12. G. L. Semenza, Drug Discov. Today 12, 853 (2007). 13. We thank members of the Fudan Molecular and Cell Biology Laboratory for valuable input; Y. Liu, X. Liu, and H. Zhu for assistance with histology; Z. Bao, L. Yang, Q. Shi, and G. Zhao for clinical samples; and S. Jackson for reading the manuscript. This work is supported by the 985 program from the Chinese Ministry of Education, State Key Development Programs of China (2009CB918401, 2006CB806700), National 863 Program of China (2006AA02A308), China NSF grants (30600112 and 30871255) and Shanghai Key Basic Research Projects (06JC14086, 07PJ14011, and 08JC1400900), and NIH grants (to K.-L.G. and Y.X.). Y. Xiong, K.-L. Guan, and S. Zhao are applying for a patent related to the work on permeable alpha-ketogluterate. Supporting Online Material www.sciencemag.org/cgi/content/full/324/5924/261/DC1 Materials and Methods Figs. S1 to S8 Table S1 15 January 2009; accepted 10 March 2009 10.1126/science.1170944 Demonstration of Genetic Exchange During Cyclical Development of Leishmania in the Sand Fly Vector Natalia S. Akopyants, 1 * Nicola Kimblin, 2 * Nagila Secundino, 2 Rachel Patrick, 2 Nathan Peters, 2 Phillip Lawyer, 2 Deborah E. Dobson, 1 Stephen M. Beverley, 1 † David L. Sacks 2 †‡ Genetic exchange has not been shown to be a mechanism underlying the extensive diversity of Leishmania parasites. We report here evidence that the invertebrate stages of Leishmania are capable of having a sexual cycle consistent with a meiotic process like that described for African trypanosomes. Hybrid progeny were generated that bore full genomic complements from both parents, but kinetoplast DNA maxicircles from one parent. Mating occurred only in the sand fly vector, and hybrids were transmitted to the mammalian host by sand fly bite. Genetic exchange likely contributes to phenotypic diversity in natural populations, and analysis of hybrid progeny will be useful for positional cloning of the genes controlling traits such as virulence, tissue tropism, and drug resistance. P arasitic protozoa of the genus Leishmania cause a spectrum of human diseases that pose serious public health challenges for prevention, diagnosis, and treatment. The diver- sity of Leishmania species, with more than 20 currently recognized, is thought to have arisen by gradual accumulation of divergent mutations rather than by sexual recombination. Tibayrenc et al. (1) have reported strong linkage disequilibrium in several Leishmania species and proposed that these parasites are essentially clonal. This notion must be reconciled, however, with the accumu- lating examples of naturally occurring strains that share genotypic markers from two recognized species and thereby provide circumstantial evi- dence for sexual recombination (2–4). Genetic exchange has been documented for the other trypanosomatids that cause human disease. Hybrid genotypes were observed in tsetse flies during cotransmission of two strains of Trypano- soma brucei ( 5) and in mammalian cells after coinfection with two clones of Trypanosoma cruzi differing in drug-resistance markers (6). Using drug resistance markers, we provide evi- dence for genetic exchange in Leishmania major and discuss the implications of these findings to Leishmania biology and experimen- tal analysis. One parental clone, LV39c5(HYG), was de- rived from strain LV39 clone 5 (MHOM/SU/59/P) and was heterozygous for an allelic replacement of the LPG5A on chromosome 24 by a hygromycin B–resistance cassette (LPG5A/LPG5A::∆HYG) (7). The second parental clone, FV1(SAT), was derived from NIH Friedlin clone V1 (MHOM/ IL/80/FN) and bore a heterozygous nourseothricin– resistance (SAT) marker, integrated along with a linked firefly luciferase (LUC) reporter gene into one allele of the ~24 rRNA cistrons located on chromosome 27 (8) (+/SSU::SAT-LUC). These strains were chosen as they are phenotypically identical to their respective parental wild-type (WT) virulent L. major ; whereas the markers were chosen because they are functionally independent (9). The target gene modifications were chosen because they caused no effect on normal growth in vitro or in mouse infections (10), and epistatic interac- tions were not anticipated between these alleles. Multiple attempts to generate hybrid para- sites resistant to both antibiotics during in vitro co- culture of the parental lines were unsuccessful ( 11). The parental clones were tested for their ability to generate parasites resistant to both drugs during coinfection in the sand fly. The growth of each parental line in Phlebotomus duboscqi, a natural vector of L. major , is shown in fig. S1. Promas- tigotes of each parent survived the initial period of blood-meal digestion and excretion (days 1 to 6) and underwent metacyclogenesis at a comparable frequency (20 to 60%), although the FV1(SAT) parent established and maintained a higher intensity of infection by a factor of 3 to 4. The parental clones were tested for their ability to generate doubly drug–resistant parasites dur- ing coinfection in the sand fly. Flies were fed through a membrane on mouse blood contain- ing 3 and 1 × 10 6 /ml of the LV39c5(HYG) and FV1(SAT) lines, respectively, each obtained from log-phase cultures and extensively washed to remove antibiotics. A total of 102 flies from four independent coinfection experiments were dis- sected 13 to 16 days postinfection; at this time, they harbored mature infections with an average of 39,400 T 14,700 promastigotes per midgut. Flies cannot be maintained under aseptic con- ditions, and more than half of the cultures es- tablished from the midgut parasites were lost to fungal contamination during the subsequent 1 to 2 weeks of culture. In the remaining cultures, 12 (26%) grew out promastigotes that were resistant to both drugs. Clonal lines were generated from nine of the doubly drug–resistant populations, and the genotypes and phenotypes of one or two clones from each culture were determined (sum- marized in Table 1). Polymerase chain reaction (PCR) tests with primers specific for the parental markers