Research Collection Doctoral Thesis Kombination von Transmissionselektronenmikroskopie und Pulverbeugungsdaten zur Lösung von komplexen Zeolithstrukturen Author(s): Gramm, Fabian Publication Date: 2007 Permanent Link: https://doi.org/10.3929/ethz-a-005466842 Rights / License: In Copyright - Non-Commercial Use Permitted This page was generated automatically upon download from the ETH Zurich Research Collection . For more information please consult the Terms of use . ETH Library
Transcript
Research Collection
Doctoral Thesis
Kombination von Transmissionselektronenmikroskopieund Pulverbeugungsdaten zur Lösung von komplexenZeolithstrukturen
1 Ziel der Arbeit Um die Eigenschaften von Materialien zu verstehen ist die Strukturinformation
unerlässlich. Liegen die Materialien in Form von Einkristallen vor ist die
Kristallstrukturbestimmung fast immer problemlos. Etwas schwieriger ist dies, wenn nur
polykristalline Proben vorhanden sind. In diesem Fall werden Röntgen-
Pulverdiffraktionsmethoden angewendet um die Kristallstruktur zu bestimmen. Das
Hauptproblem dabei ist die Überlappung von Reflexen mit ähnlichen d-Werten. Nur die
Summe der Intensitäten solcher Reflexe kann bestimmt werden, nicht aber die Intensität der
einzelnen Reflexe. Mit speziell entwickelten Methoden ist es oft möglich das
Überlappungsproblem zu umgehen. Manchmal versagen aber diese Methoden, besonders
bei komplexen Kristallstrukturen. Für solche Fälle müssen andere Verfahren entwickelt
werden. Das Ziel dieser Arbeit ist es, dieses Überlappungsproblem mittels
elektronenmikroskopischer Methoden anzugehen.
Elektronen interagieren viel stärker mit Kristallen als Röntgenstrahlen. Deshalb ist es
möglich, einzelne Kristallite aus dem polykristallinen Material als Einkristalle für
Beugungsexperimente zu verwenden. Zwei Möglichkeiten werden als vielversprechend
angeschaut. Erstens könnten die Reflexintensitäten aus dem Einkristall-
Elektronenbeugungsexperiment verwendet werden. Zweitens könnten Bilder aus der
Hochauflösungstransmissionselektronenmikroskopie (HRTEM) zur Bestimmung von
Phasen der Strukturfaktoren gebraucht werden. Diese Phasen können entscheidend zur
erfolgreichen Strukturbestimmung beitragen.
Von besonderem Interesse für diese Arbeit sind die Zeolithe, deren vielseitigen
Eigenschaften eng mit ihrer Struktur verbunden sind. Die Zeolithe werden zum Beispiel als
Katalysatoren in der Petrochemie verwendet zur Spaltung der langkettigen
Kohlenwasserstoffe im Rohöl und zur Herstellung von Superbenzin durch Umformung und
Hydrierung der gespaltenen Moleküle. Sie werden auch als Wasserenthärter in
Waschmittel, und als Futterzusatz in der Schweinemast und in der Hühnerzucht gebraucht
und als Wirt für Gastspezies in "Advanced Materials".
2
Diese industriell wichtigen und wissenschaftlich sehr interessanten Materialien sind aber
oft nur in polykristalliner Form herstellbar. In diesem Projekt soll deshalb untersucht
werden, wie Elektronenmikroskopie in Kombination mit Pulverdiffraktionsdaten die
Grenzen der Strukturbestimmung von polykristallinen Materialien, insbesondere von
Zeolithen, erweitern kann.
3
2 Hintergrund und Methoden
2.1 Zeolithe
Zeolithe sind mikroporöse Materialien, aufgebaut aus einem dreidimensionalen, über alle
Ecken verknüpften TO4-Tetraeder Gerüst. Normalerweise ist ihre Zusammensetzung TO2,
wobei T ein 4-fach koordiniertes Atom (typischerweise Si, Al, Ga, P) ist. Jedes 4-fach
koordinierte T-Atom ist mit vier weiteren T-Atomen über eine Sauerstoffbrücke verbunden.
Das Gerüst kann neutral (z.B. SiO2 oder AlPO4) oder anionisch (z.B. AlSiO4) sein und
bildet Kanäle und Poren, welche mit Kationen, organische Molekülen und/oder Wasser
gefüllt sein können.
Abbildung 2-1: Kristallstruktur von Zeolith ZSM-5
Im Allgemeinen können die Kationen ausgetauscht und die Wassermoleküle durch Erhitzen
entfernt werden. Diese entscheidenden Eigenschaften von Zeolithen machen sie für
industrielle Anwendungen interessant als Ionenaustauscher, Molekularsiebe und
formselektive Katalysatoren. Zeolithe findet man aber auch in der Natur in Basalten und
Sedimentgesteinen. Für die meisten industriellen Anwendungen werden die Zeolithe
künstlich hergestellt. Ein solcher Zeolith mit relativ komplexer Struktur ist zum Beispiel
ZSM-5 (vgl. Abbildung 2-1). Der Zeolith IM-5, der vor ca. 10 Jahren erstmals synthetisiert
4
wurde, hat ebenfalls interessante katalytische Eigenschaften. welcher in der Petrochemie
verwendet wird. Seine Kristallstruktur konnte bis jetzt aber aus den Röntgenpulverdaten
nicht gelöst werden, lediglich ein vager Vorschlag für die Einheitszelle lag vor. Es bestand
die Hoffnung, dass Elektronenkristallographie hier weiterhelfen kann.
2.2 Kristallstrukturbestimmung aus Röntgen-Pulverdaten
Wenn Kristalle gross genug (ca. 50µm Kantenlänge) und in genügender Qualität gezüchtet
werden können, werden für die Kristallstrukturbestimmung sogenannte Einkristall-
methoden angewendet. Dabei werden die Phasen ( )hϕ der Strukturfaktoren ( )hF
( ) ( ) )(hhh ϕieFF ⋅= 2-1
rekonstruiert, damit eine Elektronendichte ( )rρ :
( ) ( )∑ −⋅=h
hh )(1 ϕρ ieFV
r 2-2
berechnet werden kann, wobei V das Volumen der Einheitszelle ist. Aus den
Röntgenbeugungsexperimenten können nur die Amplituden der Reflexe ( )hF bestimmt
werden und die Kunst der Kristallographie ist es, die richtigen Phasen zu bestimmen.
Dieses Phasenproblem wird heute meistens mit den so genannten Direkten Methoden
gelöst. Basierend auf statistischen Überlegungen, können Phasen aus der
Intensitätsinformation geschätzt werden. Heute existieren viele Computerprogramme,
welche die Direkten Methoden erfolgreich anwenden, sodass für Einkristalle die Lösung
des Phasenproblems in den meisten Fällen zur Routine geworden ist. Eine andere Methode
das Phasenproblem anzugehen ist Patterson-Methode, welche nicht direkt die Positionen
der Atome in der Elementarzelle liefert, sondern interatomare Vektoren. Sie wird bevorzugt
für Strukturen eingesetzt, welche aus wenigen Schweratomen und vielen Leichtatomen
bestehen.
Manchmal können aber keine Kristalle gezüchtet werden, die gross genug sind um
Einkristallmethoden anzuwenden. Insbesondere bei Zeolithen ist das der Normalfall. Die
kleinen Kristalle liegen dann zufallsorientiert im Pulver vor.
5
2.2.1 Überlappende Reflexe
Die Beugungsreflexe eines Einkristalls sind dreidimensional im Raum verteilt. Das
Beugungsbild einer Pulverprobe hingegen ist nur eindimensional. Es besteht aus der
Überlagerung von den Einkristallbeugungsbildern der einzelnen Kristallite im Pulver
(Abbildung 2-2) welche die charakteristischen Pulverringe bilden. Es fallen Reflexe,
welche beim Einkristallexperiment im reziproken Raum dreidimensional verteilt sind, im
Pulverexperiment aufeinander. Von der Überlappung betroffen sind die Reflexe, welche
den gleichen oder nahezu gleichen Abstand vom Ursprung (d.h. ähnliche d-Werte) haben.
Die Intensitäten der einzelnen überlappenden Reflexen können deshalb nicht aus dem
Pulverdiagramm bestimmt werden.
Auch die Indizierung eines Pulverdiagramms wird durch die Überlappungen oft erschwert.
Wenn zu viele Reflexe überlappen ist eine Strukturbestimmung sehr schwierig, wenn nicht
gar unmöglich.
In Pulverdiagrammen von grossen, komplexen Zeolithstrukturen wie TNU-9 (Gramm,
2006), ist die Peaküberlappung besonders gross. Das macht die Aufteilung der Intensitäten
überlappender Peaks schwierig. Trotzdem müssen die Reflexintensitäten, auch jene der
Abbildung 2-2: Kristallanordnung und entsprechendes Beugungsbild für (A) einen, (B) zwei und (C) viele Kristalle. In C) sind die typischen Ringe der Pulverdiffraktion und die vielen Reflexüberlagerungen sichtbar.
6
überlappenden Peaks, so gut wie möglich aus dem Pulverdiagramm extrahiert werden, denn
falsch bestimmte Reflexintensitäten können die Strukturbestimmung verunmöglichen. Je
kleiner die Halbwertsbreite der Peaks im Pulverdiagramm ist, desto weniger
Überlappungen wird es geben. Synchrotrondaten ermöglichen wegen der geringeren
Peakbreite eine bessere Extraktion der Reflexintensitäten.
2.2.2 Extraktion der Intensitäten
Um die direkten Methoden anwenden zu können, müssen die einzelnen Intensitäten der
Reflexe bekannt sein. Durch ein Vollprofil-Fit können die Intensitäten der Reflexe aus
einem Pulverdiagramm extrahiert werden, aber die überlappenden Reflexe müssten
irgendwie aufgeteilt werden. Die einfachste Aufteilungsmethode ist die
Äquipartitionierung, bei der jeder Reflex der Gruppe den gleichen Anteil an Intensität
bekommt. Wenn also n Reflexe in einer Gruppe überlappen, wird jedem der Reflexe 1/n der
Gesamtintensität zugeordnet. Heute werden zum Bestimmen der Intensität vor allem zwei
Ansätze verwendet. Das sind einerseits die Extraktion nach Pawley (Pawley, 1981) und
andererseits diejenige nach LeBail (LeBail, 1988). Während beim LeBail-Algorithmus die
Reflexintensitäten in einem iterativen Prozess angenähert werden, wird beim Pawley-
Algorithmus die Methode der kleinsten Fehlerquadrate benutzt, um der Reflexintensitäten
zu verfeinern. Beide Algorithmen wurden in verschiednen Programmen implementiert. Der
LeBail-Algoritmus wird zum Beispiel in GSAS (von Dreele, 1990), FULLPROF
(Rodriguez-Carvajal 1990), und EXTRAC (Baerlocher, 1990) verwendet und der Pawley-
Algorithmus zum Beispiel in DASH (David und Shankerland, 1998), EXPOL (Prokic,
2004), TOPAS (Bruker AXS, 2007) und TOPAS academic (Coelho, 2006).
Mit zunehmendem Überlappungsgrad werden die eingeführten Fehler aber zu gross, und
die extrahierten Intensitäten genügen nicht mehr für eine erfolgreiche Anwendung der
Direkten Methoden. Deshalb wurden Methoden entwickelt welche die Aufteilung
verbessern. Sivia und David (1994) benutzten Bayesian-Statistik, um die Pawley-Extraktion
durch Vermeidung negativer Intensitäten zu verbessern. Ein Vorteil der Methode ist, dass
Standardabweichungen für die Intensität von jedem Reflex berechnet werden können. Eine
weitere Methode ist, die Information von anderen Reflexen im Datensatz zu verwenden, um
die Amplituden überlappender Reflexe abzuschätzen, wie zum Beispiel mit der Methode
7
„Maximum Entropie“ (Gilmore, 1996) oder über Ansätze der Direkten Methoden (Jansen,
Peschar und Schenk 1992, Cascarno, Favia und Giacovazzo, 1992, Dorset 1997). Bei der
"Fast Iterative Patterson Squaring" (FIPS)-Methode (Estermann und Gramlich, 1993),
werden die überlappenden Reflexe durch einen iterativen Prozess über eine
Pattersonsynthese neu aufgeteilt. Dabei wird die Pattersonkarte quadriert. Die
Fourierkoeffizienten dieser quadrierten Pattersonkarte werden dann benutzt um die
Intensitäten der überlappenden Reflexe neu aufzuteilen. Die nicht überlappenden Reflexe
bleiben unverändert und werden dazu benutzt die Güte der Neuaufteilung zu beurteilen.
2.2.3 Einbezug chemischer Information
Ein anderer Ansatz um das Problem überlappender Reflexe zu lösen, ist die Verwendung
von chemischer Information über die gesuchte Struktur. So können zum Beispiel die
Koordinationspolyeder der Atome bekannt sein (z.B. SiO4-Tetraeder in Zeolithen), aber
ihre Position und Orientierung nicht, oder das Molekül ist bekannt (z.B. in organischen
Verbindungen), aber seine Konformation und die Kenntnis seiner Lage und Orientierung in
der Einheitszelle fehlt. Für solche Fälle ist die Strukturlösung im direkten Raum besonders
geeignet. Aufgrund der chemischen Informationen können im direkten Raum verschiedene,
zufällige Strukturen generiert werden. Von diesen Strukturen kann das Pulverdiagramm
berechnet und mit dem experimentell gemessenen verglichen werden. Durch Anwendung
von Optimierungsmethoden werden die Parameter der Strukturen variiert bis das
gerechnete Pulverdiagramm der Struktur möglichst den experimentellen Daten entspricht.
Es existieren verschiedene Methoden um diese Parameter zu variieren. Dafür wurde eine
Vielzahl von Computerprogrammen entwickelt, die auf Zufallsalgorithmen basieren und
verschiedene Optimierungsmethoden anwenden. Deem und Newsam (1992) entwickelten
ein Programm, welches ein "Simulated Annealing"-Algorithmus anwendet um
Zeolithstrukturen mit Hilfe von Pseudo-Potentialen zu bestimmen. Dies wurde später von
Falconi und Deem (1999) mit einem "Parallel Tempering"-Algorithmus erweitert. Für
Molekularstrukturen verwendeten Kariuki, et al. (1999) genetische Algorithmen (EAGER),
während LeBail (2001) in ESPOIR, Pagola und Stephens (2000) in PSSP, David und
Shankland (2001) in DASH und Andreev et al. (1997,1998) MonteCarlo-Methoden
und/oder "Simulated Annealing"-Algorithmen benutzten. In ENDEAVOUR führt
Putz (1999) Energieminimierung als Zusatzinformation ein. Im Jahr 2002 schrieben Favre-
8
Nicolin und Černý das allgemein verwendbare Programm FOX, welches auf "Simulated
Annealing" mit "Parallel Tempering" basiert. Die immer schnelleren Computer
ermöglichen eine immens grössere Anzahl verschiedener Strukturen in kürzerer Zeit zu
würfeln und immer mehr Parametern zu variieren. Trotzdem können so nicht beliebig
grosse Strukturen gelöst werden, da die benötigte Rechenleistung exponentiell mit der
Anzahl freier Parametern steigt.
Einen anderen Ansatz verfolgt das Programm FOCUS (Grosse-Kunstleve et. al., 1997). Es
wurde speziell für Zeolithe entwickelt. Zu den extrahierten Intensitäten aus dem
Pulverdiagramm wird ein zufälliger Satz von Phasen generiert. Daraus wird eine
Elektronendichtekarte berechnet, deren Maxima als Siliziumatome interpretiert werden. In
den gefundenen Positionen werden Zeolithgerüste gesucht, welche gesetzten
Wenn ein grobes Modell der Kristallstruktur vorliegt, wird meistens die Rietveldmethode
(Rietveld, 1969) angewendet um aus dem Pulverdiagramm genauere Information über die
Atompositionen zu erhalten. Bei dieser Methode wird das Pulverdiagramm als Ganzes
berücksichtigt und nicht einzelne extrahierte Intensitäten, weshalb das Problem der
Peaküberlapppung umgangen werden kann. Anstatt Intensitäten von einzelnen
beobachteten Reflexen oder Reflexgruppen zu vergleichen wird das ganze berechnete
Pulverdiagramm mit dem gemessenen Pulverdiagramm verglichen. Die Aufteilung der
10
Intensität einer Gruppe von Reflexen wird gar nicht erst versucht. Mit der Rietveldmethode
können Strukturen erstaunlich gut verfeinert werden, sofern ein Startmodell vorhanden ist.
2.3 Elektronenkristallographie
Im Begriff Elektronenkristallographie sind verschiedene Elektronenmikroskopische
Methoden vereint, welche zusätzliche Information wie Reflexintensitäten oder gar Phasen
liefern können. Unter der Annahme kinematischer Bedingungen und damit, dass die
Elektronenbeugungsintensitäten direkt proportional zum Quadtrat der Strukturfaktor-
amplituden sind, löste Cowley (1953a, 1953b, 1953c und 1955) die ersten Strukturen
basierend auf Elektronenbeugungsintensitäten. Dorset und Hauptman (1976) wendeten
dann die direkten Methoden auch für Elektronendaten an. In aktuelleren Arbeiten zeigen
Zou et al. (2003) wie mit Hilfe von Reflexintensitäten aus der Elektronenbeugung (SAED)
und der Phaseninformation aus den Hochauflösungsbildern (HRTEM) eine
dreidimensionale Potentialverteilung rekonstruiert werden kann. Aus dieser
Potentialverteilung konnte die Struktur des Quasikristallapproximanten (ν-ALCrFe-Phase)
bestimmt werden. Damit wurde auch demonstriert welches Potential in den
elektronenkristallographischen Techniken steckt, auch für Strukturen mit grosser
Einheitszelle.
Sowohl mit HRTEM, wie auch mit SAED ist es möglich selbst bei Pulverproben mit sehr
kleinen Kristalliten, Beugungsbilder und Hochauflösungsbilder von einzelnen Kristallen zu
machen. Meistens ist es aber aus experimentellen Gründen (beschränkte Kippwinkel des
Probenhalters, Vorzugsorientierung in der Probe etc.) nicht möglich, einen kompletten
Datensatz zu sammeln, was wiederum hinderlich ist für eine ab initio Strukturbestimmung.
Diese Einkristalldaten könnten aber eine ideale Ergänzung sein für die Pulvermethoden.
Weil Elektronen geladene Teilchen sind, ist die Interaktion von Elektronen mit Materie
wesentlich stärker als diejenige von Röntgenstrahlung. Der Wirkungsquerschnitt für die
elastische Streuung von Elektronen ist wesentlich grösser als für Röntgenstrahlung.
Deshalb genügen viel kleinere Kristalle für Elektronenbeugung verglichen mit
Röntgenbeugung. Ein Einkristall für Röntgenbeugung mit Synchrotronstrahlung zum
Beispiel sollte ca. 10x10x10µm3 gross sein, während für Elektronenbeugung Kristalle nur
ca. 100x100x50nm3 (also rund 2 Millionen Mal kleiner) gross sein müssen. Für kleine
11
Kristallite, wie z.B. Zeolithe, die normalerweise als polykristallines Pulver vorliegen,
ermöglicht Elektronenbeugung deshalb Einkristalldaten von einzelnen Kristalliten des
Pulvers zu messen. Dafür müssen die Kristallite aber genügend transparent sein für
Elektronen, das bedeutet sie sollten dünner als 50nm sein.
Wenn in der Elektronenmikroskopie von Elektronenbeugung gesprochen wird, sind damit
vor allem zwei Techniken gemeint:
(1) SAED (Selected Area Electron Diffraction) und
(2) CBED (Convergent Beam Electron Diffraction)
Bei CBED wird, wie der Name schon sagt, mit einem konvergenten Elektronenstrahl
gearbeitet. Der Strahl wird mit den Kondensorlinsensystem des TEM (Transmissions
Elektronen Mikroskop) auf die Probe fokussiert. Für Zeolithe ist dies aber problematisch,
weil sie im Elektronenstrahl nicht stabil sind. Wird der ganze Elektronenstrahl auf einen
Punkt innerhalb der Probe fokussiert, ergibt das eine grosse Elektronenstromdichte in der
Probe, was dazu führt, dass die Probe innert kürzester Frist zerstört wird. CBED wäre
interessant, weil die Methode genaue Informationen über die Raumgruppe liefern kann,
aber für elektronenstrahlempfindliche Proben wie Zeolithe, ist CBED aufgrund der kurzen
Lebensdauer der Proben schlecht geeignet. Deshalb wird in dieser Arbeit für Elektronen-
beugungsexperimente ausschliesslich SAED verwendet.
2.4 SAED
2.4.1 Einführung
Bei der SAED-Methode (vgl. Abbildung 2-3) wird mit einer Blende sozusagen ein kleiner
Teil eines Kristalliten ausgewählt, um von diesem ein Elektronenbeugungsbild
aufzunehmen. Die Methode wird auch in verschiedenen Textbüchern (z.B. Williams und
Carter, 1996) beschrieben. Um im TEM ein SAED-Bild zu erzeugen wird die Probe mit
einem parallelen, kohärenten Elektronenstrahl beleuchtet. Weil in der Probenebene keine
Blende plaziert werden kann, wird ein Zwischenbild von der Probe erzeugt. In diesem
Zwischenbild wird mit der SAED-Blende das Gebiet ausgewählt, von welchem ein SAED-
Bild erzeugt wird. Mit der Zwischenlinse wird ein Beugungsbild vom ausgewählten
Bereich erzeugt und dieses mit der Projektionslinse auf den Bildschirm vergrössert.
12
Abbildung 2-3: Schematische Abbildung eines Elektronenmikroskops. links: Strahlengang für das Abbilden der Probe, rechts: Strahlengang für Elektronenbeugung. Die Pfeile symbolisieren die Probe und deren Abbildung. Als Symbol für das Beugungsbild sind Punktreihen eingezeichnet.
13
2.4.2 Kalibrierung
Beugungsbilder liefern Informationen über die Abmessung und die Form der Einheitszelle,
sowie die Symmetrieelemente in der Einheitszelle. Die Abmessung der Einheitszelle kann
durch die Abstände zwischen den Reflexen bestimmt werden.
Um die Abstände zwischen den Reflexen möglichst präzise bestimmen zu können, wird der
Abbildungsmassstab kalibriert. Dafür wird zum Beispiel ein Goldpulver verwendet, dessen
Gitterparameter bekannt sind. Diese Kalibrierung muss für jede Kameralänge (virtueller
Abstand zwischen Probe und Beugungsbild) separat durchgeführt werden.
Das Beugungsbild wird im Abstand L (Kameralänge) zur Probe aufgenommen. Durch die
Projektionslinse, wird es auch, um den Faktor M vergrössert. Die geometrischen
Verhältnisse zwischen reziprokem Raum und dem Beugungsbild sind in Abbildung 2-4
ersichtlich und können durch die folgende Gleichung beschrieben werden, wenn λ die
Wellenlänge der Elektronen und d der Abstand der Netzebenen im Kristall ist:
LMRd
⋅= :1:1λ
2-3
Was auch geschrieben werden kann als:
LMRd ⋅⋅=⋅ λ 2-4
Für Beugungsbilder die unter gleichen Bedingungen aufgenommen wurden, ist LM ⋅⋅λ
eine Konstante für alle Reflexe im Beugungsbild. Mit einem Material dessen d -Werte
bekannt sind, kann deshalb die MLλ -Konstante bestimmt werden. Die Konstante kann
dann verwendet werden um über die umgeformten Beziehung 2-4
RLMd ⋅⋅
=λ
2-5
die d-Werte von unbekannten Materialien zu berechnen.
14
2.4.3 Einheitszelle bestimmen aus Kippserien
SAED-Bilder liefern einen zweidimensionalen Schnitt durch den reziproken Raum.
Einsehbar ist jeweils nur die nullte Lauezone. Höhere Lauezonen können im Normalfall
nicht beobachtet werden. Um die Einheitszelle bestimmen zu können, benötigt man
Informationen aus allen drei Dimensionen des reziproken Raumes. Ein Beugungsbild
genügt deshalb nicht. Es müssen daher mehrere SAED-Bilder entlang unterschiedlicher
Richtungen aufgenommen werden, um einen dreidimensionalen Datensatz zu erhalten. Mit
einem Doppelkipphalter (Abbildung 2-5) ist es möglich, die Probe im TEM um zwei in der
Probenebene liegende, senkrecht zueinander stehende Achsen zu kippen. Damit kann ein
Kristallit um eine beliebige, in der Probenebene liegende Achse gekippt werden, sodass
Beugungsbilder verschiedener Orientierungen von der gleichen Stelle eines Kristalliten
Abbildung 2-4: Kalibrierung an einem Gold Pulverdiagramm. M ist die Vergrösserung, L die Kameralänge, R der Radius des Ringes mit d-Wert d.
15
aufgenommen werden können. Wenn die Kippwinkel α zwischen den Orientierungen
bekannt sind, kann der dreidimensionale reziproke Raum wie in Abbildung 2-6
rekonstruiert werden. Das wurde zum Beispiel von Zou et. al. (1993) für den Quasikristall
Approximanten ν-AlFeCr gezeigt.
Abbildung 2-5: Doppelkipphalter mit den zwei Kipprichtungen α und β.
Der Doppelkipphalter hat aus konstruktiven Gründen einen begrenzten Kippwinkel.
Deshalb kann nicht jede Orientierung des Kristalls beobachtet werden und daher auch nicht
der gesamte reziproke Raum aus Beugungsdaten von einem einzigen Kristallit rekonstruiert
werden. Haben die Kristalle in der Probe eine Vorzugsorientierung, können ebenfalls
einzelne Richtungen nicht beobachtet werden. Das ist zum Beispiel bei nadeligen
Kristalliten der Fall, da die Nadeln meistens flach liegen und nicht auf der Spitze stehen.
Um entlang der Nadel schauen zu können müsste die Nadel durch kippen der Probe
aufgestellt werden. Das ist aufgrund des eingeschränkten Kippwinkels normalerweise nicht
möglich. Zudem sind die Nadeln meistens viel zu lang (länger als 50nm), um entlang der
Längsrichtung noch elektronentransparent zu sein. Aber auch von einem unvollständigen
Datensatz kann meistens eine Elementarzelle ausfindig gemacht werden. Für die
Rekonstruktion des reziproken Raumes und dessen Visualisierung kann die Software
TRICE (Zou et. al., 2004) benutzt werden. Das Programm schlägt auch Einheitszellen für
die rekonstruierten Daten vor.
16
2.4.4 M
Mehrfac
Röntgen
interagie
Röntgen
(nur Str
Mehrfac
AbbildunSAED Bi
16
Mehrfachbe
chbeugung h
nbeugung, w
eren. Bei
nstrahlen und
reuung, ohn
chbeugung v
g 2-6: Kippserldern kann der
eugung
hat bei Elek
weil Elektro
dieser Inte
d sie können
ne Energiev
verfälscht.
rie. Die SAED-r reziproke Rau
ktronenbeugu
onen viel s
eraktion w
n auch Energ
verlust) Date
-Bilder (von Zeum rekonstruie
ung eine we
stärker als
werden die
gie verlieren
en sozusage
eolith IM-5) w
ert werden.
esentlich grö
Röntgenstr
Elektronen
. Dadurch w
en durch dy
wurden um die a
össere Bedeu
ahlen mit
n stärker g
werden die ki
ynamische
a*-Achse gekip
utung als be
der Materie
gestreut al
inematischen
Effekte und
ppt. Aus diesen
ei
e
s
n
d
n
17
Für eine ab initio Strukturbestimmung werden aber Reflexintensitäten benötigt die unter
kinematischen Bedingungen aufgenommen wurden. Aus dynamischen
Elektronenbeugungsdaten können Kristallstrukturen nur bestimmt werden, wenn bereits ein
