53
CS5238 Combinatorial methods in bioinformatics Topic: Gene Finding – Promoter Recognition Cen Cen, Er Inn Inn, Miao Xiaoping, Piyush Kanti Bhunre, Yin Jun 1 November 2002
| Date post: | 13-Dec-2015 |
| Category: |
Documents |
| Upload: | imogen-stanley |
| View: | 219 times |
| Download: | 0 times |

CS5238 Combinatorial methods in bioinformatics
Topic: Gene Finding –
Promoter Recognition
Cen Cen, Er Inn Inn, Miao Xiaoping,
Piyush Kanti Bhunre, Yin Jun1 November 2002

Outline of Presentation Biological Background Gene Finding Promoter Recognition Dragon Promoter Finder Open Problem and Future Research New Algorithm Conclusion
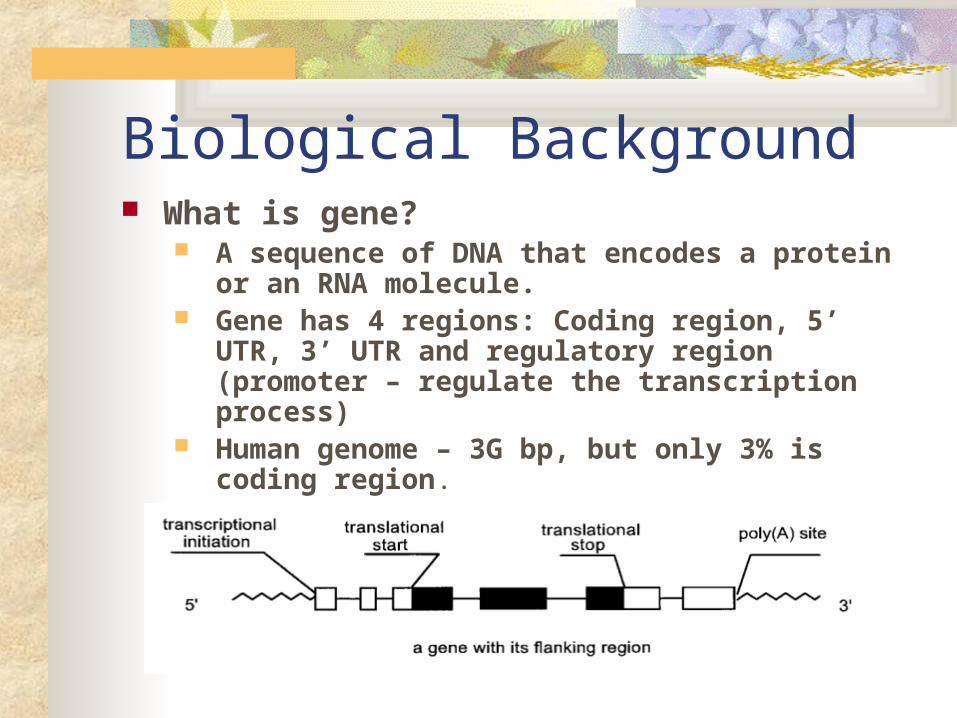
Biological Background What is gene?
A sequence of DNA that encodes a protein or an RNA molecule.
Gene has 4 regions: Coding region, 5’ UTR, 3’ UTR and regulatory region (promoter – regulate the transcription process)
Human genome – 3G bp, but only 3% is coding region.

Central Dogma Central Dogma- process where DNA
sequence generates a protein Transcription & Translation
Promoter – responsible for initiation and regulation of transcription
RNA-polymerase binds to a TATA base sequence in promoter region

Central Dogma

Promoter Region Core Promoter –
TATA-box Initiator (Inr) Downstream promoter element
3 types of core promoter TATA-box TATA-less, Inr-containing Inr + DPE
Upstream promoter elements TSS -where transcription starts
on DNA
The biology of eukaryotic promoter prediction – a review by Pedersen, A.G. et. al.

Outline of Presentation Biological Background Gene Finding Promoter Recognition Dragon Promoter Finder Open Problem and Future Research New Algorithm Conclusion

What is Gene Finding? Generate predictions of gene locations from
primary genomic sequence (DNA sequence) by computational methods.
Task of gene finding – separate the coding regions, non-coding regions and intergenic regions. Input: A seq of DNA, X = x1x2x…xn, where xi
belongs to {A, C, G, T} Output: Correct labeling of each element in X as a
belonging to CR, NCR, Intergenic Region

Gene Finding 3 major kinds of gene finding strategies:
Content-based – overall properties of the sequence when making predictions
Site-based – make use of presence or absence of a specific sequence, pattern or consensus
Comparative – sequence homology (database searching)
Combinatorial approach - GeneMachine GRAIL, FGENEH, MZEF, GenScan, GeneID,
GeneParser, HMMgene and so on.

