See discussions, stats, and author profiles for this publication at: http://www.researchgate.net/publication/44616550 Dephosphoryiation of purified brain tyrosine hydroxylase by rat striatal extracts ARTICLE in NEUROCHEMISTRY INTERNATIONAL · JANUARY 1983 Impact Factor: 2.65 · DOI: 10.1016/0197-0186(83)90016-5 · Source: PubMed CITATIONS 2 DOWNLOADS 15 VIEWS 58 3 AUTHORS: Mitchell A Lazar University of Pennsylvania 292 PUBLICATIONS 33,204 CITATIONS SEE PROFILE Joachim Raese Kaweah Delta Health Care District 61 PUBLICATIONS 821 CITATIONS SEE PROFILE Jack D. Barchas Cornell University 338 PUBLICATIONS 10,798 CITATIONS SEE PROFILE Available from: Joachim Raese Retrieved on: 13 September 2015
0022-3565/81/2163-0647$02.OO/OTHE Jouaa*i. OF P,u.ucacol.ooY AND Exi’uuau�smL TsaaapxuTicaCopyright C 1981 by The American Society for Pharmacology and Experimental Therapeutics
Tyrosine Hydroxylase: Studies on the Phosphorylation of aPurified Preparation of the Brain Enzyme by the Cyclic AMP-Dependent Protein Kinase1
ARTHUR M. � JOACHIM D. RAESE, MITCHELL A. LAZAR and JACK 0. BARCHAS4
Nancy Pritzker Laboratory of Behavioral Neurochemistry, Department of Psychiatry and Behavioral Sciences, Stanford University School ofMedicine, Stanford, California
Accepted for publication December 1 0, 1980
Edelman, Arthur M., Joachim D. Raese, Mitchell A. Lazarand Jack D. Barchas: Tyrosine hydroxylase: Studies on thephosphorylation of a purified preparation of the brain enzymeby the cyclic AMP-dependent protein kinase. J. Pharmacol.Exp. Ther. 21 6: 647-653, 1 981 .
Tyrosine hydroxylase [L-tyrosine, tetrahydropteridine: oxygenoxidoreductase (3-hydroxylating); EC 1 .1 4.1 6.2] (TH) was pu-rified from bovine corpus striatum. The purification involvedsequential DEAE cellulose, hydroxylapatite and CM SephadexC-50 chromatography, followed by glycerol density gradientcentrifugation. Final preparations appeared to be 90 to 100%pure as judged by polyacrylamide gel electrophoresis under
denaturing conditions in acetic acid-urea. The enzyme wasestimated to have a minimum molecular weight of approxi-mately 60,000 daltons. Purified TH could be activated in vitroby incubation with magnesium adenosine triphosphate and thecatalytic subunit of cyclic AMP-dependent protein kinase(ATP/protein phosphotransferase; EC 2.7.1 .37). When thefinal purified preparation of TH was incubated under theseconditions utilizing [y-32P]ATP, it was found to incorporate 0.7to 0.9 mol of phosphorus/mol of protein. These results suggestthat the activation of TH in the presence of phosphorylatingconditions is due to its phosphorylation by cyclic AMP-depen-dent protein kinase.
Tyrosine hydroxylase (TH) catalyzes the initial and most
probably rate-limiting step of catecholamine biosynthesis (Na-
gatsu et al., 1964; Levitt et al., 1965). Knowledge of the regu-
lation of this enzyme is crucial, therefore, for an understanding
of catecholamine synthesis in the adrenal gland and in the
peripheral and central nervous systems.
TH is subject to multiple modes of regulation. The enzyme
is activated in vitro by phospholipids (lloyd and Kaufman,1974; Raese et al., 1976) and polyanions such as heparin
(Kuczenski and Mandell, 1972; Katz et al., 1976) and is inhibited
by the catecholamine end products (Nagatsu et al., 1964). TH
is also activated by phosphorylation reactions catalyzed by the
cyclic AMP-dependent protein kinase (Harris et al., 1975; Lloyd
and Kaufman, 1975; Lovenberg et al., 1975; Morgenroth et al.,
1975; Raese et al., 1977) and by a cyclic nucleotide independent
protein kinase (Raese et al., 1979b). We have previously pre-
sented evidence using a partially purified preparation of TH
Received for publication December 12, 1978.
1 � work was supported by National Institutes of Mental Health Program-
Project Grant MH 23861 as well as by National Institutes of Drug Abuse GrantDA 01207 and the ONR. A preliminary report of portions of this work waspresented at the Fourth International Catecholamine symposium in PacificGrove, CA, Sept. 17-22, 1978.
2 Present address: Department of Pharmacology, SJ-30, University of Wash-ington &hool of Medicine, Seattle, WA 98195.
3 Held a National Institutes of Mental Health Biosciences Fellowship from
Grant MH 15147.
4 Recipient of Research Scientist Development Award MH 24161.
that the enzyme can be phosphorylated in vitro (Raese et aL,
1977). We here present further data indicating the capability
for phosphorylation of bovine corpus striatal TH by cyclic
AMP-dependent protein kinase. Some of these results have
been previously reported in preliminary form (Edelman et al.,
1978; Raese et al., 1979a,b).
