Page 1
ORIGINAL ARTICLE
Differential effects on nitric oxide synthase, heat shock proteinsand glutathione in human endothelial cells exposed to heat stressand simulated diving
Lise Fismen • Astrid Hjelde • Asbjørn M. Svardal •
Rune Djurhuus
Received: 6 July 2011 / Accepted: 4 November 2011 / Published online: 24 November 2011
� Springer-Verlag 2011
Abstract Decompression sickness (DCS) may result from
damage to the endothelium caused by the gas bubbles
formed during decompression and may be related to nitric
oxide (NO) production by nitric oxide synthase (NOS).
Heat stress prior to diving has been shown to protect ani-
mals from DCS, and by simulating this treatment in human
endothelial cells (HUVEC) we have shown that a simulated
dive performed subsequent to a heat stress potentiated the
heat-induced expression of HSP70 and increased the level
of the antioxidant glutathione (GSH). Since operational
saturation diving is performed at an increased oxygen level,
HUVEC have been exposed to heat stress and simulated
diving at 40 kPa O2, comparing the response on HSP70,
HSP90 and GSH level to the effects previously observed at
20 kPa O2. In addition, we wanted to investigate the effect
on both endothelial NOS (eNOS) protein and enzymatic
activity. The present results showed that a heat stress (45�C,
1 h) decreased the NOS activity and the protein markedly.
Hyperoxia (40 kPa) alone or a dive either at 20 or 40 kPa
O2,had no effects on NOS activity or protein. At 40 kPa
O2 a simulated dive after heat stress potentiated the
HS-induced HSP70 response, whereas the heat-induced
HSP90 response decreased. GSH levels were found to be
inversely related to NOS activity and protein expression,
and might be explained by a possible post-translational
regulation by glutathionylation of eNOS protein. The
results add to the limited knowledge of these critical factors
in cellular defence mechanisms that can prevent injury
during decompression.
Keywords Endothelial cells � Diving � Decompression �Nitric oxide synthase � Heat shock protein � Glutathione
Introduction
Decompression sickness (DCS) may result from damage to
the endothelium caused by the gas bubbles formed during
decompression (Brubakk et al. 2007). For this reason, the
endothelium is thought to represent an important target for
preventing DCS. The biochemical mechanisms involved
are still unresolved; however, heavy physical exercise 20 h
before a dive has been shown to significantly reduce the
number of vascular bubbles and confer protection against
DCS in man and animals (Dujic et al. 2004; Wisloff et al.
2003). Heat stress (42�C) for 1 h, 24 h prior to a dive
conferred protection from decompression injury in rat and
was accompanied by an increase in the production of heat
shock protein 70 (HSP70; Kampinga et al. 2009) but did
not affect bubble formation (Medby et al. 2008). Huang
and colleagues (Huang et al. 2003) observed that heat stress
prior to a dive reduced air bubble-induced lung injury and
that occurrence of DCS was associated with increased
HSP70 expression in rats. We have recently shown that a
simulated dive at 250 msw performed subsequent to a heat
stress (45�C) had a potentiating effect on the heat-induced
expression of HSP70 in human endothelial cells (HUVEC)
Communicated by Guido Ferretti.
L. Fismen � R. Djurhuus (&)
NUI AS, 5848 Bergen, Norway
e-mail: [email protected]
L. Fismen � A. M. Svardal
Institute of Medicine, University of Bergen,
5021 Bergen, Norway
A. Hjelde
Department of Circulation and Medical Imaging,
Norwegian University of Science and Technology (NTNU),
7491 Trondheim, Norway
123
Eur J Appl Physiol (2012) 112:2717–2725
DOI 10.1007/s00421-011-2241-4
Page 2
(Djurhuus et al. 2010). Experiments on rats exposed to
rapid decompression indicated an association between DCS
and increased gene expression of small heat shock proteins
in brain and lung, but surprisingly HSP70 was not signif-
icantly affected (Montcalm-Smith et al. 2007).
Exercise is known to be an inducer of endothelial-derived
nitric oxide (NO) in the vascular system (Green et al. 2002;
Kingwell et al. 1997). NO is a potent vasodilator and the
smallest cell signalling molecule known. It has several
physiological functions including regulation of vascular
tone (Lefroy et al. 1993), blood pressure (Rees et al. 1989)
and inflammation (Gross and Wolin 1995). NO dilates the
blood vessels by stimulating soluble guanylyl cyclase,
which elevates cyclic GMP causing smooth muscles to
relax. The NADPH-dependent oxidation from L-arginine to
NO and L-citrulline is catalyzed by nitric oxide synthase
(NOS). NOSs occur in three isoforms: the neuronal (nNOS),
the endothelial (eNOS) and the inducible (iNOS) form.
All the three isoforms require a number of cofactors
including heme protein, the flavins FAD and FMN, NADPH
and tetrahydrobiopterin (BH4) to function normally.
nNOS and eNOS also require calcium and calmodulin
(CaM) (Pollock et al. 1991; Snyder and Bredt 1991).
Endothelial NOS is regulated by and binds directly to heat
shock protein 90 (HSP90; Kampinga et al. 2009), indicating
that HSP90 is involved in NO generation (Fontana et al.
2002; Garcia-Cardena et al. 1998). eNOS synthesis requires
tight regulation at multiple levels, including transcription
and post-translational modifications (Fulton et al. 2001).
eNOS may produce NO or generate the superoxide anion
radical O2-, a process known as NOS uncoupling (Pou et al.
1992). The dominating pathway is dependent upon avail-
ability of its substrate, L-arginine, and its cofactor, BH4
(Vasquez-Vivar et al. 1998; Wever et al. 1997). BH4 has
been shown to be highly susceptible to oxidation, and when
oxidized causes NOS-derived superoxide generation rather
than NO formation. The cellular effects on signalling and
function exerted by O2- are rather different and usually
opposite to those of NO (Cardounel et al. 2005). Inhibiting
NO synthesis resulted in increased bubble formation and
reduced survival in sedentary, but not exercised rats fol-
lowing decompression (Wisloff et al. 2003). Conversely,
administration of a NO-releasing chemical resulted in
reduced bubble formation and increased survival in rats
following an identical dive (Wisloff et al. 2004). These
findings indicate that NO may play a role in bubble for-
mation and endothelial dysfunction.
The cellular antioxidant glutathione (GSH), a major
determinant of intracellular redox state, might be linked to
NOS activity by serving as a necessary reducing cofactor
(Laursen et al. 2001). We have recently shown that heat
stress (45�C, 1 h) prior to a simulated dive at 250 msw
resulted in an approximately 62% increase in the level of
intracellular glutathione in HUVEC (Djurhuus et al. 2010).
