Distinct Role of Calmodulin and Calmodulin-dependentProtein Kinase-II in Lipopolysaccharide and TumorNecrosis Factor-�-mediated Suppression of Apoptosis andAntiapoptotic c-IAP2 Gene Expression in HumanMonocytic Cells*□S
Received for publication, May 5, 2005, and in revised form, September 8, 2005 Published, JBC Papers in Press, September 9, 2005, DOI 10.1074/jbc.M504971200
Sasmita Mishra‡1, Jyoti P. Mishra‡1, Katrina Gee‡2, Dan C. McManus§3, Eric C. LaCasse§, and Ashok Kumar‡¶�4
From the Departments of ¶Pathology and Laboratory Medicine and ‡Biochemistry, Microbiology, and Immunology, University ofOttawa, Ottawa, Ontario K1H 8M5, �Infectious Disease and Vaccine Research Centre, Research Institute, Children’s Hospital ofEastern Ontario, Ottawa, Ontario K1H 8L1, and §Aegera Therapeutics Inc., Ottawa, Ontario K1H 8L1, Canada
Exposure of phagocytic cells to bacterial endotoxin (lipopolysac-charide; LPS) or inflammatory cytokines confers antiapoptotic sur-vival signals; however, in the absence of the appropriate stimulus,monocytes are programmed to undergo apoptosis. Macrophagesurvival may thus influence inflammatory and immune responsesand susceptibility to microbial pathogens. Herein, we demonstratethat LPS and the proinflammatory cytokine, tumor necrosis fac-tor-� (TNF-�), enhance monocytic cell survival through the induc-tion of the antiapoptotic c-IAP2 gene in a human promonocyticTHP-1 cell line.We also investigated the role of upstream signalingmolecules including the mitogen-activated protein kinases, phos-phatidylinositol 3-kinase, and the calcium signaling pathways in theregulation of c-IAP2 expression and eventual survival of monocyticcells. Our results suggest that LPS and TNF-�-induced c-IAP2expression was regulated by calmodulin (CaM) through the activa-tion of calmodulin-dependent protein kinase-II (CaMKII). In addi-tion, CaM and CaMKII regulated c-IAP2 expression in LPS- andTNF-�-stimulated cells through NF-�B activation. Moreover, theCaM/CaMKII pathway also regulated LPS- and TNF-�-mediatedinhibition of apoptosis in these cells. Taken together, these resultssuggest that LPS- and TNF-�-induced c-IAP2 expression and itsassociated antiapoptotic survival signals in THP-1 cells are regu-lated selectively by CaM/CaMKII through NF-�B activation.
Apoptosis is a fundamental event in developmental and homeostaticprocesses of multicellular organisms. Caspase activation is required toaccomplish apoptosis by either extrinsic death receptor or intrinsicmitochondria mediated pathways (1, 2). One of the major regulators ofthe caspases and suppressors of apoptosis are the inhibitors of apoptosis
proteins (IAPs).5 Initially discovered in baculovirus system, there arenow eight mammalian homologs of IAPs: XIAP (hILP), NAIP, c-IAP1(HIAP2), c-IAP2 (HIAP1), livin (ML-IAP/KIAP), survivin, Ts-IAP(hILP2), and Apollon (Bruce) (3–8). The IAP family of proteins is dis-tinguished by the presence of 1–3 baculovirus IAP repeat domains at theN-terminal region (6, 7). The c-IAP1 and c-IAP2 genes lie in tandem atchromosome location 11q22, a locus associated with the developmentof leukemia and lymphoma (9). Recently, high levels of c-IAP1 andc-IAP2 expression have been associated with esophageal, cervical, lung,and colorectal cancer (8, 10, 11).Mononuclear phagocytes play a central role in both innate and
acquired immunity and are a major source of inflammatory/growthcytokines following exposure to bacterial endotoxins/lipopolysacchar-ides (LPS). Activation of macrophages by cytokines or a mild bacterialinfection is shown to confer antiapoptotic survival signals (12, 13); how-ever, in the absence of appropriate stimulation, monocytes are pro-grammed to undergo apoptosis (14). Macrophage survival may thusinfluence inflammatory and immune responses and susceptibility tomicrobial pathogens. The molecular mechanism by which LPS confersantiapoptotic survival signals in monocytic cells is poorly understood.Recently, LPS was shown to induce the expression of the antiapoptoticgene, c-IAP2, in PMA-differentiated human monoblastic U-937 cells;however, its role in suppressing apoptosis inmonocytic cells is not clear(15).LPS may rescue monocytic cells from apoptosis directly through the
activation of the CD14�Toll-like receptor complex or indirectly via theinduction of cytokines such as TNF-� in an autocrine manner (13, 14),suggesting a key role for TNF-� in monocytic cell survival. TNF-� gen-erates two opposing signals: one that triggers apoptosis and another thatinhibits apoptosis (16). The outcome of TNF-�-mediated effects isdetermined by the balance between these two signals. It has been sug-gested that TNF-�-induced cell death is mediated by TNF receptor 1(TNF-RI), whereas bothTNF-RI andTNF-RII are required to transducesignals for antiapoptotic activity primarily through NF-�B activation(17, 18). The protective role of NF-�B against apoptosis is believed to bemediated by the induction of antiapoptotic genes including c-IAP2
* This work was supported by grants from the Canadian Institutes of Health Research (toA. K.). The costs of publication of this article were defrayed in part by the payment ofpage charges. This article must therefore be hereby marked “advertisement” inaccordance with 18 U.S.C. Section 1734 solely to indicate this fact.
□S The on-line version of this article (available at http://www.jbc.org) contains supple-mental Figs. 1– 4.
1 Supported by scholarships from the OGSST and OGS.2 Supported by fellowships from the Ontario HIV Treatment Network (OHTN) and Stra-
tegic Areas of Development, University of Ottawa.3 Supported by a Natural Sciences and Engineering Research Council-Industrial Research
Fellowship.4 Recipient of the Career Scientist Award from the OHTN. To whom correspondence
should be addressed: Division of Virology, Research Institute, Children’s Hospital ofEastern Ontario, University of Ottawa, 401 Smyth Rd., Ottawa, Ontario K1H 8L1, Can-ada. Tel.: 613-737-7600 (ext. 3920); Fax: 613-738-4825; E-mail: [email protected].
5 The abbreviations used are: IAP, inhibitor of apoptosis protein; LPS, lipopolysaccharide;MAPK, mitogen-activated protein kinase; CaM, calmodulin; CaMK, calmodulin-de-pendent protein kinase; CaMKII, calmodulin-dependent protein kinase-II; ER, endo-plasmic reticulum; TRAF, TNF receptor-associated factor; PMA, phorbol 12-myristate13-acetate; TNF-�, tumor necrosis factor; TNF-R, TNF receptor; ERK, extracellular sig-nal-regulated kinase; PI3K, phosphatidylinositol 3-kinase; CysA, cyclosporine A; CAPE,caffeic acid phenethyl ester; DN, dominant negative; MOPS, 4-morpholinepropane-sulfonic acid; PI, propidium iodide; 2APB, (2-aminoethoxy) diphenylborate.
(19–22). It has been suggested that TNF-� stimulation of Jurkat T cellsinduced the expression of the c-IAP2 gene through the activation ofNF-�B, and conversely, c-IAP2 activated NF-�B via an I�B�-targetingmechanism to further enhance c-IAP2 expression (21). c-IAP2 wasshown to enhance NF-�B activity following its recruitment to theTNF-R via its association with the adaptor proteins, TNF-R-associatedfactors: TRAF-1 and TRAF-2 (4). Moreover, c-IAP1 and c-IAP2 wereoriginally identified as TRAF-1 and TRAF-2 binding partners (4).c-IAP2 exerts its antiapoptotic activity by directly binding and inhibit-ing the downstream protease caspase-3, -7, and -9 (7). Interestingly,TRAF-1, TRAF-2, c-IAP1, and c-IAP2, the molecules involved in anti-apoptotic activity, are all components of the TNF-RI and -RII complex,and TRAF-1 and c-IAP2 are also transcriptionally regulated by NF-�B(23). Overall, these observations suggest that NF-�B plays a central rolein the induction of a group of genes that function cooperatively to sup-press apoptosis by inhibiting the activity of caspases.Expression of c-IAP2 has been suggested to be regulated by multiple
regulatory elements in its promoter region. In addition toNF-�B (19, 21,24), c-IAP2 induction was recently shown to be regulated through aputative glucocorticoid response element in A549 human lung cancercells in response to stimulation with dexamethasone (25). In anotherstudy, the cAMP-responsive element was shown to be vital for c-IAP2induction in T84 colon cancer cells (26). However, the upstream signal-ing molecules involved in the activation of these transcription factorsregulating c-IAP2 expression are not well understood. Recently, c-IAP2expression was shown to be regulated by the extracellular signal-regu-lated kinases (ERKs), p38 mitogen-activated protein kinases (MAPK),and protein kinase C-� in human colon cancer cells (27, 28). In addition,phosphatidylinositol 3-kinase (PI3K) and ERKMAPKs were implicatedin endoplasmic reticulum (ER) stress-induced cell death (29),whereas the JAK2-STAT-3 pathway was suggested to regulate gran-ulocyte colony-stimulating factor-stimulated c-IAP2 expression inhuman neutrophils (30).Herein, we investigated the molecular mechanism by which LPS and
the proinflammatory cytokine TNF-� induce antiapoptotic survival sig-nals in humanmonocytic cells by employing promonocytic THP-1 cellsas a model system.We demonstrate for the first time that both LPS andTNF-� induce survival of humanmonocytic cells through the inductionof the c-IAP2 gene. Furthermore, LPS induced c-IAP2 expression, atleast in part, through the endogenous production of TNF-� in an auto-crine manner. The molecular mechanism involved in the up-regulationof c-IAP2 following stimulation of monocytic cells either with LPS orTNF-� is not known. We investigated the role of upstream signalingmolecules including the members of the MAPK, PI3K, and calciumsignaling pathways involved in the regulation of c-IAP2 expression andconsequent survival of monocytic cells. Our results suggest that LPS-and TNF-�-induced c-IAP2 expression in THP-1 cells and their sur-vival are regulated selectively by calmodulin-dependent proteinkinase-II (CaMKII) through the activation of NF-�B.