showed that all doubly drug–resistant clones tested con- tained both the HYG and SAT drug-resistance genes (Fig. 1A, Table 1, and table S3). Controls showed that the marker loci had not rearranged during hybrid formation but had maintained their location within the LPG5A or SSU rRNA loci, respectively (fig. S2). These data indicated that the doubly drug–resistant clonal lines were genetic hybrids. To confirm that hybrid forma- tion was compatible with transmission to the mam- malian host, coinfected sand flies were allowed to bite and to induce lesions in BALB/c mice. Two doubly drug–resistant clonal lines, desig- nated 6.16.E8 and 6.14.F9, were recovered from the eight dermal lesions examined and were found to be similar to those directly recovered from insects, as discussed below (Table 1). Co- injection of both parental lines into mouse ears by needle never led to the recovery of doubly drug–resistant parasites from the 20 dermal le- sions examined, each containing >2.4 × 10 7 amastigotes, which suggested that the hybrids selected after transmission by the coinfected sand flies were generated in the fly and not in the mammalian host. We examined the segregation of loci not linked to chromosomes 24 and 27, the location of the HYG and SAT markers, respectively. We used single-nucleotide polymorphisms (SNPs) developed from comparisons of the terminal ~30-kb chromosomal regions encompassing the SCG genes located on L. major chromosomes 2, 1 Department of Molecular Microbiology, Washington Univer- sity School of Medicine, St. Louis, MO 63110, USA. 2 Labora- tory of Parasitic Diseases, National Institute of Allergy and Infectious Diseases, NIH, Bethesda, MD 20892, USA. *†These authors contributed equally to this work. ‡To whom correspondence should be addressed. E-mail: [email protected]www.sciencemag.org SCIENCE VOL 324 10 APRIL 2009 265 REPORTS on April 10, 2009 www.sciencemag.org Downloaded from
Transcript
12. G. L. Semenza, Drug Discov. Today 12, 853 (2007).13. We thank members of the Fudan Molecular and Cell
Biology Laboratory for valuable input; Y. Liu, X. Liu, andH. Zhu for assistance with histology; Z. Bao, L. Yang,Q. Shi, and G. Zhao for clinical samples; and S. Jacksonfor reading the manuscript. This work is supported by the985 program from the Chinese Ministry of Education,State Key Development Programs of China
(2009CB918401, 2006CB806700), National 863Program of China (2006AA02A308), China NSF grants(30600112 and 30871255) and Shanghai Key BasicResearch Projects (06JC14086, 07PJ14011, and08JC1400900), and NIH grants (to K.-L.G. and Y.X.).Y. Xiong, K.-L. Guan, and S. Zhao are applying fora patent related to the work on permeablealpha-ketogluterate.
Supporting Online Materialwww.sciencemag.org/cgi/content/full/324/5924/261/DC1Materials and MethodsFigs. S1 to S8Table S1
15 January 2009; accepted 10 March 200910.1126/science.1170944
Demonstration of Genetic ExchangeDuring Cyclical Development ofLeishmania in the Sand Fly VectorNatalia S. Akopyants,1* Nicola Kimblin,2* Nagila Secundino,2 Rachel Patrick,2 Nathan Peters,2Phillip Lawyer,2 Deborah E. Dobson,1 Stephen M. Beverley,1† David L. Sacks2†‡
Genetic exchange has not been shown to be a mechanism underlying the extensive diversity ofLeishmania parasites. We report here evidence that the invertebrate stages of Leishmania arecapable of having a sexual cycle consistent with a meiotic process like that described for Africantrypanosomes. Hybrid progeny were generated that bore full genomic complements from bothparents, but kinetoplast DNA maxicircles from one parent. Mating occurred only in the sand flyvector, and hybrids were transmitted to the mammalian host by sand fly bite. Genetic exchangelikely contributes to phenotypic diversity in natural populations, and analysis of hybrid progenywill be useful for positional cloning of the genes controlling traits such as virulence, tissuetropism, and drug resistance.
Parasitic protozoa of the genus Leishmaniacause a spectrum of human diseases thatpose serious public health challenges for
prevention, diagnosis, and treatment. The diver-sity of Leishmania species, with more than 20currently recognized, is thought to have arisen bygradual accumulation of divergent mutations ratherthan by sexual recombination. Tibayrenc et al.(1) have reported strong linkage disequilibriumin several Leishmania species and proposed thatthese parasites are essentially clonal. This notionmust be reconciled, however, with the accumu-lating examples of naturally occurring strains thatshare genotypic markers from two recognizedspecies and thereby provide circumstantial evi-dence for sexual recombination (2–4). Geneticexchange has been documented for the othertrypanosomatids that cause human disease.Hybrid genotypes were observed in tsetse fliesduring cotransmission of two strains of Trypano-soma brucei (5) and in mammalian cells aftercoinfection with two clones of Trypanosomacruzi differing in drug-resistance markers (6).Using drug resistance markers, we provide evi-dence for genetic exchange in Leishmaniamajor and discuss the implications of thesefindings to Leishmania biology and experimen-tal analysis.
One parental clone, LV39c5(HYG), was de-rived from strain LV39 clone 5 (MHOM/SU/59/P)and was heterozygous for an allelic replacement ofthe LPG5A on chromosome 24 by a hygromycinB–resistance cassette (LPG5A/LPG5A::∆HYG)(7). The second parental clone, FV1(SAT), wasderived from NIH Friedlin clone V1 (MHOM/IL/80/FN) and bore a heterozygous nourseothricin–resistance (SAT) marker, integrated along with alinked firefly luciferase (LUC) reporter gene intoone allele of the ~24 rRNA cistrons located onchromosome 27 (8) (+/SSU::SAT-LUC). Thesestrains were chosen as they are phenotypicallyidentical to their respective parental wild-type (WT)virulent L. major; whereas the markers were chosenbecause they are functionally independent (9). Thetarget gene modifications were chosen becausethey caused no effect on normal growth in vitroor in mouse infections (10), and epistatic interac-tions were not anticipated between these alleles.
