Page 1
ORIGINAL PAPER
Effects of Selenium on Calcium Signaling and Apoptosisin Rat Dorsal Root Ganglion Neurons Induced by Oxidative Stress
Abdulhadi Cihangir Uguz • Mustafa Nazıroglu
Received: 20 February 2012 / Revised: 8 March 2012 / Accepted: 16 March 2012 / Published online: 3 April 2012
� Springer Science+Business Media, LLC 2012
Abstract Ca2? is well known for its role as crucial sec-
ond messenger in modulating many cellular physiological
functions, Ca2? overload is detrimental to cellular function
and may present as an important cause of cellular oxidative
stress generation and apoptosis. The aim of this study is to
investigate the effects of selenium on lipid peroxidation,
reduced glutathione (GSH), glutathione peroxidase (GSH-
Px), cytosolic Ca2? release, cell viability (MTT) and
apoptosis values in dorsal root ganglion (DRG) sensory
neurons of rats. DRG cells were divided into four groups
namely control, H2O2 (as a model substance used as a
paradigm for oxidative stress), selenium, sele-
nium ? H2O2. Moderate doses and times of H2O2 and
selenium were determined by MTT test. Cells were pre-
terated 200 nM selenium for 30 h before incubatation with
1 lM H2O2 for 2 h. Lipid peroxidation levels were lower
in the control, selenium, selenium ? H2O2 groups than in
the H2O2 group. GSH-Px activities were higher in the
selenium groups than in the H2O2 group. GSH levels were
higher in the control, selenium, selenium ? H2O2 groups
than in the H2O2 group. Cytosolic Ca2? release was higher
in the H2O2 group than in the control, selenium, sele-
nium ? H2O2 groups. Cytosolic Ca2? release was lower in
the selenium ? H2O2 group than in the H2O2. In conclu-
sion, the present study demonstrates that selenium induced
protective effects on oxidative stress, [Ca2?]c release and
apoptosis in DRG cells. Since selenium deficiency is a
common feature of oxidative stress-induced neurological
diseases of sensory neurons, our findings are relevant to the
etiology of pathology in oxidative stress-induced neuro-
logical diseases of the DRG neurons.
Keywords Calcium ion release � Selenium �Oxidative stress � Dorsal root ganglion neurons � Apoptosis
Introduction
Oxidative stress represents an imbalance status between
excessive production of reactive oxygen species (ROS),
oxygen-derived radical species and biological system’s
scavenging ability to detoxify the reactive agents. Most of
the ROS are formed as a result of the mitochondrial
respiratory chain pathways but can also be formed exoge-
nously [1]. Antioxidants such as glutathione (GSH), glu-
tathione peroxidase (GSH-Px) and related enzymes are
believed to play critical roles in protecting cells from
hazardous oxygen species [2]. GSH-Px, one of the major
intracellular antioxidant enzymes, detoxifies hydrogen
peroxide (H2O2) to water and also scavenge other perox-
ides [3].
Selenium is an essential dietary trace element which
plays an important role in a series of biological function. It
has been previously demonstrated that selenium plays an
important role in the continuation of the physiological
functions of the nervous system such as signal transduction
and development [4]. It also acts as a cofactor for the GSH-
Px enzyme and is also incorporated into the selenoproteins
that are involved in antioxidant defenses [5]. Selenium is
incorporated in mammalian proteins as selenocysteine or
selenomethionine, both of which are dietary forms of
selenium, although selenomethionine is the major form [6].
A. C. Uguz � M. Nazıroglu (&)
Department of Biophysics, Faculty of Medicine, Suleyman
Demirel University, Dekanlık Binasi, 32260 Isparta, Turkey
e-mail: [email protected]
A. C. Uguz � M. Nazıroglu
Neuroscience Research Center, Suleyman Demirel University,
Isparta, Turkey
123
Neurochem Res (2012) 37:1631–1638
DOI 10.1007/s11064-012-0758-5
Page 2
Even though at high concentrations it can be toxic to the
biological systems, at low concentration of selenium is
implicated as neuronprotective agents in several neuronal
diseases including epilepsy [4, 7] and pain [6]. The neu-
roprotecive effects of selenium are attributed to its ability
to inhibit apoptosis [8, 9] and to modulate Ca2? influx
through ion channels [5, 9].
Dorsal root ganglions (DRG) are nodules on a dorsal
root that contains cell bodies of neurons in afferent spinal
nerves. They transducers noxious stimuli into electric
impulses, conduct impulses and modulate the synaptic
transmission in central nervous system [10]. Sensory neu-
ronopathies and neuropathic pain are common chronic
clinical conditions that affect million of people all around
the world. Sensory neuronopathies are caused by nerve
injury or diseases such as diabetes and cancer which
damage peripheral nerves. As a result of this damage DRG
neurons become more excitable. Sensory neuronopathies
are proposed to being characterised by the primary
degeneration of DRG neurons [11, 12].
