60
fully accredited since 2006 June 18 & 20, 2013 eConsent for Research: Considerations in Implementation and IRB Review
| Date post: | 20-Aug-2015 |
| Category: |
Health & Medicine |
| Upload: | quorum-review-independent-review-board |
| View: | 429 times |
| Download: | 0 times |

fully accredited since 2006
June 18 & 20, 2013
eConsent for Research: Considerations in Implementation and IRB Review

• Questions & Answers
• Feel free to submit questions at any point during the webinar using the chat box on your webinar dashboard
• Responses will be sent by the presenters following the presentation
4
WEBINAR HOUSEKEEPING

• Recording & Slide Deck
• The webinar recording and slide deck will be posted on our website within 5 business days
• We will email you a link to view the recording as soon as it is available
• Feel free to share the link with your staff and/or colleagues
5
WEBINAR HOUSEKEEPING

6
ABOUT QUORUM REVIEW IRB
Accredited Fully accredited by AAHRPP through 2014
Fully compliant with FDA and OHRP requirements
RegulatoryLeadership
6 in-house licensed attorneys providing guidance and thought-leadership
International Boards available for the review of U.S. and Canadian studies
Strong Framework
One of the largest IRBs in the U.S. with ~180 employees
Certified IRBProfessionals
(CIP)
60% of Affiliated IRB members, 40% of Regulatory staff and 20% of study management & study support positions

• 14 Board meetings each week
• 24-hour site turnaround, 36-hour amendment review, and same day site changes
• One time CV and audit documentation submission
• Support available 8am-8pm ET
• Dedicated Study Manager
7
THE QUORUM ADVANTAGE

• Secure portal with SmartForms, status reports, and approval documents
• Customized Phase I and Post-Marketing processes
• Flexible, customized process for AMCs
• 100% Quality Control on all documents
8
THE QUORUM ADVANTAGE

Quorum Review Regulatory AttorneyJ. Claire Carbary, JD, CIP
IRB Experience Joined Quorum Review IRB in September 2009 WIRB prior to Quorum CIP certification since 2010 Member of the Northwest Association for Biomedical Research (NWABR) and Public Responsibility in Medicine and Research (PRIM&R)
Legal Background Juris Doctor from Seattle University Member of the Washington State Bar Association (WSBA) Member of the Health and Corporate Law Sections of the WSBA
9
ABOUT THE PRESENTERS

ConsentSolutions President & CEOSusan G. Brink, DrPH
ConsentSolutions, Inc. Founded by Dr. Brink in 2005 To further the development of media-based approaches to informed consent Grew out of Phase II SBIR NCI funding for the development of an online informed consent process for clinical trials
Dr. Brink’s Background Served as PI on the grant for the SBIR NCI funding Authored articles on patient experience, the need for eConsent, and pathways to implementation Has presented on electronic consent at DIA, PRIM&R, ACRP, and SoCRA
10
ABOUT THE PRESENTERS

11
eConsent for Research: Considerations in Implementation and IRB Review
Considerations in Implementing eConsent 12 Introduction to eConsent 13 Paper-based vs. eConsent 17 Initiating eConsent 22 A Look at eConsent 26
IRB Review of eConsent 30 Documentation of Consent 31 Privacy and Confidentiality of Data 37 IRB Documentation of Review and Approval 41 Consent Process 45
WEBINAR OVERVIEW
Part I
Part II

