ORIGINAL ARTICLE Timothy Lovell Jian Li Louis Noodleman Density functional and electrostatics study of oxidized and reduced ribonucleotide reductase; comparisons with methane monooxygenase Received: 31 August 2001 / Accepted: 22 February 2002 / Published online: 19 April 2002 ȑ SBIC 2002 Abstract A combined broken symmetry density func- tional and electrostatics approach has been used to examine the active sites of the resting (RNR ox ) and reduced (RNR red ) forms of class I type ribonucleotide reductase in the protein and solvent environment. Active site cluster geometries and Heisenberg J values are discussed in the context of the available protein data. The total electrostatic interaction energy in the protein comprises a large reaction field component and a much smaller protein field term, the former suggest- ing strong dielectric polarization between the cluster and protein-solvent dielectrics; the latter is indicative of a very weak link to the protein environment. Decom- position of the protein field term elucidates the major electrostatic interactions between amino acid residues in the RNR R2 local environment and the active site cluster, enabling an energetic comparison of structur- ally equivalent residues with a related diiron protein, methane monooxygenase. Keywords Ribonucleotide reductase Methane monooxygenase Binuclear non-heme iron Density functional theory Protein electrostatics comparison Introduction Ribonucleotide reductases (RNRs) are a group of met- alloenzymes that play a central role in the operation of all living organisms [1, 2]. Their unique chemical function is to provide cells with the precursors required for the ini- tial step in DNA synthesis by catalyzing the reduction of ribonucleotides (NTPs) to their 2¢-deoxyribonucleotide (dNTP) counterparts [3, 4]. This pivotal step in cell proliferation has been the focus of much attention of late because of the interest associated with the development of drugs containing antitumor and antiviral agents which may be targeted towards inhibiting the function of RNR in the production of diseased tissue [5, 6, 7, 8, 9]. Many different RNRs are known, but the well-characterized forms can be grouped into one of four distinct classes. In the present work, attention is restricted exclusively to the class I RNRs found in all eukaryotes, some prokaryotes, and several viruses [10]. Within this subclass, proteins are known to consist of two dissimilar components: the first component, denoted R2, comprises a diiron-containing unit that functions to generate a tyrosine radical (Tyr122) nec- essary for chemical function to occur; the second subunit, R1, binds the NTP substrate and catalyzes the dehydroxylation of the 2¢-hydroxyl group of the ribose ring, initiated by radical abstraction of the 3¢-H atom. The biological function of RNR is often re- ferred to in terms of the activation cycle summarized by: Fe III O Fe III þ 2e þ 2H þ ! 2ðFeÞ II þ H 2 O ð1Þ 2ðFeÞ II þ O 2 þ e þ H þ ! Fe III ðl OÞOH Fe IV ð2Þ Fe III ðl OÞOH Fe IV þ Tyr122 H ! Fe III O Fe III þ Tyr122 þ H 2 O ð3Þ Although apparently simple in principle, this reaction occurs in a surprisingly complicated sequence of steps [11]. Coupled electron and proton transfer to the resting diferric form of the enzyme, RNR ox , generates the J Biol Inorg Chem (2002) 7: 799–809 DOI 10.1007/s00775-002-0358-y T. Lovell (&) J. Li L. Noodleman (&) Department of Molecular Biology TPC-15, The Scripps Research Institute, La Jolla, CA 92037, USA E-mail: [email protected]Fax: +1-858-7848896 Present address: J. Li Johnson & Johnson Pharmaceutical Research & Development LLC, Welsh & McKean Roads, PO Box 776, Spring House, PA 19477, USA E-mail: [email protected]Fax: +1-858-7848896
Transcript
ORIGINAL ARTICLE
Timothy Lovell Æ Jian Li Æ Louis Noodleman
Density functional and electrostatics study of oxidizedand reduced ribonucleotide reductase; comparisonswith methane monooxygenase
Received: 31 August 2001 /Accepted: 22 February 2002 / Published online: 19 April 2002� SBIC 2002
Abstract A combined broken symmetry density func-tional and electrostatics approach has been used toexamine the active sites of the resting (RNRox) andreduced (RNRred) forms of class I type ribonucleotidereductase in the protein and solvent environment.Active site cluster geometries and Heisenberg J valuesare discussed in the context of the available proteindata. The total electrostatic interaction energy in theprotein comprises a large reaction field component anda much smaller protein field term, the former suggest-ing strong dielectric polarization between the clusterand protein-solvent dielectrics; the latter is indicative ofa very weak link to the protein environment. Decom-position of the protein field term elucidates the majorelectrostatic interactions between amino acid residuesin the RNR R2 local environment and the active sitecluster, enabling an energetic comparison of structur-ally equivalent residues with a related diiron protein,methane monooxygenase.
Keywords Ribonucleotide reductase Æ Methanemonooxygenase Æ Binuclear non-heme iron Æ Densityfunctional theory Æ Protein electrostatics comparison
Introduction
Ribonucleotide reductases (RNRs) are a group of met-alloenzymes that play a central role in the operation of allliving organisms [1, 2]. Their unique chemical function is
to provide cells with the precursors required for the ini-tial step in DNA synthesis by catalyzing the reduction ofribonucleotides (NTPs) to their 2¢-deoxyribonucleotide(dNTP) counterparts [3, 4]. This pivotal step in cellproliferation has been the focus of much attention of latebecause of the interest associated with the developmentof drugs containing antitumor and antiviral agents whichmay be targeted towards inhibiting the function of RNRin the production of diseased tissue [5, 6, 7, 8, 9]. Manydifferent RNRs are known, but the well-characterizedforms can be grouped into one of four distinct classes. Inthe present work, attention is restricted exclusively to theclass I RNRs found in all eukaryotes, some prokaryotes,and several viruses [10].