We investigated the effects of selenium supplementation
on cytosolic Ca2? levels, apoptosis and oxidative stress
parameters in cultured DRG neurons.
Materials and Methods
Chemicals
All chemicals (cumene hydroperoxide, KOH, NaOH, thio-
barbituric acid, 1,1,3,3-tetraethoxypropane, 5,5-dithiobis-2
nitrobenzoic acid, tris-hydroxymethyl-aminomethane, glu-
tathione, butylhydroxytoluol,Triton X-100 and ethylene
glycol-bis[2-aminoethyl-ether]-N,N,N,N-tetraacetic acid
[EGTA]) were obtained from Sigma-Aldrich (St. Louis,
MO, USA) and all organic solvents (n-hexane, ethyl
alcohol) were purchased from Merck (Darmstadt, Ger-
many). Fura-2 acetoxymethyl ester was purchased from
Invitrogen (Carlsbad, CA, USA). All reagents were ana-
lytical grade. All reagents except the phosphate buffers
were prepared daily and stored at ?4 �C. Reagents were
equilibrated at room temperature for half an hour before
an analysis was initiated or reagent containers were refilled.
Phosphate buffers were stable at ?4 �C for 1 month.
APOPercentageTM assay kit was purchased from Biocolor
(Belfast, Northern Ireland). Collegenase IV was bought
from Worthington Inc. (USA).
Animals
Twenty-four female Wistar-albino rats with 21–30 days of
age were used for the experimental procedures. Rats were
allowed 1 week to acclimatize to the surroundings before
beginning any experimentation. Animals were housed in
individual plastic cages with bedding. Standard rat food
and tap water were available ad libitum for the duration of
the experiments. The temperature was maintained at
22 ± 2 �C. A 12/12 h light/dark cycle was maintained,
with lights on at 06.00, unless otherwise noted. Experi-
mental protocol of the study was approved by the ethical
committee of the Medical Faculty of Suleyman Demirel
University (Protocol Number; 2009: 16-02). Animals were
maintained and used in accordance with the Animal Wel-
fare Act and the Guide for the Care and Use of Laboratory.
Preparation of DRG Samples
The animals were anesthetized by ether asphyxiation in
accordance with SDU Experimental Animal legislation.
DRG neurons (T13-L5) were carefully dissected from
peripheral nerve root. DRG neurons were collected and
incubated in Dulbecco’s modified Eagle’s medium
(DMEM, Gibco, Istanbul, Turkey) with 1 % penicillin–
streptomycin (Sigma, Istanbul, Turkey) in 500 ml of
DMEM. The connective tissue was removed and ganglia
were treated with collegenase IV (0.28 ml in DMEM), and
tyripsin (25,000 units/ml in DMEM, for 45 min at 37 �C
and in an atmosphere containing 5 % CO2). After disso-
ciation with a sterile syringe, the cell suspension was
centrifuged at 3,500 rpm and seeded into 25 cm2 sterile
flasks.
Study Groups
DRG neurons of each animal were divided into four groups
as follows:
Group I (n = 6) was control group and the DRG neurons
were incubated (37 �C and 5 % CO2) for 24 h with
normal medium.
Group II (n = 6) was H2O2 group and the DRG neurons
were incubated with 1 lM H2O2 (37 �C, %5 CO2) for
2 h.
Group III (n = 6) was selenium (Se) group and the DRG
neurons were incubated with 200 nM Se for 30 h.
Group IV (n = 6) was Se ? H2O2 group and initially
the DRG neurons were pre-incubated with 200 nM Se
for 30 h and then they were incubated with 1 lM H2O2
for 2 h.
At the end of the experiments, half of the DRG samples
were immediately used for Ca2? signaling and apoptosis
analyses. The remaining neurons were washed with phos-
phate buffer (pH 7.4) and then frozen at -33 �C. GSH,
GSH-Px and lipid peroxidation analyses were performed
within 1 week.
1632 Neurochem Res (2012) 37:1631–1638
123
Page 3
Determination of Moderate Incubation Doses
of Selenium by Cell Viability (MTT) Assay
Cell viability was evaluated by the MTT assay based on the
ability of viable cells to convert a water-soluble, yellow
tetrazolium salt into a water-insoluble, purple formazan
product. The enzymatic reduction of the tetrazolium salt
happens only in living, metabolically active cells but not in
dead cells. DRG cells were seeded in tubes at a density of
2 9 106/tube and subsequently exposed to several con-
centrations of sodium selenite (50 nM–1 lM) and H2O2
(1 lM–1 mM) at different incubation times (1–48 h for
sodium selenite and 0.5–24 h for H2O2) at 37 �C. After the
treatments, the medium was removed and MTT was added
to each tube and then incubated for 90 min at 37 �C in a
shaking water bath. The supernatant was discarded and
DMSO was added to dissolve the formazan crystals.
Treatments were carried out in duplicate. Optical density
was measured in an spectrophotometer at 490 and 650 nm
and presented as the fold increase over the pretreatment
level (experimental/control).
