Design of Chronomodulated Drug Delivery System of Valsartan: In Vitro CharacterizationM. SOKAR*, A. HANAFY1, A. ELKAMEL AND S. EL‑GAMALDepartment of Pharmaceutics, Faculty of Pharmacy, Alexandria University, 1st El Khartoum Square, Azarita, Elmesalla, P.O. Box 21521, Alexandria, 1Department of Pharmaceutics, Faculty of Pharmacy and Drug Manufacturing, Pharos University, Canal El Mahmoudia St, Beside Green Plaza Complex. Alexandria, Egypt
Sokar, et al.: Valsartan Time‑Clock Pulsatile System
The aim of the present study was to design and evaluate a chronomodulated time‑clock pulsatile tablets of valsartan to release it after a certain lag time, independent of the gastrointestinal pH, in its absorption window to cope with the circadian rhythm of human body for blood pressure elevation. Core tablets were prepared by direct compression of a homogenous mixture of valsartan, Avicel PH101, croscarmellose sodium, magnesium stearate and Aerosil. The core tablets were then sprayed coated with a sealing layer formed of ethyl cellulose that was subsequently coated with a release‑controlling layer. Three different aqueous dispersions namely; carnauba wax or beeswax or a mixture in a ratio of 2.5:1, respectively, were used to form five time‑clock tablet formulations having the release controlling layer with different thickness {B5, B10, B20, BW5 and CW5}. Quality control testing were carried out to the core tablets. Differential scanning calorimetry was also performed to detect the possible drug excipient interaction in the core tablet formulation. The release was carried out, for the prepared time‑clock tablet formulations, in 0.1 N hydrochloric acid for the first 2 h, followed by phosphate buffer (pH 6.8) for 4.5 h. The effect of pH on valsartan release was studied through a release study in 0.1 N hydrochloric acid for 6.5 h. Two phase dissolution study was performed to the selected time‑clock tablet formulation to predict the drug permeation through the gastrointestinal tract. Stability study of the selected formula was performed at 25°/60% RH and at 40°/75% RH for 3 months. Results showed that a release‑controlling layer composed of a mixture of carnauba wax and beeswax in a ratio of 2.5:1 showed a reasonable release lag time. The release lag time of the tablets increased with the increase of the coat thickness, thus B20>B10>B5 with corresponding lag time values of 4.5, 3 and 2.5 h, respectively. Selected B5 tablet formula exhibited a reasonable lag time after which the highest, complete % drug release at pH 6.8 was obtained. In addition, a good partitioning of valsartan, between the aqueous and organic phases in a ratio of 1:7, was observed. The selected formula was stable for at least 3 months under standard long‑term and accelerated storage conditions. In conclusion, in vitro studies revealed that the novel time‑clock system could be used successfully to deliver valsartan in a pulsatile pH‑independent manner. It provided a desirable lag time followed by a rapid and complete drug release accompanied by an expected effective permeation through the biological membranes upon release in the duodenum; the window of absorption, as indicated by the two phase release study.
Key words: Time‑clock, pulsatile, pH‑independent, valsartan, lag time, carnauba wax, bees wax, permeation
Humanbody shows24hvariation in bloodpressure.There is an increase in the atrial blood pressurein the morning between 4:00 to 8:00 am and agradual decrease at night.Valsartan is angiotensinII inhibitor used in treatment of hypertension.Thepharmacokinetics of valsartan showed that valsartanpeak plasma concentration is reached in 2 to 4 hafter conventional drug dosage form administration.Valsartan shows an average elimination half-life ofabout 6 h. Conventional drug delivery system ofvalsartan is inappropriate for the delivery of drug, as
theycannotbe administered justbefore the symptomsareworsened.Bedtime dosing of conventional drugdosage of valsartan will not provide a sufficienttherapeutic plasma drug concentration at the earlyhours ofmorning.
Research Paper
This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.
Received 31 May 2014Indian J Pharm Sci 2015;77(4):470-477
July - August 2015 Indian Journal of Pharmaceutical Sciences 471
www.ijpsonline.com
The time-clock system is a system having a lagtime independent of the gastric residence time,intestinal enzymes,mechanical action of stomachor gastrointestinal pH. Pozzi et al.[1] developed thetime-clock system for delivery of salbutamol sulfateusing the conventional coating technique.Thecoatingwas made up of a hydrophobic-surfactant layer[(Carnauba wax (CW) and beeswax (BW) alongwith tween 80] applied as aqueous dispersion, towhich awater soluble polymer (HPMC)was addedto improve adhesion to the core. This coat, driedon the core during coating, retains the capacity torehydrate and redisperse in an aqueous environmentina timeproportional to thefilm thickness.Followingredispersion, the core would be available fordissolution[1].
After performing the in vitro release studies,Pozziet al.[1] evaluated the time-clock tablets in vivo in healthy volunteers via gamma-scintigraphy usingsamarium oxide which was added to the tabletformulation[1].The time-clock tablets of salbutamol[1]are targeted to the colonwhere they reached after283min.Thefirst drug conc. appeared in the bloodafterTlag of 299-393minwithmeanTmax of 305min.
Afterwards,Wilding et al.[2] tried to develop anenteric coated time-clock release system for colonictargeting. However, the primary objective in caseof drug delivery to the colonwas that the extendedgastric residence, premature mesalazine releasein the small bowel can occur[2]. To overcome thisproblem, and tomakeuse of the relatively consistentsmall intestinal transit time[3], an enteric coatingwasapplied to the formulation topreventdispersionof thehydrophobic layerwhilst in the stomach[2].
