39
Copyright © 2013 BSI. All rights reserved. Designing Compliant/Risk Appropriate Post Market Surveillance (and PMCF) Ibim Tariah October 2016

Copyright © 2013 BSI. All rights reserved.
Designing Compliant/Risk Appropriate Post Market Surveillance (and PMCF)
Ibim Tariah October 2016

Copyright © 2013 BSI. All rights reserved. 2
Agenda
• Relationship between QMS/Risk Management/draft MDR/IVDR
• PMS Requirements
• PMCF/PMPF Requirements
• A Notified Body’s NCRs Evaluation &
Analysis: QMS and MDD
• Conclusions

Copyright © 2013 BSI. All rights reserved. 3
BS EN ISO 134845:2016 • 8.2 Monitoring and measurement
• 8.2.1 Feedback • As one of the measurements of the effectiveness of the quality management
system, the organization shall gather and monitor information relating to whether the organization has met customer requirements. The methods for obtaining and using this information shall be documented.
• The organization shall document procedures for the feedback process. This
feedback process shall include provisions to gather data from production as well as post-production activities.
03/10/2016

Copyright © 2013 BSI. All rights reserved. 4
• 8.2.1 Feedback Cont’d
• The information gathered in the feedback process shall serve as potential input into risk management for monitoring and maintaining the product requirements as well as the product realization or improvement processes.
• If applicable regulatory requirements require the organization to gain specific experience from postproduction activities, the review of this experience shall form part of the feedback process.
03/10/2016

Copyright © 2013 BSI. All rights reserved. 5
Risk Management
Risk analysis Intended use/intended purpose
identification of characteristics related to the safety of the medical device
Identification of hazards Estimation of the risks for each
hazardous situation
Risk evaluation
Risk control Risk control option analysis Implementation of risk control
measure(s) Residual risk evaluation Risk/benefit analysis Risks arising from risk control
measures Completeness of risk control
Evaluation of overall residual risk acceptability
Risk management report
Production & post production information
Permission to reproduce extracts from BS EN ISO 14971:2009 is granted by BSI. British Standards can be obtained in PDF or hard copy formats from the BSI online shop: www.bsigroup.com/Shop or by contacting BSI Customer Services for hardcopies only: Tel: +44 (0)20 8996 9001, Email: [email protected].

Copyright © 2013 BSI. All rights reserved. 6 6 Copyright © 2013 BSI. All rights reserved.
Proposed Regulation Latest draft June 2016 !

Copyright © 2013 BSI. All rights reserved. 7
QMS
PMS Article 60a / 58a Vigilance Article 61-66 / 59-64 Passive PMS Active PMS PMCF / PMPF Annex XIII / XII
Post Market Surveillance

Copyright © 2013 BSI. All rights reserved. 8
Post Market Surveillance – Article 60a / Article 58a
2. For any device, proportionate to the risk class and appropriate for the type of device, manufacturers shall plan, establish, document, implement, maintain and update a post-market surveillance system which shall be an integral part of the manufacturer’s quality management system according to Article 8(6).
3. The post-market surveillance system shall be suitable to actively and systematically gather, record and analyse relevant data on the quality, performance and safety of a device throughout its entire lifetime, to draw the necessary conclusions and to determine, implement and monitor any preventive and corrective actions.

Copyright © 2013 BSI. All rights reserved. 9
Post Market Surveillance – Article 60a / Article 58a
4. Data gathered by the manufacturer’s post-market surveillance system shall in particular be used:
a) to update the benefit risk determination and risk management, the design and manufacturing information, the instructions for use and the labelling;
b) to update the clinical evaluation / performance evaluation; c) to update the summary of safety and clinical performance as referred to in Article 26
/ Article 24; d) for the identification of needs for preventive, corrective or field safety corrective
action; e) for the identification of possibilities to improve the usability, performance and safety
of the device; f) when relevant, to contribute to the post-market surveillance of other devices. g) to detect and report trends (in accordance with article 61a / 59a). The technical documentation shall be updated accordingly.

