60
De novo identification of repeat families in large genomes Alkes L. Price, Neil C. Jones and Pavel A. Pevzner June 28, 2005
| Date post: | 17-Dec-2015 |
| Category: |
Documents |
| Upload: | dwight-paul |
| View: | 216 times |
| Download: | 0 times |

De novo identification of repeat families in large genomes
Alkes L. Price, Neil C. Jones and Pavel A. Pevzner
June 28, 2005

What is a repeat family?
A repeat family is a collection of similar sequences which appear many times in a genome.
For example, the Alu repeat family has over 1 million approximate occurrences in the human genome:
Alu Alu Alu Alu Alu

Identifying repeat families: problem formulation
Alu Alu Alu Alu Alu
INPUT:
Genome containing approximate Alu occurrences
OUTPUT:
282bp Alu consensus sequence
GGCCGGGCGCGGTGGCTCACG………..GCGAGACTCCGTCTC
+ consensus sequences of all other repeat families in genome

Identifying repeat families: an easy problem?Alu Alu Alu Alu Alu
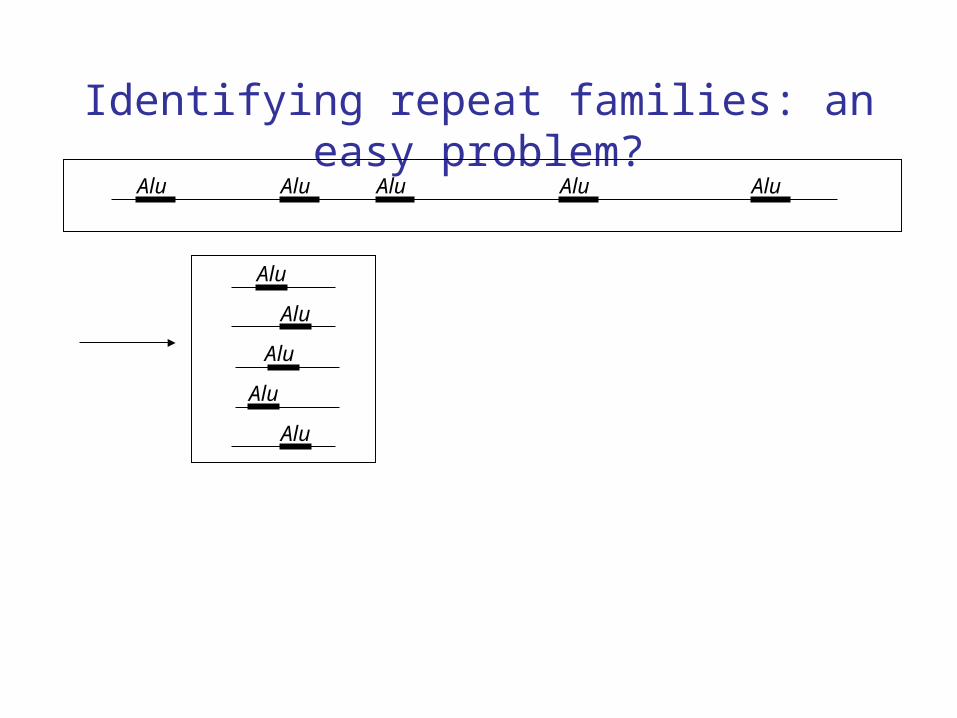
Identifying repeat families: an easy problem?Alu Alu Alu Alu Alu
Alu
Alu
Alu
Alu
Alu