Gene Finding – Open Problems Overlapping genes – no existing method
that can deal with this problem Alternative splicing, alternative
transcription/translation problem Sequencing errors Difficult to identify promoter region (PR)
& polyA (high true pos + high false pos)

Outline of Presentation Biological Background Gene Finding Promoter Recognition Dragon Promoter Finder Open Problem and Future Research New Algorithm Conclusion

Promoter Recognition Accurate PR can help to:
Detect a respective gene more easily Determine the 5’ ends of the respective gene
more precisely Localize the regions that contain numerous
different transcription control components Developing a perfect predictive model of
PR is challenging

Main Approach to PR Pattern-driven strategy
1. Collect a set of real binding sites to build characteristics definition, representation, pattern or profile from them
2. Recognition of individual potential binding sites by using their characteristic profiles
3. Assembling the candidates’ binding sites following some descriptions and rules about how these arrangements should be done.

Problem: Given a collection of known binding sites,
how to develop a representation of those sites, which is useful to search for them in new sequence? Consensus sequences Positional Weight Matrices (PWM) Hidden Markov profiles Multilayer neural networks and so on

Promoter Recognition Program Statistical approach + artificial intelligence
techniques - Dragon Promoter Finder (DPF) PromoterInspector Promoter 2.0

Accuracy Metric for PR
A common measure of prediction accuracy
Sensitivity Specificity
TP TNSE = ——— SP = ——— TP + FN TN + FP
Evaluation largely influenced by training set and test sets

Prediction of Promoter2 x 2 contingency table

Example of Prediction - DPFPromoter positions - exact positions of the TSS
2360, 2585, 4125, 5026, 5734, 7090, 8567, 10641, -2700, -12561, -12855
PREDICTED TRANSCRIPTION START SITES:gi_59865_emb_X02138.1_HEHSV1SU Herpes simplex virus type 1 _HSV1_ short unique region DNA
Sequence length: 12979 # of bases: A=2286, C=4271, G=4078, T=2344 Predicted TSS
Forward strand 4125 5733 7093 8567 10641 # of guesses = 5
Reverse complement strand -12561 -2698 # of guesses = 2

MeasurementDragon Promoter Finder, BIC-KRDL Singapore
Exp. positive negativepositive 7 4negative 0 6479
Pred.
SE = 7/11 = 0.64
SP = 6479/6479 = 1

Outline of Presentation Biological Background Gene Finding Promoter Recognition Dragon Promoter Finder Open Problem and Future Research New Algorithm Conclusion

Dragon Promoter Finder -Introduction Dragon Promoter Finder( DPF)
locates RNA polymerase II promoters in DNA sequences of vertebrates
predicts Transcription Start Site (TSS) positions. strand specific Components:
nonlinear promoter recognition models signal procession artificial neural networks (ANNs ) sensors.

Introduction (cont) The latest version
Dragon Promoter Finder Ver. 1.3
Main difference in new version models are now specialized for C+G-rich and
for C+G-poor sequences.

Structure
Overall Model comprises a collection of a number of basic
models Basic Model
made up of two sub-models, A and B trained for different ranges of system sensitivity trained separately for the best performance.
Sub-Model

Overall Model

Basic Model
A composite collection of basic models Possess identical structure Trained for narrow specificity range. Data procession in each model is analogous.

Basic Model

Sub-model

Sub-model Three Sensors
Specific functional regions of a gene: promoter, coding-exon, intron
Represented as positional distributions of overlapping pentamers
ANNs

Sensors Pentamers :
All sequences of 5 consecutive nucleotides. AAAAA,AAAAC,AAAAG…… 4^5=1024 pentamers Selected the most significant 256 pentamers from 1024
pentamers according to statistical relevance
Positional weight matrices (PWM): The positional distribution of selected pentamers Generate PWMs for each of the 3 functional groups,
promoter, exon & intron, by counting the frequencies of all selected pentamers at each position.

How to analyze the content of a data window: Sequence W=n1n2…nL-1nL, ni belongs to{A, C, G, T}
Sequence P of successive overlapping pentamers pj:
P = p1p2… pL–5pL–4.
iji
ijiij
ijij
L
iij
j
L
iij
ij
ppif
ppifffp
f
fpS
,0
,
,max
,,
4
1,
4
1,
: The jth pentamer at position i
: The frequency of the jth pentamer at position i
ijp
jif ,
S = score for each data window
The higher the s, the more likely the data window represents the respective functional region.
These scores are input to nonlinear signal processing block (SPB)
Output from SPB is then input to ANN

ANNs Inputs: scores (outputs of sensors) A multi-sensor integration. Trained by the Bayesian regularization method to
separate promoter regions from the non-promoter regions.
The threshold that best separated promoters from non promoter was selected
ANN output > threshold promoter region + TSS at a position 50bp before the data window’s end

Evaluation
Successfully recognize both CpG island-related and CpG island-nonrelated promoters.
Its performance on several large sets(A,B,and human chromosome 22) is reasonably consistent
On the average, its expected maximum sensitivities is approximately 66 percent.
In general, the DPF produces many times fewer FP predictions than comparative systems at the same sensitivity level.

