Published: August 30, 2011 r2011 American Chemical Society 12977 dx.doi.org/10.1021/la202533s | Langmuir 2011, 27, 12977–12984 ARTICLE pubs.acs.org/Langmuir Effect of Cation Size and Charge on the Interaction between Silica Surfaces in 1:1, 2:1, and 3:1 Aqueous Electrolytes Matan Dishon, Ohad Zohar, and Uri Sivan* Faculty of Physics and the Russell Berrie Nanotechnology Institute, Technion-Israel Institute of Technology, Haifa 32000, Israel b S Supporting Information ABSTRACT: Application of two complementary AFM measurements, force vs separation and adhesion force, reveals the combined effects of cation size and charge (valency) on the interaction between silica surfaces in three 1:1, three 2:1, and three 3:1 metal chloride aqueous solutions of different concentrations. The interaction between the silica surfaces in 1:1 and 2:1 salt solutions is fully accounted for by ion-independent van der Waals (vdW) attraction and electric double-layer repulsion modified by cation specific adsorption to the silica surfaces. The deduced ranking of mono- and divalent cation adsorption capacity (adsorbability) to silica, Mg 2+ < Ca 2+ < Na + < Sr 2+ <K + < Cs + , follows cation bare size as well as cation solvation energy but does not correlate with hydrated ionic radius or with volume or surface ionic charge density. In the presence of 3:1 salts, the coarse phenomenology of the force between the silica surfaces as a function of salt concentration resembles that in 1:1 and 2:1 electrolytes. Nevertheless, two fundamental differences should be noticed. First, the attraction between the silica surfaces is too large to be attributed solely to vdW force, hence implying an additional attraction mechanism or gross modification of the conventional vdW attraction. Second, neutralization of the silica surfaces occurs at trivalent cation concentrations that are 3 orders of magnitude smaller than those characterizing surface neutralization by mono- and divalent cations. Consequently, when trivalent cations are added to our cation adsorbability series the correlation with bare ion size breaks down abruptly. The strong adsorbability of trivalent cations to silica contrasts straightforward expectations based on ranking of the cationic solvation energies, thus suggesting a different adsorption mechanism which is inoperative or weak for mono- and divalent cations. ’ INTRODUCTION Hofmeister’s work 1,2 in the late 19th century that ranked salts according to their effect on protein precipitation, or “salting-out”, marked the discovery of ion-specificeffects on the interaction between charged bodies in electrolyte solution. In over a century of research, ion-specificeffects have emerged in numerous other instances from elementary physical chemistry phenomena such as surface tension, through colloidal stability and protein folding, to enzyme activity and bacterial growth. 1,3,4 Quite remarkably, the same ordering of salts found in the Hofmeister series for proteins also appeared in many seemingly unrelated phenomena. In other cases, however, the ordering was reversed or took another form. To this date, notwithstanding extensive research, neither the physics behind the original Hofmeister series nor the mechanisms underlying many of the ion-specificeffects observed in other phenomena have been fully elucidated. Among the different fields in which ion-specificeffects emerge, our research is focused on the effect of background salt type on the interaction between charged surfaces in aqueous electrolyte solution. We have previously reported 5 on the interaction between silica surfaces in different alkali chloride aqueous solu- tions studied by force versus separation measurements using the colloidal probe AFM technique. 6 In that study, we directly mea- sured the force as a function of separation between a spherical 5-μm-diameter silica bead and a flat silica surface immersed in aqueous solution, and mapped the effect of salt type and con- centration on that force. Specifically, we found that the interac- tion between silica surfaces in the presence of the three 1:1 alkali Received: July 5, 2011 Revised: August 28, 2011
Transcript
Published: August 30, 2011
r 2011 American Chemical Society 12977 dx.doi.org/10.1021/la202533s | Langmuir 2011, 27, 12977–12984
ARTICLE
pubs.acs.org/Langmuir
Effect of Cation Size and Charge on the Interaction between SilicaSurfaces in 1:1, 2:1, and 3:1 Aqueous ElectrolytesMatan Dishon, Ohad Zohar, and Uri Sivan*
Faculty of Physics and the Russell Berrie Nanotechnology Institute, Technion-Israel Institute of Technology, Haifa 32000, Israel
bS Supporting Information
ABSTRACT:
Application of two complementary AFM measurements, force vs separation and adhesion force, reveals the combined effects ofcation size and charge (valency) on the interaction between silica surfaces in three 1:1, three 2:1, and three 3:1 metal chlorideaqueous solutions of different concentrations. The interaction between the silica surfaces in 1:1 and 2:1 salt solutions is fullyaccounted for by ion-independent van der Waals (vdW) attraction and electric double-layer repulsion modified by cation specificadsorption to the silica surfaces. The deduced ranking of mono- and divalent cation adsorption capacity (adsorbability) to silica,Mg2+ < Ca2+ <Na+ < Sr2+ < K+ <Cs+, follows cation bare size as well as cation solvation energy but does not correlate with hydratedionic radius or with volume or surface ionic charge density. In the presence of 3:1 salts, the coarse phenomenology of the forcebetween the silica surfaces as a function of salt concentration resembles that in 1:1 and 2:1 electrolytes. Nevertheless, twofundamental differences should be noticed. First, the attraction between the silica surfaces is too large to be attributed solely to vdWforce, hence implying an additional attraction mechanism or gross modification of the conventional vdW attraction. Second,neutralization of the silica surfaces occurs at trivalent cation concentrations that are 3 orders of magnitude smaller than thosecharacterizing surface neutralization by mono- and divalent cations. Consequently, when trivalent cations are added to our cationadsorbability series the correlation with bare ion size breaks down abruptly. The strong adsorbability of trivalent cations to silicacontrasts straightforward expectations based on ranking of the cationic solvation energies, thus suggesting a different adsorptionmechanism which is inoperative or weak for mono- and divalent cations.
’ INTRODUCTION
Hofmeister’s work1,2 in the late 19th century that ranked saltsaccording to their effect on protein precipitation, or “salting-out”,marked the discovery of ion-specific effects on the interactionbetween charged bodies in electrolyte solution. In over a centuryof research, ion-specific effects have emerged in numerous otherinstances from elementary physical chemistry phenomena suchas surface tension, through colloidal stability and protein folding,to enzyme activity and bacterial growth.1,3,4 Quite remarkably,the same ordering of salts found in the Hofmeister series forproteins also appeared in many seemingly unrelated phenomena.In other cases, however, the ordering was reversed or tookanother form. To this date, notwithstanding extensive research,neither the physics behind the original Hofmeister series northe mechanisms underlying many of the ion-specific effectsobserved in other phenomena have been fully elucidated.