Multiple attempts to generate hybrid para-sites resistant to both antibiotics during in vitro co-culture of the parental lines were unsuccessful (11).The parental clones were tested for their ability togenerate parasites resistant to both drugs duringcoinfection in the sand fly. The growth of eachparental line in Phlebotomus duboscqi, a naturalvector of L. major, is shown in fig. S1. Promas-tigotes of each parent survived the initial periodof blood-meal digestion and excretion (days1 to 6) and underwent metacyclogenesis at acomparable frequency (20 to 60%), although theFV1(SAT) parent established and maintained ahigher intensity of infection by a factor of 3 to 4.The parental clones were tested for their ability
to generate doubly drug–resistant parasites dur-ing coinfection in the sand fly. Flies were fedthrough a membrane on mouse blood contain-ing 3 and 1 × 106/ml of the LV39c5(HYG) andFV1(SAT) lines, respectively, each obtained fromlog-phase cultures and extensively washed toremove antibiotics. A total of 102 flies from fourindependent coinfection experiments were dis-sected 13 to 16 days postinfection; at this time,they harbored mature infections with an averageof 39,400 T 14,700 promastigotes per midgut.Flies cannot be maintained under aseptic con-ditions, and more than half of the cultures es-tablished from the midgut parasites were lost tofungal contamination during the subsequent 1 to2 weeks of culture. In the remaining cultures, 12(26%) grew out promastigotes that were resistantto both drugs. Clonal lines were generated fromnine of the doubly drug–resistant populations,and the genotypes and phenotypes of one or twoclones from each culture were determined (sum-marized in Table 1).
Polymerase chain reaction (PCR) tests withprimers specific for the parental markers showedthat all doubly drug–resistant clones tested con-tained both the HYG and SAT drug-resistancegenes (Fig. 1A, Table 1, and table S3). Controlsshowed that the marker loci had not rearrangedduring hybrid formation but had maintainedtheir location within the LPG5A or SSU rRNAloci, respectively (fig. S2). These data indicatedthat the doubly drug–resistant clonal lines weregenetic hybrids. To confirm that hybrid forma-tion was compatible with transmission to the mam-malian host, coinfected sand flies were allowedto bite and to induce lesions in BALB/c mice.Two doubly drug–resistant clonal lines, desig-nated 6.16.E8 and 6.14.F9, were recovered fromthe eight dermal lesions examined and werefound to be similar to those directly recoveredfrom insects, as discussed below (Table 1). Co-injection of both parental lines into mouse earsby needle never led to the recovery of doublydrug–resistant parasites from the 20 dermal le-sions examined, each containing >2.4 × 107
amastigotes, which suggested that the hybridsselected after transmission by the coinfectedsand flies were generated in the fly and not inthe mammalian host.
We examined the segregation of loci notlinked to chromosomes 24 and 27, the locationof the HYG and SAT markers, respectively. Weused single-nucleotide polymorphisms (SNPs)developed from comparisons of the terminal~30-kb chromosomal regions encompassing theSCG genes located on L. major chromosomes 2,
1Department of Molecular Microbiology, Washington Univer-sity School of Medicine, St. Louis, MO 63110, USA. 2Labora-tory of Parasitic Diseases, National Institute of Allergy andInfectious Diseases, NIH, Bethesda, MD 20892, USA.
*†These authors contributed equally to this work.‡To whom correspondence should be addressed. E-mail:[email protected]
www.sciencemag.org SCIENCE VOL 324 10 APRIL 2009 265
7, 21, 25, 31, 35, and 36 in the WT parent ofLV39c5(HYG). SCGs make up a family of poly-morphic telomeric galactosyltransferases (12),but genes internal to these showed SNPs occur-ring at an overall frequency of ~0.15%, con-sistent with other estimates of strain variation(13, 14). For each chromosome, SNPs from oneto two loci, located from 8.5 to 23 kb inwards ofthe telomeric SCGs (12) (tables S1 and S2), wereanalyzed by a combination of SNP-CAPS (15)[SNP genotyping combined with cleaved ampli-fied polymorphic site analysis (CAPS)] and/ordirect sequencing. Each parent was homozygousfor every marker tested, and all 18 progeny showedclearly that they had inherited both parental al-leles (Fig. 1B and Table 1). This provides evi-dence that each progeny clone inherited a fullset of chromosomes from each parent and werethus full genome hybrids.
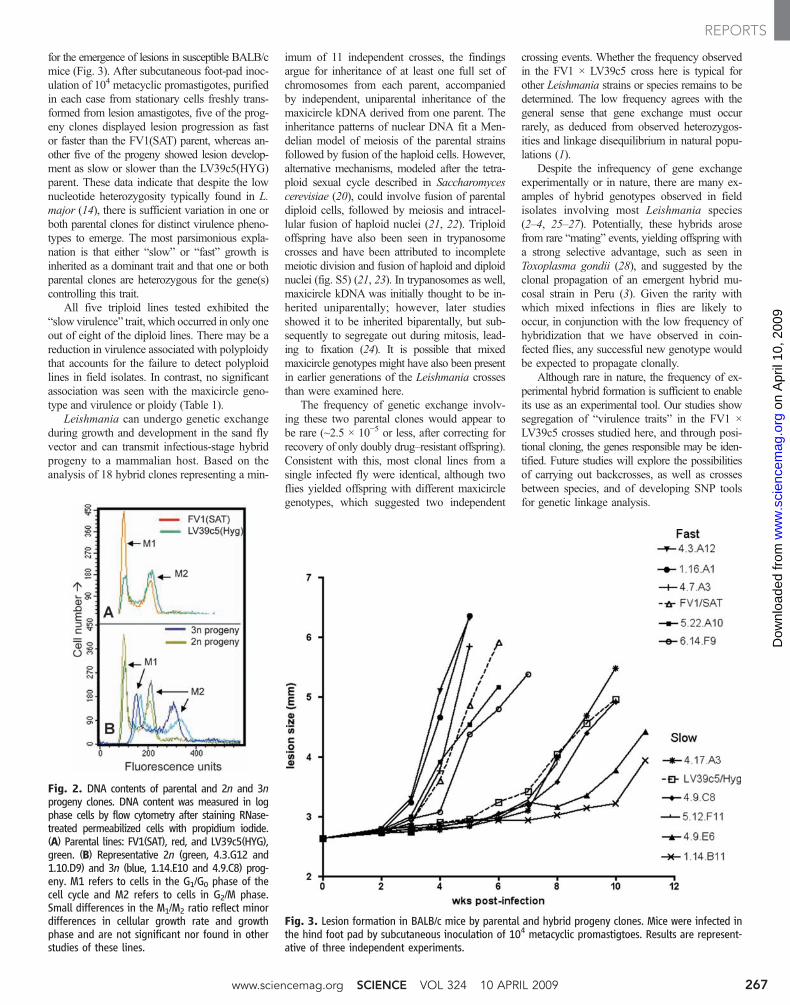
