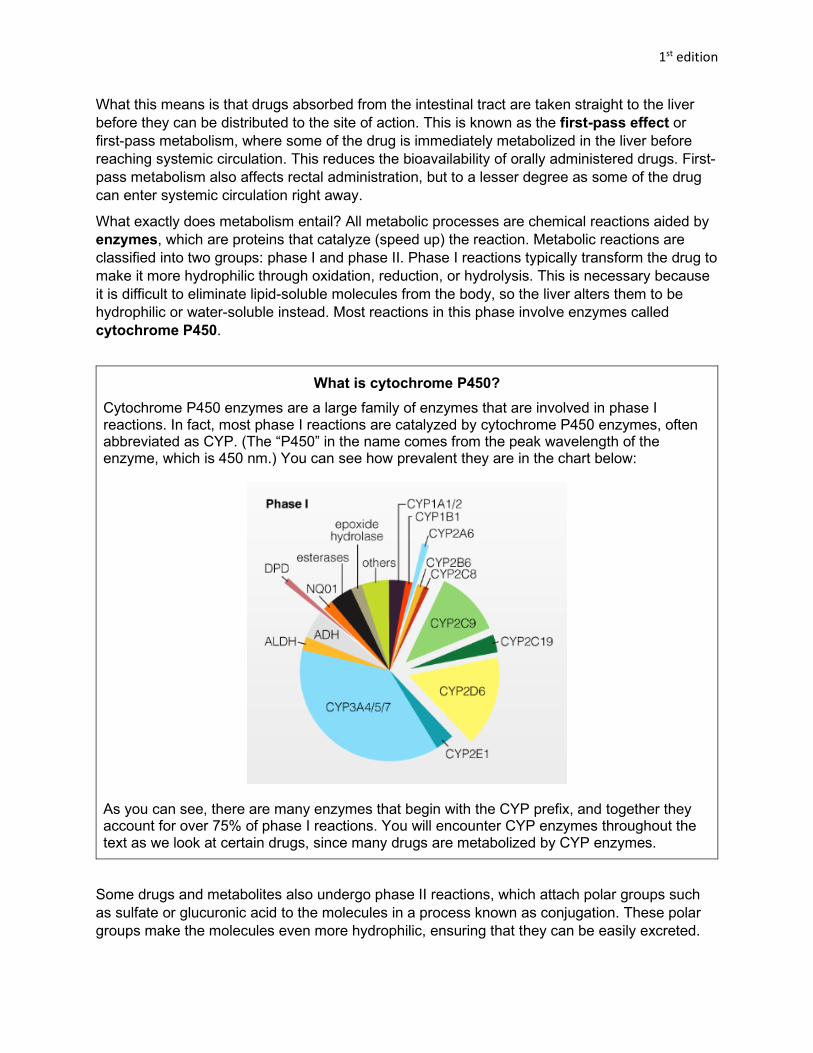
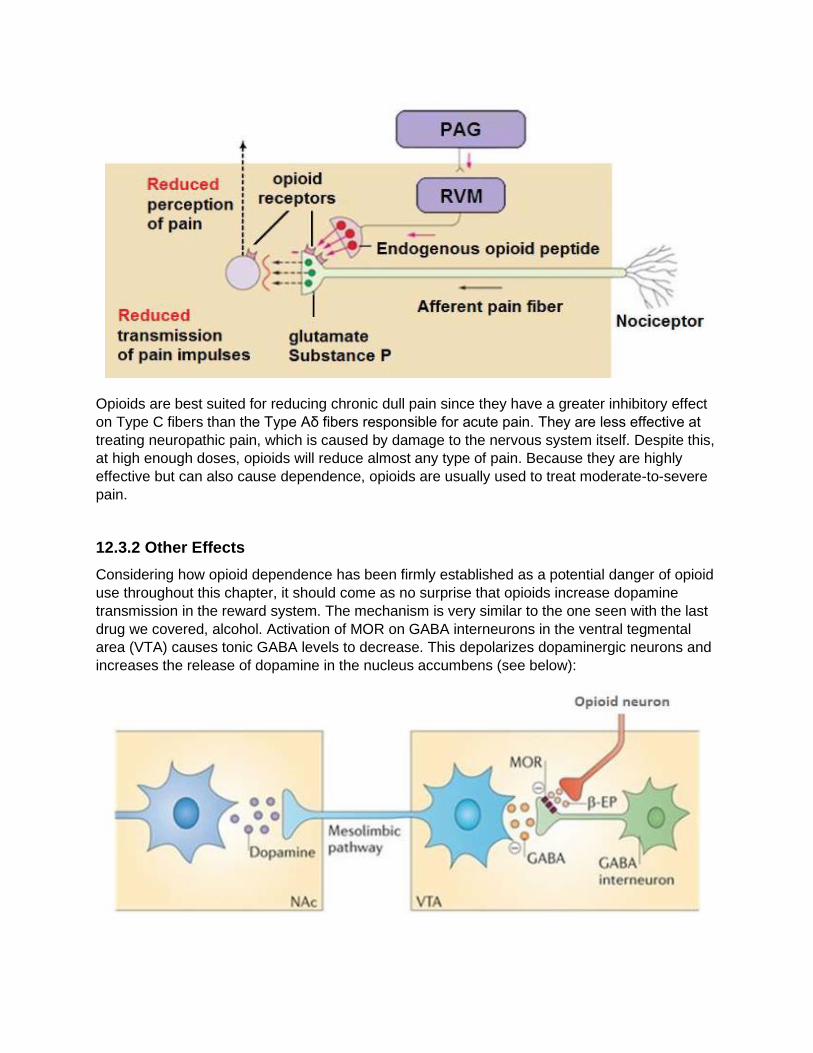
Discovering Psychology Series Drugs and Behavior 1 st edition Abigail Brewer, Justin Nathaniel Knutson, and Raymond M. Quock Washington State University Version 1.00 January 2021 Contact Information about this OER: 1. Dr. Ray Quock, Professor of Psychology – [email protected]2. Dr. Lee Daffin, Associate Professor of Psychology – [email protected]
Transcript
Discovering Psychology Series
Drugs and Behavior
1st edition
Abigail Brewer, Justin Nathaniel Knutson, and Raymond M. Quock
• Chapter 1: Introduction to Psychoactive Drugs 1-1
• Chapter 2: Neuroanatomy 2-1
• Chapter 3: Nerve Cell Physiology 3-1
• Chapter 4: Neurotransmission 4-1
• Chapter 5: Pharmacokinetics 5-1
• Chapter 6: Pharmacodynamics 6-1
• Chapter 7: Reward and Reinforcement 7-1
Part 2
• Chapter 8: High-Efficacy Stimulants 8-1
• Chapter 9: Low-Efficacy Stimulants 9-1
• Chapter 10: CNS-Depressants 10-1
• Chapter 11: Alcohol 11-1
1st edition
iii
Part 3
• Chapter 12: Opioids 12-1
• Chapter 13: Cannabinoids 13-1
• Chapter 14: Psychedelics 14-1
Part 4
• Chapter 15: Antidepressants 15-1
• Chapter 16: Anxiolytics 16-1
• Chapter 17: Antipsychotics 17-1
• Chapter 18: ADHD and Alzheimer’s Drugs 18-1
Glossary Back-1
References Back-
Index Back-
1st edition
iv
Record of Changes Edition As of Date Changes Made
1.0 January 2021 Initial writing; feedback pending
1st edition
Part 1
1st edition
Chapter 1: Introduction to Psychoactive Drugs
Welcome to Biopsychological Effects of Alcohol and Other Drugs! This course, and its accompanying text, will provide a basic introduction to the biopsychological effects of the major classes of psychoactive drugs. We’ll discuss the effects of various drugs at the neurochemical and behavioral level. The first few chapters of this text will lay the groundwork for the later chapters where we will discuss specific types of drugs. The material may be difficult, but I encourage you to stick with it and ask for help if you get stuck. Falling behind early on will make later chapters harder to follow.
Before we begin, a note about this text: This text is an Open Education Resource (OER) designed for an online platform. In the text, there will be links to outside resources like articles, infographics, videos, and interactive media. We encourage you to explore these resources, but not every resource is required. Important resources will be pointed out in the text, as will entirely optional ones. If you have trouble accessing a resource, notify your instructor—it’s possible that it was taken down recently. Rest assured that all of the critical information is contained in this text, though. If you have any feedback, please send it to [email protected].
For this first chapter, we will be introducing psychoactive drugs: what they are, how they are used and misused, how they are classified, how drug laws have changed, and what modern drug development looks like.
Chapter Outline: 1.1 Psychoactive Drugs: Use and Misuse
1.1.1 Drug Nomenclature
1.1.2 Drug Use, Misuse, Dependence, and Addiction
1.1.3 Determinants of the Drug Experience
1.2 Drug Laws in the United States 1.2.1 Legal Classification of Drugs
1.2.2 Schedule of Controlled Substances
1.2.3 History of Drug Laws
1.3 Modern Drug Development 1.3.1 Drug Development Process
First, we need to define what we mean when we use the word drug. A drug is a chemical substance (excluding food) that can influence physiological function in order to restore, maintain, or enhance physical or mental health. The study of drugs and their actions and effects on living systems is called pharmacology.
As this is a psychology course, we are primarily interested in drugs that can bring about changes in behavior. These drugs are known as psychoactive drugs and studying how they affect behavior is a subdiscipline of pharmacology called psychopharmacology.
By the end of this section, you should be able to:
• Distinguish between therapeutic and recreational use of drugs. • Distinguish between chemical, generic, and brand names for drugs. • Explain the differences between drug use and drug misuse. • Describe drug dependence and drug addiction. • Explain pharmacological and non-pharmacological determinants of the drug experience.
1.1.1 Drug Nomenclature Drugs can be used in two major ways: recreationally and therapeutically. Recreational use means taking the drug to experience its effects, and this is where street names like coke and ecstasy come from. In comparison, therapeutic use involves taking the drug to treat a physical or mental ailment.
You might not have noticed, but we often refer to a drug by multiple names. We can say that we need an ibuprofen or an Advil®. Users might refer to cocaine as coke. You may have no idea what 3,4-methylenedioxymethamphetamine is, even if you’ve heard of ecstasy before.
Most therapeutic drugs have a generic name and one or more brand names. A generic name is a name created by the drug developer and accepted by a scientific body that indicates the drug’s classification. In the United States, generic names are approved by the U.S. Adopted Name (USAN) Council. While generic names can be used by anyone, a brand name is a proprietary name used by the company marketing the drug. Advil® and Motrin® are sold by different companies under different brand names, but both can be referred to by the generic name ibuprofen.
Another way of referring to drugs is by their chemical name, which refers to the molecular structure of the drug. These names are defined by standards set by the International Unition of Pure and Applied Chemistry (IUPAC) and can end up being quite the mouthful! Take the IUPAC chemical name for ibuprofen: (RS)-2-(4-(2-methylpropyl)phenyl)propanoic acid. Obviously, it’s much easier to refer to it as ibuprofen. But many generic and brand names are derived from the drug’s chemical name. An example is acetaminophen (trade name Tylenol®), shown in the table below.
1st edition
Name Definition Example
Chemical Drug’s atomic and molecular structure N-acetyl-para-aminophenol
Generic Name assigned by the drug developer and accepted by a scientific body
Acetaminophen from N-acetyl-para-acetylaminophenol
Brand Proprietary name patented by the company that markets the drug
Tylenol®
from N-acetyl-para-acetylaminophenol
The process for selecting generic and brand names is a long and involved one, but for the time being, you just need to be able to know the difference between the three types of names listed above. If you are interested in the process, consider reading this article from Popular Science: FYI: How Does A Drug Get Its Name?
1.1.2 Drug Use, Misuse, Dependence, and Addiction Earlier we made a distinction between recreational use of a drug and therapeutic use. Another way of classifying drug use is by distinguishing use from misuse. In this context, drug use means taking a drug properly in its correct dosage. Drug use does not give rise to health or behavioral problems that can harm the user or the people around them. Most people have used drugs this way before, such as by taking an aspirin to relieve a headache or drinking a beer.
Improper or excessive usage of a drug is drug misuse and can potentially harm the user or the people around them. Misuse can be intentional or accidental. All of the following examples constitute drug misuse:
• Injecting heroin to get high • Taking prescription opioids to experience relaxation instead of to relieve pain • Prematurely stopping an antibiotic treatment • Taking twice the recommended dose because the regular amount isn’t effective enough • Using someone else’s prescription medicine
You may be more familiar with the phrase drug abuse and notice similarities between it and how we have defined drug misuse. Despite that, the terms are used to refer to different things.
Drug Misuse vs. Drug Abuse Drug misuse and drug abuse—what’s the difference? Although definitions vary, a 2013 review found that the main difference between the two is therapeutic intent. Both involve improper use of drugs, but misuse occurs when someone intends to treat a symptom, while abuse involves taking a drug to achieve a pleasurable sensation. The difference might not matter though, as the term abuse is on the decline. The National Institute on Drug Abuse (NIDA) does not make a distinction and prefers using misuse over abuse because the latter can be shaming and contribute to stigma (NIDA, 2020a). Likewise,
the fifth edition of The Diagnostic and Statistical Manual of Mental Disorders (DSM-5), a manual used widely by clinicians for diagnostic guidelines, removed references to substance abuse and now uses the broader term substance use disorder instead. Some sites still differentiate between the two, so it is worth being aware of the difference. In this text, abuse will only be used in specific contexts where it is necessary to delineate intent, such as in discussing drug scheduling; elsewhere misuse will refer to any improper drug use, regardless of intent.
What are some of the risks of drug misuse? It largely depends on the drug and how it is used, but a variety of symptoms are possible. Nausea, vomiting, and blackouts from binge drinking is one of many examples. Other changes like irritability or impaired decision-making can negatively impact the people around the user. There are also risks associated with repeated use of a drug. When a person uses the same drug over and over, it can cause physiological changes that result in drug dependence. What does dependence look like? In short, the body adapts to the drug, which literally works its way into how the body functions. More of the drug is required for the same effect (tolerance), and suddenly stopping the drug can cause the body to malfunction (withdrawal). You will learn more about tolerance and withdrawal in later chapters.
Chronic drug use can also lead to drug addiction, or compulsive drug use that continues despite harmful consequences. Addiction and dependence are not the same thing—it is possible to become dependent on a drug without being addicted. For example, a hospital patient taking opioids for chronic pain will become dependent and exhibit withdrawal if medication is discontinued. But the patient would not be considered an addict since they are not compulsively taking opioids to get high.
Before continuing, ask yourself: is addiction a choice that people make, or is it more like a disease that people suffer? How would you distinguish between the two? Once you have thought about this, read the following except from the National Institute on Drug Abuse:
Do people choose to keep using drugs? The initial decision to take drugs is typically voluntary. But with continued use, a person's ability to exert self-control can become seriously impaired. This impairment in self-control is the hallmark of addiction. Brain imaging studies of people with addiction show physical changes in areas of the brain that are critical to judgment, decision-making, learning and memory, and behavior control. These changes help explain the compulsive nature of addiction.
Source: Drug Misuse and Addiction (NIDA, 2020b)
As the passage indicated, the compulsive nature of drug addiction is caused in part by how the drug changes the body. Many drugs that are classified as having a high potential for abuse are self-reinforcing, meaning they make it more likely that you will take the drug again. The exact mechanism will be explored in more detail in subsequent chapters, but in simple terms, the drug makes the user feel good, so the user seeks out the drug again in the future.
Although the phrase drug addiction is used in this text, it’s worth noting that the DSM-5 does not refer to the disorder this way. Instead, there is a single category called substance use disorder that contains a list of diagnostic criteria and can be diagnosed as either mild, moderate, or severe. The criteria are beyond the scope of this text, but if you are interested, you may read more about them in this article from Verywell Mind.
1.1.3 Determinants of the Drug Experience What determines whether a single act of drug misuse turns into a full-fledged addiction? Take a look at the following infographic:
Source: Drug Misuse and Addiction (NIDA, 2020b)
As you can see, there are multiple factors that can influence the experience of taking a drug. The obvious factors involve the drug being used, and these are what we call pharmacological factors. These are the effects of the drug, the dosage, the route of administration, and any other properties inherent to the drug, such as price or availability. For instance, inhaling or injecting a drug is more likely to create an immediate rush of pleasure, increasing the chances of addiction compared to an experience with a slower onset and longer duration.
There are other factors that don’t have to do with the drug itself, and these are the non-pharmacological factors. We can split the non-pharmacological factors into two main groups: personal and environmental factors. Personal factors are related to the person taking the drug. A person’s reaction to taking a drug depends on their genetics as well as their psychological mindset at the time. Someone who is eagerly looking to escape will perceive the pleasure differently compared to someone who is already well-adjusted. These can also tie into environmental factors, such as home and family life, the attitudes of peers and the community one lives in, the setting in which the drug is used, and so on. All of these can influence a person’s response to taking a drug, which in turn determines whether the experience has addictive potential or not.
1.2 Drug Laws in the United States
Drug use in in the U.S. predates even precolonial days. Native Americans on the east coast had many uses for tobacco, while Southwestern cultures used hallucinogens like peyote in religious rituals. In more modern times, our society has often seen drug use define entire generations. Watch this short three-minute video to get a quick overview of some of the drugs of choice in different decades:
100 Years Of Drugs In America: From Coffee To Heroin [3:17]
Throughout this long history, attempts at federal regulation have only occurred in the past century. In this section, we will look at drugs from a legal perspective. We will begin with how drugs are classified today, then review some of the historical changes that led to our current system.
By the end of this section, you should be able to:
• Explain the definitions of legend vs. non-legend drugs, controlled vs. non-controlled drugs, and licit vs. illicit drugs
• Explain the DEA Schedule of Controlled Substances • Explain how drug laws have changed over the past century in the US
1.2.1 Legal Classification of Drugs To explain drug laws, we will need to start by discussing how drugs are legally classified in the United States. Examine the following chart and refer back to it as you progress through this section, as it should help you keep all of these different categories organized:
The first main category of drugs are legend drugs. A legend drug is one that is approved by the U.S. Food and Drug Administration (FDA) and by state or federal law can only be dispensed to the public with a prescription from a licensed physician or other licensed provider. The legend is a label on the drug container that usually says, “Federal law prohibits dispensing without a prescription from a licensed healthcare provider.” Because of this, they are also known as prescription drugs. In order to acquire these drugs, you must have a prescription filled out by a medical doctor or other licensed professional. The drug itself must be approved by the FDA through the New Drug Application process, which we will cover in more detail later in this chapter.
Drugs that do not belong to this first category are called non-legend drugs, and as you would imagine, you do not need a prescription to purchase them. Drugs in this category are considered safe to use without the supervision of a health care provider, so you can simply walk into a store and buy them. As such these are also called over-the-counter or OTC drugs. These would include your typical pain relievers, cough suppressants, and antihistamines. To compare prescription and OTC drugs, read this short FAQ by the FDA.
Legend drugs can be further separated into controlled and non-controlled drugs. A controlled drug or substance is one that is considered to have some potential for abuse. These drugs are subject to the Controlled Substances Act and regulated by the Drug Enforcement Administration (DEA), which categorizes them into five different schedules. We will examine the schedules in detail in the next section, but some examples include heroin, LSD, opioids, steroids, and certain sleeping pills. In comparison, non-controlled drugs are deemed to have no potential for abuse and are not regulated by the DEA. Examples include antibiotics, diabetes medications, heart medications, and asthma inhalers.
Finally, controlled substances can be either licit or illicit. A licit drug is a legal drug—you need to obtain a prescription for it from a physician or other professional licensed by the DEA. These drugs have a therapeutic use but are still liable to be misused, such as opioid painkillers,
sedative hypnotics, and cough syrups that contain codeine. Likewise, an illicit drug is an illegal drug. Examples of illicit drugs are heroin, cocaine, LSD, and ecstasy.
1.2.2 Schedule of Controlled Substances Now let’s take a closer look at the drug schedules determined by the DEA. The following table is simplified but shows the main differences between each schedule along with notable examples.
Schedule Description Examples
I No accepted medical use High abuse potential (++++) heroin, LSD, MDMA (ecstasy), marijuana
II Accepted medical use High abuse potential (++++)
III Accepted medical use Moderate abuse potential (+++) ketamine, testosterone, anabolic steroids
IV Accepted medical use Low abuse potential (++) Xanax®, Valium®, Ambien®, tramadol
V Accepted medical use Low abuse potential (+)
Lyrica®, Lomotil®, many codeine-containing cough syrups
As you can see, the schedules are sorted according to abuse or dependency potential. Drugs that have a greater potential to create severe psychological or physical dependence are placed in more restrictive schedules. The most restrictive schedule is Schedule I, which also requires the substances to have no accepted medical use.
You may have noticed that marijuana is italicized. According to Federal law, marijuana is classified as a Schedule I drug, meaning it is considered to have a high risk of dependency and no accepted medical use. This is a point of contention, as evidenced by the numerous states that have legalized medical and sometimes recreational marijuana use, including Washington State. The question of whether marijuana should be legalized or rescheduled will be revisited in the chapter on cannabinoids.
1.2.3 History of Drug Laws To understand why the DEA drug schedules were created, it is necessary to understand how drug laws have changed over the years. This section will provide a very brief overview of the major changes to U.S. drug laws—not every change will be included. If you are interested in a more in-depth look at drug legislation, the first half of this report put out by the Congressional Research Service is a comprehensive read. Doing so is entirely optional; for this class, you will only need to know what is covered below.
For most of U.S. history, there were no federal regulations or restrictions on the use of drugs. Drugs like opium and cocaine were freely prescribed and used. The earliest attempts at
restricting drug use were mostly at the city or state level; it wasn’t until 1906 when the first federal legislation concerning drugs was passed. The Pure Food and Drug Act enforced labeling ingredients, regulated the contents of food and drugs, and created The Food and Drug Administration to enforce the changes. It did not place any restrictions on drug use.
The first real restrictions arrived with the Harrison Narcotics Tax Act of 1914. The Harrison Act was passed in response to growing levels of drug abuse and required importers, manufacturers, and distributors of opium and cocaine to register with the Department of Treasury, pay taxes, and record transactions. Physicians were still allowed to prescribe these drugs, but the interpretation of the law meant that many persons who used narcotics for non-medical purposes were prosecuted, effectively criminalizing the drugs covered under the act.
In 1920, the 18th Amendment was ratified. Also known as the National Prohibition Act or Volstead Act, it banned the sale and distribution of alcohol. Consumption was still legal, but nevertheless driven underground to speakeasies supplied by bootleggers. The amendment was eventually repealed by the 21st Amendment in 1933.
Until 1937, the growth and use of marijuana was legal. The Marihuana Tax Act ended this by requiring a high-cost tax stamp for every sale of marijuana. The stamps were rarely issued however, and states soon made the possession of marijuana illegal after the act was passed.
In the 1960s, support for severe punishment of drug abuse started to decrease. The Presidential Commission on Narcotics and Drug Abuse of 1963 encouraged Congress to support medical approaches to treating drug dependency. At the same time, the Presidential Commission endorsed strong enforcement of drug laws which eventually lead to a war on drugs.
Perhaps the most significant change occurred in 1970, when the Controlled Substances Act (CSA) was passed. The CSA placed control of certain drugs under the federal government and was pushed by President Richard Nixon as part of his War on Drugs. The CSA outlined the five schedules and later gave rise to the DEA in 1973, which still regulates controlled substances to this day.
Although the CSA has persisted for the past 50 years, drug laws have continued to change. The state legalization of marijuana mentioned earlier is one such example. Although it is important to understand the law as it currently stands, it is also important to recognize that the law will continue to change and adapt in response to increased research, shifts in public sentiment, and potential future health crises.
Below is a table summarizing the important drug laws mentioned above:
1st edition
Year Legislation Purpose
1906 Pure Food and Drug Act Created the FDA to regulate ingredients and enforce labeling of food and drugs.
1914 Harrison Narcotics Act Effectively criminalized the prescription and use of narcotics like opium and cocaine.
1920 18th Amendment (National Prohibition Act)
Banned the sale and distribution of alcohol and drove consumption underground. Repealed in 1933.
1937 Marihuana Tax Act Effectively criminalized the possession and growth of marijuana.
1963 Presidential Commission on Narcotics and Drug Abuse
Advised for medical treatments to addiction and strong enforcement of drug laws.
1970 Controlled Substances Act Placed certain drugs in schedules and created the DEA to regulate them.
1.3 Modern Drug Development
Earlier when discussing legend drugs, we mentioned the FDA’s New Drug Application process. Compared to OTC medication, prescription drugs need to go through a lengthy process before being approved for marketing in the U.S. This is a consequence of the various laws that have been instated to regulate drug contents and ensure safety and effectiveness in drugs marketed to the public. In this section, we will discuss the process for introducing a new drug to the market, as well as the laws that shape how drugs are priced and sold.
By the end of this section, you should be able to:
• Explain the process of drug development, including pre-clinical trials, the different phases of clinical testing, and phase 4 surveillance
• Explain drug patent rights and the difference in costs between originally patented drugs and generic drugs
1st edition
1.3.1 Drug Development Process As mentioned previously, the process to develop and review a new drug is a long one. How long? Consider the following graphic:
From drug discovery to FDA approval, the entire process can take over ten years. In addition, the overall success rate is very low. Numerous compounds will be synthesized and analyzed, but only a small amount will reach preclinical trials. Of those, only a handful will progress to clinical trials, and fewer still will go on to receive FDA approval. In the graph above, out of 5,000-10,000 new compounds discovered by the pharmaceutical industry, only 250 will advance to preclinical studies, and only five enter into clinical trials, of which, on average, only one is approved by the FDA. What is happening at each stage that takes so long and results in so few successes?
Once a compound has been identified as a potential therapeutic drug, it begins the process in the preclinical stage. Lab studies test the drug on cells, organs, and animal subjects at this stage. Most drugs at this stage either prove to be too toxic or fail to demonstrate a strong enough effect to continue research. Only drugs that meet standards for safety and efficacy are allowed to proceed to the next stage of trials. Instead of animals, human subjects are used during clinical trials.
Clinical trials consist of three phases. Phase 1 emphasizes safety and determines side effects and how the drug is metabolized and excreted in a small healthy population. Phase 2 emphasizes effectiveness instead, using a large sample to check whether the drug has the desired effect in patients with the intended ailment. Treatment patients are compared against a control group that is provided placebos or a different drug. Phase 3 expands the sample size further and gathers additional information about how the drug performs in different populations, at different doses, and when combined with other drugs. At any point if a drug appears to be unsafe or not effective enough, research will stop and the pharmaceutical company will have to start again with a new drug.
1st edition
Assuming the drug makes it through all three phases of clinical trials, the drug will finally be ready for FDA approval. Even after the drug is approved and manufacturing proceeds, testing is still not over. Once a drug enters the market it begins Phase 4 (post-marketing surveillance), where it continues to be monitored for any safety issues. If new risks are found, labeling and information will be updated, and, in rare cases, the drug may even have to be withdrawn from the market.
To review this process, check out this infographic from the FDA. All the steps of the drug approval process are clearly laid out, so be sure to check it out and make sure you understand it.
How much does all this research and development cost? It’s hard to pin down exact numbers, but suffice to say, the answer is “a lot.” One study from 2020 reported that the estimated research and development cost per drug was $985 million. In addition, most drugs do not make it out of preclinical or clinical stages, and in those cases the company sees zero return on investment. Add it all up, and pharmaceutical research is an extremely costly endeavor.
1.3.2 Patent Rights and Generic Drugs So how do pharmaceutical companies recoup those costs? For the most part, it comes from the few successes that make it through the research pipeline and go to market. To understand why a single successful new drug can be so lucrative, it’s important to understand how patents work.
A patent is a property right issued by the United States Patent and Trademark Office (USPTO). In exchange for public disclosure of the invention, the inventor is granted the right to “exclude others from making, using, offering for sale, or selling the invention throughout the United States” for a limited time. This period is usually 20 years from the date the patent application was filed. During this period, the original manufacturer is the only one that can sell the product in the U.S.
At first glance, it may seem like the pharmaceutical company would have 20 years then to sell the new drug without competition. However, that is not quite the case. Companies will usually apply for a patent from the USPTO earlier on during the research and development process, so the actual time that the company can take advantage of the patent may only be 12 years or less. This is why new drugs can be so expensive when they first come out—the company is trying to recoup all the costs of developing the drug, as well as the costs of developing the drugs that failed and did not make it out of the preclinical or clinical stages. As a result, the company has to charge a high amount over a short period of time.
You may be wondering why the pharmaceutical company can’t just keep selling the drug after the patent expires. The company certainly will, but it won’t be as profitable once the patent expires because other companies can sell competing versions of the drug. These versions use the generic name of the drug, not the brand name that the original drug was marketed under, which is why these drugs are called generic drugs. Generic versions tend to be priced much cheaper than brand name drugs, and the lower price eventually drives the price of the brand name drugs down as well, otherwise it won’t be able to compete. Once the patent expires, the company that developed the drug will find it much harder to turn a profit.
So does that mean the obscenely high prices of drugs is justified? Perhaps not. Recent investigative journalism has pointed to a large discrepancy between spending on research and
development versus advertising. An episode of Last Week Tonight with John Oliver from 2015 revealed that 9 out of 10 of the top pharmaceutical companies spent more on marketing than research. While the companies disagree with the numbers, they nevertheless continue to pour billions of dollars into drug ads and marketing to doctors, as this Ars Technica article from 2019 points out. This practice can create conflicts of interest in health care professionals and may be contributing to medicalization and an increase in demand for prescription remedies.
If you’re interested in watching the original piece by John Oliver on the subject, you can find it on YouTube: Marketing to Doctors: Last Week Tonight with John Oliver (HBO) [17:12] [TV-MA]
Chapter Summary and Review In this chapter, we introduced the main focus of this course: psychoactive drugs. We discussed how these drugs are named, the various ways they can be used, and what factors influence the drug experience. We also covered a history of drug laws in the United States, including how to legally classify drugs and the different schedules of controlled substances. Finally, we explored the modern drug development process and how prices are influenced by patents and generic drugs.
Make sure to check your understanding before moving on. At the end of each chapter will be a list of practice questions. These are adapted from the learning objectives from each section and are meant to help you identify any gaps in your knowledge. They are not comprehensive or part of a required assignment. Test yourself on these items, either by creating study materials like flash cards or answering the questions out loud; if you struggle with any of them, use that feedback to direct your studies and ask your instructor questions.
Practice Questions
• What is the difference between recreational and therapeutic drug use? • What is the main difference between a generic and brand name? Provide an example of
each. • What is the difference between drug use and misuse? • Explain what drug dependence is. • What are some things that might affect whether someone has a good or bad experience
with a drug? Provide three examples: a pharmacological one, a personal one, and an environmental one.
• What are legend drugs sometimes called? What about non-legend drugs? • What is the difference between licit and illicit? • How many schedules are there? Define the criteria for each schedule. • Name six important changes in the history of drug laws in the United States. What were
the key changes from each? • How many phases of drug development are there? Name each. • How long are drug patents in the US? When does this period start?
Last chapter we introduced psychoactive drugs, or drugs that can influence our behavior and physiological function. It’s important to realize that a drug cannot make the body do something it is incapable of doing. Drugs cannot give you the ability to see through walls or fly. Instead, they interact with the systems that regulate our bodily functions, causing us to feel awake (caffeine), lose inhibition (alcohol), or experience less pain (opioids).
This means that to understand how drugs work, you must learn how the body works normally. The next few chapters focus on this and examine the human nervous system at three different levels: the overall structure, the individual nerve cells, and neurotransmitters. This crash course in neuroscience is simplified and streamlined for this course, but it can be a lot of information to take in, so make sure to give yourself time to process it.
In this chapter, we will cover the basic structure of the nervous system and how it is organized. We will also highlight a few regions that are relevant to drug use and the development of drug dependence. This is a short chapter, but a lot of terminology will be introduced. Make sure to test yourself on the divisions of the nervous system and regions of the brain until you are able to differentiate between them and define their functions.
Chapter Outline: 2.1 Overview of the Nervous System
2.1.1 The Structure of the Nervous System
2.1.2 The Divisions of the Peripheral Nervous System
2.2 The Human Brain 2.2.1 Cerebral Cortex
2.2.2 Thalamus and Limbic System
2.2.3 Cerebellum and Brainstem
1st edition
2.1 Overview of the Nervous System If you’re taking this class, you’ve likely studied the nervous system before as part of biology. To refresh, the nervous system is responsible for transmitting signals and coordinating activity between different parts of the body. Understanding what the nervous system is and how it functions is important for this class because all of the drugs we will study influence the body through the nervous system.
To start, we will provide a basic overview of the nervous system and its major divisions. To prepare for this, please watch this brief video that summarizes the upcoming information in a concise and easy-to-understand format:
Alila Medical Media - Overview of the Nervous System [4:10]
By the end of this section, you should be able to:
• Explain the role of the nervous system. • Define nerve and distinguish it from a neuron. • Distinguish between central and peripheral nervous systems, afferent and efferent
nerves, and the somatic and autonomic nervous systems. • Explain the how the sympathetic and parasympathetic nervous systems work. • Describe dual innervation.
2.1.1 The Structure of the Nervous System In vertebrates like humans, the nervous system can be divided into two main parts.
The first is the central nervous system (CNS), which consists of the brain and spinal cord. The CNS is “central” because it responsible for the coordination half of the nervous system—it receives signals from different parts of the body, integrates and processes the information, then coordinates a response. In comparison, the peripheral nervous system (PNS) deals with transmission, bringing signals to and from the CNS to different parts of the body. It is involved in everything outside (the periphery) of the CNS.
The PNS consists of ganglia and nerves, which are bundles of fibers that transmit electrical impulses. Nerves can contain either afferent nerve fibers, efferent nerve fibers, or a mix of afferent and efferent nerve fibers. The afferent nerve fibers convey sensory information from the body to the CNS, while efferent nerve fibers convey motor commands from the CNS to various muscles and glands; mixed nerves contain both types. (A good way to remember the difference is that afferent nerves arrive at the CNS, while efferent nerves exit the CNS.) Each nerve fiber is part of a nerve cell called a neuron, which we will cover in detail next chapter. Neurons are also present in the CNS, forming a web of connections that can process information and coordinate sophisticated responses. We will explore the CNS in the second half of the chapter, but before that we will discuss the PNS.
2.1.2 The Divisions of the Peripheral Nervous System The PNS itself can be further separated into two main divisions. The somatic nervous system is associated with voluntary movement. It transmits sensory information from the body (soma in Greek, hence the name) and conveys motor commands to skeletal muscles. This is done with
the afferent and efferent nerve fibers mentioned above. The autonomic nervous system, as its name suggests, is associated with automatic or unconscious functions and is always active. It transmits sensory information from internal organs (also called viscera, which is why this system is sometimes called the visceral nervous system) and controls the heart, smooth muscles, and glands.
The autonomic nervous system has two branches. The sympathetic nervous system is our “fight or flight” system; when we are stressed or perceive danger, this system increases our heart rate, dilates our pupils, and inhibits digestion, among other things. The parasympathetic nervous system is our “rest and digest” system; in times of relaxation, it slows our heart rate, constricts our pupils, stimulates digestion, and so forth. As you can see, the two branches tend to have opposing effects, working together to adapt your body to different situations. The sympathetic nervous system allows for quick mobilization and response, while the parasympathetic nervous system brings the body back to its default state.
Why is it called the “sympathetic” nervous system? Although we usually use the word sympathy to mean feeling pity or sharing feelings with someone else, the sympathetic nervous system is not related to this concept. Instead the word sympathetic was used in the past because it was noted that organs were responding in the same way to the same thing, as if they were working together (Ackerknecht, 1974). It is from this less common definition of the word sympathy (“feeling or responding together”) that the term sympathetic nervous system comes from.
Because most autonomic tissues are connected to both the sympathetic and parasympathetic nervous systems, they are said to be controlled through dual innervation. An example would be the heart, which can be told to beat faster by the sympathetic nerve or beat slower by the parasympathetic nerve. Having both systems allows the heart to shift quickly from one state to another, much like how the accelerator and brake in your car allows you to adjust its speed. While most dual innervation is opposing or antagonistic, it’s important to note that there are exceptions; sexual arousal and urination for instance are caused by complementary effects triggered by both branches of the autonomic nervous system.
Before moving on to the next section, it may be a good idea to review the material covered so far. Below is a chart showing the various divisions of the nervous system and their main functions. You may also want to watch this short video [2:00] by 2-Minute Neuroscience that summarizes this information.
2.2 The Human Brain Here we will take a closer look at the central nervous system, or CNS. Recall that the CNS consists of the brain and spinal cord and is responsible for coordinating activity across the entire body. For this discussion, we will focus on the brain, but don’t forget that the spinal cord is part of the CNS too.
Why is the spinal cord part of the CNS? The spinal cord is more than just a bundle of nerves heading to the brain. While it does transmit signals, it is also responsible for coordinating certain reflexes independent of the brain. An example is pulling your hand back from a hot stove—the withdrawal reflex happens before your brain is even aware of the pain.
The human brain is the most complex organ in existence, comprised of approximately 80-100 billion neurons, each connected to on average a thousand other neurons, resulting in nearly 100 trillion different connections. Electrical signals propagating through these connections give rise to reflexes, movement, and higher intellectual function. Different regions of the brain are associated with different functions; this section will highlight some of them, although be aware that there are many, many more regions than just these.
