Aust. J. Chem. 2010, 63, 578–588 www.publish.csiro.au/journals/ajc
Crystal Engineering Studies on Ionic Crystals of Pyridineand Carboxylic Acid Derivatives ContainingAmide Functional Groups
Lalit Rajput,A Ramkinkar Santra,A and Kumar BiradhaA,B
ADepartment of Chemistry, Indian Institute of Technology, Kharagpur-721302, India.BCorresponding author. Email: [email protected]
Seven crystal structures of pyromellitic acid or trimesic acid salts of molecules that contain pyridine and amide function-alities were determined and their structures were analyzed in detail in terms of various intermolecular interactions. Thepresence of multiple functionalities (acid, pyridine, amide, and hydroxy groups) in these structures resulted in diversifiedsupramolecular architectures. Amide-to-amide hydrogen bonds are not observed in any of these structures because ofinterference by the anions, water molecules, or pyridinium cations. The symmetry of the components was found to beimportant in determining the resultant supramolecular synthon and, therefore, the overall architecture. The pyromellitateanions exhibited four types of geometries, which differ in valencies and intramolecular hydrogen bonding, and theseanions also exhibit self stacks when they have planar geometries.
Manuscript received: 28 July 2009.Manuscript accepted: 3 September 2009.
Introduction
Crystal engineering studies with molecules that contain multiplefunctional groups provides an excellent opportunity to study andunderstand the hierarchy of intermolecular interactions betweenthe various functional groups.[1] The disruption of a robust syn-thon of a particular functional group by the presence of anotherfunctional group has been termed as ‘interaction interference’.[2]
For example, the presence of pyridine and carboxylic acid in amolecule leads to the formation of heteromeric synthons II–IVas the conventional synthon I of –COOH becomes disrupted bythe pyridine (Scheme 1).[3] Although these synthons are similarin terms of geometry, they alter significantly the chemical andphysical properties, such as melting point, solubility, conductiv-ity, dissolution rate, etc., of the material. Here it is important tonote that the recent database survey of Aakeröy et al. on pyridineand carboxylic acid derivatives reveals that the structural pre-dictability is more in co-crystal (II) than in ionic solids (IV).[4]
We have recently explored the interference of pyridine func-tionalities in amide-to-amide hydrogen bonding by studying aseries of molecules that contain pyridine and secondary amidefunctionalities.[5] These studies show that geometrical restric-tions play a crucial role in determining the final outcome ofsupramoleuclar aggregation in crystalline solids.
However, the crystal engineering studies on multi-componentsystems (co-crystals or salts) are of importance because oftheir functional properties in pharmaceutical industries.[6] Themolecules that contain COOH or OH functional groups wereshown in several instances to be good co-crystal or salt for-mers with pyridine-containing molecules.[3,7] The occurrenceof synthons II–IV was often attributed to �pKa value, whichis defined as pKa (PyNH+) − pKa (COOH). Synthon II formspreferably when the �pKa < 0; synthon III[8] forms when0 < �pKa < 3.75, and synthon IV forms when �pKa > 3.75.[9]
However, in practice the pKa values of acids and pyridines varybased on the solvents and other accompanying components inthe reaction. Therefore, it is not easy to predict the nature of theresultant crystals. Here we intend to use H3TMA (trimesic acid,benzene-1,3,5-tricarboxylic acid) and H4PMA (pyromelliticacid, benzene-1,2,4,5-tetracarboxylic acid) as linkers betweenthe molecules 1–5 (Scheme 2) to build supramolecular architec-tures with predefined network geometries. These two acids wereshown to be good building blocks in numerous varieties of exam-ples to form predictable and interesting supramolecular archi-tectures with characteristic features such as interpenetration,supramolecular isomerism, and guest inclusion.[3,10]
H4PMA or H3TMA are generally expected to form dianionswhen mixed with pyridine-containing compounds, for examplemolecules 1–5, because of their high pKa1 (2.43 for H4PMAand 3.16 for H3TMA) and pKa2 (3.13 for H4PMA and 3.98 forH3TMA) values. When complexed with molecules 1–5, thesedianions have the ability to link the pyridine units by syn-thons II–IV, whereas the amide groups interact with themselves.Molecules 1 and 2 are linear and expected to produce a lineararray of molecules in a two-dimensional (2D) layer. Molecules3–5 were chosen based on our previous studies in which severalderivatives of 3 were shown to form co-crystals with H3TMA bya new supramolecular synthon V between acid and amide func-tional groups (Scheme 3).[11] In these co-crystals, the –COOHprefers to interact with secondary amide functionalities over thepyridine functionalities. The violation of the hydrogen bond-ing hierarchy rule in these systems prompted us to investigatethe role of molecular three-fold symmetry in the formation ofsynthon V. Therefore, H4PMA and bis-amides 4 and 5 were con-sidered as they do not possess a three-fold symmetry. With theseintentions we have synthesized molecules 1–5 and complexedthem with H3TMA and H4PMA. Our studies here show that the
Crystal Engineering Studies on Ionic Crystals of Pyridine and Carboxylic Acid Derivatives 579
C
O
O N
H
HC
O
O� N�
H
H
C
O
O N
H
H
I II IIIC
O
O O
C
O
H
H
IV
Scheme 1.
COOHHOOC
HOOC COOH
COOH
HOOC COOH
O
N
O
N
H
HN
N
OH
OH
O
N
H
N
O
N
H
N
H3TMA H4PMA
12
R1
R2R3
3 R1 � R2 � R3 � -CONH-CH2-2-pyridyl
4 R1 � H; R2 � R3 � -CONH-CH2-2-pyridyl
5 R1 � H; R2 � R3 � -CONH-CH2-3-pyridyl
Scheme 2.
ON
H
O O
H
ON
H
V
Scheme 3.
crystal structures of these ionic crystals are highly unpredictableand may not always follow the rule of hierarchy of interactions(Scheme 3).
Results and Discussion
Compound 1 was prepared by coupling the methyl ester ofsuccinic acid with 4-aminopyridine in the presence of NaH.Compound 2 was prepared by condensing tartaric acid with3-aminopyridine, whereas 3–5 were prepared by the reactionof corresponding amines with their respective acid chloridesof H3TMA or isophthalic acid. The reaction of 1 and 2 withH3TMA and H4PMA, respectively, resulted in the crystals ofionic compounds [H21][HTMA]·2H2O, 6 and [H22][H2PMA],7. The reaction of 3 with H3TMA and H4PMA resultedin the co-crystals of [H3TMA·3]2·anisole·2H2O, 8[9] and[H23]2[H2PMA-H-PMAH2][H3PMA]·10H2O, 9, respectively.The reaction of H4PMA with 4 and 5 resulted in the crys-tals of [H24][H2PMA], 10 and [H25][H2PMA], 11, respectively.The crystallographic parameters for all the crystal structures aregiven in Table 1, the CO bond lengths of –COOH or –COO− aretabulated in Table 2 and hydrogen bond parameters are tabulatedin Table 3.