The lag time is the time needed for thewaxes’ coatto emulsify and erodeby thehelpof surfactants suchaspolyoxyethylene sorbitanmonooleate.The lag timeis proportional to the film thickness[1,2].The aim ofthepresent studywas to preparevalsartan time-clockpulsatile tablets to release the drug after certainlag time in its absorptionwindow to copewith thecircadian rhythmof human body for blood pressureelevation.
MATERIALS AND METHODS
Valsartan (a gift from European EgyptianPharmaceutical Industries Company,Alexandria,
Preparation of pulsatile valsartan tablets, core tablet:Valsartan core tablets were prepared as describedbySokar et al.[4].Valsartan (80mg),Avicel PH-101(22.5 mg) and CCNa (5 mg) were mixed bygeometric addition technique.Aerosil 130 (0.5mg)andmagnesium stearate (2mg)were then added tothemixedpowders.All thepowdersusedwerepassedthrough sieve no. 45 separately. Finally, 110mg ofthe blendwasweighed and compressed by a singlepunch tablet press machine (Royal artist, India),equippedwith6mmconcave-facedpunches.
Preparation of time-clock tablets:The time-clock tabletswere prepared as stated byPozzi et al.[1] with somemodifications.An extrasealing layerwas added between the core tablet andtheouter release-controlling layer.
Core tabletswereweighed accurately before coatingthen arrangedmanually in a tray and sprayedwithECethanolic solution (3%).Thenumberof sprayingsand distance between the sprayer and tabletswerefixed. The sprayed core tablets were dried usinghot air oven (Human-lap, Korea) adjusted at 60°,then allowed to cool in ambient air. Half-sprayedcore tablets were reciprocated in the tray and theother sideswere sprayed asmentioned previously.After cooling, tabletswere reweighed.The sprayedcore tabletswere coatedwithCW,BWandHPMC.These polymerswere previously ground, sieved anda size range of (90-180µm)was selected. Threedifferent aqueous dispersionswere prepared,whosecompositionwas listed inTable1.All thedispersionswerepreparedusing the same technique.
Tween 80wasmixedwithwater then heated to 80°andHPMCwas sprinkled slowly. Dispersion was
472 Indian Journal of Pharmaceutical Sciences July - August 2015
www.ijpsonline.com
allowed tocoolandwaxeswereaddedgraduallywhilemixing using amagnetic stirrer (Daihan ScientificCo.,Korea).Core tablets, previously sprayedwith thesealing layer,were arranged in the tray and sprayedusing thepreviouslymentioned technique.
After spraying the core tablets with the aqueousdispersion, theywereallowed todry inambientairandweighed todetermine the total%weight increase.Thecoating stepswere repeated until the desiredweightwasgained.Fivebatchesof the time-clock tabletswerepreparedwithdifferentcoat thicknesses (Table2)usingthepreviouslypreparedaqueousdispersions.
Differential scanning calorimetry:Differential scanning calorimetry (DSC) wasperformed using aDSC-6 (Perkin-Elmer, England).The samples [valsartan, components of tablet coreformulation as aphysicalmixture (PM), and thefinalcompressed tablet core form (C)]were investigated.The instrumentwas calibratedwith indium standard.Valsartanpowder (10mg) and14mg samplesofPMandgroundCcontainingan equivalent amountof thedrugwereweighed andplaced in a closed aluminumsample pans with pin hole under nitrogen flow(30ml/min) at a scanning rate of 5°/min from35 to150°.Theheatflowas a functionof temperaturewasmeasured for all samples[4].
Quality control testing of the prepared core tablets:Disintegration timeof the core tabletswasdeterminedusing thedisintegration tester (Copley,ScientificLtd,
UK) in900mlwater, 0.1NHClandphosphatebuffer(pH6.8), in triplicates.Tabletswere also subjected tofriability test using the tablet friability test apparatus(Copley,UK).Tablet breaking force of 10 randomlyselected tablets was determined using the tablethardness tester (Dr. Schleuniger PharmatronAG,Switzerland).
Testswere performed according to the officialUSPmethod[5].Theweight variation testwas carried outas stated in theBP[6].Theprepared core tabletsweresubjected to uniformity of thickness and diameterusing aVernier caliper (For-broEngineers,Mumbai,India). The testswere carried out on 20 randomlyselected tablets[4].
In vitro drug release studies:The drug release testing from the time-clock tabletswas carried out usingUSP dissolution apparatus II(Hanson Research Corporation, USA), in 730ml0.1NHCl for2h afterwhich thepHof themediumwas changed to 6.8 by adding 270 ml of 0.2MNa3PO4.12H2O solution
[7].The studywas performedat 37±0.5° and a stirring rate of 50 rpm.At differenttime intervals, 5ml samplewaswithdrawn, filteredthrough a cellulose acetatemembrane filter (0.45µm) and analyzed usingUV/Vis spectrophotometerat 250 nm (Shimadzu Corporation, Japan). Eachwithdrawn samplewas compensatedwith 5ml offresh correspondingmedium.All release experimentswere carried out in triplicates.Validation parametersof the spectrophotometric assay was limited tolinearity, inter-day precision and intra-day precisiondata.
Modeling of release profiles:To determine Valsartan release kinetics fromthe selected formulation, the release data wastreated according to Higuchi, Korsmeyer-Peppas,Hixson-Crowell andWeibullmodels alongwith zeroandfirstorderpatterns[8] using theAdd-In ‘DDSolver’software[9].