Before reading the next section, watch this video from National Geographic that covers some of the upcoming information and will help you start thinking about the structure and function of the brain:
Brain 101 | National Geographic [3:58]
By the end of this section you should be able to:
• Describe the cerebral cortex and explain the different functions of the frontal, parietal, temporal, and occipital lobes.
• Describe the roles of the thalamus and limbic system, including the amygdala, hippocampus, nucleus accumbens, and hypothalamus.
• Explain the functions of the cerebellum, medulla oblongata, pons, and the ascending reticular activating system.
2.2.1 Cerebral Cortex The cerebral cortex is the outer layer of brain tissue and is probably what comes to mind when you think about or see images of the brain. It is the largest section of the brain and is folded in on itself, which gives rise to the characteristic ridges (gyri) and grooves (sulci) that you can see from the outside. Although you do not need to know it for this class, when combined with subcortical regions it is called the cerebrum or telencephalon (from the Greek for têle [far from] and enképhalos [brain]) and is part of the forebrain.
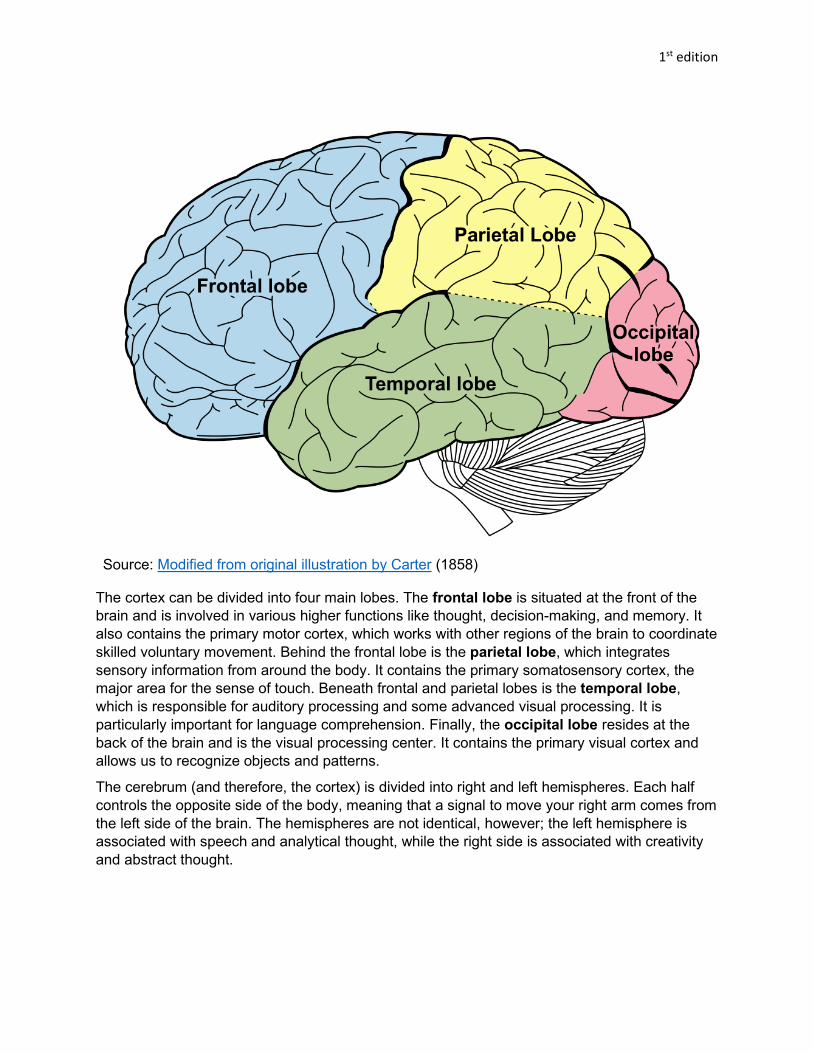
Source: Modified from original illustration by Carter (1858)
The cortex can be divided into four main lobes. The frontal lobe is situated at the front of the brain and is involved in various higher functions like thought, decision-making, and memory. It also contains the primary motor cortex, which works with other regions of the brain to coordinate skilled voluntary movement. Behind the frontal lobe is the parietal lobe, which integrates sensory information from around the body. It contains the primary somatosensory cortex, the major area for the sense of touch. Beneath frontal and parietal lobes is the temporal lobe, which is responsible for auditory processing and some advanced visual processing. It is particularly important for language comprehension. Finally, the occipital lobe resides at the back of the brain and is the visual processing center. It contains the primary visual cortex and allows us to recognize objects and patterns.
The cerebrum (and therefore, the cortex) is divided into right and left hemispheres. Each half controls the opposite side of the body, meaning that a signal to move your right arm comes from the left side of the brain. The hemispheres are not identical, however; the left hemisphere is associated with speech and analytical thought, while the right side is associated with creativity and abstract thought.
2.2.2 Thalamus and Limbic System In the interior of the brain, hidden by the cerebral cortex, are various important regions. These regions are a part of the diencephalon (diá [through] + enképhalos [brain]), which rests above the midbrain and is the other part of the forebrain. For this class, we will focus on two main regions of it: the thalamus; and the limbic system.
The thalamus is a large mass in the center of the brain and is the main relay center of the brain. It connects various parts of the cerebral cortex and other areas and relays information between them like a sort of hub. All sensory information (with the exception of olfaction, or smell) passes through the thalamus.
Source: Unattributed; retrieved from Standard of Care
The limbic system encircles the thalamus and is deeply connected to our emotions and motivations, influencing our behavior to ensure our survival. It is not a single discrete structure but an interconnected network of many structures, only a handful of which are relevant to this class. A few of them are the amygdala, which is associated with emotional responses such as fear and aggression; the hippocampus, which plays a critical role in storing memories; and the nucleus accumbens, which is an area that is critical in reward, pleasure, and the development of addiction.
Another such structure is the hypothalamus, which is situated below the thalamus and is an important regulatory center in the brain. Its main function is to maintain homeostasis, which is the stable equilibrium of the internal state of the body. It works with the autonomic nervous system to control body temperature, metabolism, hunger and thirst, and fatigue. It also works with the nearby pituitary gland to secrete various hormones.
2.2.3 Cerebellum and Brainstem Beneath the forebrain and midbrain is the hindbrain, which consists of the cerebellum and brainstem. The cerebellum is near the back of the brain beneath the cerebral cortex and is smaller and more tightly folded. As such, the name cerebellum is from the Latin for “little brain.” It is primarily responsible for balance, posture, and coordinating movement. To accomplish this, it receives sensory information from the visual and auditory system, as well as messages from the cortex and other nerves.
Source: Lynch & Jaffe (2006)
The brainstem joins the spinal cord at the medulla oblongata, which is a regulatory center like the hypothalamus. It oversees breathing, coughing, vomiting, heart rate, blood pressure, and other basic functions. Above the medulla is another brainstem structure called the pons that regulates various functions such as sleep and bladder control and relays signals between the cerebellum and thalamus. A pathway called the ascending reticular activating system goes through the medulla and pons and controls the arousal level of the brain, influencing consciousness, alertness, and the sleep/wake cycle.
Chapter Summary and Review In this chapter, we began our exploration into neuroscience by examining different structures in the human nervous system and how it can be divided into functional groups. We then took a closer look at the brain in particular and discussed the functions of different areas in the cerebral cortex, the thalamus and limbic system, and the cerebellum and brainstem. We will continue our study of the nervous system next chapter, so make sure to study the terms in this chapter and check your understanding with the practice questions below before moving on.
Practice Questions
• What is the role of the nervous system? • Explain the difference between a nerve and a neuron. • Describe the divisions of the nervous system using six key terms. Be sure to explain the
function of each division and how they relate to one another. • What is dual innervation, and where is it used? • How many lobes are there in the cerebral cortex? Give a brief description of the function
of each. • What is main role of the thalamus in the brain? • Name four of the structures in the limbic system and their function. • What are two of the functions of the cerebellum? • Name two structures located in the brainstem. What pathway passes through those
structures?
1st edition
References
2-Minute Neuroscience. (2014, August 8). 2-Minute Neuroscience: Divisions of the nervous
system [Video]. YouTube. https://www.youtube.com/watch?v=q3OITaAZLNc
Ackerknecht, E. H. (1974). The history of the discovery of the vegetative (autonomic) nervous
system. Medical History, 18(1), 1–8. https://doi.org/10.1017/s0025727300019189
Alila Medical Media. (2019, September 3). Overview of the nervous system, animation [Video].
Now that you have a sense of the human nervous system, it’s time to take a closer look at the individual cells that comprise the nervous system. This might seem less important than understanding large structures in the brain, but drugs affect the body on the cellular level. Understanding how cells in the nervous system work is the first step towards understanding why different drugs cause different psychoactive effects.
In this chapter, we will examine the types of cells found in the nervous system and how an individual nerve cells conducts a signal. We will look at the different properties of this signal as well as how signals jump from one cell to the next. Some basic knowledge in biology and chemistry is required to understand this chapter, so it may help to brush up if you feel rusty.
Chapter Outline:
3.1 Nerve Cell Structure 3.1.1 Neurons: Signal Transmission Cells
3.1.2 Glia: Neuron Support Cells
3.2 Nerve Conduction 3.2.1 Polarity of the Nerve Membrane
3.2.2 Conducting Electrical Signals: The Action Potential
3.2.3 Postsynaptic Potentials
1st edition
3.1 Nerve Cell Structure Before we get into specific types of cells, we should do a quick refresher on basic biology. Almost all tissues and organs in your body are made up of cells, which are the smallest unit of life. Each cell has a nucleus that contains genetic data and is surrounded by a cell membrane. The membrane is important because it allows the cell to control what comes in and what goes out. In order to live, cells must take in nutrients from outside the cell while removing waste that builds up inside the cell. Keep this principle in mind as you read through this chapter. Neurons, more than any other cell in our body, must keep the inside of the neuron different from the outside and control the movement of substances across the membrane.
By the end of this section, you should be able to:
• Describe parts of a typical neuron and their function, including the soma, dendrites, axon, and axon terminals.
• Describe glial cells, including Schwann cells and oligodendrocytes, and explain their role in the formation of the myelin sheath.
• Distinguish between white matter and grey matter.
3.1.1 Neurons: Signal Transmission Cells Last chapter, we mentioned that nerve fibers emanate from nerve cells called neurons. Neurons are present everywhere in the nervous system—the brain, spinal cord, and nerves are all made up of neurons. This is because the neuron is the cell responsible for carrying electric signals throughout your body. Neurons have a unique structure that helps them do this. Examine the following diagram:
Here you can see the cell body or soma on the left. (Recall that soma is from the Greek for body, which is how we got the term somatic nervous system.) The soma branches off into multiple dendrites that are responsible for receiving signals from other neurons. The name comes from the Greek word for tree, déndron, reflecting their tree-like shape and branches. To the right, there is the axon, which is a long fiber that extends away from the soma. Each neuron can have many dendrites but only one axon; signals are usually received by the dendrites, passed through the soma, and sent down the axon. The end of the axon splits into many branches, the ends of which are called axon terminals. It is here that the signal will be passed on to other cells.
Neurons in the CNS connect to other neurons, while neurons in the PNS carry signals to and from various peripheral tissues. The length of the axon depends on the location of the neuron—axons can range from a thousandth of a millimeter to over a meter in some of the longest nerves in the human body. Because axons can be so long, it’s important that the electrical signal travels quickly down the length of the axon. Because of this, many axons are wrapped in a myelin sheath that acts similar to insulation around a wire. To discuss the myelin sheath, however, we will need to address the other types of cells in the nervous system.
3.1.2 Glia: Neuron Support Cells Neurons serve a critical role in our bodies. In order to function properly, they need the support of other cells, called glia or glial cells. There are many types of glial cells that serve different functions, but you won’t need to name all of them for this class. Some provide physical support and direct neuron growth; others provide neurons with nutrition, clear away waste, and maintain the environment around the neuron; some even monitor for threats like our immune cells do in the rest of our body.
One important type of glia that you do need to know are Schwann cells, which wrap around axons in the PNS and form the myelin sheath mentioned above. They can also help nerves regenerate by removing damaged parts of the axon and guiding regrowth. These cells do not cover the whole axon; between individual Schwann cells there are small gaps where the axon is exposed. These gaps are called nodes of Ranvier and are very important for signal conduction along the axon. Keep this in mind during the next section, where we will discuss how the nerve conducts signals in the first place.
Are axons covered in myelin sheathes in the CNS? You may have noticed in the previous section that Schwann cells are limited to the PNS. Despite this, some axons in the brain and spinal cord are indeed covered in myelin. These areas are called white matter—the name comes from the fatty content of myelin, which appears white after preservation. In comparison, grey matter contains more unmyelinated axons and appears darker as a result. You can see the difference in the picture of a dissected brain below:
1st edition
Source: John A. Beal (2005)
Cells that produce the myelin sheath in the CNS are called oligodendrocytes, and have a few differences compared to the Schwann cells in the PNS. Unlike Schwann cells, which can only wrap around a single axon, oligodendrocytes can extend and wrap around multiple axons at once (see image below). They also cannot help axons regrow the way Schwann cells can, which is why even mild brain or spinal cord damage can be very dangerous and cause irreversible harm.
Source: Holly Fischer (2013)
Glia are important cells that have only recently been studied in depth. Although we won’t focus on them in this class, glial cells may play important roles in memory and learning, as well as neurodegenerative diseases like Alzheimer’s. If you are interested in learning more about their potential, I suggest reading this 2013 article from NPR: To Make Mice Smarter, Add A Few Human Brain Cells
3.2 Nerve Conduction So far, we have consistently described neurons and nerves as capable of carrying signals around the body. You may have been able to imagine nerves as long wires that carry electrical signals, but this analogy can only get you so far. After all, neurons are not made out of metal (if they were, we would always set off metal detectors). So what exactly are these signals, and how are neurons able to conduct them? These are the questions we will be answering..
By the end of this section, you should be able to:
• Explain how neurons are polarized and describe the state of a neuron at rest. • Describe the action potential and define resting potential, threshold potential,
depolarization, hyperpolarization, and refractory period. • Define saltatory conduction and explain what causes it. • Explain what a synapse is and provide examples of types of synapses. • Define excitatory and inhibitory postsynaptic potentials.
3.2.1 Polarity of the Nerve Membrane Recall that at the start of the previous section, we mentioned how neurons, like all cells, must keep their cell interior different from the exterior. This is very important for neurons, because they need to conduct electrical signals. Neurons are only able to do this because they are polarized; in other words, the electrical charge inside a neuron is different than the charge outside a neuron. The neuron accomplishes this using chemical ions, which are atoms that have an electrical charge. Watch the following video to see how this works in action:
Membrane Potential, Equilibrium Potential and Resting Potential [4:14]
Okay, there was a lot information in that video, but hopefully it helped you visualize why neurons are polarized. Let’s go over the important details here. Neurons have a resting potential of -70 mV, meaning they are more negatively charged inside than outside. They also keep two types of positive ions in a gradient across the membrane—sodium ions (Na+) want to get into the cell, while potassium ions (K+) want to get out. To remember the state of a neuron at rest, use the acronym INK: Inside the cell, Negative charge, K (potassium). This resting state is maintained by the neuron, which constantly consumes energy pumping sodium out and potassium in to maintain the gradient.
3.2.2 Conducting Electrical Signals: The Action Potential So what happens when a neuron is stimulated and told to fire? In most cases, this causes sodium ion channels to open up. There is much more sodium outside the neuron than inside, so the positively charged sodium ions flow in, causing the inside of the cell to become less negative. This decrease in charge is called depolarization because the neuron is less polarized than before.
Eventually, if enough channels are opened and enough sodium ions enter, the charge inside the neuron reaches a critical value called the threshold potential. In most neurons, this is at about -55 mV. This value is important because there are additional ion channels that will open once this voltage is reached. They are fittingly called voltage-gated ion channels, and once they are
opened, there is no going back. The neuron has now entered a runaway process that will result in an action potential—a reversal of the polarity that will travel across the neuron. This is the signal we have been referring to. To see it in action, watch this video explaining it (start at the 2:24 mark):
Action Potential in Neurons [6:30]
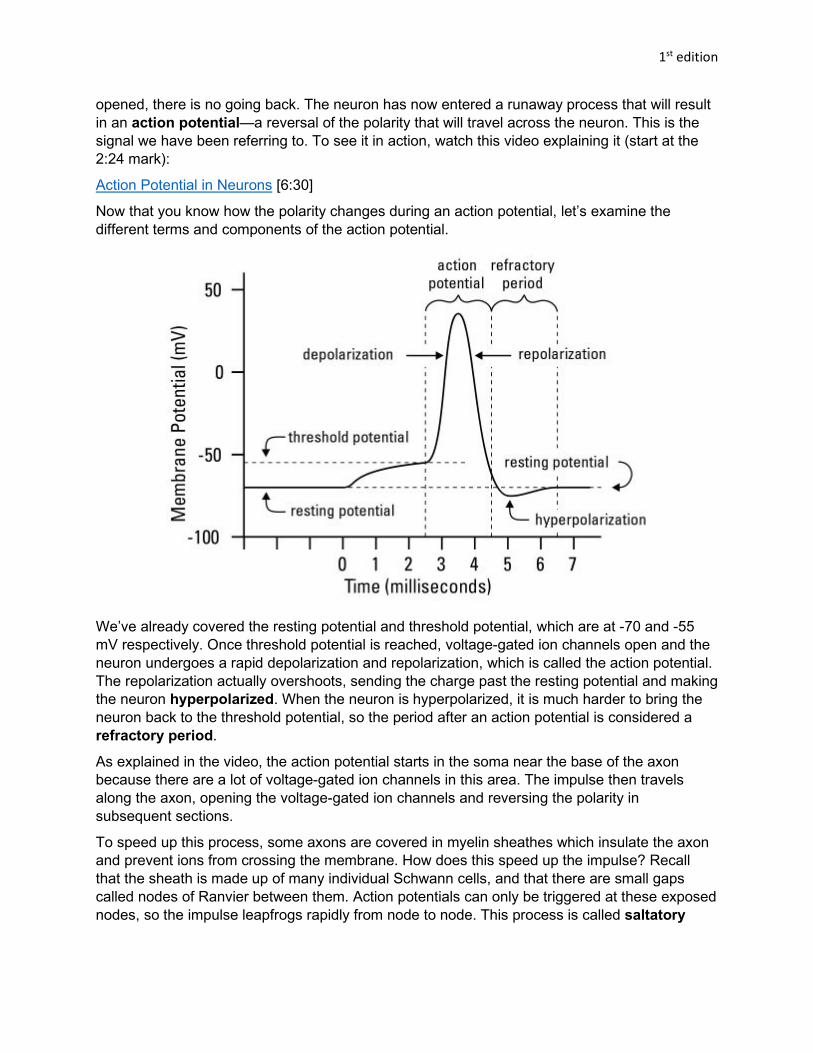
Now that you know how the polarity changes during an action potential, let’s examine the different terms and components of the action potential.
We’ve already covered the resting potential and threshold potential, which are at -70 and -55 mV respectively. Once threshold potential is reached, voltage-gated ion channels open and the neuron undergoes a rapid depolarization and repolarization, which is called the action potential. The repolarization actually overshoots, sending the charge past the resting potential and making the neuron hyperpolarized. When the neuron is hyperpolarized, it is much harder to bring the neuron back to the threshold potential, so the period after an action potential is considered a refractory period.
As explained in the video, the action potential starts in the soma near the base of the axon because there are a lot of voltage-gated ion channels in this area. The impulse then travels along the axon, opening the voltage-gated ion channels and reversing the polarity in subsequent sections.
To speed up this process, some axons are covered in myelin sheathes which insulate the axon and prevent ions from crossing the membrane. How does this speed up the impulse? Recall that the sheath is made up of many individual Schwann cells, and that there are small gaps called nodes of Ranvier between them. Action potentials can only be triggered at these exposed nodes, so the impulse leapfrogs rapidly from node to node. This process is called saltatory
conduction and is why myelinated axons conduct signals faster than unmyelinated ones. Check out this short video to see it animated:
Continuous and Saltatory Propagation [0:52]
3.2.3 Postsynaptic Potentials Eventually, the nerve impulse travels all the way to the end of the neuron at the axon terminal. How does the neuron transfer the signal to another cell? If the axon terminal were touching the other cell, it could transfer ions directly, continuing the impulse. While these types of connection are possible, they are very rare in humans. Instead, there tends to be a gap between the axon terminal and the receiving cell. This gap between two neurons and the structure that surrounds it is called a synapse, and we can call the receiving cell the postsynaptic cell. Most synapses connect axon terminals to dendrites (called axo-dendritic), but others connect to the soma (axo-somatic) or to another axon (axo-axonic) as shown in the illustration below.
Source: Cornell (2016)
When the action potential reaches the axon terminal, it causes the terminal to release certain chemicals called neurotransmitters into the synapse. (We will learn more about them in the next chapter.) These chemicals can bind to receptors on the postsynaptic cell, opening or closing various ion channels. We will cover this process in detail next chapter. For the time being, it’s important to know that some of those chemicals can increase the polarity of the postsynaptic cell, while others can decrease the polarity. The effect depends on the chemical released by the axon terminal, as well as the types of receptors on the postsynaptic cell.
Take the above image for an example. In the first example, the axon terminal releases glutamate, which causes sodium ion channels to open in the postsynaptic neuron. This causes it to depolarize, bringing it closer to the threshold potential. This is called an excitatory postsynaptic potential (EPSP) because it “excites” the postsynaptic neuron, or makes it easier for it to fire. In comparison, the axon terminal in the second example releases GABA, which opens chloride ion channels in the neuron. Since chloride ions are negatively charged, this further polarizes the neuron, i.e., causes the inside of the neuron to become even more negative, moving it farther away from the threshold potential. This is called an inhibitory postsynaptic potential (IPSP) since it inhibits the postsynaptic neuron and makes it less likely to fire.
Often, a single EPSP may not be enough to cause the postsynaptic neuron to fire. But a single neuron can receive signals from many different neurons at once. In this way, multiple EPSPs can add up to reach the threshold potential, as in the image below:
1st edition
A single neuron may have postsynaptic receptors connected to hundreds of axon terminals, any number of which may be sending excitatory or inhibitory signals to the neuron. This complex interaction is what drives all of the integration, processing, and coordination that occurs in your brain.
1st edition
Chapter Summary and Review In this chapter, we took a closer look at the individual cells that make up the nervous system: the neurons that are responsible for transmitting signals and the glia that support the neurons. We then thoroughly explored how signals can be transmitted down the length of a neuron through an action potential, which is a reversal of the polarity inside the cell, and how signals can jump from neuron to neuron using postsynaptic potentials. Make sure you understand this chapter before moving on to the next, since we’ll be covering how neurons communicate with each other over the synapse in detail and will be using a lot of the terminology and concepts established in this chapter. Check your understanding with the practice questions and ask your instructor for help if you need it.
Practice Questions
• Draw a picture of a neuron and label the soma, dendrites, axon, and axon terminals. • What types of cells are Schwann cells and oligodendrocytes? Where are they located,
and how do the two differ? • What is the difference between white matter and grey matter? • State whether each of these ions are positively or negatively charged: sodium,
potassium, and chloride. • What is the typical electrical charge of a neuron at rest? Give the answer in millivolts
(mV). • Explain depolarization and hyperpolarization. Which makes it easier for a neuron to fire? • Explain how saltatory conduction works. • What are the two types of postsynaptic potentials? Which causes depolarization, and
which causes hyperpolarization?
1st edition
References
Alila Medical Media. (2018, April 23). Membrane potential, equilibrium potential and resting
potential, animation [Video]. YouTube.
https://www.youtube.com/watch?v=MplWXZTOk6o
Beal, J. A. (2005). Human brain right dissected lateral view [Photograph]. Wikimedia Commons.
In the previous chapter, we learned how electrical signals called action potentials propagate through neurons. At the end of the chapter, we briefly mentioned how a single neuron can transfer a signal to a postsynaptic neuron by releasing certain chemicals into the synapse. The focus of this chapter is on these chemicals—what they are, how they are released, and how they can alter cell physiology.
In this chapter, we will explore the process of neurotransmission, including how it was discovered, how it works, and how drugs can interact with the process. We will then cover a number of neurotransmitters and receptors to see examples of what effects they can have on human functioning. Just like the chapter on the nervous system, many terms are introduced that will be used throughout the course, so make sure to practice and test yourself until you are comfortable with the terminology.
Chapter Outline: 4.1 Overview of Neurotransmission
4.1.1 The Discovery of Neurotransmitters
4.1.2 The Process of Neurotransmission
4.1.3 Ligands and Receptors
4.1.4 How Drugs Alter Neurotransmission
4.2 Neurochemical Transmitters and Receptors 4.2.1 Acetylcholine
4.2.2 Norepinephrine, Epinephrine, and Dopamine
4.2.3 Serotonin and Histamine
4.2.4 Glutamate, GABA, and Glycine
4.2.5 Endorphins and Substance P
4.2.6 Nitric Oxide
4.2.7 Transmitters and Receptors Review
1st edition
4.1 Overview of Neurotransmission
By now, you have probably figured out that a neurotransmitter is a chemical substance that is released into the synapse once an action potential reaches an axon terminal. As the name suggests, it transmits a signal across a synapse. You may be wondering: why though? Why bother with synapses and neurotransmitters at all? Why not just have the neuron transmit the signal directly?
It’s certainly not an intuitive idea, but using chemicals to transmit the signal indirectly comes with distinct advantages. This section is meant to not only explain what neurotransmission is, but why it occurs in the first place, and perhaps most importantly, how drugs can influence it.
By the end of this section, you should be able to:
• Describe the concept of neurochemical transmission, how it was discovered, and why it is important.
• Explain the process of neurotransmission and define all relevant terms. • Differentiate between ionotropic and metabotropic receptors and describe ligand affinity
and efficacy. • Explain how drugs can influence function by altering neurotransmission.
4.1.1 The Discovery of Neurotransmitters How exactly did the idea come about? By the 20th century, scientists knew that the nervous system was comprised of neurons and that there were gaps between them. But researchers weren’t sure how the signals crossed the synapse. It could have been entirely electrical, since electrical impulses could cause cell firing and responses like muscle contraction. But certain chemicals could achieve similar effects. With the technology of the time, there was no easy way to determine which method neurons used.
The person who would finally settle the debate was a German pharmacologist named Otto Loewi. In 1921, he came up with an experiment that would prove chemical transmission occurred. His experiment used two frog hearts. For the first heart, Loewi stimulated the vagus nerve, which was known to slow the heart rate when stimulated. He then extracted fluid from the first heart and applied it to the second heart, which had its vagus nerve removed. This caused the heart rate to slow on the second heart as well. Because only the fluid was transferred, it ruled out electrical conduction—there had to be some chemical substance in the fluid that carried the signal.
1st edition
Source: Nrets at Wikimedia Commons (2005)
Loewi called this substance vagusstoff, literally meaning “vagus substance” in German. It was the first neurotransmitter discovered and proved that neurons communicated over the synapse chemically rather than electrically. Loewi published his results and would later win the Nobel Prize in Physiology or Medicine in 1936 for his discovery.
4.1.2 The Process of Neurotransmission To understand the process, let’s return to the synapse. Last chapter we mentioned that when an action potential reaches an axon terminal, it causes the terminal to release certain chemicals. In a resting neuron, these neurotransmitters are stored in little bubbles within the cell. The bubbles are made out of the same material as the cell membrane and are called synaptic vesicles. You can see them in the diagram below:
Even when the neuron is at rest, there is spontaneous release of a small amount of neurotransmitter into the synaptic cleft. The amount is insufficient to cause a postsynaptic potential, though, and most of the neurotransmitters stay in the vesicles. To release enough of the neurotransmitters to trigger a response, an action potential must reach the axon terminal. Watch this video to see how the process works:
How Neurotransmission Works [1:34]
Let’s go over each step covered in that video in more detail. When an action potential arrives, various voltage-gated ion channels are opened. Along with the usual sodium and potassium ion channels, calcium ion (Ca2+) channels are opened. Calcium ions flow into the cell and cause the synaptic vesicles to fuse with the cell membrane, releasing the stored neurotransmitters into the synaptic cleft.
Neurotransmitters travel across the synapse and activate receptors on the postsynaptic neuron, which are protein structures that respond to neurotransmitters. The type of response depends on the type of receptor, but the simplest is a sodium ion channel that opens when activated, allowing sodium ions into the postsynaptic neuron, depolarizing it.
Eventually, the neurotransmitters are released from the receptors and return to the synaptic cleft. At this point, they need to be removed from the cleft somehow; otherwise, they will continue to activate receptors. Most neurotransmitters are returned to the presynaptic cell by
transport proteins in a process called neuronal reuptake or, simply, reuptake. Once returned inside the cell, they are repackaged into vesicles for recycling or destroyed by enzymes. A few neurotransmitters are destroyed in the synaptic cleft instead.
4.1.3 Ligands and Receptors Although we’ve been talking about how neurotransmitters can bind to and activate receptors, not every chemical that can do so is a neurotransmitter. Instead, we use the more general term ligand to refer to any chemical that can bind to a receptor. Ligands can occur naturally in our body (as in the case of neurotransmitters and hormones) or be introduced from outside (like certain types of drugs). We use the term endogenous to refer to the former and exogenous to refer to the latter. (The prefixes endo- and exo- mean inside and outside, respectively.)
When looking at receptors, there are two major types in the human body. The first are ionotropic receptors. As you can probably guess by the name, they have something to do with ions. The suffix -tropic in this context means “affecting,” so these are receptors that affect ion channels. Another name for them is ligand-gated ion channels because the channels can be opened or closed by ligands. (Compare this with the voltage-gated ion channels that create action potentials.) Take a look at the following illustration:
Source: Khan Academy
The drawing shows how neurotransmitters bind to the surface of the ion channels, causing them to open. The effect depends on the type of ion channel. Sodium ion (Na+) channels depolarize the postsynaptic neuron, while chlorine channels (Cl-) hyperpolarize it. Ionotropic receptors tend to be very fast—they activate almost immediately after the neurotransmitter binds to them, and they close quickly once the neurotransmitter is removed from the synaptic cleft.
The second type of receptor is the metabotropic receptor. The metabo- prefix is the same one you’ll find in words like metabolism, and both refer to chemical processes. Compared to ionotropic receptors, they open ion channels indirectly through secondary messengers. These messengers communicate with other parts of the cell through a series of steps, eventually opening or closing ion channels. You can see this in the following diagram (compare with the ionotropic one):
Source: Khan Academy
As you can see, the neurotransmitter does not directly bind to the ion channel, but instead binds to the metabotropic receptor, which then opens the ion channel through a series of steps. Because of the cascade of steps, metabotropic receptors are slower to respond than ionotropic receptors. But because the secondary messengers can change other aspects of the cell’s physiology, the effects (depolarization or hyperpolarization) can last longer and be more widespread.
One type of metabotropic receptor worth noting is the G-protein coupled receptor or GPCR for short. These receptors use G-proteins as messengers, hence the name. They are involved in many important pathways and are often the target of therapeutic drugs as a result; around 34% of all FDA-approved drugs target GPCRs (Hauser et al., 2018). They are also known as seven-transmembrane (7TM) receptors because they snake across the cell membrane 7 times.
Something important to remember is that it is the receptor, not the neurotransmitter, that determines the function. The same neurotransmitter (or ligand) can cause different effects if it binds to different receptors. Additionally, ligands and receptors are not always a perfect fit. Some ligands can bind to receptors more easily than others. We would say that those ligands have a high affinity for that receptor. But just because a ligand binds to a receptor doesn’t mean it activates it. A ligand’s ability to active the receptor is its efficacy. It is possible to have ligands with a high affinity but low efficacy, meaning they easily bond to a receptor but don’t activate it. Indeed, this is how certain drugs work—by blocking certain receptors and preventing them from activating. This is analogous to a key fitting into a lock (affinity) and having the appropriate ridges and cuts to open the lock (efficacy).
4.1.4 How Drugs Alter Neurotransmission As mentioned at the start of this section, drugs influence the body by altering neurotransmission. We’ll save the exact methods for when we discuss individual drugs, but you should be able to tell that there are many ways this can occur. Some drugs mimic endogenous ligands, activating receptors directly; others interfere with reuptake; some block receptors by binding to them without activating them.
How the drug alters neurotransmission determines how our behavior changes. Often, learning the effects of a drug starts with learning what neurotransmitters it affects. Once you know that, you’ll be able to predict the kinds of physiological changes that will occur while under the drug’s influence. This pattern will show up with every type of drug we cover in this class, so it is a good idea to get comfortable with it.
1st edition
4.2 Neurochemical Transmitters and Receptors Now that you know how neurotransmission occurs, it is time to learn about some of the neurotransmitters that are used in the human body. There are many more neurotransmitters than what is included in this section (possibly up to one hundred); the ones selected are some of the most prevalent and well-researched. Most of these will be affected in some way by the drugs that we will examine later in the course.
There is a lot of material in this section, but don’t worry; at the end of the section, there will be a table that will give you an overview of all the important information. As you read this section, try to associate the name of each neurotransmitter with its function; once you reach the end, you will be able to go over all the receptors and categories again, and organizing everything will be easier once you have a sense of what each term means.
By the end of this section, you should be able to:
• Provide examples of neurochemical transmitters, their general functions, and the receptors they activate.
• Explain the classification of various types of neurotransmitters, including monoamines, amino acids, peptides, and gaseous signaling molecules.
4.2.1 Acetylcholine In this subsection, we will look at acetylcholine, the first neurotransmitter identified. This was the chemical involved in Otto Loewi’s experiment that he named vagusstoff. Acetylcholine had already been discovered in biological organisms by then, and Loewi and other researchers suspected that vagusstoff was acetylcholine, although it took a few years before this was verified.
Acetylcholine
Source: NEUROtiker on Wikimedia Commons (2007)
In the PNS, acetylcholine plays a large role in the parasympathetic nervous system. Recall that in Loewi’s experiment, the vagusstoff caused the heart rate to slow. This is one of the many effects that acetylcholine is responsible for, along with other “rest and digest” responses such as pupil constriction and innervating smooth muscle. Acetylcholine is also the neurotransmitter that activates skeletal muscle in the somatic nervous system, meaning your voluntary movements are all regulated by this neurotransmitter.
In the CNS, acetylcholine plays an important role in processing memories. In the hippocampus, damage to acetylcholine receptors is associated with the memory loss seen in people with Alzheimer’s disease. The neurotransmitter is also involved in attention and arousal.
Acetylcholine receptors are called cholinergic receptors. In this context, the -ergic suffix means “activated by,” so the term cholinergic just means “activated by choline.” (Choline is one of the chemicals that acetylcholine is made out of, which is why it’s in the name.) There are two types of cholinergic receptors: muscarinic and nicotinic. Both are named after drugs that produced a different subset of the effects that acetylcholine does. As you can probably guess, nicotinic receptors are activated by nicotine, which will be cover in detail by this class.
4.2.2 Norepinephrine, Epinephrine, and Dopamine In this subsection we will cover catecholamines, which are organic compounds that have a catechol (benzene ring with two hydroxyl side groups) connected by two carbons to a single amine (NH) group. You don’t need to memorize the molecular structure of each neurotransmitter for this class, but, by examining the chemical structures below, you can see similarities between each of the neurotransmitters in this class.
Epinephrine
Source: NEUROtiker on Wikimedia Commons (2007)
The first catecholamine we will discuss is epinephrine. In the UK and Europe, epinephrine is called adrenaline, which probably gives you a sense of what epinephrine does. We often talk about getting a rush of adrenaline in fight-or-flight situations, so as you might expect, epinephrine is involved the sympathetic nervous system. Epinephrine can dilate the pupils and increase blood flow from the heart to the muscles. It is a hormone that is produced by the adrenal glands and activates adrenergic receptors, of which there are two main types called alpha (α) and beta (β) adrenergic receptors. You might also see adrenergic receptors called adrenoreceptors, which is just a shorter name for the same thing.
A very similar chemical is called norepinephrine, which is also called noradrenaline internationally. The nor- prefix comes from the fact that in this molecule, the CH3 group is missing. Similar to epinephrine, it is involved in the sympathetic response and activates α and β adrenergic receptors.
Unlike epinephrine, which is released solely as a hormone, norepinephrine is released as both a hormone and a typical neurotransmitter. In the CNS, it plays a role in arousal and attention and is important in mood regulation. A deficiency in norepinephrine can be a component to certain types of depression, which we will cover in detail when we discuss antidepressants.
What is the difference between a hormone and neurotransmitter? We mentioned that epinephrine is a hormone secreted by the adrenal gland, but what exactly is a hormone? Hormones are produced in organs called glands and are secreted into the bloodstream. Compare this to most neurotransmitters, which are produced inside the axon terminal of neurons and released into the synaptic cleft. The big difference is range: hormones are circulated throughout the body and can reach distant target cells, while neurotransmitters are limited to the synapse. Although there is a clear line between hormones and neurotransmitters in terms of function, some chemicals can behave as neurotransmitters in some cases and hormones in others. If you are interested in learning more (it is not required for this class), a good place to start is this brief writeup by Alpana and Murari Chaudhuri: Hormones and Neurotransmitters: The Differences and Curious Similarities
The third and final catecholamine is dopamine. Dopamine is a very important component of drug dependence, as one of dopamine’s main roles in the CNS is reward and reinforcement. The addictive properties of many drugs come from how the drugs stimulate dopamine release and create long-term changes in dopamine-related pathways in the brain. Dopamine is also involved in motor control, and two well-known neurological disorders, Parkinson’s disease and Huntington’s disease, are caused by a deficiency (Parkinson’s) or excess (Huntington’s) in dopamine activity in the basal ganglia. The psychiatric disorder schizophrenia is also caused by an excess of dopamine activity in the limbic system.