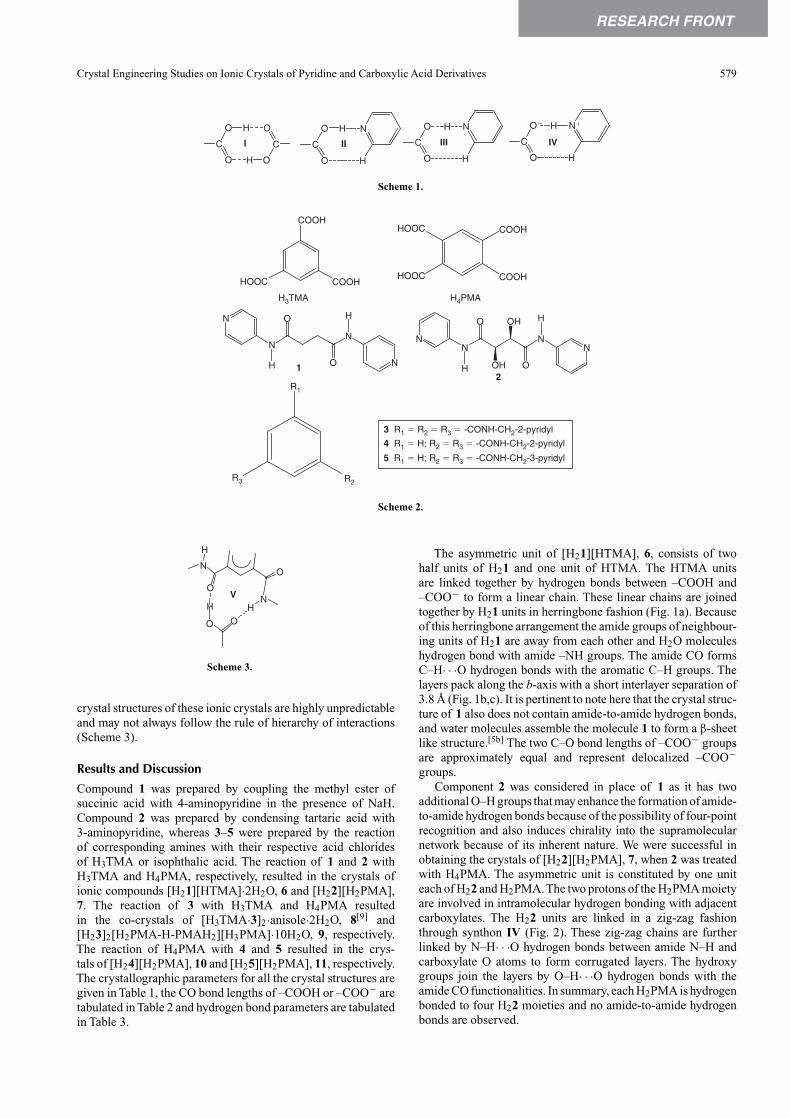
The asymmetric unit of [H21][HTMA], 6, consists of twohalf units of H21 and one unit of HTMA. The HTMA unitsare linked together by hydrogen bonds between –COOH and–COO− to form a linear chain. These linear chains are joinedtogether by H21 units in herringbone fashion (Fig. 1a). Becauseof this herringbone arrangement the amide groups of neighbour-ing units of H21 are away from each other and H2O moleculeshydrogen bond with amide –NH groups. The amide CO formsC–H· · ·O hydrogen bonds with the aromatic C–H groups. Thelayers pack along the b-axis with a short interlayer separation of3.8 Å (Fig. 1b,c). It is pertinent to note here that the crystal struc-ture of 1 also does not contain amide-to-amide hydrogen bonds,and water molecules assemble the molecule 1 to form a β-sheetlike structure.[5b] The two C–O bond lengths of –COO− groupsare approximately equal and represent delocalized –COO−groups.
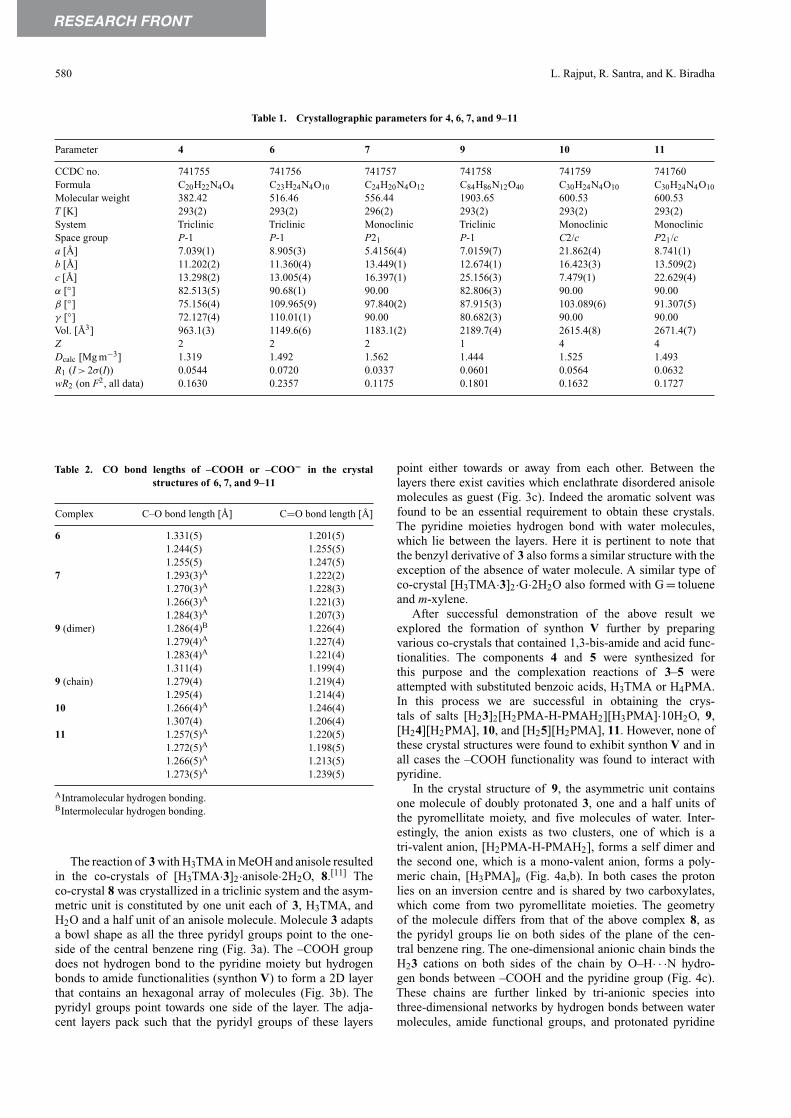
Component 2 was considered in place of 1 as it has twoadditional O–H groups that may enhance the formation of amide-to-amide hydrogen bonds because of the possibility of four-pointrecognition and also induces chirality into the supramolecularnetwork because of its inherent nature. We were successful inobtaining the crystals of [H22][H2PMA], 7, when 2 was treatedwith H4PMA. The asymmetric unit is constituted by one uniteach of H22 and H2PMA.The two protons of the H2PMA moietyare involved in intramolecular hydrogen bonding with adjacentcarboxylates. The H22 units are linked in a zig-zag fashionthrough synthon IV (Fig. 2). These zig-zag chains are furtherlinked by N–H· · ·O hydrogen bonds between amide N–H andcarboxylate O atoms to form corrugated layers. The hydroxygroups join the layers by O–H· · ·O hydrogen bonds with theamide CO functionalities. In summary, each H2PMA is hydrogenbonded to four H22 moieties and no amide-to-amide hydrogenbonds are observed.