Identifying repeat families: an easy problem?Alu Alu Alu Alu Alu
Alu
Alu
Alu
Alu
Alu
Alu
Alu
Alu
Alu
Alu
Alu consensus
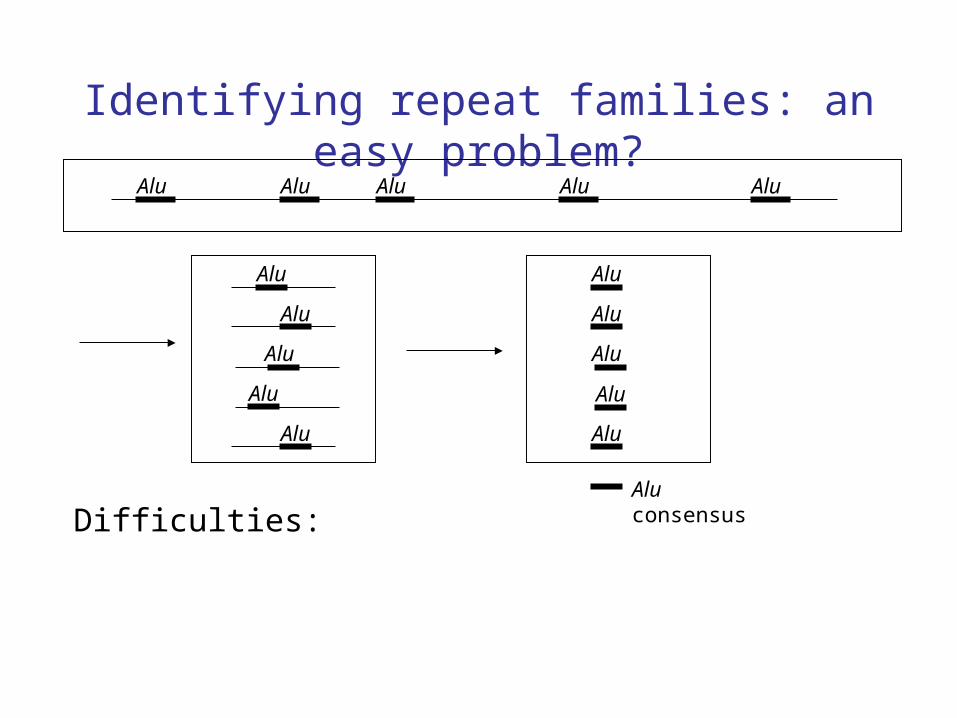
Identifying repeat families: an easy problem?Alu Alu Alu Alu Alu
Alu
Alu
Alu
Alu
Alu
Alu
Alu
Alu
Alu
Alu
Alu consensus
Difficulties:

Identifying repeat families: an easy problem?Alu Alu Alu Alu Alu
Alu
Alu
Alu
Alu
Alu
Alu
Alu
Alu
Alu
Alu
Alu consensus
Difficulties:• Regions containing repeat occurrences are not known a priori
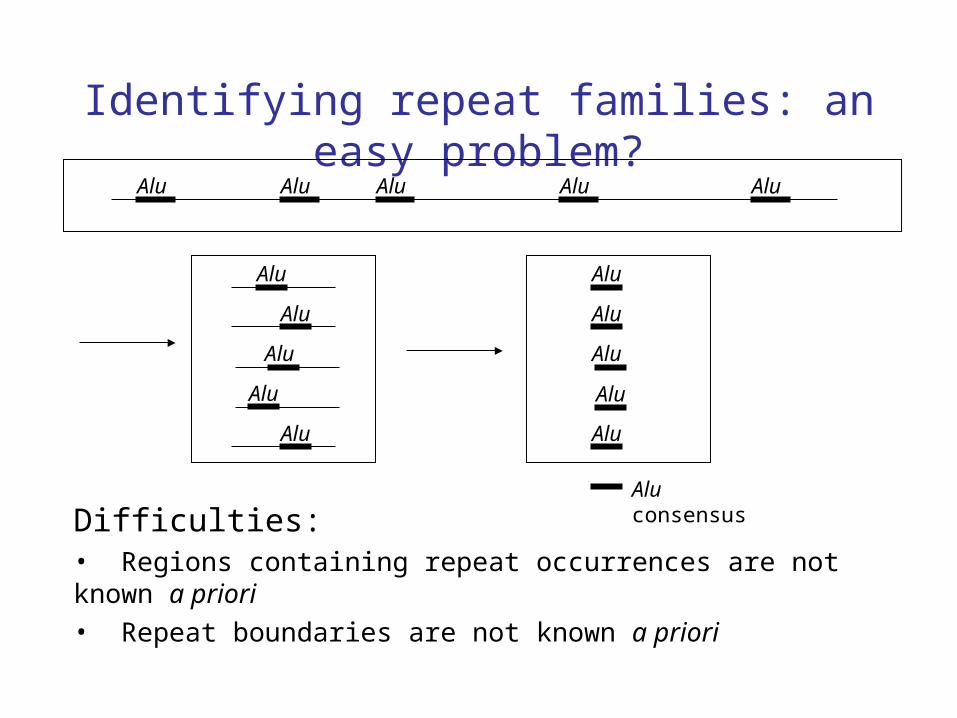
Identifying repeat families: an easy problem?Alu Alu Alu Alu Alu
Alu
Alu
Alu
Alu
Alu
Alu
Alu
Alu
Alu
Alu
Alu consensus
Difficulties:• Regions containing repeat occurrences are not known a priori
• Repeat boundaries are not known a priori
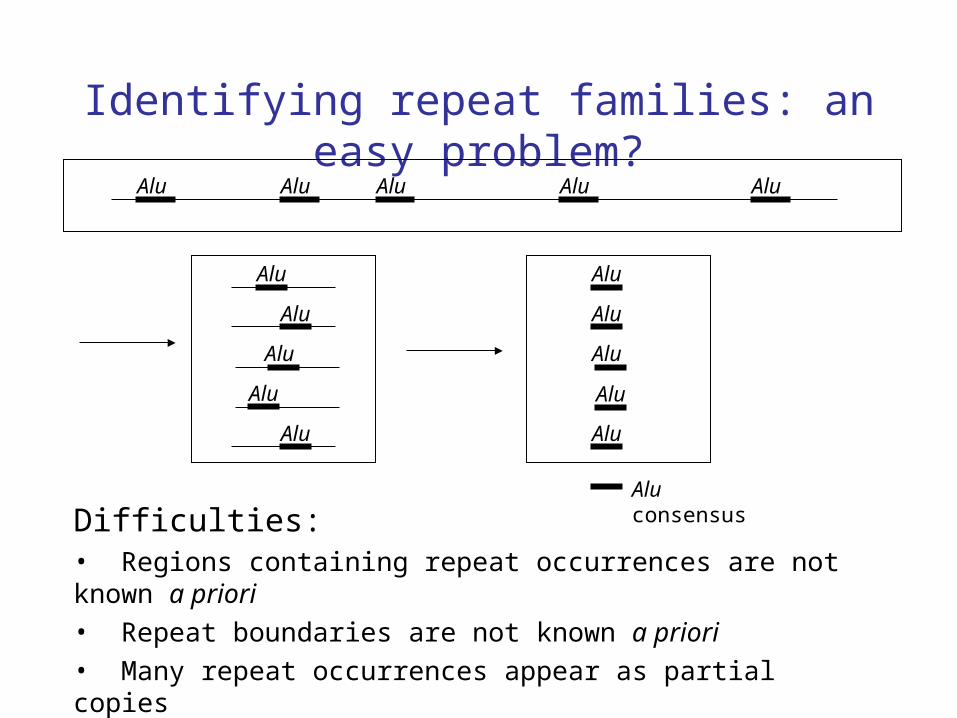
Identifying repeat families: an easy problem?Alu Alu Alu Alu Alu
Alu
Alu
Alu
Alu
Alu
Alu
Alu
Alu
Alu
Alu
Alu consensus
Difficulties:• Regions containing repeat occurrences are not known a priori
• Repeat boundaries are not known a priori
• Many repeat occurrences appear as partial copies