CONSIDERATIONS IN IMPLEMENTING eCONSENT
Part I
12

INTRODUCTION TO eCONSENT
13
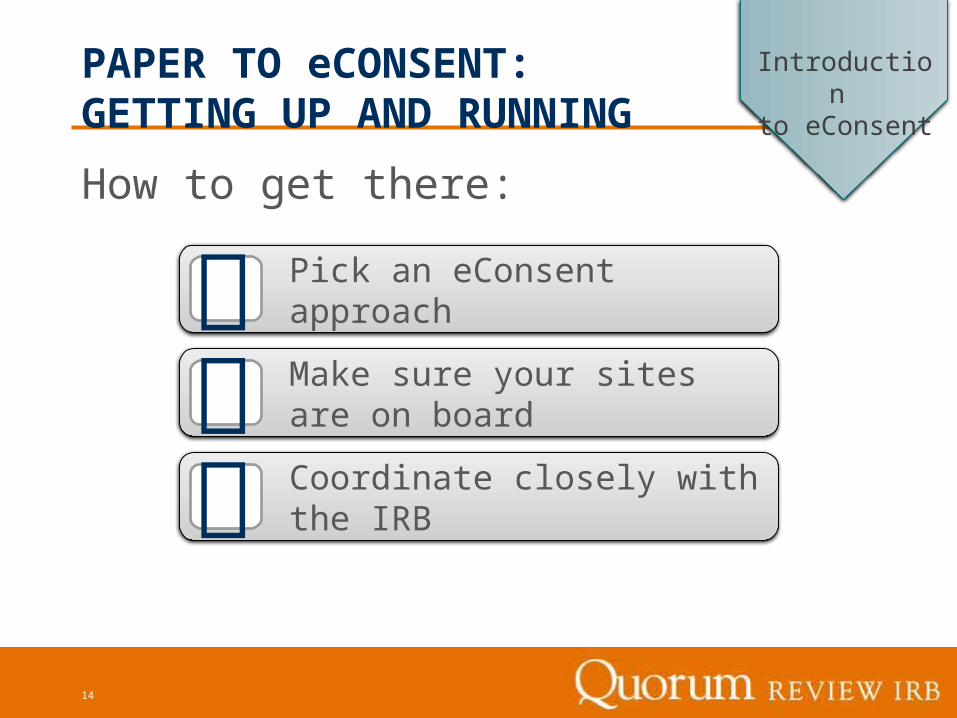
How to get there:
PAPER TO eCONSENT: GETTING UP AND RUNNING
Pick an eConsent approach
Coordinate closely with the IRB
Make sure your sites are on board
Introduction to eConsent
14
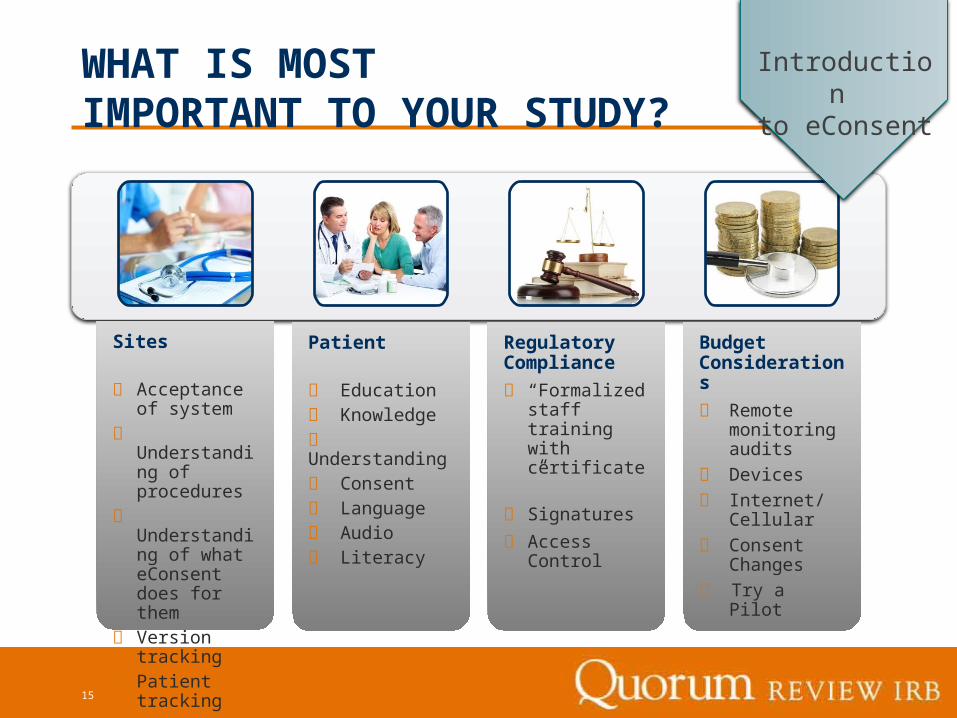
Sites
Acceptance of system
Understanding of procedures
Understanding of what eConsent does for them
Version tracking
Patient tracking
Patient
Education Knowledge Understanding Consent Language Audio Literacy
Regulatory Compliance
“Formalized staff training with certificate”
Signatures
Access Control
Budget Considerations
Remote monitoring audits
Devices
Internet/Cellular
Consent Changes
Try a Pilot
WHAT IS MOST IMPORTANT TO YOUR STUDY?
Introduction to eConsent
15

• Where and how can the system be used?
• What is needed to run the system?
E-INFORMED CONSENTIntroduction to eConsent
Internet Specific Devices
16