Dopamine activates dopaminergic receptors, of which there are at least five different subtypes.
4.2.3 Serotonin and Histamine The three catecholamines mentioned above are actually a subset of a larger group of neurotransmitters called monoamines. As their name suggests, monoamines have a single amino group, which is connected to an aromatic ring by a two-carbon chain. It is less important that you are able to explain the molecular structure of monoamines; instead, you should be able to name five neurotransmitters that are classified as monoamines—the three catecholamines listed above, the two covered below.
Serotonin
Source: NEUROtiker on Wikimedia Commons (2007)
The fourth monoamine is serotonin. The full name for serotonin is 5-hydroxytryptamine, or 5-HT for short. You will often see serotonin called 5-HT in the scientific literature, so it is worth remembering.
In the PNS, serotonin plays a large role in digestion. Most of the serotonin produced in the body is found in the digestive tract, where it regulates intestinal movement, gastric acid secretion, and mucus production. In the CNS, serotonin is linked to numerous functions, including sleep, anxiety, mood, appetite, nausea, and social and sexual behavior. Similar to norepinephrine, one type of depression is classified as being due to a deficiency in serotonin. 5-HT also plays a major role in the genesis of anxiety.
Serotonin receptors are called 5-HT receptors; they are the most abundant in the human body and contain the most subtypes out of all receptors. They are grouped into 7 main types, labeled 5-HT1 to 5-HT7, and each of those can be distinguished further into subtypes like 5-HT1A.
Histamine
Source: NEUROtiker on Wikimedia Commons (2007)
The last monoamine we will cover is histamine. (There are other monoamines, but we will focus on these five for this class.) In the PNS, histamine is a part of the inflammatory response, which helps the immune system fight pathogens. This response is what causes itching, sneezing, and a runny nose when you have a cold. In the CNS, histamine is involved in a variety of effects, the most important of which is promoting wakefulness. This is why antihistamine medication is well-known for its sleep-inducing properties. Histamine acts at four main H receptors, H1 to H4, all of which are metabotropic.
4.2.4 Glutamate, GABA, and Glycine The next group of neurotransmitters are the amino acid transmitters. You may recall learning that amino acids are the building blocks of proteins. There are a few amino acids that also act as neurotransmitters. In this subsection, we will look at the three most important ones.
Glutamate
Source: NEUROtiker on Wikimedia Commons (2007)
The first is glutamate, which is the main excitatory neurotransmitter in the CNS. It is involved in various cognitive functions like learning and memory. It is also implicated in neurological conditions such as Alzheimer’s disease, Parkinson’s disease, and epilepsy through a process called excitotoxicity, where overstimulation of glutamate receptors can result in cell degradation and eventually cell death.
The three main glutamate receptors are all ionotropic and are called AMPA, kainate, and NMDA receptors. The latter is of particular interest to this class, since NMDA receptors are the target sites for certain psychoactive substances like PCP and ketamine.
GABA
Source: NEUROtiker on Wikimedia Commons (2007)
The next neurotransmitter is called gamma-aminobutyric acid, or GABA for short, and it is very important for this class. It is the main inhibitory neurotransmitter in the brain, meaning that glutamate and GABA have opposing effects. In fact, some scientists claim that glutamate and GABA are the only two neurotransmitters in the brain; in this view, other neurotransmitters are considered neuromodulators, since they enhance or reduce glutamate and GABA transmission. GABA acts at two receptor subtypes, called GABAA and GABAB, to dampen overactive neurons. Many sedatives and tranquilizers achieve their effects by enhancing GABA transmission in some way. Because it is involved in a wide variety of CNS depressants, including alcohol, we will cover GABA and its receptors in more detail in a later chapter.
Glycine
Source: NEUROtiker on Wikimedia Commons (2007)
Finally, whereas GABA is the main inhibitory transmitter of the brain, glycine is the main inhibitory transmitter in the spinal cord, although it is also present in the brainstem and retina. It has a single receptor simply called the glycine receptor (GlyR) and aside from its role as an inhibitor like GABA, it is also involved in processing motor and sensory information.
4.2.5 Endorphins and Substance P So far, all the neurotransmitters we’ve covered are considered small molecule transmitters, since their molecular structure is relatively small. Neurotransmitters can be much larger, however. Such transmitters are called peptides or neuropeptides; peptide means that they are
comprised of a chain of amino acids, similar to proteins (although much less complex). We will cover two types of relevant neuropeptides: endorphins and Substance P.
β-Endorphin
Source: Edgar181 on Wikimedia Commons (2010)
The first type of peptides we will look at are the endorphins, of which there are three types: alpha, beta, and gamma (γ). Above you can see the structure of β-endorphin—clearly, it is much larger than all of the neurotransmitters we have looked at so far. β-endorphin consists of 31 aino acids. Endorphins act at opioid receptors, of which there are also three types: mu (μ), kappa (κ), and delta (δ). You have probably heard of opioids such a morphine, heroin, oxycodone, and fentanyl. In fact, the name endorphin comes from a contraction of endogenous morphine, since they occur naturally in the body. They are also called endogenous opioid peptides.
Like opioids, endorphins are noted for producing an analgesic (pain-blocking) effect. This is because the activation of opioid receptors inhibits the release of Substance P, which we will discuss next. In the PNS, endorphins (mostly β-endorphin) are secreted by the pituitary gland and act as hormones. In the CNS, endorphins also trigger feelings of pleasure by inhibiting GABA, which in turn increases dopamine activity in the reward center. The euphoric sensations that occur from listening to music, eating something delicious, sex, or vigorous aerobic exercise (known as a “runner’s high”) are all the result of endorphin activity.
Two other common endogenous opioid peptides are the enkephalins, which consists of five amino acids, and the dynorphins, which are 13- and 17-amino acid fragments of a larger 32-amino acid precursor.
As previous mentioned, endorphins can inhibit the release of Substance P, which is involved in transmitting pain signals to the CNS. It was the first neuropeptide ever discovered consisting of a chain of eleven amino acids. Although you might assume the P in the name stands for pain, it actually stands for powder, since it was originally discovered and purified in powder form (Hochberg et al., 2019). Substance P is released from the ends of sensory nerves and activates neurokinin-1 (NK1) receptors.
4.2.6 Nitric Oxide Aside from small molecule neurotransmitters and peptides, there is a third type of transmitter called gaseous signaling molecules or gasotransmitters. These are gas molecules that can freely permeate cell membranes—instead of binding to receptors on the surface of a cell like all of the neurotransmitters discussed previously, these molecules simply cross the membrane on their own and alter cell physiology directly inside the cell. Because they can permeate cell membranes, they cannot be stored in vesicles like other neurotransmitters and instead are produced on demand. Gaseous signaling molecules are recent discoveries in neurotransmission and a lot is currently unknown about them, so we will only look at one type that has been studied the most.
Nitric Oxide
Source: Yikrazuul on Wikimedia Commons (2008)
Nitric oxide (abbreviated NO) is a molecule comprised of a single nitrogen atom coupled to a single oxygen atom. NO is a free radical, meaning it has an unpaired electron. The “receptors” that it binds to are actually enzymes inside the cell, of which the most researched is soluble guanylate cyclase (SGC). When nitric oxide binds to the enzyme, it triggers a signal cascade
that leads to the relaxation of smooth muscle. Because of this, a common use of nitric oxide is to dilate blood vessels, increasing blood supply.
Nitric oxide also plays a role in the immune system, as it is toxic to cellular organisms and can be secreted in response to pathogens. Studies have suggested that it can inhibit the replication of certain viruses, including SARS (Åkerström et al., 2005), which has led to nitric oxide being proposed as a potential treatment to COVID-19 by mitigating pulmonary symptoms and inhibiting the replication of the virus (Adusumilli et al., 2020; Pieretti et al., 2021).
4.2.7 Transmitters and Receptors Review Phew, that was a lot to cover. If you got a little lost at times, it’s okay—there were a lot of new terms being thrown around. To learn this material and organize it in your head, you will have to start by memorizing the associations between the neurotransmitters, their receptors, and their functions. Creating flash cards is a great way to practice and form those connections.
To help, below is a chart that summarizes all of the important information from this section. You may also want to watch this video from Khan Academy, which covers most of the neurotransmitters from this section:
Types of neurotransmitters | Nervous system physiology [8:45]
Histamine H1, H2, H3, and H4 inflammatory response, wakefulness
Am
ino
Aci
ds Glutamate AMPA, kainate, and NMDA main excitatory transmitter (CNS)
GABA GABAA and GABAB main inhibitory transmitter (brain)
Glycine glycine (GlyR) main inhibitory transmitter (spinal cord)
Peptides Endorphins μ, κ, and δ opioid analgesia, euphoria
Substance P neurokinin (NK1) pain transmission
Gaseous Signaling Molecules
Nitric Oxide soluble guanylate cyclase (SGC)
smooth muscle dilation, immune response
1st edition
Chapter Summary and Review In this chapter, we took a deep dive into neurotransmitters, starting with how they were discovered and how they worked in the synapse. We explored the different types of receptors that they could activate and how other substances could interact with those receptors. Finally, we provided an overview of a handful of important neurotransmitter/receptor pairs, including how they are classified and what functions they are involved in.
Good work on making it to the end of the neuroscience chapters. This chapter contained many new terms that will be used throughout this course, so testing yourself using flash cards is highly recommended. As in previous chapters, you can check your understanding with the practice questions below, but remember that they are not comprehensive and are only meant as a starting point.
Practice Questions
• Who discovered the first neurotransmitter? What was the neurotransmitter named? • Name three different ways that neurotransmitters can be removed from the synaptic
cleft. • Describe the differences between ionotropic and metabotropic receptors. • What is the difference between a ligand that has high affinity for a receptor and one that
has a high efficacy? • What are the two types of receptors activated by acetylcholine? • Name the five monoamines covered in this chapter. Which ones are the
catecholamines? • What neurotransmitter is highly associated with reward and motivation? • What is another name for serotonin? • Name the three amino acid neurotransmitters and where each is found in the nervous
system. • Which two neurotransmitters are considered the main excitatory and inhibitory
transmitters in the brain? • What type of receptors do endorphins activate? • Describe the differences between small molecules, peptides, and gaseous signaling
molecules.
1st edition
References
Adusumilli, N. C., Zhang, D., Friedman, J. M., & Friedman, A. J. (2020). Harnessing nitric oxide
for preventing, limiting and treating the severe pulmonary consequences of COVID-19.
Nitric Oxide, 103, 4–8.
Åkerström, S., Mousavi-Jazi, M., Klingström, J., Leijon, M., Lundkvist, Å., & Mirazimi1, A.
(2005). Nitric oxide inhibits the replication cycle of severe acute respiratory syndrome
coronavirus. Journal of Virology, 79(3), 1966–1969.
Edgar181. (2011, August 31). Chemical structure of beta-endorphin [Illustration]. Wikimedia
For the past three chapters, we have looked at the human nervous system from the overall structure down to the individual synapse. For the next two chapters, we will be bringing drugs to the forefront by exploring pharmacology, which can be divided into two main branches. The first branch—pharmacokinetics—is the focus of this chapter.
Just like with neuroscience, pharmacology is a vast and complex subject that is often studied over multiple semesters. This discussion will be simplified somewhat, but there will still be many new terms and concepts to learn. Nevertheless, doing so will be vital, as these ideas will show up again and again when we transition to looking at specific types of drugs for the rest of the semester. Stay focused, and be sure to reach out to your instructor if you need help.
Chapter Outline: 5.1 Overview of Pharmacology
5.1.1 The Pharmaceutical Sciences
5.1.2 ADME
5.2 Absorption 5.2.1 Determinants of Absorption
5.2.2 Enteral Routes
5.2.3 Parenteral Injection Routes
5.2.4 Parenteral Non-Injection Routes
5.3 Distribution 5.3.1 Plasma Protein Binding
5.3.2 The Blood-Brain Barrier
5.4 Metabolism 5.4.1 Metabolism in the Liver
5.4.2 Enzyme Induction and Inhibition
5.4.3 Prodrugs
5.5 Excretion 5.5.1 Routes of Excretion
5.5.2 Elimination Kinetics
1st edition
5.1 Overview of Pharmacology As mentioned in the introduction, this chapter is the start of our exploration of pharmacology, which is the study of the actions and effects of drugs. You can easily see how such a field is relevant to a class with the words “effects of alcohol and other drugs” in its name. Pharmacology is the foundation of many health sciences and is critical to developing therapeutic drugs and drug treatments.
By the end of this section, you should be able to:
• Differentiate between pharmaceutics, pharmacokinetics, and pharmacodynamics. • Define the four different components of pharmacokinetics.
5.1.1 The Pharmaceutical Sciences Pharmacology can be broken down into two different branches: pharmacokinetics, which is the study of how the drug moves around the body, and pharmacodynamics, which is the study of how the drug changes the body. You can use these meanings to tell the two terms apart; the suffixes -kinetics [movement] and -dynamics [change] refer to how the drug moves and what the drug changes.
Pharmacology is only one of many different areas of study related to drugs. Another example is medicinal chemistry, which is the synthesis of new drug compounds. We briefly touched on it during the discussion of the New Drug Approval process in the first chapter, although not by name. There are other fields as well, each with many different subspecialties.
One area worth mentioning is pharmaceutics, or the study of how a drug is formulated and dispensed. In the past, pharmacists often dispensed drugs directly as a powder containing just the active ingredients. Nowadays, drugs are usually designed with a dosage form in mind, which is a mix of active and inactive ingredients prepared in a particular form, such as a capsule or tablet. Dosage forms allow for greater control over the dose of the drug and how it is taken.
Although we will not cover pharmaceutics in detail in this course, it is worth knowing because of the relationship between pharmaceutics and pharmacokinetics. As you can see in the diagram below, the dosage form determines how the drug is made available to the body. This influences the pharmacokinetics of the drug, which in turn influences the pharmacodynamics of the drug.
1st edition
5.1.2 ADME The focus of this chapter is pharmacokinetics, which as we just mentioned is concerned with how the drug moves throughout the body. In reality, the drug isn’t moving on its own—it’s actually being moved around by the natural systems in our body. Because of this, we can also say that pharmacokinetics is what the body does to the drug. (Next chapter we will look at pharmacodynamics, which is the opposite—what the drug does to the body.)
There are four main things that the body does to the drug: it absorbs it into the bloodstream, distributes it to various areas of the body, metabolizes it into different compounds, and excretes it from the system. A useful mnemonic that can help you remember this process is ADME—Absorption, Distribution, Metabolism, and Excretion. We will spend the rest of this chapter examining each of these in detail.
1st edition
5.2 Absorption The first factor that influences how a drug moves throughout the body is absorption. Absorption describes the movement of the drug from its site of administration to the circulatory system. For most drugs, the bloodstream is what will carry the drug to its site of action. As such, understanding how the drug gets absorbed into the bloodstream is an important component of pharmacokinetics.
By the end of this section, you should be able to:
• Define bioavailability and diffusion. • Explain how a drug’s ability to permeate membranes is critical to absorption and
describe what factors can influence this. • Describe different routes of administration and explain how they influence drug
absorption and bioavailability.
5.2.1 Determinants of Absorption Following administration, not all of the drug will be absorbed into the bloodstream, and not always at the same rate. The amount that does get absorbed is termed the bioavailability, expressed as a percentage of the amount administered. Some drugs will also be absorbed more quickly, which can increase the strength of their effects.
The bioavailability and rate of absorption depend heavily on how well the drug can diffuse from its site of administration. Diffusion simply refers to a substance spreading out, i.e., moving from an area of high concentration to low concentration. We have already encountered this idea before when discussing action potentials in chapter 3. A drug that can easily pass through membranes will diffuse faster than one that cannot.
How well the drug can permeate these membranes depends on certain properties of the drug. Larger molecules, ionized chemicals, and hydrophilic (water-loving) substances all have a harder time passing through membranes. This is because the phospholipid bilayers that make up cell membranes consist of hydrophilic heads and uncharged tails that repel hydrophilic and ionized molecules.
Aside from diffusion that occurs on its own, known as passive diffusion, drugs can also be moved via active transport mechanisms. These mechanisms, such as ion channels and transport proteins, consume energy but can move larger molecules and work against concentration gradients. By now, you should be familiar with the ion channels found in nervous tissue; similar channels exist in different cells. Active transport can allow drugs with larger molecules to pass through membranes and be absorbed.
The rate of absorption and bioavailability also depend on the route of administration, or the path that the drug takes into the body. Some paths are more direct that others—a drug that is injected directly into the bloodstream will by definition have 100% bioavailability, while drugs that pass through the gastrointestinal tract face a gauntlet of obstacles that will slow down the rate of absorption and reduce the total amount of the drug that reaches the systemic circulation. Recall that the dosage form of a drug can determine how it is taken, so the route of administration is often influenced by the drug’s pharmaceutics. Another way of looking at it is that if a certain route is preferred, the dosage form has to be changed to match.
1st edition
For the remainder of this section, we will look at various routes of administration. As we cover each one, pay close attention to how they differ in terms of absorption and bioavailability.
5.2.2 Enteral Routes There are two main categories for routes of administration. The first type is the enteral route, which refers to the routes that pass through the gastrointestinal tract. (The word enteral is from the Greek for énteron, meaning “intestine”). This is usually accomplished through oral administration, or taking the drugs by mouth. This is the method of drug-taking that you are probably the most familiar with; capsules, tablets, and liquids like cough syrups and alcohol are all taken orally (although we say we drink alcohol rather than “orally administer” it). Aside from the oral route, there is also rectal administration, which involves inserting the drug directly into the rectum, as in the case of suppositories.
Of these two routes, the rectal route is faster and simpler. Drugs taken orally must first pass through the stomach. The stomach typically absorbs drugs more slowly than the intestines, so it can take longer for the drug to be absorbed. If the stomach is full of food, the drug will spend more time in the stomach, reducing the rate of absorption even further. Finally, the dosage form of oral medication is important, because not all drugs can survive the highly acidic environment in the stomach. These drugs must be enclosed in acid-resistant capsules that delay the release of the drug until after it reaches the intestine.
Both oral and rectal routes pass through the intestinal walls, which are comprised of epithelial cells. Drugs must be able to permeate these cells in order to be absorbed; otherwise, they will simply pass through the intestines and be excreted without accomplishing anything. If a drug cannot be absorbed through the intestinal wall, it may require a different route altogether.
Even if a drug makes it past the intestinal walls and into the bloodstream, it will be taken to the liver before circulating to the rest of the body. This is significant because the liver often metabolizes drugs, which may reduce the bioavailability further. We will cover this in detail when we reach the section on metabolism. For now, it is enough to grasp that enteral routes tend to have low bioavailability and slow rates of absorption, especially in the case of oral administration. In spite of this, taking medication by mouth is generally the most convenient option, so the effort to design a drug that can be taken orally—and make it all the way to the bloodstream—is usually worth it.
5.2.3 Parenteral Injection Routes The alternative to the enteral routes is the parenteral route, which includes all the routes that do not pass through the gastrointestinal tract. This often involves an injection of some sort, although there are non-injection routes as well. We’ll start by looking at the routes that involve injection first.
First is intravenous, or IV, which involves injecting the drug into a vein. Because the drug is injected directly into the bloodstream, IV administration is fast and results in 100% bioavailability. For drugs like heroin this manifests as an immediate rush of pleasure, which is why they are often injected this way. IV therapy is also ideal for emergency use in hospitals, as it can be used for blood transfusions, fluid replacement, nutrition, and medications. The
1st edition
downside of the IV route is that it requires skill and knowledge to use, since a vein must be found and pierced with a needle. Although some users of drugs like heroin become proficient at IV injections, veins can collapse if they are used excessively.
Another common method of injection is intramuscular, abbreviated IM. As the name suggests, intramuscular medications are injected into the skeletal muscle, where they are absorbed into the bloodstream. The IM route results in high bioavailability but is somewhat slower than IV. Although many drugs can be administered intramuscularly, most people have experienced IM administration when getting vaccinated, as vaccines are typically given with an IM injection.
Aside from injecting the drug into the veins or muscles, it can also be injected below the skin, known as subcutaneous (sometimes abbreviated as SC or SQ). Compared to the IM or IV routes, absorption takes longer because there are fewer blood vessels underneath the skin. In exchange, subcutaneous injections are good for drugs that need to be absorbed for a long period of time, which is why insulin is usually administered subcutaneously.
Another method is intraosseous infusion (IO), which involves injecting directly into bone marrow. As you may recall from biology, the marrow is the part of the bone that is responsible for producing new blood cells, and, as such, has direct access to the bloodstream. In fact, IO administration is comparable to IV in terms of speed of absorption and bioavailability. IO is useful when IV access cannot be established quickly, such as with trauma patients or during cardiac arrests; in these cases, the IO route can be used to administer fluids and drugs used in resuscitation like epinephrine.
The last injection route we will look at is intrathecal, which means injecting into the theca, or the sheath of the spinal cord that contains the cerebrospinal fluid. This route is notable because it bypasses the blood-brain barrier, an impediment to distribution that we will cover in more detail in the next section. Certain anesthetics and chemotherapy drugs are administered this way.
5.2.4 Parenteral Non-Injection Routes Now we will look at routes that bypass the gastrointestinal tract without the need for a needle. First up is inhalation, which involves inhaling the drug as a vapor. This produces high bioavailability like IV administration but is actually faster because the drug enters the circulatory system at the lungs, instead of at the veins where it has to be carried back to the heart before being circulated. This makes inhalation a common method for recreational drug use, as it provides an immediate effect. Although smoking is convenient, the chemical byproducts produced by it can damage the lungs. Safer methods of inhalation are found in therapeutic drugs, such as the asthma inhalers that contain corticosteroids, or the anesthetics used during general surgery.
Another method is topical, which means applied to a certain place, often a body surface. This is typically the skin, as in the case of ointments or creams, but can also refer to things like eye drops and ear drops. Topical administration does not result in systemic effects; that is, instead of being absorbed in the bloodstream and distributed to the site of action, topical medications simply work locally at their intended site of action. As a result, they have negligible bioavailability and do not have to be concerned with distribution.
1st edition
In comparison, transdermal administration (meaning “through the skin”) does result in the drug reaching the systemic circulation, as the drug is gradually absorbed by capillaries in the skin. This process is very slow but is similar to subcutaneous injections in that it can support sustained absorption of the drug. You have probably heard of the nicotine patches used to help people quit smoking; these are an example of transdermal administration.
A similar method of administration is sublingual. This method, which means “beneath the tongue,” involves placing a tablet underneath the tongue, where it dissolves and is absorbed by the capillaries there. Sublingual medications can also be applied as a dissolvable strip or liquid drops. Nitroglycerin tablets, used to treat angina pectoris, are administered sublingually.
Finally, drugs can be administered through a nasal route. The nasal passage contains mucosal membranes that can absorb drugs into the capillaries, similar to sublingual or transdermal routes. Drugs can be applied as a liquid or powder, that latter of which dissolves inside the nasal passage. Examples of drugs that use this route are nasal decongestant sprays and some recreational drugs that are snorted (most notably cocaine).
Before moving on, take a moment to look over the table summarizing each of the routes of administration below.
1st edition
Route Site Example
Enteral Oral mouth (e.g.,
swallowed) pills, tablets, cough syrups,
alcohol
Rectal rectum suppositories
Parenteral
Inje
ctio
n
Intravenous vein many drugs (e.g., morphine, heroin)
Sublingual beneath the tongue nitroglycerin tablets
Nasal nasal passage decongestant sprays, snorting
1st edition
5.3 Distribution Once the drug enters the circulatory system, the bloodstream carries it to the site of action. This process is known as distribution. Distribution determines how much of the drug actually reaches the site of action, similar to how absorption determines how much enters the bloodstream in the first place. In this section, we will examine two factors that influence drug distribution: plasma protein binding and the blood-brain barrier.
By the end of this section, you should be able to:
• Explain plasma protein binding and how it affects drug distribution. • Describe the blood-brain barrier and explain how it affects drug distribution.
5.3.1 Plasma Protein Binding Not all of the drug that is absorbed will be free to activate receptors at the target area. Some amount of drug may be retained in the blood, unable to diffuse out of the circulatory system to the site of action. This is because the plasma in our blood contains many different proteins, some of which can reversibly bind to drugs in a process known as plasma protein binding. To see how it works and why it can complicate drug dosage, watch this video:
Protein Binding [5:22]
Let’s review the information covered in the video. The drug-binding plasma proteins act like sponges, “soaking” up the drug by binding to it. Once bound to the protein, the drug will be stuck inside the circulatory system and unable to reach the site of action. In order to activate receptors, it is necessary to first saturate the protein binding sites in the blood, meaning a larger amount of drug is required. The amount depends on how well the drug binds to the proteins. A drug that has a binding rate of 99% means that only 1% of the drug will be able to activate receptors, so the amount that needs to be absorbed is 100x that amount.
What complicates this process is that other drugs may also compete for these binding sites. If a new drug is introduced that binds to the same sites, it will displace some of the original drug, increasing the amount that reaches the target area. The magnitude of the change depends on each drug’s ability to bind to the plasma proteins. If a drug has a high binding rate (like in the 99% example above), even a small change in the amount of available binding sites can double or triple the amount of drug that reaches the site of action, which can result in severe effects. The opposite is also true: discontinuing a drug can render another drug ineffective, as seen in the example provided in the video. This is why it is dangerous to drink alcohol with some medications and why physicians need to be aware of what medications you’re taking before prescribing a new one.
5.3.2 The Blood-Brain Barrier Due to how important our brain is, our body has an extra layer of security meant to protect it from pathogens and toxins that may be carried in the blood. This defense is called the blood-brain barrier (sometimes abbreviated BBB), and it is an additional boundary that separates the circulatory system from the brain. Watch this video from 2-Minute Neuroscience that explains what it is and how it works:
As mentioned in the video, the blood-brain barrier is formed by tight junctions of endothelial cells, which are the cells that line blood vessels. Unlike in most parts of the body, where there are gaps between the cells to let substances through, the tight junctions in the blood-brain barrier limit which substances can diffuse through the capillaries. These tight junctions are formed with the help of astrocytes, which are a type of glial cell found in the brain (recall from chapter 3 how glial cells are the support cells of the nervous system).
The blood-brain barrier helps maintain a constant environment for the brain and protects it from foreign substances or neurotransmitters from other parts of the body. As a consequence, not all drugs can pass through the barrier. Recall the discussion of a drug’s ability to permeate membranes in the previous section; large, ionized, or hydrophilic drugs will find it harder to reach the brain. This means that if a drug’s intended site of action is in the brain, it needs to find some way of penetrating the blood-brain barrier, such as by being lipid-soluble or relying on active transport mechanisms.
Similar barriers exist in other parts of the body. The cerebrospinal fluid is protected by a barrier that lets in some substances that are blocked by the blood-brain barrier. The placental barrier also exists between a fetus and its mother, although this barrier is much more permeable to drugs and other substances, which is why expecting mothers are advised to abstain from drinking, smoking, or other drug use as the drugs can cross the barrier and harm the fetus.
5.4 Metabolism The human body is not limited to simply moving drugs around. After all, many substances that we consume, intentionally or not, can be toxic to us. Our bodies chemically modify these substances in a process known as metabolism or biotransformation. Metabolism can transform inert substances into nutrients or alter toxic chemicals so that they are more easily expelled from the body.
When a drug is metabolized by our body, the result is called a metabolite. If a metabolite has a physiological effect of its own, it is called an active metabolite, but sometimes drugs are transformed into inactive metabolites that have no effect on the body. The transformation of a drug into active and inactive metabolites affects all other aspects of pharmacokinetics, which is why we will be taking a closer look at the process in this section.
By the end of this section, you should be able to:
• Explain the first-pass effect and how it affects bioavailability of oral drugs. • Describe the metabolic processes that occur in the liver and explain the role of enzymes
such as cytochrome P450. • Explain how enzyme inducers and inhibitors affect bioavailability. • Describe prodrugs and explain why they are useful.
5.4.1 Metabolism in the Liver The main site where metabolism occurs is the liver. Although biotransformation occurs elsewhere in the body, we will focus on the liver for this course. The reason why the liver is so significant is because everything we eat and drink is sent to the liver first for processing. Substances are absorbed from the intestinal tract and carried directly to the liver by the portal vein; they are only able to reach other parts of the body after passing through the liver (see image below).
1st edition
What this means is that drugs absorbed from the intestinal tract are taken straight to the liver before they can be distributed to the site of action. This is known as the first-pass effect or first-pass metabolism, where some of the drug is immediately metabolized in the liver before reaching systemic circulation. This reduces the bioavailability of orally administered drugs. First-pass metabolism also affects rectal administration, but to a lesser degree as some of the drug can enter systemic circulation right away.
What exactly does metabolism entail? All metabolic processes are chemical reactions aided by enzymes, which are proteins that catalyze (speed up) the reaction. Metabolic reactions are classified into two groups: phase I and phase II. Phase I reactions typically transform the drug to make it more hydrophilic through oxidation, reduction, or hydrolysis. This is necessary because it is difficult to eliminate lipid-soluble molecules from the body, so the liver alters them to be hydrophilic or water-soluble instead. Most reactions in this phase involve enzymes called cytochrome P450.
What is cytochrome P450? Cytochrome P450 enzymes are a large family of enzymes that are involved in phase I reactions. In fact, most phase I reactions are catalyzed by cytochrome P450 enzymes, often abbreviated as CYP. (The “P450” in the name comes from the peak wavelength of the enzyme, which is 450 nm.) You can see how prevalent they are in the chart below:
As you can see, there are many enzymes that begin with the CYP prefix, and together they account for over 75% of phase I reactions. You will encounter CYP enzymes throughout the text as we look at certain drugs, since many drugs are metabolized by CYP enzymes.
Some drugs and metabolites also undergo phase II reactions, which attach polar groups such as sulfate or glucuronic acid to the molecules in a process known as conjugation. These polar groups make the molecules even more hydrophilic, ensuring that they can be easily excreted.
1st edition
5.4.2 Enzyme Induction and Inhibition Because enzymes control the rate at which drugs metabolize, changes in enzyme activity have an impact on drug bioavailability. If the amount of an enzyme increases, the metabolism of the drug will speed up and less of the drug will be available. Drugs that increase the expression of enzymes are enzyme inducers.
Enzyme inducers can come from substances other than drugs. Saint-John’s wort, which is often used as an herbal remedy, induces the enzyme CYP3A4. This can reduce the effectiveness of drugs that are metabolized by CYP3A4, such as indinavir, an anti-HIV drug. Some drugs even induce the very enzymes that metabolize them. Phenobarbital, a barbiturate used to treat epilepsy, is one such example; over time, repeated administration will result in the drug having a reduced effect due to it being metabolized at a faster rate.
Drugs can also act as enzyme inhibitors by reducing the expression of the enzyme or blocking the enzyme’s active sites. As you might expect, this slows down metabolism of the drug, increasing its bioavailability and prolonging its effects. As with inducers, this can be done by the drug itself or by another substance. A notable example is grapefruit—the juice contains compounds that inhibit CYP3A4 enzymes, which can increase the concentrations of many medications that are metabolized by CYP3A4.
If these concepts are still confusing to you, before you move on you may want to review them by watching this short video: Enzyme Inhibition and Enzyme Induction [3:19]
5.4.3 Prodrugs There is one final concept worth discussing in the context of drug metabolism. So far, we have framed metabolism as a process that tends to work against the drug. Although that is the case for most drugs, not every biotransformation reduces the effectiveness of the drug. It is possible for the metabolite to be more pharmacologically active than the drug initially administered. This is the case for prodrugs: drugs that are administered in an inactive form that only becomes active after the drug is metabolized. To learn about prodrugs and examples of prodrugs, watch this brief video:
Biotransformation: Prodrugs [1:21]
Why are prodrugs useful? The chemotherapy drug mentioned in the video is a good example—sometimes the active form is too toxic to be administered directly. There are other potential reasons as well. Some prodrugs are better at crossing cell membranes. If you recall from the previous chapter, Parkinson’s disease is caused by a deficiency of dopamine. It would be nice to administer dopamine directly to treat the disease, but dopamine cannot cross the blood-brain barrier. Fortunately, its precursor, L-DOPA, is able to pass the barrier instead. This allows it to be administered as a prodrug that is converted to dopamine in the brain.
5.5 Excretion The final step in every drug’s journey is to leave the body in some manner. Excretion is the elimination of a drug from the body, either in its unchanged form or as a metabolite. Although this may seem like an automatic process, it cannot be taken for granted. If drugs or other waste products accumulate in the body, they can cause harm, which is why energy must be constantly spent removing these substances from the body.
By the end of this section, you should be able to:
• Describe various routes of excretion for drugs. • Differentiate between first-order and zero-order kinetics and define half-life.
5.5.1 Routes of Excretion Although drugs can be excreted through a variety of routes, most drugs are excreted by the kidneys into urine. The kidneys work like filters, filtering out the waste products from the bloodstream. The exact process is complex and beyond the scope of this class, but it involves the same process of diffusion mentioned in the absorption section. Recall that lipophilic drugs need to be metabolized into hydrophilic metabolites before they can be filtered out by the kidneys, since lipid-soluble molecules would simply reenter the bloodstream. Nonionized molecules are also difficult to excrete for the same reasons. The kidneys are also unable to excrete any drug that is still bound to plasma proteins.
Because most drugs are excreted through the kidneys, drug tests usually involve taking a urine sample. By measuring the presence of a drug or its metabolites in the urine, it is possible to determine whether a drug was present in a person’s body recently. Drugs can also be excreted through the liver as bile or feces, through sweating, and even through tears, although these routes are less important when it comes to drugs. Drug tests that involve mouth swabs are testing your saliva, which is another excretory route. Another type of drug test that you have probably heard of is the breathalyzer, which measures alcohol levels in the blood just be breathing into it. This is possible because the lungs can expel certain drugs like alcohol or anesthetics from the bloodstream directly.
There is one final route that requires consideration: mother’s milk. Some drugs can be eliminated through mother’s milk, which could have adverse effects on breastfeeding infants. Although cases are rare, special caution is warranted as infants have livers and kidneys that are still developing, making them more susceptible to any toxic effects.
5.5.2 Elimination Kinetics As a drug is eliminated from the body, the amount of drug remaining decreases over time. If we were to graph how much drug remains in the bloodstream, for most drugs we would get a shape like the graph below:
1st edition
Source: Doggrell (2014)
This curved shape follows first-order kinetics, which means it is eliminated at a rate proportional to the amount of drug. More specifically, first-order kinetics refers to a drug being eliminated in half-lives. A half-life is the amount of time that must pass for the body to eliminate half of the drug. Take methadone, a drug used in therapy for patients with opioid addiction. In a typical patient, the half-life of methadone is approximately 24 hours (Berkowitz, 1976). If we start with 80 mg of methadone in the bloodstream, after 1 day has passed there will be 40 mg remaining. After 2 days, 20 mg; after 3 days, only 10 mg, and so on. Under first-order kinetics, the drug will be eliminated rapidly at the start, but the rate will taper off as the concentration of drug in the blood decreases.
Not all drugs follow this pattern, however. Some drugs such as alcohol are eliminated at a constant rate—the amount of drug eliminated is always the same no matter how much of the drug there is. This is known as zero-order kinetics; if we were to plot the concentration of a drug being eliminated under zero-order kinetics, it would look like the graph below:
Source: Doggrell (2014)
Because the drug is eliminated at a constant rate, the slope of the graph is flat instead of curved. A few drugs such as aspirin follow first-order kinetics for the most part, but once a high enough concentration of the drug is reached, the enzymes that metabolize them become saturated. When the enzymes become saturated, increased metabolism of the drug is impossible, and the elimination rate follows zero-order kinetics instead. This is rare to see in practice, however, because such levels of concentration are well above therapeutic levels.
Chapter Summary and Review In this chapter, we explored the first branch of pharmacology, pharmacokinetics. After providing an overview of the different branches of pharmaceutical sciences, we took a journey through the four components of pharmacokinetics captured in the mnemonic ADME—absorption, distribution, metabolism, and excretion. We discussed factors that affect absorption, compared different routes of administration, learned about plasma proteins and the blood-brain barrier, examined metabolic processes, enzymes, and prodrugs, and covered the pathways and rates at which drugs can be excreted from the body. This was a dense chapter, so make sure to give yourself time to digest it, and don’t forget to check your understanding with the practice questions below.
Practice Questions
• What are the different things that pharmaceutics, pharmacokinetics, and pharmacodynamics study?
• What is the bioavailability of a drug that is administered intravenously? • Rank these routes of administration from fastest absorption to slowest: intramuscular
injection, inhalation, and transdermal. • Which drug is more sensitive to competition for plasma protein binding sites: one with a
high rate of binding, or one with a low rate? • Describe what the blood-brain barrier is made up of. How is this different from a typical
blood vessel? • Which routes of administration are subject to the first-pass effect? • Does enzyme induction increase or decrease the bioavailability of a related drug? • What is a prodrug, and in what circumstances would it be useful? • Would a lipid-soluble molecule be able to be excreted by the kidneys? Explain why or
why not.
