Effects of Rifampin on TacrolimusPharmacokinetics in Healthy Volunteers
Mary F Hebert, PharmD, Richard M. Fisher, BS, Christopher L. Marsh, MD,Dawna Dressler, BS, and Ihor Bekersky, PhD, FCP
Tacrolimus is a marketed immunosuppressant used in liverand kidney transplantation. It is subject to extensive metabo-lism by CYP3A4 and is a substratefor P-glycoprotein-mediatedtransport. A pharmacokinetic interaction with rifampin, anantituberculosis agent and potent inducer of CYP3A4 andP-glycoprotein, and tacrolimus was evaluated in six healthymale volunteers. Tacrolimus was administered at doses of 0. 1mg/kg orally and 0.025 mg/kg/4 hours intravenously. Thepharmacokinetics of tacrolimus were obtained from serialblood samples collected over 96 hours, after single oral and
Tacrolimus (FK506, Prograf), a potent immunosup-pressive, was marketed in the United States for the
prevention of rejection in liver transplant patients (in1994) and kidney transplant patients (in 1997). It hasbeen used in heart and small bowel transplantation aswell."2 Tacrolimus is a substrate for CYP3A4 andP-glycoprotein.'4 With the lifelong immunosuppres-sion administered to solid organ transplant patients,the potential for infection with or reactivation of Myco-bacterium tuberculosis is a concern. Rifampin is con-sidered to be a first-line agent for the treatment oftuberculosis, but it induces CYP3A metabolism andP-glycoprotein-mediated transport and may lead tolower concentrations of coadministered drugs.5" Thepurpose of this study was to quantify the effects of
From the Departments of Pharmacy (Dr. Hebert and Mr. Fisher) and Surgery(Dr. Marsh), University of Washington, Seattle, and Fujisawa USA, Inc.,Deerfield, Illinois (Ms. Dressier and Dr. Bekersky). Supported in part by Fuji-sawa USA, Inc. Submitted for publication June 1 9, 1 998; revised versionaccepted August 25, 1 998. Address for reprints: Mary F. Hebert, PharmD,Department of Pharmocy, University of Washington, H-375 Health Sci-ences Center, Box 357630, Seattle WA 98195-7630.
intravenous administration prior to and during an 18-dayconcomitant rifampin dosing phase. Coadministration ofrifampin significantlyincreased tacrolimus clearance (36.0 ±8.1 mllhrlkg vs. 52.8 ± 9.6 ml/hr/kg; p = 0. 03) and decreasedtucrolimus bioavailability (14.4% ± 5.7% vs. 7.0% ± 2.7%;p = 0.03). Rifampin appears to induce both intestinal andhepatic metabolism of tacrolim us, most likely through induc-tion ofCYP3A and P-glycoprotein in the liverand small bowel.
rifampin on the estimated pharmacokinetic parame-ters of tacrolimus.
METHODS
The concentration-time profiles of single-dose, orally,and intravenously administered tacrolimus in thewhole blood of healthy volunteers, with and withoutcoadministered oral rifampin at the steady state, wereexamined. Pharmacokinetic parameters were esti-mated using noncompartmental techniques. The con-tribution of hepatic and intestinal metabolism to themeasured oral bioavailability of tacrolimus was evalu-ated using a previously described mathematicalmodel.5" Intravenous and oral tacrolimus (Prograf,Fujisawa USA, Inc., Deerfield, IL) and rifampin cap-sules (Rifadin, Hoechst Marion Roussel, Kansas City,MO) were supplied by Fujisawa USA, Inc. This studywas conducted at the University of Washington Clini-cal Research Center.
Subject selection. Six healthy male volunteers par-ticipated in this study after giving informed consent.
J Clin Pharmacol 1999;39:91-96 91
HEBERT ETAL
The study protocol was approved by the Investiga-tional Review Board at the University of Washington.All subjects were Caucasian, ages between 25 and 42years, weight between 73 and 93 kg, and heights be-tween 172 and 192 cm. Subjects were considered to behealthy based on physical examination, medical his-tory, and standard blood and urine laboratory tests. Allsubjects were within 20% of their estimated ideal bodyweight. Subjects with active medical problems, takingany medications, weighing in excess of 20% over theirideal body weight, and smokers were excluded fromthe study.
