47
PYRAMIDINE IMIDAZOLE AS POTENT DRUGS FOR TREATING GUILLAIN BARRE SYNDROME - GB004 Chythra Rani Chandregowda - Under the Guidance of Prof. Evegeny Vulfson
| Date post: | 18-Feb-2017 |
| Category: |
Documents |
| Upload: | chythra-rc |
| View: | 204 times |
| Download: | 0 times |

PYRAMIDINE IMIDAZOLE
AS POTENT DRUGS FOR
TREATING GUILLAIN BARRE
SYNDROME - GB004
Chythra Rani Chandregowda - Under the Guidance of Prof.
Evegeny Vulfson

1
TABLE OF CONTENTS:
Sl No. Content Page No.
1 Introduction 4
2 Drug, Medications and Treatments for GB Syndrome 6
3 Drug Target/ Medical Need 7
4 Lead Molecule 10
5 Structure Activity Relations 12
6 In-Vitro Pharmacological Profiling 16
7 On Target Toxicity 19
8 Biomarkers 23
9 Pre-Clinical Studies 26
10 Investigational New Drug Application 30
11 Phase I Clinical Trials 32
12 Phase II Clinical Trials 34
13 Phase III Clinical Trials 37
14 New Drug Application 39
15 References 43
2
List of Tables
TABLE CONTENT PAGE
NUMBER
1 List of Immunoglobulins for treatment of GB Syndrome 7
2 Properties of Lead Molecule 10
3 Different NCE’s with structure 12
4 Structure Activity Relations table 13
5 NCE-Enzyme Interactions 15
6 In-vitro studies results for different NCE’s 19
7 Summary of all assay values of NCE’s 21
8 Efficacy Studies on animal models 25
9 DRF studies on rat 26-27
10 Toxicology Data for rat 28
11 Toxicology Data for dogs 28
12 Phase I results 33
13 Phase II results 36
14 Phase III results 39

3
A BRIEF LOOK INSIDE
Guillain Barre Syndrome is a reversible peripheral poly neuropathy, which is an
autoimmune disorder caused due to infection by the C.jejuni bacteria. Gangliosides on
the myelin sheath of the neurons are molecular mimics of the cell wall LPS layer
components of C.jejuni bacteria and this triggers the auto immune attack. The cascade
of immune reaction is triggered by p-38 MAP Kinase which is responsible for the
production of cytokines which in turn is responsible for T cell differentiation and
immune attack on myelin of neurons.
GB004 are pyrimidine imidazole compounds, these bind irreversibly to the ATP binding
site ofp-38 MAP Kinase blocking its activity of phosphorylation which blocks
differentiation of the T cells hence the auto immune reaction against neurons is
aborted and the damage caused to myelin is reduced.

4
INTRODUCTION:
uillaine-Barre syndrome also called as Landry’s paralysis is an auto-immune post infection
polyneuropathy [1] which is a reversible nerve damage disorder. This disorder was first identified by Jean
Baptiste Octave Landry (1859) [2]. An infection by a bacteria (Campylobacter jejuni) or virus is a trigger
factor, due to which the body’s immune system gets provoked. It is mainly an auto immune disorder where the
body’s immune system starts acting against the peripheral nervous system. There are various forms of GB
syndrome, the most common of which is the damaged caused to the peripheral nervous system also called as the
Miller Fischer variant [3]. In this type of GB syndrome the patient’s body starts producing antibodies against the
nerve gangliosides as a result of which there is neuromuscular block which leads to partial or complete paralysis
of the body.
There are two main subtypes of this disorder:
- Acute inflammatory demyelinating polyneuropathy (AIDP).
- Acute motor axonal neuropathy (AMAN) [4].
Of these the second type is more prone to be preceded by an infection in any part of the body. Sometimes this
disorder is seen in association with AIDS, food poisoning also.
CAUSES:
Occurs after the body is exposed to an infection like gastroenteritis or respiratory tract infection [1]. The link
between occurrences of GB syndrome is very much higher when it is a Campylobacter jejuni diarrhea. Research
shows that in AMAN type occurrence the antibodies are targeted against gangliosides on the myelin sheath of the
neurons, this in turn triggers macrophages to attack the neuron at the node of Ranvier [5]. This auto immune attack
is triggered after an infection by C.Jejuni, reason being the lipopolysaccharide structure of the cell membrane in
the microorganism has similar structure of gangliosides like epitopes on their surface. As a result of this even
after the infection is cured the anti-ganglioside antibodies will target the gangliosides present on the neurons in
humans. This auto immune response is mainly triggered against the following ganglioside complexes being
GM1b, GM1 and GD1a and in the AMAN type it is GQ1b which is the major target. In some other forms of GBS
T cell mediated response is observed against myelin proteins like P2, P0, PMP2 [6] etc. Ultimately this damage
caused to the myelin sheath proteins or to the gangliosides leads to damaged nerves and hence person suffers
from partial or complete paralysis.
Fig 1: Illustration of how damaged myelin can slow down the message transfer which leads to weakness. [1].
G

5
SYMPTOMS:
The symptoms that appear are due to damage caused to the 3, 4 & 6 cranial nerves (gangliosides damage). The
major symptoms are:
1) Weakness, numbness and tingling sensation in limbs with pain.
2) Weakness in the facial muscles leads to difficulty in swallowing (Dysphagia) [7].
3) Opthalmoplegia – Loss of activity in ocular muscles.
4) Weakness of heart muscles lead to heart related problems like tachycardia, bradycardia etc.
5) 25% of the people suffer from respiratory failure hence they need to be intubated and mechanical
ventilation needs to be provided.
6) Organs start to fail in the later stages.
DIAGNOSIS:
There are various diagnostic methods available for detecting GB syndrome. Few of them are as follows:
1) Cerebrospinal fluid analysis: The cerebrospinal fluid is extracted using a needle from the vertebral
column. Elevated levels of protein and reduction in WBC’s is a positive indication [1].
2) Nerve conduction studies
3) Testing for the presence of antiganglioside antibodies in the blood.
4) MRI scanning: for detection of abnormal sizes of nerve roots.
TREATMENTS:
There are majorly two types of treatments (Immunotherapy) which are either provided separately or as a
combination. Research shows that with combined effect of the treatment patients tend to recover faster.
1) Immunotherapy: This treatment mainly aims at reducing the immune system attack on the nerve cells [1].
The two ways of immunotherapy are
Plasmapheresis, in which blood from the patient is drained out and antibodies are filtered out, the
anti-gangliosides antibodies are now administered back into the patient [8].
Intravenous administration of Immunoglobulins to diminish the effect of the anti-ganglioside
antibodies on the nerve.
During the entire course of treatment the patient is kept in intensive care unit with mechanical ventilation and
various other life supporting devices. After a long term course of treatment the patient needs to undergo
physiotherapy in order to regain full muscular activity [9].
EPIDEMIOLOGY:
Average annual findings of this disorder includes 0.4 to 1.7 per 100,000 in the world population. The
occurrence was found to be higher in females than in males and also more in older people as compared to
younger population [10]. Also the percentage rate of occurrence of different subtypes of the GB syndrome varies
in different countries for example in western countries ADIP subtype is seen in 60 to 80 % of the people
whereas AMAN subtype is 6 to 7 % only. On the contrary AMAN sub type is seen 30 to 65 % in Asian
countries [1].

6
DRUGS, MEDICATION AND TREATMENT:
As mentioned earlier Guillain-Barre syndrome is an autoimmune disorder triggered by an infection [1]. The
treatment to this disorder is mainly Immuno-modulatory therapy which includes Plasmapheresis and
administration of immunoglobulins (against anti-ganglioside antibodies) to reduce the effect of attack on the
nerves [11].
Plasmapheresis: Usually this course of treatment is considered when it is still an early course of the disease [12].
This is also called as blood plasma exchange. In this procedure patient’s blood Is withdrawn and is filtered to
separate the different components of the blood (basically different blood cells are separated and isolated from the
original blood plasma) later they are suspended in a synthetic plasma and administered back into the patient. This
way the anti-ganglioside antibodies is eliminated from the patient’s body. This procedure only works for initial
stages of the disorders and has its own disadvantages.
Immunoglobulins: Immunoglobulins are obtained by purifying plasma collected from many donors, these plasma
contain antibodies against the anti-ganglioside antibodies which neutralizes the effect. The different
Immunoglobulins drugs used are listed below in the table:
Sl. No.
DRUG MANUFACTURER ROUTE EFFICACY SIDE EFFECTS DOSAGE
1 Carimune NF CSL IV Reacts efficiently with any complexes formed by antibodies without affecting IgG’s [13].
Thrombosis, Renal dysfunction or acute renal failure.
400-800 mg/kg every 3-4 weeks.
2 Gammagard Liquid
Baxter IV or Subcutaneous
Passive transfer of antibodies (antibodies usually obtained from healthy humans).
Headache, Fatigue, Thrombosis, Renal dysfunction or acute renal failure [14].
300-600 mg/kg every 3-4 weeks.
3 Bivigam Biotest Pharmaceuticals Corporation.
IV Treats muscle weakness in auto immune disorders by transfer of antibodies.
Acute renal dysfunction and acute renal failure.
300-800 mg/kg every 3-4 weeks.
4 Octagem Medispan Limited
IV Effectively transfers the antibodies and reduces weakness of limbs.
Headache, Minor chest pain, dizziness, also harmful to the kidneys.
300-600 mg/kg for every 3-4 weeks.
Table 1: List of Immunoglobulin based drugs used to treat Guillain-Barre syndrome.