Enzyme Purifications
Methods
TH. Bovine corpora striata were freshly dissected at a local slaugh-terhouse and frozen on Dry Ice. The tissue was either homogenized onthe same day or stored at -70#{176}Cfor subsequent purifications. All ofthe following steps were performed at 4#{176}C.Up to 1 kg of tissue washomogenized in 3 volumes of a buffer containing 25 mM Tris HCI(pH 7.5), 15 mM /3-mercaptoethanol, 1 mM disodium(ethylene dini-
trilo)tetraacetic acid (EDTA), 0.05 M NaC1 and 10% glyceroL Thehomogenate was centrifuged 2.5 hr at 30,000 x g. The supernatant wasdirectly applied to a DEAE Cellulose (Sephacel; Pharmacia Fine Chem-icals, Piscataway, NJ) column equilibrated with the homogenizationbuffer. The packed bed volume of the column was approximately equalto the volume of supernatant applied. The column was washed withthe homogenization buffer until the absorbance at 280 am of theeffluent was �O.O& absorbance units (-4 column volumes). This wasfollowed by approximately 2 column volumes of this buffer containing0.1 mM cyclic AMP to elute the catalytic subunit of protein kinase(Kinzel and Kubler, 1976). TH was then eluted with the homogenization
buffer containing a linear gradient of NaCl, 0.05 to 1.0 M. The totalvolume of the gradient was 2.5 times that of the packed bed. Enzymat-
ically active fractions, eluting between 0.13 and 0.35 M NaC1, were
pooled and dialyzed exhaustively against a buffer containing 25 mMpotassium phosphate (pH 6.8), 15 mM fl-mercaptoethanol, 1 mMEDTA and 20% glyceroL Insoluble material was removed by centrifu-gation and the resultant supernatant was applied to a hydroxylapatite(Bio-Rad Laboratories, Richmond, CA) column (2.6 cm X 28 cm)equilibrated with the same buffer. The column was washed with this
buffer until the absorbance at 280 am ofthe effluent was approximately�O.O6 absorbance units. The enzyme was eluted with a linear potassium
phosphate gradient (1.4 1), 25 to 400 mM, pH 6.8, containing 15 mM
fl-mercaptoethanol, 1 mM EDTA and 20% glycerol. Active fractions,eluting between 0.05 and 0.16 M potassium phosphate, were pooled and
dialyzed exhaustively against a buffer containing 50 mM potassium
phosphate (pH 6.8), 1 mM dithiothreitol (DTT), 1 mM EDTA and 20%
glyceroL The enzyme was applied to a CM Sephadex C-50 (Pharmacia)column (2.6 cm x 60 cm) equilibrated with the same buffer. The columnwas washed with this buffer until the absorbance at 280 am was <0.04
absorbance units. The enzyme was then eluted with a linear potassium
phosphate gradient (1.5 1), 50 to 250 mM, pH 6.8, containing 1 mM
DTT, 1 mM EDTA and 20% glycerol. TH activity was eluted between
approximately 0.10 and 0.14 M potassium phosphate.Enzyme was either stored at 4#{176}Cor concentrated (approximately 20-
fold) by vacuum dialysis and purified further after exchange of buffercomponents and reduction of the glycerol concentration by density
gradient centrifugation in 9 to 34% (w/v) linear glycerol gradientscontaining 50 mM potassium phosphate (pH 6.8), 1 mM DTT and 1
mM EDTA. Centrifugation was performed in a Beckman L2-65B
ultracentrifuge at 40,000 rpm for 20.5 hr at 4#{176}Cusing a Spinco SW4O
rotor. Fractions containing TH activity (sedimenting at -9S) from theglycerol gradient were pooled and stored at 4#{176}C.
Several aspects of the purification could be modified without appar-
ent effect. DEAE Sephadex could be substituted for DEAE cellulose;DTT (0.5-2 mM) could be substituted for /3-mercaptoethanol; andEDTA could be used at either 0.1 or 1.0 mM. However, during thehydroxyl.apatite chromatography, 10% glycerolgave better performancethan 20% in terms of binding of the enzyme to the column.
Protein kinase. The catalytic subunit of cyclic AMP-dependentprotein kinase is here referred to simply as protein kinase. All stepswere performed at 4#{176}C.Active fractions from the cyclic AMP elutionfrom the DEAE column (see TH Purification) were pooled, dialyzedagainst a buffer containing 50 mM potassium phosphate (pH 6.8), 1mM DTT, 0.1 mM EDTA and 30% glycerol and applied to a CMSephadex C-50 column (2.6 cm x 15 cm) equilibrated with the same
buffer. The column was washed with this buffer and the enzyme elutedwith a buffer containing 250 mM potassium phosphate (pH 6.8), 1 mM
DTT, 0.1 mM EDTA and 30% glycerol. The specific activity of this
preparation was 180,000 pmol/min/mg of protein using histone assubstrate (see Enzyme Assays below). This source of protein kinasewas used routinely for activation experiments. Alternatively, the en-zyme after CM Sephadex chromatography was incubated at 30#{176}Cfor30 mm with 9 mM MgCl2 and 94 �tM [y-�P]ATP and chromatographedon Sephadex G100 (2.6 x 95 cm) in a buffer containing 50 mM potassium
phosphate (pH 6.8), 1 mM DTT, 1 mM EDTA and 30% glyceroL Thisprocedure allowed the identification and removal of phosphate accept-lag contaminants present in the protein kinase preparation at the CM-Sephadex stage. The removal of minor contaminants by the G-100column produced essentially no change in the specific activity of the
enzyme. Protein kinase prepared by this method was homogeneous as
judged by polyacrylamide gel electrophoresis in either acetic acid-urea(Zahler, 1974) or in the sodium dodecylsulphate (SDS)-phosphatesystem of Weber and Osbom (1975). This preparation was used in theexperiments illustrated in figures 3 to 5. The enzyme was stored at 4#{176}C.At this temperature, the rate of decay of enzyme activity was less than10% per month.
Enzyme assays. TH was assayed essentially according to themethod of Waymire et al. (1971). A typical assay mixture contained:sodium 2-[N-morpholino]ethane sulfonate (pH 6.0, 0.24 M); beef livercatalase, 860 units (Worthington Biochemical Corp., Freehold, NJ);
excess sheep liver dihydropteridine reductase in 10 mM Tris-HC1, pH,7.4, purified through the second ammonium sulfate step, 2000 units
(defined according to Method I ofCraine et al., 1972); reduced NADPH,
0.1 �imol; L-[1)4C]tyrosine (51.9 mCi/mmol), 39 �zM; tetrahydrobiop-term, 1.2 mM and 25 �tl of enzyme from the various stages of purifica-
tion. Bovine serum albumin (BSA; Sigma Chemical Company, St.Louis, MO) was added to bring the protein concentration in each tubeto at least 2 mg/mi before the start of the reaction. The volume during
this phase of the reaction was 85 4 After 25 mm at 30#{176}C,0.5 �imol of3-iodotyrosine in 5 mM HC1 was added to stop TH activity. Excesspartially purified hog kidney decarboxylase (Waymire et al., 1971) wasadded with pyridoxal phosphate (final concentration, 0.1 mM) in 0.5 Mphosphate buffer (pH 8) containing 10% glycerol to bring the volume to
160 4 ‘4C02 was trapped and counted essentially according to theprocedure of Ichiyama et al. (1970). Biopterin (Regis Chemical Co.,
Morton Grove, IL) was reduced to tetrahydrobiopterin according to theprocedure of Lloyd and Weiner (1971).