Measurement of Cytosolic Ca2? Concentration
([Ca2?]c)
The DRG cells were loaded with 4 lM Fura-2/AM in
loading buffer with 5 9 106 cells/ml for 45 min at 37 �C
in the dark, washed twice, incubated for an additional
30 min at 37 �C to complete probe de-esterification and
resuspended in loading buffer at a density of 3 9 106 cells/
ml according to a procedure published elsewhere [9]. The
four groups were exposed to H2O2 for stimulating intra-
cellular Ca2? release. Fluorescence was recorded from
2-ml aliquots of a magnetically stirred cellular suspension
at 37 �C using a spectrofluorometer (Carry Eclipse; Varian,
Sydney, Australia) with excitation wavelengths of 340 and
380 nm and emission at 505 nm. Changes in [Ca2?]c were
monitored using the Fura-2/AM 340/380 nm fluorescence
ratio and calibrated according to the method of Gry-
nkiewicz et al. [13]. In the experiments where calcium-free
medium is indicated, Ca2? was omitted and 2 mM EGTA
was added. Ca2? release was estimated using the integral
of the rise in [Ca2?]c for 40 s after addition of H2O2. Ca2?
release is expressed in nanomoles, taking a sample every
second as previously described [9].
Apoptosis Assay
The APOPercentageTM assay (Biocolor Ltd., Belfast,
Northern Ireland) was performed according to the
instructions provided by Biocolor Ltd. and elsewhere [14].
The APOPercentageTM assay is a dye-uptake assay,
which stains only the apoptotic cells with a red dye. When
the membrane of apoptotic cell loses its asymmetry, the
APOPercentage dye is actively transported into cells,
staining apoptotic cells red, thus allowing detection of
apoptosis by spectrophotometer [15].
Lipid Peroxidation (LP) Determinations
Lipid peroxidation levels in the DRG neurons cultures were
measured with the thiobarbituric-acid reaction by the
method of Placer et al. [16]. Thiobarbituric acid-reactive
substances were quantified by comparing the absorption to
the standard curve of malondialdehyde (MDA) equivalents
generated by acid-catalyzed hydrolysis of 1,1,3,3-tetra-
methoxypropane. LP values in DRG neurons were expressed
as micromoles per gram of protein.
Reduced Glutathione (GSH), Glutathione Peroxidase
(GSH-Px) and Protein Assay
The GSH content of the DRG neurons samples was mea-
sured at 412 nm using the method of Sedlak and Lindsay
[17]. The samples were precipitated with 50 % trichlor-
acetic acid and then centrifuged at 1,000g for 5 min. The
reaction mixture contained 0.5 ml of supernatant, 2.0 ml of
Tris–EDTA buffer (0.2 M; pH 8.9) and 0.1 ml of 0.01 M
5,50-dithio-bis-2-nitrobenzoic acid. The solution was kept
at room temperature for 5 min, and then read at 412 nm
using a spectrophotometer (Shimadzu UV-1800, Kyoto,
Japan). GSH-Px activities of DRG neurons samples were
measured spectrophotometrically at 37 �C and 412 nm
according to the method of Lawrence and Burk [18]. The
protein content in DRG neurons samples was measured by
the method of Lowry et al. [19] with bovine serum albumin
as the standard.
Statistical Analysis
Data are expressed as means ± SEM of the numbers of
determinations. Statistical significance was analyzed using
the SPSS program (9.05; SPSS, Inc., Chicago, IL, USA).
To compare the different treatments, statistical significance
was calculated by the Mann–Whitney U test. P \ 0.05 was
considered to indicate a statistically significant difference.