MATERIALS AND METHODS
Cell Culture and Reagents—THP-1, a promonocytic cell line derivedfrom a human acute lymphocytic leukemia patient was obtained fromthe American Type Culture Collection (Manassas, VA) (31). Cells werecultured in Iscove’s modified Dulbecco’s medium (Sigma) supple-mented with 10% fetal bovine serum (Invitrogen), 100 units/ml penicil-lin, 100 �g/ml gentamicin, 10 mM HEPES, and 2 mM glutamine. LPSderived from Escherichia coli 0111:B4 (Sigma), TNF-� (BIOSOURCE,Montreal, Canada), and anti-TNF-�-R1 antibody (R&D Systems,Minneapolis, MN) capable of neutralizing TNF-� activity were also
purchased. The following calcium signaling inhibitors were employed:EGTA (Sigma), a calcium-chelating agent; SKF-96365 hydrochloride(Calbiochem), specifically inhibiting receptor-mediated Ca2� entry(32); 2-APB (Calbiochem), inhibiting inositol 1,4,5-trisphosphate-in-duced Ca2� release from the ER (33);W-7 hydrochloride (W-7; Calbio-chem), a calmodulin antagonist; KN-93 (Calbiochem), a specific cell-permeable inhibitor of CaMKII; FK506 (AG Scientific Inc., San Diego,CA), interacting with FK506-binding protein, forming a FK506-FK506-binding protein complex, which binds to and blocks calcineurin; andcyclosporine A (CysA; Sigma), binding to cyclophilin and inhibiting theCa2�-dependent phosphatases. Caffeic acid phenethyl ester (CAPE) hasbeen shown to act as a potent inhibitor of NF-�B activation (Calbio-chem). The dominant negative (DN) mutant for the human CaMKII �
isoform in pSR� vector (pCaMK-II�B) was kindly provided by Drs.Alain Lilienbaum and Alain Isreal from the Institute Pasteur, Paris (34).The control vector pSR� was generated from pCaMK-II�B by digestingwith EcoRI. Endotoxin-free preparations of pCaMK-II�B and controlvectors were used to transfect cells.
Real TimePCR—c-IAP2mRNA levelsweremeasured using real timequantitative reverse transcription-PCR as per the Taqman method.Total RNAwas isolated using RNeasyminispin columns combinedwithDNase I treatment (Qiagen, Mississauga, Canada). The reverse tran-scribed RNA was amplified by PCR using the Taqman EZ reverse tran-scription-PCR kit (PerkinElmer Life Sciences). All of the reverse tran-scription-PCR steps were performed on an ABI Prism 7700 sequencedetector and quantified using the cycle threshold method and normal-ized to glyceraldehyde-3-phosphate dehydrogenase mRNA using PE-ABI supplied primers (600 nM) and probe (200 nM, JOE-labeled). Thethermal cycling conditions for the reverse transcription step were asfollows: 50 °C for 2 min, 60 °C for 30 min, and 95 °C for 5 min followedby 45 PCR cycles at 94 °C for 20 s and 60 °C for 1 min/cycle.
Ca2� Influx—THP-1 cells were washed with Ca2�-free phosphate-buffered saline for 5 min at room temperature and resuspended inBuffer A (RPMI 1640 containing 20 mMHEPES, pH 7.4). The cells werewashed again and resuspended in Buffer A containing 1mM Fluo-3/AM(Molecular Probes, Inc., Eugene, OR) in 1 mM Me2SO and 3.75% plu-ronic F-127 solution (Sigma) followed by incubation in dark for 45 minin a 37 °C shaking water bath. The reaction was stopped by adding anequal volume of Buffer B (BufferA containing 5% fetal bovine serum, pH7.4), followed by incubation for 15 min in a 37 °C water bath. The cellswere washed and resuspended in Buffer B at a final concentration of0.5� 106 cells/ml. The cells were washed again, aliquoted, and analyzedfor Ca2� levels by the FACScan flow cytometer (BD Biosciences)equipped with CellQuest software, version 3.2.1fl. Cell samples weremaintained at 37 °C during data acquisition. Intracellular Ca2� levels atbase line and following stimulation with LPS/TNF-� were measured.Ca2� ionophoreA23187 (20mM) and 5mMEGTA (Sigma) were used aspositive and negative controls, respectively.
Cell Stimulation andWestern Blot Analysis—Cells (1 � 106 cells/ml)were treated with the indicated concentration of inhibitors for 2 h fol-lowed by stimulationwith either LPS (1�g/ml) or TNF-� (10 ng/ml) for15–60 min for detection of kinase activation and for 24 h to determinec-IAP2 expression byWestern blot analysis as described earlier (31, 35).Briefly, total proteinswere subjected to SDS-PAGE, followed by transferonto a polyvinylidene difluoride membrane (Bio-Rad). The membraneswere probed with anti-c-IAP2 (RIAP-1) polyclonal antibody (36), fol-lowed by donkey anti-rabbit polyclonal antibodies conjugated to horse-radish peroxidase (Amersham Biosciences). To determine NFAT4phosphorylation, Western blot analysis was performed by employinganti-phospho-NFAT4 antibodies (Cell Signaling). To control for total
Suppression of Apoptosis by c-IAP2 in Monocytic Cells
NOVEMBER 11, 2005 • VOLUME 280 • NUMBER 45 JOURNAL OF BIOLOGICAL CHEMISTRY 37537
protein loading, the membranes were stripped and reprobed withmouse monoclonal antibodies specific for �-actin (Sigma). All immu-noblots were visualized by enhanced chemiluminescence (Santa CruzBiotechnology, Inc., Santa Cruz, CA).
Measurement of CaMKII Activity—The CaMKII assay was per-formed using a CaMKII kit (Upstate Biotechnology, Inc., Missisauga,Canada) as per the manufacturer’s instructions. Cells were pretreatedwith inhibitors for 2 h followed by stimulation of cells with either LPS orTNF-� for 30 min. Cell pellets were lysed with lysis buffer (50 mM
HEPES (pH 7.5), 150 mM NaCl, 10% glycerol, 1% Triton X-100, 1.5 mM
MgCl2, 100 mM NaF, 100 mM sodium orthovanadate, 1 mM EGTA (pH7.7), 10�g/ml leupeptin, 10�g/ml aprotinin, 10�g/ml pepstatin A, and10 �g/ml phenylmethylsulfonyl fluoride) followed by centrifugation for20 min at 14,000 � g, 4 °C. CaMKII activity was assayed from totalcellular proteins utilizing a peptide substrate (KKALRRQETVDAL)specific for CaMKII. Total proteins (200 �g) were added to 10 �l ofCaMK substrate, 0.4 �M each of peptide inhibitors for protein kinase Aand protein kinase C, and 100 �Ci of MgCl2-[�-32P]ATP in ADB IIbuffer (20 mM MOPS, pH 7.2, 2.5 mM �-glycerol phosphate, 1 mM
sodiumorthovanadate, 1mMdithiothreitol, and 1mMCaCl2). The reac-tion was incubated at 30 °C for 10 min. The phosphorylated substratewas separated from the residual [�-32P]ATP using P81 phosphocellu-lose paper. The papers were washed twice in 0.75% H3PO4 and once inacetone for 2 min and were placed in 24-well Wallac plates (Turko,Finland) in scintillation fluid. Radioactivity was measured by scintilla-tion counting using a Microbeta counter (Wallac, Turko, Finland).Blanks to correct for nonspecific binding of [�-32P]ATP and its break-down products to the phosphocellulose paper and controls for phos-phorylation of endogenous proteins in the sample were performed.CaMKII activity was expressed as counts/min/�g of protein.
Analysis of Cellular Apoptosis—Cells were incubatedwith staurospo-rine (2 �M) for 4 h, and then apoptotic cells with DNA fragmentationwere analyzed by flow cytometric analysis with propidium iodide (PI)staining in permeabilized cells. Briefly, cells (1� 106) werewashed twicewith phosphate-buffered saline containing 1% fetal calf serum, fixedwith methanol for 15 min at 4 °C, and treated with 1 �g/ml of RNase A,followed by staining with 50 �g/ml of PI at 4 °C for 1 h. The DNAcontent was then analyzed by FACScan, and data were analyzed usingWin-MDI version 2.8 software (J. Trotter, Scripps Institute, San Diego,CA).
PlasmidConstruction andMutagenesis—The full-length c-IAP2 pro-moter (3.5 kb) was amplified by PCR (with Pfu turboTM enzyme) from apreviously characterized bacterial artificial chromosome containing thegenomic region encompassing both c-IAP1 and c-IAP2 genes (Gen-BankTM accession number AF070674) (24, 37). The primers used wereas follows: sense, 5�-GAT GGT ACC ACT AGT ACT AGAATA ATGC-3�; antisense, 5�-GCT GAA TTC GCA TGC ACC AGC AAGGAC-3�. The underlined bases indicate restriction sites for cloning thattogetherwith the preceding bases do not correspond to sequences in thepromoter. The amplified promoter fragment was cloned into pCR2.1TOPO, sequenced, and then subcloned into the pGL3B vector. Sincetwo NF-�B sites (sites 1 and 3) are critical in induction of c-IAP2, site-specific mutagenesis of these two sites was performed with theQuikChangeTM multisite-directed mutagenesis kits (Stratagene, LaJolla, CA) as per the manufacturer’s protocol with 5�-phosphorylatedprimers: site 1, 5�-CTT TTG GGT CAT GGA AAT AGC CGA GTGGGTTTGCCAG-3�; site 3, 5�-GGTTATTACCGCTGGAGTTAACCTAAGTCC TAAAAGG-3�. The mutagenized bases are indicated(underlined and boldface type) that created convenient EcoRI restric-tion site for analysis. The primerswere used together in themutagenesis
reactions to create the double mutant. Successful mutants were identi-fied by EcoRI digests and sequencing.