RESEARCH FRONT
580 L. Rajput, R. Santra, and K. Biradha
Table 1. Crystallographic parameters for 4, 6, 7, and 9–11
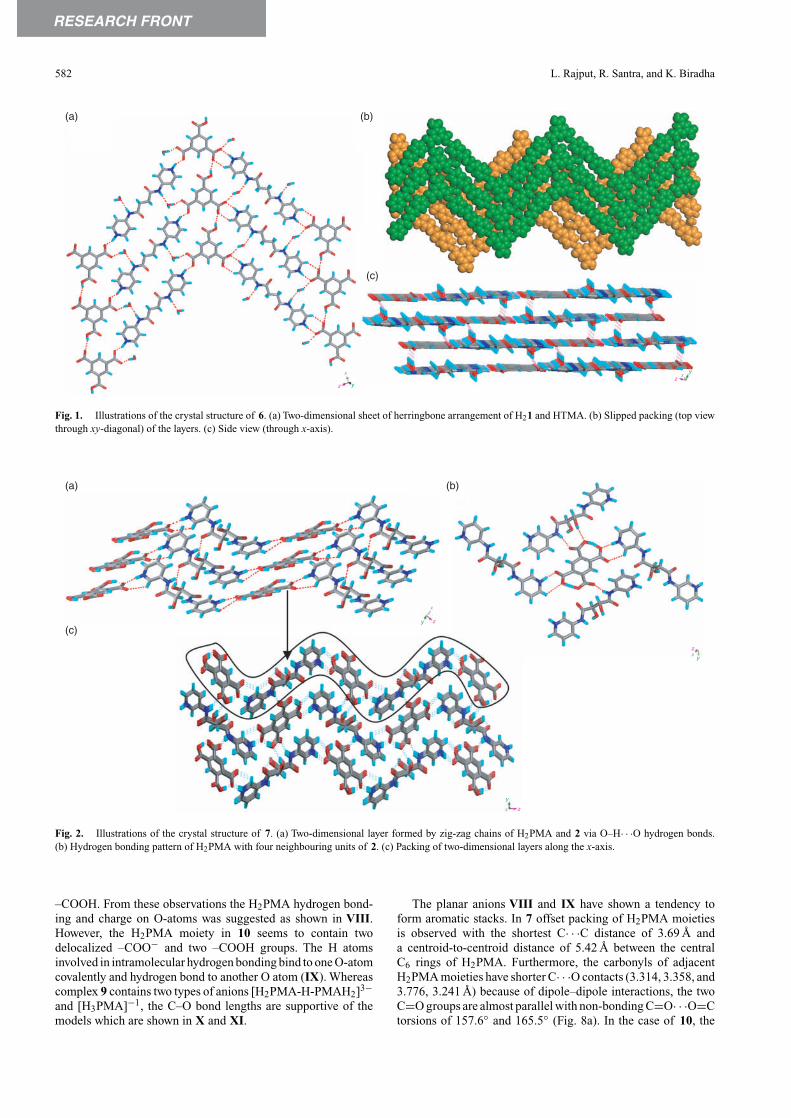
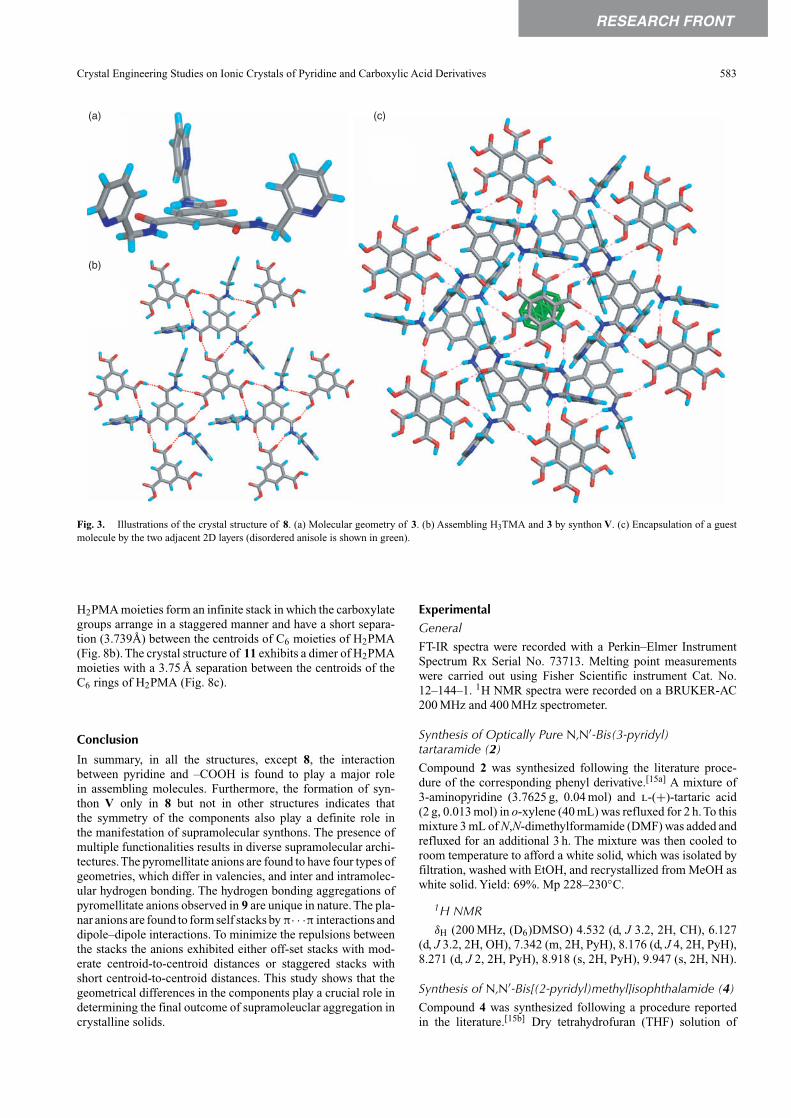
The reaction of 3 with H3TMA in MeOH and anisole resultedin the co-crystals of [H3TMA·3]2·anisole·2H2O, 8.[11] Theco-crystal 8 was crystallized in a triclinic system and the asym-metric unit is constituted by one unit each of 3, H3TMA, andH2O and a half unit of an anisole molecule. Molecule 3 adaptsa bowl shape as all the three pyridyl groups point to the one-side of the central benzene ring (Fig. 3a). The –COOH groupdoes not hydrogen bond to the pyridine moiety but hydrogenbonds to amide functionalities (synthon V) to form a 2D layerthat contains an hexagonal array of molecules (Fig. 3b). Thepyridyl groups point towards one side of the layer. The adja-cent layers pack such that the pyridyl groups of these layers
point either towards or away from each other. Between thelayers there exist cavities which enclathrate disordered anisolemolecules as guest (Fig. 3c). Indeed the aromatic solvent wasfound to be an essential requirement to obtain these crystals.The pyridine moieties hydrogen bond with water molecules,which lie between the layers. Here it is pertinent to note thatthe benzyl derivative of 3 also forms a similar structure with theexception of the absence of water molecule. A similar type ofco-crystal [H3TMA·3]2·G·2H2O also formed with G = tolueneand m-xylene.