For experiments illustrated in Figures 1 and 3, the assay conditions
employed were as follows: 0.1 to 0.2 M sodium acetate (pH 6) was thebuffer, the concentrations ofsubstrates were: tyrosine (15 �tM, fig. 1; 21
1iM, fig. 3) and tetrahydrobiopterin (160 �tM, fig. 1; 800 tiM, fig. 3). BSAwas not included. The reaction proceeded for 30 to 45 mm in a total
volume of 75 to 125 i1, after a 30-mm preincubation in the case of theexperiment shown in figure 1. The decarboxylase-pyridoxal phosphatesolution was added in 5 mM phosphate buffer (pH 7) containing 10%glycerol to bring the final volume to 150 to 200 �d. Other aspects of theassay were as outlined above.
Protein kinase was assayed according to the procedure of Witt andRoskoski (1975) with 0.12 mM ATP, 4.3 mM MgCl2 and 4.8 mg/mI ofHistone LI-A (Sigma). One unit of protein kinase activity is defined asthe amount of enzyme which catalyzes the transfer of 1 pmol of �Pfrom [y�nP]ATP to histone per rain.
Protein assays. Protein concentrations shown in table 1 were
determined by the fluorescamine procedure (Bohien et al., 1973). Insome experiments (illustrated in figs. 1-5), TH was radioactively meth-ylated using [‘4C]formaldehyde according to the procedure of Rice and
Means (1971) in order to quantitate small amounts of protein duringanalytical procedures. In general, this procedure, which involved con-
centration and dialysis against 0.2 M borate buffer (pH 9), caused
relatively minor losses of enzyme activity. ‘4C-labeling did not preventthe activation response of TH to phosphorylating conditions. For thestudies to determine the molar ratio of aP�labehag, both methylatedand unmethylated TH was utilized. For studies using unmethylatedenzyme, protein was determined by the fluorescamine procedure.
Electrophoresis in acid-urea gels. The procedure used was amodification of that of Zahler (1974). Gels containing urea and aceticacid were polymerized to be 6 or 6.5% acrylamide, using N,N’-diallyl-
tartardiamide as the cross-linking reagent (at 15% of the total acryl-amide). Samples (2-5 ,ig of protein) were prepared by dilution with anequal volume ofa dissociating solution containing 25% (v/v) acetic acid,50% (w/v) phenol, 2% (v/v) $-mercaptoethanol and 4 M urea, followedby incubation at 30#{176}Cfor 2 hr. Boiling was avoided as this tended to
produce additional bands, presumably due to acid hydrolysis. Electro-phoresis was performed as described by Zahier (1974). Staining was in0.25% (w/v) Coomassie blue in 10% (v/v) acetic acid and 10% (v/v)isopropanol for 4 to 6 hr. Gels were destained electrophoretically.
Molecular weights of enzyme subunits were estimated by a semilogplot of molecular weight (ordinate: log scale) vs. relative mobility
(abscissa: standard scale). Mobility was calculated relative to the mo-bility of deoxyribonuclease I. Molecular weights of the standards used
were taken as: rabbit muscle phosphorylase a: 92,500 pig heart fumar-am: 48,500; ovalbumin: 44,000 and deoxyribonuclease I from beefpancreas: 31,000 (Lindberg, 1967; Castellino and Barker, 1968; Klotz etal., 1975). For estimation of relative purity of TH, gels were scannedspectrophotometrically at 550 nra with a Gilford 2000 spectrophotom-eter equipped with a linear transport attachment. For liquid scintilla-
tion counting, gels were sliced into approximately 1.5 mm slices. Eachslice was digested at room temperature overnight in 1 ml of 2% (w/v)
a Picomoles of dopa per minute.b Dialyzed against homogenization buffer.
periodic acid and counted in 10 ml of Hydromix (Yorktown Research;Piscataway, NJ).
Determination ofphosphorus incorporation. The purified prep-
aration of TH (7 �g) was incubated for up to 240 rain at 30#{176}Cwith: 600units (3.6 big) of protein kinase purified through the Sephadex G-100step, 10 mM MgCl2 and 90 �M [y�P]ATP (35.3 �Ci/umol). The incu-bation volume was 0.32 ml. A 20-sal aliquot of the incubation mixturewas added to 100 �il of a solution of 2 mg/ml of BSA followed immedi-ately by 1 ml of 11% (w/v) ice-cold trichioroacetic acid (TCA). ‘4C-labeled purified TH (1 x i0� cpm, 3.0 ig) was then added for estimation
of recovery. The mixture was incubated at 0#{176}Cfor 30 rain and centri-fuged. The pellet was redissolved in 300 �il of ice-cold 0.25 N NaOH.The TCA precipitation and NaOH resolubilization was repeated twicemore. The final pellet was redissolved in 300 �l of 1 N NaOH at room
temperature. One hundred microliters were counted in 10 ml of Hydro-mix. The blank was a parallel procedure identical in all respects to thatdescribed above except that the appropriate buffer was substituted forTH. The TH used for these determinations was approximately 95%pure as judged by electrophoresis under denaturing conditions. Theminor contaminants (approximately 5% of the protein) did not detect-ably incorporate phosphorus.
Results
Table 1 illustrates a purification of bovine striatal TH. The
yield of this particular preparation was atypically low. A final
yield of 1 to 4% would be expected for this scheme. The
preparation showed good stability (approximately 30% loss per
month) when stored at 4#{176}C.