Dosing regimen. Subjects were randomized in phase Ito receive tacrolimus either as a single oral dose (ap-proximately 0.1 mg/kg, adjusted to commercially avail-able units) or as an intravenous dose (0.025 mg/kg over4 hours) on day 1. After a 7-day washout period, a sec-ond dose of tacrolimus was administered by the alter-nate route. In phase II of the study, thepharmacokinetics of tacrolimus were estimated in thepresence of the inducer, rifampin. The dose and orderfor the route of tacrolimus administration were thesame as in phase I. To achieve maximum inductive ef-fects of rifampin before tacrolimus dosing in phase II,administration of rifampin (600 mg orally at bedtimefor 18 days) began on study day 15. On study day 22,subjects received either oral or intravenous tacrolimuswith the alternate route on study day 29. Tacrolimusand rifampin doses were separated by approximately10 hours to avoid a direct chemical interaction in thegut. The lower weight between the subject's actual andideal body weight was used for the calculation of tacro-limus dosing. Subjects were asked to record on a calen-dar the actual time of rifampin administration eachnight during phase II of the study. Rifampin pill countswere conducted prior to each tacrolimus administra-tion to evaluate compliance.
Subjects were asked to abstain from ethanol, grape-fruit, and caffeine-containing foods and beveragesthroughout the entire study period. All subjectsreceived the same diet at the same time on all 4 tacro-limus administration days to control for the effects offood on tacrolimus pharmacokinetics.'"' Serial bloodsamples were collected from an indwelling venouscatheter at 0, 0.5, 1, 2, 3, 4, 5, 6, 8, 10, 12, and 14 hoursafter tacrolimus oral administration or the initiation ofthe intravenous infusion. In addition, serial blood sam-ples were collected at 24, 36, 48, 72, and 96 hours fol-lowing tacrolimus dosing by separate peripheral veinneedle sticks. Intravenous infusions of tacrolimuswere administered through a separate catheter in theopposite arm.
Compliance. Serum rifampin concentrations weremeasurable for at least 10 hours following rifampindosing in subjects 2, 3, 4, and 6 for all phase II studydays. Subject 1 had measurable rifampin serum levelsfor at.least 10 hours following rifampin dosing on 5 ofthe 10 phase II study days. (There were 5 days of bloodsampling with each tacrolimus dose administered.)Subject 1 did not miss any rifampin doses based on thesubject's rifampin dosing calendar and investigator-conducted pill counts. Subject 5 had measurable ri-fampin serum concentrations for at least 10 hours on 9of the 10 phase II study days and was noted to havemissed one rifampin dose based on the rifampin pillcount. Both subject 1 and subject 5 had similar changesin estimated pharmacokinetic parameters with the co-administration of rifampin, as observed with the othersubjects (see Table I).
Analysis of blood samples. Whole blood was frozenat -200C and later analyzed by enzyme-linked immu-nosorbent assay (ELISA) for tacrolimus concentra-tions.'2 The lower limit of quantitation of the assay was0.5 ng/ml. Rifampin serum concentrations were ana-lyzed by a HPLC method.'13.14 The lower limit of quanti-tation of the assay was 0.3 R.g/ml.
Data analysis. The total area under the observed tac-rolimus concentration-time curve (AUC) was calcu-lated using the linear trapezoidal rule for the ascendingconcentrations and the log trapezoidal rule for the de-scending concentrations. AUC values were extrapo-lated from the 96-hour concentration to infinity byCl,st/ke.hml, in which Calst was the last measurable tacro-limus concentration and keli was the terminal elimina-tion rate constant, as determined by log-linearregression. Area under the moment curves (AUMC)was calculated by the linear trapezoidal rule for boththe ascending and descending portions of the curveand were extrapolated to infinite time."5 Intravenoustacrolimus mean residence time (MRT = [AUMC/AUC]- [Infusion duration/2]), intravenous tacrolimus clear-ance (CL = Dose/AUCiv), steady-state volume of distri-bution (Vss = CL x MRT), and observed oralbioavailability (FM = [AUCo,raI/AUCVI x[Doseiv/Doseo,all) were also calculated."5 The hepatic ex-traction ratio (ERH = CL/Q) was estimated from intra-venous data (assuming that nonhepatic clearance afteran intravenous dose is negligible), with hepatic bloodfloW (QH) assumed to be 1500 ml/min/70 kg.
Assuming that tacrolimus elimination is primarilyhepatic once the parent drug reaches the systemic cir-culation,"b it follows that the measured oral bioavail-ability of tacrolimus (F,r,,, is a product of the fraction
92 * J Clin Pharmacol 1999;39:91-96
EFFECTS OF RIFAMPIN ON TACROLIMUS PHARMACOKINETICS
co A
CZcO
4"E
CD
a)
CO
CO
CO -a)-
r44C
ca)
AZ -CO*. ~ .