Among the different fields in which ion-specific effects emerge,our research is focused on the effect of background salt type onthe interaction between charged surfaces in aqueous electrolytesolution. We have previously reported5 on the interactionbetween silica surfaces in different alkali chloride aqueous solu-tions studied by force versus separation measurements using thecolloidal probe AFM technique.6 In that study, we directly mea-sured the force as a function of separation between a spherical5-μm-diameter silica bead and a flat silica surface immersed inaqueous solution, and mapped the effect of salt type and con-centration on that force. Specifically, we found that the interac-tion between silica surfaces in the presence of the three 1:1 alkali
chloride salts, NaCl, KCl, and CsCl, could vary significantly. In∼100 mM concentration, for instance, we saw that the forcebetween the silica surfaces was repulsive in NaCl while attractivein KCl and CsCl. By analyzing the results in the framework ofDLVO7 (Derjaguin�Landau�Verwey�Overbeek), that is, as asum of electric double-layer (edl) overlap and van der Waals(vdW) interactions, we established that the underlying reason forion specificity of the interaction between the silica surfaces isalteration of the silica surface charge by specific adsorption ofpositive ions from solution onto the initially negatively chargedsilica surfaces. The possibility that cation-specific ionization ofsilanol groups on the silica surfaces plays a major role in thealteration of surface charge could be ruled out in our pH range(pH 5.5), as can be appreciated by analyzing Figure 1 of Doveet al.8 Cation-specific adsorption hence resulted in modificationof the edl repulsion, which combined with ion-independent vdWattraction to yield an ion-specific interaction. As shown in theSupporting Information, in a subsequent study comparing theresults in NaCl and in NaBr electrolyte solutions we haveexcluded halide anion-specific effects on this interaction.
The tendency of cations to adsorb (adsorbability) to silica couldbe parametrized by the respective salt-specific concentration atwhich full neutralization of the silica surface charge took place. Atthis concentration, the electrostatic repulsion vanished and vdWattraction remained the sole interaction between the surfaces. AtpH 5.5, these concentrations were >1 M for NaCl, 0.2�0.5 M forKCl, and ∼0.1 M for CsCl. As a common chloride anion wasmaintained, these concentrations allowed ranking of the alkalications based on their adsorbability to silica: Na+ < K+ < Cs+.
Curiously, at higher concentrations of CsCl and also of KCl,reemergence of repulsion was brought about by overcharging ofthe silica surfaces. Adsorption of excess K+, and more intenselyCs+, led to charge reversal of the silica surfaces and, hence, torecurrence of edl repulsion. Charge reversal in the presence of 1:1salts was previously characterized by zeta-potential measurements.9
A compilation of experimental data published by us and othergroups and discussion of the different observations can be foundin ref 5.
At the conclusion of our previous study, we have learned that,although the interaction between silica surfaces was profoundly
salt dependent, the phenomenology of the interaction as a func-tion of salt concentration was common to these three electro-lytes. Specifically, at sufficiently low salt concentrations the silicasurfaces have born enough negative surface charge, due todeprotonated silanols, to produce a considerable edl repulsionthat overcame vdW attraction at all separations. Surface chargeneutralization of silica by cation adsorption and, hence, vanishingof edl repulsion and domination of vdW attraction, was achievedat a certain salt-specific concentration. Subsequently, at elevatedconcentrations of certain salts, overcharging of silica surfaces byexcess adsorption of cations induced charge reversal of thesurfaces and, consequently, reemergence of repulsion betweenthe two positively charged silica surfaces.
Interestingly, the cation adsorbability series that emerged inthat study followed cation bare size (three first rows in Table 1);the larger the cation was, the more it adsorbed to silica. Not-withstanding the value of this empirical observation, it wasdifficult to clarify the rules governing ion-specific interaction,as well as ion-adsorption to silica based on such homogeneous(alkali only) series. For example, other ionic properties such asvolume and surface charge density,10 hydrated radius11 as ex-tracted from conductivity measurements, and various thermo-dynamic data, such as the free energy of solvation12 conformed tothe same ordering.
Attempting to elucidate the combined effect of ionic chargeand size on ion-specific interaction between silica surfaces, weextend our scope in the present study to electrolytes of divalentand trivalent cations in a broad range of ion concentrations, from0.5 mM to 1 M. The normal force�separation measurementsupon tip approach are augmented withmeasurements of the pull-off force13 required for detaching the silica surfaces upon tipretraction. Overall, we study, using both techniques, nine salts,the same three 1:1 salts, NaCl, KCl, and CsCl; three 2:1salts, MgCl2, CaCl2, and SrCl2; and three 3:1 salts, LaCl3,[Co(NH3)6]Cl3, and AlCl3.
Just like ion bare size, ion valency is a fundamental parameter,which affects other ionic properties. Hydrated radius, for in-stance, usually grows with ion valency while the bare radiusshrinks. On the third row of the periodic table, for example, Na+
hydrated radius is smaller than that ofMg2+, which is smaller thanthat of Al3+.11 The bare radii obey an opposite ordering.10 Forcemeasurements in electrolytes of multivalent cations, maintaininga common chloride anion, thus allowed study and disentanglingthe roles of different cationic properties in these cation-specificeffects, thus revealing the relevant parameters for these cation-specific interactions.