grobes Strukturmodell vorliegt. Ist ein solches vorhanden wäre eine Möglichkeit dieses so
zu verfeinern, dass die simulierten Daten möglichst die experimentellen widerspiegeln.
Zusätzlich zu den üblichen Parametern (Atompositionen, Skalenfaktor und Debye-Waller
Faktor usw.) einer klassischen Strukturverfeinerung müssen für den dynamischen Fall auch
die Kristalldicke und Absorptionsparameter verfeinert (Jansen et al., 1998) werden.
Eine andere Möglichkeit besteht darin, Bedingungen zu finden, unter welchen dynamische
Effekte und die Mehrfachstreuung vernachlässigbar klein sind, das heisst wo die
Beugungsbilder also quasikinematisch sind. Folgende Faktoren sind dabei in Betracht zu
ziehen:
Kristalldicke: Je dünner der Kristall ist, desto kleiner ist die Wahrscheinlichkeit für
Mehrfachbeugung. An einem Zeolithen zeigten Pan und Crozier (1993), mit 200Å dicken
ZSM-5 Kristalliten, dass bei 400kV Beschleunigungsspannung gemessene und simulierte
Elektronenbeugungsdaten gut übereinstimmen.
Beschleunigungsspannung: Je höher die Beschleunigungsspannung, desto kürzer ist die
Wellenlänge und desto weniger Mehrfachstreuung findet statt. Der Anteil an
Mehrfachbeugung wird zwar geringer bei hohen Beschleunigungsspannungen (z.B.
1000kV), verschwindet aber nicht gänzlich wie Honjo und Kitamura (1957) und Dorset
(1976) zeigten.
Kristallorientierung: Leichtes Wegkippen des Kristalls von der gesuchten Orientierung
kann verhindern, dass die Atome säulenartig, exakt übereinander liegen. Mehrfachbeugung
kann dadurch teilweise vermieden werden (Fultz und Howe, 2001).
Präzession des Elektronenstrahls: Auch durch Präzession des Elektronenstrahls (Vincent
und Midgley, 1994) kann Mehrfachbeugung reduziert werden, weil über verschiedene
Einstrahlrichtungen gemittelt wird. An einem Schwermetalloxid zeigten
Weirich et al. (2006), dass mit der Präzessionsmethode gemessene Reflexintensitäten
erfolgreich für die ab initio Strukturbestimmung eingesetzt werden können.
18
2.4.5 Raumgruppenbestimmung aus SAED
Durch Kippserien kann, wie in Abschnitt 2.4.3 beschrieben, der reziproke Raum
rekonstruiert werden. Aufgrund der ausgelöschten Reflexe kann auf die Raumgruppe
geschlossen werden. Mehrfachbeugung kann dabei aber Schwierigkeiten verursachen.
Durch Mehrfachbeugung können Reflexe, die eigentlich ausgelöscht sind, trotzdem
Intensität bekommen und im Elektronenbeugungsbild beobachtet werden. Werden im
SAED-Bild aber Auslöschungen gefunden, können damit Auslöschungsregeln definiert
werden. Es muss aber immer in Betracht gezogen werden, dass ausgelöschte Reflexe
aufgrund von Mehrfachbeugung oder dynamischen Effekten trotzdem vorhanden sein
können. Wie bei Röntgenbeugungsexperimenten kann aufgrund dieser Auslöschungsregeln
In Beugungsexperimenten geht die Phase des Strukturfaktors verloren. Diese Phasen
werden aber benötigt um die Elektronendichte bestimmen zu können. Ganz anders sieht das
bei der Hochauflösungs-Transmissionselektronenmikroskopie (HRTEM) aus. Im HRTEM-
Bild werden sowohl die Phasen, wie auch die Intensitäten von Reflexen übertragen und
können aus dem fouriertransformierten Bild unter bestimmten Voraussetzungen extrahiert
werden. Das Hochauflösungsbild stellt eine Projektion des Kristalls, respektive der
Kristallstruktur dar. Verschiedene Parameter beeinträchtigen aber das Hochauflösungsbild,
so dass es nicht direkt interpretierbar ist. Klug (1978) zeigte an einer Serie unterschiedlich
fokussierter Hochauflösungsbilder an einem anorganischen Kristall, dass aus jedem der
Bilder ein Abbild der korrekten Projektion des Kristalls berechnet werden kann, wenn die
Bilder mit der Kontrast-Transfer-Funktion korrigiert werden. Genau genommen ist das nur
für dünne Proben möglich. Voraussetzung ist, dass die Probe als schwaches Phasen-Objekt
(Anhang A) betrachtet werden kann.
19
2.5.2 Die Abbildung in der Hochauflösungs-Transmissionselektronenmikroskopie
Jeder Punkt von der Probe sollte im Idealfall im Hochauflösungsbild als Punkt abgebildet
werden. Aufgrund von Abbildungsfehlern der magnetischen Linsen und wegen mangelnder
Kohärenz des Elektronenstrahls ist die Abbildungsleistung aber beeinträchtigt. Ein Punkt in
der Probe wird deshalb nicht als Punkt abgebildet sondern als Kreisfläche, wobei die
Flächen im Bild überlappen. Wenn )(rf die Probe beschreibt so kann die Abbildung im
direkten Raum ( )(rg ) mit folgender Faltungsfunktion beschrieben werden:
)'()()( rrhrfrg −⊗= 2-6
Wobei )'( rrh − beschreibt, welcher Punkt wie stark zum Bild beiträgt. Wenn )(uG , )(uF
und )(uH die Fouriertransformierten von )(rg , )(rf und )(rh sind, wird mit:
)()()( uFuHuG ⋅= 2-7
die Abbildung im reziproken Raum beschrieben. Die Funktion )(uH wird durch Parameter
des Mikroskops bestimmt und kann in drei verschiedene Anteile ( )(),( uBuA und )(uE )
unterteilt werden. Die Bedeutung der drei Anteile wird in diesem Abschnitt kurz erklärt.
Mit der Objektivblende im Mikroskop werden in der rückwärtigen Brennebene die
gebeugten Elektronenstrahlen ausgewählt, welche zum Bild beitragen sollen. Die Wirkung
dieser Blende wird mit der Blendenfunktion )(uA beschrieben. Magnetische Linsen sind bei
weitem nicht perfekt, sie weisen Abbildungsfehler auf. Zum Beispiel wird der
Elektronenstrahl im Zentrum und in den Randbereichen der Linse nicht gleich stark
abgelenkt, was zur so genannten sphärischen Abberation führt. Auch Elektronen
unterschiedlicher Wellenlänge erfahren nicht die gleiche Ablenkung in der magnetischen
Linse, was als chromatische Abberation bezeichnet wird. Diese sphärische und die
chromatische Abberation werden durch die Abberationsfunktion )(uB ausgedrückt.
Weitere Effekte beeinflussen die Auflösung in Hochauflösungsbildern, wie zum Beispiel
die Kohärenz der Elektronenquelle. Diese Effekte werden in der umhüllenden
Dämpfungsfunktion )(uE zusammengefasst. Die Funktion )(uH kann unter
Berücksichtigung dieser Effekte wie folgt geschrieben werden:
20
)()()()( uEuBuAuH ⋅⋅= 2-8
Die drei Funktionen (Blendenfunktion, Aberrationsfunktion und umhüllende
Dämpfungsfunktion) werden im Folgenden kurz beschrieben. Die Blendenfunktion soll 0
sein dort wo die Strahlen durch die Blende ausgeblendet werden und 1, dort wo die
Strahlen durch das Loch der Blende durchtreten können:
=derBlendeausserhalbu wenn 0
Blendeder innerhalbu wenn 1)(uA 2-9
Die Abberationsfunktion )(uB wird (z.B. Williams und Carter, 1996) wie folgt beschrieben:
( ))(e)( uixpuB χ= 2-10
432
21)( uCuu sλππελχ += 2-11
)(uB ist demzufolge abhängig vom Defokus ε dem reziproken Gittervektor u , wie auch
von der Wellenlänge λ der Elektronen (d.h. von deren Beschleunigungsspannung), und der
sphärischen Abberationskonstante sC des Elektronenmikroskopes.
Inkohärenz der Elektronen führt zu geringerer Auflösung im Hochauflösungsbild. Es sind
vor allem die chromatische Abberation und die Konvergenz des Elektronenstrahls, welche
die Kohärenz des Elektronenstrahls beeinflussen und somit die Auflösung des
Elektronenmikroskops begrenzen. Die umhüllende Dämpfungsfunktion wird
dementsprechend in zwei Teile aufgeteilt:
( ) ( ) ( )uEuEuE α⋅= ∆ 2-12
wobei die Funktion ( )uE ∆ den Anteil der chromatischen Abberation beschreibt und ( )uEα
den Anteil der Konvergenz. Der Elektronenstrahl ist um so kohärenter, je kleiner die
Streubreite der Energie der emittierten Elektronen ist und je stabiler die Linsenströme sind.
Schwankungen des Linsenstromes führen dazu, dass sich die Brennebene der Linse
verschiebt. Elektronen mit unterschiedlicher Energie werden verschieden von den Linsen
im TEM abgelenkt. Für Elektronen mit höherer kinetischer Energie wird die Brennebene
21
näher bei der Linse liegen als für solche mit geringerer kinetischer Energie. Der Effekt
chromatischer Abberation kann deshalb durch Variation des Fokus der Objektivlinse
modelliert werden. Wenn ε der Defokus der Linse ist und die Fokuseinstellung im Bereich
∆−ε und ∆+ε variiert, ist
( )
∆−=∆
4222
21exp uuE λπ 2-13
Die Konvergenz des Elektronenstrahls kann durch den halben Konvergenzwinkel α des
Elektronenstrahls beschrieben werden.
Für die Dämpfungsfunktion aufgrund der Strahlkonvergenz ergibt sich folgende Beziehung:
( ) ( )( )222222exp uCuuE sλεαπα +−= 2-14
Um die Funktion )(uH beschreiben zu können müssen deshalb zusätzlich zu den
Parametern aus Gleichung 2-11, noch der halbe Konvergenzwinkel α und die
Defokusstreubreite ∆, bekannt sein.
2.5.3 Die Schwaches-Phasenobjekt-Approximation
Die Schwaches-Phasenobjekt-Approximation wird in verschiedenen Textbüchern
(Williams und Carter (1996), Spence (2003), Alexander (1997)) ausführlich beschrieben,
eine Zusammenfassung davon befindet sich im Anhang A. Damit die Schwaches-
Phasenobjekt-Approximation angewendet werden kann, muss einerseits ein Phasenobjekt
vorliegen und dessen projiziertes Potential muss klein sein, so dass das Potential der Probe
durch dessen Projektion ersetzt werden kann. Ein Phasenobjekt liegt vor, wenn keine
Absorption stattfindet, das heisst die Elektronenwelle läuft durch das Objekt, erfährt eine
Phasenverschiebung, aber die Amplitude bleibt unverändert. Beide Voraussetzungen für die
Schwaches-Phasenobjekt-Approximation können nur erfüllt werden, wenn die Probe sehr
dünn ist.
Wenn die Schwaches-Phasenobjekt-Approximation angewendet werden kann, besteht ein
linearer Zusammenhang zwischen dem projizierten Potential der Probe und der Amplitude
der Austrittswelle.
22
2.5.4 Die Kontrasttransferfunktion (CTF)
Wenn die Schwaches-Phasenobjekt-Approximation gilt, beschreibt die Funktion )(uH den
Zusammenhang, zwischen dem Bildkontrast und dem projizierten Potential der Probe.
Deshalb wird die Funktion )(uH in diesem Fall Kontrasttransferfunktion (CTF) )(uT
genannt und wird dann beschrieben durch:
( ) ( ) )(sin2)( uuEuAuT χ= 2-15
Die CTF beschreibt welche Frequenzen, ausgedrückt durch den reziproken Gittervektor u ,
auf welche Art ins Hochauflösungsbild übertragen werden. Die CTF ist abhängig von
optischen Parametern des Mikroskops (Abbildung 2-7 und Abbildung 2-8). Die
Informationsübertragung ist unterschiedlich für jede übertragene Frequenz je nach
Einstellung des Mikroskops (Beschleunigungsspannung, Defokus etc.). Ein Hoch-
auflösungsbild ist deshalb nicht direkt interpretierbar, sondern muss zuerst mit der CTF
korrigiert werden. Dies ist auch ersichtlich wenn die Bedeutung der CTF genauer betrachtet
wird. Ändert das Vorzeichen der CTF bedeutet das eine Umkehr des Kontrastes. Ist die
CTF gar 0 wird dieser Frequenzbereich gar nicht ins Hochauflösungsbild übertragen.
Aus dem Hochauflösungsbild kann aber das projizierte Potential der Probe dann
rekonstruiert werden, wenn die Schwaches-Phasenobjekt-Approximation erfüllt und die
CTF bekannt ist (Zou, 1995).
2.5.5 Der Scherzer-Defokus
Die CTF ist abhängig vom Defokus (Gleichung 2-13). Deshalb hängt auch der Kontrast im
HRTEM-Bild vom Defokus ab. Man kann sich nun fragen ob es einen optimalen
Defokuswert gibt, bei welchem der Kontrast im HRTEM-Bild besonders gut und das
HRTEM-Bild einfach interpretierbar ist. Dazu wird am besten von der CTF ausgegangen.
Damit ein Bild direkt interpretierbar wird, müsste die CTF am besten alle Frequenzen
gleich übertragen. Das ist der Fall wenn die CTF eine Konstante ist. Das ist aber mit keiner
Defokuseinstellung möglich. Es gibt aber eine Situation, bei welcher die CTF im Bereich
niedriger Frequenzen keine Nulldurchgänge hat und relativ konstant verläuft bis zum ersten
Nulldurchgang (Abbildung 2-9).
23
Diese Situation wird Scherzerfokus genannt und man findet ihn bei folgender
Fokuseinstellung:
λε sScherzer C2.1−= 2-16
Wird ein Hochauflösungsbild im Scherzerdefokus aufgenommen, kann es direkt
interpretiert werden. Ein weiterer Vorteil des Scherzerdefokus ist, dass die Doppelbeugung
von Elektronen nicht signifikant zum Hochauflösungsbild beiträgt (Zou, 1995).
Abbildung 2-7: Kontrasttransferfunktion T(u) mit der Umhüllenden E(u) und der Phasenverschiebungsfunktion χ(u), für Defokus ε = -100nm, U= 300kV, Cs=1.0mm, ∆=20nm, α=0.0045°.
u [Å-1]
E(u)
T(u)
χ(u)
24
Abbildung 2-9: Kontrasttransferfunktion T(u) für Scherzer-Defokus ( Scherzerε =53.26nm), für U= 300kV, Cs=1.0mm, ∆=20nm, α=0.0045°. Die Kontrasttranseferfunktion verläuft bis 4.2 Å-1 relativ konstant, ohne Nulldurchgang. Das HRTEM- Bild ist in diesem Fall direkt interpretierbar, weil alle Frequenzen gleich übertragen werden.
Abbildung 2-8: Kontrasttransferfunktion T(u) mit der Umhüllenden E(u) und der Phasenverschiebungsfunktion χ(u), für Defokus ε = -30nm, U= 300kV, Cs=1.0mm, ∆=20nm, α=0.0045°.
u [Å-1]
u [Å-1]
E(u)
T(u)
χ(u)
E(u)
T(u)
χ(u)
25
2.5.6 Der Astigmatismus
Wird der Elektronenstrahl im TEM von den elektromagnetischen Linsen nicht in allen
Richtungen gleich behandelt, so wird der Strahl verzerrt, es entsteht Astigmatismus. Das
Hochauflösungsbild wird dann ebenfalls verzerrt abgebildet. Am besten wird der
Astigmatismus am TEM selbst korrigiert. Es ist jedoch möglich den Astigmatismus auch
nachträglich zu korrigieren. Das Fouriertransformierte HRTEM-Bild (reziproker Raum) ist,
wie das HRTEM-Bild selbst, auch beeinträchtigt von der CTF. Weil bei den
Nulldurchgängen der CTF keine Information übertragen wird, erscheinen diese schwarz. Ist
das HRTEM-Bild frei von Astigmatismus ist auch die CTF in alle Richtungen gleich. Ist
hingegen Astigmatismus vorhanden, ist die CTF richtungsabhängig. Das bedeutet, dass im
astigmatismusfreien, fouriertransformierten Bild, die Nulldurchgänge der CTF als
konzentrische schwarze Kreise sichtbar werden. Ist Astigmatismus vorhanden sind die
Nulldurchgänge aber als Ellipsen oder Hyperbeln sichtbar (siehe Abbildung 2-10). Wird die
CTF in diesem Fall in Abhängigkeit der Richtung bestimmt und das HRTEM-Bild mit
dieser CTF korrigiert, dann ist auch der Astigmatismus im Bild korrigiert.
Abbildung 2-10: Fouriertransformation eines Bildes von amorphem Kohlestoff. Leichter Astigmatismus ist erkennbar an den elliptischen schwarzen Ringen (Defokus ε =-57.3nm).
26
2.5.7 Bestimmung des Defokus
Die Nulldurchgänge der CTF sind im fourier-transformierten Bild vom amorphen
Bereichen einer Probe besonders gut sichtbar. Die amorphen Bereiche werden als diffuser
Untergrund abgebildet, während bei den Nulldurchgängen der CTF natürlich auch hier
keine Information übertragen wird. Dies führt wiederum zu schwarzen Kreisen, Ellipsen
oder in speziellen Fällen auch zu Hyperbeln welche im weissen Untergrund sichtbar
werden. Aus den Nullpunkten der CTF kann der Defokus bestimmt werden (Krivanek,
1976). Ein Nulldurchgang der CTF liegt vor, wenn:
( ) ( ) 0)(sin2)( == uuEuAuT χ 2-17
was der Fall ist wenn:
221)( 432 πλππελχ ⋅=+= nuCuu s 2-18
wobei n=0, ±1, ±2, ist für den nullten, ersten und zweiten Nulldurchgang. Für jeden
sichtbaren Ring im Fourier-transformierten Bild kann u bestimmt werden. Sind λ und sC
bekannt, kann aus Gleichung 2-16 der Defokus ε berechnet werden:
222 uC
un
sλλε −= 2-19
Ist sC nicht bekannt, müssen zwei Nulldurchgänge sichtbar sein im Fourier-transformierten
Bild, damit mit Gleichung 2-16 sowohl ε , wie auch sC berechnet werden können.
2.5.8 Anwendung von Symmetrie
Das HRTEM-Bild kann weiter verbessert werden, wenn zusätzliche Information
eingebracht wird. Oft ist die Symmetrie einer Kristallstruktur bereits bekannt, auch wenn
die Struktur selbst noch nicht gelöst wurde. Ist dies nicht der Fall beschreibt Zou (1995) ein
Verfahren, wie Ursprung und Symmetrie bestimmt werden können. Dazu wird verglichen
wie gut die im HRTEM-Bild gefundenen Phasen der Strukturfaktoren die Vorgaben der
verschiedenen planaren Symmetrien erfüllen. Die Symmetrie bei welcher die Phasen am
27
wenigsten Abweichungen zur Vorgabe durch die Symmetrie aufweisen, wird dann als die
wahrscheinlichste Symmetrie betrachtet.
Ist die planare Symmetrie der Projektion bekannt, kann im Fourier-transformierten Bild
über die Amplituden symmetrieäquivalenter Strukturfaktoren gemittelt und durch
Rücktransformation ein HRTEM-Bild mit angewandter Symmetrie berechnet werden.
Fehler wie zum Beispiel kleine Fehlorientierungen des Kristalls können durch Anwendung
der Symmetrie teilweise korrigiert werden. Das rücktransformierte Bild entspricht dann
besser dem projizierten Potential.
28
29
3 Probenpräparation
3.1 Ziele der Präparation
Für ein Experiment im Elektronenmikroskop muss eine geeignete Probe vorbereitet (z.B.
zerkleinert) und auf den Probenträger transferiert werden. Als Probenträger wird ein
Kupfergitterchen mit 3mm Durchmesser verwendet, welches mit einer löchrigen
Kohlestoffolie überzogen wurde.
3.1.1 Probendicke
Wie anfangs erwähnt, ist es für HRTEM wie auch für SAED wichtig, dass die Kristallite
möglichst dünn sind. Nur mit dünnen Kristalliten können kinematische Bedingung in der
Elektronenbeugung überhaupt erreicht werden. Für HRTEM-Bilder sollte die Schwaches-
Phasenobjekt Näherung (Anhang A) erfüllt sein, um aus HRTEM-Bildern Amplituden und
Phasen der Strukturfaktoren berechnen zu können. Die Proben müssen
elektronentransparent sein, was normalerweise bedeutet, dass diese dünner als 100nm sind
und am besten so dünn wie möglich.
3.1.2 Vorzugsorientierung
Neben der Dicke der Probe spielt auch ihre Vorzugsorientierung eine wichtige Rolle. Um
den reziproken Raum aus SAED-Bildern so vollständig wie möglich rekonstruieren zu
können, ist es wichtig, dass in der Probe viele Orientierungen beobachtet werden können.
Aus den HRTEM-Bildern können Amplituden und Phasen der Strukturfaktoren berechnet
werden, so dass auch hier ein kompletter Phasensatz und damit Aufnahmen von möglichst
vielen Orientierungen erwünscht sind.
Oft liegt es in der Morphologie der Probe begründet, dass nur vorzugsorientierte Proben
präpariert werden können. Nadelige Kristalle werden zum Beispiel horizontal auf dem
Probenträger liegen und sich nicht senkrecht darauf stellen. Um die Blickrichtung entlang
der Nadeln einsehen zu können, müssen darum Querschnitte der Nadeln präpariert werden.
Für die Querschnittpräparation ist dann natürlich eine gute Vorzugsorientierung erwünscht,
damit im präparierten Querschnitt die gesuchte Orientierung (z.B. die Blickrichtung entlang
eines nadelförmigen Kristalls) oft angetroffen wird.
30
3.1.3 Verteilung des Probenmaterials
Ideal für SAED und HRTEM ist es, wenn die Kristalle einzeln auf dem Probenträger
liegen. Zeolithkristalle, die gern agglomerieren, werden daher bei der Präparation
dispergiert, um eine möglichst gute Separation der Kristalle zu erhalten. Beim Kippen der
Probe kann damit ein Überlappen der Nachbarkristalle vermieden werden. Um dünne, gut
dispergierte Kristalle mit möglichst vielen Orientierungen zu erhalten, können verschiedene
Methoden angewendet werden. Die benutzten Methoden werden im Folgenden vorgestellt.
3.2 Präparationsmethoden
3.2.1 Transferieren aus einer Suspension
Die Zeolithe liegen normalerweise als Pulverproben vor. Die Kristallite sind zwar klein,
aber meistens nicht klein genug, respektive nicht dünn genug, damit sie für SAED und
HRTEM direkt verwendet werden können. Um kleine, dünne Bruchstücke zu produzieren,
wurden die Proben deshalb zuerst in einem Achatmörser zermahlen. Für hartnäckige
Proben, deren Kristalle trotz ausgiebiger Mahlprozedur im Achatmörser nicht in
Bruchstücke zerfielen, wurden zwei spezielle Methoden verwendet.
Einerseits wurde mit dem Stössel auf die Proben eingehämmert (vgl. Abbildung 3-1-b)
anstatt gemahlen. Durch das Hämmern werden mehr und kleinere Splitter gebildet, als beim
Mahlen. Andererseits wurde ein besonders kleiner Mörser (Abbildung 3-1-c) mit sehr
glatter Oberfläche verwendet. Besonders Kristalle mit Durchmessern kleiner als 10µm,
liessen sich meistens nur in diesem kleinen Mörser in Bruchstücke weiter zersplittern.
Die Kristalle wurden trocken gemahlen oder "gehämmert" anschliessend mit Ethanol im
Mörser aufgeschlämmt. Diese Suspension wurde mit einer Pipette in ein Reagenzglas
transferiert und während 20-40 Sekunden im Ultraschallbad behandelt, um die Kristalle
möglichst gut zu dispergieren. Längere Ultraschallbehandlungen bewährten sich nicht, weil
die Kristalle nicht besser dispergierten und die Kristallstruktur der Proben bei langer
Ultraschallbehandlung (mehrere Minuten) zum Teil zerstört wurde.
31
Nach der Ultraschallbehandlung wurde das Reagenzglas mit der Suspension in einen Halter
gestellt, um die Suspension während 2-3 Minuten sedimentieren zu lassen. Anschliessend
wurde mit einer Pasteurpipette aus der oberen Hälfte der Suspension ein bis vier Tropfen
der Suspension auf ein TEM-Probenträger (Kohlestoffnetz mit Löchern) transferiert.
Danach wurde die Probe an der Luft getrocknet.
Bei dieser Präparation agglomerierten die Zeolithkristalle. Auch die Zugabe von
Detergenzien konnte die Bildung der Agglomerate nicht verhindern, sodass oft nur nach
langem Suchen und nur vereinzelt alleinstehende Kristallite in diesen Präparaten gefunden
werden konnten. Wenn trotz intensiven Suchens kein einzelner Kristallit gefunden werden
konnte, wurde versucht aus dem Agglomerat herausragende Kristallteile für die
Experimente zu benutzen. Das Einkippen dieser Kristalle ist aber schwierig, weil sie von
störenden Kristallen umgeben sind und die herausragenden Teile normalerweise klein
waren. Es ist deshalb grosse Erfahrung nötig, um trotzdem gute Resultate erzielen zu
können.
Die beschriebene Präparationsmethode ist einfach schnell und auch kostengünstig.
Nachteile treten vor allem bezüglich Vorzugsorientierung von nadel- und
Abbildung 3-1 Zerkleinern der Proben a) mahlen in einem Achatmörser b) hämmern in einem Achatmörser, führt zu kleineren Splittern c) mahlen im kleinen Achatmörser, ermöglicht auch von Körnern mit kleinem Durchmesser Bruchstücke zu erhalten.
32
plättchenförmigen Kristallen auf da diese wie erwähnt auf dem Kohlestoffnetz liegen und
nicht auf ihrer Spitze respektive ihrer Kante stehen.
Jedoch, auch ein unvollständiger Datensatz hilft weiter, insbesondere wenn die
Strukturfaktorphaseninformation aus dem Hochauflösungsbild genutzt werden soll. Jede
zusätzliche Phaseninformation verbessert die Erfolgschancen bei der Strukturlösung
(Kapitel 5.3). Damit die Intensitäten überlappender Reflexe im Pulverdiagramm neu
aufgeteilt werden können, ist aber ein vollständiger Datensatz von Vorteil.
Um einen möglichst vollständigen Datensatz zu erhalten, wurden deshalb von den
Zeolithkristallen zusätzlich mit zwei verschiedenen Methoden Querschnittpräparate
hergestellt.
3.2.2 Querschnittpräparationen mit Ionenätzung
Die Querschnittpräparation wurde gemacht, wie sie von Müller und Krumeich (2000)
beschrieben wurde. Das Probenmaterial wurde mit wenig G1 Epoxy Harz (Gatan)
vermischt und zwischen zwei Si-Wafer eingebettet.
Die Si-Wafer wurden gegeneinander verschoben, so dass sich die nadeligen Kristallite
parallel zu Oberfläche der Si-Wafer und parallel zur Verschiebungsrichtung der Si-Wafer
ausrichten (Abbildung 3-2B). Eine andere Möglichkeit ist, die Si-Wafer einfach
zusammenzupressen (Abbildung 3-2C), wobei sich die Kristalle radial ausrichten. Dieses
"Sandwich" (Si- Wafer - Epoxyharz mit Probe – Si-Wafer) wurde mit dem G1 Epoxy Harz
Abbildung 3-2: Präparation von Vorzugsorientierten Proben mit Sandwich-Technik. Durch verschieben (mitte) oder gegeneinander drücken zweier Platen soll sich die Probe ausrichten.
33
in ein Kupferröhrchen eingebettet (Abbildung 3-3). Dieses Röhrchen soll der Probe im
anschliessenden Schleifprozess mehr Stabilität verleihen.
Aus dem entstandenen Kupferrohrzylinder wurden mit einer Diamantdrahtsäge Scheibchen
von 250µm Dicke abgeschnitten. Diese Scheibchen wurden auf einer Schleifscheibe bis auf
100 µm hinunter geschliffen. In der Mitte dieser 100µm-dicken Scheibe wurde
anschliessend mit einem Schleifrächen ein kleines Grübchen geschliffen, so dass die Probe
an der dünnsten Stelle gerade noch 20 µm dick war. Diese Stelle wurde beidseitig mit Ar-
Ionen gedünnt bis ein kleines Loch in der Probenmitte entstand. Für diese Ionenätzung
