Drug Development Stages, Departments & Careers Clinical trials: The various Roles & Obligations; SOP for a Clinical trial Patrick Brunel, MD, MSc, Cardiologist Fundamentals in Clinical Research – Module 4 Clinical trial Conduct & Data Handling, 10-Jan-2015, Beirut, Lebanon
Transcript
Drug Development Stages,
Departments & Careers
Clinical trials: The various Roles &
Obligations; SOP for a Clinical trial
Patrick Brunel, MD, MSc, Cardiologist
Fundamentals in Clinical Research – Module 4 Clinical trial
Conduct & Data Handling,
10-Jan-2015, Beirut, Lebanon
Drug Development Stages
A new path to drug discovery will be essential:traditional discovery & development takes a long time
10’000 compounds in the beginning
1 new Medicine on the market
10 compounds in the clinic
Preclinical
Phase incl.
Research
Clinical
Phase I
1’000 compounds in vitro testing
~14 years
Clinical
Phase II
Clinical
Phase IIIRegistration
ICH E8 Guidelines(Federal Register. 29540, May 30, 1997)
“ The studies may be classified according to their objectives.
The concept that clinical drug development is comprised of
four temporal phases, I through IV, is widely used. It is
important to appreciate that this is a description not a set of
requirements, and that for some drugs and development
programs the typical sequence will not be appropriate or
necessary.”
Phases in Drug Development
Discovery
• New compounds are evaluated for their potential to treat disease
• Limited preclinical safety studies are conducted
• A route of synthesis is established
Pre-clinical
• Determines the efficacy and tolerability in animal models
• Successful candidates are tested to substantiate safety
• Pharmacokinetic parameters are defined
• Preliminary formulation is developed
Clinical Phase I
• Tolerability in humans
• Successful products are tested in an initial Proof of Concept study to demonstrate activity in man
• Pharmacokinetics and metabolisms are evaluated in humans (usually healthy volunteers)
• Formulation is refined
• Animal safety studies required for longer term treatment are conducted
• A Pre-GPT is formed
Phases in Drug Development
Clinical Phase II
• Phase in which dosage and efficacy in patients is determined
• Some preliminary data provided on the effectiveness of the drug for a particular indication or indications in patients with a disease or condition
• This phase helps determine the common short-term side effects and risks associated with the drug
• Safety and clinical pharmacology studies also take place
Clinical Phase III
• Large-scale clinical trials for safety and efficacy in large patient populations
• Controlled and uncontrolled trials are conducted in this phase to gain additional information about effectiveness and safety needed to evaluate the overall benefit-risk relationship of the drug
Phases in Drug Development
Registration
• Product registration occurs
• Regulatory agencies review documents and decide about approval
• Marketing authorization is granted
• Additional studies, both post-marketing requirements and profiling studies (Phase IV,) are set in motion
Launch/Post-Launch
• Physicians prescribe the product to patients
• Phase IV studies continue to profile the product, and to comply with regulatory requirements
• Additional indications and/or formulations may be developed (life cycle management)
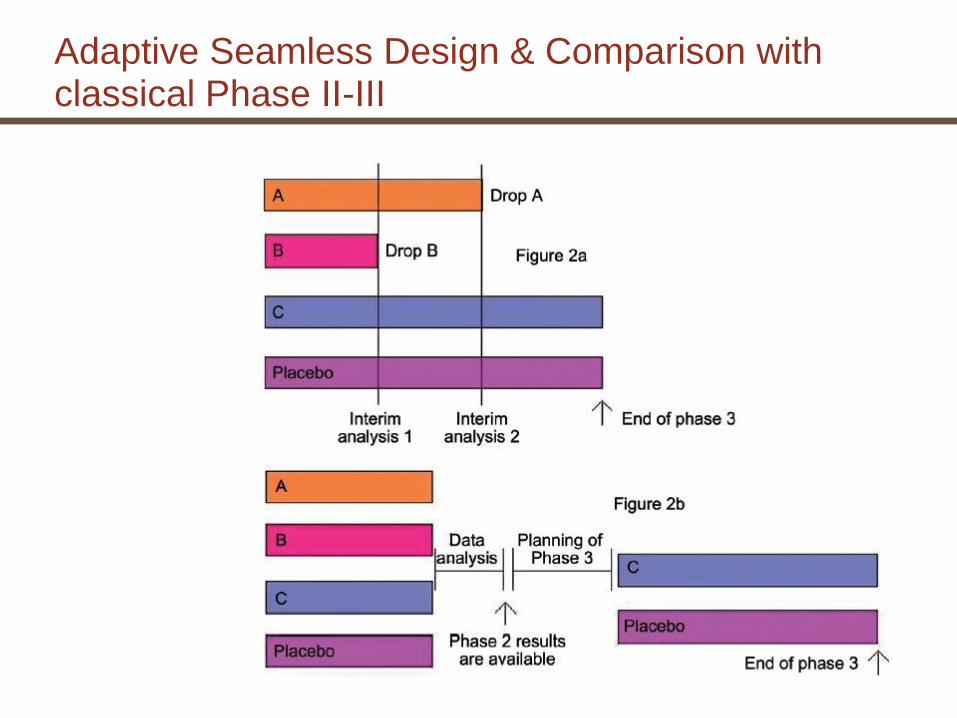
Flexible Designs (Definitions)
• Seamless designs join two distinct subsequent trials in a drug program into a single trial without combining the information.