Using force versus separation measurements upon tip ap-proach, we quickly encountered an experimental limitation in thecase of 2:1 salt solutions, as ion-specificity first appeared onlyabove∼50 mM and became substantial at 100�200 mM. Over-charging in these electrolytes, if it existed, was thus expected ateven higher concentrations. Since the screening length of 2:1electrolyte solutions at these concentrations was well below1 nm, ion-specific effects on the interaction, and specificallycharge inversion and its accompanying short-range repulsion,turned hard to discern by normal force measurements upon tipapproach. Moreover, as shown in our previous study, whenstrong attraction commences separations below ∼2 nm becomeinaccessible due to cantilever snap-in to the surface taking placeas soon as the derivative of the measured force with respect toseparation exceeds the cantilever’s spring constant (∼0.5 N/m).We have therefore chosen to work around this problem here by
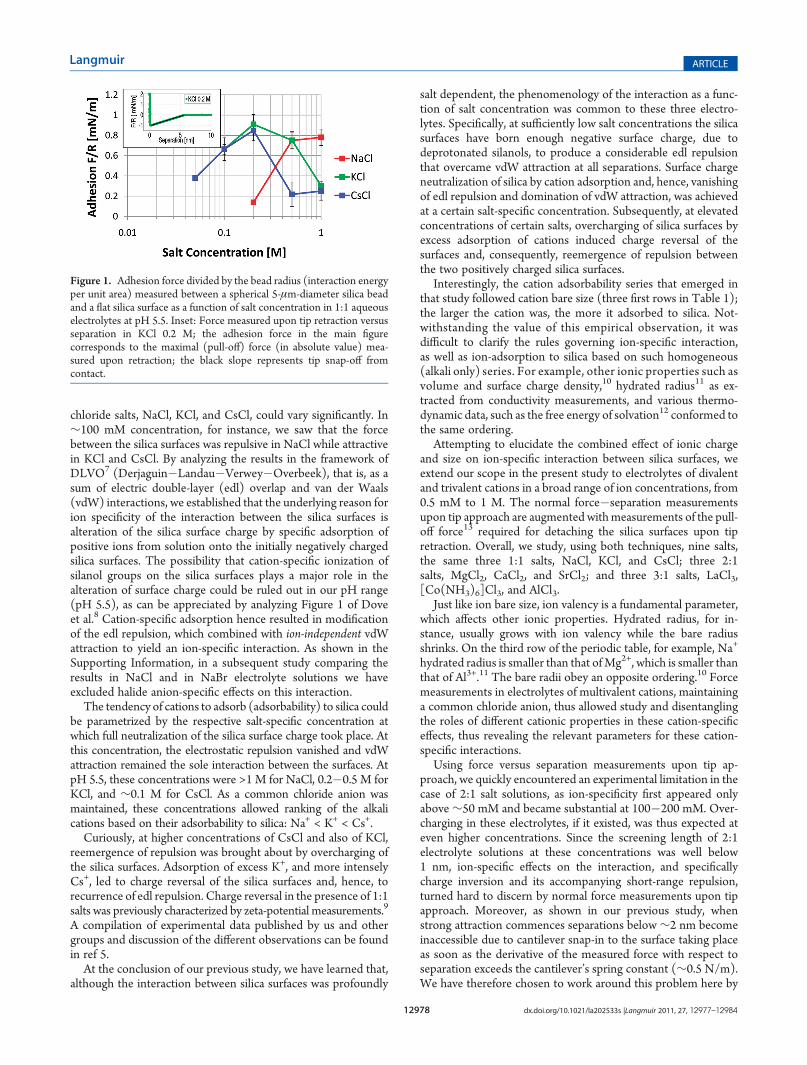
Figure 1. Adhesion force divided by the bead radius (interaction energyper unit area) measured between a spherical 5-μm-diameter silica beadand a flat silica surface as a function of salt concentration in 1:1 aqueouselectrolytes at pH 5.5. Inset: Force measured upon tip retraction versusseparation in KCl 0.2 M; the adhesion force in the main figurecorresponds to the maximal (pull-off) force (in absolute value) mea-sured upon retraction; the black slope represents tip snap-off fromcontact.
considering the adhesion between the silica bead and the silicasurface as measured upon tip retraction from the surface,13 anduse these data to characterize the ion-specific effects. In additionto being a fundamental and important physical property on itsown, the pull-off force also complements the force versusseparation data with an additional point at zero separation, whichwas missing in most force vs distance measurements due to thecantilever snap-in effect described above. Adhesion provided aninsightful measurement for studying ion-specific effects andovercharging in 2:1 salt solutions, which were otherwise hiddenbeyond certain salt concentrations.
The article is outlined in the following way: Following a shortdescription of the experimental techniques, we present adhesionmeasurements in 1:1 salt solutions. Comparing these results withdata from force�separation measurements upon tip approach,5
we establish that adhesion measurements provide an alternativemeans to study ion-specific effects on the interaction between thesilica surfaces. Specifically, we show that as a function of saltconcentration the phenomenology of the interaction as drawnfrom adhesion data is similar to that concluded in ref 5 and thatestimates of the salt-specific concentration at which surfaceneutralization occurs are practically identical for the two meth-ods. Having established that adhesion force measurementsindeed provide a parallel method for studying salt-specific effectson the interaction between silica surfaces, we present a study ofthe interaction between silica surfaces in 2:1 electrolytes based onthis method. We show that as a function of salt concentration theinteraction between silica surfaces in these electrolytes exhibitsthe same phenomenology as in 1:1 salt solutions. Moreover, thedata can be explained in the same framework as in the 1:1 salt caseleading to ranking of divalent cation adsorbability. By analyzingthe adhesion data as a function of ionic strength, rather than saltconcentration, we unfold that co-ranking of monovalent anddivalent cation adsorbability correlates with bare ion size, but notwith volume or surface charge density nor with hydrated radius.We then turn to 3:1 salts and find, that notwithstanding theresemblance of the phenomenology to that in 1:1 and 2:1electrolytes, the interaction between silica surfaces can no longerbe explained solely in terms of summation of edl overlap andvdW interactions. This result affirms a previous study14 reportingthat the attraction mechanism between silica surfaces in[Co(NH3)6]Cl3 (hexaminecobalt(III) chloride) aqueous solu-tion cannot be attributed to conventional vdW alone. Finally, wereport that the adsorbabilities to silica of the three trivalentcations studied here are considerably larger than those character-izing mono- and divalent cations. Accordingly, when trivalent cations
are added to our cation adsorbability series, the correlation withion bare size breaks down abruptly. We conclude with discussionof the results and hypothesize on the rules governing these ion-specific effects in all three metal chloride families.
’EXPERIMENTAL DETAILS
We used the same experimental setup as in our previous studies.5,14
Briefly, following the colloidal probe AFM technique,6 a spherical 5-μm-diameter silica bead (Bangs Laboratories) was glued to the AFM tipusing glass bond (Loctite). After UV curing of the glue, the tip and avirgin silicon (100) substrate were placed in oxygen plasma (AxicMultimode HF-8200, 50 mTorr, 100 W) for 50 min and immediatelyintroduced into the AFM (Veeco Multimode III) fluid cell. The AFMwas placed in an acoustic hood and its piezoelectric crystal was driven byan external low noise synthesizer (HP3325B) followed by a low passfilter with a 6 Hz roll-off frequency. Deflection signal and piezoelectricdriving signal were recorded using an external DAQ (National Instru-ments PCI-6289) and later analyzed using Matlab code. This arrange-ment yielded superior data compared with the commercial AFMelectronics, especially at small separations. Spring constants of the gold-coated silicon nitride cantilevers (Veeco model MLCT) varied between0.3 and 0.8 N/m, as measured by analysis of their thermal fluctuations.
Force versus separation was measured upon tip approach. The distancesbetween the tip and the surface were calibrated with a conversion factorbetween the bias applied to the piezoelectric crystal and the tip deflectionfound at “constant compliance”, that is, when the silica beadwas resting on thesubstrate. Approach velocities were kept below ∼100 nm/s to avoidhydrodynamic effects.
Adhesionor pull-off forcewasmeasured as the force required for detachingthe silica bead from the silica surface upon tip retraction, and was calibratedwith a conversion factor found at constant compliance upon retraction.
All solutions were prepared from 18MΩDI water and analytic-gradesalts. A constant pH 5.5 was maintained in most of the study by naturalbuffering of atmospheric CO2. However, in the case of Al
3+, which acts asa Lewis acid, pH 3.8 was measured already at 1 mM.We thus limited ourstudy in this case to concentrations up to 1 mM and maintained aconstant pH 3.8 by titrating HCl at lower salt concentrations. Each set ofmeasurements began and ended by measuring the force in a calibrationsolution (NaCl 1 mM), confirming sample stability over the wholeexperiment. Rare cases where the force at the end of the measurementsdid not coincide with the initial curve were discarded. We were careful tocompare data sets measured on the same sample and were careful tomonitor the solution pH before each experiment.