Transient Transfection and Luciferase Assay—Cells were transientlytransfected with plasmids containing the c-IAP2 promoter by Lipo-fectamine 2000 (Invitrogen) as described previously (31, 35). Briefly, 5�g of the test plasmid and 3 �g of pSV-�-galactosidase vector (Pro-mega) were incubated for 30 min at room temperature with 16 �l ofLipofectamine reagent in 200 �l of OPTI-MEM I medium to allowformation of DNA-liposome complexes. These complexes were thenadded to the cell suspension (2 � 106 cells/ml) for 24 h followed bystimulation either with LPS or TNF-� in the presence or the absence ofthe indicated inhibitors. The cells were harvested and assayed for lucif-erase and �-galactosidase activity by using luciferase assay and �-galac-tosidase assay kits (both from Promega) in a Bio Orbit 1250 Luminom-eter (Fisher) and spectrophotometer, respectively. THP-1 cells werealso transfected with either 2–5 �g of antisense oligonucleotides forc-IAP2 (5�-GAU GTT TTG GTT CTT CUU C-3�) or control oligonu-cleotides (5�-CUU CTT CTT GGT TTT GUA G-3�).
Electrophoretic Mobility Shift Assays—Electrophoretic mobility shiftassays were performed as described earlier (31, 35). Briefly, cells werestimulated either with LPS or TNF-� for 45–60 min in the presence orthe absence of the indicated inhibitors. The nuclear proteins (5�g)weremixed with 32P-labeled NF-�B oligonucleotide probes for 20 min, andthe resulting complexes were separated on a 5% nondenaturing gel. Theoligonucleotide probes contained sequences corresponding to theNF-�B sites 1 and 3 in the c-IAP2 promoter as follows: site 1, sense(5�-ATG GAA ATC CCC GA-3�) and antisense (5�-TCG GGG ATTTCC AT-3�); site 3, sense (5�-GCT GGA GTT CCC CT-3�) and anti-sense (5�-AGG GGA ACT CCA GC-3�). To determine specificity ofNF-�B probes, parallel electrophoretic mobility shift assay reactionswere incubated with 50–200-fold excess of unlabeled specific and non-specific oligonucleotide probes (Egr-1) for 20 min prior to the additionof labeled probe. Supershift experiments were also performed by usingmouse anti-NF-�B p50 and p65 monoclonal antibodies (Santa CruzBiotechnology).
Statistical Analysis—Means were compared by the two-tailed Stu-dent’s t test. The results are expressed as mean � S.D.
RESULTS
LPS- and TNF-�-induced Suppression of Apoptosis in THP-1 Cells IsMediated by c-IAP2 Induction—To determine the role of c-IAP2 inLPS-induced suppression of apoptosis, we first demonstrated that LPSinduced the expression of c-IAP2 in THP-1 cells as determined byWestern blot (Fig. 1A) and real time reverse transcription-PCR analysis(Fig. 1B). c-IAP2 protein expression was detectable as early as 4 h, andmaximum induction to the extent of 20-fold was observed 24 h follow-ing LPS stimulation compared with the unstimulated cells (Fig. 1A).Since TNF-� is produced in response to LPS stimulation in THP-1 cells(38), we investigated whether LPS-induced c-IAP2 expression is medi-ated by endogenously produced TNF-� by employing neutralizing anti-bodies specific for TNF-�-R1 as described previously (38). The resultsshow that anti-TNF-�-R1 antibodies at concentrations of 20 �g/mlinhibited LPS-induced c-IAP2 expression to almost undetectable levels(Fig. 1C). In addition, TNF-� induced c-IAP2 expression in THP-1 cellsas determined by both Western blot and real time PCR analysis (Fig. 1,A and B). Similar to the results obtained with LPS, c-IAP2 proteinexpression in response to TNF-� was detectable as early as 4 h, andmaximum induction to the extent of 15-fold was observed at 24 h com-pared with the unstimulated cells (Fig. 1A). It may be noted that cells
Suppression of Apoptosis by c-IAP2 in Monocytic Cells
37538 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 280 • NUMBER 45 • NOVEMBER 11, 2005
expressed c-IAP1 constitutively that was not inducible by either LPS orTNF-�.
THP1 cells are resistant to the Fas- or FasL-induced apoptosis (39).Therefore, staurosporine has been used to induce apoptosis in thesecells (26, 27). To examine the role of c-IAP2 in LPS- and TNF-�-in-duced inhibition of apoptosis, we determined whether LPS and TNF-�stimulation could prevent staurosporine-induced apoptosis in THP-1cells. LPS- and TNF-�-stimulated cells were treated with staurosporinefor 4 h followed by determination of apoptosis by PI staining. Treatmentof unstimulated THP-1 cells with staurosporine resulted in �30% celldeath. Stimulation of cells with either LPS or TNF-� resulted in signif-icant inhibition of staurosporine-induced apoptosis from �30 to 10%(Fig. 1E).To determine whether c-IAP2 is involved in LPS- or TNF-�-induced
antiapoptotic cell survival, cells were transfected with c-IAP2 antisenseor control oligonucleotides prior to stimulation with either LPS orTNF-� for 24 h followed by determination of c-IAP2 expression andstaurosporine-induced apoptosis. Antisense c-IAP2 oligonucleotidessignificantly decreased by 4-fold the LPS- as well as the TNF-�-inducedexpression of c-IAP2 as compared with the cells treated with controloligonucleotides (Fig. 1D). Furthermore, treatment of LPS- andTNF-stimulated cells with antisense c-IAP2 oligonucleotidesenhanced staurosporine-induced apoptosis compared with the cells
treated with control oligonucleotides (Fig. 1E). However, antisensec-IAP2 oligonucleotides did not affect staurosporine-induced apoptosisin unstimulated cells compared with the cells treated with control oli-gonucleotides (data not shown). It may be noted that abrogation ofc-IAP2 expression by antisense oligonucleotides may not be possiblebecause of low transfection efficiency in monocytic cells. These resultssuggest that stimulationwith either LPS or TNF-� enhancedmonocyticcell survival that was mediated by c-IAP2 induction.
LPS- and TNF-�-induced c-IAP2 Expression Is Selectively Regulatedby the Ca2� Signaling Pathway in Monocytic Cells—To elucidate theupstream signaling pathways involved in the regulation of c-IAP2expression, we first investigated the role ofMAPKs and PI3K in LPS andTNF-�-stimulated THP-1 cells by employing their specific inhibitors.The results show that c-IAP2 expression was not affected by any of theinhibitors specific for p38, ERK, and c-Jun N-terminal kinase MAPKs(SB202190, PD98059, and SP600125) or PI3K (wortmannin andLY294002) at any concentration (supplemental text and supplementalFigs. 1–3). Subsequently, we investigated the role of the calcium signal-ing pathway, since changes in intracellular Ca2� concentrations play amajor role in the regulation of transcription, protein synthesis, and apo-ptosis (40). We first determined whether LPS and TNF-� activatedcalcium signaling by examining calcium influx by flow cytometry usingFluo-3 as a Ca2� binding dye. Both LPS and TNF-� enhanced calcium
FIGURE 1. LPS- and TNF-�-induced inhibition ofapoptosis is mediated by c-IAP2 expression inTHP-1 cells. A and B, LPS and TNF-� induce c-IAP2expression. Cells (106/ml) were stimulated witheither LPS (1 �g/ml) or TNF-� (10 ng/ml) for 0 –24h. c-IAP2 expression was determined by Westernblot (A) and real time reverse transcription-PCRanalysis (B). The experiments shown are represent-ative of three different experiments. C, LPS-in-duced c-IAP2 expression is mediated by endog-enously produced TNF-�. Cells (106/ml) werestimulated with LPS (1 �g/ml) in the presence andthe absence of anti-TNF-�-R1 (10 –20 �g/ml) orisotype-matched control antibodies followed bydetermination of c-IAP2 expression by Westernblot analysis. D and E, LPS and TNF-�-induced inhi-bition of apoptosis is mediated by c-IAP2 expres-sion. Cells (106/ml) were transfected with eitherantisense (AS) c-IAP2 or control oligonucleotidesfollowed by stimulation with LPS (1 �g/ml) orTNF-� (10 ng/ml) for 24 h followed by determina-tion of c-IAP2 expression by Western blot analysis(D). Stimulated cells were also treated with stauro-sporine (Stauro) (2 �M) for 4 h, and DNA content ofcells was analyzed by PI staining (E). The valuesshown in the histograms indicate percentages ofapoptotic cells. The experiments shown are repre-sentative of three different experiments.
Suppression of Apoptosis by c-IAP2 in Monocytic Cells
NOVEMBER 11, 2005 • VOLUME 280 • NUMBER 45 JOURNAL OF BIOLOGICAL CHEMISTRY 37539
influx at 12 and 8 min poststimulation, respectively. To determine theinvolvement of calcium, cells were treated with EGTA after stimulationwith either LPS or TNF-�. EGTA inhibited LPS- and TNF-�-inducedCa2� influx to the basal level (Fig. 2A). To determine the role of Ca2� inthe regulation of c-IAP2 expression, we analyzed c-IAP2 expression inTHP-1 cells treated with EGTA for 2 h prior to stimulation with eitherLPS or TNF-� for 24 h. LPS-induced as well as TNF-�-induced c-IAP2expression was inhibited in a dose-dependent manner. EGTA at con-centrations of 10 mM decreased the expression of c-IAP2 to undetect-able levels following LPS stimulation and by 6-fold following TNF-�stimulation (Fig. 2B), suggesting the involvement of the Ca2� signalingpathway in LPS- and TNF-�-induced expression of c-IAP2.Elevations in cytoplasmic Ca2� concentrations occur following stim-
ulation by diverse stimuli that activate voltage- or ligand-gated Ca2�
channels in the plasma membrane or following release of Ca2� presentin intracellular stores, mainly in the ER (40, 41). To determine whethercalcium release from the ER regulates c-IAP2 expression, we used theinositol 1,4,5-trisphosphate receptor inhibitor, 2-APB, which inhibitsthe release of calcium from the ER by blocking inositol 1,4,5-trisphos-phate receptor-gated Ca2� channels (33). 2-APB did not inhibit eitherLPS- or TNF-�-induced c-IAP2 expression (Fig. 2C). Subsequently, weinvestigated the role of receptor-mediated entry of extracellular Ca2� byemploying SKF-96365, a specific inhibitor for receptor-mediated Ca2�
entry (32). SKF-96365 treatment at concentration of 100 �M signifi-cantly reduced both LPS- andTNF-�-induced c-IAP2 expression by 11-and 7-fold, respectively, and in a dose-dependent manner (Fig. 2D).SKF-96365 and 2-APB were biologically active, since both of theseagents inhibited TNF-�-induced CD44 expression (data not shown).These results suggest that receptor-mediated Ca2� entry rather thanthe Ca2� release from the ER may be involved in LPS- and TNF-�-induced c-IAP2 expression.