Results
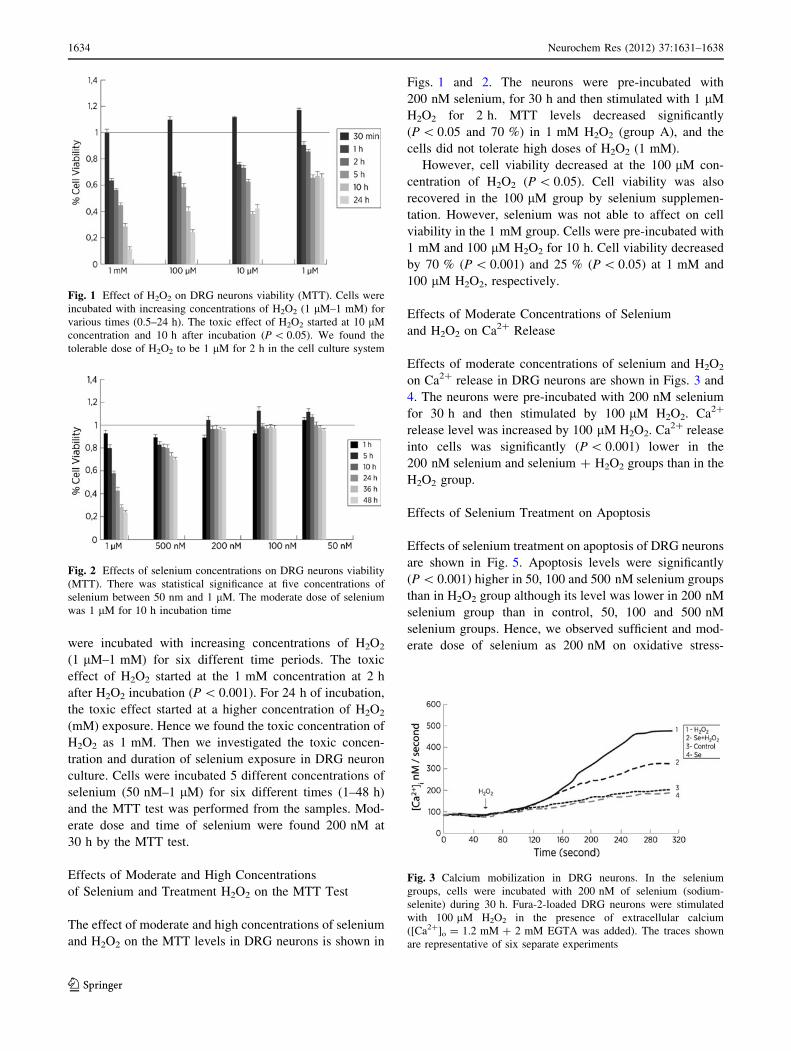
Determination of Moderate Doses of H2O2
and Selenium in DRG Neurons by Cell Viability (MTT)
Test
The effects of H2O2 and selenium on the MTT in DRG
neurons were shown in Figs. 1 and 2, respectively. Cells
Neurochem Res (2012) 37:1631–1638 1633
123
Page 4
were incubated with increasing concentrations of H2O2
(1 lM–1 mM) for six different time periods. The toxic
effect of H2O2 started at the 1 mM concentration at 2 h
after H2O2 incubation (P \ 0.001). For 24 h of incubation,
the toxic effect started at a higher concentration of H2O2
(mM) exposure. Hence we found the toxic concentration of
H2O2 as 1 mM. Then we investigated the toxic concen-
tration and duration of selenium exposure in DRG neuron
culture. Cells were incubated 5 different concentrations of
selenium (50 nM–1 lM) for six different times (1–48 h)
and the MTT test was performed from the samples. Mod-
erate dose and time of selenium were found 200 nM at
30 h by the MTT test.
Effects of Moderate and High Concentrations
of Selenium and Treatment H2O2 on the MTT Test
The effect of moderate and high concentrations of selenium
and H2O2 on the MTT levels in DRG neurons is shown in
Figs. 1 and 2. The neurons were pre-incubated with
200 nM selenium, for 30 h and then stimulated with 1 lM
H2O2 for 2 h. MTT levels decreased significantly
(P \ 0.05 and 70 %) in 1 mM H2O2 (group A), and the
cells did not tolerate high doses of H2O2 (1 mM).
However, cell viability decreased at the 100 lM con-
centration of H2O2 (P \ 0.05). Cell viability was also
recovered in the 100 lM group by selenium supplemen-
tation. However, selenium was not able to affect on cell
viability in the 1 mM group. Cells were pre-incubated with
1 mM and 100 lM H2O2 for 10 h. Cell viability decreased
by 70 % (P \ 0.001) and 25 % (P \ 0.05) at 1 mM and
100 lM H2O2, respectively.
Effects of Moderate Concentrations of Selenium
and H2O2 on Ca2? Release
Effects of moderate concentrations of selenium and H2O2
on Ca2? release in DRG neurons are shown in Figs. 3 and
4. The neurons were pre-incubated with 200 nM selenium
for 30 h and then stimulated by 100 lM H2O2. Ca2?
release level was increased by 100 lM H2O2. Ca2? release
into cells was significantly (P \ 0.001) lower in the
200 nM selenium and selenium ? H2O2 groups than in the
H2O2 group.