1st edition
References
Berkowitz B. A. (1976). The relationship of pharmacokinetics to pharmacological activity:
morphine, methadone and naloxone. Clinical Pharmacokinetics, 1(3), 219–230.
https://doi.org/10.2165/00003088-197601030-00004
Doggrell, S. A. (2014). Metabolism and kinetics [Graph]. Queensland University of Technology.
Now that we have covered what the body does to the drug, it is time to examine what the drug does to the body. As you may recall, pharmacodynamics is the study of the actions and effects of drugs on the body. Over the course of this chapter, we will explore the different ways drugs bring about their effects and how we can compare the effects of different doses or drugs. Before starting, it may be useful to review the contents of Chapter 4, as neurotransmission plays a large role in pharmacodynamics.
Chapter Outline: 6.1 Mechanisms of Drug Action
6.1.1 Altering Neurotransmission
6.1.2 Direct Agonists and Antagonists
6.1.3 Indirect Agonists and Antagonists
6.1.4 Drug Interactions
6.2 Evaluating Drug Effects 6.2.1 The Relationship Between Dose and Effect
6.2.2 Analyzing Dose-Response Curves
6.2.3 Measuring Drug Safety
1st edition
6.1 Mechanisms of Drug Action
Pharmacodynamics in its simplest form asks, “What do drugs do?” We have touched on this topic before in previous chapters. As stated then, drugs cannot make the body do anything that it doesn’t normally do. All they do is change the rate of preexisting physiological processes. Drugs accomplish this by altering neurotransmission, a process that we studied extensively in Chapter 4. Understanding what a drug does means knowing where and how a drug affects neurotransmission.
By the end of this section, you should be able to:
• Differentiate between the different types of receptors and explain their roles in neurotransmission.
• Explain the different ways in which drugs can influence neurotransmission and define all relevant terms.
• Distinguish between the three different types of drug interaction and describe potential causes.
6.1.1 Altering Neurotransmission First, it is necessary to pinpoint exactly what is changing when a drug alters neurotransmission. Across all drugs, what ultimately changes is the degree of postsynaptic receptor activation. Remember that postsynaptic receptors are the receptors on the receiving side of the synapse. When these receptors are activated, they open or close ion channels in the postsynaptic neuron, giving rise to excitatory postsynaptic potentials (EPSPs) or inhibitory postsynaptic potentials (IPSPs) and making it either more or less likely to trigger an action potential All of this information was covered in chapters 3 and 4 and should already be familiar to you.
In addition to the postsynaptic receptors, there are also receptors near the synapse on the presynaptic neuron. These receptors modulate the production or release of neurotransmitters by the presynaptic neuron and can be classified as one of two types.
1st edition
Autoreceptors regulate the neurotransmitter that activates them. This serves as a negative feedback loop—the release of a neurotransmitter into the synapse activates its autoreceptors, which tell the neuron to stop releasing the neurotransmitter. Heteroreceptors are activated by neurotransmitters released from other neurons and can increase or decrease the release of neurotransmitters from the presynaptic neuron. The hetero- prefix means “different”.
Drugs can affect postsynaptic receptors by binding to them directly, but they can also bind elsewhere, such as to autoreceptors or heteroreceptors on the presynaptic neuron. This indirectly changes the activity of the postsynaptic receptors by altering how much of the neurotransmitter is released. We will look at both direct and indirect methods, starting with the former.
6.1.2 Direct Agonists and Antagonists Some drugs can activate the postsynaptic receptors directly. These drugs are called agonists. Remember from Chapter 4 that a drug (or ligand) can have both affinity and efficacy for binding to a receptor site. Agonists have both affinity and efficacy; that is, they are able to bind to the site and activate the receptor. Some agonists are more effective than others. If the drug is able to produce the maximum response possible, it is called a full agonist. Likewise, if the drug has reduced efficacy, it is called a partial agonist. Partial agonists can actually reduce the overall receptor activity if they are competing with full agonists for the same sites. An example of a full agonist would be morphine, which activates the same receptors as endorphins (remember that the term endorphin originated from “endogenous morphine” because morphine was discovered first). An example of a partial agonist would be buprenorphine, which activates the same receptors as morphine but with reduced efficacy, meaning it produces weaker effects than morphine.
In comparison, if a drug has affinity for the receptor but no efficacy, it will bind to the receptor but not activate it. A drug that does this is called an antagonist because it works against agonists (the ant- prefix is the same as anti-, meaning “against”). This reduces the number of receptors available to be activated, thus decreasing the effect of the neurotransmitter. Because of how they work, they are also called receptor blockers. See the image below to compare agonist and antagonist drugs:
1st edition
Source: Dolleyj on Wikimedia Commons (2014)
Antagonists can be either competitive or noncompetitive. A competitive antagonist binds to the same active site as the neurotransmitter. Because the antagonistic drug and agonistic neurotransmitter will compete for the same active site, it is possible to increase the amount of neurotransmitter and “crowd out” the drug, reducing the antagonistic effect. A noncompetitive antagonist avoids this by binding to the active site irreversibly or by binding to a site on the receptor that is different from the active site and preventing activation of the receptor. In these cases, increasing the amount of agonist will not outcompete the antagonist or surmount the noncompetitive antagonist.
Many receptors have a baseline level of activity. Agonists enhance this, while antagonists keep the activity at the baseline. Some drugs, however, can actually reduce the activity of the receptor below its resting state. These drugs are called inverse agonists because they have the opposite effect that agonists have on the receptor. Many drugs that were classified as antagonists are being relabeled as inverse agonists because we learn that they impede the resting activity of receptors—antihistamines that target H1 receptors are one such example, as H1 receptors are somewhat active even in the absence of a bound ligand.
One final term you should familiarize yourself with is allosterism. An allosteric modulator is a drug or ligand that alters receptor activity by binding to a site other than the active site (i.e., the one that an endogenous agonist would bind to.) This is done by changing the shape of the receptor protein; the term allosteric is derived from this (allo- [other] + steros [shape]). Some
noncompetitive antagonists are allosteric as mentioned above, but allosteric modulators can also increase agonist affinity or efficacy.
6.1.3 Indirect Agonists and Antagonists As we mentioned before, not all of the action has to occur on the postsynaptic receptors. Some drugs influence neurotransmission at different steps in the process. An indirect agonist is a drug that enhances receptor activity without binding directly to the receptor. One way to accomplish this is to induce the release of a neurotransmitter, such as by binding to a heteroreceptor on the presynaptic neuron. This may cause the cell to produce more neurotransmitter or release it more easily.
One very common method of indirect agonism is to increase the amount of neurotransmitter in the synaptic cleft by blocking the mechanisms meant to remove it. A reuptake inhibitor does this by blocking the presynaptic neuron’s ability to reclaim the neurotransmitter from the cleft. This increases the amount of neurotransmitter available to activate receptors (see image below).
Reuptake inhibitors are a common type of drug and can be found in many therapeutic medications, such as in selective serotonin reuptake inhibitors (SSRIs) and selective norepinephrine reuptake inhibitors (SNRIs). We will cover these in greater detail when we discuss antidepressants in Chapter 16.
If the neurotransmitter is broken down by enzymes within the synaptic cleft or after reuptake, an enzyme inhibitor can interfere with this process by inhibiting the enzymes responsible. The result is similar to reuptake inhibitors; by preventing the breakdown of released neurotransmitter, the amount available to activate receptors increases. An example are the monoamine oxidase inhibitors (MAOIs) used to treat some forms of depression by preventing monoamine oxidase (MAO) from breaking down monoamine neurotransmitters (see diagram below).
1st edition
It is also possible to have indirect antagonistic effects. By inhibiting the production or release of neurotransmitters, a drug can indirectly reduce overall receptor activity by decreasing the amount of neurotransmitter available. A presynaptic regulator binds to the autoreceptors on the presynaptic neuron and inhibits neurotransmitter release (see diagram below).
Ultimately, drugs can target any step in the neurotransmission process; what is important to remember is that the effect of the drug is defined by the end effect it has on the target receptor. Of course, many ligands act at multiple receptors. Part of the difficulty in developing a new drug has to do with finding a more selective compound that acts at the desired site without activating other receptors and triggering adverse side-effects.
6.1.4 Drug Interactions The effects of a drug can also be influenced by the presence of other drugs or substances. Drug interactions can result in the enhancement or reduction of a drug’s effect. When two drugs with similar effects are used together, the result can be additive, meaning the total response is what you would expect if you added the independent effects of the drugs together. A synergistic interaction, meanwhile, is greater than you would expect—the two drugs interact
1st edition
in a way that multiplies their effect. Finally, the drugs could reduce the effectiveness of each other, which would be considered antagonistic.
Causes of drug interactions are numerous. We covered many possible causes in the previous chapter on pharmacokinetics. Competition for plasma protein binding sites is one such cause, as this can change the bioavailability of the drug; other drugs or substances alter drug metabolism to similar effect.
Drugs can also interact in a pharmacodynamic manner by competing over certain receptors or pathways. Usually this is unintended and can result in severe consequences, but there are cases where this is desirable. Naloxone, a competitive opioid antagonist, is administered in the case of an opioid overdose because it has an antagonistic interaction with other opioids. By blocking opioid receptors, the effects of opioid overdose (most importantly, respiratory failure, which is life-threatening) can be reversed.
1st edition
6.2 Evaluating Drug Effects
Once you know where a drug acts, it is necessary to evaluate the drug’s effect. Does it produce a strong response? How much drug is needed? As the amount of drug changes, how does the effect change? And most importantly, is the drug safe to use? To answer these questions, pharmacodynamics provides a set of tools to evaluate and compare the effects of drugs. In the second half of this chapter, we will learn about these tools and how to use them.
By the end of this section, you should be able to:
• Describe dose-response curves and explain how they can be used to compare drugs. • Explain how dose-response curves can be used to measure drug safety. • Differentiate between therapeutic and certain safety indices.
6.2.1 The Relationship Between Dose and Effect In pharmacology, it is important to know how strong the response will be for a given dose. If we administer 1.0 mg of Drug X, will it produce the desired effect? What about 5.0 mg, or 10 mg, or 100? In reality, what we are interested in is what the effect of a drug will be at any dose and how that effect changes. This will allow us to select the proper dosage for the drug and determine whether this drug is safe and useful.
By taking measurements at a few key points (and understanding the mechanism of action), it is possible to produce a graph showing the intensity of effects across a range of doses. This graph is called a dose-response curve or a dose-effect curve. You can see an example below:
On a dose-response curve, the effect or response that we are interested in goes on the y-axis; this ranges from 0% (no effect) to 100% (maximum possible effect). The dose of the drug goes on the x-axis. Because we want to see a wide range of doses, we actually plot them in log scale
1st edition
to compress the graph; note how the intervals do not go 1, 2, 3, 4, 5, but rather 0.01, 0.1, 1.0, 10, 100. (The graph is called a semi-log graph because only one axis is in logarithmic scale.)
Most drugs produce a graph shaped like the one above. The S-shaped curve is called a sigmoidal curve, from the Greek letter sigma (S). It is important to recognize that the S-shape is a result of the log scale used on the x-axis. If we were to plot the graph linearly, you would see the steepest part at the lowest values, while the slope would gradually decrease. This checks with the intuitive understanding you might have of increasing the dose; when the dose is very small, a small change can cause a large increase in activity because all of the drug is being used. At very high concentrations though, increasing the dose has diminishing returns since the target site is already saturated in the drug. Without the dose scale in log, the shape of the curve would be hyperbolic.
Not all dose-response curves are sigmoidal. Some curves are biphasic, meaning the greatest effect occurs at a moderate dose. Alcohol is one such example; small doses provide a stimulating effect (a “buzz”), but drinking more causes a depressive effect. (The term biphasic literally means “two phases.”) We will return to this when we cover alcohol in Chapter 12, but we will stick to sigmoidal curves for the examples in this chapter as they are the most common.
6.2.2 Analyzing Dose-Response Curves On every dose-response curve there are a few key values of note. Take a look at the graph below:
The threshold dose is the lowest dose that produces any measurable response. Doses beneath the threshold will cause no effect. Once the graph levels off, it reaches its maximum response, also called maximum effect or ceiling response/effect. Increasing the dose beyond this point will not increase the effect of the drug. It is also important to note that the ceiling for a drug is not always at 100%; some drugs are only capable of producing a partial response at most, in which case the ceiling would be lower. (We will revisit this shortly.) Finally, the dose
1st edition
that produces half of the maximum response would be the median effective concentration, commonly referred to as EC50 This is the dose that produces half of the maximum response; if the drug has a maximum response of 80%, this would be the dose that produces a 40% response.
These values are useful when comparing different drugs. One way drugs can differ is in terms of potency. The potency of a drug is defined as the dose required to produce a response of a given intensity. The more potent a drug is, the lower the dose required to produce an effect. Examine the example below:
Source: Mohmmed Laique on Wikimedia Commons (2018b)
As you can see, drugs with curves that are closer to the left side of the graph are more potent than those further to the right. You can also see that each drug has the same maximum response and EC50, but the more potent drugs reach these values at lower doses.
In comparison, the efficacy of a drug is determined by the maximum response produced by the drug. As mentioned previously, not every drug can reach a 100% response. A drug with a lower efficacy has a reduced maximum response and EC50, which flattens the sigmoidal curve. You can see this in the graph below showing four drugs with the same potency but different efficacies:
Source: Mohmmed Laique on Wikimedia Commons (2018a)
Finally, it is worth noting the slope of the dose-response curve. The effects of a drug with a steep slope are more sensitive to small changes in dose. Another way of thinking about it is that the threshold and maximum response doses are closer together on a steep graph compared to a shallow one. Although most research will focus on the potency or efficacy of a drug, the slope of the dose-response curve is another important factor that has implications for the relative safety of different drugs.
6.2.3 Measuring Drug Safety How exactly do we determine whether a drug is safe? Part of the issue is that not everyone responds to the same drug in the same way. To reflect this, it is common to construct a slightly different dose-response curve. Instead of plotting the intensity of the effect on the y-axis, we instead plot the percentage of the population that shows the desired effect. On this version, 50% response does not mean “half of the maximum effect possible;” instead, it means “half of the population shows the desired effect.” A dose that produces a response is called an effective dose, and the dose at which 50% of the population responds is called the effective dose, 50%, or simply ED50.
We can also plot toxic effects, such as lethality, on a similar graph. A lethal dose is similar to an effective dose, except the effect being measured is lethality. Similar to the effective dose, we can define the lethal dose, 50% or LD50 as the dose necessary to produce lethal effects in 50% of the population. (If we’re interested in a toxic effect other than lethality, we can use the terms
toxic dose and TD50 instead.) Take a look at this graph showing effective and lethal doses for the drug phenobarbital:
As shown by the graph on the right, at low doses, phenobarbital can produce therapeutic effects (in this case, sleeping). However, at high doses, the drug can lead to lethal effects such as respiratory failure. Obviously, for a drug to be therapeutic, the effective doses need to be smaller than the lethal doses. In fact, the distance between effective dose and lethal dose or toxic dose is very important in drug safety. Compare the following two graphs:
1st edition
The first graph shows a large distance between ED50 and TD50, whereas the second graph shows a much smaller distance. The drug represented by the first graph is a much safer choice because the therapeutic and toxic effects do not overlap. We can quantify this difference by calculating the therapeutic index (abbreviated TI), defined as TD50 (or LD50) divided by ED50. A higher therapeutic index means that there is a greater distance between the two curves and thus a reduced chance of experiencing toxic effects.
Although the therapeutic index is widely used, it is not a perfect measure of drug safety. The therapeutic index ignores the slope of the dose-response curves, which may lead to two drugs with the same therapeutic index having very different levels of safety. Take the following example:
1st edition
Although drugs A and B have the same therapeutic index, the therapeutic and toxic effects overlap much more in the case of the latter. By the time you reach a dose that is likely to produce the desired therapeutic effect, there is already a small chance that a toxic or lethal effect will occur.
A stronger measure of safety would compare the dose required to near-guarantee a therapeutic response to the threshold dose for a toxic effect. This is what the certain safety index (CSI), also called the certain safety factor (CSF) or margin of safety (MOS), measures. Instead of dividing TD50 by ED50, the CSI divides the TD1 by ED99. (As with the therapeutic index, lethal dose can be used instead of toxic dose.) You can see how this compares to the therapeutic index in the graphs below:
1st edition
Chapter Summary and Review In this chapter, we finished up our coverage of pharmacology by examining pharmacodynamics, or what the drug does to the body. We explained how drugs can alter neurotransmission in different ways and the ways in which drugs can interact with each other. We also learned how to evaluate drug effects using dose-response curves, including different ways to measure the safety of a drug. That’s it for this chapter. Next we will be moving on to the behavioral side of drug use. Make sure to check your understanding and reach out to your instructor if you have any questions.
Practice Questions
• Where are autoreceptors located, and what neurotransmitters activate them? • Explain the difference between competitive and noncompetitive antagonists. • What is an allosteric modulator? • Name three different types of drugs that can indirectly change receptor activation. • What are some possible causes for drug interactions? Provide a pharmacokinetic
example and a pharmacodynamic one. • Are all drug-response curves sigmoidal in shape? • Differentiate between potency and efficacy. • Define EC50, ED50, TD50, and LD50. What does the “50” stand for in each term? • For any given drug, would you expect the therapeutic index or certain safety index to be
Now that we’ve covered the basics of neuroscience and pharmacology, it should be apparent how drugs interact with our bodies on a cellular scale. What may be less obvious is how this culminates in the experiences and behaviors we see at a human scale. What makes drug use so pleasurable? How does an addiction develop, and what can we do to stop it? Believe it or not, the answers to these questions are rooted in the subjects we have already covered. To wrap up this unit, we will explain the rewarding aspect of drug use and the way it can change our behavior over time. This should help you integrate the topics we have covered so far with what you already know or have seen about drug use.
Chapter Outline: 7.1 Learning: How Experience Shapes Behavior
7.1.1 Introduction to Behaviorism
7.1.2 Operant Conditioning
7.1.3 Operant Conditioning and Drug Use
7.2 Biological Basis of Reinforcement 7.2.1 Dopamine in the Brain
7.2.2 The Reward System
7.3 Consequences of Repeated Drug Use 7.3.1 Drug Dependence
7.3.2 Addiction
7.3.3 Treating Substance Use Disorders
1st edition
7.1 Learning: How Experience Shapes Behavior
To understand how drugs affect behavior, we need to cover basic principles of learning. You may think of learning as something that primarily happens in school. But in psychology, learning is defined as a change in behavior caused by experience. We experience things all the time, and the nature of those experiences influences our actions in the future. We not only learn from our teachers, but also from the consequences of our actions.
This concept applies to drug use as well. In short, people use drugs because they learn that using them feels good or provides relief. This is as true for someone addicted to cocaine as it is for habitual coffee-drinkers. In this section, we’ll explore how psychologists talk about learning and use those ideas to explain how and why drugs change our behavior.
By the end of this section, you should be able to:
• Differentiate between classical and operant conditioning. • Explain operant conditioning and describe various factors that determine how drugs
change behavior.
7.1.1 Introduction to Behaviorism In many cases, studying learning theory involves studying behaviorism, a discipline of psychology that was founded in the 1910s by John Watson and later championed by B. F. Skinner. The reason why behaviorism is so important is because much of the terminology we will use to explain changes in behavior was coined by behaviorists.
In behaviorism, a behavior is seen as a response to some sort of stimulus. Sometimes the response is involuntary, or a reflex. For instance, a dog will salivate when presented with food. We can pair a reflex-inducing stimulus with an unrelated stimulus such as the ring of a bell. Normally, a bell won’t cause a dog to salivate. But if you ring the bell each time you feed the dog, it will eventually learn that the bell is associated with food and will start to salivate once you ring it. This process, in which a stimulus is paired with a reflexive behavior, is a form of learning called classical conditioning (see image below).
1st edition
Source: Salehi.s on Wikimedia Commons (2016)
Although classical conditioning is powerful, it is limited to reflexes and other involuntary behaviors. It cannot explain how you decide what to wear in the morning or whether you choose to keep eating a new dish. For that, we have to turn to operant conditioning, which describes how voluntary behaviors are changed by their consequences. (To distinguish between classical conditioning and operant conditioning, remember that classical conditioning involves reflexes, while operant conditioning involves voluntary behaviors.)
7.1.2 Operant Conditioning The idea behind operant conditioning is probably intuitive to you. If you went to a restaurant and had a great time, you would probably be more likely to return there in the future. On the other hand, if the food and service were terrible, you might avoid that place. In both of these cases, your behavior (going to a restaurant) was changed by its consequence (a good or bad experience).
If a behavior is strengthened—that is to say, if it becomes more likely to occur in the future—then we say that the behavior was reinforced. A good dining experience reinforces deciding to eat at that place. In comparison, is a behavior is weakened or decreases, we say that it is punished. A bad dining experience would punish our decision.
There are two main ways that we can reinforce a behavior. We can add something good, or we can take away something bad. We call the former positive reinforcement and the latter negative reinforcement. What are some examples? Well, if you ever received an allowance for taking out the trash, that would be positive reinforcement—the money was something rewarding that was added. If you take out the trash when it starts to smell though, that’s negative reinforcement—the bad smell was removed. In both cases, the behavior (taking out the trash) became more likely to happen again in the future.
We can do the same thing with punishment. Adding something bad will result in positive punishment, while removing something good is negative punishment. Getting a ticket after you speed will make you less likely to break the speed limit in the future (positive punishment), but so will having your license revoked (negative punishment).
It is easy to get these four terms mixed up. Remember that the words positive and negative refer to whether something is being added or removed, not whether the thing is good or bad. (Think of the math symbols + and – if you have to.) In comparison, reinforcement and punishment always refer to whether the behavior increases or decreases.
Here is a table defining each of these terms:
1st edition
Something
“Bad” (aversive)
Something “Good” (rewarding)
Giving (positive)
Positive Punishment
(behavior is weakened)
Positive Reinforcement
(behavior is strengthened)
Taking Away (negative)
Negative Reinforcement
(behavior is strengthened)
Negative Punishment
(behavior is weakened)
7.1.3 Operant Conditioning and Drug Use So, how do we use this terminology to describe drug use? If using a drug makes us more likely to use it again in the future, then we would say that the drug is a reinforcer. In fact, drugs are considered primary reinforcers because they are intrinsically rewarding, similar to food or sex. This is contrasted with a secondary reinforcer, which is only rewarding because it has some sort of learned value, such as money. A $100 bill isn’t a reward for a toddler, but, as we grow older, we learn that money can help us get primary reinforcers like food and learn to value it on its own.
Of course, not everyone will become addicted to a drug after the first use. In fact, sometimes a first experience will turn someone off from a drug and discourage them from using it. This is an example of punishment; if you cough a lot the first time you smoke a cigarette and decide smoking is not for you, then your drug-taking behavior is reduced. The truth is, the reinforcing power of a drug is reliant on a variety of conditions.
First is satiation, which is essentially the state of being “full” or satiated. In the context of food, this is obvious: food is more rewarding when you are hungry. In the context of drug use, this can extend to the user’s overall situation. Many people use drugs because they lack other sources of pleasure in their lives. The rush of pleasure from drug use will be more rewarding if you can’t get that same feeling from relationships, work, or hobbies in your life. It can also refer to the particular effect of the drug. Caffeine, for instance, is a lot more rewarding when you are tired and need a boost compared to when you are already brimming with energy.
Two other factors are immediacy and contingency. Immediacy refers to how quickly the response occurs; contingency describes how reliably the consequence follows the behavior. Immediate, reliable responses are more effective reinforcers. A drug that produces an effect immediately is more addictive than one with a delayed response. This can also influence the
1st edition
route of administration, where routes that absorb the drug faster (such as IV injection or snorting) may be preferred.
Finally, the size of the stimulus determines its effect. The more potent the stimulus, the more reinforcing it becomes. This has clear implications for drug use, as certain drugs provide more pleasurable effects than others. As we discuss different types of drugs in the coming chapters, keep these factors in mind.
1st edition
7.2 Biological Basis of Reinforcement
In the previous section, we stated that drugs are primary reinforcers because they are intrinsically rewarding. Why is that the case? Although drugs have a variety of different effects, almost all drugs of abuse target the same structures in the brain that are responsible for handling reward and motivation. By examining these structures, we can learn how our brain determines what behaviors to reinforce and how that reinforcement happens.
By the end of this section, you should be able to:
• Describe the three main brain dopamine pathways that are related to reward. • Describe the circuitry of the reward system and briefly explain the functions of the
various structures involved in it.
7.2.1 Dopamine in the Brain When discussing neurotransmitters in Chapter 4, we mentioned how dopamine is related to reward and reinforcement. (As you may recall it is also related to motor control, but that is not the focus of this chapter.) What this really means is that dopamine is the neurotransmitter used in a handful of important pathways in our brain that control reward and motivation. These pathways connect various structures that play a role in determining which stimuli to attend to and which behaviors to reinforce.
Although dopaminergic (dopamine-releasing) neurons can be found throughout the brain, there are two major locations where they are clustered together; one is the substantia nigra and the other is the ventral tegmental area (VTA). Both are located in the midbrain at the top of the brainstem and project axons to multiple different regions of the brain. The name substantia nigra literally means “black substance” and refers to its color; meanwhile, ventral tegmental simply refers to the location of the area (the underside of the tegmentum, a part of the midbrain).
These areas connect to different structures in the brain to form dopaminergic pathways. There are three major pathways related to reward, although there are other dopamine pathways not mentioned here that are involved in other processes. The three reward-related pathways are the mesolimbic, mesocortical, and nigrostriatal pathways.
Although the names may look confusing, they are actually fairly simple since they describe the areas each pathway connects. The mesolimbic pathway connects the tegmentum of the midbrain (meso-, meaning “middle” in Greek) to the nucleus accumbens and limbic system. The mesocortical pathway follows the same pattern, with cortical referring to the cortex. Finally, the nigrostriatal pathway connects the substantia nigra to the striatum.
All three pathways are implicated in reward, but the main focus of this text will be on the two pathways originating from the VTA: the mesolimbic and mesocortical ones. Together they form a circuit of neurons called the reward system.
1st edition
7.2.2 The Reward System The reward system is a collection of structures that are responsible for reward and reinforcement. Because it involves the mesolimbic and mesocortical pathways, it is sometimes called the mesocorticolimbic (MCL) system instead. To get an overview of some of the structures involved and the roles they play, watch this video from Khan Academy:
Reward pathway in the brain | Processing the Environment [8:25]
Let’s review the structures mentioned in the video. We have already learned about the VTA, which contains a cluster of dopaminergic neurons. Many of these neurons project to the nucleus accumbens, which contains many dopamine receptors. Reward and pleasure are consistently associated with the activation of these receptors, which is why dopamine is sometimes called the feel-good neurotransmitter.
Neurons from the VTA and nucleus accumbens also connect to certain structures in the limbic system. In particular, they connect to the amygdala, which handles emotional responses, and the hippocampus, which forms long-term memories. These areas work together to create positive memories of pleasurable experiences, making it more likely that we will remember the stimulus and how to get it in the future.
Part of the mesolimbic pathway connects to the prefrontal cortex, which is also connected to the VTA through the mesocortical pathway. The prefrontal cortex is front part of the frontal lobe and is responsible for many executive functions such as planning, attention, and motivation. Activity here can direct our attention to rewarding stimuli and cause us to seek such stimuli out.
The circuitry of the reward system is very complex, but its overall purpose is to respond to things that are important to our survival and to reinforce the behaviors that helped us obtain those things. This is usually oriented towards natural rewards such as food, sex, or sleep. Drugs can interfere with this process though, stepping in at some point and activating the reward system directly. Some drugs activate dopaminergic VTA neurons; others increase dopamine production and release; others might block dopamine reuptake at dopamine receptors. The end result is the same—the reward system is activated and we register the experience of taking the drug as pleasurable and desirable.
Now that we know why drugs are reinforcing, it is time to learn about how drugs change our behavior over a long period of time. This is beyond the immediate increase in drug use that occurs as a result of reinforcement and instead focuses on the consequences of consistent and repeated drug use. Although our discussion of dependence and addiction will bring to mind illicit drugs, keep in mind that the same processes can occur with licit drugs as well.
By the end of this section, you should be able to:
• Describe tolerance and withdrawal and how they influence drug-taking behavior. • Explain how addiction can hijack the reward system in the brain. • Describe the treatment of substance use disorders.
7.3.1 Drug Dependence The main consequence of repeated drug use is developing drug dependence. This term was introduced and defined in Chapter 1, but to refresh, it refers to the physiological changes wherein the body adapts to the drug over the course of repeated use.
Drug dependence will usually lead to tolerance to the drug, meaning higher doses are required to produce the same effect. Tolerance can arise from changes in pharmacokinetics or pharmacodynamics. If chronic drug use results in an increased rate of metabolism of the drug (pharmacokinetic tolerance), this would reduce the amount of drug that reaches the site of action. Chronic drug use can also result in the desensitization of target receptors (pharmacodynamic tolerance), reducing the effect of the drug but not the amount of drug reaching the site of action.
Why would the receptors change? The answer lies in homeostasis, or the body’s attempt to keep itself in a steady equilibrium. If a certain type of receptor is being constantly overactivated, the body may try to compensate for this activity by reducing the number of receptors available or making them less responsive.
One reason why overdoses occur is because tolerance for different effects develops at different rates. As tolerance for a desired effect increases, the dose taken increases, but the dose required for a toxic or lethal effect stays the same. Recall the dose-response curves from the previous chapter—this is analogous to the therapeutic index of a drug becoming narrower as the effective dose curve shifts further right, except in this case it usually represents a desired recreational effect. Eventually, the curves will overlap, and the dose necessary to produce the desired effect will be dangerously close to the lethal dose.
There are other forms of tolerance. Cross tolerance occurs when tolerance develops for a similar drug. For instance, someone who is tolerant to heroin will also be somewhat tolerant to morphine as well, since both drugs have similar biological actions. People can also develop behavioral tolerance, in which the user becomes accustomed to the effects of the drug and learns to compensate for them (such as acting less disinhibited after drinking alcohol).
Tolerance has many other downsides besides needing to take more of the drug. Tolerance typically is tied to withdrawal, which is a severe reaction to a sudden drop or cessation of drug
1st edition
use. Because the body has attempted to adapt to the drug being present, when the drug is absent, this balance is thrown off. Withdrawal involves many different symptoms depending on the type of drug; all are generally unpleasant, and some may even be life-threatening. To avoid the effects of withdrawal, people with drug dependence will feel compelled to take the drug as it will provide a temporary relief from the symptoms (a form of negative reinforcement).
Another consequence is the possibility of a conditioned compensatory response, which is an automatic response that is conditioned (learned) by repeated drug use. The response attempts to compensate for the effects of the drug by recognizing familiar stimuli associated with administration of the drug (such as the sight of a needle or a particular location), then preparing the body for the drug’s effects. This is another potential overdose cause, as taking a drug in an unfamiliar context can produce greater effects even when the dose is kept the same, since the cues to trigger a compensatory response are not present.
7.3.2 Addiction Also described in Chapter 1, addiction is characterized by compulsive drug use despite harmful consequences. If you recall from that chapter, we mentioned how continued drug use can interfere with self-control and the ability to stop taking the drug. This is because drug addiction hijacks the reward system in the brain. What this short video from the Addiction Policy Forum to learn more:
ADDICTION || The Hijacker [3:17]
As mentioned in the video, the reward system becomes impaired with chronic drug use. One of the main ways this happens is through the desensitization of dopamine receptors in the reward system. As mentioned previously, in order to preserve homeostasis, the body may reduce the number or responsiveness of receptors that are overactive. Chronic drug use floods synapses in the reward system with dopamine, resulting in the desensitization of dopamine receptors. This causes natural rewards such as food, shelter, and companionship become less rewarding. The drug becomes the sole source of pleasure, which compels the user to continue to seek the drug and neglect other parts of their life.
There are various models of addiction. Originally, addiction was thought of as a moral failing. Although this view is still common, it is increasingly being challenged by the disease model of addiction, which views addiction as a kind of disease that interferes with the normal functioning of the body. As we have learned above, there is evidence in support of this model, since chronic drug use does, indeed, cause physiological changes. According to the disease model, the goal is to manage and treat addiction like any other disease.
Another model is drive theory, which states that the body has innate drives (such as hunger) that increase and intensify until they are temporarily met. In this model, the repeated use of drugs generates a drive to seek the reinforcing effect of the drug. Under this theory, the motivation for drug-seeking behaviors is always to gain positive reinforcement.
A related model is derived from the opponent-process theory, which, in essence, states that the effects of the drug are opposed by the actions of the body. This is again due to homeostasis as the body tries to reach a relative equilibrium to maintain normal functioning. As tolerance for the drug increases, the opponent processes also increase and result in withdrawal. In this
model, the motivation shifts back and forth between seeking the pleasurable effects of the drug (positive reinforcement) and avoiding withdrawal (negative reinforcement).
Finally, another modern model is the incentive salience model. In this model, the drug is designated as something that should be desired; in other words, it becomes salient (commands our attention) and we are incentivized to pursue it. This model differentiates between merely enjoying a drug (pleasure) and being motivated to obtain it (compulsion).
These four models each take a different approach to describing the mechanisms of addiction, but each views addiction as the consequence of natural physiological processes being altered, accelerated, or interfered with in some way. While addiction is not included as a disorder in the DSM-5 by name, it is referred to as a substance use disorder instead.
7.3.3 Treating Substance Use Disorders By now it should be clear that addiction and substance use disorders are dangerous and destructive. Luckily, they can be treated, and normal functioning can be reclaimed. Treatment is a long process and involves multiple strategies, many of which may need to be combined to result in a successful treatment. The same treatment plan will not work for every drug and every person, but there are some common elements of each.
The first step is typically to remove the drug from the system, a process known as detoxification or simply detox. The next step is to manage withdrawal symptoms, as withdrawal is unpleasant and can increase drug-taking compulsions. This may involve drug therapies where weaker agonists or partial agonists are administered in a safe and controlled manner to replace the effects of the drug and reduce withdrawal. Common examples include nicotine patches or lozenges that are designed to replace smoking.
Therapy is also used to help prevent relapse and reestablish order in life. Psychotherapies such as cognitive-behavioral therapy and multidimensional family therapy can help patients identify and avoid triggers for relapse, as well as address and cope with other issues in their lives that may contribute to drug use.
In some cases, individuals might undergo short inpatient treatment or stay in longer-term residential treatment facilities, such as therapeutic communities or halfway houses. These places help patients transition back to normal life by helping them develop life skills or seek employment. People recovering from substance use disorders may also be referred to various other programs and services, such as 12-step programs and support groups.
According to the National Survey of Substance Abuse Treatment Services, in 2014, around 22.5 million people needed treatment for an illicit drug or alcohol use problem, but only 18.5% of those people received any substance use treatment (SAMHSA, 2014). Although treatment is possible, there are disparities in access to resources and other barriers to treatment such a stigma around addiction that prevent people from getting the help they need.
1st edition
Chapter Summary and Review In this chapter, we explored how drug use affects behavior through reward and reinforcement. We started our discussion by covering operant conditioning and the different factors that lead drugs to be reinforcing. We then moved on to the reward system, a collection of dopamine pathways that serves as the biological basis for reinforcement. Finally, we described how chronic drug use can lead to dependence and addiction, and covered methods for treating substance use disorders.
This is the final chapter for the first unit of this textbook. From this point on, the remainder of the text will be about different types of drugs and their specific mechanisms, effects, and treatments. We will kick off the next week by looking at CNS stimulants first. Make sure that you have a strong grasp of the concepts from these first seven chapters, as they will come up again and again in every chapter moving forward.
Practice Questions
• John decides to take a shortcut through a park on his walk home but ends up getting mud all over his shoes. He makes a note to avoid the route in the future. What type of reinforcement or punishment occurred?
• Frederic finds Gatorade incredibly tasty after a hard workout but enjoys it less when he isn’t thirsty. What principle is at play here?
• Name two areas that are populated with large amounts of dopaminergic neurons. • Which structures does the mesocortical pathway connect? • Name three ways that drugs can increase dopamine in the reward system. • How can tolerance lead to an overdose? • Name four models of addiction and briefly describe each. • What are some ways that drug therapies can be used in treating substance use
disorders?
1st edition
References
Salehi.s. (2016, November 23). A diagram demonstrating classical conditioning [Illustration].
Now that we have covered the fundamentals in the first unit, it is time for us to examine specific types of drugs. For the remainder of this book, each chapter will introduce a new type of psychoactive drug and focus on a few key examples. Be prepared to apply the concepts covered in the first unit when learning about each drug. In this unit, we will examine stimulants and depressants, starting with high-efficacy stimulants.
Chapter Outline: 8.1 CNS Stimulants
8.1.1 Stimulant Overview
8.1.2 High-Efficacy vs. Low-Efficacy Stimulants
8.2 Cocaine 8.2.1 Drug History and Overview
8.2.2 Administration and Pharmacokinetics
8.2.3 Mechanisms of Action and Effects
8.3 Amphetamine 8.3.1 Drug History and Overview
8.3.2 Administration and Pharmacokinetics
8.3.3 Mechanisms of Action and Effects
1st edition
8.1 CNS Stimulants
The first class of drugs we will be looking at are stimulants. A stimulant is any drug that increases the activity of the central nervous system. For this reason, they are also known as CNS stimulants. Before examining any specific drugs, we will first go over some properties that are common across all stimulants.
By the end of this section, you should be able to:
• Explain how sympathomimetic drugs enhance CNS activity. • Differentiate between high-efficacy and low-efficacy stimulants.