Within this subclass, proteins are known to consistof two dissimilar components: the first component,denoted R2, comprises a diiron-containing unit thatfunctions to generate a tyrosine radical (Tyr122) nec-essary for chemical function to occur; the secondsubunit, R1, binds the NTP substrate and catalyzesthe dehydroxylation of the 2¢-hydroxyl group of theribose ring, initiated by radical abstraction of the 3¢-Hatom. The biological function of RNR is often re-ferred to in terms of the activation cycle summarizedby:
FeIII�O� FeIII þ 2e� þ 2Hþ ! 2ðFeÞII þ H2O
ð1Þ
2ðFeÞII þ O2 þ e� þ Hþ ! FeIIIðl �OÞOH FeIV
ð2Þ
FeIIIðl �OÞOH FeIV þ Tyr122�H
! FeIII�O� FeIII þ Tyr122� þ H2O ð3Þ
Although apparently simple in principle, this reactionoccurs in a surprisingly complicated sequence of steps[11]. Coupled electron and proton transfer to the restingdiferric form of the enzyme, RNRox, generates the
T. Lovell (&) Æ J. Li Æ L. Noodleman (&)Department of Molecular Biology TPC-15,The Scripps Research Institute, La Jolla, CA 92037, USAE-mail: [email protected]: +1-858-7848896
Present address: J. LiJohnson& Johnson Pharmaceutical Research &Development LLC,Welsh & McKean Roads, PO Box 776,Spring House, PA 19477, USAE-mail: [email protected]: +1-858-7848896
reduced diferrous form, RNRred, with protein structuresavailable to reasonable resolution for both forms (Fig. 1and Eq. 1) ([12] and references therein).
Following the binding, reduction, and cleavage ofdioxygen by RNRred, the active site acquires an extraelectron from the protein matrix to give a structurally ill-characterized Fe(III)Fe(IV) intermediate (Eq. 2), capa-ble of abstracting an electron (and a proton) fromTyr122 to yield a neutral Tyr radical and the activeFe(III)Fe(III) form (Eq. 3). Substrate reactions at R1are then initiated by a complicated sequence of long-range electron transfer over a distance of 30 A, wherebythe radical character is transferred back and forth asrequired from Tyr122 to Cys439, the latter being the site
of substrate binding and chemical transformation. Thereduction of all four NTPs then begins the process ofDNA synthesis.
Soluble methane monooxygenase (MMO) is anotherprotein belonging to the select class of binuclear non-heme iron enzymes capable of activating dioxygen forfurther oxidation chemistry [13, 14, 15, 16]. The bio-logical function of MMO is also well established andexemplified by its catalytic reaction in which methane isoxidized by oxygen to form methanol and water underambient conditions:
FeIII�ðOHÞ2�FeIII þ 2e� þ 2Hþ ! 2ðFeÞII þ 2H2O
ð4Þ
2ðFeÞII þ O2 ! FeIVðl�OÞ2FeIV ð5Þ
FeIVðl�OÞ2FeIV þ R�H þ H2O ! FeIII�ðOHÞ2
�FeIII þ R�OH ð6Þ
Fig. 1. The active sites of (a) RNRox and (b) RNRred from E. coli.Second-shell hydrogen-bonding partners are also included forreference, as are the active sites of (c) MMOHox and (d) MMOHred
from Methylococcus capsulatus for comparison. Atoms are identi-fied by color: Fe, green; N, blue; O, red; C, gray; H, white
800
Even though the diiron active sites of RNR and MMOare, for all intents and purposes, identical, the elec-tronically very different goals of ribonucleotide reduc-tion and methane hydroxylation suggest their functionalroles are breathtakingly different (see Eqs. 1, 2, 3 and 4,5, 6 which serve to highlight the similarity in chemicalreactions). A major source of appeal in the study ofbinuclear non-heme iron enzymes is the stark contrast infunctionality of RNR and MMO and the principalquestion, which as yet remains unanswered, is centeredaround elucidating the subtle differences in active sitestructures and local protein environments that arecausal. To this end, spectroscopy coupled with theoret-ical methods have proved very valuable in probing themore fundamental, microscopic elements of the elec-tronic and geometric structure of the active site. Theresults of several independent groups have been instru-mental in guiding subsequent experimental studies [12].Mutagenesis techniques have undertaken the equallydifficult objective of understanding how the protein se-quence and the underlying structure of the local mac-roscopic environment surrounding the diiron coreregulate active-site structure-reactivity relationships andhave provided a wealth of information testable by the-oreticians [17, 18, 19, 20, 21]. A theoretical approachthat interpolates between the spectroscopic-theoreticaland genetics-based approaches, and thereby incorpo-rates elements of the microscopic electronic structure ofthe active site with the macroscopic effects of the proteinenvironment, can also make a valuable contribution. Inthis respect, the characterized resting (MMOHox) andreduced (MMOHred) states of the hydroxylase (MMOH)component of MMO have recently been examined in theprotein environment using a combined density func-tional (DF) and electrostatics methodology [22, 23]. Theactive site structures were optimized using broken sym-metry (BS) DF methods and then embedded in anelectrostatics/dielectric representation of the protein andsolvent environment. This approach circumvented theinherent problem of the protein and solvent environ-ment being too large to be treated by an appropriatequantum mechanical or hybrid quantum mechanics/molecular mechanics (QM/MM) approach and enableda comparison of trends in calculated and experimentalactive site structures and exchange coupling constants.Furthermore, pKa calculations [23] in the protein affor-ded the protonation state of the various bridging sol-vent-derived ligands, and the decomposition of theprotein field term into individual residues provided sig-nificant insight into the intimate interaction between thelocal amino acids and the active site (Fig. 2).
From the protein field analysis of MMOH, the activesite was noted to be anchored largely to a crescent-shaped base of anionic and polar residues that preventedany movement for the lower portion of the diiron site,while the upper half or the cluster was less constrainedby the protein environment, consistent with ideasregarding the interconversion of one intermediate to thenext throughout the MMO catalytic cycle.
In this paper, the combined DF/electrostatics meth-odology is now applied to the structurally characterizedforms of RNR in the Escherichia coli protein environ-ment. The protein folds defining the diiron sites (locatedin a bundle of four a-helices within the active domain) ofRNR and the structures of the diiron clusters themselvesare remarkably similar to those of MMOH. Further-more, the partial matching in both the amino acid se-quence alignment of the MMOH and RNR proteins [24]and the position of structurally equivalent residuessuggests additional similarities may also persist betweenthese two systems; to what extent these similarities ex-tend to the interaction of the diiron cluster with proteinenvironment is also of interest. An analysis of the de-composition of the protein field interaction energywithin or around the motif of four a-helices to individualresidues for RNR with that noted previously forMMOH enables a comparison beyond that of the simpleactive site treated in vacuo, and may be of relevance tothose that utilize site-directed mutagenesis tools toexamine the structure-function relationship betweenfamilies of related proteins.