After successful demonstration of the above result weexplored the formation of synthon V further by preparingvarious co-crystals that contained 1,3-bis-amide and acid func-tionalities. The components 4 and 5 were synthesized forthis purpose and the complexation reactions of 3–5 wereattempted with substituted benzoic acids, H3TMA or H4PMA.In this process we are successful in obtaining the crys-tals of salts [H23]2[H2PMA-H-PMAH2][H3PMA]·10H2O, 9,[H24][H2PMA], 10, and [H25][H2PMA], 11. However, none ofthese crystal structures were found to exhibit synthon V and inall cases the –COOH functionality was found to interact withpyridine.
In the crystal structure of 9, the asymmetric unit containsone molecule of doubly protonated 3, one and a half units ofthe pyromellitate moiety, and five molecules of water. Inter-estingly, the anion exists as two clusters, one of which is atri-valent anion, [H2PMA-H-PMAH2], forms a self dimer andthe second one, which is a mono-valent anion, forms a poly-meric chain, [H3PMA]n (Fig. 4a,b). In both cases the protonlies on an inversion centre and is shared by two carboxylates,which come from two pyromellitate moieties. The geometryof the molecule differs from that of the above complex 8, asthe pyridyl groups lie on both sides of the plane of the cen-tral benzene ring. The one-dimensional anionic chain binds theH23 cations on both sides of the chain by O–H· · ·N hydro-gen bonds between –COOH and the pyridine group (Fig. 4c).These chains are further linked by tri-anionic species intothree-dimensional networks by hydrogen bonds between watermolecules, amide functional groups, and protonated pyridine
RESEARCH FRONT
Crystal Engineering Studies on Ionic Crystals of Pyridine and Carboxylic Acid Derivatives 581
Table 3. Hydrogen bonding parameters for the crystals structures of6–11
groups. The anionic aggregations observed in this complexare unique and, to our knowledge, so far no such examplesof aggregation for pyromellitate anions are reported in theliterature.
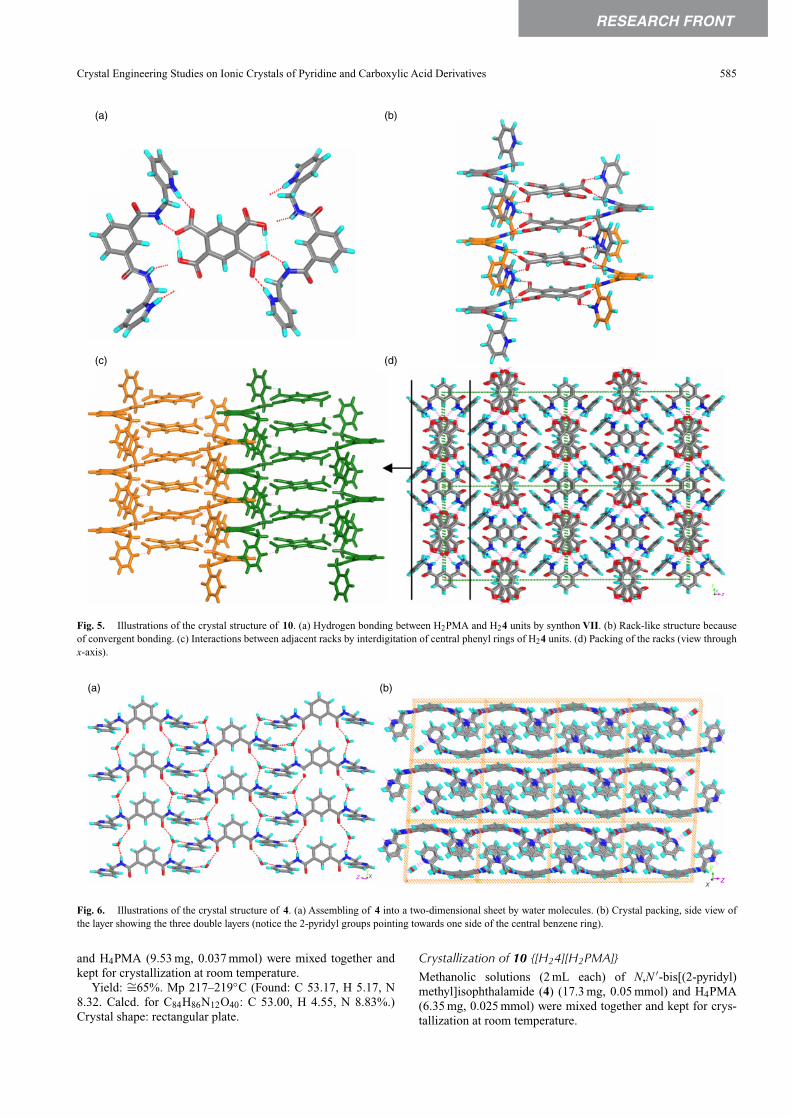
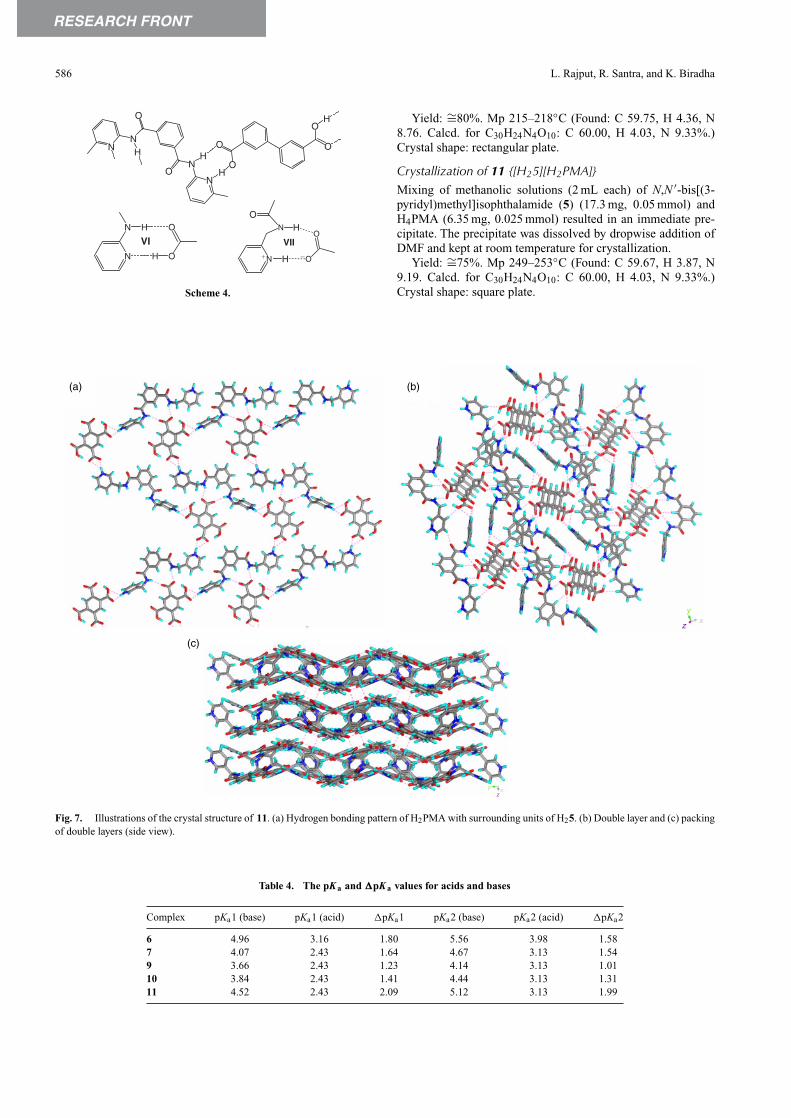
Molecule 4 forms crystals of 10 with H4PMA, the crystalstructure has an interesting supramolecular aggregation, whichresembles the co-crystals of the 2-pyridyl analogue of 4 with sev-eral di-carboxylic acids (Scheme 4). Previously, the 2-pyridylanalogue of 4 was shown to form one-dimensional zig-zagchains with di-carboxylic acids by synthonVI.[12] We anticipatedthat the presence of a –CH2– group between pyridine and amideN–H groups does not allow the formation of a synthon similarto VI in the present complex. However, the crystal structure of10 also contains synthon VII, which is an expanded version ofsynthon VI. The presence of flexibility in 4 changed the zig-zaggeometry of the chain (divergent binding) to a linear geometry(convergent binding) to form a rack like structure (Fig. 5). TheH2PMA moiety acts as a plank of the rack, whereas the