Ecophysiology of FreshwaterVerrucomicrobia Inferred fromMetagenome-Assembled Genomes
Shaomei He,a,b Sarah L. R. Stevens,a Leong-Keat Chan,d Stefan Bertilsson,c
Tijana Glavina del Rio,d Susannah G. Tringe,d Rex R. Malmstrom,d
Katherine D. McMahona,e
Department of Bacteriology, University of Wisconsin—Madison, Madison, Wisconsin, USAa; Department ofGeoscience, University of Wisconsin—Madison, Madison, Wisconsin, USAb; Department of Ecology andGenetics, Limnology and Science for Life Laboratory, Uppsala University, Uppsala, Swedenc; DOE JointGenome Institute, Walnut Creek, California, USAd; Department of Civil and Environmental Engineering,University of Wisconsin—Madison, Madison, Wisconsin, USAe
ABSTRACT Microbes are critical in carbon and nutrient cycling in freshwater eco-systems. Members of the Verrucomicrobia are ubiquitous in such systems, and yettheir roles and ecophysiology are not well understood. In this study, we recovered19 Verrucomicrobia draft genomes by sequencing 184 time-series metagenomesfrom a eutrophic lake and a humic bog that differ in carbon source and nutrientavailabilities. These genomes span four of the seven previously defined Verrucomi-crobia subdivisions and greatly expand knowledge of the genomic diversity offreshwater Verrucomicrobia. Genome analysis revealed their potential role as(poly)saccharide degraders in freshwater, uncovered interesting genomic featuresfor this lifestyle, and suggested their adaptation to nutrient availabilities in their en-vironments. Verrucomicrobia populations differ significantly between the two lakes inglycoside hydrolase gene abundance and functional profiles, reflecting the autoch-thonous and terrestrially derived allochthonous carbon sources of the two eco-systems, respectively. Interestingly, a number of genomes recovered from thebog contained gene clusters that potentially encode a novel porin-multiheme cy-tochrome c complex and might be involved in extracellular electron transfer inthe anoxic humus-rich environment. Notably, most epilimnion genomes have largenumbers of so-called “Planctomycete-specific” cytochrome c-encoding genes, whichexhibited distribution patterns nearly opposite to those seen with glycoside hydro-lase genes, probably associated with the different levels of environmental oxygenavailability and carbohydrate complexity between lakes/layers. Overall, the recoveredgenomes represent a major step toward understanding the role, ecophysiology, anddistribution of Verrucomicrobia in freshwater.
IMPORTANCE Freshwater Verrucomicrobia spp. are cosmopolitan in lakes and rivers,and yet their roles and ecophysiology are not well understood, as cultured freshwa-ter Verrucomicrobia spp. are restricted to one subdivision of this phylum. Here, wegreatly expanded the known genomic diversity of this freshwater lineage by recov-ering 19 Verrucomicrobia draft genomes from 184 metagenomes collected from aeutrophic lake and a humic bog across multiple years. Most of these genomes repre-sent the first freshwater representatives of several Verrucomicrobia subdivisions.Genomic analysis revealed Verrucomicrobia to be potential (poly)saccharide degrad-ers and suggested their adaptation to carbon sources of different origins in the twocontrasting ecosystems. We identified putative extracellular electron transfer genesand so-called “Planctomycete-specific” cytochrome c-encoding genes and identifiedtheir distinct distribution patterns between the lakes/layers. Overall, our analysisgreatly advances the understanding of the function, ecophysiology, and distribution
Received 28 June 2017 Accepted 5September 2017 Published 27 September2017
Citation He S, Stevens SLR, Chan L-K, BertilssonS, Glavina del Rio T, Tringe SG, Malmstrom RR,McMahon KD. 2017. Ecophysiology offreshwater verrucomicrobia inferred frommetagenome-assembled genomes. mSphere2:e00277-17. https://doi.org/10.1128/mSphere.00277-17.
Editor Steven J. Hallam, University of BritishColumbia
Verrucomicrobia are ubiquitous in freshwater and exhibit a cosmopolitan distributionin lakes and rivers. They are present in up to 90% of lakes (1), with abundances that
are typically between �1% and 6% of total microbial communities (2–4) and as high as19% in a humic lake (5). Yet Verrucomicrobia have received less attention than otherfreshwater bacterial groups, such as members of the Actinobacteria, Cyanobacteria, andProteobacteria phyla, and their functions and ecophysiology in freshwater are not wellunderstood.
As a phylum, Verrucomicrobia was first proposed relatively recently, in 1997 (6).Together with Planctomycetes, Chlamydiae, and sister phyla such as Lentisphaerae, theycomprise the Planctomycetes-Verrucomicrobia-Chlamydiae (PVC) superphylum. In addi-tion to being cosmopolitan in freshwater, Verrucomicrobia have been found in oceans(7, 8), soil (9, 10), wetlands (11), rhizosphere (12), and animal guts (13, 14) as free-livingorganisms or symbionts of eukaryotes. Verrucomicrobia isolates are metabolically di-verse and include aerobes, facultative anaerobes, and obligate anaerobes, and they aremostly heterotrophs, using various mono-, oligo-, and polysaccharides for growth (6, 7,11, 14–20). Not long ago, an autotrophic verrucomicrobial methanotroph (Methylacid-iphilum fumariolicum SolV) was discovered in acidic thermophilic environments (21).
Verrucomicrobia are also ubiquitous in marine environments (22) and have beensuggested to have a key role as polysaccharide degraders (23, 24). Genomic insightsgained through sequencing single cells (24) or extracting Verrucomicrobia bins frommetagenomes (25) have revealed high abundances of glycoside hydrolase (GH) genes,providing more evidence for their critical roles in carbon (C) cycling in marine envi-ronments.
In freshwater, Verrucomicrobia have been suggested to degrade glycolate (26) andpolysaccharides (24). The abundance of some phylum members was favored by highnutrient availabilities (27, 28), cyanobacterial blooms (29), low pH, high temperature,high hydraulic retention time (30), and more-labile dissolved organic carbon (DOC)(5). To date, very few freshwater Verrucomicrobia have been isolated, including Verru-comicrobium spinosum (31) and several Prosthecobacter spp. (6). Physiological studiesshowed that they are aerobes, primarily using carbohydrates but not amino acids,alcohols, or organic acids for growth. However, those few cultured isolates representonly a single clade within subdivision 1. In contrast, 16S rRNA gene-based studiesdiscovered a much wider phylogenic range of freshwater Verrucomicrobia, includingspecies representing subdivisions 1, 2, 3, 4, 5, and 6 (3–5, 24, 32). Due to the very fewcultured representatives and few available genomes from this freshwater lineage, theecological functions of the vast uncultured freshwater Verrucomicrobia are largelyunknown.
In this study, we sequenced a total of 184 metagenomes in a time-series study oftwo lakes with contrasting characteristics, particularly differing in C sources, nutrientavailabilities, and pH. We recovered a total of 19 Verrucomicrobia draft genomesspanning subdivisions 1, 2, 3, and 4 of the seven previously defined Verrucomicrobiasubdivisions. We inferred their metabolisms, revealed their adaptation to C and nutrientconditions, and uncovered some interesting and novel features, including a novelputative porin-multiheme cytochrome c (porin-MHC) system that may be involved inextracellular electron transfer (EET). The insights that were gained advanced ourunderstanding of the ecophysiology, potential roles, and ecological niches of thisubiquitous freshwater bacterial group.
RESULTS AND DISCUSSIONComparison of the two lakes. The two studied lakes exhibited contrasting char-
acteristics (Table 1). The most notable differences are the primary C sources and
nutrient availabilities. Mendota is an urban eutrophic lake, with most of its C beingautochthonous (produced in-lake through photosynthesis). In contrast, Trout Bog is anutrient-poor dystrophic lake, surrounded by temperate forests and sphagnum matsand thus receiving large amounts of terrestrially derived allochthonous C that is rich inhumic and fulvic acids. Trout Bog features higher DOC levels than Mendota but is morelimited in nutrient availability, with much higher DOC/total nitrogen (TN) and DOC/totalphosphorus (TP) ratios (Table 1). Nutrient limitation in Trout Bog is even more extremethan is revealed by these ratios because much of the N and P is tied up in complexdissolved organic matter. In addition, Trout Bog has lower levels of oxygenic photo-synthesis due to decreased levels of photosynthetically active radiation (PAR) as a resultof absorption by DOC (33). Together with the consumption of dissolved oxygen byheterotrophic respiration, oxygen levels decrease quickly with depth in the watercolumn in Trout Bog. Dissolved oxygen is below detectable levels in the hypolimnionnearly year-round (34). Due to these contrasts, we expected to observe differences inbacterial C and nutrient use, as well as differences between these two lakes reflectingthe electron acceptor conditions. Hence, the retrieval of numerous Verrucomicrobiadraft genomes in the two lakes not only allows the prediction of their general functionsin freshwater but also provides an opportunity to study their ecophysiological adap-tation to the local environmental differences.
Retrieval of Verrucomicrobia draft genomes and their distribution patterns. Atotal of 184 metagenomes, including 94 from the top 12 m of Mendota (mostlyconsisting of the epilimnion layer and therefore referred to here as “ME”), 45 from TroutBog epilimnion (“TE”), and 45 from Trout Bog hypolimnion (“TH”), were generated fromsamples collected across multiple years. Three combined assemblies were generated bycoassembling reads from all metagenomes within the ME, TE, and TH groups, respec-tively. Using binning facilitated by tetranucleotide frequency data and relative abun-dance patterns determined over time, a total of 19 Verrucomicrobia metagenome-assembled genomes (MAGs) were obtained, including 8 from the combined assemblyof ME, 3 from the combined assembly of TE, and 8 from the combined assembly of TH(Table 2). The 19 MAGs exhibited a clustering of their tetranucleotide frequency datalargely based on the two lakes (see Fig. S1 in the supplemental material), suggestingdistinct overall genomic signatures associated with each system.