7
MEDICAL NEED FOR A NEW DRUG:
It is explained before that GB syndrome has two treatments which are provided to the patients either separately
or in a combination for a long period of time after which the patient needs to undergo physiotherapy for complete
recovery. It is noticed that the only medication provided is either in the form of Immunoglobulins or treatments
like Plasmapheresis. Instead why not discover a drug that can act on the anti-ganglioside antibodies or any factors
involved in the pathology of the syndrome and the medical need for this is as follows:
1) When a patient is diagnosed with the disorder in the initial stages, when the concentration of the anti-
ganglioside antibodies is still not alarmingly high, it is better to treat it by using a drug rather than to
subject the patient to a tedious and complicated process like Plasmapheresis which has its own
disadvantages like formation of blood clots etc.
2) All immunoglobulins provided are through Intra venous administration, instead if a drug is discovered
against the anti-ganglioside antibodies then this can be administered in the form of a pill.
This way the treatment process, the route of administration can be simplified and time required for the effective
eradication of the anti-ganglioside antibodies can be reduced.
DRUG TARGET:
My target for treating GBS is p-38 MAP Kinase, which are the enzymes involved in production of cytokines
involved in proliferation of T helper cells during immune response [15].
IMPORTANCE OF IFN- γ IN GBS
The gangliosides on myelin sheath in the neurons are molecular mimics of certain structures on the
lipopolysaccharide layer of the C.jejuni bacteria. During the duration of the infection human body produces
antibodies against the bacteria whose memory is stored by the body’s immune system. Since the gangliosides
present on the myelin sheath share similar structures to the LPS of the bacteria an auto immune response is
triggered against them. IFN- γ and IL-12 are cytokines produced by the Th1 cells during an immune response, of
these IFN- γ is majorly involved in the induction of cell mediated autoimmune response [16]. Once the APC cells
interact with the CD4+ T cells the T cells gets activated and become naïve T helper cells [17], due to further
interactions between the CD28 on the CD4+ T cells and proteins on APC’s Th cells proliferate into Th0 cells.
The Th0 cells produce IFN- γ, IL-2 and IL-4 (cytokines) which promotes conversion of Th0 cells to Th1 cells
which further produces cytokines to produce Th2 cells. The Th2 cells further differentiate into effector Th cells,
memory Th cells and regulatory Th cells. Effector Th cells will produce more of the IFN- γ cytokines to increase
the BNB permeability, and to activate macrophages to attack on the compliment formed by the Anti-ganglioside
antibody and the gangliosides on the myelin sheath resulting in myelin sheath damage. Figure 2 gives a brief
picture of the immune mechanism in GBS. In this overall process we notice that IFN- γ is mainly involved in the
following events during the immune response:
1) Induces the differentiation of Th0 cells to Th1 leading to production of more cytokines.
2) Activates and enhances the expression of MHC complex II and increases the antigen binding capacity of
the macrophages [18] (Activating macrophages).
3) Increasing the permeability of BNB by allowing more macrophages to migrate towards the myelin sheath.

8
Mainly in the immune response of the human body against GBS, IFN- γ has immuno stimulatory and immuno
modulatory responses [19].
ROLE OF p-38 MAP KINASE
The CD4+ Th cells (Naïve) recognize specific MHC peptide complexes on APC through T cell receptor
complexes [15]. This is called the TcR mediated signal for T cell differentiation. Apart from this there is another
signal that plays an important role in the differentiation of naïve Th cells, being the co stimulatory signal which
is produced by the APC. A combination of these two signals induces the clonal expansion of T cells to produce
armed Th cells which triggers the immune response. This is a brief description of the T cell differentiation process.
Although there is selective differentiation of precursor CD4+ T cells into Th1 and Th2 cells. This process occurs
due to certain factors like:
1. Cytokine environment
2. Concentration of antigen – the costimulatory signal.
Off the two the first factor plays a major role and cytokines like IFN- γ which are produced by the NK and the
Th1 cells require p-38 MAP Kinase for its production. The p-38 MAP kinase is involved in regulation of
expression of specific cytokine genes. So it is involved in altering the production of these cytokines.
Fig2: Role of p-38 MAP Kinase in the differentiation of Th cells [15].

9
Fig3: The immune response against the gangliosides and the roles and functions of Th cells in GBS (T cell
mediated auto immune response) [19].
HYPOTHESIS:
By inhibiting p-38 MAP kinase the following therapeutic effects can be obtained:
1) Prevent differentiation of Th0 cells to Th1 cells, which is a major step in the T cell mediated auto immune
response.
2) Preventing the activation of macrophages which attack on the Anti-ganglioside antibody and the
compliment formed on the myelin sheath.
3) Can also prevent the increase in the permeability if the BNB.
Technically by preventing the p-38 MAP Kinase we prevent the production of cytokines like IFN- γ and hence
prevent the differentiation of T cells which mediate the auto-immune response.
LINK TO THE MEDICAL NEED:
It was mentioned in the medical need that instead of only immunotherapy for the patient which involves prolonged
tedious procedures, a drug against the anti-ganglioside antibodies or any factors involved in the pathology of GBS
can be discovered. From the available resources it is evident that IFN- γ and other cytokines plays a major role in
triggering the immune response in GBS, hence drugs targeting the factors necessary for the production of these
cytokines might give positive results and serve the purpose of avoiding immune suppression as a treatment for
GBS. Research are still going on discovering drugs against p-38 MAP Kinase for treating auto immune disorders
and there are not much drugs that are released into the market [17], hence competition in the market is also less.

10
LEAD MOLECULE
The lead molecule of my choice for inhibiting the cytokine production is Benzylsulfanyl Imidazole [20]. Imidazole
pyrimidines are new class of chemical molecules that exhibit cytokine release inhibitory role. Some of the
important properties of the lead molecule is listed below.
Sl no. Property Value
1 Structure
2 Chemical name 2-benzylsulfanyl-4,5-dihydro-1H-imidazole
3 Chemical formula C10H12N2S
3 Molecular Weight 192.28068 g/mol
4 Hydrogen Bond Donor Count 1
5 Hydrogen Bond Acceptor Count 2
6 Log P 1.7
7 N+O count 2
Table 2: Properties of the drug molecule. [21]
NCE (New Chemical Entity):
The lead molecule being 2-benzylsulfanyl-4,5-dihydro-1H-imidazole has a core imidazole ring [23] through which
hydrogen bond is formed with the lysine amino acid residue of one of the active site pockets of the JNK (p-38
MAP kinase) enzyme and the benzene side chain makes its way into the phosphate binding pocket of the enzyme.
Although these interaction do bring about certain amount of the changes required in the enzyme (since the
phosphate binding site itself is blocked) we still need to optimize the lead molecule to make these bonds stronger.
The lead molecule structure is as below:

11
Fig4: structure of the lead molecule indicating the positions for addition of functional groups for optimization.
The different NCE’s designed are tabulated below:
COMPOUND R1 R2 R3 STRUCTURE [24].
NCE(1) F-Phe 4-Pyr - Addition of F-Phe to fit the hydrophobic pocket and 4-Pyr
to interact with methionine.
NCE(2) F-Phe 4-Pyr S-CH3 S-CH3 is added to extend the link between the glycine
amino acid and the lead.

12
Table3: The different NCE’s and their structures.
SAR (Structure Activity Relationship):
COMPOUND LogP [24]. MW (g/mol) Assay data, IC50 nM
Lead 1.99 192.28 11
NCE(1) 4.028 363.46 9
NCE(2) 4.430 409 5.5
NCE(3) 4.692 437.56 40 nM
NCE(4) 3.873 428.55 10 nM
Table4: Assay data of the lead and the different NCE’s.
NCE(3) F-Phe 4-Pyr S-O-
CH3
O is bonded to S-CH3 because there is a stronger bond
between O and H of the glycine.
NCE(4) F-Phe Imidazole
ring
S-O-
CH3
4-phe is replaced by an imidazole ring so that there are
two bonds formed in the methionine binding pocket which
is stronger.

13
The table mainly indicates the assay data obtained for different NCE’s. The assay chosen for determining the
inhibition capacity of the NCE’s was the p-38 MAP kinase assay [25]. According to which the assay data was
interpreted based on the OD obtained after ELISA. The OD was directly proportional to the cytokine produced
and also the OD is used to calculate the IC50 value, if the p-38 MAP kinase (JNK) was to be inhibited then there
is less or no color developed which in turn means no or less cytokine produced and the OD will be less as a result
the IC50 also reduces. Hence from the tabular column of assay data it is evident that OD produced by NCE (4) is
way less than those produced by the rest of the compounds hence the IC50 value of NCE (4) is lesser than the
rest. Taking this into consideration we can arrange the kinase inhibition activity of these compounds in the
increasing order as below:
Lead < NCE (1) < NCE (2) < NCE (3) < NCE (4).
ACTIVE SITE POCKETS:
Fig5: Active site pockets in the enzyme [23].
There are four important binding site pockets available in the kinase enzyme and they are:
1) Lysine pocket: The drug compounds will react with the hydrogen of the amide group of Lys 55.
2) Phosphate binding pocket: This is the main active site where the phosphate group or ATP binds when the
kinase is involved in transfer of phosphate from ATP to the compound involved. This site has a glycine
amino acid residue. The lead molecule is to react with the H on the alpha carbon of glycine.
3) The hydrophobic pocket: mainly contains a Threonine residue.
4) Another additional binding pocket available which has Methionine residue which provides additional
interaction.