8.1.1 Stimulant Overview As mentioned, stimulants tend to increase CNS activity. A common method for doing so involves enhancing the effects of the sympathetic nervous system. Recall that this is the division of the autonomic nervous system that is responsible for our fight-or-flight response. When our sympathetic nervous system is activated, we enter a state of heightened arousal; our body prepares for action by increasing our heart rate, dilating pupils, constricting of blood vessels, and causing various other physiological changes.
The primary neurotransmitters used in the sympathetic nervous system are the catecholamines: epinephrine, norepinephrine, and dopamine. Many stimulants, including the ones in this chapter, possess direct or indirect agonist activity at adrenergic receptors. These drugs are called sympathomimetic drugs because they mimic the effects of the sympathetic nervous system.
Most stimulants cause euphoria and agitation while increasing energy, alertness, confidence, and sexual desire. Many stimulants have therapeutic uses, although the pleasurable effects and addictive properties of the drugs mean that they are also used frequently for recreational purposes. They are sometimes colloquially referred to as “uppers” due to their stimulating effects.
8.1.2 High-Efficacy vs. Low-Efficacy Stimulants Not all stimulants produce the same level of effect. Think back to the dose-response curves that you studied in Chapter 7 and how the height of the curve showed the maximum possible effect of the drug. Some drugs have lower ceilings than others. This is the case for stimulants, which we can group into two main divisions—high-efficacy stimulants and low-efficacy ones.
1st edition
Some stimulants, called high-efficacy stimulants, increase CNS activity by a significant amount. The drugs we will look at in this chapter fall in this category, as do many other sympathomimetic drugs. In comparison, low-efficacy stimulants like nicotine and caffeine produce modest effects. We will explore the differences between the two in greater depth when covering low-efficacy stimulants in the next chapter. For now, let’s examine the two most notable high-efficacy stimulants: cocaine and amphetamine.
1st edition
8.2 Cocaine Cocaine, often informally referred to as coke, is one of the most well-known illicit drugs. Aside from its common depictions in media, it is also the second-most used illicit drug in the U.S. behind marijuana (National Survey of Drug Use and Health, 2019). In the following section, we will explore its role in culture and medicine and outline the exact mechanisms through which it causes its stimulatory effects.
By the end of this section, you should be able to:
• Explain the history and uses of cocaine. • Describe the pharmacokinetic properties of cocaine and compare cocaine hydrochloride
to cocaine freebase. • Explain cocaethylene. • Describe the pharmacodynamic properties of cocaine and the effects of long-term use.
8.2.1 Drug History and Overview Cocaine is a psychoactive substance extracted from the leaves of the coca plant. Coca leaves have been chewed for thousands of years in South America, where coca is natively found. Its use spread to other parts of the world with the help of Spanish conquistadors, who found its ability to make workers eat less and work longer appealing.
Chemists began to try to isolate the alkaloid responsible for these effects from the plant in the 1800s. In 1860, graduate student Albert Niemann isolated the alkaloid and named it cocaine, after the coca plant. Interest in the drug’s potential medicinal uses quickly followed; ophthalmologist Karl Koller demonstrated its properties as a local anesthetic, while Sigmund Freud advocated for its use as a stimulant and pain reliever.
Use of cocaine was popularized throughout the late 1800s. In Europe and America, it was readily available and advertised for its therapeutic effects. In fact, the original recipe for Coca-Cola included coca leaves, hence the name. Cocaine was purchasable as an over-the-counter drug up until 1916, not long after it became regulated under the Harrison Narcotics Act. By then, reports of addiction and dependence had become more common and governments moved to regulate cocaine’s use.
Although cocaine is still used as a local anesthetic, it has mostly been replaced by safer alternatives such as lidocaine and procaine (aka Novocain®), which derive their -caine suffixes from cocaine. Despite this, it is still used occasionally in dentistry and cardiology, as cocaine is unique in also inducing vasoconstriction, or constricting nearby blood vessels. Because of this, it is classified as a Schedule II controlled substance (accepted medical use with a high potential for abuse).
8.2.2 Administration and Pharmacokinetics Cocaine, like many stimulants, comes in multiple forms. The salt form, cocaine hydrochloride (HCl), appears as a white powder that is typically snorted intranasally or dissolved and injected directly into a vein. Cocaine can be taken orally, but its effects are heavily reduced as stomach
1st edition
acid tends to leave it inactive, so oral administration is uncommon. It can also be used topically as a local anesthetic.
Removing the HCl from cocaine salt turns it into cocaine freebase, which can be easily smoked. (Technically, cocaine salt can be smoked as well, but the resulting fumes are much more unpleasant.) Cocaine freebase can be made by heating the drug with volatile chemicals (freebasing) or by mixing it with baking soda in water and drying the crystals to produce crack cocaine. See the image below to compare cocaine HCl (left) with crack cocaine (right):
Source: Drug Enforcement Agency (2014)
Cocaine freebase has a higher bioavailability and onset of action compared to its salt form. It also produces a stronger effect due to its increased lipid solubility. As a result, smoking cocaine freebase provides a strong and immediate rush that is more addictive than using cocaine HCl. (This should make sense to you, as last chapter we discussed how the size and immediacy of a reinforcer increases its reinforcing effect.) Take a look at the following chart comparing various pharmacokinetic properties of cocaine HCl and cocaine freebase:
Onset of Action 10-30 sec (snorting) 1-2 sec (smoking)
Time to peak levels in brain 10 min (snorting) 8 sec (smoking)
Duration of action 2 hours 5-15 min
As you can see the absorption rate depends on the route of administration. Once the drug is in the bloodstream, it is actively hydrolyzed by enzymes and has a relatively short half-life of approximately 1 hour. Cocaine is capable of crossing the blood-brain barrier, which is how it reaches its target receptors in the CNS.
When taken with alcohol, the two drugs interact to produce a toxic metabolite called cocaethylene in the liver, which has a lower LD50 than cocaine. Cocaethylene is psychoactive and has a similar mechanism of action to cocaine, but it has a longer half-life and enhances the effects of both cocaine and alcohol, making it a synergistic interaction.
1st edition
8.2.3 Mechanisms of Action and Effects Cocaine produces its effects by blocking transporter proteins responsible for the neuronal reuptake of monoamines. To refresh, these are the pumps that remove excess transmitters from the synapse and return it to the presynaptic cell. Cocaine blocks the transporter proteins for monoamines, in particular, norepinephrine, dopamine, and serotonin, preventing them from being reabsorbed and increasing the overall amount present in the synapse.
The norepinephrine activates α-adrenergic receptors, which are responsible for the sympathomimetic effects such as increased wakefulness and attention and reduced appetite. Cocaine also increases dopamine levels that activate dopaminergic neurons in the reward system, which produces the euphoria and pleasure associated with the high obtained from cocaine.
Tolerance to the euphoric effects can develop over time due to downregulation (reduction in the number) of dopamine receptors. Cross-tolerance is also common between different stimulants that share similar mechanisms. Withdrawal symptoms involve drug cravings, lethargy, dysphoria, sleep disturbance, and an increased appetite.
At high doses, people may experience psychosis and hallucinations. A common hallucination involves feeling like bugs are crawling around under the skin, called formication. Overdose may cause abnormal blood pressure, heart rate, or other cardiopulmonary symptoms, which may sometimes be fatal.
It is worth noting the mechanism for cocaine’s anesthetic effects. Cocaine blocks sodium channels at sensory neurons, which prevents EPSPs and the generation of an action potential, thus blocking signal transmission along sensory nerves. You can see this in the diagram below:
1st edition
Cocaine is used recreationally as a party drug most often due to the increase in energy and euphoria it provides. Because of this, its use often correlates with other party drugs such as MDMA (ecstasy) and amphetamines or other drugs such as heroin. The presence of alcohol in such environments can be particularly dangerous due to the synergistic interaction between the two mentioned earlier.
1st edition
8.3 Amphetamine Amphetamine is another major stimulant that sees therapeutic and recreational use. Its chemical structure is the basis for its own group of drugs called substituted amphetamines, which are drugs that have similar structures to amphetamine and include cathinone, bupropion, and MDMA. The most notable of these is methamphetamine (often called meth or “speed”), which is an even more potent stimulant. We will focus on amphetamine and methamphetamine in this section, but keep in mind that some other drugs like cathinone have similar effects.
By the end of this section, you should be able to:
• Explain the history and uses of amphetamine and methamphetamine. • Define chirality and enantiomers and explain why they are pharmacologically relevant. • Describe the pharmacokinetic properties of amphetamine and methamphetamine. • Describe the pharmacodynamic properties of amphetamine and methamphetamine and
the consequences of long-term use.
8.3.1 Drug History and Overview Amphetamine was first synthesized in 1927 by chemist G. A. Alles, who was searching for a substitute for ephedrine to treat asthma and allergies. Ephedrine is a psychoactive ingredient found in ephedra plants, most notably má huáng in Asia, where it has been used in Chinese medicine for thousands of years.
It was initially marketed under the name Benzedrine® as a treatment for narcolepsy, obesity, mild depression, and various other medical conditions. Amphetamines were soon seen as being capable of enhancing cognition, which led to their widespread use in academics in the late 1930s. Amphetamine was also given to soldiers in World War II as energy pills. The potential for addiction was largely ignored until after the war as governments around the world started to regulate amphetamines more strictly.
Currently, amphetamine is classified as a Schedule II controlled substance, same as cocaine. The accepted medical use for amphetamine that keeps it out of Schedule I is its use in treating attention deficit hyperactivity disorder (ADHD). This was one of the earliest therapeutic applications discovered for amphetamine. and it has remained one of the primary options in treating it to this day. That said, the exact formulation used to treat ADHD has changed since the 1930s.
1st edition
Left-handed molecules: Chirality and enantiomers Are your right and left hands identical? They are certainly very similar; in fact, they are mirror images of each other. Despite this, they are not identical. You will always be able to discern a right hand from a left hand because hands are not symmetrical—the thumb is on a different side for each. In a similar fashion, certain molecules can be mirror images of each other without being perfectly identical. These molecules are chiral, meaning they have right-handed and left-handed versions. (The term chiral comes from the Ancient Greek cheir, meaning “hand”.) You can see an example of a chiral molecule below:
Source: NASA Astrobiology Institute
What does this have to do with amphetamine? Well, amphetamine is actually a chiral molecule. It has right-handed and left-handed versions called enantiomers. The right-handed version is called dextroamphetamine or D-amphetamine, while the left-handed is levoamphetamine or L-amphetamine. (This naming scheme might sound familiar—in fact, we’ve already mentioned a chiral molecule before called L-DOPA, the precursor to dopamine. As you would expect, there is a right-handed version of it called D-DOPA instead.) The reason why enantiomers are important is because they sometimes have different pharmacological properties. D-amphetamine is more centrally active, while L-amphetamine is more peripherally active. So, for a drug like amphetamine, it is important to know how much of each type is present in the mixture.
The original formula for amphetamine had equal parts L- and D-amphetamine, which is called a racemic mixture. (You don’t need to know this term for this class, but it might show up when doing further research.) Later, a drug known as Dexedrine® was developed using only the more potent D-amphetamine enantiomers. The modern amphetamine used to treat ADHD which you are probably most familiar with, Adderall®, has a 3:1 mixture of D-amphetamine to L-amphetamine.
Believe it or not, methamphetamine can also be used to treat ADHD or obesity, but this is very rare since there are safer alternatives with less toxicity and a lower potential for developing addiction. As such, methamphetamine is mostly used illicitly for recreational purposes. It is still enough for methamphetamine to be classified as a Schedule II drug because it does have an
accepted medical use. That said, its prescription and use is tracked much more intently than amphetamine.
One consequence of this is limitations being placed on the distribution of pseudoephedrine, a substance found in the same plants as ephedrine. It is an active ingredient in certain decongestants (e.g., Sudafed®), but because of its chemical structure it can be used to illegally manufacture methamphetamine. As such, decongestants containing pseudoephedrine that used to be sold as over-the-counter medicines are now more tightly regulated, with limits to purchase quantity and requiring a photo ID to buy.
8.3.2 Administration and Pharmacokinetics For therapeutic use, amphetamine is typically given orally, which allows for consistent blood concentrations over time. Amphetamine can also be administered via injection, inhalation, or intranasally, which results in a more rapid absorption. The rate of absorption is also dependent on the ionization of the drug; amphetamine is a weak base that is commonly ionized in the gut, which contributes to the slow rate of oral absorption.
Once amphetamine reaches the bloodstream, it is readily distributed throughout the body and can easily cross the blood-brain barrier. Some amphetamine (about 20%) is bound to plasma proteins. Amphetamine and methamphetamine have much longer half-lives than cocaine; the half-life is typically around 10-12 hours and changes depending on the pH level and mixture of enantiomers. In addition, the effects last longer; the high from smoking cocaine tends to last 20 to 30 minutes, while the high from methamphetamine can last from 8 to 24 hours.
Part of the reason for the increased potency and effects of methamphetamine is due to the fact that one of its active metabolites is amphetamine itself. Thus, to eliminate methamphetamine from the system, it must first be metabolized into amphetamine, which in turn must be metabolized as well. The amphetamine produced as a metabolite helps prolong the effects of the drug.
8.3.3 Mechanisms of Action and Effects Similar to cocaine, amphetamine increases the levels of monoamines such as norepinephrine, dopamine, and serotonin. Amphetamine has a similar chemical structure to monoamines, so instead of blocking transport proteins, amphetamine molecules are actually taken into the axon terminal by the reuptake mechanism.
Once inside, amphetamine increases the release of monoamines while also forcing the transport proteins to work in reverse. Instead of pumping neurotransmitters out of the synapse, more and more neurotransmitters are pushed in. In addition, amphetamines have been found to inhibit monoamine oxidase, the enzyme responsible for breaking down monoamine transmitters that have been taken back up by the presynaptic cell.
1st edition
The neurotransmitters released by amphetamine activate the same α-adrenergic and dopaminergic receptors that cocaine does, leading to the stimulating effects of the drug and euphoria. Methamphetamine has a similar mechanism of action but has a stronger effect on the dopamine transporter, which accounts for its greater addictive properties.
The tolerance and withdrawal symptoms are also very similar to those of cocaine, since they target similar receptors. High doses can result in some of the cardiopulmonary symptoms seen with cocaine intoxication, but changes in mental state such as anxiety, paranoia, and hallucinations are more common with amphetamine and methamphetamine.
Long-term use of methamphetamine is associated with numerous health risks. “Meth mouth,” or permanent degradation of teeth, can occur due to the side effects of methamphetamine, increased grinding of teeth, and poor oral hygiene. In addition, methamphetamine also results in an increased production of ceramide, which speeds up cell metabolism and results in earlier cell death and faster aging (see below). This can cause long-term health problems and permanent damage.
1st edition
Because of the various effects produced by amphetamines, they can be misused for a variety of different purposes. Common reasons include weight loss, pleasure-seeking, increased focus, or enhanced athletic performance. Amphetamine use was widely prevalent in baseball for decades as a performance-enhancing drug. If you are interested in learning more, check out this article:
Chapter Summary and Review In this chapter, we introduced our first class of drugs, stimulants, and took a detailed look at two key high-efficacy stimulants: cocaine and amphetamine. We documented the history, uses, and pharmacological properties of both drugs. This involved distinguishing between cocaine hydrochloride or cocaine salt and cocaine freebase (e.g., crack cocaine) and describing the synergistic interaction of cocaine and alcohol. We also discussed chirality in the context of amphetamine formulas and examined methamphetamine, a potent substituted amphetamine that shares similar mechanisms of action.
In the next chapter, we will move on to low-efficacy stimulants. As mentioned before, these chapters will draw from all of the topics we have covered previously, so if you notice yourself stumbling or having difficulties with a particular concept, it is not too late to go back and review.
Practice Questions
• What class of neurotransmitters do sympathomimetic drugs target? Why? • What is the therapeutic use for cocaine, and how does it achieve this effect? • What is the bioavailability of crack cocaine when smoked? • What metabolite forms when cocaine and alcohol interact? Why is it dangerous? • Why is it important to be aware of enantiomers when developing a new drug? • Describe the mechanisms of action for cocaine and amphetamine. In what ways are they
the same? How are they different? • Name two health risks associated with chronic methamphetamine use.
1st edition
References
Drug Enforcement Agency. (2014). Photographs of cocaine and crack cocaine [Photograph].
Retrieved from http://www.dea.gov/pr/multimedia-library/image-
gallery/images_cocaine.shtml
NASA Astrobiology Institute. (n.d.). Amino acid chirality with hands [Illustration]. Retrieved from
Continuing our discussion of CNS stimulants, this chapter we will turn our attention to nicotine and caffeine. These drugs are called low-efficacy stimulants because they have moderate stimulating effects compared to cocaine, amphetamines, and other sympathomimetic drugs. If you pay close attention to the mechanisms of action for these drugs, you should be able to recognize why nicotine and caffeine have a smaller stimulatory effect. Keep this in mind as you read through this chapter.
Chapter Outline: 9.1 Nicotine
9.1.1 Drug History and Overview
9.1.2 Administration and Pharmacokinetics
9.1.3 Mechanisms of Action and Effects
9.1.4 Long-Term Effects and Treatment
9.2 Caffeine 9.2.1 Drug History and Overview
9.2.2 Administration and Pharmacokinetics
9.2.3 Mechanisms of Action and Effects
1st edition
9.1 Nicotine
Most people know someone who is either a current smoker or a former smoker. It may be a friend, sibling, parent, child, or even you. Because of its prevalence, smoking is the most common addiction in the U.S. and is the leading cause of preventable death—7 million people die every year worldwide from smoking, with 480,000 deaths per year in the U.S. alone (Centers for Disease Control and Prevention [CDC], 2020). In this section we will examine nicotine, the key ingredient in cigarettes that is responsible for the pervasiveness of smoking and why the habit can be so hard to quit.
By the end of this section, you should be able to:
• Explain the history and uses of nicotine and tobacco. • Describe the pharmacokinetic properties of nicotine and compare smoking to vaping. • Describe the pharmacodynamic properties of nicotine • Explain the effects of long-term use and describe treatments for nicotine addiction.
9.1.1 Drug History and Overview Nicotine is the major psychoactive component of tobacco. For the most part, the history of nicotine is the history of tobacco. Nicotiana tabacum is the most common tobacco plant, although nicotine can be found in other tobacco species. Tobacco plants are native to the Americas and were used by Native Americans, who valued them for their psychoactive properties and used the leaves to barter and trade for goods.
Tobacco use spread to Europe in the 1500s and was studied by physicians of the time. Its use steadily increased and by the 1600s, English settlers in Jamestown were growing tobacco as a reliable source of income. Tobacco was not only imported back across the Atlantic for recreational use, it also served as an insecticide.
In the late 1800s, machines were invented that could roll cigarettes, fueling the modern smoking epidemic. Cigarette use rapidly rose in the early 1900s, but as smoking increased, so did data linking it to lung cancer. In 1965, the U.S. government began to require warning labels on packs of cigarettes, and subsequent legislation placed restrictions on its use or imposed heavy taxes.
In recent decades, smoking rates have fallen regularly in the U.S. and other developed countries, although smoking continues to increase in less developed countries as tobacco companies focus marketing on places with fewer regulations (Dani & Balfour, 2011). According to the World Health Organization (2008), by the year 2030, over 80% of the world’s deaths from tobacco use will be from developing countries.
9.1.2 Administration and Pharmacokinetics Tobacco is usually consumed by inhaling or chewing. There are various ways to smoke tobacco, such as through cigarettes, cigars, pipes, and hookahs. Each method varies in the amount of tobacco it contains and how the tobacco is prepared. A recent development is e-
1st edition
cigarettes, which allow users to inhale vaporized nicotine directly, a process known as vaping. Vaping is touted as a safer alternative to smoking, and while there are fewer recognized health risks, the discussion is more complicated because of how new e-cigarettes are. Watch this video to learn more about vaping and how it differs from traditional smoking:
Smoking vs Vaping [4:19]
Nicotine is a lipid-soluble drug, meaning it is readily absorbed by the body. Nicotine that is smoked or vaped is absorbed by the lungs, while in the case of chewed tobacco and snuff, it is absorbed through the mucous membranes in the mouth or nose. As you can see in the graphs below comparing the nicotine concentration of different methods, absorption is faster through the lungs.
Source: p. 397 of The Biology of Nicotine Dependence (Bock & Marsh, 2007)
Aside from the lungs or mucous membranes, nicotine can also be absorbed transdermally, as in the case of nicotine patches. Blood concentrations usually reach 12-16 ng/ml from tobacco products; however, smoking allows users to titrate, or adjust the dose of nicotine by inhaling more frequently or deeply. Once in the bloodstream, nicotine is capable of crossing the blood-brain barrier.
Most of the absorbed nicotine is metabolized into cotinine, an active metabolite, by enzyme CYP2A6 in the liver. The half-life of nicotine is 1-2 hours, while cotinine has a half-life of about 16 hours. Because of cotinine’s longer half-life, its presence in urine or blood is used as an indication of recent nicotine use.
9.1.3 Mechanisms of Action and Effects Recall that the neurotransmitter acetylcholine (Otto Loewi’s vagusstoff) acts on two types of cholinergic receptors: muscarinic receptors and nicotinic receptors. The latter are named after nicotine, which produces most of its effects by being a full agonist on nicotinic receptors. Cotinine, its active metabolite, is a partial antagonist. Nicotinic receptors are ligand-gated ion channels that allow sodium, calcium, and potassium ions to pass through when opened. The result is a depolarization of the postsynaptic neuron (see below):
Nicotinic receptors are present throughout the nervous system and are generally excitatory in nature. Nicotinic receptors modulate various different neurotransmitters in the CNS. The main effects of nicotine are stimulatory and anxiolytic (anxiety-reducing). Below is a list of the significant transmitters that nicotine influences and their effects:
1st edition
In the brain, nicotine increases dopamine release in the mesolimbic pathway, particularly in the ventral tegmental area and nucleus accumbens, which accounts for its addictive properties. Tobacco smoke also contains monoamine oxidase inhibitors that may contribute to dependence from chronic use. Recall that monoamine oxidase is the enzyme that breaks down monoamines such as dopamine after reuptake; inhibiting these enzymes will result in greater levels of dopamine in the synapse.
At higher doses, nicotine produces a biphasic response, showing effects of a depressant instead of a stimulant. This is due to how nicotine interacts with nicotinic receptors. After nicotine binds to the receptor, it becomes desensitized and cannot be activated again for a short period, even if another agonist binds to it during that time. With enough nicotine activity, receptors may spend most of the time desensitized. This causes the ion channels to be closed for longer than they are open, leading to functional antagonism.
Nicotine is toxic at large doses. Initial symptoms include an increase in salivation, nausea, fluid in the lungs, high blood pressure, anxiety, muscle spasms, seizures, and an elevated heart rate (see the graphic below for a full list). This is eventually followed by low blood pressure and heart rate, difficulty breathing, and paralysis. In extreme cases this can result in respiratory depression, heart failure, and death. During pregnancy, nicotine is toxic at lower doses and increases the risk of sudden infant death syndrome (SIDS).
1st edition
Source: Mikael Häggström on Wikimedia Commons (2016)
Some of the adverse effects associated with nicotine are the result of tobacco and smoking rather than the drug itself. Nicotine does not cause cancer on its own, but smoke from tobacco contains carcinogens such as nitrosamines that can cause mouth, throat, and lung cancer, among others. Smoking can also cause other pulmonary and cardiovascular diseases, as the particles are hazardous to epithelial cells. Non-smokers can also be exposed to these risks through second-hand smoke.
9.1.4 Long-Term Tolerance Effects and Treatment Chronic nicotine use leads to dependence and tolerance. Tolerance (neuroadaptation) is commonly attributed to a downregulation of nicotinic receptors in a compensatory response to overactivation in order to maintain homeostasis. Tolerance develops to some, but not all, effects of nicotine.
Development of tolerance to nicotine effects can lead to increased use of nicotine, development of physical dependence and withdrawal when nicotine use is stopped. Withdrawal symptoms include cravings, irritability, anxiety and depression, hostility, difficulty concentrating, insomnia, headaches, and increased appetite. Although rarely life-threatening, these symptoms are unpleasant and explain why smoking can be so hard to quit; during withdrawal, nicotine use will relieve these symptoms and result in negative reinforcement, strengthening the habit.
Treatment for nicotine addiction may involve nicotine replacement therapies. These allow users to gradually decrease nicotine use and avoid withdrawal symptoms while avoiding the health risks associated with smoking. Transdermal nicotine patches and nicotine-containing gums, lozenges, or nasal sprays are available. There are also certain drugs, such as varenicline
(trade name Chantix®) or bupropion (mentioned last chapter in regard to substituted amphetamines), that can mitigate the effects of withdrawal by acting at certain nicotinic receptor subtypes.
Successful treatment often involves one or more drug therapies in addition to behavioral therapies designed to help patients cope with temptations and deal with situations where smoking is habitual. Despite the large number of resources available for people who want to quit, relapse is still common. According to a 2015 survey, while 70% of current adult smokers wished to quit and 55% had attempted to do so in the past year, only 7% were successful in quitting (CDC, 2017).
9.2 Caffeine
Although it is not likely to be thought of as a drug, caffeine is a stimulant compound that produces its psychoactive effects by targeting certain receptors in the nervous system, similar to every drug we’ve discussed so far. In fact, because we consider it harmless and impose little to no regulation of it, caffeine is the most commonly used drug in the world.
By the end of this section, you should be able to:
• Explain the history and uses of caffeine. • Describe the pharmacokinetic properties of caffeine. • Describe the pharmacodynamic properties of caffeine and explain the toxic effects of
caffeine.
9.2.1 Drug History and Overview Caffeine is an alkaloid that is found in numerous plant species across the world. Some of the most notable sources of caffeine are the beans of coffee and cocoa plants, the leaves of tea plants, and kola nuts. Because of this, we can find caffeine in coffee, chocolate, tea, and many colas and other carbonated soft drinks.
Caffeine belongs to a class of stimulants called methylated xanthines, or methylxanthines for short. Methylxanthines share similar chemical structures derived from xanthine, a naturally occurring base found in many organisms, including humans. Other methylxanthines include theobromine and theophylline, which can be found in some of the same plants as caffeine. However, caffeine is the most common, so it will be the focus of this section.
The origins of caffeine consumption go back thousands of years. Caffeine has traditionally been consumed in various parts of the world, from tea in China, coffee in the Arabian Peninsula, and cocoa beans in the Americas. It is not known who first discovered the stimulant properties of caffeine, but it was first extracted in 1819 by German chemist Friedlieb Ferdinand Runge, who called it kaffebase (“coffee base”).
1st edition
9.2.2 Administration and Pharmacokinetics Caffeine is almost always administered orally in drinks, food, or pills. For beverages, the amount of caffeine administered depends on the drink, but it usually ranges from 10 to 200 mg per serving. See the table below for a list of examples:
Beverage (Serving) Caffeine
Brewed coffee (8 oz) 100-200 mg
Decaffeinated coffee (8 oz) 2-12 mg
Instant coffee (8 oz) 40-80 mg
Starbuck’s Caffè Americano (16 oz) 225 mg
Black tea (8 oz) 15-70 mg
Green tea (8 oz) 24-45 mg
Coca-Cola or Pepsi (12 oz) 25-40 mg
Red Bull (8.4 oz) 75-80 mg
Monster (24 oz) 240 mg
5-Hour Energy (2 oz) 200 mg
NoDoz Max Strength (1 tablet) 200 mg
Caffeine is absorbed by the stomach and small intestine, with 99% being absorbed within 45 minutes of ingestion (Institute of Medicine, 2001). Once absorbed, caffeine is quickly distributed throughout the body and can easily cross the blood-brain barrier. Blood concentration peaks between 30 to 120 minutes after ingestion, although effects are felt sooner (White et al., 2016).
Cytochrome P450 enzymes in the liver, in particular CYP1A2, metabolize caffeine into other methylxanthines such as theobromine and theophylline. Although they are active metabolites, they have much weaker effects than caffeine. Caffeine has a median half-life of about 5 hours, although this can very among individuals, especially in the case of pregnant women in the last trimester, where the half-life can be increased to 18 hours. Other drugs that influence CYP1A2 can increase the half-life of caffeine as well, such as the antidepressant fluvoxamine, which can extend it to over 57 hours (Culm-Merdek et al., 2005).
1st edition
9.2.3 Mechanisms of Action and Effects The primary mechanism of action for caffeine is antagonism of adenosine receptors. Adenosine was not covered in our discussion of neurotransmitters in Chapter 4, but it is a neuromodulator that inhibits neural activity throughout the nervous system. By inhibiting the release of other neurotransmitters, such as norepinephrine, adenosine can cause drowsiness.
Caffeine blocks adenosine receptors, thereby disinhibiting the release of norepinephrine and increasing feelings of wakefulness. Adenosine receptors also inhibit cholinergic and dopaminergic neurons, which helps explain why caffeine can cause jitteriness and dependence. See the diagrams below showing regular adenosine activity (top) and caffeine antagonism (bottom).
1st edition
1st edition
As a mild stimulant, caffeine can cause various physiological effects common to stimulants such as increased heart rate, reduced appetite, increased alertness, improved mood, and vasoconstriction. Chronic caffeine use can result in physical and psychological dependence. The DSM-V does not contain a specific diagnosis for caffeine use disorder but has recommended that it be studied further. Caffeine intoxication and caffeine withdrawal are recognized as disorders in DSM-V.
Tolerance will develop to effects like increased alertness and improved mood; withdrawal symptoms therefore range from anxiety, fatigue, irritability, depressed mood, and difficulty concentrating. Headaches will often occur as caffeine wears off due to the relaxation of blood vessels in the brain that were previously constricted, resulting in a sudden increase in circulation.
Although rare, it is possible to overdose on caffeine, since large doses are toxic. Doses up to around 400 mg (approximately 3 cups of coffee, 10 cans of cola, or 2 energy drinks) are mostly harmless. Above this, caffeine can cause toxic effects such as anxiety, insomnia, irritability, and increased heart rate and blood pressure. This condition is known as caffeinism.
Fatal doses of caffeine are typically well beyond normal consumption levels for caffeinated beverages, and as such death from caffeine overdose is uncommon. However, there are bottles of caffeine powder sold as supplements which contain large amounts of pure caffeine. These can potentially lead to lethal doses if the user does not measure the correct amount. Despite calls to regulate caffeine powder, the FDA has only issued warnings and recommends avoiding these products.
1st edition
Chapter Summary and Review
In this chapter, we finished up our coverage of stimulants by exploring the low-efficacy stimulants, nicotine and caffeine. We compared different methods of administrating nicotine, then explained its mechanism of action and the consequences of chronic use. We also discussed caffeine’s role as the most commonly used drug, examining its effects on adenosine receptors and how caffeine dependence and overdose can occur.
Next chapter we will move on to the flip side of stimulants—depressants. With a new class of drugs, we will soon be learning about a different neuromodulator, GABA, and its function in the CNS as an inhibitor. Make sure that you are familiar with stimulants before moving on.
Practice Questions
• Is vaping a risk-free alternative to nicotine? Why or why not? • Are nicotinic receptors ionotropic or metabotropic? • Name two specific mechanisms through which chronic smoking can lead to dependence. • Name five symptoms of nicotine poisoning. • Is nicotine a carcinogen? • What class of stimulants are caffeine and its active metabolites part of? • What is typical median half-life of caffeine? • Name three types of neurons that adenosine receptors modulate. • What is caffeinism? What are some of the symptoms of it? • Out of all the stimulants discussed so far (cocaine, amphetamine, methamphetamine,
nicotine, and caffeine), which drug is the most addictive? Which drug causes the most deaths? Which drug is used the most?
1st edition
References
Bock, G., & Marsh, J. (2007). The biology of nicotine dependence: Ciba Foundation Symposium
152. Hoboken, NJ: John Wiley & Sons.
Centers for Disease Control and Prevention. (May 21, 2020). Fast facts: Smoking & tobacco
Now that we have covered stimulants, it is time to move on to drugs that have opposing effects. In this chapter we will examine a variety of depressants and learn about how they alter neurotransmission to reduce the activity of the central nervous system.
Chapter Outline: 10.1 Depressants Overview
10.1.1 The GABA Receptor
10.1.2 Types of Depressants
10.2 Barbiturates 10.2.1 Drug History and Overview
10.2.2 Administration and Pharmacokinetics
10.2.3 Mechanisms of Action and Effects
10.3 GHB 10.2.1 Drug History and Overview
10.2.2 Administration and Pharmacokinetics
10.2.3 Mechanisms of Action and Effects
10.4 Inhalants 10.2.1 Drug History and Overview
10.2.2 Administration and Pharmacokinetics
10.2.3 Mechanisms of Action and Effects
1st edition
10.1 Depressants Overview
As you would expect from the name, depressants are the opposite of stimulants; they are drugs that reduce CNS activity. Because of this they are sometimes colloquially referred to as “downers,” in contrast to the term “uppers” being used for stimulants. Although there are many different types of depressants, most target the same site of action: the GABA receptor. Therefore, before we discuss any particular drugs, it is worth taking a closer look at this receptor and how different depressants interact with it.
By the end of this section, you should be able to:
• Describe the role of GABA and GABA receptors in neural inhibition. • Define sedative-hypnotics and provide examples of types of depressants.
10.1.1 The GABA Receptor Recall from Chapter 4 that γ-aminobutyric acid (gamma-aminobutyric acid, or GABA for short) is the brain’s main inhibitory neurotransmitter. This is because GABA receptors are inhibitory; in other words, they promote hyperpolarization of the postsynaptic cell. This inhibits the postsynaptic cell from firing and releasing other neurotransmitters, such as glutamate or norepinephrine. As a result, increasing GABA activity will, in general, reduce the activity of other neurons and transmitters.
There are two GABA receptor subtypes. The first type is the GABAA subtype. GABAA receptors are ionotropic and are part of the chloride channel receptor complex; when they are activated, chloride ions (Cl-) flow into the cell, increasing the negative charge inside the neuron. In comparison, GABAB receptors are metabotropic. Instead of opening a chloride ion channel, when an agonist binds to these receptors, a g-protein induced signal cascade increases the outward flow of potassium ions (K+), which also increases the negative charge inside the cell. To review this information, you may find it helpful to watch this short video:
2-Minute Neuroscience: GABA [1:59]
GABAA receptors are comprised of five subunits surrounding the central chloride ion pore. The most common type has two α and β subunits and one γ subunit, as seen in the diagram below. The primary binding site, also known as the orthosteric site, is where GABA normally binds to the receptor.
Aside from the main site, there also exist multiple other sites where ligands can bind. These sites are called allosteric sites; the name should be familiar to you, since we covered them in Chapter 4. To refresh, ligands that bind to these sites are called allosteric modulators. These change the function of the receptor without competing for the main binding site.
Many depressants are allosteric modulators of the GABAA receptor. When they bind to the receptor, they change its shape so that GABA has increased efficacy at the main site. Because they increase efficacy, they are known as positive allosteric modulators. Positive allosteric modulators do not increase the amount of GABA present in the synapse like reuptake inhibitors or activate the receptor on their own, as in the case of direct agonists. Instead, they change the function of the receptor so that it is more responsive to GABA binding.
10.1.2 Types of Depressants Many types of drugs produce depressant effects. Perhaps the most well-known depressant is alcohol. Because of its significance and certain unique properties, the entirety of the next chapter is devoted to covering it. Aside from alcohol, we will also find sedatives and hypnotics in this category. Sedatives calm anxiety and agitation, while hypnotics induce sleep. Since they share similar functions and many sedatives cause hypnotic effects at higher doses (and vice-versa), they are usually referred to as a single class of drugs, sedative-hypnotics.
Sedative-hypnotics include barbiturates, benzodiazepines, and nonbenzodiazepines (also called Z-drugs). We will discuss some of these in greater detail during our last unit on therapeutic drugs, but, for this chapter, we will focus on barbiturates. Other types of drugs have sedative effects through action on the GABA receptor, such as GHB, another drug we will be covering in this chapter.
1st edition
Not all CNS depressants are sedative-hypnotics; inhalants, which we will also be examining, do not have any sleep-inducing effects. At the same time, some drugs produce sedative effects through mechanisms other than the GABA receptor. Antihistamines are one such example, which act at histamine receptors and cause drowsiness as a side-effect. Although we will not be exploring them in this chapter, keep this in mind.
10.2 Barbiturates
The first depressants we will discuss are barbiturates. Barbiturates are potent sedative-hypnotics that were widely used in the early 1900s. Although their use has declined in recent decades, they remain an illustrative example of how depressants affect neurotransmission.
By the end of this section, you should be able to:
• Explain the history and uses of barbiturates. • Describe the pharmacokinetic properties of barbiturates and differentiate between
barbiturates by duration. • Define the pharmacodynamic properties of barbiturates and the consequences of
barbiturate dependence and tolerance.
10.2.1 Drug History and Overview Barbiturates are derived from barbituric acid, first synthesized in 1864. No use was found for it until 1903, when German chemists discovered the sedative-hypnotic effects of its derived compounds. The first barbiturate, barbital, was marketed by Bayer under the name Veronal that year, and barbiturate use steadily increased in the first half of the 20th century.
Barbiturates were routinely used to induce sleep in psychotic patients and were prescribed to treat insomnia and anxiety. They were also shown to reduce the number and intensity of seizures—a first, since no other drugs were effective at treating epilepsy at the time—and began to see use as anticonvulsants. In 1912, Bayer produced another barbiturate, phenobarbital, which is still used to treat epilepsy to this day.