In 7 out of 18 hybrid progeny clones, the rela-tive ratio of the parental alleles seen in SNP-CAPSdigestions differed from the expected 1:1. In-stead, in each case the most intense bands orsequencing trace peaks were those associatedwith the LV39c5 parent, a finding seen at allloci and in all lines tested (Fig. 1B, fig. S3, andTable 1). One possibility is the occurrence of trip-loid offspring, bearing two chromosomal com-plements from one parent (LV39) and one fromthe other (FV1). The DNA contents of the prog-eny clones were measured relative to those ofthe parents by flow cytometry, and the profilescompared with 2n and 4n lines studied previ-ously (16). Although the parents and most hybridsshowed 2n DNA contents, the seven hybrids
detected above showed 3n DNA content (Fig. 2and Table 1); intermediate DNA content wasnot observed. Thus, although all doubly drug–resistant lines were full genome hybrids, a sig-nificant fraction appeared to be triploid ratherthan diploid and had inherited two genomiccomplements from the LV39 and one from FV1parents. Similarly, for four of the 2n lines (1.16.A1, 1.16.C4, 5.12.D9, and 5.12.F11), the segrega-tion of chromosome 31 markers appeared to dif-fer from the expected 1:1 ratio, with the LV39c5parent again predominating in sequencing and/orSNP-CAPs analysis of the hybrids (Table 1). Thismay arise from the finding that Leishmania chro-mosomes are occasionally aneuploid (17), whichincludes tetrasomy of Leishmania chromosome31 in the parental lines studied here. Potentially,tetrasomy may affect mitotic inheritance and seg-regation, as seen in autotetraploid or allotetra-ploid species (18). This was not pursued furtherin this study.
SNP markers were identified to determine theinheritance of mitochondrial maxicircle formedof kinetoplast DNA (kDNA) (Fig. 1C, Table 1,and tables S1 to S3). In contrast to the chromo-somal DNA, maxicircle markers demonstratedclear and consistent uniparental inheritance, with6 of the progeny clones having maxicircle kDNAexclusively from the LV39c5(HYG) and 12 inher-iting maxicircle kDNA exclusively from FV1(SAT)(Table 1 and Fig. 1C). These markers allowedus to establish that two clonal lines arising fromthe same fly that had identical nuclear markerswere in fact different (4.9.C8/C6 and 4.3.A12/G12)(Table 1). kDNA minicircles were not examined.
Several studies were undertaken to comparethe progeny clones for parental phenotypic traits.Metacyclic promastigotes of the FV1 line reactwith monoclonal antibody (mAb) 3F12, whichrecognizes the abundant surface lipophospho-glycan (LPG) and other phosphoglycans express-ing mono–b-galactose–modified repeat unitsterminating with b(1,2)arabinose residues. Thesame antibody fails to bind to LV39c5 metacy-clics, owing to differences in the b-galactose chainlength (19). Metacyclic promastigotes purifiedfrom all progeny clones displayed strong reac-tivity with mAb 3F12, as determined by surfaceagglutination of live parasites (fig. S4A). Thus,the mono-galactosylated 3F12 trait appeared tobe inherited as a dominant trait. Another dominanttrait is the “clumpy” appearance of LV39c5(HYG)when grown in standard culture medium, incontrast to FV1(SAT), which grows as individ-ual cells. All hybrid offspring appeared clumpy(fig. S4B).
It is noteworthy that the parental clones differin a virulence trait defined as the time required
Fig. 1. Genotyping of hybrid Leishmania. (A) PCRfor parental selectable drug markers SAT and HYG.Samples are F, FV1(SAT); L, LV39c5(HYG); M, 1 kbplus marker (Invitrogen, Carlsbad, CA); -, no tem-plate control, 1, 1.10.B12; 2, 1.10.D9; 3, 4.3.G12;4, 4.3.A12; 5, 1.14.E10; 6, 5.12.D9; 7, 5.12.F11; 8,1.14.B11; and 9, 5.22.10. (B) SNP-CAPS analysisfor loci on chromosomes 7, 25, and 35. F, L and Mare as in (A). Mix a, b, and c, parental templatesmixed in different ratios (1:1, 3:1, or 1:3 F:L, respec-tively). DNA content analysis showed that hybrids 5and 8 are 3n (marked by an asterisk), and the re-mainder shown are 2n progeny. (C) SNP-CAPSanalysis of maxicircle. Digestion with Bfa I of theND5–divergent region PCR product is shown.Hybrids tested are 1, 4.7.A3; 2, 4.17.A3; 3, 4.9.E6;4, 4.17.F4; 5, 1.10.B12; 6, 1.10.D9; 7, 4.3.G12; 8,4.3.A12; 9, 1.14.E10; 10, 5.12.D9; 11, 5.12.F11;12, 1.14.B11; and 13, 5.22.10.
Table 1. Summary of phenotype and genotype data for parental and progeny clones. Hybrid linesnomenclature: experiment number.fly number.clone name, and for 6.14.F9 and 6.16.E8, experimentnumber.ear lesion number.clone name. Virulence profile: fast (f) or slow (s). Maxicircle inheritance: F,FV1(SAT) SNP pattern; L, LV39c5(HYG) SNP pattern. Chromosomal analysis: SEQ, ratio of parental SNPpeaks by sequence analysis; CAPS, cleaved and amplified polymorphic site analysis; and H, hybrid. Thesix chromosomes are 2, 7, 21, 25, 35, and 36. Chromosome 31 is tetrasomic.