14
INTERACTION OF THE LEAD AND NCE’s WITH THE ACTIVE SITE POCKETS:
Compo
und
SAR and Active site interaction Explanation
Lead
Here we can see two interactions:
1) The N of the imidazole ring
reacting with the H of the
amide group in the Lys 55
residue.
2) The benzene ring of the lead
molecule fits into the
phosphate binding site of the
enzyme.
Although this interaction serves the
main purpose of blocking the
phosphate binding pocket we can
notice that there are still two pockets
left unfilled and also the interaction of
the benzene and the phosphate binding
pocket is not strong enough.
NCE(1)
In addition to the interaction between
the imidazole and the Lys 55 pocket
and the Benzene – phosphate binding
pocket, in this we notice that the 4-
Pyridine group added interacts and
fills in the Met 109 pocket and the F-
Phenyl ring added fills the
hydrophobic pocket. This additional
interaction stabilizes the remaining
two active site pockets as well hence
the increase in affinity compared to the
lead molecule.

15
NCE(2)
The S-CH3 group added to the
benzene of the imidazole lead atom
provides an additional interaction of
the H of glycine and the CH3 of the
extension. Hydrogen bond interaction
mainly causes the increased affinity
since now the pocket is not only
occupied but there is some chemical
interaction.
NCE(3)
In this compound the S-CH3 is
replaced by S-O-CH3. This extension
of the bond provides a better leverage
to the binding site by occupying it
completely and also the O of the S-O-
CH3 interacts with the hydrogen of the
H of glycine residue which is a much
stronger linkage that H-H. Hence the
increased affinity compared to the
previous compound.

16
NCE(4)
This is by far the most strongly
binding compound and the reasons for
this is:
The 4-Pyridine ring had only one
interaction with the Met pocket (N-H).
By replacing it with another imidazole
ring we achieve two strong
interactions as depicted in the picture,
one being the N-H and the other being
O-H.
This along with the rest of the
interactions makes NCE (4) the most
strongly binding compound of all the
four NCE compounds.
Table 5: The interaction between NCE compounds and the Active site of the enzyme kinase
From the above table it is clearly evident that our NCE (4) has the most binding affinity and based on the
interactions and by the assay binding information from the table 4 NCE(4) has the least OD which indicates that
it has the most kinase inhibitory activity compared to the other NCE compounds.
Note: All chemical structures are drawn using molinspiration website and all the interaction was manually done using MS Office.
IN VITRO PHARMACOLOGICAL PROFILING:
In order to study the efficiency with which these molecules bind to the target and bring about the required result,
various assays were conducted in-vitro. The different assays conducted and their description is as follows:
1) CELL BASED BINDING ASSAY [26]:
Mononuclear cells are used for this assay. Dilutions of the inhibitor molecules are prepared. The
mononuclear cell suspensions are mixed with the inhibitor dilutions and incubated for 15 minutes under
room temperature. Lipopolysaccharide layer obtained from bacteria is added to the incubated samples and
incubated for 4 hours. This stimulates the mononuclear cells to produce cytokines as an immune response
against the bacterial LPS.
Expected result:
The p-38 MAP Kinase enzymes are involved in production of cytokines to trigger immune response
against the bacterial LPS layer. Hence if the inhibitor molecule have the capacity to attack the target
And inhibit the enzyme then there will be less or no production of the cytokines. This can imply that more
the concentration of the cytokines lesser is the binding affinity of the inhibitor molecule. The concentration
of the cytokine produced is then converted into IC50 values for calculating the best binding inhibitor.

17
2) KINASE ASSAY [22]:
As discussed earlier in this assay the direct binding ability of the inhibitor molecule to the enzyme can be
studied. In this assay the inhibitor dilutions are directly incubated with enzyme solution and the activity
is measured by ELISA by using ATF-2 as substrate. The color developed is measured as OD at 405 nm.
Mechanism of assay:
ATF-2 is a substrate of p38 MAP Kinase enzymes. Kinase phosphorylates ATF-2 to phospho-ATF-2.
Hence when ELISA is conducted, anti-phospho-ATP-2 antibody (primary antibody) binds to Phospho
ATP and in turn alkaline phosphatase conjugated GAR antibody (secondary antibody) binds to the
previous complex and when the substrate (4-Nitrophenolphosphate) is added there is color development
which can be measured as OD (proportional to the concentration of the cytokines produced). When the p-
38 MAP Kinase is inhibited then there is no color development since ATF-2 is not phosphorylated.
Fig 6: pictorial representation of assay
Expected result:
If the p-38 MAP kinase is blocked by the inhibitor then there is no or less color developed and hence OD
appears to be less. The OD obtained is then converted to IC50 values using certain calculations.
3) CYP INHIBITION ASSAY [27].
CYP3A4 plays an important role in drug metabolism [28], hence in this assay we concentrate on the
inhibition capacity of CYP3A4 by the inhibitor molecules. CYP3A4 enzymes are members of cytochrome
P450 family of oxidizing enzymes. These enzymes reside in the liver and are involved in metabolizing
the drug molecules. In this assay we use fluorescent substrates of CYP3A4, and these substrates have
blocked fluorescent dyes which means that they yield fluorescence signals only if the substrates are
cleaved else no. The inhibitor molecule dilutions are prepared and incubated with the enzyme (CYP3A4)
solution. To this known concentration of the fluorescent substrates are added and the amount of the
fluorescent signal obtained is recorded.
Expected Result:
If the inhibitor molecule inhibits the CYP3A4 activity then the enzyme will fail to cleave the fluorescent
substrate hence no signal will be generated, on the contrary if the inhibitor molecule does not block the
CYP3A4 enzyme then the enzyme is capable of cleaving the substrate leading to signal generation. Hence

18
the intensity of the signal generated is indirectly proportional to the CYP inhibition capacity of the
inhibitor molecule.
4) hERG INHIBITION ASSAY [29]:
For this assay human embryonic kidney cells (HEK 293) are used. These cells are then transfected with
hERG cDNA to produce hERG recombinant gene. These hERG recombinants are now expressed in HEK
293 cells which are very identical to the human cardiac cell Ikr channels. A patch clamp amplifier is used
to record the membrane current generated. This is the main parameter used in this assay to determine the
inhibition capacity. A hERG containing bath solution is prepared and this is used to generate and record
the steady state current.
hERG expressing cells are placed in a glass bath chamber into which the bath solution is supplied
continuously and in order to record the steady state current simultaneously repetitive test pulses of 0.05Hz
is applied to the set up. Now once the steady state current is recorded the bath solution is replaced by
diluted concentration of the inhibitor test molecule one after the other and the membrane current generated
is recorded.
Expected result:
Through the amount of membrane potential generated the % inhibition can be calculated. More the
inhibition lesser the membrane potential. The % inhibition value can be used to calculate the IC50 by the
following equation:
𝐼𝐶50 =100 − %𝐼𝑛ℎ𝑖𝑏𝑖𝑡𝑖𝑜𝑛
% 𝐼𝑛ℎ𝑖𝑏𝑖𝑡𝑖𝑜𝑛 𝑥 𝐶𝑜𝑛𝑐.
Conc. = is the concentration of the inhibitor used in the solution.
MW
(g/mol)
Log P Cell
assay
[IC50, µM]
Kinase assay
[IC50, µM]
CYP Inhibition assay
[IC50, µM]
hERG assay
[IC50, µM]
LEAD 192.28 1.99 11 18 2 2
NCE(1) 363.46 4.028 9 15 6 8
NCE(2) 409 4.430 5 8 9 10
NCE(3) 437.56 4.692 40 nM 35 nM 15 15
NCE(4) 428.55 3.873 10 nM 10 nM 20 22
Table 6: in- vitro studies results for different NCE’s

19
Chart1: Graphical representation of the assay data.
RESULT:
From the various assays conducted and the results obtained from the same it is evident that NCE (4) molecule is
selected as a potent inhibitor of the other compounds. The reasons being:
1) NCE (4) fits the Lipinski’s rule of four since its log P is lesser than 4, Molecular weight is very slightly
above 400 etc.
2) Its IC50 value in both the cell assay and the kinase assay is lesser than that of the remaining molecules
indicating NCE (4) to be a potent inhibitor having the best binding capacity compared to the other
molecules.
3) NCE (4) has maximum percentage inhibition of both CYP and hERG. This conclusion is drawn based on
the IC50 value obtained for the same.
ON TARGET TOXICITY
The enzyme p-38 MAP kinase is involved in many biological functions in the human body. Based on them the
various side effects (on target toxicity) can be explained as follows:
1) Immune-suppression:
The main function associated with this enzyme is initiation of cytokine production during an immune
response which leads to T cell differentiation and the further steps. If our inhibitor molecule suppresses
the activity of the enzyme, the resulting adverse effect would be Immune-suppression. Hence the patient
will be prone to minor infections like cold, cough etc. But still since these patients are usually kept in ICU
during the course of treatment, even though the person’s immune response is delayed or suppressed
chances of catching an infection is low.
0
50
100
150
200
250
300
Lead NCE(1) NCE(2) NCE(3) NCE(4)
hERG assay(%inhibition)
CYP assay (%inhibition)
Target assay (nM)
Cell assay (nM)