H24units form the frame. We have analyzed the crystal of molecule4 in order to compare its molecular geometry and aggregationwith that of 4 in 10. Molecule 4 in its crystal structure, unlikein 10, exhibits near mirror symmetry and the 2-pyridyl groupspoint to one side of the central benzene ring (Fig. 6). The watermolecules assemble the molecules into a 2D layer such that allthe pyridyl groups face to one side of the layer. These layers packin a head-to-head manner such that the pyridyl groups or the cen-tral benzene rings from the adjacent layers face each other. Ina sense, it can be regarded as a double layer assembled througharomatic interactions.
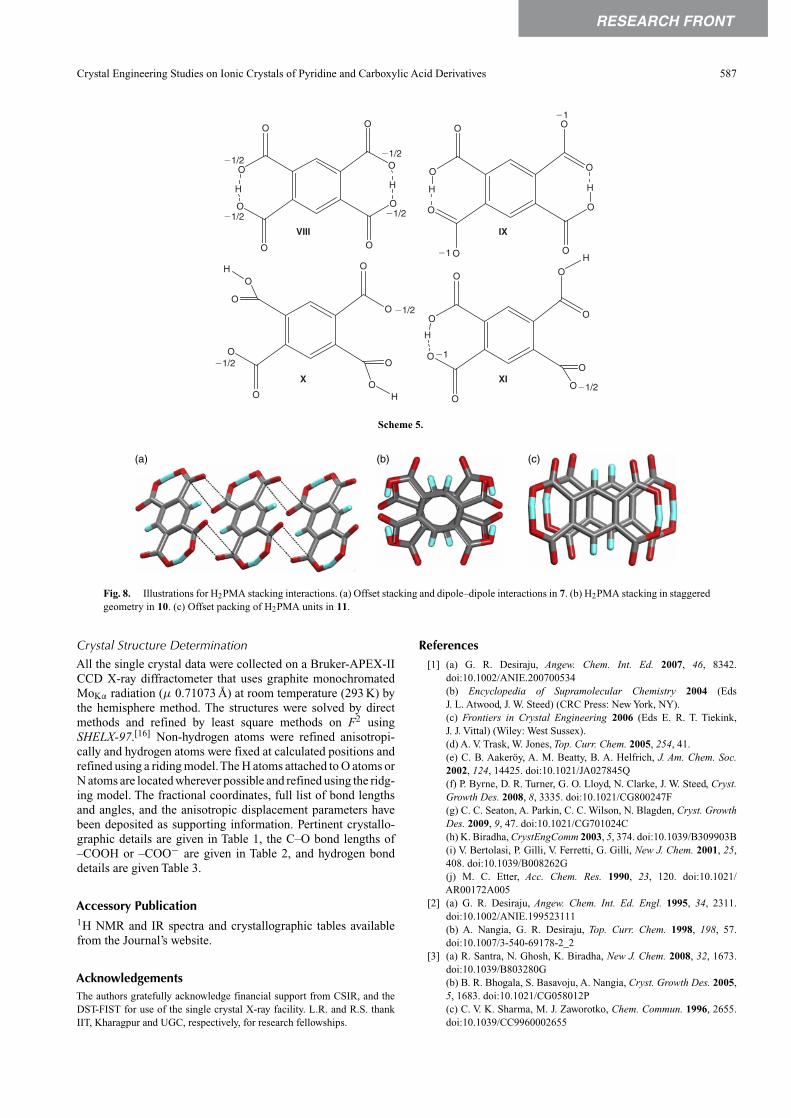
The crystal structure of 11 differs significantly from 10; itforms a highly corrugated 2D layers in which each H2PMAwas surrounded by three units of H25 by N–H· · ·O hydrogenbonds. One of the carboxylates binds to the two amide N–Hgroups of one H25 unit. The layer contains cavities that arefilled by the pyridyl groups such that a double layer is formed(Fig. 7).
�pKa Values of Acids and 1–5The dependency of proton transfer or the formation of synthonsII–IV on �pKa values was discussed in the introduction. In thepharmaceutical industry �pKa > 3 is used as a thumb rule forselecting counter ions to prepare salt forms.[13] The pKa valuesof H3TMA, H4PMA, and 1–5 are tabulated in Table 4.[14] The�pKa values of these systems are between 1.01 and 2.09, whichsuggests that synthon III, which has mixed ionization, is the mostprobable one. However, all these systems with the exception of8 contain synthon IV (salt or ionic), which is supposed to occuronly when the �pKa is above 3.0. Given the sensitivity of the pKavalues of the components to the nature of solvents, ratio of thecomponents, and concentration of the solution, the �pKa valuesmentioned here are may not have any relevance to the resultsobtained. However, it is pertinent to note that all the H2PMAmoieties except one have intra or intermolecular hydrogen bondsin which the protons are attached equally to both the O atomsrepresenting the mixed ionic state.
The Geometry and Stacking Interactionsof Pyromellitate AnionsThe C–O bond lengths of the carboxylates are given in Table 2.The geometry of the H2PMA moieties in complexes 7 and 11are similar, the H atoms which are involved in intramolecularhydrogen bonding, are shared equally between two carboxy-late O atoms (Scheme 5). The C–O distances in these twocomplexes indicate that the carboxylates are not delocalized.Furthermore, the C–O bonds involved in intramolecular hydro-gen bonding are shorter than the usual single C–O bonds of
RESEARCH FRONT
582 L. Rajput, R. Santra, and K. Biradha
(a) (b)
(c)
x
yz
xy
z
Fig. 1. Illustrations of the crystal structure of 6. (a) Two-dimensional sheet of herringbone arrangement of H21 and HTMA. (b) Slipped packing (top viewthrough xy-diagonal) of the layers. (c) Side view (through x-axis).
