The DR UV-vis spectra of TixSiBEA exhibit two main bands at around 220-230 and 265-290 nm assigned to oxygen-tetrahedral and oxygen-octahedral Ti(IV) ligand to metal charge transfer (LMCT) transitions respectively, as reported earlier for TiMCM-41 [5]. XPS and XAS investigations confirm that for low Ti content mainly framework tetrahedral Ti(IV) are present in TixSiBEA zeolites. The octahedral Ti(IV) framework and/or extra-framework are also formed whose relative amount increases with Ti content, originating from the high titanium content. Our catalytic and photocatalytic investigation show that the single tetrahedral Ti(IV) sites are more efficient than octahedral one. References [1] M. Anpo, M. Che Adv. Catal. 44 (1999) 119. [2] M. Anpo, S. Dzwigaj, M. Che Adv. Catal. 52 (2009) 1. [3] R. Hajjar, Y. Millot, P.P. Man, M. Che, S. Dzwigaj, J. Phys. Chem. C 112 (2008) 20167. [4] S. Dzwigaj, M. Che, J. Phys. Chem. B 110 (2006) 12490. [6] L. Marchese, T. Maschmeyer, E. Gianotti, S. Coluccia, J.M. Thomas, J. Phys. Chem. B 101 (1997) 8836.
Benahavís (Málaga, Spain), 21st to 23rd of October, 2009 41
Glycerol etherification over alkaline earth metal o xides
Mònica Calatayud a,*, Agnieszka M. Ruppert b,c and Bert M. Weckhuysen b
a Laboratoire de Chimie Théorique CNRS UMR 7616 Univ. P. M. Curie, 4 Pl. Jussieu case 137, 75252 Paris, France
*[email protected] bInorganic Chemistry and Catalysis group, Dpt of Chemistry, Faculty of Science, Utrecht
University, Sorbonnelaan 16, 3584 CA Utrecht, The Netherlands cInstitute of General and Ecological Chemistry Technical University of Lodz, 90-924 Łódź, ul.
śeromskiego 116, Poland
Glycerol finds application in many fields such as cosmetics, polymer additives or in the pharmaceutical industry. Recently an increasing effort is put in the development of new applications of glycerol derivatives, in order to valorize this molecule [1,2,3]. It is easily obtained from sugars or as a by-product in the biodiesel process, and might become a platform molecule in the biorefinery schemes in the near future. One possible route of its transformation is the etherification to di-, tri- or poly-glycerol. This reaction is catalyzed by both acid- [4] and base-type catalysts [5]. Alkaline earth oxides have been successfully used as basic catalysts for this reaction [5], with the conversion to products increasing with increasing catalyst basicity: MgO<CaO<SrO<BaO. In this work periodic DFT calculations are carried out to model glycerol interaction with MO (M=Mg, Ca, Sr, Ba) surfaces [6]. The role of defects has been investigated for a CaO stepped surface (see Figure). In particular, the adsorption mode and strength of glycerol interaction with the surfaces have been calculated. Different geometries have been tested for the interaction of glycerol with those materials. The results are discussed and compared with the experimental data.
Figure: glycerol in interaction with CaO regular (left) and stepped (right) surfaces. The main conclusions are:
• glycerol interacts with surface acid-base pairs. The geometry of adsorption depends on the structural parameters of the surface,
• the strength of the interaction correlates with the material basicity: MgO < CaO < SrO < BaO,
• the dissociation of glycerol increases in the series: MgO (not dissociated) < CaO (partially dissociated) < SrO (partially dissociated) < BaO (completely dissociated),
• surface defects play a key role in the adsorption process, • the results of our theoretical calculations are in very good agreement with our earlier
experimental observations of the glycerol etherification reaction over alkaline earth oxides [5].
References [1] Y. Zheng, X. Chen and Y. Shen, Chem. Rev 108 (2008) 5253. [2] M. Pagliaro, R. Ciriminna, H. Kimura, M. Rossi, C. Della Pina, Angew. Chem. Int. Ed. 46 (2007) 2 [3] F. Jérôme, Y. Pouilloux, J. Barrault, ChemSusChem 1 (2008) 586. [4] J.M. Clacens, Y. Pouilloux, J. Barrault, Appl. Catal. A : General 227 (2002) 181. [5] A.M. Ruppert, J. D. Meeldijk, B.W.M. Kuipers, B.H. Erné, B.M. Weckhuysen, Chem. Eur. J. 14 (2008) 2016. [6] M. Calatayud, A.M. Ruppert, B.M. Weckhuysen, accepted Chem. Eur. J.
Benahavís (Málaga, Spain), 21st to 23rd of October, 2009 43
Glycerol oxidation on gold catalysts supported on g roup five metal oxides –a comparative study with other metal oxide and carbon based catalysts
Izabela Sobczak *, Katarzyna Jagodzinska, Maria Ziolek
A. Mickiewicz University, Faculty of Chemistry, Grunwaldzka 6, 60-780 Poznań, Poland
* [email protected] Introduction Nowadays much attention has been devoted to applying green catalytic processes to convert biorenewable feedstock to commodity chemicals and clean fuels [1,2]. Glycerol is a potentially important biorefinery feedstock, available as a byproduct in the production of biodiesel by transesterification of vegetable oils or animal fats. Since new energy resources such as biodiesel fuel have grown in importance in recent years, new uses for glycerol need to be found. Recently, a series of novel catalytic conversion processes for glycerol transformation was reported, showing that glycerol can readily be oxidized, reduced, halogenated, etherified, and esterified to obtain valuable commodity chemicals. The focus of this work was on gold catalysts applied for liquid phase glycerol oxidation with oxygen. The main task was to apply new supports for gold (V2O5, Nb2O5, Ta2O5) and to investigate the effect of group five metal oxides on the efficiency of the oxidation of glycerol. Gold catalysts based on carbons and metal oxides from Project AuTEK (Al2O3, TiO2 and ZnO) were also tested for comparison. Our interest was to study the influence of gold-support interaction on activity and selectivity in glycerol oxidation. Moreover, the influence of preparation method and gold dispersion is considered. Experimental Commercial oxides (V2O5 – Aldrich, Nb2O5(anh) –Alfa Aesar, Nb2O5(aq) – CBMM-Brasil, Ta2O5
–Aldrich) and carbon supports (CAld – Aldrich and CPOCH- POCH) were modified by gold-sol method [3] with THPC as reducing agent and HauCl4 as a source of gold (1 wt.% of Au). Additionally, Nb2O5 oxides were modified by deposition-precipitation (DP) method using urea as reducing agent [3]. The prepared materials were calcined at 623 K for 4 h. All the materials were charcterised by the use of standard techniques, XRD, UV-Vis, TEM, XPS, test reactions. For a comparative study, industrial MINTEK catalysts, Au/Al2O3 (0.8 wt. % of Au), Au/TiO2 and Au/ZnO (1 wt. % of Au ) were used. The glycerol oxidation experiments were performed in a 300 ml batch reactor from Parr. The oxidation reactions were carried out with oxygen under pressure 6 atm, at 333 K for 5 h. NaOH (NaOH/glycerol molar ratio = 2) and 0.2 g of gold catalyst (glycerol/Au molar = 980) were added to a 1 M aqueous solution of glycerol. The quantitative analyses of the reaction mixtures were performed by high performance liquid chromatography (HPLC). Results and discussion The state of Au in the prepared catalysts was studied by XRD, TEM, UV-VIS and XPS. The results clearly indicated that metallic gold crystallites are formed on all materials and their size is determined by the chemical composition of the support and the method of Au introduction. It was found that much bigger Au agglomerates are formed on oxides prepared by deposition-precipitation method using urea than in the case when gold-sol method with the use of THPC as reducing agent is applied. TEM images allowed the estimation of Au crystallites as ~5 and ~125 nm for gold-sol and DP method, respectively. Among supports modified with the use of THPC the higher dispersion and smaller gold particle size was obtained on niobia and vanadia as well as on C ALD than Ta2O5 and C POCH. The oxidation of glycerol with oxygen was investigated using the Au-catalysts at 333 K and the results are given in Table 1. Similarly as it was shown in the literature [3], the activity of gold catalysts studied in this work is highly dependent on the Au particle size and dispersion. Au-oxides prepared by the gold-sol method that leads to a higher gold dispersion show much higher activity than the catalyst prepared by the precipitation method. It is worthy of notice that the highest activity among oxide supports, comparable with
44 Benahavís (Málaga, Spain), 21st to 23rd of October, 2009
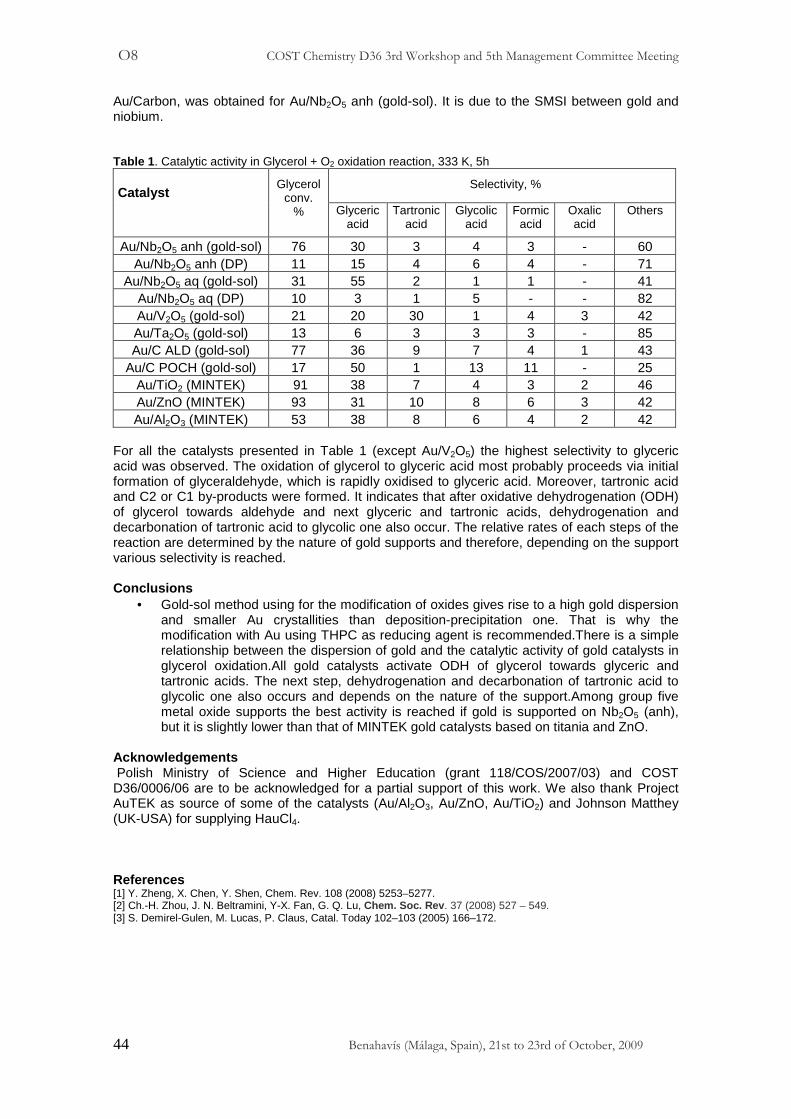
Au/Carbon, was obtained for Au/Nb2O5 anh (gold-sol). It is due to the SMSI between gold and niobium. Table 1 . Catalytic activity in Glycerol + O2 oxidation reaction, 333 K, 5h
Selectivity, % Catalyst
Glycerol conv.
% Glyceric acid
Tartronic acid
Glycolic acid
Formic acid
Oxalic acid
Others
Au/Nb2O5 anh (gold-sol) 76 30 3 4 3 - 60 Au/Nb2O5 anh (DP) 11 15 4 6 4 - 71
For all the catalysts presented in Table 1 (except Au/V2O5) the highest selectivity to glyceric acid was observed. The oxidation of glycerol to glyceric acid most probably proceeds via initial formation of glyceraldehyde, which is rapidly oxidised to glyceric acid. Moreover, tartronic acid and C2 or C1 by-products were formed. It indicates that after oxidative dehydrogenation (ODH) of glycerol towards aldehyde and next glyceric and tartronic acids, dehydrogenation and decarbonation of tartronic acid to glycolic one also occur. The relative rates of each steps of the reaction are determined by the nature of gold supports and therefore, depending on the support various selectivity is reached. Conclusions
• Gold-sol method using for the modification of oxides gives rise to a high gold dispersion and smaller Au crystallities than deposition-precipitation one. That is why the modification with Au using THPC as reducing agent is recommended.There is a simple relationship between the dispersion of gold and the catalytic activity of gold catalysts in glycerol oxidation.All gold catalysts activate ODH of glycerol towards glyceric and tartronic acids. The next step, dehydrogenation and decarbonation of tartronic acid to glycolic one also occurs and depends on the nature of the support.Among group five metal oxide supports the best activity is reached if gold is supported on Nb2O5 (anh), but it is slightly lower than that of MINTEK gold catalysts based on titania and ZnO.
Acknowledgements Polish Ministry of Science and Higher Education (grant 118/COS/2007/03) and COST D36/0006/06 are to be acknowledged for a partial support of this work. We also thank Project AuTEK as source of some of the catalysts (Au/Al2O3, Au/ZnO, Au/TiO2) and Johnson Matthey (UK-USA) for supplying HauCl4. References [1] Y. Zheng, X. Chen, Y. Shen, Chem. Rev. 108 (2008) 5253–5277. [2] Ch.-H. Zhou, J. N. Beltramini, Y-X. Fan, G. Q. Lu, Chem. Soc. Rev . 37 (2008) 527 – 549. [3] S. Demirel-Gulen, M. Lucas, P. Claus, Catal. Today 102–103 (2005) 166–172.
Benahavís (Málaga, Spain), 21st to 23rd of October, 2009 45
Etherification of Glycerol and Other Biomass-Derive d Polyols: New Routes to Valuable Bulk Chemicals
Andrei N. Parvulescu a*, Pieter C. A. Bruijnincx a, Peter J.C. Hausoul a,b, Maria Arias a,
Robertus J.M. Klein Gebbink b and Bert M. Weckhuysen a
a Inorganic Chemistry and Catalysis, Debye Institute for Nanomaterials Science, Faculty of
Science, Utrecht University, Utrecht, The Netherlands; bChemical Biology & Organic Chemistry, Debye Institute for Nanomaterials Science, Faculty of
Science, Utrecht University, Utrecht, The Netherlands; [email protected]
Objectives New catalytic routes have to be developed to transform renewable highly oxygenated platform molecules to valuable bulk and fine chemicals [1]. The polyols constitute an important class of these biomass-derived oxygenates, with glycerol being a prime example. Glycerol is produced in large quantities as a by-product of biodiesel production, i.e. for every 1000 kg of fatty acid esters around 100 kg of glycerol is formed. Although glycerol itself has a lot of commercial applications, e.g. in the cosmetic and pharmaceutical industries, new attractive applications should be introduced to improve the economics of the biodiesel process [2]. Furthermore, glycerol is regarded as a potential platform molecule since it can be directly produced from sugars or sugar alcohols, which are considered as the cornerstones of future biorefinery schemes. The development of new catalytic routes for glycerol valorization is therefore of great importance. In this respect, etherification of glycerol [3] or, indeed, other biomass-derived polyols represents an important application as the products can be used as fuel additives, intermediates in the pharmaceutical industry, agrochemicals or as non-ionic surfactants. In this contribution we will discuss the recent efforts of our group which have focused on glycerol/polyol valorization [4] through etherification with long linear alkenes. Results In our approach, we investigated the direct etherification of glycerol with a long linear alkene using as a model 1-octene over solid acid catalysts [3]. Remarkably, the catalytic etherification route of direct nucleophilic addition of glycerol to linear, long-chain olefins has been little explored even if this process may provide a direct route to long alkyl chain ethers with potential surfactant application. Therefore we decided to screen various heterogeneous acid catalysts in the etherification of neat glycerol with 1-octene. Zeolites showed modest conversions compared to Amberlyst 70 or pTSA, but superior selectivities towards the most valuable monoether products. The highest activity was obtained with H-Beta zeolites, which gave excellent selectivities for the mono and di-octyl ethers of glycerol of always > 85 %. H-Beta with a Si/Al ratio of 12.5 gave the highest conversion (15 %) and the highest selectivity to the mono-octyl ether (94 %). Several factors were found to influence both the etherification activity and the selectivity. Hydrophilic properties and porous structure of the catalyst turned out to be the critical parameters. Other parameters like reaction time, 1-octene: glycerol molar ratio, reaction temperature and the addition of an inert gas were investigated in order to improve the etherification activity of H-Beta zeolite. Catalysts deactivation due coke formation was investigated as well. In addition, the catalyst was recovered and re-used in 3 reaction cycles without any loss of activity or selectivity (Table 1). The substrate scope was successfully extended to other bio-based polyols and the results will be also discussed. Table 1. Re-use of H-Beta (37.5) zeolite in the etherification of glycerol with 1-octene.
Run Conv.(%) Sel.C8Glyc (%) Sel.C16Glyc (%) Sel.other(%)
1 14 78 14 8 2 13 80 13 7 3 14 79 12 9
Reaction conditions: 1 g of catalyst, 1-octene: glycerol 3:1, 5 h, 10 bar Ar, 140 °C.
46 Benahavís (Málaga, Spain), 21st to 23rd of October, 2009
Figure 1. Etherification of alcohols with 1-octene over H-Beta (Si/Al =12.5). Conclusions The direct etherification of glycerol and glycols with 1-octene in a solvent-less system is possible by using heterogeneous acid catalysts. The type of the solid acid used strongly influences the activity and selectivity of the etherification process. H-Beta zeolites proved to be the most selective etherification catalysts, whereas for Amberlyst-70 a high amount of by-products were formed. H-Beta (12.5) produced the C8Glyc and C16Glyc ethers with 96 % selectivity at 16 % conversion. H-Beta zeolites were succesfully applied in etherification of other bio-based polyols. These results show the potential of using heterogeneous acid catalysts for the green synthesis of valuable long alkyl mono- or di-ethers of various bio-based alcohols. Acknowledgements We would like to thank the ASPECT-ACTS Program for financial support. References [1] Gallezot, P., ChemSusChem, 2008, 1, 734-737. [2] Jérome, F., Pouilloux, Y., Barrault, J., 2008. Rational design of solid catalysts for the selective use of glycerol as a natural organic building block, ChemSusChem, 2008, 1, 586-613. [3] Ruppert, A. M.; Meeldijk, J. D.; Kuipers, B. W. M.; Erne, B. H.; Weckhuysen, B. M. Chem. Eur. J. 2008, 14, 2016-2024 [4] Ruppert, A. M.; Parvulescu A. N.; Arias, M.; Hausoul P. C.; Bruijnincx P. C. A.; Klein Gebbink, R. J. M.; Weckhuysen, B. M. J. Catal. 2009, submitted for publication
Benahavís (Málaga, Spain), 21st to 23rd of October, 2009 47
Relevance of the physicochemical properties of CaO catalyst for the methanolysis of triglycerides to obtain biodiesel
D. Martín Alonso a, F. Vila a, R. Mariscal a*, M. Ojeda a, M. López Granados a, J. Santamaría-
González b
a Instituto de Catálisis y Petroleoquímica, CSIC, C/Marie Curie 2, Campus de Cantoblanco, E-28049, Madrid, Spain. b Departamento de Química Inorgánica, Cristalografía y Mineralogía (Unidad Asociada al ICP-CSIC), Facultad de Ciencias, Universidad de Málaga, Campus de
Objective Calcium oxide is a suitable solid catalyst for the production of biodiesel (fatty acid methyl esters, FAME) via triglycerides methanolysis reaction [1]. The physicochemical properties of metallic oxides may be affected significantly by the preparation method [2]. Therefore, we have investigated a series of different CaO precursors with the final aim of improving the biodiesel yield. A detailed characterization study of all materials has been also carried out to establish a structure-activity relationship. Results Four calcium oxide samples were prepared by thermal decomposition of different calcium salts commercially available: carbonate (CaO-C), acetate (CaO-A), oxalate (CaO-O) and nitrate (CaO-N). Two additional samples were prepared by decomposition of Ca(OH)2 previously prepared by controlled addition of a NaOH solution to aqueous solutions of calcium acetate (CaO/OH-A) and nitrate (CaO/OH-N). The precursor decomposition process was characterized by EGA-MS spectrometry, which allows the determination of the minimum temperature required to obtain CaO. Accordingly, all samples were first decomposed ex-situ at 1073 K during 1 h under an O2/Ar flow (20/80 v/v). Subsequently, the samples were cooled down in N2 flow and loaded in a batch reactor avoiding any contact with ambient air to thus prevent sample hydration and carbonation.
0.0 0.5 1.0 1.5 2.0 2.5 3.00
20
40
60
80
100
FA
ME
yie
ld (
%)
Time (h)
CaO-C CaO-A CaO-O CaO-N CaO/OH-A CaO/OH-N
Figure 1. FAME yield obtained with the different CaO samples.
48 Benahavís (Málaga, Spain), 21st to 23rd of October, 2009
Figure 1 shows that there is one group of active catalysts (CaO-C, CaO-A, CaO-O and CaO/OH-A), with minor differences among them, that reach a 90 % yield to FAME in 3 h (> 80 % in 2 h). In contrast, CaO-N and CaO/OH-N depict low FAME yields (<30 % in 3 h) at the same conditions. To understand the observed catalytic performance, we have characterized the activated CaO samples by XRD and N2 adsorption-desorption isotherms. XRD patterns for all activated samples are almost identical, presenting exclusively diffraction lines assigned to the CaO phase (JCPDS 77-2376). The average crystal size (Table 1) has been determined by using the Debye-Scherrer equation to the diffraction peak at 2θ=37.4º. The smallest CaO crystallites are obtained when calcium acetate and oxalate are used, while calcium nitrate precursors lead to the larger particles. Table 1 also summarizes the textural properties (BET surface area, pore volume and pore diameter) for all samples. The most active samples (CaO-C, CaO-A, CaO-O and CaO/OH-A) show similar BET surface areas (20-27 m2.g-1) and higher than the almost inactive samples (CaO/OH-N and CaO-N). Table 1. Crystal size (XRD), textural properties (N2 isot.) and basicity (FTIR-pyrrole) of CaO samples
Sample Crystal size (nm)
BET surface
area (m2·g1)
Pore volume (cm 3·g-1)
Pore diameter
(nm)
Intensity (a.u.) at RT of 1446 cm -1
band
CaO-C 67 25.9 0.27 41 1.20
CaO-A 37 21.9 0.18 33 0.46
CaO-O 47 25.7 0.24 38 0.40
CaO-N 116 <1.0 -- -- --
CaO/OH-A
60 26.7 0.16 24 --
CaO/OH-N
93 6.8 0.05 33 --
The basicity of the catalysts was evaluated by FT-IR of adsorbed pyrrole (C4H4NH) as a probe molecule. Pyrrole is an amphoteric substance that interacts with the surface basic sites at the sample by forming a H-bond between the NH group and the framework oxygen atoms (C4H4NH-O). Therefore, it has been often used to detect and estimate the strength of base sites on oxide surfaces [3]. The most active samples (CaO-C, CaO-A, CaO-O) show a clear band centered at 1446 cm-1 associated to pyrrolate species formed on strong base sites on the solid (Table 1). Furthermore, we have noted that the intensity and thermal stability of this band (indicative of the number and strength of basic surface sites, respectively) can explain the small differences observed among the most active catalysts. Interestingly, when calcium nitrate takes part in any step of the preparation of calcium oxide, low surface areas are obtained, and consequently, the observed catalytic performance is poor. In summary, an excellent CaO catalyst for methanolysis of triglycerides to obtain biodiesel should not involved the use of calcium nitrate salt as a precursor. Moreover, the suitable preparation method should yield small CaO particles displaying elevated surface area and a high number of strong basic sites on the surface. Conclusions In all cases, our data strongly suggest that crystal size, textural properties and base character are key parameters in the catalytic behaviour of CaO materials in the triglycerides methanolysis reaction to produce biodiesel. These physicochemical properties are determined by the calcium salt used as precursor of the CaO. References [1] M. Lopez Granados, M.D. Zafra Poves, D. Martin Alonso, R. Mariscal, F. Cabello-Galisteo, R. Moreno-Tost; J. Santamaría, J.L.G. Fierro, Appl. Catal. B: Environ. 73 (2007) 317-326. [2] T. Matsuda, J. Tanabe, N. Hayashi, Y. Sasaki, H. Miura, K. Sugiyama, Bull. Chem. Soc. Jpn. 55 (1982) 990-994. [3] D. Murphy, P. Massiani, R. Frank, D. Barthomeuf; J. Phys. Chem. 100 (1996) 6731-6738.
Benahavís (Málaga, Spain), 21st to 23rd of October, 2009 49
Influence of porosity and surface groups on catalyt ic activity of carbon materials for the microwave-assisted CO 2 reforming of CH 4
B. Fidalgo, A. Arenillas, J.A. Menéndez *
Instituto Nacional del Carbón, CSIC, Apartado 73, 33080 Oviedo, Spain
[email protected] Objective The use of carbon materials as catalysts for hydrocarbon pyrolysis reactions has been extensively studied, since carbon-based catalysts offer some advantages over metal catalysts as availability, durability and low cost. Besides, they are usually good microwave receptors, which is advantageous in case of heating by using microwaves. It has been observed that the decomposition of CH4 using carbon catalysts occurs mainly in micropores, and a rapid deactivation is observed after high initial conversions as a consequence of blockage of pores by carbonaceous deposits. Microwave-assisted dry reforming of CH4 (reaction 1) has been proposed as a viable “in situ” regeneration process, thanks to the gasification of the generated deposits with CO2. Besides, high conversions can be achieved since microwave heating enhances heterogeneous reactions and heterogeneous catalytic reactions (as CO2 gasification and CH4 decomposition, respectively). Dry reforming reaction: CH4 + CO2 ↔ 2H2 + 2CO ∆H298K = +247 kJ/mol (1) On the other hand, a clear connection between catalytic activity of carbons and surface groups has not been established since contradictory results about the role of surface chemistry and CH4 decomposition can be found. The aim of the present work is to investigate factors governing catalytic activity of carbonaceous materials in the microwave-assisted CO2 reforming of CH4 reaction, focusing on textural and surface properties. Results In order to study the influence of porosity, carbon materials with different textural properties were tested as catalysts for the microwave-assisted dry reforming reaction (metallurgical coke, activated carbon and carbon xerogel). Experiments were conducted in a quartz reactor charged with the carbon used as catalyst/microwave receptor (C/MR) and heated in a single mode microwave oven.
CH4 conversion
0
20
40
60
80
100
0 25 50 75 100 125 150
Time (min)
FY5 CQ
CO2 conversion
0
20
40
60
80
100
0 25 50 75 100 125 150
Time (min)
Con
vers
ion
(%)
FY5 CQ
CH4 conversion
0
20
40
60
80
100
0 25 50 75 100 125 150
Time (min)
FY5 CQ
CO2 conversion
0
20
40
60
80
100
0 25 50 75 100 125 150
Time (min)
Con
vers
ion
(%)
FY5 CQ
Figure 1. Conversions for microwave-assisted dry reforming carried out over a commercial activated carbon (FY5) and a metallurgical coke (CQ). Operating conditions: microwave oven; T = 800 ºC; 50%CH4 – 50%CO2; VHSVtotal-FY5 = 0.32 L/g h and VHSVtotal-CQ = 0.64 L/g h. It was found that large microporosity is necessary for an acceptable catalytic activity of carbons. Thus, carbonaceous materials without development of microporosity gave rise to negligible conversions (case of metallurgical coke, CQ in Figure 1), whereas high conversions were attained using carbons with large micropores volumes as catalysts (case of the activated carbon FY5 in Figure 1). Besides, to keep high CH4 and CO2 conversions with time, the microporosity
50 Benahavís (Málaga, Spain), 21st to 23rd of October, 2009
must be preserved from being blocked, which depends on the operating conditions used, mainly the proportion of CO2. Presence of mesopores in the catalyst was also studied by testing carbon materials with controlled porosity (carbons with similar micropores volumes but different mesopores volumes). However, it was not observed so clear influence on the catalytic activity as in the case of microporosity. In order to investigate the effect on dry reforming of the presence of oxygen groups on the surface of carbon catalysts, commercial activated carbons were modified by oxidation (using a saturated solution of (NH4)2S2O8, added in a proportion of 1g of activated carbon per 10 mL of solution). Obviously, oxidized activated carbons shows higher volatile matter and oxygen content, but no important change in textural parameters regarding original carbons is observed. Oxygen surface groups reduced dramatically the catalytic activity of activated carbons, giving rise to much lower conversions than original carbons (compare FY5 in Figure 1 with conversion profiles in Figure 2). From the point of view of individual reactions, CO2 gasification seems to be more negatively affected. Actually, CO2 conversion was lower than CH4 conversion at any time, which had not been observed with non-oxidized activated carbons before.
CH4 conversion
0
20
40
60
80
100
0 25 50 75 100 125 150
Time (min)
MW EF
CO2 conversion
0
20
40
60
80
100
0 25 50 75 100 125 150
Time (min)
Con
vers
ion
(%)
MW EF
CH4 conversion
0
20
40
60
80
100
0 25 50 75 100 125 150
Time (min)
MW EF
CO2 conversion
0
20
40
60
80
100
0 25 50 75 100 125 150
Time (min)
Con
vers
ion
(%)
MW EF
Figure 2. Conversions for dry reforming carried out over oxidized activated carbon and under conventional (EF) or microwave (MW) heating. Operating conditions: FY5ox; T = 800 ºC; 50%CH4 – 50%CO2; VHSVtotal = 0.32 L/g h. Interestingly, performance of microwave-assisted dry reforming over oxidized carbons was worse than under conventional heating, contrary to the results expected, since microwave heating is known to give rise to enhanced conversions when dry reforming is carried out over original carbon materials. This could be due to trouble to heat oxidized activated carbons in the microwave device, caused by less density of delocalized π-electrons, making more difficult the generation of microplasmas. Conclusions Catalytic activity of carbon materials for dry reforming is mostly determined by their textural and surface properties. Dry reforming of CH4 over carbon materials occurs mainly in micropores. Therefore, large micropores volumes are related with high conversions and blockage of microporosity with a reduction in the catalytic activity. Addition of oxygen surface groups diminishes significantly the catalytic activity of activated carbons respect to original carbons. Microwave-assisted dry reforming is specially affected (more than conventionally heated) since oxidized activated carbons are more difficult to be heated in the microwave oven.
Benahavís (Málaga, Spain), 21st to 23rd of October, 2009 51
Niobium Oxide Species in and on Silica Materials; a Molecular Picture
Frederik Tielens * and Stanislaw Dzwigaj
UPMC Univ Paris 6, UMR 7609, Laboratoire de Réactivité de Surface, 4 Place Jussieu, 75252 Paris Cedex 05, France
CNRS, UMR 7609, Laboratoire de Réactivité de Surface, 4 Place Jussieu, 75252 Paris Cedex 05, France
[email protected] Introduction Recently [1,2], we have described the Si lattice substitution by V in a sodalite (SOD) structure using ab initio periodic DFT calculations. Different vanadium framework sites models have been proposed after a systematic theoretical study of the substitution of a T-site by vanadium atoms. The vanadium framework sites were characterized by their calculated geometrical parameters, and vibrational frequencies. The results obtained were fully consistent with experimental data reported earlier by Dzwigaj et al. [3,4] and allowed to identify the molecular structure of the vanadium sites in the zeolite framework. Beside de substitution of Si atoms in zeolites by transition metals other silica based materials have been used, such as meso porous materials MCM-41 and SBA-15. For the preparation of such materials both, co-precipitation and post-synthesis methods were applied.[5,6] The grafting of V-oxide on an amorphous silica surface has been modelled [7] and described in parallel with experimental data. In the present work we characterize the Nb substituted and grafted sites in the zeolite framework and on an amorphous silica surface. The results are related and discussed with experimental data obtained by spectroscopic techniques.
Results and discussion Different possible molecular models for active sites in niobium substituted silicate zeolites were investigated. It was found that the isomorphous substitution of niobium into the zeolite structure is slightly endothermic and the formation of Nb(III) site containing bridging hydroxyls is very seldom or even not present in the sodalite structure. The most favorable Nb(V) structure is one having a Nb(V)O-H group and Nb linked by four V-Osi bounds to the zeolitic walls (T5 site). The Nb(V)=O site is less stable than the T5 site, but when hydrated becomes similar in stability as the hydrated T5 site. The Niobium zeolites are hydrophilic but the active site’s geometry is less influenced by the presence of water located in the center of the sodalite cage. Concerning the grafted species one obtained from a first principle thermodynamic approach that the most stable species at low temperature is the mono-grafted Onb(OH)2(O-Si) complex. This configuration corresponds to the fully hydrated niobia catalyst on the silica support. An increase in temperature or a decrease in hydration stabilizes the di-grafted Onb(OH)(O-Si)2 complex as observed. High temperatures iusep the formation of the pyramidal Onb(O-Si)3 arrangement.