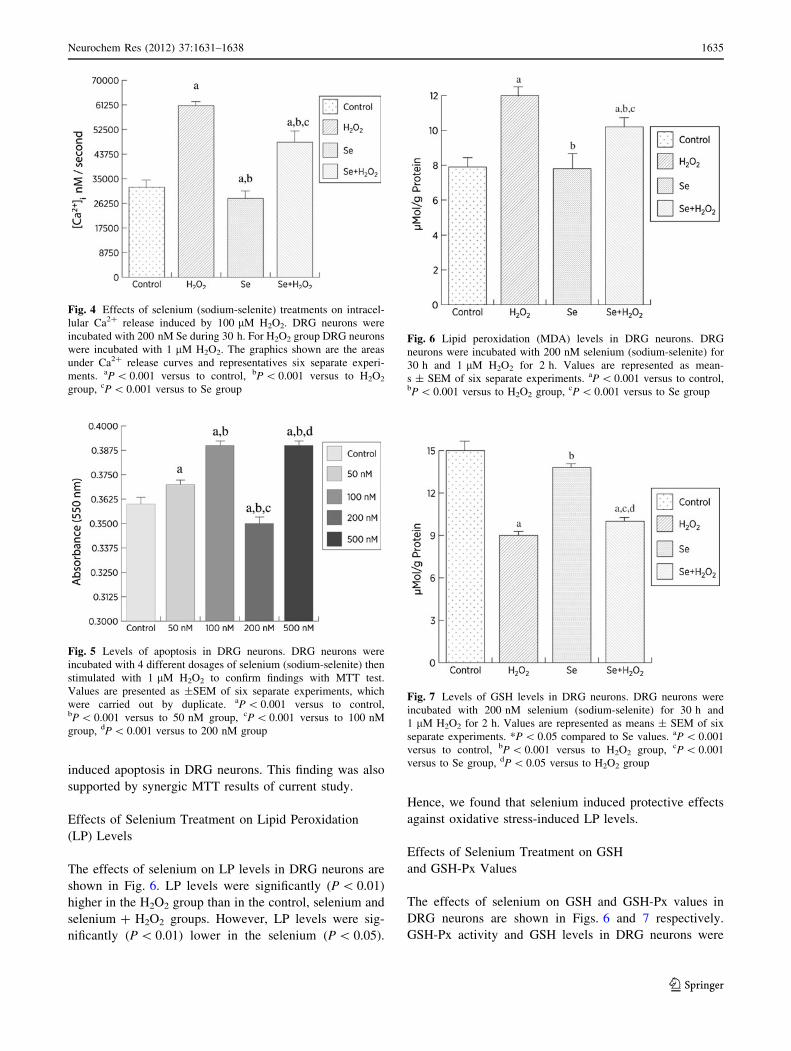
Effects of Selenium Treatment on Apoptosis
Effects of selenium treatment on apoptosis of DRG neurons
are shown in Fig. 5. Apoptosis levels were significantly
(P \ 0.001) higher in 50, 100 and 500 nM selenium groups
than in H2O2 group although its level was lower in 200 nM
selenium group than in control, 50, 100 and 500 nM
selenium groups. Hence, we observed sufficient and mod-
erate dose of selenium as 200 nM on oxidative stress-
Fig. 1 Effect of H2O2 on DRG neurons viability (MTT). Cells were
incubated with increasing concentrations of H2O2 (1 lM–1 mM) for
various times (0.5–24 h). The toxic effect of H2O2 started at 10 lM
concentration and 10 h after incubation (P \ 0.05). We found the
tolerable dose of H2O2 to be 1 lM for 2 h in the cell culture system
Fig. 2 Effects of selenium concentrations on DRG neurons viability
(MTT). There was statistical significance at five concentrations of
selenium between 50 nm and 1 lM. The moderate dose of selenium
was 1 lM for 10 h incubation time
Fig. 3 Calcium mobilization in DRG neurons. In the selenium
groups, cells were incubated with 200 nM of selenium (sodium-
selenite) during 30 h. Fura-2-loaded DRG neurons were stimulated
with 100 lM H2O2 in the presence of extracellular calcium
([Ca2?]o = 1.2 mM ? 2 mM EGTA was added). The traces shown
are representative of six separate experiments
1634 Neurochem Res (2012) 37:1631–1638
123
Page 5
induced apoptosis in DRG neurons. This finding was also
supported by synergic MTT results of current study.
Effects of Selenium Treatment on Lipid Peroxidation
(LP) Levels
The effects of selenium on LP levels in DRG neurons are
shown in Fig. 6. LP levels were significantly (P \ 0.01)
higher in the H2O2 group than in the control, selenium and
selenium ? H2O2 groups. However, LP levels were sig-
nificantly (P \ 0.01) lower in the selenium (P \ 0.05).
Hence, we found that selenium induced protective effects
against oxidative stress-induced LP levels.
Effects of Selenium Treatment on GSH
and GSH-Px Values
The effects of selenium on GSH and GSH-Px values in
DRG neurons are shown in Figs. 6 and 7 respectively.
GSH-Px activity and GSH levels in DRG neurons were
Fig. 4 Effects of selenium (sodium-selenite) treatments on intracel-
lular Ca2? release induced by 100 lM H2O2. DRG neurons were
incubated with 200 nM Se during 30 h. For H2O2 group DRG neurons
were incubated with 1 lM H2O2. The graphics shown are the areas
under Ca2? release curves and representatives six separate experi-
ments. aP \ 0.001 versus to control, bP \ 0.001 versus to H2O2
group, cP \ 0.001 versus to Se group
Fig. 5 Levels of apoptosis in DRG neurons. DRG neurons were
incubated with 4 different dosages of selenium (sodium-selenite) then
stimulated with 1 lM H2O2 to confirm findings with MTT test.