(a)
(c)
(b)
x
y z
x
y
z
xy
z
Fig. 2. Illustrations of the crystal structure of 7. (a) Two-dimensional layer formed by zig-zag chains of H2PMA and 2 via O–H· · ·O hydrogen bonds.(b) Hydrogen bonding pattern of H2PMA with four neighbouring units of 2. (c) Packing of two-dimensional layers along the x-axis.
–COOH. From these observations the H2PMA hydrogen bond-ing and charge on O-atoms was suggested as shown in VIII.However, the H2PMA moiety in 10 seems to contain twodelocalized –COO− and two –COOH groups. The H atomsinvolved in intramolecular hydrogen bonding bind to one O-atomcovalently and hydrogen bond to another O atom (IX). Whereascomplex 9 contains two types of anions [H2PMA-H-PMAH2]3−and [H3PMA]−1, the C–O bond lengths are supportive of themodels which are shown in X and XI.
The planar anions VIII and IX have shown a tendency toform aromatic stacks. In 7 offset packing of H2PMA moietiesis observed with the shortest C· · ·C distance of 3.69 Å anda centroid-to-centroid distance of 5.42 Å between the centralC6 rings of H2PMA. Furthermore, the carbonyls of adjacentH2PMA moieties have shorter C· · ·O contacts (3.314, 3.358, and3.776, 3.241 Å) because of dipole–dipole interactions, the twoC=O groups are almost parallel with non-bonding C=O· · ·O=Ctorsions of 157.6◦ and 165.5◦ (Fig. 8a). In the case of 10, the
RESEARCH FRONT
Crystal Engineering Studies on Ionic Crystals of Pyridine and Carboxylic Acid Derivatives 583
(a) (c)
(b)
Fig. 3. Illustrations of the crystal structure of 8. (a) Molecular geometry of 3. (b) Assembling H3TMA and 3 by synthon V. (c) Encapsulation of a guestmolecule by the two adjacent 2D layers (disordered anisole is shown in green).
H2PMA moieties form an infinite stack in which the carboxylategroups arrange in a staggered manner and have a short separa-tion (3.739Å) between the centroids of C6 moieties of H2PMA(Fig. 8b). The crystal structure of 11 exhibits a dimer of H2PMAmoieties with a 3.75 Å separation between the centroids of theC6 rings of H2PMA (Fig. 8c).
Conclusion
In summary, in all the structures, except 8, the interactionbetween pyridine and –COOH is found to play a major rolein assembling molecules. Furthermore, the formation of syn-thon V only in 8 but not in other structures indicates thatthe symmetry of the components also play a definite role inthe manifestation of supramolecular synthons. The presence ofmultiple functionalities results in diverse supramolecular archi-tectures.The pyromellitate anions are found to have four types ofgeometries, which differ in valencies, and inter and intramolec-ular hydrogen bonding. The hydrogen bonding aggregations ofpyromellitate anions observed in 9 are unique in nature. The pla-nar anions are found to form self stacks byπ· · ·π interactions anddipole–dipole interactions. To minimize the repulsions betweenthe stacks the anions exhibited either off-set stacks with mod-erate centroid-to-centroid distances or staggered stacks withshort centroid-to-centroid distances. This study shows that thegeometrical differences in the components play a crucial role indetermining the final outcome of supramoleuclar aggregation incrystalline solids.
ExperimentalGeneralFT-IR spectra were recorded with a Perkin–Elmer InstrumentSpectrum Rx Serial No. 73713. Melting point measurementswere carried out using Fisher Scientific instrument Cat. No.12–144–1. 1H NMR spectra were recorded on a BRUKER-AC200 MHz and 400 MHz spectrometer.
Synthesis of Optically Pure N,N′-Bis(3-pyridyl)tartaramide (2)Compound 2 was synthesized following the literature proce-dure of the corresponding phenyl derivative.[15a] A mixture of3-aminopyridine (3.7625 g, 0.04 mol) and l-(+)-tartaric acid(2 g, 0.013 mol) in o-xylene (40 mL) was refluxed for 2 h. To thismixture 3 mL of N,N-dimethylformamide (DMF) was added andrefluxed for an additional 3 h. The mixture was then cooled toroom temperature to afford a white solid, which was isolated byfiltration, washed with EtOH, and recrystallized from MeOH aswhite solid. Yield: 69%. Mp 228–230◦C.
Synthesis of N,N′-Bis[(2-pyridyl)methyl]isophthalamide (4)Compound 4 was synthesized following a procedure reportedin the literature.[15b] Dry tetrahydrofuran (THF) solution of
RESEARCH FRONT
584 L. Rajput, R. Santra, and K. Biradha
x
yz
(a) (b)
(c)
Fig. 4. Illustrations of the crystal structure of 9. (a) Hydrogen bonding pattern between H23 and H2PMA-H-PMAH2 (shown in green). (b) One-dimensionalpolymeric chain of the H3PMA unit. (c) Two-dimensional arrangement of the H23 unit and anions (compare the geometry of H23 with that of 3 in Fig. 3).
2-(aminomethyl)pyridine (1.0563 g, 0.01 mol) and triethylamine(1.9936 g, 0.02 mol) was cooled to 0◦C under nitrogen atmo-sphere, and to this solution isophthaloyl chloride (1.0 g,0.005 mol) in THF was added dropwise with continuous stirring.The solution was stirred overnight. THF was distilled off and theresulting white solid was washed several times with water anddried under vacuum. The resulting solid was crystallized frommethanol as thin needles. Yield: 85.80%. Mp 102–104◦C.
Crystallization of N,N′-Bis[(2-pyridyl)methyl]isophthalamide (4)Good quality crystals were obtained from mixture of methanoland chlorobenzene (1:1) solvents.
Compound 4·2H2O (Found: C 62.53, H 5.60, N 14.03. Calcd.for C20H22N4O4: C 62.82, H 5.80, N 14.65%.) Crystal shape:plates.
Crystallization of 6 {[H21][HTMA]·2H2O}Mixing of methanolic solutions (2 mL each) of N,N ′-bis(4-pyridyl)succinamide (1) (27 mg, 0.1 mmol) and H3TMA (14 mg,0.067 mmol) resulted in an immediate precipitate. The precip-itate was dissolved by dropwise addition of DMF and kept atroom temperature for crystallization.
Yield: ∼=80%. Mp >300◦C (Found: C 54.06, H 4.47, N 11.14.Calcd. for C23H24N4O10: C 53.49, H 4.68, N 10.85%.) Crystalshape: rectangular blocks.
Crystallization of 7 {[H22][H2PMA]}A 2:1 mixture of l-bis(3-pyridylamino)tartarate (2) (40 mg,0.132 mmol) and H4PMA (16.83 mg, 0.066 mmol) was dis-solved in 10 mL of MeOH and 5 mL of water mixture and keptfor crystallization.
Yield: ∼=40%. Mp 245–248◦C (Found: C 51.37, H 3.23, N9.69. Calcd. for C24H20N4O12: C 51.78, H 3.59, N 10.06%.)Crystal shape: white plates.