Each of the graphs presented below depict an average over 8�10sequential measurements. The vertical error bars correspond to thestandard deviation between these measurements.
1:1 Salts: NaCl, KCl, and CsCl. In this section, we presentmeasurements of adhesion between silica surfaces immersed in1:1 electrolytes, NaCl, KCl, and CsCl, and show that theobservations and conclusions regarding ion-specific effects fromsuch measurements are similar to those obtained in our previousforce�separation study.5
Figure 1 shows adhesion or pull-off force, as a function of saltconcentration, in each of the three 1:1 salt solutions. Regardlessof salt type, adhesion was not observed below ∼50 mM con-centration. Adhesion first appeared at a salt-specific concentra-tion we term critical adhesion concentration (cac) that wasmeasured to be between 20 mM and 50 mM in CsCl, between50 mM and 0.1 M in KCl, and between 0.1 and 0.2 M in NaCl.For each salt, the low limit indicates the highest measuredconcentration at which no adhesion was observed while the highlimit indicates the lowest concentration at which adhesion wasmeasured. For each of the salts, at concentrations higher than therespective cac the adhesion first grew with salt concentration. InCsCl and in KCl, the adhesion force reached a maximum at∼0.2 M. At higher concentrations, the adhesion declined rapidlyfor CsCl and more slowly for KCl. The adhesion dropped to 1/4of its maximal value at 0.5 M for CsCl and at 1 M for KCl. InNaCl, it seemed that the adhesion approached its maximum atconcentrationJ1 M. We limited our study to concentrations upto 1 M, considering the solubility limits of the salts and the factthat the local concentration of ions near the charged silicasurfaces could be significantly higher than in bulk, especiallywhen the two surfaces approach each other. Although maximaladhesion was reached at different concentrations for each salt, itsvalue, ∼0.8�1 mN/m, was practically identical for all salts,supporting the attribution of adhesion to ion-independent vdWattraction. As in our force�separation study,5 we were able toexplain the adhesion as a function of salt concentration in termsof two opposing contributions, namely, edl repulsion modifiedby specific cation adsorption and ion-independent vdW attrac-tion. At concentrations lower than the respective cac, extensivesurface-charge generated strong double-layer repulsion betweenthe two negatively charged silica surfaces which overcame vdWattraction, giving rise to a net repulsive force at contact. The risein adhesion with concentration that followed the respective cacwas attributed to the decrease of surface-charge resulting fromcation-specific adsorption to the silica and parallel shrinkage ofthe screening length. As salt concentration increased, morecations adsorbed to the initially negatively charged silica surfaces,lowering the absolute value of the surface-charge (see Figure 4 inref 5) and weakening the double-layer repulsion. VdW attraction,on the other hand, remained unaffected by the added salt5
and, hence, dominated the interaction above the cac. Maximaladhesion was observed at salt concentrations corresponding tofull neutralization of the silica surface. Note that the salt-specificconcentrations corresponding to maximal adhesion were practi-cally identical to the respective estimates of surface neutralizationconcentrations reported in our previous force versus separa-tion study.5 Since vdW attraction theoretically diverges atcontact, the finite maximal force observed in the experimentindicated that the silica surfaces maintained aminimal distance or“closest approach”, h0, probably due to surface roughness or awater layer also remaining in contact. By taking this minimaldistance into consideration in the respective force�separationcurves, we could establish that, independent of salt type, the
Hamaker constant between the silica surfaces across the aqueouselectrolytes was H = (6.6 ( 0.6) � 10�21 J and the closestapproach, corresponding to an inward displacement of the planeof origin, was h0 = (1.05( 0.1) nm (see Figure 6). The minimalapproach factor has been neglected in our previous study,5
leading to underestimation15 of the Hamaker constant.A plateau (saturation) in the adhesion force would be expected
in Figure 1 if the surface charge remained neutralized at increasedsalt concentrations. However, the KCl and more so the CsClcurves disclosed significant decline in adhesion, implying ree-mergence of edl repulsion upon charge reversal of the silicasurfaces due to excess adsorption of the respective cations.Overcharging of silica surfaces in CsCl and in KCl was studiedin our previous work and observed by others in force as well aszeta-potential measurements (ref 5 and references therein).Although the adhesion between the silica surfaces was pro-
foundly affected by background salt type, the phenomenology ofthe adhesion as a function of salt concentration was similaramong the three electrolytes and was differentiated only by theextent of cation-specific adsorption to silica. Ranking of adsorb-ability to silica based on adhesion measurements consideredseveral observable features in the adhesion versus concentrationgraphs (Figure 1). Enhanced adsorption was characterized by alow cac, a steep increase in adhesion with concentration, quicklyachieving the adhesion maximum corresponding to surfacecharge neutralization, and subsequently, a steep decline inadhesion, indicating significant overcharging. From these char-acteristic features, we deduced the same cation adsorption series(relative adsorbability) as found from force vs separation curvesin approach (ref 5)
Naþ < Kþ <Csþ ð1ÞIt was thus established that adhesion measurements comple-
mented normal force versus separation measurements. More-over, adhesion measurements yielded an additional data point ofthe force�separation curve, corresponding to contact, a pointwhich was otherwise inaccessible in certain situations due tocantilever snap-in to the surface.2:1 Salts: MgCl2, CaCl2, and SrCl2. As mentioned in the
Introduction, studying ion-specific interactions in 2:1 electro-lytes by normal force measurements upon tip approach wasimpeded by the fact that cation adsorption and, hence, ion-specific effects in these electrolytes became significant only atrelatively high salt concentrations where edl repulsion was
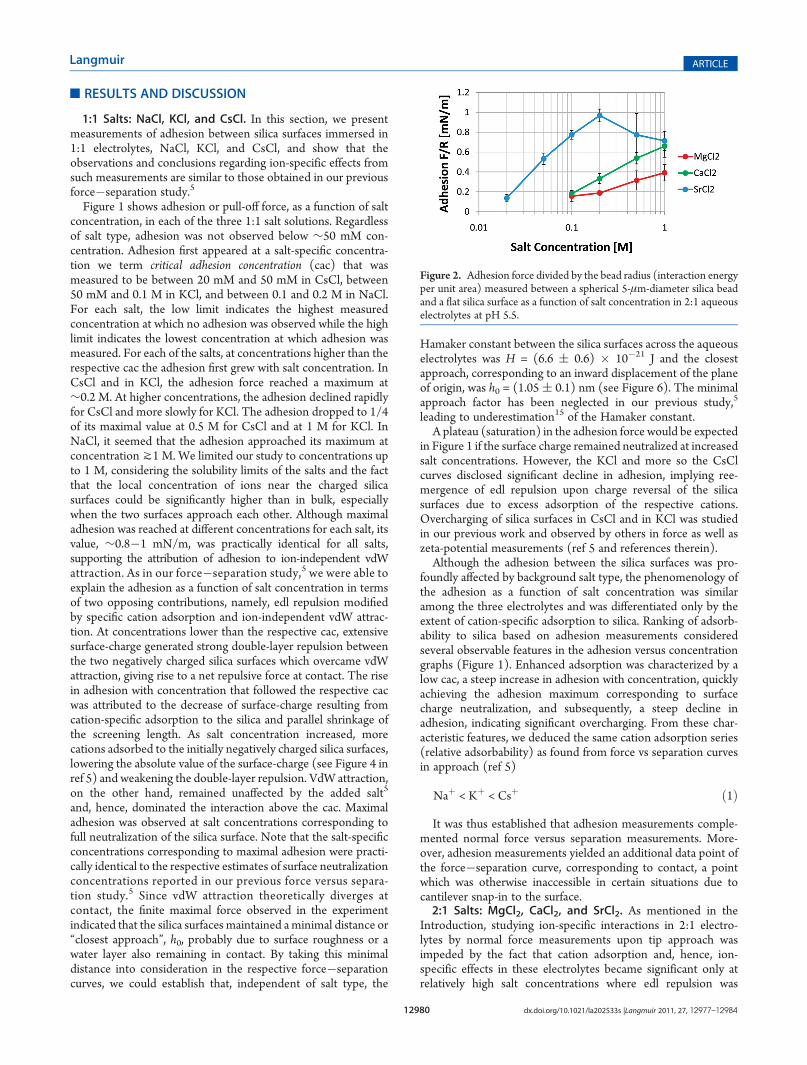
Figure 2. Adhesion force divided by the bead radius (interaction energyper unit area) measured between a spherical 5-μm-diameter silica beadand a flat silica surface as a function of salt concentration in 2:1 aqueouselectrolytes at pH 5.5.