Calmodulin (CaM) and CaMKII Regulate LPS- and TNF-�-inducedc-IAP2 Expression—CaM, a major Ca2� receptor, is present in bothcytoplasmic and nuclear compartments. The complex of Ca2�-CaMregulates several downstream targets, including many protein kinasesand protein phosphatases (42). To understand the role of CaM, weemployed its specific antagonist, W7-hydrochloride (43), which inhib-ited LPS- and TNF-�-induced c-IAP2 expression in a dose-dependentmanner. W7 at concentrations of 50 �M decreased the expression ofc-IAP2 to undetectable levels following LPS stimulation and by 4-foldfollowing TNF-�-stimulation (Fig. 3B). To gain further insight into therole of the CaM pathway, we examined the involvement of CaMKII,which is activated subsequent to the binding of Ca2� to CaM (44), byemploying its specific inhibitor KN-93 (42, 43). KN-93 inhibited bothLPS- and TNF-�-induced c-IAP2 expression by 7- and 11-fold respec-tively and in a dose-dependent manner (Fig. 3B).We also demonstratedthat stimulation of cells with either LPS or TNF-� for 30 min inducedCaMKII activity that was inhibited by bothW7 and KN-93 inhibitors ina dose-dependent manner (Fig. 3A).To confirm the involvement of CaMKII in LPS and TNF-�-induced
c-IAP2 expression, cells were transfectedwith aDNCaMKII plasmid ora control vector. c-IAP2 expression was significantly inhibited in cellstransfected with the DN CaMKII plasmid following LPS as well asTNF-� stimulation by 4-fold compared with the cells transfected withthe control vector (Fig. 3C). In addition, LPS- and TNF-�-inducedCaMKII activity was inhibited by transfecting cells with DN CaMKIIplasmid. Following transfection with the DNCaMKII plasmid, CaMKIIactivity was observed as 3 � 1 pM following stimulation with either LPSor TNF-� compared with the activity of 6.5 � 1 pM in LPS-stimulatedand 5.5 � 1 pM in TNF-�-stimulated cells transfected with the controlvector.
FIGURE 2. Involvement of receptor-mediated Ca2� entry rather than the Ca2� release from endoplasmic reticulum in LPS- and TNF-�-induced c-IAP2 expression in THP-1cells. A, stimulation of THP-1 cells with either LPS or TNF-� induces Ca2� influx. THP-1 cells (0.5 � 106/ml) loaded with Fluo-3/AM were stimulated with either LPS or TNF-�, and theresulting Ca2� influx was measured by flow cytometric analysis. Top panel, base-line Ca2� levels in unstimulated cells. Second panel, stimulation with LPS followed by the addition ofEGTA. Third panel, stimulation with TNF-� followed by the addition of EGTA. Bottom panel, stimulation with the Ca2� ionophore A23187 followed by the addition of EGTA. B–D, cells(106/ml) were pretreated either with EGTA (B), 2-APB (C), or SKF-96365 (D) for 2 h prior to stimulation with either LPS (1 �g/ml) or TNF-� (10 ng/ml) for 24 h followed by analysis ofc-IAP2 expression by Western blot analysis. To ensure equal loading of protein, the membranes were stripped and reprobed with anti-�-actin antibodies. The experiment shown isrepresentative of three different experiments.
Suppression of Apoptosis by c-IAP2 in Monocytic Cells
37540 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 280 • NUMBER 45 • NOVEMBER 11, 2005
Calcineurin is also activated by the binding of Ca2� to CaM, whichdissociates the two components and allows the catalytic site of cal-cineurin to become accessible (43, 45). To determine the role of cal-cineurin, cells were treated with CysA or FK506, the inhibitors of cal-cineurin, prior to stimulation with either LPS or TNF-�. Neither CysAnor FK506 inhibited LPS- or TNF-�-induced c-IAP2 expression at anyconcentration (Fig. 3D). The biological activity of CysA and FK506 wasdetermined by analysis of NFAT4 expression in Jurkat T cells asdescribed (46, 47). PMA and ionomycin are potent activators of cal-cineurin and cause dephosphorylation of NFAT proteins. CysA andFK506 are potent inhibitors of calcineurin phosphatase activity andrestore PMA- and ionomycin-induced NFAT dephosphorylation and,thus, NFAT-mediated gene induction (46, 47). Our results show thatNFAT4 phosphorylation was significantly reduced after stimulation ofJurkat T cells with PMA and ionomycin. Pretreatment of cells witheither CysA or FK506 prior to stimulation with PMA and ionomycinrestored NFAT4 phosphorylation (Fig. 3E). These results suggest thatLPS and TNF-�-induced c-IAP2 expression is regulated by calmodulinthrough the activation of CaMKII. It should be noted that none of these
inhibitors were found to be apoptotic at the concentrations used asdetermined by PI staining (data not shown).
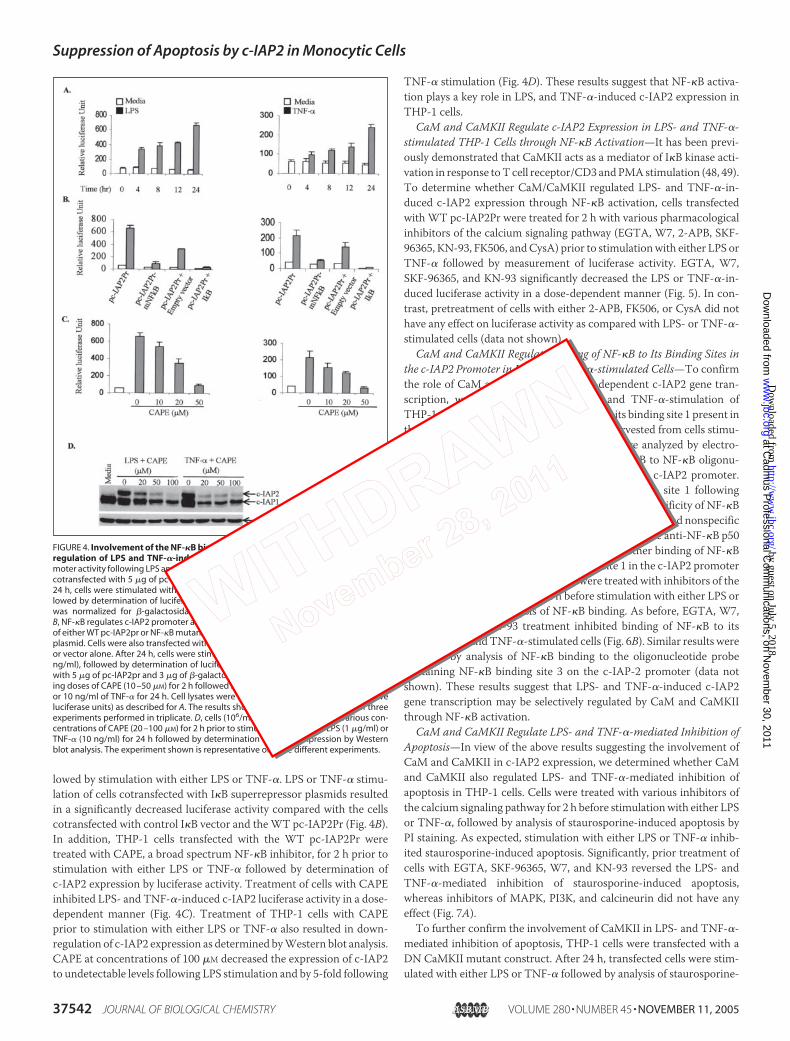
Involvement of the NF-�B Binding Sites within the c-IAP2 Promoter inthe Regulation of LPS and TNF-�-induced c-IAP2 Expression—Todetermine whether NF-�B regulates LPS- and TNF-�-induced c-IAP2expression, THP-1 cells were transfected with the human c-IAP2 pro-moter (�606 to �121 bp) linked to the luciferase reporter construct(pc-IAP2Pr) followed by stimulation with either LPS or TNF-�. Theresults show a significant 6–10-fold increase in luciferase activity at 24 hfollowing stimulation with either LPS or TNF-� compared with theunstimulated cells (Fig. 4A). Since two NF-�B sites (sites 1 and 3) (sup-plemental Fig. 4) have been shown to regulate c-IAP2 gene transcription(24), theseNF-�B sequencesweremutated by site-directedmutagenesisand cloned into the pGL3B vector (pc-IAP2Pr-mNF-�B). Stimulation ofTHP-1 cells transfected with pc-IAP2Pr-mNF-�B with either LPS orTNF-� resulted in a significant decrease in luciferase activity comparedwith cells transfectedwith thewild type pc-IAP2Pr (Fig. 4B). To confirmthe involvement of NF-�B, cells were cotransfected with either I�Bsuperrepressor or control vector along with the WT pc-IAP2Pr fol-
FIGURE 3. CaM and CaMKII regulate LPS andTNF-�-induced c-IAP2 expression. A, LPS- andTNF-�-induced CaMKII activity is inhibited by W-7and KN-93. THP-I cells were pretreated with inhib-itors for 2 h followed by stimulation of cells witheither LPS or TNF-� for 30 min. CaMKII activity wasassayed from total cell proteins utilizing a peptidesubstrate (KKALRRQETVDAL) specific for CaMKII.The results shown represent the mean � S.D. ofthree independent experiments. B, LPS- and TNF-�-induced c-IAP2 expression is mediated by CaMand CaMKII. THP-1 cells (106/ml) were treated witheither W-7 or KN-93 for 2 h prior to stimulationwith either LPS (1 �g/ml) or TNF-� (10 ng/ml) for24 h followed by determination of c-IAP2 expres-sion by Western blot analysis. C, cells were trans-fected with either DN CaMKII or control vector andcultured for 24 h, followed by stimulation witheither LPS (1 �g/ml) or TNF-� (10 ng/ml) foranother 24 h. c-IAP2 expression was determinedby Western blot analysis. D, CysA and FK506 do notinduce LPS- or TNF-�-mediated c-IAP2 expression.THP-1 cells (106/ml) were treated with various con-centrations of either CysA or FK506 for 2 h prior tostimulation with either LPS (1 �g/ml) or TNF-� (10ng/ml) for 24 h followed by determination ofc-IAP2 expression by Western blot analysis. E, Jur-kat T cells (106/ml) were treated with either CysAor FK506 for 2 h prior to stimulation with PMA (P)and ionomycin (I) for 5 min followed by determi-nation of NFAT4 phosphorylation by employinganti-phospho-NFAT4 antibodies by Western blotanalysis. To ensure equal loading of protein, themembranes were stripped and reprobed withanti-�-actin antibodies. All of the experimentsshown above are representative of three differentexperiments.