Genome completeness of the 19 MAGs ranged from 51% to 95%, as determined bycheckM (35). Phylogenetic analysis of these MAGs using a concatenated alignment oftheir conserved genes indicates that they span a wide phylogenetic spectrum anddistribute in subdivisions 1, 2, 3, and 4 of the seven previously defined Verrucomicrobiasubdivisions (5, 21, 36) (Fig. 1), as well as three unclassified Verrucomicrobia MAGs.
Presently available freshwater Verrucomicrobia isolates are restricted to subdivision1. The recovered MAGs allow the inference of metabolisms and ecology of a consid-
TABLE 1 Lakes included in this studya
Lake parameter
Result
Mendota Trout Bog
GPS location 43.100°N, 89.405°W 46.041°N, 89.686°WLake type Drainage lake Seepage lakeSurface area (ha) 3,938 1.1Mean depth (m) 12.8 5.6Max depth (m) 25.3 7.9pH 8.3 5.2Primary carbon source Phytoplankton Terrestrial subsidiesDOC (mg/liter) 5.0 20.0Total N (mg/liter) 1.5 1.3Total P (�g/liter) 131 71DOC/N 3.3 15.6DOC/P 38.0 281.9Trophic state Eutrophic DystrophicaData are from NTL-LTER (https://lter.limnology.wisc.edu) and were averaged from the study years. DOC, dis-solved organic carbon; GPS, Global Positioning System; N, nitrogen; P, phosphorus.
erable diversity within uncultured freshwater Verrucomicrobia. Notably, all MAGs fromsubdivision 3 were recovered from TH, and all MAGs from subdivision 1, except TH2746,were from the epilimnion (either ME or TE), indicating differences in phylogeneticdistributions between lakes and between layers within a lake.
We used normalized coverage depths of MAGs within individual metagenomescollected at different sampling time points and in different lakes/layers to compara-tively infer relative population abundances across time and space (see detailed cover-age depth estimations in Text S1 in the supplemental material). Briefly, we mappedreads from each metagenome to MAGs with a minimum identity of 95% and used thenumber of mapped reads to calculate the relative abundance for each MAG based oncoverage depth per contig and several normalization steps. Thus, we assumed thateach MAG represents a distinct population within the lake layer from which it wasrecovered (37, 38). This estimate does not directly indicate the actual relative abun-dances of these populations within the total community per se; rather, it allows us tocompare the levels of abundance of populations from different lakes and samplingoccasions within the set of 19 MAGs. This analysis indicates that Verrucomicrobiapopulations in Trout Bog were proportionally more abundant and persistent over timethan those in Mendota in general (Table 2). Verrucomicrobia populations in Mendotaboosted their abundances once to a few times during the sampling season anddiminished to extremely low levels for the remainder of the sampling season (generallyMay to November), as reflected by the low median coverage depth of Mendota MAGsand their large coefficient of variation (Table 2).
Saccharolytic lifestyle and adaptation to different C sources. Verrucomicrobia
isolates from different environments are known to grow on various mono-, oligo-, andpolysaccharides but are unable to grow on amino acids, alcohols, or most organic acids(6, 7, 11, 14–20, 39). Culture-independent research suggests that marine Verrucomicro-bia are candidate polysaccharide degraders with large numbers of genes involved inpolysaccharide utilization (23–25).
TABLE 2 Summary of Verrucomicrobia MAGsa
GenomeIMG taxonOID Subdivision
RecoveredMAG size(Mbp)b
Genomecompletenessestimate (%)c
Genomecontaminationestimate (%)c
GCcontent(%)
Codingbase(%)
Genecount
Normalized coveragedepthd
Median MeanCoefficient ofvariation (%)
ME3880 2582580573 1 1.6 70 2 58 90.9 1,585 0.2 2.9 217TH2746 2582580664 1 6.5 81 3 62 86.7 5,430 3.3 4.7 82ME12612 2582580523 1 2.2 79 3 59 89.0 2,335 0.0 0.9 261ME12173 2582580521 1 2.1 63 3 52 91.3 2,070 0.0 0.8 583TE4605 2582580638 1 4.7 91 0 59 91.1 4,380 1.0 4.8 198ME6381 2582580593 1 2.4 62 0 57 92.5 2,221 0.0 0.4 285ME8366 2582580607 2 3.6 87 5 63 87.4 3,450 0.0 1.2 326TH2747 2582580665 3 5.2 93 8 58 89.6 4,846 1.8 2.8 99TH3004 2582580668 3 4.5 93 6 57 91.4 3,798 1.9 5.8 139TH0989 2556921153 3 7.2 91 8 62 90.3 5,583 6.1 6.3 61TH2519 2593339181 4 1.8 69 2 42 94.3 1,654 6.2 6.7 60TE1800 2593339189 4 2.2 84 2 42 94.3 1,998 10.8 11.3 77TH4590 2582580688 4 3.3 87 1 65 90.7 3,132 2.4 3.6 99ME2014 2582580546 4 1.9 77 5 66 93.7 1,700 1.0 3.3 174ME12657 2582580524 4 1.9 81 7 68 94.0 1,838 0.0 0.8 344TE1301 2582580616 4 2.0 95 0 54 94.7 1,943 4.1 14.2 187TH4093 2582580682 Unclassified 4.7 77 6 48 86.5 3,982 4.3 3.9 61ME30509 2582580559 Unclassified 1.2 51 2 63 92.3 1,160 0.0 0.5 479TH4820 2582580691 Unclassified 3.0 56 3 63 86.7 2,794 1.0 2.1 110aMAGs from Lake Mendota are indicated in boldface.bRecovered MAG size data represent the sum of the length of all contigs within a MAG.cGenome completeness and contamination were estimated with checkM using Verrucomicrobia-specific marker gene sets.dNormalized coverage depths of MAGs were calculated from the 94, 45, or 45 individual ME, TE, or TH metagenomes, respectively, and were used to comparativelyinfer relative population abundances at the different sampling points. In addition to the median and mean coverage depths, the coefficient of variation is also shownto indicate variation among the sampling points.
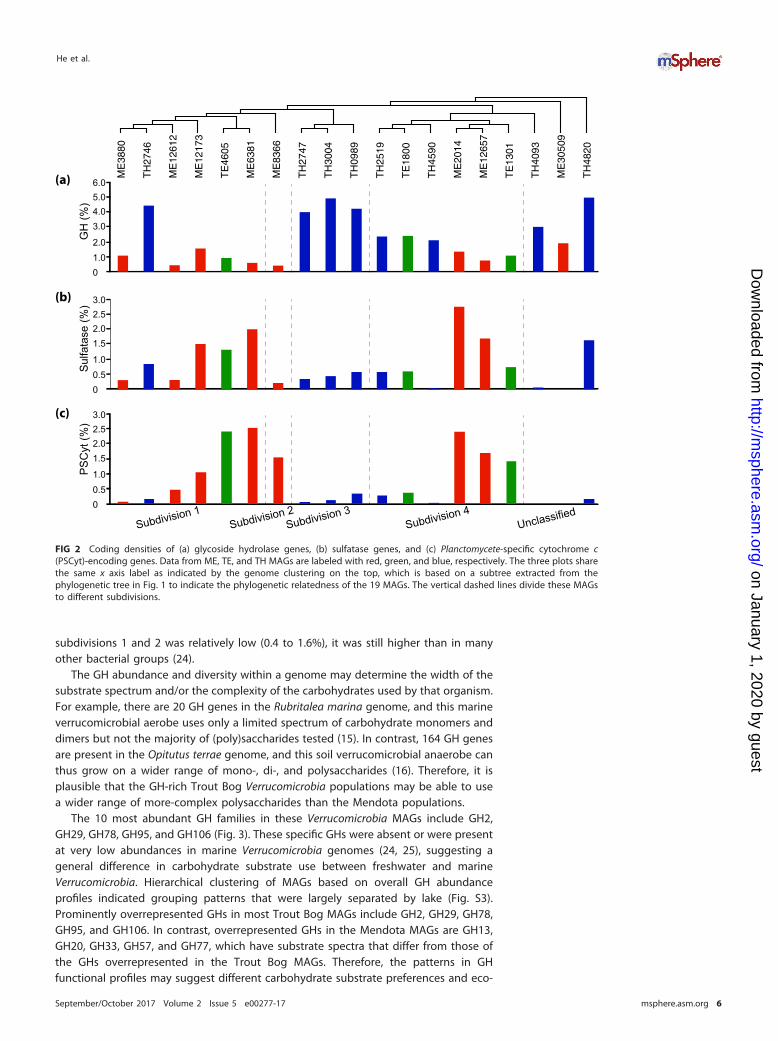
In the 19 Verrucomicrobia MAGs, we observed rich arrays of GH genes, representinga total of 78 different GH families acting on diverse polysaccharides (Fig. S2). Althoughthese genomes have different degrees of completeness, genome completeness wasnot correlated with the number of GH genes recovered (correlation coefficient � 0.312,P value � 0.194) or with the number of GH families represented in each MAG (i.e., GHdiversity; correlation coefficient � 0.278, P value � 0.250). To compare GH abundanceamong MAGs, we normalized GH occurrence frequencies by the total number of genesin each MAG to estimate the percentage of genes annotated as GHs (i.e., GH codingdensity) to account for the different genome sizes and completeness levels. Thisnormalization method assumes that GH genes are randomly distributed between therecovered and the missing parts of the genome, and it allows us to make some generalcomparisons among these MAGs. GH coding density ranged from 0.4% to 4.9% forthese MAGs (Fig. 2a) and, in general, was higher in Trout Bog MAGs than in MendotaMAGs. Notably, six TH MAGs had extremely high (~4%) GH coding densities (Fig. 2a),with each MAG harboring 119 to 239 GH genes, representing 36 to 59 different GHfamilies (Fig. 3 and Fig. S2). Although the GH coding density in most ME genomes in
FIG 1 Phylogenetic tree constructed with a concatenated alignment of protein sequences from five conserved essential single-copygenes (represented by TIGR01391, TIGR01011, TIGR00663, TIGR00460, and TIGR00362) that were recovered in all Verrucomicrobia MAGs.ME, TE, and TH MAGs are labeled with red, green, and blue, respectively. Genome ID in IMG or NCBI is indicated in the bracket. Theoutgroup is Kiritimatiella glycovorans L21-Fru-AB, which was initially assigned to subdivision 5, but this subdivision was recently proposedas a novel sister phylum to Verrucomicrobia (67).
subdivisions 1 and 2 was relatively low (0.4 to 1.6%), it was still higher than in manyother bacterial groups (24).