TO
OO
Si
Si
Si
Si
O
O
O
OOH
Figure 2. Phase diagram (surface energy vs. temperature) showing the stability ranges for the different grafting geometries.
Figure 1 . Most stable configuration for Nb incorporated in a zeolite framework
52 Benahavís (Málaga, Spain), 21st to 23rd of October, 2009
Conclusions Niobium containing zeolite BEA was successfully synthesized. The characterization of this material indicated that niobium is incorporated into the zeolite framework. First principle calculation techniques were performed to investigate the nature of the Nb site in the zeolite framework. It was found that the Nb site is a Nb(V) having a Nb(V)O-H group and Nb linked by four Nb-Osi bounds to the zeolitic walls. The grafting geometry is depending on the degree of hydration, however the Onb(OH)(O-Si)2 complex is found to have the largest domain of stability as a function of the temperature. In general it is found that substitution or grafting of Nb oxide species do not have the same molecular structure. Acknowledgements The authors thank GENCI project x20090812022 and the CINES, IDRIS and CCRE (Université Pierre et Marie Curie) for providing the computation facilities. Polish Ministry of Science and Higher Education (grant 118/COS/2007/03) and COST D36/0006/06 are to be acknowledged for a partial support of this work. References [1] F. Tielens, M. Trejda, M. Ziolek, S. Dzwigaj, Catal. Today 139 (2008) 221. [2] F. Tielens, M. Calatayud, S. Dzwigaj, M. Che. Micropour. Mesopour. Mater. 119 (2009) 137. [3] S. Dzwigaj, E.M. Ei Malki, M.J. Peltre, P. Massiani, A. Davidson, M. Che, Topics Catal. 11/12 (2000) 379. [4] R. Hajjar, Y. Millot, P.P. Man, M. Che, S. Dzwigaj, J. Phys. Chem. C 112 (2008) 20167. [5] X. Gao, I.E. Wachs, M.S. Wong, J. Y. Ying, J. Catal., 203 (2001) 18-24. [6] M. Ziolek, I. Nowak, Zeolites, 18 (1997) 356-360. [7] M.M. Islam, D. Costa, M. Calatayud, F. Tielens, J. Phys. Chem. C, 113, 10740 (2009).
Benahavís (Málaga, Spain), 21st to 23rd of October, 2009 53
Study of Nanoporous Catalysts in the Selective Cata lytic Reduction of NO x
María José Orellana Rico, Ramón Moreno Tost, Antoni o Jiménez López, Enrique Rodríguez Castellón *
Departamento de Química Inorgánica, Cristalografía y Mineralogía (Unidad Asociada
al ICP-CSIC), Facultad de Ciencias, Universidad de Málaga, Campus de Teatinos, 29071 Málaga (Spain) [email protected]
Objective The SCR of NO with ammonia is an effective, selective and available industrial process to control the emissions of NO from stationary sources. On the other hand, its application remains limited to the control of NOx emissions from stationary sources and mobile sources such as trucks, diesel locomotives or maritime transport, but it is not viable for light vehicles. In addition, the current catalyst based on V2O5-WO3(MO3)-TiO2 suffers processes of deactivation for sulfur and heavy metals compounds [1] and, moreover, the issues related to vanadium toxicity. The discovery of catalysts based on zeolite active in the SCR process using hydrocarbons as reducing agents, has focused the interest of the researchers on non-zeolite materials. Thus, siliceous SBA-15 materials have been proposed as supports for the SCR of NO with hydrocarbons [2], in which high dispersion of the active metal and good performance have been attained. The main goal of this work is the study of a mesoporous silica with SBA-15 (Si) structure prepared by means of a low cost synthetic route [3] as support of copper catalysts (ca. 1-6wt%). It has been also synthesized an alumina grafted on the mesoporous silica (SiAl) with a Si/Al molar ratio of 10. Copper catalysts have been characterized and tested in the SCR of NO with propane in excess of oxygen. Results The textural parameters of Si and SiAl supports show that the aluminum is incorporated mainly inside of the pores of the mesoporous silica since a reduction in the mean pore diameter and pore volume took place as well as an increase of the width of the pore walls. Moreover, the XRD patterns of the Si and SiAl supports show that the hexagonal structure of SBA-15 is maintained after grafting with alumina. The copper was incorporated into the Si and SiAl supports by means of incipient wetness method. The XRD patterns of copper catalysts show that for a same copper loading, the presence of aluminum improves the active phase dispersion over the support. On the other hand, XPS analysis of catalysts points out that copper is present as CuO and a spinel-like structure in the SiAl_3 and SiAl_6 catalysts as well. The copper dispersion over the supports has been studied by means of N2O decomposition showing that the aluminum exerts a favorable effect on the copper dispersion and therefore, over the metal surface exposed. These catalysts have been tested in the SCR of NO with propane in the presence of oxygen. Figure 1 depicts the catalytic activity of Si_x and SiAl_x respectively. Si_1 catalyst shows the highest conversion of NO among the Si_x catalysts. This fact is due to the highest dispersion of copper since the particle size (1.9 nm) is lower than those observed for Si_3 and Si_6, 8.8 and 10.9 nm, respectively. In addition, these particles are more reducible than the larger ones of Si_3 and Si_6, respectively. On the other hand, SiAl_3 catalyst displays the higher conversion of NO for the catalysts tested.
54 Benahavís (Málaga, Spain), 21st to 23rd of October, 2009
0
5
10
15
20
250 350 450 550
Temperature (ºC)
NO
Con
vers
ion
(%
)
Si_6 Si_3 Si_1
0
5
10
15
20
25
250 350 450 550
Temperature (ºC)
NO
Co
nver
sio
n (%
)
SiAl_6 SiAl_3 SiAl_1
Figure 1. Catalytic performance of Si_x and SiAl_x. Catalytic conditions:1000ppm NO, 1000ppm C3H8, 2.5vol%O2, balance He and total flow = 75 mL/min. In this case, the copper loading increase over SiAl supports does not involve an increase of the particle size (1.0 and 1.8 nm for SiAl_1 and SiAl_3, respectively). However, the loading increase from 3wt% to 6wt% (6.8 nm) is high enough to produce larger copper particles, which do not produce an improvement in the catalytic activity. Furthermore, it has been studied the effect of NO, C3H8, O2 and space velocity in the catalytic performance of SiAl_6 catalyst 450ºC with. As it was expected, the parameter whose influence was greater in the catalytic activity was the space velocity since the NO conversion reached 90% when the flow was reduced up to 50mL/min, however if the total flow was increased up to 150 mL/min the NO conversion was 15%. SiAl_3 catalyst has been tested in presence of H2O and SO2 in the feed. The Figure 2 shows the catalytic results showing that the presence of SO2 improves the NO conversion while the presence of H2O exerts a negative effect on the catalytic performance.
The beneficial effect of SO2 has been already reported by the authors [4] in the case of mordenite zeolites exchanged with copper. This effect was attributed to the presence of superficial CuSO4 active in the SCR reaction.
Figure 2. Effect of SO2 and H2O in the SCR of NO with propane
Acknowledgements The authors are grateful to financial support from CICYT (project NAN20004-09267-C01) and Junta deAndalucía (PO6-FQM-01661).RMTwould like to thanks the Ministry of Science and Innovation (Spain) for the financial support under the Program Ramón y Cajal (RYC-2008-03387). References [1] G. Busca, M. A. Larrubia, L. iusep, G. Ramis, Catal Today 107-108 (2005) 139 [2] N. El Hassan, A. Davidson, Patrick Da Costa, G. Djéga-Mariadassou, Catal Today 137 (2008) 191. [3] M. Gómez Cazalilla, J.M. Mérida Robles, A. Gurbani, E. Rodríguez Castellón, A. Jiménez López, J. Solid State Chem. 180 (2007) 1130. [4] R. Moreno-Tost, J Santamaría-González, E. Rodríguez-Castellón, A. Jiménez-López., M. A. Autié, E. González, M. Carreras Glacial, C. De las Pozas, Appl. Catal. B 50 (2004) 279
Benahavís (Málaga, Spain), 21st to 23rd of October, 2009 55
New HDS catalysts supported on thiol functionalized mesoporous silica
Valeria La Parola a, Brindusa Dragoi b, Anna Maria Venezia a*
aIstituto per lo Studio dei Materiali Nanostrutturati (ISMN-CNR) via Ugo La Malfa, 153, Palermo, I-90146; bTechnical University, Faculty of Chemical Engineering,Laboratory of Catalysis, 71A D.
A great part of the research for hydrotreament is nowadays looking at new materials as catalysts support, among these, ordered mesoporous silicas (MCM-41, HMS, SBA-15) have recently attracted much interest due to their high surface area and controlled porosity [1-2]. With respect to the CoMo or NiMo type of HDS catalysts, an important role is played by the active species particle sizes. In the present study, aiming to increase the dispersion and consequently the activity of the supported active phase, siliceous HMS and SBA-15 were functionalised with mercaptosilanes by direct synthesis [3-4]. The silica oxides were then functionalized with –C3H6SH groups by co-condensation of tetrahetylorthosilicate (TEOS) and 3 mercaptopropyltriethoxisilane (3-MPTS) in the presence of the appropriate surfactant, either dodecylammine for the synthesis of HMS and Pluronic P123 for the SBA-15. The functionalised silica were then used as supports for CoMo catalysts. The CoMo catalysts were obtained by co-impregnation of aqueous solution of (NH4)6Mo7O24·4H2O (Mo= 7wt%) and Co(NO3)2 6H2O (Co=1.7wt%), followed by 2 h drying at 120°C and 4 h calcinations at 400°C. The materials were characterized by N2 physisorption, XRD (SAXS and WAXS), TEM and XPS. Their catalytic activity was tested in the HDS of thiophene. An effect of the support morphology was observed with the HMS giving rise to more active catalysts. According to TEM and XPS analyses, the functionalization of the support did not increase the metal dispersion. However, the thiol modified supports contributed to an increased activity of the supported CoMo catalysts. According to the XPS results the main effect of the thiol group consisted in a decrease of the Mo(VI) and Co(II) reducibility and less sulphides formation. References [1] A. Corma, A. Martinez, V. Martinez-Soria, J. Catal. 169 (1997), 480. [2] T. A. Zepeda, B. Pawelec, J. L. G. Fierro, A. Olivas, S. Fuentes, T. Halachev, Micropor. Mesop. Mater. 111 (2008) 157. [3] N. Marin-Astorga, G. Pecchi, T. J. Pinnavaia, G. Alvez-Manoli, P. Reyes, J. Mol. Catal. A 247 (2006) 145. [4] D. Zhao, J. ius, Q. Huo, N. Melosh, G.H. Fredrickson, B.F. Chmelka, G.D. Stucky, Science 279 (1998) 548-552.
Benahavís (Málaga, Spain), 21st to 23rd of October, 2009 57
Structural flexibility in ~SbVO 4
Angel R. Landa Cánovas *,a, F: Javier García-García b and Staffan Hansen c
aInstituto de Ciencia de Materiales de Madrid, CSIC, E-28049, Madrid, Spain.
bLehrstuhl für Festkörperchemie, Institut für Physik, UniVersität Augsburg, UniVersitätsstrasse 1, D-86159 Augsburg (Germany).
c Division of Polymer and Materials Chemistry, Department of Chemistry, Lund University, Chemical Center, P.O. Box 124, SE-221 00 Lund, Sweden
Objective ~SbVO4 is a well known catalyst used for the ammoxidation of propane to acrylonitrile. Vanadium site isolation as well as cooperation with Sb2O4 is considered to be very important for the catalysis. However, very little importance is given to the structural flexibility exhibited by ~SbVO4 phase. The purpose of this communication is to highlight the high structural flexibility exhibited by this phase accommodating the changes in the oxidation state of vanadium which is accompanied by the introduction of cation vacancies. Results ~SbVO4 presents rutile-type structure with antimony in 5+ oxidation state while vanadium ranges from 3+ to 4+ depending on the reaction conditions. ~SbVO4 exhibits a wide range of non-stoichiometry [1] since substitution of V3+ by V4+ requires the introduction of cation vacancies, see reaction 1 in Figure 1. Besides, Sb5+ can substitute vanadium according reaction 2 or according reaction 3 in Figure 1. The whole existence interval of ~SbVO4 is given by the general expression Sb5+
8/9 –yV4+
2/9+3x+2yV3+
8/9-4x-yxO4 where 0 ≤ x ≤ 2/9 and 0 ≤ y ≤ 8/9-4x. In this way, up to almost 1/8 of the total cation positions can be vacant. However, ~SbVO4 rutile phase can accommodate such a big amount of cation vacancies in a very soft way, through vacancy population waves which give rise to incommensurate structural modulations, without the collapse of the structure as it usually happens in other rutile phases through crystallographic shear defects. In Figure 2 we show a high resolution TEM micrograph showing bright white dots due to the rutile cation positions, with their intensity scanned in the upper right graph, and wider dark bands generated by the soft accommodation of cation vacancies in the rutile structure with their intensity scanned in the lower right graph.
Figure 1.- Diagram showing the existence margins for the rutile-type SbVO4.
58 Benahavís (Málaga, Spain), 21st to 23rd of October, 2009
Figure 2 -a. High resolution TEM image of oxidized SbVO4 phase showing bright white dots which correspond to the cationic rutile positions and wide dark bands generated by cationic vacancies. Intensity line scan graphics of the lattice rutile positions (-b) and of the cation vacancy waves (-c). Reference [1] A.R. Landa-Cánovas, J. Nilsson, S. Hansen, K. Ståhl and A. Andersson. J. Solid State Chem. 116, 369-377 (1995)
Objective Among the different methods of synthesis of nanoparticles (NPs) for catalytic purposes, chemical synthesis is the most used strategy [1]. Electrochemical synthesis exhibits some advantages when NPs are going to be used for electrocatalysis [2,3]. The main improvement is that NPs are strongly attached to the electrode in only one step. The size, shape and number of deposited nanoparticles depend dramatically on the electrode material. Conducting polymers (CP) have been used as support for catalysts and are promising materials because CP are good dispersing material for NPs and introduce a high porosity and roughness, which generate a large surface area. However, the method used for the CP and NPs synthesis has a deep influence on the nanocomposite catalytic properties. The main objective of this work is to electrosynthesize Pt nanoparticles with high catalytic activity for methanol electrooxidation. We present in this communication the synthesis of poly(3,4-ethylenedioxythiophene) (PEDOT), as support for Pt NPs (PEDOT/Pt), in aqueous media without using any surfactant. Results Electropolymerization of 3,4-ethylenedioxythiophene (EDOT) was performed in absence of surfactants by applying an anodic potential high enough to oxidize the monomer and form a polymer film on a glassy carbon electrode. Monomer concentration, time and applied potential are key parameters to generate a film with good properties for hosting and dispersing catalytic particles. Pt Nanoparticles were formed on PEDOT modified electrode using cyclic voltammetry in a PtCl6
2- solution. In this case, PtCl62- concentration, potential, number of cycles and scan rate are
the most important parameters to control and form high catalytic nanostructures. The synthesis of the nanocomposites has been followed using in-situ spectroelectrochemistry. Changes in the reflectance of the electrode provide valuable information on the deposition process. The catalytic materials were also characterized using in-situ Raman spectroscopy at open circuit potential. Raman spectra of PEDOT/Pt NPs composites were very similar to that of PEDOT films [4,5]: the oxyethylene ring deformation, symmetric C-S-C deformation, C-O-C deformation, Cα-Cα’ stretching and symmetric Cα=Cβ(-O) stretching were observed in the two materials.
The nanocomposites were used as catalyst for the oxidation of methanol. When appropriate synthesis conditions were used, the typical voltammogram of methanol was obtained. A forward oxidation peak appears around +0.70 V vs. Ag/AgCl, and a backward oxidation peak appears around +0.57 V. Figure shows a SEM image of one of the PEDOT/Pt composites generated on the glassy carbon electrode
60 Benahavís (Málaga, Spain), 21st to 23rd of October, 2009
Conclusions It is possible to synthesize electrochemically a PEDOT/Pt NPs composite in two consecutive steps: potentiostatic polymerization of EDOT and potentiodynamic reduction of PtCl6
2- on the PEDOT film. UV-Visible spectroelectrochemistry and Raman spectroscopy using a 785 nm laser have demonstrated to be very useful techniques to characterize these materials. The voltammetric reduction of PtCl6
2- on PEDOT film generates Pt nanoparticles homogeneously distributed on the conducting polymer matrix. Raman spectra confirm that PEDOT film is obtained successfully in absence of any kind of surfactant in aqueous media and it is modified by Pt nanoparticles. The electrosynthesized composite is a good catalyst for the oxidation of methanol. Acknowledgements Support from University of Burgos, Caja de Burgos, Ministerio de Ciencia e Innovación (MAT2006-13875), Junta de Castilla y León (GR71, BU006A09, BU012A09), COST Action D36 (WG D36-0005-06), Academy of Finland (V.R., Academy Research Fellowship) is acknowledged. References [1] Drillet, J. F.; Dittmeyer, R.; Jüttner, K.; Li, L.; Mangold, K. M. Fuel Cells 6, (2006) 432. [2] Kuo, C. W.; Huang, L. M.; Wen, T. C.; Gopalan, A. J. Power Sources 160 (2006) 65. [3] Patra, S.; Munichandraiah, N. Langmuir 25 (2009) 1732. [4] Garreau, S. ; Louarn, G. ; Buisson, J.P. ; Froyer, G. ; Lefrant, S. Macromolecules 32, (1999) 6807. [5] Zhang, L. ; Peng, H. ; Kilmartin, P.A. ; Soeller, C. ; Travas-Sejdic, J. Macromolecules 41, ( 2008) 7671.
Summary and Objectives Colloidal based synthetic methods provide delicate control over size, composition and shape of metallic and bi-metallic nanostructures. These developments have allowed unravelling fascinating catalytic properties, particularly in system involving Au and Pd nanoparticles [1]. The present contribution focuses on the electrochemical properties of ultrathin Pd shells grown on Au nanoparticles towards hydrogen loading. The key objectives of the work include:
• Synthesis and characterisation of Pd@Au nanostructures by seeding-growth methods • Two and three dimensional assemblies of the nanostructures via electrostatic layer-by-
layer adsorption • Study of the evolution of voltammetric responses associated with hydrogen adsorption
and absorption as a function of the nanoparticle dimensions and the electrode potential The experimental results demonstrate that layer-by-layer adsorption employing electrochemically inactive polyelectrolytes generates three dimensional networks of electrically connected metal nanostructures. The intrinsic high surface area of the 3D assemblies promotes a significant enhancement of surface sensitive electrochemical responses in comparison to extended surfaces. This property has lead to the observation of unexpected electrochemical properties associated with the hydrogen storage in Pd nanoshells. Contrary to experimental work carried out at thin Pd layers on flat Au surfaces, our results show a large hydrogen supersaturation for Pd layers of less than 10 nm. Results and Conclusions The synthesis of Au-Pd nanostructures involves a two step process initiated by the nucleation of Au particles with an average diameter of 19.3±1.2 nm employing sodium citrate as reductant and stabiliser. The second step involves the nucleation of Pd onto as-grown Au colloids by reduction of PdCl4
2- in the presence of ascorbic acid [2]. By controlling the amount of the Pd precursor added in the second step, the thickness of the Pd layer can be tuned as shown in table 1. Pd nanoparticles were also synthesised following a similar protocol as for the Au nanoparticles, yielding an average diameter of 10.1±1.8 nm. Characterisation of the monometallic and core-shell nanostructures by HRTEM-EDX, electron diffraction and XRD and will be presented in this contribution. The results in table 1 also show that the mass ratio of core-shell nanostructures as expected from the synthesis conditions (first column) closely matches with the values obtained from EDX. The sequential electrostatic adsorption of poly-l-lysine (PLL) and the negatively charged metal nanoparticles leads to the generation of 2D and 3D assemblies as illustrated in figure 1. This process allows controlling the number density of nanoparticles [3] and the effective surface roughness of the electrode [4]. Previous studies have demonstrated strong electronic coupling between nanostructures and metal electrodes linked via ultrathin PLL films [5].
62 Benahavís (Málaga, Spain), 21st to 23rd of October, 2009
Table 1 . Dimensions and composition of the synthesised Au-Pd nanostructures
Mass ratio (synthesis)
Diameter/nm Pd thickness/nm
Mass ratio (EDX)
Au 19.3 ± 1.2 --- --- Pd20 Au80
21.8 ± 1.1 1.3 ± 0.9 18.7 ± 2.4 81.3 ± 2.4
Pd40 Au60
24.7 ± 1.3 2.7 ± 1.0 38.1 ± 1.1 61.9 ± 1.1
Pd60 Au40
29.5 ± 1.2 5.1 ± 0.9 57.8 ± 0.9 42.2 ± 0.9
Pd80 Au20
38.9 ± 1.5 9.9 ± 1.1 82.1 ± 0.71 17.9 ± 0.71
Pd 10.1 ± 1.8 ---- ----
A
B
Figure 1. Two (A) and three (B) dimensional assemblies of citrate stabilised Au nanoparticles generated by electrostatic layer-by-layer adsorption employing poly-L-lysine. The electrochemical responses associated with hydrogen loading into the Pd domains in 3D nanostructured assemblies were investigated in acid solutions. Responses arising from H adsorption were effectively deconvoluted from the volume dependent absorption. As expected, the charge originating from H absorption increases as the thickness (volume) of the Pd shell increases. However, detailed analysis of the hydrogen loading charges vs. the effective surface roughness revealed unexpectedly high H/Pd ratios which have not been previously reported for either nanostructured or extended surfaces. The origin of the high H-loading will be discussed in terms of lattice stress at the Au-Pd boundary. The structure of the ultrathin Pd layer also affects the energetics associated with the electrochemical formation of atomic layers, e.g. Te underpotential deposition. Acknowledgement We gratefully acknowledge the support by the Mexican National Council for Science and Technology (CONACYT) and the ESF-COST Action D36/005/06. References 1. (a) Weijiang Zhou, J. Y. L. Electrochem. Comm. 2007, 9, 1725–1729, (b) A. Sarkany, O. Geszti, G. Safran. App. Cat. A – General 2008, 350, 157. 2. L. Lehui, H. Wang, X. Shiquan and H. Zhang. J. Mat. Chem. 2002, 12, 156. 3. (a) Zhao J., Bradbury C.R., Huclova S., Potapova I., Carrara M. and Fermín D.J. J. Phys. Chem. B 2005, 109, 22985, (b) F. Li, I. Ciani, P. Bertoncello, P.R. Unwin, J. Zhao, C.R. Bradbury and D.J. Fermin. J. Phys. Chem C, 2008, 112, 9686. 4. M. Carrara, J.J. Kakkassery, J.P. Abid, D.J. Fermín. ChemPhysChem, 2004, 5, 571. 5. J. Zhao, C.R. Bradbury and D.J. Fermín, J. Phys. Chem. C, 2008, 112, 6832
Benahavís (Málaga, Spain), 21st to 23rd of October, 2009 63
One More Step Closer to Realizing the Dream of the Polymeric RGB Electrochromics
Merve Đçli a, Fatih Algı b, Atilla Cihaner c*, Ahmet M. Önal a
a Department of Chemistry, Middle East Technical University, TR-06531 Ankara, Turkey.
b Laboratory of Organic Materials, Çanakkale Onsekiz Mart University, TR-17100 Çanakkale, Turkey.c Chemistry Group, Faculty of Engineering, Atılım University, TR-06836 Ankara,Turkey. :
*[email protected] Design, synthesis, and properties of a novel series of donor-acceptor type conducting materials, namely poly(4,7-di-2-thienyl-2,1,3-benzoselenadiazole) (PTSeT), poly(4,7-di-2,3-dihydrothieno[3,4-b][1,4]dioxin-5-yl-2,1,3-benzoselenadiazole) (PESeE) and poly(4,7-bis(3,3-didecyl-3,4-dihydro-2H-thieno[3,4-b][1,4]dioxepin-6-yl)-2,1,3-benzoselenadiazole) (PPSeP) were highlighted (Chart 1). The role of donor units on the electronic and optical properties of the conducting polymers was investigated. The unique combination of benzoselenadiazole unit and electron rich donor units provides an ambipolar (n- and p-doping processes) low band gap polymers between 1.46 and 1.05 eV and they are exceptionally stable (even after prolonged standing at ambient conditions). PPSeP is the first processable neutral state green polymer with three absorption bands that can simultaneously be controlled by an applied potential. Furthermore, PESeE and PPSeP show electrochromic behaviour with a color change from green to highly transparent color in an extremely low switching time(0.6 s) during oxidation with a high coloration efficiency (208 cm2/C).
NSe
N
SS
NSe
N
S
OO
C10H21 C10H21
S
OO
C10H21 C10H21
NSe
N
ESeE
SS
OO OO
TSeT PSeP
Chart 1. 2,1,3-Benzoselenadiazole based D-A systems. The voltammogram of PseP which was recorded in 0.1 M tetrabutylammonium hexafluorophosphate (TBAH)/CH2Cl2 (DCM) solution exhibited an irreversible oxidation peak
( oxamE , ) at 0.50 V vs. Fc/Fc+ in the positive side, which was attributed to the oxidation of external
P units, and a reversible reduction peak ( redmE 2/1, ) at -1.64 V in the negative side, which was
ascribed to the radical anion formation from the central 2,1,3-benzoselenadiazole scaffold, during the anodic and cathodic scans, respectively. These values indicated that the electronic
nature of PseP is between TseT ( oxamE , = 0.75 V and red
mE 2/1, = -1.62 V) and EseE ( oxamE , = 0.35 V
and redmE 2/1, = -1.72 V), as expected when considering the nature of the D-units.1
The polymerization of PseP to get PPSeP was performed via electrochemically and after repetitive anodic scans, a new reversible redox couple appeared, which clearly indicated the formation of an electroactive polymer film on the electrode surface. Also, an increase in the thickness of the polymer film was confirmed by intensified current after each successive cycle. The as-prepared polymer film was both green in its neutral state and highly soluble in common organic solvents such as DCM, CHCl3, diethylether, since processability is vital for the applications of the neutral state green polymers to electrochromic devices and displays. It is noteworthy that this rational design where judicious selection of A-parts, in this case P units, provided access to a unique solution processable neutral state green PEC bearing 2,1,3-benzoselenadiazole scaffold. PPSeP exhibited a single and well-defined reversible redox
64 Benahavís (Málaga, Spain), 21st to 23rd of October, 2009
couple ( oxpE 2/1, = 0.16 V vs. Fc/Fc+) which was consistent with the redox behaviours of PTSeT
( oxpE 2/1, = 0.43 V) and PESeE ( ox
pE 2/1, = -0.59 V).
PPSeP exhibits three absorption bands at 343 nm (3.62 eV), 425 nm (2.92 eV) and 715 nm (1.73 eV), two of which (<500 nm) absorb the red color and the latter (>700 nm) absorbs blue (Fig. 1). The band gap (Eg) value for PPSeP on the basis of the low energy end of π-π* transitions at 715 nm was found to be 1.37 eV which is between the Eg values of PESeE (1.05 eV) and PTSeT (1.46 eV).
A novel processable neutral state green polymer (PPSeP) exhibiting low switching time and exceptional redox stability is highlighted. The polymer film shows electrochromic behavior: a color change from green to highly transparent color in an low switching time with high coloration efficiency. Furthermore, it is exceptionally stable even after prolonged standing under ambient conditions. Studies to get novel neutral state green polymeric electrochromics are under way and will form the basis of future reports, while the synthesis and engineering of derivatives with modulated intrinsic properties will also be pursued. References [1] A. Cihaner, F. Algı, Adv. Funct. Mater. 18 (2008) 3583. [2] Jayakannan, M.; Hal, P. A. V.; Janssen, R. A. J. J. Polym. Sci., Part A: Polym. Chem. 2002, 40, 251.
Benahavís (Málaga, Spain), 21st to 23rd of October, 2009 65
Modelling of Au/FeO x interface by in situ Sum Frequency Generation Tech nique
Z.Pászti 1, and L. Guczi 1,2
Chemical Research Center, Institute of Nanochemistry and Catalysis, Hungarian Academy of Sciences, P. O. Box 17, H1525 Budapest, Hungary, Institute of Isotopes, Hungarian Academy
of Sciences, P. O. Box 77, H-1525 Budapest, Hungary Abstract In this work a complex surface analytical unit developed by connection of a multi-technical surface analysis system and sum frequency generation vibrational spectroscopy (SFG), provides a unique technique to study interfaces under in situ condition. Preliminary results of CO adsorption experiments on gold and iron-oxide thin layers grown on gold were presented as application examples of the experimental setup. In our previous studies it has been established that the Au/oxide interface played a prevailing role in the CO oxidation both in model Au/FeOx sytems. It was suggested that the CO activation occurred at the perimeter of the gold-oxide interface [1-3]. Deeper understanding the processes occurring at interfaces between solid materials and their environment in situ or even operando investigation techniques, especially in connection with catalysis related solid-gas interfaces is required [4], where optical spectroscopic methods seem to provide particularly useful information about adsorption and transformation of the adsorbed species. Due to its inherent properties, (SFG) offers new possibilities for exploring gas adsorption processes and reactions important from the catalytic point of view in model catalytic systems. In our laboratory we developed a very promising experimental setup by connecting the SFG spectrometer to a multi-technique surface analysis system via a suitably designed chamber capable of experiments at elevated pressures equipped a heating/cooling system. Such a complex instrument allows sample preparation and characterization according to the standards of traditional surface science as well as in situ determination of the gas adsorption/transformation properties of the sample at ambient pressure. In this apparatus we have investigated CO adsorption properties of iron-oxide thin films deposited on gold substrates (film or single crystal). SFG is a second order nonlinear optical spectroscopy technique with monolayer sensitivity. The method is inherently surface/interface specific in most practical cases, as second order nonlinear optical processes are forbidden in the bulk of centro-symmetric media (e.g. metals, liquids, gases, etc.), therefore the sum frequency signal is generated only at interfaces, where the centro-symmetry is necessarily broken. SFG provides characteristic vibration frequencies resulting from the interfacial species, using two energetic laser beams which have to be overlapped temporally and spatially at the surface/interface of the sample. One of the exciting
light beams is a tunable IR beam ( )IRω , the other has a fixed frequency ( )Visω in the visible.