Dependence and overdose were identified as severe problems soon after the drug was synthesized. Despite this, barbiturates continued to be prescribed up until the 1950s and 1960s, when increased reports and greater visibility of barbiturate misuse led to significant change. Perhaps the most well-known instance of barbiturate overdose was Marilyn Monroe’s death in 1962. By 1970, barbiturates were considered controlled substances and physicians were prescribing them at much lower rates.
Currently, most barbiturates are classified as Schedule III controlled substances, although some types, such as phenobarbital, are Schedule IV instead. Barbiturates have mostly been replaced with benzodiazepines and Z-drugs for treatment of insomnia and anxiety because they have fewer issues with dependence and overdose. They remain in use as anticonvulsants, general anesthetics, and antagonists to the effects of certain stimulants.
1st edition
10.2.2 Administration and Pharmacokinetics Barbiturates can be classified by their duration of action. Long-acting barbiturates such as phenobarbital have low lipid solubility and are absorbed slowly. In exchange for a delayed onset (about 1 hour), effects can last for up to 12 hours. This makes them useful as anticonvulsants, since fewer doses are required to maintain the drug’s level in the body.
Intermediate- and short-acting barbiturates like pentobarbital have moderate lipid solubility. They are absorbed faster and have an onset of about 30 minutes, but effects do not last as long (up to 8 hours). The faster onset means these are used most often as sedative-hypnotics.
Ultrashort-acting barbiturates such as thiopental have the highest lipid solubility out of all barbiturates. Time to action can be mere minutes, although effects only last for around half an hour. Drugs like these are more suited for serving as general anesthetics for short surgical procedures.
Because they are weak acids, barbiturates are readily absorbed after oral administration. Other routes include rectal or intravenous. The method chosen depends on the intended use and recipient. Ultrashort-acting barbiturates are usually administered by IV, while long-acting anticonvulsant medications may be taken through suppository.
Here is a table summarizing the information above:
Long-acting Intermediate- and short-acting Ultrashort-acting
Onset 1 hour 30 minutes 1-5 minutes
Duration 12 hours 8 hours 30 minutes
Main Use anticonvulsant sedative-hypnotic general anesthetic
Example phenobarbital pentobarbital thiopental
10.2.3 Mechanisms of Action and Effects Barbiturates are positive allosteric modulators of GABAA receptors. By binding to areas other than the main site of the receptor, they enhance GABA activity. In particular, they increase the amount of time that the chloride ion channel remains open when GABA binds to the receptor. At high concentrations, barbiturates can also bind to the main site as direct agonists.
At the same time, barbiturates are also antagonists to certain glutamate receptors. Recall that glutamate is an excitatory neurotransmitter. By blocking these glutamate receptors—AMPA and ainite—barbiturates further reduce CNS activity. This accounts for the strong effects of barbiturates compared to other sedative-hypnotics; barbiturates not only enhance inhibition but also block excitation.
1st edition
The effects of barbiturates are dose-dependent. At lower doses, they produce sedation and hypnosis; at higher doses, they can induce anesthesia, coma, and even death. These effects tend to follow one another in sequence as you increase the dose (see image below):
Intermediate-acting barbiturates used as sedative-hypnotics can induce sleep. Specifically, they reduce the time needed to fall asleep, increase the time spent asleep, and reduce the occurrence of rapid eye movement (REM) sleep.
At high doses, barbiturates can result in generalized CNS depression. Symptoms include loss of muscle coordination, difficulty thinking and speaking, and shallow breathing. These symptoms often result in behavior similar to that exhibited by someone who is drunk. Eventually, these symptoms can worsen and lead to respiratory depression, coma, and death.
There are no antagonists for barbiturate intoxication, so in the case of overdose the emphasis is to maintain breathing, blood pressure, and kidney function. Another antidotal measure involves speeding up the elimination of the drug from the kidneys. Recall that for a drug to be excreted, it is often useful to metabolize it to a more ionized form as this will prevent the drug from being reabsorbed as easily.
A similar effect can be achieved by adjusting the pH of the urine in the kidneys. Because barbiturates are weak acids, they can be more rapidly eliminated from the body by acidifying the urine with ammonium chloride. On the other hand, drugs that are weak bases such as amphetamine can be more rapidly cleared by alkalinizing the urine with sodium bicarbonate. In both cases, the increased elimination rate means that less of the drug is available in the bloodstream to activate target receptors.
As mentioned earlier, barbiturate dependence is noted to be a considerable problem. Tolerance to the sedative-hypnotic effects of barbiturates will develop with repeated use, but the same cannot be said for toxic effects such as respiratory depression. This means that over time, the therapeutic index for barbiturates grows smaller and smaller as the dose-response curve shifts to the right (see below):
1st edition
Withdrawal symptoms include anxiety, insomnia, nausea and vomiting, muscle weakness, abdominal cramps, and increased heart rate. At high levels of dependence, these symptoms are exacerbated, and withdrawal may involve convulsions, hallucinations, delirium, cardiovascular collapse, and death.
Treatment for barbiturate dependence involves detoxification and gradual reduction in symptoms of dependence. Fortunately, the withdrawal symptoms can be suppressed by safer sedative-hypnotic drugs. Benzodiazepines are the antidotes of choice; however, in an emergency, even ethanol (alcohol) can be used to suppress withdrawal from barbiturates.
1st edition
10.3 GHB
The next depressant we will examine is γ-hydroxybutyric acid, or GHB. It is an endogenous substance that can also be taken as a medication or used recreationally. Although it primarily acts as a depressant, it actually causes biphasic effects, with stimulatory effects occurring at low doses or for a short time initially. Because of this, it is primarily used as a club drug.
By the end of this section, you should be able to:
• Explain the history and uses of GHB. • Describe the pharmacokinetic properties of GHB. • Describe the pharmacodynamic properties of GHB and the role of GHB and GABAB
receptors.
10.3.1 Drug History and Overview As mentioned at the start of this section, GHB is found naturally in the body. It is a precursor to the neurotransmitters GABA, glutamate, and glycine and is a neurotransmitter itself. Contrary to what you might expect, its endogenous receptor, the GHB receptor, is excitatory rather than inhibitory. (The inhibitory effects come from GHB’s activity at GABAB receptors, which we will cover in more detail shortly.)
It was first studied in depth in the 1960s for its potential use in treating narcolepsy and alcoholism. Although there was little support for its use in treating alcoholism, the salt form of GHB, sodium oxybate, is still used for the treatment of narcolepsy to this day under the brand name Xyrem®. In the 1990s, GHB was marketed as a dietary supplement and found some use among athletes as a performance-enhancing drug, despite lack of any actual evidence for performance-enhancing effects.
GHB found its main use as a club drug or party drug because of its euphoric effects at low doses. It is also easier to manufacture than most other club drugs, making it an attractive alternative. GHB is also occasionally used as a date-rape drug due to the drug’s ability to induce unconsciousness and amnesia. It is colorless and odorless and can be easily poured into a drink without notice. Although its use as a date-rape drug has been highly publicized, it is difficult to know how frequently it is used this way.
In 2000, GHB was classified as a Schedule I controlled substance, with the exception of sodium oxybate, which is classified as Schedule III. Some of the chemicals that can be used to manufacture GHB, such as gamma-butyrolactone (GBL), are also classified Schedule I substances.
10.3.2 Administration and Pharmacokinetics GHB is typically taken orally as a dissolved powder or a solution. It is absorbed quickly and reaches peak concentration in the blood at around 45 minutes. Effects can begin to show as early as 20 minutes after administration and last up to 2 hours.
1st edition
GHB is metabolized rapidly and has a short half-life of about 30 minutes. Because of this, GHB is eliminated from the body faster than most drugs and can only be detected for 8-12 hours after its administration. This is part of the reason why GHB’s use as a date-rape drug is hard to track; a urine sample would need to be analyzed within a day of the suspected administration for there to be any chance of getting a positive result.
When GHB and alcohol are combined, the sedative and depressant effects are amplified, and GHB may reduce the rate at which alcohol is eliminated from the system. This synergistic interaction can lead to unexpected respiratory failure and death.
10.3.3 Mechanisms of Action and Effects GHB is both an endogenous substance as well as an exogenous chemical. At low physiological concentrations such as those released from nerve terminals, GHB preferentially activates GHB receptors. These receptors enhance glutamate activity and stimulate dopamine and serotonin release. This is where the stimulatory effects of GHB come from. The release of dopamine as a consequence of GHB receptor activity also contributes to the addictive properties of the drug.
Exogenous GHB has affinity and efficacy for inhibitory GABAB receptors. When GHB is abused as a drug, it attains higher concentrations in the brain than endogenous GHB. Because GABAB receptors are metabotropic, their activation causes slower but longer-lasting changes to postsynaptic firing, which contributes to the expression of stimulatory effects at low doses before most of the drug has been absorbed.
The earliest effects of GHB consist of stimulation, relaxation, euphoria, and increased energy. Some users report hallucinations and aphrodisiac effects. As time goes on, users begin to exhibit symptoms similar to alcohol intoxication, including relaxed inhibitions, impaired motor coordination, and slurred speech. At high doses, toxic effects such as nausea and vomiting, slowed heart rate, low blood pressure, convulsions, coma, and respiratory failure can occur. After use, people will experience fatigue, amnesia, confusion, and anxiety.
GHB is highly addictive and repeated use can lead to a rapid development of tolerance. This is somewhat undercut by the drug’s short half-life; users would have to take the drug very frequently, making addiction somewhat rare. Despite this, it is still possible to develop dependence from constant use. Withdrawal symptoms can be very dangerous and include tremors, seizures, and insomnia, in addition to the amnesia, confusion, and anxiety that occurs after GHB use.
1st edition
10.4 Inhalants
For the last section of our chapter on depressants, we will cover a type of drug that many people might overlook. Inhalants are solvents or other materials that produce vapors that elicit psychoactive effects. While a wide variety of products can be used as inhalants, most induce CNS depression through similar mechanisms of action.
By the end of this section, you should be able to:
• Define inhalants and describe types of inhalants and the prevalence of inhalant use. • Describe the pharmacological properties of inhalants. • Differentiate between different methods of inhaling. • Describe the four stages of inhalant-induced CNS depression and explain sudden
sniffing death syndrome. • Describe the use and misuse of nitrous oxide.
10.4.1 Drug History and Overview Inhalant is an umbrella term that refers to numerous chemicals that can be inhaled to produce intoxication. These chemicals can be found in various household goods and cleaning supplies such as glues, aerosol sprays, paint thinner, nail polish remover, gasoline, whipped cream, and felt-tip markers. A notable inhalant is nitrous oxide, a gas used as an anesthetic in surgeries and dentistry. These substances are typically unregulated and can be easily purchased or found in products around the house.
1st edition
Unlike other psychoactive drugs, inhalants are most commonly used by children and adolescents. Most users are adolescents beneath the 8th grade; by then, 1 in 4 students in America have misused a household product to get high. Studies have shown that as users age, they tend to use inhalants less often. Because of their widespread use by children, inhalants are the fourth-most misused substance after alcohol, tobacco, and marijuana.
10.4.2 Administration and Pharmacokinetics As indicated by the name, inhalants are administered by inhaling the substance over a period of time at high concentrations. There are multiple ways the chemicals can be inhaled. Sniffing involves inhaling the vapors off of a cloth or small container. Huffing entails placing the cloth directly over the mouth and nose to inhale. Bagging, meanwhile, involves inhaling the compound from a paper or plastic bag.
Most inhalants are lipid-soluble and absorbed very quickly, with concentrations in the blood peaking close to the time of administration. The combination of fast absorption and taking in the drug through the lungs results in an immediate rush and noticeable effects. Metabolism and excretion vary depending on the chemical in question, but half-lives tend to be very short; nitrous oxide, for instance, is exhaled almost entirely through the lungs unchanged, resulting in a half-life of about 5 minutes.
1st edition
10.4.3 Mechanisms of Action and Effects Because inhalants are a collection of chemicals, all with different mechanisms, it can be difficult to summarize drug actions. In general, however, there are two common mechanisms. Inhalants often are allosteric modulators of GABAA receptors as well as antagonists to glutamate NMDA receptors. Both actions result in decreased CNS activity and depressant effect.
The exact effects of inhalants also vary, but they typically follow four stages. The first stage is the excitatory stage, where the user experiences euphoria and agitation. This turns into early CNS depression, which is characterized by slurred speech and hallucinations. In the third stage, medium CNS depression, the user experiences confusion, delirium, and impaired muscle coordination (ataxia). The final stage is late CNS depression, which can cause stupor, seizure, coma, and death.
The vapors in inhalants compete with oxygen, which means users of inhalants may have a low oxygen supply. Coupled with slowed breathing or respiratory failure, this may result in a delayed death. Inhalants can also cause immediate arrhythmias and cardiac arrest, known as sudden sniffing death syndrome. This can occur after a single misuse and be fatal.
Worth taking a closer look at is nitrous oxide. As mentioned earlier, nitrous oxide is used as a general anesthetic. It also has analgesic (pain-relieving) and anxiolytic effects. Nitrous oxide is often misused because it is unregulated and produces euphoria and giddiness, which is why it is also called laughing gas. It can also lower inhibitions and cause dissociation, unconsciousness, dizziness, and loss of motor function.
Despite its safe use in medical practice, recreational use of nitrous oxide is dangerous because it can cause a loss of blood pressure or heart attack if used without an adequate oxygen supply. Chronic exposure can also lead to a loss of bone marrow and neurotoxicity in the CNS.
1st edition
Chapter Summary and Review
In this chapter, we learned about how depressants reduce CNS activity through GABA and examined a few different types of depressants. We started with barbiturates and learned about their sedative-hypnotic effects and risks associated with dependence. We then moved on to GHB and discussed its biphasic effects. Finally, we discussed the effects and misuse of inhalants such as nitrous oxide.
That will be all for this chapter. For the last chapter in this unit, we will take a detailed look at alcohol, the most infamous depressant of all. Much of the terminology used to describe alcohol’s effects will have already been introduced in this chapter, so make sure you are comfortable with this chapter’s material before moving on.
Practice Questions
• Both GABAA and GABAB receptor subtypes cause depolarization. What are the two different ways that they achieve this?
• What is a positive allosteric modulator? How does this compare to a direct agonist? (Bonus question: What do you think a negative allosteric modulator does?)
• Methohexital is a barbiturate with a rapid onset of action that is typically used for anesthesia. What type of barbiturate is it classified as?
• Why is barbiturate dependence likely to result in an eventual overdose? • Are GHB receptors excitatory or inhibitory? • Describe the biphasic effects of GHB. What are some symptoms from both phases? • Classify the following drugs by their schedule: barbital, phenobarbital, GHB, and sodium
oxybate. • Does inhalant use increase as users get older? • List the four stages of CNS depression from inhalant use. Provide at least one symptom
for each stage.
Chapter 11: Alcohol
To close out this unit, we will spend the entirety of this chapter looking at alcohol. Alcohol is a CNS depressant, though it has many other pharmacological actions as well. This chapter will go into greater detail than usual to explore its unique properties and cover the breadth of alcohol research. Stay focused, and don’t forget to take breaks when you need them.
Chapter Outline: 11.1 Alcohol Overview
11.1.1 History and Legislation
11.1.2 Prevalence of Alcohol Use
11.1.3 Ethanol Properties and Concentrations
11.2 Absorption and Distribution 11.2.1 Absorption of Ethanol
11.2.2 Distribution of Ethanol
11.2.3 Blood Alcohol Concentration
11.3 Metabolism and Excretion 11.3.1 Metabolism of Ethanol
11.3.2 Genetic Polymorphisms of ADH and ALDH
11.3.3 Microsomal Ethanol-Oxidizing System
11.4 Ethanol Pharmacodynamics 11.4.1 Receptors and CNS Effects
11.4.2 Ethanol Dependence
11.4.3 Effects on Other Systems
11.5 Adverse Health Risks 11.4.1 Alcohol-Related Cancers
11.4.2 Wernicke-Korsakoff Syndrome
11.4.3 Fetal Alcohol Syndrome
11.1 Alcohol Overview Alcohol is a drug that has a long history and is extensively used throughout the world. Before exploring the pharmacology of alcohol, we will start by learning more about its use, prevalence, legislation, chemical composition, and how it is measured.
By the end of this section, you should be able to:
• Explain the history of alcohol use and legislation. • Explain how prevalent alcohol use and alcohol use disorder are. • Describe how ethanol is measured and define standard drink.
11.1.1 History and Legislation It is estimated that humans have been consuming alcohol for around 9000 years. Wine and beer existed in ancient Egyptian and Sumerian civilizations, with records dating as far back as 3200 B.C. Ancient Greeks used wine in religious ceremonies around 800 B.C., and Roman culture involved heavy drinking that persisted until the fall of Rome in 476 A.D. Distilled liquor first appeared around 1250 A.D., eventually followed by brandy, gin, and rum.
Alcohol is created by fermentation of sugars from variety of sources, such as grapes (wine), grains (beer), honey (mead), and sugarcane (rum). Methods of production spread throughout the world and contributed to its popularity. Alcoholic beverages were seen as more nutritious than water. Its consumption was also tied to social interaction, religious ceremonies, and celebrations, which increased its cultural importance.
Opposition to alcohol use has existed for as long as the recorded history of alcohol. In the United States, the movement to prohibit alcohol started to pick up steam in the 1800s. After the Civil War, traction for the movement increased, fueled largely by the Women’s Christian Temperance Union. The National Prohibition Act, also known as the Volstead Act, was passed in 1920 and added to the Constitution as the 18th Amendment. It banned the sale and distribution, but not the consumption, of alcohol across the U.S.
During the Prohibition era, consumption of alcohol was driven underground to speakeasies. At the same time, widespread smuggling of alcohol, known as bootlegging, arose to meet demand. Support for prohibition steadily dropped, and 13 years later, it was repealed by the 21st Amendment in 1933. Although this marked the end of national prohibition, some counties and towns remained “dry” and continue to prohibit the sale of alcohol to this day. Individual states were assigned responsibility for regulating alcohol and setting their own legal drinking ages. The National Minimum Drinking Age Act, passed in 1984, ensures that sale was prohibited to anyone under the age of 21.
11.1.2 Prevalence of Alcohol Use Alcohol is one of the most commonly used drugs. According to the 2019 National Survey on Drug Use and Health (NSDUH), about 55% of Americans over 18 reported that they drank in the past month. This widespread use of alcohol also applies to risky behaviors. Binge drinking involves drinking large amounts of alcohol in a short time span (usually 4–5 drinks in about 2
hours) and carries higher risk than normal drinking. 23% of people over 18 reported binge drinking in the past month (SAMHSA, 2019).
Driving while intoxicated is another common consequence of alcohol use. Vehicle collisions involving alcohol-impaired driving accounted for 29% of all driving fatalities in the U.S. Alcohol consumption also imparts considerable health risks to the user, similar to smoking. Alcohol is the third-most preventable cause of death in the U.S, after smoking and poor diet (Mokdad et al., 2004).
Underage drinking is also common. According to the 2019 NSDUH, nearly 40% of people ages 12-20 reporting drinking alcohol. Although young people drink less often than adults, they also engage in riskier behaviors. More than 90% of all alcoholic drinks consumed by young people involved binge drinking (NIAAA, 2020). Statistics show that at least 60% of undergraduate students acknowledge past-month consumption of alcohol.
Alcohol use disorder (AUD) is a substance use disorder outlined in the Diagnostic and Statistical Manual of Mental Disorders (DSM). It is characterized by compulsive alcohol used despite negative consequences. The DSM-5 defines 11 criteria for diagnosis, with the severity of AUD (mild, moderate, or severe) depending on the number of criteria met. Nearly 15 million people have AUD, with AUD being more common among men (9.0 million) than women (5.5 million) (SAMHSA, 2019).
11.1.3 Ethanol Properties and Concentrations The scientific name for the drug is ethyl alcohol or ethanol. An ethanol molecule is comprised of an ethyl group (CH3−CH2−) linked to a hydroxyl (OH) group, as you can see in the images below. Because of this, the molecule is often abbreviated as EtOH.
Sources: Jü on Wikimedia Commons (2017); Benjah-bmm27on Wikimedia Commons (2007)
Alcohol or Ethanol? Although the terms alcohol and ethanol are mostly interchangeable in this chapter, we will prefer the term ethanol when talking about the drug. In chemistry, alcohol can refer to various compounds that have similar molecular structures to ethanol. You won’t usually see this meaning of the word used in the context of drugs—ethanol is the only alcohol widely consumed by humans, so the term alcohol is regularly used in scientific literature—but it’s worth being aware of the distinction. Other alcohols include isopropyl alcohol (rubbing alcohol) and methanol (wood alcohol), but these cannot be safely consumed.
Ethanol is an organic compound with a variety of applications. It is used as a disinfectant, such as in hand sanitizers or medical wipes. It is also a universal solvent and has many uses in the manufacture of other compounds. Ethanol is a suitable fuel source and is used in alternative fuels. It is also, of course, the key ingredient in alcoholic drinks.
These are multiple ways to measure the amount of ethanol in an alcoholic drink. The simplest approach is to describe the percentage of ethanol content. This is used by the standard measure of alcohol by volume, abbreviated ABV. Instead of ABV, you might also see alcohol proof used instead. The proof of a drink is defined as twice the percentage of ABV, and so ranges from 0–200.
An absolute or anhydrous solution of ethanol is 100% ethanol. Another common specification is outlined by the U.S. Pharmacopeia (USP) as 95% ethanol solution. Both of these concentrations are intended more for use in research, medicine, and industry. Alcoholic drinks tend to have concentrations in the 1–20% for beers and wines, with distilled spirits usually having around 40% ABV. Consult the table below to view typical percent ABV for various drinks:
Drink ABV
Beer 4–6%
Cider 4–6%
Wine 10–12%
Mead 10–14%
Sake 16%
Fortified Wine 15–22%
Tequila 38–40%
Rum 40%
Vodka 40%
Whiskey 40–50%
Source: Nutrients Review (2016)
A common shorthand for determining alcohol consumption is a standard drink, defined as about 14 grams of pure ethanol (NIAAA, n.d.). This would be approximately equivalent to a 12-oz can of beer, a 5-oz glass of wine, or a 1.5-oz shot of distilled spirits. Three standard drinks will usually put you over the legal limit for driving, though even a single drink can cause significant impairment.
The pharmacokinetics of ethanol start with its absorption and distribution. These topics are important to more than just pharmacology majors; understanding how alcohol is absorbed is necessary for responsible drinking, and laws written to punish drunk driving reference the distribution of ethanol in the blood.
By the end of this section, you should be able to:
• Describe how ethanol is absorbed and what factors influence the rate of absorption. • Explain how ethanol is distributed throughout the body and how individual characteristics
can influence blood concentrations. • Define blood alcohol concentration and describe the effects of alcohol at different BACs.
11.2.1 Absorption of Ethanol Ethanol is extremely soluble in both water and fat, making it readily absorbed through tissue. Ethanol is typically ingested orally through alcoholic drinks. These can also be administered rectally, although the practice is uncommon and not advised. Rectal administration results in faster absorption and higher blood concentrations because it bypasses the stomach.
When taken orally, 75–80% is absorbed through the small intestines; the rest is absorbed through the stomach, which contains enzymes that can metabolize alcohol. The presence of food in the stomach slows absorption and reduces peak blood concentration because ethanol spends more time in the stomach being metabolized and slowly absorbed before reaching the small intestine. This is the reason why eating food while drinking is advised. Refer to the graph below to see the effect of food on ethanol absorption:
On an empty stomach, 50% of ethanol is absorbed in 15 minutes, with maximum blood levels being reached in 20 minutes. Most of the ethanol (80–90%) is absorbed within an hour or less of administration. The more food in the stomach, the less ethanol absorbed, although there is a gender difference; women have fewer stomach enzymes than men, so more ethanol will be absorbed.
11.2.2 Distribution of Ethanol Ethanol is miscible with water, meaning that when the two are mixed, ethanol is distributed evenly throughout the solution. This means that once ethanol is absorbed, it is found in approximately equal concentrations in water throughout the body. This includes not only blood, but also the water in tissues and other fluids. Altogether, this is known as the total body water. The amount of total body water varies depending on age and gender, which influences the concentration of ethanol. Women tend to have a higher percentage of body fat and thus lower percentage of body water than men. Total body water also tends to decrease with age. The lower the total body water, the higher the concentration of ethanol, so the same amount of alcohol will produce greater effects in women and older people. Compare the percentage of total body water in different types of people in the table below:
Male Female
18–40 61% 52%
Over 60 51% 46%
Because it is both water- and fat-soluble, ethanol can also cross barriers very easily. Ethanol crosses the blood-brain barrier to reach synapses in the brain. It is also able to cross the placenta in pregnant women to reach the fetus, which can result in developmental disorders. We will discuss this in more detail later in this chapter in the section on adverse health risks.
11.2.3 Blood Alcohol Concentration The concentration of ethanol in the body is measured as blood alcohol concentration (BAC), also called blood alcohol content or blood alcohol level (BAL). BAC is defined as the number of grams of ethanol per 100 mL of blood. For example, a BAC of 0.05 is equivalent to 0.05 grams of ethanol per 100 mL of blood.
The legal limit for BAC while driving is 0.08, except in Utah where it is 0.05. For drivers under 21 years of age, most states have penalties for BAC above 0.02, although some states have stricter laws. BAC can be estimated by breathalyzer tests, which measure the amount of ethanol expelled through respiration. Although this is not identical to actual blood alcohol concentration, it is reasonably close in most circumstances and can be used as basis for arrest.
Below is a table of the effects of alcohol at different BACs. As you can see, although the legal limit is 0.08, impairment occurs at lower concentrations. Take a moment to study the contents of the table before moving on.
BAC Physical and Behavioral Effects
0.03 Relaxation, happiness, mild motor impairment
0.05 Lowered alertness, euphoria
0.10 Slowed reaction time, impaired motor function, slurred speech
0.15 Impaired balance, movement, judgment, and perception; blurred vision
0.20 Depressed sensory and motor capacity, difficulty keeping eyes open, extremely slurred speech, double vision
0.30 Stupor, confusion, inability to stay awake
0.40+ Respiratory depression, circulatory collapse, coma, death
11.3 Metabolism and Excretion
Ethanol is metabolized in the stomach before absorption and the liver afterwards. Ethanol metabolism is a complex topic, so we will devote more time than usual to address it. There will be a lot of new terms in this section, so make sure to pause and check your understanding every so often.
By the end of this section, you should be able to:
• Explain the kinetics of ethanol elimination. • Describe how ethanol is metabolized and discuss differences in metabolism by gender. • Explain genetic polymorphisms and describe the effects and prevalence of atypical ADH
and ALDH enzymes. • Explain how disulfiram is used in the treatment of alcohol use disorder. • Describe the microsomal ethanol-oxidizing system and its effect on ethanol kinetics.
11.3.1 Metabolism of Ethanol Most of the ethanol ingested (around 90–95%) is metabolized by the body. The remainder is excreted as-is in urine, feces, and other secretions. Some of the non-metabolized ethanol escapes via the lungs, which is why breathalyzers can measure BAC indirectly.
Ethanol is eliminated by zero-order kinetics. All the drugs we have covered so far followed first-order kinetics and have half-lives. Ethanol does not have a half-life because a fixed amount of it is metabolized per unit of time (note the relatively flat and parallel slopes of the graphs below). Therefore, drinking more alcohol does not speed up metabolism—with a few notable exceptions that we will get to later in this section.
Metabolism of ethanol follows two important steps. The first step involves converting ethanol into acetaldehyde. Acetaldehyde is a toxic substance that contributes to the negative symptoms associated with hangovers and is a known carcinogen. The enzyme responsible for the reaction is alcohol dehydrogenase (ADH) and is found in the stomach and liver. The activity of ADH is rate-limited, making ADH the reason why ethanol follows zero-order kinetics.
Acetaldehyde is then converted to acetic acid by aldehyde dehydrogenase (ALDH), which is not rate-limited and metabolizes acetaldehyde rapidly. The metabolite produced, acetic acid, is much less harmful than acetaldehyde and is eventually broken down into carbon dioxide and water.
Organizing key terms in ethanol metabolism Before moving on, let’s address a practical matter: distinguishing between these terms can be difficult without a background in chemistry. Still, this is key information that needs to be memorized. To distinguish the terms and remember their meanings, it may be useful to use logic to organize them and place them in order. First, remember that alcohol is metabolized by alcohol dehydrogenase. This should be simple because they both have refer to alcohol by name. Next, note how acetaldehyde is metabolized by aldehyde dehydrogenase. Acetaldehyde is an aldehyde, which is why it is included is both terms. In a similar vein, acetic acid is a metabolite of acetaldehyde. Finally, remember that ADH comes before ALDH in the process because it has fewer letters. These patterns and mnemonics should help you link these terms together.
Ethanol metabolism differs by gender. Overall, men metabolize more ethanol than women. In men, ADH is present in the stomach lining and can reduce ethanol absorption by up to 30%. In comparison, women have little to no ADH in their stomach walls. Women have less ADH in the liver as well. Between these differences and the differences in total body water mentioned last section, women get higher BACs than men when ingesting the same amount of ethanol.
11.3.2 Genetic Polymorphisms of ADH and ALDH In biology, you probably learned about DNA, genes, and mutations. Mutations occur when a section of DNA gets randomly changed. The change may be beneficial, harmful, or even fatal, but mutations are rare occasions. A genetic polymorphism is like a mutation, except the changes appear more often in the population (at least 1%). One type of polymorphism, called a single nucleotide polymorphism (SNP), occurs when a single unit of DNA, known as a nucleotide, is substituted with a different nucleotide.
Genes, or functional sections of your DNA, are responsible for enzyme expression. In this context, you can think of a gene as a set of instructions for how to manufacture a particular enzyme. When a gene has a polymorphism, some of the instructions are substituted for different instructions. If the polymorphism is a SNP, only one instruction (nucleotide) is substituted. This results in an enzyme with a slightly different characteristic.
The ADH enzyme actually comes in 7 different types in humans, labeled ADH1, ADH2, etc. Each enzyme is encoded by its own gene. The gene that encodes ADH2 has SNPs that produce variants of the ADH2 enzyme. The typical enzyme is listed as ADH2*1, while the variants are ADH2*2 and ADH2*3. Examine the table below to see how they compare:
Enzyme Metabolism Rate Ethnic Differences
ADH2*1 Normal
ADH2*2 Fast Very common in Asians (90%); some Jewish people (20%)
ADH2*3 Fast Some African-Americans (15–25%)
As you can see, the variants metabolize ethanol at a faster rate than normal. Although this sounds like an advantage, it results in a buildup of acetaldehyde. This can cause flushed skin, headaches, and a sensation of warmth. Because there is less of the desired ethanol and more of the toxic acetaldehyde, people with fast-acting ADH2 tend to drink less and have a reduced risk of AUD.
Similar to ADH, there are multiple ALDH enzymes in humans—a whopping 19 have been discovered, some with names like ALDH1L2. Fortunately for us, we only care about the ALDH2 enzyme. The gene encoding this enzyme is polymorphic, with two variants (see table below):
As you can see, the atypical ALDH2*2 enzyme metabolizes acetaldehyde very slowly. In fact, it is so slow that it is nearly inactive. Similar to the atypical ADH enzymes, this causes a buildup of acetaldehyde and the associated symptoms, except significantly worse. As a result, people with ALDH2*2 tend to have very low rates of alcohol use and AUD.
Comparing the two tables above, you may notice that Asians are at an increased risk of both ADH2*2 and ALDH2*2. They are prone to exhibiting the “Asian flush” when consuming alcohol. Because of this, Asians as a whole tend to exhibit lower rates of alcohol use and AUD, and those that have both experience extreme acetaldehyde toxicity. Of course, Asians are a broad group, so there are some differences by region and culture as well.
One final note about ALDH: some drugs, such as disulfiram (Antabuse®), are capable of blocking ALDH enzymes. When these drugs are administered, they cause the same effects as ALDH2*2, causing a surge in acetaldehyde levels and unpleasant hangover symptoms. As such, these drugs can be used in treatment of AUD to punish relapse and make it less likely to occur again in the future.
Enzyme Metabolism Rate Ethnic Differences
ALDH2*1 Normal
ALDH2*2 Very slow Common in Asians (40–50%)
11.3.3 Microsomal Ethanol-Oxidizing System There is one final complication to ethanol metabolism that we need to cover. (I know—as if it wasn’t complicated enough already, right?) So far, when we have talked about ethanol metabolism, we have assumed that all alcohol is metabolized by ADH. This is not always true. When ADH is saturated, the microsomal ethanol-oxidizing system or MEOS is activated.
The MEOS uses part of the cytochrome P450 enzyme family, in particular, CYP2E1, to oxidize excess ethanol into acetaldehyde, same as ADH (see image below). Unlike ADH, however, the MEOS follows first-order kinetics. This means that as BAC increases, more and more ethanol is oxidized by the MEOS, and overall metabolism is increasingly first-order. ADH is still functioning at maximum capacity during this time, so the rate is a mix of zero- and first-order kinetics.
11.4 Ethanol Pharmacodynamics
Now that we have covered the pharmacokinetics of ethanol, it is time to move on to its pharmacodynamics. Ethanol has a wide range of effects beyond just CNS depression. We will begin this section by examining its mechanisms of action before moving on to dependence and effects on other systems.
By the end of this section, you should be able to:
• Describe the pharmacodynamics of ethanol and its effect on the central nervous system. • Explain ethanol tolerance, dependence, and withdrawal. • Describe the effects of ethanol on other systems in the body.
11.4.1 Receptors and CNS Effects Ethanol is considered a “dirty drug,” an informal term meaning it affects many different receptors in the body and can produce a wide range of effects. The primary depressant mechanism of ethanol occurs at the GABAA receptor. The exact place that ethanol binds to is unknown, as is its exact effect on chloride ion channels, but it enhances the effects of GABA and causes hyperpolarization.
CNS depression may also be the result of ethanol binding to NMDA receptors for glutamate and reducing the flow of sodium and calcium ions into the cell. This lowers the excitatory effect of glutamate on NMDA receptors and enhances neuronal inhibition in the CNS. See the images below to compare these two mechanisms:
The effects of ethanol are biphasic. At low doses, consumption of alcohol can elicit relaxation, giddiness, and euphoria. This is known as a buzz and is what makes initial drinking feel good. As BAC rises, depressant effects start to kick in. Motor coordination is impaired, speech becomes slurred, and sensory input is diminished. At high doses, ethanol can induce stupor, sleep, respiratory depression, coma, and death. (For a full list of effects at different BACs, check the table under the BAC subsection 11.2.3 above.)
Ethanol interacts with many drugs due to its widespread affinity for different receptors and pharmacological properties. You may have noticed that many of the drugs we have covered so far have adverse interactions with ethanol. Indeed, it is the most important drug interaction in the CNS and often causes side effects when paired with medications and recreational drugs. Some of the common effects of drug interactions with ethanol are dizziness, nausea and vomiting, coma, and death.
11.4.2 Ethanol Dependence Chronic ethanol use results in both tolerance and dependence. Tolerance develops in multiple ways. Standard pharmacodynamic tolerance occurs, with involves changes in target receptor regulation (neuroadaptation). Long-term exposure to ethanol causes the receptors to downregulate and become less responsive.
Pharmacokinetic tolerance also develops with heavy drinking. Recall that when ADH is saturated, the MEOS kicks in and CYP2E1 starts oxidizing excess ethanol. Unlike ADH, CYP2E1 and other cytochrome P450 enzymes can be induced, which results in faster metabolism of ethanol. This only occurs when ADH is saturated however, so this type of tolerance only affects binge drinkers and other heavy users.
Finally, becoming familiar with the effects of inebriation leads to behavioral tolerance. This is a conditioned response to the drug that allows the user to compensate for its behavioral effects. People with behavioral can appear sober even after drinking a significant amount of alcohol and may have better control over their speech, movement, appearance, and emotional state compared to inexperienced drinkers.
Like other drugs, ethanol causes dependence by altering dopamine transmission in the reward system. Normally, dopaminergic neurons in the VTA are under tonic inhibition by GABA interneurons. This means that ambient GABA activity somewhat inhibits these neurons from firing and releasing dopamine in the nucleus accumbens.
When ethanol is ingested, β-endorphin is released. This activates μ-opioid (mu-opioid) receptors on the GABA interneurons and inhibits the tonic release of GABA. This in turn depolarizes the dopaminergic neurons and increases dopamine release in the nucleus accumbens. See the image below for the full process:
Ethanol withdrawal can be very dangerous and last for days. Because chronic use decreases the number of GABAA receptors and increases NMDA receptors, CNS activity is significantly increased during withdrawal. Symptoms can include seizures, agitation, anxiety, increased heart rate and blood pressure, insomnia, nausea and vomiting, and paranoia. During withdrawal, delirium tremens may occur, which involves shaking, confusion, and hallucinations and may lead to death. Delirium tremens usually occurs a few days after the onset of withdrawal and may last for two to three days.
Because ethanol withdrawal is so dangerous, treatment is usually intensive and requires hospitalization. Sedatives such as benzodiazepines are administered to mitigate withdrawal symptoms. Of those with severe symptoms, up to 15% may die from withdrawal (Simpson, Wilson, & Nordstrom, 2016).