The GH abundance and diversity within a genome may determine the width of thesubstrate spectrum and/or the complexity of the carbohydrates used by that organism.For example, there are 20 GH genes in the Rubritalea marina genome, and this marineverrucomicrobial aerobe uses only a limited spectrum of carbohydrate monomers anddimers but not the majority of (poly)saccharides tested (15). In contrast, 164 GH genesare present in the Opitutus terrae genome, and this soil verrucomicrobial anaerobe canthus grow on a wider range of mono-, di-, and polysaccharides (16). Therefore, it isplausible that the GH-rich Trout Bog Verrucomicrobia populations may be able to usea wider range of more-complex polysaccharides than the Mendota populations.
The 10 most abundant GH families in these Verrucomicrobia MAGs include GH2,GH29, GH78, GH95, and GH106 (Fig. 3). These specific GHs were absent or were presentat very low abundances in marine Verrucomicrobia genomes (24, 25), suggesting ageneral difference in carbohydrate substrate use between freshwater and marineVerrucomicrobia. Hierarchical clustering of MAGs based on overall GH abundanceprofiles indicated grouping patterns that were largely separated by lake (Fig. S3).Prominently overrepresented GHs in most Trout Bog MAGs include GH2, GH29, GH78,GH95, and GH106. In contrast, overrepresented GHs in the Mendota MAGs are GH13,GH20, GH33, GH57, and GH77, which have substrate spectra that differ from those ofthe GHs overrepresented in the Trout Bog MAGs. Therefore, the patterns in GHfunctional profiles may suggest different carbohydrate substrate preferences and eco-
01.02.0
3.04.05.06.0
GH
(%)
00.51.0
1.52.02.53.0
PS
Cyt
(%)
00.51.0
1.52.02.53.0
Sul
fata
se (%
)
Subdivision 1Subdivision 2
Subdivision 4Subdivision 3
Unclassified
(a)
(b)
(c)
ME
3880
TH
2746
ME
1261
2
ME
1217
3
TE
4605
ME
6381
ME
8366
TH
2747
TH
3004
TH
0989
TH
2519
TE
1800
TH
4590
ME
2014
ME
1265
7
TE
1301
ME
3050
9
TH
4820
TH
4093
FIG 2 Coding densities of (a) glycoside hydrolase genes, (b) sulfatase genes, and (c) Planctomycete-specific cytochrome c(PSCyt)-encoding genes. Data from ME, TE, and TH MAGs are labeled with red, green, and blue, respectively. The three plots sharethe same x axis label as indicated by the genome clustering on the top, which is based on a subtree extracted from thephylogenetic tree in Fig. 1 to indicate the phylogenetic relatedness of the 19 MAGs. The vertical dashed lines divide these MAGsto different subdivisions.
logical niches occupied by Verrucomicrobia, probably reflecting the different carbohy-drate compositions derived from different sources between Mendota and Trout Bog.
Overall, GH diversity and abundance profile may reflect the DOC availability andchemical variety and complexity and may suggest microbial adaptation to different Csources in the two ecosystems. We speculate that the rich arrays of GH genes andpresumably broader substrate spectra of Trout Bog populations partly contribute totheir higher abundance and persistence over the sampling season (Table 2), as they areless likely impacted by fluctuations of individual carbohydrates. In contrast, Mendotapopulations with fewer GHs and presumably more-specific substrate spectra may relyon autochthonous C and therefore exhibit a “bloom-and-bust” abundance pattern(Table 2) that might be associated with cyanobacterial blooms as previously suggested(29). On the other hand, bogs experience seasonal phytoplankton blooms (40, 41) thatintroduce brief pulses of autochthonous C to these otherwise allochthonously drivensystems. Clearly, much remains to be learned about the routes through which C ismetabolized by bacteria in such lakes, and comparative genomics is a novel way to usethe organisms to tell us about C flow through the ecosystem.
Other genome features of the saccharide-degrading lifestyle. Seven Verrucomi-crobia MAGs spanning subdivisions 1, 2, 3, and 4 possess genes needed to constructbacterial microcompartments (BMCs), which are quite rare among studied bacteriallineages. Such BMC genes in planctomycetes are involved in the degradation of plantand algal cell wall sugars and are required for growth on L-fucose, L-rhamnose, andfucoidans (42). Genes involved in L-fucose and L-rhamnose degradation cluster withBMC shell protein-coding genes in the seven Verrucomicrobia MAGs (Fig. 4). This isconsistent with the high abundance of �-L-fucosidase or �-L-rhamnosidase GH genes(represented by GH29, GH78, GH95, and GH106) in most of these MAGs (Fig. 3),suggesting the importance of fucose- and rhamnose-containing polysaccharides forthese Verrucomicrobia populations.
Total counts of all GH genes 17 239 10 32 40 13 14 193 186 235 39 48 66 23 14 21 119 22 138
Total number of GH families represented 12 48 7 23 23 7 10 53 49 58 19 21 26 10 10 13 35 15 45
FIG 3 Gene counts for the top 10 most abundant GH families, total gene counts for all GH families, and the number of GH families represented by these genes.MAGs are ordered as in the clustering in Fig. 2. Data were shaded in grayscale according to the value, with the largest value shaded with the darkest color (black)and the smallest value with the brightest color (white) to highlight the difference. The shading scale was applied among the counts of the top 10 GHs, as wellas among total counts of all GHs and among total number of GH families.
TonB-dependent receptor (TBDR) genes were found in Verrucomicrobia MAGs andare present at over 20 copies in TE1800 and TH2519. TBDRs are located on the outercellular membrane of Gram-negative bacteria, usually mediating the transport of ironsiderophore complex and vitamin B12 across the outer membrane through an activeprocess. More recently, TBDRs were suggested to be involved in carbohydrate transportacross the outer membrane by some bacteria that consume complex carbohydrates,and in their carbohydrate utilization (CUT) loci, TBDR genes usually cluster with genesencoding inner membrane transporters, GHs, and regulators for efficient carbohydratetransportation and utilization (43). Such novel CUT loci are present in TE1800 andTH2519, with TBDR genes clustering with genes encoding inner membrane sugartransporters, monosaccharide utilization enzymes, and GHs involved in the degradationof pectin, xylan, and fucose-containing polymers (Fig. 5). Notably, most GHs in the CUTloci are predicted to be extracellular or outer membrane proteins (Fig. 5), catalyzingextracellular hydrolysis reactions to release mono- and oligosaccharides, which aretransported across the outer membrane by TBDR proteins. Therefore, such CUT loci may
L-fucose/L-rhamnose
fucolose-1~P/rhamnulose-1~P
lactaldehyde
1,2-propanediol
lactyl~CoA
lactyl~P lactate pyruvate
ME12173(ME12173DRAFT_00626)
ME3880(ME3880DRAFT_00619)
TH2746(TH2746DRAFT_03121, TH2746DRAFT_01404)
ME8366(ME8366DRAFT_02738)
TH4590(TH4590DRAFT_01817, TH4590DRAFT_00413)
TH2747(TH2747DRAFT_01686)
TH3004(TH3004DRAFT_00386)
FIG 4 Gene clusters encoding bacterial microcompartments (BMCs) involved in L-fucose and L-rhamnose degradation. The verticalline indicates the end of a contig, and IMG gene locus tag for the first gene in each presented gene cluster is indicated inparentheses. The BMC is schematically represented by a hexagon with the two building blocks labeled in red and green,respectively. The two building blocks and reactions inside the BMC are indicated with colored labels corresponding to the colorsused with the encoding genes on the left side.
Carbohydrate active enzyme
TBDR
Sugar transporter
Hexose metabolism
Other functions or unknown
Extracellular or outer membrane protein
Legend
TH2519 / TE1800GH92
TBDRGH78
GH28Putative GH
GH65
(TH02519DRAFT_00824 / TE01800DRAFT_01394)
TH2519 / TE1800GH106
GH28Rhamnose transport
LIpaseRhamnose isomerase
TBDRSugar tra
nsport
GH28GH2 Rhamnose mutarotase
(TH02519DRAFT_00400 / TE01800DRAFT_01044)
TE1800Sugar tra
nsport
GH29GH28
Fucose permease
acetyl xylan esterase
TBDRGH43
TBDRGH28
GH28Fucose isomerase
TBDRPectin lyase
(TE01800DRAFT_00147)
FIG 5 Gene clusters encoding putative tonB-dependent carbohydrate utilization (CUT) loci. The IMG gene locus tag for the first gene in each presented genecluster is indicated in parentheses. The horizontal solid lines below the gene designations indicate predicted extracellular or outer membrane proteins.
allow these verrucomicrobial populations to coordinately and effectively scavenge thehydrolysis products before they diffuse away.