The frequency (photon energy) of the coherently generated outgoing signal beam ( )SFGω is
equal to the sum of the two exciting frequencies (photon energies) ( VisIRSFG ωωω += ). The
sum-frequency intensity is resonantly enhanced when the IR frequency matches the frequency of a molecular vibration. 100 nm polycrystalline gold film was evaporated onto a Si(100) wafer covered by 100 nm SiO2. The sample was cleaned by cyclic argon ion bombardment treatments (3 keV, 10 min) in the analytical chamber of the UHV system at room temperature. The cleanliness of the surface was checked by UPS and XPS measurements. Iron-oxide layers were formed on the cleaned support by electron-beam evaporation from an iron rod on 4x10-7 mbar O2 at room temperature. The layers were characterized by XPS and UPS measurements. On CO adsorption on polycrystalline gold in vacuum gives a featureless but relatively intensive signal was observed, which corresponds to the non-resonant background of the metallic Au
66 Benahavís (Málaga, Spain), 21st to 23rd of October, 2009
substrate. Exposure to 5 mbar CO results in a broad negative going band around 2035 cm-1 and two narrow bands at 2105 and 2180 cm-1. While the intensity of the 2035 cm-1 band remains unchanged up to 10 mbar, the narrow bands become much more pronounced at the higher pressure. If the chamber is evacuated, all CO related signal disappears. If the temperature is increased, very little change can be observed even up to room temperature. Since the gas phase infrared spectrum of molecular CO shows two strong absorption peaks around 2115 and 2175 cm-1, it is very probable that the two narrow bands observed in our spectra are due to the decrease of the intensity of the infrared excitation in those spectral regions caused by gas phase absorption and are not related to the sum frequency generation process (the SFG intensity is proportional to the product of the intensities of the visible and infrared excitations). This assignment is confirmed by the fact that the intensity of these bands is closely related to the pressure of CO. In the other hand, the 2035 cm-1 band is certainly due to adsorbed CO. Although this frequency is significantly lower than that usually reported for CO on supported Au nanoparticles, it is not very far from the value reported for CO on Au(111) in Ref. [5] (2060 cm-1), which was assigned to chemisorbed CO on top sites. In the same work shifts in the CO frequency to even lower values is predicted for certain crystal planes like (331) [6]. The gold supported iron-oxide thin film was studied by XPS prior to SFG measurements. A quantitative evaluation of the intensities of the Fe, O and Au lines performed by the XPS MultiQuant software indicated that the thickness of the films is around 2 and 8 nm, respectively. Preliminary SFG results for CO interaction with the 2 and 8 nm iron-oxide films show that peaks around 2090 cm-1 and a narrower one around 2175 cm-1. The pressure dependence of this band confirms its assignation: it is already obviously present at 1 mbar, where the gas phase absorption related contribution is hardly noticeable. Measurements of the SFG spectra of the CO stretching region on the 8 nm iron-oxide film were similarly conducted as described for the bare gold sample. If CO is present in the chamber, the spectra contain the doublet at 2105 and 2175 cm-1, with higher intensity for higher pressures. If CO is removed, practically no signal is observable even at low temperatures. Our data thus indicate that the CO adsorption properties of the 2 and 8 nm iron-oxide layers are different. Nevertheless, it still remains a question if this difference is connected to the thickness or the composition of the films, which is in the focus of our current research efforts. In the future work the single crystal will be investigated. References [1] [L. Guczi, D. Horváth, Z. Pászti, L.Tóth, Z. E. Horváth, A. Karacs and G. Petı, J. Phys. Chem B., 104, 3183 (2000), [2] László Guczi, Gábor Petı, Andrea Beck, Krisztina Frey, Olga Geszti, György Molnár and Csaba Daróczi, J. Am. Chem. Soc. 125, 4332 (2003) [3] László Guczi, Krisztina Frey, Andrea Beck, Gábor Petõ, Csaba Daróczi, Norbert Kruse and Sergey Chenakin, Appl. Catal. A., 291, 116 (2005)] [4] G. Rupprechter, Catal. Today 126, 3 (2007) [5] L. Piccolo, D. Loffreda, F.J. Cadete Santos Aires, C. Deranlot,Y. Jugnet, P. Sautet, J.C. Bertolini, Surf. Sci. 566-568, 995 (2004) [6] T. Keszthelyi, Z. Pászti,T. Rigó,O. Hakkel, J. Telegdi, L. Guczi, J. Phys. Chem. B 110, 8701 (2006)
Benahavís (Málaga, Spain), 21st to 23rd of October, 2009 67
The effect of the Mo precursor on the nanostructure and activity of PtRuMo electrocatalysts for Proton Exchange Membrane Fuel Cells
N. Tsiouvaras, M.V. Martínez-Huerta, J.L.G. Fierro and M.A. Peña
Instituto de Catálisis y Petroleoquímica, CSIC, 28049 Madrid, Spain
Introduction The state of the art anodic catalysts for Proton Exchange Membrane Fuel Cells (PEMFC), feed with H2/CO or methanol in Direct Methanol Fuel Cells (DMFC), are binary catalysts based on carbon supported PtRu nanoparticles. However, these bimetallic systems are costly and not efficient enough for its implementation [1, 2]. Exploring ternary PtRuMo catalysts is some of the most interesting approach for improving their performance. Mo is a transition metal with several advantages as it has been described extensively in literature in binary PtMo systems, although the Mo role has yet to be fully determined. Structural characteristics as compositions, chemical state, degree of alloying, particle size and the stability of Mo in ternary PtRuMo/C systems are not clear and more studies are necessary in order to further comprehend their full effect in the CO and methanol electrooxidation. In the present work the effect of the Mo precursor on the activity for CO electrooxidation has been investigated. Different Mo precursor as MoCl5, MoO3
and (NH4)6Mo7O24 leads to the incorporation of different Mo phases as Mo (V), Mo (VI) and Mo (V)/Mo (VI) mixed phases, respectively. Consequently can alter the surface chemistry of nanoparticles and affect their electroactivity. Experimental For the catalyst preparation a two step procedure has been employed [3]. In a first step the carbon support was impregnated with the respective Mo precursor in MoO/C, MoNH/C and MoCl/C supports (see Table 1). In a second step 20 wt. % of PtRu (1:1) has been incorporated to the Mo/C supports following a colloidal technique. In order to study a representative Mo phases from each precursor during electrochemical measurements, double amount of Mo was used with MoO3 precursor (20 wt. %) with respect to other supports (10 wt.%) due to fact that crystal phases of MoO3 was easily dissolved into the electrolyte (0.5 M H2SO4). Electrocatalysts as well as support characterization has been carried out through various physicochemical (XRD, HRTEM, XPS, TPR, TXRF and TG) and electrochemical techniques as cyclic voltammetries and current-time curves.
Table 1 . Prepared materials
Results and discussion XRD analysis of Mo/C supports presents no crystal phases in MoCl/C and MoNH/C supports. However, MoO/C presents diffraction peaks typical of crystalline MoO3. XRD patterns of the ternary samples show the characteristic diffraction lines of Pt0 metal with a low degree of crystallinity and the absence of Mo, Ru and crystalline metals oxides. The average particle size was estimated from XRD patterns (Table 1) and also from TEM images and histograms, and they are in the range 2-3 nm for all catalysts. XPS results point that the incorporation of PtRu over different Mo/C supports generate mainly Mo phases with oxidation states between Mo(V)-Mo(VI), and Mo(VI) phases with lower contributions in PRMCl and PRMNH catalysts. PRMO show the highest percentage of Mo (VI) (> 80%).
68 Benahavís (Málaga, Spain), 21st to 23rd of October, 2009
TPR analysis of the catalysts presented in Figure 1 show different profiles depending of the precursor used. The incorporation of Pt and Ru to these supports affects even more greatly their reduction due to metal interaction phenomena like H2 spill over from the active platinum sites. In general, TPR peaks between 30 and 70 °C a re due to partially oxidized platinum, and reduction peaks between 99 and 131 °C correspond to a RuOxHy species. The other peaks in PRMO and PRMCl are assigned to molybdenum oxide species. In the case of PRMNH the reduction peaks at different temperatures could indicate particular interaction between nanoparticles and carbon support with respect to other catalysts
These metal interactions in PRMNH catalyst also cause a shift to lower onset potentials of about 0.1V of the COads oxidation to CO2 compared with PRMCl (Figure 2). That means that the activity in the CO electrooxidation is higher when ammonium molybdate is used as precursor. The presence of higher percentage of Mo(VI) in PRMO catalyst decreases dramatically the activity in the CO oxidation
Conclusions These results clearly demonstrate the importance of the Mo precursor in the preparation procedure of ternary electrocatalyst PtRuMo/C and the fact that highly oxidized molybdenum phases are not desirable in this kind of systems.
Acknowledges This research was funded by the Ministry of Science and Innovation, Spain (Project ENE2007-67533-C02-01). M.V.M.-H. acknowledges the Ramon y Cajal program of the Ministry of Science and Innovation of Spain for financial support. References [1] Antolini, E. Journal of Applied Electrochemistry 34 (2004) 563 [2] Wee,J.H. and Lee,K.Y. Journal of Power Sources 157 (2006) 128 [3] Martinez-Huerta, M.V., Rodriguez, J.L., Tsiouvaras, N., Peña, M.A., Fierro, J.L.G. and Pastor E., Chemistry of Materials 20 (2008) 4249
Benahavís (Málaga, Spain), 21st to 23rd of October, 2009 69
Electrocatalytic activity of polypyrrole films inco rporating palladium particles
Ana Mourato, Joana S.Cabrita, Luisa M. Abrantes *
CQB,Departamento de Química e Bioquímica, Faculdade de Ciência da Universidade de Lisboa, Campo Grande, 1749-016 Lisboa, Portugal
*luisa.abrantes@ fc.ul.pt Objective Displaying a high surface area and providing an efficient route for the flow of electronic charges to and from the catalyst centres, conducting polymer films have been employed as supporting matrices for the immobilization of metal particles. For this purpose, chemical or electrochemical procedures [1,2] can be pursued, including the electroless precipitation, already reported for the preparation of Polyaniline-Pd composites [3]. This approach, here exploited for the incorporation of palladium in polypyrrole (Ppy) films, relies on the spontaneous Ppy oxidation and simultaneous reduction of the metal ions, taking place when the polymer is immersed in a solution of appropriate pH; the process is auto-sustained as far as the film is exposed to the metal ion containing solution. The present work addresses essential aspects in the preparation of electrocatalytic Ppy-Pd modified electrodes, namely for observing a suitable dispersion of the metal particles deposited on the polymer matrix. Results The polymer features e.g. electroactivity, thickness, porosity and morphology, are imparted by the electrosynthesis conditions as revealed by the electrochemical (cyclic voltammetry (CV), electrochemical quartz crystal microbalance (EQCM)) and microscopic (SEM) characterization of Ppy layers prepared on Pt, under potentiostatic and potentiodynamic control. The rate and efficiency of the Pd up-take process is determined by the properties of Ppy films and by the nature and the pH of the palladium ion containing media, as illustrated by the open circuit potential measurements and EQCM data obtained from PdSO4 and PdC12 solutions. In the latter case, X-ray photoelectron spectroscopy (XPS) analysis of the loaded polymers shows that the acidity is crucial to hinder the predominance of Pd(II) species over Pd(0) in the polymeric matrix. The electrocatalytic activity of the modified Ppy films towards the anodic oxidation of hydrazine in KCl solution is also analysed. Conclusions The electroless precipitation of Pd on Ppy occurs mainly on the surface of the polymer; although the size and distribution of Pd particles correlate to the film porosity, an increase in the amount of polymer is not replicated on the deposited Pd. For pristine Ppy, under the studied conditions and within 0.0 to 0.6 V vs. SCE potential range, the response to hydrazine oxidation is featureless, whereas for Ppy-Pd films important catalytic anodic currents are observed. Notwithstanding care must be taken to avoid a disadvantageous increase on the number and size of the Pd particles since the best behaviour is attained with Ppy matrices bearing uniform dispersions of sub-micron Pd particles. References [1] M. Ilieva, V. Tsakova, W. Erfurth, Electrochim. Acta, 52 (2006) 816–824. [2] M. Ocypa, M. Ptasinska, A. Michalska, K. Maksymiuk, E.A.H. Hall, J. Electroanal. Chem., 596 (2006) 157–168. [3] A. Mourato, A.S. Viana, J.P. Correia, H. Siegenthaler, L.M. Abrantes, Electrochim. Acta, 49 (2004) 2249–2257.
Benahavís (Málaga, Spain), 21st to 23rd of October, 2009 71
Bio-inspired electrochemistry: Hydrogen evolution a nd oxygen reduction at soft interfaces
Hubert H.Girault – WP7
LEPA, Ecole Polytechnique Fédérale de Lausanne, Lausanne, CH-1015 (Switzerland)
[email protected] Abstract As biomembranes, polarised liquid-liquid interfaces provide a spatial separation of reactants and products and can be the locus of different types of charge transfer reactions such as ion transfer, acid-base or heterogeneous electron transfer reactions.
Charge transfer at a polarised liquid-liquid interface: a) ion transfer reaction, b) assisted proton transfer reaction and c) hetero-geneous electron transfer reactions between an aqueous acceptor O1 and a lipophilic donor R2 As with biomembranes, all those different reactions are coupled. By controlling the polarisation of the interface, and to a certain extent the potential distribution across the interface, we have an electrochemical control of the different reactions. Electrochemistry at the Interface between Two Immiscible Electrolyte Solutions (ITIES) has developed over the last decades as a rather well characterised field of research. From a charge transfer viewpoint, most of the electrochemical methodologies developed to study charge transfer reactions at solid electrodes have been transposed taking in to account the different types of mass transfer processes. Recently within the project D36-WP7, we have developed the concept of electrocatalysis at ITIES using porphyrins as electrocatalysts for oxygen reduction [1]. In particular, we shall discuss proton coupled electron transfer reactions at ITIES that are relevant for energy research. Results with different porphyrins including free base porphyrins will be presented.
Oxygen reduction catalysed by CoTPP with electro-chemical proton pumping
Finally we have also shown that the protonation of decamethylferrocene at ITIES leads to the production of hydrogen in anaerobic conditions [2], and to the production of hydrogen peroxide
72 Benahavís (Málaga, Spain), 21st to 23rd of October, 2009
in the aerobic case (see below) [3]. We shall show how voltammetry at ITIES can be used to elucidate the reaction mechanism.
Oxygen reduction by decamethylferrocene (DMFc) control-led by proton transfer reaction. The liquid|liquid interface is pola-rised by the choice of the hydrophilic salt lithium tetra(pentafluoro-phenyl)-borate (LiTB) and of the lipophilic salt bis(triphenylphos-phoranylidene)ammonium tetra(pentafluorophenyl)borate (BATB) References [1] R.P. Nia, B. Su, F. Li, C.P. Gros, J.-M. Barbe and H.H. Girault, Chem. Eur. J., 2009, 15, 2335 [2] I. Hatay, B. Su, F. Li, R. Partovi-Nia, H. Vrubel, X.L. Hu, M. Ersoz and H. H. Girault, Angew. Chem., 2009, 48, 5139 [3] B. Su, R.P. Nia, F. Li, M. Hojeij, M. Prudent, C. Corminboeuf, Z. Samec, and H. H. Girault, Angew. Chem., 2008, 47, 4675
Objectives There is a need to develop electrocatalytic materials appropriate for oxygen insertion reactions to double bonds [1]. Specific reactivity could be achieved using metal nanostructured electrodes. Experimental studies indicated the preferable carbon dioxide formation in the case of the oxidation of ethylene and propylene on platinum, the same processes on gold surface yields a mixture of partially oxidized product. The current understanding of these reactions is, at present, rather limited. Considering these limitations of the state of the art, the research program for investigation of fundamental aspects of reactivity at bulk gold and platinum nanostructured electrodes is desirable. Theoretical calculations of reactivity and the potential energy curves for different reactant and product conformations would be done. This work addresses theoretical aspects of the electronic structure of reaction intermediates, ionic distribution at the cluster – solution interface and the more fundamental aspects of looking for transition states in order to map the reaction coordinate of reactions that takes place in ethylene oxidation at metal nanoparticles, modeled by Aun and Ptn (n=10-25) clusters. Results
DFT method was used for the modeling of gold and platinum clusters up to size Au25 and Pt25 and their interaction with organic molecules. The mechanisms of the O and C2H4 adsorption at the metal clusters of varying size, the formation of Mx-Et-O intermediates and the possible intermediate transfer under the influence of electric field or electrode reaction
were studied. During the geometry optimization and transition structure search the geometry of clusters was fixed, the remaining part was fully optimized. By the interaction of the adsorbed oxygen with ethylene the stable surface oxametallacycle intermediates are formed analogously to ones described in the case of ethylene oxide reaction at Ag(111) surface [2]. The interaction with the surface depends on the type of the cluster, its size and the reaction site (the plane steps or edges). Figure shows the oxametallacycle intermediate on Au22 (left) and Pt21 (right). In the case of catalysis on gold, the transition state leading to oxirane lies 11.7 kcal/mol above the energy of the intermediate, the formation of acetaldehyde is barrierless. The results of the DFT calculations are in accordance with experimental results of electrocatalytic oxidation of ethylene on both gold and platinum. The on-line differential electrochemical mass spectrometry (DEMS) identifies in the case of oxidation on gold preferential formation of acetaldehyde (with carbon dioxide forming a minor reaction product); in the case of Pt electrodes the reaction produces solely the carbon dioxide. In both cases the oxidation processes gets inhibited by the metal surface oxidation and is accompanied with metal dissolution. The geometry of the intermediates is only slightly influenced by the static electric field corresponding to the electrochemical potential around 1 V or by the influence of solvent cavity. External effects more strongly influence transition states geometries and energetics.
74 Benahavís (Málaga, Spain), 21st to 23rd of October, 2009
Conclusions The anodic oxidation of ethylene on gold nanostructures leads to a formation of acetaldehyde as a major product. In the case of platinum the ethylene oxidation proceeds carbon dioxide is almost quantitatively formed. DFT calculations support experimental findings and indicate possible reaction mechanisms of catalytic reactions. DFT calculations points to the different reactivity on individual types of surfaces and different types of clusters. Acknowledgements This work was supported by COST D36 action and the Academy of Sciences of the Czech Republic (grant KAN100400702). References [1] S. Otsuka, I. Yamanaka, Catalysis Today 41 311 (1998). [2] S. Linic, M.A. Barteau, J. Am. Chem. Soc. 124 310 (2002).
Objective In this work, functional materials with catalytic interest in the electrooxidation of ethanol are studied by operando infrared techniques. A series of supported binary (Pt-Sn) and ternary (Pt-Sn-Rh) nanostructured materials supported on carbon were studied by Single Potential Alteration Infrared Reflectance Spectroscopy (SPAIRS) and Substractively Normalized Interfacial FTIR Spectroscopy (SNIFTIRS). This techniques allows the study of the double layer at the metal/solution interface. Results are then correlated with surface composition obtained by XPS and structural characterization (XRD and analytical microscopy). Addition of Rh to PtSn/C containing the Pt3Sn alloy phase seems to enhance ethanol electrooxidation at low potential provided Rh content is kept around 1%wt. Experimental Catalyst preparation PtRh/C and PtSn/C samples were prepared by conventional impregnation-reduction method. An aging step is required to avoid loss of volatile tin chlorides during the thermal treatments. Ternary samples were prepared by successive impregnation of the PtSn/C sample with a water solution of rhodium trichloride and subjected to the same thermal treatment. Total metal loading is 40%wt. Atomic Pt/Sn = 3:1, and Rh content is 3%wt in PtRh/C sampe and between 1 and 3 %wt in ternary samples. Electrochemical experiments and In situ IR reflectance spectroscopy measurements IR reflectance spectra in the wavenumber region 1000-3000 cm-1 were collected by a Fourier transform infrared spectrometer (Bruker IFS 66vs) with an incidence angle of 65º, after passing through the IR window (CaF2) of a conventional thin layer spectrochemical cell, using a Reversible Hydrogen Electrode (RHE), a gold wire and a gold disk as reference, counter and working electrodes, respectively. This apparatus was equipped with a spectral reflectance device allowing the observation of reflectance spectra of the electrode-electrolyte interface with the IR light beam passing entirely through a chamber under vacuum. Electrode reflectivity REi was recorded at different potentials, Ei, each separated by 50 mV at a sweep rate of 1 mV·s-1. A positive absorption band indicates the consumption of species and a negative absorption band means the production of species. Results Bimetallic PtRh/C samples resulted much less active than PtSn/C for the electrooxidation of ethanol. The addition of Rh to PtSn/C resulted in an increase in the performance of the catalyst at potentials less positive than 0.6 V [1,2]. Figure 1a shows forward scans of the linear voltammograms of the catalysts in 0.1 M HclO4 and 0.1 M EtOH. The current densities recorded for the ternary samples are the highest of the series. The performance of PtSn-1Rh/C is the best of the series at every recorded potentials, while that of the ternary sample with higher Rh content, PtSn-3Rh/C drops below that of PtSn/C at E ≥ 0.6 V. We used SPAIRS technique to identify reaction intermediates and products of ethanol electrooxidation. Figure 1b shows a set of representative SPAIR spectra of the species resulting of the oxidation of ethanol on PtSn-1Rh/C. The band at 1115 is attributed to the adsorption of the electrolyte. Bands at around 1293, 1393 and 2620 cm-1 correspond to the coupling between the elongation vibration ν(C-O) of
76 Benahavís (Málaga, Spain), 21st to 23rd of October, 2009
the C-O bond, the deformation mode δ(OH) of the –COOH group of acetic acid, and the vibration νCH, respectively. The intense band at 1715 cm-1 is assigned to the stretching mode ν(CO) from the carbonyl group [3]. The presence of this band is associated to the existence of intermediates adsorbed over the catalyst similar to acetaldehyde. The bands at 2343 and 2048 cm-1 are due to CO2 and CO, respectively. These two bands are more clearly seen in the SNIFTIRS spectra in Figure 1c.
Figure 1. a) Forward potential scan at 0.1 mV/s in 0.1 M HclO4 and 0.1 M EtOH solution of PtSn/C (black solid), PtRh/C
(gray solid), PtSn-1Rh/C (black dashed) and PtSn-3Rh/C (gray dashed); b) SPAIRS spectra recorded from the PtSn-1Rh/C at the same conditions that in Figure 1a; SNIFTIRS spectra of a selected region of the spectra comprising Co and CO2 bands obtained from the electrooxidation of ethanol with: c) PtSn/C, d) PtSn-1Rh/C, e) PtSn-3Rh/C, and f)
PtRh/C, recorded at the same conditions that Figure 1a. Conclusions Addition of Rh enhances the performance of PtSn/C for ethanol electrooxidation. According to SPAIRS data, Rh may promote the appearance of carbonyl species on the surface of the catalyst, while tin oxidize CO species. References [1] S. García-Rodríguez, F. Somodi, I. Borbáth, J.L. Margitfalvi, M.A. Peña, J.L.G. Fierro, S. Rojas, Appl. Catal. B, In press. [2] A. Kowal, M.Li, M. Shao, K. Sasaki, M.B. Vukmirovic, J. Zhang, N.S. Marinkovic, P. Liu, A.I. Frenkel, R.R.Adzic, Nature Materials, 8 (2009) 325. [3] F.L.S. Purgato, P. Olivi, J.-M. Léger, A.R. de Andrade, G. Tremiliosi-Filho, E.R. Gonzalez, C. Lamy, K.B., Kojoh, J. Electroanal. Chem. 628 (2009) 81.
Objective Diamond like carbon (DLC) layers are suggested to reduce essential non-selective protein adsorption of polymer materials used as medical implants. We investigated the protein adsorption on several DLC layers produced under different conditions (without and with N2 and Ar plasma treatment and sputtered with Ti) to gain different hydrophilic and hydrophobic surfaces. The DLC layers were applied to glass and PET surfaces. Contact angle, fluorescent spectroscopy, electrokinetic methods, fluorescent microscopy and QCM quartz micro balance were applied to characterize the surface properties and the protein adsorption kinetics. Results The protein adsorption is an entropy and enthalpy driven two step processes. A first fast and reversible process is followed by a slow conformation change leading to an irreversible adsorption. The adsorption kinetics is not much influenced by DLC coating compared to glass or PET surfaces but the amount of adsorbed protein is reduced by a factor 2,5 by the DLC coating independent of the substrate. The electrokinetic experiments show that both, the DLC layers as well as the proteins are negatively charged at pH 7.3. Nevertheless the electrostatic repulsion, screened by high ionic strength does not suppress adsorption. Hydrophobic interactions of non-polar segments are the driving forces. Proteins with a strong inner structure (hard proteins, Lysocyme) show less adsorption on hydrophilic then on hydrophobic surfaces, soft proteins (BSA) adsorb on all surfaces independent on the surface thermodynamics. This difference is caused by the entropy increase due to conformational changes. The protein adsorption and the rigidity of the adsorbed film were measured using fluorescent microscopy and QCM. The obtained results show the following characteristics: Extremely hydrophilic DLC-surfaces having a contact angle between 16° to 20° show an IEP of 2,5 and the smallest protein affinity of 10% compared to reference glass (5mg/cm2). The protein adsorption of less hydrophilic DLC-surfaces with contact angles between 25° to 30° causes a shift of the IEP towards 4.4. The amount of adsorbed protein is between 15 – 25% of that of reference glass. In the case of hydrophobic DLC layers (contact angle between 55° to 80°) an IEP shift to 4.8 is observed and the amount of adsorbed protein is 55% to 70% of the reference surfaces.
78 Benahavís (Málaga, Spain), 21st to 23rd of October, 2009
Conclusions: Changes of spectroscopic parameters in the solution are almost not observable due to the small amount adsorbed on the solid phase. A direct observation of the amount of adsorbed protein is not possible in the case of soft proteins due to their structural changes after adsorption. The adsorption is directly observable using streaming potential and QCM. The isoelectric point is significantly shifted with increasing adsorption. QCM experiments describe the amount as well as the adsorbed layers structural properties.
Figure. Zetapotential vs. pH of DLC- layers after adsorption of BSA in 67 mM Phosphat, pH 7.4, different protein concentrations
Benahavís (Málaga, Spain), 21st to 23rd of October, 2009 79
Amino Acid-Based Biocompatible Surfactants
Mª Rosa Infante, Lourdes Perez, Aurora Pinazo, MªCa rmen Moran, Ramon Pons, Mª Teresa Garcia, Mª Pilar Vinardell 1
IQAC-CSIC, Jordi Girona 18. 08008 Barcelona
1Facultad de Farmacia. Universidad de Barcelona There is a pressing need for developing efficiently surfactants that are biodegradable and biocompatible. Surfactant molecules from renewable raw materials that mimic natural lipoamino acids are one of the preferred choices for food, pharmaceutical and cosmetic applications. Given their natural and simple structure they show low toxicity and quick biodegradation. The value of amino acids and vegetable oil derivatives as raw materials for the preparation of surfactants was recognized as soon as they were discovered early in the last century. The combination of polar amino acids/peptides (hydrophilic moiety) and non-polar long-chain compounds (hydrophobic moiety) for building up the amphiphilic structure has produced molecules with high surface activity. Our group has a wide experience in synthesis (chemical, enzymatic or, usually, by a combination of both methodologies) of amino acid-based surfactants obtained from the combination of natural saturated fatty acids, alcohols and amines with different amino acid head groups through ester and amide linkages. Thus, saturated single-chain, double-chain, and iusep surfactants of different ionic character have been found to be in all cases highly biodegradable, with low toxicity, ecotoxicity and irritation effects. Water solubility and self-aggregation properties were directly associated with the chemical structure of the molecule and only cationic lipoamino acids possessed antimicrobial activity. Their multifunctional performance makes amino acid based surfactants a valuable high added value compounds from renewable raw materials for potential multipurpose new applications in bio/nano materials and biomedicine.
Department of Chemistry, Coimbra University, Coimbra, PORTUGAL Cationic polymers and surfactants are efficient in compacting DNA and can also be efficient transfection agents. These systems are also characterized by a strong associative phase separation. In attempts to mimic the DNA-histone interactions in chromatin, the phase behaviour and aggregate structure in different aqueous mixtures of DNA and a cationic protein were investigated. We also describe the preparation of covalent DNA gels and describe their swelling-deswelling behaviour. It is found that covalent gels offer novel opportunities for monitoring DNA-cosolute interactions. Based on the associative phase separation, the preparation of novel DNA particles by mixing DNA and surfactant solutions can be achieved; the size of the gel particles can be controlled from ca. 100 nm and upwards. The properties and DNA release characteristics of these particles are also described. A local phase separation in covalent gels can lead the formation of a surface phase, a “skin”. The different types of studies are performed for both double- and single-stranded DNA. Throughout, a stronger interaction is observed with denatured DNA. On the basis of these results and other observations it is found useful to view DNA as an amphiphilic polymer self-assembling by hydrophobic interactions.
Benahavís (Málaga, Spain), 21st to 23rd of October, 2009 83
Symmetry-asymmetry effects on the self-assembly of ion-paired surfactant systems
Eduardo F. Marques 1, Bruno Silva 1, Rodrigo Brito 1, Ulf Olsson 2
1Centro de Investigação em Química, Department of Chemistry, Faculty of Science, University
of Porto. 2Physical Chemistry, Centre for Chemistry and Chemical Engineering, Lund University,
Sweden. In this talk, we will discuss the effect of chain length symmetry or asymmetry on the aqueous bulk behaviour of mixtures of cationic and anionic surfactants (ion-paired systems), through a brief revision of systems recently studied in our group. Both conventional amphiphiles of commercial origin and novel amino acid based amphiphiles will be considered. For salt-free alkyltrimethylammonium alkylsulfonates of the type Cm
+Cn-, we will see that they
can be water soluble at or near room temperature if the chain length difference between Cm+
and Cn- is high [1], i.e. if m>>n or n>>m. This is a consequence of the solubility mismatch that
originates a variable surface charge density in the aggregates, according to a charge regulation mechanism[2]. We compare different binary C16
+Cn-/water systems where n is varied from 6 to
10. A peculiar consequence of this concentration-dependent charge density is the coexistence of a dilute and a concentrated lamellar phases. At low concentration, stable vesicles are formed and when temperature is changed, an unusual vesicle-to-micelle transition occurs, involving in both directions the formation of lamellar domains as an intermediate structure. With respect to more bio-friendly systems, based on lysine- and serine-derived surfactants, two pair systems were studied, one symmetric with C12/C12 chains and one asymmetric, with C8/C16 chains (where the Ser-derived surfactant has the longest chain) [3]. Different vesicle size and mechanisms of the vesicle-to-micelle transition have been found. The results are discussed on the basis of models for the micelle-vesicle transitions and the equilibrium stabilization of vesicles (spontaneous curvature energy, bending moduli and translational entropy). References [1] Silva, B. F. B.; Marques, E. F.; Olsson, U., Langmuir 2008, 24, 10746. [2] Silva, B.F.B; Marques, E.F.; Linse, P; Olsson U., J. Phys. Chem. B, 2009, 113, 10230. [3] Marques, E.F.; Brito, R.O.; Silva, S.G.; Rodríguez-Borges, J.E.; Vale, M.L.; Gomes, P.; Araújo M.J., Langmuir 2008, 24, 11009.
Benahavís (Málaga, Spain), 21st to 23rd of October, 2009 85
Formic Acid as a Hydrogen Source for Vapor Phase Ca talytic Reactions
Dmitri A. Bulushev* and Julian R.H. Ross
Charles Parsons Initiative, CES, University of Limerick, Limerick (Ireland) *[email protected]
Introduction There is currently a great deal of interest in the development of so-called “Second Generation” bio-refining processes, an example being the Biofine Process. This transforms cellulosic biomass feedstock by acid catalyzed hydrolysis to give a mixture of levulinic acid and formic acid (FA) together with a “char” residue [1]. The levulinic acid can be used for the production of chemicals and transport fuel additives. We are currently examining potential uses of the FA produced. For example, FA can be decomposed over a variety of different catalysts to give hydrogen and CO2; the aim is to find catalysts to optimize hydrogen production with a minimum of CO production. As part of our study, we have examined the possibility of using formic acid as a direct source of hydrogen in hydrogenation reactions, thus avoiding the separate step of hydrogen production. To test this idea, we have examined the use of FA in the hydrogenation of ethylene and propylene over a number of catalysts (reactions 1 and 2): HCOOH + C2H4 CO2 + C2H6 (1) HCOOH + C3H6 CO2 + C3H8 (2) Materials and Methods Several different materials (for example, 10 wt% Pd/C (Degussa), 1 wt% Au/C (WGC), 1 wt% Au/TiO2 (WGC), 1 wt% Pt/ZrO2) were tested for both FA decomposition and the direct hydrogenation of olefins (C2H4, C3H6) by FA. The catalysts were placed in the quartz tubular reactor of a microreactor system, then pretreated in a 1% H2/Ar mixture at 573 K for 1 h and cooled in He to reaction temperature. The reaction products were analyzed by GC. Results and Discussion Good results were obtained with a number of catalysts and will be reported. However, only the results for the Pd/C catalyst will be shown here since this material gives the best results obtained for both FA decomposition and the hydrogenation of the olefins. A very small amount of this catalyst (6 mg) gave decomposition of FA to hydrogen and CO2 at temperatures as low as 358-433 K. Furthermore, C2H4 (see Fig. 1) as well as C3H6 (not shown) could be hydrogenated effectively by FA over the same range of temperatures. No deactivation was observed. Complete conversion of the FA was achieved at about 433 K. The undesirable CO production was almost completely eliminated, the selectivity to CO2 being very high (Fig. 1). Generally, the products of the Biofine process contain significant amounts of water and so that it is important that the FA reactions will occur in the presence of water iusep. It was found that water iusep has a small positive effect on both FA decomposition (not shown) and C2H4 hydrogenation by FA (Fig. 1). It is suggested that the mechanism involves two important steps: the formation of adsorbed hydrogen from FA and its consumption by the olefin. That the second step is probably fast was shown by the observation that the olefins examined could both be hydrogenated by hydrogen with 100% conversion even at 313 K over the Pd/C catalyst. Thus, hydrogenation by molecular hydrogen is more effective than that by FA, the rate of the latter reaction being determined by the rate of production of surface hydrogen.
86 Benahavís (Málaga, Spain), 21st to 23rd of October, 2009
Conclusions It has been shown that olefins can be hydrogenated effectively in the iusep phase by using FA instead of hydrogen. This means that the separate step of hydrogen production as well as the need for hydrogen storage and transportation can both be eliminated. It may also be possible to use FA as a hydrogen source for some of the hydrogenation steps in the conversion of levulinic acid to fuel additives and this is being examined. The method may also have applications in other hydrogenation reactions.
0
0.2
0.4
0.6
0.8
1
0 50 100 150 200
time, min
HC
OO
H c
onve
rsio
n,
HCOOHHCOOH+H2O
HCOOH+H2O+C2H4
HCOOH+C2H4
HCOOH CO
2 se
lect
ivity
,
Figure 1. Comparison of the HCOOH conversion and CO2 selectivity with different gas compositions: (2.4% HCOOH,
2.4% HCOOH/2.3% H2O, 2.4% HCOOH/2.3% H2O/1% C2H4, 2.4% HCOOH/1% C2H4, 2.4% HCOOH, balance He) over a 10 wt% Pd/C catalyst (total flow rate 51 ml/min, reactor temperature 388 K)
References [1] Hayes, D. J.; Fitzpatrick, S.; Hayes, M. H. B.; Ross, J. R. H., The Biofine process - Production of Levulinic Acid, Furfural, and Formic Acid from Lignocellulosic Feedstocks. In Biorefineries-Industrial Processes and Products, Kamm, B.; Gruber, P. R.; Kamm, M., Eds. Wiley-VCH: Weinheim, 2006; Vol. 1, pp 139-164.