Identifying repeat families: a difficult problem
“The problem of automated repeat sequence family classification is inherently messy and ill-defined and does not appear to be amenable to a clean algorithmic attack.”
Bao and Eddy, 2002
In this talk, we present a simple and efficient algorithm for solving this problem.

Why is identifying repeat families important?
• Genome rearrangements (Kazazian, 2004)
• Drift to new biological function (Kidwell and Lisch, 2001)
• Increased rate of evolution under stress (Capy et al, 2000)
1. Repeats are biologically meaningful
Repeats are drivers of genome evolution (Kazazian, 2004) which can play a beneficial (rather than parasitic) role (Holmes, 2002). In particular, repeats have been implicated in

Why is identifying repeat families important?
• Repeats need to be masked prior to performing most single-species or multi-species analyses.
“Every time we compare two species that are closer to each other than either is to humans, we get nearly killed by unmasked repeats.”
Webb Miller (personal communication)
2. Repeat masking

Why is identifying repeat families important?
• Repeats need to be masked prior to performing most single-species or multi-species analyses.
GENE1
GENE2

Why is identifying repeat families important?
• If repeat families are known, repeats can be masked using RepeatMasker (http://www.repeatmasker.org).
GENE1
GENE2

Why is identifying repeat families important?
•If repeat families are known …
GENE1
GENE2

Identifying repeat families: manual approaches
• For widely studied genomes such as human and mouse, libraries of repeat families have been manually curated:– Repbase Update library (http://www.girinst.org)– RepeatMasker library (http://www.repeatmasker.org)

Identifying repeat families: algorithmic approaches
• Many, many new genomes are being assembled. How to identify the repeat families present in these genomes? Clearly, algorithmic approaches are needed.