10 APRIL 2009 VOL 324 SCIENCE www.sciencemag.org266
for the emergence of lesions in susceptible BALB/cmice (Fig. 3). After subcutaneous foot-pad inoc-ulation of 104 metacyclic promastigotes, purifiedin each case from stationary cells freshly trans-formed from lesion amastigotes, five of the prog-eny clones displayed lesion progression as fastor faster than the FV1(SAT) parent, whereas an-other five of the progeny showed lesion develop-ment as slow or slower than the LV39c5(HYG)parent. These data indicate that despite the lownucleotide heterozygosity typically found in L.major (14), there is sufficient variation in one orboth parental clones for distinct virulence pheno-types to emerge. The most parsimonious expla-nation is that either “slow” or “fast” growth isinherited as a dominant trait and that one or bothparental clones are heterozygous for the gene(s)controlling this trait.
All five triploid lines tested exhibited the“slow virulence” trait, which occurred in only oneout of eight of the diploid lines. There may be areduction in virulence associated with polyploidythat accounts for the failure to detect polyploidlines in field isolates. In contrast, no significantassociation was seen with the maxicircle geno-type and virulence or ploidy (Table 1).
Leishmania can undergo genetic exchangeduring growth and development in the sand flyvector and can transmit infectious-stage hybridprogeny to a mammalian host. Based on theanalysis of 18 hybrid clones representing a min-
imum of 11 independent crosses, the findingsargue for inheritance of at least one full set ofchromosomes from each parent, accompaniedby independent, uniparental inheritance of themaxicircle kDNA derived from one parent. Theinheritance patterns of nuclear DNA fit a Men-delian model of meiosis of the parental strainsfollowed by fusion of the haploid cells. However,alternative mechanisms, modeled after the tetra-ploid sexual cycle described in Saccharomycescerevisiae (20), could involve fusion of parentaldiploid cells, followed by meiosis and intracel-lular fusion of haploid nuclei (21, 22). Triploidoffspring have also been seen in trypanosomecrosses and have been attributed to incompletemeiotic division and fusion of haploid and diploidnuclei (fig. S5) (21, 23). In trypanosomes as well,maxicircle kDNA was initially thought to be in-herited uniparentally; however, later studiesshowed it to be inherited biparentally, but sub-sequently to segregate out during mitosis, lead-ing to fixation (24). It is possible that mixedmaxicircle genotypes might have also been presentin earlier generations of the Leishmania crossesthan were examined here.
The frequency of genetic exchange involv-ing these two parental clones would appear tobe rare (~2.5 × 10−5 or less, after correcting forrecovery of only doubly drug–resistant offspring).Consistent with this, most clonal lines from asingle infected fly were identical, although twoflies yielded offspring with different maxicirclegenotypes, which suggested two independent
crossing events. Whether the frequency observedin the FV1 × LV39c5 cross here is typical forother Leishmania strains or species remains to bedetermined. The low frequency agrees with thegeneral sense that gene exchange must occurrarely, as deduced from observed heterozygos-ities and linkage disequilibrium in natural popu-lations (1).
Despite the infrequency of gene exchangeexperimentally or in nature, there are many ex-amples of hybrid genotypes observed in fieldisolates involving most Leishmania species(2–4, 25–27). Potentially, these hybrids arosefrom rare “mating” events, yielding offspring witha strong selective advantage, such as seen inToxoplasma gondii (28), and suggested by theclonal propagation of an emergent hybrid mu-cosal strain in Peru (3). Given the rarity withwhich mixed infections in flies are likely tooccur, in conjunction with the low frequency ofhybridization that we have observed in coin-fected flies, any successful new genotype wouldbe expected to propagate clonally.
Although rare in nature, the frequency of ex-perimental hybrid formation is sufficient to enableits use as an experimental tool. Our studies showsegregation of “virulence traits” in the FV1 ×LV39c5 crosses studied here, and through posi-tional cloning, the genes responsible may be iden-tified. Future studies will explore the possibilitiesof carrying out backcrosses, as well as crossesbetween species, and of developing SNP toolsfor genetic linkage analysis.
Fig. 3. Lesion formation in BALB/c mice by parental and hybrid progeny clones. Mice were infected inthe hind foot pad by subcutaneous inoculation of 104 metacyclic promastigtoes. Results are represent-ative of three independent experiments.
Fig. 2. DNA contents of parental and 2n and 3nprogeny clones. DNA content was measured in logphase cells by flow cytometry after staining RNase-treated permeabilized cells with propidium iodide.(A) Parental lines: FV1(SAT), red, and LV39c5(HYG),green. (B) Representative 2n (green, 4.3.G12 and1.10.D9) and 3n (blue, 1.14.E10 and 4.9.C8) prog-eny. M1 refers to cells in the G1/G0 phase of thecell cycle and M2 refers to cells in G2/M phase.Small differences in the M1/M2 ratio reflect minordifferences in cellular growth rate and growthphase and are not significant nor found in otherstudies of these lines.
www.sciencemag.org SCIENCE VOL 324 10 APRIL 2009 267
References and Notes1. M. Tibayrenc, S. Ben Abderrazak, F. Guerrini, A. Banuls,
Arch. Inst. Pasteur Tunis 70, 375 (1993).2. J. M. Kelly, J. M. Law, C. J. Chapman, G. J. Van Eys,
D. A. Evans, Mol. Biochem. Parasitol. 46, 253(1991).
3. D. Nolder, N. Roncal, C. R. Davies, A. Llanos-Cuentas,M. A. Miles, Am. J. Trop. Med. Hyg. 76, 573 (2007).
4. C. Ravel et al., Int. J. Parasitol. 36, 1383 (2006).5. L. Jenni et al., Nature 322, 173 (1986).6. M. W. Gaunt et al., Nature 421, 936 (2003).7. A. A. Capul, T. Barron, D. E. Dobson, S. J. Turco,
S. M. Beverley, J. Biol. Chem. 282, 14006 (2007).8. S. Martinez-Calvillo et al., Mol. Biochem. Parasitol. 116,
147 (2001).9. P. B. Joshi, J. R. Webb, J. E. Davies, W. R. McMaster,
Gene 156, 145 (1995).10. A. A. Capul, S. Hickerson, T. Barron, S. J. Turco,
S. M. Beverley, Infect. Immun. 75, 4629 (2007).11. Materials and methods are available as supporting
material on Science Online.12. D. E. Dobson, L. D. Scholtes, P. J. Myler, S. J. Turco,
S. M. Beverley, Mol. Biochem. Parasitol. 146, 231(2006).
13. J. Lukes et al., Proc. Natl. Acad. Sci. U.S.A. 104, 9375(2007).
14. A. C. Ivens et al., Science 309, 436 (2005).15. T. Thiel, R. Kota, I. Grosse, N. Stein, A. Graner, Nucleic
Acids Res. 32, e5 (2004).16. A. K. Cruz, R. Titus, S. M. Beverley, Proc. Natl. Acad. Sci.