Solubility studies:Valsartan solubilitywas investigated in 0.1NHCl,phosphate buffer (pH 6.8) and n-octanol by addinga known excess amount of valsartan to a flaskcontaining 25ml of eachmedium.The flaskswereshaken for 24 h in a water bath at 37° and thentheywere left to attain equilibrium for another 24h.
TABLE 1: COMPOSITIONS OF DIFFERENT RELEASE- CONTROLLED LAYER AQUEOUS DISPERSIONSIngredients Aqueous dispersion composition (g%)
July - August 2015 Indian Journal of Pharmaceutical Sciences 473
www.ijpsonline.com
Solutionswere filtered, diluted and analyzed usingUV/Vis spectrophotometer (ShimadzuCorporation,Japan) at thepredeterminedλmaxin eachmedium
[10,11].
Two-phase dissolution study:A two-phase dissolution studywas performed forthe selected time-clock tablet formulation in USPdissolution apparatus II using a 150 ml vesselcontaining 50ml 0.1NHCl as the lower aqueousphaseand50mln-octanolas theupperorganicphase.The temperature was adjusted at 37±0.5° and thestirring ratewas50 rpm[12].The studywasperformedin duplicates.
The dissolution vesselwas filledwith the aqueousphase followed by addition of one tablet. Then,n-octanolwas slowly added over the aqueous phase(being careful to prevent the contact of n-octanolwith the tablet).After2h, thepHof the lowerphasewas changed to 6.8 by adding 18.5 ml of 0.2MNa3PO4.12H2O solution
[7].At different time intervals,1ml samplewaswithdrawn fromeach layer, filteredthrough a cellulose acetatemembrane (0.45µm) (forsamples withdrawn from the aqueous phase) or aPTFEsyringefilter (0.45µm,ABIMED,Deutschland)(for sampleswithdrawn from the organic phase) andanalyzed using UV/Vis spectrophotometer at thepredeterminedλmax for each layer. Eachwithdrawnsample was compensated with 1 ml of the freshcorrespondingmedium.
Stability studies:The formula of choicewas tested for its physicalstability according to the ICHguidelines[13]. Standardlong-term stability studywas performed at 25° and60%RHfor3months.Accelerated stability studywascarried out at 40° and 75%RH for 3months[14].Atthe specified time intervals, the tabletswereexaminedfor theirphysical appearanceand in vitro drug release.
Statistical analysis:Results were depicted as mean values±SD.Allstatistical comparisons were performed usingMicrosoftExcel 2007.Theobtaineddata of repeatedmeasurementswas subjected toStudent’s t-test and a P value≤0.05was considered as significant.
RESULTS AND DISCUSSION
The core tablets were compressed using concavepunches tominimize edges’ space so as to obtain
homogenous coating[15].EC sealing layerwas appliedon the core tablets during the preparation to actas an insoluble barrier protecting the core fromearly disintegration. Itwas added in a single layerthickness; so theweight increase of core tabletswasnegligible.
Pozziet al.[1] prepared the coatingaqueousdispersionusingwaxesandHPMC inconcentrationshigher thanthat used in this study. Similar high concentrationsresulted in an impractically viscous dispersion andnozzle blockage. Consequently, the concentrationsof the coating aqueous dispersion usedmentioned inTable1 to facilitate the sprayingprocess.HPMCwasincorporated to help the adherence and cohesivenessofwaxes to the core as they exhibit poormechanicalpropertiesbecauseof their lackof cohesive structuralintegrity[16].
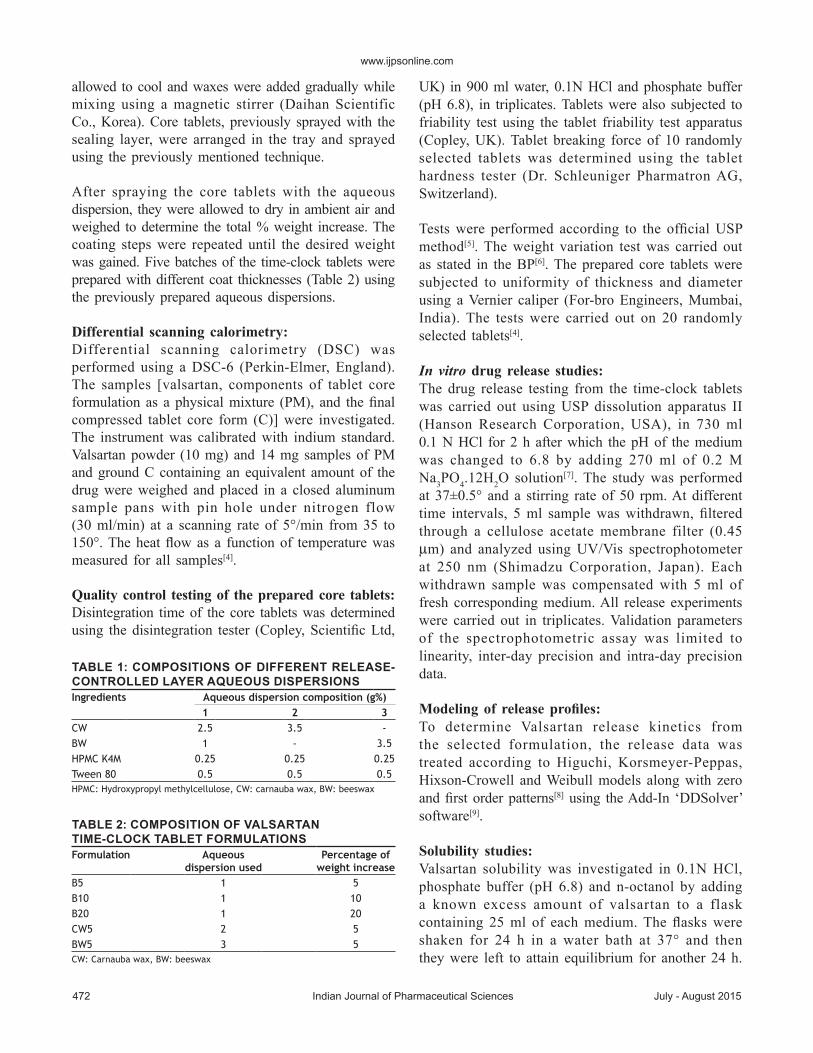
Five formulations were prepared (Table 2) withthe following%weight increase; B5 (4.85±0.2%),B10 (10.31±0.2%), B20 (19.81±0.5%), CW5(5.13±0.2%) andBW5 (4.92±0.3%). InDSC studies,the thermogramof valsartan showed a clearmeltingendothermic peak at 100.8°. Valsartan peak inPM thermogram was slightly shifted to appearat 95.31° and in C thermogram it was shifted to96.07°.Valsartan peak intensities in bothPMandC
Fig. 1: DSC thermograms.DSC thermograms of valsartan, physical mixture of core tablet components (PM) and their compressed form (C).