Identifying repeat families: algorithmic approaches
All existing algorithms for de novo identification of repeat families rely on a set of pairwise similarities:
• Single-linkage clustering (Agarwal and States, 1994)• REPuter (Kurtz et al., 2000)• RepeatFinder (Volfovsky et al., 2001)• RECON (Bao and Eddy, 2002)• RepeatGluer (Pevzner et al., 2004)• PILER (Edgar and Myers, 2005)

Identifying repeat families: algorithmic approaches
Disadvantages of using pairwise similarities:
• Computational intractability human genome: ~106 Alus => ~1012 pairwise alignments
• Difficulty defining repeat boundaries “Local sequence alignments do not usually correspond to the biological boundaries … Difficulty in defining element boundaries causes problems in clustering related elements into families.” Bao and Eddy, 2002

Identifying repeat families: algorithmic approaches
Disadvantages of using pairwise similarities:
• Computational intractability
• Difficulty defining repeat boundaries
Our RepeatScout algorithm uses an efficient method of similarity search which enables a rigorous definition of repeat boundaries.

RepeatScout: the main idea
Consider a repeat family with many occurrences in a genome:
Equivalently, we have:
TAGCACCTTAGGGCGTCTCGCAACGTCTGCCCACGAACGTTAATCAGTAA
GATTATCATGAAGCGCTTCGCAACGTCTGCAGCTGTCCAGACCGCTGTCA
TATATCCGGTAATCGCCCCGCAACGTCTGCTAACGGGCGTACGGTCGAAT
TGACCTGCTCAGGAGCCTTGCAACGTCTGCTCGCGGATGTGTATGCACGC
ATCCATGCTCGGTATGAATCCAACGTCTGCTCATGGACATCTCATGACGT
CGATCCTCTGAGGCACCTCACAACGTCTGCTCACTGACGCACGGTTGCTG

RepeatScout: the main ideaTAGCACCTTAGGGCGTCTCGCAACGTCTGCCCACGAACGTTAATCAGTAA
GATTATCATGAAGCGCTTCGCAACGTCTGCAGCTGTCCAGACCGCTGTCA
TATATCCGGTAATCGCCCCGCAACGTCTGCTAACGGGCGTACGGTCGAAT
TGACCTGCTCAGGAGCCTTGCAACGTCTGCTCGCGGATGTGTATGCACGC
ATCCATGCTCGGTATGAATCCAACGTCTGCTCATGGACATCTCATGACGT
CGATCCTCTGAGGCACCTCACAACGTCTGCTCACTGACGCACGGTTGCTG
Consensus: ?

RepeatScout: the main ideaTAGCACCTTAGGGCGTCTCGCAACGTCTGCCCACGAACGTTAATCAGTAA
GATTATCATGAAGCGCTTCGCAACGTCTGCAGCTGTCCAGACCGCTGTCA
TATATCCGGTAATCGCCCCGCAACGTCTGCTAACGGGCGTACGGTCGAAT
TGACCTGCTCAGGAGCCTTGCAACGTCTGCTCGCGGATGTGTATGCACGC
ATCCATGCTCGGTATGAATCCAACGTCTGCTCATGGACATCTCATGACGT
CGATCCTCTGAGGCACCTCACAACGTCTGCTCACTGACGCACGGTTGCTG
Consensus: ?

RepeatScout: the main ideaTAGCACCTTAGGGCGTCTCGCAACGTCTGCCCACGAACGTTAATCAGTAA
GATTATCATGAAGCGCTTCGCAACGTCTGCAGCTGTCCAGACCGCTGTCA
TATATCCGGTAATCGCCCCGCAACGTCTGCTAACGGGCGTACGGTCGAAT
TGACCTGCTCAGGAGCCTTGCAACGTCTGCTCGCGGATGTGTATGCACGC
ATCCATGCTCGGTATGAATCCAACGTCTGCTCATGGACATCTCATGACGT
CGATCCTCTGAGGCACCTCACAACGTCTGCTCACTGACGCACGGTTGCTG
Consensus: CAACGTCTGC
Idea: greedily extend 1 bp at a time from short l-mer seed

RepeatScout: the main ideaTAGCACCTTAGGGCGTCTCGCAACGTCTGCCCACGAACGTTAATCAGTAA
GATTATCATGAAGCGCTTCGCAACGTCTGCAGCTGTCCAGACCGCTGTCA
TATATCCGGTAATCGCCCCGCAACGTCTGCTAACGGGCGTACGGTCGAAT
TGACCTGCTCAGGAGCCTTGCAACGTCTGCTCGCGGATGTGTATGCACGC
ATCCATGCTCGGTATGAATCCAACGTCTGCTCATGGACATCTCATGACGT
CGATCCTCTGAGGCACCTCACAACGTCTGCTCACTGACGCACGGTTGCTG
Consensus: CAACGTCTGCT
Idea: greedily extend 1 bp at a time from short l-mer seed