Crystallization of 9 {[H23][H2PMA-H-PMAH2][H3PMA]·10H2O}Methanolic solutions (2 mL each) of N,N ′,N ′-tris[(2-pyridyl)methyl]benzene-1,3,5-tricarboxamide (3) (24 mg, 0.05 mmol)
RESEARCH FRONT
Crystal Engineering Studies on Ionic Crystals of Pyridine and Carboxylic Acid Derivatives 585
y
zx
(a) (b)
(c) (d)
Fig. 5. Illustrations of the crystal structure of 10. (a) Hydrogen bonding between H2PMA and H24 units by synthon VII. (b) Rack-like structure becauseof convergent bonding. (c) Interactions between adjacent racks by interdigitation of central phenyl rings of H24 units. (d) Packing of the racks (view throughx-axis).
(a) (b)
z
y
xZ
YX
Fig. 6. Illustrations of the crystal structure of 4. (a) Assembling of 4 into a two-dimensional sheet by water molecules. (b) Crystal packing, side view ofthe layer showing the three double layers (notice the 2-pyridyl groups pointing towards one side of the central benzene ring).
and H4PMA (9.53 mg, 0.037 mmol) were mixed together andkept for crystallization at room temperature.
Yield: ∼=65%. Mp 217–219◦C (Found: C 53.17, H 5.17, N8.32. Calcd. for C84H86N12O40: C 53.00, H 4.55, N 8.83%.)Crystal shape: rectangular plate.
Crystallization of 10 {[H24][H2PMA]}Methanolic solutions (2 mL each) of N,N ′-bis[(2-pyridyl)methyl]isophthalamide (4) (17.3 mg, 0.05 mmol) and H4PMA(6.35 mg, 0.025 mmol) were mixed together and kept for crys-tallization at room temperature.
RESEARCH FRONT
586 L. Rajput, R. Santra, and K. Biradha
NO
O
NH H
N
N
O
O
H
O
O
H
N
N H O
OH
VI�N
NO
�OH
HO
VII
Scheme 4.
xy
z
xyz
(a)
(c)
(b)
Fig. 7. Illustrations of the crystal structure of 11. (a) Hydrogen bonding pattern of H2PMA with surrounding units of H25. (b) Double layer and (c) packingof double layers (side view).
Table 4. The pK a and �pK a values for acids and bases
Yield: ∼=80%. Mp 215–218◦C (Found: C 59.75, H 4.36, N8.76. Calcd. for C30H24N4O10: C 60.00, H 4.03, N 9.33%.)Crystal shape: rectangular plate.
Crystallization of 11 {[H25][H2PMA]}Mixing of methanolic solutions (2 mL each) of N,N ′-bis[(3-pyridyl)methyl]isophthalamide (5) (17.3 mg, 0.05 mmol) andH4PMA (6.35 mg, 0.025 mmol) resulted in an immediate pre-cipitate. The precipitate was dissolved by dropwise addition ofDMF and kept at room temperature for crystallization.
Yield: ∼=75%. Mp 249–253◦C (Found: C 59.67, H 3.87, N9.19. Calcd. for C30H24N4O10: C 60.00, H 4.03, N 9.33%.)Crystal shape: square plate.
RESEARCH FRONT
Crystal Engineering Studies on Ionic Crystals of Pyridine and Carboxylic Acid Derivatives 587
O
O
O
O
O
O
O
O
H
H
XI
O
O
O
O
O
O
O
OH
H
X
O
O
O
O
O
O
O
O
H H
�1/2
�1/2 �1/2
�1/2
�1/2
�1/2
�1
�1
�1�1/2
VIII
O
O
O
O
O
O
O
O
H H
IX
Scheme 5.
(a) (b) (c)
Fig. 8. Illustrations for H2PMA stacking interactions. (a) Offset stacking and dipole–dipole interactions in 7. (b) H2PMA stacking in staggeredgeometry in 10. (c) Offset packing of H2PMA units in 11.
Crystal Structure DeterminationAll the single crystal data were collected on a Bruker-APEX-IICCD X-ray diffractometer that uses graphite monochromatedMoKα radiation (µ 0.71073 Å) at room temperature (293 K) bythe hemisphere method. The structures were solved by directmethods and refined by least square methods on F2 usingSHELX-97.[16] Non-hydrogen atoms were refined anisotropi-cally and hydrogen atoms were fixed at calculated positions andrefined using a riding model.The H atoms attached to O atoms orN atoms are located wherever possible and refined using the ridg-ing model. The fractional coordinates, full list of bond lengthsand angles, and the anisotropic displacement parameters havebeen deposited as supporting information. Pertinent crystallo-graphic details are given in Table 1, the C–O bond lengths of–COOH or –COO− are given in Table 2, and hydrogen bonddetails are given Table 3.
Accessory Publication1H NMR and IR spectra and crystallographic tables availablefrom the Journal’s website.
AcknowledgementsThe authors gratefully acknowledge financial support from CSIR, and theDST-FIST for use of the single crystal X-ray facility. L.R. and R.S. thankIIT, Kharagpur and UGC, respectively, for research fellowships.
References[1] (a) G. R. Desiraju, Angew. Chem. Int. Ed. 2007, 46, 8342.