Genes encoding inner membrane carbohydrate transporters are abundant in Ver-rucomicrobia MAGs (Fig. S4). The Embden-Meyerhof pathway for glucose degradation,as well as pathways for degrading a variety of other sugar monomers, includinggalactose, rhamnose, fucose, xylose, and mannose, were recovered (in complete orpartly complete form) in most MAGs (Fig. 6). As these sugars are abundant carbohy-drate monomers in plankton and plant cell walls, the presence of these pathways,together with that of GH genes, suggests that these Verrucomicrobia populations mayuse plankton- and plant-derived saccharides. Machineries for pyruvate degradation toacetyl-coenzyme A (acetyl-CoA) and for the tricarboxylic acid (TCA) cycle are present inmost MAGs. These results are largely consistent with their hypothesized role in carbo-hydrate degradation and previous studies on Verrucomicrobia isolates.
Notably, a large number of genes encoding proteins belonging to a sulfatase family(pfam00884) are present in the majority of MAGs (Fig. 2b), which is similar to the highrepresentation of these genes in marine Verrucomicrobia genomes (24, 25). Sulfataseshydrolyze sulfate esters, which are rich in sulfated polysaccharides. In general, sulfatedpolysaccharides are abundant in marine algae and plants (mainly in seaweeds) (44) buthave also been found in some freshwater cyanobacteria (45) and plant species (46).Sulfatase genes in our Verrucomicrobia MAGs were often located in the same neigh-borhood as genes encoding extracellular proteins with a putative pectin lyase activity,proteins with a carbohydrate-binding module (pfam13385), GHs, and proteins with
FIG 6 Completeness estimates of key metabolic pathways. Data were shaded in grayscale according to the value, with “1” shaded with the darkest color (black)and “0” with the brightest color (white) to highlight the difference. A completeness value of “1” indicates that a pathway is complete; “0” indicates that no geneswere found in that pathway; and “(0)” indicates that, although some genes in a pathway were present, the pathway was likely absent both because signaturegenes for that pathway were not found in that draft genome and because signature genes were missing in more than two-thirds of all draft genomes.
planctomycete-specific cytochrome c (PSCyt) domains (as shown in Fig. 2c and dis-cussed below). Their genome context lends support to the idea of the participation ofthese genes in C and sulfur cycling by degrading sulfated polysaccharides, which canserve as an abundant source of sulfur for cell biosynthesis as well as C for energy andgrowth.
Previously, freshwater Verrucomicrobia were suggested to use the algal exudateglycolate in humic lakes, based on the retrieval of genes encoding subunit D (glcD) ofglycolate oxidase, which converts glycolate to glyoxylate (26). However, these recov-ered genes might not be bona fide glcD genes due to the lack of other essentialsubunits as revealed in our study (see Text S1). Among the MAGs, only TE4605possesses all three essential subunits of glycolate oxidase (glcDEF) (Fig. S5). However,genetic context analysis suggests that TE4605 likely uses glycolate for amino acidassimilation instead of for energy generation (Fig. S5 and Text S1). These results areconsistent with the absence of the glyoxylate shunt in the 19 Verrucomicrobia MAGs,and especially the absence of the malate synthase, which converts glyoxylate to malateto be used through the TCA cycle for energy generation (Fig. S6). Therefore, Verruco-microbia populations represented by the 19 MAGs are not likely key players in glycolatedegradation but are more likely important (poly)saccharide degraders in freshwater, assuggested by the high abundances of GH, sulfatase, and carbohydrate transportergenes, metabolic pathways for degrading diverse carbohydrate monomers, and othergenome features adapted to the saccharolytic lifestyle.
Nitrogen (N) metabolism and adaptation to different N availabilities. MostVerrucomicrobia MAGs in our study did not appear to reduce nitrate or other nitroge-nous compounds, and they seemed to take up and use ammonia (Fig. 6), and occa-sionally amino acids (Fig. S4), as an N source. Further, some Trout Bog populations mayhave additional avenues to generate ammonia, including genetic machineries forassimilatory nitrate reduction in TH2746, nitrogenase genes for nitrogen fixation, andurease genes in some of the Trout Bog MAGs (Fig. 6), probably as adaptions toN-limited conditions in Trout Bog.
Although Mendota is a eutrophic lake, N can become temporarily limiting duringthe high-biomass period when N is consumed by large amounts of phytoplankton andbacterioplankton (47). For some bacteria, when N is temporarily limited while C is inexcess, cells convert and store the extra C as biopolymers. For example, the verruco-microbial methanotroph Methylacidiphilum fumariolicum SolV accumulated a largeamount of glycogen (up to 36% of the total dry weight of cells) when the culture wasgrown under conditions of N limitation (48). Similarly to this verrucomicrobial metha-notroph, genes in glycogen biosynthesis were present in most MAGs from Mendotaand Trout Bog (Fig. 6). Indeed, a glycogen synthesis pathway is also present in mostgenomes of cultivated Verrucomicrobia in the public database (data not shown),suggesting that glycogen accumulation might be a common feature for this phylum tocope with the changing pools of C and N in the environment and to facilitate theirsurvival when either is temporally limited.
Phosphorus (P) metabolism and other metabolic features. Verrucomicrobia pop-ulations represented by these MAGs may be able to survive under low-P conditions, assuggested by the presence of genes responding to P limitation, such as the two-component regulator (phoRB), alkaline phosphatase (phoA), phosphonoacetate hydro-lase (phnA), and the high-affinity phosphate-specific transporter system (pstABC)(Fig. 6). Details of P acquisition and metabolism and other metabolic aspects, such asacetate metabolism, sulfur metabolism, oxygen tolerance, and the presence of thealternative complex III and cytochrome c oxidase genes in the oxidative phosphoryla-tion pathway, are discussed in Text S1 and Fig. S6.
Anaerobic respiration and a putative porin-multiheme cytochrome c system.Respiration using alternative electron acceptors is important for overall lake metabo-lism in the DOC-rich humic Trout Bog, as the oxygen levels decrease quickly with depthin the water column. We therefore searched for genes involved in anaerobic respiration
and found that genes involved in the dissimilatory reduction of nitrate, nitrite, sulfate,sulfite, dimethyl sulfoxide (DMSO), and trimethylamine-N-oxide (TMAO) are largelyabsent in all MAGs (Fig. S6 and Text S1). Compared to those anaerobic processes, genesfor dissimilatory metal reduction are less well understood. In the more extensivelystudied cultured iron [Fe(III)] reducers, outer surface c-type cytochromes (cytc), such asOmcE and OmcS in Geobacter sulfurreducens, are involved in Fe(III) reduction at the cellouter surface (49). Further, a periplasmic multiheme cytochrome c (MHC; e.g., MtrA inShewanella oneidensis and OmaB/OmaC in G. sulfurreducens) can be embedded in aporin (e.g., MtrB in S. oneidensis and OmbB/OmbC in G. sulfurreducens), forming aporin-MHC complex as an extracellular electron transfer (EET) conduit to reduceextracellular Fe(III) (50, 51). Such outer surface cytc and porin-MHC systems involved inFe(III) reduction were also suggested to be important in reducing the quinone groupsin humic substances (HS) at the cell surface (52–54). The reduced HS can be reoxidizedby Fe(III) or oxygen; thus, HS can serve as electron shuttles to facilitate Fe(III) reduction(55, 56) or as regenerable electron acceptors at the anoxic-oxic interface or over redoxcycles (57).
Outer surface cytc or porin-MHC systems homologous to the ones in G. sulfurredu-cens and S. oneidensis are not present in Verrucomicrobia MAGs. Instead, we identifieda novel porin-coding gene clustering with MHC genes in six MAGs (Fig. 7). These porinswere predicted to have at least 20 transmembrane motifs, and their adjacent cytc werepredicted to be periplasmic proteins with eight conserved heme-binding sites. Inseveral cases, a gene encoding an extracellular MHC is also located in the same genecluster. As their gene organization is analogous to that of the porin-MHC gene clustersin G. sulfurreducens and S. oneidensis, we hypothesize that these genes in Verrucomi-crobia may encode a novel porin-MHC complex involved in EET.
As these porin-MHC gene clusters are novel, we further confirmed that they areindeed from Verrucomicrobia. Their containing contigs were classified to Verrucomicro-bia based on the consensus of the best BLASTP hits for genes on these contigs. Notably,the porin-MHC gene cluster was observed only in MAGs recovered from the HS-richTrout Bog, especially from the anoxic hypolimnion environment. Searching the NCBIand IMG databases for the porin-MHC gene clusters homologous to those in Trout Bog,we identified homologs in genomes within the Verrucomicrobia phylum, including
Porin with n transmembraneregions
Multiheme cytochrome c (MHC) with m heme-binding sites
Other functions or unknown
Outer membrane
Legend
Periplasmic
Extracellular
m
n
EA(ox)
Porin
MHCe
e
OM
IM
MHC
EA(red)
EA(ox) EA(red)
ET in IM
208TH2519(TH02519DRAFT_01527)
268 1TH2746(TH2746DRAFT_03829)
208TE1800(TE01800DRAFT_01857)
2889TH2747(TH2747DRAFT_01547)
2789TH3004(TH3004DRAFT_00154)
2688TH0989(TBH0989DRAFT_03834)
248TH0989(TBH0989DRAFT_03575)
Periplasm
Extracellular
FIG 7 Gene clusters encoding putative porin-multiheme cytochrome c complex (PCC). The IMG gene locus tag for the first gene in each presented genecluster is indicated in parentheses. The vertical line indicates the end of a contig, and horizontal lines below gene designations indicate predicted cellularlocations of their encoded proteins. These putative PCC genes are in 18.1-, 9.0-, 6.1-, 18.4-, 70.0-, 10.6-, and 10.8-kbp-long contigs. A hypothesized modelof extracellular electron transfer is shown on the right, with yellow arrows indicating electron flows. “IM” and “OM” refer to inner and outer membranes,respectively, “ET in IM” refers to electron transfer in the inner membrane, and “EA(ox)” and “EA(red)” refer to oxidized and reduced forms of the electronacceptor, respectively.