RepeatScout: the main ideaTAGCACCTTAGGGCGTCTCGCAACGTCTGCCCACGAACGTTAATCAGTAA
GATTATCATGAAGCGCTTCGCAACGTCTGCAGCTGTCCAGACCGCTGTCA
TATATCCGGTAATCGCCCCGCAACGTCTGCTAACGGGCGTACGGTCGAAT
TGACCTGCTCAGGAGCCTTGCAACGTCTGCTCGCGGATGTGTATGCACGC
ATCCATGCTCGGTATGAATCCAACGTCTGCTCATGGACATCTCATGACGT
CGATCCTCTGAGGCACCTCACAACGTCTGCTCACTGACGCACGGTTGCTG
Consensus: CAACGTCTGCTC
Idea: greedily extend 1 bp at a time from short l-mer seed

RepeatScout: the main ideaTAGCACCTTAGGGCGTCTCGCAACGTCTGCCCACGAACGTTAATCAGTAA
GATTATCATGAAGCGCTTCGCAACGTCTGCAGCTGTCCAGACCGCTGTCAAGCTGTCCAGACCGCTGTCA
TATATCCGGTAATCGCCCCGCAACGTCTGCTAACGGGCGTACGGTCGAAT
TGACCTGCTCAGGAGCCTTGCAACGTCTGCTCGCGGATGTGTATGCACGC
ATCCATGCTCGGTATGAATCCAACGTCTGCTCATGGACATCTCATGACGT
CGATCCTCTGAGGCACCTCACAACGTCTGCTCACTGACGCACGGTTGCTG
Consensus: CAACGTCTGCTCA
Idea: greedily extend 1 bp at a time from short l-mer seedDiscard a sequence after it stops aligning to consensus

RepeatScout: the main ideaTAGCACCTTAGGGCGTCTCGCAACGTCTGCCCACGAACGTTAATCAGTAA
GATTATCATGAAGCGCTTCGCAACGTCTGCAGCTGTCCAGACCGCTGTCAAGCTGTCCAGACCGCTGTCA
TATATCCGGTAATCGCCCCGCAACGTCTGCTAACGGGCGTACGGTCGAAT
TGACCTGCTCAGGAGCCTTGCAACGTCTGCTCGCGGATGTGTATGCACGC
ATCCATGCTCGGTATGAATCCAACGTCTGCTCATGGACATCTCATGACGT
CGATCCTCTGAGGCACCTCACAACGTCTGCTCACTGACGCACGGTTGCTG
Consensus: CAACGTCTGCTCAC
Idea: greedily extend 1 bp at a time from short l-mer seedDiscard a sequence after it stops aligning to consensus

RepeatScout: the main ideaTAGCACCTTAGGGCGTCTCGCAACGTCTGCCCACGAACGTTAATCAGTAA
GATTATCATGAAGCGCTTCGCAACGTCTGCAGCTGTCCAGACCGCTGTCAAGCTGTCCAGACCGCTGTCA
TATATCCGGTAATCGCCCCGCAACGTCTGCTAACGGGCGTACGGTCGAAT
TGACCTGCTCAGGAGCCTTGCAACGTCTGCTCGCGGATGTGTATGCACGC
ATCCATGCTCGGTATGAATCCAACGTCTGCTCATGGACATCTCATGACGT
CGATCCTCTGAGGCACCTCACAACGTCTGCTCACTGACGCACGGTTGCTG
Consensus: CAACGTCTGCTCACG
Idea: greedily extend 1 bp at a time from short l-mer seedDiscard a sequence after it stops aligning to consensus

RepeatScout: the main ideaTAGCACCTTAGGGCGTCTCGCAACGTCTGCCCACGAACGTTAATCAGTAA
GATTATCATGAAGCGCTTCGCAACGTCTGCAGCTGTCCAGACCGCTGTCAAGCTGTCCAGACCGCTGTCA
TATATCCGGTAATCGCCCCGCAACGTCTGCTAACGGGCGTACGGTCGAAT
TGACCTGCTCAGGAGCCTTGCAACGTCTGCTCGCGGATGTGTATGCACGC
ATCCATGCTCGGTATGAATCCAACGTCTGCTCATGGACATCTCATGACGT
CGATCCTCTGAGGCACCTCACAACGTCTGCTCACTGACGCACGGTTGCTG
Consensus: CAACGTCTGCTCACGG
Idea: greedily extend 1 bp at a time from short l-mer seedDiscard a sequence after it stops aligning to consensus