The trials should not differ operationally (essentially the same CRF, centers, indication, …) and be defined in a single protocol.
• Adaptive designs allow for initial uncertainties in trial design to be confirmed/adapted during the trial.
The integrity of the trial is maintained and the evidence for the same hypothesis before and after the adaptation is combined.
• Adaptive seamless designs (ASD) take advantage of both: Information gathered in the first stage (IIb) of the trial is used to adapt the design for the next stage (III), which seamlessly follows; the information from the learning stage will contribute evidence to the overall conclusions
Examples of Adaptations in Adaptive Designs
• Re-estimation of sample size
• Dropping or adding or changing treatment arms.
• Sample size allocation to treatment arms
• Patient population (selection, exclusion or enrichment)
• Statistical testing strategy
Adaptive Seamless Design & Comparison with classical Phase II-III
11 | Presentation Title | Presenter Name | Date | Subject | Business Use Only
Advantages of Structured Development
Clear defined goals and data collection.
Operational convenience and simple.
Decision process well defined.
• In control of the sponsor with advice on development from broad external experts with evidence (safety and efficacy).
• Little or no pre-definition of decisions required prior to each phase completion.
Easier regulatory framework with meetings and advice at clearly defined milestones.
Higher probability of success.
Rationale for Flexibility
Treatment less toxic.
Therapeutic efficacy manifested due to different mechanisms than historical standard
Accumulating evidence from the current trial or other trials.
Availability of target patient population.
• Each patient is valuable resource.
May require less patients (not always!)
Allows for initial uncertainties in trial design.
Conclusions
Structured development works reasonably well in most cases.
Combining Phase I and II should be considered only where applicable and not be considered as “default”.
• Saving time and resources should be adequately justified.
Combining Phase II and III should be considered only in rare (orphan) diseases.
• Exceptions should be clearly justified.
• In all cases, prior regulatory acceptance is strongly recommended.
Innovative approaches should be considered more in exploratory phases to accelerate development.
Clinical Trial Team:
The various Roles and their Obligations
Clinical trial Team Mission
The Clinical Trial Team (CTT) has the mandate to:
• plan, implement and execute a clinical trial as outlined in the Clinical Development Plan (CDP) to ultimately deliver the clinical study report
• provide leadership, overarching and cross-functional strategic guidance and coordination
• ensure efficient and timely execution of the clinical trial
• align Line Functions (LFs) represented on the CTT with trial goals and objectives
• facilitate and encourage open, honest and objective communication and transparency within and across all LFs, “top down and bottom up”
• anticipate potential risks and issues, come up with mitigation strategies, and make prompt decisions
• provide periodic progress report to the International Clinical Team (ICT)
• ensure detailed project timelines are defined, documented, and maintained taking into account cross-functional dependencies for achievement of intermediate and major milestones
The CTT has the global responsibility to drive the clinical trial forward and to ensure that the objectives set in the CDP and yearly goals are met. The ultimate goal is to deliver the clinical study report on time, on budget with high quality.
Clinical Trial Team Working Principles
Continuity
• Assume day-to-day responsibilities for the clinical trial, encourage communication between team members, LFs and make decisions and take prompt action.
Teamwork
• Lead by the Clinical Trial Head (CTH) who has global accountability for the trial and serve as the central contact person for coordination of all aspects of the clinical trial.
• The progress of the clinical trial is reviewed on a regular basis.
• Issues should be discussed and resolved at the CTT level whenever possible. Line Functions (LF) members on the CTT will provide expertise and guidance supporting the decision making and/or issue resolution process.
• Each team member is empowered by his LF and expected to represent a consensus of his/her respective LF to the CTT.
• If activities are outsourced to a CRO, the CRO representative becomes a CTT member in addition to the LF representative.
CTT Working Principles
Communication
• Good communication between all team members is key to the success of the CTT.
• Teams may rely on a range of tools including using available electronic team tools, one-to-one discussions, and team meetings.
• All team members are expected to initiate interactions and discussions within the team
• The CTH/CTT is responsible for on the study progress. Deviations from the planned study timelines and trial issues that may impact other trials or the overall project should be brought promptly to the attention of ICT.