AC0O
CO a,S) D
aC
,,E
H n1
.0>o z;
0
CI)11
DRUG INTERACTIONS
O.. co oLIS COC6) . 4 to
4_ -Cl -
C) cO IC) C) cO CO N CDC£ t-o AD C 06 t- t, Oc) C) C) ) C) C) C)
0 0o CO N H 1 NC I) - ) C) -/"4 IC
- - Cl "
o Lo T-O nH0 C)
L) 0
TV C tt m NJ n m
N LOI: r- Tv CIAN O6 Q C) O Ic)CO otz Cs'OCO LO LCOCl IC) n CIA 'a
LO C) CO CO C N C) CCcn 6 Cl 't C) CO C0co co C) cnO N LO ClLO CD It LO) LO) C. Cl
CON C) Cl l co CO Co 0) 0
COoCD O IC) m CD cn 0 o
CI C ) ) Ct)C C) C)
LO ~oCOLOltzv O LO CDO
d N O Ca) CO [s0N(6 11 Un 14 C Lln ts Cli 6
LO 0 C LO) T - LO LO CDcl H H q 4 ; O6
CO C co CLO CL
LO 0 CO - 1o oC'
tO LO ": CO CO "C) to t - cl t C)
t C) o Cli CltL -O C) Co CO Cco CD) -
c) '11 d1 N CO 00OLOC CO CO' CO C1) Cl
2 tCo
CO CO J4t n It: n Iv RT
COCD O(D O'
Cl
C:O
LO 0
OCD
(1) ~co C CIO :
co H-l Cl co Co 2 COn to )
4 1~~
93
HEBERT ETAL
of the dose absorbed from the gut lumen (F,bs) multi-plied by the fraction of the dose available across the gut(Fg,8t) multiplied by the fraction of the dose availableacross the liver (FH)' or Fn=e FH x F 5.917 Val-ues for FH before and with rifampin were determinedfrom the measured ERH (FH = 1 - ERH), allowing calcu-lation of intestinal bioavailability (F,1,, x Fg,1t). This cal-culation assumes that rifampin does not affect liverblood flow to any significant degree, which has beenshown by Reichel et al.'8 Therefore, changes in FH andF,,, x F,,,t should reflect the induction of tacrolimusmetabolism or the induction of tacrolimus efflux byP-glycoprotein. All results are reported as mean values± standard deviations before and with coadministra-tion of rifampin. Differences in the pharmacokineticparameters before and with rifampin were analyzedusing the Wilcoxon signed rank test and were consid-ered to be significant at p < 0.05.
RESULTS
The estimated pharmacokinetic parameters of tacro-limus in the blood for individual subjects are shownbefore and with the coadministration of rifampin(phase I and II, respectively) in Table I. The averagevalue for the extrapolated AUC beyond 96 hours wasapproximately 12% of the total AUC. With coadminis-tration of rifampin, a statistically significant increasewas observed in clearance (CL; 36.0 ± 8.1 ml/hr/kg vs.52.8 ± 9.6 ml/hr/kg; p = 0.03), decrease in oral bioa-vailability (F,,,e,,s; 14.4% ± 5.77% vs. 7.0% ± 2.7%; p =0.03), and intestinal bioavailability (F,,b x Fg,,t; 14.8%± 5.9% vs. 7.3% ± 2.9%; p = 0.03). Hepatic bioavail-ability was relatively unchanged (Fe,; 97.2%/O ± 0.6% vs.95.9% ± 0.7%;p = 0.03), even though there was a largechange in tacrolimus clearance. This was expectedsince the clearance of tacrolimus is considerably lessthan hepatic blood flow (FH = 1 - CL/QH). Steady-statevolume of distribution for intravenously administeredtacrolimus was unchanged (1.4 ± 0.2 L/kg for phase Ivs. 1.5 ± 0.5 L/kg for phase II). In addition, each sub-ject's mean residence time for intravenous tacrolimusdecreased (41.8 ± 9.7 hours vs. 28.5 ± 8.2 hours; p =0.03). The time to reach maximal concentration afteroral tacrolimus dosing was unchanged in the presenceof rifampin (1.7 ± 1.2 hours vs. 1.5 ± 1.2 hours).