The final step in the purification scheme involved density
gradient centrifugation as is ifiustrated in figure 1. TH from the
CM-Sephadex step centrifuged in a linear 5 to 20% sucrose
density gradient yielded a sedimentation coefficient (S�,w) of
8.9S corresponding to a molecular weight by reference to known
standards (Schachman, 1959; Martin and Ames, 1961; Sober,
1968) of approximately 180,000. This will be referred to as the
9S form. A smaller peak of enzyme activity at 48 was also
observed. We have previously presented evidence that this
represents the dissociation of TH into subunits (Raese et al.,
1977). The size of the 4S form (45-50,000) suggested a tetra-
meric structure for the enzyme. The sedimentation behavior of
TH in linear 9 to 34% glycerol gradients was not detectably
different from that in linear 5 to 20% sucrose gradients. Glycerol
gradients were routinely used in the purification.
The purity of the 9S form of the purified preparation of TH
was evaluated by polyacrylamide gel electrophoresis in acetic
acid-urea. Figure 2 illustrates an enzyme preparation which was
apparently homogeneous. For this particular preparation, a
second density gradient was employed to remove minor con-
taminants. We have subsequently found that a second gradient
is effective to only a slight degree in this regard and therefore
this procedure was not included in subsequent preparations (for
example, table 1). The minimum molecular weight of the pun-
fled preparation determined by electrophoresis (as described in
“Methods”) was found to be approximately 60,000. The purity
of each preparation was quantified by spectrophotometric scan-
ning of the stained gels at 550 run. By this criterion, most
preparations were about 90% pure. Polyacrylamide gel electro-
phoresis was also performed according to the SDS-phosphate
system of Weber and Osborn (1975). This technique produced
similar results as the acid-urea gels.
Fig. 1 . Sucrose density gradient centrifugation of TH and activation byphosphorylating conditions. TH from the CM-Sephadex step was radio-actively methylated using [14C]formaldehyde (Rice and Means, 1971).The enzyme (70 ,.tg) was centrifuged in a 5 to 20% (w/v) linear sucrosedensity gradient. The gradient contained 50 mM potassium phosphate(pH 6.8), 0.1 mM EDTA, 1 mM DTT and 0.1 5 M KCI. Aliquots ofgradient fractions were preincubated with 1 25 U of protein kinase,purified through the CM-Sephadex step (see � ‘Methods’ ‘), 50 �M ATPand 2 mM MgCI2 for 30 mm at 30#{176}Cand then assayed for TH activity(#{149}.left ordinate) as described in “Methods.” The same fractionsincubated without protein kinase, ATP and MgCI2 served as control(A, left ordinate). Protein (0, right ordinate) was determined by the ‘4C-content of the fractions.
TABLE 1
Purification of bovine corpus striatal TH
Fraction Volume Protein Activity� Specific Activity Fold Purification Yield
ml mg units units/mg %
I. Supernatant of crude 2420 1 3794.0 765,688 56 1 .0 100.0homogenateb
A representative result of a labeling experiment with the
purified preparation is shown in figures 4 and 5. Figure 4
illustrates the incorporation of 32P into the purified TH prepa-
ration after incubation with protein kinase, Mg� and [-y-32PJ-
ATP. In addition, a fainter second protein band was phos-
phorylated. As is shown in a control gel (fig. 5), this represented
an autophosphorylation of the protein kinase present in the
reaction mix. This autophosphorylation of the catalytic subunit
of protein kinase occurred in a nonstoichiometric fashion (max-
imally 0.2 mol ofphosphorus per mol ofprotein). The low molar
ratio of protein kinase autophosphorylation could theoretically
be explained by the formation ofan acid-labile phosphohistidine
bond which has been reported to occur with the brain enzyme
by Kochetkov et al. (1976). In some gels (for example, fig. 4),
there was nonmigrating, apparently precipitated, material on
top of the gel which is characteristic of this electrophoretic
system (Zahler, 1974). The amount of this material varied
between electrophoretic runs of the same protein fraction and
did not represent proteins aside from the two major bands.
In order to determine the molar ratio of 32P incorporation,
the purified preparation of TB was incubated with [‘y-32P]ATP
and protein kinase and incubation conditions were adjusted so
that the reaction went to completion. The amount of phospho-
fl’s incorporated was determined by TCA precipitation as de-
scribed in “Methods” (Determination of Phosphorus Incorpo-
ration). Assuming a MW of 60,000, the purified enzyme prepa-
ration was found to incorporate 0.7 to 0.9 mol of phosphorus
per mol of protein. Under similar conditions but with a different
preparation of TB, a value of 0.7 was obtained.
Discussion
Fig. 2. Acid-urea polyacrylamide gel electrophorsis of the purifiedpreparation of TH. TH from the CM-Sephadex step was radioactivelymethylated and centrifuged in a linear glycerol gradient as describedin � Methods. � � The gradient contained 50 mM sodium phosphate (pH
6.8), 1 mM EDTA and 1 mM DTT. The 9S protein peak was againsubjected to glycerol gradient centrifugation in the same manner (after
removal of glycerol and exchange of buffer components by dialysis).
The second gradient contained 20 mM Tris-HCI (pH 8.5), 1 mM EDTA
and 1 mM DTT. Peaks of enzyme activity at 9S and 4S were obtained.Fractions were stored at - 20#{176}Cbefore electrophoresis. The 9S peakfrom the second gradient with a specific activity of approximately 5000pmol of dopa formed per mm/mg of protein was electrophoresed in
acid-urea gels as described in � � Methods. �
As illustrated in Figure 1, the final preparation ofTH retained
its ability to be activated by incubation with MgATP and the
catalytic subunit of cyclic AMP-dependent protein kinase. We
have noticed no systematic changes in the extent of this acti-
vation as a function of the stage of purification of TH. The
mechanism of the activation seems to involve an increase in the
affmity of the enzyme for the reduced pterin cofactor without
a change in Vmax (Lloyd and Kaufman, 1975; Lovenberg et al.,
1975; Edelman et al., 1978). By contrast, Joh et al. (1978) found
a Vmax increase without any Km changes for the rat striatal
enzyme.