In macrophages, glutathione has been shown to be required
for maximal activity by the inducible nitric oxide synthase
(iNOS) (Stuehr et al. 1990). Whereas some investigators
have indicated that glutathione does not seem to have
significant impact on NOS (Huang et al. 2001), others have
indicated a correlation between GSH level and NOS
activity (Laursen et al. 2001). Based on the observations
above, we hypothesize that there could be a link between
NOS, GSH, HSP70, HSP90 and the possible endothelial
injury that may follow decompression.
Endothelial cells line the inside of blood vessels forming
the endothelium, and are thus the first cell layer that comes
in contact with the vascular gas bubbles formed during
decompression. In our model system, isolated HUVEC are
exposed to both heat and simulated diving conditions in a
pressure chamber. Heat stress has in several aspects similar
effects in vitro as physical exercise has in vivo and is a
known inducer of defence mechanisms such as HSPs.
Different stressors, such as hyperthermia, hypoxia, hyper-
oxia and exercise are all known to induce HSPs (Kregel
2002). Exposing bovine aortic endothelial cells (BAECs)
for heat shock (42�C, 1 h) resulted in no change in HSP90
and HSP70 protein content 24 h later (Harris et al. 2003).
However, a more severe heat shock (45�C, 1 h) caused an
eight-fold increase in HSP70, indicating that 42�C treat-
ment was too mild to produce a heat shock response. In the
present study, heat stress at 45�C is used to imitate the heat
stress used in several animal experiments and in vitro
experiments as indicated above and to induce stress
responses similar to those induced by physical exercise in
humans.
A main goal of our research is to study biochemical
mechanisms that may prevent adverse effects of decom-
pression stress in saturation diving. These mechanisms are
most likely activated prior to the decompression stress
itself, as indicated from both the animal and human studies
cited above. Accordingly, we expose our model system to
simulated dives at pressures relevant for saturation diving.
However, decompression of divers in saturation at, for
example, 250 msw would last for approximately 12 days
(Djurhuus et al. 2006), making such a decompression
profile incompatible with a cell model system. Moreover,
an important point is to preserve the biochemical param-
eters of interest, so that they as far as possible reflect the
situation immediately prior to decompression. Conse-
quently, we use a very rapid decompression profile of only
5 min. Major changes in parameters like HSPs and eNOS
are not likely to be expressed during such a short time
implying that the changes observed are not due to the
decompression, but rather to the compression, the pressure
per SE or the composition of the gas during the simulated
dive. Moreover, pilot studies at our laboratory have shown
2718 Eur J Appl Physiol (2012) 112:2717–2725
123
Page 3
that such rapid decompression had no apparent adverse
effects on the endothelial cells.
In a recent study, we exposed HUVEC to heat stress and
a subsequent simulated dive at 250 msw. The results
showed that the dive potentiated the heat stress induced
expression of HSP70 many times, while a dive alone did
not seem to have any effects on HSP70. In contrast, the
potentiating effect of a dive was not observed for HSP90.
Furthermore, the results showed that neither heat stress nor
dive had any effect on the GSH level, while a dive per-
formed after a heat stress increased the intracellular GSH
level approximately 62% (Djurhuus et al. 2010). In order to
distinguish between the effects of pressure itself and
increased partial pressure of O2, the experiments in that
study were conducted with a ‘‘normal’’ oxygen partial
pressure of 20 kPa. However, in operational saturation
diving the oxygen concentration is considerably higher,
usually from 35 to 80 kPa, depending on the different
phases of a dive. An elevated oxygen level may well-alter
redox status in the cells and affect the generation of NO,
either by altering the functionality of the NOS enzyme or
by changing the availability of essential cofactors like BH4
or molecular chaperones like HSP90.
It is important to investigate how the endothelial
NO-generating system responds to diving conditions at a
more realistic oxygen level. The present study is therefore
carried out by exposing HUVEC to heat stress and simulated
diving at 40 kPa O2, comparing the response on HSP70,
HSP90 and GSH level to the effects previously observed at
20 kPa O2. In addition, we wanted to investigate the effect on
both the eNOS protein and enzymatic activity after simulated
diving at both 20 and 40 kPa O2. The capability of NOS to
function after a dive has to our knowledge not been reported
previously.
Materials and methods
Cells and culture conditions
Human umbilical vein endothelial cells (HUVEC; Ameri-
can Type Culture Collection (ATCC) no CRL-1730) were
obtained from ATCC (Manassas, VA, USA). The cells were
grown without the addition of antibiotics in MCDB-131
medium (Gibco, Invitrogen Ltd., Paisley, UK) supple-
mented with heparin (50 lg/ml), endothelial cell growth
supplement (ECGS, 50 lg/ml; Millipore, Temecula, CA,
USA), and 20% heat-inactivated foetal bovine serum
(Biochrom AG, Berlin, Germany). Cells were seeded on
standard cell culture plastic flasks or dishes, all pre-treated
with a solution of 0.2% gelatine in Dulbecco’s phosphate-
buffered saline (DPBS) for 30 min at room temperature.
Stock cultures of cells were maintained at 37�C in an
atmosphere of 5% CO2 in air and a relative humidity of 95%
(standard conditions). For experiments, the cells were
exposed in pressure chambers as indicated below.
Exposure of cells to heat and simulated dive
Unless otherwise indicated, the experiments consisted of
five groups as indicated on Fig. 1. Cells were exposed to
heat and simulated dive in two identical, 15-l stainless steel
pressure chambers as described previously (Djurhuus et al.
2010). In brief, for heat stress the cell dishes were placed in
a pressure chamber that was flushed with helium to remove
air. The chamber was then flushed with He/O2/CO2 mixture
(75/20/5; mixed from 6.0, 5.5 and 5.0 qualities, respec-
tively, Yara Industrial AS, Bergen, Norway), isolated at
atmospheric pressure and 100% relative humidity at a
temperature of 45�C for 1 h. After exposure, the cell dishes
were returned to standard conditions in the incubator. For
the initial experiments with 20 kPa O2, the cell dishes were
left at the standard conditions for 48 h until harvesting. For
the experiments conducted with 40 kPa O2, the cell dishes
exposed to heat stress were subsequently left at standard
conditions for 24 h and then placed in a pressure chamber
that was flushed with a He/O2/CO2 mixture (55/40/5; mixed
from 6.0; 5.5 and 5.0 qualities, respectively, Yara Industrial
AS, Bergen, Norway), isolated at atmospheric pressure and
100% relative humidity at a temperature of 37�C for 24 h
until harvesting. For the simulated dive, the cell dishes were
placed in the other pressure chamber that was flushed with
He/O2/CO2 mixture (either (75/20/5 or 55/40/5). The
chamber was then pressurized with helium (6.0 quality,
Gardner Cryogenics A/S, Sandnes, Norway) to an absolute
pressure of 2.6 MPa and isolated with 100% relative
humidity at 37�C for 24 h. Following a rapid decompres-
sion (10 kPa/s), the cells were harvested immediately for
analysis as described below. An outline of the experimental
design is shown in Fig. 1.