Opitutus terrae PB90-1 isolated from rice paddy soil, Opitutus sp. strain GAS368 isolatedfrom forest soil, “Ca. Udaeobacter copiosus” recovered from prairie soil, Opititae-40 andOpititae-129 recovered from freshwater sediment, and Verrucomicrobia bacteriumIMCC26134 recovered from freshwater; some of their residing environments are alsorich in HS. Therefore, based on the occurrence pattern of porin-MHC among Verruco-microbia genomes, we hypothesize that such porin-MHCs might participate in EET toHS in anoxic HS-rich environments and that HS may further shuttle electrons to poorlysoluble metal oxides or be regenerated at the anoxic-oxic interface, thereby divertingmore C flux to respiration instead of fermentation and methanogenesis, which couldimpact the overall energy metabolism and greenhouse gas emission in the bogenvironment.
Occurrence of planctomycete-specific cytochrome c and domains. One of theinteresting features of Verrucomicrobia and its sister phyla in the PVC superphylum isthe presence of a number of novel protein domains in some of their member genomes(58, 59). These domains were initially identified in the marine planctomycete Rhodop-irellula baltica (58) and were therefore referred to as Planctomycete specific, althoughsome of them were later identified in other PVC members (59). In our VerrucomicrobiaMAGs, most genes encoding planctomycete-specific cytochrome c domains (PSCyt1 toPSCyt3) also encode other planctomycete-specific domains (PSD1 through PSD5) withvarious domain combinations and arrangements (Fig. 8 and Fig. S7a). Further, PSCyt2-encoding and PSCyt3-encoding genes are usually next to each of two differentfamilies of unknown genes (Fig. S7b). Such conserved domain architectures andgene organizations, as well as their high frequencies of occurrence in some of theVerrucomicrobia MAGs, are intriguing, and yet nothing is known about their func-tions. However, some of the PSCyt-encoding genes also encode protein domainsidentifiable as carbohydrate-binding modules (CBMs), suggesting a role in carbo-hydrate metabolism (see detailed discussion in Text S1).
PPIPSCyt1
PSCyt1 PSCyt2 PSD1
PSCyt1 PSCyt2 PSD1PPI
PSCyt1 PSCyt2 PSD1CBM
PSCyt1 PSCyt2 PSD1 CBM
PSCyt1 PSCyt2 PSD1CBM
PSCyt2 PSD1
PSCyt2 PSD1PPI
PSCyt2 PSD1PPI
PSCyt1 PSD3 PSD5 PSD4 PSCyt3 PSD2
cbb3-III PSD3 PSD5 PSD4 PSCyt3 PSD2
PSD3 PSD5 PSD4 PSCyt3 PSD2
PSD3 PSD5 PSD4 PSCyt3 PSD2CBM
cbb3-III PSD5 PSD4 PSCyt3CBM
PSCyt1 PSCyt2 PSD1PPI
cbb3-III PSD4 PSCyt3
I
II
III
PSD1 PSD4 PSD5PSD2 PSD3Planctomycete-specific domainsPSCyt2 PSCyt3PSCyt1Planctomycete-specific cytochrome c domains
cbb3-type cytochrome c oxidase, subunit III cbb3-III
FIG 8 Domain architecture and occurrence of PSCyt-encoding genes. On the basis of combinations of specific PSCyt and PSD domains, these domainstructures can be classified into three groups (indicated as I, II, and III). “CBM” refers to carbohydrate-binding modules, which include pfam13385(Laminin_G_3), pfam08531 (Bac_rhamnosid_N), pfam08305 (NPCBM), pfam03422 (CBM_6), and pfam07691 (PA14). “PPI” refers to protein-proteininteraction domains, which include pfam02368 (Big_2), pfam00400 (WD40), and pfam00754 (F5_F8_type_C).
The coding density of PSCyt-encoding genes indicates that they tend to be moreabundant in the epilimnion (either ME or TE) genomes (Fig. 2c), and their codingdensities exhibit an inverse correlation with the GH coding density (r � �0.62).Interestingly, sulfatase-coding genes are often in the neighborhood of PSCyt-encodinggenes in ME and TE genomes, whereas sulfatase-coding genes often neighbor with GHgenes in TH genomes. The genomic context suggests that PSCyt-encoding genefunctions somewhat mirror those of GH genes (although their reaction mechanismslikely differ fundamentally). However, these PSCyt-encoding gene products were pre-dicted to be periplasmic or cytoplasmic proteins rather than extracellular or outermembrane proteins. Hence, if they are indeed involved in carbohydrate degradation,they likely act on mono- or oligomers that can be transported into the cell. Further, thedistribution patterns of GH-encoding genes versus PSCyt-encoding genes between theepilimnion and hypolimnion may reflect the differences in oxygen availability andcarbohydrate substrate complexity between the two layers, suggesting some nichedifferentiation within Verrucomicrobia in freshwater systems. Therefore, we suggest thata combination of carbohydrate composition, electron acceptor availability, and Caccessibility drives gene distributions in these populations.
Summary. The recovery of Verrucomicrobia MAGs from the two contrasting lakesgreatly expanded the known genomic diversity of freshwater Verrucomicrobia andrevealed the ecophysiology and some interesting adaptive features of this ubiquitousand yet less-understood freshwater lineage. The overrepresentation of GH, sulfatase,and carbohydrate transporter genes, the genetic potential to use various sugars, andthe presence of microcompartments for fucose and rhamnose degradation suggestthat they are potentially (poly)saccharide degraders in freshwater. Most of the MAGsencode machineries to cope with the changing availability of N and P and can survivenutrient limitation. Despite these generalities, these Verrucomicrobia differ significantlybetween lakes in the abundance and functional profiles of their GH genes, which mayreflect different C sources in the two lakes. Interestingly, a number of MAGs in TroutBog possess gene clusters potentially encoding a novel porin-multiheme cytochrome ccomplex, which might be involved in extracellular electron transfer in the anoxichumus-rich environment. Intriguingly, large numbers of planctomycete-specific cyto-chrome c-encoding genes are present in MAGs from the epilimnion, exhibiting distri-bution patterns nearly opposite to those seen with GH genes. Future studies areneeded to elucidate the functions of these novel and fascinating genomic features.
In this study, we focused on using genome information to infer the ecophysiologyof Verrucomicrobia. The rich time-series metagenome data set and the many diversemicrobial genomes recovered in these two lakes also provide an opportunity for thefuture study of Verrucomicrobia population dynamics in the context of the totalcommunity and their interactions with environmental variables and other microbialgroups.
As some of the MAGs analyzed here represent the first genome representatives ofseveral Verrucomicrobia subdivisions from freshwater, an interesting issue is whetherthe populations represented by the MAGs were native aquatic residents and active inaquatic environment or were merely present after having been washed into the lakefrom surrounding soil. Previous studies on freshwater Verrucomicrobia were largelybased on analysis of 16S rRNA genes, and yet 16S rRNA genes were not recovered inmost MAGs, making it difficult to directly link our MAGs to previously identifiedfreshwater Verrucomicrobia. Notably, our MAGs were related only distantly to theubiquitous and abundant soil Verrucomicrobia species “Ca. Udaeobacter copiosus” (10)(Fig. 1). In addition, Verrucomicrobia were abundant in Trout Bog and other bogs froma 5-year bog lake bacterial community composition and dynamics study (60), withaverage relative abundances of 7.1% and 8.6% and maximal relative abundances of25.4% and 39.5% in Trout Bog epilimnion and hypolimnion, respectively. Since theMAGs were presumably from the most abundant Verrucomicrobia populations, theywere not likely soil immigrants, given their high abundance in the aquatic environment.
To confirm their aquatic origin, future experiments should be designed to test theiractivities and physiology in the aquatic environment on the basis of the genomicinsights gained in this study.
MATERIALS AND METHODSStudy sites. Samples for metagenome sequencing were collected from Lake Mendota and Trout Bog
Lake, two temperate lakes in Wisconsin in the United States, during the ice-off period (May to November)of each year. Mendota is an urban eutrophic lake with most of its C being autochthonous (producedin-lake), whereas Trout Bog is a small, acidic, and nutrient-poor dystrophic lake with mostly terrestriallyderived (allochthonous) C. General lake characteristics are summarized in Table 1.
Sampling. For Mendota, we collected depth-integrated water samples from the surface 12 m (mostlyconsisting of the epilimnion layer) at 94 time points from 2008 to 2012; those samples are referred to as“ME” (38). For Trout Bog, we collected samples from the integrated hypolimnion layer at 45 time pointsfrom 2007 to 2009 and samples from the integrated epilimnion layer at 45 time points from 2007 to 2009,and those samples are referred to as “TH” and “TE,” respectively (37). All samples were filtered through0.22-�m-pore-size polyethersulfone filters and stored at �80°C until extraction. DNA was extracted fromthe filters using a FastDNA kit (MP Biomedicals) according to the manufacturer’s instructions with someminor modifications as described previously (34).