TB from the CM-Sephadex step was incubated with protein
kinase and [‘y-32P]ATP, exhaustively dialyzed to remove labeled
ATP and centrifuged in a 9 to 34% glycerol gradient, a procedure
which separates TB from the bulk of the protein present at the
CM Sephadex step, from protein kinase, and from any residual
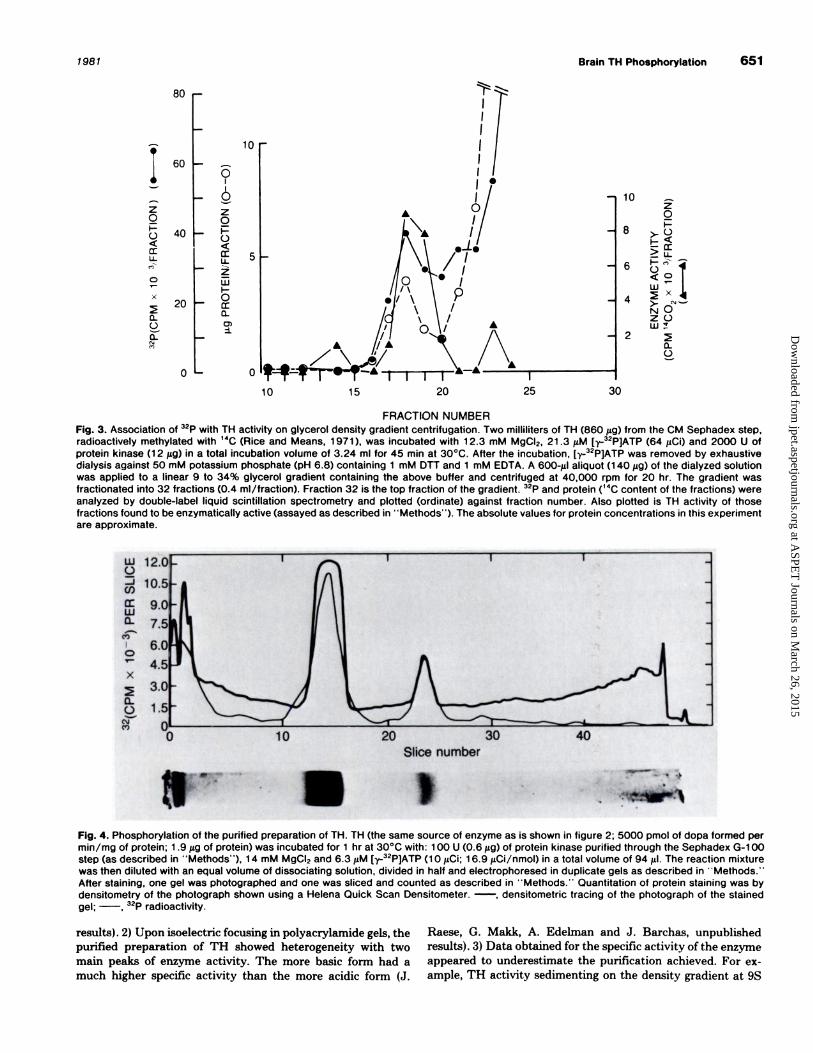
traces of [-y-32P]ATP. The result, shown in figure 3, was that
32P was associated with TH activity at 9S, indicating phos-
phorylation of the enzyme.
On density gradient centrifugation, bovine corpus striatal TB
may be present in catalytically active multiple forms depending
on the buffer composition in the gradient (Raese et al., 1977).
All forms retained the ability to be activated by phosphorylat-
ing conditions. The smallest form (4S) obtained had a MW of
45 to 50,000 by reference to the sedimentation of known stan-
dards included in the gradient and has been hypothesized to
represent monomeric TB. The data presented in this commu-
nication indicate that the 9S form had a minimum MW of
about 60,000 as estimated by gel electrophoresis under dena-
turing conditions. The 4S form, however, had the same electro-
phoretic mobility as the 9S form on SDS gels (data not shown).
This indicates that, although the methods do not precisely
agree with respect to subunit molecular weight, the 4S form
apparently represents a monomeric form of TB and not a
proteolytic product.
Several lines ofevidence indicate the possibility that a certain
portion of the TB molecules are recovered in the final prepa-
ration in an inactive state. 1) On density gradients, the protein
and TB activity peaks were not precisely superimposable. The
small protein peak at 9S reproducibly had a slightly greater
sedimentation coefficient than the enzyme activity peak (fig. 1
and 3). For example, the ratio of enzyme activity to protein
concentration calculated for the fractions through the 9S peak
of figure 1 is (taking fraction 14 as equal to 1.0 in arbitrary
units): 14, 1.0; 15, 1.0; 16, 2.4; and 17, 5.8. SDS gels run on the
density gradient fractions through the 9S peak area yielded
essentially identical staining patterns. That is, no extra protein
bands were observed whose presence could have accounted for
the lack of completely constant specific activity on the density
gradient (A. Edelman, J. Raese and J. Barchas, unpublished
Fig. 3. Association of 32P with TH activity on glycerol density gradient centrifugation. Two milliliters of TH (860 �sg) from the CM Sephadex step,radioactively methylated with 14C (Rice and Means, 1 971 ), was incubated with 1 2.3 mM MgCI2, 21 .3 1�M [y.-32P]ATP (64 1�Ci) and 2000 U ofprotein kinase (1 2 f.Lg) in a total incubation volume of 3.24 ml for 45 mm at 30#{176}C.After the incubation, [-y-32P]ATP was removed by exhaustivedialysis against 50 mM potassium phosphate (pH 6.8) containing 1 mM OTT and 1 mM EDTA. A 600-gil aliquot (1 40 �tg) of the dialyzed solutionwas applied to a linear 9 to 34% glycerol gradient containing the above buffer and centrifuged at 40,000 rpm for 20 hr. The gradient wasfractionated into 32 fractions (0.4 mI/fraction). Fraction 32 is the top fraction of the gradient. 32P and protein (14C content of the fractions) were
analyzed by double-label liquid scintillation spectrometry and plotted (ordinate) against fraction number. Also plotted is TH activity of thosefractions found to be enzymatically active (assayed as described in � � Methods’ ‘). The absolute values for protein concentrations in this experiment
are approximate.
12.(
&� 10!
�6.C
�4Ix
a.
C.) 1!
