Effects of Voluntary Physical Activity and Endurance Training on Cardiac Mitochondrial Function of Rats Sub-Chronically Treated with Doxorubicin Dissertation submitted to the Faculty of Sports, University of Porto to obtain the 2 nd cycle in Physical Activity for Elderly, under the decree-law no. 74/2006 of 24 March Supervisors. Professor Doutor António Ascensão Professor Doutor José Magalhães Diogo Nuno Mariani Felix Porto, June 2013
Transcript
Effects of Voluntary Physical Activity and Endurance Training on Cardiac Mitochondrial Function of Rats
Sub-Chronically Treated with Doxorubicin
Dissertation submitted to the Faculty of Sports,
University of Porto to obtain the 2nd cycle in Physical
Activity for Elderly, under the decree-law no. 74/2006
of 24 March
Supervisors. Professor Doutor António Ascensão
Professor Doutor José Magalhães
Diogo Nuno Mariani Felix
Porto, June 2013
Mariani, D. (2013). Effects of Voluntary Physical Activity and Endurance
Training on Cardiac Mitochondrial Function of Rats Sub-Chronically Treated
with Doxorubicin. Porto: D. Mariani. Master thesis presented to the Faculty of
Magalhães et al. 2006). The observed morphological alterations consisted of
mitochondrial damage with extensive degeneration and loss of cristae, swelling
33
and abnormal size and shape, intramitochondrial vacuoles and notorious myelin
figures that probably resulted in the formation of secondary lysosomes. All of
these alterations were attenuated in trained animals treated with DOX
(Ascensão, Magalhães et al. 2006).
At functional level, different authors reported the protective effects of chronic
exercise. In fact, Hydock et al. (2008) suggested that exercise training in rats
before DOX treatment attenuated DOX-induced cardiac dysfunction, through
the maintenance of fractional shortening, developed pressure and contractility.
Also, Chicco et al. (2005) reported that both low intensity exercise training and
an endurance training protocol with gradually increased intensity attenuated the
adverse effects of DOX by preventing DOX-induced decline in cardiac function
through maintenance of left ventricular diastolic pressure, rate of left ventricular
pressure development and rate of left ventricular relaxation.
At biochemical level, as depicted on table 2, several authors had proved the
beneficial effects of chronic exercise. Importantly, some of the most studied
biochemical parameters associated with exercise and DOX are the antioxidant
capacity, oxidative stress markers and apoptotic susceptibility (for refs see
Ascensao, Oliveira et al. 2012). As previous referred, HSP have an important
role as antioxidants molecules contributing to normal cellular integrity and are
overexpressed after endurance training. However, Chicco et al (2006) and
Kavasis et al. (2010) also showed that exercise had no influence on HSP or that
it is not determinant on cardioprotection being that exercise in cold vs. normal
temperatures may also display other types of differences regarding alteration of
mitochondrial physiology, besides alteration in the expression of HSPs.
Furthermore, the upregulation of mitochondrial manganese superoxide
dismutase (MnSOD) seems contribute for cardioprotection. In fact, as
mitochondrial DOX toxicity has been largely attributed to increased oxidative
stress, increased antioxidant activity may be important to explain how
endurance training counteracts some of DOX-induced myocardial damage.
Chicco et al. (2006) associated low-intensity treadmill exercise training-induced
cardioprotection to the inhibition of apoptotic signaling and the increased activity
34
of GPX. Also, the effect of training on preventing activation of cardiac apoptotic
pathways has been described being that, training decreased the susceptibility of
appearance of apoptotic markers in the hearts of DOX-treated animals, as
increased mitochondrial Bax, Bax-to-Bcl2 ratio and tissue caspase 3 activity
(Ascensao, Magalhaes et al. 2005). In fact, it has been suggested that chronic
exercise stimulation may also afford protection against the increased
susceptibility to the MPTP as the deleterious effects of Ca2+ on heart
mitochondrial respiration of DOX-treated animals were attenuated in trained
group treated with DOX (Ascensao, Magalhaes et al. 2005). As previous
described, MPTP is related to oxidative damage and therefore, it is possible that
increased resistance of cardiac mitochondria from trained animals to the MPTP
can be related to increased antioxidant defenses. Accordingly, the higher levels
of reduced sulfhydryl groups in trained mitochondria than in sedentary groups
may be indicative of enhanced antioxidant capacity and/or of more elevated
sulfhydryl-donors, such as GSH, in mitochondria from trained animals (for refs
see Ascensao, Oliveira et al. 2012). However, further studies are necessary in
order to better understand this issue.
