ELSEVIER 0305-4179(95)00096-S Bums Vol. 22, No. 2, 101-106, 1996 pp. Copyright Q 1996 Elsevier Saence Ltd for ISBI Printed in Great Britain. All rights reserved 0305-4179/96 $15.00 + 0.00 Early effects of smoke inhalation on alveolar macrophage functions A. Bidani, C. Z. Wang and T. A. Heming Departments of Internal Medicine, and Physiology and Biophysics, University of Texas Medical Branch and Shriners Bums Institute, Galveston, Texas, USA - Alveolar macrophage (AM) dysfunctions have been implicated in the pathogenesis of smoke inhalation lung injury. We investigated the early (within 70 minj effectsof smoke inhalation on AM. The cells were recovered by bronchoalveofar lavage from rabbits ventilated with coffon smoke for 5 min followed by OJroom air for 60 min (smoke- exposed) or with room air in place of smoke (control). Smoke injury caused arterial blood carboxyhaemoglobin levels to increase I I-fold and reduced arterial blood PO, (measured N I h postinjuyj by x per cent. Scanning electron micrographs revealed denudation of plasmalemmal pseudopods in smoke-exposed AM. Smoke exposure suppressed both AM adherence to plasfic and phagocytosis of opso- nized bacteria. Basal superoxide (O,- ) production was elevated in smoke-exposed AM, compared with controls, whereas PMA- stimulated O,- production was unaffected. Smoke-exposed AM had reduced basal secretion of fumour necrosis factor-a (TNF-a), but dis- played a greater TNF response to stimulation with LPS than did control cells. UPS-stimulated TNF-a releasesfrom control and smoke- exposed AM were suppressed by phosphodiesferase inhibifors penfoxi- fylline and theophylline, and were enhanced by the lipoxygenase inhi- bifor. MK886. The early responses of AM to smoke inhalation lung ifrjury are consistent with activation of O,- production and priming of TNF-a release. concurrent with a funclional down regulation of phagocytosis. Burns, Vol. 22, No. 2, 101-106, 1996 Introduction Smoke inhalation lung injury is a major determinant of mortality in burn patients1*2. There are four stagesin the clinical course of patients with smoke-inhalation lung injUry',3-6: asphyxia and acute poisoning, acute respiratory insufficiency, bronchopneumonia and recovery. In the first stage, carbon monoxide poisoning, hypoxia and thermal injuries occur at the scene of the fire. The second stage encompasses the initial 24-48 h postinjury and is charac- terized by acute airway obstruction, tracheobronchitis, pulmonary oedema and atelectasisbecause of damage to the airway and/or alveolar epithelia. Inhalation of toxic gases (e.g. hydrogen chloride, oxides of sulphur and nitro- gen, and aldehydes) and thermal injury are primaril) responsiblefor this damage.In addition, the lung injury is augmented by release of inflammatory mediators from damaged tissues and inflammatory cells, both resident (macrophages, mast cells) and trapped (neutrophils) in the lungs. The bronchopneumonia stage usually begins at 24 h postinjury and is a result of alterations in local and systemic defense mechanisms. Most inhalation injury-related deaths occur during the second and third stages. Patients that survive these complications enter the recovery stage. Alveolar macrophages (AM) are the major resident phagocyte on the air-exchange surface of the lung and serve as the primary cellular defence mechanism of the lung’. AM have an important regulatory role in the immune response of the lung and their efficient functioning is essential for controlling respiratory tract infections. They also produce cytokines and growth factors that regu- late the proliferation of other lung cells8and so play an important role in maintaining normal lung structure. How- ever, many macrophage products also have the potential to exacerbate the inflammatory responseto lung injury. For example, AM produce oxygen radicals (e.g. superox- ide anion, hydrogen peroxide and hydroxyl radical) and proteases (e.g., cathepsins D and E, elastase and col- lagenase)8,9. These cytotoxic agents can directly damage lung tissues. AM alsorelease chemotactic agents (e.g., C5a, LTB4 and IL-S) that activate, attract and induce migration of neutrophils, which can lead to bystander cell injury’“‘“. AM are a major source of procoagulant activity that con- tributes to microvascular thrombosis12. Furthermore, AM produce systemic inflammatory mediators (e.g., IL-I, IL-6 IL-8 and tumour necrosis factor) that are thought to be responsible for many pathophysiological changesseen in acute lung injury8,9013,14. Thus, AM are involved in the entire cascade of processes that lead from smoke-induced lung injury to subsequent lung repair and recovery. Know- ledge about the role of AM in the pathogenesis of smoke- induced lung injury and about smoke-inducedmodulation of AM functions should benefit the management of bum patients with smokeinhalation lung injury. The effects of smoke inhalation lung injury on AM are not entirely understood. Disparate results have been reported about the cellular profile of bronchoalveolar lavage fluid (BALf) fo11 owing smokeinhalation lung injury and about the effects of smoke exposure on AM functions15-19. To a certain degree, these differences prob- ably reflect heterogeneity among animal models of smoke injury, BALf cell subpopulations and specific experimental designs. An early clinical assessment of the responses of human AM to smoke injury has been difficult to obtain, because the critical condition of bum patients immediately postinjury often precludesbronchoalveolar lavage.