limited due to a short screening length and nonspecific vdWattraction led to cantilever snap-in in the relevant separationrange. Having established that adhesion measurements comple-ment normal force versus separation measurements, we wereable to expand our scope and study the interaction between silicasurfaces in MgCl2, CaCl2, and SrCl2, mainly using the formermethod.Figure 2 shows adhesion or pull-off force between a spherical
5-μm-diameter silica bead and a flat silica surface as a function ofsalt concentration in each of the three 2:1 salt solutions. For eachof the 2:1 salts, a cac was observed, after which the adhesionraised with salt concentration. In SrCl2, displaying the lowest cac,between 10 mM and 20 mM, maximal adhesion was achieved at∼0.2 M, followed by a small decline in adhesion. The cac valuesin CaCl2 andMgCl2 were found to be practically similar, between50 mM and 0.1 M, with no adhesion maxima reached for eithersalt. Again, we did not go beyond 1 M considering the highconcentration of ions captured between the surfaces and solubi-lity limits of the salts. An important observation was that themaximal adhesion force observed in SrCl2, ∼1 mN/m, waspractically identical to that measured in 1:1 salts. This findingsupported the attribution of attraction to ion-independent vdWinteraction. Evidently, the phenomenology of the adhesion as afunction of salt concentration was similar to that observed in 1:1salts. Moreover, it appeared that the interaction between silicasurfaces in 2:1 salt solutions could be fully explained in the sameframework as in the case of 1:1 electrolytes, that is, by vdWattraction and edl repulsion modified by cation-specific adsorp-tion to silica. Estimates of the relative adsorbability of divalentcations were based on the same observables characteristic ofcation adsorption as mentioned in the case of monovalentcations. The fact that adhesion grew more slowly with concen-tration in MgCl2 compared with CaCl2 indicated that theadsorption of Mg2+ is even smaller than that of Ca2+, whichwas clearly smaller than that of Sr2+. In fact, as we reason below,the adsorbability of Ca2+ was even smaller than that of Na+. Thesmall decline beyond the maximal adhesion in the SrCl2 curveindicated just mild overcharging of silica surfaces by adsorptionof Sr2+ cations. The minor adsorption of Mg2+ and Ca2+ and thegreater adsorption of Sr2+ as implied by the adhesion measure-ments were consistent with results of normal force measure-ments upon tip approach, as seen in Figure 3 for 50 mM saltconcentrations. At this concentration, a sufficiently long screen-ing length allowed noticeable ion-specific effects on the forceupon tip approach.
Analyzing the adhesion data, as well as force�separationmeasurements where possible, the following order of divalentcation adsorbability to silica emerged:
Mg2þ <Ca2þ < Sr2þ ð2Þ
Evidently, adsorbability again followed ion bare size,10 but asbefore, for the homogeneous series (alkaline earth metals only) italso correlated with volume and surface charge density,10 hy-drated radius,11 and solvation energy.12 As pointed out in theIntroduction for the case of 1:1 salts, a cation-specific ionizationof the silica silanols by the divalent cations is unlikely to play amajor role in the alteration of surface charge in our pH range(pH 5.5), as implied by Figure 1 in Dove et al.8
Comparison of Adhesion in 1:1 and 2:1 Electrolyte Solu-tions. The Hofmeister series for the various phenomena men-tioned in the Introduction rank ions according to their effect on asingle property such as protein precipitation or surface tension.Force measurements, on the other hand, reveal multiple featuresand could therefore lead, at least in principle, to different rank-ings based on the same set of data. One series can be definedaccording to the cac, another, by the salt concentration corre-sponding to maximal adhesion, and a third series with respect tothe decrease in adhesion past the maximal value. In the compar-ison between cations of a common column of the periodic table(Figure 1 and Figure 2, separately), the three criteria yieldedidentical rankings from which unequivocal ranking of cationadsorbability to the silica surfaces was deduced. However, thecomparison between cations across the two columns, depicted in
Figure 3. Force measured in approach divided by the bead radius vsseparation curves between a spherical 5-μm-diameter silica bead and aflat silica surface in 2:1 aqueous electrolytes at 50 mM salt concentra-tions and pH 5.5.
Figure 4. (a) Compilation of data from Figures 1 and 2 showingadhesion in 1:1 and 2:1 aqueous electrolytes as a function of saltconcentration. (b) The same curves shown as a function of thecorresponding ionic strengths of the solutions; curves of 2:1 salts wereshifted on the concentration axis by a factor of 3, in order to comparecurves at similar ionic strengths and, hence, at similar screening lengthsas in 1:1 salts.