The CTT, under the leadership of the CTH, has to set yearly CTT goals to be endorsed by each LF member and their management. These objectives must be in line with the ICT/GPT objectives, reviewed at least yearly and when significant changes arise, and are expected to be reflected in individual team members’ personal objectives.
They are considered as a contractual agreement between the CTT and the Line Functions.
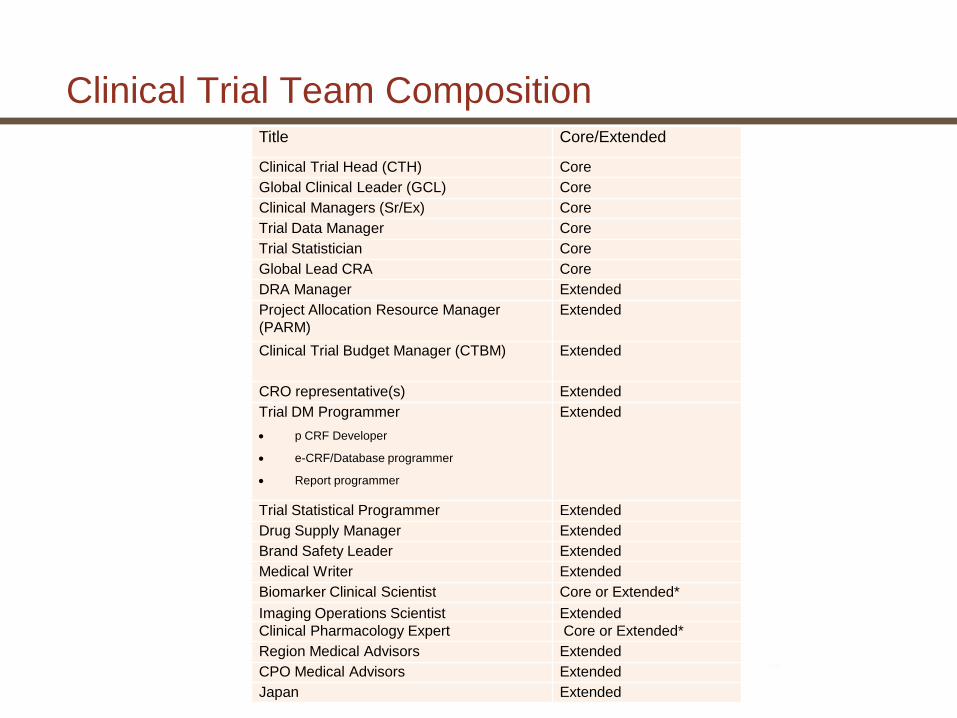
Clinical Trial Team CompositionTitle Core/Extended
Clinical Trial Head (CTH) Core
Global Clinical Leader (GCL) Core
Clinical Managers (Sr/Ex) Core
Trial Data Manager Core
Trial Statistician Core
Global Lead CRA Core
DRA Manager Extended
Project Allocation Resource Manager
(PARM)
Extended
Clinical Trial Budget Manager (CTBM) Extended
CRO representative(s) Extended
Trial DM Programmer
p CRF Developer
e-CRF/Database programmer
Report programmer
Extended
Trial Statistical Programmer Extended
Drug Supply Manager Extended
Brand Safety Leader Extended
Medical Writer Extended
Biomarker Clinical Scientist Core or Extended*
Imaging Operations Scientist Extended
Clinical Pharmacology Expert Core or Extended*
Region Medical Advisors Extended
CPO Medical Advisors Extended
Japan Extended
Roles and Responsibilities of CTT members
Clinical Trial Head (CTH)
• Chair CTT meetings
• Lead and matrix manage the global multidisciplinary CTT to ensure that all trial deliverables are met according to timelines, budget, quality standards and operational procedures
• Report study progress and issues with their resolution plan to ICT
Global Clinical Leader (GCL)
• Responsible for the achievement of indication-specific objectives and goals as defined by the CDP
• Responsible for providing medical leadership to the protocol, clinical components of regulatory documents/registration dossier and brand related medical information, clinical communication and publications relating to this trial
Roles and Responsibilities of CTT members
Clinical Manager (CM)
• Supports the CTH with deliverables for the study
• Assists with meeting organization, and drafting agenda and minutes
DRA Manager
• Responsible for providing guidance on regulatory issues/questions/strategy
Project Allocation Resource Manager
• Represents all GMO functions at the CTT serving as an interface between the CTT and field monitors
• Responsible for trial allocation and executing the monitoring plan
• Responsible for negotiating country level budget and monitoring resources
• Prepares the global recruitment plan, provides information on recruitment status/issues and implements contingency plans
Roles and Responsibilities of CTT members
Trial Data Manager (TDM)
• Leads CRF development, contributes to the review of protocol and contributes to the monitoring plan
• Responsible for chairing Validation & Planning meeting and finalizing VAP documents
• Informs CTH of key data issues from all sources including CRF data entry/query resolution status and data quality
• Shares responsibility with CTH for trial design, protocol and CRF development
• Responsible for randomization scheme
• Chairs Report and Analysis Plan meeting and responsible for finalization of RAP documents
• Contributes to monitoring plan, VAP, data review and trial report shell
Roles and Responsibilities of CTT members
Clinical Trial Budget Managers
• Prepare in collaboration with CTT the TTG
• Evaluates the impact of CTT decisions on the clinical trial budget
CRO Representative(s)
• The CRO representative(s) serves in addition to the LF that is outsourced on the CTT, under the responsibility of the line function who has contracted out this activity with regards to compliance to the company quality and timelines standards.