Transcript
ELSEVIER
0305-4179(95)00096-S
Bums Vol. 22, No. 2, 101-106, 1996 pp. Copyright Q 1996 Elsevier Saence Ltd for ISBI
Printed in Great Britain. All rights reserved
0305-4179/96 $15.00 + 0.00
Early effects of smoke inhalation on alveolar macrophage functions
A. Bidani, C. Z. Wang and T. A. Heming Departments of Internal Medicine, and Physiology and Biophysics, University of Texas Medical Branch and Shriners Bums Institute, Galveston, Texas, USA -
Alveolar macrophage (AM) dysfunctions have been implicated in the pathogenesis of smoke inhalation lung injury. We investigated the early (within 70 minj effects of smoke inhalation on AM. The cells were recovered by bronchoalveofar lavage from rabbits ventilated with coffon smoke for 5 min followed by OJroom air for 60 min (smoke- exposed) or with room air in place of smoke (control). Smoke injury caused arterial blood carboxyhaemoglobin levels to increase I I-fold and reduced arterial blood PO, (measured N I h postinjuyj by x per cent. Scanning electron micrographs revealed denudation of plasmalemmal pseudopods in smoke-exposed AM. Smoke exposure suppressed both AM adherence to plasfic and phagocytosis of opso- nized bacteria. Basal superoxide (O,- ) production was elevated in smoke-exposed AM, compared with controls, whereas PMA- stimulated O,- production was unaffected. Smoke-exposed AM had reduced basal secretion of fumour necrosis factor-a (TNF-a), but dis- played a greater TNF response to stimulation with LPS than did control cells. UPS-stimulated TNF-a releases from control and smoke- exposed AM were suppressed by phosphodiesferase inhibifors penfoxi- fylline and theophylline, and were enhanced by the lipoxygenase inhi- bifor. MK886. The early responses of AM to smoke inhalation lung ifrjury are consistent with activation of O,- production and priming of TNF-a release. concurrent with a funclional down regulation of phagocytosis.
Burns, Vol. 22, No. 2, 101-106, 1996
Introduction Smoke inhalation lung injury is a major determinant of mortality in burn patients 1*2. There are four stages in the clinical course of patients with smoke-inhalation lung
injUry',3-6: asphyxia and acute poisoning, acute respiratory insufficiency, bronchopneumonia and recovery. In the first stage, carbon monoxide poisoning, hypoxia and thermal injuries occur at the scene of the fire. The second stage encompasses the initial 24-48 h postinjury and is charac- terized by acute airway obstruction, tracheobronchitis, pulmonary oedema and atelectasis because of damage to the airway and/or alveolar epithelia. Inhalation of toxic gases (e.g. hydrogen chloride, oxides of sulphur and nitro- gen, and aldehydes) and thermal injury are primaril) responsible for this damage. In addition, the lung injury is augmented by release of inflammatory mediators from damaged tissues and inflammatory cells, both resident (macrophages, mast cells) and trapped (neutrophils) in the
lungs. The bronchopneumonia stage usually begins at 24 h postinjury and is a result of alterations in local and systemic defense mechanisms. Most inhalation injury-related deaths occur during the second and third stages. Patients that survive these complications enter the recovery stage.
Alveolar macrophages (AM) are the major resident phagocyte on the air-exchange surface of the lung and serve as the primary cellular defence mechanism of the lung’. AM have an important regulatory role in the immune response of the lung and their efficient functioning is essential for controlling respiratory tract infections. They also produce cytokines and growth factors that regu- late the proliferation of other lung cells8 and so play an important role in maintaining normal lung structure. How- ever, many macrophage products also have the potential to exacerbate the inflammatory response to lung injury. For example, AM produce oxygen radicals (e.g. superox- ide anion, hydrogen peroxide and hydroxyl radical) and proteases (e.g., cathepsins D and E, elastase and col- lagenase)8,9. These cytotoxic agents can directly damage lung tissues. AM also release chemotactic agents (e.g., C5a,
LTB4 and IL-S) that activate, attract and induce migration of neutrophils, which can lead to bystander cell injury’“‘“. AM are a major source of procoagulant activity that con- tributes to microvascular thrombosis12. Furthermore, AM produce systemic inflammatory mediators (e.g., IL-I, IL-6 IL-8 and tumour necrosis factor) that are thought to be responsible for many pathophysiological changes seen in acute lung injury8,9013,14. Thus, AM are involved in the entire cascade of processes that lead from smoke-induced lung injury to subsequent lung repair and recovery. Know- ledge about the role of AM in the pathogenesis of smoke- induced lung injury and about smoke-induced modulation of AM functions should benefit the management of bum patients with smoke inhalation lung injury.
The effects of smoke inhalation lung injury on AM are not entirely understood. Disparate results have been reported about the cellular profile of bronchoalveolar lavage fluid (BALf) f o 11 owing smoke inhalation lung injury and about the effects of smoke exposure on AM functions15-19. To a certain degree, these differences prob- ably reflect heterogeneity among animal models of smoke injury, BALf cell subpopulations and specific experimental designs. An early clinical assessment of the responses of human AM to smoke injury has been difficult to obtain, because the critical condition of bum patients immediately postinjury often precludes bronchoalveolar lavage.
102 Burns: Vol. 22, No. 2, 1996
The present study investigated the early (within 70 min) responses of rabbit AM to smoke inhalation lung injury. AM were recovered by bronchoalveolar lavage from a rabbit model of smoke inhalation lung injuryzo. Since cotton materials are common combustion sources in house fires, cotton towelling was used to generate the smoke. The results show that smoke inhalation lung injury activated O,- production and primed the AM for TNF-a release, while simultaneously depressing phagocytosis.
Materials and methods Animals and smoke inhalation protocol Adult male New Zealand White rabbits (1.8-2.5 kg, n = 27; Ray Nichols Rabbitry, Tyler, TX, USA) were main- tained and tested at the University of Texas Medical Branch in accordance with institutional and federal guidelines for animal care and use. The rabbits were ana- esthetized (35 mg/kg ketamine i.m. and 5 mg/kg xylazine i.m.) and intubated. Fourteen animals were insufflated with cotton smoke. as described previously“‘. The smoke was generated by the combustion of cotton towelling. The chemical composition of similar cotton smoke has been published previouslyzl. The smoke inhalation protocol consisted of 5 cycles of O,-smoke insufflation (ventilation frequency = 4.5 breathslmin, tidal volume = 25 ml). Each cycle began with 0, insufflation for I min, followed by smoke insufflation for I min. After the fifth cycle, the animals were ventilated with 0, for 10 min and then with room air for .5Omin. The temperature of the insufflated smoke was monitored by the use of a Swan-Ganz thermis- tor placed near the inlet of the endotracheal tube and did not exceed 40°C. Control animals (n = 13) were insufflated with room air in place of the smoke. All animals were sacrificed by injection of ketamine (200 mg i.m.), followed by thoracotomy at 60min after completion of the insuf- flation protocol.