474 Indian Journal of Pharmaceutical Sciences July - August 2015
www.ijpsonline.com
thermogramsweredecreasedandbroadenedmarkedly;fig. 1.The decreased and broadened peaks indicatethe less ordered crystallinity, rather than completelyamorphous state[4].This change could be due to theadsorption ofAerosil particles on valsartanwhichchanged its crystalline nature to an amorphouspowder[17].Mixing valsartan withAvicel PH-101,CCNaorMg stearate did not seem to be responsiblefor this change[17,18].
The average weight the core tablets was107.31±0.89 mg. Core tablets showed auniform thickness (3.32±0.08 mm) and diameter(6.06±0.02 mm). The core tablet hardness was3.87±0.4 kg/cm2. Concerning the friability, it waswithin theofficial limits as thepercentageweight losswas0.59%.Thedisintegration timeof thecore tabletswas instantaneous in 900mlwater, 0.1NHCl (pH1.2) andphosphate buffer (pH6.8)[4].
Validation parameters of the spectrophotometricassaywas limited to linearity, inter-dayprecision andintra-day precision data.Valsartan showed a linearrelationship in 0.1NHCl and pH 6.8 over a conc.range of (1-3mg%) in bothmediawith a value ofcoefficient of determination (R2) 0.998 and 0.9995,respectively. Inter and intra-day precision datawerereasonable and showedmaximumvalue for%CVof6.44%.The results are similar to those obtained bySokaret al.[4].
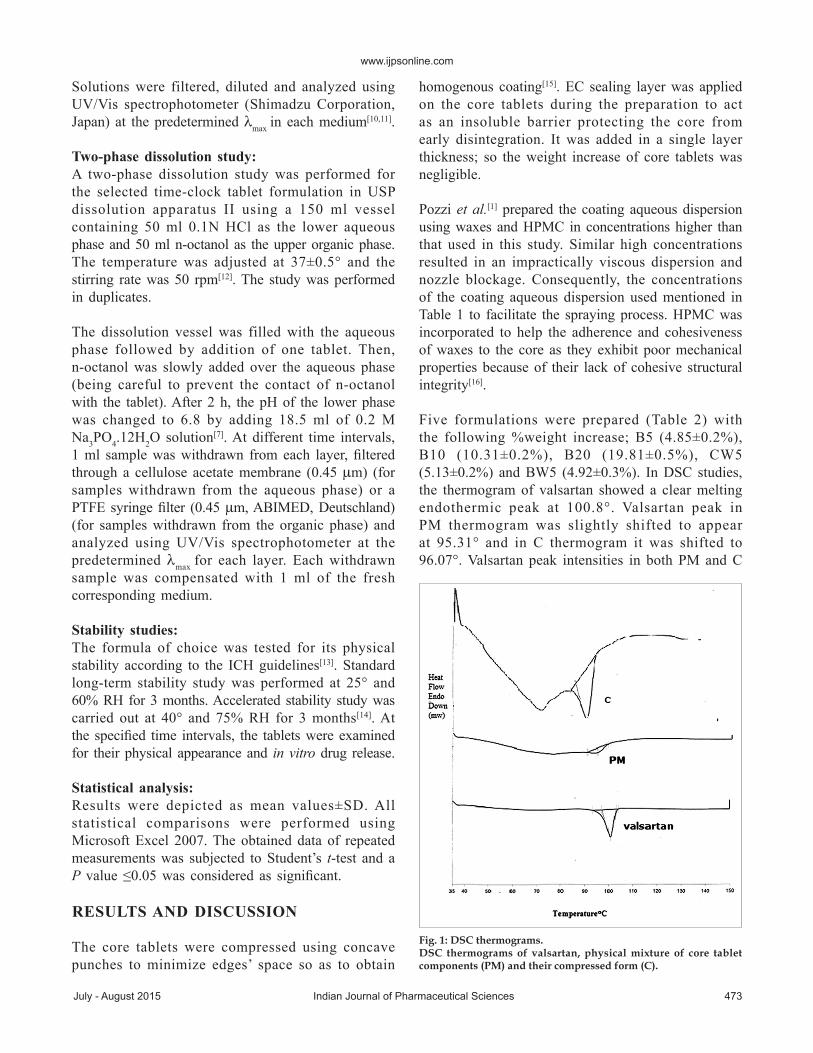
The drug release from the time-clock system issurfactant-based. The release-controlling coat wasformed of waxes and a surfactant (tween 80)[1].Upon contactwithwater, tween 80 acts to emulsifythe waxes gradually thus dissolving the coat.Asthe coat thickness increases, the time necessaryfor its complete dissolution increases. Therefore,the experiments showed that B5, B10 and B20formulations demonstrated a lag time of 2.5, 3 and4.5 h, respectively, depending on theweight gainedby the core; fig. 2.Their ascending lag timeswerein accordancewith their ascending coating thickness.The lag timewasdefinedas the timeuntil10%of thedrughas been released[19].