Benahavís (Málaga, Spain), 21st to 23rd of October, 2009 89
Silylation of functionalized commercial silica for the direct synthesis of hydrogen peroxide solution
G. Blanco Brieva 1, J. M. Campos-Martin 1, M. P. de Frutos 2 and J. L. G. Fierro 1
1 Instituto de Catálisis y Petroleoquímica, CSIC, Marie Curie, 2, Cantoblanco, 28049 Madrid, Spain
2 Centro de Tecnología Repsol YPF, A-5, Km. 18, 28931 Móstoles, Spain Introduction Hydrogen peroxide (H2O2) is a clean oxidizing and bleaching agent that is used for water purification/wastewater treatments, as whitening agent, disinfectant and as basic product of the chemical industry [0]. Efforts are therefore being made to replace H2O2 production by the standard anthraquinone process because of its high cost does not allow the H2O2 to be used for the production of bulk organic chemicals. Thus, the synthesis of H2O2 by direct reaction between H2 and O2 appears quite advantageous to conventional process. Acids are often incorporated into the reaction medium to delay or prevent the decomposition of H2O2, which indeed takes places in the presence of bases [0]. To yield H2O2 formation, some halides are often added to delay water production with the subsequent increase in H2O2 selectivity [0,0]. Recently, we have shown that the liquid-phase direct synthesis of hydrogen peroxide can be achieved using palladium nanoparticles deposited on sulphonic acid functionalized silica catalysts [0]. A recent report emphasized the strong effect of surface modification on the selectivity to hydrogen peroxide [0]. Based on these previous works, this work was undertaken with the aim to study the effect of silylation of surface modification of sulphonic acid functionalized by silylation, use these modified supports to prepare based Pd catalysts for direct synthesis of H2O2 in non acidic solutions. This surface modification can produce a more hydrophobic surface which can avoid secondary reactions. Experimental section Firstly, to a suspension of functionalized commercial silica (Silycicle Tosic Acid) (10 g.) in toluene (100 ml), Cl-Si-(CH3)3 or CF3(CF2)5-(CH2)2Si(CH3)2Cl was added drop by drop. The suspension was stirred for 6 h under reflux. Then the remaining solution was filtered off and the solid obtained was washed with toluene (50 ml) followed by air-drying at 373 K for 12 h. A reaction scheme of the silylation procedure and sample labelling is shown in Figure 1.
OH
SO3H
+ HClSi
Cl-Si-(CH3)3
O O O
O
Si
O O O
Si
SiCoCl
SiCo
R
C10H10ClF13Si
Si
O
RSi
O O O
SiCoPF
CH2-(CF2)5-CF3
Figure 1 Schematic modification of the silica functionalized support.
Modified support (10 g) was stirred with 125 ml of acetone. To this suspension, a palladium (II) acetate (Johnson Matthey) solution in acetone (50 ml) was added drop by drop. The suspension was stirred for 1 h. The remaining solution was filtered off and the solid obtained was washed and air-dried at 333 K for 2 h. Supports and catalysts were characterized by several techniques, N2 isotherms, TGA, FTIR and XPS. Catalysts were tested in the direct synthesis of H2O2. In a typical run, 1.6 g of the catalyst was put inside an autoclave with 150 g of methanol and HBr as promoter. The reactor was pressured with N2 to 9.5 Mpa. The mixture was heated to 313 K. Then, the reaction gas mixture was feed (H2:O2:N2 (3.6:46.4:50)) with a total flow of 2500 mlN min-1 without stirring, and then stirring was started up (1500 rpm) to initiate the reaction.
90 Benahavís (Málaga, Spain), 21st to 23rd of October, 2009
Results and Discussion Thermogravimetric profiles of SiCo, SiCoCl and SiCoPF show that water desorption occurs at temperatures lower than 383 K and the extent of water desorption is smaller in SiCoCl and SiCoPF samples whose surface has been made hydrophobic. Nitrogen adsorption-desorption isotherms show that the addition of silane precursors led to a decrease in adsorption capacity respect the silica reference (SiCo). The reduction of the adsorption capacity of functionalized silica samples is clearly illustrated by the decrease in specific area. Pore volume increases due to a good sililation, with the exception of SiCoPF because of the size of the silane precursor employed does not allow the surface to be effectively silylated. Both chemical state and relative abundance of palladium species of the catalysts was determined by XPS. The Pd 3d core-level spectra present the characteristic spin–orbit splitting of Pd 3d levels. By applying peak fitting procedures each component of the Pd 3d doublet, two palladium components were obtained: one at 336.5 eV, is characteristic of PdO clusters, and another at 338.2 eV, which corresponds to PdII ions interacting with the –SO3H groups of the silica. It has been demonstrated that the catalyst containing a higher amount of PdII ions interacting with –SO3H groups than PdO (amount of these, depend on Pd content) afford a high selectivity and conversion of H2O2 in liquid phase [0,0]. Pd-SiCoCl and Pd-SiCoPF show less amount of PdII ions interacting with –SO3H than Pd-SiCo. Pd-SiCoPF exhibit a large percentage of PdO clusters (85%). The concentration profiles of H2O2, Figure 2, are almost linearly dependent on the time of reaction, indicating that reaction proceeds at a constant rate. The H2O2 production rate was very high and clearly higher for PdSiCo than the other two catalysts. The activity data are in good agreement with the nature of the Pd species, as revealed by XPS. The catalyst with the lowest proportion of PdO, and hence with the highest amount of PdII ions interacting with –SO3H groups (PdSiCo), afford the highest selectivity and highest concentration of H2O2 in the liquid phase. A similar effect, although less marked, was noted for PdSiCoCl catalyst in which activity is lower than in PdSiCo even though the slopes of the straight display are similar.
0 20 40 60 80 100 1200
1
2
3
4
5
6
7
8
9
Wt %
H2O
2
Time (min)
PdSiCo Pd-SiCoCl Pd-SiCoPF
0 20 40 60 80 100 1200
10
20
30
40
50
60
70
80
% S
elec
tivity
to H
2O
2
Time (min)
PdSiCo Pd-SiCoCl Pd-SiCoPF
Figure 2 H2O2 concentration and selectivity profiles versus reaction time along the direct reaction of hydrogen and oxygen at 313 K. PdSiCoCl catalyst shows the lowest selectivity because, in this case, silylation becomes highly effective and transport phenomena between reagents (H2 and O2) and the catalyst surface take place. Finally, it is concluded that silylation of the catalyst surface reduces to some extent both productivity and selectivity to hydrogen peroxide. References [1] G. Blanco-Brieva, J. M. Campos-Martin and J. L. G. Fierro, Angew. Chem. Int. Ed., 2006, 45, 6962. [2]R. Burch and P. R. Ellis, Appl. Catal. B: Environ., 2003, 42(2), 203. [3] G. Blanco-Brieva, E. Cano-Serrano, J. M. Campos-Martin and J. L. G. Fierro EP2000205A1 (2008), assigned to Repsol Química, S.A. [4] J. K. Edwards, B. Solsona, E. Ntainjua, A. F. Carley, A. A. Herzing, C. J. Kiely, G. J. Hutchings, Science 2009, 323, 1037 [5] G. Blanco-Brieva, J. M. Campos-Martin and J. L. G. Fierro, Chem iuse., 2004, 1184. [6] G. Blanco-Brieva, M. C. Capel-Sanchez, M. P. de Frutos, J. M. Campos-Martin and J. L. G. Fierro, Ind. Eng. Chem. Res., 2008, 47, 8013.
Objective Experimental propylene oxide (PO) TPD data from PO (R or S) adsortbed on Pt surfaces with different (S)-1-(1-naphthyl)ethylamine (NEA) coverages [1] are shown in Figure 1. The behavior seen in these TPD spectra is quite complex, with two well-separated groups of peaks indicating the presence of two energetically distinct desorption states. The most surprising feature of these TPD spectra is that, as the NEA coverage on the surface increases steadily, the PO coverage does not follow the expected uniform decrease: it does start decreasing at low NEA coverage, but then increases suddenly at intermediate coverage and reaches a local coverage maximum before decreasing again. We refer to this behavior as the “regression effect,” and note that it occurs only for PO(S), i.e. PO of the same chirality of the NEA modifier. This regression effect can be seen clearly in Figure 1 (a), and is responsible for the enantioselectivity peaks shown in Figure 1 (b). Much effort was placed in simulating this behavior on the basis of pair-wise interactions between PO and NEA molecules on the surface, but those failed in reproducing even the crudest features of the experimental results. Therefore, it was concluded that the observed behavior must be the result of a cooperative effect: under the presence of some configurations of NEA and PO the surface energetics is proposed to be locally modified in such a way as to become selective toward the (S)-PO enantiomer. In addition, the creation of forbidden sites for the adsorption of PO is absolutely necessary in order to produce the observed regression effect in TPD spectra (see below). With these considerations in mind, some models are proposed and discussed as an initial step toward the understanding of the complex chiral phenomenon seen experimentally at a molecular level.
Figure 1 . Integrated coverages and enantioselectivity for the system PO/NEA/Pt(111).
92 Benahavís (Málaga, Spain), 21st to 23rd of October, 2009
Results Two models are proposed, and were tested. Model 1: A square lattice was considered. A) A site surrounded by up to two adsorbed NEAs among near-neighbor sites becomes selective and strong for (S)-PO adsorption; b) a site surrounded by four near-neighbor NEAs becomes selective and strong for I-PO adsorption; c) a site surrounded by 2 or more NEAs up to 4th order neighbors, which is not a selective site, is forbidden for PO (R or S) adsorption. The results predicted by this model are shown in Figure 2. As it can be seen, integrated coverages and selectivity are only very roughly reproduced. Model 2: the surface is considered as an effective lattice composed of intertwined two square sub-lattices with 3 types of sites: sites A (intersections of two lines in the first sub-lattice), weak sites for PO; sites B (intersections of two lines in the second sub-lattice), strong sites for PO; sites C (intersections of both sub-lattices), weak sites for PO. The adsorption energies for PO on these sites (A, B and C) were fixed at W(B) = - 13.75 kcal/mol and W(A,C) = - 11.9 kcal/mol. Both PO and NEA were assumed to occupy a single site on the effective lattice. At low coverage NEA is adsorbed randomly on A sites. Once a coverage of 25% is reached, when those sites are all full, NEA can then adsorb randomly on C sites, maybe in a “tilted” state occupying less surface area, as also suggested in [1].
The second model is based on the following rules: Rule 1. If a PO is adsorbed at a site surrounded by 3 or more next-near-neighboring NEAs, then two near-neighbor sites (chosen at random) and all next-near and 3rd-order neighbor empty sites become enatioselective for (S)-PO. In addition, the PO adsorbed on that site becomes strongly bounded to the surface. Rule 2. If a site is surrounded by 2 adsorbed NEAs up to 4th-order neighbors, then it is forbidden for PO adsorption. Rule 3. If a site is surrounded by 5 or more near and next-near neighbor NEAs, then it is forbidden for PO adsorption. Predictions of this model, shown in Figure 3, reproduce the of the experimental system. Conclusions Model 1 is very simple and can be physically rationalized, but only reproduce experimental data crudely. Model 2, on the other hand, reproduces well the experimental behavior, but its rules are difficult to be rationalized. Consequently, the present work represents a very first step toward the understanding of this chiral system at a molecular level, and may help to develop a better model where the advantages of Model 1 and Model 2 could be combined. References Lee, Z. Ma, S. Kaneko and F. Zaera, JACS 130 (2008) 14597.
Figure 2. Predictions of Model 1. Figure 3 . Predictions of Model 2.
1 Departament de Tecnologia Química i de Tensioactius, Institut de Química Avançada de Catalunya, Jordi Girona 18-26, 08034 Barcelona, Espanya
2 Dipartimento di Chimica, Università degli Studi “La Sapienza”, Piazzale Aldo Moro 5, I-00185 Rome, Italy
In search for new antimicrobial compounds to be used in food, pharmaceutical, and cosmetic applications, a novel class of cationic arginine glyceride conjugates was developed. Preliminary results on their physicochemical and biological properties showed that such novel compounds combine the advantages of glycerides and lipoamino acids [1-4]. The chemical structures are show in Figure 1. Figure1. Chemical structures of the cationic and anionic amphiphiles, Nα –lauroylarginine methyl ester hydrochloride (LAM), 1,2 myristoyl-rac-glycero-3-O(Nα-acetyl-L-arginine) hydrochloride (1414Rac), 1,2 lauroyl-rac-glycero-3-O(Nα-acetyl-L-arginine) hydrochloride (1212Rac), 1 myristoyl-rac-glycero-3-O(Nα-acetyl-L-arginine) hydrochloride (140Rac), 1,2-dipalmitoyl-sn-glycero-3-phosphate monosodium salt (DPPA). At present renewed interest has been shown for amphiphilic systems containing both cationic and anionic surfactants. These studies involve both theoretical and practical aspects. With the purpose of deepening on the fundamental properties of new surfactants as well as the possible application of these as formulated products this communication reports on the formulation of novel cationic monoacyl and diacyl glycerol arginine–based surfactants with an anionic diacyl phospholipid, 1,2-dipalmitoyl-sn-glycero-3-phosphate monosodium salt (DPPA), being part of the rich family of pseudo triple-chain and pseudo-tetra-chain catanionic mixtures respectively [ , ]. Vesicle size and ζ-potencial was measured at several mixing ratios. Additional information on counterion binding, vesicle size, and integrity was obtained from ion selective electrode, SAXS and Cryo-TEM measurements. References [1] Pérez L., Pinazo A., Vinardell M.P., Clápes P., Angelet M., Infante M.R., New J. Chem. 26, 1221 (2002). [2] Morán M.C., Infante M.R., Clápes P., J. Chem. Soc. Perkin Trans. 1, 1124 (2002) [3] Pérez L., Infante M.R., Angelet M., Clápes P., Pinazo A., Prog. Colloid Polym. Sci. 123, 210 (2004). [4] Infante M.R., Pinazo A., iusep J., Colloids Surf. A, 123, 49 (1997). [5] Marques E., Brito R., Wang Y., Silva B., J. Colloid Interface Sci., 294, 240 (2006). [6] Karukstis K., Zieleniuk C.A., Fox M.J., Langmuir 19, 10054 (2003).
Benahavís (Málaga, Spain), 21st to 23rd of October, 2009 95
Lysine-based cationic surfactants: synthesis and st udy of the effect of the polar group on their biological properties
Pérez, L. 1, Pinazo, A. 1, Vinardell, Mª.P 2., Mitjans, M. 2, Infante, M.R. 1, Ribosa, I. 1, García,
M.T. 1, Manresa, A. 3 and Colomer, A. 1
1Departamento de Tecnología de Tensioactivos, IQAC/CSIC. C/Jordi Girona 18-26, 08034 Barcelona España.
2Laboratori de Fisiología Facultat de Farmacia, UB. Av. Joan XXIII, 08028 Barcelona, España. 3Laboratori de Microbiología, Facultat de Farmacia, UB. Av. Joan XXIII, 08028 Barcelona,
España. Surfactants are surface active compounds that can adversely affect the environment. At present the main driving force behind the development of novel surfactants and emulsifiers is the search for environmentally friendly products. Due that, preparation of surfactants which mimic the structure of natural compounds such as lipoaminoacids is currently increasing because of their unique physicochemical and biological properties1,2,3,4. We report on the synthesis, biological and physico-chemical properties of five different novel lysine-based surfactants. All of them have dodecyl fatty chains, two have a monocatenary structure and three have a gemini structure (See Figure 1). The synthesis is a classical amino-acid condensation. The surfactant behaviour has been characterized by measuring the critical micelar concentration (CMC). CMC values of monocatenary surfactants are similar to those of commercial cationic surfactants. Concerning gemini surfactants CMCs are two orders of magnitude smaller than those of monocatenaries surfactants. We have studied a number of biological properties. The hemolytic activity of monocatenary compounds is smaller than that of their gemini counterparts. Both monocatenary and gemini compounds are active against Gram-positive bacteria but not noticeable activity has been observed when bacteria are Gram-negative. After 28 days, the compounds biodegradation5 ratio amounts to 60% thus they fall within the category of readily biodegradable surfactants.
Figure 1. Synthetic lysine-based surfactants structure. References 1. Pinazo, A., Wen, X., Pérez, L., Infante, M.R., Franses, E., “Langmuir”, 1999, 15, 3134-3142. 2. Pérez, L., Pinazo, A., García, M.T., Angelet, M., Lozano, M., Vinardell, P., Mitjans, M., Pons, R., Manresa, A., Infante, M.R., “Eu. J. of Med. Chem.”, 2009, 1-9. 3. Pérez, L., Pinazo, A., Vinardell, P., Clapés, P., Angelet, M., Infante, M.R., “New. J. Chem.”, 2002, 26, 1221-1227. 4. Infante, M.R., Molinero, J. Erra, P., Julia, R., García J.J., “Fette Seifen Anstrichmittel”, 1983, 87(8), 309-313. 5. OECD Chemical group Ready biodegradability. Modified Screening test Method 301D, OECD revised guidelines for ready biodegradability, OECD Paris, France, 1993].
Benahavís (Málaga, Spain), 21st to 23rd of October, 2009 97
Mono Acyl Lysine based surfactants: self-aggregatio n
Ramon Pons, Lourdes Pérez, Marina Lozano, M. Rosa I nfante, Aurora Pinazo
Departament de Tecnologia Química i de Tensioactius, IQAC-CSIC, Jordi Girona, 18-26, 08034, Barcelona, Spain, [email protected]
Self-aggregation and surface properties of new acyl lysine based surfactants have been studied as a function of hydrophobic chain length. Their general structure is presented in the figure. These surfactants are biodegradable, biocompatible and present some antimicrobial activity. The critical micellar concentration values are close to the expected for ionic surfactants with the same chain length. The aggregates were characterised by NMR difussometry and the results suggest notable anisometry as the chain length is increased. At higher concentration gel and liquid crystalline phases have been identified by optical microscopy and SAXS. Lamellar and cubic phases are present at high concentration.
Benahavís (Málaga, Spain), 21st to 23rd of October, 2009 99
Gold supported on ceria doped by Me3+ (Me=Al and Sm) for water gas shift: influence of dopant and preparatio n method
Ivanov a, R. Nedyalkova a, L. Ilieva a, J. W. Sobczak b, W. Lisowski b, D. Andreeva a
aInstitute of Catalysis, Bulgarian Academy of Sciences, 1113 Sofia, Bulgaria
bInstitute of Physical Chemistry, PAS, Kasprzaka 44/52, 01-224 Warsaw, Poland
Introduction Gold catalysts based on ceria are very promising for various applications, among them one very important is the production of pure hydrogen via water gas shift (WGS) reaction. Metal/ceria catalysts are several orders of magnitude more active than metal/alumina or other oxide supports for a number of redox reactions. Metal-modified ceria has a higher oxygen capacity and enhanced reducibility than pure ceria. The addition of metal dopants to CeO2 with valences lower than (4+) leads to the formation of oxygen vacancies in ceria, this should increased the oxidation activity. In this study we present the comparable results obtained for gold catalysts based on both ceria undoped and ceria doped by Al and Sm and prepared by two different methods. The influence of dopants’ nature and preparation techniques will be discussed. Experimental The supports were prepared by 2 different methods: by co-precipitation (CP) and by mechanochemical activation (MA). The amount of the dopant was 10 wt.%. Gold was introduced by deposition-precipitation method. The catalysts were tested in WGS reaction, the catalytic activity was expressed as CO conversion. The samples were characterized by XRD, HRTEM, Raman spectroscopy, XPS and TPR. Results The experimentally determined values of the SBET, lattice parameters of ceria and average size of gold and ceria particles are presented in Table 1. In Figure 1 are presented the WGS activities of the catalysts, compared to undoped gold supported on ceria. Table 1. BET surface area, lattice parameters of ceria and average size of gold and ceria particles
Figure 1 . Temperature dependence of CO conversion of the studied catalysts
The MA catalysts exhibited higher activity than the CP catalysts and undoped ceria (AuCe). A significant difference in the catalytic activity has been observed for CP catalysts doped by Al and Sm. The CP catalyst modified by Sm was highly active, much more than Al doped CP catalyst, and its WGS activity even increased after a long period of operation and reactivation in air at 200 oC. There is a big difference between the catalysts CP and MA modified by Al. For the corresponding samples doped by Sm this difference is smaller and after reactivation even negligible. The Raman spectroscopy data showed also a significant difference between Al doped CP and MA catalysts, while for the Sm doped samples this difference was insignificant.
100 Benahavís (Málaga, Spain), 21st to 23rd of October, 2009
One possible explanation is, that in Sm doped catalyst the oxygen vacancies are located around Sm. Ceria seems to be quite well ordered and its re-oxidation during catalytic operation is enhanced. The XPS analysis of the fresh catalysts revealed an additional Au 4f XPS state at higher BE, which we assigned to positively charged gold species. The contribution of such species seems to be relatively higher for Sm doped catalyst than for corresponding catalyst modified by Al. After catalytic operation only metallic Au was registered in all samples. The hydrogen consumption, registered by TPR measurements, both of fresh and re-oxidized samples showed higher oxygen capacity as compared to the non-doped ceria. There was no distinct correlation between reducibility and WGS activity. The addition of the Sm and Al dopant to ceria increases the stability of gold/ceria catalysts. Conclusions Gold catalysts supported on ceria doped by Me3+ were synthesized. Higher WGS activity was found for the catalysts based on doped by Sm ceria in comparison to the catalysts, doped by alumina. Generally the catalysts prepared by mechanochemical activation exhibit higher activity than those prepared by co-precipitation, but the differences between WGS activities for Sm doped samples are much smaller than that in the case of ceria-alumina catalysts. There are no big differences in the gold particle size (2-3 nm) for the samples prepared by the two methods. There is a big difference in the behaviour of the catalysts by the Raman spectroscopy data. In the Sm doped catalysts a larger number of oxygen vacancies for the MA samples than for the corresponding CP ones was observed. Most probably the oxygen vacancies are adjusted around dopant and the ceria structure seems to be better ordered than in the case of alumina doped ceria. There is no distinct correlation between reducibility and WGS activity. The surface concentration of partially positively charged gold particles in fresh Sm doped samples, registered by XPS, is higher than that of the samples doped by Al, but after catalytic operation only metallic gold is observable. This is in agreement with the model of active sites of gold/ceria catalysts for WGS reaction [1]. Acknowledgements This work is supported by Bulgarian National Fund, Ministry of Sciences and Education, project TK-X-1709.
References [1] D. Andreeva et al., Topics in Catal., 44 (2007)173.
Benahavís (Málaga, Spain), 21st to 23rd of October, 2009 101
Redox activity of gold-molybdena catalysts: influen ce of the preparation method
Petya Petrova*, Lyuba Ilieva, Donka Andreeva
Institute of Catalysis, Bulgarian Academy of Sciences, 1113 Sofia, Bulgaria, *[email protected]
Introduction The detoxification of hydrocarbons pollutants is one of the global environmental problems. Considerable efforts have been made to design highly efficient catalysts for complete oxidation of hydrocarbons. The interesting properties of ceria as a support for the noble metals catalysts are well known mainly to concern its function as oxygen buffer. The synthesis of nanosized ceria is an important factor for the preparation of highly active gold supported catalysts. The addition of alumina to ceria increases the oxygen vacancies concentration, respectively oxygen capacity and redox activity of this type of catalysts. We have also studied the effect of the introduction of vanadia and molybdena as promoters to gold catalysts on the properties and catalytic activity in complete benzene oxidation (CBO) [1-4]. The accent in this study is on the influence of the preparation method on the structure, properties and reduction activity of gold-molybdena catalysts supported on ceria-alumina. Nonpromoted and promoted by molybdena gold catalysts in the reaction of CBO are studied. The role of oxygen vacancies concerning the redox properties and catalytic activity is also the object of the discussion.
Experimental The applied supports were prepared by two different methods: by coprecipitation (CP) or by mechanochemical activation (MA). Amount of alumina was 10 or 20 wt% (signed as cipher after Al). 3 wt% of gold was introduced by deposition-precipitation technique. Molybdena was introduced by impregnation from a solution of (NH4)6Mo7O24. The catalysts were characterized by XRD, Raman spectroscopy, XPS and TPR. The activity of the catalysts was determined in CBO.
Results and discussion The catalytic behaviour of the samples is quite different depending on the preparation method applied. The gold catalysts promoted by molybdena exhibit higher activity in comparison to the corresponding nonpromoted ones in the low temperature (LT) interval. At higher temperatures the opposite behaviour is observed. Depending on the alumina content, the temperature of the cross-point of the two activity curves is different. This temperature is higher for the sample with higher alumina content. The catalysts containing only molybdena showed negligible activities at the studied temperatures. In Fig. 1 are compared the catalytic activities of the Au-Mo samples, prepared by the both applied preparation techniques for the samples containing 10 wt% of alumina (A) and for the samples, containing 20 wt% of alumina (B). One can see that in the LT region the MA catalysts are more active, while in the HT region the activities are almost equal, a little higher being that of CP ones. The redox activity of the samples was commented on the bases of TPR measurements of fresh samples as well as after their re-oxidation. The results obtained by TPR as well as by Raman spectroscopy confirmed the predominant surface modification of ceria in the case of MA preparation method while CP techniques caused oxygen vacancies formation deeper in ceria structure. Interesting XPS results were obtained with Au-Mo spent catalysts – supplementary to metallic gold both Auδ+ and Auδ- were obtained after catalytic operation. The XPS results also supported the role of Ce3+ in the formation of active sites for the redox processes.
102 Benahavís (Málaga, Spain), 21st to 23rd of October, 2009
140 160 180 200 220 240 260 280 3000
102030405060708090
100
AuMoCeAl10CP AuMoCeAl10MA
(A)
Con
vers
ion,
%
Temperature,0C
140 160 180 200 220 240 260 280 3000
102030405060708090
100
AuMoCeAl20CP AuMoCeAl20MA
(B)
Con
vers
ion,
%
Temperature,0C
Figure 1. Temperature dependence of the CBO conversion over gold-molybdena catalysts: (A) containing 10 wt% alumina and (B) containing 20 wt% alumina.
It was supposed that depending on the reaction temperature as well as on the method of catalysts’ preparation, two factors are of great importance for the high redox activity – lattice oxygen mobility and the enhanced electron transfer with the participation of nanosized gold particles and surface oxygen vacancies in close contact with them and Ce3+. The both factors are connected to the oxygen vacancies formation caused by the addition of gold and alumina as well as to the average size of gold and ceria. Conclusions The study of CBO activity over these complex catalytic systems manifested that the different factors should be of crucial importance. In the LT region the effect of the electron transfer with the participation of nano gold particles is prevailing and this is in agreement with significantly higher activity of Au-Mo samples MA. In the HT region obviously the predominant role plays the oxygen mobility, related to the oxygen vacancies in ceria structure. Acknowledgements This work was supported by the National Scientific Fund of Bulgaria, project MY-X-1603. References [1] D. Andreeva, T. Tabakova, L. Ilieva, A. Naydenov, D. Mehanjiev, M.V. Abrashev, Appl. Catal. A: Gen., 209 (2001) 291. [2] D. Andreeva, R. Nedyalkova, L. Ilieva, M. Abrashev, Appl. Catal. A: Gen. 246 (2003) 29. [3] D. Andreeva, R. Nedyalkova, L. Ilieva, M. Abrashev, Appl. Catal. B: Envir. 52 (2004) 157. [4] D. Andreeva, P. Petrova, J.W. Sobczak, L. Ilieva, M.V. Abrashev, Appl. Catal. B: Envir. 67 (2006) 237; 77 (2008) 364.
Benahavís (Málaga, Spain), 21st to 23rd of October, 2009 103
FTIR accessibility studies of 2,6 DTBPy adsorption on FCC catalysts
A.C. Psarras 1,2, E.F. Iliopoulou 2* and A.A. Lappas 2
1 Dept. of Chemical Engineering, Aristotle University of Thessaloniki. 2CPERI/CERTH 6th km Harilaou-Thermi Road, P.O. Box 60361-Thermi, GR-57001 Thessaloniki, Greece.
[email protected] Introduction-Objective Due to energy saving policies the refineries today tend to upgrade more and more of heavier or resid fractions into lighter, desirable distillates via catalytic cracking. Larger molecules in fractions of increasing boiling point mainly account for the difficulties in processing heavy oils. In this study we try to measure the acid sites of FCC catalysts that are accessible, when processing cumbersome molecules such as those of residual petroleum fractions. Vibrational spectroscopy of adsorbed probe molecules is a powerful tool for catalytic acidity assessment. Artificially and commercially deactivated FCC catalysts were studied using pyridine adsorption and FTIR analysis [1], a technique not usually applied on complete FCC catalysts. Pyridine is a strong base and easily gives rise to the formation of H-bonded and pyridinium species with weak and strong Brönsted acid sites, respectively, and to coordinated species on Lewis acid sites. For the characterization of the accessible acid sites 2,6-Di-Tert-Butyl-Pyridine is employed. This probe molecule does not adsorb on Lewis sites due to steric hindrance effects. Thus, DTBPy is suitable only for the measurement of the “useful” Brönsted acid sites. A debate exists for the characteristic band of DTBPY adsorption for quantification purposes. The bands at 3370, 1616 and 1530 cm-1 are reported as characteristics of the DTBPyH+ ion [2].The scope of the present study is to investigate the suitable characteristic band of DTBPY adsorption as well as the most appropriate adsorption temperature. Experimental Experimental apparatus and procedure for acidity measurement using Pyridine is described elsewhere [1]. Initially, the same procedure was used for the DTBPY adsorption. Interpretation of the results using the Beer-Lambert law was realized as suggested in the literature [2] due to the lack of molar extinction coefficients for this probe molecule. Besides β-zeolite we also used a mesoporous Al-MCM-41 reference sample to ensure that all sites are accessible to both pyridine molecules. Two commercially (E-cat1 & E-cat2) and two artificially (D-cat1 & D-cat2) deactivated FCC catalysts were the samples under study. The zeolitic components (Zeo1 & Zeo2) of the FCC catalysts were also investigated. Results and Discussion All experiments were realized more than once for repeatability reasons. All three characteristic bands were included in the study in order to select the most appropriate one. The percentage of the high accessible acid sites is presented in Table 1. The utilization of the band at 1530 cm-1 presents acceptable results with the best repeatability for the FCC catalysts. The band at 3370 cm-1, not present at the normal pyridine spectrum appears in the region of amines and is attributed to the vibration of the N-H+ bond. Such a band is a consequence of the higher basicity of the DTBPy in a way that the bond N-H+ is short enough to be similar to an amine. Thus, the contribution of the acid sites to this band is strongly related to their strength and not all of them contribute to this band. The results of the zeolitic samples using the band at 3370 cm-1 resemble the results using the band at 1530 cm-1 with a slightly better repeatability. This observation can be attributed to the fact that the fresh zeolitic samples contain high acidity with great strength, supporting our assumption about the 3370 cm-1 band. Thus, the band at 3370 cm-1 can be utilized, when fresh zeolitic samples are under research due to the great strength of their acid sites. However, when deactivated FCC catalysts are under research, the band at 1530 cm-1 should be taken into account. The band at 1530 cm-1 is attributed to the ring vibration of the pyridinium ion, thus all the acid sites contribute to this band independently of their strength. The band at 1616 cm-1 indicates also the ring vibrations of the pyridinium ion but it is considered less suitable, as the physisorbed molecules may also contribute to it (same case with the normal pyridine adsorption). The coverage of bridged hydroxyls calculated by integrating the negative bands at 3630 cm-1 and 3540 cm-1 on the Zeo1 sample is more than 91%
104 Benahavís (Málaga, Spain), 21st to 23rd of October, 2009
and on Zeo2 is 88%. This indicates that the results when using Al-MCM-41 as the reference sample and the 1530 cm-1 band are more realistic, although utilization of the β-zeolite as reference sample gives a slightly better repeatability. Table 1: Percentage of accessible Brönsted sites using all characteristic bands for all samples Band at 3370 cm-1 Band at 1616 cm-1 Band at 1530 cm-1
D-cat1 0.897 1.392 1.086 0.713 0.525 0.647 D-cat1 (2nd) 1.267 1.228 1.553 0.577 0.526 0.623 D-cat1 (3rd) 0.818 1.269 1.014 0.665 0.538 0.666 E-cat1 2.575 2.278 2.211 0.732 0.746 0.881 E-cat1 (2nd) 1.395 1.742 1.484 0.753 0.669 0.811 Zeo1 0.396 0.631 0.532 0.360 0.688 0.853 Zeo1(2nd) 0.513 0.840 0.581 0.406 0.685 0.852 Zeo1(3rd) 0.526 0.816 0.602 0.395 0.668 0.826 D-cat2 1.339 1.846 1.356 0.775 0.750 0.918 D-cat2 (2nd) 1.261 1.861 1.194 0.739 0.697 0.858 E-cat2 2.681 2.394 1.982 0.664 0.735 0.869 E-cat2 (2nd) 1.880 2.230 1.621 0.773 0.751 0.906 Zeo2 0.618 0.834 0.762 0.424 0.710 0.867 Zeo2(2nd) 0.446 0.887 0.503 0.441 0.589 0.748 Zeo2(3rd) 0.621 0.821 0.754 0.410 0.669 0.815 The temperature effect of the DTBPy adsorption was investigated on the zeolitic sample Zeo1. Additional adsorption trials at 50, 100 and 200 oC, (besides the standard 150 oC), were realized. This study was motivated by the appearance of double peaks at 1530 cm-1 and 3370 cm-1 when the 150 oC was applied. It was observed that the single peaks related to the DTBPyH+ ion is present only at 50 oC until equilibration. After the equilibrium is reached, a decrease is observed at both bands with a simultaneous appearance of shoulders at 1540 cm-1 and 3350 cm-1. These shifted bands are present from the beginning of the adsorption when a higher temperature is applied (≥100°C). This observation can be related to the reac tion of the DTBPy probably towards a branched pyridine smaller than DTBPy and larger than pyridine. The bands are reaching equilibrium at the elevated temperatures, suggesting that this transformation of DTBPy is not complete and only affected by the temperature. This gives validity to our former results, but the accessibility refers to less bulky molecules. The study at room temperature will possibly provide more accurate results about the highly accessible Brönsted acid sites according to the size of the DTBPy.