Copyright © 2013 BSI. All rights reserved. 10
Post Market Surveillance – Article 60a / Article 58a
6. If in the course of the post-market surveillance a need for preventive and corrective action is identified, the manufacturer shall implement the appropriate measures and, where applicable, inform the notified body and the competent authorities concerned. When a serious incident is identified or a field safety corrective action is implemented, this shall be reported in accordance with Article 61.
Article 2: • ‘serious incident’ means any incident that directly or indirectly led, might have led or
might lead to any of the following: • death of a patient, user or other person, • temporary or permanent serious deterioration of the patient's, user's or other person's
state of health, • serious public health threat;

Copyright © 2013 BSI. All rights reserved. 11
• The 4 W’s of Vigilance: • Who to report?
• What to report?
• When to report?
• Where to report
03/10/2016
Vigilance – Article 61-66 / Article 59-64

Copyright © 2013 BSI. All rights reserved. 12
Vigilance – Article 61-66 / Article 59-64
Question Answer
Who reports Manufacturers of devices, other than investigational / performance evaluation devices.
What to report (a) any serious incident involving devices made available on the Union market, except expected side-effects / erroneous results which are clearly documented in the product information and quantified in the technical documentation and are subject to trend reporting pursuant to Article 61a / 59a;
(b) any field safety corrective action in respect of devices made available on the Union market, including any field safety corrective action undertaken in a third country in relation to a device which is also legally made available on the Union market, if the reason for the field safety corrective action is not limited to the device made available in the third country.
Periodic summary reports – if agreed with Competent Authorities

Copyright © 2013 BSI. All rights reserved. 13
Vigilance – Article 61-66 / Article 59-64
Question Answer
When to report As a general rule, the time period for reporting shall take account of the severity of the serious incident.
Serious Incident
• immediately after the manufacturer has established the causal relationship with their device or that such causal relationship is reasonably possible, and not later than 15 days after they have become aware of the event.
Death or unanticipated serious deterioration in state of health
• immediately after the manufacturer established or suspected a causal relationship between the device and the event but not later than 10 days following the date of awareness of the event.
Serious Public Health Threat
• immediately, and not later than 2 days after awareness by the manufacturer of this threat.

Copyright © 2013 BSI. All rights reserved. 14
Vigilance – Article 61-66 / Article 59-64
Question Answer
When to report Where necessary to ensure timely reporting, the manufacturer may submit an initial incomplete report followed up by a complete report.
If after becoming aware of a potentially reportable incident there is still uncertainty about whether the event is reportable, the manufacturer shall submit a report within the timeframe required for that type of incident.

Copyright © 2013 BSI. All rights reserved. 15
Vigilance – Article 61-66 / Article 59-64
Question Answer
Where to report http://ec.europa.eu/growth/sectors/medical-devices/contacts/index_en.htm#vcp
How Implementing Act in the future
Article 66a - Electronic system on vigilance – EUDAMED a) reports by manufacturers on serious incidents and field safety corrective
actions referred to in Article 61 / 59 and Article 63 / 61; b) periodic summary reports by manufacturers referred to in Article 61 / 59; c) reports by competent authorities on serious incidents referred to in the second
subparagraph of Article 63 / 61; d) reports by manufacturers on trends referred to in Article 61a / 59a; e) periodic safety update reports referred to in Article 60c / 58c; f) field safety notices by manufacturers referred to in Article 63 / 61; g) information to be exchanged between the competent authorities of the
Member States and between them and the Commission in Article 63 / 61.

Copyright © 2013 BSI. All rights reserved. 16
• Post-market clinical follow-up, hereinafter: PMCF, is a continuous process to update the clinical evaluation..……..and shall be part of the manufacturer's post-market surveillance plan. To this end, the manufacturer shall proactively collect and evaluate clinical data from the use in or on humans of a device which bears the CE marking, and is placed on the market or put into service within its intended purpose as referred to in the relevant conformity assessment procedure, with the aim of confirming the safety and performance throughout the expected lifetime of the device, the continued acceptability of identified risks and to detect emerging risks on the basis of factual evidence.
03/10/2016
PMCF Requirements – Annex XIII, Part B

Copyright © 2013 BSI. All rights reserved. 17
• The PMCF shall be performed pursuant to a documented method laid down in a PMCF plan. • The PMCF plan shall specify the methods and procedures to proactively
collect and evaluate clinical data with the aim of • (a) confirming the safety and performance of the device throughout its
expected lifetime, • (b) identifying previously unknown side-effects and monitoring the identified
side-effects and contra-indications, • (c) identifying and analysing emergent risks on the basis of factual evidence, • (d) assuring the continued acceptability of the benefit/risk ratio referred to in
Sections 1 and 5 of Annex I, and • (e) identifying possible systematic misuse or off-label use of the device with a
view to verify the correctness of its intended purpose. 03/10/2016
PMCF Requirements – Annex XIII, Part B

Copyright © 2013 BSI. All rights reserved. 18
• The PMCF plan shall include at least: • the general methods and procedures of the PMCF to be applied
• the specific methods and procedures of PMCF to be applied, such as
evaluation of suitable registers or PMCF studies;
• a rationale for the appropriateness of the methods and procedures referred to in points (a) and (b);
• a reference to the relevant parts of the clinical evaluation / performance
report…and to the risk management
03/10/2016
PMCF Requirements – Annex XIII, Part B