Suppression of Apoptosis by c-IAP2 in Monocytic Cells
NOVEMBER 11, 2005 • VOLUME 280 • NUMBER 45 JOURNAL OF BIOLOGICAL CHEMISTRY 37541
lowed by stimulation with either LPS or TNF-�. LPS or TNF-� stimu-lation of cells cotransfected with I�B superrepressor plasmids resultedin a significantly decreased luciferase activity compared with the cellscotransfected with control I�B vector and theWT pc-IAP2Pr (Fig. 4B).In addition, THP-1 cells transfected with the WT pc-IAP2Pr weretreated with CAPE, a broad spectrum NF-�B inhibitor, for 2 h prior tostimulation with either LPS or TNF-� followed by determination ofc-IAP2 expression by luciferase activity. Treatment of cells with CAPEinhibited LPS- and TNF-�-induced c-IAP2 luciferase activity in a dose-dependent manner (Fig. 4C). Treatment of THP-1 cells with CAPEprior to stimulation with either LPS or TNF-� also resulted in down-regulation of c-IAP2 expression as determined byWestern blot analysis.CAPE at concentrations of 100 �M decreased the expression of c-IAP2to undetectable levels following LPS stimulation and by 5-fold following
TNF-� stimulation (Fig. 4D). These results suggest that NF-�B activa-tion plays a key role in LPS, and TNF-�-induced c-IAP2 expression inTHP-1 cells.
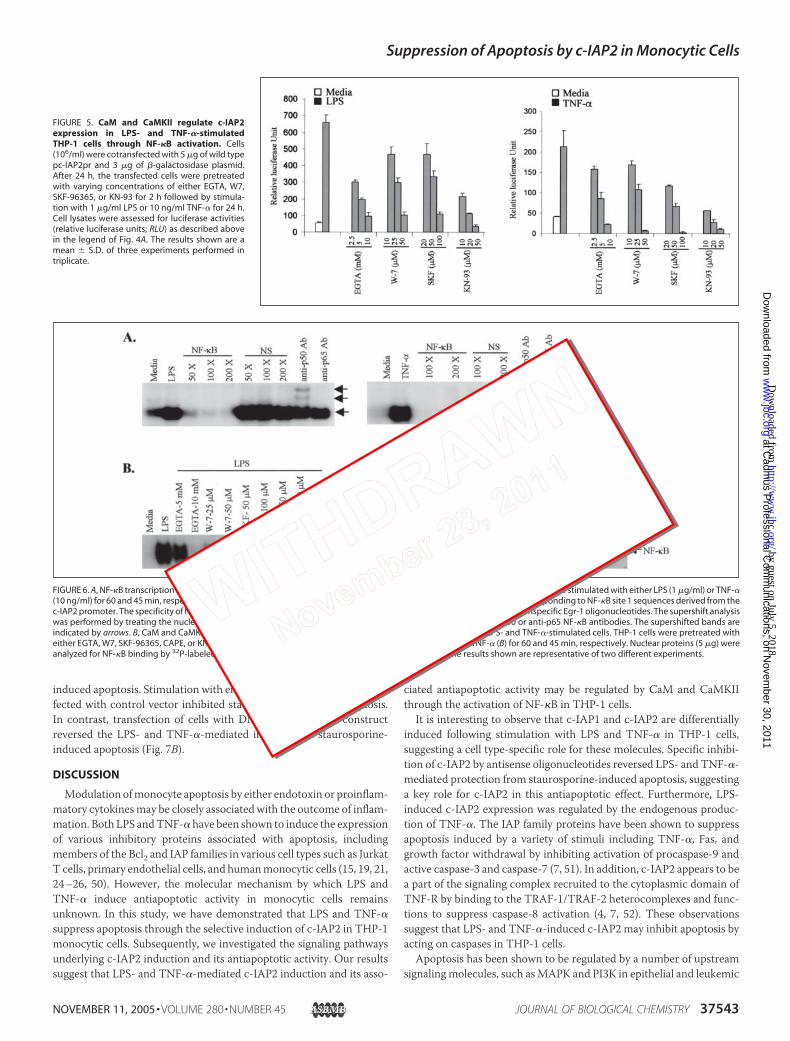
CaM and CaMKII Regulate c-IAP2 Expression in LPS- and TNF-�-stimulated THP-1 Cells through NF-�B Activation—It has been previ-ously demonstrated that CaMKII acts as a mediator of I�B kinase acti-vation in response toT cell receptor/CD3 andPMAstimulation (48, 49).To determine whether CaM/CaMKII regulated LPS- and TNF-�-in-duced c-IAP2 expression through NF-�B activation, cells transfectedwith WT pc-IAP2Pr were treated for 2 h with various pharmacologicalinhibitors of the calcium signaling pathway (EGTA, W7, 2-APB, SKF-96365, KN-93, FK506, andCysA) prior to stimulationwith either LPS orTNF-� followed by measurement of luciferase activity. EGTA, W7,SKF-96365, and KN-93 significantly decreased the LPS or TNF-�-in-duced luciferase activity in a dose-dependent manner (Fig. 5). In con-trast, pretreatment of cells with either 2-APB, FK506, or CysA did nothave any effect on luciferase activity as compared with LPS- or TNF-�-stimulated cells (data not shown).
CaM and CaMKII Regulate Binding of NF-�B to Its Binding Sites inthe c-IAP2 Promoter in LPS- and TNF-�-stimulated Cells—To confirmthe role of CaM and CaMKII in NF-�B-dependent c-IAP2 gene tran-scription, we investigated whether LPS and TNF-�-stimulation ofTHP-1 cells induced the binding ofNF-�B to its binding site 1 present inthe c-IAP2 promoter. The nuclear extracts harvested from cells stimu-lated with either LPS or TNF-� for 0–4 h were analyzed by electro-phoretic mobility shift assay for binding of NF-�B to NF-�B oligonu-cleotide probes corresponding to their sites in the c-IAP2 promoter.The results show a significant binding of NF-�B to site 1 followingstimulation of cells with either LPS or TNF-�. The specificity of NF-�Bbindingwas demonstrated by competitionwith specific and nonspecificoligonucleotides and by supershift analysis with mouse anti-NF-�B p50and p65 antibodies (Fig. 6A). To determine whether binding of NF-�Btranscription factor to the NF-�B binding site 1 in the c-IAP2 promoterwas regulated by CaM/CaMKII, cells were treated with inhibitors of thecalcium signaling pathway for 2 h before stimulation with either LPS orTNF-� followed by analysis of NF-�B binding. As before, EGTA, W7,SKF-96365, and KN-93 treatment inhibited binding of NF-�B to itsprobe in LPS- andTNF-�-stimulated cells (Fig. 6B). Similar results wereobtained by analysis of NF-�B binding to the oligonucleotide probecontaining NF-�B binding site 3 on the c-IAP-2 promoter (data notshown). These results suggest that LPS- and TNF-�-induced c-IAP2gene transcription may be selectively regulated by CaM and CaMKIIthrough NF-�B activation.