Table 2. Summary of some described mitochondrial-related alterations associated with DOX-induced cardiotoxicity and the modulation effect afforded by physical exercise against DOX (adapted from Ascensao, Oliveira et al. 2012).
DOX effect Exercise effect against DOX
ROS production ↑ ↓
Oxidative damage markers
Lipid peroxidation ↑ ↓
Protein oxidation ↑ ↓
DNA oxidation ↑
Aconitase activity ↓ or = ↑
Apoptotic signaling
35
(↑) – increase; (↓) – decrease; (=) – no alterations
Despite the extensive number of studies on this topic, the effects of both
endurance treadmill training and voluntary free-wheel running activity performed
Bax-Bcl-2 ratio ↑ ↓
Cytochrome c release ↑
Caspase 9 activation ↑ ↓
Respiratory endpoints
State 3 ↓ ↑ or =
State 4 ↑ or = or ↓ = or ↓
RCR ↓ = or ↑
ADP/O ratio = or ↓
Uncoupled respiration ↓ ↑
Creatine-stimulated respiration ↓
Maximal ΔΨ ↓ or = ↑
Ca2+-induced MTPT ↑ ↓
ANT content and functioning ↓
Mitochondrial chaperones ↑ ↑
Mitochondrial antioxidants
Thiols ↓ ↑
Vitamin E ↓ or =
Enzymes = or ↑ ↑
Coenzyme Q isoenzymes =
ETC complex activity
Complex I ↓ ↑
Complex II ↓
Complex III =
Complex IV = or ↓
Complex V ↓ ↑
36
before and during sub-chronic DOX treatment schedule on cardiac
mitochondrial bioenergetics are yet to be elucidated.
This is of particular importance in the context of exercise-induced protection
against DOX-related cardiac mitochondriopathy, as cancer patients undergoing
DOX treatment may be advised to exercise for many reasons including to
counteract physical fatigue and to improve performance, and also to mitigate
cardiac damage as a result of chemotherapy.
This master sports science course in which this work is inserted is the context of
physical activity and elderly. Despite developed with adult rats, the present work
can contribute to extend the knowledge in this particular area representing
preliminary findings in adult population and considering that DOX-based
chemotherapeutic treatments against several types of malignances are more
prevalent with increasing age.
37
3. Aim
The aim of the present study was to analyze the effect of two types of physical
exercise (treadmill endurance training (TM) and free-wheel voluntary physical
activity (FW)) against heart mitochondrial dysfunction induced by sub-chronic
treatment of DOX.
We can define as specific purposes of this work the analysis of the adaptations
induced by both types of exercise on heart mitochondria from DOX treated
The animals from FW groups were housed in a polyethylene cage equipped
with a running wheel [perimeter=1,05m, Type 304 Stainless steel (2154F0106-
1284L0106) Technicplast, Casale Litta, Italy)]. The rats were allowed to
exercise ad libitum with an unlimited access to the running wheel 24h/day.
Running distance was recorded using ECO 701 from Hengstler (Lancashire,
U.K.).
4.4 Doxorubicin treatment
After the 5th week of endurance training or free wheel exercise, the animals
were sub-chronically treated with DOX (2 mg/Kg of body weight) or sterile saline
solution NaCl 0.9% (SAL, 2 mg/kg of body weight) intraperitoneal injection/week
during 7 weeks. The animals assigned to the TM groups received DOX or SAL
injections during the weekend in a day-off training.
4.5 Animal sacrifice, heart and soleus extraction
Forty-eight hours after the last TM exercise bout, non-fasted rats were
euthanized by cervical dislocation between 9:00 and 10:00 AM to eliminate
possible effects due to diurnal variation. After quickly opening the chest cavity,
rat hearts were rapidly excised, rinsed, carefully dried, and weighed. Right
soleus muscle was also rapidly extracted and weighed. Portions of
approximately 50 milligrams (mg) of one soleus muscle were homogenized in
homogenization buffer (200 minimolar (mM) Tris, 137 mM NaCl, 0.2 mM EDTA,
0.5 mM EGTA, 1% triton X-100, tissue: buffer ratio of 100 mg/mL, pH 7.4) using
a Teflon pestle on a motor driven Potter-Elvehjem glass homogenizer at 0–4°C
three to five times for 5 s at speed low setting, with a final burst at a higher
speed setting. Homogenates were centrifuged (2 min at 3000 xg, 4°C, in order
to eliminate cellular debris) and the resulting supernatants were stored at −80°C
42
for later determinations, as detailed bellow. Protein content from soleus
homogenates were spectrophotometrically determined using the biuret method
and bovine serum albumin as standard (Gornall, Bardawill et al. 1949).