Since the activity of NOS had not been determined at
similar conditions previously, we first exposed the cells to
simulated dive at 20 kPa O2, and accordingly group 2
(Fig. 1) was omitted.
Determination of NOS activity
Harvesting of cells
HUVEC were grown until confluence on 10 cm diameter
culture dishes. The medium was aspirated off and the cells
washed with pre-heated (37�C) PBS. The cells were
detached by trypsination (0.25% trypsin–0.5 mM EDTA)
for 5 min at room temperature (RT) before trypsin was
inactivated by the addition of basal Eagle’s medium
containing 10% foetal calf serum. Detached cells were
Eur J Appl Physiol (2012) 112:2717–2725 2719
123
Page 4
centrifuged at 120g for 10 min before resuspended in cold
PBS and centrifuged again. PBS was carefully aspirated off
and the pellets snapfrozen in a mixture of dry ice and ethanol
before stored at -80�C.
Enzyme activity
The release of NO generated by NOS was measured by
monitoring the conversion of radio-labelled L-[14C]-argi-
nine to L-[14C]-citrulline following the procedures devel-
oped by Knowles and Salter (1998) and Weissman and
Gross (2001) with some modifications. The formation of
L-citrulline is stoichiometric with the synthesis of NO, hence
the production of L-citrulline was assumed to correspond to
NO synthesis. In brief, pellets containing approximately
3–4 9 106 cells were resuspended in 120 ll ice-cold
20 mM HEPES buffer pH 7.4 containing 1 mM EDTA,
1 mM DTT, 0.25% Brij35 (Sigma-Aldrich Corp., St.
Louis, MO, USA), 10 mM ascorbic acid, 25 lM BH4,
80 lM FeCl2 and a cocktail of protease inhibitors (Com-
plete Mini, Roche Diagnostics GmbH, Mannheim, Ger-
many). Cells were left to lyse on ice for about 30 min
before 20 ll aliquots of cell suspension were placed in
tubes and mixed together with 55 ll assay buffer, 20 ll
L-[14C]-arginine assay buffer and 5 ll lysis buffer to make up
a total reaction volume of 100 ll. The cell suspension was
not centrifuged, because the majority of NOS in endothelial
cells is membrane associated, and so each sample was
drawn immediately after vortexing. The reaction mixture
contained (final concentrations) 45 mM Tris-HEPES, pH
7.4, 1.25 mM DTT, 0.37 mM CaCl2, 1 mM NADPH,
56 lM BH4, 1 lM FAD, 1 lM FMN, 60 mM L-valine,
0.02 lg/ll calmodulin (bovine brain, Calbiochem, Merck
KGaA, Darmstadt, Germany) and 100 lM L-arginine
(0.1 lCi L-[14C (U)]-arginine; PerkinElmer, Inc., Boston,
MA, USA).
After incubation at 37�C for 30 min, the reactions were
terminated by the addition of 1 ml ice-cold stop buffer
(50 mM HEPES, pH 5.5; 5 mM EDTA). L-citrulline pro-
duced was separated from the L-arginine by cation-exchange
chromatography using 2 ml 50% slush of Dowex resin
(50WX8, 100–200 mesh, ion-exchange, Acros organics,
New Jersey, USA) converted to the Na? form and pre-
equilibrated in stop buffer. 3.4 ml of water were added and
each sample mixed, after 30 min the tubes were spun lightly
(30g for 2 min) in a centrifuge. 4.5 ml (82% of total sample)
of L-[14C]-citrulline supernatant was aspirated off, 5 ml
scintillation fluid was added and the samples counted in a
liquid scintillation counter. Background values obtained
from complete samples, but with cell extracts replaced by
lysis buffer were subtracted from all results.
Protein was determined by the assay of Lowry et al.
(1951) using the Bio-Rad DC protein assay kit and bovine
IgG as a standard. Each experiment consisted of three
separate dishes per group and samples in triplicate were
drawn from each dish for NOS measurements. The NOS
activity was expressed as pmol/min/mg protein and pre-
sented as activity relative to control.
Determination of intracellular HSP70, HSP90
and eNOS
Harvesting and determination of viable cells
HUVEC were grown until confluence on 6 cm culture
dishes. Cells were trypsinated as described previously and
Exposure
group 0 24 48
1: Control Harvest
2: Control 40 kPa O2 Harvest
3: HS HS 45˚C, 1 h Harvest
20 kPa O2
4: Dive Harvest
5: HS + Dive HS 45˚C, 1 h Harvest
20 kPa O2
20 or 40 kPa O2, 100 kPa 24 h
Dive 24 h, 2.6 MPa (250 msw), 20 or 40 kPa O2
Hours
Std. conditions, 37 ˚C, 100 kPa, 24 h
Std. conditions, 37 ˚C, 100 kPa (1 bar)
Std. conditions, 37 ˚C, 100 kPa Dive 24 h, 2.6 MPa (250 msw), 20 or 40 kPa O2
Std. conditions, 37 ˚C, 100 kPa 40 kPa O2, 100 kPa 24 h
Std. conditions, 37 ˚C, 100 kPa, 24 h
Fig. 1 Exposure design. Overview of exposure to heat stress (HS,
45�C, 1 h) and subsequent simulated diving at 37�C. Standard
conditions were in an incubator with 5% CO2 in air, 100% humidity
and 37�C. Where indicated a second control included exposure to
40 kPa (0.4 bar) O2 and 5 kPa (0.05 bar) CO2 in He at atmospheric
pressure in parallel with the dive exposure. Heat stress was performed
in an atmosphere of 20 kPa O2 and 5 kPa CO2 in He at atmospheric
pressure, and simulated diving were in an atmosphere of 20 kPa
(0.20 bar) or 40 kPa O2 and 5 kPa CO2 in He
2720 Eur J Appl Physiol (2012) 112:2717–2725
123
Page 5
counted in an electronic cell counter (CASY model TT,
Innovatis AG, Reutlingen, Germany). The cells were cen-
trifuged at 120g for 10 min and washed by the addition of
1.5 ml DPBS at RT, gently resuspended, transferred to
microtubes and spun in a microcentrifuge at 300g for
10 min. The cell pellets were then snapfrozen and stored at
-20�C for later analysis.