Blood analyses Arterial blood carboxyhaemoglobin (HbCO) levels were determined with a CO-oximeter (model 282, Instrumenta- tion Laboratory Inc., Lexington, MA, USA) immediately after completion of the smoke inhalation protocol. Arterial blood oxygen tension PUO, was analysed with a pH blood gas analyser I:model 1302, Instrumentation Laboratory Inc.) immedial-ely before anaesthesia and 5 min before animal sacrifice.
Collection of alveolar macrophages AM were collected by bronchoalveolar lavage imme- diately after the animals were killedz2. The recovered cells were washed twice with a HEPES-buffered saline (compo- sition in mM: 135 NaCl, 5 KCl, I CaCl,, 1 MgSO,, 2 K- phosphate, 5 o-glucose and 6 HEPES; pH = 7.4). The cells were then suspended in a complete medium (RPMI-1640) containing 25 mM HCO,-, 10 per cent fetal bovine serum, 100 U/ml penicillin and 100 pg/ml streptomycin (pH = 7.4). The resulting cell suspension (- lo6 cells/ml) contained > 95 per cent viable cells (trypan blue exclu- sion), of which > 94 per cent were macrophages by Papanicolaou staining, Giemsa staining and positive stain- ing for non-specific esterase activity.
Macrophage adherence AM were incubated in sterile multiple well plates for 2 h at 37°C in a humidified atmosphere containing 5 per cent CO,. The supematants were collected and the adherent
cells were washed with RPMI-1640. Non-adherent cells in the pooled supernatants and wash fluids were enumerated using a haemocytometer. Adherent cells were removed with a rubber policeman and similarly counted. Cell adherence was expressed as a ratio of the number of adher- ent cells to the total cell count (adherent plus non- adherent)“.
Release of TNF-c( AM were incubated in sterile multiple well plates for 2 h (5 per cent CO,, 37°C). The supernatants and non-adherent cells were removed and discarded. The adherent cells were washed, covered with RPMI-1640 containing 0 or 100 rig/ml LPS (Es c erlc ia coli serotype 055 : B5), and incu- h h bated for 24h (5 per cent CO,, 37°C). The AM- conditioned supematants then were collected and stored at -- 80°C pending analysis of TNF-a.
The bioactivity of TNF-ct in AM-conditioned media was assessed using a standard cytotoxicity assay with a TNF-sensitive L929-fibroblast cell line (ATCC CCL I; American Type Culture Collection, Rockville, MD)23. Each assay included an internal standard of recombinant murine TNF-cr (specific activity = 40 U/rig). All AM-conditioned media were assayed in duplicate.
Measurement of superoxide anion production Production of O,- was measured as the superoxide dismutase-inhibitable reduction of cytochrome cz4. AM were suspended in phosphate-buffered saline (composi- tion: 127 mM NaCl, 5 mM KCI, 11 mM Na-phosphate, 5 mM b-glucose, 2 mM NaN,, 80 PM cytochrome c, O--60 pg/rnl superoxide dismutase and O-l PM phorbol Id-myristate 13-acetate; pH = 7.4) and incubated in a sterile microcentrifuge tube for 1 h (37°C). Cell-free super- natants then were collected and analysed spectrophotome- trically for reduced cytochrome c (absorbance at 550nm) (spectrophotometer model DMS 300, Varian, Walnut Creek, CA, USA). Superoxide production was calculated using an absorption coefficient for reduced cytochrome c of 21 mM-’ cm-’ 24.
Assay of F, receptor-mediated phagocytosis Phagocytosis was quantified using fluorescein-labelled E. coli similar to the protocol described by Oben and Fore- man”. The E coli were opsonized with rabbit polyclonal IgG, washed twice, and resuspended in RPMI-1640. AM were incubated with the opsonized bacteria (ratio AM : bacteria = 1: 50) in RPMI-1640 in sterile centrifuge tubes for 30 min at 1 and 3 7°C (pH = 7.4). Phagocytosis was stopped by the addition of 10 volumes of ice-cold saline (composition in mM: 137 NaCl, 2.7 KCl, and 8.1 K-phosphate; pH = 7.4). AM were washed three times in the saline and then enumerated using a Coulter Multisizer II (Hialeah, FL, USA). Finally, the AM were lysed with 10 per cent Triton X-100 and the fluorescence of the lysate was measured at an excitation wavelength of 500 nm and emission wavelength of 525 nm (spectrofluorometer model F-2000, Hitachi Instruments Inc., San Jose, CA, USA) at room temperature. In order to correct for adherent but non-internalized bacteria, the fluorescence intensity values obtained from cells incubated at 1°C were sub- tracted from paired data obtained from cells at 37°C. The resulting phagocytic index was expressed as arbitrary fluorescence units/lo4 AM.
Measurement of cellular ATP concentration Cellular [ATP] was measured with a luminometer (Mono-
Bidani et al.: Smoke effects on lung macrophages 103
light 2010, Analytical Luminescence Laboratory, San Diego, CA) using a luciferin-luciferase assayz6.