Values are presented as ±SEM of six separate experiments, which
were carried out by duplicate. aP \ 0.001 versus to control,bP \ 0.001 versus to 50 nM group, cP \ 0.001 versus to 100 nM
group, dP \ 0.001 versus to 200 nM group
Fig. 6 Lipid peroxidation (MDA) levels in DRG neurons. DRG
neurons were incubated with 200 nM selenium (sodium-selenite) for
30 h and 1 lM H2O2 for 2 h. Values are represented as mean-
s ± SEM of six separate experiments. aP \ 0.001 versus to control,bP \ 0.001 versus to H2O2 group, cP \ 0.001 versus to Se group
Fig. 7 Levels of GSH levels in DRG neurons. DRG neurons were
incubated with 200 nM selenium (sodium-selenite) for 30 h and
1 lM H2O2 for 2 h. Values are represented as means ± SEM of six
separate experiments. *P \ 0.05 compared to Se values. aP \ 0.001
versus to control, bP \ 0.001 versus to H2O2 group, cP \ 0.001
versus to Se group, dP \ 0.05 versus to H2O2 group
Neurochem Res (2012) 37:1631–1638 1635
123
Page 6
significantly (P \ 0.05) lower in the H2O2 group than in
the control, selenium and selenium ? H2O2 groups. GSH-
Px and GSH values were significantly higher in the sele-
nium and selenium ? H2O2 groups than in the H2O2
(P \ 0.01) and control (P \ 0.05) groups (Fig. 8).
Discussion
The H2O2 is the endogen source of the free radicals and
causes oxidative stress in cellular mechanisms [1]. Sele-
nium is a co-factor in GSH-Px enzyme. GSH is used as a
substrate to synthesis the GSH-Px [6]. If the free radical
production increases proportional to the consumption of
GSH-Px enzyme activities, GSH levels are also declining.
Recently, we observed decrease in Ca2? release in GSH
depleted DRG neurons [20] although N-acetylcysteine
incubation induced protective effects on Ca2? release and
oxidative stress in GSH depleted DRG neurons [21].
Hence, selenium may also protective effects on the oxi-
dative values, Ca2? release and apoptosis in the DRG
neurons. In the current study, we aimed to investigate the
effects of selenium on cytosolic Ca2? release, GSH, GSH-
Px, LP on DRG neurons. According to our knowledge this
is the first study which clarifies the effects of selenium on
DRG neurons against H2O2 induced oxidative stress model.
Selenium administrations both in various experimental
models, in animals and humans, caused an increased level
of GSH and GSH-Px activity [5, 9, 21]. Oxidative stress
represents an imbalance status between excessive produc-
tion of ROS, other radical species and biological system’s
scavenging ability to detoxify the reactive intermediates.
Most of the ROS are formed as a result of the mitochon-
drial respiratory chain pathways but can also be formed
exogenously [1]. Hence GSH and GSH-Px and related
enzymes are believed to play critical roles in protecting
cells from hazardous oxygen species [2].
We found that LP values were increased in H2O2 group
than in control, selenium, selenium ? H2O2 groups
although GSH levels were lower in H2O2 group than in
control, selenium, selenium ? H2O2 groups. With neu-
ropathy progression, antioxidants potentially decrease and
LP levels increase. Excessive production of free radicals
due to oxidative stress may primarily stimulate the voltage-
gated Ca2? channels. Stimulation of many other ion
channels by oxidative stress causes flow of calcium ions
into cytosol and will cause depolarization in mitochondria.
Depolarization of mitochondria triggers free radical gen-
eration [23, 24].
In the current study, we confirm that DRG neurons
exhibit Ca2? ion selective channel-dependent ROS gener-
ation and Ca2? influx following cytosolic GSH depletion,
as previously reported [20, 21]. In addition, we present
evidence showing that cytosolic GSH depletion generates
lipid peroxidation and decreases GSH-Px activity. It is well
known that increase in cytoplasmic Ca2? may jointly
enhance mitochondrial ROS generation through depolar-
ization of mitochondria [22]. In response to increase in
cytosolic Ca2? through activation of Ca2? cation channels,
may incur Ca2?-induced respiratory impairment, potenti-
ating free radical generation, inflicting structural damage to
mitochondria and ultimately apoptotic cell death if the
Ca2? influx is not inhibited by antioxidants. We observed
that, [Ca2?]c release was higher in H2O2 group than in
selenium and selenium ? H2O2 groups. This situation, in
the same correlation line with other studies which per-
formed with H2O2, in H2O2 triggered apoptotic pathways
with antioxidant property, selenium induced protective
effect on apoptotic pathways [25–27].
Similarly, Koistinaho et al. [25] reported that, incuba-
tion of DRG cells with selenium will cause reduction in the
LP levels which will delay age dependent malformation.