Materials and methods
Protein structures and active site models
X-ray crystallographic coordinates have been reported for wild-type and mutant forms of RNR. Our calculations were based onthe structures of the 3D structures of wild-type RNRox (PDB code:1RIB, 2.2 A) [25] and RNRred (PDB code: 1XIK, 1.7 A) [26] fromE. coli. For each protein, hydrogen atoms were added assuming apH of 7 (His sidechains coordinated through Nd1 to Fe sites wereprotonated at N�2). The positions of the hydrogens were optimizedusing standard InsightII (Discover module, BioSym/MSI) [27]procedures, using formal metal charges (3+/2+) assigned to theiron sites, while the total charge for each single amino acid residuewas assigned as a sum of the partial atom charges. During thehydrogen atom optimization, all heavy atom positions were keptfixed.
Fig. 2. Important cluster-residue interactions for the 2OH– formof the MMOHox active site in chain D of M. capsulatus
801
Class I bacterial ribonucleotide reductase from E. coli exists asan a2b2 tetramer that comprises two non-identical protein subunits,a2 and b2. The smaller b subunit is heart-shaped and contains thecatalytically active diiron centers that function to generate thenecessary tyrosine radicals required for dNTP synthesis. The largera2 protein contains the allosteric regulatory sites, its function beingto bind the ribonucleotide substrate and catalyze the dehydroxy-lation of the ribose ring. The b subunits are related by a non-crystallographic axis of two-fold symmetry. For both the oxidizedand reduced proteins the b subunits are identical and made up oftwo protomers, chains A and B, which each contain a binucleariron site. The internal distance between the binuclear iron sites atboth oxidation levels is very similar, approximately 25 A. The ac-tive domains used in our calculations derived from chain A of theoxidized (Fig. 3a) and reduced proteins (Fig. 3b). Each chaincomprises 341 amino acid residues in total.
The quantum cluster models used to represent the active sitesare depicted in Fig. 1a for RNRox and Fig. 1b for RNRred. Hy-drogen bonding partners not included in each quantum model areindicated by a dashed line to the relevant second-shell residue. Forthe resting form (Fig. 1a), the six protein sidechain ligands boundto the iron sites derive from two histidine residues, His118 andHis241, and three glutamate residues, Glu115, Glu204, andGlu238, and one aspartate residue, Asp84. Solvent water mole-cules, one coordinated to each Fe, complete the approximatelyoctahedral environment around each metal. In addition to the di-iron core, the redox-active Tyr122 residue lies within 5.3 A of oneof the Fe sites (Fe1). Tyr122 is the key residue during catalysis andis also incorporated into our active-site model. During enzymeturnover, the diferric cluster is reduced to the diferrous state bycoupled electron and proton addition followed by substantialstructural rearrangement. The protons presumably bind to the O2–
bridge prior to or concerted with the mechanism by which theGlu238 carboxylate shifts from a terminal to a l-1,3 bidentatebridging mode. The six protein sidechains still remain coordinated,but Glu204 and Asp84 undergo substantial changes in their coor-dination mode (Fig. 1b): Glu204 switches from an anti to a synconformation; Asp84 reorients to its other rotamer that enables ahydrogen bond to Tyr122, and consequently, Tyr122 moves closer(4.8 A) to Fe1. As part of this RNRox to RNRred conversionmechanism, the addition of two protons to the l-O2– bridge gen-erates a third water molecule. Subsequent elimination of threewater molecules then produces either two four-coordinate Fe sitesor one four-coordinate Fe site and one five-coordinate Fe site. Thefifth coordination site at one Fe is suspected to arise from either thesecond oxygen of Glu204 or a solvent-bound water molecule, withthe current view from spectroscopy favoring the former [11, 28].
For all our active-site cluster models, within the first coordi-nation shell, histidine residues were approximated as neutral imi-dazole rings, tyrosine as phenol, and glutamate residues werereplaced by charged acetate groups. The active-site model was re-garded as a small part of the protein, terminating with one H atomin place of the appropriate linking C atom. The Cb atoms of thehistidine and glutamate residues were therefore replaced by Hbatoms to saturate the quantum clusters. These atoms lie at theinterface of the quantum and classical regions and thus weretreated in an appropriate manner to conserve charge [29]. Startinggeometries were taken from the crystal structure data followingoptimization of the hydrogen-bonding network of the protein whilethe final geometries were computed by the DF methods describedin the following section.
Computational methodology
The in-depth theoretical background of the various methods ap-plied previously to MMOH are identical to those employed hereand a detailed discussion can be found elsewhere [22, 23]. In brief,the calculation is a multi-step procedure. First, DF calculations arecarried out to optimize the geometries of the active-site clustermodel and to compute the gas phase energetics of the corre-sponding optimized structures. Second, a set of atomic pointcharges for the active-site cluster is fitted to best represent themolecular electrostatic potential generated by the DF calculations.Third, the optimized active-site cluster is docked back to the pro-tein structure and then a continuum-electrostatics approach isemployed to calculate the protein field (using assigned protein atomcharges) and reaction field energies.
The Amsterdam DF package (ADF, version 2.3) was used tocompute the geometries and energies of the active site clusters [30].Basis set IV was used to model Fe, C, N, O, and H and correspondsto uncontracted triple-zeta Slater-type orbitals (STOs) for the 4s,4p, and 3d valence orbitals of Fe, triple-zeta STOs for 2s and 2pvalence orbitals of C, N, and O augmented with a 3d polarizationorbital, and triple-zeta STOs for 1s of H with a 2p polarizationorbital [31, 32]. The inner core orbitals were treated by the frozencore approximation. An auxiliary charge density fit set, that con-sists of s, p, d, f, and g STO functions, accompanies the corre-sponding orbital basis sets IV and III, and was adopted to fit themolecular density and used to calculate the Coulomb and exchangepotentials [33]. The numerical integration scheme was the polyhe-dron method developed by te Velde and co-workers [34, 35]. Ge-ometry optimization was carried out using an analytical gradientmethod implemented by Versluis and Ziegler [36]. Optimizationsused the Newton-Raphson method and the Hessian was updatedwith the Broyden-Fletcher-Goldfard-Shanno scheme [37]. Con-vergence criteria were set to 0.001 A in coordinates and0.01 Hartree/A in the norm of all gradient vectors. Geometry op-timizations were done at the LDA level [Vosko-Wilk-Nusair(VWN) parametrization] [38] with the generalized gradient cor-rection terms introduced by Perdew and Wang (PW91) to the ex-change and correlation [39]. To deal with the spin polarization inthese active-site clusters, the spin-unrestricted method was used.The BS approach [40] enabled us to model the antiferromagnetic(AF) state while Heisenberg exchange coupling constants werecalculated from the energy difference of the high-spin state (HS)and the BS state using spin-projection methods [41].