Metagenome sequencing and assembly and draft genome recovery. Details of metagenomesequencing, assembly, and binning were described by Bendall et al. (37) and Hamilton et al. (61).Briefly, shotgun Illumina HiSeq 2500 metagenome libraries were constructed for each of the DNAsamples. Three combined assemblies were generated by coassembling reads from all metagenomeswithin each of the ME, TE, and TH groups. Binning was conducted on the three combined assembliesto recover “metagenome-assembled genomes” (MAGs) based on the combination of contig tetranucle-otide frequency and differential coverage patterns across time points using MetaBAT (62). Subsequentmanual curation of MAGs was conducted to remove contigs that did not correlate well with the mediantemporal abundance pattern of all contigs within a MAG, as described by Bendall et al. (37).
Genome annotation and completeness estimation. MAGs were submitted to the Department ofEnergy (DOE) Joint Genome Institute’s Integrated Microbial Genome (IMG) database for gene predictionand function annotation (63). The IMG taxon object identifiers (OIDs) for Verrucomicrobia MAGs arelisted in Table 2. The completeness and contamination of each MAG were estimated using checkMwith both the lineage-specific and Verrucomicrobia-specific workflows (35). The Verrucomicrobia-specific workflow provided more-accurate estimates (i.e., higher genome completeness and lowercontamination) than the lineage-specific workflow in testing performed on 11 complete genomes ofVerrucomicrobia isolates available at IMG during our method validation. We therefore reported onlythe estimates from Verrucomicrobia-specific workflow (Table 2). MAGs with an estimated complete-ness level lower than 50% were not included in this study.
Taxonomic and phylogenetic analysis. A total of 19 MAGs were classified in the Verrucomicrobiaphylum based on taxonomic assignment by PhyloSift using 37 conserved phylogenetic marker genes(64), as described by Bendall et al. (37). A phylogenetic tree was reconstructed from the 19 Verrucomi-crobia MAGs and 25 reference genomes using an alignment concatenated from individual proteinalignments of five conserved essential single-copy genes (represented by TIGR01391, TIGR01011,TIGR00663, TIGR00460, and TIGR00362) that were recovered in all Verrucomicrobia MAGs. Individualalignments were first generated with MUSCLE (65), concatenated, and trimmed to exclude columns thatcontained gaps for more than 30% of all sequences. A maximum likelihood phylogenetic tree wasconstructed using PhyML 3.0 (66), with the LG substitution model and the gamma distribution parameterestimated by PhyML. Bootstrap values were calculated based on 100 replicates. Kiritimatiella glycovoransL21-Fru-AB was used as an outgroup in the phylogenetic tree. This bacterium was initially designated thefirst (and so far the only) cultured representative of Verrucomicrobia subdivision 5. However, thissubdivision was later proposed as a novel sister phylum associated with Verrucomicrobia (67), making itan ideal outgroup for this analysis.
Estimate of metabolic potential. IMG provides functional annotation based on KO (KEGG orthol-ogy) term, COG (cluster of orthologous group), pfam, and TIGRfam data. To estimate metabolic potential,we primarily used KO terms due to their direct link to KEGG pathways. COG, pfam, and TIGRfam were alsoused when KO terms were not available for a function. Pathways were primarily reconstructed accordingto KEGG modules, and the MetaCyc pathway was used if a KEGG module was not available for a pathway.As these MAGs are incomplete genomes, a fraction of genes in a pathway may be missing due togenome incompleteness. Therefore, we estimated the completeness of a pathway as the fraction ofrecovered enzymes in that pathway (e.g., a pathway is 100% complete if all enzymes in that pathway areencoded by genes recovered in a MAG). As some genes are shared by multiple pathways, signaturegenes specific for a pathway were used to indicate the presence of a pathway. If signature genes for apathway were missing in all MAGs, that pathway was likely absent in all genomes. Based on this, weestablished criteria for estimating pathway completeness in each MAG. If a signature gene in a pathwaywas present, we report the percentage of genes in the pathway that we found. If a signature gene wasabsent in a MAG but was present in at least one-third of all MAGs (i.e., �7), we still report the pathwaycompleteness for that MAG in order to account for genome incompleteness. Otherwise, we consideredthe pathway to be absent (i.e., completeness is 0%).
Glycoside hydrolase identification. Glycoside hydrolase (GH) genes were identified using thedbCAN annotation tool (http://csbl.bmb.uga.edu/dbCAN/annotate.php) (68) using hmmsearch against
hidden Markov models (HMMs) built for all GHs, with an E value cutoff of 1e�7, except GH109, for whichwe found that the HMM used by dbCAN is pfam01408. This pfam is a small domain at the N terminusof GH109 proteins but is not specific for GH109. Therefore, to identify verrucomicrobial GH109, BLASTPwas performed using the two GH109 sequences (GenBank accession numbers ACD03864 and ACD04752)from verrucomicrobial Akkermansia muciniphila ATCC BAA-835 listed in the CAZy database (http://www.cazy.org), with E value cutoff of 1e�6 and a query sequence coverage value cutoff of 50%.
Other bioinformatic analyses. Protein cellular location was predicted using CELLO v.2.5 (http://cello.life.nctu.edu.tw) (69) and PSORTb v.3.0 (http://www.psort.org/psortb) (70). The beta-barrel structureof outer membrane proteins was predicted using PRED-TMBB (http://bioinformatics.biol.uoa.gr//PRED-TMBB) (71).
SUPPLEMENTAL MATERIALSupplemental material for this article may be found at https://doi.org/10.1128/
mSphere.00277-17.TEXT S1, DOCX file, 0.2 MB.FIG S1, PDF file, 0.7 MB.FIG S2, PDF file, 0.1 MB.FIG S3, PDF file, 0.6 MB.FIG S4, PDF file, 0.1 MB.FIG S5, PDF file, 0.4 MB.FIG S6, PDF file, 0.1 MB.FIG S7, PDF file, 0.2 MB.
ACKNOWLEDGMENTSWe thank the North Temperate Lakes Microbial Observatory 2007–2012 field crews,
University of Wisconsin (UW)-Trout Lake Station, the UW Center for Limnology, and theGlobal Lakes Ecological Observatory Network for field and logistical support. We givespecial thanks to past McMahon laboratory graduate students Ashley Shade, RyanNewton, Emily Read, and Lucas Beversdorf. We acknowledge efforts by many McMahonlaboratory undergrads and technicians related to sample collection and DNA extrac-tion, particularly Georgia Wolfe. We personally thank the individual program directorsand leadership at the National Science Foundation for their commitment to continuedsupport of long-term ecological research.
The project was supported by funding from the United States National ScienceFoundation Microbial Observatories program (MCB-0702395) (to K.D.M.), the Long-termEcological Research Program (NTL-LTER DEB-1440297) (to K.D.M.), and an INSPIREaward (DEB-1344254) (to K.D.M.). This material is also based on work that was sup-ported by funding from the National Institute of Food and Agriculture, U.S. Departmentof Agriculture (Hatch Project 1002996) (to K.D.M.). The work conducted by the U.S.Department of Energy Joint Genome Institute, a DOE Office of Science User Facility, issupported by the Office of Science of the U.S. Department of Energy under contractDE-AC02-05CH11231.
REFERENCES1. Zwart G, van Hannen EJ, Kamst-van Agterveld MP, Van der Gucht K,
Lindström ES, Van Wichelen J, Lauridsen T, Crump BC, Han SK, DeclerckS. 2003. Rapid screening for freshwater bacterial groups by using reverseline blot hybridization. Appl Environ Microbiol 69:5875–5883. https://doi.org/10.1128/AEM.69.10.5875-5883.2003.
2. Newton RJ, Jones SE, Eiler A, McMahon KD, Bertilsson S. 2011. A guide tothe natural history of freshwater lake bacteria. Microbiol Mol Biol Rev75:14 – 49. https://doi.org/10.1128/MMBR.00028-10.
3. Eiler A, Bertilsson S. 2004. Composition of freshwater bacterial commu-nities associated with cyanobacterial blooms in four Swedish lakes. EnvironMicrobiol 6:1228–1243. https://doi.org/10.1111/j.1462-2920.2004.00657.x.
4. Parveen B, Mary I, Vellet A, Ravet V, Debroas D. 2013. Temporal dynamicsand phylogenetic diversity of free-living and particle-associated Verru-comicrobia communities in relation to environmental variables in amesotrophic lake. FEMS Microbiol Ecol 83:189 –201. https://doi.org/10.1111/j.1574-6941.2012.01469.x.
5. Arnds J, Knittel K, Buck U, Winkel M, Amann R. 2010. Development of a16S rRNA-targeted probe set for Verrucomicrobia and its application for
fluorescence in situ hybridization in a humic lake. Syst Appl Microbiol33:139 –148. https://doi.org/10.1016/j.syapm.2009.12.005.
6. Hedlund BP, Gosink JJ, Staley JT. 1997. Verrucomicrobia div. nov., a newdivision of the bacteria containing three new species of Prostheco-bacter. Antonie Van Leeuwenhoek 72:29 –38. https://doi.org/10.1023/A:1000348616863.
7. Yoon J, Yasumoto-Hirose M, Katsuta A, Sekiguchi H, Matsuda S, Kasai H,Yokota A. 2007. Coraliomargarita akajimensis gen. nov., sp. nov., a novelmember of the phylum “Verrucomicrobia” isolated from seawater inJapan. Int J Syst Evol Microbiol 57:959 –963. https://doi.org/10.1099/ijs.0.64755-0.
8. Yoon J, Matsuo Y, Katsuta A, Jang JH, Matsuda S, Adachi K, Kasai H, YokotaA. 2008. Haloferula Rosea gen. nov., sp. nov., Haloferula harenae sp. nov.,Haloferula phyci sp. nov., Haloferula helveola sp. nov. and Haloferula sar-gassicola sp. nov., five marine representatives of the family Verrucomicro-biaceae within the phylum “Verrucomicrobia”. Int J Syst Evol Microbiol58:2491–2500. https://doi.org/10.1099/ijs.0.2008/000711-0.