Copyright © 2013 BSI. All rights reserved. 19
• The PMCF plan cont’d:
• the specific objectives to be addressed by the PMCF
• an evaluation of the clinical data related to equivalent or similar devices
• reference to relevant Common Specifications, standards and guidance on PMCF
• a detailed and adequately justified time schedule for PMCF activities….to be undertaken by the manufacturer
03/10/2016
PMCF Requirements – Annex XIII, Part B

Copyright © 2013 BSI. All rights reserved. 20
• The manufacturer shall analyse the findings of the PMCF and …..the report shall be part of the clinical evaluation report and the technical documentation.
• The conclusions of the PMCF evaluation report shall be taken into account for
the clinical evaluation….and in the risk management. If through the PMCF the need for preventive and/or corrective measures has
been identified, the manufacturer shall implement them.
03/10/2016
PMCF Requirements – Annex XIII, Part B

Copyright © 2013 BSI. All rights reserved. 21 21 Copyright © 2013 BSI. All rights reserved.
A Notified Body’s NCRs Evaluation & Analysis: QMS and MDD

Copyright © 2013 BSI. All rights reserved. 22
Objectives
• Breakdown of the top nonconformities from ISO 13485 assessments by Clause • Understand some of the specific reasons for these nonconformities • Breakdown of the top nonconformities from Technical File assessments (Class
IIa and Class IIb) per specific Articles, Annex's or Essential Requirements of the Directive
• Understand some of the specific reasons for these nonconformities • Major and Minor nonconformities
10/3/2016

Copyright © 2013 BSI. All rights reserved. 23
Methodology
• Searched the data base of BSI nonconformities from 2014 and repeated in 2015 • Identified over 3600 nonconformities relating to ISO 13485 assessments • Identified over 1700 nonconformities relating to assessments against the
Medical Devices Directive
10/3/2016
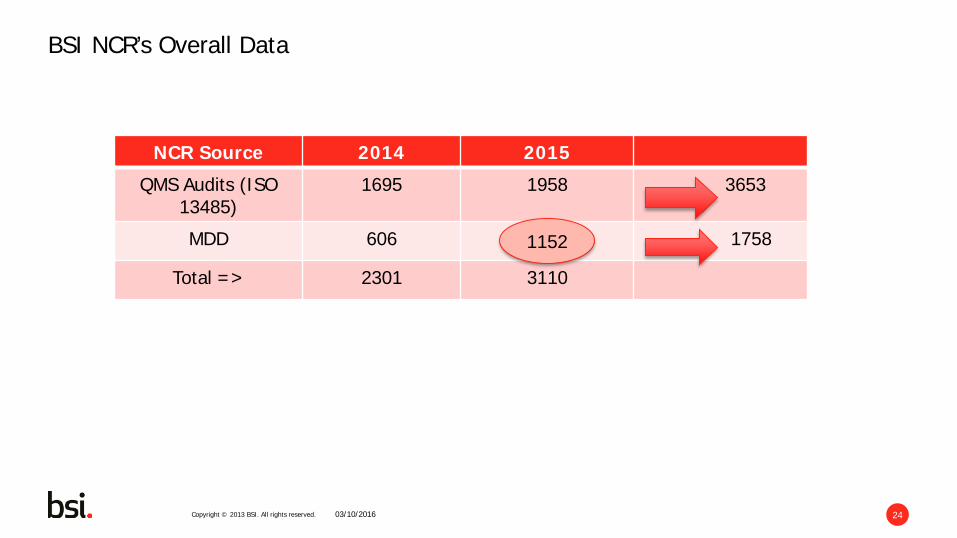
Copyright © 2013 BSI. All rights reserved. 24
BSI NCR’s Overall Data
NCR Source 2014 2015
QMS Audits (ISO 13485)
1695 1958 3653
MDD 606 1152 1758
Total => 2301 3110
03/10/2016
1152

Copyright © 2013 BSI. All rights reserved. 25
16%
15%
12%
9% 9%
8%
8%
8%
8%
7% Control of documents (4.2.3)
Internal audit (8.2.2)
Control of monitoring & measuring(7.6)Corrective action (8.5.2)
Purchasing process (7.4.1)
Documentation: General (4.2.1)
Infrastructure (6.3)
Control of records (4.2.4)
Planning of product realization(7.1)Training (6.2.2)
Top ISO 13485 Nonconformities
10/3/2016