Spectrofluorometric determination of intracellular
PH Intracellular pH (PHi) was measured using the pH-sensitive fluorescent probe, 2’,7’-biscarboxyethyl-5,6-carb- oxyfluorescein (BCECF), as described previouslyzz,z6. The fluorescence intensity of BCECF-loaded AM was moni- tored at 37°C using excitation wavelengths of 504nm (peak fluorescence) and 430nm (isosbestic point), and an emission wavelength of 530 nm (spectrofluorometer model F-2000, Hitachi Instruments Inc.).
Scanning electron microscopy AM were fixed with 2.5 per cent glutaraldehyde in an 80 mM cacodylate buffer containing 6.4 per cent sucrose (356 mOsm) for N 2 h. The cells were postfixed with 1 per cent 0~0, in an 80 mM cacodylate buffer containing 4.7 per cent sucrose, dehydrated with ethanol and, finally, critical-point dried. The fixed cells were mounted and coated with 15 nm gold for examination using a scanning electron microscope (model ISI-DS130, International Scientific Instruments, Inc., Pleasanton, CA, USA).
Chemicals and reagents HEPES was purchased from Research Organics (Cleveland, OH, USA). RPMI-1640 was purchased from Gibco (Grand Island, NY, USA). Actinomycin D was a kind gift from Merck, Sharpe & Dohme (West Point, PA, USA). Recom- binant murine TNF-a was purchased from Genzyme (Cambridge, MA, USA). Fluorescein-labelled E. coli and BCECF were purchased from Molecular Probes (Eugene, OR, USA). All other chemicals and reagents were pur- chased from Sigma (St Louis, MO, USA).
Statistical analysis The data are presented as arithmetic means f SEM. The data were statistically analysed using paired or unpaired Student’s f-tests, as appropriate. A probability (P) value of 0.05 was used to evaluate statistical significance.
Results
Neither smoke nor air insufflation was acutely lethal to any of the test animals. Smoke exposure caused a significant increase (N lx-fold) in arterial blood COHb levels and a significant decrease in Pao,, compared with control animals (Table r). Pathological examination at 60 min post- exposure found tracheobronchial foamy fluid and exudate in the upper airways of smoke-exposed animals, but none in control animals.
Smoke exposure had no detectable effect on the via- bility of BALf cells recovered within 70min of the initial injury (> 95 per cent by trypan blue exclusion) or on the relative proportion of macrophages in BALf ( > 94 per cent by Papanicolaou staining, Giemsa staining, and positive staining for non-specific esterase activity). However, the absolute number of AM recovered per animal was IV three-fold greater in smoke-exposed rabbits than in con- trols (35 f 8 vs. 1 I f 3 million cells, smoke-exposed and control animals, respectively). For our purposes, all macro- phages recovered in BALf were termed alveolar macro- phages without regard to their actual site of origin in the lung (i.e. alveolar versus airway).
Smoke exposure altered AM morphology. Scanning
Table I. Arterial blood carboxyhaemoglobin (COHb) and PO, (Pao,) levels after air or smoke exposure
Pao, (Torr) __-
COHb Baseline 60 min Condition 6) (pre-exposure) (postexposure)
Air (control) 6.6 f 0.4 115f2 102*3 Smoke exposed 75.4 f 2.3’ 114f3 77f4’
Dataaremeans+SEM (n=1>14). ‘Significantly different from corresponding control value (unpaired Student’s t-test, P< 0.05).
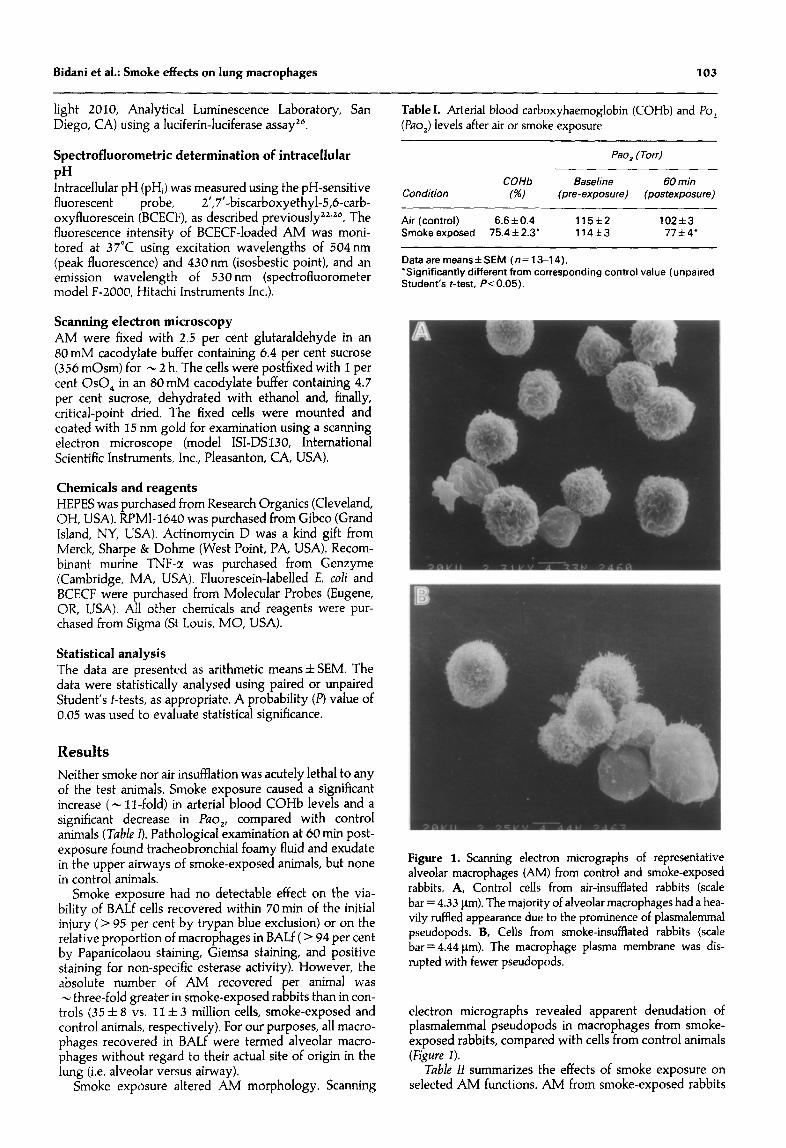
Figure 1. Scanning electron micrographs of representative alveolar macrophages (AM) from control and smoke-exposed rabbits. A, Control cells from air-insufflated rabbits (scale bar = 4.33 pm). The majority of alveolar macrophages had a hea- vily ruffled appearance due to the prominence of plasmalemmal pseudopods. B, Cells from smoke-insufflated rabbits (scale bar= 4.44 pm). The macrophage plasma membrane was dis- rupted with fewer pseudopods.