wurde ein PIPS (precision ion milling system) von Gatan verwendet. Der Winkel zwischen
Probenoberfläche und Ar-Ionen Strahl wurde auf 2° eingestellt. Durch den kleinen Winkel
verringert sich die Eindringtiefe der Argonionen. Das ist für die strahlempfindlichen
Zeolithe wichtig, damit diese bei dieser Prozedur möglichst nicht zerstört werden. Weil
durch das flache einfallen des Ar-Strahls weniger Material abgetragen wird, verlängert sich
aber die Ätzzeit. So wurden Ätzzeiten zwischen 24 und 72 Stunden benötigt bis zum
Durchbruch des Argon-Strahls.
Eine solche Querschnittpräparation mit Ionenätzung wurde mit dem Zeolithen IM-5,
durchgeführt. In aus Suspension transferierten Proben von IM-5 konnten nur Aufnahmen
entlang der Zonenachse [1 0 0] und von einigen schiefen Richtungen gemacht werden. Ziel
war es den Elektronenbeugungsdatensatz mit Informationen aus mindestens einer der
Zonen [ 0 1 0 ] und [ 0 0 1 ], zu ergänzen.
Abbildung 3-3: Probenvorbereitung für die Ionenätzung. Das Sandwich (vgl. Abbildung 3-2) wird in ein Kupferröhrchen eingebettet. Ein Scheibe von dem Kupferrohr mit Sandwich wird abgesägt. In diese Scheibe wird eine Grübchen geschliffen. Mit dem Ar-Ionenstrahl wird im Grübchen geätzt bis zum Durchbruch.
34
Bei der Präparation von IM-5 tauchten verschiedene Probleme auf. Die IM-5 Kristalle
agglomerieren stark. Wird viel Probenmaterial eingebettet liegt dieses nur in Form von
Agglomeraten vor. Damit einzeln liegende Kristalle überhaupt vorlagen, musste die
Probenmenge reduziert werden, was wiederum dazu führte, dass nur wenige Kristalle im
elektronentransparenten Bereich der Probe zu finden waren.
Abbildung 3-4 IM-5 -Kristalle querschnittpräpariert mit Ionenätzung (A) Einzelner IM-5 Kristall mit (B) zugehörigem Elektronenbeugungsbild. Die relativ schwachen Reflexe und auf dem starken, diffusen Untergrund lassen vermuten, dass der Kristall bereits durch den Argonstrahl teilweise amorphisiert wurde. (C) Agglomerat von IM-5 Kristallen. Die Dicke der Probe nimmt im Bild von rechts nach links schnell zu, erkennbar an der kontinuierlichen, deutlichen Abnahme der Helligkeit. (D) Die Dicke der Probe nimmt von unten nach oben massiv zu. Die Kristalle liegen zwar einzeln, aber im dicken Bereich der Probe.
35
Es war schwierig in der eingebetteten querschnittpräparierten Probe einzeln liegende
Kristalle zu finden, welche gut eingekippt werden konnten. In Abbildung 3-4C ist am
Helligkeitsverlauf vom hellen rechten Bildrand zum dunklen linken Bildrand erkennbar,
dass die Probendicke massiv zunimmt. Es bleibt nur ein Bereich von ca. 1µm Breite, der für
Elektronenbeugungs- und Hochauflösungsaufnahmen, aufgrund dessen Dicke überhaupt in
Frage kommt. Einzeln liegende Kristalle wie in Abbildung 3-4D, wurden nur bei geringer
Probenmenge gefunden. Im nur 0.5-1µm breiten Bereich, welcher dünn genug ist konnten
jeweils nur einzelne, im Normalfall ein bis zwei, Kristalle gefunden werden die
Das Hauptproblem in der Präparation liegt aber in der Empfindlichkeit der Zeolithe
bezüglich des Ionenstrahls. Trotz des sehr flach einfallenden Ar-Ionenstrahls beim
Ionenätzen wurden die Kristallstruktur der meisten Kristalle zerstört.
Nur bei wenigen Kristallen waren auf den Elektronenbeugungsbildern überhaupt schwache
Reflexe auf diffusem Untergrund erkennbar wie sie im in Abbildung 3-4B gezeigt werden.
Der diffuse Untergrund im Elektronenbeugungsbild zeigt, dass ein Teil der Probe bereits
amorphisiert wurde. Ob die gewünschte Vorzugsorientierung der Probe vorlag kann nicht
gesagt werden, weil aufgrund der geringen Ausbeute zu wenige Kristalle untersucht werden
konnten. Jedenfalls konnte in den einzelnen nicht zerstörten Kristallen, keine
Beugungsbilder entlang der Zoneachse [0 1 0] gefunden wurden. Auch andere
Blickrichtungen, welche bisher nicht einsehbar waren konnten keine gefunden werden.
36
3.2.3 Querschnittpräparation mit Ultramikrotomie
Als Alternative zur oben beschriebenen Ionenätzung, wurde für die Querschnittpräparation
auch Ultramikrotomie probiert. Für die Ultramikrotomie werden die Proben in ein
Epoxyharz eingebettet und mit einem Glas- oder einem Diamantmesser sehr dünn
geschnitten. Der Vorteil von Ultramikrotomie ist, dass die Proben im Vergleich zur
Ionenätzung keiner Strahlenbelastung ausgesetzt sind, welche die Kristallstruktur zerstören
könnte.
Abbildung 3-5: Mit Ultramikrotomie präparierte Proben von Zeolith IM-5. links: Hellfeldbild der in Polystyrol eingebetteten Kristalle. Rechts: SAED-Aufnahme eine Kristalliten. Schwache Reflexe sind kaum erkennbar, aufgrund des hohen Anteils diffuser Intesität.
Die Proben wurden mit starker Vorzugsorientierung in Polystyrol eingebettet. Um beim
Einbetten gute Vorzugsorientierung zu erhalten, wurde ein Gemisch von Kristallen und
Polystyrol gelöst in Tetrahydrofuran auf einen Glasobjektträger aufgetropft und mit einem
zweiten Glasobjektträger zugedeckt. Dieses Sandwich wurde dann langsam
zusammengepresst und mit einer Schraubzwinge während 12 Stunden fixiert. Durch das
Zusammendrücken des Polymers fliesst das Polystyrol vom aufgetragenen Tropfen aus in
radialer Richtung davon und bildet eine dünne Schicht zwischen den Glassplatten. Durch
das Fliessen des Polymers richten sich die nadeligen Kristallite entlang der Fliessrichtung
aus. Nach dem Trocknen der Polymerschicht wurde diese in flüssigem Stickstoff vom
Glasträger getrennt. Aus der Polymerschicht wurde ein Stück herausgeschnitten und mit
geeigneter Orientierung in einen zylindrischen Polymerblock eingebettet. Dieser Block
37
wurde angespitzt und von der Spitze wurden mit einem Diamantmesser dünne
Probestückchen abgeschnitten, welche eine Dicke von etwa 60-80nm aufweisen sollten.
Auf den TEM-Bildern ist am Kontrast zu erkennen, dass die Proben relativ dick waren.
Eine Vorzugsorientierung war nicht erkennbar. Die eingebetteten Kristalle bildeten
Agglomerate. Die IM-5 Zeolithkristalle sind relativ klein (250-500nm lang). Aufgrund der
Agglomeratbildung und der Grösse der Kristalle war es sehr schwierig und zum Teil auch
unmöglich die eingebetteten Kristalle für ein Beugungsexperiment zu orientieren. Wegen
der Polymermatrix und der Dicke der Probe enthielten die SAED-Bilder grosse Anteile an
diffuser Intensität, so dass die schwachen Reflexe kaum mehr sichtbar waren. Das
erschwerte das Einkippen der Kristalle zusätzlich. SAED-Bilder entlang einer vorher nicht
einsehbaren Zonenachse konnten in diesen Präparaten, keine gefunden werden.
Eine weitere Möglichkeit orientierte Querschnitte zu präparieren wäre die Kristalle
einzubetten und mit "Focused Ion Beam" (FIB) eine Lamelle herauszuschneiden
(Liu, 2005). Diese Methode stand hier allerdings nicht zur Verfügung.
38
39
4 Testfall ZSM-5
4.1 Elektronenbeugung an ZSM-5
4.1.1 Können Elektronenbeugungsintensitäten das Peaküberlappungsproblem im
Pulverdiagram lösen?
Wie bereits erwähnt, ist das Hauptproblem bei der Strukturbestimmung aus
Pulverdiffraktionsdaten die Überlappung der Reflexe im Pulverdiagramm. Deshalb wurde
untersucht, ob Elektronenbeugungsexperimente an einzelnen Kristalliten des Pulvers die
relativen Intensitäten solcher überlappenden Reflexe liefern können.
Bei zu hoher Elektronendosis wird die Kristallstruktur zerstört und sie wird amorph. Die
stabilsten Zeolithe sind diejenigen mit hohen Si/Al Verhältnissen. Deshalb werden für die
folgenden Untersuchungen der Zeolith ZSM-5 mit praktisch SiO2 Zusammensetzung
verwendet.
Die Einheitszelle von ZSM-5 ist orthorhombisch mit den Zellkonstanten a=20.060Å,
b=19.936Å und c=13.407Å und kann in der Raumgruppe Pnma beschrieben werden.
Ausgewählt wurde ZSM-5 für diesen Vergleich, weil die Struktur mit 12 T-Atomen in der
asymmetrischen Einheit relativ kompliziert ist. Die Elementarzelle ist relativ gross und
zudem sind die Gitterparameter a und b der Einheitszelle fast identisch. Beides führt zu
überlappenden Reflexen im Pulverdiagramm. Mit dem Überlappungsfaktor ÜLK wird
definiert wie stark die Reflexe überlappen müssen, damit sie als überlappend bezeichnet
werden.
Als überlappend werden zwei Reflexe mit den Indizes hkl respektive ''' lkh betrachtet
wenn:
( ) '''''' 2221
lkhhkllkhhklÜL fwhmfwhmK θθ −>+⋅⋅ 4-1
wobei für den Reflex mit den Indizes hkl , hklθ dessen Beugungswinkel ist und hklfwhm
dessen Halbwertsbreite. Dementsprechend sind ''' lkhθ und ''' lkhfwhm Beugungswinkel und
Halbwertsbreite des Reflexes ''' lkh .
40
Die Intensitäten von Röntgen- und Elektronenbeugungsexperimenten sind nicht direkt
vergleichbar weil die atomaren Streufaktoren für Röntgen- und Elektronenstrahlung
unterschiedlich sind. Ein Vergleich der Silizium- und der Sauerstoffstreufaktoren
(Abbildung 4-1) zeigt, dass der Streufaktor von Sauerstoff mit zunehmendem sin(θ)/λ
langsamer abnimmt als der von Silizium. Das Verhältnis von Röntgen- zu
Elektronenstreufaktoren .f/f ElRö wird grösser mit zunehmendem sin(θ)/λ (vgl. Abbildung
4-2). Dabei liegt kein einfacher Zusammenhang vor zwischen dem Verhältnis .f/f ElRö und
sin(θ)/λ. Um trotzdem verifizieren zu können, ob und wie gut Intensitäten aus
Röntgenbeugungsexperimenten mit denjenigen aus der Elektronenbeugung vergleichbar
sind, wurden von der bekannten ZSM-5 Zeolithstruktur Strukturfaktoramplituden, sowohl
mit Elektronenstreufaktoren, wie auch mit Röntgenstreufaktoren berechnet und diese
verglichen.
Abbildung 4-1: Vergleich der Streufaktoren von Silizium und Sauerstoff für Röntgen- und Elektronenstrahlung
41
Werden für die ZSM-5 Zeolithstruktur Reflexe bis zu einer Auflösung von 1.3Å betrachtet,
überlappen im Pulverdiagramm, bei hoch aufgelösten Synchrotrondaten ( )θ205.0~ ⋅fwhm
728 von 1343 vorhandenen Reflexen. Diese überlappenden Reflexe ( )3.0=ÜLK bilden 361
Reflexgruppen. Mit dem Programm XRS-82 wurden für ZSM-5 als erstes die
Reflexintensitäten sowohl für Röntgenbeugung, wie auch für Elektronenbeugung
berechnet, und die entsprechenden Pulverdiagramme simuliert (Abbildung 4-3). Bereits aus
den Pulverdiagrammen ist zu erkennen, dass die Differenzen der Reflexintensitäten eher
klein sind. Nach dieser eher qualitativen Betrachtung sollen nun die
Strukturfaktoramplituden einer genaueren quantitativen Betrachtung unterzogen werden.
Simulierte Strukturfaktoramplituden wurden wie bereits die Reflexintensitäten zuvor mit
dem Programm XRS berechnet.
Abbildung 4-2: Verhältnis Elektronen- und Röntgenstreufaktoren in Abhängigkeit von sin(θ)/λ für Sauerstoff und Siliziumatome.
42
Es wurden die Strukturfaktoramplituden für Elektronenbeugung EMF , sowie die
Strukturfaktoren für Röntgenbeugung RÖF berechnet und die experimentellen, aus dem
Pulverdiagramm extrahierten Strukturfaktoren EXTF bestimmt. Die Reflexe wurden in
Gruppen aufgeteilt, wobei überlappende Reflexe ( )3.0=ÜLK jeweils eine Gruppe bilden.
Die Aufteilung der im Pulverdiagramm gemessen Gesamtintensität einer Gruppe
überlappender Reflexe ist dann möglich, wenn die Verhältnisse der Intensitäten, respektive
der Strukturfaktoramplituden für Röntgenbeugung und für Elektronenbeugung gleich oder
sehr ähnlich sind. Der Anteil )(hklV , eines Reflexes an der Summe der
Strukturfaktoramplitude einer Reflexgruppe, wurde aus der extrahierten Gesamtintensität
und dem simulierten Strukturfaktor )(hklFsim sowohl für Röntgenbeugung wie auch
Elektronenbeugung berechnet:
∑=
Gruppesim
sim
hklFhklF
hklV)(
)()( 4-2
Abbildung 4-3: Simuliertes Pulverdiagramm (FWHM=0.05) für Röntgen- und Elektronenstrahlung von Zeolith ZSM-5 für λ=1.10126Å und in einem Streuwinkelbereich 2θ von 5 bis 25°.
43
Abbildung 4-4: Ausschnitt mit überlappenden Reflexen aus einem Pulverdiagramm von Zeolith ZSM-5 aufgenommen mit Wellenlänge λ=1.10176Å. Die Strukturfaktoramplituden für Röntgen- und Elektronenstrahlung sind in grau und schwarz eingetragen.
Abbildung 4-5: Tabelle der simulierten Strukturfaktoramplituden (
EMSF ,RÖSF ) aus Abbildung 4-4, der
Reflexe hkl, mit Beugungswinkel 2θ. Die 15 Reflexe bilden 6 Reflexgruppen (ÜLK =0.3).
44
Um einen guten Schätzwert für die Strukturfaktoramplitude im Röntgenbeugungs-
experiment zu erhalten, wurde die aus dem Pulverdiffraktogramm extrahierte
Gesamtamplitude jeder Gruppe ∑Gruppe
EXTF bestimmt. Diese Gesamtamplitude wurde auf die
einzelnen Reflexe der Gruppe aufgeteilt, gemäss den Verhältnissen )(hklV der simulierten
Strukturfaktoramplituden EMF für Elektronenbeugung, respektive der RÖF für
Röntgenbeugung.
∑=
GruppeEXT
S hklFhklVhklF
)()()( 4-3
Diese auf die extrahierte Gesamtintensität der Gruppe normierten F-Werte werden mit
einem zusätzlichen S (für Schätzwert) als SF , respektive als EMSF für Elektronenbeugung
und als RÖSF für Röntgenbeugung bezeichnet.
Als Beispiel ist in Abbildung 4-4 ein Ausschnitt des simulierten Pulverdiagramm von
Zeolith ZSM-5 gezeigt. Die Sturkturfaktoramplituden der Reflexe sind sowohl für Röntgen-
wie auch für Elektronenbeugung angegeben. Auffallend ist in diesem Ausschnitt, dass die
Strukturfaktoramplituden für Röntgen- und Elektronenbeugung fast identisch sind. In
Abbildung 4-5 sind diese Strukturfaktoramplituden in tabellarischer Form angegeben.
Für den Zeolithen ZSM-5 zeigt Abbildung 4-6, dass die Differenzen von )(hklVEMS und
)(hklVRÖS klein sind für überlappende Reflexe. Das bedeutet, dass die Verhältnisse der
Strukturfaktoren innerhalb einer Gruppe überlappender Reflexe für Röntgenbeugung und
Elektronenbeugung sehr ähnlich sind. SAED-Intensitäten können deshalb verwendet
werden für die Aufteilung der Röntgenintensitäten überlappender Reflexe, sofern das
SAED-Bild unter kinematischen Bedingungen aufgenommen werden kann.
45
Abbildung 4-6: Differenz )()( hklVhklV RÖSEMS − der überlappenden ( ÜLK =0.3) Reflexe des Zeolithen ZSM-5.
4.1.2 Experimentelle Elektronenbeugungsdaten für die Intensitätsaufteilung
Um kinematische SAED-Bilder aufnehmen zu können, sollten diese an möglichst dünnen
Stellen der Probe aufgenommen werden. Die Dicke des Kristalls kann im TEM aber nicht
einfach bestimmt werden.
Die ZSM-5 Kristalle sind plättchenförmig und deshalb dünn in einer Richtung. Dass der
Kristall in Abbildung 4-7 relativ dünn ist, ist daran zu erkennen, dass der Probenträger
durch den Kristall hindurch schimmert. In der unteren Kristallhälfte ist am weissen Saum
zu erkennen, dass der Kristall nicht flach auf dem Probenträger liegt und deshalb in diesem
Bereich nicht im Fokus liegt. Für das Beugungsbild wurde deshalb mit der SAED-Blende
ein kleiner Bereich aus dem oberen Kristallteil ausgewählt.
46
AbbildunProbenprä
AbbildunKamera eB: Besser
g 4-7: Dünnäparation entst
g 4-8: Elektroneines Phillips Cres SAED-Bild
ner ZSM-5 tanden. Der Kr
nenbeugungsbCM30, bei 300d mit unterschie
Kristall mit reis zeigt die G
ilder der Zone0kV. A: das schedlichen Intens
Bruchstücken
Grösse der SAE
e [ 1 0 1 ] von Zhlechtere SAEsitäten,
n welche beED-Blende.
Zeolith ZSM-5 ED-Bild mit un
eim Mörsern
aufgenommenniformer Intens
während de
n mit der CCDsitätsverteilung
er
-g.
47
Eine kleine SAED-Blende mit 80nm Durchmesser wurde eingesetzt um die SAED-Bilder
zu erzeugen. Dadurch sollten weniger dynamische Anteile das SAED-Bild beeinflussen.
Zum Beurteilen, ob das SAED-Bild von Mehrfachbeugung betroffen ist kann die Probe im
TEM leicht hin und her gekippt werden. Dadurch werden die Atomsäulen leicht verkippt,
was bereits genügt um Mehrfachbeugung zu vermeiden. War Mehrfachbeugung vorhanden,
werden sich durch das leichte verkippen die Intensitäten verändern. Erfahrungsgemäss sind
SAED-Bilder mit uniformen Reflexintensitäten wesentlich stärker von dynamischen
Effekten und Mehrfachbeugung beeinflusst als die SAED-Bilder mit sehr unterschiedlichen
Reflexintensitäten (Abbildung 4-8).
In Kapitel 4.1.1 wurde gezeigt, SAED-Intensitäten im kinematischen Fall verwendet
werden können um die Gesamtintensität überlappender Reflexe korrekt auf die einzelnen
Reflexe aufzuteilen. Nun muss überprüft werden, ob die Anteile an Mehrfachbeugung und
die dynamischen Anteile der experimentellen SAED-Intensitäten klein genug sind, um
diese als kinematisch betrachten zu dürfen. Dazu wurden in mehreren SAED-Bildern mit
der Software CRISP die Reflexintensitäten extrahiert und die Strukturfaktoren berechnet.
Diese experimentellen Strukturfaktoren wurden mit den simulierten
Röntgenbeugungsstrukturfaktoren von ZSM-5 verglichen. Abbildung 4-8 zeigt die zwei
SAED-Bilder der Zone [ 1 0 1 ] welche für den Vergleich verwendet wurden. Während im
linken Bild die Gesamtintensität uniform über die Reflexe verteilt ist, unterscheiden sich
die Reflexintensitäten im rechten SAED-Bild deutlich. Der Einfacheit halber wird vorerst
einmal angenommen, dass die oben erwähnte Erfahrung stimmt und Abbildung 4-8A
schlechtere Intensitäten für die Strukturbestimmung liefert, als Abbildung 4-8B. Deshalb
wird im folgenden Abbildung 4-8A als 'schlechteres' SAED-Bild benannt und Abbildung
4-8-B als 'besseres' SAED-Bild.
Um die Differenzen zwischen den Strukturfaktoren zu quantifizieren wurden R-Werte
berechnet:
∑∑ −
=− )()()(
hklFhklFhklF
RRÖS
RÖSEMSRÖSEMS 4-4
48
Abbildung 4-9: Normierte Differenz simulierter und experimenteller bestimmter Strukturfaktoramplituden für Zeolith ZSM-5, bestimmt aus Abbildung 4-8-A. Dargestellt sind die 63 Reflexe, welche im Pulverdiagramm überlappen und deren Intensität aus dem SAED-Bild bestimmt werden konnte.
Abbildung 4-10: Simulierte Strukturfaktoramplituden für Röntgenstrahlung (schwarz) und für Elektronenstrahlung (dunkelgrau), sowie experimentell aus Abbildung 4-8-A ('schlechteres' SAED-Bild) bestimmte Strukturfaktoramplituden (hellgrau), welche mit dem Faktor 0.22 skaliert wurden. Zwecks besserer Übersicht wird nur der 2θ-Bereich zwischen 10° und 20° gezeigt.
49
Abbildung 4-11: Normierte Differenz simulierter und experimenteller bestimmter Strukturfaktoramplituden für Zeolith ZSM-5, bestimmt aus Abbildung 4-8-B. Dargestellt sind die 71 Reflexe, welche im Pulverdiagramm überlappen und deren Intensität aus dem SAED-Bild bestimmt werden konnte.
Abbildung 4-12: Simulierte Sturkturfaktoramplituden für Röntgenstrahlung (schwarz) und für Elektronenstrahlung (dunkelgrau), sowie experimentell aus Abbildung 4-8-B ('besseres' SAED-Bild) bestimmte Strukturfaktoramplituden (hellgrau), welche mit dem Faktor 0.20 skaliert wurden. Zwecks besserer Übersicht wird nur der 2θ-Bereich zwischen 10° und 20° gezeigt.
50
∑∑ −
=− )()()(
hklFhklFhklF
RRÖS
RÖSEMRÖSEM 4-5
Wobei RÖSEMSR − der R-Wert für die berechneten Strukturfaktoren und RÖSEMR − für diejenigen
aus dem SAED-Experiment bestimmten Strukturfaktoren ist.
In Abbildung 4-9 und Abbildung 4-11 sind die normierten Differenzen von Elektronen- und
Röntgenstrukturfaktoren aufgezeichnet. Verglichen werden die normierten Differenzen von
experimentell aus SAED-Bildern ermittelten EMF mit den entsprechenden Differenzen der
simulierten EMSF . Normiert wird bezüglich der simulierten Strukturfaktoramplitude für
Röntgenbeugung. Die Differenzen sind sehr gross. Dies ist auch in Abbildung 4-10 und
Abbildung 4-12 ersichtlich, wo ein Teil der simulierten und experimentell bestimmten
Strukturfaktoramplituden aufgezeichnet sind.
Die Qualität der experimentell bestimmten Elektronenstrukturfaktoren kann auch anhand
der berechneten R-Werte, RÖSEMSR − für die simulierten EMSF und RÖSEMR − für die
experimentell bestimmten EMF , beurteilt werden. Für die EMF aus dem 'schlechteren'
SAED-Bild ist 052.2=−RÖSEMR und für diejenigen aus dem 'besseren' SAED-Bild ist
914.0=−RÖSEMR . Für die entsprechenden simulierten Strukturfaktoramplituden ( RÖSF ) ist
der R-Wert ( RÖSEMSR − ) 0.057 für die Reflexe im 'schlechteren', respektive 0.084 für die
Reflexe aus dem 'besseren' SAED-Bild, wobei sich besser und schlechter auf Qualität des
zugehörigen SAED-Bildes bezieht. Der Strahlfänger deckt unterschiedliche Reflexe in den
SAED-Bildern ab. Deshalb sind nicht in beiden Bildern dieselben Reflexe vorhanden. Das
erklärt, weshalb die R-Werte der simulierten RÖSF unterschiedlich ausfallen. Diese R-Werte
bestätigen, die anfangs genannte Erfahrung, dass SAED-Bilder mit uniformer
Intensitätsverteilung schlechtere Intensitäten liefern für die Strukturbestimmung, als SAED-
bilder mit unterschiedlichen Intensitäten. Die grosse Differenz zwischen RÖSEMSR − und
RÖSEMR − unterstreicht, was bereits visuell in Abbildung 4-9 und Abbildung 4-11 deutlich
sichtbar ist. Die experimentell bestimmten EMF , weichen oft um ein Vielfaches von den
simulier
erheblic
Struktur
Zusätzli
entlang
und Abb
Fall, wi
(kinema
mit −EMR
abweich
Abbildunmit der C
rten EMSF u
ch. Für d
rfaktorampli
ich wurden d
der Zonenac
bildung 4-15
iederum star
atisch) aussie
950.0=− RÖS n
hen.
g 4-13: ElektrCD-Kamera an
und RÖSF ab
die schwa
tuden bestim
die Struktur
chse [010] a
5 zeigen, da
rk von den s
eht. Das bes
nur geringfü
ronenbeugungsn einem Phillip
b. Auch im
chen Refl
mmt, 47% de
rfaktoren aus
aufgenomme
ass die ermit
simulierten
stätigen auc
ügig von jen
sbild von Zeolps CM30, bei 3
'besseren' S
lexe wurde
er Reflexe w
s einem SAE
en wurden, g
ttelten Struk
Werten abw
ch die R-We
nen aus dem
lith ZSM-5 au300kV.
SAED-Bild
en bis z
wichen mehr
ED-Bild (Ab
genauer ange
kturfaktoram
weichen obw
erte, welche
besseren SA
ufgenommen en
sind die Ab
zu 30-mal
als 50% ab.
bbildung 4-
eschaut. Abb
mplituden auc
wohl das SA
mit −RÖSEMSR
AED-Bild de
ntlang der Zon
51
bweichungen
zu hohe
13), welche
bildung 4-14
ch in diesem
AED-Bild gu
059.0= und
er Zone [ ]1 0 1
nenachse [010
1
n
e
s
4
m
ut
d
,
0]
52
Abbildung 4-15: Simulierte Sturkturfaktoramplituden für Röntgenstrahlung (schwarz) und für Elektronenstrahlung (dunkelgrau), sowie experimentell aus Abbildung 4-13 bestimmte
Abbildung 4-14: Normierte Differenz simulierter und experimentell bestimmter Strukturfaktoren (aus Abbildung 4-13) für Zeolith ZSM-5. Sichtbar sind die 62 Reflexe, welche im Pulverdiagramm überlappen und deren Intensität aus dem SAED-Bild bestimmt werden konnte.
51
53
Strukturfakoramplituden (hellgrau), welche mit dem Faktor 0.09 skaliert wurden. Zwecks besserer Übersicht wird nur der 2θ-Bereich zwischen 10° und 20° gezeigt.
Die grossen Differenzen zwischen simulierten und experimentellen Strukturfaktoren sind
vermutlich auf Mehrfachbeugung und dynamische Effekte in den SAED-Bildern
zurückzuführen, was sich offensichtlich auch bei guten Aufnahmebedingungen (dünne
Probe, kleine Blende, etc.) nicht vermeiden lässt. Solch grosse Abweichungen
verunmöglichen eine gute Aufteilung der Intensität im Pulverdiagramm auf die
überlappenden Reflexe.
Aus einem SAED-Bild kann nur ein Bruchteil aller Reflex bestimmt werden. Weil einige
Richtungen nicht einsehbar sind aufgrund der Vorzugsorientierung der Probe kann kein
vollständiger Datensatz aufgenommen werden. Deshalb fehlt in einer Gruppe
überlappender Reflexe oft die Information zu einzelnen Reflexen. Eine gute Aufteilung der
Gesamtintensität auf die Reflexe anhand der gemessenen SAED-Intensitäten ist in solchen
Fällen nicht möglich. Anhand der beobachteten Intensitätsverhältnisse könnte aber
abgeschätzt werden welche Reflexe stark, mittel oder schwach sind. Die Intensitäten der
nicht überlappenden Reflexe im Pulverdiagramm könnten mit den im SAED-Bild
gemessenen Intensitäten verglichen werden, um die Qualität des SAED-Bildes zu
beurteilen.
54
55
5 Die Kombination von HRTEM-Phaseninformation mit Pulverdaten
5.1 Das Konzept von FOCUS
FOCUS (Grosse-Kunstleve R.W., 1997 & 1999) ist ein Programm, welches für die
Strukturlösung von komplexen Zeolithstrukturen mittels Pulverdaten entwickelt wurde.
FOCUS startet mit aus dem Pulverdiffraktionsexperiment extrahierten Strukturfaktoren. In
Abbildung 5-1 ist der Ablauf von FOCUS schematisch dargestellt. Den extrahierten
Strukturfaktoren weist das Programm zufällige Phasen zu, wobei diese konsistent mit der
Symmetrie sind. Aus diesem Datensatz wird eine Elektronendichtekarte berechnet. Zur
Interpretation dieser (zufälligen) Elektronendichtekarte werden strukturelle und chemische
Information verwendet. Für Gerüstsilikate wird zum Beispiel nach einem dreidimensional