All calculations were performed on either SGI Power Challenge(12CPU, R10000 chip, 195 MHz) or SGI Origin Series (16CPU,R10000 chip, 250 MHz) workstations at The Scripps ResearchInstitute. Calculations were done without symmetry (NoSym) usingthe parallelized version of the ADF code on four nodes for optimalscaling efficiency. For geometry optimizations, all bond lengths andangles for atoms bonded to Fe were optimized. First-shell coordi-nated water or hydroxide molecules were allowed all degrees offreedom. With the exception of the atoms ligated to the Fe centers,the internal geometries of coordinated protein residues (imidazoleand tyrosine rings, methyl groups) and torsion angles within thefirst sphere were constrained to the experimental parameters. Thisprevented any unnecessary oscillation of the energy during SCF
Fig. 3. The active domains of the (a) RNRox and (b) RNRred
proteins used in the calculations. The location of the active site isshown in each case. The figure was produced using MOLSCRIPT[64]
802
convergence, maintained reasonable fidelity with the experimentalstructures, and represented the effects of second-shell protein andsolvent hydrogen-bonding effects without the need to expand tooverly large quantum clusters. Continual rotation about the metal-ligand bonds was also avoided, ensuring that the orientation ofboth the imidazole and tyrosine rings and glutamate residues(methyl group rotors) was such that the position of the linking Cbatoms at the quantum/protein interface was preserved in an ap-proximate way. This was important for docking the optimizedactive cluster back into the protein environment accurately.
Results and discussion
Optimized structures and J values of RNRox
and RNRred
Calculated bond lengths, energies for the different spinalignment states, and exchange coupling constants aregiven in Table 1 for RNRox and RNRred model clusters,along with the accompanying X-ray structural data forthe characterized proteins. A ferromagnetic spin align-ment (RNRox, Smax=5; RNRred, Smax=4) is referred to
as high spin (HS); the corresponding antiferromagneticalignment (spin-flipped, MS=0) as broken symmetry(BS). BS geometries are RMSD matched [42] against theactive sites from the E. coli data (excluding the hydrogenatoms from the fitting algorithm). Heisenberg exchangecoupling constants (J) are calculated using EHS(Smax=S1+S2)–EBS(MS=S1–S2)=–4JS1S2 (derived fromH ¼ �2J*
s1 �*s2) where S1 and S2 are the site spin vectors
on the adjacent subunits, formally associated with theferric (S1, S2=5/2) or ferrous (S1, S2=4/2) ions and EHSand EBS are the gas phase energies of the HS and BSstates, respectively [22].
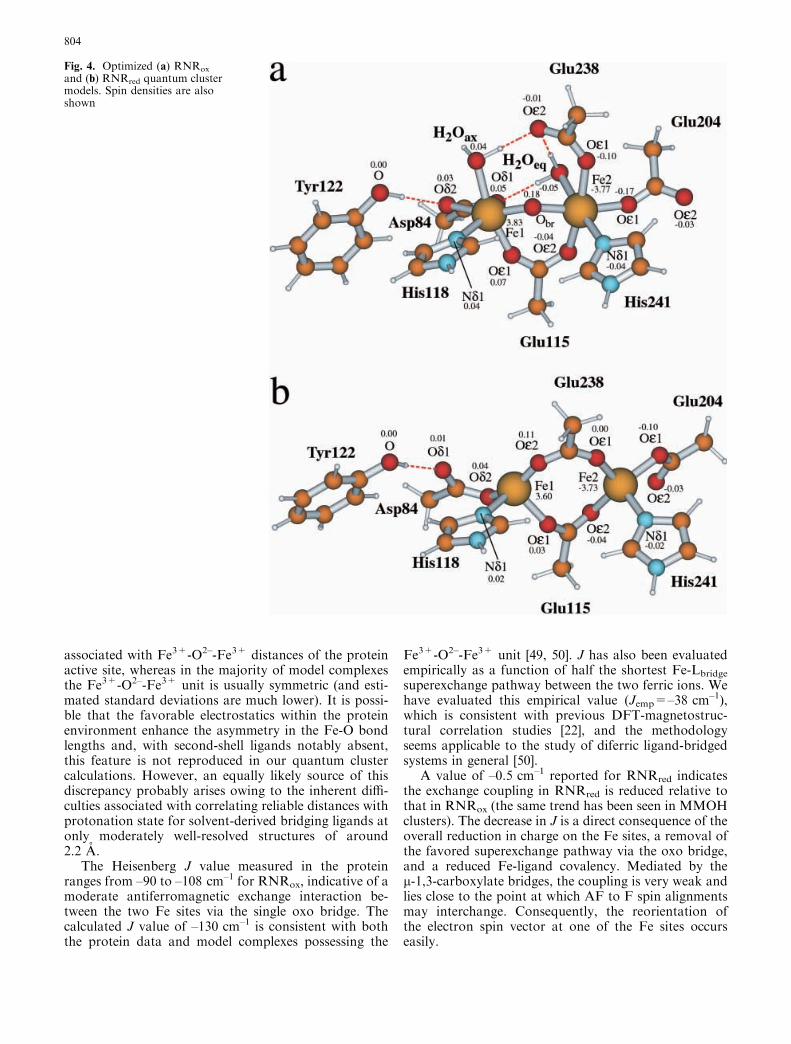
Optimized geometries and net spin densities of thequantum active sites are shown in Fig. 4 and comparewell with the known crystal data from E. coli [25, 26].Geometries for the BS and HS states give identicalRMSD fits to the experimental structures for RNRox
(0.9 A) and RNRred (0.69 A), respectively, and suggestthe geometry of the active site is relatively insensitiveto the reorientation (or flipping) of electron spins at oneof the Fe sites. Calculated Fe1-Fe2 distances for RNRox
(3.41 A) and RNRred (4.16 A) are reasonable, althoughin RNRred the Fe1-Fe2 distance is a little long comparedto experiment (3.9 A), but similar to that observed in therelated stearoyl acyl carrier protein D9-desaturase (Fe-Fe=4.22 A) [24]. The distance of Tyr122 from Fe1 isalso well reproduced, although slightly overestimatedfrom the calculations in RNRred (by 0.2 A). CalculatedFe-H2Oax and Fe-H2Oeq distances match those of theprotein X-ray data and also agree with the range ofdistances typically associated with Fe3+-H2O bonds(2.1–2.3 A) from model complexes.