Trial DM Programmer (Based on roles)
• Responsible for CRF development/database specification/design.
• Responsible for development of data transfer specifications for third party data
Roles and Responsibilities of CTT members
Trial DM Programmer (Con’d)
• Produces exception reports, monitoring data listings, and Patient Profiles for data cleaning and validation efforts
• Provides input to Protocol Deviation (PD) Specification and Development of PD programs.
• Contributes to protocol review and VAP documents
Trial Statistical Programmer
• Responsible for derived analysis data set specifications
• Performs dry runs
• Produces summary tables and data listings for the Clinical Study Report
• Contributes to CRFs, VAP and RAP documents
Roles and Responsibilities of CTT members
Safety Leader (SL)
• Represents Drug Safety and Epidemiology
• Responsible for assessment and interpretation of safety and pharmacovigilance activities
• Contributes to protocol preparation and the safety sections of the Clinical Study Report
Global Lead CRA (GLCRA)
• Represents field monitoring
• Responsible for preparing the monitoring plan
Drug Supply Manager (DSM)
• Contributes to protocol development
• Forecast and coordination of drug supplies
Roles and Responsibilities of CTT members
Medical Writer (MW)
• Coordinates the editing and/or writing of trial documents (i.e., CSR, SAE narratives, summary reports)
• Contributes to the AP
Biomarker Clinical Scientist (BCS)
• Provide input for all aspects of biomarker samples (consent, lab SSWs, data management, CRFs, data transfers, site/monitor training).
Imaging Operations Scientist
• Imaging Operations Scientists provide support to clinical teams on all aspects of imaging related activities including ISSW and selection imaging CRO vendor, study start up activities (input to imaging lab manuals etc), and provide input to clinical trial documents
Roles and Responsibilities of CTT members
Clinical Pharmacology Expert (CPE)
• Responsible for providing clinical pharmacology leadership to the protocol, clinical components of regulatory documents/registration dossier and brand related medical information, clinical communication and publications relating to this trial
• Shares responsibility with CTH and TS or CPS for trial design, protocol and CRF development
• Provides input to the clinical pharmacology and pharmacokinetics objectives and analyses in the protocol
• Co-ordinates activities related to exploratory and modeling and simulation analyses of the study; convenes as needed members of the “quantitative circle” (i.e. CPE, TS and/or CPS, Modeling and Simulation Expert, Biomarker Clinical Scientist)
Region, Country, Japan Medical Advisors
• Provide input from their respective countries/regions with regards to the preparation and conduct of the trial locally
| Change Advocate Onboarding | DEV OD | June 2012 | Fran/GCO Better Development | Business Use Only27 27
What is RACI?A tool used for clarifying ownership & decision making
Responsible“The Doer”
Individual who:
actually completes task or supports
task completion with other Rs or A
Responsible for action / implementation
Degree of Responsibility is determined by the
Accountable individual
Accountable
Individual who:
is accountable to ensure it happens
Signs-off on work completed: answerable for the
correct & thorough completion of the deliverable or task
Only one “A” can be assigned to a decision, task, or
deliverable
Informed
Individual who:
needs to be informed after a decision
or action
One-way communication
Kept up-to-date on progress, typically at task or
deliverable completion
Consulted
Individual who:
is consulted during decision-making
process or action
Two-way communication
A strong role, includes right to escalate
Typically Subject-Matter Experts
“The Expert”
“The Notified”“The Approver”
| Change Advocate Onboarding | DEV OD | June 2012 | Fran/GCO Better Development | Business Use Only28
Clinical trial Team charter
Clinical Development Plan
| Change Advocate Onboarding | DEV OD | June 2012 | Fran/GCO Better Development | Business Use Only30
Involve :• Therapeutic Area Head
• Global Project Leader
• Clinical trial Head
• ...as appropriate
Consult :• External scientific experts
• Health Authorities: FDA (IND meeting), EMEA (Scientific advice)
Include :• Clinical studies (Phases I-III)
• Supportive studies (Specific populations, drug interaction,
population PK etc.)
Clinical Development Plan
| Change Advocate Onboarding | DEV OD | June 2012 | Fran/GCO Better Development | Business Use Only31
CDP – Points to consider
Defining the unmet need and assessing the benefit-risk
• Is the unmet medical need established in the intended population and are the benefit-risk assumptions realistic?