electron micrographs revealed apparent denudation of plasmalemmal pseudopods in macrophages from smoke- exposed rabbits, compared with cells from control animals (Figtlre I).
Table II summarizes the effects of smoke exposure on selected AM functions. AM from smoke-exposed rabbits
104 Burns: Vol. 22, No. 2, 1996
800
u-a 600 22 c 3
u basal
LPS
* r-
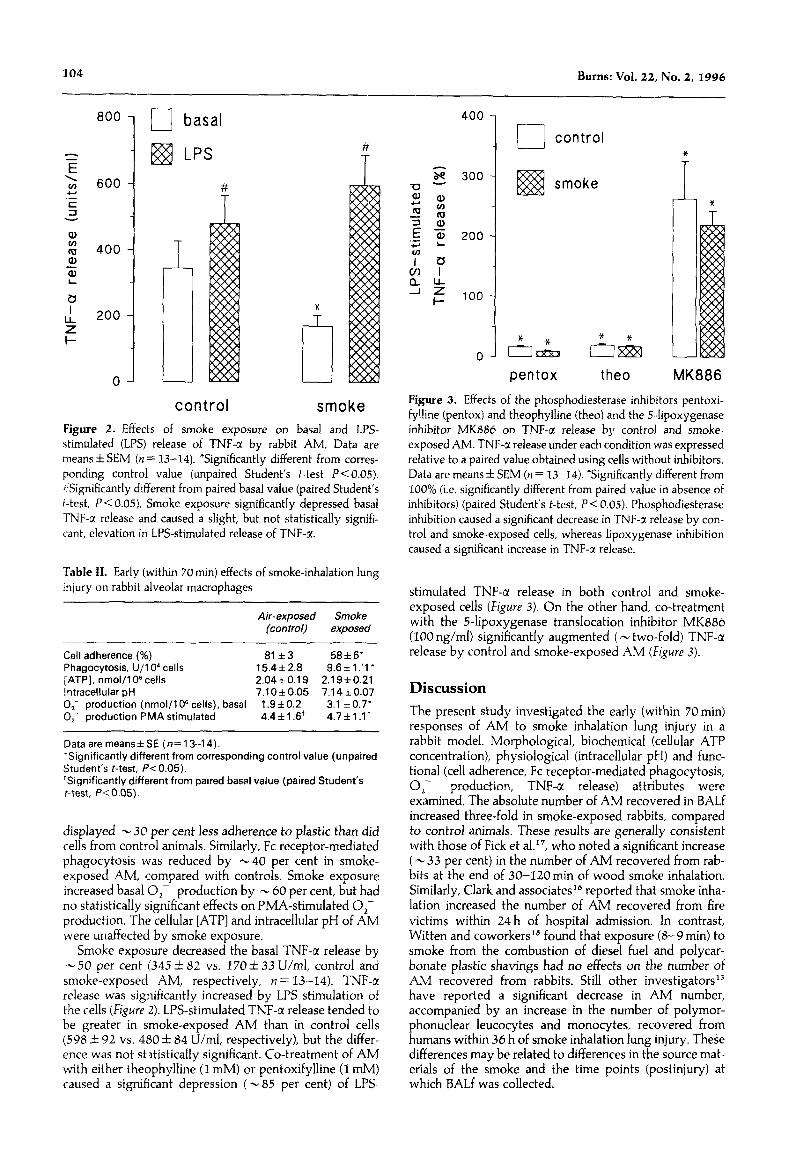
control smoke Figure 2. Effects of smoke exposure on basal and LPS- stimulated (LPS) release of TNF-@ by rabbit AM. Data are means f SEM (n = 13-14). ‘Significantly different from corres- ponding control value (unpaired Student’s f-test PC 0.05). &ignificantly different from paired basal value (paired Student’s t-test, P< 0.05). Smoke exposure significantly depressed basal TNF-a release and caused a slight, but not statistically signifi- cant, elevation in LPS-stimulated release of TNF-a.
Table II. Early (within 70 min) effects of smoke-inhalation lung injury on rabbit alveolar macrophages
Air-exposed Smoke (control) exposed
Cell adherence (X) 81*3 58*6’ Phagocytosis, U/l 0” cells 15.4~ 2.8 9.6+1.‘1’ [ATP], nmol/l O6 cells 2.04*0.19 2.19+0.21 Intracellular pH 7.10~0.05 7.14*0.07 0; production (nmol/l O6 cells), basal 1.9 f 0.2 3.1 f 0.7’ O,- production PMA stimulated 4.4 f 1.6’ 4.7fl.l’
Dataaremeans*SE(n=13-14). ‘Significantly different from corresponding control value (unpaired Student’s t-test, P< 0.05). ‘Significantly different from paired basal value (paired Student’s r-test, P< 0.05).
displayed - 30 per cent less adherence to plastic than did cells from control animals. Similarly, Fc receptor-mediated phagocytosis was reduced by -40 per cent in smoke.. exposed AM, compared with controls. Smoke exposure increased basal O,- production by - 60 per cent, but had no statistically significant effects on PMA-stimulated O,- production. The cellular [ATP] and intracellular pH of AM were unaffected by smoke exposure.