9. Sangwan P, Kovac S, Davis KE, Sait M, Janssen PH. 2005. Detection and
cultivation of soil Verrucomicrobia. Appl Environ Microbiol 71:8402– 8410. https://doi.org/10.1128/AEM.71.12.8402-8410.2005.
10. Brewer TE, Handley KM, Carini P, Gilbert JA, Fierer N. 2016. Genomereduction in an abundant and ubiquitous soil bacterium “CandidatusUdaeobacter copiosus”. Nat Microbiol 2:16198. https://doi.org/10.1038/nmicrobiol.2016.198.
11. Qiu YL, Kuang XZ, Shi XS, Yuan XZ, Guo RB. 2014. Terrimicrobiumsacchariphilum gen. nov., sp. nov., an anaerobic bacterium of the class“Spartobacteria” in the phylum Verrucomicrobia, isolated from a ricepaddy field. Int J Syst Evol Microbiol 64:1718 –1723. https://doi.org/10.1099/ijs.0.060244-0.
12. da Rocha UN, van Elsas JD, van Overbeek LS. 2010. Real-time PCRdetection of Holophagae (Acidobacteria) and Verrucomicrobia subdivi-sion 1 groups in bulk and leek (Allium porrum) rhizosphere soils. JMicrobiol Methods 83:141–148. https://doi.org/10.1016/j.mimet.2010.08.003.
13. Wertz JT, Kim E, Breznak JA, Schmidt TM, Rodrigues JLM. 2012. Genomicand physiological characterization of the Verrucomicrobia isolateDiplosphaera colitermitum gen. nov., sp. nov., reveals microaerophilyand nitrogen fixation genes. Appl Environ Microbiol 78:1544 –1555.https://doi.org/10.1128/AEM.06466-11.
14. Derrien M, Vaughan EE, Plugge CM, de Vos WM. 2004. Akkermansiamuciniphila gen. nov., sp. nov., a human intestinal mucin-degradingbacterium. Int J Syst Evol Microbiol 54:1469 –1476. https://doi.org/10.1099/ijs.0.02873-0.
15. Scheuermayer M, Gulder TA, Bringmann G, Hentschel U. 2006. Rubritaleamarina gen. nov., sp. nov., a marine representative of the phylum“Verrucomicrobia”, isolated from a sponge (Porifera). Int J Syst EvolMicrobiol 56:2119 –2124. https://doi.org/10.1099/ijs.0.64360-0.
16. Chin KJ, Liesack W, Janssen PH. 2001. Opitutus terrae gen. nov., sp. nov.,to accommodate novel strains of the division “Verrucomicrobia” isolatedfrom rice paddy soil. Int J Syst Evol Microbiol 51:1965–1968. https://doi.org/10.1099/00207713-51-6-1965.
17. Otsuka S, Suenaga T, Vu HT, Ueda H, Yokota A, Senoo K. 2013. Brevifollisgellanilyticus gen. nov., sp. nov., a gellan-gum-degrading bacterium ofthe phylum Verrucomicrobia. Int J Syst Evol Microbiol 63:3075–3078.https://doi.org/10.1099/ijs.0.048793-0.
18. Sangwan P, Chen X, Hugenholtz P, Janssen PH. 2004. Chthoniobacterflavus gen. nov., sp. nov., the first pure-culture representative of subdi-vision two, Spartobacteria classis nov., of the phylum Verrucomicrobia.Appl Environ Microbiol 70:5875–5881. https://doi.org/10.1128/AEM.70.10.5875-5881.2004.
19. Hedlund BP, Gosink JJ, Staley JT. 1996. Phylogeny of Prosthecobacter,the fusiform caulobacters: members of a recently discovered division ofthe bacteria. Int J Syst Bacteriol 46:960 –966. https://doi.org/10.1099/00207713-46-4-960.
20. Otsuka S, Ueda H, Suenaga T, Uchino Y, Hamada M, Yokota A, Senoo K.2013. Roseimicrobium gellanilyticum gen. nov., sp. nov., a new memberof the class Verrucomicrobiae. Int J Syst Evol Microbiol 63:1982–1986.https://doi.org/10.1099/ijs.0.041848-0.
21. Pol A, Heijmans K, Harhangi HR, Tedesco D, Jetten MS, Op den Camp HJ.2007. Methanotrophy below pH 1 by a new Verrucomicrobia species.Nature 450:874 – 878. https://doi.org/10.1038/nature06222.
22. Freitas S, Hatosy S, Fuhrman JA, Huse SM, Welch DB, Sogin ML, MartinyAC. 2012. Global distribution and diversity of marine Verrucomicrobia.ISME J 6:1499 –1505. https://doi.org/10.1038/ismej.2012.3.
23. Cardman Z, Arnosti C, Durbin A, Ziervogel K, Cox C, Steen AD, Teske A.2014. Verrucomicrobia are candidates for polysaccharide-degrading bac-terioplankton in an arctic fjord of Svalbard. Appl Environ Microbiol80:3749 –3756. https://doi.org/10.1128/AEM.00899-14.
24. Martinez-Garcia M, Brazel DM, Swan BK, Arnosti C, Chain PS, ReitengaKG, Xie G, Poulton NJ, Lluesma Gomez M, Masland DE, Thompson B,Bellows WK, Ziervogel K, Lo CC, Ahmed S, Gleasner CD, Detter CJ,Stepanauskas R. 2012. Capturing single cell genomes of active polysac-charide degraders: an unexpected contribution of Verrucomicrobia.PLoS One 7:e35314. https://doi.org/10.1371/journal.pone.0035314.
25. Herlemann DP, Lundin D, Labrenz M, Jürgens K, Zheng Z, Aspeborg H,Andersson AF. 2013. Metagenomic de novo assembly of an aquaticrepresentative of the verrucomicrobial class Spartobacteria. mBio4:e00569-12. https://doi.org/10.1128/mBio.00569-12.
26. Paver SF, Kent AD. 2010. Temporal patterns in glycolate-utilizing bacte-rial community composition correlate with phytoplankton populationdynamics in humic lakes. Microb Ecol 60:406 – 418. https://doi.org/10.1007/s00248-010-9722-6.
27. Haukka K, Kolmonen E, Hyder R, Hietala J, Vakkilainen K, Kairesalo T,Haario H, Sivonen K. 2006. Effect of nutrient loading on bacterioplanktoncommunity composition in lake mesocosms. Microb Ecol 51:137–146.https://doi.org/10.1007/s00248-005-0049-7.
28. Lindström ES, Vrede K, Leskinen E. 2004. Response of a member of theVerrucomicrobia, among the dominating bacteria in a hypolimnion, toincreased phosphorus availability. J Plankton Res 26:241–246. https://doi.org/10.1093/plankt/fbh010.
29. Kolmonen E, Sivonen K, Rapala J, Haukka K. 2004. Diversity of cyano-bacteria and heterotrophic bacteria in cyanobacterial blooms in LakeJoutikas, Finland. Aquat Microb Ecol 36:201–211. https://doi.org/10.3354/ame036201.
30. Lindström ES, Kamst-Van Agterveld MP, Zwart G. 2005. Distribution oftypical freshwater bacterial groups is associated with pH, temperature,and lake water retention time. Appl Environ Microbiol 71:8201– 8206.https://doi.org/10.1128/AEM.71.12.8201-8206.2005.
31. Schlesner H. 1987. Verrucomicrobium spinosum gen. nov., sp. nov.: afimbriated prosthecate bacterium. Syst Appl Microbiol 10:54 –56. https://doi.org/10.1016/S0723-2020(87)80010-3.
32. Zwart G, Huismans R, Agterveld MP, Peer Y, Rijk P, Eenhoorn H, MuyzerG, Hannen EJ, Gons HJ, Laanbroek HJ. 1998. Divergent members of thebacterial division Verrucomicrobiales in a temperate freshwater lake.FEMS Microbiol Ecol 25:159 –169. https://doi.org/10.1111/j.1574-6941.1998.tb00469.x.
33. Read JS, Rose KC. 2013. Physical responses of small temperate lakes tovariation in dissolved organic carbon concentrations. Limnol Oceanogr58:921–931. https://doi.org/10.4319/lo.2013.58.3.0921.
34. Shade A, Jones SE, McMahon KD. 2008. The influence of habitat heter-ogeneity on freshwater bacterial community composition and dynamics.Environ Microbiol 10:1057–1067. https://doi.org/10.1111/j.1462-2920.2007.01527.x.
35. Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, Tyson GW. 2015.CheckM: assessing the quality of microbial genomes recovered fromisolates, single cells, and metagenomes. Genome Res 25:1043–1055.https://doi.org/10.1101/gr.186072.114.
36. Schlesner H, Jenkins C, Staley J. 2006. The phylum Verrucomicrobia: aphylogenetically heterogeneous bacterial group, p 881–896. In Dworkin M,Falkow S, Rosenberg E, Schleifer K-H, Stackebrandt E (ed), The prokaryotes.Springer, New York, NY. https://doi.org/10.1007/0-387-30747-8_37.
37. Bendall ML, Stevens SLR, Chan LK, Malfatti S, Schwientek P, Tremblay J,Schackwitz W, Martin J, Pati A, Bushnell B, Froula J, Kang D, Tringe SG,Bertilsson S, Moran MA, Shade A, Newton RJ, McMahon KD, MalmstromRR. 2016. Genome-wide selective sweeps and gene-specific sweeps innatural bacterial populations. ISME J 10:1589 –1601 https://doi.org/10.1038/ismej.2015.241.