• Is there alignment with the assumptions made in the TPP?
• Are the decision criteria for Go/No-Go adequately defined?
• Are the critical safety and other project risks adequately prioritized with mitigation steps in place?
• Has modeling and simulation been engaged and is there a plan to evaluate dose response and the minimally effective dose?
| Change Advocate Onboarding | DEV OD | June 2012 | Fran/GCO Better Development | Business Use Only32
CDP – Points to consider
Optimizing Response/Facilitating Market Access• Have potential active comparators been well delineated?• Have novel or innovative clinical trial designs been considered?• Can the program be executed within the defined timeline (encountering
regulatory feedback, operational aspects, etc.)?• Does the strategy utilize profiling/biomarkers? Has development and timing of a
companion diagnostic been considered, if applicable?• Is there a plan for development and validation of other clinical outcome
assessments, if applicable?• Does the plan include clinical trials needed in “real world settings” to address
market access needs at the time of registration?• Have special populations been adequately considered and reflected?• Is the regional strategy adequately represented?• Is there a plan for payor engagement prior to approval and registration?
| Change Advocate Onboarding | DEV OD | June 2012 | Fran/GCO Better Development | Business Use Only33
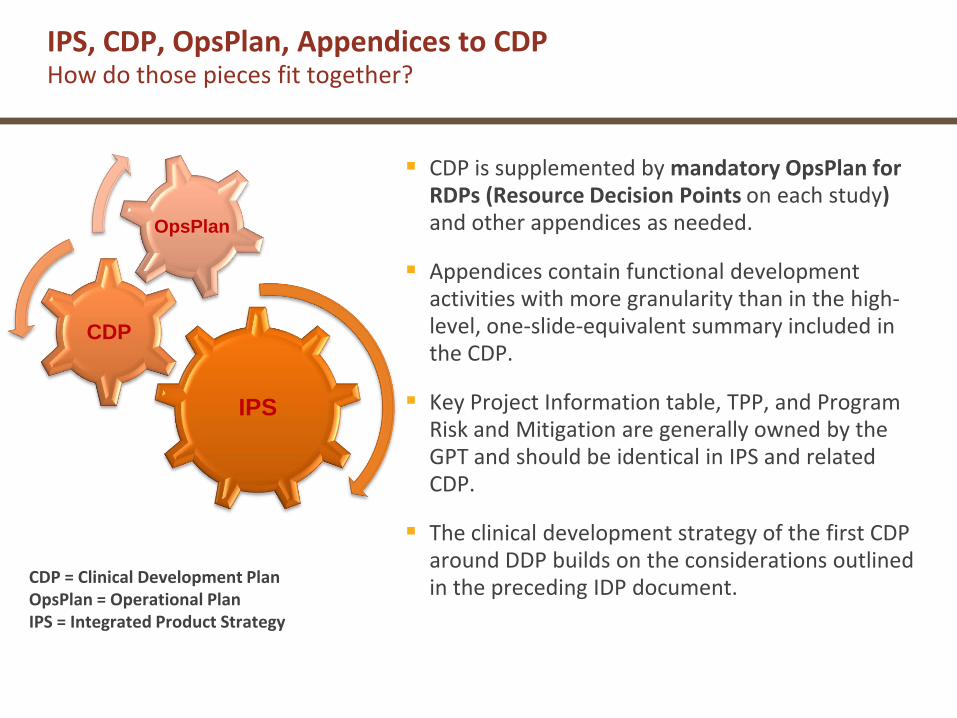
IPS
CDP
OpsPlan
CDP is supplemented by mandatory OpsPlan for RDPs (Resource Decision Points on each study)and other appendices as needed.
Appendices contain functional development activities with more granularity than in the high-level, one-slide-equivalent summary included in the CDP.
Key Project Information table, TPP, and Program Risk and Mitigation are generally owned by the GPT and should be identical in IPS and related CDP.
The clinical development strategy of the first CDP around DDP builds on the considerations outlined in the preceding IDP document.
IPS, CDP, OpsPlan, Appendices to CDPHow do those pieces fit together?