Smoke exposure decreased the basal TNF-a release by -50 per cent (345 & 82 vs. 170*33 U/ml, control and smoke-exposed AM, respectively, n = 13-14). TNF-c( release was significantly increased by LPS stimulation of the cells (Figure 2). LPS-stimulated TNF-a release tended to be greater in smoke-exposed AM than in control cells (598 X+Z 02 vs. 480 f 84 U/ml, respectively), but the differ- ence was not statistically significant. Co-treatment of AM with either theophylline (I mM) or pentoxifylline (I mM) caused a significant depression (- 85 per cent) of LPS-
400
1 cl control
2 300 - u- Q) Q, * u3 m 5 ti E .- w
p 200 -
‘; 0 w I au- -J Z
l- 100 -
smoke
0 1 &A cs&
pentox theo MK886
Figure 3. Effects of the phosphodiesterase inhibitors pentoxi- fylline (pentox) and theophylline (thee) and the Hipoxygenase inhibitor MKBS6 on TNF-a release by control and smoke- exposed AM. TNF-a release under each condition was expressed relative to a paired value obtained using cells without inhibitors. Data are means f SEM (n = 13-14). *Significantly different from 100% (i.e. significantly different from paired value in absence of inhibitors) (paired Student’s t-test, P < 0.05). Phosphodiesterase inhibition caused a significant decrease in TNF-a release by con- trol and smoke-exposed cells, whereas lipoxygenase inhibition caused a significant increase in TNF-a release.
stimulated TNF-a release in both control and smoke-
exposed cells (Figure 3). On the other hand, co-treatment with the 5-lipoxygenase translocation inhibitor MK886 (100 rig/ml) significantly augmented ( - two-fold) TNF-a release by control and smoke-exposed AM (Figure 3).
Discussion
The present study investigated the early (within 70mi1-1) responses of AM to smoke inhalation lung injury in a rabbit model. Morphological, biochemical (cellular ATP concentration), physiological (intracellular pH) and func- tional (cell adherence, Fc receptor-mediated phagocytosis, O,- production, TNF-ct release) attributes were examined. The absolute number of AM recovered in BALf increased three-fold in smoke-exposed rabbits, compared to control animals. These results are generally consistent with those of Fick et al.“, who noted a significant increase ( - 33 per cent) in the number of AM recovered from rab- bits at the end of 30-120min of wood smoke inhalation. Similarly, Clark and associates’6 reported that smoke inha- lation increased the number of AM recovered from fire victims within 24 h of hospital admission. In contrast, Witten and coworkers I5 found that exposure (8-9 min) to smoke from the combustion of diesel fuel and polycar- bonate plastic shavings had no effects on the number of AM recovered from rabbits. Still other investigators’” have reported a significant decrease in AM number, accompanied by an increase in the number of polymor- phonuclear leucocytes and monocytes, recovered from humans within 36 h of smoke inhalation lung injury. These differences may be related to differences in the source mat- erials of the smoke and the time points (postinjury) at which BALf was collected.
Bidani et al.: Smoke effects on lung macrophages 105
The number of AM recovered in BALf is expected to be affected by migration of macrophages into the airspaces and/or by changes in the adherence properties of those macrophages present in the airspaces. Previous studies have found that smoke inhalation and bum injury alters the chemotactic responses of human AM but do not agree on whether migration was decreased*’ or increased”. Fick and coworkers’7 reported that exposure to wood smoke (30-120 min) reduced both cell adherence to glass and the phagocytic capabilities of rabbit AM. The present data confirm that the early responses of rabbit AM to smoke inhalation lung injury include decrements in cell adherence (to plastic) and phagocytosis. Macrophage migration, adherence and phagocytosis are dependent on the integ- rity of the plasma membrane and intracellular cytoskel- eton. Thus, it is conceivable that the macrophage plasma membrane represents a site of action for smoke toxicants. This conclusion is supported by the observation that smoke-exposed AM appeared to be denuded of plasmalemmal pseudopods, compared with control cells.
The present smoke inhalation protocol had no effects on the cellular [ATP] or intracellular pH of AM. These data suggest that smoke inhalation lung injury has no early effects on the biochemical energy status of AM or the setpoint for intracellular pH.
Exposure (5 min) to cotton smoke increased the basal O,- production of rabbit AM. This suggests that smoke exposure activated the NADPH-oxidase system that is pri- marily responsible for O,- generation in phagocytes. Riyami et alI9 have suggested that AM activation may contribute to development of the pathophysiological changes seen in patients with smoke inhalation lung injury. Activation Iof pulmonary phagocytic cells in fire victims can lead to functional down-regulation of complement receptors . 27 This provides a plausible link between the effects of smoke inhalation lung injury on O,- production (i.e. early activation of smoke-exposed AM) and receptor- mediated phagocytosis (i.e. early depression in smoke- exposed AM).
The bronchopneumonia stage of smoke inhalation lung injury begins within 24 h postinjury and reflects the sup- pression of both local and systemic defense mech- anisms”5.6. To evaluate the potential effects of smoke inhalation lung injury on the capacity of AM to mount an oxidative burst, control and smoke-exposed cells were stimulated with a maximum dose of PMA and production of o,- was measured. PMA-stimulated O,- production was not altered significantly by smoke exposure (see Table 10. Thus, the total ‘oxidative burst’ capability of rabbit AM was not affected by smoke inhalation lung injury, at least during the first 70 min postinjury. None the less, it is pos- sible that smoke inhalation and thermal injury have delayed effects on AM functions. This is suggested by evidence that thermal injury (26-28 per cent body surface scald) of rats causes a time-dependent depression of the respiratory burst of AM recovered at 4 and 24 h post- injury? the magnitude of the effect increased two- to three-fold from the 4-h time point to the 24-h time point. In addition, the effects of smoke inhalation lung injury on the oxidative burst capabilities of pulmonary phagocytes are probably intluenced by the specific exposure conditions. For example, Witten et al. Is found that PMA-stimulated O,- production by rabbit AM was significantly depressed immediately following exposure (8-9 min) to smoke from diesel fuel and polycarbonate plastic shavings.