RepeatScout: the main ideaTAGCACCTTAGGGCGTCTCGCAACGTCTGCCCACGAACGTTAATCAGTAA
GATTATCATGAAGCGCTTCGCAACGTCTGCAGCTGTCCAGACCGCTGTCAAGCTGTCCAGACCGCTGTCA
TATATCCGGTAATCGCCCCGCAACGTCTGCTAACGGGCGTACGGTCGAAT
TGACCTGCTCAGGAGCCTTGCAACGTCTGCTCGCGGATGTGTATGCACGC
ATCCATGCTCGGTATGAATCCAACGTCTGCTCATGGACATCTCATGACGT
CGATCCTCTGAGGCACCTCACAACGTCTGCTCACTGACGCACGGTTGCTG
Consensus: CAACGTCTGCTCACGGA
Idea: greedily extend 1 bp at a time from short l-mer seedDiscard a sequence after it stops aligning to consensus

RepeatScout: the main ideaTAGCACCTTAGGGCGTCTCGCAACGTCTGCCCACGAACGTTAATCAGTAA
GATTATCATGAAGCGCTTCGCAACGTCTGCAGCTGTCCAGACCGCTGTCAAGCTGTCCAGACCGCTGTCA
TATATCCGGTAATCGCCCCGCAACGTCTGCTAACGGGCGTACGGTCGAAT
TGACCTGCTCAGGAGCCTTGCAACGTCTGCTCGCGGATGTGTATGCACGC
ATCCATGCTCGGTATGAATCCAACGTCTGCTCATGGACATCTCATGACGT
CGATCCTCTGAGGCACCTCACAACGTCTGCTCACTGACGCACGGTTGCTG
Consensus: CAACGTCTGCTCACGGAC
Idea: greedily extend 1 bp at a time from short l-mer seedDiscard a sequence after it stops aligning to consensus

RepeatScout: the main ideaTAGCACCTTAGGGCGTCTCGCAACGTCTGCCCACGAACGTTAATCAGTAA
GATTATCATGAAGCGCTTCGCAACGTCTGCAGCTGTCCAGACCGCTGTCAAGCTGTCCAGACCGCTGTCA
TATATCCGGTAATCGCCCCGCAACGTCTGCTAACGGGCGTACGGTCGAAT
TGACCTGCTCAGGAGCCTTGCAACGTCTGCTCGCGGATGTGTATGCACGC
ATCCATGCTCGGTATGAATCCAACGTCTGCTCATGGACATCTCATGACGT
CGATCCTCTGAGGCACCTCACAACGTCTGCTCACTGACGCACGGTTGCTG
Consensus: CAACGTCTGCTCACGGACG
Idea: greedily extend 1 bp at a time from short l-mer seedDiscard a sequence after it stops aligning to consensus

RepeatScout: the main ideaTAGCACCTTAGGGCGTCTCGCAACGTCTGCCCACGAACGTTAATCAGTAA
GATTATCATGAAGCGCTTCGCAACGTCTGCAGCTGTCCAGACCGCTGTCAAGCTGTCCAGACCGCTGTCA
TATATCCGGTAATCGCCCCGCAACGTCTGCTAACGGGCGTACGGTCGAAT
TGACCTGCTCAGGAGCCTTGCAACGTCTGCTCGCGGATGTGTATGCACGC
ATCCATGCTCGGTATGAATCCAACGTCTGCTCATGGACATCTCATGACGT
CGATCCTCTGAGGCACCTCACAACGTCTGCTCACTGACGCACGGTTGCTG
Consensus: CAACGTCTGCTCACGGACGT
Idea: greedily extend 1 bp at a time from short l-mer seedDiscard a sequence after it stops aligning to consensus

RepeatScout: the main ideaTAGCACCTTAGGGCGTCTCGCAACGTCTGCCCACGAACGTTAATCAGTAATAATCAGTAA
GATTATCATGAAGCGCTTCGCAACGTCTGCAGCTGTCCAGACCGCTGTCAAGCTGTCCAGACCGCTGTCA
TATATCCGGTAATCGCCCCGCAACGTCTGCTAACGGGCGTACGGTCGAATACGGTCGAAT
TGACCTGCTCAGGAGCCTTGCAACGTCTGCTCGCGGATGTGTATGCACGCGTATGCACGC
ATCCATGCTCGGTATGAATCCAACGTCTGCTCATGGACATCTCATGACGTCTCATGACGT
CGATCCTCTGAGGCACCTCACAACGTCTGCTCACTGACGCACGGTTGCTGACGGTTGCTG
Consensus: CAACGTCTGCTCACGGACGT
Idea: greedily extend 1 bp at a time from short l-mer seedDiscard a sequence after it stops aligning to consensusStop extending when most sequences no longer align