11.4.3 Effects on Other Systems Ethanol has considerable effects on systems in the body outside of the CNS. The first and foremost is the liver. The majority of ethanol is metabolized in the liver, which leads to a buildup of acetaldehyde in the liver. As mentioned before, acetaldehyde is toxic and can cause damage to the liver. Consuming large amounts of alcohol regularly will lead to a gradual deterioration of liver functioning known as alcoholic liver disease.
The initial stages of liver disease involve a buildup of excess fat in the liver, known as fatty liver disease or hepatic steatosis. Fatty liver disease can be reversed with changes to alcohol consumption and diet, but if consumption continues, scar tissues may form in a process known as fibrosis. The scar tissue interferes with normal liver functioning and is permanent. When large amounts of scar tissue develop, the person now has cirrhosis; at this stage, the scar tissue has grown and is killing healthy liver cells.
Ingestion of ethanol also affects the gastrointestinal system in a variety of ways. Low doses of ethanol accelerate gastric emptying, while moderate consumption stimulates digestive juices and acid secretion. At high doses, ethanol damages the lining of the stomach and causes inflammation, while also reducing the digestive system’s ability to absorb nutrients.
The endocrine system, which is a series of glands that secrete hormones, is also altered by ethanol use. Ethanol increases blood sugar levels, which eventually results in the over-production of insulin and a subsequent decrease in blood sugar. Disruption in calcium supply
results in decreases in bone density, called osteoporosis. Inhibition of release of vasopressin, a pituitary hormone that promotes water retention by the body, can result in excessive amounts of urine. Additionally, in men, a loss of testosterone can result in erectile dysfunction.
Finally, excessive ethanol consumption may suppress the immune system. This manifests as a decrease in T cells, increased memory T cells, and higher rates of induced cell death. A weakened immune system causes increased susceptibility to bacterial infections such as pneumonia, tuberculosis, hepatitis, and septicemia.
1.5 Adverse Health Risks
To wrap up this chapter, we will consider some of the adverse health risks associated with ethanol use. Of course, some of the effects mentioned in the previous section are related to the risks mentioned here. While this is not a comprehensive list, it should give you some idea of how alcoholism can have far-reaching consequences beyond addiction.
By the end of this section, you should be able to:
• Explain what cancers are associated with alcohol use. • Describe Wernicke encephalopathy and Korsakoff syndrome. • Describe the causes and symptoms of fetal alcohol syndrome.
11.5.1 Alcohol-Related Cancers As mentioned before, acetaldehyde is a carcinogen. Countless studies have linked the consumption of alcohol to increased risks for developing various cancers. This link is mediated by the level of exposure to acetaldehyde caused by the metabolization of ethanol.
The risk of cancer increases in all areas of the gastrointestinal tract, such as the mouth and upper throat, esophagus, stomach, and bowels. The larynx is also at an increased risk for developing cancer, as is the liver (which should not be a surprise given how it is exposed to acetaldehyde). Finally, compared to women who don’t drink, women who have at least three drinks per week have a 15% higher risk of breast cancer.
11.5.2 Wernicke-Korsakoff Syndrome One of the common side effects of alcoholism is vitamin deficiency. People who drink heavily often have nutritional deficits on top of the effects of ethanol. A particularly common deficit is thiamine (vitamin B1) deficiency. This can be caused by malnutrition, decreased absorption of thiamine by the gastrointestinal tract, a reduction in stored thiamine due to liver disease, or a combination of these factors.
Thiamine deficiency is linked to two separate but related neurological disorders. The first is Wernicke encephalopathy (encephalopathy literally means “disease of the brain”). There are three characteristic symptoms associated with Wernicke encephalopathy: confusion,
ophthalmoplegia, and ataxia. Ophthalmoplegia refers to paralysis of eye muscles (the -plegia suffix is also used in paraplegic), while ataxia is general lack of motor control. These symptoms are the result of degenerative changes to the thalamus and hypothalamus.
The second disorder is Korsakoff syndrome, sometimes referred to as Korsakoff psychosis. The main symptoms are amnesia (both retrograde and anterograde) and confabulation, or distorted memories that the patient is confident in despite being bizarre or false. As you would expect, these symptoms are caused by damage to the hippocampus and other areas of the brain associated with memory.
Mnemonic for Wernicke-Korsakoff syndrome Having trouble keeping track of the different symptoms for Wernicke encephalopathy and Korsakoff syndrome? Just remember these two words: COAT RACK. The first three letters in COAT stand for the key symptoms of Wernicke encephalopathy: confusion, ophthalmoplegia, and ataxia. The T stands for thiamine, the vitamin that is deficient in both disorders. Likewise, the first three letters in RACK stand for the symptoms of Korsakoff syndrome: retrograde amnesia, anterograde amnesia, and confabulation. The K lets you know that these apply to Korsakoff syndrome (and makes the mnemonic stick).
Although both are related to thiamine deficiency, it is possible to have one and not the other. Wernicke encephalopathy can occur in people without a history of alcohol use, and people can develop Korsakoff syndrome without having ever exhibited the former. Still, the two disorders do often overlap, in which cases they are simply referred to together as Wernicke-Korsakoff syndrome. Generally, the symptoms related to Wernicke encephalopathy will appear first, then subside as the symptoms of Korsakoff syndrome emerge. If left untreated, 75% of people suffering from Wernicke-Korsakoff syndrome end up with permanent brain damage and about 20% die.
11.5.3 Fetal Alcohol Syndrome Recall that in a previous section, it was noted that ethanol can cross the placenta. What happens if this occurs? In the worst case, assuming the fetus does not die, it may be born with a significant developmental disorder called fetal alcohol syndrome or FAS.
Fetal alcohol syndrome is part of a larger category of fetal alcohol spectrum disorders (FASD) and is the most severe disorder. It is estimated that 2–5% of school-aged children were born with FAS. FAS can be identified by distinct facial characteristics such as wide-set eyes with narrow openings, a short nose, thin upper lip, and a smooth philtrum (the ridge above the upper lip). Growth is delayed, resulting in reduced weight, height, and head circumference. There may be deformities in joints, limbs, and fingers as well as defects in the heart, kidneys, or bones.
FAS results in neurological underdevelopment as well. People born with FAS may have difficulty with learning and memory and have a higher risk of psychiatric problems, especially ADHD and
conduct disorders. These challenges may serve as significant obstacles to normal living; 80% of individuals with FAS reported having difficulties with employment and independent living.
What causes FAS? A fetus has an undeveloped liver that is not properly equipped to metabolize ethanol. As a result, even very small concentrations of ethanol can build up and cause toxic effects. At the same time, the gestation period is essential for development, so even small amounts of toxins can cause lasting and irreparable harm. There is no safe level of alcohol consumption during pregnancy—abstinence is necessary to ensure normal development. As such, alcoholic beverages sold in the U.S. must contain a label warning that pregnant women should not drink alcoholic beverages due to the risk of birth defects.
Chapter Summary and Review
In this chapter, we took a deep dive into alcohol, also known as ethanol. We described its history, legislation, prevalence, and how it is measured. We learned about the pharmacokinetics of alcohol, starting with its absorption and distribution before exploring factors that influence its metabolism. We examined its mechanisms of action, effects in the CNS and elsewhere, and certain heath risks associated with its use.
That’s all for this chapter and this unit. There was a lot of information in this chapter, so make sure to take your time to study it properly. Best of luck, and don’t hesitate to reach out to your instructor if you have any questions.
Practice Questions
• Do adults engage in riskier drinking behaviors than adolescents? • If an alcoholic beverage is 90 proof, what is its percent ABV? • On average, how many standard drinks will put you over the legal driving limit? • Name three reasons why women tend to have higher BACs from the same amount of
alcohol compared to men. • At what BAC does mild motor impairment begin? • Jamie is out drinking and says that he is seeing double. What is his BAC likely at? • What percentage of ethanol is metabolized in the body? • Which enzyme polymorphism metabolizes acetaldehyde at a near-inactive rate? • What does disulfiram do, and how does it work? • Annette says that alcohol always follows zero-order kinetics. How would you respond to
her? • What are the three types of tolerance to alcohol that one can develop? • Explain how the ingestion of ethanol ends up increasing dopamine levels in the nucleus
accumbens. • What are the stages of liver disease? • Name five parts of the body that see an increased risk of cancer due to alcohol use. • What are the symptoms of Wernicke-Korsakoff syndrome? Be sure to distinguish
between the two disorders. • Is it possible to safely drink alcohol during pregnancy?
References
Benjah-bmm27. (2007, January 21). Ethanol molecule model [Illustration]. Wikimedia
For this chapter, we will discuss a class of drugs that shares many similarities to opioids. Both opioids and cannabinoids are drugs that are extracted from plants with a long history of use, both act on endogenous receptor systems named after the exogenous drugs, and both have largely inhibitory effects on neural activity. Both have also been under intense scrutiny in recent decades, although for different reasons. Whereas opioids have invaded the public awareness through the opioid epidemic, most people are probably familiar with cannabis, or marijuana, because of modern efforts to legalize the drug in the U.S. In this chapter, we will examine the key substances in cannabis and learn about where the effects of marijuana come from, as well as how its medical and recreational use has been changing in recent years.
Chapter Outline: 13.1 Cannabinoids and Overview
13.1.1 The Cannabis Plant
13.1.2 History and Legislation
13.1.3 The Endocannabinoid System
13.2 Pharmacology 13.2.1 Administration and Pharmacokinetics
13.2.2 Mechanisms of Action and Effects
13.2.3 Tolerance, Withdrawal, and Long-Term Effects
13.2.4 Medical Marijuana and Cannabinoid Medications
1st edition
13.1 Cannabinoids Overview
A cannabinoid is a biologically active compound found in cannabis. There are over 100 such substances that can be isolated from cannabis, only some of which are psychoactive. In this chapter, we will focus on two of the most common cannabinoids and compare the effects of cannabis containing them in different amounts.
By the end of this section, you should be able to:
• Describe the cannabis plant and identify its main active ingredients. • Differentiate between hemp and marijuana. • Compare and contrast Δ9-tetrahydrocannabinol (THC) and cannabidiol (CBD). • Describe the history of marijuana. • Explain the endocannabinoid system and define its primary receptors and transmitters.
13.1.1 The Cannabis Plant All cannabinoids originate from the resin produced by the Cannabis genus of plants, which is often differentiated into three variations. Cannabis sativa strains tend to have long, thin leaves and can grow up to three meters tall in hot climates. In comparison, the Cannabis indica variety has wider leaves and shorter, bushier plants that grow better in cold climates. Cannabis ruderalis is the shortest variety and is noted for being heartier and easier to grow.
1st edition
Source: Craft Sense (2018)
Although these variations are often presented as distinct species, in reality, there is a large amount of overlap. Most strains of cannabis are hybrids have undergone crossbreeding to enhance particular properties (e.g., to produce a stronger high, or to be easier to grow indoors). For example, Cannabis ruderalis is notable for being autoflowering, meaning that it will flower according to age and takes a shorter period of time before harvesting. Cannabis sativa and Cannabis indica are photoperiod flowering, dependent on the light cycle, and take a longer period of time before harvesting. Despite its low THC content, Cannabis ruderalis is frequently crossbred with other strains to transfer the autoflowering trait and increase the yield. As a result, it is often more useful to distinguish between different strains by examining the type and concentration of cannabinoids each contains.
Perhaps the most notable cannabinoid is delta-9-tetrahydrocannabinol, also referred to as Δ9-THC or simply THC. (Although there are other tetrahydrocannabinols, when referring to THC in this chapter we will be specifically talking about Δ9-THC). This is the main psychoactive component found in cannabis and is typically sought after for recreational use.
Another well-known cannabinoid is cannabidiol or CBD. Unlike THC, CBD is non-psychoactive; although it can still interact with the nervous system, it does not produce psychoactive effects. In fact, its presence may actually counteract some of the effects of THC (more on that later). Because of this, CBD is used more often in therapeutic medications.
1st edition
As THC is the main psychoactive cannabinoid, another way of classifying cannabis strains is by THC content. Cannabis with low THC content (less than 0.3%) is also called hemp and is non-psychoactive and federally legal. Hemp has many industrial uses; its fibers are used in rope, clothing, and textiles, and it can also be processed into foods, bird seed, and even biofuel. If the THC content is higher than 0.3%, it is, instead, called marijuana. Because it is psychoactive, marijuana is only legal in some states and is used primarily for its psychoactive affects. See the table below for a comparison of the two.
Hemp Marijuana
Less than 0.3% THC More than 0.3% THC
Non-psychoactive Psychoactive
Federally legal Only legal in some states
CBD > THC THC > CBD
Available online and in many places
Available at licensed dispensaries in legal states
Used in clothing, textiles, foods, bird seed, and more
Used for recreational and therapeutic purposes
The THC content of marijuana is not fixed and has, in fact, increased in recent decades. In the 1970s, the average THC content was 1–4%, but this has risen to 10–15% in recent years, with some strains containing as high as 30% THC content. This increase in potency is caused by changes in growing methods that selectively breed strains with higher THC content that produce more resin.
One final thing worth noting is that it is possible to artificially manufacture substances that produce similar effects as cannabinoids. These compounds are known as synthetic cannabinoids and can have significantly different effects compared to cannabis. To distinguish the two types, cannabinoids extracted from plants are called phytocannabinoids (the phyto- prefix means “plant”).
13.1.2 History and Legislation The history of cannabis is much like the history of opium poppies and coca leaves. The earliest written record of cannabis use occurred in 2700 BC, when Chinese emperor Shen Nung (or Shennong) included it in a book of medicinal herbs. Cannabis originated in central Asia and India, but its cultivation and use spread throughout the world. In 200 BC, cannabis was used in ancient Greece as a remedy for earaches, edema, and inflammation. From 800–1500 AD, cannabis was used as medicine in the Arabian Peninsula, where it was also used as hashish for its mood-changing properties.
1st edition
The history of cannabis in the U.S. dates all the way back to the 17th century, where it was brought over by settlers and grown in the colonies for hemp products. Marijuana saw use as a medicine in the 1800s and early 1900s and was an ingredient in many medicines and tinctures at the time. Marijuana was also commonly smoked for recreational purposes at the time.
The legal status of marijuana changed in 1937 with the passage of the Marihuana Tax Act. This act effectively outlawed the recreational use of marijuana and placed restrictions on its trade. It also legitimized the term marijuana to refer to cannabis products, as it was previously only a slang term for cannabis. Because of the tax imposed by the act, medical use of marijuana also declined.
In 1970, the Controlled Substances Act classified marijuana as a Schedule I drug with no accepted medical use, meaning it was illegal to prescribe it for medical treatment. The initial placement of the drug in Schedule I was intended to be provisional because of the lack of studies on its safety, but in 1972, the Nixon administration refused to reschedule marijuana despite the report of a national commission recommending its decriminalization.
The Schedule I status of marijuana has been repeatedly challenged in subsequent decades, although to this date it is still federally classified as an illegal drug. Despite this, some exceptions have been carved out over the years, and state legislatures have passed laws legalizing its use. California was the first state to legalize medical cannabis in 1996, while Washington and Colorado were the first to legalize recreational use in 2012. In 2018, the Hemp Farming Act formally differentiated between hemp and marijuana using the THC content threshold of 0.3% mentioned above, effectively legalizing certain cannabinoids at the federal level for the first time.
13.1.3 The Endocannabinoid System Similar to opioids, the effects of cannabis were well-established long before we knew how and why cannabis was capable of interacting with the brain. The eventual discovery of an endogenous system of cannabinoid receptors and ligands, referred to as the endocannabinoid system, did much to further our understanding of how cannabis achieves its effects.
Two types of cannabinoid receptors have been identified: the cannabinoid receptor type 1 (CB1) and cannabinoid receptor type 2 (CB2). Both are G-protein coupled receptors. CB1 receptors are found throughout the CNS and peripheral tissues, while CB2 receptors are largely located outside of the nervous system on immune cells. The pharmacological effects of cannabinoids are mainly determined by their affinity and efficacy for CB1 receptors, although there may be additional types of cannabinoid receptors not yet identified that account for other effects of cannabinoids.
Endogenous ligands for these receptors are called endocannabinoids. The first endocannabinoid discovered was named N-arachidonylethanolamine (AEA) or anandamide after the Sanskrit word Ananda, meaning joy or bliss. The other primary endocannabinoid is 2-arachidonoylglycerol, or 2-AG. Anandamide is an agonist of the CB1 receptor, while 2-AG is an agonist of the CB2 receptor, although the two have similar chemical structures and can interact with both receptor types.
Anandamide and 2-AG are synthesized on demand and released almost immediately into the synaptic space. They tend to have short half-lives and are quickly metabolized by enzymes.
1st edition
Anandamide is metabolized by fatty acid hydrolase (FAAH) into arachidonic acid, while 2-AG is metabolized by monoacylglycerol lipase (MAG-L) into fatty acids and glycerol.
Endocannabinoids act as retrograde neurotransmitters at many synapses; instead of being released by the presynaptic neuron, endocannabinoids are released by the postsynaptic neuron, whereupon they travel across the synapse in the opposite direction of most neurotransmitters and bind to CB receptors on the presynaptic neuron (see image below). This reduces neurotransmitter release, causing an inhibitory effect.
As such, the endocannabinoid system is an important method of neuromodulation. Although our understanding of the endocannabinoid system is still evolving, preliminary research suggests that the system is involved in many functions, such a regulating appetite, sleep, mood, and pain. Scientists believe that the main role of the endocannabinoid system is the maintenance of homeostasis. The endocannabinoid system also seems to play an important role in forgetting by suppressing long-term potentiation in the hippocampus.
1st edition
13.2 Pharmacology
As mentioned in the previous section, cannabinoids are any biologically active substances found in cannabis. Because there are numerous cannabinoids with varying properties, in this section, we will look at THC and CBD in particular. When talking about the psychoactive effects of cannabis or marijuana, remember that this is due to THC, its psychoactive component, and not due to CBD, which is non-psychoactive.
By the end of this section, you should be able to:
• Describe the pharmacokinetics of marijuana. • Describe the physiological and behavioral effects of marijuana. • Discuss tolerance and dependence on marijuana. • Describe long-term adverse and toxic effects of marijuana. • Define medical marijuana and explain the use and origin of Charlotte’s Web. • Describe the approved cannabinoid drugs and their clinical uses.
13.2.1 Administration and Pharmacokinetics Cannabis is most often inhaled or ingested. Cannabis can be rolled into a cigarette or joint, or it can be smoked with the aid of hookahs or bongs. Second-hand exposure to the smoke can result in a contact high. Oral administration occurs when eating hash oil or preparations that contain cannabis, such as brownies.
When smoked, about 50% of the THC content is absorbed, with the remainder being released into the environment. Once absorbed into the bloodstream by the lungs, onset of effects and peak concentrations of THC occur within minutes, with maximal effects occurring within half an hour. The duration of the effects usually last for 1–3 hours depending on the amount administered.
In comparison, oral ingestion results in a higher rate of absorption of THC (about 90–95%), but due to first-pass metabolism in the liver, an overall lower bioavailability of around 6–20%. In addition, the effects are much more delayed compared to smoking; onset occurs after 30–90 minutes, while peak effects may not occur until 2–3 hours after administration. Duration is prolonged, with effects lasting for at least 4 hours. Compare the two routes below:
Inhalation Ingestion
Absorption 50% THC 90–95% THC
Bioavailability 10–35% THC 6–20% THC
Onset of Effects < 10 min 30–90 min
Maximal Effect 15–30 min 2–3 hr
Duration 1–3 hr 4+ hr
1st edition
Because it is lipid-soluble, THC can cross the blood-brain barrier and is readily throughout body tissue. Metabolism of THC occurs primarily in the liver through cytochrome P450 enzymes, in particular CYP2C9, which converts Δ9-THC into 11-OH-Δ9-THC (often referred to simply as 11-hydroxy-THC). An active metabolite itself, 11-hydroxy-THC has similar effects to THC and is, in turn, metabolized into glucuronide conjugates and other inactive metabolites before being excreted.
THC tends to have a half-life of around 20–50 hours, although it can be detected for days or up to a month after administration. This is in part because it can accumulate in fat cells and is gradually rereleased into the bloodstream over time. Most of the drug (more than 65%) is excreted in the feces, with about 20% excreted through the urine.
13.2.2 Mechanism of Action and Effects The effects of cannabinoids are largely mediated by the CB1 receptor. THC is a partial agonist with a high affinity for the CB1 receptor, similar to the endocannabinoid anandamide. Some synthetic cannabinoids have a much higher affinity for the CB1 receptor compared to anandamide and THC. As mentioned previously, there may be additional cannabinoid receptors responsible for some of the effects of THC that are being investigated or yet to be discovered.
CBD is unique in that it may counteract the effects of THC. This is because CBD is a negative allosteric modulator of the CB1 receptor. In other words, when CBD binds to the CB1 receptor, it changes the shape of the receptor so that THC cannot bind to it as easily. At the same time, CBD also interacts with the receptors for other neurotransmitters, such as serotonin, and may enhance the effects of THC in some cases.
Because the endocannabinoid system is found throughout the CNS and peripheral tissue, administration of THC produces a variety of effects. Perhaps the most well-known physiological effect is an increase in appetite, also known as the munchies. THC also increases heart rate and blood pressure, reduces motor activity, and dilates the airways and blood vessels in the conjunctiva, a part of the eye. It can also cause drowsiness, dizziness, dry mouth and throat, and reduce restful sleep by suppressing REM sleep.
1st edition
Source: Mikael Häggström (2009)
Cannabis also has effects on the endocrine system and the release of sex hormones. In men, the release of testosterone is decreased, which can result in erectile dysfunction and reduced sperm count and motility. In women, a decrease in progesterone can result in reduced sexual pleasure, disruption of menstrual cycles, reduced fertility, and an increased risk of premature birth.
Subjective effects of marijuana are dose-dependent and generally follow three stages. The buzz occurs at low doses and is characterized by feelings of dizziness, light-headedness, and tingling sensations. This is usually followed by the high, which may entail euphoria, exhilaration, and disinhibition. In first-time users or at very high doses, this may also result in anxiety, paranoia, and panic attacks. The effects on mood may be influenced by the setting, with pleasant settings causing positive effects and unpleasant settings causing negative effects. Finally, the stoned phase involves relaxation and altered perception; users often report enhanced senses and the feeling that time passes more slowly.
Marijuana also interferes with judgment and short-term memory, which can increase risky behaviors and make it difficult to retain information. High doses of marijuana can cause paranoia and psychosis. Marijuana causes dose-dependent impairment of psychomotor activity, which results in decreased alertness, concentration, coordination, and reaction time. At low doses there is relatively little effect, but the impairment increases with larger doses. In addition, combining marijuana with alcohol produces an additive effect, which can cause severe impairment and increase the risk of a car crash.
13.2.3 Tolerance, Withdrawal, and Long-Term Effects Regular use of cannabis can cause tolerance to the behavioral and subjective effects of cannabis. If used frequently, withdrawal can occur upon stopping. Compared to other drugs we have examined, cannabis withdrawal is usually mild and rarely life-threatening. Symptoms include craving, irritability, anxiety, lack of appetite, sleep disturbance, and depression. Some users may also experience physical symptoms such as tremors, sweating, headache, stomach pain, and fever. Withdrawal tends to last for up to two weeks, with peak symptoms occurring 2–6 days after cessation.
Activation of CB1 receptors causes an increase in dopamine release in the nucleus accumbens, although the exact mechanism through which this occurs is poorly understood at the time. This increase in dopamine is responsible for some of the euphoric effects of cannabis. Although dependence is possible, it is not common, and some researchers believe that the dependence is psychological, rather than physiological in nature. Tolerance and dependence are more likely to develop with frequent use of strains that are high in THC content.
Although dependence and addiction are uncommon, they do occur. The DSM-V describes cannabis use disorder as a type of substance use disorder and lists 11 criteria to evaluate its severity. Similar to other substance use disorders, cannabis use disorder is characterized by cravings, preoccupation with obtaining cannabis, compulsive use despite negative consequences, and relapse after stopping use.
Frequent or heavy use of cannabis can cause many negative long-term effects, especially if use begins in adolescence. Addiction occurs in 9% of overall users; this number increases to 17% in those who began in adolescence and 25–50% of those who use cannabis daily. Early use is also associated with changes to brain development and cognitive impairment, as well as diminished life satisfaction and educational achievement.
Chronic cannabis use also increases the risks of certain respiratory diseases, such as emphysema and bronchitis. Although burning cannabis for smoking releases similar carcinogens to smoking cigarettes, cannabis use is not as strongly associated with an increase in lung cancer. This is because some compounds in marijuana may suppress tumor growth. Smoking cannabis can also increase cardiovascular problems if the user has a pre-existing heart problem.
13.2.4 Medical Marijuana and Cannabinoid Medications Although marijuana is most well-known for being a recreational drug, it is also sometimes used to treat diseases or other health conditions as medical marijuana. Although there have been many reports of marijuana’s potential therapeutic effects, the FDA has not approved marijuana as a medicine and the National Institute on Drug Abuse (NIDA) does not recommend using marijuana joints for therapeutic purposes. Some of the reasons cited are that it is an unpurified plant with cognitive impairing properties that, when smoked, is a known hazard to health.
Despite this, marijuana is often used to treat a variety of problems, and many states have legalized medical marijuana use. Common uses for marijuana include treatment of intraocular pressure in glaucoma (due to vasodilation of blood vessels in the conjunctiva), suppression of nausea and vomiting during chemotherapy, stimulation of appetite in cachexia (a type of ongoing muscle loss), and relief from multiple sclerosis symptoms. One of the most commonly
1st edition
reported uses of medical marijuana is for pain relief, especially neuropathic pain that is not as easily treated by traditional pain relievers.
Because cannabis that is low in THC is non-psychoactive, many medical uses of cannabis use strains that contain high amounts of CBD. Strains that are CBD-rich have equal or more amounts of CBD when compared to THC, while CBD-dominant strains or products have very little THC content. After the passage of the Hemp Farming Act in 2018, cannabis that contains less than 0.3% THC is considered hemp, not marijuana, and is federally legal. Despite this, CBD derived from marijuana is still classified as a Schedule I drug.
Certain conditions shave been reported to be responsive to CBD treatment, including schizophrenia, inflammatory disorders, social anxiety disorder, mental depression, and nausea and vomiting induced by chemotherapy. One of the first demonstrations of therapeutic CBD use was an oil extract called Charlotte’s Web, which was named after Charlotte Figi, a five-year-old child with Dravet syndrome (a rare and severe type of epilepsy) that was treated with it. To learn more about Charlotte Figi and her story, watch the video below:
CNN documentary on Charlotte's Web, medical marijuana treating seizure disorders [7:44]
Charlotte’s Web contained 17% CBD and only 0.3% THC. Although it was never an officially approved medical treatment for seizures, in 2018 the FDA approved Epidiolex®, an extract mixture of 99% CBD and less than 0.1% THC, for treatment of Dravet syndrome and another severe form of epilepsy, Lennox-Gastaut syndrome. Epidiolex® was the first FDA-approved drug containing a substance derived from marijuana.
Other cannabinoid medications have been released. Dronabinol (trade name Marinol®) was a synthetic THC approved in 1985 by the FDA as an appetite stimulant and antiemetic (anti-vomiting) drug. Another cannabinoid, nabilone (Cesamet®), was approved by the FDA in 2006 to treat neuropathic pain alongside its antiemetic properties; it is also a synthetic cannabinoid that mimics THC. Finally, in the early 2010s, several European nations approved nabiximols (Sativex®), an oral spray with approximately equal amounts of THC and CBD, for use in treating pains related to neuropathy, cancer, and multiple sclerosis. Nabiximols has not yet been approved by the FDA for use in the U.S. but has been studied in several Phase 3 clinical trials for treatment of cancer pain.
In this chapter, we learned about the cannabis plant and its two main components, THC and CBD, and compared the two. We covered the history of cannabis and marijuana use before moving on to the endocannabinoid system, where endocannabinoids use retrograde transmission to inhibit neural activity. We then covered the pharmacological properties of THC and described its pharmacokinetics, mechanism of action, effects, tolerance, and withdrawal. Finally, we discussed medical marijuana and the few cannabinoid medications that are FDA-approved.
Practice Questions
• What are the three variations of Cannabis plants? Briefly describe each. • How much THC content is required for cannabis to be classified as hemp? • What schedule is marijuana? • Describe retrograde transmission. How does this differ from typical neurotransmission? • Which route of administration has a higher bioavailability: inhalation or ingestion? • What effects do THC and CBD have on the CB1 receptor? • Name at least four ways that THC alters or impairs normal cognition. • THC content in cannabis has steadily increased in recent decades. Explain why this
could cause an increase in dependency and addiction. • Is smoking cannabis strongly associated with lung cancer? • Name three FDA-approved cannabinoid medicines and their clinical uses.
1st edition
References
Craft Sense. (2018, September 17). Sativa, Indica…Ruderalis?
For the final chapter of this unit, we will turn our attention to a broad group of drugs known as psychedelics. Although the so-called Psychedelic Revolution began in the 1960s, hallucinogenic substances have been used for thousands of years. In this chapter, we will examine three different types of psychedelics and some of the most notable examples of each.
Chapter Outline: 14.1 Hallucinogens
14.1.1 History and Overview
14.1.2 Administration and Pharmacokinetics
14.1.3 Mechanisms of Action and Effects
14.2 Mixed Stimulant-Psychedelics 14.2.1 History and Overview
14.2.2 Administration and Pharmacokinetics
14.2.3 Mechanisms of Action and Effects
14.3 Dissociatives 14.3.1 History and Overview
14.3.2 Administration and Pharmacokinetics
14.3.3 Mechanisms of Action and Effects
1st edition
14.1 Hallucinogens
The focus of this chapter is psychedelics, or drugs that cause altered states of consciousness. Psychedelics can be grouped into three broad categories, the first of which is hallucinogens, or drugs that mainly produce hallucinations. Some of the most well-known psychedelics are classified as hallucinogens, including magic mushrooms, mescaline, and LSD.
By the end of this section, you should be able to:
• Define psychedelics and hallucinogens. • Briefly discuss the history of natural and synthetic hallucinogens. • Describe the pharmacokinetic properties of LSD and other hallucinogens. • Differentiate between true hallucinations and pseudohallucinations. • Define the pharmacodynamic properties and effects of LSD and other hallucinogens.
14.1.1 History and Overview Hallucinogens have been used for thousands of years. Native American cultures used peyote, a type of cactus containing the psychoactive alkaloid mescaline, over 5000 years ago (El-Seedi et al., 2005). Other examples of early hallucinogens include psilocybin mushrooms (aka “magic mushrooms”) and plants containing dimethyltryptamine (DMT). Hallucinogens were been valued as a component of religious ceremonies because of their effects.
The modern history of hallucinogens is largely defined by the discovery of lysergic acid diethylamide, more commonly known as LSD. The psychoactive effects of LSD were accidentally discovered by Swiss chemist Albert Hofmann, who was isolating alkaloids in ergot, a type of fungus that grows on wheat, rye, and barley. While synthesizing ergot derivatives in 1943, Hofmann accidentally absorbed LSD through his fingers and came to learn of its hallucinogenic effects.
LSD was marketed as a psychiatric drug and soon became the subject of research. In the 1950s, the Central Intelligence Agency (CIA) distributed LSD to hospitals, prisons, and other institutions in order to study its effects, sometimes without the patients’ consent. The CIA was interested in LSD (and other drugs) as a potential interrogation tool to force confessions, although nothing came of the program.
Although psychiatric use of LSD declined due to reports of negative effects, recreational use increased. LSD was strongly associated with the counterculture movement of the 1960s and saw extensive use. Around the same time, psilocybin was being researched in experiments at Harvard University. Figures such as Timothy Leary and Aldous Huxley began to advocate for the use of hallucinogens to enhance consciousness and spirituality.
Throughout the late 1960s and early 1970s, public concerns about psychedelic drugs led to all hallucinogens being classified as schedule I drugs with no accepted medical use. As a result, research on LSD, mescaline, and other hallucinogens was stifled. These restrictions have been relaxed in recent years, however, and ongoing research on LSD has indicated potential therapeutic uses of the drug as a treatment for addiction, depression, and anxiety. If you are interested, the following video details some of these advances in our understanding of hos LSD works:
1st edition
The first modern images of a human brain on LSD [3:33]
14.1.2 Administration and Pharmacokinetics LSD, mescaline, and psilocybin are typically ingested orally, although LSD can be absorbed through the skin (as was the case when it was first discovered by Hofmann). DMT is typically smoked or administered orally by dissolving it in a drink.
When taken orally, LSD reaches peak concentrations a 30-40 minutes after ingestion with the duration of effects lasting from 6 to 12 hours. LSD is metabolized in the liver by cytochrome P450 enzymes into 2-oxo-3-hydroxy-LSD. The half-life is about 3 hours, matching the long duration of effect.
Compared to other hallucinogens, LSD is extremely potent. While the threshold dose of mescaline is about 100 mg, the threshold dose for LSD is only 20 micrograms, or 5000 times smaller. Finally, while we will not cover the pharmacokinetics of other hallucinogens in detail, it is worth noting that psilocybin is actually a prodrug; it is inactive but rapidly metabolized into psilocin, which is active and is responsible for the effects commonly attributed to psilocybin.
14.1.3 Mechanisms of Action and Effects Similar to many other psychedelic drugs, LSD produces its main effects by binding to serotonin (5-HT) receptors. Because it has a similar chemical structure to serotonin, LSD has affinity for various receptor subtypes, the most important being the 5-HT2A receptor. When binding to this receptor, LSD creates a sort of lid over the binding site that ensures LSD remains bound to the active site for a long time, which contributes to the drug’s long-lasting effects.
Other hallucinogens act at the same 5-HT2A receptor and produce similar effects to LSD; these include mescaline, psilocybin, and DMT. Compared to other 5-HT hallucinogens, LSD is unique in that it is also an agonist for dopaminergic receptors, in particular D2, which may play a role in its psychotic effects.
The main effects of hallucinogens are visual and auditory hallucinations and illusions, the experience of which is commonly referred to as a trip. Trips can involve sensory distortions, synesthesia, altered time perception, and out-of-body sensations. The experience is dependent on mood and setting and can be pleasant or unpleasant, leading to good or bad trips, respectively.
True hallucinations vs. pseudohallucinations Strictly speaking, the true definition of a hallucination is a perception that it totally unreal, such as hearing a voice that does not exist or seeing something that is not present. These are true hallucinations and are associated with psychosis and schizophrenia more often than drug use. Psychedelics tend to produce pseudohallucinations instead, which merely involve altered perceptions of real stimuli. Colors may appear more vibrant, for instance, and sounds may take on unique distorted qualities. Despite this, hallucinogens may cause true hallucinations, which tend to be associated more strongly with bad trips.
LSD is also capable of producing sympathomimetic effects. Recall from our previous discussion of stimulants that these are effects that mimic the sympathetic nervous system, such as increased heart rate and blood pressure, dilation of the pupils, and reduced appetite. LSD may also cause nausea, sweating, and ataxia (muscle weakness).
1st edition
Source: Mikael Häggström (2009)
There are no reports of direct fatalities due to high doses of LSD, meaning the major risk is that of a bad trip, which can cause paranoia and panic attacks. This can have long-term consequences, as users may experience flashbacks to good or bad trips years after LSD use. When such flashbacks are pervasive and negative, it is known as hallucinogen persisting perception disorder (HPDD). Tolerance for the drug’s effects develops after 3–6 days of regular use and results in reduced intensity of hallucinations and other effects. Cross-tolerance will also develop to other 5-HT hallucinogens, which includes psilocybin, mescaline, and DMT. There is no evidence for physical dependence, which means that there is also no withdrawal syndrome following discontinuation of drug use. Some users may become psychologically dependent on the effects of LSD, but chronic use does not result in cravings or addiction.
In cases where antidotal therapy is required, the effects of LSD can be reversed with the administration of chlorpromazine, an antipsychotic medication. As you may expect, it is an antagonist at 5-HT2A and D2 receptors. We will examine chlorpromazine in more detail when discussing antipsychotic medications in Chapter 17.
The next category of psychedelics that we will cover are the mixed stimulant-psychedelics. As the name suggests, these are drugs that cause both stimulant and psychedelic effects. Although there are many drugs in this category, the most notable one is MDMA, which will be the focus of this section.
By the end of this section, you should be able to:
• Define mixed stimulant-psychedelics, empathogens, and entactogens. • Describe the pharmacokinetic properties of MDMA. • Describe the pharmacodynamic properties and effects of MDMA.
14.2.1 History and Overview Mixed stimulant-psychedelics are also called empathogens or entactogens. An empathogen is that enhances empathy, while an entactogen increases feelings of connectedness with others. Both terms were coined to describe MDMA and similar drugs that caused elevated levels of empathy and can be used interchangeably. These drugs are actually substituted amphetamines, similar to methamphetamine but have more prominent empathogenic effects..
MDMA was first developed in 1912 by the drug company Merck, although it was not popularized until the 1970s, when psychopharmacologist Alexander Shulgin developed easier methods to synthesize MDMA. Shulgin advocated for the use of MDMA in therapy, and throughout the late 1970s and early 1980s, use of MDMA by therapists and psychiatrists increased as did recreational use. MDMA became popular as a club drug because its empathogenic effects enhanced the experience of raves and dance parties, especially in the electronic dance music scene.