TNF-r is I-hought to be responsible for many pathophy-
siological alterations observed in acute lung injuryL9-31. Exposure to cotton smoke significantly reduced basal TNF-u production by rabbit AM and caused a slight, but not statistically significant, increment in the TNF response to LPS. These data suggest that inhalation of cotton smoke primed AM for TNF-or release. This pattern is similar to that reported previously for other attributes of smoke- exposed AM. Specifically, AM obtained from patients with smoke inhalation lung injury have a significant reduc- tion in basal chemiluminescence but an increased response to opsonized staphylococci-mediated stimulation”. Simi- larly, Williams et al. 32 have recently shown that thermal injury enhances the sensitivity of AM to LPS. An enhanced response of AM to endotoxin may be an important factor in the pathogenesis of acute respiratory failure after ther- mal and smoke inhalation injuries.
The mechanisms that regulate TNF-a production at the cellular and molecular levels are not entirely understood. Several investigations have suggested a regulatory role for cyclic nucleotides33-35. Pentoxifylline, a dimethylxanthine-derived inhibitor of phosphodisester- ases, has been shown to suppress LPS-induced TNF-a release by murine macrophages34, presumably via increased levels of cellular nucleotides. Doherty et al.33 have demonstrated that pentoxifylline inhibits endotoxin- mediated TNF-a production in thioglycolate-elicited peri- toneal macrophages (murine) by inhibiting endotoxin- induced transcription of the TNF-a gene. Similarly, the related phosphodiesterase inhibitor theophylline has been shown to inhibit TNF-c( release by ‘normal’ rat AM and blood monocytes35. Our results confirm a major suppres- sive effect of pentoxyfilline and theophylline on TNF-a release by rabbit AM (control and smoke-exposed). These data suggest that pentoxifylline or theophylline may have salutary effects in smoke inhalation lung injury, as has been postulated previously for acute lung injury secondary to sepsis29-3*.
Production of TNF-a was significantly increased by co- treatment of rabbit AM (control and smoke-exposed) with the 5-lipoxygenase translocation inhibitor MK886 (see Figwe 3). These in vitro results are consistent with the in vivo data of Ferrandiz and Foster‘lb. The latter authors studied the role of eicosanoids in TNF-u production by the use of a rat airpouch model of inflammation. They were able to dissociate leukotriene generation and TNF-a pro- duction experimentally, and concluded that 5- lipoxygenase products are not involved in regulation of TNF-a production in vivo.
In summary, the early (within 70 min) effects of cotton smoke on rabbit AM are consistent with direct and imme- diate effects of smoke toxicants on AM. Smoke inhalation injury stimulated O,- production and primed AM for TNF-c( release. Appropriate upregulation of AM effector functions is essential for recovery and repair of lung injury. However, hyperreactivity of AM with excessive O,- pro- duction and TNF-a release can exacerbate lung injury. Intense activation of pulmonary phagocytes may also lead to functional downregulation of complement receptors” and consequent suppression of receptor-mediated phago- cytosis. Smoke exposure reduced Fc receptor-mediated phagocytosis in rabbit AM. Such responses would undoubtedly contribute to the development of bron- chopneumonia (stage 3) after smoke inhalation. Overall, the data suggest that early intervention to modulate AM responses, specifically to upregulate phagocytosis while preventing hyperreactivity of oxidant and cytokine
106 Burns:Vol.22,No.2,1996
release, may benefit the management of patients with smoke inhalation lung injury.
Acknowledgements Dr Julie Horton (Scaly Center for Molecular Science) gen- erously provided access to the Coulter Multisizer. Techni- cal assistance was provided by Serina Flores, Jessie George and Tammy Wheeler. This work was supported in part by a grant from the Shriners Hospital for Crippled Children.
References 1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
Hemdon DN. Langner F, Thompson P, Linares HA, Stein M, Traber DL. Pulmonary injury in burned patients. Surg Clin Norfk Am 1987; 67: 31-46. Thompson PB, Hemdon DH, Traber DL, Abston S. Effect on mortality of inhalation injury. J Trauma 1986; 26: 163-165. Traber DL, Linares HA, Hemdon DN, Prien T. The patho- physiology of inhalation injury - a review. Burns 1988; 14: 357-364.
23
Barrow RE, Morris SE, Basadre JO, Herndon DN. Selective permeability changes in the lungs and airways of sheep after toxic smoke inhalation. J Appl Pkysioll990; 68: 2165-2170. Bidani A, Brown SES, Heming TA, Gurich R, DuBose TD Jr. Cytoplasmic pH in pulmonary macrophages: recovery from acid load is Naf independent and NEM sensitive. Am J Pkysiol 1989; 257: C65bC76. Flick DA, Gifford GE. Comparison of in vitro cell cytotoxic assays for tumor necrosis factor. J lmmunol Methods 1984; 68:167-175.
24
25
Stone HH, Martin JD. Pulmonary injury associated with thermal burns. Srnrg Gyrrecol Obsfet 1969; 129: 1242-1246. Stone HH, Rhame DW, Corbitt JD* Given KS, Martin JD. Respiratory bums: a correlation of clinical and laboratory results. Ann Surg 1967; 165: 157-168. Pruitt BA Jr, Erickson DR, Morris A. Progressive pulmonary insufficiency and other complications of thermal injury. J Trauma 1975; 15: 369-379. Green GM. In defense of the lung. Am Rev Resp Dis 1970; 1023691-703.