Comparison

Outline of Presentation Biological Background Gene Finding Promoter Recognition Dragon Promoter Finder Open Problem and Future Research New Algorithm Conclusion

Open Problem & Future Research Open problem:
Lack of biological information on transcription process
Characteristics of promoter -> low ratio of accuracy
Future research work: Designing specific algorithm for either classes of
promoters or species-specific promoters Comparative sequence analysis Combinatorial approach Data mining tools

Outline of Presentation Biological Background Gene Finding Promoter Recognition Dragon Promoter Finder Open Problem and Future Research New Algorithm Conclusion

Gene Recognition Algorithm
Using Dynamic Programming Approach Presented by: Yin Jun

Dynamic Programming Algorithm
Existing Dynamic Programming Algorithm for Gene Finding
Snyder and Stormo’s method GeneParser
Solovyev et al’s method FGENEH
MORGAN’s DP algorithm

Goal of those Algorithm
1. Divide DNA sequence into alternate intron and exon regions.
2. Define a score for each kind of division. Try to find a kind of division which has the maximum score. The higher the score, the better the division.

Advantage and Disadvantage of Snyder and Stormo’s algorithm Advantage
the donor and the acceptor site HMM hidden status
Disadvantage Cannot recognize promoter 3-mer based

Our Algorithm
Combine the ideas of “Dragon Promoter Finder” and “Snyder and Stormo’s algorithm”
Can deal with promoters Use pentamer instead of 3-mer, more
efficient Dynamic Programming

Training Phase
Pentamer – 5 consecutive bases For example: “ACGGT” There are 45=1024 different kind of
pentamers Divide a DNA sequence into pentamers From training data, we can obtain the
probability for each kind of pentamer to become a promoter, an intron or an exon

Probability Table
Pentamer promoter intron Exon
A: ACGGT 0.13 0.20 0.67
B: CGATA 0.10 0.44 0.46
C: AUGCC 0.87 0.07 0.06
D: TAGTG 0.24 0.49 0.27

Principle of Division (1) Good (red: promoter; green: intron; blue:
exon)
Bad (low sum of probability)
C AA B B C BC D D D
C AA B B C BC D D D

Principle of Division (2) Good (red: promoter; green: intron; blue:
exon)
Bad (too frequent mutation)C AA B B C BC D D D
C AA B B C BC D D D

Mutation Penalty M(x, x) should be 0, x {1, 2, 3}∈
1: promoter 2: intron 3: exon
Example
To
From1 2 3
1 0 4.1 4.4
2 8 0 2.6
3 7.1 3.2 0

Notation P(p, r) – Probability for pentamer p belongs
region r Obtain from training data
M(s, t) – Mutation penalty Parameters to specify
pi (1≤i≤n) – The i th pentamer in the DNA sequence Input data (testing data)
a(pi) – Region assignment result; a(pi) {1, 2, 3}∈ Output data

Score Function For division assignment a, its score is
We use dynamic programming algorithm to find the best division assignment, whose score is the highest
n
iii
n
iii papaMpapPaS
21
1
))(),(())(,()(

Bases Let F(i, j, s, t) be the optimal score for the
consecutive segment of pentamers from i th to j th, where i th pentamer is assigned region s, j th pentamer is assigned region t
Bases
0),,1,( tsiiF
),(),,,( spPssiiF i

Recursive Definition Recursive Definition
Finally, we get F(1, n, s, t) where s, t {1, ∈2, 3}
Pick up the highest score from the 9 scores
)),(),,,1(),,,((max),,,(,,
vuMtvjkFuskiFtsjiFvuk

Time Complexity There are 9n2/2=O(n2) entries in the
dynamic programming table Filling each entry needs average n/2=O(n)
time The total time complexity is O(n3)

Outline of Presentation Biological Background Gene Finding Promoter Recognition Dragon Promoter Finder Open Problem and Future Research New Algorithm Conclusion

Conclusion Significant achievement in promoter
recognition technique & algorithms contributes to major advances in gene finding.
There is still room for improvement in promoter recognition.
A new algorithm is proposed for gene recognition.

