� C10 20 30 40
Slice number
h;#{149}� �
� :
Fig. 4. Phosphorylation of the purified preparation of TH. TH (the same source of enzyme as is shown in figure 2; 5000 pmol of dopa formed permm/mg of protein; 1 .9 �zg of protein) was incubated for 1 hr at 30#{176}Cwith: 1 00 U (0.6 �zg) of protein kinase purified through the Sephadex G-1 00
step (as described in ‘ ‘Methods’ ‘), 1 4 mM MgCI2 and 6.3 �M [‘y-32P]ATP (1 0 DCi; 1 6.9 �iCi/nmol) in a total volume of 94 il, The reaction mixturewas then diluted with an equal volume of dissociating solution, divided in half and electrophoresed in duplicate gels as described in � Methods,”After staining, one gel was photographed and one was sliced and counted as described in �‘Methods. ‘‘ Quantitation of protein staining was bydensitometry of the photograph shown using a Helena Quick Scan Densitometer. -, densitometric tracing of the photograph of the stained
gel; , 32P radioactivity.
results). 2) Upon isoelectnic focusing in polyacrylamide gels, the
purified preparation of TB showed heterogeneity with two
main peaks of enzyme activity. The more basic form had a
much higher specific activity than the more acidic form (J.
Raese, G. Makk, A. Edelman and J. Barchas, unpublished
results). 3) Data obtained for the specific activity ofthe enzyme
appeared to underestimate the purification achieved. For ex-
ample, TB activity sedimenting on the density gradient at 9S
Fig. 5. Autophosphorylation of protein kinase, Incubation was as described in the legend to figure 4, except that the appropriate buffer wassubstituted for TH. Processing of duplicate gels was as described in figure 4. -, densitometric tracing of the photograph of the stained gel;
, 32P radioactivity.
40
652 Edelman et al. Vol. 216
was usually cleanly separated from the bulk of the protein
sedimenting at 4S (fig. 1), suggesting that a much greater
purification was achieved by this procedure than that indicated
by the calculated increase in specific activity (Table 1). In
addition, the fmai specific activity is lower than that reported
for preparations from other tissues (Joh et al., 1978; Vulliet et
al., 1980). For example, Joh and co-workers give a specific
activity of approximately 252 nmol/min/mg for TH they purify
from rat striatum.
There are a number of possible explanations to account for
these observations. One would be that contaminating protein(s)
have not been adequately removed during the purification.
Another is the possibility of a lower turnover number for the
bovine stnatal enzyme as compared with TB isolated from
other tissues. Another report of a lower specific activity for the
bovine striatal enzyme was that of Uoyd and Kaufman (1974)
whose 40% pure preparation of TB from bovine caudate had a
specific activity of 0.95 to 2.2 nmol of dopa per mg/mm. Finally,
it may be that the enzyme as isolated contains a fraction of
denatured molecules (perhaps due to covalent modification for
example, by carbohydrate attachment or partial phosphoryla-
tion or perhaps due to lability under the conditions of the
purification) and a fraction of “native” molecules. We have in
fact observed an increased lability of TB after exposure to
phosphorylating conditions. That is a fraction of isolated TB
molecules may be inactive due to their endogenous phos-
phorylation. The molecular weights of these two forms are
expected to be the same; however, the sedimentation coeffi-
cients (perhaps due to a change in partial specific volume) and
isoelectric points could differ (points 1 and 2 above).
The association of 32P with TB activity and the phosphoryl-
ation of the purified preparation of TB by protein kinase are
illustrated in figures 3 and 4. Since the molar ratio was found
to be 0.7 to 0.9 mol of phosphorus per mol of protein, it is
therefore unlikely that a minor contaminating protein could
account for the observed phosphorylation. The molar ratio of
32P-labeling was similar with methylated or unmethylated TB
indicating that methylation with [‘4C] formaldehyde did not
artifactually introduce an ability of TB to be phosphorylated.
Bylund and Krebs (1975) observed that, with some proteins,
denaturating treatment resulted in a greater susceptibility to
phosphorylation while, with other proteins, the susceptibility
to phosphorylation was reduced. That TB might belong to the
latter class is suggested by the report of Markey et al. (1980)
who found that pheochromocytoma TB lost its ability to be
phosphorylated concomitant with loss of activity upon storage.
This suggests that the phosphorylation reported here with the
striatal preparation cannot be simply explained as a nonspecific
effect due to denaturation.
Although initial reports were negative (Lovenberg et al.,
1975; lloyd and Kaufman, 1975), evidence has now accumulated
from a number of laboratories that TH from a variety of species
and tissues is capable of being directly phosphorylated (Le-
tendre et al., 1977a,b; Raese et al., 1977, 1979a; Edelman et al.,
1978; Joh et al., 1978, 1979; Vulliet et al., 1979, 1980; Waymire
et al., 1979; Yamauchi and Fujisawa, 1978, 1979a,b; Markey et
al., 1980; Edelman et al., this communication). Most of these
reports presented evidence for phosphorylation specifically by
the cyclic AMP-dependent protein kinase. Letendre and co-
workers reported (1977a,b) that TB immunoprecipitated from
adrenals and superior cervical ganglia cultured in the presence
of 32P1 incorporated radioactive phosphate. The possibility ex-
ists that in vivo phosphorylation may not exclusively be cata-
lyzed by the cyclic AMP-dependent protein kinase. Data of
Yamauchi and Fujisawa (1979) indicated that in bovine adrenal
meduilary cytosol, the cyclic AMP-dependent protein kinase
was involved. A recent report (Raese et al., 1979b), however,
demonstrating phosphorylation in vitro by a cyclic nucleotide
independent kinase raises the possibility of regulatory controls
other than cyclic AMP being involved in TB phosphorylation.
An as yet unresolved issue is that ofthe subunit stoichiometry
of TB. The report of Joh et al. (1978) suggested that TH was
composed of three nonidentical subunits only one of which
could be phosphorylated. Similarly, Markey et al. (1980) re-
ported that one (offour) subunit is phosphorylated. Preliminary
data from our laboratory using a better resolving electropho-
retic system than in this communication suggest that the sub-
units of TB are closely related in size but nonidentical (J. D.