20
2) Hyperglycemia:
P-38 MAPK is known to inhibit Glucocorticoid Receptors (GR) and reduce its availability for the
glucocorticoids. The enzyme phosphorylates the GR and inhibits its transcription activity [30]. For
glucocorticoids (GC) to perform its function it needs to first bind to the GR. One of GC’s main functions
are maintaining glucose metabolism i.e. enhancing gluconeogenesis. Hence by inhibiting p-38 MAPK
GR’s are made available for GC and hence there is over expression of the same. This means that the
gluconeogenesis process is enhanced and hence high glucose level can be observed in the blood, which is
termed as Hyperglycemia.
3) Hypertension:
Also GCs are released by the adrenaline glands as a response to stress, during which the enzyme prepares
the body for fight or flight. The major changes observed in the body during a stress response are
hypertension [31]. Hence by inhibiting the enzyme p-38 MAPK, GC expression is enhanced and hence
chances of the patient acquiring hypertension are high.
4) Cardiovascular effects:
As we all know, Ischemia one of the major causes for damage of myocardium. Research work explains
that during ischemic damage caused to the heart -38 MAPK pathway is stimulated which positively
prevents the damage caused to the heart [32]. Hence by inhibiting the enzyme patients might be prone to
cardiovascular disorders caused due to damage of myocardium.
OFF TARGET:
One of the isoforms of p-38 MAPK is p-38 β [33]. It is involved in few important biological functions and the
related side effects are listed below:
1) Hypoglycemia:
P-38 β is involved in metabolism i.e. inhibition of GS (Glycogen synthase) activity by inhibiting its
activity by phosphorylating it [34]. GS is involved in conversion of glucose to glycogen for storage. Hence
by inhibiting the activity p-38 MAPK, GS will not be inhibited as a result of which there will be less
concentration of glucose in the blood and hence Hypoglycemia.
2) P-38 β are involved in keratinocyte differentiation. These cells are involved in functions like formation of
new skin layer, hair growth. Based on these the probable adverse side effects can be as follows:
Delayed wound healing.
Cessation of hair growth.
ADRENERGIC ALPHA 2C RECEPTORS [35]:
I choose the above mentioned receptor for pharmacological profiling of my drug because this receptor is very
closely associated with majority of the side effects caused by the drug molecule as explained before such as
hypertension and cardiac ischemia, hyperglycemia etc. Since the drug molecule has high chances of leading to

21
these safety concerns and Aα2c receptors are highly associated with these side effects I choose this receptor from
the research paper provided [36].
ASSAY FOR PROFILING:
Fluorescence Based Binding assay:
Assay involves binding a fluorescent molecule [fluorophore] to the drug molecule to be tested. This fluorescently
conjugated drug molecule is incubated with Aα2c expressing cells. If the molecule effectively bind to the receptor
and bring about its effect of inhibiting the receptor then the fluorescence intensity is reduced. Intensity of
fluorescence is measured confocal microscope through which confocal images. The intensity of fluorescence is
used to calculate IC50 values.
Cell assay
[IC50, µM]
Kinase
assay
[IC50,
µM]
CYP Inhibition
[IC50, µM]
hERG
Inhibition
[IC50, µM]
Aα2c
Inhibition
[IC50, µM]
p-38 β
[IC50, µM]
LEAD 11 18 2 2 2 17
NCE(1) 9 15 6 8 5 15
NCE(2) 5 8 9 10 10 13
NCE(3) 40 nM 35 nM 15 15 13 12
NCE(4) 10 nM 10 nM 20 22 21 9
Table 7: Summary of all assay values for different NCE’s.
As explained earlier the IC50 values of the NCE (4) molecule has way more positive implications in all the assays
than the rest.
ANIMAL MODELS:
GBS being a poly neuropathy disorder is associated with sensory and motor impairment related symptoms. For
clinical trials there have been two models which replicate the symptoms of GBS and they are as follows:
1) RABBIT MODELS WITH T CELLS SENSITIZED AGAINST GM1 GANGLIOSIDES [37]:
GBS is an autoimmune disorder triggered by C.jejuni bacterial infection, due to the molecular mimicry
between the LPS of the bacteria and the gangliosides the myelin sheath of the nerve cells, due to which
IGg auto antibodies are developed against the gangliosides resulting in the degeneration of the myelin.
Keeping this concept in mind, in order to create diseased rabbit models, the rabbits were sensitized with
bovine brain gangliosides mixture that included GM1 [38] (since GM1 are the auto antigens in this
disorder). After the administration of the GM1 into the rabbit’s system IGg anti-GM1 antibodies were
observed in the animal. The production of the same led to the onset of symptoms in the animal like flaccid
limb, weakness with a monophasic course. On investigating for the reason behind the symptoms it was
found that the peripheral nerves had Wallerian like degeneration.
These pathological findings were very similar to those of the human AMAN type GBS.

22
Fig7: a) Symptoms of flaccid limb due to motor axonal neuropathy;
b) Electron micrograph of the nerve indicating macrophage infiltration.
2) L31/CD4-/- MICE MODEL:
B7.2 is a type of peripheral protein found in APC’s that trigger immune response due to the co stimulatory
signal generation. Transgene derived constitutive expression of co stimulator signal of B7.2 on antigen
presenting cells leads to neurological disorders [39]. It was observed that depletion of CD4-/- cells in L31
mice accelerated the symptoms associated with GBS. According to this research C.jejuni infection leads
to autoimmunity disorders in mice through co stimulatory activity of APC which triggers the expansion
of the T cells to act against the neurons. L31 mice were genetically modified to deplete the CD4+ Tcells
which led to the onset of the disease due to constitutive expression of B7.2 protein [40]. 2-6 month old mice
were used for this study. 49 L31/CD4-/- mice and 18 control mice were tested and it was observed that
there was an increased expression of B7.2 in macrophages of L31/CD4-/-. These mice in the study showed
the following symptoms during the test are motor impairment, weakness in the limb due to which the
mice were not able to walk properly, numbness in the limb observed as lack of pain, myelin loss
were observed in symptomatic L31/CD4-/- leading to axonal damage in peripheral nerves.
In conclusions these genetically modified mice shows over expression B7.2 proteins on APC as a result
of which these mice show mimics of clinical and pathological aspects on GBS in humans. Based on this
research a cascade of events was proposed to the following mouse model:
1) Transgene derived over expression of B7.2 on APC’s sensitizes APC’s in nervous system.
2) T cells which are specific to PNS recognize the B7.2 on APC as antigens and gets activated.
3) This is followed by infiltration of macrophages, expansion of the T cells due to production of
cytokines.
4) This immune reaction is directed against the neurons leading to demyelination and hence the
symptoms.
Of the two models described above I choose L31/CD4-/- mice model for efficacy testing trials due to the following
reasons:

23
1) This model has a clear explanation of the cascade of events which occurs during the auto immune reaction
which is similar to those observed in GBS.
2) It emphasizes on the fact that the symptoms are seen due to the T cell activation which is important in our
trials since my drug molecule is targeted towards the p-38 MAPK required for expansion of T cells.
3) Mice are easy to handle.
BIOMARKERS:
The major source of biomarkers associated with Guillain Barre Syndrome is the Cerebrospinal Fluid (CSF).
During GB syndrome, when the peripheral nerves get damaged during auto immune attack, this leads to swelling
of nerve root which in turn leads to disturbance of micro circulation hence plasma proteins and other factors are
released into the CSF [41]. These factors act as biomarkers for this disorder. Two of these are listed below:
1) Axonal damage marker: Axonal damage can be indicated by the neurofilaments, these are axoskeletal
proteins which are heteropolymers with four subunits. They are abbreviated as Nfs. Research shows that
during GB syndrome due to axonal damage the Nfs concentration is increased in the CSF. Hence elevated
level of Nfs in the CSF indicates damage to proximal axon parts. Study shows that patients with GBS
have > 0.73 ng/ml concentration of Nfs in the CSF [42]. Through high throughput analysis of Nfs is possible
using ELISA technique. These act as efficacy biomarkers since the subsequent decrease in the
concentration of Nfs in the CSF indicate the efficacy of the drug.
2) Chemokines: These are small cytokines or signaling protein that are secreted by the macrophages and T
cells during an immune attack [43]. CSF levels of these chemokines seems to be increased in GBS
indicating their role in the neural inflammatory process [44]. Since my drug molecule aims at targeting the
enzyme p-38 MAPK which prevents the production of cytokines, hence the decrease in the concentration
of the chemokines like IP-10 indicates that the drug is effectively inhibiting the enzyme form producing
cytokines. Hence this can be termed as a target biomarker.
CSF is extracted from the animals through the lumbar region of the spinal cord.
ENDPOINT ASSAYS:
Since the main symptom associated with GBS is weakness in the limb which leads to movement impairment the
main endpoint for this would be ability of the mice to walk which can be measured by the following assay:
3) Rotorod Assay [39]:
This assay mainly measures the motor coordination of the symptomatic mice. In this task the mouse was
made to walk on a speed ramp of speed 0 to 30 rpm, followed by a maximal speed for 240s. The time
taken by each animal is used to relate ant amount of motor impairment caused. An increase in walking
mice is suggestive of repair of the motor impairment.
4) Neuropathy impairment score [45]:
NIS is a complete clinical scoring system that is used to assess the severity of peripheral neuropathy. It
mainly quantifies the findings of various neurological examination that include testing of components like