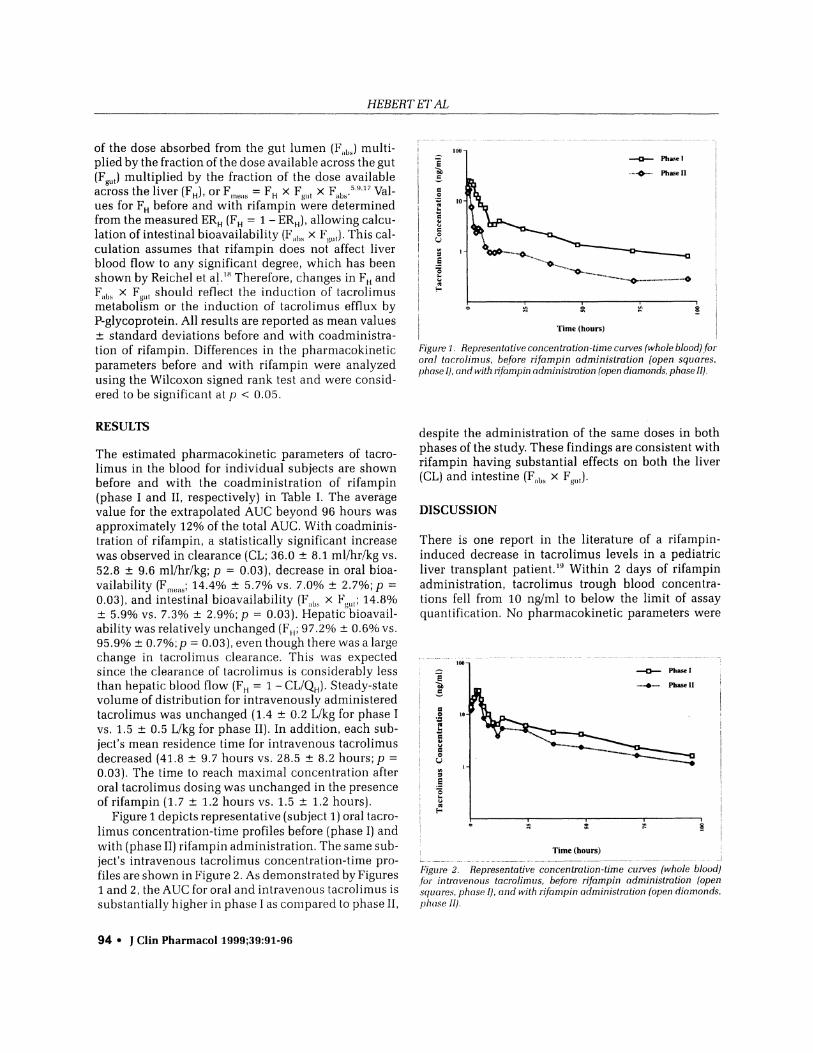
Figure 1 depicts representative (subject 1) oral tacro-limus concentration-time profiles before (phase I) andwith (phase II) rifampin administration. The same sub-ject's intravenous tacrolimus concentration-time pro-files are shown in Figure 2. As demonstrated by Figures1 and 2, the AUC for oral and intravenous tacrolimus issubstantially higher in phase I as compared to phase II,
i _ Phas~~~~~~~~~~~~~t e I2 = y ~~~~~~~~~ -wo_ ~~~~~~~................. .I ~
'Time (hours)
Figure 1. Representative concentration-time curves (whole blood) fororal tacrolim us, before rifampin administration (open squares,phase I), and with rlfampin administration (open diamonds, phase II).
despite the administration of the same doses in bothphases of the study. These findings are consistent withrifampin having substantial effects on both the liver(CL) and intestine (F,11s x F813.
DISCUSSION
There is one report in the literature of a rifampin-induced decrease in tacrolimus levels in a pediatricliver transplant patient.'" Within 2 days of rifampinadministration, tacrolimus trough blood concentra-tions fell from 10 ng/ml to below the limit of assayquantification. No pharmacokinetic parameters were
100
_~~~~~~~~~~~~~~ Phase I0
._
f~~~~~~~~~~~~~~~~~Pa °1
to
Time (hours)
Figure 2. Representative concentration-time curves (whole blood)for intravenous tacrolimus, before rifampin administration (opensquares, phase I), and with rifanipin administration (open diamonds,pha(ise 11).
94 * J Clin Pharmacol 1999;39:91-96
EFFECTS OF RIFAMPIN ON TACROLIMUS PHARMACOKINETICS
reported. Based on this case report, as well as resultsfrom previous work with cyclosporine (also a substratefor CYP3A and P-glycoprotein) and rifampin, thisstudy was conducted to evaluate the effects of rifampinon the estimated pharmacokinetic parameters oftacrolimus.