Raese, 0. Makk and J. D. Barchas, manuscript submitted). The
prediction from the work of Joh and co-workers and that of
Markey et al., however, would be that 0.2 to 0.3 mol of phos-
phorus per mol of protein (assuming subunits are present in
equimolar ratios) should have been obtained here rather than
0.7 to 0.9. Our results are in agreement with those of Vulliet et
al. (1980). The cause of this discrepancy is, at present, unclear.
To summarize, our results show that a purified preparation
of striatal TB can be phosphorylated in vitro by cyclic AMP-
dependent protein kinase which also activates the enzyme.
Furthermore, this is the first report on the stoichiometry of
phosphorylation of TH from brain. Although the purification
we describe results in low yield, it should allow for the routine
preparation of several milligrams ofthe striatal enzyme purified
to 90 to 100% of electrophoretic homogeneity. This method
should prove a useful tool for the study of the biochemical
properties of brain TH.
References
Bom.sa�i, P., STEIN, S., D�unai*.i�i, W. *i�D UDENFRIF.ND, S.: Fluorometric assayof proteins in the nanogram range. Arch. Biochem. Biophys. 155: 213-220,1973.
BYLUND, D. B. �D Kazas, E. G.: Effect of denaturatlon on the susceptibility ofproteins to enzymic phosphorylation. J. Biol. Chem. 250: 6355-6361, 1975.
CASTELLINO, F. J. AND B*iuc.zR, R: Examination ofthe dissociation of multichainproteins in guanidine hydrochloride by membrane osmometry. Biochemistry1: 2207-2217, 1968.
Ciuirz, J. E., � E. S. �.sD K�uFMA]�i, S.: The isolation and characterizationof dihydropteridine reductase from sheep liver. J. Biol. Chem. 247:6682-8091,1972.
EDELMAN, A. M., RAESE, J. D., L*z�n, M. A. AND B�nca�s, J. D.: In vitrophosphorylation of a purified preparation of bovine corpus striatal tyrosinehydroxylase. Commun. Psychopharmacol. 2: 461-465, 1978.
HARRIs, J. E., B�LDass4�amI, R J., MORGENROTH, V. H. ifi *i�D Ham, R H.:Activation by cyclic 3’:5’-adenosine monophosphate of tyrosine hydroxylase in
the rat brain. Proc. Natl. Acad. Sci. U.S.A. 72: 789-793, 1975.Icmy*it�, A�, N�.K�.Mua&, S., Nismzux�, Y. AND HAYAI5m, 0.: Enzymic studies
on the biosynthesis ofserotonin in mammalian brain. J. Biol. Chem. 245: 1699-1709, 1970.
Joa, T. H., P�nx, D. H. *i�D Rais, D. J.: Direct phosphorylation ofbrain tyrosine
hydroxylase by cyclic AMP-dependent protein kinase: Mechanism of enzymeactivation. Proc. Natl. Acad. Sci. U.S.A. 75: 4744-4748, 1978.
JO0, T. H., PARK, D. H. AND REIS, D. J.: Direct phosphorylation of tyrosinehydroxylase by cyclic AMP-dependent protein kinase: Mechanism of enzymeactivation. In Catecholamines: Basic and Clinical Frontiers, ed. by E. Usdin, LKopin and J. Barchas, pp. 43-45, Pergarnon, New York, 1979.
KATZ, I. R., YAMAUCHI, T. �im KAUFMAN, S.: Activation of tyrosine hydrozylaseby polyanions and salts An electrostatic effect. Biochim. Biophys. Acts 429:
84-95, 1976.KINZEL, V. AND KUBLER, D.: Single step purification of the catalytic subunit(s)
of cyclic 3’,5’-adenosine monophosphate-dependent protein kinases from rat
KL0Tz, L, D*.ns*.u�, D. AND LANGERMAN, N.: Quaternary structure of proteins.In The Proteins, voL I, ed. by H. Neurath and R. L Hill, pp. 293-411, AcademicPress, New York, 1975.
KocHETKov, S. N., BULARGINA, T. V., SASHCHENKO, L P. AND Szvzsus, E. S.:Role of a histidine residue in the active site of cyclic AMP-dependent histonekinase. FEBS Lett. 71: 212-214, 1976.
Kuczra�szi, R T. AND MANDELL, A. J.: Regulatory properties of soluble andparticulate rat brain tyrosine hydroxylase. J. BioL Chem. 247: 3114-3122, 1972.
LETENDRE, C. H., MAcDONNELL, P. C. AND GuRor�, G.: The biosynthesis ofphosphorylated tyrosine hydroxylase by organ cultures of rat adrenal medullaand superior cervical ganglia. Biochem Biophys. Baa. Commun. 74: 891-897,
1977a.LETENDRE, C. H., MACDONNELL, P. C. AND GUROFF, 0.: The biosynthesis of
phosphorylated tyrosine hydroxylase by organ cultures of rat adrenal medullaand superior cervical ganglia A correction. Biochem. Biophys. Baa. Commun.
76: 615-617, 197Th.Lzvrrr, M., Spzcron, S., Sjoams�, A. A�iD UDENPRIEND, S.: Elucidation of
the rate-limiting step in norepinephrine biosynthesis in the perfused guinea-pigheart. J. PharmacoL Exp. Ther. 148: 1-9, 1965.
LINDBERG, V.: Molecular weight and amino acid composition of deoxyribonucle-see I. Biochemistry 8: 33�-342, 1967.
LLOYD, T. �z�iD KAUFMAN, S.: The stimulationofpartiallypurifled bovine caudatetyrosine hydroxylase by phosphatidyl-L-serine. Biochem. Biophys. Res. Com-mun. 59: 1262-1269, 1974.
LLOYD, T. AND KAUFMAN, S.: Evidence for the lack of direct phosphorylation ofbovine caudate tyrosine hydroxylase following activation by exposure to enzy-matic phosphorylating conditions. Biochem. Biophys. Baa. Commun. 68: 907-913, 1975.
LLOYD, T. AND WEINER, N.: Isolation and characterization of a tyrosine hydrox-ylase cofactor from bovine adrenal medulla. Mol. PharmacoL 7: 569-580, 1971.