verknüpften Netzwerk gesucht, indem die Siliziumatome vierfach mit ihren benachbarten
Siliziumatomen verknüpft sind. Die zwischen den Siliziumatomen liegenden
Sauerstoffatome werden nicht gesucht. Findet FOCUS Teile eines sinnvollen
(Bindungslängen, Verknüpfung) Netzwerkes wird dieses verwendet um einen neuen
Phasensatz zu den Strukturfaktoramplituden zu berechnen. Damit wird wiederum die
Elektronendichtekarte berechnet und erneut nach einem sinnvollen Netzwerk gesucht.
Dieses "Fourierrecycling" wird mehrmals (typischerweise 5 bis 15-mal) wiederholt. Findet
FOCUS ein sinnvolles, vollständiges Netzwerk wird dieses klassifiziert und in die
Ausgabedatei geschrieben. Egal ob während dieses gesamten Zyklus ein komplettes
Netzwerk gefunden wurde oder nicht, wird ein neuer Zyklus mit einem neuen zufälligen
Phasensatz gestartet. Das am häufigsten gefundene Netzwerk ist meistens die richtige
Lösung. FOCUS arbeitet sowohl im direkten (Modellsuche) wie auch im reziproken
(Phasenzuweisung) Raum. Zusätzliche Informationen können deshalb in beiden Räumen
sehr einfach integriert werden.
5.2 Vorgeben von Phaseninformation in FOCUS
Aus den HRTEM-Bildern können Phasen der Strukturfaktoren berechnet werden. Diese
Information kann in FOCUS als Startphasen anstatt der gewürfelten Phasen vorgegeben
werden. FOCUS würfelt dann nur noch für die nicht bekannten Phasen einen Startwert.
56
Beim "Fourierrecycling" werden die vorgegebenen Phasen aber nicht festgehalten, sondern
auch entsprechend dem gefundenen Modell neu zugeteilt.
Mit der vorgegebenen Phaseninformation wird lediglich ermöglicht, einen Startphasensatz
zu berechnen, welcher besser ist als ein zufällig generierter Phasensatz. Dass die
vorgegebenen Phasen auch falsche Phasen enthalten können ist aber kein Problem, weil die
Abbildung 5-1: Flussdiagramm von FOCUS
57
vorgegebenen Phasen beim "Fourierrecycling" verändert werden können. Damit ist FOCUS
im Prinzip in der Lage falsche Phaseninformation zu korrigieren.
5.3 Was bringt die zusätzliche Information in FOCUS?
Um diese Frage zu beantworten wurden Tests mit den Zeolithstrukturen ITQ-22 (Corma, et
al., 2003) und ZSM-5 gemacht. ITQ-22 (16 T-Atome) und ZSM-5 (12 T-Atome) wurden
ausgewählt, weil beide relativ grosse, komplexe Zeolithstrukturen sind. In beiden
Einheitszellen sind zudem zwei Gitterkonstanten fast gleich gross, was zu massiven
Peaküberlappungen im Pulverdiagramm führt und die Strukturlösung mit Pulverdaten
erschwert. Mit den Tests sollten optimale Parameter gefunden werden für die
Strukturbestimmung mit FOCUS. Vor allem wurde auch getestet was ein "Structure
Envelope" für die Strukturbestimmung bringt und wie stark das Vorgeben von Phasen
Vorteile bringt.
5.3.1 Vergleiche mit simulierten Einkristalldaten
Für den Vergleich, welche Zusatzinformationen wie viel Erfolg bringen können bei der
Strukturlösung von komplexen Zeolithstrukturen, wurde zuerst ITQ-22 verwendet. Mit dem
Programm CrystalDiffract wurden von der Struktur (Abbildung 5-2) die Amplituden und
Phasen der Strukturfaktoren berechnet. Zur Simulation von Pulverdaten wurden die
Je grösser die Zeolithstruktur ist, desto mehr Versuche muss FOCUS rechnen, bis eine
korrekte Lösung gefunden wird. Um den Erfolg von FOCUS quantifizieren zu können,
kann zum Beispiel angegeben werden, wie viele korrekte Lösungen FOCUS in einer
vorgegebenen Zeit oder einer vorgegebenen Anzahl Versuche findet. Für ITQ-22 benötigte
FOCUS mit den simulierten Pulverdaten (ohne zusätzliche Information) zum Beispiel 31
Tage, um 1 korrekte Lösung zu finden. Es ist natürlich vom Zufall abhängig, wann FOCUS
eine Lösung findet. Deshalb muss eine minimale Anzahl Versuche gerechnet werden, um
die Erfolgsaussichten quantifizieren zu können. Es muss solange gerechnet werden bis die
Anzahl gefundener Lösungen pro Zeit oder pro Versuche nicht mehr ändert. Für ITQ-22
dauert das zu lange.
Um trotzdem testen zu können, unter welchen Bedingungen gute Erfolgsausssichten
bestehen, wurden für die ersten Tests simulierte Einkristalldaten anstatt der simulierten
58
Pulverdaten verwendet. Mit den Einkristalldaten findet FOCUS wesentlich mehr Lösungen
pro Versuch als mit äquipartitionierten Reflexintensitäten.
Erste Erfahrungen mit HRTEM-Bildern der Zeolithstrutkur ZSM-5 zeigten, dass jeweils die
Phasen der 13 Reflexe mit der grössten Strukturfaktoramplitude aus diesen Bildern korrekt
bestimmt werden konnten. Die Phasen der schwächeren Reflexe waren teilweise falsch. Es
wurde angenommen, dass sich dies auch für andere Strukturen ähnlich verhält. Deshalb
wurden im folgenden Vergleich jeweils 12 Phasen als zusätzliche Information für FOCUS
verwendet.
Mit den simulierten Einkristalldaten für ITQ-22 wurde verglichen wie sich die Vorgabe
eines "Structure Envelopes" und die Vorgabe von Phasen auf die Erfolgsaussichten von
FOCUS auswirken.
Abbildung 5-2: Stereoabbildung der Kristallstruktur von Zeolith ITQ-22. Dargestellt ist das Netwerk der Si-Atome. Die O-Atome wurden weggelassen zwecks besserer Übersicht.
59
Title ITQ-22 F's calculated with CrystalDiffract SpaceGroup Pbam UnitCell 41.6908 12.7128 12.7114 AtomType + Node Si 112 AtomType + NodeBridge O 224 Chemistry MinDistance Node Si Node Si 2.6 Chemistry MinDistance Node Si NodeBridge O 1.4 Chemistry MinDistance NodeBridge O NodeBridge O 2.3 MaxPotentialAtoms 168 MaxRecycledAtoms 168 FwSearchMethod FwTracking MaxPeaksFwSearch 336 MaxPeaksFwFragmentSearch 336 MinNodeDistance 2.6 MaxNodeDistance 3.6 MinSymNodes 0 MaxSymNodes 168 NodeType 4 * -6 -3 -1 4 6 MinLoopSize 3 MaxLoopSize 24 EvenLoopSizesOnly Off Check3DimConnectivity On IdealT_NodeDistance 3.1 CheckTetrahedralGeometry Normal RandomInitialization 1129646940 FeedBackCycles 1 1 1 1 1 1 1 1 1 1 FeedBackBreakIf PhaseDiff < 5.00 % and DeltaR < 3.00 % Grid_xyz 140 40 40 eDensityCutOff 1 % MinPfI 17 CatchDistance 0.5 eD_PeaksSortElement Grid_eD Lambda 1.54056 FobsMin_d 1.3 FobsScale 1.00 SigmaCutOff 0 OverlapFactor 0.15 OverlapAction EqualMF2 ReflectionUsage 65 % # h k l F_cal sigma fwhm 0 0 1 765.94 * 0.05 4 0 0 769.84 * 0.05 2 0 0 249.18 * 0.05 2 1 0 359.1 * 0.05 … … …
Abbildung 5-3: FOCUS Eingabedatei für Zeolith ITQ-22.
60
Um vergleichbare Resultate zu erhalten wurde der Einfluss von einzelnen Parametern
getestet. Grosse-Kunstleve (1996) beschreibt die Schlüsselwörter in der FOCUS
Eingabedatei ausführlich.
Für die Tests mit ITQ-22 wurden einige Parameter so gewählt, dass später auch mit anderen
Strukturen vergleichbare Rechnungen durchgeführt werden können. In der Eingabedatei
werden mit verschiedenen Schlüsselwörtern diese Parameter definiert. Die wichtigsten
Parameter welche für die folgenden Rechnungen benutzt wurden werden hier kurz
beschrieben. Mit AtomType wird angegeben, wie viele Atome von welchem Typ die
Einheitszelle enthält. Dabei wird unterschieden zwischen Knotenpunkten im Netzwerk,
welche mit Node bezeichnet werden (z.B. vierfachverknüpftes Si) und Brückenatomen
zwischen den Knotenpunkten, welche mit NodeBridge bezeichnet werden (z.B.
Sauerstoffbrücken zwischen den Si-Atomen). MaxPotentialAtoms bezeichnet die
maximale Anzahl Peaks aus der Elektronendichtekarte, welche berücksichtigt werden im
Zuordnungsalgorithmus. MaxPotentialAtoms wurde jeweils 1.5-mal grösser gewählt
als die erwartete Anzahl Siliziumatome in der Einheitszelle (112 x 1.5=168 für ITQ-22).
MaxRecyledAtoms gibt die maximale Anzahl Peaks in der Einheitszelle an, welche für
das Recycling benutzt werden und wurde für die folgenden Tests jeweils gleich gross wie
MaxPotentialAtoms gewählt. MaxPeaksFwSearch bestimmt die maximale Anzahl
Maxima aus der Einheitszelle, welche für das Fourierrecycling verwendet werden. Die
maximale Anzahl Knotenpunkte bei der Suche nach einem Netzwerk wird durch
MaxSymNodes begrenzt. MaxSymNodes wurde ebenfalls 1.5-mal grösser als die
erwartete Anzahl Siliziumatome in der Einheitszelle gewählt. Welche Reflexe FOCUS,
benützt wird in Prozent der Gesamtintensität angegeben mit dem Schlüsselwort
ReflectionUsage. Zum Beispiel für ReflectionUsage 65%, wählt FOCUS
beginnend beim stärksten Reflex, Reflexe aus bis deren aufsummierte Intensität 65% der
Gesamtintensität aller Reflexe erreicht.
Abbildung 5-4: Die Tabelle gibt an bei welchem Prozentsatz der Gesamtintensität, wie viele Reflexe verwendet werden aus dem Einkristalldatensatz ( mind =1.3Å) von ITQ-22..
61
In Abbildung 5-3 ist eine Eingabedatei für FOCUS zu sehen, wie sie verwendet wurde in
den folgenden Tests. Wie sich der ReflectionUsage-Parameter auf die Anzahl
benutzter Reflexe auswirkte wird in Abbildung 5-4 gezeigt.
Es wurden folgende 9 FOCUS-Tests mit simulierten ITQ-22 Einkristalldaten durchgeführt:
(1) nur Reflexamplituden, ohne Zusatzinformation
(2) Phasen der 12 Reflexe mit der grössten Strukturfaktoramplitude (Abbildung 5-5)
(3) Phasen der 12 hk0-Reflexe mit der grössten Strukturfaktoramplitude (Abbildung 5-6)
(4) Phasen von 12 niedrig indizierten Reflexen mit grosser Strukturfaktor-
amplitude (Abbildung 5-9)
(5) Phasen von 12 niedrig indizierten hk0-Reflexe mit grosser
Strukturfaktoramplitude (Abbildung 5-10)
(6) "Structure Envelope" aus den 12 Reflexen mit den grössten Strukturfaktor-
amplituden (Abbildung 5-7)
(7) "Structure Envelope" aus den 12 hk0-Reflexen mit der grössten
Strukturfaktoramplitude (Abbildung 5-8)
(8) "Structure Envelope" generiert aus 12 niedrig indizierten Reflexen mit der grosser
Strukturfaktoramplitude (Abbildung 5-11)
(9) "Structure Envelope" generiert aus 12 niedrig indizierten hk0-Reflexen mit der
grosser Strukturfaktoramplitude (Abbildung 5-12)
Die Vorgabe von Phasen von hk0-Reflexen, soll zeigen ob es besonders wichtig ist, einen
dreidimensionalen Phasensatz vorzugeben, oder ob ein zweidimensionaler Phasensatz mit
Phasen von Reflexen aus einer reziproken Ebene auch genügen könnten. Für einen
zweidimensionalen Phasensatz genügt ein HRTEM-Bild um die Phaseninformation zu
berechnen. Für einen dreidimensionalen Phasensatz werden mehrere HRTEM-Bilder von
unterschiedlichen Blickrichtungen benötigt. Besonders bei vorzugsorientierten Proben sind
die zusätzlichen Blickrichtungen nur schwer oder zum Teil gar nicht zugänglich. Alle Tests
wurden als Funktion der "ReflectionUsage" von 25% bis 75% in Schritten von 10%
Abbildung 5-9: ITQ-22 Phasensatz mit den 12 niedrig indizierten, starken Reflexen.
Abbildung 5-10: ITQ-22 Phasensatz mit den 12 niedrig indizierten, starken hk0-Reflexen.
Test 8
Test 9
Abbildung 5-11: ITQ-22 Struktur mit einem "Structure Envelope" aus den 12 niedrig indizierten starken Reflexen .
Abbildung 5-12: ITQ-22 Struktur mit einem "Structure Envelope" aus 12 niedrig indizierten starken hk0-Reflexen
64
Der Erfolg von FOCUS wird mit der Anzahl gefundener ITQ-22 Lösungen pro 10'000
Versuche angegeben. Diese Grösse ist unabhängig von der Geschwindigkeit des Rechners.
Mindestens so wichtig ist aber die Anzahl gefundener ITQ-22 Lösungen pro Zeit. Dies ist
zwar von der Rechnergeschwindigkeit abhängig, aber es ist die entscheidende Grösse, ist es
doch das Ziel, in möglichst kurzer Zeit eine Lösung zu finden. Weil alle Rechnungen auf
einem MAC G5 X-Serve ausgeführt wurden, sollte die Rechnergeschwindigkeit für den
Vergleich keine Rolle spielen. Deshalb wurden die Erfolgsaussichten von FOCUS für diese
Tests anhand der gefundenen Lösungen pro Zeit beurteilt.
Die Erfolgsaussicht in Abhängigkeit von "ReflectionUsage" ist in Abbildung 5-13
aufgezeichnet. Am meisten ITQ-22 Lösungen werden gefunden, wenn diese verwendeten
Reflexe 45%, 50% oder 55% der Gesamtintensität ausmacht, unabhängig von der
verwendeten Zusatzinformation.
Am erfolgreichsten war FOCUS (vgl. Abbildung 5-13), bei der Vorgabe der Phasen der 12
stärksten Reflexe (Test 2 und 3), wobei die Vorgabe der 12 stärksten hk0-Reflexe sogar
besser abschnitt, als diejenige der stärksten hkl-Reflexe. Die Vorgabe von 12 Phasen von
niedrig indizierten Reflexen (Test 4) bringt nur minimal mehr Erfolg. Bei Vorgabe von
hk0-Reflexen sind die Erfolgsaussichten leicht erhöht, wenn die Reflexe mit 50%-55% der
Gesamtintensität verwendet werden. Leichte Vorteile können auch bei der Vorgabe eines
"Structure Envelopes" ausgemacht werden. Das "Structure Envelope" aus 12 hk0-Reflexen
(Test 7) ermöglicht FOCUS 1.8 bis 2.4 mal mehr Lösungen pro Stunde zu finden,
verglichen mit einem FOCUS-Prozess ohne zusätzliche Vorgaben. Das "Structure
Envelope" aus 12 hkl-Reflexen (Test 6) bringt nur Vorteile, wenn mehr als 50% der
Reflexe verwendet werden. Dann ist FOCUS 1.2x bis 1.4x schneller. Die Vorgaben von
niedrig indizierten Reflexen in Form von Phaseninformation oder als "Structure Envelope"
bringen nur einen geringen Geschwindigkeitsgewinn. Das "Structure Envelope", welches
aus 12 starken, niedrig indizierten Reflexen berechnet wurde führt sogar zu einem leichten
Geschwindigkeitsverlust.
65
Abbildung 5-13: Erfolgsaussicht von FOCUS in Abhängigkeit der verwendeten Reflexe (in % der Gesamtintensität) für die Strukturlösung von ITQ-22 mit simulierten Einkristalldaten. Die Erfolgsaussicht wird angegeben in Anzahl Lösungen pro 10'000 Zyklen, rsp. pro Stunde Rechenzeit. Die Datensätze sind beschriftet mit (fo) für FOCUS ohne Zusatzinformation. Für die übrigen Datensätze wurde folgende Zusatzinformation verwendet: (12sF) Phasenvorgabe der 12 stärksten Reflexe, (12sF_hk0) Phasenvorgabe der 12 stärksten hk0-Reflexe, (surf_12sF) Vorgabe eines "Structure Envelope" generiert aus den Phasen der 12 stärksten Reflexe, (surf_12sF_hk0) Vorgabe eines "Structure Envelope" generiert aus den Phasen der 12 stärksten hk0-Reflexe. Analog sind die Bezeichnungen mit (..hd..) wobei bei diesen Datensätzen nur niedrig indizierte Reflexe, verwendet wurden.
66
5.3.2 Vergleiche an ITQ-22 mit simulierten Pulverdaten
Nach dieser ersten Übersicht mit Einkristalldaten wurden weitere Tests mit simulierten
Pulverdaten gemacht. Dazu wurden die Einkristalldaten äquipartitioniert. Als
Zusatzinformation wurden die Phasen der stärksten (grösste Strukturfaktoramplitude)
Reflexe vorgegeben. FOCUS rechnete jeweils 100'000 Versuche mit 124, 62 und 31
vorgegebenen Phasen. Ohne vorgegebene Phasen und mit 31 vorgegebenen Phasen mussten
mehr als 100'000 Versuche gerechnet werden um überhaupt eine ITQ-22 Lösung zu finden.
Je mehr Phasen vorgegeben wurden, desto mehr korrekte Lösungen wurden pro Versuch
Abbildung 5-14: Erfolg von FOCUS mit 124, 62, 31 vorgegebenen Phasen und von FOCUS ohne Zusatzinformation für simulierte Pulverdaten.
Die ITQ-22 Struktur konnte auch ohne vorgegebene Phasen gelöst werden, der Zeitgewinn
durch Vorgabe von Phasen ist jedoch markant. Die ITQ-22 Struktur wurde bei Vorgabe von
31 Phasen 113-mal öfter gefunden pro Zeiteinheit, als ohne Phasenvorgabe, mit Vorgabe
von 62 Phasen wurde ITQ-22 sogar 343-mal öfter gefunden und bei 124 vorgegebenen
Phasen 40000-mal öfter.
Wie bereits für die Einkristalldaten, sollten auch für Pulverdaten weitere Tests zeigen wie
viel Reflexe verwendetet werden sollen, um die grössten Erfolgsaussichten zu haben. Es
wurde auch getestet ob die Erfolgsaussichten durch vorgeben eines "Structure Envelopes"
oder durch vorgeben von Phasen besser sind.
67
Abbildung 5-15: Erfolgsaussicht in Abhängigkeit der verwendeten Reflexe ("ReflectionUsage") für die Strukturlösung von ITQ-22 mit simulierten Pulverdaten (äquipartitionierte Einkristalldaten). Der Erfolg wird angegeben in Anzahl Lösungen pro 100'000 Zyklen, rsp. pro Stunde Rechenzeit. Die Datensätze sind beschriftet mit (fo) FOCUS ohne Zusatzinformation, (62sF) Phasenvorgabe der 62 stärksten Reflexe, (surf) Vorgabe eines "Structure Envelope" generiert aus den Phasen der 62 stärksten Reflexe, (surf & 62sF) der Phasen der 62 stärksten Reflexe und zusätzlich auch Vorgabe des "Structure Envelopes", welches aus diesen Reflexen erzeugt wurde.
68
Für diese Tests wurden die Phasen der 62 stärksten Reflexe vorgegeben, respektive aus
diesen Reflexen mit ihren Phasen ein "Structure Envelope" erzeugt. Die simulierten
Einkristalldaten wurden äquipartitioniert und Reflexe mit einem d-Wert kleiner als 1.3 Å
wurden verwendet.
Jeweils 100'000 Versuche wurden gerechnet. Innerhalb dieser 100'000 Versuche konnte
ohne zusätzliche Vorgaben keine Lösung von ITQ-22 gefunden werden (vgl. Abbildung
5-15). Auch mit Vorgabe des "Structure Envelopes" wurde keine ITQ-22 Lösung gefunden
innerhalb der 100'000 Versuche. Erst nach über 4 Millionen Versuchen (entspricht ca. 1
Monat Rechenzeit) konnte eine ITQ-22-Lösung gefunden werden. Am meisten Lösungen
wurden gefunden mit den 62 vorgegeben Phasen. Das sind 982-mal mehr Lösungen
verglichen mit den Versuchen ohne Zusatzinformation. Am erfolgreichsten war aber die
Kombination von "Structure Envelope" und Vorgabe der 62 Phasen der stärksten Reflexe.
Die Erfolgsaussicht war mit ReflectionUsage 75%, sowohl bei der Vorgabe der
Phasen wie auch bei der Kombination von Phasen und "Structure Envelope" am grössten.
Mit dieser Kombination von Zusatzinformationen fand FOCUS 3415-mal mehr Lösungen
pro Zeiteinheit, verglichen mit FOCUS ohne Zusatzinformation. Bereits mit
ReflectionUsage 35% liefert FOCUS gute Resultate, während mit
ReflectionUsage 45% eine leichte Verschlechterung zu verzeichnen war, arbeitet
FOCUS mit ReflectionUsage 55% bis 75% kontinuierlich besser.
Verschiedenste Parameter von FOCUS haben Einfluss auf die Laufzeit von einem Versuch.
Interessant wäre die Laufzeit für einen Versuch zu verkürzen, ohne den Erfolg von FOCUS
wesentlich zu beeinträchtigen. Besonders Zeitaufwändig ist die Suche eines Netzwerkes in
der Elektronendichtekarte. FOCUS liest dazu aus der Elektronendichtekarte, welche aus
den verwendeten Reflexen und deren Phasen berechnet wurden, die lokalen Maxima
heraus. In der Eingabedatei für FOCUS kann bestimmt werden wie viele lokale Maxima,
beginnend beim grössten Maximum, FOCUS aus der Elektronendichtekarte heraus liest, als
potentielle Atompositionen für das Gerüst des Netzwerkes. Je mehr Positionen
herausgelesen werden, desto mehr Abstände und Winkel zwischen all diesen möglichen
Atompositionen muss FOCUS berechnen, um ein mögliches Netzwerk oder ein mögliches
Fragment davon zu finden.
69
Abbildung 5-16: Erfolg von FOCUS in Abhängigkeit der Anzahl verwendeten Maxima aus der Elektronendichtekarte am Beispiel von ITQ-22. Der Erfolg wird ausgedrückt in der gefundenen Anzahl ITQ-22 Lösungen pro Stunde, respektive die gefundene Anzahl pro 100'000 Zyklen, immer unter Vorgabe der Phasen der 62 stärksten Reflexe, mit Reflexen mit d-Werten bis zu 1.3Å, unter Verwendung der stärksten Reflexe, welche 65% der Gesamtintensität ausmachen.
Abbildung 5-17: Erfolgsaussichten in Abhängigkeit der Auflösung der Pulverdaten. Der Erfolg wird ausgedrückt in der gefundenen Anzahl ITQ-22 Lösungen pro Stunde, respektive die gefundene Anzahl pro 100'000 Zyklen, immer unter Vorgabe der Phasen der 62 stärksten Reflexe, unter Verwendung der stärksten Reflexe, welche 65% der Gesamtintensität ausmachen.
70
Das wird extrem zeitaufwändig, wenn viele Atompositionen vorliegen. Deshalb wurde
getestet, wie die Erfolgsausichten aussehen in Abhängigkeit der Anzahl Maxima welche
aus der Elektronendichtekarte herausgelesenen werden.
In Abbildung 5-16 ist zu erkennen, dass wenn mehr Maxima für die Netzwerksuche
verwendet wurden, auch mehr ITQ-22 Lösungen pro Versuch gefunden wurden. Die
Laufzeit pro Versuch nimmt aber gleichzeitig massiv zu. Um den Erfolg zu beurteilen
wurde deshalb wieder die Anzahl gefundener ITQ-22 Lösungen pro Zeit betrachtet. Mit
2.41 ITQ-22 Lösungen pro Stunde, bei 270 verwendeten Maxima aus der
Elektronendichtekarte war FOCUS am erfolgreichsten. ITQ-22 hat in der Einheitszelle 112
T-Atome. Die 270 verwendeten Maxima sind demzufolge das 2.4-fache der vorhandenen
T-Atome.
Ebenfalls getestet wurde wie hoch die Auflösung der Pulverdaten sein sollte, damit FOCUS
erfolgreich ist. Dazu wurden in FOCUS aus dem simulierten Pulverdatensatz nur Reflexe
bis zu einem bestimmten minimalen d-Wert ( mind ) für die Struktursuche zugelassen. Je
höher die Auflösung der Pulverdaten war desto mehr Lösungen wurden gefunden bei den
100'000 Versuchen. Erst ab 1.3 Å Auflösung konnte FOCUS überhaupt ITQ-22 Lösungen
finden. Die Erfolgsaussichten sind am grössten mit 1 Å und 1.1 Å Auflösung. Für diese
Auflösung wurden je 19.9 ITQ-22 Lösungen pro Stunde gefunden, während mit einer
Auflösung von 1.2 Å noch 3.9 und mit 1.3 Å noch 1.6 Lösungen pro Stunde gefunden
wurden.
71
5.3.3 Vergleiche mit experimentellen Röntgenbeugungsdaten
Nach den Tests mit den simulierten Einkristall und Röntgendaten Daten folgt nun der Test
mit experimentellen Daten. Für diesen Test wurde die Zeolithstruktur ZSM-5 ausgewählt,
weil einerseits Röntgenbeugungsdaten (Abbildung 5-18) von einer Synchrotronmessung
vorlagen und andererseits, weil ZSM-5 mit 12 T-Atomen in der asymmetrischen Einheit
auch eine grössere Zeolithstruktur ist, welche aber trotzdem kleiner ist als ITQ-22 mit
16 T-Atomen. ZSM-5 ist eine Zeolithstruktur welche 1998, mit den damaligen Computer
Rechengeschwindigkeiten, von FOCUS gerade noch lösbar war. Für die Strukturlösung von
ZSM-5 mit FOCUS wurden mit XRS-82 (Baerlocher, 1982) extrahierte Synchrotron-
pulverdaten verwendet. Die extrahierten Daten wurden mit der FIPS-Methode (Estermann
und Gramlich, 1993) behandelt, um eine bessere Intensitätsverteilung überlappender Peaks
zu bekommen.
Abbildung 5-18: Teil des Pulverdiagramm von Zeolith ZSM-5, gemessen an der SNBL (Swiss Norwegian Beamline) am ESRF in Grenoble (Frankreich). Die Reflexpositionen sind mit senkrechten Strichen unter dem Pulverdiagramm angegeben.
Von dem ZSM-5 Kristall in Abbildung 5-19 wurden HRTEM-Bilder aufgenommen. Aus
dem HRTEM-Bild in Abbildung 5-20A wurde von 11 starken Reflexen mit dem Programm
CRISP die Phasen bestimmt. Der Defokus von -650Å des Bildes wurde anhand von
amorphen Anteilen des fouriertransformierten Bildes berechnet. Das HRTEM-Bild wurde
CTF korrigiert und über die planare Symmetrie pgg gemittelt (Abbildung 5-20C). Ein
Vergleich des korrigierten HRTEM-Bildes mit dem simulierten Bild (Abbildung 5-20D)
zeigt, dass die Hauptmerkmale gut übereinstimmen.
72
Abbildung 5-19: Sehr dünner (Randbereich ca. 10nm dick) ZSM-5 Kristall auf einer Kohlestoffolie mit Löchern.
Abbildung 5-20: (A) HRTEM-Bild eines ZSM-5 Zeolithkristallites entlang der Zoneachse [0 1 0 ], aufgenommen, (B) SAED-Bild von ZSM-5, (C) HRTEM-Bild korrigiert mit CTF (Cs=1.4mm, ∆f=70nm, α=1.0mrad) und symmetriegemittelt, (D) simuliertes HRTEM-Bild für Probendicke 10nm und Defokus -65nm.
73
Neben dem Vergleich mit der Simulation zeigt auch die Überlagerung der ZSM-5 Struktur
mit dem HRTEM Bild (Abbildung 5-22), dass das korrigierte HRTEM-Bild die
Hauptmerkmale, wie die Kanäle und die Schmetterlingsstruktur korrekt wiedergibt und
deshalb gut verwendet werden kann um die Phasen zumindest der starken Reflexe zu
berechnen.
Der Phasensatz mit den 11 Reflexen wurde durch einen ursprungsbestimmenden Reflex
ergänzt. Dazu wurde der 011-Reflex mit der willkürlichen Phase 180° ausgewählt. Dieser
Phasensatz mit 12 Reflexen (Abbildung 5-23) wurde vorgegeben. Aus denselben 12
Reflexen wurde auch ein "Structure Envelope" berechnet. Das "Structure Envelope" wurde
mit dem Programm SayPerm (Brenner, 1999): erzeugt. Um das "Structure Envelope"
(Abbildung 5-24) zu erzeugen, wurden sowohl die Reflexamplitude, wie auch die
Phaseninformation aus dem Hochauflösungsbild verwendet. Dagegen wurden für die
Strukturlösung, die aus den Pulverdiffraktionsdaten extrahierten Reflexintensitäten
verwendet.
Die FOCUS-Bedingungen für gute Erfolgsaussichten bei der Strukturlösung, mit den
experimentellen Daten von ZSM-5 (vgl. Abbildung 5-25) unterschieden sich von
denjenigen für die simulierten Daten von ITQ-22 (Kapitel 5.3.2). Mit den experimentellen
Daten wurden am meisten ZSM-5 Lösungen gefunden, wenn die stärksten Reflexe