Copyright © 2013 BSI. All rights reserved. 26
Typical Nonconformity
• Control of Documents (16%): • Incorrect version referenced either in the procedure or on the production floor • Improper control of documents of external origin (e.g., Guidance) • Incorrectly completed change request (missing signatures, incorrect signatories)
• Internal Audit (15%): • Conflict of interest of internal auditors (e.g., auditing own department or personnel) • Failure to follow up on open audit action items (open CAPAs) • Audit does not cover the scope specified in the procedure (MDD, CMDR not covered) • Audits not conducted per plan
• Control of Monitoring and Measuring devices (12%): • Equipment used in production not properly identified and/or not calibrated • Equipment was found to be out of calibration; also absence of corrective actions • Gauges used to calibrate equipment were not NIST traceable • Calibration records do not contain data (as found values, as measured values)
10/3/2016

Copyright © 2013 BSI. All rights reserved. 27
Typical Nonconformity
• Corrective Action (9%): • Inadequate root cause identified (Restatement of problem, No root cause) • Effectiveness checks not completed or verified • CAPAs not closed with specified timeframe
• Purchasing (9%): • Supplier is not on the Approved Supplier List • Missing supplier approval documentation (e.g., agreement, evaluation, audits, surveys) • No evidence of monitoring or requalification of suppliers
• Documentation: General (8%) • Missing required procedures (Technical file, Clinical evaluation, Risk, Canadian regulations) • Incorrect version of standards cited in Quality Manual or procedures (e.g., ISO 14971:2007) • Quality manual does not correctly reference exclusions and Not applicable clauses
10/3/2016

Copyright © 2013 BSI. All rights reserved. 28
Typical Nonconformity
• Infrastructure (8%): • Preventative maintain for a given piece of equipment has not been defined (or been conducted) • Pest control records are incomplete • Process for bio-contamination control/environmental monitoring is not effective
• Control of records (8%): • Records retention period not defined or not followed (no electronic back-up) • Incomplete records (missing records, missing required signatures, record not complete)
• Planning of product realization (8%) • Process for risk management is not fully effective (incomplete documentation, risk a action level) • Risk management process is not in compliance with ISO 14971:2012 (Annex ZA)
• Training (7%) • No process for determination/demonstration of required training per job description • Training records not developed • Individual has not be training on a particular procedure
10/3/2016

Copyright © 2013 BSI. All rights reserved. 29
NCR’s from ISO 13485 Audits
03/10/2016
0%
2%
4%
6%
8%
10%
12%
14%
2015
2014

Copyright © 2013 BSI. All rights reserved. 30
Top MDD Nonconformities
10/3/2016
27%
20%
12%
9%
8%
7%
6%
4% 4% 3%
Annex X & ER 6a: Clinical
ER 1 & ER 2: Risk Management
Article 3 & Annex I: ERs
Annex IX: Classification
ER 13: Label and IFU
Annex II, Section 3.2 (c):Documentation
ER 5: Packaging & Transportation
ER 3: Performance
ER 7.1: Biocompatibility

Copyright © 2013 BSI. All rights reserved. 31
Typical Nonconformity
• Clinical (27%): • Literature review not incompliance with MEDDEV 2.7.1 (no search terms/criteria, no critical analysis,
no conclusion) • Device equivalency has not be established (limited number of devices, basis is vague) • Clinical evaluation report has not be updated (with data from the field) • No post market clinical follow-up or justification for no plan
• ER 1 and 2 – Risk (20%) • Risk management is not in compliance with EN ISO 14971:2012 (IFU used for mitigation, use of
ALARP) • Missing risk management documents (plans for devices) • Device usability has not be addressed (EN 62366)
• Article 3 & Annex I – ERs (12%) • The ER Checklist was not complete (Missing ERs, reference appropriate standards, no rationale for
non-applicability) • Device is also a “Machine”; Machinery Directive has not been considered
10/3/2016

Copyright © 2013 BSI. All rights reserved. 32
Typical Nonconformity
• Classification (9%): • Missing classification rationale (device appears to be correctly classified) • Accessories have not been classified “in their own right separately from the device”
• ER 13 - Labels and IFU (8%): • Failure to comply with eIFU Regulation 207/2012 (website incorrectly referenced, Not addressed in
Risk management) • Incorrect use of or missing symbols (not harmonized, No EC rep) • No CE mark on label
• Annex II, Section 3.2 (c) (7%): • Risk management documentation is not complete • Missing documentation demonstrating compliance with device standards (EN60601-1-2, EN 62304) • No Design inputs, No Design verification
10/3/2016