Analysis of HSP70, HSP90 and eNOS
Cell pellets were lysed in the lysis buffer according to man-
ufacturer’s instructions. HSP70 (EKS-700B) and HSP90a
(EKS-895) ELISA kits were obtained from Stressgen Bior-
eagents (Victoria, Canada). Human eNOS Quantikine ELISA
kit was from R&D Systems Europe Ltd, Abingdon, England.
All ELISA kits were quantitative sandwich immunoassays
containing a monoclonal antibody against the actual protein
as the primary antibody and determine a coloured product
formed by a horseradish peroxidase conjugated to a second-
ary antibody. Colour formation was measured in a microplate
reader at 450 nm, and HSP or eNOS were quantified from a
standard curve generated with standard protein provided in
the kit. Concentrations were calculated in nanograms per 106
cells and presented as concentration relative to the control
group.
Determination of intracellular glutathione
HUVEC were grown until confluence on 6 cm culture
dishes. The analysis of glutathione was performed as
described previously (Djurhuus et al. 1991). In brief,
medium was removed, and the culture dishes were imme-
diately placed on ice and gently washed twice with 5 ml
ice-cold DPBS. The cells were extracted with 300 ll 5%
ice-cold sulphosalicylic acid containing 50 lM DTE using
a rubber policeman to scrape the cells off the dish, trans-
ferred to a microtube and frozen at -20�C. For analysis,
the samples were thawed, centrifuged and the supernatants
were used for determination of reduced and total glutathi-
one as described below.
Total free glutathione which represents the sum of
GSH, GSSG and soluble mixed disulphides (GSSR), and
reduced glutathione (GSH) were determined in the acid
extracts according to a modification (Mansoor et al. 1992)
of a chromatographic procedure described previously
(Svardal et al. 1990). Each experiment consisted of three
separate dishes per group and two parallel samples were
drawn from each dish for both reduced and total gluta-
thione measurements. Another three dishes in each group
were used for determination of viable cell number.
The results were expressed as glutathione equivalents in
nmol/106 cells, and presented as concentration relative to
control.
Statistical analysis
All results are calculated as the average ± SD of all indi-
vidual dishes seeded from the same stock culture and
undergoing the same exposure. The results were evaluated
by the analysis of variance (ANOVA) with the Tukey–
Kramer procedure for multiple comparisons. Effects were
considered significant when p \ 0.05.
Results
NOS enzyme activity and eNOS protein
Pilot experiments showed that the rate of NO production in
HUVEC was linear with regard to time for 30 min (data
not shown). The typical NOS activity for control cells
incubated at standard conditions was approximately
85 pmol/min/mg protein. A heat stress (45�C, 1 h) alone
decreased NOS activity 48%, while HS and a subsequent
exposure to a simulated dive decreased NOS activity 46%,
both compared to control at 20 kPa O2 (p \ 0.001, Fig. 2).
A dive alone at 20 kPa O2 had no significant effects on the
enzymatic activity. Increasing pO2 to 40 kPa had a slight
but not significant increasing effect on the NOS activity
(Fig. 3). Exposure to HS alone decreased the activity 54%,
while HS and a subsequent exposure to a simulated dive at
40 kPa O2 decreased NOS activity 51% (p \ 0.001 vs.
control 40 kPa O2). Exposing cells to 40 kPa O2 during the
dive did not affect enzyme activity. The results of NOS
activity at 20 and 40 kPa O2 showed similar patterns,
indicating that the elevated level of O2 during simulated
dive did not influence the enzyme’s capability for NO
production.
Determination of eNOS protein demonstrated a rather
similar pattern to the NOS activity. Increasing pO2 to
40 kPa had a slight but not significantly increasing effect
on the eNOS protein level (Fig. 3). HS decreased the
amount of eNOS protein 41% (p \ 0.01 vs. control 40 kPa
O2) (Fig. 3). Similarly, HS prior to simulated dive
decreased eNOS protein 52% (p \ 0.001 vs. control
40 kPa O2). No significant difference was observed
between eNOS protein in the control (40 kPa O2) and the
dive group.
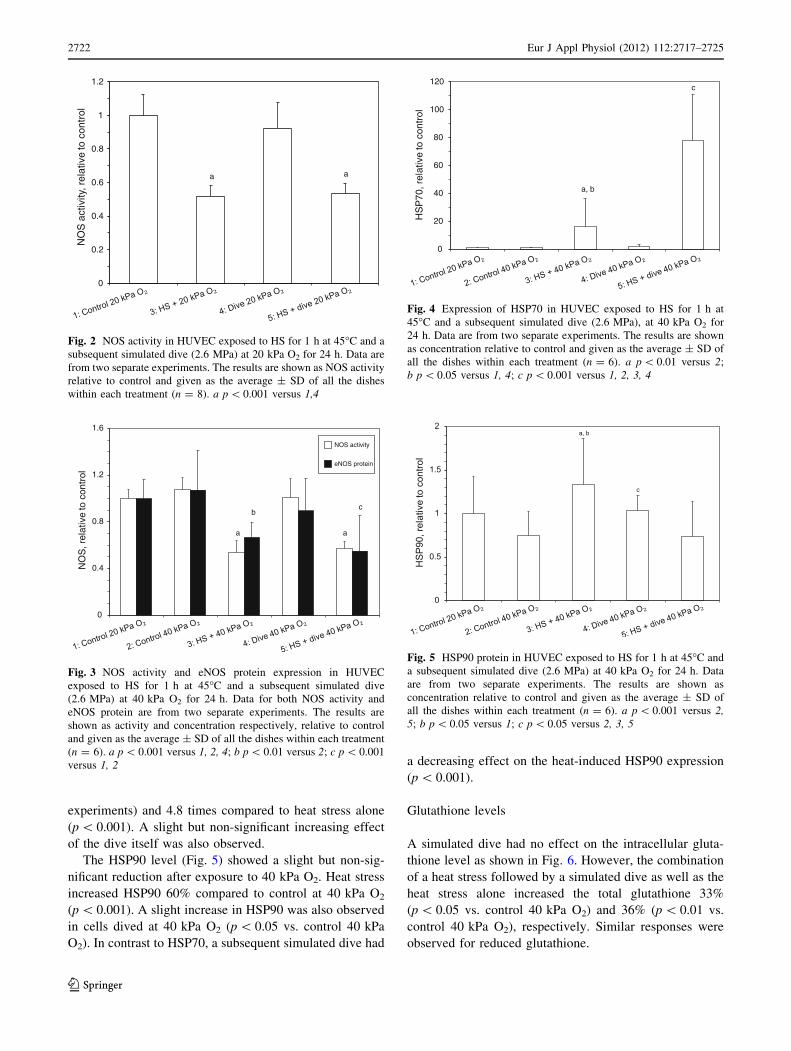
Expression of HSP70 and HSP90
The results shown in Fig. 4 demonstrated that heat stress
increased HSP70 many fold (p \ 0.01 vs. control 40 kPa
O2) and that a dive performed after heat stress had a
potentiating effect on the HSP70 expression increasing its
concentration on average 79 times compared to control
at 40 kPa O2 (p \ 0.001; data from two independent
Eur J Appl Physiol (2012) 112:2717–2725 2721
123
Page 6
experiments) and 4.8 times compared to heat stress alone
(p \ 0.001). A slight but non-significant increasing effect
of the dive itself was also observed.