Molecular and cellular pathways of pain are not well elu-
cidated yet [28]. There is growing consensus, driven by
both clinical and laboratory studies, demonstrate that
excessive free radical production and oxidative stress may
play a critical role in pathophysiology of DRG neurons
[28, 29]. In vivo studies demonstrated that free radicals
directly join exitotoxicity mechanisms and antioxidants
will be useful in exitotoxicity induced neuronal diseases
[20, 21, 30]. Mitochondrial dysfunction and oxidative
stress are both reason and consequence of the neuropathy
in DRG neurons [31]. DRG neurons are important for
pathophysiology of pain mechanism. The pathogenesis of
Fig. 8 GSH-Px activity in DRG neurons. DRG neurons were
incubated with 200 nM selenium (sodium-selenite) for 30 h and
1 lM H2O2 for 2 h. Values are represented as means ± SEM of six
separate experiments. *P \ 0.05 compared to Se values. aP \ 0.001
versus to control, bP \ 0.001 versus to H2O2 group, cP \ 0.001
versus to Se group
1636 Neurochem Res (2012) 37:1631–1638
123
Page 7
most common two disorders of the peripheral nervous
system, namely neuropathic pain and diabetic polyneu-
ropathy, has been associated with aberrant Ca2? channel
expression and function [32].
The activity of selenium is strictly dependent on its
serum and tissue concentrations; while the lower concen-
trations induce cell growth, the higher concentrations
inhibit growth and induce cell death [6]. Uguz et al. [9]
have also investigated the effects of different selenium
concentrations in HL-60 cells, where they have demon-
strated that at low concentrations (200 nM) selenium
induces a mild endoplasmic reticulum (ER) stress whereas
this stress is much more severe at higher concentrations
(1 lM). Uezono et al. [33] reported that treatment of cul-
tured bovine adrenal chromaffin cells with selenium for
24 h caused decreased in neurotransmitter contents and
Ca2? release, association with cell damage, at concentra-
tions over 30 and 300 lM. They observed also that sele-
nium even at higher concentrations (1 mM) did not affect
any stimulus-induced Ca2? influx. These studies have
demonstrated the dose dependent effects of selenium in
mediating oxidative stress via modulating the Ca2? release
from the ER [8].
In conclusion, we demonstrated that DRG cells, in
oxidative stress model created by H2O2, selenium induced
protective effect against oxidative stress and suppressed
apoptosis of the neurons through regulation of cytosolic
Ca2? release. Furthermore, we observed that oxidative
stress and irregularities in intracellular Ca2? release caused
by H2O2 should be improved by selenium. Our study
supports the neuroprotective effect of selenium. As anti-
oxidant element, selenium may useful in the treatment of
DRG neurons dependent pain and disorders.
Acknowledgments MN and ACU formulated the present hypothe-
sis. MN made critical revision to the manuscript. ACU was respon-
sible to make the analysis and writing report. The study was partially
supported by the Scientific Research Project Unit of Suleyman
Demirel University (1881-D-09). The abstract of the study was sub-
mitted in 14th International Symposium on Trace Elements in Man
and Animals, Enshi, Hubei, China.
References
1. Davies MJ (2003) Singlet oxygen-mediated damage to proteins
and its consequences. Biochem Biophys Res Commun 305:
761–770
2. Kockar MC, Nazıroglu M, Celik O et al (2010) N-acetylcysteine
modulates doxorubicin-induced oxidative stress and antioxidant
vitamin concentrations in liver of rats. Cell Biochem Funct
28:673–677
3. Nazıroglu M, Kilinc F, Uguz AC et al (2010) Oral vitamin C and
E combination modulates blood lipid peroxidation and antioxi-
dant vitamin levels in maximal exercising basketball players. Cell
Biochem Funct 28:300–305
4. Wirth EK, Conrad M, Winterer J et al (2010) Neuronal seleno-
protein expression is required for interneuron development and
prevents seizures and neurodegeneration. FASEB J 24:844–852
5. McKenzie RC, Arthur JR, Beckett GJ (2002) Selenium and the
regulation of cell signaling, growth, and survival: molecular and
mechanistic aspects. Antioxid Redox Signal 4:339–351
6. Schweizer U, Brauer AU, Kohrle J, Nitsch R, Savaskan NE
(2004) Selenium and brain function: a poorly recognized liaison.
Brain Res Rev 45:164–178
7. Nazıroglu M (2009) Role of selenium on calcium signaling and
oxidative stress-induced molecular pathways in epilepsy. Neu-
rochem Res 34:2181–2191
8. Savas S, Briollais L, Ibrahim-zada I, Jarjanazi H, Choi YH,
Musquera M, Fleshner N, Venkateswaran V, Ozcelik H (2010)
A whole-genome SNP association study of NCI60 cell line panel
indicates a role of Ca2? signaling in selenium resistance. PLoS
ONE 5:e12601
9. Uguz AC, Naziroglu M, Espino J et al (2009) Selenium modu-
lates oxidative stress-induced cell apoptosis in human myeloid
HL-60 cells through regulation of calcium release and caspase-3
and -9 activities. J Membr Biol 232:15–23
10. Wang W, Gu J, Li YQ et al (2011) Are voltage-gated sodium
channels on the dorsal root ganglion involved in the development
of neuropathic pain? Mol Pain 7:16
11. Sghirlanzoni A, Pareyson D, Lauria G (2005) Sensory neuron
diseases. Lancet Neurol 4:349–361
12. Devor M (2009) Ectopic discharge in abeta afferents as a source
of neuropathic pain. Exp Brain Res 196:115–128
13. Grynkiewicz C, Poenie M, Tsien RY (1985) A new generation of
Ca2? indicators with greatly improved fluorescence properties.