verwendet wurden, welche zusammen 35% der Gesamtintensität ausmachen. Für ITQ-22
war dies mit 75% der Reflexe der Fall. Noch unterschiedlicher fällt der Vergleich aus,
wenn die Erfolgsaussichten anhand der Anzahl gefundenen Lösungen pro Zeit betrachtet
werden. Bei ZSM-5 werden am meisten Lösungen gefunden, wenn nur die stärksten
Reflexe mit total nur 15% der Gesamtintensität verwendet wurden.Das ist reproduzierbar
und unabhängig Startwert des Zufallsgenerators. Werden weniger Reflexe verwendet
rechnet FOCUS wesentlich schneller. Werden weniger Reflexe als die 35% benutzt sind die
Erfolgsaussichten pro Versuch zwar kleiner, dies wird aber durch die Geschwindigkeit
kompensiert, sodass die Erfolgsaussicht gemessen an der Rechenzeit trotzdem grösser ist.
Abbildung 5-21: Die Tabelle gibt an bei welchem Prozentsatz der Gesamtintensität (ReflectionUsage), wie viele Reflexe verwendet werden Datensatz von ZSM-5. Bei Daten mit einer Auflösung ( mind ) von 1.3Å.
74
Abbildung 5-22: Projektion der ZSM-5 Struktur in Blickrichtung [010] über dem CTF-korrigierten und symmetriegemittelten HRTEM-Bild.
Abbildung 5-23: ZSM-5 Phasensatz mit den 11 stärksten aus dem HRTEM Bild der Orientierung (0 1 0), zusätzlichen mit dem ursprungs-bestimmenden 0 1 1 – Reflex ergänzt.
Abbildung 5-24: ZSM-5 Struktur mit "Structure Envelope" berechnet aus 12 Phasen, welche in Abbildung 5-23 aufgelistet sind.
75
Abbildung 5-25: Erfolgsaussicht von FOCUS in Abhängigkeit der verwendeten Reflexe (ReflectionUsage) für die Strukturlösung von ZSM-5 mit experimentellen Pulverdaten. Der Erfolg wird angegeben in Anzahl Lösungen pro 100'000 Versuchen, rsp. pro Stunde Rechenzeit. Die Datensätze sind beschriftet mit (fo) FOCUS ohne Zusatzinformation, (12sF) Phasenvorgabe der 12 stärksten Reflexe, (surf) Vorgabe eines "Structure Envelope" generiert aus den Phasen der 12 stärksten Reflexe, (surf & 12sF) der Phasen der 12 stärksten Reflexe und zusätzlich auch Vorgabe des "Structure Envelopes", welches aus diesen Reflexen erzeugt wurde.
76
Die Erfolgsausichten sind höher, wenn weniger Reflexe verwendet werden. Eine Erklärung
dafür könnte sein, dass die Struktur von ZSM-5 wesentlich kleiner und damit auch
einfacher zu lösen ist als diejenige von ITQ-22. Für die etwas einfachere Struktur von
ZSM-5 genügen offenbar die stärksten Reflexe um die Struktur zu lösen, während für
komplexere Strukturen mehr Reflexe benötigt werden.
Werden auch Reflexe geringerer Intensität verwendet, sind das meist auch höher indizierte
Reflexe. Damit steht auch Information mit höherer Auflösung zur Verfügung.
Im Fall von ZSM-5 schneiden das "Structure Envelope, welches aus den Phasen der 12
stärksten Reflexe berechnet wurde und das "Structure Envelope" in Kombination mit der
Vorgabe der Phasen dieser 12 Reflexe am besten ab. Werden die Reflexe mit 15% der
Gesamtintensität verwendet, ist die Erfolgsaussicht bei der Vorgabe der 12 Phasen 1.26-mal
höher als ohne Zusatzinformation.
Die Kombination von "Structure Envelope" und Vorgabe der Phasen bringt FOCUS eine
1.16-fachen Geschwindigkeitsgewinn. Die Zusatzinformationen bringen also weit weniger
als bei der komplexeren ITQ-22 Struktur.
5.3.4 Qualität der Phaseninformation
Die Phaseninformation, die hier als Vorgabe verwendet wurde, stammt aus einem HRTEM-
Bild. Ein Teil dieser Phasen kann auch falsch sein. Am Beispiel des HRTEM-Bildes von
ZSM-5 konnten festgestellt werden, dass die Phasen der 11 stärksten Reflexe korrekt
waren. Mit den simulierten Röntgenbeugungsdaten von ITQ-22 wurde deshalb auch
getestet, welchen Einfluss falsche Phaseninformation auf die Erfolgsaussichten von
FOCUS hat. Im vorgegebenen Phasensatz mit 124 Phasen, wurden gezielt falsche Phasen
eingefügt. Weil die Phasen der starken Reflexe in der Regel eher korrekt sind, wurden die
falschen Phasen vor allem den schwachen Reflexen zugeordnet. Für welche der 124
Reflexe, falsche Phasen gesetzt werden, wurde deshalb mit einer gewichteten Zufallszahl
ausgewählt. Das Gewicht für den Reflex mit den Indizes hkl wurde aus der
Strukturfaktoramplitude ( )hklF , der Strukturfaktoramplitude des stärksten Reflexes
( )hklFmax und der Strukturfaktoramplitude des schwächsten Reflexes ( )hklFmin der 124
Reflexe, wie folgt bestimmt:
77
( )( )( )( )
21
max
min
max
1
1
−
−=
hklFhklFhklF
hklF
w 5-1
Mit diesem Modell (Abbildung 5-26) wurden Datensätze mit 10%, 20%, 25 % und 30%
falschen Phasen erzeugt. Es wurden für jeden Prozentsatz vier Datensätze erzeugt. In jedem
Datensatz wurden die Reflexe deren Phase falsch gesetzt wurde, mit dieser Gewichtung
(Gleichung 5-1) zufällig ausgewählt. In jedem der Datensätze waren demzufolge andere
Reflexe mit falschen Phasen versehen. Mit diesen Phasensätzen wurde in FOCUS jeweils
100'000 Versuche gerechnet. Dazu wurden die stärksten Reflexe verwendet, welche 65%
der Gesamtintensität ausmachen und Reflexe bis zu einem d-Wert von 1.3Å. Über die
Resultate der vier Datensätze mit gleichem Prozentsatz falscher Reflexe wurde am Ende
gemittelt.
Abbildung 5-26: Gewichtung in Abhängigkeit der Strukturfaktoramplitude: Gewichtung der Zufallszahl für die falsch zu setzenden Reflexe im Phasensatz der 124 vorgegebenen Reflexe. Die Phase des stärksten Reflexes wird mit der Gewichtung 0, also nie falsch gesetzt werden, während die Phasen der schwachen Reflexe mit zunehmender Wahrscheinlichkeit falsch sein können.
78
Abbildung 5-27: Erfolg von FOCUS mit Phasensätzen unterschiedlicher Qualität (unterschiedlicher Anteil der 124vorgegebenen Phasen ist falsch) am Beispiel von ITQ-22.
Abbildung 5-28: Zeitgewinn von FOCUS bei Verwendung von Phaseninformation unterschiedlicher Qualität. Mit dem Zeitgewinn wird ausgedrückt wievielmal schneller die ITQ-22 findet, wenn 0, 10, 20, 25 und 30% der 124 vorgegeben Phasen falsch sind, im Vergleich zur Verwendung von FOCUS ohne Phaseninformation.
79
Die Resultate in Abbildung 5-27 zeigen, dass wie erwartet, falsche Phasen die
Erfolgsaussichten wesentlich beeinträchtigen. Bereits mit 10% falscher Phaseninformation,
wurden bei 100'000 Versuchen nur noch 227 richtige Lösungen gefunden. Mit einem
Phasensatz ohne falsche Phaseninformation wären dies mit 2248 Lösungen immerhin fast
10-mal mehr. Mit 20% falscher Phaseninformation wurden noch 14 und mit 30% nur noch
3 Lösungen gefunden. Betrachtet man den Erfolg in Abhängigkeit der Zeit, dann sieht das
ganz ähnlich aus. Mit 10% falschen Phasen wurden 20 Lösungen pro Stunde gefunden, mit
20% noch 1.2 und mit 30% noch 0.045. FOCUS ist demzufolge 9-mal langsamer mit 10%
falschen Phasen, 151-mal langsamer mit 20% falschen Phasen und sogar 4122-mal
langsamer mit 30% falschen Phasen.
Vergleicht man jedoch diese Resultate mit denjenigen ohne Verwendung von vorgegebenen
Phasen, so ist FOCUS auch mit 30% falschen Phasen immer noch 10-mal schneller
(vgl. Abbildung 5-28). Mit anderen Worten, auch mit etwas "zweifelhaften" Phasen, wie
man sie von einem HRTEM-Bild erhalten kann, erhöhen sich die Chancen für eine Lösung
dramatisch.
80
81
6 TNU-9
6.1 Zeolith TNU-9, als echter Test des Verfahrens
Die Synthese von TNU-9 wurde von Hong et al. (2004) mit Hilfe des Templates 1,4-bis(N-
methypyrrolidinum)butan gemacht. Nach der Kalzinierung ist TNU-9 ein Zeolith mit der
Zusammensetzung [ ]384182.79.39.3 OSiAlH . Hochaufgelöste Pulverdaten wurden von
Paul A. Wright (School of Chemistry, University of St. Andrew, UK) von "as synthesized"
TNU-9 am ESRF in Grenoble gemessen. Die Daten wurden aufgenommen mit einer
Wellenlänge von 0.80124Å und gemessen wurde der 2θ-Bereich von 1 bis 50° ( mind
=0.95Å) mit extrem hoher Auflösung (fwhm=0.0083°). Als Ergänzung zu den
Pulverdiffraktionsdaten wurden von Zheng Liu, Tetsu Ohsuna und Osamu Terasaki vom
Arrhenius Labor der Universität Stockholm (Schweden) mit einem JEM-3010
Transmissionselektronenmikroskop Hochauflösungsbilder und die entsprechenden SAED-
Bilder aufgenommen.
6.2 Indizierung TNU-9
In Pulverdiagrammen von der Zeolithstruktur TNU-9, ist die Peaküberlappung besonders
gross. Das macht die Aufteilung der Intensitäten überlappender Peaks schwierig. Trotzdem
müssen die Reflexintensitäten, auch jene der überlappenden Peaks, so gut wie möglich aus
dem Pulverdiagramm extrahiert werden, denn falsch bestimmte Reflexintensitäten können
die Strukturbestimmung verunmöglichen.
Je kleiner die Halbwertsbreite der Peaks im Pulverdiagramm ist, desto weniger
Überlappungen wird es geben. Synchrotrondaten ermöglichen wegen der geringeren
Peaküberlappungen eine bessere Extraktion der Reflexintensitäten. Eine Indizierung anhand
der Pulverdaten von TNU-9 alleine war zunächst trotz hoher Auflösung nicht eindeutig
möglich. Bei jedem Einheitszellenvorschlag waren einige klar sichtbare Peaks nicht
indizierbar. Wie sich später herausstellte stammten diese Peaks aus einer Verunreinigung.
Zur Verifizierung der Indizierung des Pulverdiagramms und für die Bestimmung der
Symmetrie wurden daher die SAED-Aufnahmen zu Hilfe genommen. Aus dem SAED-Bild
in Abbildung 6-2 konnten die Gitterkonstanten a=28.2 Å und b=20.0Å und deren
Zwischenwinkel γ zu 90° ermittelt werden. In der SAED-Aufnahme von Abbildung 6-3
kommt wiederum der Gittervektor *ar
vor.
82
Abbildung 6-1: Pulverdiagramm von Zeolith TNU-9, aufgenommen von P.A. Wright (School of Chemistry, University of St. Andrew, UK), am ESRF in Grenoble bei einer Wellenlänge von 0.80124Å. Der 2θ-Bereich zwischen 20° und 50° wurde mit Faktor vier skaliert, um die Details besser sichtbar zu machen.
Abbildung 6-2: (A) HRTEM-Bild entlang der Zonenachse [0 0 1] eines TNU-9 Zeolithkristallites (B) SAED-Bild von TNU-9 (C) HRTEM-Bild korrigiert mit CTF (Cs=0.6mm, ∆f=90nm, α=0.5mrad) und symmetriegemittelt, (D) simuliertes HRTEM-Bild für die Probendicke 27nm und von 60nm Unterfokus. Die experimentellen Aufnahmen stammen von O. Terasaki.
2θ
83
Eine nahe liegende Möglichkeit war, die Richtung, welche auf der SAED-Aufnahme mit
dem Gittervektor *ar
einen Zwischenwinkel von 88° bildet, als dritten Gittervektor *cr zur
Komplettierung der Einheitszelle zu verwenden. In der SAED-Aufnahme der Zone [0 0 1]
ist eine Auslöschung für die Reflexe mit h+k=2n+1 erkennbar, was auf eine C-Zentrierung
schliessen lässt. Im HRTEM-Bild welches entlang der Zonenachse [0 0 1] aufgenommen
wurde sind zudem zwei Spiegelebenen (planare Raumgruppe C2mm) zu erkennen. Im
HRTEM-Bild welches entlang der Zoneachse [0 1 0] aufgenommen wurde ist eine
zweizählige Achse (planare Raumgruppe p2) erkennbar. Diese Symmetrieelemente (in den
Projektionen) kommen im monoklinen Kristallsystem zusammen nur in der Raumgruppe
C2/m vor. Mit der so gewählten Einheitszelle konnte die vermutete Zelle eindeutig bestätigt
und das Pulverdiagramm mit relativ grosser Sicherheit in der Raumgruppe C2/m indiziert
werden.
Abbildung 6-3: (A) HRTEM-Bild entlang der Zonenachse [0 1 0 ] eines TNU-9 Zeolithkristallites (B) SAED-Bild von TNU-9 (C) HRTEM-Bild korrigiert mit CTF (Cs=0.6mm, ∆f=90nm, α=0.5mrad) und symmetriegemittelt, (D) simuliertes HRTEM-Bild für Probendicke 8nm und 46nm Unterfokus. Die experimentellen Aufnahmen stammen von O. Terasaki.
84
6.3 Extraktion
Das Pulverdiagramm wurde danach auch verwendet, um die Einheitszelle zu verfeinern.
Die Gitterkonstanten der verfeinerten Einheitszelle vor der Strukturbestimmung waren
a=28.216Å, b=20.009Å, c=19.489Å und β=92.320Å. Die Gitterparameter sind eng
verwandt miteinander, so ist ba 2~ und cb ~ . Das führt dazu, dass auch Reflexe ohne
symmetriebedingte Abhängigkeit sehr ähnliche d-Werte aufweisen. Deshalb überlappen
( )3.0=ÜLK von den 3705 Reflexen ( mind =1.17Å) 3154 Reflexe, trotz des sehr gut
aufgelösten Synchrotrondatensatzes. Die 3154 überlappenden Reflexe bilden dann 637
Gruppen.
Mit dem Programm EXPOL (Prokić, S. 1999) wurden 1482 Reflexeintensitäten ( =mind1.3Å) extrahiert. EXPOL verwendet einen Pawley-Algorithmus für die Extraktion der
Intensitäten, was die extrahierten Intensitäten weniger empfindlich bezüglich der
Verunreinigung macht. Um möglichst gute Reflexintensitäten zu bekommen wurden die
Intensitäten mit der FIPS (fast iterative Patterson squaring)-Methode (Estermann, M.A. und
Gramlich, V., 1993) neu aufgeteilt.
Die gefundene Einheitszelle hat ein Volumen von 10993.6ų. Da in Zeolithen
typischerweise zirka 16 T-Atome pro 1000ų vorkommen, wurden in der Raumgruppe
C2/m, 24 T-Atome pro asymmetrische Einheit erwartet. Bisher war noch keine solch
komplexe Zeolithstruktur bekannt. Die komplexeste war ITQ-22 mit 16 T-Atomen in der
asymmetrischen Einheit.
6.4 Strukturbestimmung
Für die Strukturbestimmung von ITQ-22, hätte FOCUS ohne zusätzliche Information 31
Tage benötigt bis eine erste Lösung gefunden worden wäre. Das zeigt, dass für grössere
Strukturen die Strukturlösung mit FOCUS, basierend auf Pulverdaten in vernünftiger Zeit
kaum machbar ist.
Die beiden SAED-Aufnahmen, welche entlang der Zonenachse [0 1 0] und [0 0 1]
aufgenommen wurden (Abbildung 6-2 und Abbildung 6-3), wurden bereits für die
Indizierung zu Hilfe genommen. Die dritte SAED-Aufnahme konnte mit der Software
85
CRISP (Zou, X.D et al. ,1996), basierend auf der obigen Einheitszelle eindeutig indiziert
und der Zone [−1 1 0] zugeordnet werden (Abbildung 6-4).
Für diese Hochauflösungsbilder wurde die CTF berechnet und die Bilder entsprechend
korrigiert. Zusätzlich wurde die planare Symmetrie in das CTF-korrigierte Bild
implementiert, indem über die Intensitäten der symmetrieäquivalenten Reflexe gemittelt
wurde. Für all diese Schritte wurde die Software CRISP verwendet.
Für jede der drei Zonen konnte auf diese Weise ein Phasensatz ermittelt werden.
Abbildung 6-4: A) HRTEM-Bild entlang der Zonenachse [-1 1 0] eines TNU-9 Zeolithkristallites B) zugehörige SAED-Aufnahme ebenfalls C) HRTEM-Bild korrigiert mit CTF (Cs=0.6mm, ∆f=90nm, α=0.5mrad) und symmetriegemittelt, D) simuliertes HRTEM-Bild für Probendicke 10nm und 50nm Unterfokus. Die experimentellen Aufnahmen stammen von O. Terasaki.
Symmetriebedingt ausgelöschte Reflexe, welche in der Elektronenbeugung auf Grund von
Mehrfachbeugung trotzdem Intensität erhalten können, wurden aus dem Phasensatz
entfernt.
Aus diesen bereinigten Phaseninformationen wurden vorerst fünf verschiedene
Phasensätze (1)-(5) zusammengestellt und mit FOCUS für die Strukturlösung verwendet.
86
(1) Aus Abbildung 6-2extrahierte Phaseninformation.
(2) Aus Abbildung 6-3 extrahierte Phaseninformation.
(3) Aus Abbildung 6-4 extrahierte Phaseninformation.
Damit die Phasensätze zusammengesetzt werden konnten wurde der passende Ursprung
anhand der Phasen der gemeinsamen Reflexe aus zwei SAED-Bildern bestimmt. Der dritte
Phasensatz würde dann auf den Ursprung der ersten beiden Phasensätze angepasst, sofern
nötig.
(4) Aus jedem HRTEM-Bild die acht stärksten Reflexe ausgewählt, weil angenommen
wurde dass vor allem die Phasen der starken Reflexe korrekt bestimmt werden
können. Deren Phasen wurden zu einem Phasensatz kombiniert, sodass ein
Phasensatz aus 24 Reflexen entstand.
(5) Die Phasen von Reflexen mit grossen d-Werten sollte ebenfalls sicherer bestimmt
werden können als jene von Reflexen mit kleinem d-Wert.Deshalb wurde ein
Phasensatz mit der Phaseninformation von den 111 Reflexen mit dem grössten d-
Wert erstellt.
Mit jedem dieser Phasensätze rechnete FOCUS mindestens 5 Millionen Versuche
(entspricht ca. 1 Monat Rechenzeit auf einem Mac G5 X-Serve). Auch die Parameter in
FOCUS wurden variiert (ReflectionUsage 50% und 65%, und dmin 1.2Å und 1.3 Å). Mit all
diesen verschiedenen Vorgaben konnte aber keine Strukturlösungsvorschlag gefunden
werden. Um nun möglichst viel Phaseninformation zu verwenden, anstatt auf die Qualität
der Phaseninformation zu achten, wurden nun die drei Phasensätze die aus den drei
HRTEM-Bildern resultierten zu einem Satz zusammengeführt. Dieser Phasensatz enthielt
nun Phasen von 258 Reflexen aus den drei Zonen (siehe Anhang C) mit d-Werten von
19.497Å bis zu 1.810Å.
Ein spezieller Datensatz, welcher nur die in FOCUS aktiven Reflexe enthielt (basieren auf
ReflectionUsage 65%) wurde für FOCUS zusammengestellt. Zu diesem Datensatz
wurden alle Reflexe aus dem Phasensatz hinzugefügt und ReflectionUsage auf 100%
gesetzt. So wurden alle vorhandenen Phasen verwendet (auch diejenigen von schwachen
Reflexen), ohne andere schwache Reflexe mit unbestimmten Phasen einzubeziehen.
87
Mit diesen Vorgaben und den Parametern, wie sie in Abbildung 6-5 in der Eingabedatei
aufgelistet sind, war FOCUS nach 2.4 Millionen Zyklen (ca. 16 Tage Rechenzeit auf einem
Mac G5 X-Serve) erfolgreich. Ein vollständiges Netzwerk von vierfachverknüpften
Silizium mit 24 Si-Atomen in der asymmetrischen Einheit wurde gefunden. Dieses
Title TNU9, synchrotron data measured by P.A. Wright at ESRF in Grenoble extrated with Expol, ETHZ Zürich SpaceGroup C 2/m UnitCell 28.2139 20.0094 19.4894 90.000 92.320 90.000 AtomType + Node Si 192 AtomType - NodeBridge O 384 Chemistry MinDistance Node Si Node Si 2.6 Chemistry MinDistance Node Si NodeBridge O 1.4 Chemistry MinDistance NodeBridge O NodeBridge O 2.3 MaxPotentialAtoms 288 MaxRecycledAtoms 192 FwSearchMethod FwTracking MaxPeaksFwSearch 422 MaxPeaksFwFragmentSearch 422 MinNodeDistance 2.6 MaxNodeDistance 3.6 MinSymNodes 0 MaxSymNodes 350 NodeType 4 * -6 -3 -1 4 6 MinLoopSize 3 MaxLoopSize 24 Check3DimConnectivity Off IdealT_NodeDistance 3.1 CheckTetrahedralGeometry Normal RandomInitialization Time FeedBackCycles 1 1 1 1 1 1 1 1 1 1 FeedBackBreakIf PhaseDiff < 5.00 % and DeltaR < 3.00 % Grid_xyz 96 68 68 eDensityCutOff 1% MinPfI 17 CatchDistance 0.5 eD_PeaksSortElement Grid_eD Lambda 0.80124 FobsMin_d 1.2 FobsScale 1.00 SigmaCutOff 0 OverlapFactor 0.15 OverlapAction NoAction ReflectionUsage 100% # h k l Fobs Sigma HalfWd -2 0 1 370.41 0.03 0.015 -1 1 2 132.88 0.07 0.017 ... ... End
Abbildung 6-5: FOCUS Eingabedatei für Zeolith TNU-9, die Eingabeparameter sind beschrieben in Grosse-Kunstleve, R.W. (1996).
88
Siliziumgerüst wurde mit den Sauerstoffbrücken zwischen den Siliziumatomen
vervollständigt.
Als Test, ob es die zusätzliche Phaseninformation war, welche es ermöglichte die Struktur
zu lösen, wurden unter denselben Bedingungen FOCUS ohne zusätzliche Information und
FOCUS mit einem "Structure Envelope" gestartet, wobei das aussergewöhnliche
"Structure Envelope" mit den 258 Reflexen aus dem Phasensatz generiert wurde, welcher
zur erfolgreichen Lösung führte. Nach 10 Mio. Zyklen (ca. 160 Tage auf einem Mac G5 X-
Serve) in FOCUS brachte weder FOCUS alleine noch FOCUS mit dem "Structure
Envelope" eine korrekte Lösung hervor.
Die vorgeschlagene Struktur von TNU-9 wurde von Lynne McCusker durch eine
Rietveldverfeinerung bestätigt. Für die Rietveldverfeinerung (Abbildung 6-6) wurden
Puvlerdiffraktionsdaten von einer kalzinierten Probe verwendet, welche am Synchrotron in
Daresbury durch Paul A. Wright aufgenommen wurden.
Die R-Werte konvergierten zu FR =0.057 und wpR =0.156. Die grösseren Differenzen
zwischen dem berechneten Pulverdiagramm der verfeinerten Struktur und dem gemessenen
Pulverdiagramm werden durch eine Verunreinigung verursacht, welche später als NU-87
identifiziert wurde. Die Atomkoordinaten der TNU-9 Struktur sind in Anhang C aufgelistet.
Mit den verfeinerten TNU-9-Strukturdaten wurden die HRTEM-Bilder mit dem Programm
JEMS (Stadelmann A., 1987) simuliert (Abbildung 6-2 bis Abbildung 6-4). Die simulierten
Bilder zeigen zwar mehr Details als das HRTEM-Bild, aber die Hauptmerkmale stimmen
jeweils überein. Im HRTEM-Bild der Zone [0 1 0] scheint die Probe leicht verkippt
aufgenommen worden zu sein, weshalb die grossen runden Kanäle in elliptischer Form
abgebildet wurden. Dass die simulierten HRTEM-Bilder mit den experimentellen gut
übereinstimmen ist ein zusätzlicher Hinweis für die Richtigkeit der verfeinerten
TNU-9 Struktur.
89
Abbildung 6-7: Stereoskopisches Bildpaar einer Schicht der TNU-9-Struktur. Zur besseren Übersicht wurden die Sauerstoffatome weggelassen. Die beiden Typen von Kanälen entlang der [0 1 0]-Richtung sind mit A und B gekennzeichnet und in Abbildung 6-8A respektive Abbildung 6-8B grösser zu sehen. Ein zusätzlicher Kanal (schwarz markiert) verläuft in der xz-Ebene
Abbildung 6-6: Rietveldverfeinerung von TNU-9. Um die Details besser erkennen zu können wurde der 2θ-Bereich zwischen 20-50° mit Faktor vier skaliert und der 2θ-Bereich von 10°-20°, in welchem die Peaks der Verunreinigung gut zu erkennen (grosse Differenz) sind, wurde herausgeschnitten und vergrössert. In dunkelgrau ist das beobachtete, in hellgrau das berechnete Pulverdiagramm und in schwarz deren Differenz dargestellt.
90
Abbildung 6-8: Stereoskopisches Bild der beiden Kanaltypen entlang der [0 1 0]-Richtung. Die Bildbezeichnung A und B entspricht den Markierungen in Abbildung 6-7.
Die Projektion entlang [0 1 0] (Abbildung 6-7) ist jener von ZSM-5 (MFI Gerüsttyp) sehr
ähnlich, die Projektionen entlang [1 0 0] und [0 0 1] sind aber offensichtlich unterschiedlich
zu denjenigen von ZSM-5. Die Struktur weist ein dreidimensionales Kanalsystem auf. Zwei
Typen von gerade durchgehenden Kanälen (Typ A und Typ B in Abbildung 6-7), welche
durch Ringe aus 10 T-Atomen gebildet werden, verlaufen entlang der [0 1 0 ]-Richtung.
Die beiden Kanaltypen sind in Abbildung 6-8 separat gezeichnet. Ein weiterer Kanal
verläuft im Zick-Zack (dunkel markiert in Abbildung 6-7), innerhalb der in Abbildung 6-7
gezeigten Schicht von TNU-9.
6.5 Qualität der Phaseninformation
Viele Faktoren (Linsenfehler, Probendrift, Probendicke, Instabilität von Zeolithen im
Elektronenstrahl, etc.) können die Qualität der Phaseninformation beeinträchtigen. Die
Phasen welche aus den HRTEM-Bildern berechnet wurden, werden deshalb nur teilweise
korrekt sein.
Weil die Phaseninformation wesentlich zur erfolgreichen Strukturlösung beitrug, ist es
interessant zu wissen wie gut die Qualität der Phaseninformation war. Anhand der
Atomkoordinaten der verfeinerten Struktur wurden deshalb mit dem Programm
CrystalDiffract die Amplituden und Phasen der Strukturfaktoren berechnet und mit den
91
Phasen aus den HRTEM-Bildern (vgl. Abbildung 6-9) verglichen. Dazu wurden die
Reflexe anhand des Streuwinkels 2θ in Klassen von jeweils 2.5° eingeteilt. Die Klasse 2.5°
enthält demzufolge die Reflexe mit 0°<2θ<=2.5°, die Klasse 5° jene Reflexe mit
2.5°<2θ<=5° und so weiter. Für die Beugungswinkel 2θ kleiner als 12.5° waren in jeder
Klasse mehr als 75% der Phasen korrekt, in den Klassen bei grösseren Streuwinkeln (2θ >
12.5°) waren nur noch zwischen 55% und 67% der Phasen korrekt.
Werden nur die Daten bis zu einem maximalen Streuwinkel betrachtet, wird im Folgenden
von einem limitierten Datensatz gesprochen. Ein limitierter Datensatz mit maximalem
Streuwinkel von 10° würde also nur die Reflexe zwischen 0° und 10° enthalten. In einem
limitierten Datensatz mit Streuwinkeln 2θ kleiner als 12.5° waren noch 78.9% der Phasen
korrekt. Wurden alle Phaseninformation aus den HRTEM-Bildern berücksichtigt waren nur
noch 65.1% der Phasen korrekt, wobei die Streuwinkel 2θ kleiner als 30° waren für alle
Reflexe von denen Phaseninformation vorlag.
Abbildung 6-9: Qualität der Phasen in Abhängigkeit des Beugungswinkels (λ=0.80124Å).
92
Die Qualität der Phasen wurde auch in Abhängigkeit der Röntgenstrukturfaktoramplitude
betrachtet. Die Phasen der 12 stärksten Reflexe waren korrekt. Die F -limitierten
Datensätze, in welchem nur die starken Reflexe, bis zu einem vorgegebenen Grenzwert
vorkommen, zeigen ein eindeutiges Bild (Abbildung 6-10). Werden nur die Reflexe mit
Strukturfaktoramplituden über 900 einbezogen sind noch alle Phasen der Reflexe korrekt.
Wird die Limite bei einer Strukturfaktoramplitude von 500 gesetzt sind noch 82.1% der
Phasen korrekt und wenn alle Reflexe des Phasensatzes einbezogen werden sind noch
65.1% der Phasen korrekt.
Es sind also die starken Reflexe von welchen korrekte Phaseninformation vorlag. Obwohl
35% der Phasen falsch waren, hat die Phaseninformation wohl entscheidend zur
