The majority of the calculated parameters thereforereproduce the experimental structure quite well. InRNRox, Fe1 has a coordination environment that hasbeen commonly described as octahedral, with bothcarboxylate oxygen atoms of Asp84 bound to Fe1. Theprotein data suggest Fe1-Asp84.Od2 is 2.5 A, to whichestimated standard deviations of 0.2 A are applicable.Our calculated distance of 2.5 A agrees with the X-raydata and suggests trigonal bipyramidal coordinationmay be more appropriate.
The bridging Fe1-Obr and Fe2-Obr distances are cal-culated to be 1.86 A and 1.91 A, respectively. The O2–
ligand is calculated to be evenly positioned between thetwo Fe sites at distances consistent with those observedin model complexes (1.7–1.9 A) [43] and other calcula-tions [22] of systems containing the Fe3+-O2–-Fe3+
species. The X-ray data, however, indicate much longerbond lengths of 2.17 A (to Fe1) and 2.06 A (to Fe2).These distances would be more typical of Fe3+-H2O(2.1–2.3 A) and Fe3+-OH– (1.9–2.1 A) bonds, respec-tively [44, 45, 46]. Fe1-Obr is long, even after allowingfor estimated standard deviations of 0.2 A or more.Resonance Raman spectroscopy has provided strongevidence that the bridging oxygen species is O2– [47, 48]and so the protonation state of the bridging oxygenligand is well defined. There is also some asymmetry
Table 1. Comparison of calculated and experimental bond dis-tances (A) for the BS (MS=0) states of RNRox and RNRred. Theenergies (eV) of the BS and HS states as well as Heisenberg ex-change coupling constants of analogous MMOH clusters (cm–1)are also shown
aJ=Ae–BP (A=8.763·1011 cm–1, B=12.663 A–1, P=half theshortest superexchange pathway)bCalculated in the Fe3+-(OH–)2-Fe
3+ formcExperimental value from the Methylococcus trichosporium protein
803
associated with Fe3+-O2–-Fe3+ distances of the proteinactive site, whereas in the majority of model complexesthe Fe3+-O2–-Fe3+ unit is usually symmetric (and esti-mated standard deviations are much lower). It is possi-ble that the favorable electrostatics within the proteinenvironment enhance the asymmetry in the Fe-O bondlengths and, with second-shell ligands notably absent,this feature is not reproduced in our quantum clustercalculations. However, an equally likely source of thisdiscrepancy probably arises owing to the inherent diffi-culties associated with correlating reliable distances withprotonation state for solvent-derived bridging ligands atonly moderately well-resolved structures of around2.2 A.
The Heisenberg J value measured in the proteinranges from –90 to –108 cm–1 for RNRox, indicative of amoderate antiferromagnetic exchange interaction be-tween the two Fe sites via the single oxo bridge. Thecalculated J value of –130 cm–1 is consistent with boththe protein data and model complexes possessing the
Fe3+-O2–-Fe3+ unit [49, 50]. J has also been evaluatedempirically as a function of half the shortest Fe-Lbridgesuperexchange pathway between the two ferric ions. Wehave evaluated this empirical value (Jemp=–38 cm–1),which is consistent with previous DFT-magnetostruc-tural correlation studies [22], and the methodologyseems applicable to the study of diferric ligand-bridgedsystems in general [50].
A value of –0.5 cm–1 reported for RNRred indicatesthe exchange coupling in RNRred is reduced relative tothat in RNRox (the same trend has been seen in MMOHclusters). The decrease in J is a direct consequence of theoverall reduction in charge on the Fe sites, a removal ofthe favored superexchange pathway via the oxo bridge,and a reduced Fe-ligand covalency. Mediated by thel-1,3-carboxylate bridges, the coupling is very weak andlies close to the point at which AF to F spin alignmentsmay interchange. Consequently, the reorientation ofthe electron spin vector at one of the Fe sites occurseasily.
Fig. 4. Optimized (a) RNRox
and (b) RNRred quantum clustermodels. Spin densities are alsoshown
804
The calculated J values for oxidized and reduced RNRcan be compared with those calculated for the equivalentMMOH structures. An interesting pattern emerges inDFT-calculated J values as the nature and coordinationmode of the bridging ligand changes: the strength of theAF coupling decreases in accord with the sequencel-O2–(–130)<l-OH–(–35)<l-1,3-COO–(+13)<l-1,1-COO–(+32) (the first two values are for the RNR andMMOH diferric systems; the latter two for RNR andMMOH diferrous species). The trend is consistent withthe corresponding experimental measurements (–90 to–108 for RNRox, –4 to –10 for MMOHox, –0.5 forRNRred, +0.35 for MMOHred) [12]. The calculatedJ values for these binuclear non-heme iron systems arecloser to experiment compared to J values we have cal-culated previously for other spin-coupled systems [51, 52]and it appears that the PW91 exchange-correlationfunctional is particularly promising in this respect, espe-cially when one considers the small energy differencesinvolved between the BS and HS states.