U.S.A. 90, 1599 (1993).17. C. Ravel, P. Dubessay, P. Bastien, J. M. Blackwell,
A. C. Ivens, Parasitol. Today 14, 301 (1998).18. M. E. Moody, L. D. Mueller, D. E. Soltis, Genetics 134,
649 (1993).19. D. E. Dobson et al., J. Biol. Chem. 278, 28840 (2003).20. M. D. Rose, Annu. Rev. Cell Dev. Biol. 12, 663 (1996).21. W. Gibson, L. Garside, M. Bailey, Mol. Biochem. Parasitol.
51, 189 (1992).22. J. Heitman, Curr. Biol. 16, R711 (2006).23. M. Hope et al., Mol. Biochem. Parasitol. 104, 1 (1999).24. C. M. Turner, G. Hide, N. Buchanan, A. Tait, Exp.
Parasitol. 80, 234 (1995).25. J. C. Dujardin et al., Acta Trop. 59, 293 (1995).26. J. M. Schwenkenbecher et al., Int. J. Parasitol. 36, 237
(2006).27. I. L. Mauricio, M. K. Howard, J. R. Stothard, M. A. Miles,
Parasitology 119, 237 (1999).
28. M. E. Grigg, S. Bonnefoy, A. B. Hehl, Y. Suzuki,J. C. Boothroyd, Science 294, 161 (2001).
29. This research was supported in part by the IntramuralResearch Program of the NIH, National Institute ofAllergy and Infectious Diseases (NIAID), and in part byNIAID grant support (S.M.B., D.E.D., and N.S.A.A1029646 and A1020941). We thank K. Owens forinventorying and shipping parasite strains; K. Beacht formouse infection studies; and M. Grigg, L. Miller, andA. Sher for critical review of the manuscript. Maxicirclesequences have been submitted to GenBank and hadbeen assigned accession numbers FJ349262 to FJ349263(12S rRNA) and FJ349264 to FJ349265 (Divergentregion).
Supporting Online Materialwww.sciencemag.org/cgi/content/full/324/5924/265/DC1Materials and MethodsFigs S1 to S5Tables S1 to S3References
8 December 2008; accepted 18 February 200910.1126/science.1169464
Green Evolution and DynamicAdaptations Revealed by Genomes ofthe Marine Picoeukaryotes MicromonasAlexandra Z. Worden,1* Jae-Hyeok Lee,2† Thomas Mock,3†‡ Pierre Rouzé,4† Melinda P. Simmons,1†Andrea L. Aerts,5 Andrew E. Allen,6 Marie L. Cuvelier,1,7 Evelyne Derelle,8 Meredith V. Everett,7Elodie Foulon,9 Jane Grimwood,5,10 Heidrun Gundlach,11 Bernard Henrissat,12 Carolyn Napoli,13Sarah M. McDonald,1 Micaela S. Parker,3 Stephane Rombauts,4 Aasf Salamov,5 Peter Von Dassow,9Jonathan H. Badger,6 Pedro M. Coutinho,11 Elif Demir,1 Inna Dubchak,5 Chelle Gentemann,14Wenche Eikrem,15 Jill E. Gready,16 Uwe John,17 William Lanier,18 Erika A. Lindquist,5Susan Lucas,5 Klaus F. X. Mayer,10 Herve Moreau,8 Fabrice Not,9 Robert Otillar,5Olivier Panaud,19 Jasmyn Pangilinan,5 Ian Paulsen,20 Benoit Piegu,19 Aaron Poliakov,5Steven Robbens,4 Jeremy Schmutz,5,10 Eve Toulza,21 Tania Wyss,22 Alexander Zelensky,23Kemin Zhou,5 E. Virginia Armbrust,3 Debashish Bhattacharya,18 Ursula W. Goodenough,2Yves Van de Peer,4 Igor V. Grigoriev5
Picoeukaryotes are a taxonomically diverse group of organisms less than 2 micrometers indiameter. Photosynthetic marine picoeukaryotes in the genus Micromonas thrive in ecosystemsranging from tropical to polar and could serve as sentinel organisms for biogeochemical fluxes ofmodern oceans during climate change. These broadly distributed primary producers belong to ananciently diverged sister clade to land plants. Although Micromonas isolates have high 18Sribosomal RNA gene identity, we found that genomes from two isolates shared only 90% of theirpredicted genes. Their independent evolutionary paths were emphasized by distinct riboswitcharrangements as well as the discovery of intronic repeat elements in one isolate, and inmetagenomic data, but not in other genomes. Divergence appears to have been facilitated byselection and acquisition processes that actively shape the repertoire of genes that are mutuallyexclusive between the two isolates differently than the core genes. Analyses of the Micromonasgenomes offer valuable insights into ecological differentiation and the dynamic natureof early plant evolution.
Ancestral green algae were of fundamentalimportance to the eukaryotic greeningthat shaped the geochemistry of our plan-
et. This process began over a billion years agowhen a cyanobacterium was captured by a het-erotrophic protist and incorporated as an en-dosymbiont, giving rise to the first eukaryoticalga (1). The extant Prasinophytae retain charac-teristics that are believed to have been present in
the last common ancestor of green algae (chloro-phytes) and land plants (streptophytes, includ-ing charophyte algae) (2). Most prasinophyteswithin the monophyletic marine order Mamiel-lales (Fig. 1A and fig. S1), such asMicromonas,are tiny (≤2 mm in diameter) and known as pico-eukaryotes.Micromonas is a motile unicell, witha single chloroplast and mitochondrion (Fig. 1A,inset), first reported as a dominant phytoplankter
in the 1950s (3) and now recognized as having aglobal distribution (Fig. 1B) (4).