24
sensation, reflexes and muscle weakness. The scoring system starts from 0(normal) to 88(total
impairment).
DOSE SELECTION:
Previously I had presented the assay information of all the NCE molecules. Considering the data from this we
have the following information about NCE (4)
IC50 = 10 nM
Molecular Weight= 428.55 g/mol.
Calculation:
IC50 = 10 nM
Hence IC90= 100 nM (Approximately 10x of IC50)
Assuming the bioavailability of the drug to be 20% (ignoring protein binding)
We get IC90 = 100 / 0.2 = 500 nM
Drug dose= Drug available x MW of NCE.
= (500 x 428.55) nM*g/mol
= 214275 x 10-6 mg/kg.
= 0.214 mg/kg.
Similarly dose at IC50 = 0.02 mg/kg and dose at IC10 = 0.002 mg/kg.
The dose range selected for this study was 0 – 1.0 mg/kg. The reading obtained in various tests conducted and the
biomarkers were observed and tabulated as follows:
Dose (mg/kg) No. Of Animals
used in testing.
Rotorod
Assay
(sec)
NIS
Score
IP-10
ng/ml
Nfs
ng/ml
0 10 0 60 50 1.0
0.002 10 5 50 48 0.9
0.01 10 10 40 35 0.8
0.02 10 30 30 20 0.75
0.1 10 45 20 15 0.6
0.21 10 50 0 8 0.5
0.5 10 50 0 8 0.5
1.0 10 48 10 10 0.7
Table 8: Efficacy studies on animal models
Description:
25
Since we are using diseased mouse models to check for the efficacy of the drug it is expected that the mouse with
no dose provided (control) would fail in the rotorod assay and it’s NIS due to motor impairment. As the drug is
administered in the doses tabulated due to its effect we see a progressive repair of the impairment which concludes
that the drug is efficient in stopping the nerve damage caused due to the auto immune reaction, by aborting the T
cell action against the neurons. Of all the doses the most efficient dose is 0.21 mg/kg since at this dose we find
NIS to be 0 which implies that the motor impairment is completely cured. We also observe that the animal seems
to be efficiently performing in the rotorod assay as well. Even though the mouse shows improvement in motor
functions at doses above 0.21 we observe side effects and hence I prefer to select 0.21 mg/kg as the best dose for
further studies.
The concentration of biomarkers in the CSF was tabulated for the respective doses. It is seen that the concentration
of both IP-10 and Nfs is decreasing with the increase in the dose administered indicating that the drug is working
effectively in the animal model. And it decreases to the normal level at an efficacious dose of 0.21 mg/kg.
Graphical representation of the tabulated column:
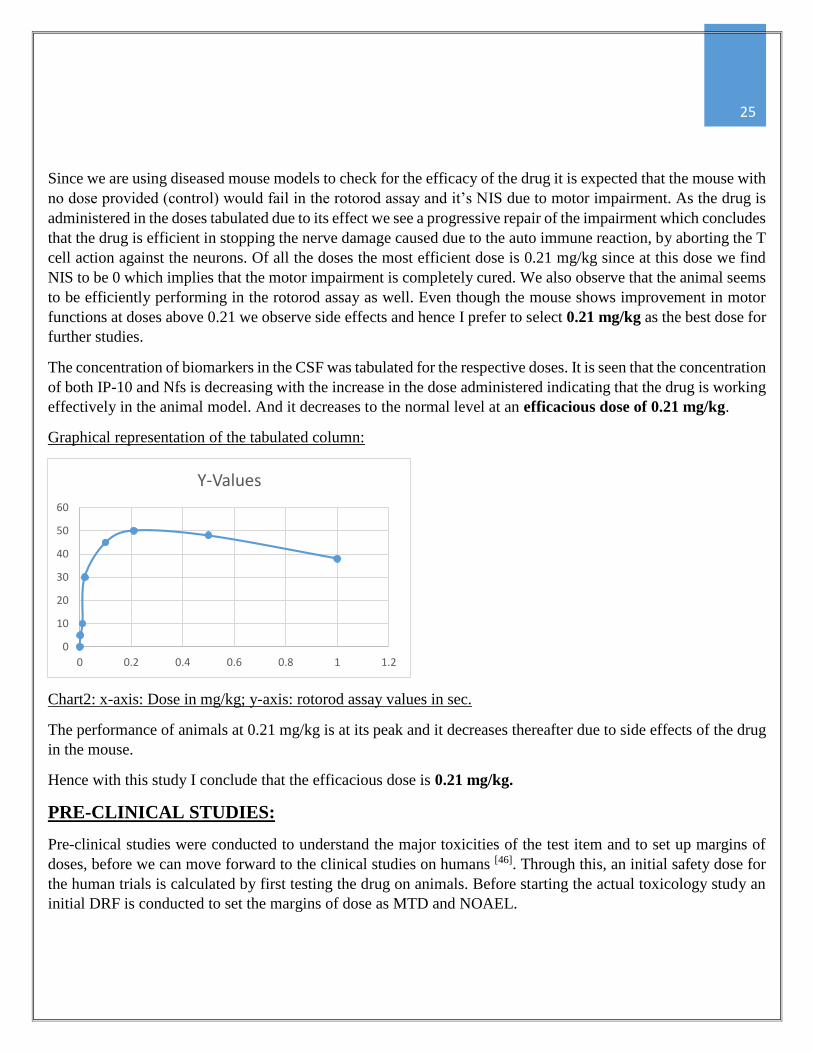
Chart2: x-axis: Dose in mg/kg; y-axis: rotorod assay values in sec.
The performance of animals at 0.21 mg/kg is at its peak and it decreases thereafter due to side effects of the drug
in the mouse.
Hence with this study I conclude that the efficacious dose is 0.21 mg/kg.
PRE-CLINICAL STUDIES:
Pre-clinical studies were conducted to understand the major toxicities of the test item and to set up margins of
doses, before we can move forward to the clinical studies on humans [46]. Through this, an initial safety dose for
the human trials is calculated by first testing the drug on animals. Before starting the actual toxicology study an
initial DRF is conducted to set the margins of dose as MTD and NOAEL.
0
10
20
30
40
50
60
0 0.2 0.4 0.6 0.8 1 1.2
Y-Values

26
DOSE RANGE FINDING
DRF studies are conducted to establish a dose response and to provide the appropriate dose margins for the
following regulatory studies. I have used rat as the animal model for DRF studies. The starting dose and the side
effects associated with it are represented in table 9
DOSE
mg/kg
No. of animals Adverse effects
0.21 10 No
0.6 10 No
2 10 No
5.61 10 <10% weight loss
17.01 10 >10% weight loss
Less food consumption
51 10 >20% weight loss, anorexia (complete absence of
food intake)
150.0 10 2 animals had to be euthanized.
Table 9: Dose Range Finding Studies
Illustration of the data:
The starting dose 0.21 mg/kg is taken from the previous efficacy studies since at this level no side effects were
observed. The subsequent doses are taken at half log intervals. From the above data we see that at dose = 1.89
mg/kg there is still no adverse effects observed, but immediately after this, at the administration of the next dose
AE’s are observed. Hence this dose is taken as the NOAEL (No Adverse Effect Level). As we increase the dose
beyond this level at the maximum dose of 150 mg/kg 2 animals needed to be euthanized. Hence the dose
immediately before it is selected as the MTD (Maximum Tolerated dose), since at this level we do find adverse
effects but it is tolerable.

27
REGULATORY TOXICOLOGY STUDIES
The two animal species used for the safety studies are:
1) Rat
2) Adult Beagle Dogs.
DRF studies provided us with the NOAEL and MTD using which we select intermediate doses between the two
for our regulatory studies for both rat and dogs.
ANIMAL MODEL 1 : RAT
DOSE
mg/kg/day
No. OF ANIMALS ADEVRSE EFECTS
0 (Control) 10 No
2 10 No
7 10 <10% weight loss
18 10 >10% weight loss
Less food consumption
50 10 >20% weight loss, anorexia (complete absence of
food intake)
Table 10: Toxicology data for rat
Table illustration:
We select the first dose based on the DRF studies to be 2 mg/kg and the MTD to be 50 mg/kg (lesser than the
actual MTD). Two intermediate doses are administered. All doses are administered to the rat once a day
throughout the month and the adverse effects are also tabulated.
Conclusion of DRF Studies:
NOAEL: 2 mg/kg
MTD: 51 mg/kg

28
ANIMAL MODEL 2 : ADULT BEAGLE DOG
DOSE
mg/kg/day
No. OF ANIMALS ADEVRSE EFECTS
0 (Control) 4 No
8 4 No
24 4 <10% weight loss
65 4 >15% weight loss
Less food consumption with vomiting
Table 11: Regulatory toxicity studies in dogs.
The NOAEL being 8 mg/kg and the MTD being 65 mg/kg is used as margins between which two intermediate
doses are selected to be administered to dogs.
Note: All doses are administered once a day for a period of one month.
HUMAN EQUIVALENT DOSE:
The NOAEL of each of the species used is converted into a dose equivalent to be administered in humans based
on a scaling factor formulated by FDA as indicated below:
This conversion is based on the normalization of doses in relation to the body surface area which is indicated as
a Body Surface Area-Conversion Factor (BSA-CF) [47], this is obtained from the guidelines and standards set by
FDA. Calculation of the HED is indicated below:
SPECIES BSA-CF
HUMANS 37
RAT 6
DOGS 20

29
For rat:
As per prior results we have NOAEL as 1.89 mg/kg
Hence HED = 2 x (6/37)
= 0.324 mg/kg
For dogs
As per prior results we have NOAEL for dogs as 15 mg/kg
Hence HED = 8 x (20/37)
= 4.32 mg/kg.
Although the HED of rat is lesser, I chose the HED value of dogs for further dose calculations because of the
following reasons:
1) The HED dose for dogs is higher than that of rats hence it would provide a much better Therapeutic index
for further calculations.
2) The ADME routes of dogs are very well known and is more similar to humans
3) Dogs show more similar symptoms when they are induced with peripheral neuropathy, unlike rats which
shows only limb flaccidity.
MAXIMUM RECOMMENDED STARTING DOSE:
MRSD can be defined as the highest amount of the test item that can be safely administered in humans without
complications to the patient while it is maintaining its maximum efficiency. For calculations of MRSD a safety
factor of atleast 10 is used in order to provide better protection to humans. Hence
MRSD ≤ 0.1 (HED)
RESULTS OF REGULATORY TOXICOLOGY STUDIES
FOR RAT: FOR DOGS:
NOAEL: 1.89 mg/kg NOAEL: 8 mg/kg
MTD: 50 mg/kg MTD: 65 mg/kg
HED: 0.324 mg/kg HED: 4.32 mg/kg