CaM and CaMKII Regulate LPS- and TNF-�-mediated Inhibition ofApoptosis—In view of the above results suggesting the involvement ofCaM and CaMKII in c-IAP2 expression, we determined whether CaMand CaMKII also regulated LPS- and TNF-�-mediated inhibition ofapoptosis in THP-1 cells. Cells were treated with various inhibitors ofthe calcium signaling pathway for 2 h before stimulationwith either LPSor TNF-�, followed by analysis of staurosporine-induced apoptosis byPI staining. As expected, stimulation with either LPS or TNF-� inhib-ited staurosporine-induced apoptosis. Significantly, prior treatment ofcells with EGTA, SKF-96365, W7, and KN-93 reversed the LPS- andTNF-�-mediated inhibition of staurosporine-induced apoptosis,whereas inhibitors of MAPK, PI3K, and calcineurin did not have anyeffect (Fig. 7A).To further confirm the involvement of CaMKII in LPS- and TNF-�-
mediated inhibition of apoptosis, THP-1 cells were transfected with aDN CaMKII mutant construct. After 24 h, transfected cells were stim-ulated with either LPS or TNF-� followed by analysis of staurosporine-
FIGURE 4. Involvement of the NF-�B binding sites within the c-IAP2 promoter in theregulation of LPS and TNF-�-induced c-IAP2 expression. A, kinetics of c-IAP2 pro-moter activity following LPS and TNF-� stimulation. THP-1 cells (106/ml) were transientlycotransfected with 5 �g of pc-IAP2pr-GL3B and 3 �g of �-galactosidase plasmid. After24 h, cells were stimulated with either 1 �g/ml LPS or 10 ng/ml TNF-� for 0 –24 h, fol-lowed by determination of luciferase and �-galactosidase activities. Luciferase activitywas normalized for �-galactosidase activity to give relative luciferase units (RLU).B, NF-�B regulates c-IAP2 promoter activity. Cells (106/ml) were cotransfected with 5 �gof either WT pc-IAP2pr or NF-�B mutant pc-IAP2pr-mNF-�B, and 3 �g of �-galactosidaseplasmid. Cells were also transfected with either the I�B superrepressor gene in pcDNA3or vector alone. After 24 h, cells were stimulated with either LPS (1 �g/ml) or TNF-� (10ng/ml), followed by determination of luciferase activity. C, cells (106/ml) cotransfectedwith 5 �g of pc-IAP2pr and 3 �g of �-galactosidase plasmid were pretreated with vary-ing doses of CAPE (10 –50 �M) for 2 h followed by stimulation with either 1 �g/ml of LPSor 10 ng/ml of TNF-� for 24 h. Cell lysates were analyzed for luciferase activity (relativeluciferase units) as described for A. The results shown above are a mean � S.D. of threeexperiments performed in triplicate. D, cells (106/ml) were pretreated with various con-centrations of CAPE (20 –100 �M) for 2 h prior to stimulation with either LPS (1 �g/ml) orTNF-� (10 ng/ml) for 24 h followed by determination of c-IAP2 expression by Westernblot analysis. The experiment shown is representative of three different experiments.
Suppression of Apoptosis by c-IAP2 in Monocytic Cells
37542 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 280 • NUMBER 45 • NOVEMBER 11, 2005
induced apoptosis. Stimulation with either LPS or TNF-� of cells trans-fected with control vector inhibited staurosporine-induced apoptosis.In contrast, transfection of cells with DN CaMKII mutant constructreversed the LPS- and TNF-�-mediated inhibition of staurosporine-induced apoptosis (Fig. 7B).
DISCUSSION
Modulation ofmonocyte apoptosis by either endotoxin or proinflam-matory cytokinesmay be closely associated with the outcome of inflam-mation. Both LPS andTNF-�have been shown to induce the expressionof various inhibitory proteins associated with apoptosis, includingmembers of the Bcl2 and IAP families in various cell types such as JurkatT cells, primary endothelial cells, and humanmonocytic cells (15, 19, 21,24–26, 50). However, the molecular mechanism by which LPS andTNF-� induce antiapoptotic activity in monocytic cells remainsunknown. In this study, we have demonstrated that LPS and TNF-�suppress apoptosis through the selective induction of c-IAP2 in THP-1monocytic cells. Subsequently, we investigated the signaling pathwaysunderlying c-IAP2 induction and its antiapoptotic activity. Our resultssuggest that LPS- and TNF-�-mediated c-IAP2 induction and its asso-
ciated antiapoptotic activity may be regulated by CaM and CaMKIIthrough the activation of NF-�B in THP-1 cells.It is interesting to observe that c-IAP1 and c-IAP2 are differentially
induced following stimulation with LPS and TNF-� in THP-1 cells,suggesting a cell type-specific role for these molecules. Specific inhibi-tion of c-IAP2 by antisense oligonucleotides reversed LPS- and TNF-�-mediated protection from staurosporine-induced apoptosis, suggestinga key role for c-IAP2 in this antiapoptotic effect. Furthermore, LPS-induced c-IAP2 expression was regulated by the endogenous produc-tion of TNF-�. The IAP family proteins have been shown to suppressapoptosis induced by a variety of stimuli including TNF-�, Fas, andgrowth factor withdrawal by inhibiting activation of procaspase-9 andactive caspase-3 and caspase-7 (7, 51). In addition, c-IAP2 appears to bea part of the signaling complex recruited to the cytoplasmic domain ofTNF-R by binding to the TRAF-1/TRAF-2 heterocomplexes and func-tions to suppress caspase-8 activation (4, 7, 52). These observationssuggest that LPS- and TNF-�-induced c-IAP2 may inhibit apoptosis byacting on caspases in THP-1 cells.Apoptosis has been shown to be regulated by a number of upstream
signalingmolecules, such asMAPK and PI3K in epithelial and leukemic
FIGURE 5. CaM and CaMKII regulate c-IAP2expression in LPS- and TNF-�-stimulatedTHP-1 cells through NF-�B activation. Cells(106/ml) were cotransfected with 5 �g of wild typepc-IAP2pr and 3 �g of �-galactosidase plasmid.After 24 h, the transfected cells were pretreatedwith varying concentrations of either EGTA, W7,SKF-96365, or KN-93 for 2 h followed by stimula-tion with 1 �g/ml LPS or 10 ng/ml TNF-� for 24 h.Cell lysates were assessed for luciferase activities(relative luciferase units; RLU) as described abovein the legend of Fig. 4A. The results shown are amean � S.D. of three experiments performed intriplicate.
FIGURE 6. A, NF-�B transcription factor binds to the site 1 of NF-�B on the c-IAP2 promoter in LPS- and TNF-�-stimulated cells. Cells were stimulated with either LPS (1 �g/ml) or TNF-�(10 ng/ml) for 60 and 45 min, respectively. Nuclear proteins (5 �g) were incubated with 32P-labeled oligonucleotide probes corresponding to NF-�B site 1 sequences derived from thec-IAP2 promoter. The specificity of NF-�B binding was determined by incubating nuclear proteins with unlabeled NF-�B or nonspecific Egr-1 oligonucleotides. The supershift analysiswas performed by treating the nuclear proteins with oligonucleotide probes in the presence or the absence of anti-p50 or anti-p65 NF-�B antibodies. The supershifted bands areindicated by arrows. B, CaM and CaMKII regulate binding of NF-�B to its binding sites on the c-IAP2 promoter in LPS- and TNF-�-stimulated cells. THP-1 cells were pretreated witheither EGTA, W7, SKF-96365, CAPE, or KN-93, for 2 h followed by stimulation with 1 �g/ml LPS (A) or 10 ng/ml TNF-� (B) for 60 and 45 min, respectively. Nuclear proteins (5 �g) wereanalyzed for NF-�B binding by 32P-labeled oligonucleotide probes corresponding to site 1 sequences. The results shown are representative of two different experiments.
Suppression of Apoptosis by c-IAP2 in Monocytic Cells
NOVEMBER 11, 2005 • VOLUME 280 • NUMBER 45 JOURNAL OF BIOLOGICAL CHEMISTRY 37543
cell systems (29, 53–56). Recently, c-Jun N-terminal kinase was showntomediate the antiapoptotic activity of XIAP (53), whereas p38 and ERKMAPKs were shown to regulate c-IAP2 expression in colon cancer celllines (27). Although both LPS and TNF-� induced the activation of p38,ERK, and c-Jun N-terminal kinase MAPK and PI3K in this study, noneof these kinases were involved in LPS- or TNF-�-induced c-IAP2expression in monocytic cells. Because of the lack of involvement ofthese major signaling pathways, we investigated the role of upstreamCa2� signaling proteins, which are important intracellular messengersin many biological processes, including apoptosis (40, 41). Influx ofCa2� through ligand and voltage-gated calcium channels in the plasmamembrane, together with Ca2� release from ER stores, results in com-plex calcium signaling cascades (40, 41). Several mechanisms may con-
trol Ca2� entry in response to external stimuli, including membranedepolarization, activation of intracellular messengers, and depletion ofintracellular calcium storage (42). The release of Ca2� from internalstores (ER) is controlled by Ca2� itself or by an expanding group ofmessengers. For example, the inositol 1,4,5-trisphosphate, produced inresponse to a signal from the membrane lipid phosphatidyl inositol,triggers Ca2� release from the ER after binding to the inositol 1,4,5-trisphosphate receptor (42).Calcium signaling has been suggested to play a key role in LPS- and
TNF-�-induced regulation of several genes (57–63). Both LPS andTNF-� have been shown to induce calcium flux (57–59, 64–67). LPShas also been shown to increase phosphatidylinositol 1,4,5-triphosphate(64, 65). Furthermore, the lipid A component of LPS was suggested tomarkedly enhance free intracellular calcium (64). There is also evidenceto suggest that the sources of increased cytosolic calcium in LPS-stim-ulated cells are extracellular calcium as well as stored calcium from theendoplasmic reticulum (68). Our results also suggest that both LPS andTNF-� induced calcium influx into the cytosol, although TNF-� actedmore quickly than LPS. This is consistent with the induction of TNF-�by LPS playing a role in c-IAP2 expression and antiapoptotic activity.Although LPS has been found to increase intracellular calcium, and thelipid A component has been specifically found to trigger calcium flux(64, 69), the precise mechanism by which LPS and TNF-� impact cal-cium signaling is still not well understood.Calmodulin, a key signaling protein responsible for integrating the
Ca2� signal with transcription factor activation, is known to regulatecell cycle and related cytoskeletal functions and ion channel activity (42,70, 71). Following binding to Ca2�, CaM undergoes a conformationalchange that renders it active and able to recognize and bind target pro-teins with high affinity (42, 71). Among the possible downstream targetsof CaM are calcineurin and CaMKII (72–74). As with other kinases,CaMKII undergoes autophosphorylation on a threonine residue con-tained in a phosphopeptide common to its � and � subunits and con-verts it into a Ca2�/CaM-independent enzyme (24). The resultsobtained by employing specific inhibitors suggested that CaMKII mayact as a key link in LPS- andTNF-�-inducedCaMactivation and c-IAP2expression.Intracellular mobilization of Ca2� triggered by various stimuli is
known to act as a key second messenger necessary for the induction ofNF-�B activity (43, 72, 74). It has been shown that two NF-�B elementsare required for c-IAP2 promoter activity, and they function coopera-tively in inducing c-IAP2 expression (24). The results of this study sug-gest that both NF-�B binding sites are involved in the up-regulation ofLPS- and TNF-�-induced c-IAP2 expression in monocytic cells. Fur-thermore, receptor-mediated Ca2� entry into the cells rather than Ca2�
release from the ER may regulate LPS- and TNF-�-induced NF-�Bactivity in the c-IAP2 promoter and c-IAP2 transcription. This identi-fied a calcium-triggered signaling cascade, which may stimulate p50/p65 NF-�B-transactivating potential, eventually leading to c-IAP2 geneexpression. Analysis of c-IAP2 promoter and NF-�B activities in thepresence of specific inhibitors of NF-�B and the calcium signaling path-way revealed that LPS- and TNF-�-induced c-IAP2 expression may beregulated by NF-�B through intracellular calcium mobilization andsubsequent activation of CaM/CaMKII. CaMKII has also been previ-ously shown to act as a mediator of I�B kinase activation specifically inresponse to T cell receptor/CD3 and PMA stimulation (48, 49). Overall,these observations suggest that CaM and CaMKII establish a linkbetween receptor-mediated intracellular Ca2� mobilization on onehand and activation of downstream p50/p65 NF-�B on the other toregulate c-IAP2 expression.