| Change Advocate Onboarding | DEV OD | June 2012 | Fran/GCO Better Development | Business Use Only34
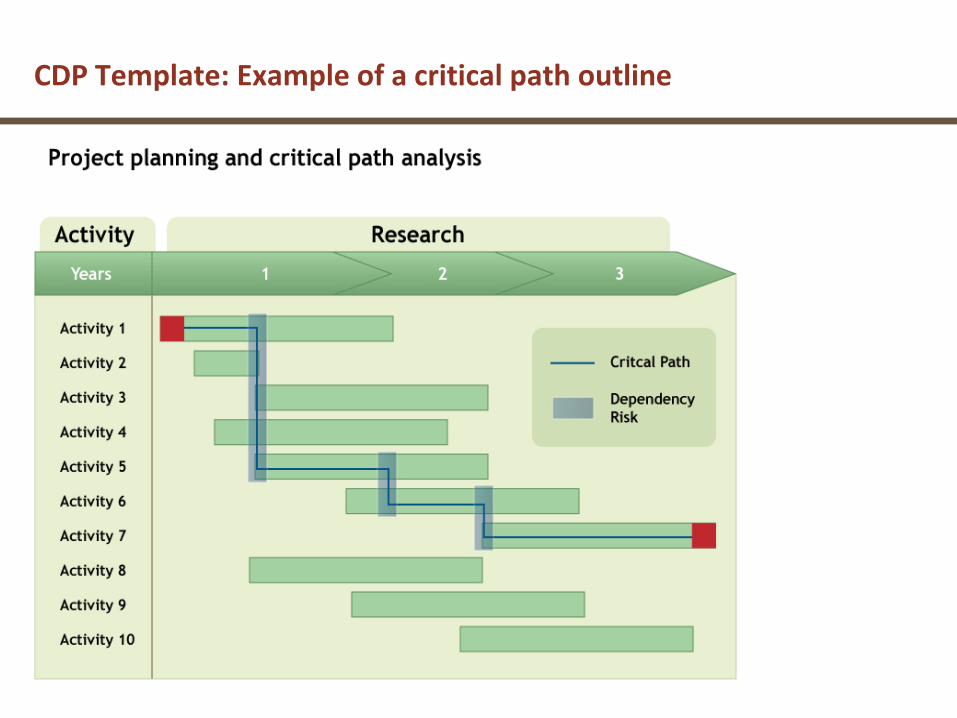
CDP Template: Example of a critical path outline
Clinical Study Set-up
| Change Advocate Onboarding | DEV OD | June 2012 | Fran/GCO Better Development | Business Use Only36
| Change Advocate Onboarding | DEV OD | June 2012 | Fran/GCO Better Development | Business Use Only37
Study set-up Roadmap (Clinical trial Toolkit – NIH)
| Change Advocate Onboarding | DEV OD | June 2012 | Fran/GCO Better Development | Business Use Only38
EU Clinical Trial Directive General
Came into force in May 2004 and has been implemented into
National Legislation in all EU countries. A Clinical Trial
Directive update is expected to be published in 2014.
Substantial differences across countries
Transposition
Country specific requirements
Scope of the Directive:
•Applicable for all interventional Clinical trials with Medicinal products
-Phase I (patient & healthy volunteers), II, III and IV studies
-Bio-availability and bio-equivalence studies
•Applicable to industry as well as academia sponsored clinical research
Directive National Legislation
| Change Advocate Onboarding | DEV OD | June 2012 | Fran/GCO Better Development | Business Use Only39
EudraCT Databasehttps://eudract.ema.europa.eu/
Purpose of this database:▪ The registry of all clinical trials in EU (contains for each trial: HA&EC
authorisation, protocol, amendment, end of trial….)
▪ To facilitate communication on clinical trials between authorities (accessible
by National HA, EMA, EU Commission)
▪ To enhance protection of subjects and patients receiving Investigational
Medicinal Product (IMP)
▪ To be linked to the EudraVigilance database (containing SUSAR database)
▪ Each clinical trial protocol is identified by a unique mandatory reference
number (EudraCT Number)
European database of all Clinical Trials within the EU
EudraCT Number format: YYYY-NNNNNN-CCYYYY: Year in which the number is issued
NNNNNN: six digit sequential number
CC: check digit39 _ Clinical trials in the EU_ June 2013_ Business Use Only
| Change Advocate Onboarding | DEV OD | June 2012 | Fran/GCO Better Development | Business Use Only40
CTA Dossier
Standard CT
Application Form
including
EudraCT Number
•Protocol & amendments
•Informed Consent Form
•Case report Form
Clinical Trial
Information
•Quality data
•Non-clinical pharmacology & toxicology data
•Clinical trial & previous human experience data
•Benefit/risk assessment
IMPD
(Core Dossier)
•Subject related
•Protocol related
•Facilities & staff related
•Finance related
Member State
Specific
Information
Changes to: protocol and/or key evolving data
impact on CTA documents Amendment
Not applicable in all countries
| Change Advocate Onboarding | DEV OD | June 2012 | Fran/GCO Better Development | Business Use Only41