PAPER-BASED VS. eCONSENT
17

COMPARISON: PAPER-BASED VS. eCONSENT
SignatureSite Staff
Involvement
Paper-based vs. eConsent
ComparisonPaper-based
eConsent
Researchers want to find out more about an investigational drug called XYZ. An investigational drug is a drug that is being tested and is not approved for sale in the United States by the U.S. Food and Drug Administration (FDA).
IRB-approved Language
SignatureSite Staff
InvolvementIRB-approved
Language
18
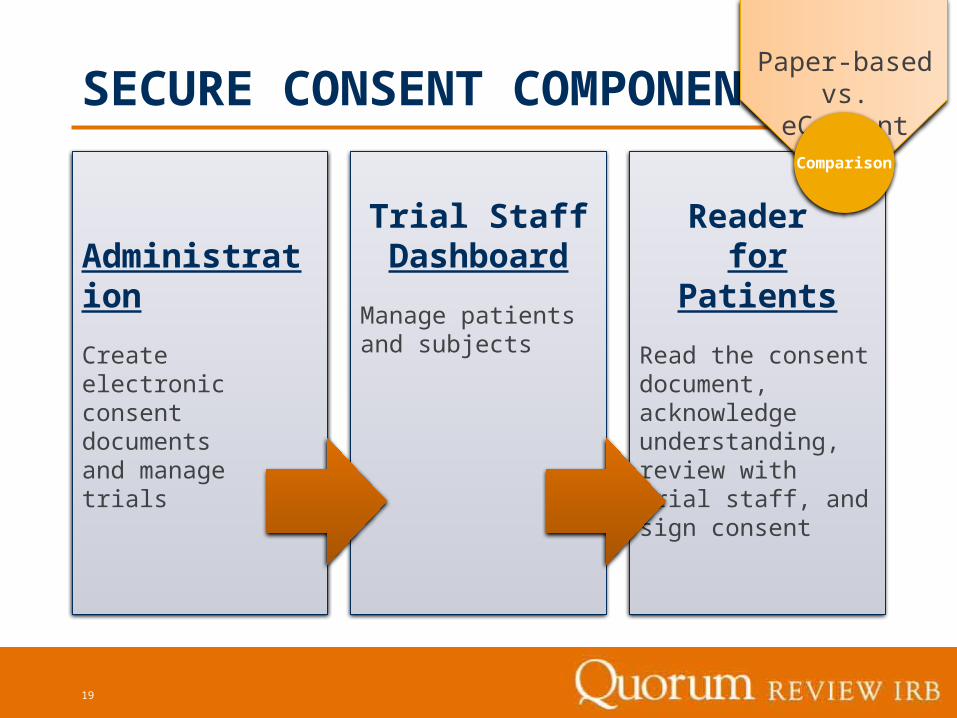
SECURE CONSENT COMPONENTS
Trial Staff Dashboard
Manage patients and subjects
Reader for Patients
Read the consent document, acknowledge understanding, review with trial staff, and sign consent
Administration
Create electronic consent documents and manage trials
Paper-based vs. eConsent
Comparison
19

Paper Consent
Page numbers
No education
Must be read by patients, orread aloud by staff to patient
Handwritten signature, dated by signer
eConsent System
Presented by section
Just-in-time Media-based education accessible during consent
Audio track of exact consent text
Digital handwritten signature, system assigns date and time
APPROACHES - DIFFERENCESPaper-based vs. eConsent
Comparison
20

Paper Consent
Black box as to what patient does
Version control can be minimal
Maintained in paper files/scanned to EMR
Electronic Secure Consent System
Timeline by date and time of all actions; analytics on patient consent actions
Robust Version control/revision tracking and management
Stored on server; can accommodate transfer of consent information to other systems
COMPARISONPaper-based vs. eConsent
Comparison
21

INITIATING eCONSENT
22

Have the eConsent contractor review draft consent before sending to IRB (maybe even before internal compliance)
ENGAGE WITH eCONSENT PROVIDER EARLY
Initiating eConsent
Moving from an unstructured to structured system
Changing consent/approval workflow
Embedded education takes time
IRB education and review take time
23

What are the special characteristics for the study? Trial population?
SPECIAL CHARACTERISTICS
Initiating eConsent
Ask eConsent Provider
Ask Your Team
Which sites are open to an eConsent process?
Which device(s) will the sites use?
What do we want to assure patient knows?
What are the constraints on the system?
24

IRB PROCESSInitiating
eConsent
Does the IRB already have the process in place for review of eConsents?
Yes No
What does the eConsent provider need to do?
Who will talk with the IRB?
What does our preferred IRB need to know?
Who do they talk to at the IRB?
What does the IRB require?
Do they have guidelines for eConsent submission?
25