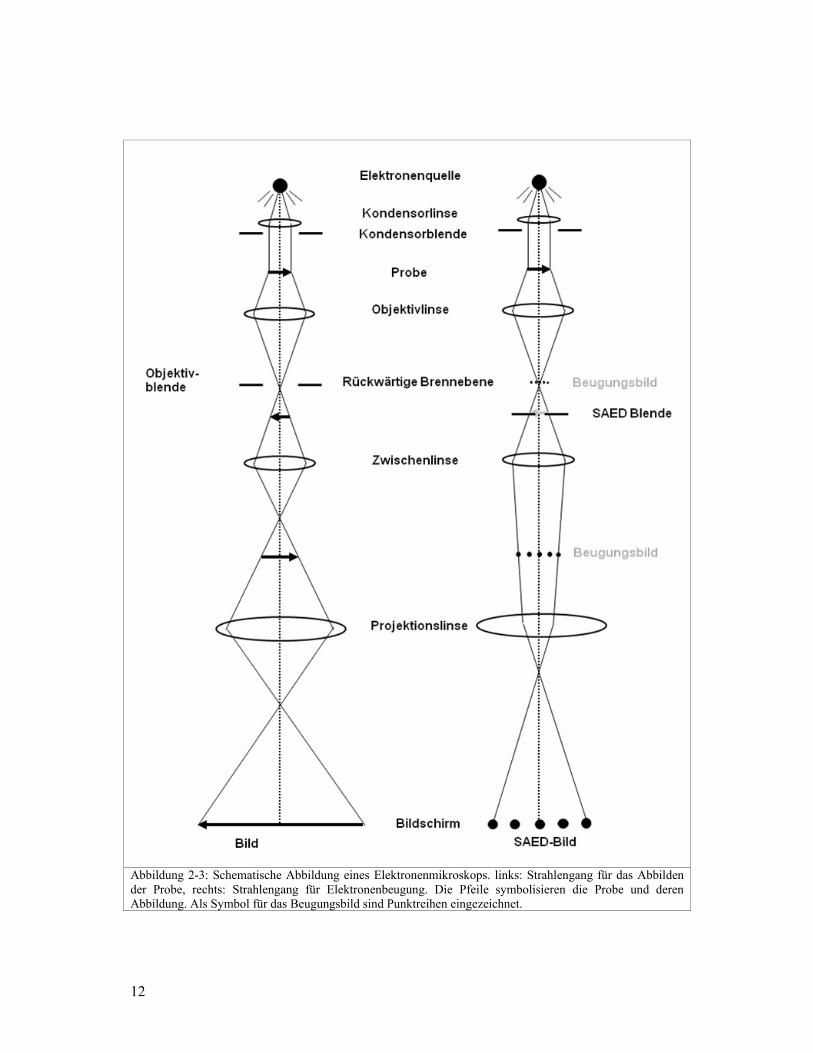










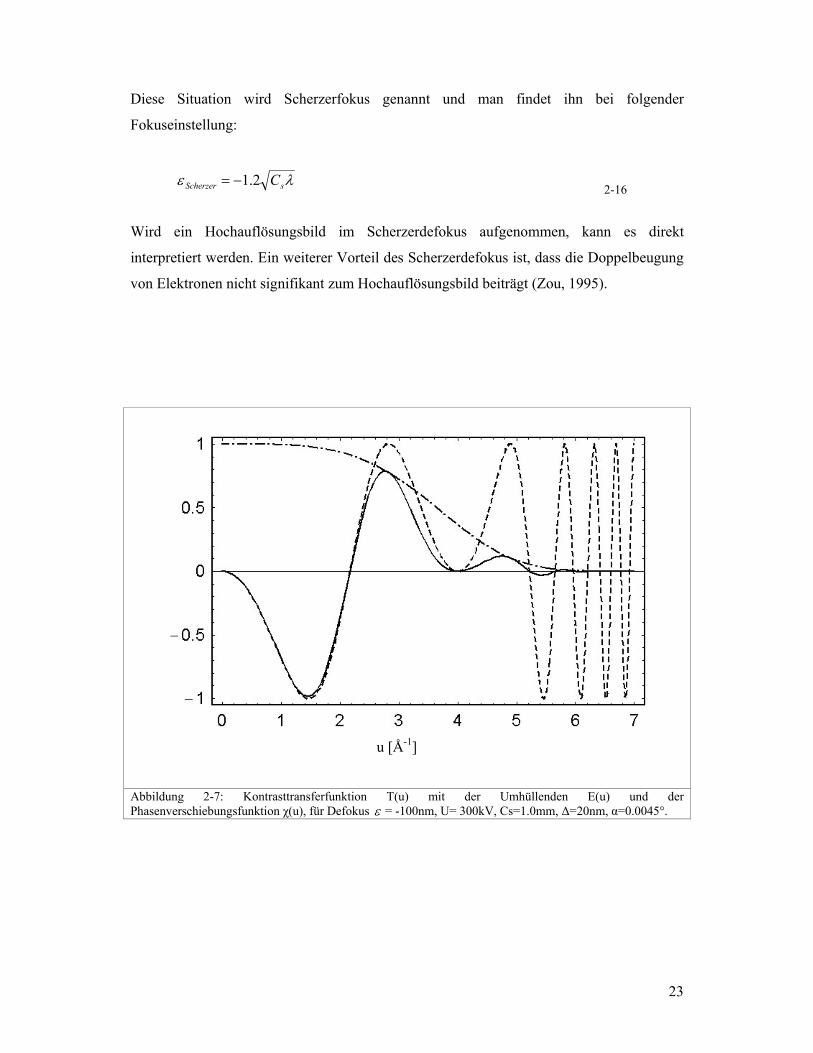
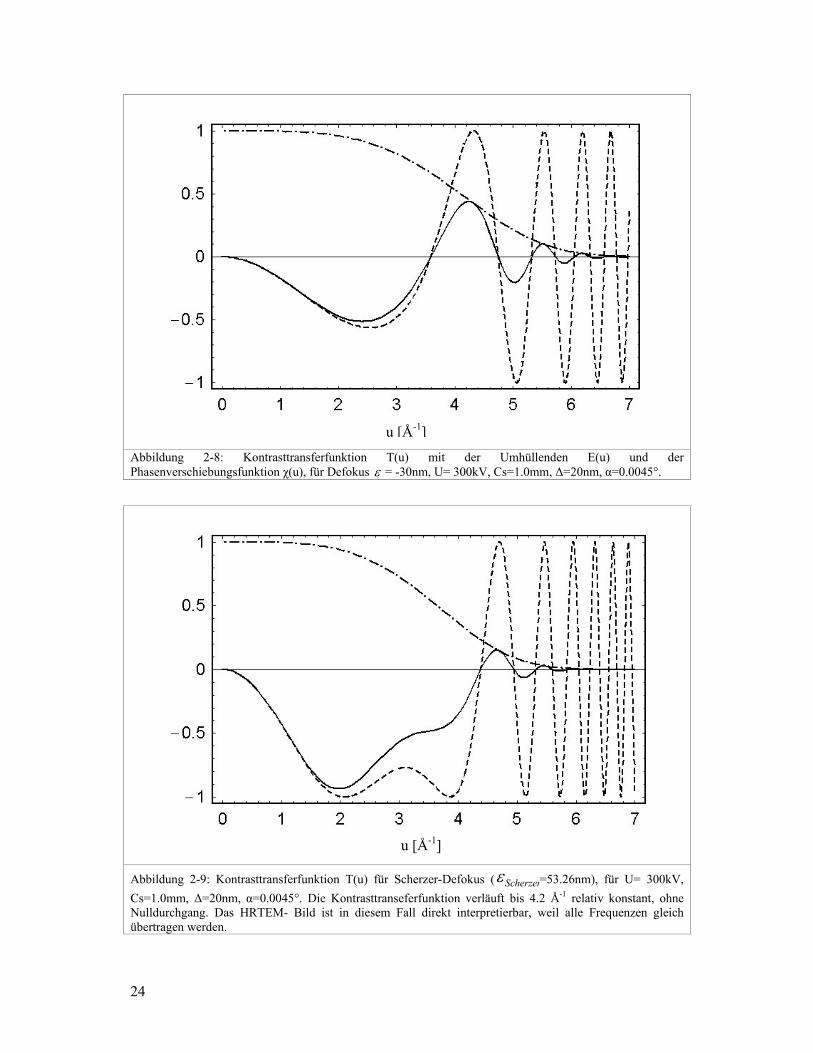
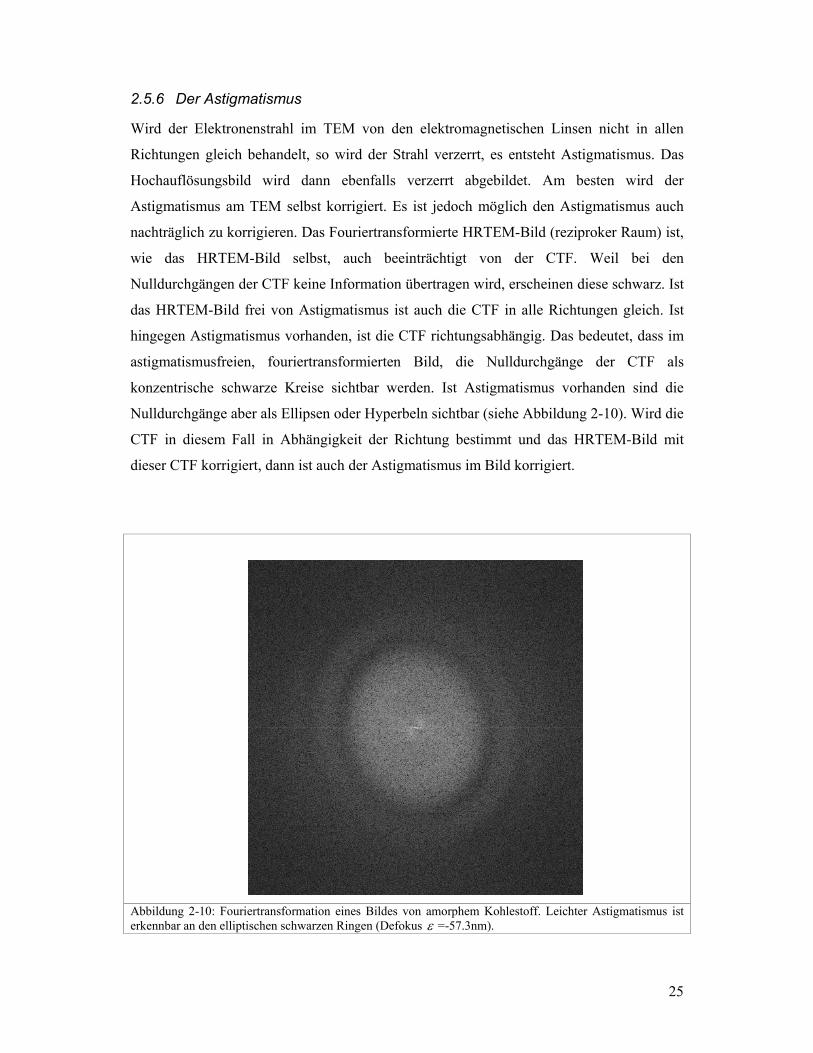














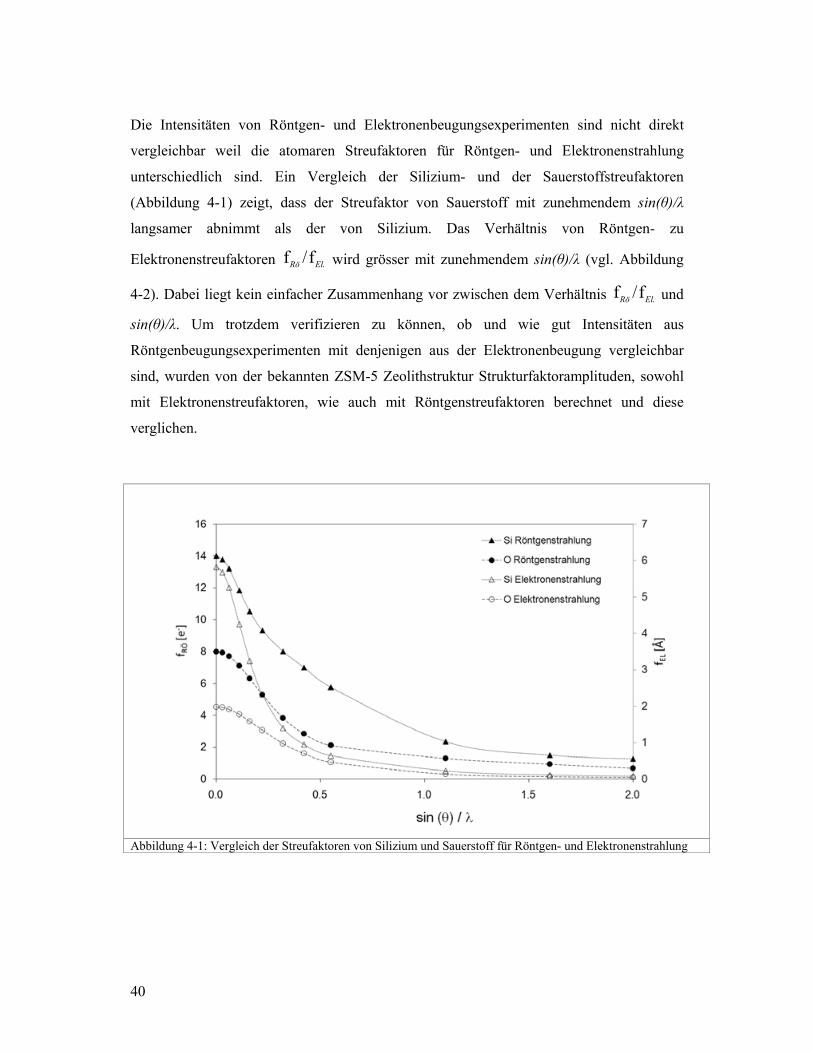
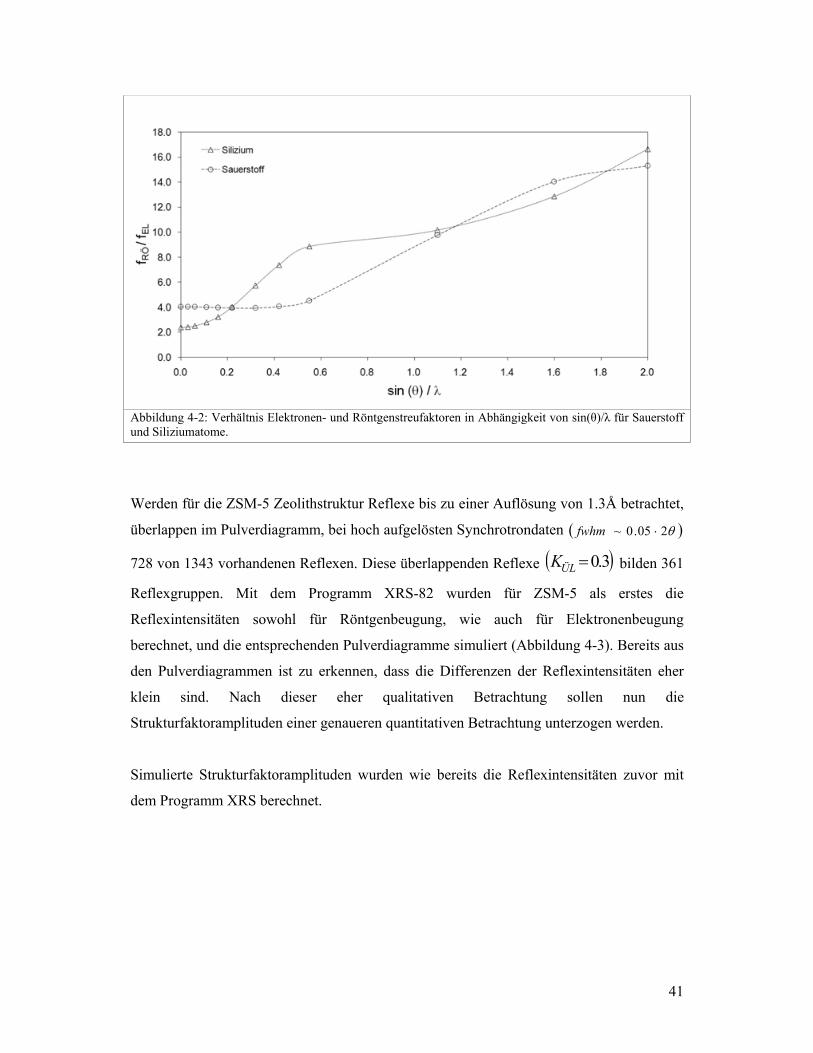

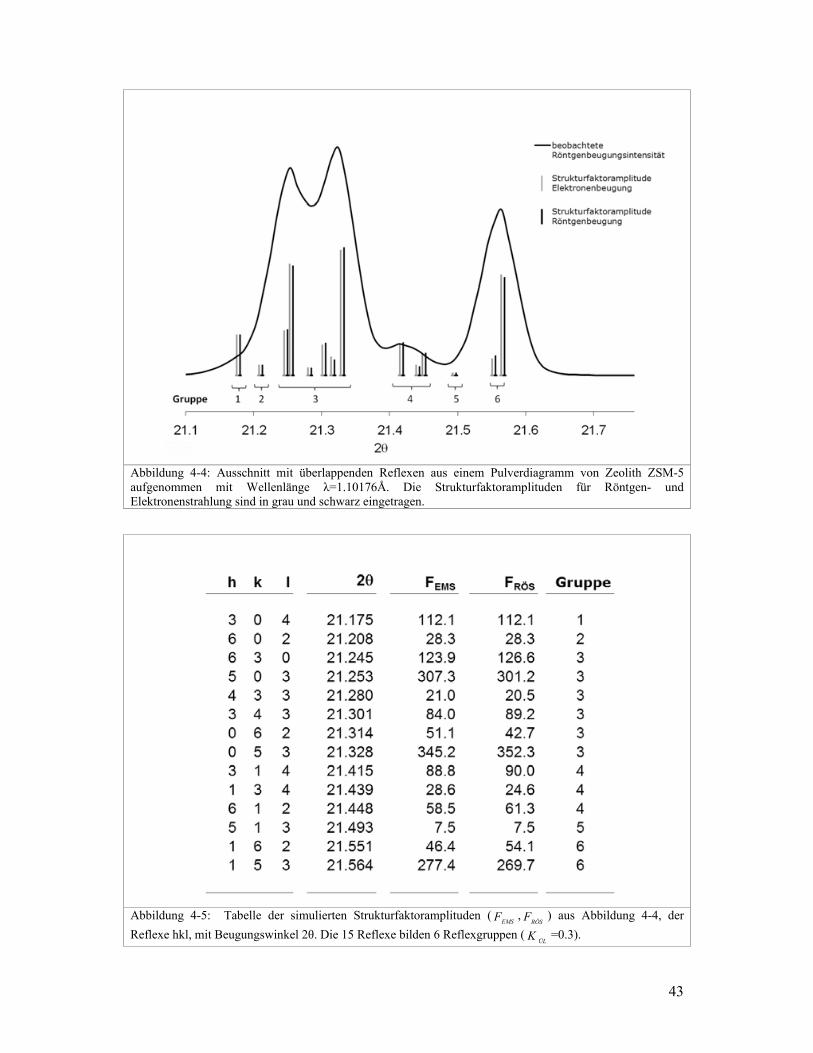

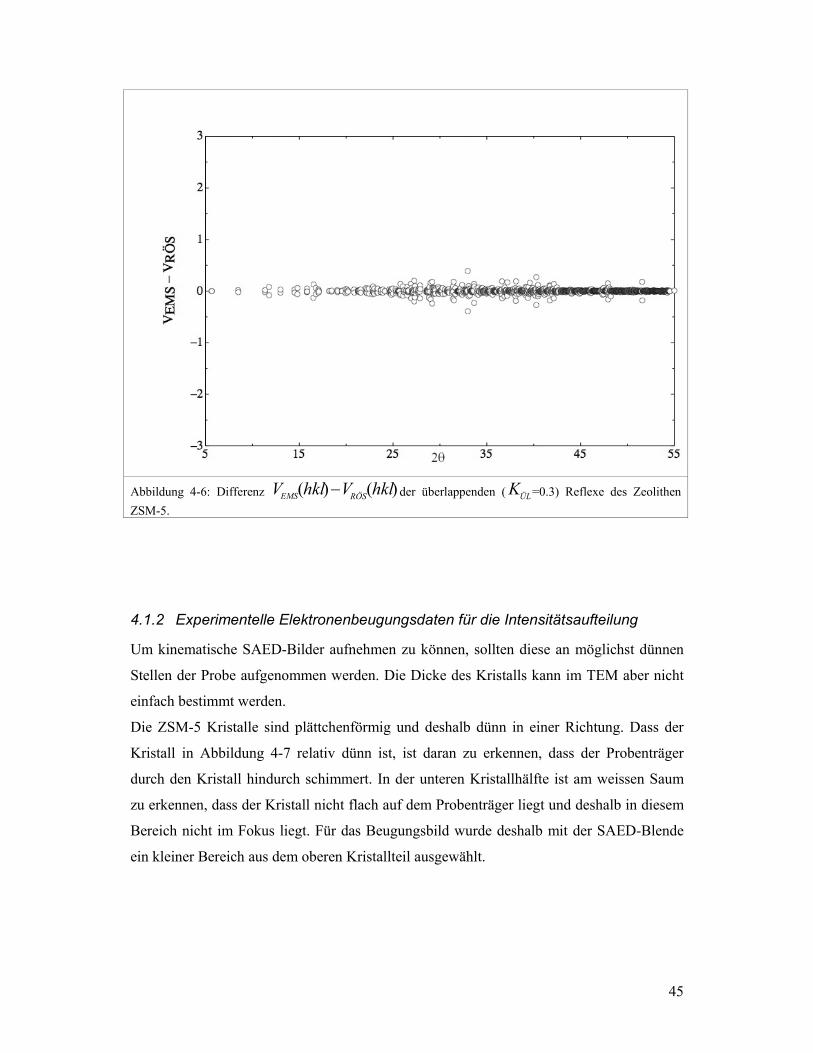
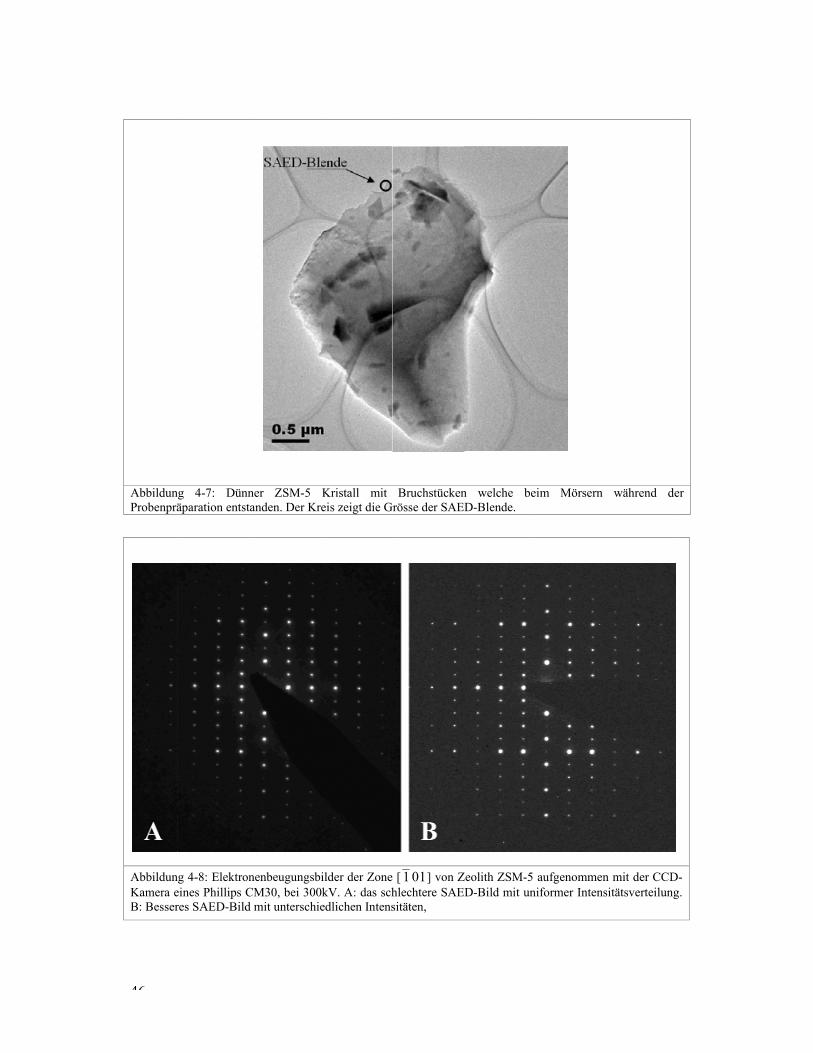