CTA
Dossier
1.4 List of Competent Authorities
1.4.1 List of countries
…1- General
2.3 Arrangements for
Recruitment of Subjects2- Subject
Related
3.1.2 Case Report Form
3.2 Summary of the Protocol
in National Language…
3- Protocol
Related
4.2.2 Non clinical
4.2.3 Clinical trial & human exposure
4.3 SmPC…
4- IMP
Related
5.3 CV of Each Investigator
Responsible for the Conduct of a
Trial in a Site in the MS Concerned
5.4 Information about Supporting Staff
5- Facilities
&Staff related
6.6 Agreement between the
Investigators and the Trial Site
…
6- Finance
Related
1.1 EudraCT Number
1.2 Cover letter
1.3 Application form
2.1 Subject Information
and Informed Consent Form
2.2 Subject Information Leaflet
3.1 Protocol
with all Current Amendments
&Post-text supplements
4.1 Investigator’s Brochure
4.2 Investigational
Medicinal Product Dossier (IMPD)
4.2.1 Quality Data
5.1 Facilities for the Trial
5.2 CV of the Coordinating
Investigator in the MS Concerned
6.3 Compensations to Investigators
6.4 Compensations to Subjects
6.5 Agreement between Sponsor
and the Trial Site
IMPD/CTA preparation Detailed content of the CTA Dossier
| Change Advocate Onboarding | DEV OD | June 2012 | Fran/GCO Better Development | Business Use Only42
Preparation of
“global”
documents
Compilation
&
Publishing
QC
Dispatch
Through regulatory
Link to
Country Resp
Addition of
local
documentation
HQ CPO
IMPD/CTA preparation General process
| Change Advocate Onboarding | DEV OD | June 2012 | Fran/GCO Better Development | Business Use Only43
Submit a CTA dossier to
Health Authority (HA)
Positive vote from EC
Start Clinical Trial
Initiate Trial
Submission
parallel
or
sequential:
country
specific
Approval of clinical trials is the responsibility of individual EU Member States
Maximum 60 days assessment period (incl. validation) + Question/Answer period
Review/Validation
Authorisation from HA
Voluntary Harmonization Procedure (VHP) as an optional regulatory
pathway for multinational clinical trials in EU
Review/Validation
Submit a CTA dossier to
Ethics Committee (EC)
| Change Advocate Onboarding | DEV OD | June 2012 | Fran/GCO Better Development | Business Use Only44
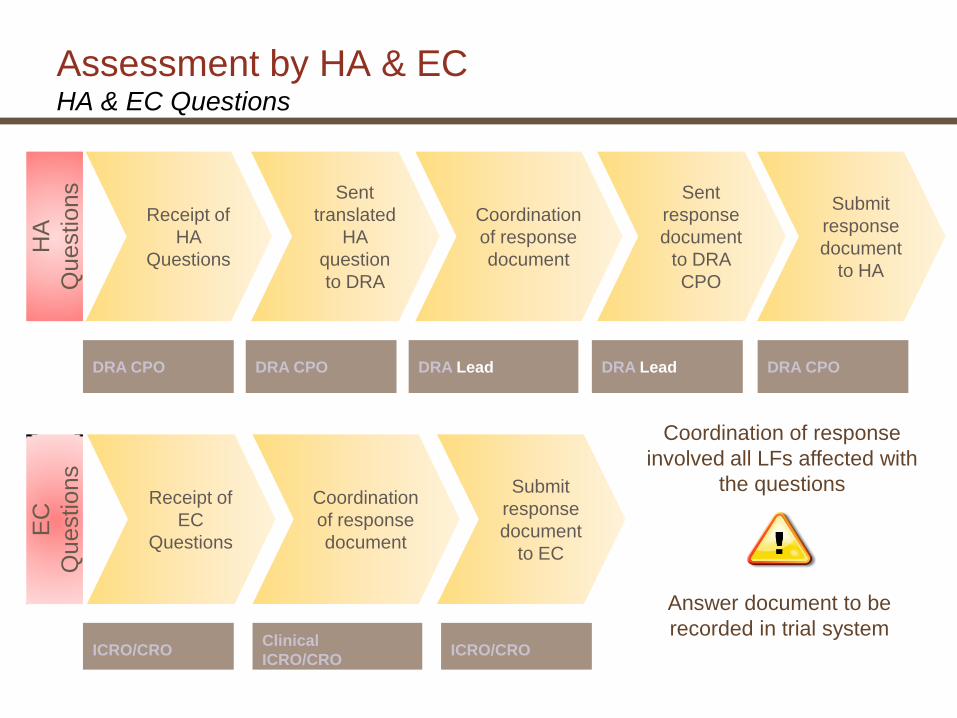
Assessment by HA & EC HA & EC Questions
HA
Questions
Coordination
of response
document
Submit
response
document
to HA
Sent
response
document
to DRA
CPO
DRA CPO DRA CPO DRA Lead DRA Lead DRA CPO
ICRO/CROClinical
ICRO/CROICRO/CRO
EC
Questions
Coordination of response
involved all LFs affected with
the questions
Sent
translated
HA
question
to DRA
Receipt of
HA
Questions
Receipt of
EC
Questions
Coordination
of response
document
Submit
response
document
to EC
Answer document to be
recorded in trial system
Standard Operating Procedures
for a Clinical Study
What are SOPs?