FIGURE 7. A, CaM and CaMKII regulate LPS- and TNF-�-mediated inhibition of apoptosisin THP-1 cells. Cells (106/ml) were pretreated with indicated doses of either EGTA, W7,SKF-96365, KN-93, FK506, SP600125, PD98059, or LY294002 for 2 h prior to stimulationwith either LPS (1 �g/ml) or TNF-� (10 ng/ml). After 24 h, cells were treated with stauro-sporine (2 �M) for 4 h followed by analysis of apoptotic cells by PI staining and flowcytometry. The results shown are mean � S.D. of three different experiments. B, DNCaMKII reverses LPS- and TNF-�-mediated inhibition of staurosporine-induced apopto-sis in THP-1 cells. Cells (106/ml) were transfected with either 5 �g of DN-CaMKII plasmidor control vector and cultured for 24 h followed by stimulation with either LPS (1 �g/ml)or TNF-� (10 ng/ml) for another 24 h. Stimulated cells were treated with staurosporine(Stauro) (2 �M) for 4 h, following which DNA content of cells was analyzed by PI staining.The values shown in the histograms indicate percentage of apoptotic cells. The experi-ment shown is representative of three different experiments.
Suppression of Apoptosis by c-IAP2 in Monocytic Cells
37544 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 280 • NUMBER 45 • NOVEMBER 11, 2005
At present, the role of c-IAP1 in LPS- and TNF-�-mediated anti-apoptotic effect is not clear. Both c-IAP1 and c-IAP2 have been sug-gested to contribute toward apoptotic resistance of different cancers(23). The genes encoding c-IAP1 and c-IAP2 share 75% homology at thelevel of nucleotide and amino acid sequence and are thought to havearisen from a gene duplication event (37). The molecular mechanisminvolved in the regulation of c-IAP1 expression is not clear at present.Herein, we show that c-IAP1 is expressed constitutively and is notinduced by either LPS or TNF-�. Furthermore, constitutive expressionof c-IAP1may be regulated selectively by the calcium pathway, since itsexpression levels were down-regulated by EGTA, SKF, andW-7. How-ever, c-IAP1 does not seem to be regulated by theCaMKII pathway as itsinhibitor KN93 inhibited LPS/TNF-�-induced expression of c-IAP2alone. Since expression levels of c-IAP1 are not affected by antisensec-IAP2 oligonucleotides, LPS- and TNF-�-induced antiapoptotic activ-ity may not be regulated by c-IAP1. It is likely that c-IAP1 may have arole in spontaneous survival of THP-1 cells. Furthermore, a role of otherinhibitory proteins including Bcl2 and other members of the IAP familyin LPS- and TNF-�-induced antiapoptotic activity cannot be ruled outand requires further investigation.In summary, this is the first study that demonstrates the involvement
of c-IAP2 in LPS and TNF-�-induced antiapoptotic survival of humanmonocytic cells. Our results clearly suggest that LPS- and TNF-�-in-duced c-IAP2 expression and its associated antiapoptotic activity areregulated distinctly by calmodulin/CaMKII through the activation ofthe NF-�B pathway. Since c-IAP2 is one of the antiapoptotic genes, theresults suggest that strategies based on suppression of c-IAP2 inductionby agents known to inhibit the calcium/NF-�B signaling pathways maybe helpful in controlling infection with pathogenic organisms andinflammatory responses.
Acknowledgments—The technical assistance of Martin St-Jean and CharlesLefebvre (Aegera Oncology Inc.) is gratefully acknowledged. We thank Dr. S.Wong for the BAC clone. Drs. M. Kryworuchko and M. Pinkosky are alsoacknowledged for critically reading themanuscript.We are thankful to Drs. A.MacKenzie and R. Korneluk for help in the project.
REFERENCES1. Herr, I., and Debatin, K. M. (2001) Blood 98, 2603–26142. Waterhouse, N. J., Goldstein, J. C., von Ahsen, O., Schuler, M., Newmeyer, D. D., and
Green, D. R. (2001) J. Cell Biol. 153, 319–3283. Liston, P., Roy, N., Tamai, K., Lefebvre, C., Baird, S., Cherton-Horvat, G., Farahani, R.,
McLean, M., Ikeda, J. E., MacKenzie, A., and Korneluk, R. G. (1996) Nature 379,349–353
4. Rothe, M., Pan, M. G., Henzel, W. J., Ayres, T. M., and Goeddel, D. V. (1995) Cell 83,1243–1252
5. Roy, N., Mahadevan, M. S., McLean, M., Shutler, G., Yaraghi, Z., Farahani, R., Baird,S., Besner-Johnston, A., Lefebvre, C., Kang, X., Salih, M., Aubry, H., Tamet, K., Guan,X., Ioannou, P., Crawford, T. O., de Jong, P. J., Surh, L., Ikeda, J., Korneluk, R. G., andMackenzie, A. E. (1995) Cell 80, 167–178
6. Salvesen, G. S., and Duckett, C. S. (2002) Nat. Rev. Mol. Cell Biol. 3, 401–4107. Deveraux, Q. L., Roy, N., Stennicke, H. R., Van, A. T., Zhou, Q., Srinivasula, S. M.,
Alnemri, E. S., Salvesen, G. S., and Reed, J. C. (1998) EMBO J. 17, 2215–22238. Deveraux, Q. L., and Reed, J. C. (1999) Genes Dev. 13, 239–2529. Rajcan-Separovic, E., Liston, P., Lefebvre, C., andKorneluk, R.G. (1996)Genomics 37,
404–40610. Imoto, I., Yang, Z. Q., Pimkhaokham, A., Tsuda, H., Shimada, Y., Imamura, M., Ohki,
M., and Inazawa, J. (2001) Cancer Res. 61, 6629–663411. Imoto, I., Tsuda, H., Hirasawa, A., Miura, M., Sakamoto, M., Hirohashi, S., and In-
azawa, J. (2002) Cancer Res. 62, 4860–486612. Flad, H. D., Grage-Griebenow, E., Petersen, F., Scheuerer, B., Brandt, E., Baran, J.,
Pryjma, J., and Ernst, M. (1999) Pathobiology 67, 291–29313. Manna, S. K., and Aggarwal, B. B. (1999) J. Immunol. 162, 1510–151814. Mangan, D. F., and Wahl, S. M. (1991) J. Immunol. 147, 3408–341215. Cui, X., Imaizumi, T., Yoshida, H., Tanji, K., Matsumiya, T., and Satoh, K. (2000)
Biochim. Biophys. Acta 1524, 178–182
16. Ashkenazi, A., and Dixit, V. M. (1998) Science 281, 1305–130817. Chen, G., and Goeddel, D. V. (2002) Science 296, 1634–163518. Hsu, H., Shu, H. B., Pan, M. G., and Goeddel, D. V. (1996) Cell 84, 299–30819. Stehlik, C., de Martin, R., Kumabashiri, I., Schmid, J. A., Binder, B. R., and Lipp, J.
(1998) J. Exp. Med. 188, 211–21620. Shu, H. B., Takeuchi, M., and Goeddel, D. V. (1996) Proc. Natl. Acad. Sci. U. S. A. 93,
13973–1397821. Chu, Z. L., McKinsey, T. A., Liu, L., Gentry, J. J., Malim, M. H., and Ballard, D. W.
(1997) Proc. Natl. Acad. Sci. U. S. A. 94, 10057–1006222. Beg, A. A., and Baltimore, D. (1996) Science 274, 782–78423. Wang, C. Y., Mayo, M. W., Korneluk, R. G., Goeddel, D. V., and Baldwin, A. S., Jr.
(1998) Science 281, 1680–168324. Hong, S. Y., Yoon, W. H., Park, J. H., Kang, S. G., Ahn, J. H., and Lee, T. H. (2000)
J. Biol. Chem. 275, 18022–1802825. Webster, J. C.,Huber, R.M.,Hanson, R. L., Collier, P.M.,Haws, T. F.,Mills, J. K., Burn,
T. C., and Allegretto, E. A. (2002) Endocrinology 143, 3866–387426. Nishihara, H., Kizaka-Kondoh, S., Insel, P. A., and Eckmann, L. (2003) Proc. Natl.