LOVENBERG, W., BRUCKWICK, E. A. AND HANBAUER, I.: ATP, cyclic AMP, andmagnesium increase the affinity of rat striatal tyrosine hydroxylase for itscofactor. Proc. NatL Aced. Sd. U.SA. 72: 2955-2958, 1975.
MA.RKEY, K. A., K0ND0, S., SHENEMAN, L AND GOLDSTEIN, M,: Purification andcharacterization of tyrosine hydroxylase from a clonal pheochromocytoma cellline. Mol. Pharmacol. 17: 79-85, 1980.
MARTIN, R. G. AND AMES, B. N.: A method for determining the sedimentationbehavior of enzymes: Application to protein mixtures. J. BioL Chem, 236:1372-1379, 1961.
MORGENROTH, V. H. 111, HEGSTRAND, L R., ROTH, R. H. �i�D GREENGARD, P.:Evidence for involvement of protein kinase in the activation by adenosine 3’:5’-monophosphate of brain tyrosine 3-monooxygenase. J. Biol. Chem. 250: 1946-1948, 1975.
NAGATSU, T., Lzvrrr, M. s.riD UDENFRIEND, S.: Tyrosine hydroxylase, the initial
step in norepinephrine biosynthesis. J. Biol. Chem. 239: 2910-2917, 1964,RAE5E, J. D., EDELMAN, A. M., L�zsa, M. A. AND B�aca�s, J. D.: Bovine striatal
tyrosine hydroxylase: Multiple forms and evidence for phosphorylation bycyclic AMP-dependent protein kinase. In Structure and Function of Mono-
amine Enzymes, ad. by E. Usdin, N. Weiner and M. B. H. Youdim, pp.383-400,Marcel Dekker, New York, 1977.
RAESE, J. D., EDzu,s�, A. M., L�z�ut, M. A. �sD B�acHA�s, J. D.: Tyrosinehydroxylase from brain Phosphorylation of the enzyme by cyclic AMP-depen-
dent protein kinase. In Catecholamines: Basic and Clinical Frontiers, ad. by E.Usdin, I. Kopin and J. Barchas, pp. 46-48, Pergamon, New York, 1979a.
RAE5E, J. D., Ei�u�u�i, A. M., M*xx, G., BRUCKWICK, E. A., L0vENBERG, W.AND B�itcir*.s, J. D.: Brain striatal tyrosine hydroxylase: Activation of theenzyme by cyclic AMP-independent phosphorylation. Commun. Psychophar-macoL 3: 295-301, 1979b.
RAE5E, J., PATRICK, R. L. AND BARCHA5, J. D.: Phospholipid-induced activationof tyrosine hydroxylase from rat brain striatal synaptosomes. Biochem. Phar-macel. 25: 2245-2250, 1976.
RIcE, R. H. AND MEANS, G.: Radioactive labeling of proteins in vitro. J. Biol.Chem. 246: 831-832, 1971.
SCHAcHMAN, H. K.: Ultracentrifugation in Biochemistry, Academic Press, NewYork, 1959.
SOBER, H. A. (EDIToR): Handbook of Biochemistry, Chemical Rubber Co.,Cleveland, 1968.
VULLIET, P. R, LANGAN, T. A. �r�iD WEINER, N.: Tyrosine hydroxylase: Asubstrate for cyclic AMP-dependent protein kinase in vitro. In Catecholamines:Basic and Clinical Frontiers, ed. by E. Usdin, I. Kopin and J. Barchas, pp. 94-96, Pergamon, New York, 1979.
VULLIET, P. R, LANGAN, T. A. AND WEINER, N.: Tyrosine hydroxylase: Asubstrate ofcyclic AMP-dependent protein kinase. Proc. Natl. Acad. Sci. U.S.A.77: 92-96, 1980.
WAYMIRE, J. C., BJUR, R. AND WEINER, N.: Assay of tyrosine hydroxylase bycoupled decarboxylation of DOPA formed from 1-’4C-L-tyrosine. Anal. Bio-chem. 43: 588-600, 1971.
WAYMIRE, J. C., HAYCOCK, J. W., MELIGENI, J. A. �.siD BROWNING, M. D.:Activation and phosphorylation of tyrosine hydroxylase by cAMP. In Cate-cholamines: Basic and Clinical Frontiers, ed. by E. Usdin, I. Kopin and J.Barchas, pp. 40-42, Pergamon, New York, 1979.
WEBER, K. AND OsBoas, M.: Proteins and sodium dodecyl sulfate: Molecularweight determination on polyacrylamide gels and related procedures. In TheProteins, voL I, ed. by H. Neurath and R. L Hill, pp. 179-223, Academic Press,New York, 1975.
Wrrr, J. J. AND RosKosx!, R., JR.: Rapid protein kinase assay using phosphocel-
lulose-paper absorption. Anal. Biochem. 66: 253-258, 1975.YAMAUCHI, T. AND FUJISAWA, H.: Evidence for phosphorylation ofbovine adrenal
tyrosine hydroxylase by cyclic AMP-dependent protein kinase. Biochem. Bio-phys. Res. Commun. 82: 514-517, 1978.
1�AMAUCHI, T. AND FUJISAWA, H.: In vitro phosphorylation of bovine adrenaltyrosine hydroxylase by adenosine 3’:5’-monophosphate-dependent protein Id-rinse. J. Biol. Chem. 254: 503-507, 1979a.
Y*.atauciu, T. AND FUJISAWA, H.: Regulation of bovine adrenal tyrosine 3-monooxygenase phosphorylation-dephosphorylation reaction, catalyzed byadenosine 3’:5’-monophosphate dependent protein kinase and phosphoproteinphosphatase. J. Biol. Chem. 254: 6408-6413, 1979b.
ZARLER, W. L.: Analytical polyacrylamide gel electrophoresis and molecularweight determination. In Methods in Enzymology, ed. by S. Fleischer and L.Packer, pp. 70-81, Academic Press, New York, 1974.
Send reprint requegts to: Arthur M. Edelman, Ph.D., Department of Phar.macology, SJ-30, University of Washington School of Medicine, Seattle, WA98195.