26
27
Stein M, Keshav S, Gordon S. Macrophage activation assays. In: Blackwill FR, ed. Cyfokines. New York: IRL Press, 1991; pp 245-252. Oben JA, Foreman JC. A simple quantitative fluorometric assay of in vitro phagocytosis in human neutrophils. JImmunolMefkods 1988; 112:99-103. Bidani A, Brown SES. ATP-dependent pHi recovery in lung .macrophages: evidence for a plasma membrane H+ -ATPase. .AmJPkysiol 1990; 259: C586-C598. Davies CF, Moore FD, Rodrick ML, Fearon DT, Mannick JA. Neutrophil activation after bum injury: contributions of the classic complement pathway and of endotoxin. Surgery 1987; X02:477-484.
28
Takemura R, Werb Z. Secretory products of macrophages and their physiological functions. Am J Pkysiol 1984; 246: Cl-c9.
29
Demling RH. Current concepts on the adult respiratory dis- tress syndrome. Circ Shock 1990; 30: 297-309. Ogle CK, Ogle JD, Keynton L, Nagy H, Alexander JW. The effect of TP-5 on the production of C3, PGE2, and TXB2 by macrophages obtained from burned guinea pigs. J Btirn Care Rekabill989; 10:146-150. Bonney RJ, Opas EE, Humes JL. Lipoxygenase pathways of macrophages. Fed Proc 1985; 44: 2933-2936. Maier RV, Hahnel GB. Microthrombosis during endotoxe- mia: potential role of hepatic versus alveolar macrophages. JSurg Res 1984;36: 362-370.
30
31
Loose LD, Turinsky J. Depression of the respiratory burst in alveolar and peritoneal macrophages after thermal injury. infection lmmunify 1980; 30: 718-722. Lilly CM, Sandhu JS, Ishizaka A et al. Pentoxifylline prevents tumor necrosis factor-induced lung injury. Am Rev Resp Dis 1989; 139:1361-2368. Schade UF. Pentoxifylline increases survival in murine endotoxin shock and decreases formation of tumor necrosis factor. Circ Shock 1990; 31: 171-181. Harada H, Ishizaka A, Yonemaru M et al. The effects of aminophylline and pentoxyfylline on multiple organ damage after Esckerickia coli sepsis. Am Rev Resp Dis 1989; 140: 974-980.
32
Elias JA, Gustilo K, Freundlich B. Human alveolar macro- phage and blood monocyte inhibition of fibroblast prolifer- ation. Evidence for synergy between interleukin-1 and tumor necrosis factor. Am Rev Resp dis 1988; 138: 1595-1603. Rich EA, Tweardy DJ, Fujiwara H, Ellner JJ. Spectrum of immunoregulatory functions and properties of human alve- olar macrophages. Am Rev Resp Dis 1987; 136: 258-265. Doherty GM, Jensen JC, Alexander HR. Buresh CM, Norton JA. Pentoxifylline suppression of tumor necrosis factor gene transcription. Surgery 1991; 110: 192-198. Clark CJ, Pollock AJ, Reid WH, Campbell D. Role of pulmon- ary alveolar macrophage activation in acute lung injury after bums and smoke inhalation. Lancet 1988; ii: 872-874. Fick RB Jr, Paul ES, Merrill WW, Reynolds HY, Loke JSO. Alterations in the antibacterial properties of rabbit pulmon- ary macrophages exposed to wood smoke. Am Rev Resp Dis 1984; 129: 76-81.
33
Williams JG, Bankey PE, Mine JP. Burn injury enhances alveolar macrophage endotoxin sensitivity. Proc Am Burn ~tisoc 1994; 26: 32 (abstr). Strieter RM, Remick DG, Ward PA et al. Cellular and mol- ecular regulation of tumor necrosis factor-alpha production by pentoxifylline. Biockem Biopkys Res Commun 1988; 155: 1230-1236.
34
35
36
Spatafora M, Chiappara G, Merendino AM, Amico DD, Bellia V and Bonsignore G. Theophylline suppresses the release of tumor necrosis factor-a by blood monocytes and alveolar macrophages. Eur Resp J 1994; 7: 223-228. Demarest GB, Hudson LD, Altman LC. Impaired alveolar rnacrophage chemotaxis in patients with acute smoke inhala- tion. Am Rev Resp Dis 1979; 119: 279-286. Ferrandiz ML, Foster SJ. Tumor necrosis factor production in a rat airpouch model of inflammation: role of eicosanoids. Agents Actions 1991; 32: 289-294.
Paper accepted after revision 20 June 1995.
Witten ML, Lantz RC, Grad R et al. Effect of smoke inhala- Correspondence should be addressed to: A. Bidani, Department of tion on immediate changes in lung chemical mediators. Res Internal Medicine, University of Texas Medical Branch, Gal- Commun Ckem Pafkol Pkarmacol 1991; 74: 259-272. veston, TX 77555-0561. USA.
19
20
21
22
Riyami BMS, Kinsella J, Pollok AJ et al. Alveolar macrophage chemotaxis in fire victims with smoke inhalation and bums injury. Eur J Clin Invesf 1991; 21: 485-489. Wang CZ, Hemdon DN, Mifflin RC, Yang SF, Barrow RE. Triiodothyronine enhances surfactant protein C mRNA expression after smoke inhalation. J Trauma 2993; 35: 172 (abstr).








![Activation of Alveolar Macrophage Tumoricidal …...[14C]DSPC. All counts were quench-corrected before calculation of in corporation or recovery. Freeze-dried liposomes were prepared](https://static.documents.pub/doc/80x56/5f22ac6bc2a49c46796e8cca/activation-of-alveolar-macrophage-tumoricidal-14cdspc-all-counts-were-quench-corrected.jpg)