4.6 Isolation of heart mitochondria
Heart mitochondria were daily prepared using conventional methods of
differential centrifugation (Bhattacharya, Thakar et al. 1991) as follows. Briefly,
the animals were sacrificed as stated above and the heart was immediately
excised and finely minced in an ice-cold isolation medium containing 250 mM
sucrose, 0.5 mM EGTA, 10 mM HEPES (pH 7.4), and 0.1% defatted bovine
serum albumin (BSA, Sigma, cat. no. A-7030). The minced blood-free tissue
was then resuspended in 40 mL of isolation medium containing 0,75 mg/mL
protease subtilopeptidase A Type III (Sigma P-5380) and homogenized with a
tightly fitted homogenizer (Teflon: glass pestle). The suspension was incubated
for 1 min (4°C) and then re-homogenized. The homogenate was then
centrifuged at 13,000 xg for 10 min. The supernatant was decanted and the
pellet, essentially devoid of protease, was gently re-suspended with a loose-
fitting homogenizer. The suspension was centrifuged at 750 xg for 10 min and
the resulting supernatant was centrifuged at 12,000 xg for 10 min. The pellet
was re-suspended using a paintbrush and re-pellet at 12,000 xg for 10 min.
EGTA and defatted BSA were omitted from the final washing medium.
Mitochondrial protein content was determined by the Biuret method calibrated
with BSA (Gornall, Bardawill et al. 1949). The isolation procedures were
performed within approximately 1 h at 0–4°C. Aliquots of isolated mitochondria
were separated and frozen at −80°C for later determination of oxidative
damage. The remaining mitochondrial were used within 2–3 h after the excision
of the heart and was maintained on ice (0–4 ºC) throughout this period.
43
4.7 Mitochondrial respiratory activity
Mitochondrial respiratory function was measured polarographically at 30ºC
using a Biological Oxygen Monitor System (Hansatech Instruments) and a
Clarktype oxygen electrode (Hansatech DW1, Norfolk, UK). The reactions were
conducted in a 0.75 mL closed, thermostatted and magnetically stirred glass
chamber containing 0.5 mg/mL of mitochondrial protein in a respiration buffer
containing 50 mM KCl, 130 mM sucrose, 2.5 mM KH2PO4, and 0.5 mM Hepes,
pH 7.4. After 1-min equilibration period, mitochondrial respiration was initiated
by adding glutamate/malate (G/M) to a final concentration of 5 and 2.5 mM,
respectively. State 3 respiration was determined after adding ADP (150 nmol);
state 4 was measured as the rate of oxygen consumption after ADP
phosphorylation. The RCR (state 3/state 4) and the ADP/O (the number of nmol
ADP phosphorylated by atom of oxygen consumed) ratios were calculated
according to Estabrook (1967).
4.8 Mitochondrial transmembrane electric potential
Mitochondrial transmembrane electric potential (∆ψ) was monitored indirectly
based on the activity of the lipophilic cation tetraphenylphosphonium (TPP+)
using a TPP+ selective electrode prepared in our laboratory as previously
described (Ascensao, Lumini-Oliveira et al. 2011). Reactions were carried out in
1 mL of reaction buffer containing 50 mM KCl, 130 mM sucrose, 2.5 mM
KH2PO4, and 0.5 mM Hepes, pH 7.4 supplemented with 3 µM TPP+ and 0.5
mg/mL of mitochondrial protein. For the measurements of ∆ψ with complex I-
linked substrates, energization was carried out with G/M (5 mM and 2.5 mM,
respectively) and ADP phosphorylation was achieved by adding 150 nmol ADP.
The lag phase, which reflects the time needed to phosphorylate the added ADP,
was also measured during experiments.
44
4.9 Mitochondrial osmotic swelling during MPTP induction
Mitochondrial osmotic volume changes were followed by monitoring the classic
decrease of absorbance at 540 nm with a Jasco V-630 spectrophotometer.