To investigate the effect of pH on the lag time ofB5, B10 andB20 tablets, a release studywas alsoperformed in 0.1NHCl for 6.5 h using dissolutionapparatus II, at 37±0.5° anda stirring rateof50 rpm.B5,B10 andB20 tablets demonstrated a lag time of
2.5, 3 and >6.5 h, respectively.Although the pH ofthemediumwas not shifted from pH1.2 to pH 6.8as described above, B5,B10 andB20 formulationsshowed nearly the same lag time as in case of thepreviouslymentioned pH change release procedure.Thus, the pH of themedium has no effect on thedrug release. Therefore, the time-clock system ispH-independent and themain factor controlling thedrug release is the coat thickness.
It isworthmentioning that in 0.1NHCl the drugrelease after the lag timewas slow in comparisonwith the release after changing thepHof themediumto 6.8. This could be due to the low intrinsicsolubility of valsartan in acidicmedium.
B5 tablet formula (having the smallest%weightincrease among the previouslymentioned time-clockformulae) exhibited reasonable lag time (2.5h).After2.5h, the tabletwill bedelivered to theupperpart ofthe intestine (duodenum) at pH 6.8.At this pH thedrugwillbe released instantaneously.Consequently,B5formulationwas selected for further stability studies.
Up to our knowledge, all previous time-clockstudies, a mixture of BW and CWwith differentconcentrations for coatingwas used.To investigatethe effect of using of a single wax type on thedrug release and lag time, two time-clock tabletformulations were prepared involving CW orBW solely as the coating wax (CW5 and BW5,respectively). Thus, aqueous dispersions (2) and(3)were preparedwith the compositionmentionedpreviously inTable 1.
Fig. 2: In vitro release profiles of different formulations.In vitro release profiles of valsartan from different time-clock tablet formulations ( B5, B10 and B20) in 0.1 N HCl for 2 h then phosphate buffer (pH 6.8) for 4.5 h at 37±0.5° and 50 rpm.
July - August 2015 Indian Journal of Pharmaceutical Sciences 475
www.ijpsonline.com
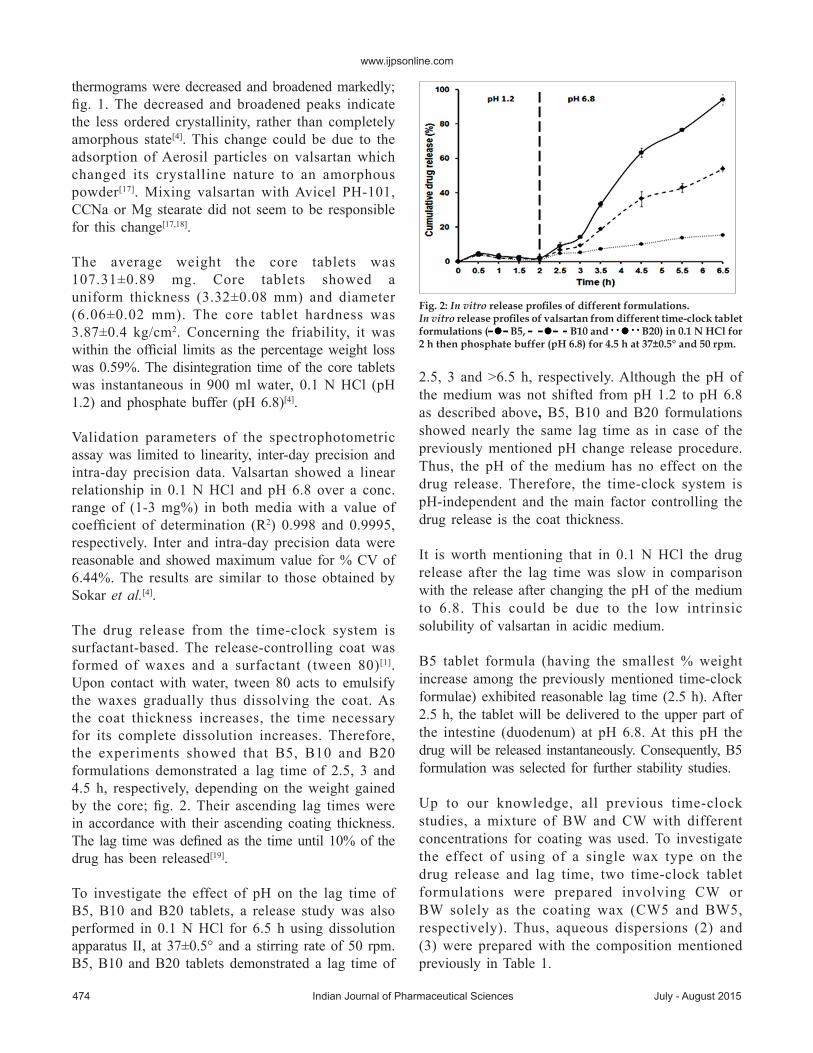
Asshown infig.3,CW5andBW5showed2and3hlag time, respectively.Although thedrug release fromCW5 seems faster than that fromB5, this differenceis considered insignificant (P>0.05).BW5 showed abit slower release thanB5but the difference in drugreleasewas insignificant too (P>0.05).