The HSP90 level (Fig. 5) showed a slight but non-sig-
nificant reduction after exposure to 40 kPa O2. Heat stress
increased HSP90 60% compared to control at 40 kPa O2
(p \ 0.001). A slight increase in HSP90 was also observed
in cells dived at 40 kPa O2 (p \ 0.05 vs. control 40 kPa
O2). In contrast to HSP70, a subsequent simulated dive had
a decreasing effect on the heat-induced HSP90 expression
(p \ 0.001).
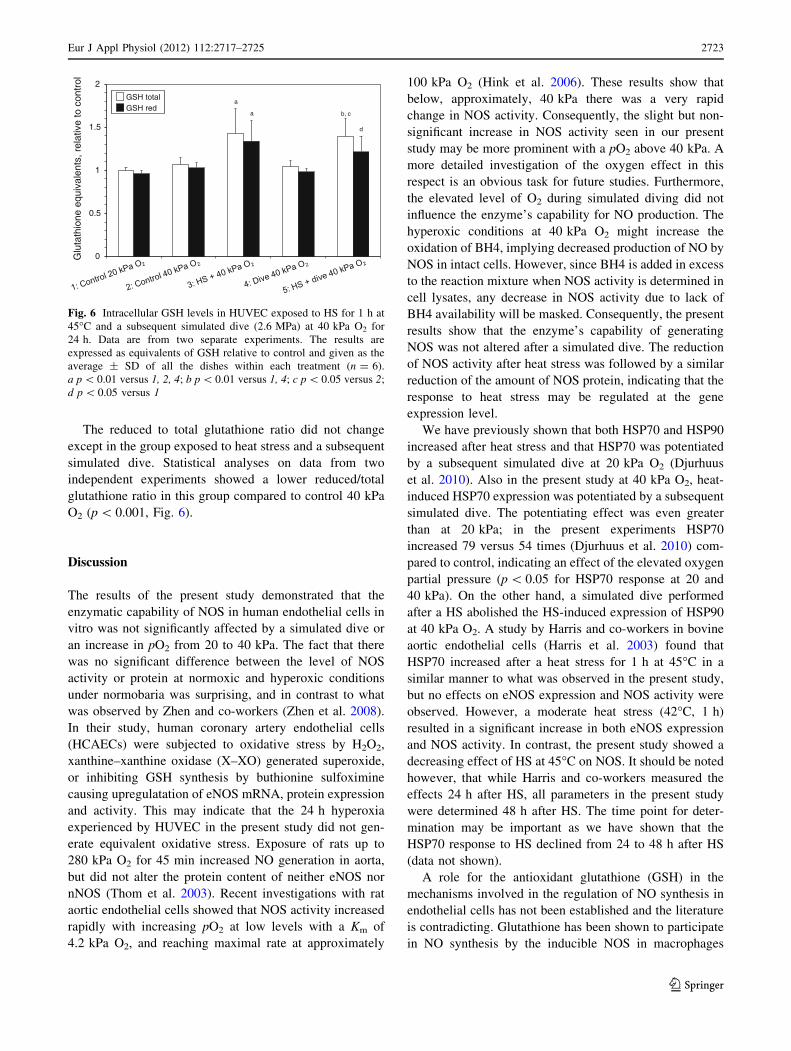
Glutathione levels
A simulated dive had no effect on the intracellular gluta-
thione level as shown in Fig. 6. However, the combination
of a heat stress followed by a simulated dive as well as the
heat stress alone increased the total glutathione 33%
(p \ 0.05 vs. control 40 kPa O2) and 36% (p \ 0.01 vs.
control 40 kPa O2), respectively. Similar responses were
observed for reduced glutathione.
0
0.2
0.4
0.6
0.8
1
1.2N
OS
act
ivity
, rel
ativ
e to
con
trol
a a
Fig. 2 NOS activity in HUVEC exposed to HS for 1 h at 45�C and a
subsequent simulated dive (2.6 MPa) at 20 kPa O2 for 24 h. Data are
from two separate experiments. The results are shown as NOS activity
relative to control and given as the average ± SD of all the dishes
within each treatment (n = 8). a p \ 0.001 versus 1,4
0
0.4
0.8
1.2
1.6
NO
S, r
elat
ive
to c
ontr
ol
NOS activity
eNOS protein
a a
bc
Fig. 3 NOS activity and eNOS protein expression in HUVEC
exposed to HS for 1 h at 45�C and a subsequent simulated dive
(2.6 MPa) at 40 kPa O2 for 24 h. Data for both NOS activity and
eNOS protein are from two separate experiments. The results are
shown as activity and concentration respectively, relative to control
and given as the average ± SD of all the dishes within each treatment
(n = 6). a p \ 0.001 versus 1, 2, 4; b p \ 0.01 versus 2; c p \ 0.001
versus 1, 2
0
20
40
60
80
100
120
HS
P70
, rel
ativ
e to
con
trol
a, b
c
Fig. 4 Expression of HSP70 in HUVEC exposed to HS for 1 h at
45�C and a subsequent simulated dive (2.6 MPa), at 40 kPa O2 for
24 h. Data are from two separate experiments. The results are shown
as concentration relative to control and given as the average ± SD of
all the dishes within each treatment (n = 6). a p \ 0.01 versus 2;
b p \ 0.05 versus 1, 4; c p \ 0.001 versus 1, 2, 3, 4
0
0.5
1
1.5
2H
SP
90, r
elat
ive
to c
ontro
la, b
c
Fig. 5 HSP90 protein in HUVEC exposed to HS for 1 h at 45�C and
a subsequent simulated dive (2.6 MPa) at 40 kPa O2 for 24 h. Data
are from two separate experiments. The results are shown as
concentration relative to control and given as the average ± SD of
all the dishes within each treatment (n = 6). a p \ 0.001 versus 2,5; b p \ 0.05 versus 1; c p \ 0.05 versus 2, 3, 5
2722 Eur J Appl Physiol (2012) 112:2717–2725
123
Page 7
The reduced to total glutathione ratio did not change
except in the group exposed to heat stress and a subsequent
simulated dive. Statistical analyses on data from two
independent experiments showed a lower reduced/total
glutathione ratio in this group compared to control 40 kPa
O2 (p \ 0.001, Fig. 6).