Conclusions The band at 1530 cm-1 is more reliable for the quantification of accessible Brönsted sites on FCC catalysts when 2,6-DTBPy is used as a probe molecule, although the band at 3370 cm-1 can be utilized for pure fresh zeolitic samples due to their high and rather homogeneous acidity strength. The mesoporous Al-MCM-41 gives more realistic results when used as a reference sample. The temperature of adsorption is affecting the results by enhancing the reaction (probably dealkylation) of DTBPy. The adsorption equilibrium validates the results at 150 oC, but the percentage of the really highly accessible Brönsted acid sites is revealed by the adsorption study at room temperature.
References [1] A.C. Psarras, E.F. Iliopoulou, K. Kostaras, A.A. Lappas and C. Pouwels, Micropor. Mesopor. Mater. 120 (2009) 141-146 [2] A. Corma, V. Fornés, L. Forni, F. Márquez, J. Martinez-Triguero and D. Moscotti, J. Catal. 179 (1998) 451-458
* [email protected] In the last years attention was focused to experience the experimental conditions leading from simple association colloids, i.e. surfactant-, or lipid-based, micelles and liquid crystalline phases, to hierarchically more complex structures, such as vesicles, cubosomes, nanoplatelets and nanorods. All such systems are relevant in biologically-oriented advanced technologies, because of the strong similarities the aforementioned objects have with structures occurring in vivo. That’s why strong efforts are oriented to use such items in biomedical application fields, including protein and DNA transfection technologies. Experiments performed on dilute regimes give strong evidence on the formation of vesicles and nanorods, whereas other in concentrated systems allow preparing vesicle-based entities from lamellar structures ordered by shear or application of mechanical stresses. Vesicles and nanorods are stable for indefinitely long times and were characterized by many different experimental methods, spanning from DLS to electrophoretic mobility, from SAXS to TEM or SEM, and so forth. Such supramolecular organization modes are ascribed to the combination of different effects, namely hydrophobic, electrostatic, hydrogen-bond, etc. The resulting structures and stability are governed by a delicate balance of all such effects, which are also responsible for the interactions with biomacromolecules (DNA or Proteins). Some biochemically intended applications are briefly discussed. As to concentrated regimes, it has been demonstrated that applied shear favors the formation of transient vesicular, tubular or rod-like (spring roll) geometries, whose stability is essentially governed by composition, the system rheology and working temperature. Such transient structures were investigated and rationalized by combining rheological methods with multinuclear NMR, Rheo-NMR, NMR Imaging and PFGSE-NMR methods. Efforts were made to link the properties of structures observed in dilute with those occurring in concentrated regimes, to determine what are the main forces responsible for the stability of such entities, and to determine whether the resulting stabilization is of kinetic or thermodynamic origin. References. [1] C. Letizia, P. Andreozzi, A. Scipioni, C. La Mesa, A. Bonincontro, E. Spigone J. Phys. Chem. B, 2007, 111, 898-908. [2] A. Bonincontro, C. La Mesa, C. Proietti, G. Risuleo Biomacromolecules, 2007, 8, 1824-1829. [3] A. Bonincontro, M. Falivene, C. La Mesa, G. Risuleo, M. Ruiz-Pena Langmuir, 2008, 24, 1973-1978. [4] M. Alvarez Alcalde, A. Jover, F. Meijide, L. Galantini, N.V. Pavel, A. Antelo, J. Vasquez Tato Langmuir, 2008, 24, 6060-6066. [5] V.H. Soto Tellini, A. Jover, F. Meijide, J.V. Tato, L. Galantini, N.V. Pavel Adv. Mater., 2007, 19, 1752-1756. [6] C. Moran, M.R. Infante, L. Perez, A. Pinazo, L. Coppola, M. Youssry, I. Nicotera Colloids Surf. A: Physicochem. Eng. Aspects, 2008, 327, 111-121. [7] M. Youssry, L. Coppola, E.F. Marques, I. Nicotera J. Colloid Interface Sci., 2008, 324, 192-198. [8] M. Youssry, L. Coppola, I. Nicotera, C. Moran J. Colloid Interface Sci., 2008, 321, 459-467.
bUniversité catholique de Louvain, Unité de Catalyse et Chimie des Matériaux Divisés, Croix du Sud 2/17, 1348 Louvain-la-Neuve, Belgium
[email protected] Introduction In the last years many researchers focused on catalysts containing niobium, vanadium and tantalum as an active species. For the preparation of such materials both, co-precipitation and post-synthesis methods were applied. Moreover, different kinds of the supports were used, among them oxides [1] and mesostructured oxides [1,2] were studied. However, there are only few data concerning the incorporation of niobium into crystalline structure, e.g. into zeolite framework. The same is true in the case of two other elements belonging to the group five of periodic table, i.e. vanadium and tantalum. Exemplary, such an attempt was done by Kevan et al., who substituted silicon atoms by niobium during the synthesis of MFI structured zeolites [3]. Tantalum was also introduced via co-precipitation method into the same zeolite structure, i.e. MFI, by Ko et al. [4].The post-synthesis method was applied by Dzwigaj et al. for the introduction of vanadium into BEA structure [5]. In our work we focused on the incorporation of niobium, vanadium and tantalum species into faujasite framework, namely into Y zeolite. The structure and Si/Al ratio of Y type zeolite significantly differ from those of MFI and BEA zeolites studied before. Therefore, we expected to obtain zeolites characterised by new surface properties and catalytic activity. For the incorporation of mentioned elements the co-precipitation method was applied. Experimental The preparation of G5 elements containing FAU zeolites based on the modified two-steps procedure reported originally by Ginter et al. [6]. In the first stage the so-called seed gel was prepared with the composition of: 10.67 Na2O : Al2O3 : 10 SiO2 : 180 H2O and left to age at the room temperature for one day. In the second stage so-called feedstock gel was prepared with the composition of 4.3 Na2O : Al2O3 : 10 SiO2 : 180 H2O. The metal source was added both to the seed and feedstock gel. The assumed Si/T ratio (T – V, Nb or Ta) was 64. Afterwards, a part of the seed gel was added to the feedstock gel and the mixture was stirred vigorously for at least 20 minutes. After stirring the gel was put to a polypropylene bottle and heated in oven for 5 hours in 373 K. The final product was washed with distilled water. The last step of preparation was drying in 383 K for 12 hours. The vanadium containing zeolite was also prepared without the addition of this metal into seed gel.The zeolites prepared in this study were characterised using XRD, XRF, XPS, ICP, FTIR, UV-vis techniques. The catalytic tests for acidity/basicity and redox properties of the samples were performed (e.g. acetonylacetone cyclisation). Results and discussion Vanadium zeolites The XRD patterns of synthesized vanadium containing zeolits (Na-VY1 and Na-VY2) indicated the faujasite structure. Any peaks related to extraframework vanadium oxide phase were observed. The SEM images showed well defined crystal structure and similar morphology. The addition of vanadium into seed gel was found to be important for metal incorporation into the zeolite. Nevertheless, the Si/V ratio in the final material was 15 times less that the assumed value even in the case when vanadium was added to the seed gel (Table 1, Na-VY2). The UV-vis spectra pointed out that vanadium species incorporated via co-precipitation method into Y zeolite show the tetrahedral coordination of V5+, which is typical for elements inside the zeolie framework. However, further estimation of vanadium state by XPS analysis was not possible due to the low concentration of this element in the final sample. The significant role of vanadium species on surface properties of the material was shown in acid-base test reaction, i.e. acetonylacetone cyclisation. The Brønsted basicity observed for this sample was assigned to OH groups connected to V5+ in the skeleton.
108 Benahavís (Málaga, Spain), 21st to 23rd of October, 2009
Niobium zeolites The XRD pattern of Na-NbY zeolite showed the characteristic peaks for crystalline FAU structure indicating the successful synthesis of this kind of material. The extraframework niobium oxide species were not found on the material surface, suggesting the presence of niobium in the zeolite framework. Similar information was given by UV-vis spectra, which showed only the tetrahedral coordinated niobium and no band corresponding to Nb2O5. The assumed
Si/Nb ratio for Na-NbY differed from this in the final sample. The amount of introduced niobium was 2.5 times less than the assumed value. However, the efficiency of niobium incorporation was much higher than those observed for vanadium zeolites. The increase of baseline in the XRD pattern between 2 theta ca. 20-40o suggested that a part of the obtained material is also present in the amorphous form. This was confirmed by SEM images, which showed amorphous phase also beside crystalline one. To examine the location and state of niobium species the XPS spectra were recorded. It was found that all niobium exhibit oxidation state +5. Moreover, the biding energy for niobium (3d5/2) was higher (207.9 eV) than that typical for niobium bulk oxide (207.3 eV). The increase of the binding energy was attributed to the different surrounding of niobium species, e.g. in the Si-O-Nb link. Such a link is formed when niobium is incorporated into the zeolite framework. Tantalum zeolites The synthesis of tantalum containing zeolite (Na-TaY) was also succeded as evidenced by XRD pattern. Moreover, the prepared sample exhibited well defined crystals and morphology. Inspite of high efficency of tantalum incorporation (Table 1), i.e. relatively high metal concentration, no extraframework Ta2O5 phase was detected. The UV-vis spectrum of Na-TaY sample showed very intens and symmetric band at ca. 221 nm, which is characteristic of tanatlum in tetrahedral coordination. This spectrum was completely different from the other registered for the Y zeolite with tanatalum oxide in the extraframework position (reference sample). It strongly suggested the incorporation of tanatalum into the zeolite framework. Similary to niobium, the XPS spectra indicated that all tantalum is present on oxidation state +5. Moreover, the binding energy registered for tantalum, which was higher than that typical for bulk tantalum oxide, suggested the incorporation of this element into zeolite structure. Conclusions Niobium, vanadium and tantalum containing Y zeolites were successfully synthesized. The characterisation of these materials indicated that G5 elements are incorporated into the zeolite framework. The efficiency of the group five elements incorporation into faujasite framework can be given in the following order: Na-TaY > Na-NbY > Na-VY. The synthesised materials showed the different surface properties, Na-TaY and Na-VY Brønsted basic whereas Na-NbY acidic character. Acknowledgements Polish Ministry of Science and Higher Education (grant 118/COS/2007/03) and COST D36/0006/06 are to be acknowledged for a partial support of this work. References [1] X. Gao, I.E. Wachs, M.S. Wong, J. Y. Ying, J. Catal., 203 (2001) 18-24. [2] M. Ziolek, I. Nowak, Zeolites, 18 (1997) 356-360. [3] L. Kevan, A.M. Prakash, J. Am. Chem. Soc., 120 (1998) 13148-13155. [4] Y.S. Ko, W.S. Ahn, Microporous Mesoporous Mat., 30 (1999) 283-291. [5] S. Dziwigaj, Current Opinion in Solid State and Materials Science, 7 (2003) 461-470. [6] D.M. Ginter, A.T. Bell, C.J. Radke, in Synthesis of Microporous Materials, Vol. 1, Molecular Sieves, M. L. Occelli, H. E. Robson (eds.), Van Nostrand Reinhold, New York, 1992, p 6.
Benahavís (Málaga, Spain), 21st to 23rd of October, 2009 109
Lignin-based electrospun carbon microforms
R. Ruiz-Rosas 1*, J. Bedia 1, M. Lallave 2, A. Barrero 4, I.G. Loscertales 3, J. Rodríguez-Mirasol 1, T. Cordero 1.
1 Chemical Engineering Department, University of Malaga, 29071 Málaga (Spain)
2 YFLOW-Sistemas y Desarrollo S.L., PTA, 29050 Málaga (Spain) 3 Fluids Mechanics Department, University of Malaga, 29071 Málaga (Spain) 4 Fluids Mechanics Department, University of Sevilla, 41092 Sevilla (Spain)
Objectives Electrospinning is a simple and versatile method for generating fibrilar and spherical structures from a rich variety of materials. This technique requires the use of a high voltage electrostatic field to charge the surface of a solution droplet and thus to induce the ejection of a liquid jet through a spinneret. Carbon submicroforms have attracted enormous attention due to their excellent mechanical, chemical and thermal properties. Lignin is one of the most abundant polymers in nature and constitutes an underutilized by-product of the papermaking industry. This work is aimed to the production of carbon spheres, tubes, hollow and filled fibers of interesting surface properties through electrospinning of lignin-based solutions.
Results Figure 1 shows SEM and TEM images from the microforms carbonized at 900ºC. Change between spherical (Fig. 1.a) or fibrilar (Figures 1.b to 1.d) form is achieved modifying the feed rate and viscosity of the lignin solution in the electrospraying process. Hollow spheres or fibers (Figures 1.a to 1.c) can be prepared using an oil template, which is inserted through the inner needle of a tri-axial syringe electrospinning configuration. Selecting oils of different viscosity allows moulding the shape of the fibers. The green bean appearance of the carbon fibers is conferred by droplets of low viscosity template oil inside them and can be seen in the TEM image detail of Figure 1.b. The use of higher viscosity oils conducted to formation of carbon tubes. It was confirmed by transmission electron micrographs, which show tubes of diameters in the range of 1-2 microns and shell widths from 200 to 600 nm, Figure 1.c and detail. Filled carbon fibers of diameters around 400 nm were prepared by co-axial electrospinning. The TEM on Figure 1.d shows similar carbon fibers with well dispersed platinum particles of sizes around 10 nm, which were directly added to lignin solution as a platinic salt. This simple method to prepare platinum supported carbon fibers points out the flexibility of this technique.
Table 1 presents the textural properties of the carbonized fibers. The high BET areas and the lack of mesoporous structure demonstrates that these materials are mainly microporous. Similar values of micropore volume measured by means of nitrogen and carbon dioxide indicates the narrow character of this microporosity.
Table 1. Porous structural parameters of the carbon fibers.
110 Benahavís (Málaga, Spain), 21st to 23rd of October, 2009
Figure 1. SEM and TEM images of carbonized a) hollow spheres (SEM bar: 2 µm), b) hollow greenbean-like fibers, c)
tubes (SEM bar: 1000 nm) and d) Filled fibers (SEM bar: 500 nm).
Table 2 presents the XPS and ultimate analysis for the lignin-based tubes. The stabilization process increases the oxygen content of the tubes, which improve their glass transition temperatures avoiding the fusion during the subsequent carbonization process. Carbonization of the stabilized tubes produces an increase in the carbon content due to the removal of oxygen surface groups. Similar trend is observed for the other fibrilar morphologies.
Table 2. Mass concentration for tubes at different preparation stages. XPS Ultimate Analysis
Conclusions Electrospinning is a suitable technique for obtain carbon materials of several forms from lignin. The flexibility of the technique makes possible the tailoring of the material morphology just modifying a few of the electrospinning variables, even allowing the preparation of catalysts through the incorporation of metallic salts during the initial stage. Carbon submicroforms of sizes well below the micron, with total surface areas of more than 1000 m2/g and of different quantities and types of oxygen superficial groups have been obtained using this method. Acknowledgements This work was supported by the Spanish Ministry of Education and Science under grants NAN2004-09312-C03-03 and CTQ 2006/11322.
Objective The objective of this study is the preparation and characterization of carbon based catalysts obtained by chemical activation of olive stone waste for the catalytic decomposition of 2-propanol. The activation was carried out with different activating agents of acidic (H3PO4 and H2SO4) and basic (Ca(OH)2 and Ba(OH)2) character. Results The carbon based catalysts were denoted with PAC, SAC, BaAC and CaAC when H3PO4, H2SO4, Ca(OH)2 and Ba(OH)2 were used activating agents, respectively, followed by the activation temperature in degrees Celsius. The porous structure of the carbons was characterized by N2 adsorption-desorption at -196 ºC. All the carbons but the obtained by activation with H3PO4 (PAC) show type I N2 isotherms (not shown) characteristic of microporous materials. PAC carbon show a type IV isotherm characteristic of a microporous structure with a significant contribution of the mesoporosity. The carbons obtained show apparent surface areas between 68 m2/g for the carbon obtained by activation with Ca(OH)2 to 580 m2/g for the obtained by activation with H3PO4. The activity of the carbon based catalysts was analyzed for the catalytic conversion of 2-propanol. Figure 1 represents the steady state conversion of 2-propanol on the different carbon based catalysts in the absence of oxygen. The acidic carbons PAC-500, SAC-600 and SAC-900 show significantly higher steady state conversions than those corresponding to the basic carbons BaAC-700 and CaAC-900. The highest conversions were obtained using PAC-500 as catalyst. The activation of lignocellulosic residues with phosphoric acid has proven to yield acid carbon catalysts in a single step with a high activity in the alcohol decomposition [1]. Figure 1. Steady state conversion of 2-propanol on the different carbon based catalysts in the absence of oxygen (Po =
0.0185 atm, W/Fo = 0.0.073 g·s/µmol).
The conversion of 2-propanol has been related to the presence of both acid and basic sites on the surface of catalysts. In the broad outline, 2-propanol dehydrates to propylene or diisopropyl ether over acid catalysts and dehydrogenates to acetone over basic catalysts [2]. In absence of oxygen acidic carbon based catalysts (PAC and SAC) yield only propylene as dehydration product, while basic carbon catalysts (CaAC and BaAC) mainly dehydrogenate 2-propanol to acetone although a significant amount of propylene is also obtained (Figure 2). In the presence of oxygen (21%vol), the acidic carbons show higher conversion values and part of the 2-propanol suffer oxidative dehydrogenation yielding acetone as well as propylene, although at
112 Benahavís (Málaga, Spain), 21st to 23rd of October, 2009
high 2-propanol conversion values only propylene is obtained as can be seen in Figure 3, which represents the 2-propanol steady state conversion on PAC-500 carbon based catalyst.
Conclusions Acidic and basic carbon based catalysts were prepared by chemical activation of olive stone waste. The activation was carried out with different activating agents of acidic (H3PO4 and H2SO4) and basic (Ca(OH)2 and Ba(OH)2) character. In absence of oxygen acidic carbon based catalysts dehydrate 2-propanol to propylene and basic carbon catalysts mainly dehydrogenate 2-propanol to acetone although propylene is also obtained. In the presence of oxygen, the acidic carbons show higher conversion values and part of the 2-propanol suffer oxidative dehydrogenation yielding acetone as well as propylene, although at high 2-propanol steady state conversion values only propylene is obtained. Acknowledgements We gratefully acknowledge to the Spanish DGICYT, Projects PPQ2003-07160 and CTQ2006-11322. J.B. acknowledges the assistance of the Ministry of Science and Education of Spain for the award of a FPI grant. References [1] J Bedia, J M Rosas, J Márquez, J Rodríguez-Mirasol, T Cordero. Carbon 47 (2009) 286-94. [2] MA Aramendia, V Borau, C Jiménez, JM Marinas, A Porras, FJ Urbano. J Catal 161 (1996) 829–38.
Figure 2 . Steady state 2-propanol conversion and selectivities for BaAC-700 carbon in the absence of oxygen (Po = 0.0185 atm, W/Fo = 0.0.073 g·s/µmol).
Figure 3. Steady state 2-propanol conversion and selectivities for PAC-500 carbon in the presence of oxygen (Po = 0.0185 atm, W/Fo = 0.0.073 g·s/µmol).
Benahavís (Málaga, Spain), 21st to 23rd of October, 2009 113
Catalytic and non-catalytic carbonization of hemp: the carbonaceous product
M.J. Valero *, A. Gallardo, J. Bedia, J. Rodríguez-Mirasol, T. C ordero
Department of Chemical Engineering, School of Industrial Engineering, University of Málaga, 29071 Málaga, Spain
[email protected] Objective The study of new ways of biomass conversion into useful materials, such as carbon materials with controllable micro and nanostrucutures has been an appealing topic in materials chemistry because of their many important applications as adsorbents, filter materials, catalyst supports, electrode materials, energy-storage materials and stationary phases in liquid chromatography. Hemp is currently being used in the textile, paper and plastics industries, which generates a significant amount of residue. The development of recycling processes of such residues and research into new methods for obtaining high-value materials are generating great interest. Hydrothermal carbonization (HTC) or hydropyrolysis is a convenient way to convert biomass at moderate conditions into carbonaceous nanostructures and/or liquid oil. The objective of this work is the analysis of the carbonaceous nanostructures and the liquid oil obtained from the uncatalyzed and catalyzed hydrothermal carbonization of hemp residues at different reaction conditions. Results The hydropyrolysis of hemp (canes and fibers) biomass was performed in a Teflon-lined stainless steel autoclave at moderate conditions (temperature up to 250ºC). The catalysts used were NaOH and FeCl3 at different concentrations. The effect of the catalyst, the reaction temperature and the residence time was studied. The hydropyrolysis of hemp results in carbonaceous nanoparticles, coexisting with some larger structures and liquid mixtures containing mainly acetic acid with lower amounts of methanol, 2-furaldehyde, guaiacol and acetone among other products. A significant increase of both biomass conversion and liquid product yields were observed using FeCl3 as catalyst. Figure 1 shows some of the carbonaceous structures obtained using hemp as raw material. Suitable conditions of pH, reaction time and temperature were essential for the synthesis of carbonaceous spheres. The HTC of hemp canes and fibres resulted in different amounts of these carbon spheres. The presence of FeCl3 and the uncatalized HTC resulted in a massive formation of carbonaceous microstructures, while the presence of an alkaline solution of NaOH restrained their formation. Hemp canes (HC) and solid products obtained by HTC of the HC with and without catalyst (WC) were carbonized at 500 and 900 ºC under continuous N2 flow. The mass surface concentrations of the carbonized samples, determined by XPS quantitative analysis, are reported in Table 1. The main elements found on the surfaces of the carbonized samples were carbon, oxygen and lower amounts of phosphorus, calcium and silicon, characteristic of biomass materials. Other elements such as nitrogen, magnesium or chlorine were also detected, but at very low concentrations. A significant amount of sodium and iron were observed on the surface of the carbonized solid products from the catalytic process with NaOH and FeCl3, respectively. The treatment at 900 ºC eliminated almost completely the sodium content from the surface by volatilization. However, the iron content on the surface decreases only slightly with carbonization at 900 ºC.
114 Benahavís (Málaga, Spain), 21st to 23rd of October, 2009
Figure 1 : SEM image of the solid product obtained from HTC at 250ºC of (a) hemp canes (bar length: 1 µm) and (b) of hemp fibers (bar length: 2 µm). Table 1 . Mass surface concentration (%) determined by XPS quantitative analysis of the hemp canes and HTC solid products carbonized at 500 and 900ºC.
Conclusions HTC process is a promising method for the synthesis of interesting well-defined carbonaceous micro and nanostructures using hemp biomass as carbon precursors. The influence of reaction time, temperature and the use of catalysts on the carbonaceous nanostructures and/or liquid oil produced during HTC of hemp residues have been studied.
Acknowledgements This work was supported by the Spanish Ministry of Education and Science under grants NAN2004-09312-C03-03 and CTQ 2006/11322.
Benahavís (Málaga, Spain), 21st to 23rd of October, 2009 115
Surface chemistry modification of carbon supported chromium catalysts after NO reduction by XPS analyses
J.M. Rosas, J. Rodríguez-Mirasol, T. Cordero
Chemical Engineering Department. School of Industrial Engineering, University of Málaga.
Campus de El Ejido, s/n, 29013 Málaga, Spain
Introduction X-ray photoelectron spectroscopy (XPS) allows the analysis of the atomic surface composition of a solid and also provides the oxidation state of their components. This technique can give us surface information about both the modification of atomic surface composition after a reaction and the reactive and products bound to the solid surface. In this sense, XPS can be helpful for mechanistic studies of different reactions, used with other complementary techniques. This method was used to study the nitric oxide reduction on carbon supported chromium catalyst with different gas atmospheres. Experimental The catalyst, CAC-Cr, was obtained by pore-volume impregnation of a dried activated carbon (prepared by chemical activation of citrus skin with phosphoric acid, CAC) with an aqueous solution of Cr(NO3)3·9H2O, corresponding to 10 wt% of the most stable oxide (Cr2O3). After drying, the active phase (Cr2O3) was obtained by heating at 400 ºC in a flow of nitrogen. The reduction experiments were performed at atmospheric pressure and different temperatures (300-600 ºC), in a fixed bed reactor with 4 mm of internal diameter, using 300 mg of sample (80 mg of catalyst, diluted with 220 mg of SiC). The total flow rate was 200 cm3 STP/min, for different concentrations of NO ranging from 200 to 800 ppm of NO, 1% CO, 2000 ppm C3H6, 2000 ppm SO2 and 3% O2. X-ray photoelectron spectroscopy (XPS) analyses were obtained using a 5700C model Physical Electronics apparatus with MgKα radiation (1253.6 eV). Results and Discussion CAC-Cr catalyst fresh and after NO reduction reactions was analyzed by XPS. Table 1 shows the mass surface concentrations of the carbon-supported chromium catalyst before an after reaction with 200 ppm NO and different gases, at temperatures between 300-600 ºC, except for the reaction in the presence of oxygen that was carried out at 350 ºC. The results show an increase of the surface nitrogen amounts after direct NO reduction and reduction in the presence of CO, compared to that of the fresh catalyst, while a similar nitrogen content is observed after the reaction NO-C3H6 .Figure 1 shows the XPS spectrum of N 1s for the catalyst CAC-Cr at the same previous conditions. The results evidence the formation of nitrogen surface complexes with the reaction of NO, NO-CO and NO-SO2. The presence of oxygen produces the formation of oxygen surface complexes that iusep the NO reduction to N2, avoiding the formation of stably surface nitrogen complexes, as reported by Suzuki et al.[1]. Different authors suggest the presence of pyridinic complexes at 398.7 eV, pyrrolic complexes at 400.2 eV and quaternary nitrogen complexes with a positive formal charge at 401.4 eV.[2]. A higher contribution of the peak obtained at lower binding energies is observed with the NO reaction, associated to pyridinic complexes. We have previously observed by transient kinetic experiments that the reducing agent must be necessary adsorbed on the catalyst surface in order to reduce nitric oxide. A similar surface nitrogen content of the carbon-supported chromium catalyst before an after NO-C3H6 reaction suggests that a Eley-Rideal mechanism may be taken place. Specifically, for CO as reducing agent, the formation of carbon-nitrogen complexes, as pyrrolic and pyridinic groups, during the reduction of NO, indicates, preferentially, a Langmuir-Hinshelwood mechanism, where both NO and CO must be adsorbed on the catalyst surface.
395396397398399400401402403404405Binding energy (eV)
N(E
)/E
CAC-Cr
NO (300-600 ºC)
NO+CO+O2 (350 ºC)
NO+SO2 (300-600 ºC)
NO+CO (300-600 ºC)
NO+C3H6+O2 (350 ºC)
NO+C3H6 (300-500 ºC)
Figure 1. XPS N1s spectra of fresh CAC-Cr and after reaction with 200 ppm NO and different gases at temperatures between 300-600 ºC. Conclusions XPS analyses show an increase of the nitrogen content for both NO direct reduction and in the presence of CO, as pyridinic and pyrrolic complexes. However, no formation of nitrogen complexes is observed in the presence of propylene as reductant agent. In based of these results, an Eley-Rideal mechanism can be taken place more probably, for NO reduction, where propylene is adsorbed on the active sites. While, for CO as reducing agent, a Langmuir-Hinshelwood mechanism, where both NO and CO are adsorbed could be likely obtained. Acknowledgements The authors thank the Ministry of Education and Science of Spain for financial support (Project CTQ2006/11322). References [1] Suzuki, T.; Kyotani, T.; Tomita, A. Ind. Eng. Chem. Res. 1994, 33, 2840-2845. [2] Pels, J.R.; Kapteijn, F.; Moulijn, J.A.; Zhu, Q.; Thomas, J.M. Carbon 1995, 33, 1641-1653
Pd-based catalysts for methane oxidation at low temperatures suffer from deactivation due to particle sintering and exposure to sulfur compounds. Recent studies had shown a remarkable effect of the mesoporous HMS silica, limiting the PdO agglomeration, lessening the SO2 poisoning and allowing easy reactivation of the catalyst [1]. When amorphous composite sol-gel oxides, TiO2-SiO2, with a small amount of titania and with high surface area were used as supports, an additional increase of the catalyst oxidation activity and also an enhancement of the sulfur tolerance was obtained [2]. Aiming to further improve the catalytic performance of the PdO catalysts the use as supports of mesoporous incorporating titania, is here explored.
Results
Mesoporous silicas (SBA-15 and HMS) containing 5 and 10 wt% of TiO2 were prepared and characterized by XPS, XRD, BET and the acidity of the supports evaluated on the basis of ammonia TPD. The 1 wt% Pd was supported by incipient wet impregnation from aqueous solution of Pd(NO3)2 (Aldrich). The methane oxidation activity was measured in lean conditions at WHSV= 60 000 ml g−1 h−1 in absence and in the presence of 10 vol. ppm SO2. Stability tests were performed after 16 h at 600°C under the reacti ng mixture. Mesoporous silicas enhanced the palladium activity as compared to amorphous silica, to an extent which depended on the particular mesoporous structure. Moreover, as shown in Fig.1, for the SBA-15 supported catalysts, the presence of TiO2 improved the sulfur tolerance and most importantly it favored the regeneration of the catalyst in the following SO2-free run. The opposite effect was observed on the HMS samples, see Table 1. In fact, by increasing the Ti loading in the Pd-HMS catalysts, an increase of the T50 (temperature of 50% CH4 conversion) values was observed in both cycles, with and without SO2.
200 300 400 500 6000
20
40
60
80
100
full symbol =1st run with SO2
open symbol =2nd run SO2 -free
CH
4 co
nver
sion
(%
)
Temperature (°C)
Figure. 1 . Methane conversion for PdSBA15 (black), Pd/Ti5SBA (red) and Pd/Ti10SBA (green).
Conclusions The presence of TiO2 improves the activity and the regeneration of the catalyst in the SO2-free run in case of SBA-15 samples whereas exactly the opposite trend was observed on HMS samples. The catalytic behaviour could be explained in terms of structural properties and acidity. Acknowledgements European Community through (NoE) IDECAT and COST D36 action is acknowledged for financial support. References [1] A. M. Venezia, R. Murania, G. Pantaleo, G. Deganello, J. Catal. 251 (2007) 94. [2] A. M. Venezia, G. Di Carlo, G. Pantaleo, L. F. Liotta, G. Melaet, N. Kruse, Appl. Catal B. 88 (2008) 430
1 Latvian Institute of Organic Synthesis, 21 Aizkraukles Str., Riga LV-1006, Latvia
* [email protected] 2 Institute of Catalysis, Bulgarian Academy of Sciences, 1113 Sofia, Bulgaria
Objective It is known that acetone and other ketones as highly reactive compounds find wide application in synthesis of polymers, pesticides, and pharmaceuticals, as well as in utilization as solvents and extractives. We have established earlier, that at temperatures below 400ºC the main products of aliphatic aldehydes and alcohols transformation over zink chromite catalyst are ketones [1, 2]. The aim of the present work is to gain deeper insight into the behavior of Zn-Cr oxide catalyst in the conversion of aliphatic acids. Results Zn-Cr oxide catalyst was prepared by coprecipitation from salt solution. X-ray diffraction analysis established that by mixing of saturated ZnSO4 and (NH4)2CrO4 solutions and addition of equimolar amount of NH4OH, zinc ammonium oxychromate ZnOHNH4CrO4 is precipitated. The dried precipitate upon heating at 290-300ºC formed the catalyst possessing disordered zinc chromite spinel structure, with equimolar quantity of ZnO being dissolved in it. The TPR method showed that Cr+6 is completely reduced to Cr3+ during calcination of catalyst precursor by NH3, evolved in this process. It is supposed that catalyst deactivation above 400ºC is connected with ordering of the spinel structure as a result of thermal processing. BET surface area of the obtained catalyst is 57 m2/g. The catalysts have been tested for ketonization of acetic, propionic and butyric acids. The effects of temperature, catalyst loading, and the molar ratio of acid:water were investigated. The results of catalytic tests performed in the presence of water in molar ratio acid:water=1:2) in the temperature range 300-400оС are represented in Table 1. The acids conversion into ketones was the highest (87-96%) at 325-350оC. Table 1. Conversion of aliphatic carboxylic acids to ketones over zinc chromite catalyst in accordance with temperature.