It is preferred to use a mixture of CW and BWfor tablet coating to combine the advantages ofboth waxes. CW is incorporated in the aqueousdispersion so as to obtain tablets of good lusterwithout rubbing, increase the coat strength andfacilitate swallowing.Whilst,BW is used to increasethe elasticity of the coat but due to its lowmeltingpoint (61°) itmay result in softer andmore elasticcoats[20]. Therefore, a mixture of both waxes wasused in the reported ratio to improve the propertiesand appearance of the coat. Therefore, B5 waschosen as the selected valsartan time-clock tabletformulation for further studies.
After treating the release data of valsartan fromB5formulation, according to thedifferent releasepatternspreviouslymentionedunder section2.5. Itwas foundthat B5 fits intoWeibullmodel[21] in both releasestudies (0.1NHCl for 2 h then pH 6.8 for 4.5 h)and (0.1NHCl for 6.5 h) as indicatedby the valuesofR2 (coefficientof determination); 0.992 and0.934,respectively.As b, the shape parameter, is ˃1 (1.869and 1.062, respectively), the shape of the curve getssigmoidalwith a turning point.The sigmoidal curveshape is indicative of complex releasemechanismsi.e. the rateof releasedoesnot changemonotonically.
In fact, the release rate initially increasednonlinearlyup to the inflection point and thereafter decreasedasymptotically[22].
Because of the lipid nature of biologicalmembranes,drug lipophilicity has long been consideredan important determinant of absorption. Theoctanol/water partition coefficient (P) at a selectedpH is a key indicator of drug lipophilicity andcould predict the drug permeation through thegastrointestinalmembrane[23].Valsartan log P valueis 1.499, indicating that the compound has a ratherhydrophilic character at physiological pH[24]. Thus,valsartan is classified, according toBCS, asClass IIIdrugwith lowpermeability andhigh solubility[25].
Asmaintaining ‘sinkcondition’ throughout the releasestudyofpoorly solubledrugs canbedifficult, in vitro data obtained from the single-phase dissolutionsystemmaynot satisfactorily reflect the in vivo drugabsorption characteristics[26].
The two-phase dissolution systemmaintains sinkconditionall the time. Inaddition, the transferofdrugbetween the twophasesmimics the entire process ofdrug release, dissolution and absorptionwithin theGIT.Thus, it couldbeused topredict the formulation in vivo behavior[27].
The choice of n-octanol as the organic phasewasbased on its desirable physicochemical properties.It is less dense than water (specific gravity of
Fig. 3: In vitro release profiles of coated tablets.In vitro release profiles of valsartan from different coated time-clock tablet formulations ( B5, CW5 and BW5) in 0.1 N HCl for 2 h then phosphate buffer (pH 6.8) for 4.5 h at 37±0.5° and 50 rpm.
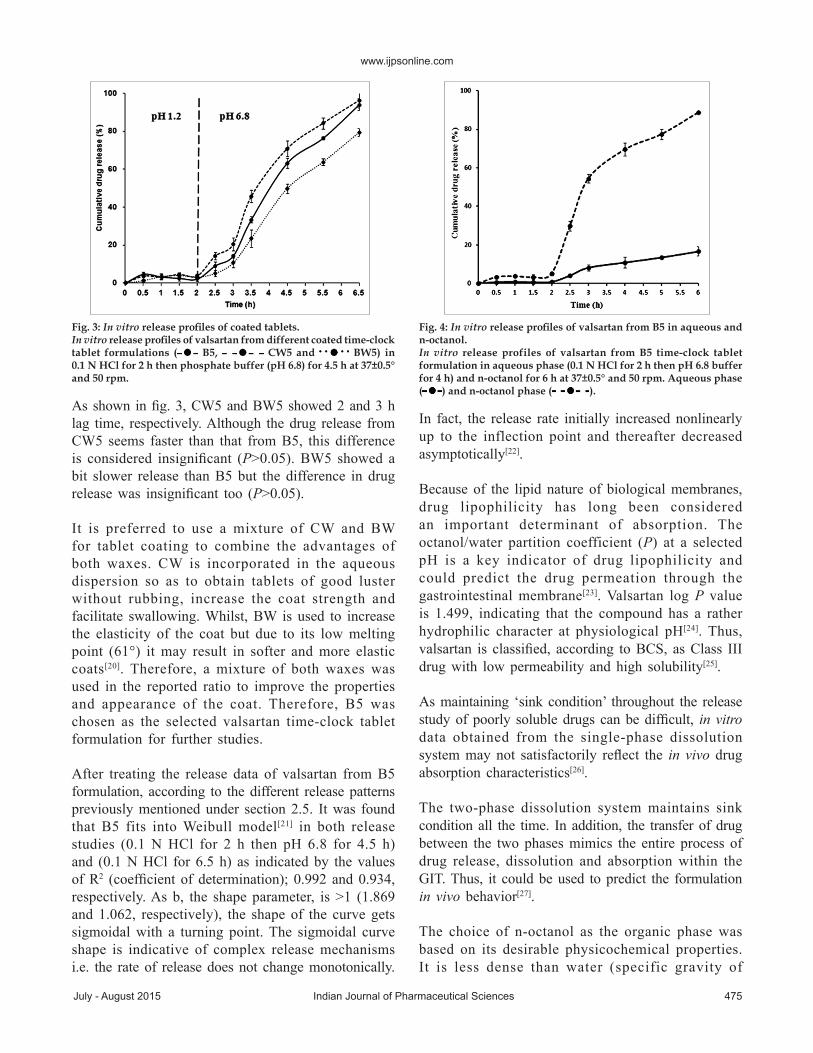
Fig. 4: In vitro release profiles of valsartan from B5 in aqueous and n-octanol.In vitro release profiles of valsartan from B5 time-clock tablet formulation in aqueous phase (0.1 N HCl for 2 h then pH 6.8 buffer for 4 h) and n-octanol for 6 h at 37±0.5° and 50 rpm. Aqueous phase ( ) and n-octanol phase ( ).