RepeatScout: the main ideaTAGCACCTTAGGGCGTCTCGCAACGTCTGCCCACGAACGTTAATCAGTAATAATCAGTAA
GATTATCATGAAGCGCTTCGCAACGTCTGCAGCTGTCCAGACCGCTGTCAAGCTGTCCAGACCGCTGTCA
TATATCCGGTAATCGCCCCGCAACGTCTGCTAACGGGCGTACGGTCGAATCGAAT
TGACCTGCTCAGGAGCCTTGCAACGTCTGCTCGCGGATGTGTATGCACGCGTATGCACGC
ATCCATGCTCGGTATGAATCCAACGTCTGCTCATGGACATCTCATGACGTCTCATGACGT
CGATCCTCTGAGGCACCTCACAACGTCTGCTCACTGACGCACGGTTGCTGTGCTG
Consensus: CAACGTCTGCTCACGGACGTACGGT
Idea: greedily extend 1 bp at a time from short l-mer seedDiscard a sequence after it stops aligning to consensusStop extending when most sequences no longer alignNote: pairwise alignment is a poor boundary criteria.

RepeatScout: the main ideaTAGCACCTTATAGCACCTTAGGGCGTCTCGCAACGTCTGCCCACGAACGTTAATCAGTAATAATCAGTAA
GATTATCATGGATTATCATGAAGCGCTTCGCAACGTCTGCAGCTGTCCAGACCGCTGTCAAGCTGTCCAGACCGCTGTCA
TATATCCGGTTATATCCGGTAATCGCCCCGCAACGTCTGCTAACGGGCGTACGGTCGAATACGGTCGAAT
TGACCTGCTCTGACCTGCTCAGGAGCCTTGCAACGTCTGCTCGCGGATGTGTATGCACGCGTATGCACGC
ATCCATGCTCGGTATGAATCATCCATGCTCGGTATGAATCCAACGTCTGCTCATGGACATCTCATGACGTCTCATGACGT
CGATCCTCTGCGATCCTCTGAGGCACCTCACAACGTCTGCTCACTGACGCACGGTTGCTGACGGTTGCTG
Consensus: AGGCGCCTCGCAACGTCTGCTCACGGACGT
Idea: greedily extend 1 bp at a time from short l-mer seedDiscard a sequence “after it stops aligning to consensus”Stop extending “when most sequences no longer align”First extend right, then extend left in similar manner

Repeat boundaries: the objective function Let S1, …, Sn be strings containing occurrences of a repeat family which share a short l-mer seed.
We define the consensus sequence Q of the repeat family to be the sequence which maximizes
A(Q; S1, …, Sn) = ∑k a(Q, Sk) where
a(Q, Sk) is a fit-preferred alignment score

Repeat boundaries: the objective function Let S1, …, Sn be strings containing occurrences of a repeat family which share a short l-mer seed.
We define the consensus sequence Q of the repeat family to be the sequence which maximizes
A(Q; S1, …, Sn) = ∑k a(Q, Sk) – c |Q| where
a(Q, Sk) is a fit-preferred alignment score
c is a repeat frequency threshold

Repeat boundaries: the objective function
A(Q; S1, …, Sn) = ∑k a(Q, Sk) – c |Q|
Optimizing the objective function:
• Start with Q = short l-mer seed
• Greedily extend Q to the right (left) 1 bp at a time. Stop when + many consecutive iterations fail to improve upon the optimal Q.
The optimal Q defines the consensus sequence of the repeat family.
This provides a rigorous definition of repeat boundaries.

Repeat boundaries: the objective function
TAGCACCTTATAGCACCTTAGGGCGTCTCGCAACGTCTGCCCACGAACGTTAATCAGTAATAATCAGTAA
GATTATCATGGATTATCATGAAGCGCTTCGCAACGTCTGCAGCTGTCCAGACCGCTGTCAAGCTGTCCAGACCGCTGTCA
TATATCCGGTTATATCCGGTAATCGCCCCGCAACGTCTGCTAACGGGCGTACGGTCGAATACGGTCGAAT
TGACCTGCTCTGACCTGCTCAGGAGCCTTGCAACGTCTGCTCGCGGATGTGTATGCACGCGTATGCACGC
ATCCATGCTCGGTATGAATCATCCATGCTCGGTATGAATCCAACGTCTGCTCATGGACATCTCATGACGTCTCATGACGT
CGATCCTCTGCGATCCTCTGAGGCACCTCACAACGTCTGCTCACTGACGCACGGTTGCTGACGGTTGCTG
Consensus: AGGCGCCTCGCAACGTCTGCTCACGGACGT
Greedily extend right/left to optimize A(Q, S1, …, Sn)

RepeatScout: finding all repeat families
To find all repeat families in a genome, we could apply this procedure to extend all frequent l-mers.