30
≤ 0.1 x 4.32 mg/kg
≤ 0.432 mg/kg
THERAPEUTIC INDEX
TI can be defined as a comparision of the amount of a therapeutic agent that can cause the therapeutic effect to
the amount that causes toxicity. It can be calculated as follows:
TI = NOAEL/ Efficacious dose
Efficacious dose (from efficacy studies) = 0.21 mg/kg
Hence TI = 8/0.21
= 38.09
The entire pre-clinical studies was conducted for a duration of one year.
INVESTIGATIONAL NEW DRUG APPLICATION:
Drug GB004 [NCE (4)]
MW 428.55 g/mol
Log P 3.873
Mechanism of Action The S-OCH3 extension in the molecule binds to the
phosphate binding pocket of the enzyme making the space unavailable for the phosphate, hence the
enzyme will not be able to phosphorylate ant substrates that are involved in cytokine production. The two
hydrophobic pockets being Imidazole ring and F-phenyl bind to the other regions on the enzyme making
the binding strong.
Route of administration Oral administration
SUMMARY OF PRE-CLINICAL STUDIES:
1) Human Equivalent Dose: 4.32 mg/kg
2) MRSD ≤ 0.432.
3) Therapeutic Index: 38.09

31
Clinical Protocol and investigator’s information The protocols for clinical studies were documented and is submitted along with this and also the
information of the investigator was also provided.
Chemistry, Manufacturing, Control The drug substance with its properties, name and address of the manufacturer, manufacturing protocols is
submitted. Stability tests was conducted for one year. The ADME characteristics of the drug was also
tested and information is provided.
Pharmacokinetics and Pharmacodynamics study Efficacy tests were conducted and two animal models being rats and adult beagle dogs and the efficacious
dose from this was determined to be 0.21 mg/kg.
During these tests it was also tested and determined that the drug remains in the animals system for 12
hours and reaches Cmax at the 6 to 8 hours of administration.
In-vitro studies Various assays were conducted in-vitro to determine how efficiently the drug binds to the target and to
determine its inhibitory property. The two in-vitro assays conducted were cell binding assay and p-38 map
kinase assays. The tabulated information of these two assays is provided.
Toxicology studies In order to study the toxicology of the drug safety studies was conducted on healthy mouse models and
the results obtained is provided.
Regulatory toxicology studies were conducted on both rat and adult beagle dogs for a period of one year
and an MTD of 50 mg/kg for rats and 65 mg/kg for dogs was determined.
These pre-clinical data predicts the drug’s safety in using them on humans also all the pre-clinical activities
are conducted under GLP.
Safety Pharmacology Studies Various safety tests were conducted in-vitro and in-vivo and are as follows:
- CYP inhibition assay: The drug was found to be minimally inhibiting CYP.
- hERG inhibition assay: It was found that the drug caused negligible side effects on the heart and
the hERG inhibition assay values are also provided.
- On target toxicity studies and off target toxicity studies were conducted.
- Genotoxicity studies were conducted by in-vitro AMES test and the drug was proved to not to be
a mutagen.
First In Human Dose Through regulatory toxicology studies the Human Equivalent Dose was calculated to be: 4.32 mg/kg and
the Maximum Recommended Starting Dose calculated to be: 0.432 mg/kg which will used as the
initial dose in Phase I clinical trials.
The IND application was submitted to the F.D.A for approval.

32
INVESTIGATOR’S BROCHURE:
The document was submitted to Institutional Review Board (IRB) for review. A copy of this as per requirement
was submitted to FDA with IND application. This includes all clinical, pharmacological information, toxicology
studies of the drug GB004 [NCE (4)].
PHASE I CLINICAL TRIALS IN HUMANS:
Purpose: To determine the safe dose range, clinical safety signals and to determine the hMTD.
Condition: Guillain-Barre syndrome
Primary purpose: To formulate a safe dose range and to test toxicity of the drug in humans.
Drug: GB004 (NCE4)
Starting Dose: Concentration of 0.432 mg/kg/day.
STUDY DESIGN
Primary purpose: Toxicology study
Allocation: Randomized [48]
Endpoint: Safety and efficacy study
Intervention model: Parallel assignment
Masking: Single Blind
Number of participants: 112 in cohorts of 16.
Healthy Volunteers: Yes
Title: Phase1, randomized, single blind, single ascending dose study to investigate the safety and efficacy of
GB004 (NCE4).
Inclusion criteria:
Healthy volunteers of age group 18 to 65 years old.
Both male and females can participate.
Exclusion criteria:
Should not have a previous record of cardiovascular disorders.
Should not be pregnant or lactating women.
Volunteers under medication for other disorders are not allowed.
Clinical trials were conducted for a period of 6 months. Volunteers were administered with a dose every day and
the various adverse effects were observed. This was conducted for a period of one week before the next dose
could be administered. The dose was incremented by 2X, 4X etc. for the consecutive weeks. The results of the
phase one trials are as indicated in table 12:

33
Drug dose mg/kg No. of subjects No. of subjects with
adverse effects
Adverse effects
0 (Control Placebo) 16 0 None
0.43 (MRSD) 16 0 None
0.86 16 0 None
1.72 16 0 None
3.44 16 0 None
6.88 16 3 Gastrointestinal problems,
Hair fall
13.76 16 9 Gastrointestinal Issues, Kidney issues,
Hair fall
Table 12: Doses and its adverse effects in the phase 1 trial.
DISCUSSION:
From the information available in the table we can see that starting from the MRSD 0.43 mg/kg till 3.4 mg/kg
there are no adverse effects observed. At 6.8 mg/kg 3 out of 16 patients showed signs of AE but since it is optimal
and not as bad as the AE indicated in the drug doses afterwards. The last dose administered shows that 9 of 16
volunteers (more than 33%) showed adverse effects. This is way higher than the number of subjects showing
adverse effects in the dose range 6.8 mg/kg (approximately equal to 408 mg as per the avg. weight of human
beings taken as 60 kg) hence this dose can be considered as hMTD for further studies since less than 33%
of volunteers show adverse effects.
IND UPDATE
Phase I Clinical Trial Summary:
Phase I clinical trials was conducted on 112 volunteers in cohorts of 16.
Initial dose of 0.43 mg/kg (MRSD) was given.
No fatal toxicological side effects observed and the hMTD was determined to be 6.8 mg/kg
Major side effects were GI problems and hyperglycemia which could be controlled by insulin injection
(prevailed only during course of drug administration).
The entire study was conducted for a duration of 6 months.
Pharmacokinetic & Pharmacodynamics Studies
During Phase I trials on collecting the blood samples of patients and observed for the concentration of
the drug, it was noticed that the drug remained in the human system for a period of 6 hours and the Cmax
was observed at 2 to 3 hours of administration, hence for further studies drug needs to be administered
twice a day.
QT prolongation did not prevail in volunteers.

34
Non-Clinical Activities:
Further testing of toxicity was conducted on animals, for a period of one year the drug was safe for further
usage.
Single and repeat dose toxicity studies were conducted for a duration of 6 months
Chemistry, Manufacturing, Control
Drug Manufacturing protocols have been provided.
Stability tests for the drug were conducted for a period of one year and the drug is stable for clinical use.
The physical, chemical and biological characteristics of the drug was provided.
All assays and methods used to detect the purity, quality of the drug is provided.
INVESTIGATOR’S BROCHURE UPDATE:
An updated copy of the document was submitted to Institutional Review Board (IRB) for review. A copy of this
as per requirement was submitted to FDA with IND application. This includes all clinical, pharmacological
information, toxicology studies of the drug GB004 [NCE (4)].
PHASE II CLINICAL TRIALS
Purpose: To study the efficacy and safety of GB004 drug in the treatment of GB syndrome on diseased patients
and to find an optimal dose for Phase III.
Study Type: Interventional
Study Design:
Allocation Randomized
Endpoint Classification Efficacy/Safety study
Intervention mode Crossover Assignment
Masking Double masking
Enrollment 240

35
Primary Endpoints Measured:
Neuropathy Impairment Score (NIS) [49]: The main symptom associated with GB syndrome is weakness,
numbness in the limbs of the patients due to the damage caused to peripheral nerves. NIS is a clinical
scoring system which is used to study the severity of the peripheral neuropathy. Scoring is provided as a
cumulative score which includes various components like sensation, reflexes, limb movement and muscle
weakness. The scoring system lies between 0(total recovery) to 80(total impairment).
Secondary Endpoints Measured:
Cytokine concentration in CSF: The drug mainly is designed to act on p-38 MAPK which is involved in
cytokine production which in turn reduces the differentiation of T cells. Hence the efficacy of drug action
can be measured by the concentration of cytokines in the CSF i.e. lesser the concentration more efficient
the drug.
Cerebrospinal Fluid biomarker concentration as explained earlier- the CSF is extracted from patients
through lumbar puncture from the spinal cord.
Patient Eligibility:
Age group: 19 – 75 years.
Genders: Both
Healthy Volunteers: No
Inclusion Criteria:
Patients must be of age group 19 to 75 years only.
Patient should have neurological impairment due to GBS only which has been stable for less than 12
months.
Patient must comply with the therapy and protocols provided during the study.
Exclusion Criteria:
Subjects must not be involved in any other medication.
Pregnant and lactating women are not allowed to participate.
Heart patients are not allowed to participate.
Subjects must not be enrolled in any other trial.
Study Title [50]:
Assessment of efficacy/safety of GB004 in GB syndrome patients.
This trial was conducted on a total of 240 patients suffering from GB Syndrome. Patients were orally administered
with the GB004 drug in cohorts of 80. Initial dose was 50 mg twice a day the dose was increased in an order for
the subsequent cohorts. The dose range selected does not exceed the hMTD deduced in the previous phase. The
different endpoints and the biomarker assays were conducted every week to check the progress of treatment in
patients. The results obtained were tabulated as follows. Placebo was used as a control to measure the results