doi:10.1002/ANIE.200700534(b) Encyclopedia of Supramolecular Chemistry 2004 (EdsJ. L. Atwood, J. W. Steed) (CRC Press: New York, NY).(c) Frontiers in Crystal Engineering 2006 (Eds E. R. T. Tiekink,J. J. Vittal) (Wiley: West Sussex).(d) A. V. Trask, W. Jones, Top. Curr. Chem. 2005, 254, 41.(e) C. B. Aakeröy, A. M. Beatty, B. A. Helfrich, J. Am. Chem. Soc.2002, 124, 14425. doi:10.1021/JA027845Q(f) P. Byrne, D. R. Turner, G. O. Lloyd, N. Clarke, J. W. Steed, Cryst.Growth Des. 2008, 8, 3335. doi:10.1021/CG800247F(g) C. C. Seaton, A. Parkin, C. C. Wilson, N. Blagden, Cryst. GrowthDes. 2009, 9, 47. doi:10.1021/CG701024C(h) K. Biradha, CrystEngComm 2003, 5, 374. doi:10.1039/B309903B(i) V. Bertolasi, P. Gilli, V. Ferretti, G. Gilli, New J. Chem. 2001, 25,408. doi:10.1039/B008262G(j) M. C. Etter, Acc. Chem. Res. 1990, 23, 120. doi:10.1021/AR00172A005
[2] (a) G. R. Desiraju, Angew. Chem. Int. Ed. Engl. 1995, 34, 2311.doi:10.1002/ANIE.199523111(b) A. Nangia, G. R. Desiraju, Top. Curr. Chem. 1998, 198, 57.doi:10.1007/3-540-69178-2_2
[3] (a) R. Santra, N. Ghosh, K. Biradha, New J. Chem. 2008, 32, 1673.doi:10.1039/B803280G(b) B. R. Bhogala, S. Basavoju, A. Nangia, Cryst. Growth Des. 2005,5, 1683. doi:10.1021/CG058012P(c) C. V. K. Sharma, M. J. Zaworotko, Chem. Commun. 1996, 2655.doi:10.1039/CC9960002655
RESEARCH FRONT
588 L. Rajput, R. Santra, and K. Biradha
(d) T. R. Shattock, P. Vishweshwar, Z. Wang, M. J. Zaworotko, Cryst.Growth Des. 2005, 5, 2046. doi:10.1021/CG0501985(e) B. R. Bhogala, A. Nangia, New J. Chem. 2008, 32, 800.doi:10.1039/B800293B
[4] C. B. Aakeröy, M. E. Fasulo, J. Desper, Mol. Pharm. 2007, 4, 317.doi:10.1021/MP060126O
[5] (a) M. Sarkar, K. Biradha, Cryst. Growth Des. 2006, 6, 202.doi:10.1021/CG050292L(b) L. Rajput, S. Singha, K. Biradha, Cryst. Growth Des. 2007, 7,2788. doi:10.1021/CG0706417(c) L. Rajput, P. Sanphui, K. Biradha, Cryst. Growth Des. 2007, 7,1872. doi:10.1021/CG0705557(d) L. Rajput, K. Biradha, J. Mol. Struct. 2008, 876, 339. doi:10.1016/J.MOLSTRUC.2007.07.006
[6] (a) L. Jie, R. Sohrab, Curr. Med. Chem. 2009, 22, 884.(b) M. Pop, P. Sieger, P. W. Cains, J. Pharm. Sci. 2009, 98, 1820.doi:10.1002/JPS.21531(c) P. M. Bhatt, Y. Azim, T. S. Thakur, G. R. Desiraju, Cryst. GrowthDes. 2009, 9, 951. doi:10.1021/CG8007359(d) M. K. Stanton, S. Tutekcic, C. Morgan,A. Bak, Cryst. Growth Des.2009, 9, 1344. doi:10.1021/CG8005019(e) A. V. Trask, Mol. Pharm. 2007, 4, 301. doi:10.1021/MP070001Z(f) A. P. Vishweshwar, J. A. McMahon, J. A. Bis, M. J. Zaworotko,J. Pharm. Sci. 2006, 95, 499. doi:10.1002/JPS.20578(g) L. S. Reddy, N. J. Babu, A. Nangia, Chem. Commun. 2006, 1369.doi:10.1039/B515510J
[7] (a) R. Santra, K. Biradha, CrystEngComm 2008, 10, 1524.doi:10.1039/B813721H(b) L. R. MacGillivray, J. L. Reid, J. A. Ripmeester, J. Am. Chem. Soc.2000, 122, 7817. doi:10.1021/JA001239I(c) X. Gao, T. Frišcic, L. R. MacGillivray,Angew. Chem. Int. Ed. 2004,43, 232. doi:10.1002/ANIE.200352713
[8] The occurrence of synthon-III is rarely reported in the literature dueto the difficulty in the determination of H atom positions. However,there are some reports that exist:(a) S. L. Childs, G. P. Stahly, A. Park, Mol. Pharmaceutics 2007, 4,323. doi:10.1021/MP0601345(b) Z. J. Li,Y. Abramov, J. Bordner, J. Leonard, A. Medek, A. V. Trask,J. Am. Chem. Soc. 2006, 128, 8199. doi:10.1021/JA0541332(c) B. R. Bhogala, S. Basavoju, A. Nangia, CrystEngComm 2005, 7,551. doi:10.1039/B509162D
[9] (a) S. L. Johnson, K. A. Rumon, J. Phys. Chem. 1965, 69, 74.doi:10.1021/J100885A013(b)Y. B. Men, J. Sun, Z. T. Huang, Q.Y. Zheng, CrystEngComm 2009,11, 978. doi:10.1039/B822936H
(c) L. S. Reddy, S. J. Bethune, J. W. Kampf, N. R. Hornedo, Cryst.Growth Des. 2009, 9, 378. doi:10.1021/CG800587Y
[10] (a) K. Biradha, M. J. Zaworotko, Cryst. Eng. 1998, 1, 67.doi:10.1016/S0025-5408(98)00036-1(b) N. Shan, W. Jones, Tetrahedron Let. 2003, 44, 3687. doi:10.1016/S0040-4039(03)00685-3(c) K. K. Arora, V. R. Pedireddi, J. Org. Chem. 2003, 68, 9177.doi:10.1021/JO034434Z(d) S. H. Dale, M. R. J. Elsegood, M. Hemmings, A. L. Wilkinson,CrystEngComm 2004, 6, 207. doi:10.1039/B404563G(e) C. Ruiz-Pérez, P. A. L. Luis, M. H. Molina, M. M. Laz, P. Gili,M. Julve, Cryst. Growth Des. 2004, 4, 57. doi:10.1021/CG034133I(f) M. Du, Z. H. Zhang, X. J. Zhao, Cryst. Growth Des. 2005, 5, 1247.doi:10.1021/CG0495680(g) M. Du, Z. H. Zhang, X. G. Wang, H. F. Wu, Q. Wang, Cryst. GrowthDes. 2006, 6, 1867. doi:10.1021/CG0601812
[11] L. Rajput, K. Biradha, Cryst. Growth Des. 2009, 9, 40. doi:10.1021/CG801132R
[12] (a) J.Yang, E. Fan, S. J. Geib, A. D. Hamilton, J. Am. Chem. Soc. 1993,115, 5314. doi:10.1021/JA00065A061(b) F. Garcia-Tellado, S. J. Geib, S. Goswami, A. D. Hamilton, J. Am.Chem. Soc. 1991, 113, 9265. doi:10.1021/JA00024A036(c) I. L. Karle, D. Rangnathan, V. Haridas, J. Am. Chem. Soc. 1997,119, 2777. doi:10.1021/JA9623121
[13] (a) W. Tong, G. Whitesell, Pharm. Dev. Technol. 1998, 3, 215.doi:10.3109/10837459809028498(b) T. R. Shattock, K. K. Arora, P. Vishweshwar, M. J. Zaworotko,Cryst. Growth Des. 2008, 8, 4533. doi:10.1021/CG800565A(c) S. M. Berge, L. D. Bighley, D. C. Monkhouse, J. Pharm. Sci. 1977,66, 1. doi:10.1002/JPS.2600660104(d) B. Fritz, J. L. Lach, L. D. Bighley, J. Pharm. Sci. 1971, 60, 1617.doi:10.1002/JPS.2600601104
[14] The pKa values for acids H3TMA and H4TMA were takenfrom the website: http://www.petrik.com/PUBLIC/library/misc/acid_base_pk.htm. The pKa values for components 1–5 were calcu-lated using the MarvinSketch (version 5.2.2) program, http://www.chemaxon.com.
[15] (a) W. Chen, Y. Liu, Z. Chen, Eur. J. Org. Chem. 2005, 1665.doi:10.1002/EJOC.200400670(b) H.-J. Liu, Y.-H. Hung, C.-C. Chou, C.-C. Su, Chem. Commun.2007, 495. doi:10.1039/B613583H
[16] G. M. Sheldrick, SHELX-97, Program for the Solution and Refinementof Crystal Structures 1997 (University of Göttingen: Germany).