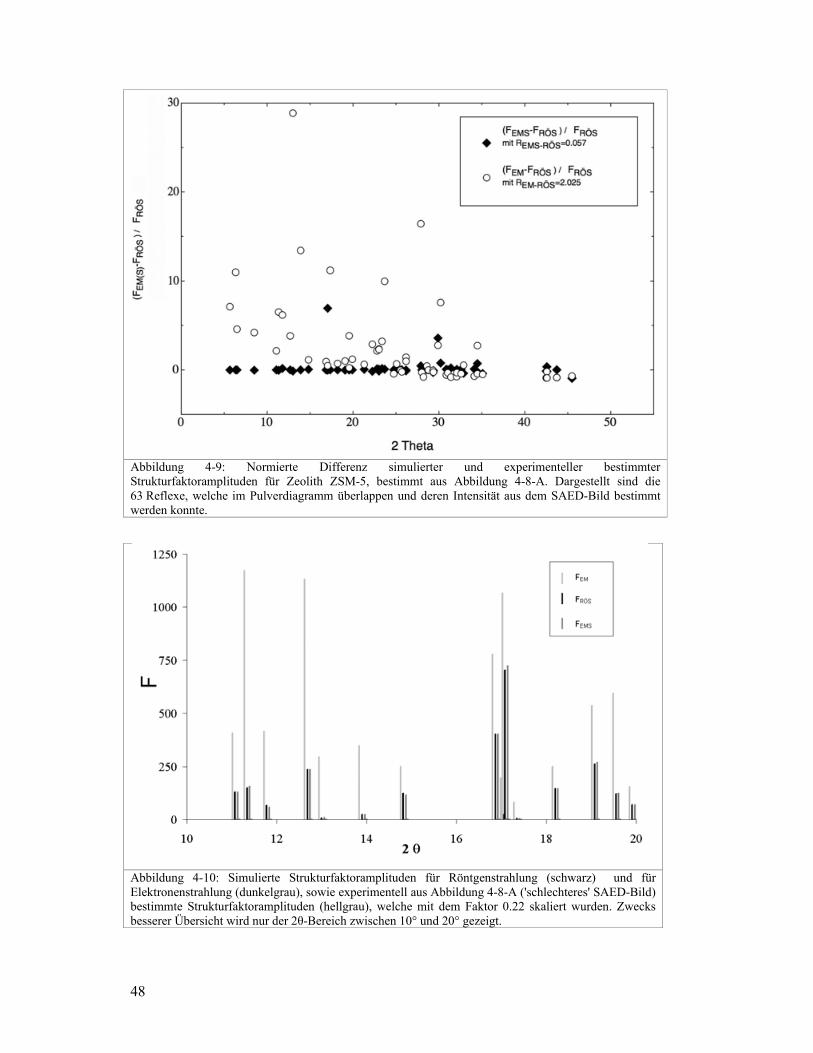
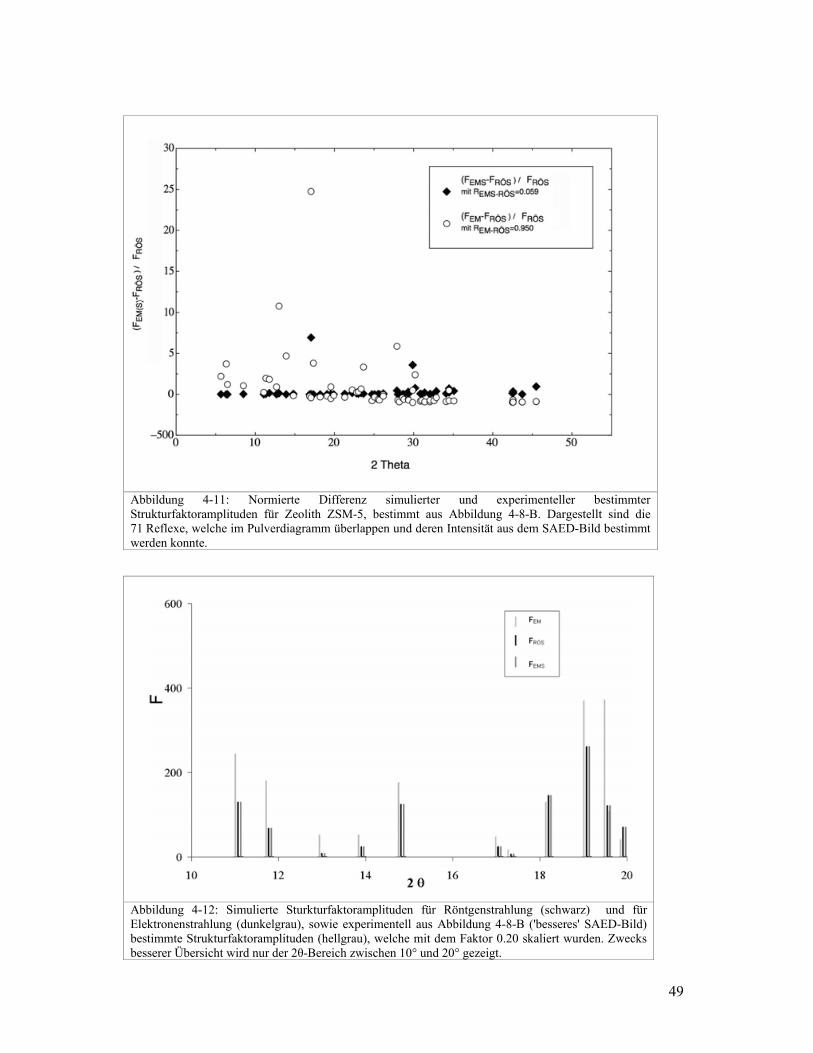


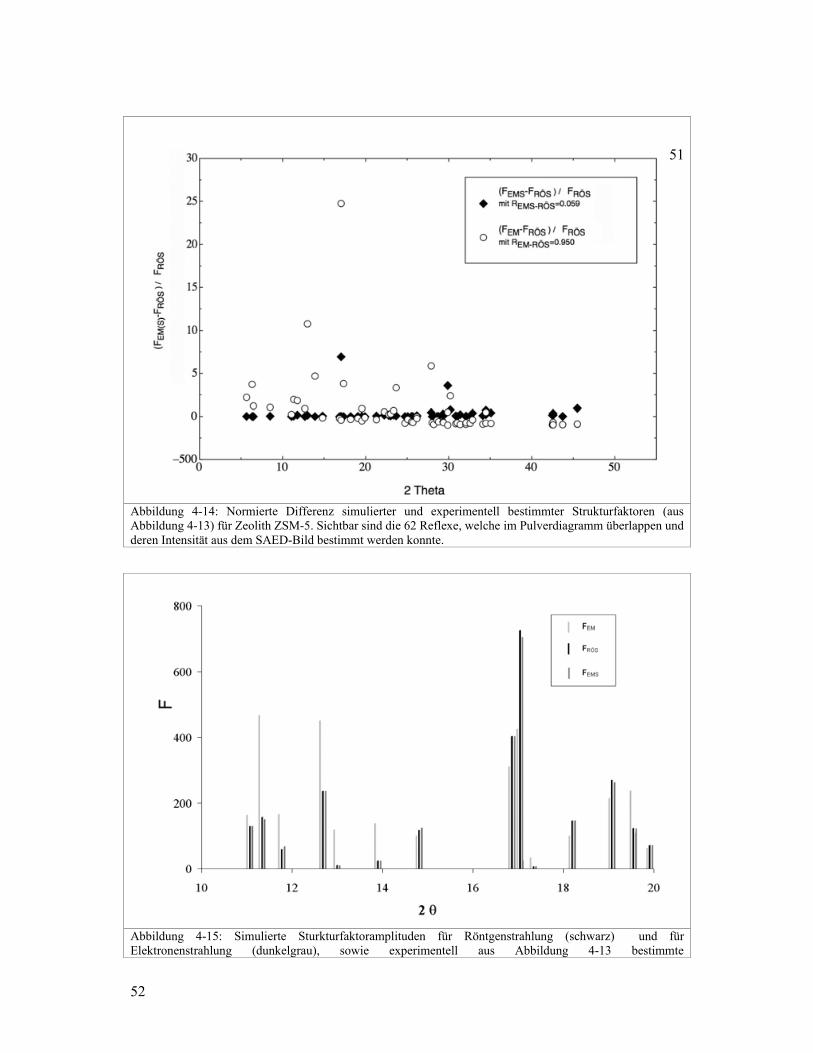



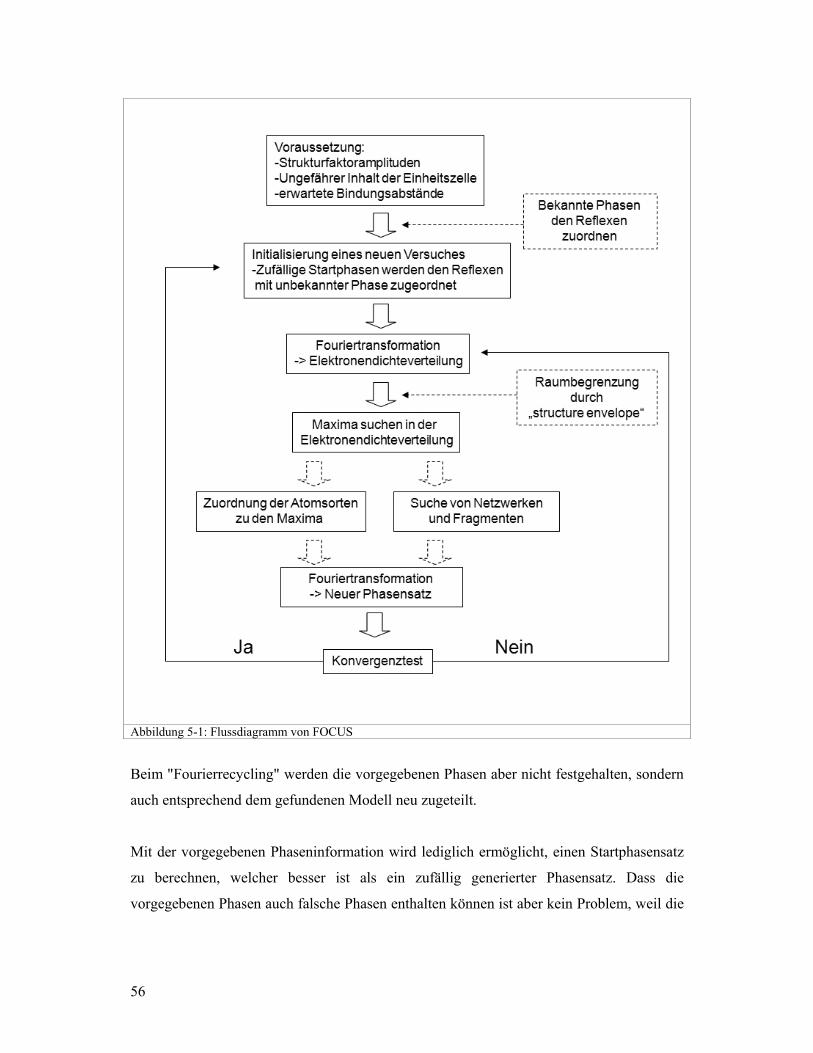



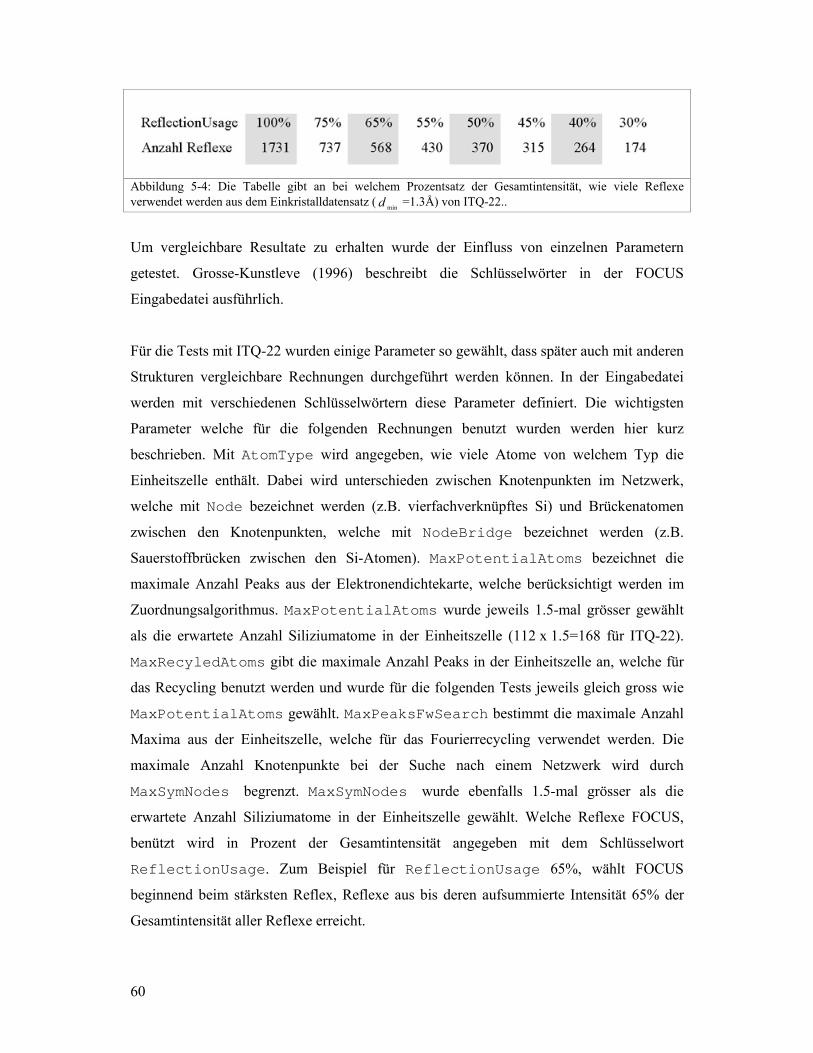


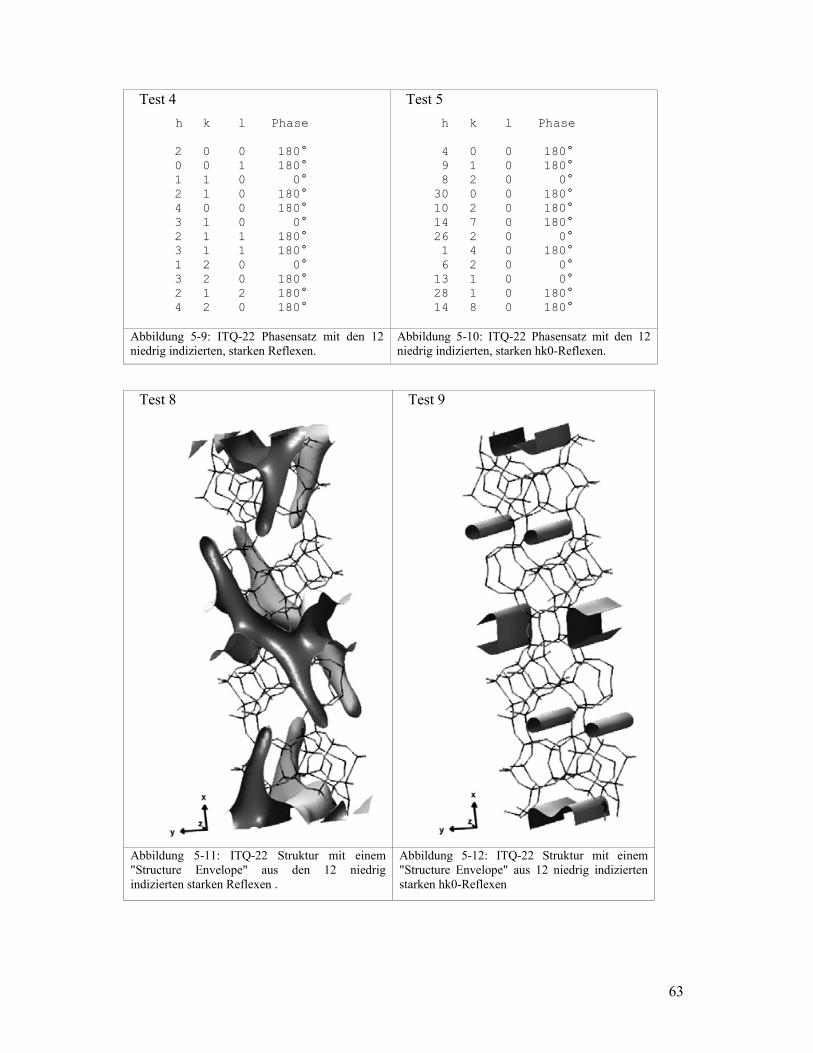

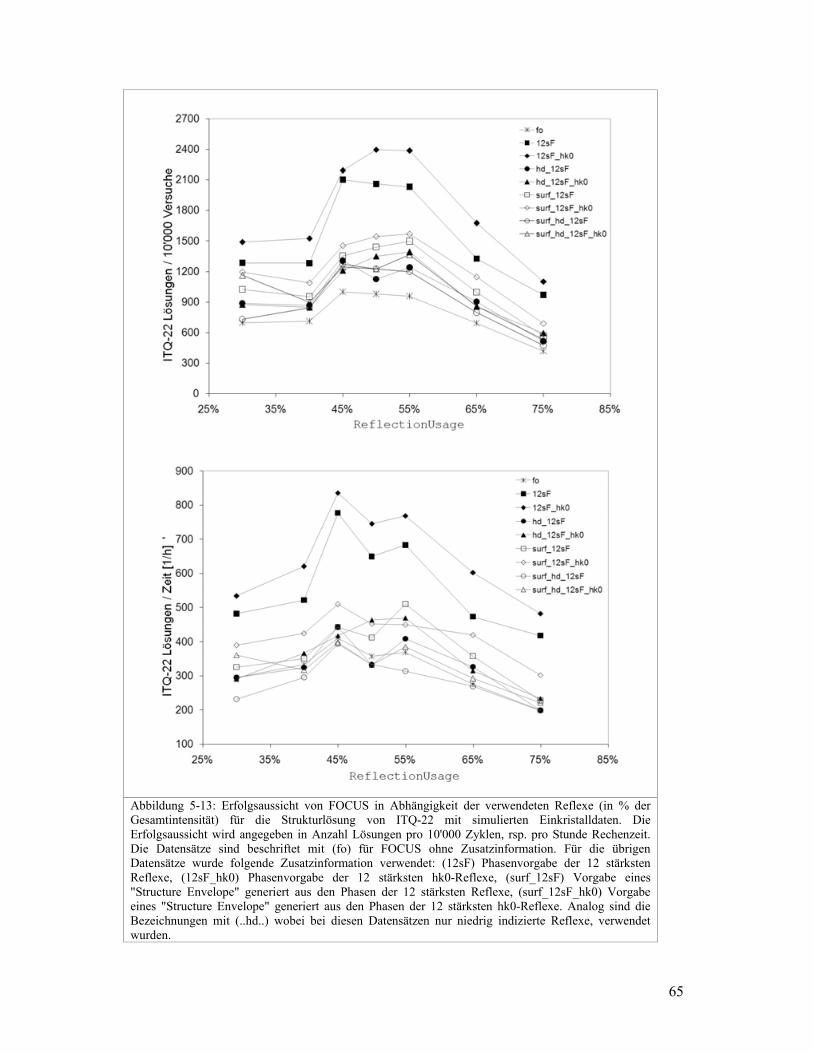
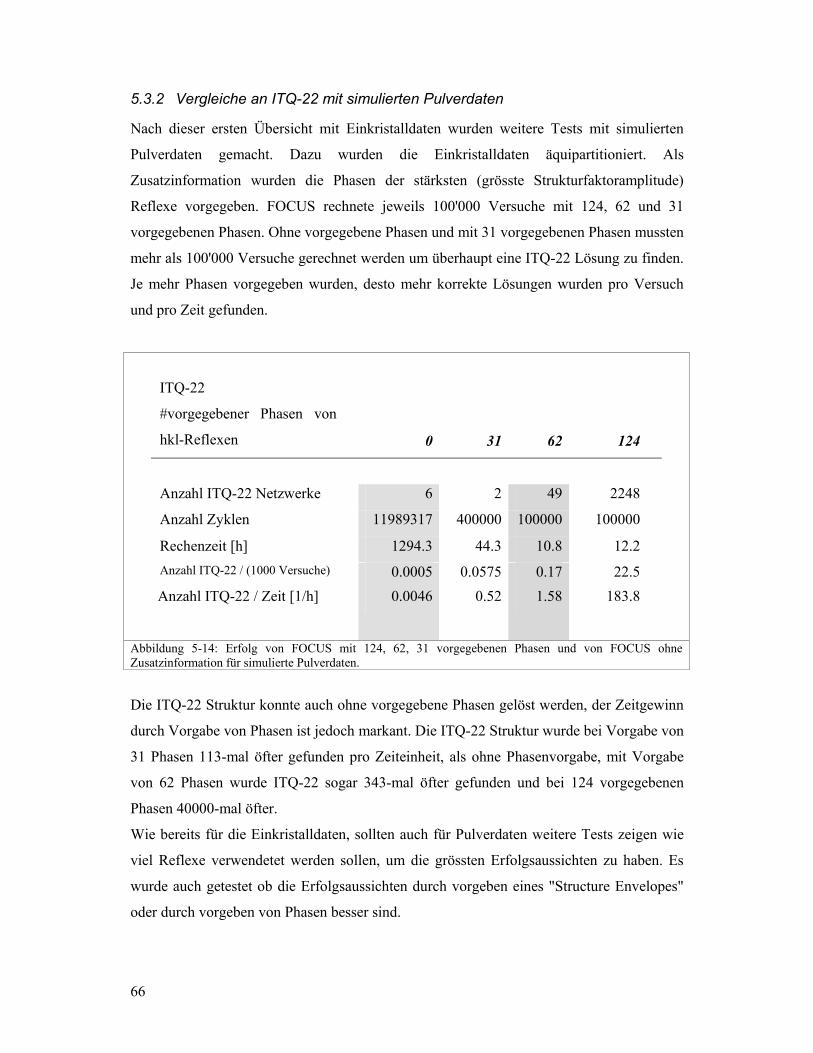
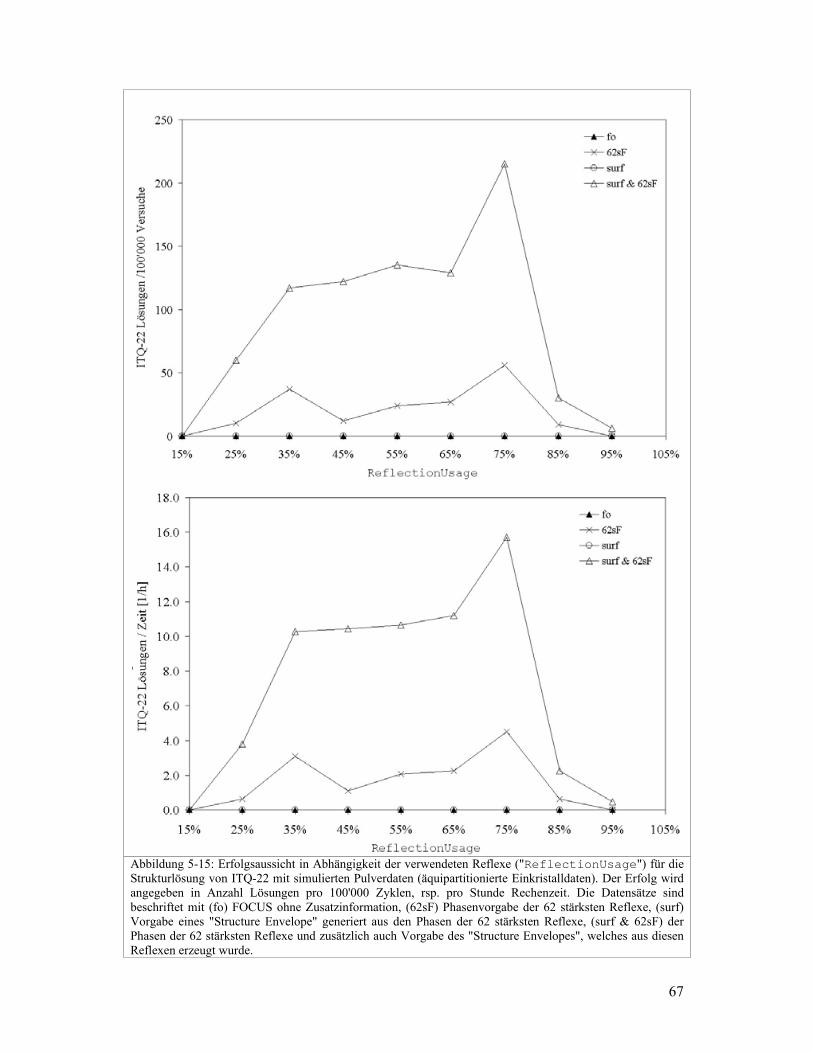

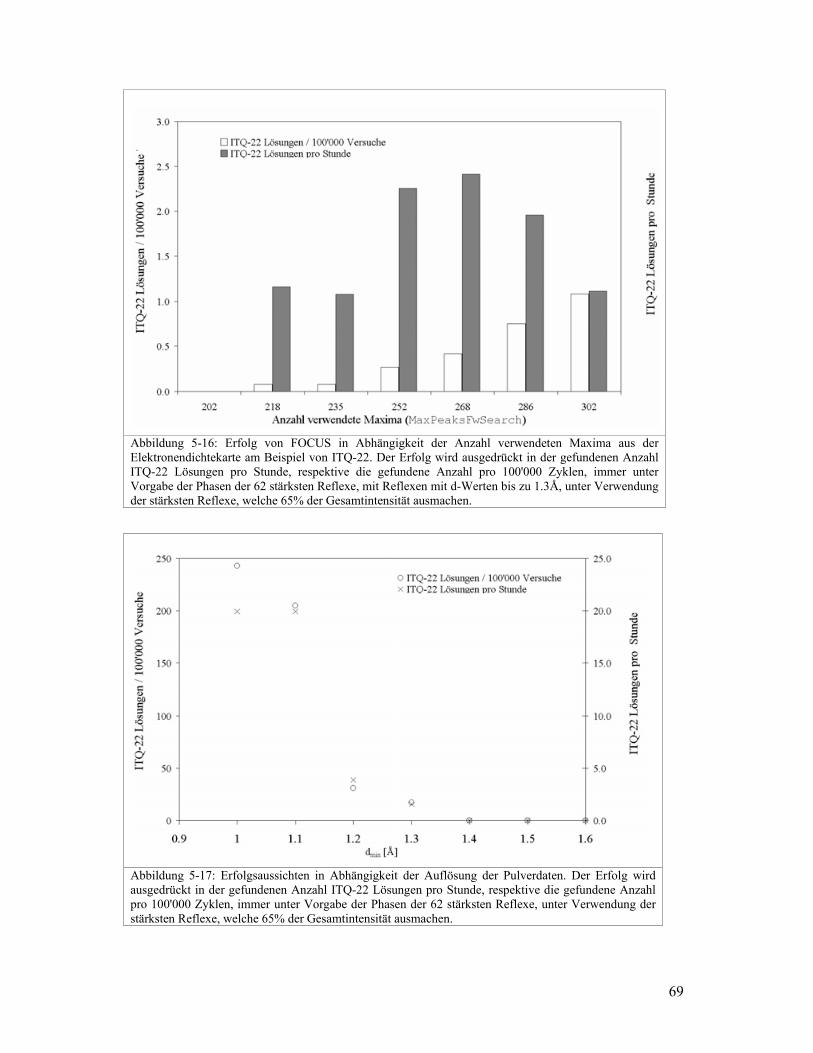

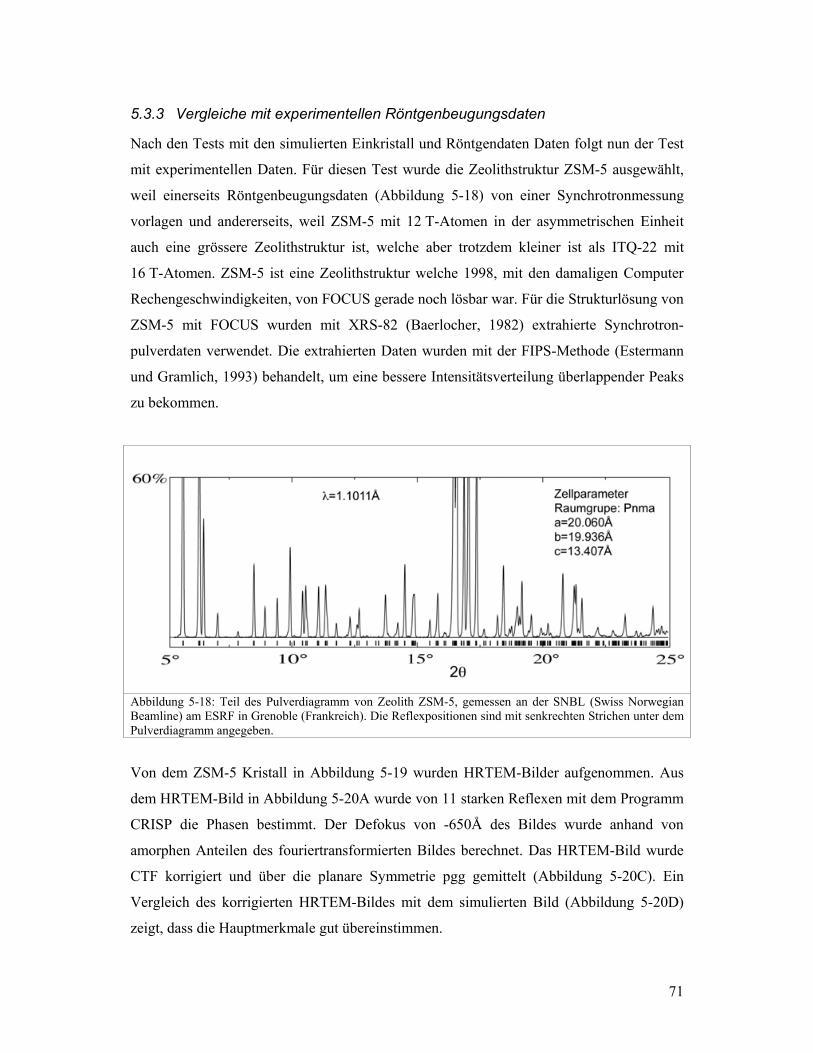
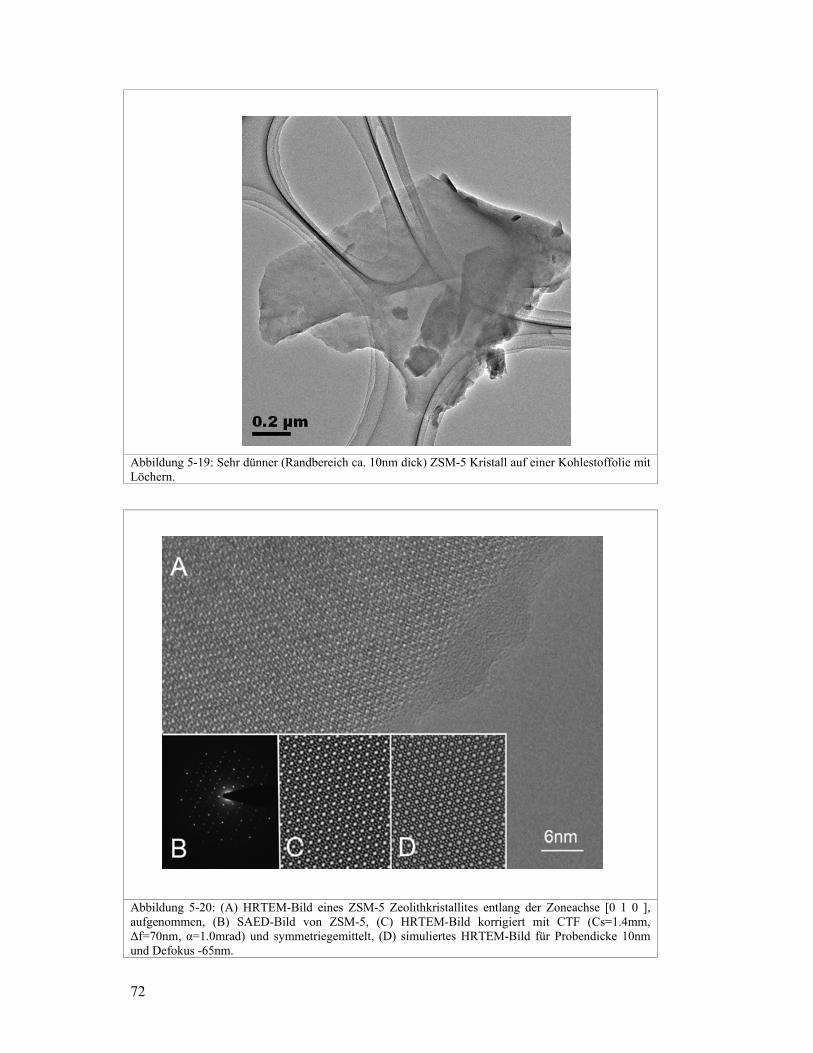