In 1985, faced with rising levels of recreational use, the DEA classified MDMA as a Schedule I substance with no accepted medical use in spite of protests from some therapists, psychiatrists, and researchers at the time. The drug remains a Schedule I substance to this day, although in 2017 the FDA approved its use in research for treatment of PTSD.
14.2.2 Administration and Pharmacokinetics MDMA is typically administered orally in the form of tablets, pills, or capsules. The tablet or pill form is also called ecstasy, while the capsules are referred to as molly (a slang term referring to the molecular-crystalline powder form of MDMA found in the capsules). These are street names, however, and similar to other drugs such as heroin or cocaine, drugs sold as ecstasy or molly may be cut with other substances like caffeine, amphetamine, ketamine, or GHB. Some preparations may not even include MDMA at all.
After absorption, peak concentrations occur about 2 hours after administration. MDMA has a long half-life of about 9 hours and is metabolized in the liver by cytochrome P450 enzymes, in particular CYP2D6 and CYP1A2. Although the former metabolizes most of the drug, the latter converts MDMA into an active metabolite, methylenedioxyamphetamine, or MDA. MDA is a psychedelic drug in its own right and a common adulterant of MDMA.
1st edition
Drug interactions with MDMA can also occur. Certain antidepressants, such as SSRIs (selective serotonin reuptake inhibitors, which we will discuss in a later chapter) can enhance the effects of MDMA. These drugs inhibit the activity of CYP2D6, which slows down the metabolism of MDMA and prolongs its effects.
14.2.3 Mechanisms of Action and Effects Knowing that MDMA is a mixed stimulant-psychedelic, you may infer that the primary neurotransmitters affected by the drug would be norepinephrine (stimulant) and serotonin (psychedelic), and if so, you would be right. MDMA’s main mechanism of action is reversing monoamine transport. This causes reuptake transporters to pump transmitters out into the synapse instead, raising the levels of serotonin and norepinephrine. Dopamine transport is also affected, although to a much smaller extent. See the image below:
In addition, MDMA also inhibits the movement of monoamine molecules into storage vesicles. This increases the number of monoamines present in the intracellular fluid, which the reversed transporters can then easily pump out of the cell. This further increases serotonin and norepinephrine levels in the synapse.
As mentioned before, increased norepinephrine transmission results in sympathomimetic effects, while increased serotonin transmission results in psychedelic effects. At low doses, MDMA produces increased perception and mild hallucinations. At higher doses, the physiological effects of MDMA are similar to those of amphetamine, such as increased energy and blood pressure.
Probably the most unique effect is the enhanced empathy, which appears to be sex-linked; women generally report more profound effects compared to men (Liechti, Gamma, & Vollenweider, 2001). Similar to LSD, subjective effects of MDMA depend on the setting and mood in which the drug is used. Bad trips, true hallucinations, and paranoia are all possible, and MDMA is also capable of causing flashbacks and HPPD.
Common risks of use are hyperthermia and dehydration. MDMA causes a prolonged increase in body temperature, which can result in hyperthermia (heat stroke), especially when MDMA is used in raves. This can cause users to drink excessive amounts of water without replenishing electrolytes, leading to an extremely low sodium concentration in the blood known as
1st edition
hyponatremia. This in turn can lead to multiple organ failure, seizures, stroke, and heart attacks. Although toxic effects usually occur with high doses or multiple doses in quick succession, low doses can trigger toxic effects in susceptible individuals.
Chronic exposure to MDMA also causes damage to serotonergic neurons in the brain. This neurotoxicity decreases serotonin levels and appears to be semi-permanent, with even partial recovery taking years. Although this hasn’t been confirmed directly in humans, tests on primates have demonstrated long-term neurodegeneration (see image below), and heavy MDMA users have low serotonin levels.
Source: Hatzidimitriou, McCann, & Ricaurte (1999)
Normal MDMA use causes rebound effects due to depletion of serotonin and dopamine stores. Synthesis of new serotonin and dopamine takes several days, during which users may feel depressed or lethargic. As is the case with LSD, tolerance to the subjective effects can develop. Repeated use can cause deficits in memory and attention, which may be partially permanent (Brown, McKone, & Ward, 2010).
Evidence for dependence is mixed. Although MDMA lacks a well-characterized withdrawal experience, rebound effects cause withdrawal-like symptoms, and long-term users may demonstrate compulsive use despite negative consequences, a hallmark of addictive behaviors (Cottler et al., 2001). Heavy use may result in changes to the brain’s reward system, although MDMA is less addictive when compared to stimulants like amphetamine or cocaine.
1st edition
14.3 Dissociatives
The final category of psychedelics that we will be looking at this chapter are dissociatives. As the name suggests, these are drugs that cause dissociation, or feeling disconnected from the body and environment. Being psychedelics, they are also capable of producing hallucinations or altered perceptions. They also tend to exhibit depressant effects, making them sort of the counterparts to mixed stimulant-psychedelics. Dissociative drugs include phencyclidine, ketamine, and dextromethorphan.
By the end of this section, you should be able to:
• Define dissociatives and dissociation. • Describe the pharmacokinetic properties of PCP and ketamine. • Describe the pharmacodynamic properties and effects of PCP and ketamine.
14.3.1 History and Overview Although there are many different dissociatives, the two most well-known are phencyclidine (PCP) and ketamine. Both drugs were originally developed as anesthetics in the 1950s and 1960s, but reports of adverse side effects such as emergence hallucinations, nightmares, anxiety, and delirium resulted in their use in humans being discontinued. Ketamine continues to be used in veterinary medicine and small children (Prus, 2018).
After use of PCP and ketamine was largely abandoned, use of these drugs went in two different ways. Recreational use began in the 1960s despite the negative side effects, with both becoming club drugs due to their psychedelic effects. At the same time, PCP and ketamine found applications in psychiatric research, as use of the drugs produces a temporary state that resembles schizophrenia. This allowed researchers to model schizophrenia symptoms (Jentsch & Roth, 1999) and gave rise to the glutamate hypothesis of schizophrenia, which will be covered in Unit 4.
Another dissociative mentioned earlier was dextromethorphan (sometimes abbreviated DXM), an ingredient found in over-the-counter cough syrups like Robitussin®. It is an opioid derivative and enantiomer of levomethorphan, an opioid analgesic. At therapeutic doses it has antitussive effects while lacking other opioid effects, but when taken in high doses, it functions as a dissociative similar to PCP or ketamine.
PCP is currently classified as a Schedule II drug, while ketamine is under Schedule III. Dextromethorphan is not listed as a controlled substance, although some states and retailers have implemented restrictions on its sale.
14.3.2 Administration and Pharmacokinetics PCP can be administered via injection, snorting, or inhalation. In the 1960s, PCP was typically taken orally, although modern use of the drug has shifted to a powder form. PCP may also be taken alongside other drugs such as cannabis. Orally ingested PCP has poor bioavailability due to first-pass metabolism and has a duration of action of 15-60 minutes (Bey and Patel, 2007). A more common route of administration is to smoke it, usually applied to a marijuana or tobacco
1st edition
cigarette. Smoking PCP leads to an onset of effects within 1–5 minutes, with peak effects occurring 5–30 minutes after administration and lasting 4-6 hours. Snorting the drug causes a faster onset of effect at under a minute. PCP has a long half-life of about 18 hours but can last as long as 51 hours in some cases.
In comparison, ketamine is typically injected IV, although it can also be administered orally, intranasally, sublingually, or intramuscularly. IV injection is preferred for therapeutic use because it allows for precise dosage and concentrations. Ketamine has high water and lipid solubility and is rapidly distributed throughout the body. It is metabolized primarily in the liver by cytochrome P450 enzymes and has a half-life of about 3 hours.
14.3.3 Mechanisms of Action and Effects The psychedelic effects of PCP are largely mediated through the same mechanisms as LSD and MDMA. Not only does PCP reverse serotonin reuptake, it also acts as an agonist at 5-HT2A and D2 receptors. This accounts for altered perceptions and hallucinations.
The primary action of most dissociatives, including PCP and ketamine, is antagonism of the NMDA receptor, an ionotropic glutamate receptor (see image below). Since glutamate is an excitatory neurotransmitter, inhibition of it causes CNS depression and anesthetic effects. At high concentrations, PCP also acts as a noncompetitive antagonist for nicotinic receptors. This can change the activity of the autonomic nervous system and cause muscle contractions.
PCP’s activity at the NMDA receptors interferes with long-term potentiation, a type of synaptic plasticity important for learning and memory. Chronic use can lead to inhibition of glutamatergic transmission in the hippocampus and other regions of the brain. Users may forget any experiences they have while under the influence of the drug and suffer problems with short- and long-term memory (Morgan & Curran, 2012).
Behavioral effects of dissociatives are dose-dependent. At low doses, users exhibit a state similar to drunkenness, sometimes accompanied with numbing of the extremities. At moderate doses, numbing sensations may spread through the whole body due to the CNS depressant effects. Users also experience the disconnectedness from the body that is emblematic of dissociatives. Persons appear as if in a trance with open eyes and a fixed glaze.
1st edition
At high doses, PCP produces psychostimulant effects similar to amphetamine. This leads to increased heart rate and blood pressure, increased respiration, and higher body temperature. Toxic effects may also occur. Users may experience a state similar to catalepsy, where muscle movement becomes rigid and fixed. In addition, PCP can induce a schizophrenia-like psychosis that may last for days or weeks after use.
Tolerance to the effects of PCP and ketamine can occur with regular use, although it is sometimes slow to develop because infrequent use is more common. For the same reason, dependence on PCP or ketamine is rare, although both have been shown to have reinforcing properties. When dependence does occur, it is mostly psychological in nature, and withdrawal symptoms include cravings, lethargy, and depression.
1st edition
Chapter Summary and Review
In this chapter, we covered three different categories of psychedelics: hallucinogens; mixed stimulant-psychedelics; and dissociatives. We examined examples for each and their varying pharmacological properties; in particular, we learned about LSD, MDMA, PCP, and ketamine. This is the final chapter for the third unit. For the fourth and final unit, we will be examining psychotherapeutic drugs: antidepressants; anxiolytics; antipsychotics; and others.
Practice Questions
• Name four hallucinogens. • Which receptors does LSD act on? • Describe the difference between a true hallucination and pseudohallucination. • What does HPPD stand for, and what does it refer to? • What is another term for mixed stimulant-psychedelics? • What is the main active metabolite of MDMA? • Define hyponatremia and its consequences. How could MDMA use lead to it? • What is one of the most significant long-term risks associated with MDMA use? • Describe how PCP and ketamine have been used in the past and present. • How can PCP use lead to memory loss? • Which of these drugs is most potent: LSD, MDMA, or PCP? • Which drug has a shorter half-life, PCP or ketamine?
1st edition
References
Bey, T., & Patel, A. (2007). Phencyclidine intoxication and adverse effects: A clinical and
pharmacological review of an illicit drug. California Journal of Emergency Medicine,
8(1), 9-14.
Brown, J., McKone, E., & Ward, J. (2010). Deficits of long-term memory in ecstasy users are
related to cognitive complexity of the task. Psychopharmacology, 209(1), 51–67.
https://doi.org/10.1007/s00213-009-1766-2
Cottler, L. B., Womack, S. B., Compton, W. M., & Ben-Abdallah, A. (2001). Ecstasy abuse and
dependence among adolescents and young adults: applicability and reliability of DSM-IV
criteria. Human Psychopharmacology: Clinical and Experimental, 16(8), 599–606.
https://doi.org/10.1002/hup.343
El-Seedi, H. R., De Smet, P. A., Beck, O., Possnert, G., & Bruhn, J. G. (2005). Prehistoric
peyote use: Alkaloid analysis and radiocarbon dating of archaeological specimens of
Lophophora from Texas. Journal of Ethnopharmacology, 101(1–3), 238–242.
Hatzidimitriou, G., McCann, U. D., & Ricaurte, G. A. (1999). Altered serotonin innervation
patterns in the forebrain of monkeys treated with (+/-)3,4-
methylenedioxymethamphetamine seven years previously: Factors influencing abnormal
recovery. The Journal of Neuroscience, 19(12), 5096–5107.
For the fourth and final unit of this course, we will be covering psychotherapeutic drugs. These
are drugs that are used to treat mental disorders and physiological conditions that cause
behavioral, cognitive, and personality changes. We will start with the affective disorders, namely
depression and bipolar disorder, as well as the drugs used to treat them.
Chapter Outline:
15.1 Depression
15.1.1 Types of Mood Disorders
15.1.2 Monoamine Hypothesis of Depression
15.1.3 Pharmacological Treatment
15.1.4 Non-Pharmacological Treatments
15.2 Antidepressant Drugs
15.2.1 Overview of Antidepressants
15.2.2 Monoamine Oxidase Inhibitors
15.2.3 Tricyclic Antidepressants
15.2.4 Selective Serotonin Reuptake Inhibitors
15.2.5 Other Atypical Antidepressants
15.3 Bipolar Disorder
15.3.1 Symptoms of Bipolar Disorder
15.3.2 Mood Stabilizers
1st edition
15.1 Depression
Although we often use the term as a stand-in for sadness, in a clinical setting, depression
refers to a negative change in mood that is more severe or persistent than normal. In this
section, we will define depression and explore some of its causes and treatments.
By the end of this section, you should be able to:
• Define the major types of mood disorders.
• Differentiate between the diagnostic criteria of major depressive disorder and persistent
depressive disorder.
• Describe the neurochemical causes of mental depression.
• Differentiate between depressions caused by deficiencies in norepinephrine and
serotonin.
• Describe the goal of pharmacological treatment of depression.
• Provide two possible explanations for the delayed effect of antidepressants.
• Discuss the use of psychotherapy and electroconvulsive therapy in the treatment of
depression.
15.1.1 Types of Mood Disorders
Depression is a type of mood disorder, also known as an affective disorder. It is normal for
people to have shifts in mood. At times, people may even dip into moments of depression, such
as after the loss of a job or relationship, or moments of heightened excitement and activity,
known as mania. These changes are usually mild though and do not last for long periods of
time. Disorders occurs when mood changes are severe or persistent.
There are two main types of affective disorders: depression and bipolar disorder. We will
discuss bipolar disorder in detail in a later section. For now, we will focus on depression, which
comes in two main forms.
The first is major depressive disorder or MDD. The critical symptoms of MDD are depressed
mood and anhedonia, or a loss of interest or pleasure. These symptoms must last for at least
1st edition
two weeks and be accompanied by at least three additional symptoms. Consult the following
table for a full list, but note that the most important symptoms are the two described above:
Signs of Major Depressive Disorder
Depressed mood* Anhedonia*
Weight gain/loss Sleep disturbance
Loss of energy Low self-esteem
Poor concentration Suicidal thoughts
Feelings of hopelessness
Psychomotor agitation
*Required for positive diagnosis of major depressive disorder
The other major classification of depression is persistent depressive disorder, which is also
sometimes referred to as dysthymia. The only critical symptom here is depressed mood, but
instead of two weeks, the symptoms must occur on most days for at least two years. Persistent
depressive disorder may also be accompanied by some of the signs mentioned above.
Compare the diagnostic criteria for the two below:
Major Depressive Disorder Persistent Depressive Disorder
Required symptoms
Depressed mood Anhedonia
Depressed mood
Additional signs
At least 3 additional signs At least 1 additional sign
Duration At least 2 weeks Most days for at least 2 years
Depression is one of the most common mental illnesses in the U.S., with nearly 7.1% of all
adults experiencing at least one episode of major depression in 2019. Depression is more
common among women than men, with women being nearly twice as likely to develop
depression (Albert, 2015). The exact symptoms shown varies and is influenced by gender, age,
and the presence of other mental illnesses such as anxiety.
In addition, while we will not cover them for this class, there are other types of depression that
you may see mentioned in the media or scientific literature. An example is postpartum
depression, or the “baby blues” that can occur after childbirth. Another common example is
seasonal affective disorder (SAD), where depression is related to the change in seasons. These
1st edition
forms of depression share similar symptoms to the two main types described above but can
have unique causes.
15.1.2 Monoamine Hypothesis of Depression
What are the causes of depression? There are many different possible answers to this question.
For instance, many major depressive episodes show a clear precipitating event, such as the
death of a loved one. Not all cases are like this, however, and some people are at a higher risk
than others for developing depression. In the search for the underlying causes of depression, it
was discovered that monoamine levels may form a biological basis for depression.
In particular, it was noted that low levels of norepinephrine and/or serotonin in the limbic system
corresponded to symptoms of depression. The monoamine hypothesis of depression states
that depression is caused by low levels of these monoamines. Take a look at the diagram
below:
The brain on the left shows a healthy brain where levels of norepinephrine (NE) and serotonin
(5-HT) are normal. These transmitters are involved in ascending pathways that start in the
midbrain and project to different areas of the forebrain and cortex. These pathways and
structures are involved in mood regulation in the brain.
Low levels of norepinephrine activity in these regions can lead to slow thinking and actions
(psychomotor retardation), symptoms commonly seen in those with major depression.
Depression involving these symptoms is sometimes referred to as retarded depression (the
“retarded” refers to the psychomotor retardation mentioned previously).
At the same time, low levels of serotonin activity corresponded to anxiety, irritability, racing
thoughts, and restlessness. This type of depression is sometimes referred to as agitated
depression. Because there is substantial overlap between various types of depression, it is
sometimes diagnosed as depression with mixed features instead. Over ⅔ of major depressive
disorders also meet criteria for anxiety disorder (agitated depression).
1st edition
15.1.3 Pharmacological Treatment
Pharmacological treatment of depression largely follows the monoamine hypothesis of
depression. In other words, the goal of pharmacological treatment is to increase the amount of
norepinephrine and/or serotonin transmission in the limbic system. The antidepressant drugs
that we will discuss in the next section accomplish this through various methods, but the goal is
always to increase the level of some monoamine transmitter.
A potential challenge to the monoamine hypothesis is the fact that clinical improvement in mood
is much slower than the increase in transmitter levels. When taking an antidepressant
medication, the amount of neurotransmitter in the synapses increases within days. Despite this,
the onset of improved mood can be slow and take up to 2–4 weeks. What might explain this
latency?
One possible explanation is that the real cause of improved mood is reduced receptor
sensitivity. Although the amount of synaptic transmitter increases rapidly, it takes some time for
the body to adjust to the new level and downregulate the receptors in response. This decrease
in receptor sensitivity correlates more closely with clinical improvement in mood, as indicated in
the chart below.
Another explanation is hippocampal neurogenesis. Recall that the hippocampus is a structure
in the limbic system associated with learning and memory. Neurogenesis is the formation of
new neurons in the brain. Although neurogenesis largely ceases once humans reach adulthood,
the hippocampus is one of the few regions where neurogenesis continues into adulthood.
The hippocampus is particularly susceptible to stress. Although mild to moderate levels of stress
can be tolerated, strong or chronic stress can disrupt the functioning of the hippocampus and
lead to reduced attention, perception, and perception. In severe cases, such as the levels of
stress seen in depression, this may lead to reduced neurogenesis and neurotoxicity.
Hippocampal neurogenesis is relevant to depression because studies have shown that
antidepressants increase neurogenesis in the hippocampus. The time it takes for neurogenesis
to increase is 2–4 weeks, similar to the clinical improvement of mood. As such, increased
1st edition
hippocampal neurogenesis may be the cause for increased mood in people taking
antidepressants.
15.1.4 Non-Pharmacological Treatments
There are a variety of non-pharmacological treatments for depression. A common choice is
psychotherapy, which can be used on its own or in conjunction with antidepressant medications.
Psychotherapy can help patients understand the causes for depression, feel more in control of
their lives, and teach patients coping skills.
There are various approaches to psychotherapy for the treatment of depression. Individual
therapy between the patient and therapist is most common, but other options include group
therapy, couples therapy, and family therapy. Therapy may involve others in order to assure the
patient that they are not alone in their experiences, or to help the patient build a support
network.
Another form of treatment is electroconvulsive therapy or ECT. In ECT, an electrical current is
passed between two electrodes on each side of the brain to induce a brief seizure. The patient
is under anesthesia during the treatment and side effects are generally limited to mild memory
loss. Although the modern procedure is humane and consent is required, ECT is typically
reserved as a last resort because of its negative portrayal in films such as One Flew Over the
Cuckoo’s Nest.
Scientists are not entirely certain why ECT is effective at treating severe depression, although it
is hypothesized that the induced seizure acts to reset the brain somehow. Treatment involves
giving ETC 2–3 times per week until a clinical improvement in mood is noted, which may take a
anywhere between 3–15 sessions. ECT has a higher success rate than any other form of
antidepressant treatment besides esketamine (which we will discuss later) and can produce
dramatic and life-saving results.
1st edition
15.2 Antidepressant Drugs
Now that we understand the basis for pharmacological treatment of depression, it is time to
examine specific types of antidepressant drugs.
By the end of this section, you should be able to:
• Differentiate between first- and second-generation (atypical) antidepressant drugs.
• Describe the main side effects of antidepressants and define antidepressant
discontinuation syndrome.
• Discuss the mechanism of action and drug interactions of monoamine oxidase inhibitor
(MAOI) antidepressants.
• Discuss the mechanism of action of tricyclic antidepressants.
• Discuss the mechanism of action and drug interactions of selective serotonin reuptake
inhibitor (SSRI) antidepressants.
• Briefly discuss other atypical depressants and the significance of esketamine.
15.2.1 Overview of Antidepressants
Antidepressants can largely be split into two groups. First-generation antidepressants include
monoamine oxidase inhibitors and some tricyclic antidepressants. These drugs were discovered
earlier and tend to have greater side effects. In comparison, second-generation
antidepressants were discovered more recently and have fewer side effects because they are
more selective than first-generation antidepressants. Drugs in this group are also referred to as
atypical antidepressants because they do not fit into the typical categories of first-generation
antidepressants.
Although there are many types of antidepressants, almost all of them share the same general
effects and side effects. Perhaps the most noteworthy effect of antidepressants is that they only
elevate mood if it is depressed. Unlike stimulants like amphetamine, which produce stimulation
followed by a compensatory depression, antidepressants do not cause rebound depression
when stopped. Because of this, antidepressants are not habit-forming and are typically not
misused (although there are exceptions—more on that later).
Are antidepressants stimulants?
Because we have previously framed CNS stimulants and depressants as opposing classes of drugs, it is reasonable to assume that anti-depressants are CNS stimulants. Unfortunately, this is wrong—in fact, antidepressants themselves are CNS depressants and can produce effects such as sedation.
The confusion here is that there are two uses of the word depression here. When referring to CNS depression or drugs like barbiturates, depression refers to an inhibition of CNS activity. In comparison, the use of depression in the term antidepressant refers to depressed mood. Notably, depressed mood and inhibition of CNS activity are not the same thing, which is why some antidepressant drugs can also inhibit CNS activity.
1st edition
Common side effects of antidepressants include sedation, anticholinergic effects, orthostatic
hypotension, sexual dysfunction, and weight gain. Sedation and weight gain are attributed to
blockade of histamine H1 receptors. Anticholinergic refers to blocking acetylcholine receptors,
which means that anticholinergic effects can include constipation, dry mouth, and increased
heart rate. Orthostatic hypotension is caused by blockade of vascular alpha receptors and
describes a sudden drop in blood pressure upon standing up.
Antidepressants vary in terms of which side effects are expressed and to what degree. This is
important because side effects—in particular, sexual dysfunction and weight gain—can be
highly undesirable to patients and cause them to stop taking their medication resulting in a
remission of their depression. As such, the decision of which type of antidepressant to use is
often determined by the side effects.
Finally, although antidepressants do not cause withdrawal, stopping antidepressant use can
cause antidepressant discontinuation syndrome. This occurs in about 20–50% of patients
following discontinuation after at least one month of use, with increased risks for longer
treatment durations or medications with a shorter half-life. Symptoms include dizziness, nausea,
headaches, irritability, and insomnia. It can be mitigated by gradually reducing the dose at the
end of treatment, a process known as tapering, rather than abruptly stopping.
15.2.2 Monoamine Oxidase Inhibitors
We will start with the earliest antidepressants discovered, the monoamine oxidase inhibitors
(MAOIs). We have mentioned monoamine oxidase, or MAO, before. It is an enzyme that breaks
down amine neurotransmitters, such as norepinephrine, serotonin, and dopamine, within the
presynaptic cell. After reuptake of amine transmitters, a portion will be deactivated by MAO,
while the remainder will be repackaged into storage vesicles. These recycled neurotransmitters
will eventually be released into the synaptic space again.
MAOIs increase the amount of amine neurotransmission by blocking the MAO enzyme. As a
result, almost all of the reclaimed anime transmitters will be repackaged into vesicles after
reuptake. This increases the amount of neurotransmitter released into the synapse. Although all
amine transmitters may be affected, MAOIs accomplish their antidepressant effects primarily
through changes to norepinephrine and serotonin transmission. See the full process in the
diagram below:
1st edition
Above: Effect of MAOIs on norepinephrine (left) and serotonin (right) transmission. Normally, MAO breaks down amine transmitters
that have been taken back up into the presynaptic cell (top). By blocking these enzymes, MAOIs increase the amount of presynaptic
neurotransmitter available for release, thus leading to increased activation of postsynaptic receptors (bottom).
1st edition
Although MAOIs were the first antidepressants discovered, nowadays they are rarely prescribed
and are typically reserved for patients who are nonresponsive to other types of antidepressants.
The main reason for this is that MAOIs are hepatotoxic (i.e., they damage the liver) and can
cause serious and potentially lethal drug or dietary interactions. MAOIs inhibit enzymes in the
blood that are responsible for metabolizing other medications. This can cause dangerous
interactions if MAOIs are taken alongside other drugs. In particular, indirect-acting
sympathomimetic drugs such as pseudoephedrine (Sudafed®) should also be avoided.
MAOI treatment also requires dietary restrictions because of an interaction with the substance
tyramine. Tyramine is an amine naturally found in foods such as cheese, beer, beef, pepperoni,
dried fruits, and many others. Normally, tyramine is metabolized by MAO, but when under the
effects of MAOIs, tyramine levels can increase quickly. At high levels, tyramine acts as a weak
amphetamine, inducing the release of large amounts of norepinephrine. This can lead to an
acute hypertensive crisis, or a severe increase in blood pressure that can be life-threatening.
15.2.3 Tricyclic Antidepressants
Tricyclic antidepressants are part of the first-generation antidepressants along with MAOIs
and were discovered around the same time. They are often abbreviated as TCAs or simply
referred to as tricyclics. The tricyclic in the name refers to their chemical structure, which you
can see in the two examples below:
Tricyclic antidepressants are reuptake inhibitors of norepinephrine and serotonin. TCAs can be
divided into two subgroups: secondary (2º) amine TCAs and tertiary (3º) amine TCAs. The
two types differ in terms of which neurotransmitter they preferentially inhibit reuptake of;
secondary amine TCAs tend to increase norepinephrine levels more than serotonin, while
tertiary anime TCAs have a greater effect on serotonin levels. Compare the two in the table and
diagram below:
2º amine TCA 3º amine TCA
Preference Norepinephrine Serotonin
Example Desipramine (Norpramin®)
Amitriptyline (Elavil®)
1st edition
Above: Effect of TCAs on neurotransmission. Under normal reuptake, most of the neurotransmitter is taken back into the presynaptic
neuron (top). TCAs inhibit reuptake, leading to greater amounts of transmitter in the synapse (bottom). 2º amine TCAs like
desipramine have a greater effect on norepinephrine (left), while 3º amine TCAs like amitriptyline have a greater effect on serotonin
(right).
1st edition
TCAs do not have the same food and drug interactions as MAOIs, which made them a safer
alternative and the main type of antidepressant prescribed after MAOIs fell out of favor.
Compared to second-generation antidepressants, however, TCAs tend to have greater side
effects. As you may recall, some of these side effects (especially sexual dysfunction and weight
gain) can cause noncompliance, which leads to newer antidepressants being prescribed more
often. Despite this, TCAs are still a good choice for patients with depression that is resistant to
other medications.
15.2.4 Selective Serotonin Reuptake Inhibitors
The first type of second-generation antidepressant discovered were selective serotonin
reuptake inhibitors (SSRIs). As the name suggests, these drugs selectively inhibit serotonin
reuptake, unlike TCAs (see image below). Because they have weak affinity for norepinephrine
transporters, SSRIs are more useful for treating agitated depression compared to retarded
depression.
1st edition
Perhaps the most significant SSRI is fluoxetine, marketed under the trade name Prozac®.
Although it was not the first SSRI developed or sold, it had the largest impact and remains one
of the most prescribed medications in the U.S. to this date (ClinCalc, 2021). Other commonly
used SSRIs include sertraline (Zoloft®), citalopram (Celexa®), and escitalopram (Lexapro®).
When used as treatment for depression, SSRIs have a similar effectiveness as TCAs
(Anderson, 2000). The reason why SSRIs were considered a step-up was because they had
fewer adverse side effects, making it more likely that patients would continue medication use.
They also have a greater margin of safety because the toxic dose is higher compared to TCAs.
SSRIs are not without their complications, however. In the 1990s, increasing reports of suicidal
thoughts and behaviors under SSRI treatment led to FDA investigations and hearings on the
matter. Over time, it was found that SSRIs and other antidepressant drugs could cause
increased suicidality, especially in children and adolescents. This led to a public warning about
the risk by the FDA in 2004, as well as the requirement to include a black box warning on
antidepressant package inserts.
What is a black box warning?
A black box warning, also referred to as a boxed warning, is the strongest warning that the FDA can require. The name comes from the fact that the warning must be formatted with a black border around the text. Black box warnings are designed to call attention to “serious or life-threatening risks” and must appear on the prescription’s label or package insert (FDA, 2012). Below you can see an example for Prozac®:
What might account for the increased suicidality after taking SSRIs? One explanation is the
activating effect of SSRIs and other antidepressant drugs. This effect is an increase in energy,
comparable to taking caffeine, that occurs rapidly after starting the drug treatment. Importantly,
this effect occurs before clinical improvement in mood. Compare the onset of the two effects in
the graph below.
1st edition
Because of this, during the first few weeks of treatment, patients may see an increase in the
energy to act on suicidal impulses. Because the improvement in mood lags behind the
activating effect, suicidality may increase for a brief period before decreasing. This effect is
found in many SSRIs, especially fluoxetine (Prozac®).
Earlier, we mentioned how antidepressants are not habit-forming and are not usually misused,
although there are some exceptions. You may recall that we have mentioned one such example
before, in the previous chapter on psychedelics. SSRIs are sometimes paired with MDMA in the
rave scene. This is because the cytochrome P450 enzymes responsible for metabolizing MDMA
are inhibited by SSRIs. This slows down the metabolism of MDMA and prolongs its effects.
One final topic related to SSRIs that we should discuss is serotonin syndrome. This is a
potentially life-threatening condition caused by an excessive accumulation of serotonin and
overactivation of serotonin receptors throughout the body. Serotonin syndrome can occur from
use of any antidepressant medication that affects serotonin, not just SSRIs, although the fact
that SSRIs are selective to serotonin transporters makes them one of the most common causes.
In mild cases, serotonin syndrome consists of increased blood pressure and heart rate,
sweating, and pupil dilation (mydriasis). These symptoms intensify in moderate cases and are
accompanied by agitation, disorientation, tremors, and increased reflexes. In severe cases,
symptoms include hyperthermia, hypertension, delirium, and muscle rigidity. Antidotal therapy
may involve benzodiazepines to relieve agitation, or administration of a serotonin antagonist in
extreme cases.
1st edition
15.2.5 Other Atypical Antidepressants
Following the successful development of SSRIs, many other types of atypical antidepressants
were created. Similar to SSRIs, these drugs were designed to be selective to certain types of
receptors. Most are reuptake inhibitors, but some are antagonists of presynaptic autoreceptors.
Recall that autoreceptors are part of a negative feedback loop that inhibits further release of a
neurotransmitter, so blocking autoreceptors increases the amount of transmitter in the synapse.
An up-and-coming antidepressant drug type is the serotonin/norepinephrine reuptake inhibitor
(SNRI), which compares favorably with SSRIs. Representative SNRIs include venlafaxine
(Effexor XR), desvenlafaxine (Pristiq®), and duloxetine (Cymbalta®). SNRIs and SSRI account
for the lion’s share of antidepressant drug prescriptions.
The table below contains a list of atypical antidepressants. It is not necessary to memorize all of
them for this class; however, it is worth noting how the targets of antidepressants have
changed. Newer antidepressants may be designed to interact with dopamine receptors, since it
is hypothesized that dopamine plays a role in depression like norepinephrine and serotonin. You
may also recognize bupropion, which is also used to help people quit smoking and was
Is forgetful in daily activities Interrupts or intrudes on others
To be diagnosed with one of the two types, the individual must show at least five of nine
possible symptoms for that subtype for at least six months. These symptoms should also be
inconsistent with normal development and have a negative effect on social or
academic/occupational activities. If the criteria for both types are met, then the individual can be
diagnosed with the combined type of ADHD.
1st edition
18.1.2 Causes of ADHD
As mentioned in the video at the start of this section, the exact causes of ADHD are currently
unknown. Genetics are involved, since people with relatives who have been diagnosed with
ADHD have an increased risk for developing the disorder themselves (Faraone, 2003). The
connection is likely complex, though, and may involve multiple genes and epigenetic
interactions.
Because genetics cannot fully account for the development of ADHD, environmental factors are
expected to play a role. There is a lot of misinformation and false beliefs about what might
contribute to ADHD in children. Some causes, such as eating sugary foods, watching television,
parenting styles, and social factors have been shown to be unrelated to the development of
ADHD.
Factors that interfere with early neurodevelopment, such as drug use during pregnancy,
premature birth, exposure to toxins, or brain injuries are linked with an increased risk (NIMH,
2016). Recent studies have also linked prenatal exposure to acetaminophen to a higher risk of
ADHD and other developmental disorders such as autism (Ystrom et al., 2017; Ji et al., 2020).
Part of the difficulty in determining the causes of ADHD lies with the fact that there are many
other problems that can occur in childhood that give rise to ADHD-like symptoms. For instance,
children with hearing or vision problems may exhibit some of the behavioral tendencies of
children with ADHD because they are unable to focus on a teacher’s voice or presentation.
18.1.3 Neurochemistry of ADHD
As in the case of the disorders discussed in previous chapters, ADHD is thought to be caused in
part by a chemical imbalance in the brain. The two neurotransmitters implicated in ADHD are
norepinephrine and dopamine. It is thought that low levels of norepinephrine and dopamine
activity in the limbic system and prefrontal cortex cause the impulsivity and inattentiveness seen
in ADHD. Examine the diagram below:
1st edition
By now you should be familiar with how dopamine neurons in the ventral tegmental area (VTA)
project to the limbic system and prefrontal cortex. This is part of the reward system, which
regulates motivation and involves areas that are responsible for our emotional responses and
judgments. It is thought that low levels of dopamine activity in the limbic system contributes to
the symptoms seen in predominantly hyperactive-impulsive type ADHD, since these low levels
mean our brain is constantly seeking stimulation and experiencing stronger impulses than
normal.
Norepinephrine neurons in the locus coeruleus (Latin for “blue spot”) project along similar
pathways as the dopamine neurons discussed above. These neurons reach areas in the
prefrontal and frontal cortex that are responsible for executive functions, cognitive processes
that include attention, working memory, and the ability to tune out distractions or suppress
impulses. As you might suspect, disruption of these executive functions due to low
norepinephrine activity is associated with inattentive type ADHD. Compare the two in the
diagram below:
1st edition
Above: Role of norepinephrine (left) and dopamine (right) in ADHD. In normal cases, a regular amount of norepinephrine and
dopamine is released, leading to normal activation of adrenergic and dopaminergic receptors (top). Low levels of norepinephrine
activity are associated with inattentive type ADHD (bottom left), while low levels of dopamine activity are associated with hyperactive-
impulsive type ADHD (bottom right).
1st edition
18.2 ADHD Treatment
Now that we understand the neurochemical basis for ADHD symptoms, we can discuss the
different types of drugs used to treat ADHD. Although there are some exceptions, the most
common medications are CNS stimulants such as amphetamine.
Stimulants for hyperactivity?
At first glance, giving stimulants to children who already have problems sitting still and focusing seems like it would only make the problem worse. However, remember that stimulants increase levels of norepinephrine and dopamine, which is why they can have a therapeutic effect on ADHD symptoms, which are caused by low levels of these transmitters.
If you are still confused by the idea, consider watching the video below by Hank Green of SciShow:
Why Stimulants Help ADHD [6:08]
It should be noted that aside from medications, ADHD can also be addressed with certain
behavioral therapies. Our discussion in this section, however, will be limited to the drugs used in
pharmacotherapy.
By the end of this section, you should be able to:
• Explain the mechanisms of action and effects of drugs used to treat ADHD.
• Discuss the adverse effects and misuse of ADHD medications.
18.2.1 Stimulants
The classic treatment of ADHD involves stimulants. You may remember the discussion of
Adderall®, a non-racemic amphetamine mixture, from the chapter on high-efficacy stimulants.
You might also recall that methamphetamine can also be used to treat ADHD; it is sold under
the brand name Methedrine®. These medical uses keep amphetamine and methamphetamine
Schedule II drugs, rather than Schedule I.
The actions of Adderall® and Methedrine® are the same as what was covered in that chapter, so
we will only quickly go over the main points again. Amphetamine and methamphetamine
reverse the transport proteins responsible for reuptake of monoamines, causing them to pump
out more neurotransmitter into the synapse. This increases the activation of norepinephrine and
dopamine receptors, reducing ADHD symptoms (see below).