Interaction with the protein environment
Table 2 quantifies the total electrostatic interaction en-ergy of the active-site cluster in the protein (Epr), whichcan be broken down into contributions from a reactionfield component (Er) and protein field component (Ep)
1.Er represents the energy term due to the polarization ofthe protein and solvent dielectric induced by the chargedistribution in the active-site cluster in the protein en-vironment. Large values of Er are indicative of a fairlystrong electrostatic interaction between the active-sitecluster and the protein and solvent dielectric polariza-tion. Similarly, Ep represents the energy contributionarising from the charge distribution in the protein resi-dues acting on the diiron cluster. Also shown for com-parison in Table 2 is the equivalent analysis for theneutral MMOHox [Fe
3+-(OH–)2-Fe3+] and MMOHred
clusters in chain D of Methylococcus capsulatus.Let us first consider the energetic effects associated
with RNR. Compared to Er, the Ep contribution to Epris small (22.4% for RNRox, 10.1% for RNRred), sug-gesting the active site is only weakly connected to thelocal amino acid environment. The active site geometries
are not fixed; rather, they are easily transformed intoother conformations and they do not have to overcomelarge energetic barriers associated with strong bonds tothe protein environment to do so. By comparison, Ercontributes 77.6% (RNRox) and 89.9% (RNRred) to Epr,indicating that the electrostatic interaction between theprotein/solvent dielectrics and the active site must befairly strong. An Ep contribution of similar magnitude isalso noted for the MMOH clusters and the same trendof stabilization in Er appears evident on conversion fromMMOHox to MMOHred, although the effect is reduced(compared to RNR).
Following generation of the reduced form of the en-zyme, the subsequent step in the RNRred catalytic cycleis the reaction of the diiron cluster with O2. RNRred isknown to react with O2 directly, while MMOHred re-quires component B (MMOB) to bind for a more fa-vorable rate of reaction with O2 (via a three-foldenhancement in rate) [53, 54]. The reduction in the Epterm that accompanies the RNRox to RNRred conver-sion means that the protein field effect on the active sitebecomes weaker; the reaction field for RNRred is morestabilizing than for RNRox in the protein. Given that thefolds of the local protein environment for RNR andMMOH are similar, and assuming the two proteinsdisplay similar modes of operation, then one also mightanticipate the same type of energetic trend in Er and Epaccompanying the MMOHox to MMOHred conversion.The equivalent trend in the Er and Ep terms cannot beseen in the MMOH clusters. One reason for this ap-parent difference might be that the active sites ofRNRred and MMOHred are structurally different. Wespeculate that the same type of energetic trend may onlybecome apparent after the docking of MMOB toMMOHred, which is known to modify the structure ofMMOHred in a manner yet to be established.
Comparison of active site-peptide interactionswith MMO
The effect of the protein environment on the RNRox
active site is shown in Fig. 5, where the total Ep is de-composed to individual residues, the majority of whichlie within and around the bundle of four a-helices thatencompass the active site. The breakdown to individualresidues outlined in Table 2 is arranged in order of in-creasing amino acid residue number extracted from thesequence of the RNR protein. Negative interaction en-ergies are stabilizing; positive interaction energies aredestabilizing. For each residue in RNR, its cognateresidue in MMOH is also shown where matching basedon sequence alignment and structural comparison per-mits. Given that the active sites in both proteins arefound within a bundle of four helices and seem to dis-play similar local environments, the most apparent en-ergetic differences would appear due to the directreplacement of one residue in the RNR protein by itscognate residue in MMOH. The sequence matching of
1The ESP charges of the active site polarize the protein and solventdielectric regions and produce a reaction field that interacts with allcharges at the atom centers of the active-site cluster, giving rise tothe (Er) energy term. The protein charges also interact with thecluster atom charges by generating a protein field potential.The Coulomb field generated by the protein charges is screened bythe protein and solvent dielectrics, giving rise to the Ep energy term.The total interaction energy (Epr) of the active-site cluster in theprotein and solvent environment is equal to the sum of reactionfield and protein field contributions, Epr=Er+Ep. In a pure solventenvironment, the protein field term is reduced to zero and, from acontinuum solvent description alone, the energy term that arises(Es) equals the reaction field energy term. This is an appropriatereference energy from which one can evaluate the energetic effectsof the local protein field term
805
the RNR and MMOH proteins therefore enables anenergetic comparison of structurally equivalent residues;by assuming a degree of structural similarity between thetwo proteins, an approximate measure of how mucheach residue replacement costs energetically can begauged.
From the electrostatics profile for RNRox (Fig. 5),the residues with the largest stabilizing interaction en-ergy with the active site are, in order of decreasing in-teraction: Asp237 (–3.37 kcal/mol), Gln43 (–1.83),Trp111 (–1.77), Ser114 (–1.68), Phe208 (–1.52), andSer121 (–1.12). The effects of these residues are derivedprimarily through the interaction of their respectiveamino acid sidechains with the active site. Backbonecontributions are minor. The most prominent destabili-zation arises from the backbone component of His118(+1.95). Because of its incorporation in the active sitequantum cluster, the sidechain component is 0 by con-struction. On conversion to RNRred, only three of thesix stabilizing interactions noted in RNRox remain:Asp237 (–2.96), Trp111 (–2.71), and Gln43 (–1.42).Tyr122 (–1.13) also makes a backbone contribution tothe overall stabilization, consistent with Tyr122 beingcloser to the active site in RNRred (the Tyr122 sidechainis part of the quantum active site so its sidechain Ep termis 0). While the destabilization effect associated withHis118 (+1.95) appears unaffected, an additional sourceof destabilization comes from Gln87 (+1.32). The originof the destabilization is traced to the orientation of the
Gln87 sidechain, which has rotated around its Cd-Ccbond by 180� to form its other rotamer, enhancingbackbone C=O repulsion with the partially unsaturatedGlu238 sidechain.
The amino acid sidechains surrounding the RNRox
active site cluster are shown in Fig. 6 and the location ofeach residue is depicted by the following convention:(f) signifies the residue appears in front of the mean
Table 2. Breakdown of the protein field energy (kcal/mol) to individual residues based on sequence alignment of structurally equivalentresidues in RNR (E. coli) and MMOH (M. capsulatus). Only the major residues are shown
Ea Residue RNRox RNRred Residue MMOHox MMOHred
Total Side Back Total Side Back Total Side Back Total Side Back
aEp=total protein field component; Er=total reaction field component; Epr=total electrostatic interaction energy in protein. See maintext and footnote 1 for a detailed discussion of terms
Fig. 5. Decomposition of the protein field interaction energy toindividual residues for RNRox in chain A of E. coli
806
Fe-O-Fe-Glu115 plane, (b)=behind the plane, and(p)=approximately in this plane. The overall distribu-tion bears some resemblance to that seen previously forMMOH [23]. The lower half of the active site appearsembedded in a crescent of charged and polar residues,which serve to limit the mobility of the bottom portionof the diiron site. It therefore seems unlikely that Glu115directly participates in catalysis since surrounding resi-dues His118 and His241 are anchored to the protein(Fig. 1a and b) by hydrogen bonds to Asp237, Ser114,and Gln43. By contrast, the density of protein residuesaround the upper portion of the cluster is relativelysmall. Few residues are therefore capable of constrainingthe upper portion of the cluster, particularly aroundTyr122, Asp84, and Glu238. This type of electrostaticpicture would be consistent with speculation regardingRNR function: the position of Tyr122 changes in re-sponse to the needs of the redox state of the diironcluster, Asp84 is capable of using either of its Od atomsto mediate coupled electron and proton transfer betweenTyr122 and the diiron site, and Glu238 carboxylateshifting from one coordination mode to anotherswitches on or off the interaction between Tyr122 andAsp84.