476 Indian Journal of Pharmaceutical Sciences July - August 2015
www.ijpsonline.com
n-octanol is 0.825 g/cm3 at 20°) and practicallyinsoluble inwater (0.05%w/w). It has lowvolatility(boilingpoint=195°) hence, n-octanolwill not readilyevaporate at 37°.Thus, a relatively constant upperphase volume could bemaintained throughout theexperiment[27].
As solubility of valsartan in n-octanol is 17 g/l, sinkcondition is guaranteed. Thus, the dissolved drugis continuously removed from the aqueous phaseinto the organic phase of the dissolutionmedium,mimicking the process of absorption in the systemiccirculation[27].
As shown in fig. 4, during the first 2 h, therewasalmost no drug release observed in both the aqueousand organicmedia.After changing the pH of theaqueous phase to 6.8; at 2.5 h from the beginningof the experiment, drug release was initiated andvalsartanwas partitioned between the aqueous andorganicphases in a ratio of 1:7, respectively.
Thus, valsartan favors beingdissolved in the organiclayer rather than the aqueous layer.Consequently, itis supposed that upondrug release in the duodenum,thewindowof absorption, valsartanwould permeateeffectively through thebiologicalmembranes.
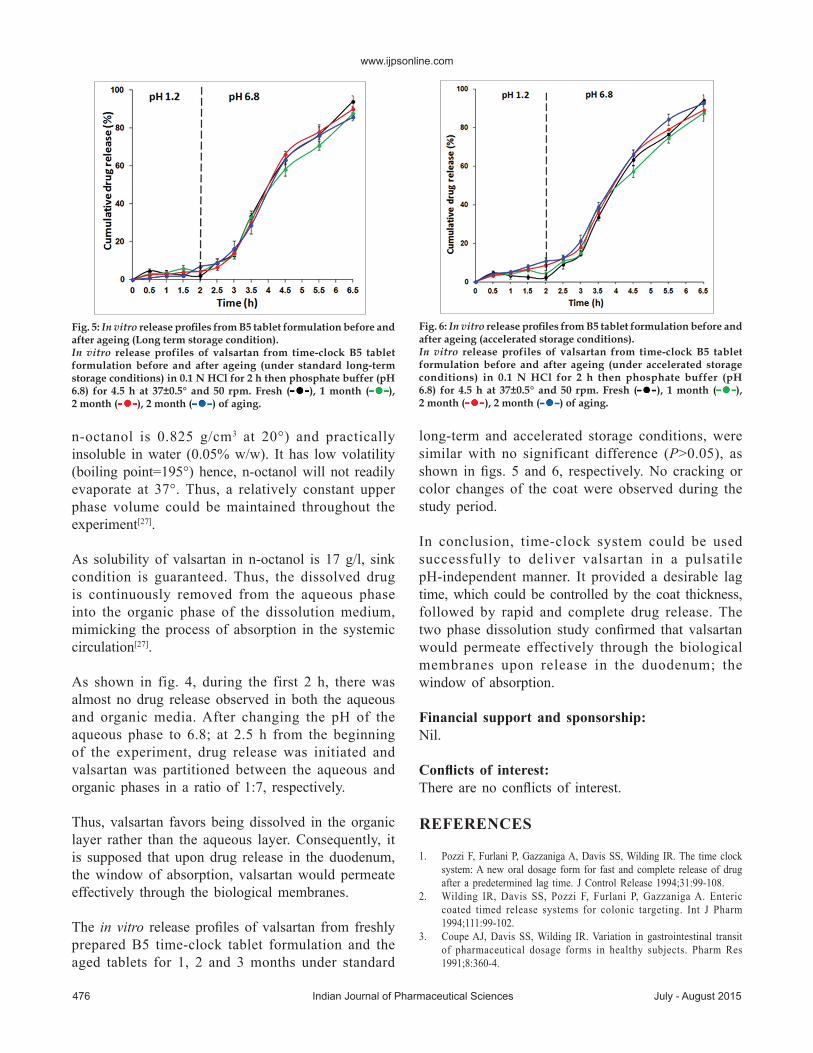
The in vitro releaseprofilesof valsartan from freshlypreparedB5 time-clock tablet formulation and theaged tablets for 1, 2 and 3months under standard
long-term and accelerated storage conditions,weresimilarwith no significant difference (P>0.05), asshown infigs. 5 and 6, respectively.No cracking orcolor changes of the coatwere observed during thestudyperiod.
In conclusion, time-clock system could be usedsuccessfully to deliver valsartan in a pulsatilepH-independentmanner. It provided a desirable lagtime,whichcouldbecontrolledby thecoat thickness,followed by rapid and complete drug release.Thetwo phase dissolution study confirmed that valsartanwould permeate effectively through the biologicalmembranes upon release in the duodenum; thewindowof absorption.
Financial support and sponsorship:Nil.
Conflicts of interest:There are no conflicts of interest.