Swelling amplitude and rate of decreased absorbance upon Ca2+ addition were
considered as MPTP susceptibility indexes. The reaction was continuously
stirred and the temperature was maintained at 25 ºC. The assays were
performed in 1 ml of reaction medium containing 200 mM sucrose, 10 mM
HEPES, 5 mM KH2PO4, 10 µM EGTA , pH 7.4, supplemented with 1.5 µM
rotenone, 8 mM succinate and a single pulse of 80 nmol of Ca2+ with 0.5 mg/ml
protein. Control trials were performed by using 1 µM of cyclosporin-A, the
selective MPTP inhibitor (Broekemeier, Dempsey et al. 1989).
4.10 Mitochondrial oxidative damage
Before analysis, mitochondrial membranes were disrupted by several freeze–
thawing cycles to allow free access to substrates. The extent of lipid
peroxidation in heart mitochondria was determined by measuring MDA contents
by colorimetric assay, according to a modified procedure described previously
(Buege and Aust 1978). Suspended mitochondria were centrifuged at 12,000 xg
for 10 min and re-suspended in 150 µL of a medium containing 175 mM KCl
and 10 mM Tris-HCl, pH 7.4. Subsequently, mitochondria from the six groups
were mixed with 2 volumes of trichloroacetic acid (10%) and 2 volumes of
thiobarbituric acid (1%). The mixtures were heated at 80–90 ºC for 10 min and
re-cooled in ice for 10 min before centrifugation (4,000 xg for 10 min). The
supernatants were collected and the absorbance measured at 535 nm. The
amount of MDA content formed was expressed as nanomoles of MDA per
milligram of protein (ε535=1.56 x 10−5 M−1 cm−1)
The basal mitochondrial content of oxidative modified -SH groups, including
GSH and other -SH containing proteins, was quantified by spectrophotometric
measurement according to Hu (1990). Briefly, a mitochondrial suspension
45
containing 5 mg/mL protein was mixed with 0.25 M Tris buffer pH 8.2 and 10
mM DTNB and the volume was adjusted to 1 mL with absolute methanol.
Subsequently, the samples were incubated for 30 min in the dark at room
temperature and centrifuged at 3000 xg for 10 min. The colorimetric assay of
supernatant was performed at 414 nm against a blank test. Total -SH content
was expressed in nanomoles per milligrams of mitochondrial protein (ε414=13.6
mM−1 cm−1).
4.11 Soleus citrate synthase activity
Soleus CS activity was measured using the method proposed by Coore et al.
(1971). The principle of assay was to initiate the reaction of acetyl-CoA with
oxaloacetate and link the release of CoA-SH to 5,5-dithiobis (2-nitrobenzoate)
at 412 nm.
4.12 Statistical analysis
All data are expressed as the mean±SEM (Standard Error of the Mean).
Statistical analyses were performed using GraphPad Prism (version 6.0) or
Statistical Package for the Social Sciences (SPSS version 21.0). Three-way
repeated-measures ANOVA for body weight and distance cover by exercised
groups to verify the effect of exercise and treatment over time. Two-way
analysis of variance ANOVA were used to examine possible effect of treatment
and/or exercise. To determine specific group differences, the two-way ANOVA
were followed by Bonferroni post-hoc tests. In all cases, the significance level
was set at p≤0.05.
47
5. Results
5.1. Characterization of animals and exercise protocols
Body weight alterations and distances covered by the animals during the entire
protocol are shown in figure 3. No significant differences in the mean body
weight of the animals from the beginning of the protocol until the 5th week, when
sub-chronical DOX treatment was initiated, were found. Body weights of DOX
treated animals were lower than SAL counterparts at the end of the protocol
(DOX+SED vs. SAL+SED; DOX+TM vs. SAL+TM; DOX+FW vs. SAL+FW;
p≤0.05). No differences in body weight between exercised groups, were found.
TM and FW decreased body weight at 12th and 9th week, respectively (SAL+TM
and SAL+FW vs. SAL+SED; p≤0.05). DOX treatment combined with TM
decreased body weight from the 5th week (DOX+TM vs. DOX+SED; p≤0.05),
whereas no significant differences were found between FW and SED treated
groups (Figure 3A). After the 5th week DOX treated groups consumed less food
than their SAL counterparts (data non shown, p≤0.05). Water consumption
increased in FW groups (SAL and DOX) compared with SAL+SED and
DOX+SED groups (data non shown, p≤0.05).