International Conference on Harmonization (ICH) defines a SOP as “Detailed, written instructions to achieve uniformity of the performance of a specific function.” (ICH
GCP 1.55)
In simple terms a SOP is…
• A written process
• A way for the clinical site to perform a task the same way each time it is completed.
SOPs are used to:
Identify the responsible person for each task.
Describe actions (what is to be completed).
Train staff.
Monitor site performance.
Are SOPs Required by Law/Regulations?
SOPs are not specifically mentioned in the FDA regulations
• However there is guidance and regulations that infer responsibility and SOPs formalize investigator responsibilities.
• 21 CFR312.53 the investigator will “ensure that all associates, colleagues, and employees assisting in the conduct of the study (ies) are informed of their obligations in meeting the above commitments.”
Additionally, SOPs are mentioned repeatedly in the ICH GCP Guidelines.
ICH GCP 2.13 -“Systems with procedures that assure the quality of every aspect of the trial should be implemented
Benefits of a SOP?
Ensures that all research conducted within the clinical site follows federal regulations, ICH GCP, and institutional policies to protect the rights and welfare of human study participants.
Provides autonomy within the clinical site.
Improves the quality of the data collected, thereby improving the science of the study.
Utilized as a reference and guideline as to how research will be conducted within the clinical site
Excellent training source for new employees and/or fellows
SOP Topics
Preparing and Submitting Initial IRB Documents
Preparing and Submitting Continuing Review IRB Documents
Preparing and Submitting Amendment IRB Documents
Establishing and Training the Clinical Study Team, and Delegating Responsibilities
Establishing Study Files
Establishing Source Documents
SOP Topics
Study Subject Recruitment Plan
Contacting and Scheduling Potential Study Subjects for an Initial Visit
Obtaining Informed Consent from a Potential Study Subject
Enrolling a Subject
Recording Subject Data
Making Corrections on Study Documents
SOP Topics
Monitoring Subject Compliance During a Study
Responding to a Clinical Hold Order
Receiving and Storing Investigational Drugs
Drug and Study Supply Transfer Between Sites
Dispensing Study Drugs to Study Subjects
Identifying and Reporting Adverse Events
Packing, Labeling, and Shipping Samples
Identifying and Reporting Protocol Deviations
SOP Topics
Contact with potential sponsors
Budget review and determinations
Pre-study study site visit
Initiation Site Visit
Monitor Visits
Audits
Data management
Study closure
Long term storage
Writing SOPs
Develop a template for the SOP to be used throughout the document.
Potential elements of the SOP
• Header – title, original version date, revision date, effective date, approved by
• Purpose – why one has the policy
• Responsibilities – who the policy pertains to
• Instruction/Procedures – how to accomplish the items of the policy
• References – what the policy is based on
• Appendix – source documents/case report forms
Process Mapping for Writing SOPs
Determine which clinical site task needs mapping.
Lay out all the steps currently used to complete that task.
“Mapping” involves taking each step in the task and making it more efficient and easier to follow.
Process Mapping for Making a Cup of Coffee
Primary Step
Ensure the coffee maker is ready
Add the water
Turn on the machine
Serve the coffee
Add the coffee
Woodin, K. (2004) The CRC’s Guide to Coordinating Clinical Research p. 60-65. Centerwatch
Process Mapping for Making a Cup of Coffee
Ensure the coffee maker is ready
Add the coffee
Add the water
Turn on the machine
Serve the coffee
Secondary Step
Ensure the machine is plugged in
Ensure carafe is empty and clean
Place a filter in the basket
Measure the coffee
Use the carafe to measure the water
Place the carafe on the heating element
Wait until the coffee has stopped dripping
Process Mapping for Writing SOPs
Once you have finished mapping, convert your process map to an outline for easy use.
Once a task has been mapped, it should be tested.
Implementing and Monitoring SOPs
SOPs should be introduced gradually.
Prioritize most relevant SOPs and present them first.
Principle Investigator should approve all SOPs and designate an effective date.
SOPs should be reviewed on a regular basis (usually annually) to ensure policy based regulations are up-to-date.
Previous versions of SOPs should be retained.
SOP Training
All staff should have SOP training.
Training should be documented.
SOP should be accessible to staff.
What is the difference between a SOP and a Manual of Procedures (MOP)?
These terms have been used interchangeably.
Both provide a standardization of a process.
SOP provides general information that is to be utilized throughout any research study.
• How as a clinical site we will assess delegation of duties.
MOP is specifically written for a particular research study which will incorporate elements of the SOP.
MOP
The MOP should be written so that anyone in your clinical site can follow the procedures for that study and find all relevant materials.
The MOP should be extremely detailed.
SOP Resources Examples from National Cancer Institute