A LOOK AT eCONSENT
26

Staff Dashboard Patient list Single patient
timeline
Patient Interface Consent section Signature page
EXAMPLES OF eCONSENT
A Look at eConsent
27

• Dashboard with list of patients
• Individual Timeline
• Monitor view during study
• FDA view during inspection/audit
MONITORING AND AUDITINGA Look at eConsent
28

Questions to ask the provider of the eConsent application:
QUESTIONS TO ASK
Which IRBs have you worked with?What support/training do you offer the sites? Is the software 21 CFR Part 11 compliant?Has the software been used in FDA-regulated clinical studies?How is system access controlled?Is there a robust back-up process?
A Look at eConsent
29

IRB REVIEW OF eCONSENT
Part II
30

DOCUMENTATION OF CONSENT
31

REQUIREMENT FOR DOCUMENTATION OF CONSENT
Citation: 45 CFR 46.109(c); 21 CFR 56.109(c)
Documentation of Consent
An IRB shall require documentation of informed consent…” “
FDA Guidance
32

21 CFR Part 11 applies to records in electronic form that are created, modified, maintained, archived, retrieved, or transmitted under any
records requirementsset forth in the FDA regulations.
ELECTRONIC SIGNATURES UNDER PART 11
Documentation of Consent
33

FDA regulations, at 21 CFR Part 11 , establish the criteria for acceptance by FDA of electronic records, electronic signatures, andhandwritten signatures executed to electronic
records as equivalent to paper records and handwritten signatures executed on paper.
VALIDITY OF ELECTRONIC SIGNATURES
Documentation of Consent
34

PART 11 COMPLIANCE requires both procedure controls (notification, training, SOPs, administration) and administrative controls to be put in place in addition to the technical controls that exist in the system.
These include: Each electronic signature shall be unique to one individual
and shall not be reused by, or reassigned to, anyone else The organization must verify the identity of an individual
before an electronic signature may be utilized Certification must be provided to FDA that the electronic
signature is intended to be the legally binding equivalent of a traditional handwritten signature
ELECTRONIC SIGNATURES UNDER PART 11
Citation: 21 CFR 11.200
Documentation of Consent
FDA Guidance
35

Required Controls for ID Codes and Passwords:
ELECTRONIC SIGNATURES UNDER PART 11
Citation: 21 CFR 11.200
Documentation of Consent
Maintenance of unique combined ID codes and passwordsPeriodic checking of code and passwords (to cover password aging)Loss management procedures to de-authorize lost, stolen, missing or otherwise compromised passwordsTransaction safeguards to prevent unauthorized use of passwordsTesting of devices that bear or generate ID code or password information
FDA Guidance
36

PRIVACY & CONFIDENTIALITY OF DATA
37

PRIVACY & CONFIDENTIALITY
Citation: 45 CFR 46.111(7); 21 CFR 56.111(7)
Privacy and Confidentiality
The IRB must determine that there are adequate provisions to protect the privacy of subjects and to maintain the confidentiality of data.” “
FDA Guidance
38

Evaluating Privacy and Confidentiality in the Paper World
How are records stored and protected?
Who has access to the records?
THE PAPER WORLDPrivacy and
Confidentiality
39

Evaluating Privacy & Confidentiality Protections with eConsent:
PROTECTIONS WITH eCONSENTPrivacy and
Confidentiality
Is the system Part 11 Compliant?What type of hardware and platform will be used?Is the technology Web-based or app-based? If web-based what type of encryption is used?Does it interface with existing EHR?How do users get access?Does the technology use location or other tracking features?
40

IRB DOCUMENTATION OF REVIEW AND APPROVAL
41

IRB RECORDS & DOCUMENTING APPROVAL
Citation: 45 CFR 46.115(a), 46.109(d) & 21 CFR 56.115 & 56.109(e)
IRB Documentation
The IRB is required to…
“Prepare and maintain adequate documentation of IRB activities, including…copies of all…approved sample consent documents.”
“Notify investigators…in writing of its decision to approve…the proposed research activity.”
FDA Guidance
42

The IRB must consider compatibility for both storage and access:
Screenshots Video files of consent presentation Archived web-pages
DOCUMENTATION OF APPROVED eCONSENT
IRB Documentation
43

DOCUMENTATION OF APPROVED eCONSENT
Stamp a screenshot Refer to the version, date, of the
electronic version in approval letters or documentation
IRB Documentation
In the paper world the IRB places a “stamp”
of approval on the finalized paper copy
In the electronic world, what do
you stamp?
Paper World Electronic World
OPTIONS
44