39. Shieh WY, Jean WD. 1998. Alterococcus agarolyticus, gen.nov., sp.nov., ahalophilic thermophilic bacterium capable of agar degradation. Can JMicrobiol 44:637– 645. https://doi.org/10.1139/w98-051.
40. Kent AD, Jones SE, Yannarell AC, Graham JM, Lauster GH, Kratz TK,Triplett EW. 2004. Annual patterns in bacterioplankton community vari-ability in a humic lake. Microb Ecol 48:550 –560. https://doi.org/10.1007/s00248-004-0244-y.
41. Kent AD, Yannarell AC, Rusak JA, Triplett EW, McMahon KD. 2007.Synchrony in aquatic microbial community dynamics. ISME J 1:38 – 47.https://doi.org/10.1038/ismej.2007.6.
42. Erbilgin O, McDonald KL, Kerfeld CA. 2014. Characterization of a planc-tomycetal organelle: a novel bacterial microcompartment for the aero-bic degradation of plant saccharides. Appl Environ Microbiol 80:2193–2205. https://doi.org/10.1128/AEM.03887-13.
43. Blanvillain S, Meyer D, Boulanger A, Lautier M, Guynet C, Denancé N,Vasse J, Lauber E, Arlat M. 2007. Plant carbohydrate scavenging throughtonB-dependent receptors: a feature shared by phytopathogenic andaquatic bacteria. PLoS One 2:e224. https://doi.org/10.1371/journal.pone.0000224.
44. Jiao G, Yu G, Zhang J, Ewart HS. 2011. Chemical structures and bioac-tivities of sulfated polysaccharides from marine algae. Mar Drugs9:196 –223. https://doi.org/10.3390/md9020196.
45. Filali Mouhim R, Cornet J-F, Fontane T, Fournet B, Dubertret G. 1993.Production, isolation and preliminary characterization of the exopoly-
saccharide of the cyanobacterium Spirulina platensis. Biotechnol Lett15:567–572. https://doi.org/10.1007/BF00138541.
46. Dantas-Santos N, Gomes DL, Costa LS, Cordeiro SL, Costa MS, TrindadeES, Franco CR, Scortecci KC, Leite EL, Rocha HA. 2012. Freshwater plantssynthesize sulfated polysaccharides: heterogalactans from water hya-cinth (Eicchornia Crassipes). Int J Mol Sci 13:961–976. https://doi.org/10.3390/ijms13010961.
47. Beversdorf LJ, Miller TR, McMahon KD. 2013. The role of nitrogen fixationin cyanobacterial bloom toxicity in a temperate, eutrophic lake. PLoSOne 8:e56103. https://doi.org/10.1371/journal.pone.0056103.
48. Khadem AF, van Teeseling MC, van Niftrik L, Jetten MS, Op den Camp HJ,Pol A. 2012. Genomic and physiological analysis of carbon storage in theverrucomicrobial methanotroph �Ca. Methylacidiphilum fumariolicum�SolV. Front Microbiol 3:345.
49. Mehta T, Coppi MV, Childers SE, Lovley DR. 2005. Outer membranec-type cytochromes required for Fe(III) and Mn(IV) oxide reduction inGeobacter sulfurreducens. Appl Environ Microbiol 71:8634 – 8641.https://doi.org/10.1128/AEM.71.12.8634-8641.2005.
50. Liu Y, Wang Z, Liu J, Levar C, Edwards MJ, Babauta JT, Kennedy DW, ShiZ, Beyenal H, Bond DR, Clarke TA, Butt JN, Richardson DJ, Rosso KM,Zachara JM, Fredrickson JK, Shi L. 2014. A trans-outer membrane porin-cytochrome protein complex for extracellular electron transfer by Geo-bacter sulfurreducens PCA. Environ Microbiol Rep 6:776 –785. https://doi.org/10.1111/1758-2229.12204.
51. Shi L, Fredrickson JK, Zachara JM. 2014. Genomic analyses of bacterialporin-cytochrome gene clusters. Front Microbiol 5:657. https://doi.org/10.3389/fmicb.2014.00657.
52. Bücking C, Piepenbrock A, Kappler A, Gescher J. 2012. Outer-membranecytochrome-independent reduction of extracellular electron acceptorsin Shewanella oneidensis. Microbiology 158:2144 –2157. https://doi.org/10.1099/mic.0.058404-0.
53. Shyu JBH, Lies DP, Newman DK. 2002. Protective role of tolC in efflux ofthe electron shuttle anthraquinone-2,6-disulfonate. J Bacteriol 184:1806 –1810. https://doi.org/10.1128/JB.184.6.1806-1810.2002.
54. Voordeckers JW, Kim BC, Izallalen M, Lovley DR. 2010. Role of Geobactersulfurreducens outer surface c-type cytochromes in reduction of soilhumic acid and anthraquinone-2,6-disulfonate. Appl Environ Microbiol76:2371–2375. https://doi.org/10.1128/AEM.02250-09.
55. Lovley DR, Blunt-Harris EL. 1999. Role of humic-bound iron as an elec-tron transfer agent in dissimilatory Fe(III) reduction. Appl Environ Micro-biol 65:4252– 4254.
56. Lovley DR, Coates JD, Blunt-Harris EL, Phillips EJP, Woodward JC. 1996.Humic substances as electron acceptors for microbial respiration. Nature382:445– 448. https://doi.org/10.1038/382445a0.
57. Klüpfel L, Piepenbrock A, Kappler A, Sander M. 2014. Humic substancesas fully regenerable electron acceptors in recurrently anoxic environ-ments. Nat Geosci 7:195–200. https://doi.org/10.1038/ngeo2084.
58. Studholme DJ, Fuerst JA, Bateman A. 2004. Novel protein domains andmotifs in the marine planctomycete Rhodopirellula baltica. FEMS Micro-biol Lett 236:333–340. https://doi.org/10.1016/j.femsle.2004.06.007.
59. Kamneva OK, Knight SJ, Liberles DA, Ward NL. 2012. Analysis of genomecontent evolution in PVC bacterial super-phylum: assessment of candi-date genes associated with cellular organization and lifestyle. GenomeBiol Evol 4:1375–1390. https://doi.org/10.1093/gbe/evs113.
60. Linz AM, Crary BC, Shade A, Owens S, Gilbert JA, Knight R, McMahon KD.2017. Bacterial community composition and dynamics spanning fiveyears in freshwater bog lakes. mSphere 2:e00169-17. https://doi.org/10.1128/mSphere.00169-17.
61. Hamilton JJ, Garcia SL, Brown BS, Oyserman BO, Moya-Flores F, Ber-tilsson S, Malmstrom RR, Forest KT, McMahon KD. 2007. High-throughput metabolic network analysis and metatranscriptomics of acosmopolitan and streamlined freshwater lineage. bioRxiv https://doi.org/10.1101/106856.
62. Kang DD, Froula J, Egan R, Wang Z. 2015. MetaBAT, an efficient tool foraccurately reconstructing single genomes from complex microbial com-munities. PeerJ 3:e1165. https://doi.org/10.7717/peerj.1165.
63. Markowitz VM, Chen I-MA, Palaniappan K, Chu K, Szeto E, Pillay M, RatnerA, Huang J, Woyke T, Huntemann M, Anderson I, Billis K, Varghese N,Mavromatis K, Pati A, Ivanova NN, Kyrpides NC. 2014. IMG 4 version ofthe integrated microbial genomes comparative analysis system. NucleicAcids Res 42:D560 –D567. https://doi.org/10.1093/nar/gkt963.
64. Darling AE, Jospin G, Lowe E, Matsen FA, Bik HM, Eisen JA. 2014.PhyloSift: phylogenetic analysis of genomes and metagenomes. PeerJ2:e243. https://doi.org/10.7717/peerj.243.
65. Edgar RC. 2004. MUSCLE: a multiple sequence alignment method withreduced time and space complexity. BMC Bioinformatics 5:113–113.https://doi.org/10.1186/1471-2105-5-113.
66. Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O.2010. New algorithms and methods to estimate maximum-likelihoodphylogenies: assessing the performance of PhyML 3.0. Syst Biol 59:307–321. https://doi.org/10.1093/sysbio/syq010.
67. Spring S, Bunk B, Spröer C, Schumann P, Rohde M, Tindall BJ, Klenk HP.2016. Characterization of the first cultured representative of Verrucomi-crobia subdivision 5 indicates the proposal of a novel phylum. ISME J10:2801–2816. https://doi.org/10.1038/ismej.2016.84.
68. Yin Y, Mao X, Yang J, Chen X, Mao F, Xu Y. 2012. dbCAN: a Web resourcefor automated carbohydrate-active enzyme annotation. Nucleic AcidsRes 40:W445–W451. https://doi.org/10.1093/nar/gks479.
69. Yu CS, Chen YC, Lu CH, Hwang JK. 2006. Prediction of protein subcellularlocalization. Proteins 64:643– 651. https://doi.org/10.1002/prot.21018.
70. Yu NY, Wagner JR, Laird MR, Melli G, Rey S, Lo R, Dao P, Sahinalp SC, EsterM, Foster LJ, Brinkman FSL. 2010. PSORTb 3.0: improved protein subcel-lular localization prediction with refined localization subcategories andpredictive capabilities for all prokaryotes. Bioinformatics 26:1608 –1615.https://doi.org/10.1093/bioinformatics/btq249.
71. Bagos PG, Liakopoulos TD, Spyropoulos IC, Hamodrakas SJ. 2004. PRED-TMBB: a Web server for predicting the topology of beta-barrel outermembrane proteins. Nucleic Acids Res 32:W400 –W404. https://doi.org/10.1093/nar/gkh417.