Discussion
The results of the present study demonstrated that the
enzymatic capability of NOS in human endothelial cells in
vitro was not significantly affected by a simulated dive or
an increase in pO2 from 20 to 40 kPa. The fact that there
was no significant difference between the level of NOS
activity or protein at normoxic and hyperoxic conditions
under normobaria was surprising, and in contrast to what
was observed by Zhen and co-workers (Zhen et al. 2008).
In their study, human coronary artery endothelial cells
(HCAECs) were subjected to oxidative stress by H2O2,
xanthine–xanthine oxidase (X–XO) generated superoxide,
or inhibiting GSH synthesis by buthionine sulfoximine
causing upregulatation of eNOS mRNA, protein expression
and activity. This may indicate that the 24 h hyperoxia
experienced by HUVEC in the present study did not gen-
erate equivalent oxidative stress. Exposure of rats up to
280 kPa O2 for 45 min increased NO generation in aorta,
but did not alter the protein content of neither eNOS nor
nNOS (Thom et al. 2003). Recent investigations with rat
aortic endothelial cells showed that NOS activity increased
rapidly with increasing pO2 at low levels with a Km of
4.2 kPa O2, and reaching maximal rate at approximately
100 kPa O2 (Hink et al. 2006). These results show that
below, approximately, 40 kPa there was a very rapid
change in NOS activity. Consequently, the slight but non-
significant increase in NOS activity seen in our present
study may be more prominent with a pO2 above 40 kPa. A
more detailed investigation of the oxygen effect in this
respect is an obvious task for future studies. Furthermore,
the elevated level of O2 during simulated diving did not
influence the enzyme’s capability for NO production. The
hyperoxic conditions at 40 kPa O2 might increase the
oxidation of BH4, implying decreased production of NO by
NOS in intact cells. However, since BH4 is added in excess
to the reaction mixture when NOS activity is determined in
cell lysates, any decrease in NOS activity due to lack of
BH4 availability will be masked. Consequently, the present
results show that the enzyme’s capability of generating
NOS was not altered after a simulated dive. The reduction
of NOS activity after heat stress was followed by a similar
reduction of the amount of NOS protein, indicating that the
response to heat stress may be regulated at the gene
expression level.
We have previously shown that both HSP70 and HSP90
increased after heat stress and that HSP70 was potentiated
by a subsequent simulated dive at 20 kPa O2 (Djurhuus
et al. 2010). Also in the present study at 40 kPa O2, heat-
induced HSP70 expression was potentiated by a subsequent
simulated dive. The potentiating effect was even greater
than at 20 kPa; in the present experiments HSP70
increased 79 versus 54 times (Djurhuus et al. 2010) com-
pared to control, indicating an effect of the elevated oxygen
partial pressure (p \ 0.05 for HSP70 response at 20 and
40 kPa). On the other hand, a simulated dive performed
after a HS abolished the HS-induced expression of HSP90
at 40 kPa O2. A study by Harris and co-workers in bovine
aortic endothelial cells (Harris et al. 2003) found that
HSP70 increased after a heat stress for 1 h at 45�C in a
similar manner to what was observed in the present study,
but no effects on eNOS expression and NOS activity were
observed. However, a moderate heat stress (42�C, 1 h)
resulted in a significant increase in both eNOS expression
and NOS activity. In contrast, the present study showed a
decreasing effect of HS at 45�C on NOS. It should be noted
however, that while Harris and co-workers measured the
effects 24 h after HS, all parameters in the present study
were determined 48 h after HS. The time point for deter-
mination may be important as we have shown that the
HSP70 response to HS declined from 24 to 48 h after HS
(data not shown).
A role for the antioxidant glutathione (GSH) in the
mechanisms involved in the regulation of NO synthesis in
endothelial cells has not been established and the literature
is contradicting. Glutathione has been shown to participate
in NO synthesis by the inducible NOS in macrophages
0
0.5
1
1.5
2G
luta
thio
ne e
quiv
alen
ts, r
elat
ive
to c
ontr
ol
GSH total GSH red
a
a b, c
d
Fig. 6 Intracellular GSH levels in HUVEC exposed to HS for 1 h at
45�C and a subsequent simulated dive (2.6 MPa) at 40 kPa O2 for
24 h. Data are from two separate experiments. The results are
expressed as equivalents of GSH relative to control and given as the
average ± SD of all the dishes within each treatment (n = 6).
a p \ 0.01 versus 1, 2, 4; b p \ 0.01 versus 1, 4; c p \ 0.05 versus 2;
d p \ 0.05 versus 1
Eur J Appl Physiol (2012) 112:2717–2725 2723
123
Page 8
(Stuehr et al. 1990). Decrease in GSH level has also been
found to impair the ability of cultured hepatocytes to
synthesize NO (Harbrecht et al. 1997). Laursen and co-
workers (2001) concluded that GSH is a necessary cofactor
for endothelial NO synthesis, and in conditions of low GSH
availability, such as during oxidative stress, GSH levels
may be a rate-limiting factor in the synthesis of NO in
endothelial cells from rat. In contrast, several investigators
have suggested that GSH-depleting agents had no effect on
NO synthesis in endothelial cells (Huang et al. 2001;
Murphy et al. 1991). In the present study GSH seemed to
be inversely related to the effect on NOS, demonstrating
approximately 35% increase in total GSH after heat stress
with or without a subsequent dive at 40 kPa O2. This is in
contrast to our previous results at 20 kPa O2 where only the
combination of heat and a subsequent dive gave an increase
in GSH level (Djurhuus et al. 2010). Elevated GSH content
might protect against oxidation of BH4, and thus help to
maintain the NO production. Hence, there could be a role
for GSH in maintaining BH4 in its reduced form to pre-
serve eNOS function. An interesting explanation may be
found in a recent work by Chen and co-workers (Chen et al.