CONSENT PROCESS
45

IRB REVIEW OF THE CONSENT PROCESS
Citation: FDA Information Sheet, “A Guide to Informed Consent”, available at: http://www.fda.gov/RegulatoryInformation/Guidances/ucm126431.htm.
Consent Process
The informed consent process is more than just a signature… it is a process of information exchange… IRBs, clinical investigators, and research sponsors all share responsibility for ensuring that the informed consent process is adequate… the consent document should be the basis for a meaningful exchange between the investigator and the subject.”
“
FDA Guidance
46

The IRB should be aware of:
REQUIREMENTS FOR CONSENT PROCESS AND IRB REVIEW
Citation: FDA Information Sheet, “A Guide to Informed Consent,” available at: http://www.fda.gov/RegulatoryInformation/Guidances/ucm126431.htm.
Who will conduct the consent interviewThe timing of obtaining informed consent (any waiting period between informing and obtaining consent)The amount of time a patient is given to consider participationAdditional agreements they are asked to sign (Terms of Use)
Consent Process
FDA Guidance
47

IRB REVIEW OF THE CONSENT PROCESS
Citation: 45 CFR 46.116; 21 CFR 50.20
An investigator shall seek…consent only under circumstances that provide the prospective subject or the representative sufficient opportunity to consider whether or not to participate…”
“
Consent Process
FDA Guidance
48

IRB REVIEW OF THE CONSENT PROCESS
Citation: 45 CFR 46.116; 21 CFR 50.20
NO informed consent, whether oral or written, may include any exculpatory language through which the subject or the representative is made to waive or appear to waive any of the subject’s legal rights, or releases or appears to release the investigator, the sponsor, the institution, or its agents from liability for negligence.
Consent Process
FDA Guidance
49

A copy of the consent must be provided to subjects:
FDA & HHS do not require a signed copy be provided to subjects (or LAR)
ICH does require a signed and dated copy be provided to subjects (or representative)
REQUIREMENTS FOR CONSENT PROCESS AND IRB REVIEW
Citations: 45 CFR 46.117(a); 21 CFR 50.27(a); ICH E6 4.8.11
Consent Process
FDA Guidance
50

COMPLYING WITH CONSENT PROCESS REQUIREMENTS
Who will be obtaining the consent?What is the involvement of the PI?Where will the consenting take place?Are subjects asked to sign a Terms of Use or other type of agreement?How will copies be provided? (Electronic vs. Paper)
Consent Process
51

FDA, HHS, and ICH require the consent (where appropriate) to indicate that—
REQUIREMENT TO PROVIDE NEW INFORMATION
Citation: 45 CFR 46.116(b)(5); 21 CFR 50.25(b)(5)
significant new findings developed during the course of the research which may relate to the subject’s willingness to continue participation will be provided to the subject.”
“
Consent Process
FDA Guidance
52

• Flexibility is important if information may need to be given to subjects immediately
(A hybrid paper and e-process may be needed)
• A web-based eConsent could deliver content faster than paper
UPDATING THE eCONSENT
Consent Process
53

• There will be a learning curve when first submitting an electronic consent to IRBs for review
• Propose a meeting with staff and/or Board members prior to review to discuss how things should be submitted, talk about what the review process will entail, and get information about the review timeline
• Refer to Quorum’s List of eConsent Questions for an IRB to assist with the process
SUMMARY
54

55
• You may submit questions during our webinar survey, or
• You may email your questions to:[email protected]
• We will do our best to follow-up individually or answer your questions in the Q&A we post on our website
ADDITIONAL QUESTIONS

56
• The webinar Recording, Slide Deck, and Q&A will be posted on our website
• We will email you a link to view these items as they become available
• We value your opinion – please take our SURVEY and provide us with feedback
WEBINAR FOLLOW-UP

ConsentSolutions is a developer of internet-based multimedia systems for informed consent in the clinical trial industry.
To learn more, contact ConsentSolutions at: [email protected] 240-575-1918 www.consentsolutions.com
CONSENTSOLUTIONS
57

CONNECT WITH QUORUM!
plus.google.com/105052192707896902777
@quorumreview
youtube.com/quorumreview
linkedin.com/company/quorum-review
58
facebook.com/QuorumReview
slideshare.net/QcustomerR
www.QuorumReview.com

THANK YOU FOR ATTENDING!
59

fully accredited since 2006
June 18 & 20, 2013
eConsent for Research: Considerations in Implementation and IRB Review