Today’s oceans contain a polyphyletic diver-sity of algae, some with plastids that shareancestry with land plants (green algae) and oth-ers (chromalveolates) that are derived from redalgae through secondary or tertiary (eukaryotic-eukaryotic) endosymbioses (5, 6). Unlike mostepisodic chromalveolate bloomers and the fresh-water green alga Chlamydomonas (7), theMamiellales have reduced genomes, as firstshown in Ostreococcus (8, 9). Ostreococcus hasa narrower environmental distribution thanMicro-monas (Fig. 1B) and a smaller genome (12 to 13Mb containing only ~8000 genes). Open-oceanbacteria, including SAR11 and Prochlorococcus(10, 11), show similar patterns of cell size andgenome minimization. Conditions favoring pico-phytoplankton growth, such as increased stratifica-tion, lessmixing, and reduced nutrient concentrationsin ocean surfacewaters, are predicted climate changeoutcomes, and thus picoeukaryote dynamics maybe useful ecosystem indicators.
We sequenced the nuclear genomes of Mi-cromonas isolates RCC299 and CCMP1545(Table 1 and figs. S2 and S3) (12). These isolatesare from distant ocean provinces and fall intodistinct phylogenetic clades that can co-occur(Fig. 1) (12, 13) but are generally considered asingle species (Micromonas pusilla). Transmis-sion electron microscopy revealed no morpho-logical differences (12), and 18S ribosomal DNA(rDNA) identity was high (97%). Surprisingly,only 90% of their 10,056 (RCC299) and 10,575(CCMP1545) predicted genes (table S1) wereshared (Fig. 2A). In contrast, Ostreococcuslucimarinus and O. tauri share 97% of catalogedgenes (12), and yeast genera can share ~95% ofhomologs (14). The divergence we observed be-tween the Micromonas isolates supports theirclassification as distinct species.
Synteny, GC content, and codon usage pointedto a shared evolutionary history for RCC299 and
10 APRIL 2009 VOL 324 SCIENCE www.sciencemag.org268
Demonstration of Genetic Exchange During Cyclical Development of Leishmania in the Sand Fly Vector
Natalia S. Akopyants, Nicola Kimblin, Nagila Secundino, Rachel Patrick, Nathan Peters, Phillip Lawyer, Deborah E. Dobson, Stephen M. Beverley, David L. Sacks*
*To whom correspondence should be addressed. E-mail: [email protected]
Published 10 April 2009, Science 324, 265 (2009)
DOI: 10.1126/science.1169404
This PDF file includes
Materials and Methods Figs. S1 to S5 Tables S1 to S3 References
Page 1
Supporting Online Material Materials and Methods Parasites, hybrid selections. and phenotype analyses. The L. major lines and clones were
grown in M199 medium as described (1), and containing 25 ug/ml hygromycin B (Sigma, St.
Louis) and/or 100 µg/ml nourseothricin (Jena Bioscience, Jena, Germany), as necessary. For
selection of double drug resistant lines in vitro, cultures were initiated by seeding log-phase cells
of each parent at 1x106/ml, and the two antibiotics were added at days 3, 7, or 11. All cultures
were static by 2-4 days, and all parasites were dead by 10-24 days following addition of the
antibiotics. For selection of double drug resistant lines during co-infection in the mammalian
host, BALB/c mice were co-inoculated in the ear dermis with 104 metacyclic promastigotes of
each parental strain. Four weeks after infection, ear dermal cells were prepared and aliquots
serially diluted without antibiotics to quantify the number of amastigotes per lesion, as described
(1). The remaining cells from each ear were cultured in 2 ml media containing both antibiotics.
For selection of double drug resistant lines during co-infection in the sand fly vector, P. duboscqi
sand flies were infected and used for transmission of L. major by bite as described (1). Briefly, 2
to 4-day-old P. duboscqi females were infected by feeding through a chick skin membrane on
heparinized mouse blood containing 3 and 1 x 106 / ml logarithmic phase promastigotes of the
LV39c5(HYG) and FV1(SAT) lines, respectively. Midguts were dissected 13-16 days post-
infection, and individual midguts were transferred directly to 96 well plates containing 0.1 ml
media without antibiotics. After 2 days of growth, the parasites were transferred to 2 ml media
containing both antibiotics. For selection of hybrid lines and clones from mouse lesions initiated
by infected sand fly bites, 13 day co-infected flies were permitted to feed on the ear pinnae of
BALB/c mice, 10 flies per ear. After 4-6 weeks, the ear tissue homogenates were prepared as
described (1), and cultured in M199 medium containing both antibiotics. Hybrid clones were
Page 2
generated by distribution in 96 well blood agar plates in 0.1 ml M199 containing both antibiotics.
Poisson analysis was used to determine the percentage probability of clonality, and was in each
case >95%. Animal virulence tests were performed in BALB/c mice by s.c footpad inoculation
of 104 metacyclic promastigotes, purified as described (2) from stationary phase promastigotes
freshly transformed from lesion amastigotes. Agglutination of metacyclic promastigotes with
monoclonal antibody 3F12 was performed as previously described (3).
DNA content analysis. DNA contents were determined by flow cytometry following staining of
106 permeabilized RNAse-treated cells with propidium iodide, as previsouly described (4). L.
major promastigotes (5×106) were washed with PBS and fixed with 90% methanol–10% PBS for
15 min at 4°C. Parasites were pelleted, washed twice with PBS, resuspended in 1 ml of PBS
containing 20 µg/ml propidium iodide and 200 µg/ml RNase A, and incubated at 25 °C for 1
hour. Data were acquired on a FACS flow cytometer (Becton Dickinson), counting at least
10,000 cells per sample, and analyzed using CellQuest 3.1 (BD Bioscience) software.
Maxicircle sequencing. PCR primers (Supplemental Table 3) were designed using L. major
FV1 maxicircle sequences found in a shotgun sequencing survey previously (5). Single pass
sequencing was done using dye terminators and AmpliTaq DNA polymerase and analyzed using
Lasergene software v.7.1.0 (DNASTAR, Inc, USA). Sequences for parental lines were submitted
to the GenBank (NCBI) (http://www.ncbi.nlm.nih.gov) with accession numbers FJ349262-
FJ349263 (12SrRNA) and FJ349264- FJ349265 (Divergent region). SNP positions, insertions
and deletions are listed in Supplementary Table 1.