REFERENCES
1. PozziF,FurlaniP,GazzanigaA,DavisSS,Wilding IR.The timeclocksystem:Aneworal dosage form for fast and complete release of drugafter a predetermined lag time. JControlRelease1994;31:99-108.
2. Wilding IR, Davis SS, Pozzi F, Furlani P, GazzanigaA. Entericcoated timed release systems for colonic targeting. Int J Pharm1994;111:99-102.
3. CoupeAJ,Davis SS,Wilding IR.Variation in gastrointestinal transitof pharmaceutical dosage forms in healthy subjects. Pharm Res1991;8:360-4.
Fig. 5: In vitro release profiles from B5 tablet formulation before and after ageing (Long term storage condition).In vitro release profiles of valsartan from time-clock B5 tablet formulation before and after ageing (under standard long-term storage conditions) in 0.1 N HCl for 2 h then phosphate buffer (pH 6.8) for 4.5 h at 37±0.5° and 50 rpm. Fresh ( ), 1 month ( ), 2 month ( ), 2 month ( ) of aging.
Fig. 6: In vitro release profiles from B5 tablet formulation before and after ageing (accelerated storage conditions).In vitro release profiles of valsartan from time-clock B5 tablet formulation before and after ageing (under accelerated storage conditions) in 0.1 N HCl for 2 h then phosphate buffer (pH 6.8) for 4.5 h at 37±0.5° and 50 rpm. Fresh ( ), 1 month ( ), 2 month ( ), 2 month ( ) of aging.
July - August 2015 Indian Journal of Pharmaceutical Sciences 477
www.ijpsonline.com
4. SokarM,HanafyA, El-KamelA, El-Gamal S. Pulsatile core-in-cupvalsartan tablet formulations: In vitro evaluation.Asian J PharmSci2013;8:234-43.
5. United States Pharmacopoeial Commission. United StatesPharmacopoeia 35 andNational Formulary 30. 1st ed. Rockville:UnitedStatesPharmacopoeialCommission; 2012.
6. HerMayesty’s Stationary Office. British Pharmacopeia. Vol. IV.London:HMSO;2013.
8. Dash S,Murthy PN, Nath L, Chowdhury P. Kineticmodeling ondrug release from controlled drug delivery systems.Acta Pol Pharm2010;67:217-23.
9. ZhangY, HuoM, Zhou J, ZouA, LiW,Yao C, et al. DDSolver:An add-in program formodeling and comparison of drug dissolutionprofiles.AAPS J 2010;12:263-71.
10. MbahCJ. Physicochemical properties of valsartan and the effect ofethyl alcohol, propylene glycol and pH on its solubility. Pharmazie2005;60:849-50.
11. ChellaN, Shastri N,TadikondaRR.Use of the liquisolid compacttechnique for improvement of the dissolution rate of valsartan.ActaPharmSinB2012;2:502-8.
12. ShiY, Gao P, GongY, Ping H.Application of a biphasic testfor characterization of in vitro drug release of immediate releaseformulationsof celecoxib and its relevance to in vivo absorption.MolPharm2010;7:1458-65.
13. Branch SK. Guidelines from the International Conference onHarmonisation (ICH). JPharmBiomedAnal 2005;38:798-805.
14. Bajaj S, Singla D, Sakhuja N. Stability testing of pharmaceuticalproducts. JApplPharmSci 2012;2:129-38.
16. Gontard N, Marchesseau S, Cuq JL, Guilbert S.Water vapour
permeability of edible bilayer films ofwheat gluten and lipids. Int JFoodSciTechnol 1995;30:49-56.
17. ShrivastavaAR, Ursekar B, Kapadia CJ. Design, optimization,preparation and evaluation of dispersion granules of valsartan andformulation into tablets.CurrDrugDeliv 2009;6:28-37.
18. CaoQR, LiuY, XuWJ, Lee BJ,YangM, Cui JH. Enhanced oralbioavailabilityofnovelmucoadhesivepelletscontainingvalsartanpreparedbyadrypowder-coating technique. Int JPharm2012;434:325-33.
19. Nayak UY, Shavi GV, Nayak Y, Averinen RK, Mutalik S,ReddySM, et al.Chronotherapeutic drug delivery for earlymorningsurge in blood pressure:A programmable delivery system. JControlRelease2009;136:125-31.
26. MudieDM,ShiY,PingH,GaoP,AmidonGL,AmidonGE.Mechanisticanalysis of solute transport in an in vitro physiological two-phasedissolutionapparatus.BiopharmDrugDispos2012;33:378-402.
27. HeigoldtU, Sommer F,Daniels R,WagnerKG. Predicting in vivo absorption behavior of oralmodified release dosage forms containingpH-dependentpoorly solubledrugsusing anovelpH-adjustedbiphasic in vitro dissolution test.Eur JPharmBiopharm2010;76:105-11.








![Bimodal Gastroretentive Drug Delivery Systems of ......a gastroretentive floating drug delivery system[12]. The drug concentrations can be controlled by formulating bimodal drug delivery](https://static.documents.pub/doc/80x56/5e6f0293269d113bd9170da6/bimodal-gastroretentive-drug-delivery-systems-of-a-gastroretentive-floating.jpg)


![Intelligent drug delivery system - pgsitecdn.persiangig.com/dl/9MZwnq/student Intelligent drug delivery syste… · Table 2. Marketed technologies of pulsatile drug delivery [31]](https://static.documents.pub/doc/80x56/5f3dc762b8577c0d041fed9b/intelligent-drug-delivery-system-intelligent-drug-delivery-syste-table-2-marketed.jpg)