Acad. Sci. U. S. A. 100, 8921–892627. Nishihara, H., Hwang, M., Kizaka-Kondoh, S., Eckmann, L., and Insel, P. A. (2004)
J. Biol. Chem. 279, 26176–2618328. Wang, Q., Wang, X., and Evers, B. M. (2003) J. Biol. Chem. 278, 51091–5109929. Hu, P., Han, Z., Couvillon, A. D., and Exton, J. H. (2004) J. Biol. Chem. 279,
49420–4942930. Hasegawa, T., Suzuki, K., Sakamoto, C., Ohta, K., Nishiki, S., Hino, M., Tatsumi, N.,
and Kitagawa, S. (2003) Blood 101, 1164–117131. Ma, W., Lim, W., Gee, K., Aucoin, S., Nandan, D., Kozlowski, M., Diaz-Mitoma, F.,
and Kumar, A. (2001) J. Biol. Chem. 276, 13664–1367432. Merritt, J. E., Armstrong,W. P., Benham, C. D., Hallam, T. J., Jacob, R., Jaxa-Chamiec,
A., Leigh, B. K., McCarthy, S. A., Moores, K. E., and Rink, T. J. (1990) Biochem. J. 271,515–522
33. Peppiatt, C. M., Collins, T. J., Mackenzie, L., Conway, S. J., Holmes, A. B., Bootman,M. D., Berridge, M. J., Seo, J. T., and Roderick, H. L. (2003) Cell Calcium 34,97–108
34. Nghiem, P., Saati, S. M., Martens, C. L., Gardner, P., and Schulman, H. (1993) J. Biol.Chem. 268, 5471–5479
35. Ma, W., Gee, K., Lim, W., Chambers, K., Angel, J. B., Kozlowski, M., and Kumar, A.(2004) J. Immunol. 172, 318–330
36. Holcik, M., Lefebvre, C. A., Hicks, K., and Korneluk, R. G. (2002) BMCGenomics 3, 537. Young, S. S., Liston, P., Xuan, J. Y.,McRoberts, C., Lefebvre, C. A., andKorneluk, R. G.
(1999)Mamm. Genome 10, 44–4838. Gee, K., Kozlowski, M., and Kumar, A. (2003) J. Biol. Chem. 278, 37275–3728739. Kim, K. M., Lee, K., Hong, Y. S., and Park, H. Y. (2000) Exp. Mol. Med. 32,
246–25440. Berridge,M. J., Bootman,M. D., and Roderick, H. L. (2003)Nat. Rev.Mol. Cell Biol. 4,
517–52941. Kasri, N. N., Holmes, A. M., Bultynck, G., Parys, J. B., Bootman, M. D., Rietdorf, K.,
Missiaen, L., McDonald, F., De, S. H., Conway, S. J., Holmes, A. B., Berridge,M. J., andRoderick, H. L. (2004) EMBO J. 23, 312–321
42. Stull, J. T. (2001) J. Biol. Chem. 276, 2311–231243. Lilienbaum, A., and Israel, A. (2003)Mol. Cell Biol. 23, 2680–269844. Braun, A. P., and Schulman, H. (1995) Annu. Rev. Physiol. 57, 417–44545. Aramburu, J., Rao, A., and Klee, C. B. (2000) Curr. Top. Cell Regul. 36, 237–29546. Feske, S., Draeger, R., Peter, H. H., Eichmann, K., and Rao, A. (2000) J. Immunol. 165,
297–30547. Schreiber, S. L., and Crabtree, G. R. (1992) Immunol. Today 13, 136–14248. Hughes, K., Edin, S., Antonsson, A., and Grundstrom, T. (2001) J. Biol. Chem. 276,
36008–3601349. Hughes, K., Antonsson, A., and Grundstrom, T. (1998) FEBS Lett. 441, 132–13650. Zou, T., Rao, J. N., Guo, X., Liu, L., Zhang, H.M., Strauch, E. D., Bass, B. L., andWang,
J. Y. (2004) Am. J. Physiol. 286, C1009–C101851. Liston, P., Fong, W. G., and Korneluk, R. G. (2003) Oncogene 22, 8568–858052. Roy, N., Deveraux, Q. L., Takahashi, R., Salvesen, G. S., and Reed, J. C. (1997) EMBO
J. 16, 6914–692553. Sanna, M. G., Duckett, C. S., Richter, B. W., Thompson, C. B., and Ulevitch, R. J.
(1998) Proc. Natl. Acad. Sci. U. S. A. 95, 6015–602054. Schwenger, P., Alpert, D., Skolnik, E. Y., andVilcek, J. (1998)Mol. Cell Biol. 18, 78–8455. Yu, C., Rahmani, M., Dai, Y., Conrad, D., Krystal, G., Dent, P., and Grant, S. (2003)
Cancer Res. 63, 1822–183356. Hu, X., Haney, N., Kropp, D., Kabore, A. F., Johnston, J. B., and Gibson, S. B. (2004)
J. Biol. Chem. 280, 9498–950857. Mishra, J. P., Mishra, S., Gee, K., and Kumar, A. (2005) J. Biol. Chem. 280,
26825–2683758. Han, H., Stessin, A., Roberts, J., Hess, K., Gautam, N., Kamenetsky,M., Lou, O., Hyde,
E., Nathan, N., Muller, W. A., Buck, J., Levin, L. R., and Nathan, C. (2005) J. Exp. Med.202, 353–361
Suppression of Apoptosis by c-IAP2 in Monocytic Cells
NOVEMBER 11, 2005 • VOLUME 280 • NUMBER 45 JOURNAL OF BIOLOGICAL CHEMISTRY 37545
59. Richter, J., Ng-Sikorski, J., Olsson, I., and Andersson, T. (1990) Proc. Natl. Acad. Sci.U. S. A. 87, 9472–9476
60. Amrani, Y., Chen, H., and Panettieri, R. A., Jr. (2000) Respir. Res. 1, 49–5361. Sutterwala, F. S., Noel, G. J., Clynes, R., and Mosser, D. M. (1997) J. Exp. Med. 185,
1977–198562. Lo, C. J., Garcia, I., Cryer, H. G., and Maier, R. V. (1996) Arch. Surg. 131, 44–5063. McLeish, K. R., Dean,W. L.,Wellhausen, S. R., and Stelzer, G. T. (1989) Inflammation
13, 681–69264. Prpic, V., Weiel, J. E., Somers, S. D., DiGuiseppi, J., Gonias, S. L., Pizzo, S. V., Hamil-
ton, T. A., Herman, B., and Adams, D. O. (1987) J. Immunol. 139, 526–53365. Qu, J. M., Leaver, H. A., Yap, P. L., andWilson, N. H. (1991) Biochem. Soc. Trans. 19,
(suppl.) 95S66. Schumann,M. A., Gardner, P., and Raffin, T. A. (1993) J. Biol. Chem. 268, 2134–214067. Wilkinson,M. F., Earle, M. L., Triggle, C. R., and Barnes, S. (1996) FASEB J. 10, 785–791
68. Portoles,M. T., Ainaga,M. J.,Municio, A.M., and Pagani, R. (1991)Biochim. Biophys.Acta 1092, 1–6
69. Letari, O., Nicosia, S., Chiavaroli, C., Vacher, P., and Schlegel, W. (1991) J. Immunol.147, 980–983
70. Bultynck, G., Vermassen, E., Szlufcik, K., De Smet, P., Fissore, R. A., Callewaert, G.,Missiaen, L., De Smet,H., and Parys, J. B. (2003)Biochem. Biophys. Res. Commun. 311,1181–1193
71. Colbran, R. J., and Brown, A. M. (2004) Curr. Opin. Neurobiol. 14, 318–32772. Kim, Y., Moon, J. S., Lee, K. S., Park, S. Y., Cheong, J., Kang, H. S., Lee, H. Y., and Kim,
H. D. (2004) Biochem. Biophys. Res. Commun. 314, 695–70373. Meffert, M. K., Chang, J. M., Wiltgen, B. J., Fanselow, M. S., and Baltimore, D. (2003)
Nat. Neurosci. 6, 1072–107874. Antonsson, A., Hughes, K., Edin, S., and Grundstrom, T. (2003) Mol. Cell Biol. 23,
1418–1427
Suppression of Apoptosis by c-IAP2 in Monocytic Cells
37546 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 280 • NUMBER 45 • NOVEMBER 11, 2005
Ashok KumarSasmita Mishra, Jyoti P. Mishra, Katrina Gee, Dan C. McManus, Eric C. LaCasse andApoptosis and Antiapoptotic c-IAP2 Gene Expression in Human Monocytic Cells
-mediated Suppression ofαLipopolysaccharide and Tumor Necrosis Factor-Distinct Role of Calmodulin and Calmodulin-dependent Protein Kinase-II in
doi: 10.1074/jbc.M504971200 originally published online September 9, 20052005, 280:37536-37546.J. Biol. Chem.
10.1074/jbc.M504971200Access the most updated version of this article at doi:
Alerts:
When a correction for this article is posted•
When this article is cited•
to choose from all of JBC's e-mail alertsClick here
Distinct role of calmodulin and calmodulin-dependentprotein kinase-II in lipopolysaccharide and tumornecrosis factor-�-mediated suppression of apoptosisand antiapoptotic c-IAP2 gene expression in humanmonocytic cells.Sasmita Mishra, Jyoti P. Mishra, Katrina Gee, Dan C. McManus, Eric C. LaCasse,and Ashok Kumar
Activation of JNK-dependent pathway is required forHIV viral protein R-induced apoptosis in humanmonocytic cells. INVOLVEMENT OF ANTIAPOPTOTIC BCL2AND c-IAP1 GENES.Sasmita Mishra, Jyoti P. Mishra, and Ashok Kumar
Cyclosporin A and FK506 inhibit IL-12p40 productionthrough the calmodulin/calmodulin-dependent proteinkinase-activated phosphoinositide 3-kinase inlipopolysaccharide-stimulated human monocytic cells.Wei Ma, Sasmita Mishra, Katrina Gee, Jyoti P. Mishra, Devki Nandan,Neil E. Reiner, Jonathan B. Angel, and Ashok Kumar
804 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 287 • NUMBER 1 • JANUARY 2, 2012
ADDITIONS AND CORRECTIONS
Authors are urged to introduce these corrections into any reprints they distribute. Secondary (abstract) services are urged to carry notice ofthese corrections as prominently as they carried the original abstracts.