LV39 sequencing and SNP identification. A cosmid library of LV39c5 DNA was prepared in
the vector cLHYG (6). SCG-bearing cosmids were identified by colony hybridization using a
generic SCG universal coding region probe (7), and restriction mapping and comparisons to the
Page 3
SCG loci of the sequenced L. major Friedlin genome were used to assign individual cosmids as
SCG1-7. Representative cosmids encompassing the intact SCG genes and extending ~25-35 kb
inward were subjected to random shotgun sequencing, and assembled independently and with the
Friedlin genome as a scaffold. Sequence comparisons revealed numerous SNPs, and several
from each SCG-containing chromosome were studied further in the parental and progeny lines
generated in this work (Supplementary Table 1).
Comparative SNP-CAPS analysis of parental and progeny lines. The parental lines were
screened for polymorphisms on the basis of single nucleotide differences between the lines, and
SNP markers on seven chromosomes at nine different loci were used in this work. The parental
sequences were compared by BLAST analysis (http://www.genedb.org/genedb/leish/index.jsp).
Blast output then was uploaded in its entirety to the BlastDigester website (8)
(http://www.bar.utoronto.ca/ntools/cgi-bin/ntools_blast_digester.cgi) in order to identify suitable
cleaved amplified polymorphic sequence markers (CAPS). SNP comparison for parental
genomes is presented in Supplementary Table 2. PCR primers for SNP-CAPS analysis were
generated manually (Supplementary Table 3). PCR amplification of specific fragments was
carried in 100 µl volume using 20 ng genomic DNA (9). KlentaqLA polymerase and 40 pmoles
of each primer. The reactions were performed in a thermocycler (PTC-200, MJ Research, MA)
programmed for 35 cycles as follows: 93°C, 30 sec; 50°C, 45 sec and then 68°C, 2 min. After
amplification, DNA products were ethanol precipitated, the pellets were washed with 75%
ethanol, resuspended in 40 µl of sterile nuclease-free water, and 4 µl of product was
electrophoresed on a 1.5% agarose gel to ensure homogeneity and yield. In all cases PCR
amplification resulted in a homogeneous DNA fragment of the size expected (Supplementary
Table 2). Usually 10 µl of product was digested with 5 to 10 U of restriction enzyme for 16
Page 4
hours in the buffer recommended by the supplier (NE Biolabs, MA), electrophoresed on 1.5%
agarose gel and visualized by ethidium bromide staining.
DNA sequencing and trace analysis. DNA sequencing was carried out as follows: 1 µl of
purified PCR template (~50 ng DNA) was combined with 1 µl of 10 µM primer, 4 µl of 2.5X
sequencing buffer (200 mM Tris, pH 9.0, 5 mM MgC12) and 10 µl with 10 mM Tris/0.01 mM
EDTA, pH 8.0. This mixture was heat-denatured for 5 min at 98°C in a thermocycler (PTC-200,
MJ Research, MA), placed on ice and 4 µl of the ABI Prism Big Dye Terminator Cycle
Sequencing Ready Reaction Kit mix was added (v. 3.1; Applied Biosystems, CA). The
amplification reactions were performed in a PTC-200 thermocycler for 40 cycles (96°C for 10
sec, 50°C for 5 sec, and 60°C for 2 min). Excess dye was removed by gel filtration on a 96-well
filter plate filled with Sephadex G-50 beads. The samples were then heat-denatured for 2 min at
95°C and electrophoresed on an ABI3700 genetic analyzer (Applied Biosystems, CA) under
conditions recommended by the manufacturer. Prior to sequencing reactions 1:1 mix of parental
templates were prepared as a technical control. Trace peaks at SNP positions for 1:1 mix control
sequencing reaction showed equal height. These data along with the parental traces were
compared to all sequencing data generated for progeny clones and analyzed using Lasergene
software (Supplementary Fig. S2).
References
1. N. Kimblin et al., Proc Natl Acad Sci U S A 105, 10125 (Jul 22, 2008).
2. G. F. Spath, S. M. Beverley, Exp Parasitol 99, 97 (Oct, 2001).
3. D. L. Sacks, R. P. da Silva, J Immunol 139, 3099 (1987).
4. A. K. Cruz, R. Titus, S. M. Beverley, Proc Natl Acad Sci U S A 90, 1599 (1993).
Page 5
5. N. S. Akopyants et al., Mol Biochem Parasitol 113, 337 (Apr 6, 2001).
6. K. A. Ryan, S. Dasgupta, S. M. Beverley, Gene 131, 145 (Sep 6, 1993).
7. D. E. Dobson et al., J Biol Chem 278, 15523 (May 2, 2003).
8. K. Ilic, T. Berleth, N. J. Provart, Trends Genet 20, 280 (Jul, 2004).
9. E. Medina-Acosta, G. A. Cross, Mol Biochem Parasitol 59, 327 (Jun, 1993).
Page 6
Page 7
Page 8
Page 10
Page 11
Supplementary Table 1. SNPs in L. major FV1 (F) relative to LV39c5 (L) used in this work.
30/F=A, L=T; 129, 270, 350/F=T, L=C; 313/F=C, L=A; 354, 370/F=C, L=T; 31, 248/insertion of A in F; 293/insertion of AATATGTTATA in F; 268/insertion of G in L
2
a ~ 10 kb between two ORFs b SNP location in GeneDB ver. 5.2 coordinates
Page 12
Supplementary Table 2. Comparative SNP-CAPS patterns for the two parental genomes.
Chromosome ORF ID PCR product (bp) Enzyme FV1 (SAT)
Predicted fragment (bp) LV39c5(HYG)
Predicted fragment (bp)
LmjF02.0060 557 Not I 361; 196 557
Hae III HaeIII: 399;178;136;99;97;13
HaeIII: 313;178;136;99;97;86;13
2 LmjF02.0085 922
Fsp I Fsp I: 693; 229 Fsp I: 653;229;40 7 LmjF07.1150 984 Dde I Dde I: 619;292;73 Dde I: 580;258;107;39