36
against. This trial was conducted for a time period of a year. The different values obtained for respective doses
and the results are shown in table 13.
Dose
(mg)
No. Of
Patients
No. Of
Patients
with AE
NIS Score
Cytokine
Conc.
(ng/ml)
Biomarkers
Nfs (ng/ml) IP-10 (ng/ml)
Placebo=0 60 1 78 200 0.73 11
50 60 10 70 180 0.60 9
150 60 15 30 100 0.30 5
250 60 21 15 80 0.18 3
Table 13: Phase II trial results
Adverse effects:
The adverse effects were minimal and tolerable. The two main adverse effects observed with these patients were
Hyperglycemia: Normal blood glucose levels in humans before food needs to be 70-99 mg/dL. The
patients in the clinical trials who were administered with the drug showed slight elevation in the glucose
level. This elevation in glucose level was constant with increase in drug doses. Precautions were taken to
administer insulin injections if glucose level exceeded alarmingly.
Weight Loss
CONCLUSION OF PHASE II TRIAL:
From the study it was determined that dose of 150 mg twice a day for the following reasons:
- The number of patients showing adverse effects are less than 33% unlike the very next dose where it
exceeds 33%.
- NIS score and cytokine concentration drops to optimum levels.
- Decrease in concentration of the biomarkers Nfs and IP-10 indicate the effectiveness of the drug.
- Even though the very next dose shows decrease in biomarker concentration in the CSF this dose cannot
be selected for further calculations since more than 33% of the patients show adverse effects.
IND UPDATE
- PHASE II Clinical Trial Summary
An optimum dose of 150 mg twice a day was concluded for the further phase III trials in humans.
Phase II trials was conducted for a duration of 2 years.
- Product Release information:
The dose of administration and the route of administration and other information related to the drug
release in provided.
- Chemistry, Manufacture and cost:
Stability tests are conducted for a time period of 2 years and the drug is stable for clinical use.

37
Details about protocol and materials required for manufacturing is provided.
Chemical, physical and biological properties of the drug is provided.
Labeling information and packaging information has been updated and the information is
provided.
Cost margin for the manufacture of the drug is provided.
- All impurities are identified, qualified and quantified and the information is provided.
- Further non clinical studies on animals were conducted for a period of one year, to test the toxicology of
the drug before entering phase III trials.
INVESTIGATOR’S BROCHURE UPDATE:
An updated copy of the document was submitted to Institutional Review Board (IRB) for review. A copy of this
as per requirement was submitted to FDA with IND application. This includes all clinical, pharmacological
information, toxicology studies of the drug GB004 [NCE (4)].
PHASE III CLINICAL TRIALS:
Study Objective: To test for the safety and effectiveness of GB004 drug in treating patients suffering from
Guillain Barre Syndrome.
Study Type: Intervention
Study Design [51]:
Allocation Randomized
Intervention Model Single group assignment
Masking Open label
Endpoint Classification Efficacy/Safety study
Purpose Treatment
Enrollment 2000
Eligibility:
Age group: 18 years and above.
Gender: Both
Healthy Volunteers: No

38
Inclusion Criteria:
It must have been atleast 2 weeks since the start of symptoms associated with GB Syndrome.
Patients must have an NIS score greater than or equal to 70.
Exclusion Criteria:
Patients with complete paralysis are not allowed to participate
Should not be involved in any other trials or in any other medications
Pregnant and lactating women are not allowed to participate.
Patients should not be treated with Immunoglobulin therapy prior to this study.
Patients with liver, cardiovascular diseases are not allowed.
Primary and Secondary endpoints mentioned is as same as the one performed in phase II.
Title: A randomized, open label study for testing safety and effectiveness of GB004 drug in treating patients with
GB Syndrome.
The study involved 2000 patients divided into cohorts of 1000 each. Placebo was selected as the Gold standard
against which the safety and efficacy of the drug would be measured. The first cohort was administered with the
placebo and the second cohort was administered with 150 mgs of the drug twice a day. The study was conducted
for a duration of one year. The trial outcome was tabulated as follows:
Trial Arm
Number
of
Patients
Number of
patients with
AE
NIS Score Cytokine Concentration
ng/ml
Placebo 1000 50 70 190
GB004
150 mg
1000 100 5 80
Table 14: Phase III trial outcomes.
Adverse Effects
The major side effects seen were fever, headache and slight increase in blood glucose level (not alarming). These
side effects were optimal and tolerable. Paracetemol tablets could be administered if fever persisted for more than
24 hours. Headache also if persistent can be treated with pain meds.
CONCLUSION:
In the first 1000 patients treated with Placebo it was seen that more than 700 people had NIS of 75 and
cytokine concentration of 190 ng/ml indicating the ineffectiveness of the placebo in treating the disease.
In the next set of 1000 patients treated with GB004 900 subjects showed a drop in the NIS score from 75
to 5, which seemed to be a drastic change in the motor functions of these patients. Also the cytokine
concentration in the blood dropped to 80 ng/ml indicating the effectiveness of drug.

39
This proves that the drug is positively inhibiting p-38 MAPK (cytokine concentration drop) as a result of
which the autoimmune attack on the nerves are inhibited which is reflected by the drop in NIS indicating
patients regaining their motor functions.
The number of people showing adverse effects when the dose of 150 mg is lesser than 33% hence it shows
the safety of the drug.
The points indicated above shows that the drug at 150 mg twice a day is effective in treating the disease and is
also safe for patients.
The total duration on phase III trials was 2 years
NEW DRUG APPLICATION (NDA)
Pyramidine Imidazole molecules are used as potent drugs to treat GB Syndrome.
Drug: GB004
Mechanism of Action: These are oral drugs that are p-38 MAP Kinase inhibitors. P-38 MAPK are responsible
for cytokine production which leads to T cell differentiation which leads to immune response against the neurons.
GB004 bind irreversibly to the enzyme, blocks the active site of the enzyme for substrate binding and hence
inhibits the immune reaction.
All three phases of the clinical trial show positive results and is proof that the drug is safe, effective and tolerable
with minimal side effects. An efficacious dose of 150 mg twice a day has been deduced from phase 2 n 3 and the
results for the same is provided with the trial details. 408 mg was deduced as the hMTD during phase 1 clinical
trial, hence administration of 150 mg twice a day (making it 300 mg) is way less than the hMTD value this
indicates the safety of the drug.
The medical need for the discovery of GB004 was that till date there has been no such oral drugs used for treating
GB Syndrome patients, IV administration of antibodies and Plasmapheresis were the only two treatments
available. GB004 is an oral drug which is proved to treat GB Syndrome with a dose of 150 mg twice a day. With
this research it is evident that the drug full fills the medical need it was built on.
Hence the approval of this drug would avoid the painful and tedious treatments like Plasmapheresis or
Immunoglobulin administration, instead patients with GB Syndrome can just orally consume GB004 at specified
dose. Patients will not have to be bed ridden for the entire course of treatment.

40
A Summary of the entire clinical trial activities is provided below in the form of a flow chart indicated
below.
IND APPLICATION
DRUG GB004 [NCE (4)]
Route of Administration Oral
CMC Stability tests for 1 year
Manufacturer’s information
Manufacturing protocol
PK-PD Studies Efficacious dose: 0.21 mg/kg
Cmax at 6 to 8 hours after administration
Safety Pharmacology and Toxicology Studies CYP and hERG inhibition assay
On target and off target toxicity studies
Genotoxicity
Regulatory toxicology studies
First In Human Dose MSRD: 0.432 mg/kg for human trials
PHASE I CLINICAL TRIALS:
Purpose Safety & Efficacy study
Number of patients 112 in cohorts of 16
Healthy Volunteers Yes
Total duration 6 months
hMTD Determined 6.88 mg/kg = 408 mg
IB PROVIDED

41
IND UPDATE:
Non clinical toxicity studies for one year.
Stability tests for 1year.
CMC information provided
IB provided.
PHASE II CLINICAL TRIALS:
Purpose Safety and initial efficacy studies
Enrollment 240 in cohorts of 60
Healthy Volunteers No
Starting dose 50 mg twice a day
Adverse Effects Hyperglycemia, weight loss
Efficacious dose 150 mg twice a day
Total duration of study 1 year
IB
IND UPDATE:
Optimum dose for next trial 150 mg twice a day
Non clinical toxicity studies for one year.
Stability tests for 2year.
CMC
IB provided.
IB

42
FILING NDA
NEW DRUG APPLICATION was filed and submitted to the FDA for approval.
PHASE III CLINICAL TRIALS:
Purpose To Test the Effectiveness of the drug
Enrollment 2000 in cohorts of 1000
Healthy Volunteers No
Gold Standard Placebo
Dose administered 150 mg twice a day
Total Duration 2 years