The detailed energetics associated with the residueshighlighted in Table 2 for RNR have been comparedand contrasted with the energetics associated withequivalent residues in MMOH (see Fig. 1). Positive ornegative energy terms are indicated in parentheses. Inthe context of the diiron site of RNR, the single unco-ordinated Glu204 oxygen is hydrogen bonded to Trp111(–); in MMOHox, the same hydrogen-bonding role isfulfilled by Gln140 (–), although in MMOHred theequivalent hydrogen bond to Glu209 is inherently veryweak. Recent findings for MMOHred support a struc-tural water as the more appropriate hydrogen bonding
partner to Glu209 [55]. The energetic effects of Trp111and Gln140 are both similarly stabilizing and replace-ment of one by the other should be of little energeticconsequence. In RNR, Ser114 (–) directly replacesAsp143 (–) in MMOH. Asp143 (through sidechain andbackbone interactions) is one of the residues in MMOHresponsible for hydrogen bonding and the subsequentorientation of the imidazole rings of the first-shell Hisresidues. Asp242 in MMOH hydrogen bonds to theother imidazole and is analogous to Asp237 in RNR.The imidazole rings have been suggested as one possibleroute through which electrons access the active site and aspecific orientation of the histidine sidechains may becritical in this respect. In RNR, the hydrogen bond toHis241.N�2 that orients the imidazole ring uses tworesidues, Ser114 (–) and Gln43 (–). In MMOH, both theAsp143 sidechain and backbone of Asp242 (–) appearnecessary. The summed energy stabilization of Gln43and Ser114 in RNRox (–3.51) and RNRred (–1.89) issomewhat equivalent to Asp143 and Asp242 in MMO-Hox (–2.69) and MMOHred (–4.30).
Of all the amino acid residues in the near vicinity ofthe active site, with its role in the production andpropagation of the key radical, Tyr122 (–) would appearthe most critical. The cognate residue in MMOH isCys151 (+). Cysteine residues are equally capable ofproducing stable amino acid radicals [2, 56]. UnlikeTyr122, whose relative position changes in response tothe redox state of the diiron cluster, the position ofCys151 (which is over 7 A from the nearest Fe of theactive site) appears insensitive to the redox state ofMMOH and not involved in electrostatic interactionswith the cluster. By virtue of the fact that it is closer,Tyr122 stabilizes the active site of RNR more thanCys151 does in MMOH.
From the calculated protein field decomposition,Phe208 (–) in RNR is the direct analogue for Thr213[(+) in MMOHox, (–) in MMOHred] in MMOH, and thelatter has been implicated as important for the deliveryof protons to the MMOHox active site [23, 57] (necessaryto generate MMOHred). While Phe208 may not be im-portant for proton access in RNR, its location appearscritical in gating access to the active site of RNR (Figs. 1and 6). Its hydrophobic properties would suggest itprevents the Glu238 sidechain from being tied up inhydrogen bonds to solvent molecules that may try topenetrate the active site or to other protein residues lyingclose nearby (of which there are relatively few). Phe208therefore would appear to act as the primary blockingresidue that affords Glu238 the opportunity to carb-oxylate-shift back and forth as required. Also, residuesPhe212 (–) and Ile234 (–) in combination with Phe208form a hydrophobic pocket close to the active site. Thishydrophobic pocket is similar in some respects to that inwhich an exogenous acetate becomes trapped and pro-tected from dissociation in the structure of MMOHox
from Mc.In RNR the source and mechanism of the single
electron-transfer step in which the Fe(IV)Fe(IV) species
Fig. 6. Approximate distribution of the charged and polar residueswith respect to the active site of RNRox. See text for definitions of(b), (p), and (f) terms. Atoms are identified by color: Fe, green, N,blue; O, red; C, gray; H, white. Figure produced using MOL-SCRIPT [64]
807
is reduced to the Fe(IV)Fe(III) species is a poorly un-derstood portion of the activation cycle. Electrontransfer has been proposed to be mediated by a chain ofhydrogen-bonded residues comprising the near-surfacethird-shell Trp48, a second-sphere Asp237, and the first-shell His118, and recent experimental studies supportTrp48 as the physiological electron donor [58, 59]. Fromour calculations, the total stabilization associated withthese residues in RNRox is –1.10 kcal/mol; in RNRred,their combined effect reduces to –0.61 kcal/mol. Thecognate residues in MMOH are Arg245 (based onstructural comparison of the various proteins), Asp242,and His147, the latter two residues being identified fromthe amino acid sequence comparison. The sum contri-bution of these three residues totals –1.35 kcal/mol inMMOHox and –2.64 kcal/mol in MMOHred, and is,overall, one of stabilization much like in RNR.
For the 18 cognate residues of RNR and MMOHoutlined in Table 2, their sum contribution appearsfairly constant for the oxidized and reduced states ofboth proteins (RNRox=–7.78, RNRred=–4.0, MMO-Hox=–6.81 and MMOHred=–7.55 kcal/mol). From asequence comparison of the two proteins, even thoughthe vast majority of the 18 amino acid residues in RNRdiffer from their cognate counterparts in MMOH, thecombined energetic sum of the 18 residues for bothproteins is nearly equivalent (to within a few kcal/mol).Of those residues that appear in the second shell, only 2out of 15 (Asp237 and Ile234) remain conserved acrossboth proteins (this excludes the residue sidechains di-rectly bound to the diiron site, which are mostly con-served). These two conserved residues are noted to exertsimilar energetic effects to their equivalent residues inMMOH (Asp242 and Ile239). From Table 2, the sub-stitution of several amino acids within a specific guestprotein by several different amino acids from a differentbut functionally and structurally related host proteinwould appear energetically plausible. In addition to theobvious interest from the viewpoint of further calcula-tions, the direct replacement of one or several of theresidues we have highlighted for RNR with its equiva-lent residue from MMOH (or vice versa) would be ofinterest from an experimental standpoint and may pro-vide additional insight into the relationship between theRNR R2 and MMOH proteins.