Acetic acid Propionic acid Butyric acid
Selectivity, % Selectivity, % Reac- tion
T, °C Conver-
sion, %
Selectivity to acetone,
%
Conver- sion,
% Methyl-
ethylketone Diethyl-ketone
Conver-sion,
% Acetone Methylpro-pylketone
Dipropyl-ketone
300 95.1 93.3 48.4 0.6 89.7 19.8 0.0 3.4 87.8
325 98.8 96.1 90.2 0.7 95.0 40.2 0.0 2.6 92.3
350 100.0 93.9 99.8 2.7 93.1 91.5 0.1 4.2 90.3
375 100.0 90.3 100.0 6.5 84.9 99.0 2.3 17.0 67.8
400 100.0 84.2 100.0 12.3 62.1 99.0 7.8 21.8 25.6
The comparison of ketonization processes of butyric acid, butanol and butanal showed that the selectivity to ketone decreased in the order: acid>aldehyde>alcohol (Table 2).
The gaseous products of acetic acid conversion are H2, CH4, CO, of propionic acid – H2, CH4, CO, C2H4, C2H6, of butyric acid – H2, CH4, CO, C3H6, C3H8. On the basis of above data, it was possible to conclude that transformations of acetic acid over Zn chromite catalyst proceedes according to the general equation 1 and two negligible reactions: decarboxylation (2) and oxidation (3):
Similar reactions are observed in the case of propionic and butyric acid too. We are proposing that the ketonization of aliphatic aldehydes, alcohols and acids on Zn-Cr-O catalyst proceeds according to dissociative – associative reaction mechanism, i.e., the reactants dissociate on the surface of catalyst to fragments, and these fragments combine together by association to form the reaction products. The ketonization mechanism of acetic acid was investigated by semiempirical quantum chemical AM1 method using the cluster approach. It is found that the adjacent acid-base pair of the catalytic sites provokes dissociative adsorption of the acetic acid molecules resulting in the formation of surface carboxylate species. Adsorption process proceeds spontaneously. After blocking the acid-base pairs of catalyst, the new portions of acetic acid molecules interact with active species in the gas phase, converting into acyl cations. The methyl group of adsorbed carboxylate species is attacked by the acyl cation resulting in the bimolecular electrophilic substitution reaction and formation of an acetone molecule and a carbon dioxide one. Conclusions The ketonization of aliphatic acids over a zinc chromite catalyst gives symmetric ketones at 325-350ºC with high selectivity (90-96%). Selectivity to symmetric ketones in the case of conversion of acid is higher than that of the corresponding alcohol aldehyde. At 375-400ºC, the destruction of molecules significantly rises. It is concluded that ketonization of aliphatic acids over chromite type catalysts is a perspective method for ketones production. References 1. V. Stonkus, Zh.Yuskovets, M. Shymanska, Zh. Obshch. Khim., 64 (2) (1994) 295. 2. V. Stonkus, Zh. Yuskovets, M. Shymanska, Latv. Khim. Zh., N4 (1993) 460.
Benahavís (Málaga, Spain), 21st to 23rd of October, 2009 121
Supported gold catalysts for Preferential oxidation (PROX) of CO in the presence of excess H 2
L.F. Liotta *, G. Di Carlo, G. Pantaleo and A. M. Venezia
Istituto per lo Studio di Materiali Nanostrutturati, CNR, Via Ugo La Malfa, 90146 Palermo (Italy). * [email protected]
Objective Developing efficient catalysts for the selective oxidation of carbon monoxide in the presence of excess hydrogen is a challenge for the commercial application of low-temperature proton exchange membrane fuel cells (PEMFCs), where power is generated from the electrochemical oxidation of hydrogen over an anode electrocatalyst, generally, Pt/C. The production of hydrogen by steam reforming of methanol or by partial oxidation of liquid hydrocarbons followed by water gas shift reaction generates gas effluents containing 0.3-1% of CO in an excess of H2
(40-75%) and 20-25% of CO2. Since the CO molecule present in the stream would poison the Pt anode, it is important to reduce its level below 10 ppm. The preferential oxidation (PROX) of CO is a possible way to remove such contaminant from the gas stream [1]. Catalysts based on noble-metals such as Pt, Rh and Ru are generally used for this process, however, quite recently, gold has received much attention due to its ability to oxidize CO at high rates in the temperature range of the operating PEMFCs [2]. Several factors such as supported particle size, gold oxidation state and type of carrier affect the catalytic performance of such catalysts. In this study the role played by different supports, such as “reducible” (CeO2, TiO2) and “inert” (γ-Al2O3 and SiO2) oxides was investigated in the CO oxidation in the presence of hydrogen. With this purpose, catalysts with 1.5 wt% gold loading were prepared by deposition precipitation method. The morphology, the structure and the electronic properties were determined by X-ray diffraction (XRD) and X-ray photoelectron spectroscopy (XPS). For the catalytic reaction the reagent gas mixture consisting of 1% of CO + 0.5% of O2 + 70% of H2 in He was used, with a weight hourly space velocity (WHSV) of 60 000 mL h-1g-1. Results Commercial oxides, CeO2, TiO2, γ-Al2O3 and SiO2, with specific surface area ranging between 79 and 546 m2/g were used as supports. The gold catalysts listed in Table 1, containing 1.5 wt% gold, were prepared by deposition precipitation method using urea for the silica supported catalyst and Na2CO3 solution (0.1 M) for the others. The pH of the solution was optimized for the different supports. The obtained samples were dried overnight at 120°C and tested without further treatment. The alumina and the silica supported catalysts gave XRD detectable particle sizes, while highly dispersed gold crystallites (dAu less than 4 nm) were obtained over ceria and titania. According to the XPS binding energies, Au0 was present on alumina, silica and titania supported catalysts, Au1+ was found only on the ceria supported one and Au3+ was present in both, titania and ceria catalysts. The catalytic results in terms of CO conversion and CO selectivity are reported in Figure 1 for the different catalysts. In accord with our recent results [3], the gold on ceria showed the highest CO conversion and most important, at variance with the other supported catalysts, it exhibited a plateau for both, conversion and selectivity, from 90°C up to the highest measured temperatures. Gold on titania was less performing, giving at 50 °C a maximum of CO conversion equal to 40% with a selectivity slightly higher than 40%, however, both values abruptly decreased by increasing the reaction temperature. The selective oxidation of CO was further weakened on gold over alumina, showing at 70 °C 30% of CO conversion and comparable low selectivity. Gold over silica was practically inactive.
122 Benahavís (Málaga, Spain), 21st to 23rd of October, 2009
Table 1. Specific surface area (SBET), average Au particle size (d), XPS Au 4f 7/2 binding energy, XPS atomic ratio Au/M. (M n+ = Al 3+, Si 4+, Ti 4+, Ce 4+) and corresponding derived Au loading. Catalyst
Figure 1 . CO conversion % and CO selectivity % as a function of temperature for the gold supported catalysts. Conclusions Gold on ceria exhibited the best catalytic performance with the highest CO conversion and CO selectivity which reach a plateau in the range between 90°C and 150°C. Moving to the other supported catalysts, the CO oxidation in presence of hydrogen increased in the order Au/SiO2< Au/Al2O3< Au/TiO2. The comparison among the present catalysts highlights the important role in the CO oxidation played by gold ionic species (Au3+, Au1+) which are better stabilized by cerium oxide, likely due to a strong metal support interaction. On the other hand, the presence of metallic gold particles over an “inert” support, like alumina and silica, has proven to give poor catalytic activity. Acknowledgements European Community through (NoE) IDECAT and COST D36 action is acknowledged for financial support. References 1. S. H. Lee, J. Han, K. –Y. Lee, J. Power Sources 109 (2002) 394. 2. D. Cameron, R. Holliday, D. Thompson, J. Power Sources 118 (2003) 298. 3. A. M. Venezia, G. Pantaleo, A. Longo, G. Di Carlo, M. P. Casaletto, F. L. Liotta, G. Deganello, J. Phys. Chem. B 109 (2005) 2821.
Benahavís (Málaga, Spain), 21st to 23rd of October, 2009 123
Structure-Reactivity Relationships in ElectronTrans fers of Helical Polyaromatic Dications
Lubomír Pospíšil *ab, Miroslav Gál a, Michal Horá čeka, Filip Teplý b, Louis Adriaenssens b, Lukáš Severa b
a J. Heyrovský Institute of Physical Chemistry of ASCR, v.v.i., Prague, Czech Republic b Institute of Organic Chemistry and Biochemistry of ASCR, v.v.i., Prague, Czech Republic
* [email protected] Recent synthesis of various substituted [5]helquats containing five aromatic rings enabled us the comparison of the electron transfer rates in structures with a different sterical factors. These compounds are polyaromatics with a helical structure containing two quaternary nitrogens as heteroatoms. Their structural resemblance to well known diquat inspired us to call them helquats. Our previous communication compared the redox properties of helicenes[1] and helquats[2]. Compounds with six ([6]helquat) and seven ([7]helquat) aromatic rings were also prepared.
Helquats (H2+) in aprotic solvents are reduced in two subsequent reversible one-electron steps. The electrochemical impedance spectroscopy was used for determination of very fast heterogeneous charge transfer rates. The first redox step is slightly slower than the second electron transfer reaction. This is likely caused by larger inner reorganization energy of the reduction of the dication to a cation radical, which is much smaller for the second redox step. Good correlation of reversible potentials and LUMO energy was obtained. Cation radical generated by the first electron transfer acts as a donor H+ and forms a charge transfer complex with the starting oxidized form, the dication, which acts as an acceptor H2+. The EPR spectra measured at different concentration ratio of H+ and H2+ yield the bimolecular electron self-exchange rate. The correlation of the heterogeneous and homogeneous electron transfer rates in terms of Marcus theory is sought. Acknowledgement This research was supported by the Grant Agency of the Czech Rep. (203/09/0705, 203/08/1157 and 203/09/P502) and by the Czech Ministry of Education (COST D36 OC140, ME09114) and Institute of Organic Chemistry and Biochemistry, ASCR (Z4 055 0506) References [1] L. Rulíšek, O. Exner, L. Cwiklik, P. Jungwirth, I. iuse, L. Pospíšil, Z. Havlas, J. Phys. Chem. C 2007, 111, 14948. [2] L. Adriaenssens, L. Severa, T. Šálová, I. Císařová, R. Pohl, D. Šaman, S. V. Rocha, N. S. Finney, L. Pospíšil, P.
Slavíček, F. Teplý, Chemistry Eur. J., 2009, 15, 1072.
In-situ spectroscopy is an essential tool for the fundamental understanding of catalytic reactions. [1-2] However, while most of the applied techniques average the information over the whole sample, probing a distinct area of a catalyst particle or grain can reveal valuable information concerning the structure-function relationship during the catalytic action. For this purpose, micro-spectroscopic methods have been applied. The selective conversion of methanol into light olefins (MTO) is interesting because it enables the production of olefins while making use of oil alternative feedstocks. So far, the most promising catalysts for the MTO reaction are H-SAPO-34 and H-ZSM-5. [3] However, the reaction suffers from a fast deactivation caused by the formation of carbonaceous deposits. Here we aim to elucidate the differences in coke formation between large H-SAPO-34 and H-ZSM-5 individual crystals during the MTO reaction in a space and time resolved manner. This has been made possible by applying a high-temperature in-situ cell in combination with UV-Vis and confocal fluorescence micro-spectroscopy techniques. [4]
Figure 3. a) Optical Microphotographs of H-ZSM-5 crystals taken during the MTO reaction at 745K. b) Corresponding absorption spectra taken from a spot in the middle of the crystal.
126 Benahavís (Málaga, Spain), 21st to 23rd of October, 2009
Results and Discussion
Upon exposure of methanol vapour, the translucent crystals undergo darkening due to the formation of carbonaceous species. In H-ZSM-5, coke is initially formed at the triangular edges, where straight channel openings directly reach the external crystal surface. During the reaction, the formation of absorption bands, assigned to aromatic coke compounds and their precursors inside the crystal, is observed. Furthermore, a broad background absorption that extends over the entire visible region indicates the formation of graphite coke on the external surface of the crystal (Figure 1). Confocal fluorescence microscopy confirms these observations and shows that fluorescent carbonaceous species inside the crystal are initially formed at the near surface area. It is also observed that while the coke front gradually diffuses towards the centre of the crystal, internal intergrowth boundaries hinder the facile penetration for the more bulky aromatic
compounds (Figure 2). H-SAPO-34 crystals show two different temperature regions in which the formation of different coke species have been observed. In these crystals, the coke compounds formed
remain mainly at the near surface region of the crystal during the entire course of the reaction. Here, the formation of polyaromatic coke compounds leads to channel blockage, creating diffusion limitations for the coke front moving towards the middle of the crystal, thereby making the internal region of the crystal less accessible to the reactant molecules.
Conclusions The combination of in-situ UV-Vis and confocal fluorescence micro-spectroscopy is a valuable tool to probe coke deposits and their precursors during a catalytic reaction. In these molecular sieves it is shown in a space and time resolved manner that clear differences are observed in the rate of reaction and the three dimensional distribution of different coke species formed. The formation of two distinct coke systems i.e. aromatic hydrocarbons in the internal pores and graphitic coke at the external surface of the crystals is thereby illustrated. The differences in coke formation are explained in terms of pore architecture and intergrowth structure. References [1] J.F. Haw, In-situ spectroscopy in heterogeneous catalysis, Wiley-VCH, Weinheim, 2002 [2] B.M. Weckhuysen, In-situ spectroscopy of catalysis, American Scientific Publishers, Stevenson Ranch, 2004 [3] M. Stöcker, Micropor. Mesopor. Mater. 1999, 29, 3 [4] D. Mores, E. Stavitski, M.H.F. Kox, J. Kornatowski, U. Olsbye, B.M. Weckhuysen, Chem. Eur. J. 2008, 14, 11320-11327
Figure 4. fluorescence intensity profiles of H-ZSM-5 crystals during the MTO reaction at 660K depicted with time on stream at laser excitation (a) 488 nm (detection at 510-550 nm) and (b) 561 nm (detection at 565-635 nm). (c) Schematic representation of the slice where the confocal fluorescence measurement has been performed.
Synthesis and properties of a donor-acceptor type a soluble and stable neutral state green polymeric electrochromic material was prepared via electrochemical polymerization of a donor-acceptor hybrid monomer, namely 8-(2,3-dihydrothieno[3,4-b][1,4]dioxin-5-yl)-11-(2,3-dihydrothieno[3,4-b][1,4]dioxin-7-yl)acenaphtho[1,2-b]quinoxaline (EdAcQ) (Chart 1). The ambipolar (n- and p-doping processes) properties were attained by incorporation of donor-acceptor unit into polymer backbone. As a consequence, a low band gap (1.0 eV) material was obtained. The corresponding polymer exhibits has a green color in the neutral state and a blue color when oxidized. In addition, the polymer film dissolved in dichloromethane or acetonitrile when it is in reduced state.
N N
SS
SO
O
OO
Chart 1. acenaphtho[1,2-b]quinoxaline based D-A type monomer. The voltammogram of EdAcQ in 0.1 M tetrabutylammonium hexafluorophosphate
(TBAH)/CH2Cl2 (DCM) solution exhibited an irreversible oxidation peak ( oxamE , ) at 0.43 V vs.
Fc/Fc+ during anodic scan, which was ascribed to the oxidation of external 3,4-
ethylenedioxythiophene (EDOT) units, and a reversible reduction peak ( redmE 2/1, ) at -1.49 V
throughout the cathodic scan, which was attributed to the radical anion formation from the acenaphtho[1,2-b]quinoxaline unit. During repetitive scans a new reversible redox couple with intensifying current has formed. The observed behaviour indicated the formation of an electroactive polymer film on the electrode surface.
-1.0 -0.5 0.0 0.5 1.0 1.5 2.0
-30.0
-20.0
-10.0
0.0
10.0
20.0
30.0
40.0
i d/mA
cm-2
E / V vs. Ag wire
Figure 1. Electropolymerization of 1.0 x 10-3 M EdAcQ in 0.1 M TBAH/DCM at 100mV/s by potential scanning.
128 Benahavís (Málaga, Spain), 21st to 23rd of October, 2009
The PedAcQ in neutral state has three well-defined absorption maxima at 320 nm (3.75 eV), 415 nm (2.89 eV), and 690 nm (1.74 eV) including deep valleys controlling the hue and brightness of the green color1. The former two bands (<500 nm) absorb the red color and the latter band (>700 nm) absorbs blue (Fig. 2).
Figure 2. (a) Electronic absorption spectra of the PedAcQ at various applied potentials between -0.7 V and 1.0 V and (b) the colors of the PedAcQ on ITO at oxidized, neutral, and reduced states in 0.1 M TBAH/DCM. At low doping levels, the intensities of the three π-π* transition bands of PedAcQ decreased simultaneously and a broad absorption band centered around 810 nm started to intensify due to the polaron formation. Upon further oxidation, the bipolaron band (at 930 nm) at about 0.50 V formed, which was also monitored synchronously with cyclic voltammetry, and the polaron band started to decrease simultaneously. The polymer film caused the color transition from green to blue during oxidation. The stability of polymer film was studied between neutral and oxidized states by cyclic voltammetry technique at a scan rate of 200 mV/s. The significant change in redox response of the polymer was not observed at the end of a thousand cycles. The changes in percentage transmittance (∆T%) between the neutral (at -0.70 V) and oxidized states (at 1.0 V) were found as 21.3% for 415nm, 29.0% for 930 nm in the visible region as well as and 33.4% for 1080 nm in the NIR region. The polymer bearing electrochromic behaviour showed low switching time and high stability. The researches to obtain neutral state green polymeric electrochromics with adjusted intrinsic properties are in progress. References
G. Sonmez, C.K.F. Shen, Y. Rubin, F. Wudl, Angew. Chem. Int. Ed. Engl. 43 (2004) 1498.
Benahavís (Málaga, Spain), 21st to 23rd of October, 2009 129
Molecular structure and reactivity of MoO 3/TiO2 catalysts for ethane oxidative dehydrogenation studied by operando Raman spectrosc opy
George Tsilomelekis and Soghomon Boghosian*
Department of Chemical Engineering, University of Patras and Institute of Chemical Engineering and High Temperature Chemical Processes, Foundation of
Research and Technology-Hellas (FORTH/ICE-HT), Patras, GREECE
Objective The aim of the present study is to gain insight into the molecular structure of molybdena/titania(anatase) catalysts and into their catalytic behaviour for the oxidative dehydrogenation (ODH) of ethane at temperatures 420–500 oC. Operando Raman spectroscopy is used for exploring the surface composition of the working catalysts with simultaneous catalytic measurements. The response of the catalyst constituent MoOx species to alterations of the catalyst atmosphere is monitored by exploiting the relative Raman band intensities. The effect of catalyst composition, operating temperature, gas atmosphere, and reactant residence time on both Raman spectra and catalytic efficiency is studied. Isotopic substitution experiments with 18O2(g) are used for differentiating between mono-oxo and di-oxo configurations for the amorphous oxo-molybdenum species. Results The properties of the catalysts (synthesized by wet impregnation of anatase with aqueous solutions (pH = 4–5) using ammonium heptamolybdate as the precursor) are summarized in Table 1. Table 1. Catalyst properties
Catalyst wt% MoO 3 Surface Area (m 2/g) Mo surface density (Mo/nm 2)
35MoTi 35 86.3 17.0 TiO2 , MoO3 TiO2 (calcined) : 97.8m2/g The in situ Raman spectra obtained for all catalysts under O2 flow at 430oC are shown in Fig. 1. A sharp band at 992 cm-1 characteristic of Mo=O stretching is observed, of which the position remains stable with increasing loading and the relative intensity progressively increases up to the approximate monolayer (15MoTi, 5.9 Mo/nm2). A low presence of associated species possessing Mo–O–Mo linkages is evident for the monolayer sample (weak broad band at ~925 cm-1). Bulk MoO3 crystals are formed for coverages exceeding the monolayer (21MoTi, 35MoTi). By exploiting the Raman band intensities under oxidized and steady state reaction conditions (for various residence times) it was found that the reduction of the Mo=O sites is facilitated with increasing loading. The combined information of in situ Raman with in situ FTIR spectra, together with in situ Raman “snap-shots” (Fig. 2) of catalyst samples subjected to successive reduction/oxidation cycles with H2 and 18O2 points to a
1200 1000 800 600 400 200
35%
21%
9%
15%
6%
Inte
nsity
, a.
u.
Raman Shift ,cm -1
MoO3 / TiO
2
O2 , 430°C
3%
Figure 1. In situ Raman spectra of MoO3/TiO2 catalysts at 430oC under flowing O2(g).
130 Benahavís (Málaga, Spain), 21st to 23rd of October, 2009
mono-oxo molecular configuration, O=Mo(–O–Ti)3 for the dispersed oxo-molybdenum species. The selective reactivity to ethylene and the ethylene yield are found to increase up to the monolayer coverage and to moderately decrease at higher loadings. The apparent activity per Mo atom vs. loading goes over a sharp maximum at the approximate monolayer.
Acknowledgement Financial support from the Research Committee of the University of Patras (C. Caratheodory program/C.583) is gratefully acknowledged. References [1]. A. Christodoulakis and S. Boghosian S., J. Catal. 260 (2008) 178. [2]. Tsilomelekis, A. Christodoulakis and S. Boghosian S., Catal. Today 127 (2007) 139. [3]. A. Christodoulakis, E. Heracleous, A. A. Lemonidou and S. Boghosian, J. Catal. 242 (2006) 16.
Figure 2. In situ Raman spectra of the “monolayer” 15MoTi catalyst at 450oC after successive reduction/oxidation cycles with H2 and 18O2.
1040 1020 1000 980 960 940 920 900 880
30th
25th
21th
17th
13th
9th
5th
3rd
Inte
nsity
, a.
u.
Raman Shift , cm -1
16O2
1st
15%MoO3/TiO
2
T=450°C
0 2 4 6 8 10 12 14 16 18 20 220
10
20
30
40
50
60
70
80
90
100
3MoTi 6MoTi 9MoTi 15MoTi 21MoTi 35MoTi
Eth
yle
ne
Se
lect
ivity
, %
Conversion , %
Figure 3. Ethylene selectivity as a function of ethane conversion for all catalysts at 500oC.
Benahavís (Málaga, Spain), 21st to 23rd of October, 2009 131
Nanostructured MoVNbTeO Oxide Catalysts for Selective Oxidation Reactions
R. López-Medina a, H. Golinska b, M. Ziolek b,*, Miguel A. Bañares a, M.O. Guerrero-Pérez c,*
a Catalytic Spectroscopy Laboratory, Instituto de Catálisis y Petroleoquímica; CSIC; Marie Curie 2; E-29049-Madrid (Spain); b Adam Mickiewicz University, Faculty of Chemistry, Grunwaldzka 6,
60-780 Poznań, Poland; cDepartamento de Ingeniería Química. Universidad de Málaga; E-29071-Málaga (Spain)
Objectives The reactivity/selectivity properties of the nano catalysts are chemically probed with steady-state catalytic studies of methanol oxidation reactions to determine optimum conditions for selective oxidation of methanol using MoVNbO and MoVNbTeO supported catalysts on γ-Al2O3 containing 4, 8 and 12 atoms (Mo+V+Nb+Te)/nm2 calcinated in air and inert atmosphere. Also, the aim of the present work was to study the gas-phase conversion of acetonylacetone in these catalysts in order to test if acetonylacetone can be used as a sensitive molecule for the simultaneous characterization of acidic and basic surface properties. Results The oxidation of methanol can be used as a probe reaction to characterize the surface acidic and redox properties of catalysts according to the different products like formaldehyde and dimethoxymethane, dimethyl ether and carbon oxides indicating the presence of redox, acidic and basic sites respectively1,2. Incorporation of Te into MoVNbO improved the activity of selective oxidation of methanol to formaldehyde on redox sites.
4Mo5
V4N
b1
4Mo6
V3N
b1
4Mo5
V4N
b1
4Mo6
V3N
b1
4Mo5
V4N
b0.5
Te0
.5
4Mo6
V3N
b0.5
Te0
.5
4Mo5
V4N
b0.5
Te0
.5
4Mo6
V3N
b0.5
Te0
.5 --
0
10
20
30
40
50
60
70
80
90
100
iner
t
iner
t
iner
t
iner
t
air
air
air
% C
onve
rsio
n/S
elec
tivity
atomic ratio Mo/V/Nb/Te
Conversion Formaldehyde Dimethyl eter
air
without Te with Te
8Mo5
V4N
b1
8Mo6
V3N
b1
8Mo5
V4N
b1
8Mo6
V3N
b1
8Mo5
V4N
b0.5
Te0
.5
8Mo6
V3N
b0.5
Te0
.5
8Mo5
V4N
b0.5
Te0
.5
8Mo6
V3N
b0.5
Te0
.5 --
0
10
20
30
40
50
60
70
80
90
100
iner
t
iner
t
iner
t
iner
t
air
air
air
air
with Tewithout Te
atomic ratio Mo/V/Nb/Te
%S
elec
tivity
0
10
20
30
40
50
60
70
80
90
100
%C
onversion
Figure 1 . Catalytic activity in MeOH + O2 reaction A) submonolayers coverages catalysts, B) monolayer coverage catalysts, 0.1 g of catalyst activated in helium flow (40 ml/min) at 400°C for 2 h. Reaction conditio ns: 40 ml/min He/O2/ MeOH (88/8/4 mol%), T = 250°C.
The results of methanol oxidation reaction reveal that there are acid sites on the surface of submonolayer coverages of MoVNb(Te)O catalysts (4 atoms/nm2), at monolayer and monolayer and half coverage (8 and 12 atoms/nm2 respectively) there are redox sites increased with the coverage on the surface of the catalysts. The selectivity to redox products is around ~90% and 10 % to acidic products, without the formation of any products originating from surface basic sites. The reaction of acetonylacetone, [1,4-diketone (2,5-hexanedione)], is known to undergo both acid- and base-catalyzed intramolecular cyclizations leading to 2,5-dimethylfuran (DMF) and 3-methyl-2-cyclopenten-1-one (MCP), respectively1,3,4. Thus, the incorporation of increasing amounts of atoms on surface of MoVNb(Te)O oxide causes a continuous decrease in DMF selectivity. Moreover, with the catalysts with 12 atoms/nm2, (DMF) was produced at greater than 60 % selectivity; in contrast with a catalyst even below surface coverage for the supported MoVNb(Te)O (4 atoms/nm2), MCP was obtained with selectivities approaching to 50 % or better.
Figure 2 . Catalytic activity in acetonylacetone cyclisation reaction A) catalysts calcined in air, B) catalysts calcined in inert, C) catalysts calcined in air with Te and C) catalysts calcined in inert with Te, 0.07 g of catalyst activated in helium flow (40 ml/min) at 400°C for 2 h. Reaction condit ions: 40 ml/min He, T = 350°C.
Conclusions The results presented above show that acetonylacetone (2,5-hexanodione) conversion is a good test reaction to confirm the acid or base surface properties of typical solid acid or base catalysts. For MoVNb(Te) supported oxide systems, methanol oxidation decreased with increasing surface density MoVNb(Te)O/nm2 of an oxide supported. Acknowledgements The Ministry of Science and Innovation (Spain) funded this study under project CTQ2008-04261/PPQ. R.L.M. thanks MAEC-AECID (Spain) for his pre-doctoral fellowship. The authors express their thanks to Olaf Torno (SASOL Germany GmbH) for providing alumina support. References [1] I. Sobczak, N. Kieronczyk, M. Trejda, M. Ziolek, Catal. Today 139 (2008) 188 [2] M. Badlani, I.E. Wachs, Catal. Letters 75 3-4 (2001) 137 [3] R.M. Dessau, Zeolites 10 (1990) 205 [4] J.J. Alcaraz, B.J. Arena, R.D. Gillespie, J.S. Holmgren, Catal. Today 43 (1998) 89 .
Benahavís (Málaga, Spain), 21st to 23rd of October, 2009 133
Operando Studies of VPO catalysts in n-butane selec tive oxidation reaction. Activity,
selectivity and structure transformations.
Ewelina J. Mikolajska, Anna E. Lewandowska, Miguel A. Bañares*
Catalytic Spectroscopy Laboratory, Instituto de Catalisis y Petroleoquimica, CSIC, E-28049-Madrid, Spain * [email protected]
Vanadium phosphates (VPOs) are important catalysts for selective oxidation of alkanes. For many years they have been the only catalysts in commercial n-butane oxidation to maleic anhydride, which is a precursor of polyester resins. This selective oxidation reaction follows Mars and Van Kravelen mechanism, where each metal in different oxidation state plays its own role in the reaction. According to the mechanism metal cation is reduced by adsorbed organic molecule and subsequently reoxidized by gas phase oxygen in next step of the reaction. VPO catalysts contain V4+as (VO)2P2O7 phase and V5+ as VOPO4 phase. Vanadium (V5+) is probably responsible for formation of maleic anhydride and plays role in a rate determining step of the selective oxidation reaction of n-butane, while vanadium (V4+) is active in formation of by-products. However, vanadyl phosphate phase is detectable by X-ray photoelectron spectroscopy in vanadium hydroxide oxide phosphate, a commercial VPO precursor and the only detectable crystalline phase in commercial catalysts is vanadyl phyrophosphate containing V4+ cations, that is a major active component of the n-butane oxidation reaction [1,2]. The presence of VPO4 (V
5+) phase was also detectable by in situ studies [3].
512 514 516 518 520 522 524 526 528
V4+
522,
50
523,
90
516,
90
coun
ts p
er s
econ
d (a
.u.)
BE (eV)
515,
52
O1ssat
2Vp
VPO precursor
V5+
510 512 514 516 518 520 522 524 526 528
523,
34
coun
ts p
er s
econ
d (a
.u.)
BE (eV)
516,
15
O1ssat
V2p
VPO calcinated
V4+
Figure 1. XPS spectra of VPO precursor (left) and VPO calcinated catalyst (right)
134 Benahavís (Málaga, Spain), 21st to 23rd of October, 2009
VPO catalysts may reveal many possible crystalline phases, like (VO)2P2O7, VOPO4, αI, αII, β, γ, δ, and metastable ω, and disordered phases [2,4,5]. Their relative ratio depends on methods of preparation and catalysts precursor. The ω-VOPO4 phase is stable only at elevated temperatures and seems to be very sensitive to reactants and products of butane oxidation. It transforms rapidly to δ-VOPO4 phase on butane exposure upon reaction conditions [5]. Besides usually VPO catalysts contain considerable amount of disordered phases, what makes many difficulties to understand the nature of VPO catalysts during catalytic processes and the role of active sites. Operando Raman–MS and UV-vis-MS will be used to observe dynamic changes taking place during reaction/regeneration cycles. This approach allows us to study the phase transformations and characterize changes in activity and selectivity with variations in composition and structure. The operando methodology combines in situ spectroscopy and kinetic measurement in a single experiment and is thus very convenient for fully understanding the structure and reactivity of VPO catalysts during reaction.
References 1. I. E. Wachs, J. of Cat., 170, 55, 1997 2. J. C. Volta et al., J. of Cat., 145, 267, 1994 3. G. W. Coulston et al., Sci. 275, 191, 1997 4. J. C. Volta et al., J. of Cat., 134, 151, 1992 5. G. J. Hutchings et al., Sci. 313, 1270, 2006
2000 1500 1000 500
Raman Shift, cm -1
922
1032
1184
1134
1088
1009
1016
929
794
270
279
597
392
a
b
c
Figure 2. Raman spectra of VPO (a) and used VPO catalyst, orange molecule (b) and black molecule (c).