43
REFERENCE:
[1] “Guillain-Barre Syndrome”, Wikipedia.
[2] Landry, Jean-Baptiste (1859). "Note sur la paralysie ascendante aiguë". Gazette Hebdomadaire de Médecine
et de Chirurgie.
[3] J.B. Winer, (2001), Clin Pathol: Mol Pathol, (54), pp: 381-385.
[4] Satoshi Kuwabara MD (2007), Current Neurology and neuroscience reports, Volume 7, Issue 1, pp 57-62.
[5] Prof. Richard A.C Hughes, (2005), Guillain-Barre syndrome, (366), pp: 1653-1666.
doi: http://dx.doi.org/10.1016/S0140-6736(05)67665-9
[6] R.A.C Hughes, R.D.M Hadden, N.A Gregson & K.J. Smith, (1999), Pathogenesis of Guillain-Barre
syndrome, (100), pp: 74 – 97. doi: http://dx.doi.org/10.1016/S0165-5728(99)00195-2
[7] http://emedicine.medscape.com/article/315632-overview
[8] Hughes RA, Swam AV, Van Doorn PA, (2014), Intravenous immunoglobulin for Guillain-Barre syndrome,
the cochrane database of systemic reviews 9.
[9] Davidson I, Wilson T, Brissenden S (2009), “Physiotherapy and Guillain-Barre syndrome. Results of national
survey”. Physiotherapy 95 (3), pp 157 – 163.
[10] Mitlen Alter, Ann neurol (1990), “The epidemiology of Guillain-Barre syndrome”
[11] Dalakas MC, (1999), “Intravenous immunoglobulin in the treatment of autoimmune neuromuscular diseases:
present status and practical therapeutic guidelines. Muscle Nerve”.
[12] Gordan. R. Kelley, M.D, Stanley J. Swierzewski III, M.D, “Guillain-Barre Syndrome”, remedies
health.communities, 2 January 2000, 28 October 2014.
[13] http://www.cslbehring-us.com/s1/cs/enus/1255929751839/Web_Product_C/1151517249408/Product.html
[14] “Gammard Liquid [Immunoglobulin Infusion (Human)] 10%”, Baxter Healthcare Corporation, April 2014.
[15] Chen Dong*, Derek D. Yang*², Cathy Tournier³, Alan J. Whitmarsh³, Jie Xu*, Roger J. Davis² & Richard
A. Flavell*, (2000), “JNK is required for effector T-cell function but not for T-cell activation”, Letters to nature,
Vol 205, pp: 91-94
[16] Hong-Liang Zhang, Limin Wu, Xiujuan Wu & Jie Zhu, (2014), Expert Opin. Ther. Targets, 18(4), pp: 355-
363. doi: 10.1517/14728222.2014.882899. Epub 2014 Jan 30.
[17] “T helper cells”, “Wikipedia”, December 2007.
[18] Kishan Kumar Nyati & Kashi Nath Prasad, (2014), “Role of cytokines and Toll like receptors in the
Immunopathogenesis of Guillain-Barre syndrome”, Mediators of Inflammation, Hindwani Publishing
Corporation.

44
[19] Boehm U, Klamp T, Groot M, et al., (1997), “Cellular responses to interferon-gamma”, Annu Rev Immunol,
(15), pp: 749-95
[20] Stefan A. Laufer, Gerd K. Wagner, (2002), From Imidazoles to Pyrimidines: New Inhibitors of Cytokine
Release, Vol. 45, pp: 2733-2740.
[21] http://pubchem.ncbi.nlm.nih.gov/
[22] Forrer, P.; Tamaskovic, R.; Jaussi, R., (1998) Enzyme-Linked Immunosorbent Assay for Measurement of
JNK, ERK, and p38 Kinase Activities. Biol. Chem., 379, pp: 1101-1111.
[23] Stefan A. Laufer, Gerd K. Wagner, (2002), From Imidazoles to Pyrimidines: New Inhibitors of Cytokine
Release, Vol. 45, pp: 2733-2740.
[24] http://www.molinspiration.com/
[25] Forrer, P.; Tamaskovic, R.; Jaussi, R., (1998) Enzyme-Linked Immunosorbent Assay for Measurement of
JNK, ERK, and p38 Kinase Activities. Biol. Chem., 379, pp: 1101-1111.
[26] Donat, C.; Laufer S, (2000), In vitro screening assay to evaluate cytokine release inhibitors. Arch. Pharm.
Pharm. Med. Chem., Vol. 12, 333
[27] Dirk Theile, Walter Emil Haefeli , Johanna Weiss, (2014) Effects of adrenolytic mitotane on drug elimination
pathways assessed in vitro, Springer Science+Business Media, doi: 10.1007/s12020-014-0517-2.
[28] N.P. van Erp, H.J. Guchelaar, B.A. Ploeger, J.A. Romijn, J.D, Hartigh, H. Gelderblom, (2011) Mitotane has
a strong and a durable inducing effect on CYP3A4 activity, pp: 621–626.
[29] Stephan.R.Johnson, Hongwen Yu, Mary Lee Conder, Hong Shi, Arthur M. Doweyko, John Loyd and Paul
Levesque, (2007) Estimation of hERG inhibition of drug candidates using multivariate property and
pharmacophore SAR, Bioorganic & Medicinal Chemistry, Vol 15, pp: 6182-6192.
[30] B. Budziszewska, M. Szymanska, M. Leskiewicz1, A. Basta-Kaim, L. Jaworska-Feil, M. Kubera D. Jantas1,
W. Lason, (2010), Journal Of Physiology And Pharmacology, “The Decrease In Jnk- And P38-Map Kinase
Activity Is Accompanied By The Enhancement Of Pp2a Phosphatase Level In The Brain Of Prenatally Stressed
Rats”, Vol. 62, pp: 207-215.
[31] Pazirandeh A, Xue Y, Prestegaard T, Jondal M, Okret S, (May 2002). "Effects of altered glucocorticoid
sensitivity in the T cell lineage on thymocyte and T cell homeostasis". Vol 16, pp: 727–729.
doi:10.1096/fj.01-0891fje. PMID 11923224.
[32] Liguo New and Jiahuai Han, (1998), TCM, “The p38 MAP Kinase Pathway and Its Biological Function”,
Vol 8, pp: 220-228.
[33] Christina Böhm, Silvia Hayer, Anita Kilian, Mario M. Zaiss, Susann Finger, Andreas Hess, Klaus Engelke,
George Kollias, Gerhard Krönke, Jochen Zwerina, Georg Schett and Jean-Pierre David, (2009), The journal of
immunologyh, “The a-Isoform of p38 MAPK Specifically Regulates Arthritic Bone Loss”, Vol: 183, pp: 5938-
5947.

45
[34] Simon Rousseau, (2013), Ucsd Molecule Pages, “p38 beta MAP kinase”, Vol 2,
doi:10.6072/H0.MP.A001718.01.
[35] Steven Whitebread, Jacques Hamon,Dejan Bojanic and Laszlo Urban, (2005), “In vitro safety pharmacology
profiling: an essential tool for successful drug development”, Vol: 10, pp:1421-1433.
[36] Leigh A. Stoddart, Andrea J. Vernall, Jessica L. Denman, Stephen J. Briddon, Barrie Kellam and Stephen J.
Hill, (2012), Fragment Screening at Adenosine-A3 Receptors in Living Cells Using a Fluorescence-Based
Binding Assay, Vol: 19, pp:1105-1115
[37] Nortina Shahrizaila and Nobuhiro Yuki, (2010), Journal of Biomedicine and Biotechnology, “Guillain-Barr´e
Syndrome AnimalModel: The First Proof of MolecularMimicry in Human Autoimmune Disorder”, Vol: 2011,
Article ID: 829129.
[38] N. Yuki, M. Yamada, M. Koga et al., (2001), Annals of Neurology “Animal model of axonal Guillain-Barr´e
syndrome induced by sensitization with GM1 ganglioside,” vol. 49, pp. 712–720.
[39] Mu Yang, Anthony Rainone, Xiang Qun Shi, Sylvie Fournier and Ji Zhang, (2014), “A new animal model
of spontaneous autoimmune peripheral polyneuropathy: implications for Guillain-Barré syndrome”.
[40] Zehntner SP, Brisebois M, Tran E, Owens T, (2003), “Fournier S: Constitutive expression of a costimulatory
ligand on antigen-presenting cells in the nervous system drives demyelinating disease”, Vol:17, pp: 1910–1912.
[41] Gibson SJ, Polak JM, Bloom SR, Sebate IM, Mulderry PM, Ghatei MA, McGregor GP, Morrison JF, Kelly
JS, Evans RM, (1984), “ Calcitonin gene related peptide immunoreactivity in spinal cord of man and of eight
other species”, Journal of Neuroscience, Vol:4, pp:3101-3111.
[42] Wikipedia.com/chemokines
[43] Johannes Brettschneider, Axel Petzold, Sigurd Süssmuth &Hayrettin Tumani, (2009), “Cerebrospinal fluid
biomarkers in Guillain- Barré syndrome – Where do we stand?”, Journal of Neurology, pp: 3-12.
[44] Kastenbauer S, Koedel U, Wick M, Kieseier BC, Hartung HP, Pfister HW, (2003), “CSF and serum levels
of soluble fractalkine (CX3CL1) in inflammatory diseases of the nervous system”. Journal of Neuroimmunol, pp:
210-217.
[45] Coelho T, Maia L, Martins da Silva A, et al. Tafamidis for transthyretin familial amyloid polyneuropathy: a
randomized, controlled trial
[46] National Centre for Replacement, Refinement and Reduction of Animals in Research. “Guidance on Dose
Level Selection for Regulatory General Toxicology Studies for Pharmaceuticals”.
[47] U.S Department of Health and Human Sciences, FDA, CDER, (2005), “Guidance for Industry Estimating
the Maximum Safe Starting Dose in Initial Clinical Trials for Therapeutics in Adult Healthy Volunteers”
[48] www.clinicaltrails.gov
[49] Coelho T, Maia L, Martins da Silva A, et al. Tafamidis for transthyretin familial amyloid polyneuropathy: a
randomized, controlled trial.

46
[50] FDA Office of Orphan Products Development, “Assessment of Chronic Guillain-Barre Syndrome
Improvement with Use of 4-aminopyridine”
[51] Nihon Pharmaceutical Co. Ltd., “Phase III Clinical Trial of NPB-01 in Patients with Guillain-Barré
Syndrome”

