2010) who established that oxidized glutathione (GSSG)
induced S-glutathionylation of human eNOS resulting in
markedly decreased NOS activity. In our experiments, the
cells exposed to heat and a subsequent dive demonstrated
both an increase in the total GSH level and a decrease in
the ratio red/tot GSH. This might be due to increased
GSSG level and a resulting S-glutathionylation of eNOS,
thus explaining the decreased NOS activity. In the heat-
exposed group we did not observe significant changes in
the redox status, however, the total GSH level was
increased in both HS-exposed groups implying also an
increase in the absolute amount of GSSG. It is therefore
tempting to suggest that an increased GSSG level induced
glutathionylation of eNOS responsible for the decreased
NOS-activity observed in both HS-exposed groups. The
decrease in eNOS protein observed after exposure to heat
stress, might be due to decreased immunoreactivity of
eNOS against the monoclonal antibody due to the S-glu-
tathionylation, and not to a lower content of the total eNOS
protein. If the latter is the case, the previous assumption
that the eNOS response to HS is regulated at the gene
expression level may not be valid; rather eNOS may be
regulated at least in part by the post-translational mecha-
nisms, like S-glutathionylation. eNOS protein is also
known to be regulated by other post-translational modifi-
cations such as acylation, phosphorylation and S-nitrosy-
lation (Michel and Vanhoutte 2010).
Further studies of eNOS and the role of the cofactor
BH4 during saturation diving are in progress. The impact
of the depth is also an important aspect that should be
addressed in the future.
Conclusions
In summary, a heat stress (45�C, 1 h) decreased the NOS
activity and eNOS protein markedly. A dive alone, either at
20 or 40 kPa O2, had no effects on NOS activity or eNOS
protein. Increasing O2 levels under normobaria from 20 to
40 kPa also did not significantly affect the NOS activity or
protein. At 40 kPa O2, a simulated dive after heat stress
potentiated the HS-induced HSP70 response, but decreased
the heat-induced HSP90 response. GSH levels were found
to be inversely related to NOS activity and protein
expression, and might be explained by a possible post-
translational regulation by glutathionylation of eNOS
protein.
Our findings may have importance for understanding the
mechanisms involved in endothelial dysfunction following
decompression (stress).
Acknowledgments The authors would like to thank Mrs. Torill
Sage and Mr. Harald A. Sundland, both NUI AS, and Mrs. Torunn
Eide at Institute of Medicine, University of Bergen, for excellent
technical assistance. The present study is an extension of previous
work initiated by Professor Alf O. Brubakk at Dept. Circulation and
Medical Imaging, Norwegian University of Science and Technology
(NTNU), Trondheim, Norway. The authors are grateful for his never
ending enthusiasm and scientific support. This work was financially
supported by the Norwegian Research Council (NFR), Statoil,
ExxonMobil Norway and Gassco.
Conflict of interest Lise Fismen and Rune Djurhuus are employed
by NUI AS, a company that is 100% owned by Statoil. The other
authors declare no conflict of interest.
References
Brubakk AO, Eftedal OS, Wisløff U (2007) Endothelium and diving.
In: Aird WC (ed) Endothelial biomedicine. Cambridge Univer-
sity Press, Cambridge, pp 497–505
Cardounel AJ, Xia Y, Zweier JL (2005) Endogenous methylarginines
modulate superoxide as well as nitric oxide generation from
neuronal nitric-oxide synthase: differences in the effects of
monomethyl- and dimethylarginines in the presence and absence
of tetrahydrobiopterin. J Biol Chem 280:7540–7549
Chen CA, Wang TY, Varadharaj S, Reyes LA, Hemann C, Talukder
MA, Chen YR, Druhan LJ, Zweier JL (2010) S-glutathionylation
uncouples eNOS and regulates its cellular and vascular function.
Nature 468:1115–1118
Djurhuus R, Svardal AM, Mansoor MA, Ueland PM (1991)
Modulation of glutathione content and the effect on methionine
auxotrophy and cellular distribution of homocysteine and
cysteine in mouse cell lines. Carcinogenesis 12:241–247
Djurhuus R, Segadal K, Svardal AM (2006) Glutathione in blood cells
decreases without DNA breaks after a simulated saturation dive
to 250 msw. Aviat Space Environ Med 77:597–604
Djurhuus R, Nossum V, Lundsett N, Hovin W, Svardal AM, Havnes
MB, Fismen L, Hjelde A, Brubakk AO (2010) Simulated diving
after heat stress potentiates the induction of heat shock protein
70 and elevates glutathione in human endothelial cells. Cell
Stress Chaperones 15:405–414
2724 Eur J Appl Physiol (2012) 112:2717–2725
123
Page 9
Dujic Z, Duplancic D, Marinovic-Terzic I, Bakovic D, Ivancev V,
Valic Z, Eterovic D, Petri NM, Wisloff U, Brubakk AO (2004)
Aerobic exercise before diving reduces venous gas bubble
formation in humans. J Physiol 555:637–642
Fontana J, Fulton D, Chen Y, Fairchild TA, McCabe TJ, Fujita N,
Tsuruo T, Sessa WC (2002) Domain mapping studies reveal that
the M domain of hsp90 serves as a molecular scaffold to regulate
Akt-dependent phosphorylation of endothelial nitric oxide
synthase and NO release. Circ Res 90:866–873
Fulton D, Gratton JP, Sessa WC (2001) Post-translational control of
endothelial nitric oxide synthase: why isn’t calcium/calmodulin
enough? J Pharmacol Exp Ther 299:818–824
Garcia-Cardena G, Fan R, Shah V, Sorrentino R, Cirino G,
Papapetropoulos A, Sessa WC (1998) Dynamic activation of
endothelial nitric oxide synthase by Hsp90. Nature 392:821–824
Green D, Cheetham C, Mavaddat L, Watts K, Best M, Taylor R,