The effects of rifampin on the estimated pharma-cokinetic parameters of cyclosporine have beenreported.' Rifampin administration resulted in adecrease of 51% in Fmeas 1%in FH, and 51% in Fa,, x Fg,,tof tacrolimus compared to 64%, 12%, and 59%, respec-tively, as has been reported for cyclosporine. Rifampinproduced a substantial increase in clearance for bothtacrolimus and cyclosporine, 47% versus 39%, respec-tively. Since the clearance of tacrolimus is very smallrelative to hepatic blood flow, very little change wasexpected in FH (FH = 1 - CL/Q). In the past, it has gener-ally been assumed that agents with low hepatic clear-ances such as tacrolimus would not have significantfirst-pass metabolism. Tacrolimus is an example of anagent in which this rule does not apply. That is, eventhough tacrolimus has a relatively low clearance ascompared with hepatic blood Hlow, it still undergoesextensive first-pass metabolism in the intestine. Theclearance of cyclosporine is much larger than the clear-ance of tacrolimus. Therefore, an increase in the clear-ance of cyclosporine results in more of a decrease inthe FH for cyclosporine. Rifampin has substantialeffects on both oral and intravenous tacrolimus phar-macokinetics. The magnitude of the increase in tacro-limus clearance is similar to that of the decrease inbioavailability. However, the change in clearance is notthe reason for the decreased oral bioavailability, as canbe seen by the decrease in F,1,, x FgW, with minimalchange in F H-
Rifampin induces mRNA for CYP3A in humanhepatocyte cultures and in human enterocytes andincreases the expression of CYP3A4.721 21 It alsoincreases CYP3A activity in the human hepatocyte cul-ture, as measured by testosterone 6f-hydroxylation,2'and in the liver, as measured by the erythromycinbreath test.6 In addition to rifampin's CYP3A effects, ithas been reported to increase the expression ofP-glycoprotein.i The inductive effects of rifampin onCYP3A mRNA as well as CYP3A and P-glycoproteinexpression, although reversible, probably have a sus-tained effect for some period of time beyond that ofmeasurable rifampin serum concentrations. In addi-tion, rifampin's inductive effects are likely to take sometime to reach their maximum. This concept is consis-tent with what is seen clinically. For example, patientsreceiving cyclosporine, who require the addition ofrifampin therapy to their drug regimen, have been
noted to have a decrease in their cyclosporine levels.This effect begins a few days following the initiation ofrifampin, peaks at a week or so, and remains for severaldays to weeks following discontinuation.2224 In addi-tion, even though rifampin concentrations were belowquantitation limits (0.3 gug/ml) for at least 14 hours ofeach rifampin administration day in nearly all subjectsin this study, significant induction effects were seen inboth hepatic and intestinal metabolism in this study. Inconclusion, rifampin appears to induce both intestinaland hepatic metabolism of tacrolimus, most likelythrough the induction of CYP3A and P-glycoprotein inthe liver and small bowel. Although there was approxi-mately a 50% increase in CL and decrease in F,ne,,s fortacrolimus with concomitant rifampin, the magnitudeof the interaction was highly variable for both CL(range: 6.4% to 203% increase) and F.ne.s (range: 9.3% to68.7%/o decrease). This study documents the need forcareful monitoring of tacrolimus trough concentra-tions and corresponding dose escalation whenrifampin is added to either oral or intravenous tacro-limus therapy.
REFERENCES
1. Rinaldi M, Pellegrini C, Martinelli L, Goggi C, Gavazzi A, CampanaC, et al: FK506 effectiveness in reducing acute rejection after hearttransplantation: a prospective randomized study. J Heart Lung Trans-plant 1997;16:1001-1010.2. Asfar S, Atkison P, Ghent C, Duff J, Wall W, Williams S, et al: Smallbowel transplantation: a life-saving option for selected patients withintestinal failure. Dig Dis Sci 1996;41:875-883.3. Sattler M, Guiengerich FP, Yun C-H, Christians U, Sewing KF: Cyto-chrome P-450A enzymes are responsible for biotransformation ofFK506 and rapamycin in man and rat. Drug Metab Dispos1992;20:753-761.4. Saeki T, Ueda K, Tanigawara Y, Hori R, Komano T: Human p-glycoprotein transports cyclosporin A and FK506. I Biol Chem1993;268:6077-6080.5. Hebert MF, Roberts JP, Prueksaritanont T, Benet LZ: Bioavailabilityof cyclosporine with concomitant rifampin administration is mark-edly less than predicted by hepatic enzyme induLction. Clin Pharma-/ol 7hei 1992;52:453-457.