Concluding remarks
The optimized geometries and exchange coupling inter-actions calculated for our RNRox and RNRred modelclusters reproduce the known experimental structuresand J values in a reliable fashion. While conceivablythese simple gas phase calculations may not tell us ev-erything about Nature, they are a necessary prerequisiteto performing calculations in the full protein and solventenvironment. With respect to the latter, both RNR andMMOH are found to have similar interaction energies(in terms of reaction field and protein field components)
in the protein, consistent with the overall similarity incomposition of active site structures and local fold of thebundle of four a-helices in which each active site is foundin their respective proteins. The important residue-clus-ter interactions selected out by our electrostatics profilessuggest the similarity in protein field effects is due tocognate residues surrounding the active sites in bothproteins. The replacement of one or more residueswithin the protein amino acid sequence does not appearto adversely affect the protein field energetics. Both theRNR and MMOH protein structures suggest that sev-eral residues are quite easily interchangeable withoutany major disruptions to the structural integrity of theprotein. This would certainly rationalize the differentresidue combinations found in the four a-helices ofRNR and MMOH and may explain the prevalence ofthe four-helix bundle across the broader family of bi-nuclear non-heme iron proteins. If direct replacement ofa single residue can occur without loss of proteinstructure, and the protein structure is clearly importantfor the subsequent direction of the chemistry and theformation of different reaction intermediates, the con-version of a diiron protein initially displaying RNRfunctionality or characteristics to a diiron protein withMMO functionality or characteristics should, as such,also be a relatively facile process (providing a detailedworking knowledge of the residues required to bemodified is known). This type of residue substitutionand modification of function proposed as possible fromthis energetic analysis would be consistent with recentexperimental findings, where an altered form (W48F/D84E) of the RNRred protein affords a peroxo-type in-termediate with a spectroscopic signature characteristicof that observed in MMOH, notably different to wild-type RNR in which the spectroscopic signal is absent[17, 60, 61, 62]. Furthermore, in another mutant of R2,F208Y, hydroxylation of a Tyr208 residue to Dopa isobserved. The X-ray structure is suggestive of a possiblereaction geometry for a hydroxylation reaction cata-lyzed by a modified R2 diiron site and reminiscent of ageometry which might be relevant for the hydroxylationreaction catalyzed by MMOH [63].
Acknowledgements This work was supported by N.I.H. grantGM43278 to L.N. We thank J.M. Bollinger, Jr. for useful discus-sions and for providing preprints of manuscripts and unpublisheddata prior to publication. D. McRee is acknowledged for use of theXtalview progams, D. Bashford and V. Dillet for use of the MEADand decomposition codes, respectively, and we are grateful toE.J. Baerends and the Amsterdam group for use of the ADF codes.
References
1. Reichard P (1993) Science 260:1773–17772. Stubbe J, van der Donk WA (1998) Chem Rev 98:705–7623. Stubbe J (1990) J Biol Chem 265:5329–53324. Sjoberg B-M, Graslund A (1983) In: Theil EC, Eichorn GL,
Marzilli LG (eds) Advances in inorganic biochemistry. Else-vier, Amsterdam, pp 87–110
5. Cory JG (1988) Adv Enzyme Regul 27:437–455
808
6. Sato A, Bacon PW, Cory JG (1984) Adv Enzyme Regul22:231–241
22. Lovell T, Li J, Noodleman L (2001) Inorg Chem 40:5251–526623. Lovell T, Li J, Noodleman L (2001) Inorg Chem 40:5267–527824. Lindqvist Y, Huang W, Schneider G, Shanklin J (1996) EMBO
J 15:4081–409225. Nordlund P, Eklund H (1993) J Mol Biol 232:123–16426. Logan DT, Su X-D, Aberg A, Regnstrom K, Hajdu J, Eklund
H, Nordlund P (1996) Structure 4:1053–106427. Insight (1995) Insight II users guide. Insight, San Diego28. Pulver SC, Tong WH, Bollinger JM Jr, Stubbe J, Solomon EI
(1995) J Am Chem Soc 117:12664–1267829. Li J, Nelson MR, Peng CY, Bashford D, Noodleman L (1998)
J Phys Chem A 102:6311–632430. Free University of Amsterdam (1997) ADF 2.3.0. Department
of Theoretical Chemistry, Free University of Amsterdam31. Snijders JG, Baerends EJ, Vernooijs P (1982) At Nucl Data
Tables 6:48332. Vernooijs P, Snijders JG, Baerends EJ (1981) Slater type basis
functions for the whole periodic system; internal report. FreeUniversity of Amsterdam
33. Krijn J, Baerends EJ (1984) Fit functions in the HFS method;internal report (in Dutch). Free University of Amsterdam
34. Boerrigter PM, te Velde G, Baerends EJ (1988) Int J QuantumChem 33:87–113
35. te Velde G, Baerends EJ (1992) J Comput Phys 99:84–98
36. Versluis L, Ziegler T (1988) J Chem Phys 88:322–32837. Schlegel HB (1987) In: Lawley KP (ed) Ab initio methods in
quantum chemistry – I. (Advances in chemical physics, vol 67)Wiley, New York, pp
38. Vosko SH, Wilk L, Nusair M (1980) Can J Phys 58:1200–121139. Perdew JP, Chekavry JA, Vosko SH, Jackson KA, Perderson
MR, Singh DJ, Fioihais C (1992) Phys Rev B 46:6671–668740. Noodleman L (1981) J Chem Phys 74:5737–574341. McGrady JE, Lovell T, Stranger R (1997) J Phys Chem A
101:6265–627242. McRee DE (1999) J Struct Biol 125:156–16543. Kurtz DM Jr (1990) Chem Rev 90:535–60644. Armstrong WH, Lippard SJ (1984) J Am Chem Soc 106:4632–
463345. Zang Y, Pan G, Que L Jr, Fox BG, Munck E (1994) J Am