O’Driscoll G (2002) Effect of lower limb exercise on forearm
vascular function: contribution of nitric oxide. Am J Physiol
Heart Circ Physiol 283:H899–H907
Gross SS, Wolin MS (1995) Nitric oxide: pathophysiological
mechanisms. Annu Rev Physiol 57:737–769
Harbrecht BG, Di Silvio M, Chough V, Kim YM, Simmons RL,
Billiar TR (1997) Glutathione regulates nitric oxide synthase in
cultured hepatocytes. Ann Surg 225:76–87
Harris MB, Blackstone MA, Ju H, Venema VJ, Venema RC (2003)
Heat-induced increases in endothelial NO synthase expression
and activity and endothelial NO release. Am J Physiol Heart Circ
Physiol 285:H333–H340
Hink J, Thom SR, Simonsen U, Rubin I, Jansen E (2006) Vascular
reactivity and endothelial NOS activity in rat thoracic aorta
during and after hyperbaric oxygen exposure. Am J Physiol
Heart Circ Physiol 291:H1988–H1998
Huang A, Xiao H, Samii JM, Vita JA, Keaney JF Jr (2001)
Contrasting effects of thiol-modulating agents on endothelial NO
bioactivity. Am J Physiol Cell Physiol 281:C719–C725
Huang KL, Wu CP, Chen YL, Kang BH, Lin YC (2003) Heat stress
attenuates air bubble-induced acute lung injury: a novel mech-
anism of diving acclimatization. J Appl Physiol 94:1485–1490
Kampinga HH, Hageman J, Vos MJ, Kubota H, Tanguay RM,
Bruford EA, Cheetham ME, Chen B, Hightower LE (2009)
Guidelines for the nomenclature of the human heat shock
proteins. Cell Stress Chaperones 14:105–111
Kingwell BA, Sherrard B, Jennings GL, Dart AM (1997) Four weeks
of cycle training increases basal production of nitric oxide from
the forearm. Am J Physiol 272:H1070–H1077
Knowles R, Salter M (1998) Measurement of NOS activity by
conversion of radiolabeled arginine to citrulline using ion-
exchange separation nitric oxide protocols. Methods Mol Biol
100:67–73
Kregel KC (2002) Heat shock proteins: modifying factors in
physiological stress responses and acquired thermotolerance.
J Appl Physiol 92:2177–2186
Laursen JB, Boesgaard S, Trautner S, Rubin I, Poulsen HE,
Aldershvile J (2001) Endothelium-dependent vasorelaxation in
inhibited by in vivo depletion of vascular thiol levels: role of
endothelial nitric oxide synthase. Free Radic Res 35:387–394
Lefroy DC, Crake T, Uren NG, Davies GJ, Maseri A (1993) Effect of
inhibition of nitric oxide synthesis on epicardial coronary artery
caliber and coronary blood flow in humans. Circulation
88:43–54
Lowry O, Rosebrough J, Farr A, Randall R (1951) Protein measure-
ment with the folin phenol reagent. J Biol Chem 193:265–275
Mansoor MA, Svardal AM, Ueland PM (1992) Determination of the
in vivo redox status of cysteine, cysteinylglycine, homocysteine,
and glutathione in human plasma. Anal Biochem 200:218–229
Medby C, Bye A, Wisløff U, Brubakk AO (2008) Heat shock
increases survival in rats exposed to hyperbaric pressure. Diving
Hyperb Med 38:189–193
Michel T, Vanhoutte PM (2010) Cellular signaling and NO produc-
tion. Pflugers Arch 459:807–816
Montcalm-Smith E, Caviness J, Chen Y, McCarron RM (2007) Stress
biomarkers in a rat model of decompression sickness. Aviat
Space Environ Med 78:87–93
Murphy ME, Piper HM, Watanabe H, Sies H (1991) Nitric oxide
production by cultured aortic endothelial cells in response to thiol
depletion and replenishment. J Biol Chem 266:19378–19383
Pollock JS, Forstermann U, Mitchell JA, Warner TD, Schmidt HH,
Nakane M, Murad F (1991) Purification and characterization of
particulate endothelium-derived relaxing factor synthase from
cultured and native bovine aortic endothelial cells. Proc Natl
Acad Sci USA 88:10480–10484
Pou S, Pou WS, Bredt DS, Snyder SH, Rosen GM (1992) Generation
of superoxide by purified brain nitric oxide synthase. J Biol
Chem 267:24173–24176
Rees DD, Palmer RM, Moncada S (1989) Role of endothelium-
derived nitric oxide in the regulation of blood pressure. Proc Natl
Acad Sci USA 86:3375–3378
Snyder SH, Bredt DS (1991) Nitric oxide as a neuronal messenger.
Trends Pharmacol Sci 12:125–128
Stuehr DJ, Kwon NS, Nathan CF (1990) FAD and GSH participate in
macrophage synthesis of nitric oxide. Biochem Biophys Res
Commun 168:558–565
Svardal AM, Mansoor MA, Ueland PM (1990) Determination of
reduced, oxidized, and protein-bound glutathione in human
plasma with precolumn derivatization with monobromobimane
and liquid chromatography. Anal Biochem 184:338–346
Thom SR, Fisher D, Zhang J, Bhopale VM, Ohnishi ST, Kotake Y,
Ohnishi T, Buerk DG (2003) Stimulation of perivascular nitric
oxide synthesis by oxygen. Am J Physiol Heart Circ Physiol
284:H1230–H1239
Vasquez-Vivar J, Kalyanaraman B, Martasek P, Hogg N, Masters BS,
Karoui H, Tordo P, Pritchard KA Jr (1998) Superoxide
generation by endothelial nitric oxide synthase: the influence
of cofactors. Proc Natl Acad Sci USA 95:9220–9225
Weissman B, Gross S (2001) Measurement of NO and NO synthase.
Curr Protoc Neurosci May; Chapter 7:Unit 7.13
Wever RM, van Dam T, van Rijn HJ, de Groot F, Rabelink TJ (1997)
Tetrahydrobiopterin regulates superoxide and nitric oxide gen-
eration by recombinant endothelial nitric oxide synthase. Bio-
chem Biophys Res Commun 237:340–344
Wisloff U, Richardson RS, Brubakk AO (2003) NOS inhibition
increases bubble formation and reduces survival in sedentary but
not exercised rats. J Physiol 546:577–582
Wisloff U, Richardson RS, Brubakk AO (2004) Exercise and nitric
oxide prevent bubble formation: a novel approach to the
prevention of decompression sickness? J Physiol 555:825–829
Zhen J, Lu H, Wang XQ, Vaziri ND, Zhou XJ (2008) Upregulation of
endothelial and inducible nitric oxide synthase expression by
reactive oxygen species. Am J Hypertens 21:28–34
Eur J Appl Physiol (2012) 112:2717–2725 2725
123