J Biol Chem 260:3440–3450
14. Beales IL, Ogunwobi OO (2010) Microsomal prostaglandin E
synthase-1 inhibition blocks proliferation and enhances apoptosis
in oesophageal adenocarcinoma cells without affecting endothe-
lial prostacyclin production. Int J Cancer 126:2247–2255
15. Li GY, Fan B, Zheng YC (2010) Calcium overload is a critical
step in programmed necrosis of ARPE-19 cells induced by high-
concentration HO. Biomed Environ Sci 23:371–377
16. Placer ZA, Cushman L, Johnson BC (1966) Estimation of prod-
ucts of lipid peroxidation (malonyl dialdehyde) in biological
fluids. Anal Biochem 16:359–364
17. Sedlak J, Lindsay RH (1968) Estimation of total, protein-bound,
and nonprotein sulfhydryl groups in tissue with ellman’s reagent.
Anal Biochem 25:192–205
18. Lawrence RA, Burk RF (1976) Glutathione peroxidase activity in
selenium-deficient rat liver. Biochem Biophys Res Commun
71:952–958
19. Lowry OH, Rosebrough NJ, Farr AL et al (1951) Protein mea-
surement with the folin phenol reagent. J Biol Chem 193:
265–275
20. Nazıroglu M, Ozgul C, Cig B, Dogan S, Uguz AC (2011) Glu-
tathione modulates Ca(2?) influx and oxidative toxicity through
TRPM2 channel in rat dorsal root ganglion neurons. J Membr
Biol 242:109–118
21. Ozgul C, Nazıroglu M (2012) TRPM2 channel protective prop-
erties of N-acetylcysteine on cytosolic glutathione depletion
dependent oxidative stress and Ca(2?) influx in rat dorsal root
ganglion. Physiol Behav 106(2):122–128
22. Chakraborti T, Das S, Mondal M et al (1999) Oxidant, mito-
chondria and calcium: an overview. Cell Signal 11:77–85
23. Roy SS, Hajnoczky G (2009) Fluorometric methods for detection
of mitochondrial membrane permeabilization in apoptosis.
Methods Mol Biol 559:173–190
24. Liu X, Hajnoczky G (2009) Ca2?-dependent regulation of mito-
chondrial dynamics by the miro-milton complex. Int J Biochem
Cell Biol 41:1972–1976
Neurochem Res (2012) 37:1631–1638 1637
123
Page 8
25. Koistinaho J, Alho H, Hervonen A (1990) Effect of vitamin E and
selenium supplement on the aging peripheral neurons of the male
sprague-dawley rat. Mech Ageing Dev 51:63–72
26. Yuan H, Lan T, Lin J (2005) Modulation of nano-selenium on
tetrodotoxin-sensitive voltage-gated sodium currents in rat dorsal
root ganglion neurons. Conf Proc IEEE Eng Med Biol Soc
5:4846–4849
27. Yuan H, Lin J, Lan T (2006) Effects of nano red elemental
selenium on sodium currents in rat dorsal root ganglion neurons.
Conf Proc IEEE Eng Med Biol Soc 1:896–899
28. Vincent AM, Edwards JL, McLean LL et al (2010) Mitochondrial
biogenesis and fission in axons in cell culture and animal models
of diabetic neuropathy. Acta Neuropathol 120:477–489
29. Fischer LR, Glass JD (2010) Oxidative stress induced by loss of
Cu, Zn-superoxide dismutase (SOD1) or superoxide-generating
herbicides causes axonal degeneration in mouse DRG cultures.
Acta Neuropathol 119:249–259
30. Nazıroglu M (2011) TRPM2 cation channels, oxidative stress and
neurological diseases: where are we now? Neurochem Res
36:355–366
31. Vincent AM, Hinder LM, Pop-Busui R et al (2009) Hyperlipid-
emia: a new therapeutic target for diabetic neuropathy. J Peripher
Nerv Syst 14:257–267
32. Fernyhough P, Calcutt NA (2010) Abnormal calcium homeo-
stasis in peripheral neuropathies. Cell Calcium 47:130–139
33. Uezono Y, Toyohira Y, Yanagihara N, Wada A, Taniyama K
(2006) Inhibition by selenium compounds of catecholamine
secretion due to inhibition of Ca2? influx in cultured bovine
adrenal chromaffin cells. J Pharmacol Sci 101:223–229
1638 Neurochem Res (2012) 37:1631–1638
123