Figure 4a, revealed the ambiguity mentioned above. Rankingabsorbability according to cac yielded the series
Naþ < Kþ≈Ca2þ≈Mg2þ <Csþ < Sr2þ ð3Þ
while ranking by the concentration corresponding to maximaladhesion clearly positioned Na+ to the right of Mg2+ and Ca2+,although their cac ordering in Series 3 was opposite. Similarly,with respect to the rate upon which the adhesion decreasedbeyond its maximum, Cs+ and K+ are positioned to the right ofSr2+. Inference of adsorbability of cations across columns of theperiodic table required, hence, further consideration of thefactors affecting the measured force and particularly differencesassociated with ion valency.First, note that, within the framework of Poisson�Boltzmann
equation, the electrostatic interaction energy per unit area isproportional to the Debye�H€uckel screening length. For ex-ample, in the Debye�H€uckel approximation it is given by theexpression, U(h) = (2λσ0
2/ε) exp(�h/λ), where σ0 is the sur-face charge density, λ is the Debye�H€uckel screening length, h isthe separation, and ε is the permittivity of the solution. Bythe Derjaguin approximation, the force between a sphere and aplane is then given by F(h) = (4πλRσ0
2/ε) exp(�h/λ), showingthat the edl repulsion force, even for h = 0, depends on thescreening length. When comparing force measurements inelectrolytes of the same valency, the screening length is identicalfor different salts at the same salt concentration. However, whencomparing 1:1 with 2:1 data at a similar salt concentration it isimportant to bear in mind that λ, and correspondingly the force,is√3 times smaller in the latter case compared with the former
one. At the same time, the vdW interaction remains unaffected bythe salt type and its concentration. Second, the variation ofsurface charge with salt concentration reflects two counteractingprocesses: (a) Enhanced deprotonation of silanol groups as thescreening length shortens with added salt increases the negativesurface charge (see, e.g., Na+ curve in Figure 4 of ref 5). Thisprocess is ion-independent for salts of similar ion valency,8 but itclearly discerns between 1:1 and 2:1 salts at similar salt concen-trations. (b) Further adsorption of cations to the silica surfacereduces and even reverses surface charge.Both the edl interaction and the degree of silanol deprotona-
tion depend directly on the screening length. Therefore, cationsof different valency should be compared at similar ionicstrengths, I = 1/2cz(1 + z), rather than at identical salt concen-trations of their respective solutions. Here, c denotes the con-centration of the z:1 salt and z denotes the cationic valency. Sincethe ionic strength of 2:1 electrolyte solutions is 3 times largerthan that of 1:1 salt solutions of the same salt concentration, theadhesion curves of 2:1 salts solutions in Figure 4b were shifted to3 times larger concentrations compared with 1:1 salts.Relative adsorbability could now be estimated from Figure 4b
with two caveats: First, adsorption at a given ionic strength is nowestimated at different ionic concentrations. For that reason,surface charge neutralization achieved at the same ionic strengthfor certain 2:1 and 1:1 salts implies now better adsorbability ofthe divalent cation, since neutralization in the divalent case wasachieved at three times lower cation concentration. Second,adsorption of divalent cations modifies the surface charge twiceas much as the adsorption of the same number of monovalentcations. Surface neutralization at the same ionic concentrationhence implies better adsorbability of the monovalent cation.These two effects clearly compensate each other to large extent.
Thus, by analyzing the adhesion as a function of ionic strength interms of all parameters including the cac, the rise (slope) ofadhesion with ionic strength following the cac, the ionic strengthat which maximal adhesion takes place, or would have beenachieved (by extrapolation), and the decline in adhesion sub-sequent to maximal adhesion, a consistent picture emerged.Specifically, all rankings of these observable features now coin-cided and gave a single series of adsorbability of monovalent anddivalent cations. Integrating all aspects pointed out above, wefound, as mentioned earlier, that the adsorbability of Ca2+, andmoreso that of Mg2+, is smaller than that of Na+. Additionally, weestimated the adsorbability of Sr2+ to be just smaller than that of K+.We thus concluded the following ranking of monovalent anddivalent cation adsorbability to silica:
Mg2þ <Ca2þ <Naþ < Sr2þ < Kþ < Csþ ð4ÞFascinatingly, it turned out at this stage of the study that
cation adsorbability was fully correlated with ion bare size as wellas with cation solvation energy also across the two first columnsof the periodic table. Furthermore, it unfolded that the volumeand surface ionic charge density, as well as ionic hydrated radius,were no longer correlated with adsorbability. For instance, thevolume and the surface charge density as well as the hydratedradius of Sr2+ are larger than those of Na+, although Sr2+ clearlyadsorbed much better to silica than Na+.3:1 Salts: AlCl3, LaCl3, and Co(NH3)6Cl3. Finally, we supple-
ment our study with adhesion measurements between silica sur-faces in 3:1 electrolyte solutions: AlCl3, LaCl3, and [Co(NH3)6]Cl3.As mentioned earlier, pH 5.5 was maintained by atmosphericCO2 buffering in the case of LaCl3 and [Co(NH3)6]Cl3, whilepH 3.8 was kept constant in AlCl3. Figure 5 depicts the adhesionbetween silica surfaces in these electrolyte solutions as a functionof ionic strength, together with adhesion curves in 2:1 and 1:1salts. In all three 3:1 electrolytes, adhesion was first observed at apractically similar cac, between 0.3 mM and 0.6 mM ionicstrength (or 0.05mM to 0.1mM salt concentration). Subsequently,adhesion first prevailed and then vanished beyond ∼10 mMionic strength (corresponding to ∼2 mM concentration). Thecoarse phenomenology of the interaction between silica sur-faces as a function of salt concentration in trivalent electrolytes(especially in the case of [Co(NH3)6]Cl3) appeared to be similarto that in the presence of 1:1 and 2:1 salt solutions, albeit with alarger variance in the measured adhesion data. Yet, an importantobservation was that in all three 3:1 electrolyte solutions themaximaladhesion ∼2 mN/m, attained around ∼5 mM ionic strength(∼1 mM concentration) was twice as large as that attained in 1:1
Figure 5. Adhesion between silica surfaces as a function of ionic strength in3:1, 2:1, and 1:1 aqueous electrolytes at pH 5.5 (AlCl3 at pH 3.8).