Copyright © 2013 BSI. All rights reserved. 33
Typical Nonconformity
• ER 5 – Shelf Life, Packaging and Transportation (6%) • Shelf Life testing has not be satisfactorily completed (no testing, missing justification for articles
tested) • No transportation testing (Testing not conducted per the standards)
• ER 3 – Performance (4%): • No design verification/validation testing available (no rational for test failures) • In adequate product specification (No spec., or missing customer requirements)
• ER 7.1 – Biocompatibility (4%): • Biocompatibility testing not performed in accordance with the standard (ISO 10993-1)
• ER 4 – Device Lifetime (3%): • Device lifetime has been specified however no supporting documentation has ben presented • Product lifetime has not been defined
10/3/2016
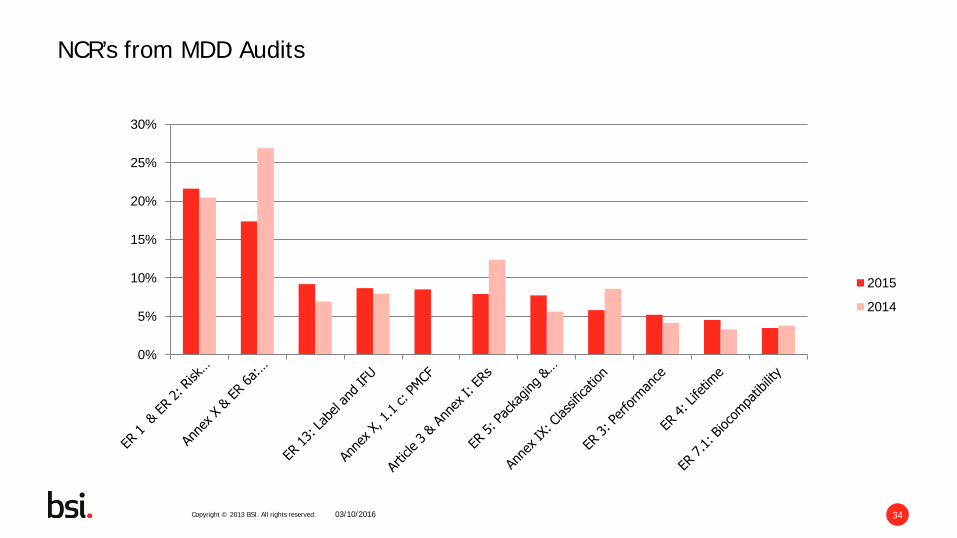
Copyright © 2013 BSI. All rights reserved. 34 03/10/2016
NCR’s from MDD Audits
0%
5%
10%
15%
20%
25%
30%
2015
2014

Copyright © 2013 BSI. All rights reserved. 35
Major and Minor Nonconformity Break Down
• Major nonconformity indicates a breakdown in the management system's ability to effectively control the processes for which it was intended. The identification of a major nonconformity places the validity of certification at risk. • BSI requires that major NC be closed out by an onsite visit
• Minor nonconformity relates to a single identified lapse in the management system. • A CAPA plan must be submitted to BSI to close out minor NC; this must include an appropriate root
cause, corrective action and effectiveness checks. • Plan will be accepted or rejected by BSI • Actions will be reviewed for affective implementation at the next assessment
10/3/2016
% of Major NC % of Minor NC
7 93

Copyright © 2013 BSI. All rights reserved. 36
Key Findings
ISO 13485 Nonconformities MDD Nonconformities
1 - Control of records: Incorrect version, No control of external documents
1 – Clinical evidence, equivalence, PMCF
2 – Internal audit: Conflict of interest, Open CAPS, Not conducted,
2 – Risk Management
3 – Control of Monitoring & Measuring devices: Equipment not identified, Out of calibration, not NIST traceable
3 – Failure to adequately address Essential Requirements
4 – Corrective Actions: Inadequate root cause, No Effectiveness checks, Not close in a timely manner
4 – Classification rationale is not specified
5 – Purchasing: Supplier is not on APL, Missing qualification documentation, No requalification
5 – Label and IFUs – Issues with eIFUS and compliance with 207/2012
10/3/2016

Copyright © 2013 BSI. All rights reserved. 37
Conclusions • Adoption of a more Holistic approach to PMS:
• A stronger link between QMS, Risk Management and Clinical Evaluation
• Regular update of Technical documentation with data from PMS
• Adoption of a cross-functional team to resolve/implement PMS issues.
• Better understanding of Vigilance reporting criteria and timelines, including FSCA requirements.
• PMCF/PMPF studies on more products
• Leverage data from International Consortia Registries?; e.g.: ICOR and ICCR

Copyright © 2013 BSI. All rights reserved. 38 03/10/2016
Questions


