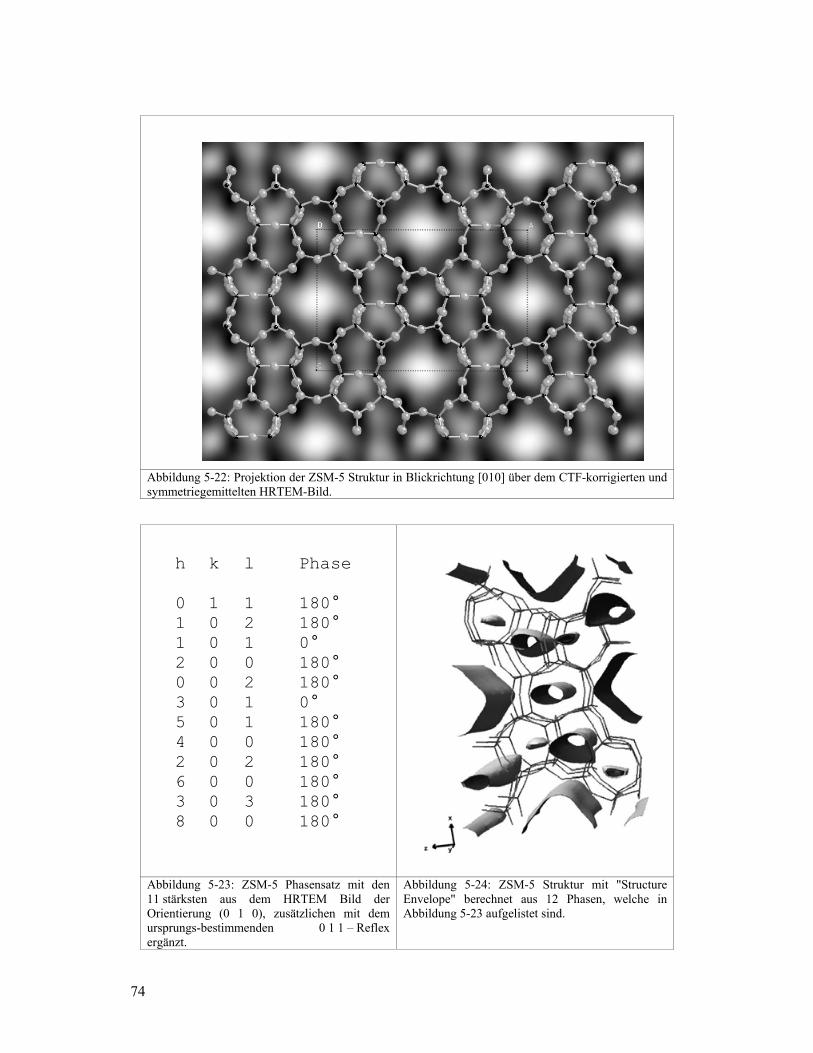

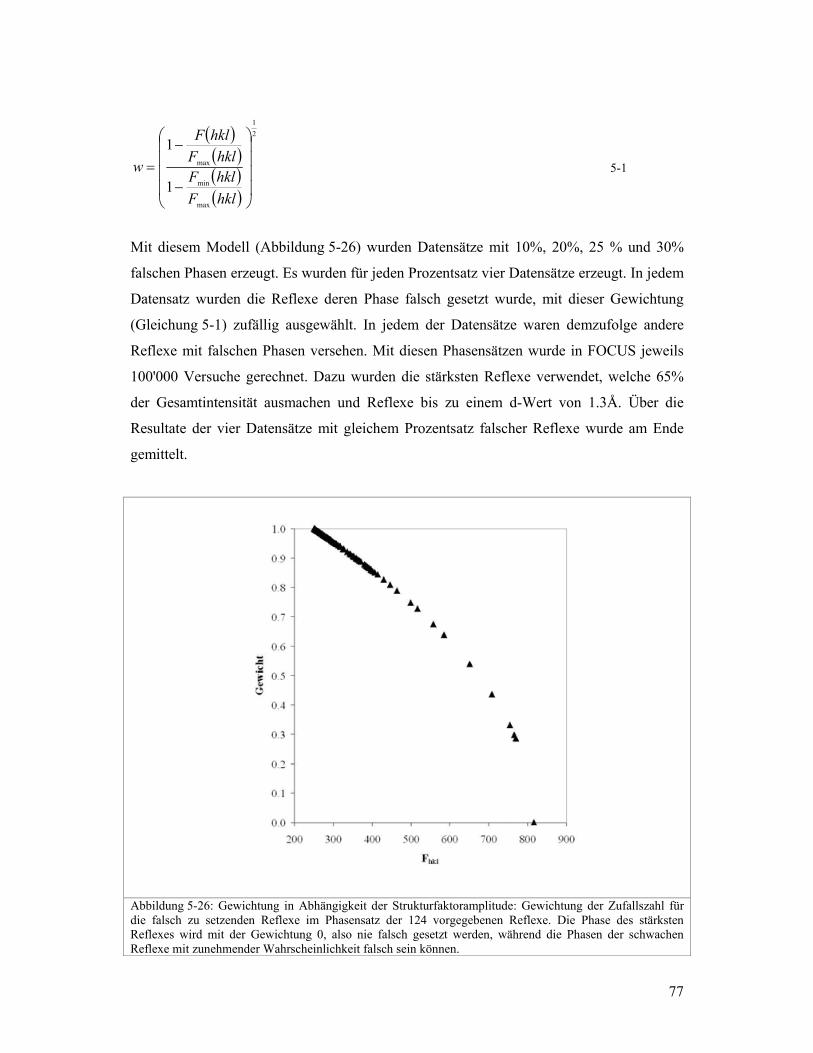
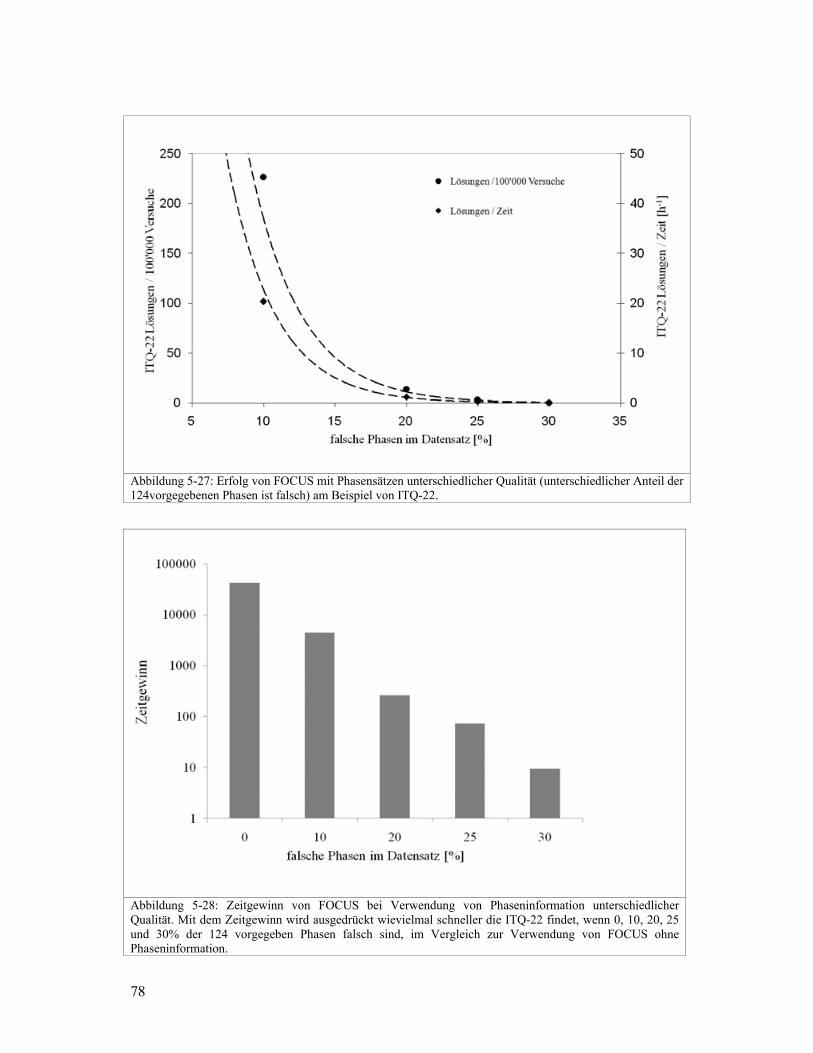



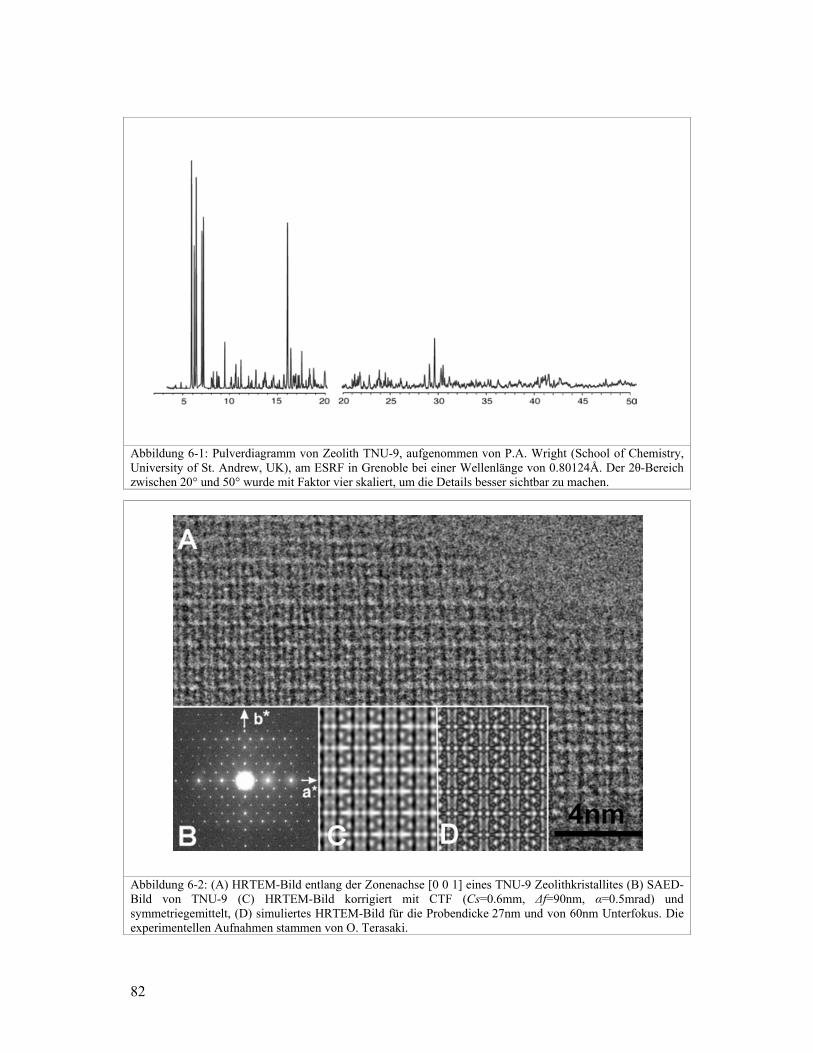






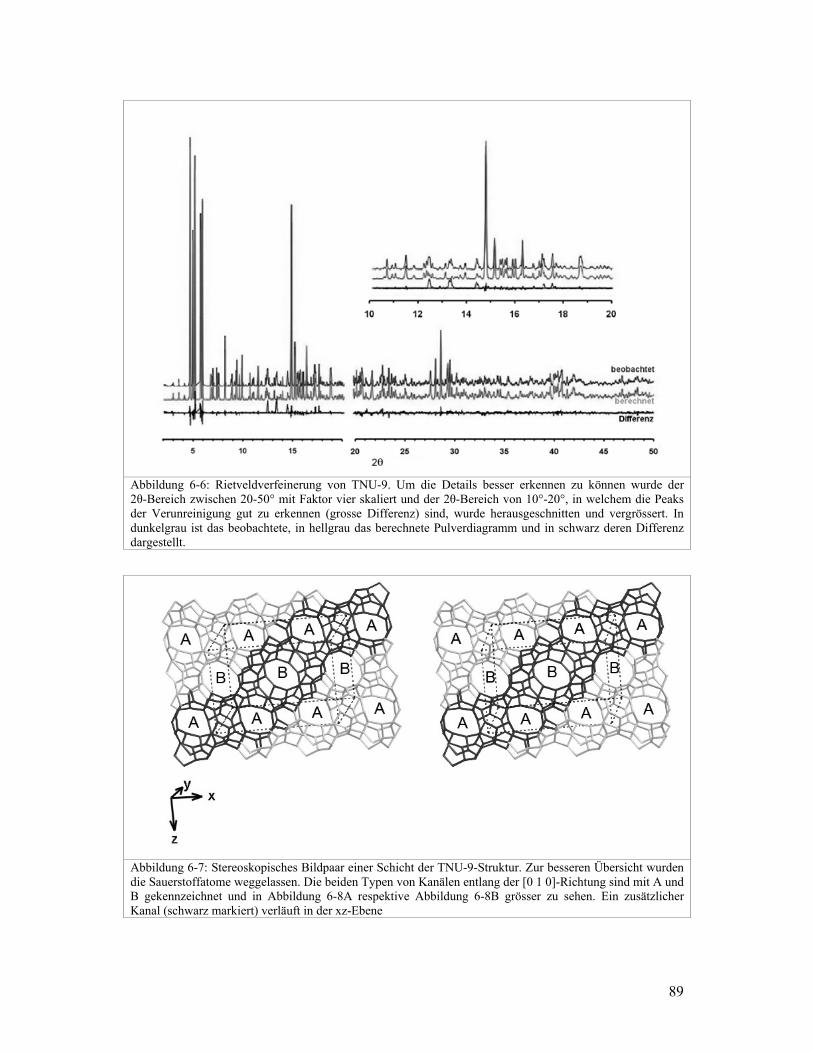

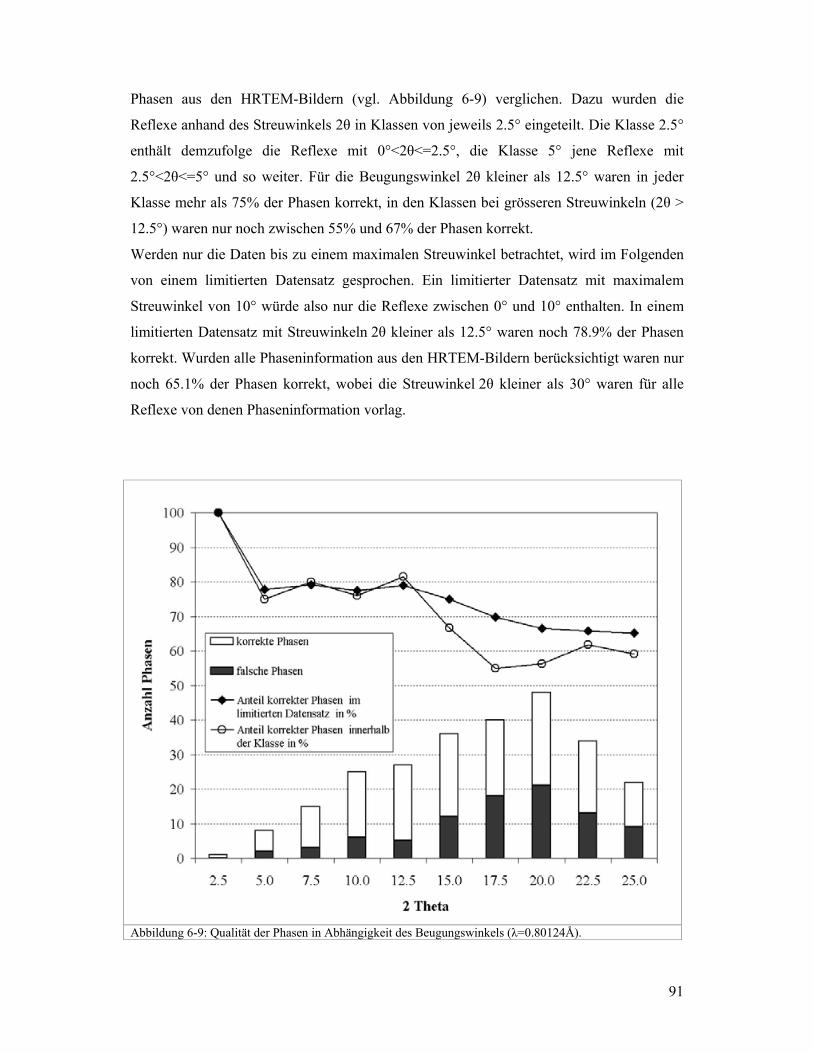
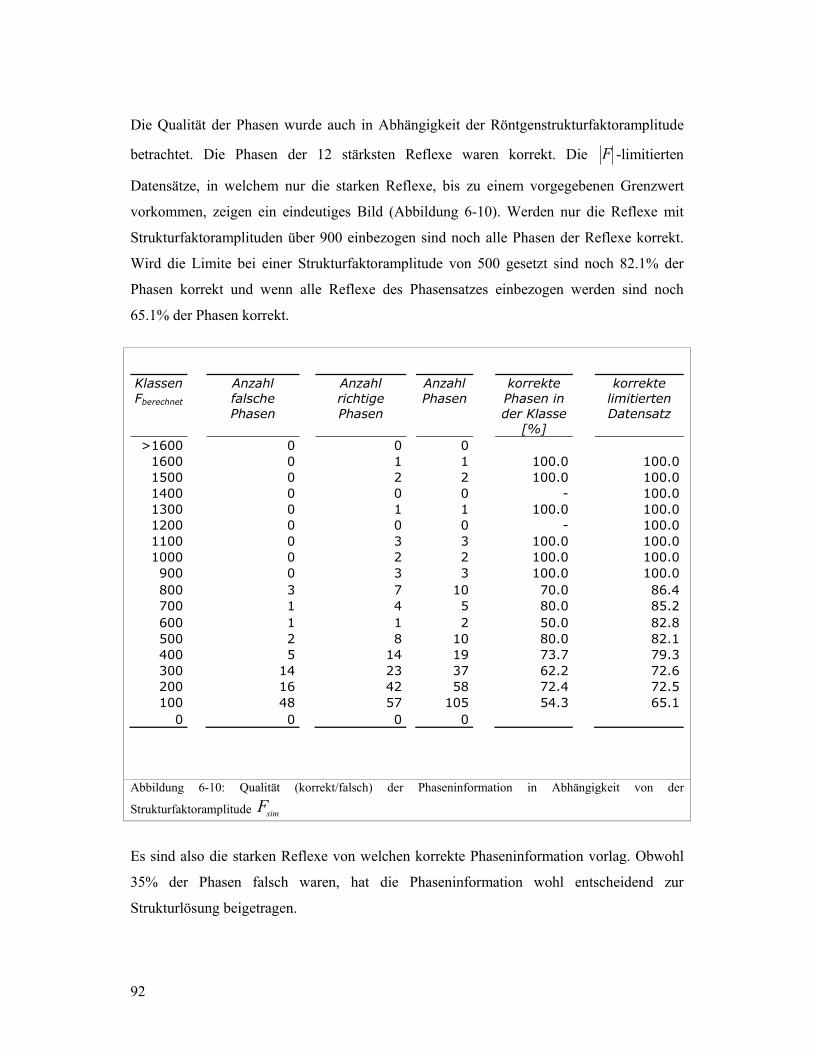
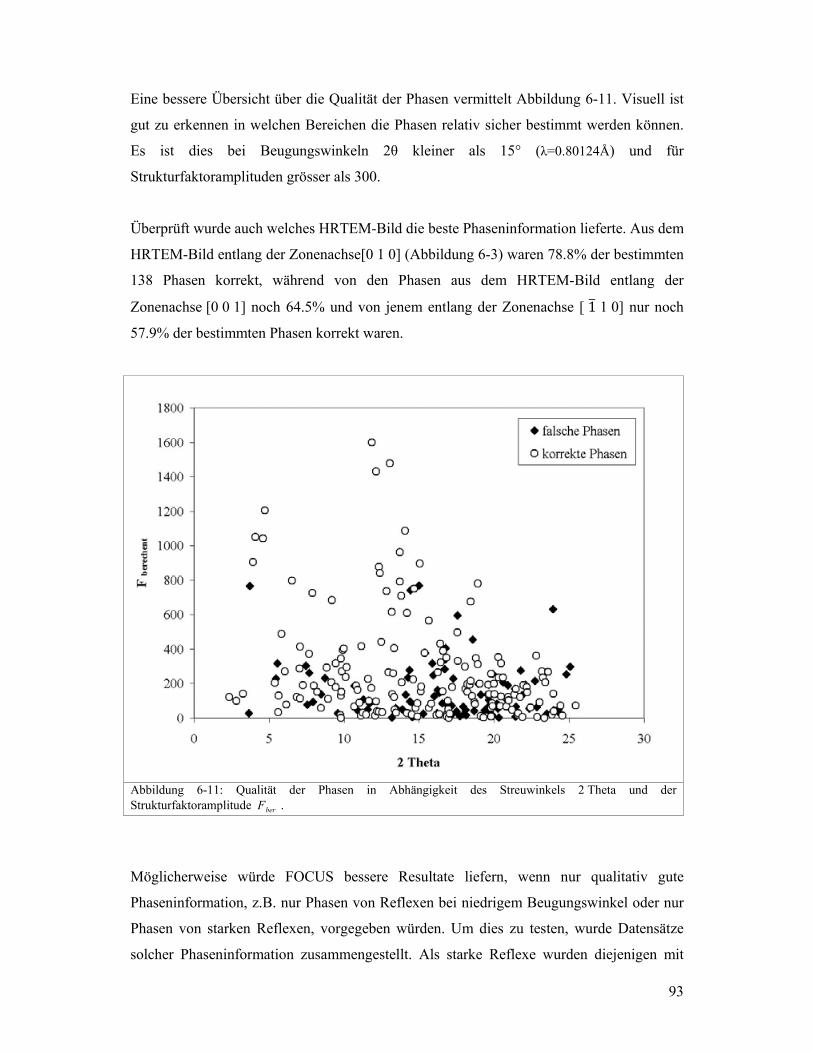







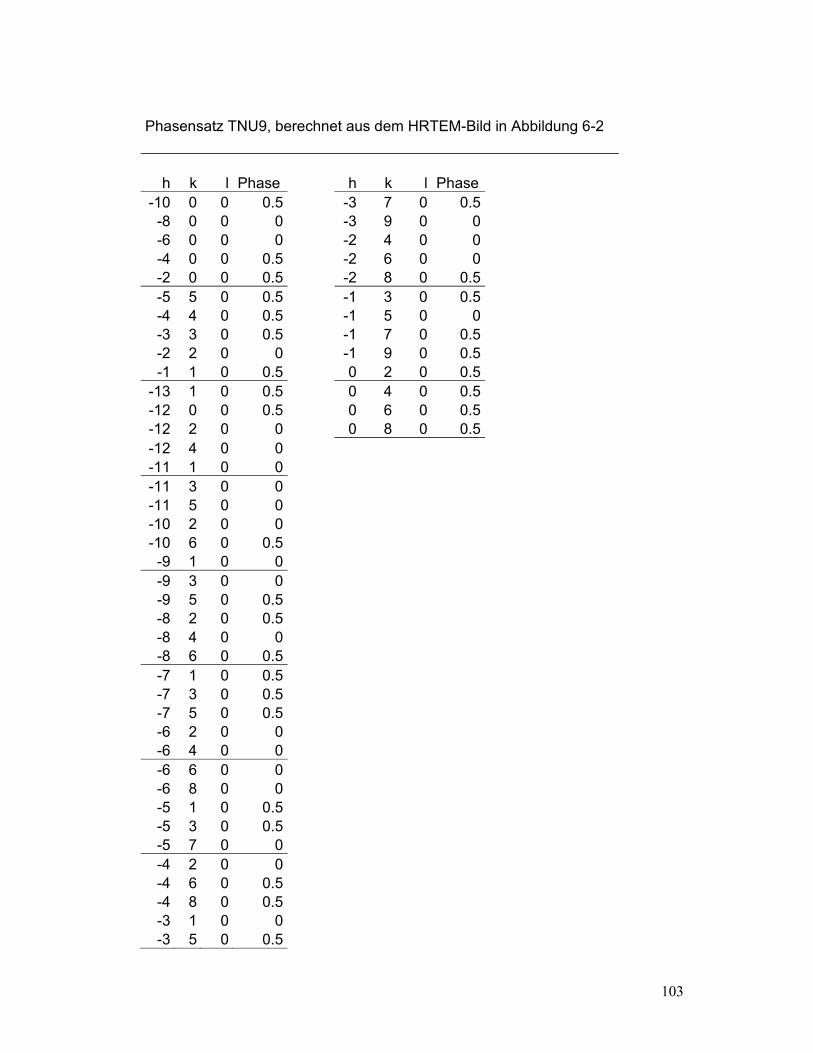



















![Tissue Intraoperative Factors€¦ · Sprüssel et al, BioTechniques 2004 B A x-fache Änderung (normalisierte Intensität) HIF-1α rel. Expression time [min] 5 8 10 12 15 20 25 30](https://static.documents.pub/doc/80x56/5ec62e150613c4741e42bbf1/tissue-intraoperative-factors-sprssel-et-al-biotechniques-2004-b-a-x-fache-nderung.jpg)



![OXER8_Manual de Usuario Advanced 4035 Reflexe [1]](https://static.documents.pub/doc/80x56/577cc4971a28aba71199d4bb/oxer8manual-de-usuario-advanced-4035-reflexe-1.jpg)