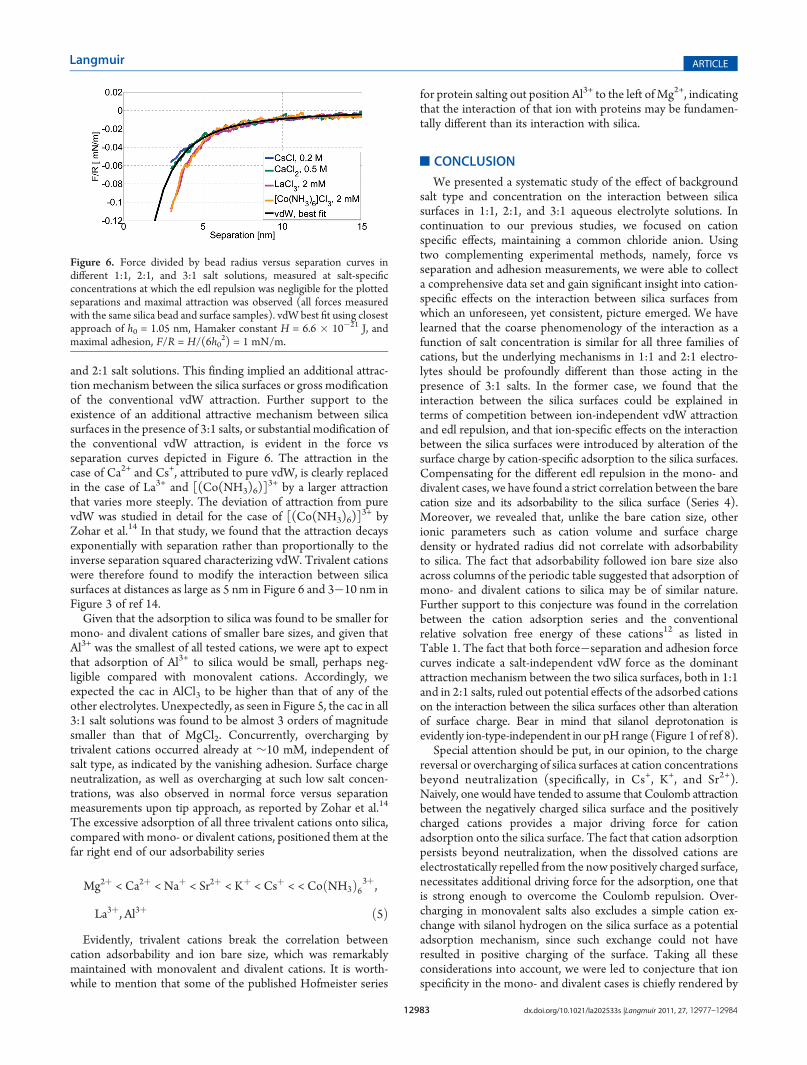
and 2:1 salt solutions. This finding implied an additional attrac-tion mechanism between the silica surfaces or gross modificationof the conventional vdW attraction. Further support to theexistence of an additional attractive mechanism between silicasurfaces in the presence of 3:1 salts, or substantial modification ofthe conventional vdW attraction, is evident in the force vsseparation curves depicted in Figure 6. The attraction in thecase of Ca2+ and Cs+, attributed to pure vdW, is clearly replacedin the case of La3+ and [(Co(NH3)6)]
3+ by a larger attractionthat varies more steeply. The deviation of attraction from purevdW was studied in detail for the case of [(Co(NH3)6)]
3+ byZohar et al.14 In that study, we found that the attraction decaysexponentially with separation rather than proportionally to theinverse separation squared characterizing vdW. Trivalent cationswere therefore found to modify the interaction between silicasurfaces at distances as large as 5 nm in Figure 6 and 3�10 nm inFigure 3 of ref 14.Given that the adsorption to silica was found to be smaller for
mono- and divalent cations of smaller bare sizes, and given thatAl3+ was the smallest of all tested cations, we were apt to expectthat adsorption of Al3+ to silica would be small, perhaps neg-ligible compared with monovalent cations. Accordingly, weexpected the cac in AlCl3 to be higher than that of any of theother electrolytes. Unexpectedly, as seen in Figure 5, the cac in all3:1 salt solutions was found to be almost 3 orders of magnitudesmaller than that of MgCl2. Concurrently, overcharging bytrivalent cations occurred already at ∼10 mM, independent ofsalt type, as indicated by the vanishing adhesion. Surface chargeneutralization, as well as overcharging at such low salt concen-trations, was also observed in normal force versus separationmeasurements upon tip approach, as reported by Zohar et al.14
The excessive adsorption of all three trivalent cations onto silica,compared with mono- or divalent cations, positioned them at thefar right end of our adsorbability series
Evidently, trivalent cations break the correlation betweencation adsorbability and ion bare size, which was remarkablymaintained with monovalent and divalent cations. It is worth-while to mention that some of the published Hofmeister series
for protein salting out position Al3+ to the left of Mg2+, indicatingthat the interaction of that ion with proteins may be fundamen-tally different than its interaction with silica.
’CONCLUSION
We presented a systematic study of the effect of backgroundsalt type and concentration on the interaction between silicasurfaces in 1:1, 2:1, and 3:1 aqueous electrolyte solutions. Incontinuation to our previous studies, we focused on cationspecific effects, maintaining a common chloride anion. Usingtwo complementing experimental methods, namely, force vsseparation and adhesion measurements, we were able to collecta comprehensive data set and gain significant insight into cation-specific effects on the interaction between silica surfaces fromwhich an unforeseen, yet consistent, picture emerged. We havelearned that the coarse phenomenology of the interaction as afunction of salt concentration is similar for all three families ofcations, but the underlying mechanisms in 1:1 and 2:1 electro-lytes should be profoundly different than those acting in thepresence of 3:1 salts. In the former case, we found that theinteraction between the silica surfaces could be explained interms of competition between ion-independent vdW attractionand edl repulsion, and that ion-specific effects on the interactionbetween the silica surfaces were introduced by alteration of thesurface charge by cation-specific adsorption to the silica surfaces.Compensating for the different edl repulsion in the mono- anddivalent cases, we have found a strict correlation between the barecation size and its adsorbability to the silica surface (Series 4).Moreover, we revealed that, unlike the bare cation size, otherionic parameters such as cation volume and surface chargedensity or hydrated radius did not correlate with adsorbabilityto silica. The fact that adsorbability followed ion bare size alsoacross columns of the periodic table suggested that adsorption ofmono- and divalent cations to silica may be of similar nature.Further support to this conjecture was found in the correlationbetween the cation adsorption series and the conventionalrelative solvation free energy of these cations12 as listed inTable 1. The fact that both force�separation and adhesion forcecurves indicate a salt-independent vdW force as the dominantattraction mechanism between the two silica surfaces, both in 1:1and in 2:1 salts, ruled out potential effects of the adsorbed cationson the interaction between the silica surfaces other than alterationof surface charge. Bear in mind that silanol deprotonation isevidently ion-type-independent in our pH range (Figure 1 of ref 8).