6. Watkins PB, MUrray SA, Winkelman LG, Heuman DM, WrightonSA, Guzelian PS: The erythromycin breath test as an assav ofglucocorticoid-inducible liver cytochromes P-450. J Clin Invest1989;83:688-697.7. Kolars JC, Schmiedlin-Ren P, Schuetz JD, Fang C, Watkins PB: Iden-tification of rifampin-inducible P450111A4 (CYP3A4) in human smallbowel enterocytes. l Clin Invest 1992;90:1871-1878.8. Schuetz EG, Beck WT, Schuetz JD: Modulators and substrates ofp-glycoprotein and cytochrome P450 3A coordinately up-regulatethese proteins in human colon carcinoma cells. Mol Pharmacol1996;49:311-318.9. Wuo CY, Benet LZ, Hebert MF, Gupta SK, Rowland M, Gomez DY, et al:Differentiation of absorption and first-pass gut and hepatic metabo-
DRUG INTERACTIONS 95
HEBERT ETAL
lism in humans: studies with cyclosporine. Clin Pharmacol Ther1995;58:492-497.10. Mekki Q, Lee C, Carrier S, Klintmalm G, Schaefer M, Burke P, et al:The effect of food oIl oral bioavailability of tacrolimus (FK506) inliver transplant patients. Clini Phaiwiacol Ther 1993;53:229.11. Dressler D, Bekersky 1, Lee J. Tracewell WG, Kisicki J, Mekki Q: l,tf-fect of ineal timiie on tac.roliniIs b)ioavailability. (/lnii Phor1nacol Ther1996;59:151.12. Lee JW, Sukovaty RL, Farmen RH, Dressler DE, Alak A, Bekersky 1:Tacrolimus (FK506): validation of a sensitive enzyme-linked immu-nosorbent assay kit for and application to a clinical pharmacokineticstudy. Ther Drug Moniit 1997;19:201-207.13. Ishii M, Ogata H: Determination of rifamnpicin and its main me-tabolites in human plasma by high-performance liquid chromatogra-phy. J Chromatogr 1988;426:412-416.14. Jamaluddin ABM, Sarwar G, Rahiim MA, Rahman MK: High-performance liquid chromatographic assay of rifampicin in huLmanserum. I Chiomnotogr 1990;525:495-497.
15. Benet LZ, Galeazzi RL: Noncompartmental determination of thesteady-state volume of distribtution. IPhorm Sci 1979;68:1071-1074.16. Paine MF, Shen DD, Kunze KL, Perkins JID, Marsh CL, McVicar JP,et al: First-pass metabolism of midazolam bv the 1lumani intestine.Clin Phormacol Ther 1996:60:14-24.
17. Beinet LZ, Wu CY, Hebert MF, Wacher VJ: Intestinal drug metabo-lism and antitransport processes: a potential paradigm shift in oraldrtug delivery. I Control Rel 1996;39:139-143.18. Reichel C, Block W, Skodra T, Traber F, Schiedermaier P, SpenglerlJ, et al: Relationship between cytochrome P-450 induction by ri-famipicin, hepatic volume and portal blood flow in Inan. Eurl Gostro-e(Iterol Hepolol 1997;9:975-979.19. Furlan V, Perello L, Jacquemin E, Debray D, Taburet A-M: Interac-tion between FK506 and rifampicin or erythromycin in pediatricliver recipients. Transplantation 1995;59:1217-1218.20. Schuetz EG, Schuetz JD, Strom SC, Thompson MT, Fisher RA,Molowa DT, et al: Regulation of human liver cytochromes P-450 infamily 3A in primary and continuous culture of human hepatocytes.Hepatology 1993;18:1254-1262.21. Williams JA, Chenery RJ, Hawksworth GM: Induction of CYP3Aenzymes in human and rat hepatocyte cultures. Biochem Soc Trans1994;22:131S.22. Coward RA, Raftery AT, Brown CB: Cyclosporine and antituber-culouis therapy. Lancet 1985;1:1342-1343.23. Daniels NJ, Dover JS, Schachter RK: Interaction between cyclo-sporine and rifampicin. Lancet 1984;2:639.24. Vandevelde C, Chang A, Andrews D, Riggs W, Jewesson P: Ri-fampin and ansamycin interactions with cyclosporine after renaltransplantation. Pharinacotherapy 1991; 11:88-89.