RepeatScout: finding all repeat families
To find all repeat families in a genome, we could apply this procedure to extend all frequent l-mers.
However, each repeat family spawns a large number of frequent l-mers and could be repeatedly rediscovered.

RepeatScout: finding all repeat families
To find all repeat families in a genome, we could apply this procedure to extend all frequent l-mers.
However, each repeat family spawns a large number of frequent l-mers and could be repeatedly rediscovered.
To address this, we dynamically adjust l-mer frequencies to exclude contributions from repeat families we have already identified.

RepeatScout: postprocessing
We discard very short “repeat families” arising from spurious frequent l-mers.
We discard repeat families with less than 10 copies.
We may further wish to distinguish between• Low-complexity repeat families• Tandem repeat families• Multicopy exon families• Segmental duplication units• Transposon families

Results: the human Alu family
Alu Alu Alu Alu Alu
Input:
Genome containing approximate Alu occurrences
Desired Output: 282bp Alu consensus sequenceGGCCGGGCGCGGTGGCTCACG………..GCGAGACTCCGTCTC

Results: the human Alu family
Alu Alu Alu Alu Alu
Input:
Genome containing approximate Alu occurrences
Desired Output: 282bp Alu consensus sequenceGGCCGGGCGCGGTGGCTCACG………..GCGAGACTCCGTCTC
RepeatScout Output (on human X chr): 282bp sequenceGGCCGGGCGCGGTGGCTCACG………..GCGAGACTCCGTCTC

Results: C. briggsaeWe benchmarked RepeatScout using the 108Mb C. briggsae genome (Stein et al., 2003), which Stein et al. analyzed using the RECON algorithm (Bao and Eddy, 2002).
We ran RepeatMasker (http://www.repeatmasker.org) using either the RECON repeat library or the RepeatScout library as input, and compared the results:

Results: C. briggsae
RECON RepeatScout library library
2.0 Mb 23.1 Mb 4.8 Mb

Results: human, mouse, ratWe ran RepeatScout on human, mouse and rat X
chromosomes. We filtered out • Low-complexity repeat families
• Tandem repeat families
• Multicopy exon families
• Known segmental duplication units
We ran RepeatMasker using either the RepeatMasker library or the RepeatScout library as input, and compared the results:

Results: human X chromosome
RepeatMasker RepeatScout library library
8.3 Mb 53.5 Mb 2.4 Mb

Results: mouse X chromosome
RepeatMasker RepeatScout library library
5.3 Mb 47.6 Mb 3.3 Mb

Results: mouse X chromosome
RepeatMasker RepeatScout library library
5.3 Mb 47.6 Mb 3.3 Mb

Results: mouse X chromosome
Repbase Update RepeatScout library library
2.7 Mb 43.2 Mb 6.4 Mb
results presented in our paper

Results: mouse X chromosome
RepeatMasker RepeatScout library library
5.3 Mb 47.6 Mb 3.3 Mb
latest results

Running times
3.0 Mb
(human)
9.0 Mb
(human)
X chr
(human)
RECON 4 hours* 39 hours* --
RepeatScout 6 min† 21 min† 8 hours†
* on a single 1.7 GHz Intel Xeon processor
† on a single 0.5 GHz DEC Alpha processor

Future Directions
• Distinguish segmental duplications from transposons
• Unify fragmented repeat families• Improve sensitivity via inexact or noncontiguous l-mer seeds
• Run RepeatScout on entire mammalian genomes

RepeatScout web site
Google search on RepeatScout• RepeatScout source code and documentation• RepeatScout repeat libraries• Slides of this talk
Google search on RepeatScout

Acknowledgements
We are grateful to
• Lincoln Stein for providing RECON C. briggsae output.
• Evan Eichler for providing segmental duplication annotations for human, mouse and rat X chromosomes.
• Arian Smit, Robert Hubley and Brian Haas for testing RepeatScout and offering numerous helpful comments and suggestions.

