Special attention should be put, in our opinion, to the chargereversal or overcharging of silica surfaces at cation concentrationsbeyond neutralization (specifically, in Cs+, K+, and Sr2+).Naively, one would have tended to assume that Coulomb attractionbetween the negatively charged silica surface and the positivelycharged cations provides a major driving force for cationadsorption onto the silica surface. The fact that cation adsorptionpersists beyond neutralization, when the dissolved cations areelectrostatically repelled from the now positively charged surface,necessitates additional driving force for the adsorption, one thatis strong enough to overcome the Coulomb repulsion. Over-charging in monovalent salts also excludes a simple cation ex-change with silanol hydrogen on the silica surface as a potentialadsorption mechanism, since such exchange could not haveresulted in positive charging of the surface. Taking all theseconsiderations into account, we were led to conjecture that ionspecificity in the mono- and divalent cases is chiefly rendered by
Figure 6. Force divided by bead radius versus separation curves indifferent 1:1, 2:1, and 3:1 salt solutions, measured at salt-specificconcentrations at which the edl repulsion was negligible for the plottedseparations and maximal attraction was observed (all forces measuredwith the same silica bead and surface samples). vdW best fit using closestapproach of h0 = 1.05 nm, Hamaker constant H = 6.6 � 10�21 J, andmaximal adhesion, F/R = H/(6h0
ion solvation properties and the free energy cost associated withpartial or full dehydration of the adsorbing cations. The drivingforce for accumulation of ions on the silica interface could reflectthe growing osmotic pressure of ions in solution at elevated saltconcentrations and its competition with ion solvation.
It is worth mentioning at this point a potentially relatedphenomenon occurring at the air/electrolyte interface. There,the surface tension is known to exhibit marked differences in thepresence of different salts,3 supposedly due to excess segregation ofanions to the interface albeit the associated electrostatic price.16
A recent publication17modeled anion-specific effects for surfacesof different hydrophobic/hydrophilic nature and surface charges.This paper found an interesting phase diagram for the Hofmeisterseries, including reversal of series orderwhen the bare surface chargepolarity changed. Our experiment does not distinguish betweenbare and adsorbed surface charge, but it is worthmentioning that wedo not observe any change in cation order when the total surfacecharge is reversed due to excess cation adsorption.
Comparing both force�separation and adhesion betweensilica surfaces in the presence of 3:1 electrolytes with those in2:1 and 1:1 electrolytes revealed two significant findings: (a) Theinteraction between the two silica surfaces in the presence oftrivalent cations takes a different shape compared with that in 2:1and 1:1 salts. (b) The adsorbabilities to silica of the three trivalentcations were much larger than those characterizing mono- anddivalent cations.
Point (a) was apparent both in the maximal adhesion force,which was approximately twice as large in the 3:1 case comparedwith the 1:1 and 2:1 cases, and in comparison of force�separa-tion measurements as depicted in Figure 6. Conventional, ion-independent vdW attraction alone could not therefore solelyaccount for the observed attraction, in accord with a previousforce�separation study in cobalt hexamine salt, [Co(NH3)6]Cl3,solutions of ref 14.
Point (b) was established from the cac in 3:1 electrolyteswhich were 3 orders of magnitude smaller than that of the bestadsorbing cation (Cs+) of the 1:1 and 2:1 families and theconcurrent enhanced overcharging. This point was also consis-tent with our previous publication, which showed surface chargeneutralization already at 0.2 mM cobalt hexamine salt concentra-tion (see Figure 2b in ref 14). Point (b) clearly implies theexistence of an additional or modified driving force for adsorp-tion of trivalent cations to silica, which is somehow insignificantor nonexistant for mono- and divalent cations. The fact that thecorrelation with the ion bare size and solvation free energy(Table 1), which was maintained with mono- and divalentcations, breaks down abruptly for trivalent cations suggests thatthe adsorption of trivalent cations to silica is of different naturethan that of mono- and divalent cations. This observationsuggests that their adsorption may be governed by particularinteractions with the silica surfaces rather than solvation. Theseinteractions need to be strong enough to overcome the large freeenergy cost associated with full or partial dehydration that islikely to accompany ion adsorption. Note, however, that, unlikethe case of monovalent cations, in the trivalent case an ionexchange process with silanol protons may lead to chargeinversion. Such a process may also provide an effective drivingforce to adsorption. The similarity among the three cationssuggests that this mechanism is common to all three trivalentcations.
Further work is needed in order to establish or disprovepotential relations between the excessive adsorption of trivalent
cations to silica and the enhanced attraction between silicasurfaces in the presence of such cations. It is evident thoughthat both properties are fundamentally different in 3:1 saltsolutions compared with their counterparts in the case ofmonovalent and divalent cations.
’ASSOCIATED CONTENT
bS Supporting Information. Force on approach divided bybead radius vs separation between a spherical 5-μm-diametersilica bead and a flat silica surface in NaCl and in NaBr aqueouselectrolytes of different salt concentrations, from 1 mM to 0.2 M,at pH 5.5. This material is available free of charge via the Internetat http://pubs.acs.org.
Financial support by the Israeli Science Foundation under grantnumber 1370/07, the German Federal Ministry of Educationand Research (BMBF) within the framework of German-IsraeliProject Cooperation (DIP), and the German-Israeli Science Founda-tion under grant I-1045-82.14/2009 are gratefully acknowledged. Wethank Prof. Roland R. Netz, Prof. David Andelman, and Dr. DanBen-Yaakov for illuminating discussions.
’REFERENCES
(1) Hofmeister, F. Arch. Exp. Pathol. Pharmakol. 1888, 24, 247–260.(2) Kunz, W.; Henle, J.; Ninham, B. W. Curr. Opin. Colloid Interface
Sci. 2004, 9, 19–37.(3) Collins, K. D.; Washabaugh, M. W. Q. Rev. Biophys. 1985, 18,
323–422.(4) Lo Nostro, P.; Lo Nostro, A.; Ninham, B. W.; Pesavento, G.;
Fratoni, L.; Baglioni, P. Curr. Opin. Colloid Interface Sci. 2004, 9, 97–101.(5) Dishon,M.; Zohar, O.; Sivan, U. Langmuir 2009, 25, 2831–2836.(6) Ducker, W. A.; Senden, T. J.; Pashley, R. M. Nature 1991,
353, 239–241.(7) Verwey, E. G. W.; Overbeck, J. J. G. Theory of the Stability of
4970.(9) Franks, G. V. J. Colloid Interface Sci. 2002, 249, 44–51.(10) Marcus, Y. J. Chem. Soc., Faraday Trans. 1991, 87, 2995–2999.(11) Nightingale, E. R. J. Phys. Chem. 1959, 63, 1381–1387.(12) Fawcett, W. R. J. Phys. Chem. B 1999, 103, 11181–11185.(13) Kappl, M.; Butt, H. J. Part. Part. Syst. Charact. 2002, 19, 129–
143.(14) Zohar, O.; Leizerson, I.; Sivan, U. Phys. Rev. Lett. 2006, 96,
2010, 12, 4863–4871.(16) Onsager, L.; Samaras, N. N. T. J. Chem. Phys. 1934, 2, 528.(17) Schwierz, N.; Horinek, D.; Netz, R. R. Langmuir 2010, 26,
7370–7379.
’NOTE ADDED AFTER ASAP PUBLICATION
This article was published on the Web on September 23, 2011,with citation errors in the Introduction, Table 1, and Results andDiscussion. The corrected version was reposted on September30, 2011.