Benahavís (Málaga, Spain), 21st to 23rd of October, 2009 135
Theoretical Study of Thiol Self Assembled Monolayer Formation on Au(111) surfaces
Frederik Tielens a,b*, Elisabeth Santos c
aUPMC Univ Paris 6, UMR 7609, Laboratoire de Réactivité de Surface, 4 Place Jussieu, 75252 Paris Cedex 05, France
bCNRS, UMR 7609, Laboratoire de Réactivité de Surface, 4 Place Jussieu, 75252 Paris Cedex 05, France
cInstitut für Theoretische Chemie, Universität Ulm, D-89081 Ulm, Germany π. [email protected]
Introduction Immersion of a gold surface in thiolate solution leads to a spontaneous adsorption of thiols[1]. These types of layers have attracted much attention because they may constitute ideal platforms for further binding or reactivity. In the frame of elaborating adjusted surface functionalities for biocompatibility, biosensor or molecular electronics, special effort was made to form two-component monolayers, one of the aims being to avoid steric hindrance of functional tail groups or disorder. Controlling the dispersion of the SAM domains enables to control the dispersion of e.g. biological systems to be attached on the SAM. Concerning the initial step in the SAM formation still some questions stay unanswered. After decades of arguing about the precise site of adsorption of the thiol chains on the gold surface one start to have a picture on the sorption process. Nevertheless since chemisorption is generally accepted above physisorption of the thiol molecule, the reaction mechanism is not known completely. Some theoretical studies have shed some light on the problem. In the present work the S-H bond breaking mechanism and the formation of the S-Au bond is investigated using DFT computational techniques. The electronic structure is analyzed and a possible reaction path is proposed. Results and discussion The SAMs are modelled using a repeated slab model for Au(111) consisting of five atomic layers and fixing the two at the bottom to the bulk positions and four thiol chains (See Fig. 1).[2-4] On the surface HS-C3H7 is adsorbed. The 2√3×2√3R30 unit cell used to build the different mixing configurations contains four thiol chains. Different configurations were considered after which the most appropriate are used to model the reaction path for the formation of H2 generated by the chemisorption process of the thiol to the surface. The transition states between the different minima on the potential energy surface are calculated using the NEB formalism. A barrier of 1.3 eV is predicted for the H2 formation. Conclusions Using first principle computational techniques a new reaction pathway is proposed for the H2 formation as a consequence of thiol chemisorption on Au(111). The thiol adsorption is discussed in relation with the Au-S bond formation/ S-H bond breaking. Acknowledgements The authors thank GENCI project x20090812022 and the CINES, IDRIS and CCRE (Université Pierre et Marie Curie) for providing the computation facilities. COST D36/0006/06 is acknowledged for a partial support of this work. References 1. Poirier, G., Chem. Rev. 97, 1117 (1997) 2. F. Tielens, V. Humblot, Claire-Marie Pradier, M. Calatayud, F. Illas, Submitted to Langmuir 3. F. Tielens, V. Humblot, C.-M. Pradier, Int. J.Quant.Chem, 108, 1792 (2008). 4. F. Tielens, D. Costa, V. Humblot, C.-M. Pradier, J. Phys. Chem. C. 112, 182 (2008).
Benahavís (Málaga, Spain), 21st to 23rd of October, 2009 137
Theoretical Investigation of the Ammonia Adsorptio n Process on (110)-VSbO 4 Surface.
Elizabeth Rojas a, Mónica Calatayud b, M. Olga Guerrero-Pérez c, Miguel A. Bañares a
aCatalytic Spectroscopy Laboratory, Instituto de Catálisis y Petroleoquímica; CSIC; Marie Curie 2; E-29049-Madrid (Spain); b Laboratoire de Chimie Theórique, Uni. Paris 06, UMR 7616 CNRS, Paris F-
75005, France bDepartamento de Ingeniería Química. Universidad de Málaga; E-29071-Málaga (Spain):
Introduction Ammonia is used as reactant in two main classes of reactions of industrial interest: (i) the ammoxidation of hydrocarbons (alkenes, alkanes, alkylaromatics) to the corresponding organic nitriles and (ii) the reduction of NO by NH, in the presence of O2. Vanadium based catalysts are efficient for hydrocarbons (amm)oxidation reactions [1-2]. A very low experimental studies have been made about the adsorption of NH3 on VsbO4 surface [3]. Even with the large experimental effort applied to the system, the nature of the NH3-derived adsorbed species has not been definitely determined. Both the molecular adsorption and dissociative adsorption as NH or NH2 have been proposed. To the best of our knowledge, no theoretical study of this system has been reported, thus, a theoretical calculation would be very valuable. In the present work, we will focus on the binding states and adsorption energies and activation of ammonia on “naked” sites present on the (110)-VSbO4 surface to understand the role of ammonia in the ammoxidation reaction. Methodology Computational Details The density functional (DFT) calculations were performed by using the Perdew−Wang exchange and correlation funtionals PW91 functional for the prediction of adsorption energies of ammonia in the (110)- VsbO4 cluster. Density functional theory using the Perdew−Wang exchange and correlation functionals PW91 functional can provide useful information about the electronic structure of transition metal oxides as well as about the interactions between the adsorbed hydrocarbon molecule and the catalyst surface. Model In our theoretical calculation the VSbO4 oxide has been modelled using a trirutile tetragonal super cell (Fig. 1), which contains the most probably metal-oxygen combinations as reported by Hansen et. Al [3]. The lattice parameters obtained are: a=b= 4,674 Å and c´= 9,373 Å (c´=3c, c=3,1243 Å). The plane (110) )- VSsbO4 used in our calculations was chosen because it appears to be one of the most stable crystal face of oxides of rutile and results from breaking the smallest number of M-O bonds [4] . The hypothetical structures, referred as SA, SB, exhibit two Sb –cations separated by one V cation, two neighboring V-cations separated by one Sb ion, respectively. Full optimization of all the constituent atoms of the adsorbate/substrate system was performed. The adsorption energy (Eads) has been calculated according to the expression:
Where E(adsorbate/substrate), Eadsorbate, and Esubstrate are the total energies of the adsorbate/substrate system, isolated adsorbate, and substrate, respectively. A negative Eads value corresponds to a stable adsorbate/substrate system. Results and Discussion Table 1 show parameters of NH3 Adsorption on the “naked” sites of (110)-VSbO4 surface on the SA and SB structures. During the NH3 adsorption on the SA and SB structure, the interaction over a “naked” surface V atom is energetically most favoured site reached the system a total energy value of -32.8 kcal/mol (Fig.1).
Ammonia adsorption on V(III) site is very exothermic, which is expected considering the electronically and coordinatively unsaturated V site. There are several possible pathways for activation of NH3, and the most favourable pathway is one where both hydrogen atoms of NH3 are transferred to the oxygen extra plane that contains the most probable Sb–V combinations formed OH species. The highest barrier for that process is 28.4 kcal/mol (see Fig. 2). These results suggest that once the reaction is initiated and V (III) sites start appearing in higher ratios, ammonia will be activated more rapidly.
Figure 1 . Geometries of ammonia adorptions in the “naked” surface sites on the (110)-VsbO4 surface. The values ahown here designate the bond lengths.
Figure 2 . Potential energy surface for activation of NH3 on V (III) sites, ∆Eads (kcal/mol).
Conclusions The calculated results of ammonia adsorption shows that the “naked” surface V atoms seen to be more reactive than that of the bulk, suggest that once the reaction is initiated and V (III) sites start appearing in higher ratios, ammonia will be activated more rapidly. Acknowledgements The Ministry of Science and Innovation (Spain) funded this study under project CTQ2008-04261/PPQ. E. Rojas thanks COST for her STMS program fellowship. References [1] Guerrero-Pérez, M.O., and Bañares, M.A., Chem. Matter.,19, 6621 (2007). [2] Guerrero-Pérez M.O., Bañares M.A., Chem. Commun. 12 , 1292 (2002). [3] Centi G., Perathoner S., Catal. Rev. Sci. Eng., 40, 175 (1998). [4] Hansen, S., Stahl, K., Nilsson, R., Andersson, A., J. Solid State Chem. 102, 340 (1993). [5] Irigoyen B., Juan A., Larrondo S., Amadeo N., Surface Science, 523, 252 (2003).
Introduction The selective catalytic reduction (SCR) is a well-established application for the removal of harmful nitrogen oxides (NOx) from stationary emission sources 1. The reductant ammonia reduces the NOx selectively to nitrogen (e.g., for NO):
4 NO + 4 NH3 + O2 4 N2 + 6 H2O The catalyst system typically used contains V2O5 and WO3 supported on TiO2 (anatase) 1. The vanadium oxide content is often ≤ 1 wt.-% to inhibit the undesired SO2 oxidation 2. WO3 (sometimes MoO3, ≈ 10 wt-%) is a promoter and stabiliser 3. These catalysts, which are considered technically mature, are very effective, with high NOx conversions and high N2 selectivities between 280°C and 400°C 4. We found, however, that activity resources can be opened up by optimising the pre-treatment conditions of the catalytic system. Thereby peculiar and complex responses of the catalytic activity to the thermal stress applied were observed. Experimental WO3 and V2O5 were deposited on TiO2 via sequential impregnation of dried titanium oxide hydrate with ammonium para tungstate and ammonium meta vanadate. The catalytic activity was investigated in a flow reactor with a feed gas mixture containing 1000 ppm NO, 1000 ppm NH3, 2 % O2 (balance – helium) at a GHSV of 100,000 h-1. NO and NH3 were determined via non-dispersive IR photometry. After appropriate treatments, the catalysts were also studied by Raman spectroscopy, by EPR and by temperature-programmed reduction (TPR). Results and discussion Thermal stress applied to the catalysts by a special calcination procedure results in remarkable changes in the catalytic activity. Up to three activity maxima can be discerned with increasing pre-treatment severity (see Fig. 1). Depending on the cata-
Figure 1: Change in catalytic activity by increasing pre-treatment duration at 750 °C, a – initial state, calci nation at 350 °C ( ), b – 2nd maximum (), c – minimum (), d – 3rd maximum (), d – final deactivation (). The first maximum is not resolved at this pre-treatment temperature. Lyst composition, the peak NO conversions in the activity maxima can be larger (cf. Fig. 1) or lower than in the initial state. The first or third maxima may escape detection (not resolved at high, not achieved at low pre-treatment temperature). Tentative explanations for this complex behaviour will be presented on the basis of char-acterization results. Raman spectra show the state of the TiO2 in the catalysts to be closer to
140 Benahavís (Málaga, Spain), 21st to 23rd of October, 2009
that of the initial oxide hydrate than to anatase: the anatase finger print is not obtained before the third activity maximum. The thermal stress results in loss of BET surface area, which constitutes a declining activity trend onto which the maxima are superimposed. In the initial state, both EPR and TPR suggest that the surface V oxide species are separated by surface W oxide species, clustered V surface oxide phases are formed only upon thermal treatment. Raman and TPR indicate that WOx species segregate upon shrinkage of the support surface whereas the VOx species appear to remain attached to the support surface. Raman spectra suggest that the development of the first activity maximum may be related to the formation of a particular monovanadate surface species. In the EPR spectra, at least two isolated V oxide species can be differentiated in the initial state and the first maximum. Upon further thermal stress, a loss of hyperfine structure is accompanied by a growing isotropic signal. The V-O-V centres indicated by this may be the origin of the second activity maximum (Fig. 1b), whereas the third maximum (Fig. 1d) is assigned to the formation of the anatase phase. Beyond this maximum, Raman spectra indicated beginning rutilisation. References
Benahavís (Málaga, Spain), 21st to 23rd of October, 2009 141
Synthesis of novel 2:1 permethylated-cyclodextrin-f ullerene conjugates
Zhu Guan a, Juan Yang a, Yali Wang a, Fathi Moussa b, Lubomir Pospíšil c, Yongmin Zhang a
a Université Pierre et Marie Curie-Paris 6, Institut Parisien de Chimie Moléculaire, UMR 7201, 4 place Jussieu, 75005 Paris, France
[email protected] b UMR CNRS 8612, Faculté de Pharmacie, Université Paris-Sud 11, 5 rue J.B. Clément, 92296
Châtenay-Malabry, France c J. Heyrovský Institute of Physical Chemistry AS CR, v.v.i., Dolejškova 3, 18223 Prague, Czech
Republic Fullerenes are currently of great interest in many areas of science and technology due to their unique chemical and physical properties. They appear to be particularly promising in the biological field, as, for example, functionalised fullerenes can be used as antioxidants and neuro-protective agents, as well as in photodynamic therapy or for the inhibition of HIV enzymes1. The predominant hydrophobic character of the spherical carbon allotrope, however, hampers solubilization in polar media such as water. Hence, looking for the possibilities to increase the solubility of fullerenes has become a central topic in synthetic fullerene chemistry. We prepared recently permethylated cyclodextrin-fullerene conjugates which were highly water-soluble2,3. However, UV and NMR spectra showed the presence of aggregates and the expected complexation between permethylated -CD and fullerene was not detected4. In order to understand the effect of linker length on the complexation between C60 and -CD, two -cyclodextrin-fullerene conjugates with shorter linkers were synthesized. The synthetic method will be presented. References: 1) S. Bosi, T. D. Ros, G. Spalluto, M. Prato, Eur. J. Med. Chem. 2003, 38, 913-923. 2) J. Yang, Y. Wang, A. Rassat, Y. Zhang, P. Sinaÿ, Tetrahedron 2004, 60, 12163-12168. 3) Y. Chen, Y. Wang, A. Rassat, P. Sinaÿ, Y. Zhao, Y. Zhang, Tetrahedron 2006, 62, 2045-2049. 4) A. Quaranta, Y. Zhang, S. Filippone, J. Yang, P. Sinaÿ, A. Rassat, R. Edge, S. Navaratnam, D. J. McGarvey, E. J. Land, M. Brettreich, A. Hirsch, R. V. Bensasson, Chem. Phys. 2006, 325, 397-403.
Benahavís (Málaga, Spain), 21st to 23rd of October, 2009 143
NRe
OC
OC
Cl
CON
R
N
N
N
NH2
Ir
N
2
+
NH2O
N
N
H
Electrochemical Functionalization of Glassy Carbon Electrode Surfaces by Organometallic Moieties
Giorgio Volpi, a Jan Fiedler, b Martina Sandroni, a Claudio Garino, a Roberto Gobetto, a Guido
Viscardi c and Carlo Nervi a
a Department of Chemistry IFM, via P. Giuria 7, 10125, Turin, Italy. Email: [email protected] b J. Heyrovský Institute of Physical Chemistry of the ASCR, v.v.i., Dolejškova 2155/3, 182 23
Prague 8, Czech Republic. c Department of General and Organic Chemistry, via P. Giuria 7, 10125 Turin, Italy.
Objective Transition metal complexes with interesting photophysical properties are currently under intense investigation for their potential applications as optically functional materials in a variety of fields such as organic light emitting diodes (OLED), light-emitting electrochemical cells (LEC), electrogenerated chemiluminescence, photoinduced hydrogen production, photoelectrochemical solar cells, and phosphorescent probes/markers for biology. On the other hand, the functionalization of surfaces is attracting interest because applications as sensors, molecular electronics, analytical detection, catalysis. The aim of our research during last years was to investigate the photophysical properties of new Os, Ru, Re and Ir organometallic complexes [1]. With this knowledge we are now attempting the functionalization of surfaces by means of organometallic moieties. The goal of the communication is to show our preliminary results following this pathway. In the literature there are several examples of molecules covalently bonded on the electrode surface, but there are very few cases that use organometallic complexes. Carbon is a very interesting material as nanotubes, fullerene, glassy carbon (GC), pyrolytic graphite, diamond, etc… Its functionalization by means of strong covalent carbon-carbon bonds found new impetus only after the work of Savéant et al [2], who used diazonium salts for facile electrochemical C-C bond formation. Another method is the oxidation of amine [3], which leads to a covalent C-N bond. Aliphatic amines has been thoroughly employed, but less attention has been paid to the use of aromatic amines, due to their less (or no) reactivity. One of the aim of this job was to test the possibility to use ligands containing aromatic amines for organometallic functionalization. Results Six Re(CO)3Cl(pip) [1] and Re(CO)3Cl(dpk) complexes (where pip=1-pyridylimidazo[1,5-]pyridine and dpk=2-dipyridyl ketone) has been synthesized and fully characterized. The electrochemical and spectroelectrochemical behaviour of the Re derivatives generally show chemically irreversible redox processes, but the spectroelectrochemical data suggests a recombination of the fragment in the OTTLE cell, which leads to a partial reformation of the starting compounds. The R part (see figure above) can be easily modified in order to include a suitable functional group (i.e. an aromatic amine) for electrochemical surface reaction. However,
one of our aims was to characterize the modified electrodes by electrochemical methods. As mentioned, after redox processes these Re complexes show weak Re-N bonds. For this reason they appears to be of limited use because the chemical reaction following redox processes might turn them into a weakly surface-bonded derivatives. Therefore, we switched to Ir and Ru complexes containing aromatic amines and bipyridine-type ligands, known to be more stable and to have a reversible electrochemical behaviour [1].
144 Benahavís (Málaga, Spain), 21st to 23rd of October, 2009
N
N
NH2
Ru
N
N
2
2+
Commercial (nitroaniline, aminophenanthroline) and synthesized (in figure) aromatic amines were oxidized at clean GC electrode. Only the latter two apparently undergoes electrode modification. Hence, their Ru and Ir complexes were synthesized, characterized and electro-chemically oxidized on GC. After sonication in pure MeCN, the Cyclic Voltammetries (CV) of the modified electrodes in MeCN containing only the supporting electrolyte show typical metal-centred redox processes of the corresponding compounds. The surface modification occurs only for the Ir complex, after the electrochemical oxidation of the amine group. The mechanism takes places via H+ elimination, and the use of collidine as base facilitates the formation of the
C-N covalent bond. In the presence of collidine also the Ru complex undergoes the GC modification. The anodic (Ru) and cathodic (Ir) CV peaks are directly proportional to the sweep rate, and integration of the peak areas (after background subtraction) allow to evaluate the electrode surface coverage, which are (Ru)=4.6×10–10 and (Ir)= 3.5×10–10 mol/cm2. These values are in agreement with the formation of a monolayer, taking in account the molecule surface area (evaluated via quantum mechanical calculation).
Conclusions We electrochemically characterized seven Re derivatives. GC surface modification by means of aromatic amines has been achieved (also with the help of collidine), and transition metal complexes having interesting photophysical properties have been covalently attached to the carbon GC electrode. In a 0.1 M TBAPF6 MeCN solution the modified electrodes show the same reversible electrochemical behaviour of the free complexes. The properties are retained even after several redox cycles, outlining the stability of the metal complexes bonded to the GC surface. Applications can be potentially found in the fields of catalysis, sensors and molecular electronics. References . [1] G. Volpi, C. Garino, L. Salassa, J. Fiedler, K.I. Hardcastle, R. Gobetto, C. Nervi, Chem.Eur.J., 2009, 15, 6415. [2] P. Allongue, M. Delamar, B. Desbat, O. Fagebaume, R. Hitmi, J. Pinson, J.-M. Savéant, J.Am.Chem.Soc., 1997, 119, 201. [3] B. Barbier, J. Pinson, G. Desarmot, M. Sanchez, J.Electrochem.Soc., 1990, 137, 1757.
Benahavís (Málaga, Spain), 21st to 23rd of October, 2009 145
0 500 5000 5500 6000-12
-10
-8
-6
-4
-2
0
2
∆f = +4.8 Hz
addition of atrazine
∆f = -9.6 Hz
addition of anti-ATZ IgG
∆f /
Hz
time / s
Self–assembled monolayers of atrazine–based thiolat es and their interaction with anti–atrazine antibody.
Magdaléna Hromadová *,a, Michèle Salmain *,b, Nathalie Fischer-Durand b, Viliam
Kolivoška a, Romana Sokolová a
a J. Heyrovský Institute of Physical Chemistry of ASCR, v.v.i., Academy of Sciences of the Czech Republic, Dolejškova 3, 18223 Prague, Czech Republic
b Laboratoire de Chimie et Biochimie des Complexes Moléculaires, UMR CNRS 7576, Ecole Nationale Supérieure de Chimie de Paris, 11 rue Pierre et Marie Curie,
As a part of our objective to build an immunosensor for the detection of pesticide atrazine (ATZ) in the environmental samples we studied the self-assembly process of atrazine derivative (ATZSSATZ) on a gold substrate.
Self-assembled monolayers of ATZSSATZ were characterized by cyclic voltammetry, ellipsometry, scanning tunneling microscopy, phase modulated IRRAS, XPS and QCM measurements. Two different time constants for the adsorption process were observed depending on the experimental method used. The QCM data reflect the adsorption kinetics of the original disulphide compound,
whereas ellipsometry and ex-situ PM-IRRAS (see Figure 1) refer to the formation of thiolate monolayers on the gold substrate (ATZS-gold).
3000 2000 1500
1.00
1.01
1.02
1.03
1.04
0 20 40 60 80 100
0
1
2
3
4
5
6PM-IRRAS
arbi
trar
y un
its
wavenumber / cm-1
area
( 3
200c
m-1 -
270
0cm
-1 )
time / min
In-situ QCM data (see Figure 2) demonstrate the suitability of such monolayers for the detection of atrazine in aqueous samples. After the preparation of thiolate monolayers an anti-atrazine antibody (anti-ATZ IgG) was added, which resulted in full monolayer coverage. Approximately half of the antibody molecules were displaced from the surface by addition of 10-5M atrazine, so the functional material is suitable for the detection of atrazine in the aqueous samples. A Grant Agency of the Academy of Sciences of the Czech Republic (IAA400400802), Grant Agency of the Czech Republic (GACR 203/08/1157 and 203/09/1607) and Ministry of Education (LC510, COST OC140) are greatly acknowledged for the financial support.
Benahavís (Málaga, Spain), 21st to 23rd of October, 2009 147
DNA gel particles from single and double-tail surfa ctants
M. C. Moran 1*, M. G. Miguel 1, B. Lindman 1,2
1 Chemistry Department, University of Coimbra, 3004-535 Coimbra (Portugal). 2 Physical Chemistry 1, University of Lund, 22100 Lund (Sweden).
* [email protected] A general understanding of DNA-oppositely charged agent interactions, and in particular the phase behaviour, has given us a basis for developing novel DNA-based materials, including gels, membranes and gel particles[1]. We have recently prepared novel DNA gel particles based on associative phase separation and interfacial diffusion. By mixing solutions of DNA (either single- (ssDNA) and double-stranded (dsDNA)) with solutions of different cationic agents, such as surfactants, proteins and polysacharides, the possibility of formation of DNA gel particles without adding any kind of cross-linker or organic solvent has been confirmed [2-6]. The adsorption strength, which is tuned by varying the structure of the cationic agent, allows to control the spatial homogeneity of the gelation process, producing either a homogeneous DNA matrix or different DNA reservoir devices. They allows for various applications in the controlled encapsulation and release of ssDNA and dsDNA, with clear differences in the mechanism. Cationic surfactants have offered a particularly efficient control of properties of DNA-based particles [2, 6]. This presentation is focused on the formation of DNA gel particles mixing DNA (either single- (ssDNA) or double-stranded (dsDNA)) with single chain (DTAB) and double chain (DDAB) surfactants. Results on the encapsulation of DNA and its release are presented, using the surfactant structure and the DNA conformation as controlling parameters. References [1] D. Costa, M. C. Morán, M. G. Miguel, B. Lindman, , Cross-linked DNA Gels and Gels Particles, in R. S. Dias and B. Lindman (Eds.) DNA Interactions with Polymers and Surfactants,Wiley Interscience, New Jersey, 2008. [2] M. C. Morán, M. G. Miguel, B. Lindman, Langmuir, 23, 6478 (2007). [3] M. C. Morán, M. G. Miguel, B. Lindman, Biomacromolecules, 8, 3886 (2007). [4] M. C. Morán, T. Laranjeira, A. Ribeiro, M. G. Miguel, B. Lindman, to appear in J. Dispersion Sci. Technol., 30 (2009). [5] M. C. Morán, A. Ramalho, A.A.C.C. Pais, M. G. Miguel, B. Lindman, Mixed protein carriers for modulating DNA release, Langmuir, accepted (2009). [6] M. C. Morán, M. R. Infante, M. G. Miguel, B. Lindman, R. Pons, Novel biocompatible DNA gel particles, Soft Matter, submitted (2009).
Benahavís (Málaga, Spain), 21st to 23rd of October, 2009 149
Adsorption of macromolecules to responsive surfaces
R.S. Dias, 1* P. Linse, 2 and A. A. C. C. Pai s 1
1 Department of Chemistry, University of Coimbra, Rua Larga, 3004-535 Coimbra, Portugal *[email protected]
2 Physical Chemistry, Centre for Chemistry and Chemical Engineering, Lund University, S-221 00 Lund, Sweden
Objective Polymer and protein adsorption onto lipid monolayers and bilayers is of fundamental importance in biology and pharmacology as well as in a large range of technological processes. However, membranes are not static, flat and homogeneous objects. The individual lipid molecules in the membrane undergo rotation, lateral diffusion, and vertical excursions out of the bilayers and into solution, the so-called protrusions. This will naturally have an impact on the structure of the membrane itself and on its interaction with biomacromolecules such as DNA and proteins. The aim of our work is to correlate the adsorption and condensation of a single polyelectrolyte, a very simplistic model of a DNA molecule, with the properties of a model membrane, namely the lateral diffusion and protrusion into the solution. It has been observed previously [1] that the lateral diffusion of the headgroups enhances significantly the adsorption and condensation of a polyanion. Adsorption appears at net neutral membranes or at weakly charged membranes, as long as the charges are mobile or frozen in a random fashion, and it is more pronounced for more flexible PA chains. These results go beyond the conventional adsorption behavior of a PA at a homogeneously charged surface. Results A very simple model was adopted to describe the interaction between a PA and a net neutral membrane carrying both cations and anions that are able to protrude into solution. The PA is described as consecutive negatively charged hard spheres connected by harmonic springs, where its intrinsic stiffness is regulated by an angular potential. More details regarding the PA model can be found in Ref. [1] and those on the membrane model and simulation details can be found in Ref. [2].
Figure 1 shows the number density of the different particles in the system along the z-coordinate, that is, perpendicular to the membrane, placed at the average position of -148.5 Å. It can be observed that both the PA beads and the respective counterions can occupy a lower position in the z-coordinate than those of systems calculated without protrusions (dashed curves). This indicates that the adsorption of the PA is more favorable in membranes with protrusions.
Figure 1. Left: Number density of the system particles vs. the z-coordinate. The (semi-flexible) polyelectrolyte beads correspond to the black line, the distribution of the membrane particles overlap (blue and green lines) and the red curves correspond to the polyanions and respective counterions. The filled curves correspond to the system with protrusions and the dashed ones to the system without protrusions. Below is a representative snapshot.
150 Benahavís (Málaga, Spain), 21st to 23rd of October, 2009
Table 1 presents some of the properties of the Pas with different flexibilities in the presence of lipid membranes with and without protrusions. Whereas the presence of protrusions does not influence substantially the adsorption of the more flexible PA, the adsorption of the semi-flexible PA is seen to increase. We recall that the more flexible PA showed improved adsorption ability in the membranes with lipid lateral diffusion due to its ability to concentrate more positive charge [1]. When lipid protrusions are also present the membrane will presumably be able to adapt to the stiffer PA, as it has been shown with a spherical macroion [2]. This will enhance the adsorption of the PA. Table 1. Average properties of Pas with different flexibilities (flexible, l0 ≈ 7 Å, and semi-flexible, l0 ≈ 22 Å)
in the presence of membranes with and without individual lipid protrusions (prot.). and
correspond to the root mean square radius of gyration based on the tails projected onto the membrane normal and based on trains and loop beads projected on the membrane plane, respectively. Nads and Ntails are the number of PA segments adsorbed at the membrane (i.e. at a distance not larger than 8 Å) and the number of monomers residing in the PA’s tails, respectively. Values are given in Å. Conclusions The details of the membrane surface are of great importance for polyion adsorption. Individual lipid protrusions enhance the adsorption of semi-flexible polyions since the membrane is able to adapt to the topology of the polyion. In the case of the more flexible PA there is no substantial improvement in the adsorption ability. Acknowledgements. R.S.D. is grateful to Fundação para a Ciência e Tecnologia (Ciência 2007). References [1] R.S. Dias, A.A.C.C. Pais, P. Linse, M.G. Miguel and B. Lindman, J. Phys. Chem. B, 109 (2005) 11781. [2] R.S. Dias and P. Linse, Biophys. J., 94 (2008) 3760.
Benahavís (Málaga, Spain), 21st to 23rd of October, 2009 151
Proton Coupled Oxygen Reduction at Liquid-Liquid In terfaces Catalyzed by Cobalt Porphine
Imren Hatay, 1,2 Bin Su, 1 Fei Li, 1 Manuel Alejandro Méndez, 1 Tony Khoury, 3 Claude P. Gros, 3 Jean-Michel Barbe, 3 Mustafa Ersoz, 2 Zdenek Samec, 4 and Hubert H. Girault *,1
1Laboratoire d’Electrochimie Physique et Analytique, Ecole Polytechnique Fédérale de
Lausanne, Station 6, CH-1015 Lausanne, Switzerland ; Department of Chemistry. 2Selcuk University, 42031 Konya, Turkey.
3 Institut de Chimie Moléculaire de l’Université de Bourgogne, ICMUB (UMR 5260), 21078 Dijon cedex, France.
4J. Heyrovsky Institute of Physical Chemistry, Academy of Sciences of the Czech Republic, Dolejskova 3, 18223 Prague 8, Czech Republic.
Objective Oxygen reduction catalyzed by porphyrins is of great interest in fields as diverse as biology, photosynthesis and electrocatalysis[1]. On the other hand, the Interface between Two Immiscible Electrolyte Solutions (ITIES) provides a physical separation of the reactants and products, and the polarization of this soft interface also allows an electrochemical control for different charge transfer reactions[2]. Herein, we show that cobalt porphine (CoP) catalyzes the reduction of O2 in biphasic systems where the aqueous phase is acidic and where the organic phase contains electron donors. Results Cobalt porphine (CoP) dissolved in the organic phase of a biphasic system is used to catalyze O2 reduction by an electron donor, ferrocene (Fc). Using voltammetry at the interface between two immiscible electrolyte solutions (ITIES), it is possible to perform this catalytic reduction at the interface as a function of the applied potential difference, where aqueous protons and organic electron donors combine to ultimately reduce O2. Thus, the current signal observed corresponds to a proton-coupled electron transfer (PCET) reaction, as no current and no reaction can be observed in the absence of either the aqueous acid, CoP, Fc or O2. Additionally and given that the oxidation product of ferrocene, namely ferrocenium, does not cross the interface in the same potential range as that of the PCET reaction and the two processes can be observed. This is the main difference with voltammetry at solid electrodes that only records electron transfer steps.
152 Benahavís (Málaga, Spain), 21st to 23rd of October, 2009
Conclusions This work shows that voltammetry at soft interfaces is a powerful tool to study proton coupled electron transfer reactions of biological interest such as the interfacial reduction of oxygen catalysed by a metalloporphyrin, e.g. Co(II) porphine. As in biosystems, the reactants can be phase–separated, the protons in the aqueous phase and the electron donors in the organic phase. In the present, we have reacted an oxygen carrier catalyst, namely cobalt porphine, aqueous protons and lipophilic electron donors, and shown that voltammetry can be efficiently used to probe the existence of such an interfacial reaction. This work is to the best of our knowledge the first voltammetric study of an electrocatalytic reaction at a soft interface, where the rate of the catalytic reaction is controlled by the interfacial polarization, i.e. by the applied potential difference. This investigation opens the way to the study of synthetic catalysts able to carry out the four-electron reduction of oxygen to water. References [1] S. Fukuzumi, S. Mochizuki, T. Tanaka, Inorg. Chem. 28 (1989) 2459-2465. [2] R. Partovi-Nia, B. Su, F. Li, C. P. Gros, J. M. Barbe, Z. Samec, H. H. Girault, Chem.--Eur. J. 15 (2009) 2335-
Benahavís (Málaga, Spain), 21st to 23rd of October, 2009 153
Synthesis of crystalline CeO 2 nanoparticles by a novel oil-in-water microemulsio n reaction method and its use as catalyst support
Margarita Sánchez-Domínguez 2, Gabriella Di Carlo 3, Magali Boutonnet 1, and Conxita
Solans 2
1Kungliga Tekniska Hogskolan (KTH), Department of Chemical Engineering and Technology, Div. Of Chemical Technology, Teknikringen 42, S-10044 Stockholm, Sweden
e-mail: [email protected], 2Consejo Superior de Investigaciones Científicas (CSIC, IIQAB)
CIBER en Biotecnología, Biomateriales y Nanomedicina (CIBER BBN) Jordi Girona 18-26, 08034 Barcelona, Spain.
Introduction Microemulsions are transparent and thermodynamically stable colloidal dispersions in which two liquids initially immiscible (typically water and oil) coexist in one phase due to the presence of a monolayer of surfactant molecules. Depending on the ratio of oil and water and on the hydrophilic-lipophilic balance (HLB) of the surfactant, microemulsions can exist as oil-swollen micelles dispersed in water (oil-in-water microemulsions), or water-swollen inverse micelles dispersed in oil (water-in-oil microemulsions). The characteristic size of microemulsion droplets is very small (typically below 10 nm).The preparation of nanoparticles using ME systems, due to its advantage of allowing high control of size, composition and structure of those particles, has been widely studied, in particular making particles for catalytic, magnetic or ceramic uses and also in polymer manufacture.The first application of water-in-oil (W/O) microemulsion for the synthesis of catalytic nanoparticles was introduced in 1982 and concerns nanoparticles of noble metals. Since this time, this method has found a wide range of applications in the field of catalysis from room temperature reactions such as iusep isomerisation to high temperature reactions such as catalytic combustion of methane [1]. Recently, we developed a novel and straightforward approach for the synthesis of inorganic nanoparticles by using oil-in-water (o/w) microemulsions[2, 3], in contrast to the typically used water-in-oil microemulsion method[4]. The new strategy implies the use of organometallic precursors, dissolved in nanometer-scale oil droplets (stabilised by surfactant), and dispersed in a continuous aqueous phase. In our preliminary work, the o/w microemulsion approach was explored as proof of concept for the synthesis of metallic and metal oxide nanoparticles. The studies revealed that metallic nanoparticles (Pt, Pd, Rh) with small diameter (3-6 nm) and narrow size distribution, as well as nanocrystalline metal oxide (cubic ceria) could be obtained in mild conditions.
Objective
The aim of this work is to explore the potential of this approach for producing nanocrystalline ceria for catalytic purposes[5]
Results The effect of organometallic precursor on the phase behaviour of several water/nonioic surfactant/oil systems was first studied in order to identify o/w microemulsion regions. A series of nano-cerias were synthesized by varying microemulsion composition and reaction conditions. Characterization studies (Figure 1) demonstrate that nanocrystalline cubic ceria was obtained in the microemulsion at room temperature (crystallite and nanoparticle size 2-5 nm), with specific surface area (SSA) from 180 up to 250 m2/g. The materials were calcined at 400ºC after which high SSA (up to 220 m2/g) and small crystallite size (~ 5 nm) were maintained, indicating good thermal stability. The potential of selected nanocerias as catalyst support was explored in the CO oxidation reaction. ‘
154 Benahavís (Málaga, Spain), 21st to 23rd of October, 2009
Conclusions
The obtained results demonstrate the feasibility of this approach for the preparation of varied nanocrystalline cerias with high SSA and small crystallite and nanoparticle size for catalytic purposes. References [1] S.Eriksson ,U. Nylen, S. Rojas, M. Boutonnet. .Preparation of catalysts from microemulsions and their applications in heterogeneous catalysis Appl Cata-Gen.2004, 265 (2): 207-219. [2] M.Sánchez-Domínguez; M.Boutonnet, C.Solans, J. Nanoparticle Research 2009, in press, online at Online FirstTM (DOI 10.1007/s11051-009-9660-8) [3] C. Solans; M.Sánchez-Domínguez; M.Boutonnet ; Patent Application in the Spanish Patent Office, Application Number: 200802114(9); Priority Date: 16 July 2008. [4] M. Boutonnet, S. Lödberg, E. E. Svensson Curr. Opin. Colloid Interface Sci., 2008, 13, 270. [5] C. Solans; M.Sánchez-Domínguez; M.Boutonnet, PCT Application in the Spanish Patent Office, Application Number PCT/ES2009/070223; Priority Date: 12 June 2009.
Figure 1 . TEM pictures and XRD spectra of ceria nanoparticles synthesized in o/w microemulsions.
Abstract Beside the classical deposition-precipitation (DP) method we have applied sol deposition technique1,2 for preparation of Au/SiO2 that was interfaced with suitable (reducible) oxides only by decoration. To fabricate a well defined catalyst one of the crucial problems is the active oxide morphology. We aim at reporting how the structure of a nanocomposite system (uniform nanosized Au particles with Au/promoter oxide perimeter) benefits the fabrication of a novel catalyst family. Gold nanoparticles were fabricated from HauCl4 reduced and stabilized by Na-citrate+tannic acid or reduced by NaBH4 and stabilised by polyvinylalcohol (PVA) or poly(diallyldimethylammonium) chloride (PDDA) to produce Au hydrosols of dAu=6-7, 2-3 and 2-5 nm, respectively. The Au sols were adsorbed on SiO2 support. The Au on SiO2 or SBA-15 was decorated by 3 different ways.3,4 TiO2 promoter was introduced using Ti-isopropoxid or Ti(IV) bis(ammoniumlactato)dihydroxide (TALH) precursors. CeO2 modified gold samples were prepared by impregnating Au/SiO2 and Au/SBA-15 by Ce(NO3)2. All samples were calcined to remove organic residues. The samples were characterized by HRTEM, XPS, XRD and the CO oxidation was employed as test reaction and the conversion vs. temperature was used to compare the activity of the various samples. In comparing the samples prepared in different methods we establish the importance of the amorphous-like TiO2 and CeO2 covering the Au/SiO2 samples5. In Fig 1. the HRTEM picture indeed shows the presence of CeO2 decorating gold particles. It is clearly indicated that the SiO2 and SBA-15 supported Au catalysts with minute amount of TiO2 or CeO2 improves the activity as compared with those deposited on pure CeO2 and TiO2 (anatase) as shown in Fig.2. The activity depends also on the length of the Au-active oxide perimeter, so its controlled formation has key importance, that is, we also studied how the surface charges and the electrostatic interactions during the colloidal preparation affect the active interface.
0 20 40 60 80 100 120 140 160 180 200 2200
10
20
30
40
50
60
70
80
90
100
CO
con
vers
ion,
%
Temperature, oC
Au-CeO2/SiO2 Au-CeO2/SBA-15
Au/CeO2 Au-TiO2/SiO2 Au/TiO2
Figure 1 HRTEM image of Au-CeO2/SBA-15 Figure 2 CO oxidation conversion curves
156 Benahavís (Málaga, Spain), 21st to 23rd of October, 2009
The active amorphous oxide decoration has an enhanced charge modification and it may stabilize the small gold particle size. The activity of the Au/MOx perimeter on SiO2 depends not only on the Au particle size, but the morphology of the oxide component. For highly active catalysts the controlled formation of the Au/active MOx nanoensembles is desirable. The formation of this interface can be efficiently favoured by proper selection of the stabilising agents of the Au colloids, precursor of promoting oxide and the pH in the preparation. Ackowledgements The financial support of National Science and Research Fund (OTKA) grants (# T-049564, # F-62481, # K-68052) is greatly acknowledged. References [1]. A. Horváth, A. Beck, A. Sárkány, Gy. Stefler, Zs. Varga, O. Geszti, L. Tóth and L. Guczi, J. Phys. Chem. B., 110 (2006) 15417 [2]. A. M. Venezia, F. L. Liotta, G. Pantaleo, A. Beck, A. Horváth, O. Geszti, A. Kocsonya and L. Guczi, Appl. Catal. A., 310 (2006) 114 [3]. L. Guczi, A. Beck, A. Horváth, A. Sárkány, Gy. Stefler, O. Geszti, Studies Surf. Sci. Catal., 172 (2007) 221 [4]. A. Beck, A. Horváth, Gy. Stefler, R. Katona, O. Geszti, Gy. Tolnai, L. Liotta, L. Guczi, Catal. Today, 139 (2008) 180 [5]. L. Guczi, A. Beck, K. Frey, Gold Bulletin, 42 (2009) 5
Benahavís (Málaga, Spain), 21st to 23rd of October, 2009 157
TiO2 and CeO 2 promoted Au/SBA-15 in propene total oxidation
T. Benkó a*, A. M. Venezia b, L. F. Liotta b, G. Pantaleo b, A.Beck a, L. Guczi a
a Institute of Isotopes, HAS, P.O. Box 77, H-1525 Budapest, Hungary b Istituto per Lo Studio dei Materiali Nanostrutturati (ISMN)-CNR, Via Ugo La Malfa, 90146
Introduction The development of active catalysts for the total combustion of volatile organic compounds (VOCs), which are recognised as major contributors to air pollution, is highly desirable from the viewpoint of environmental protection. In this study the catalytic activity of gold nanoparticles deposited on the SBA-15 and modified by oxide promoters in propene oxidation was investigated. Experimental Au-MOx (M: Ti, Ce) nanostructures supported by SBA-15 type of support were analysed. SBA-15 and alumina-modified SBA-15 as supports was prepared. Gold was deposited on the support by adsorption of colloidal gold prepared by HauCl4 reduced by NaBH4 and stabilized by PVA or PDDA. The oxide was introduced via impregnation by Ti(IV) bis(ammoniumlactato)dihydroxide (TALH) and Ce(NO3)3 precursors. The oxide is formed during the calcination.[1] The catalytic activity in propene oxidation is correlated with the composition, structural and CO oxidation properties of the catalysts. TEM, SAXS, XRD, and BET measurements were performed to confirm the structure of the catalysts. Results and Discussion The results show the mesoporous structure of both SBA-15 and AlSBA-15 support with high surface area. The size of the gold particles was investigated by TEM measurements. In the case of SBA-15 the gold nanoparticles was prevented against strong sintering during the catalytic test which suggests that the AuNPs are located predominantly inside the pores. The catalytic activity of the various catalysts was measured in propene oxidation under standard conditions. The order of the activity: AuSBACe > AuCeO2 > AuAlSBACe > AuAlSBATi > AuSBA > AuTiSBA > AuAlSBA. The presence of the oxide promoter improves the catalytic activity.
158 Benahavís (Málaga, Spain), 21st to 23rd of October, 2009
Conclusions Both CeO2 and TiO2 increase the activity of Au/SBA-15 and Au/aluminated SBA-15 in propene oxidation. CeO2 decoration leads to a higher improvement of the catalytic activity than TiO2 decoration. The promotion effect was higher in cases of AlSBA-15 supported samples. The activity order in propene oxidation of the samples differs from that obtained in CO oxidation. The active sites of the two processes should be different. References 1. A. Beck, A. Horváth, Gy. Stefler, R. Katona, O. Geszti, Gy. Tolnai, L.F. Liotta and L. Guczi, Catal. Today 139 (2008) 180. 2. A. Beck , T. Benkó, A. M. Venezia, L. F. Liotta and G. Pantaleo, L. Guczi, unpublished results, 2008.
Benahavís (Málaga, Spain), 21st to 23rd of October, 2009 159
Selective photo-oxidation of cyclohexane on TiO 2: the role of surface characteristics
M. D. Hernández-Alonso 1, A. R. Almeida 2, J. A. Moulijn 2 and G. Mul 2
1 Environmental Applications of Solar Energy, CIEMAT-PSA, Madrid, Spain 2 Catalysis Engineering, Delft University of Technology, Delft, The Netherlands
Objective
Cyclohexane is an important commercial product, used to obtain caprolactam for Nylon-6 production, and currently is obtained by liquid phase selective oxidation of cyclohexane at elevated temperatures and pressures. An alternative to this process is photocatalytic oxidation of cyclohexane over TiO2 at room conditions. Research has mainly focused on the effect of the solvent, photon flux, wavelength and particle size on product formation and selectivity. The role of the catalyst structure on the activity and selectivity to cyclohexanone has also been studied. On the contrary, catalyst stability has received relatively little attention in the literature. As has been previously demonstrated, surface carboxylates and carbonates diminish the activity of the photocatalysts [1,2]. The accumulation of these deactivating species has been proposed to be the result of consecutive oxidation of a cyclohexyl peroxide intermediate or adsorbed cyclohexanone. Thus, modification of the TiO2 surface properties could limit the accumulation of these deactivating compounds by favouring cyclohexanone desorption and/or the conversion of the cyclohexyl peroxide to the ketone. The main goal of this study is to evaluate he effect of surface modification on the behaviour of the photocatalysts in the selective photo-oxidation of cyclohexane. In situ Attenuated Total Reflectante (ATR)-FTIR spectroscopy was used to follow the reactions [3]. Results Anatase-structured Ti1-xZrxO2 materials with x = 0.00, 0.01 and 0.06, were prepared by a reverse microemulsion method, characterized, and tested as catalysts for the selective photo-oxidation of cyclohexane to cyclohexanone. The surface acidity of the materials was studied by means of ammonia adsorption microcalorimetry. In situ ATR-FTIR spectroscopy was used to evaluate the reaction. In Table 1, some physico-chemical characteristics of the samples are summarized. As can be observed, Zr incorporation into the anatase lattice enhances the surface acidity of TiO2 without causing any significant structural or electronic modification. As expected, also the stability of surface adsorbed water, i. e. the hydrophilicity, was enhanced.
Table 1. Structural and textural properties of the studied materials
160 Benahavís (Málaga, Spain), 21st to 23rd of October, 2009
Figure 1 Evolution of cyclohexane and cyclohexanone during the photocatalytic oxidation of the alkane, followed by in situ ATR-FTIR spectroscopy
Figure 1a shows the evolution of cyclohexane during the photo-oxidation reaction on the different catalysts, while in Figure 1b the total cyclohexanone production as a function of illumination time is presented. Peak area and peak height values were normalized by considering the SBET and mass of catalyst employed in the reaction. Commercial TiO2, Hombikat UV100 was also assayed for comparative reasons.
Conclusions
The increase in the Brønsted acidity, together with the higher hydrophilicity, proved to be detrimental for performance (selectivity and stability) in the selective photo-oxidation of cyclohexane. Apparently, potential intrinsic catalytic advantages of having higher acidity are outweighed by the enhanced number of water born •OH radicals, inducing non-selective reactions, and enhanced hydrophilicity leading to slow desorption and consecutive oxidation of cyclohexanone (Figure 2). References [1] C. B. Almquist, P. Biswas, Appl. Catal. A : General 214 (2001) 259. [2] P. Du, J. A. Moulijn, G. Mul, J. Catal. 238 (2006) 342. [3] M.D. Hernandez-Alonso, A.R. Almeida, J.A. Moulijn, G. Mul, Catal. Today 143 (2009) 326.
0 20 40 60 80 100
0,00
0,02
0,04
0,06
0,08
0,10
Time (min)
Cyc
lohe
xano
ne P
eak
Hei
ght (
m-2)
Hombikat TiO
2
TZ1 TZ6
b
0 20 40 60 80 100
-1,4
-1,2
-1,0
-0,8
-0,6
-0,4
-0,2
0,0
TiO2 Hombikat
TiO2
TZ1 TZ6
Cyc
lohe
xane
Pea
k ar
ea (
m-2)
Wavenumber (cm-1)
a
Figure 2 Schematic representation of the qualitative correlation between surface properties and photocatalytic performance
Benahavís (Málaga, Spain), 21st to 23rd of October, 2009 161
Porous silicon with gold nanoparticles as laser des orption/ionization mass spectrometry platform
Sergei Alekseev a,b*, Julia Romanenko b, Irina Shmygol c, Vladimir Lysenko d, Vladimir
Zaitsev b, Valeriy Pokrovsky c and Jacques Fraissard a.
aLaboratoire de Physique Quantique, University P. and M. Curie, ESPCI, 10 Rue Vauquelin,
Paris, France bDepartment of Chemistry, National Taras Shevchenko University,64 Vladimirska Str., Kiev,
Ukraine cA. A. Chuiko Institute of Surface Chemistry, NAS of Ukraine, 17 General Naumov St., 03164
Kiev, Ukraine dLyon Institute of Nanotechnologies, INL, CNRS UMR-5270, INSA de Lyon, 7 avenue Jean
Capelle, Bat. Blaise Pascal, 69621 Villeurbanne cedex, France *[email protected]
Objective Matrix assisted laser desorption ionization (MALDI) is a soft ionization mass-spectrometry technique allowing the investigation of large biomolecules [1,2]. However, introduction of the organic matrix led to the appearance of background ions in the low mass range interfering with the analysis of small analytes. To tackle this issue surface-assisted laser desorption/ionization mass-spectrometry (SALDI MS) technique was proposed. It utilizes substrates, which generally do not signicantly desorb with analytes together, to bring analytes to be desorbed and ionized. Many effective SALDI-assisted materials were investigated [3]. The porous silicon (PS) is one of the most popular among them due to its availability and advantageous characteristics (surface roughness, low thermal and high electrical conductivity, efficient light absorption) [4]. Chemical functionalization of the PS surface can make the PS surface highly selective for specific analyte capture and consecutive MS analysis [5]. Another effective SALDI assisted materials for the analysis of small organic molecules are the nanoparticles of noble metals, such as silver and gold [6]. In compare with the PS the nanoparticles (NPs) have better stability and may have enhanced LDI efficiency due to so-called plasmonic effect: the excitation of collective electron motion under laser irradiation resulting in enhanced photon absorption and huge concentration of the optical near-field in a small volume. Above mentioned effect resulted in detection of even single molecule by means of Raman spectroscopy (surface enhanced Raman, SERS). The deposition of Au NPs on the external surface of the PS should result in the combination of advantageous characteristics of both substrates, similarly as it was shown previously for the PS covered with Ag NPs layer [7] and the PS covered by Au layer by means of ion-sputtering [8]. That is why PS-Au NPs composites are promising to be a good candidate substrate for SALDI platform. Results Free layers of meso-porous silicon (approx. 65% vol. porosity, 55 µm thickness and 20 nm mean pore diameter) prepared by anodic etching of p+-doped (ρ = 10 mΩ.cm) silicon (100) wafers as previously described in [¡Error! Marcador no definido. ] were used in this study. Electroless deposition of gold was performed by means of Psi free layers treatment with ethanolic solutions of HauCl4, the reduction of Au taken place due to interaction with chemically active surface species of the PS (Scheme 1).
162 Benahavís (Málaga, Spain), 21st to 23rd of October, 2009
Depending on the reaction conditions (HauCl4 concentration and treatment time) layers of aggregated Au NPs with 10 – 100 nm mean particle diameter were obtained. The sample of PS-Au with approximately 25 nm NPs was used for following LDI MS experiments. Mass-spectrometric experiment was performed using time-of-flight Autoflex II instrument (Bruker Daltonics). Peaks of Au clusters were detected in both positive and negative mass spectra of PS-Au composites (Fig. 1, a-b)
Au-
Au+
Au2+ Au3
+ Au4+
Au5+
Au2-
Au3-
MB+
MB +
200 300 1000 500 400 700 600 800 900 m/z
285
284
a
b
c
d
Figure 1 . Negative (a) and positive (b) mass-spectra of PS-Au ionization platform; Spectra of methylene blue from PS-
Au (c) and from chemically oxidized PS (d).
The intensity of Au cluster peaks drops with increase of Au atoms quantity in the negative clusters, otherwise the intensities of odd-atom clusters in positive mass-spectrum are significantly higher, than the intensities of even-atom ones. This regularity probably takes place due to lower stability of cation-radicals (which are even-atom Aun
+ clusters) in compare with the cations without unpaired electrons (which are the odd-atom Aun
+ clusters) in the conditions of laser desorption experiment. The cationic dye methylene blue (MB) was studied as a model analyte for PS-Au ionization platform and chemically oxidized (H2O2(35%) : H2SO4, 3:7 vol., 90 oC, 15 min) porous silicon (PS-OX) taken as a reference platform. The mass-spectra of the MB were obtained only in the positive ions mode for both platforms (Fig. 1, c – d). The intensity distribution of the manifold of peaks in parent ion region (m/z = 284) is not in accordance with the isotope distribution of MB+ cation (m/z = 284), indicating the reaction of MB+ reduction/protonation to MBH+• (m/z = 285). The intensity of the reduced form peak is much higher for the PS-OX than for PS-Au. Probably, the desorption/ionization of the MB takes place on the surface of porous silicon for both studied platforms. In the case of PS-Au platform Au NPs withdraw electrons from silicon resulting in less efficient reduction of the analyte.
Conclusions The composites of porous silicon with Au nanoparticles of tunable size were obtained and their efficiency as SALDI supports was proved. Studies of biologically important small molecules (antibiotics, metabolites, antioxidant molecules, etc) desorption/ionization is in progress. References [1] K. Tanaka, H. ius, Y. Ido, S. Akita, Y. Yoshida, T. Yoshida, iuse iuse. Mass Spectrom. 1988; 2: 151. [2]. M. Karas, F. Hillenkamp, Analytical Chemistry 1988; 60: 2299. [3]. D. S. Peterson, Mass Spectrometry Reviews 2007; 26: 19. [4]. J. Wei, J. M. Buriak, G. Siuzdak, Nature 1999; 399: 243. [5]. I. V. Shmygol, S. A. Alekseev, O. Yu. Lavrinenko, N. S. Vasilyeva, V. N. Zaitsev, D. Barbier, V. A. Pokrovsky, J. Mass Spectrom. 2009; 44(8): 1234. [6]. Y. Chen, G. Luo, J. Diao, O. Chornoguz, M. Reeves, A. Vertes, J. Phys.: Conf. Ser. 2007; 59: 548. [7]. H. Yan, N. Xu, W. Y. Huang, H. M. Han, S. J. Xiao, Int. J. Mass Spectrom. 2009; 281(1-2): 1. [8]. L. C. Chen, J. Yonehama, T. Ueda, H. Hori, K. Hiraoka, J. Mass Spectrom. 2007; 42: 346.
Benahavís (Málaga, Spain), 21st to 23rd of October, 2009 163
Matrix synthesis and functionalization of ordered m esoporous carbon by palladium nanoparticles as potential sorbent for hydrogen sto rage
Vasyl Gerda a,b, Natalia Kobilinskaya b, Vladimir Zaitsev b, Jacques Fraissard a
aLaboratoire de Physique Quantique, University P. and M. Curie, ESPCI, 10 Rue Vauquelin, Paris, France
bDepartment of Chemistry, National Taras Shevchenko University, 64 Vladimirska Str., Kiev, Ukraine
Ordered mesoporous carbons (OMC) represent a new generation of materials with unique properties. A number of interesting applications for adsorption, molecular sieving and hydrogen storage systems can be imagined if carbonaceous materials are constructed with 3-D interconnected pores. The synthesis of OMC using ordered mesoporous silica MCM-48 as a matrix has been realised Here we describe the synthesis principle, structure and physical properties of OMC and the characterization of these mesoporous carbons by means of XRD, TPDMS, Raman and FTIR spectroscopies. MCM-48 was prepared by sol-gel synthesis [1] and was used as the matrix for sucrose carbonization in vacuum at 700, 900 and 1100 °C. The OMC show XRD patterns with several sharp low-angle reflections corresponding to the cubic phase. The features of step-by-step sucrose carbonization in MCM-48 pores were revealed by FTIR and TPDMS. The OMC exhibit high N2 BET specific surface area (1810 m2/g) and a large total pore volume (1.1 cm3/g). A typical OMC has uniform mesopores (3 nm) and a certain number of micropores. The structure and sorption characteristics of the OMC obtained depend markedly on the conditions of sucrose carbonizsation in the silica matrix. The OMC obtained were found to have excellent properties for the reversible adsorption of H2. The introduction of nanodispersed Pd (1 or 5 mass %) into OMC pores increases hydrogen adsorption by up to 10 %, corresponding only to the absorption by Pd particles. The resulting high-surface area carbon with uniform pores is a promising advanced material for several applications, such as adsorption of large molecules, catalyst support, capacitors and energy storage. Acknowledgment We thank COST for financial support: agreement No. D36/003/06 “Interfacial functionalization of (bi)-metallic nanoparticles to prepare highly active and selective catalysts: understanding synergy and/or promotion effect”. Reference [1] Q.G. Meng, P. Boutinaud, A.-C. Franville, et al. Microporous and Mesoporous Materials, 2003, 65, pp. 127–136.
Benahavís (Málaga, Spain), 21st to 23rd of October, 2009 165
Theoretical studies of hydrogen adsorption mechanis m on ZrO 2
Olga Syzgantseva 1,2, Monica Calatayud 1,2, Christian Minot 1,2, Mohammad Esmail
Alikhani 3,4
1 UPMC Univ Paris 06, UMR 7616, Laboratoire de Chimie Théorique, F-75005, Paris, France
2 CNRS, UMR 7616, Laboratoire de Chimie Théorique, F-75005, Paris, France 3 UPMC Univ Paris 06, UMR 7075, Laboratoire de Dynamique, Interactions et Réactivité, F-
75005, Paris, France 4 CNRS, UMR 7075, Laboratoire de Dynamique, Interactions et Réactivité, F-75005, Paris,
France
Zirconium oxide and its mixed compounds are widely applied in catalysis, fuel cells, ceramic technologies, gas sensors and photoconductive thin films. Zirconia catalyzes the hydrogenation reactions of olefins, diens, carbon monoxide, aromatic carboxylic acids, methanol synthesis from CO/H2 and CO2/H2, as well as dehydrogenation, isomerization, partial oxidation and dehydration of hydrocarbons, implying C–H bond cleavage. Photolytic decomposition of water under UV irradiation is known to be promoted by zirconia. Besides, mixed zirconia-containing oxides exhibit augmented hydrogen storage capacities. The insight in the interaction mechanism of hydrogen with zirconium oxide, as well as comprehension of the driving forces of its adsorption constitutes a challenging task. For this purpose, reaction pathway on hydrogen adsorption on ZrO2 was explored computationally on Density Functional Theory and Coupled Cluster level applying different pseudopotentials and basis sets in order to find out its impact on equilibrium geometries and energy barriers. Intermediate stages of H2 interaction with ZrO2 were revealed. It is shown that the cleavage of activated H – H bond resulting in the formation of intermediate hydride structure, which contains Zr – H and O – H groups, is followed by hydroxide Zr(OH)2 formation. Zirconium hydroxide, in its turn, can undergo water elimination to form ZrO. The rate limiting step reveals to be the formation of Zr(OH)2. The description of hydrogen adsorption on ZrO2 is coherent for all the methods, when as the water elimination is not well described by any of them, requiring the application of multi-reference approaches. As a conclusion, equilibrium geometries and energy barriers of each consecutive stage are determined. Computational results are shown to be consistent with experimental ones, available in literature. The influence of applied methodology on computational results is figured out.
166 Benahavís (Málaga, Spain), 21st to 23rd of October, 2009
BOOK OF ABSTRACTS
Section V: Index of Authors
COST Chemistry D36 3rd Workshop and 5th Management Committee Meeting Author Index
Benahavís (Málaga, Spain), 21st to 23rd of October, 2009 169
Abrantes, L.M. 69 Adriaenssens, L. 123 Alekseev, S. 161 Algı, F. 63,127 Alikhani, M.E. 165 Almeida, A.R. 159 Andreeva, D. 37, 99, 101 Andreozzi, P. 93, 105 Arenillas, A. 49 Arias, M. 45 Asaro, F. 105 Auer, G. 139 Bañares, M.A. 27,131,133, 137
Baranton, S. 75 Barbe, J.M. 151 Barrero, A. 109 Beck, A. 155, 157 Bedia, J. 109,111,113
Benkó, T. 157 Blanco-Brieva, G. 87,163 Boghosian, S. 129 Bonincontro, A. 105 Boutonnet, M. 153 Brito, R. 83 Brückner, A. 139 Bruijninck, P.C.A. 45 Bulushev, D.A. 85 Cabrita, J.S. 69 Calatayud, M. 41,137, 165 Campos-Martín, J.M. 35, 87 Capel-Sánchez, M.C. 35 Che, M. 39 Cihaner, A. 63, 127 Colina, A. 59 Colomer, A. 95 Coppola, L. 105 Cordero, T. 109,111,113,115 Costa, R. 81 de Frutos, M.P. 87 De Persiis, F. 105 Di Carlo, G. 117,121,153 Dias, R.S 81, 149 Dragoi, B. 55 Dzwigaj, S. 39, 51 Edolfa, K. 119 Ersoz, M. 151 Fermín, D.J. 61 Fernández, A.C. 59 Fidalgo, B. 49 Fiedler, J. 143 Fierro, J.L.G. 35,67,75,87 Fischer-Durand, N. 145 Fleisher, M. 119 Floch, A. 107 Fraissard, J. 25,161,163 Gaigneaux, E.M. 107 Gál, M. 123 Galantini, L. 105 Gallardo, A. 113 García, M.T. 79, 95
García-García, F.J. 57 García-Rodríguez, S. 75 Gargiulo, V. 91 Garino, C. 143 Gebbink, R.J.M.K. 45 Gerda, V. 163 Geszti, O. 155 Girault, H.H. 71, 151 Gobetto, R. 143 Golinska, H. 27, 131 Gros, C.P. 151 Grünert, W. 139 Guan, Z. 141 Guczi, L. 65,155,157 Guerrero-Pérez, M.O. 27,131,137 Hansen, S. 57 Hatay, I. 151 Hausoul, J.C. 45 Heras, M.A. 59 Hernández-Alonso, M.D. 159 Hipler, F. 139 Horáček, M. 123 Horváth, A. 155 Hromadová, M. 145 Đçli, M. 63 Ilieva, L. 37,99,101,119 Iliopoulou, E.F. 103 Infante, M.R. 79, 95, 97 Ivanov, I. 37, 99 Jagodzinska, K. 43 Jiménez-López, A. 53 Kaučič, V. 21 Khoury, T. 151 Kobilinskaya, N. 163 Kolivoška, V. 145 Kompio, P.G.W.A. 139 Krtil, P. 73 La Mesa, C. 93, 105 La Parola, V. 51 Lallave, M. 109 Landa-Cánovas, A.R. 57 Lappas, A.A. 103 Lee, I. 91 Léger, J.M. 23, 75 Leggio, C. 105 Leite, L. 119 Lewandowska, A.E. 133 Lindman, B. 81, 147 Linse, P. 149 Liotta, L.F. 117,121,157 Lisowski, W. 99 Löffler, E. 139 López Nieto, J.M. 17 López-Granados, M. 47 López-Medina, R. 27, 131 López-Palacios, J. 59 Loscertales, I.G. 109 Lozano, M. 97 Lozano, N. 93 Lundberg, D. 81
Author Index COST Chemistry D36 3rd Workshop and 5th Management Committee Meeting
170 Benahavís (Málaga, Spain), 21st to 23rd of October, 2009
Lysenko, V. 161 Manoylova, O. 139 Manrisa, A. 95 Mariscal, R. 47 Marques, E.F. 83 Martín-Alonso, D. 47 Martínez-Huerta, M.V. 67 Méndez, M.A. 151 Menéndez, J.A. 49 Mestl, G. 33, 139 Méthivier, C. 39 Miguel, M.G. 81, 147 Mikolajska, E.J. 133 Millot, Y. 39 Minot, C. 165 Mitjans, M. 95 Montes de Oca, M.G. 61 Moran, M.C. 81,147 Moran, M.C. 79 Moreno-Tost, R. 53 Mores, D. 125 Moulijn, J.A. 159 Mourato, A. 69 Moussa, F. 141 Mul, G. 159 Nedyalkova, R. 99 Nervi, C. 143 Nicotera, I. 105 Nogier, J.P. 39 Ojeda, M. 47 Oliviero-Rossi, C. 105 Olsson, U. 83 Önal, A.M. 63 Orellana-Rico, M.J. 53 Pai, A.A.C.C. 149 Pamuk, M. 127 Pantaleo, G. 37,117,121, 157
Parvulescu, A.N. 45 Pászti, Z. 65 Pavel, N.V. 105 Peña, M.A. 67, 75 Pérez, L. 79,93,95,97 Petrova, P. 101 Pinazo, A. 79,93,95,97 Pointner, B. 77 Pokrovsky, V. 161 Pons, R. 79, 93, 97 Pospíšil, L. 123, 141 Psarras, A.C. 103 Ranieri, G.A. 105 Reischl, M. 77 Ribitsch, V. 77 Ribosa, I. 95 Risuleo, G. 105 Rodríguez-Castellón, E. 53 Rodríguez-Mirasol, J. 109,111,113, 115 Rojas, E. 27, 137 Rojas, S. 75
Romanenko, J. 161 Rosas, J.M. 111,115 Ross, J.R.H. 85 Ruiz, V. 59 Ruiz-Rosas, R. 109 Ruppert, A.M. 41 Sales, J.L. 91 Salmain, M. 145 Samec, Z. 73 Sánchez-Domínguez, M. 153 Sandroni, M. 143 Santamaría-González, J. 47 Santos, E. 135 Šebera, J. 73 Severa, L. 123 Shishido, T. 39 Shmidlers, A. 119 Shmygol, I. 161 Silva, B. 83 Sobczak, I. 43 Sobczak, J.W. 99 Sokolová, R. 145 Solans, C. 153 Stana-Kleinschek, K. 77 Stavitski, E. 125 Stefler, G. 155 Stonkus, V. 119 Su, B. 151 Sullivan, J.A. 31 Syzgantseva, O. 165 Tardan, F. 105 Teplý, F. 123 Tielens, F. 51, 135 Tirkeş, S. 127 Trejda, M. 107 Tsilomelekis, G. 129 Tsiouvaras, N. 67 Valero, M.J. 113 Venezia, A.M. 37, 55, 117, 121, 157
Vera, D. 111 Vila, F. 47 Vinardell, M.P. 79, 95 Viscardi, G. 143 Volpi, G. 143 Wang, Y. 141 Weckhuysen, B.M. 41,45,125 Wojcieszak, R. 107 Wojtaszek, A. 107 Yang, J. 141 Youssry, M. 105 Zaera, F. 91 Zaitsev, V. 161, 163 Záliš S. 73 Zdenek S. 151 Zgrablich, G. 91 Zhang, Y. 141 Ziolek, M. 27,43,107,131
COST Chemistry D36 3rd Workshop and 5th Management Committee
Benahavís (Málaga, Spain), 21st to 23rd of October, 2009
López-Medina, Ricardo Instituto de Catálisis y Petroleoquímica (CSIC) Spain [email protected]
López-Nieto, José Manuel Instituto de Tecnología Química (CSIC) Spain [email protected] Mariscal, Rafael Instituto de Catálisis y Petroleoquímica Spain [email protected]
Marqués, Eduardo F. University of Porto Portugal [email protected] Martinez de la Cuesta, Pedro Universidad de Málaga Spain [email protected]
Mendez, Manuel Ecole Polytechnique Federale de Lausanne Switzerland [email protected]
Menendez, J. Ángel Instituto Nacional del Carbón Spain [email protected]
Mestl, Gerhard SÜD-CHEMIE AG Germany [email protected] Miguel, Maria Coimbra University Portugal [email protected] Mikolajska, Ewelina Joanna Instituto de Catálisis y Petroleoquímica (CSIC) Spain [email protected]