Figure 3. Effect of exercise and DOX treatment on (A) body mass over time and (B) distance covered per day by TM and FW groups during the 12 wks of protocol. Significant differences (p≤0.05) are mentioned in the text. Significant (p≤0.05) effects of Exercise (E), Treatment (T), time (t) or their interaction (E x T x t) are shown; Non Significant (NS, p>0.05).
48
As can be seen in Figure 3B, voluntary running distance decreased significantly
in DOX+FW after the 5th week and remained lower until the end of the protocol
(p≤0.05). Animals from TM group ran at the same velocity throughout the 8
weeks of the protocol. Running velocity and distance covered diminished in
DOX+TM group at 11th and 12th week compared to SED+TM (p≤0.05).
Body, heart absolute weights, heart weight and femur length to body weight
ratios, mitochondrial protein yielding as well as the activity of soleus citrate
synthase in the six groups are shown in Table 4. Final body, heart weight and
ratio of heart weight to body weight significantly decreased with DOX treatment
(SAL+SED vs DOX+SED). Both chronic exercise types decreased final body
weight, increased heart weight and the heart to body ratio (SAL+TM and
SAL+FW vs SAL+SED). DOX treatment combined with TM and FW exercise
induced a significant increase in heart weight and heart weight to body weight
ratio compared with their DOX+SED counterparts. No significant differences
were observed between groups regarding the Initial body weight, femur length
to body weight ratio and yield of mitochondria isolation. TM induced a significant
increase in the activity of soleus citrate synthase in both SAL and DOX treated
animals (SAL+SED vs SAL+TM and DOX+SED vs DOX+TM).
49
Table 4. Animal data and yield of mitochondrial protein isolation
soleus citrate synthase activity (nmol. min-1.mg-1) 10.94±2.07a 23.34±1.79b 11.22±2.65a 8.69±1.15a 22.22±1.97b 10.27±1.32a E, T
Values (mean ± SEM). Different letters are significantly different (p≤0.05). * Significant (p≤0.05) effects of Exercise (E), Treatment (T), or their interaction (E x T) are shown; Non Significant (NS, p> 0.05).
50
5.2 Heart mitochondrial oxygen consumption
Mitochondrial respiratory activity in both SAL and DOX treated groups was
measured to identify exercise-dependent effects (Figure 4). DOX treatment
decreased heart mitochondrial respiration during state 3 and increased state 4
in SED animals (DOX+SED vs SAL+SED). Importantly, TM and FW exercise
per se increased State 3 respiration in both SAL and DOX (SAL+TM and
SAL+FW vs. SAL+DOX; DOX+TM and DOX+FW vs. DOX+SED). The coupling
between oxygen consumption and ADP phosphorylation (RCR) was
significantly affected by DOX treatment (DOX+SED vs. SAL+SED). TM
significantly increased RCR in both SAL and DOX groups (SAL+TM vs.
SAL+SED; DOX+TM vs. DOX+SED). FW increased RCR in DOX treated
animals (DOX+FW vs. DOX+SED). Also, both TM and FW increased ADP/O in
DOX group (DOX+SED vs. DOX+TM and DOX+FW).
51
Figure 4. Effect of exercise and DOX treatment on (A) state 3 of heart mitochondrial respiration, (B) state 4 of heart mitochondria respiration, (C) RCR and (D) ADP/O. Data are means±SEM for heart mitochondria (0.5 mg/mL protein) obtained from different mitochondrial preparations for each experimental group. Oxidative phosphorylation was measured polarographically at 30ºC in a total volume of 0.75 mL. Respiration medium and other experimental details are provided in methods. RCR, respiratory control ratio (state 3/state 4); ADP/O, number of nmol ADP phosphorylated by atom of oxygen consumed. Different letters are significantly different (P≤0.05). Significant (p≤0.05) effects of Exercise (E), Treatment (T), or their interaction (E x T) are shown; Non Significant (NS, p>0.05).
5.3 Heart mitochondrial transmembrane electric potential
Heart mitochondrial variations in maximal ∆ψ and during ADP phosphorylation
were determined using G/M as substrates. DOX treatment significantly affected
the maximal ∆ψ, repolarization and ADP lag-phase (Figure 5). FW but not TM,
increased maximal ∆ψ and repolarization, whereas both types of exercise
decreased the ADP lag phase (SAL+TM and SAL+FW vs. SAL+SED). Both
exercise protocols were able to counteract the DOX harmful effect normalizing
maximal ∆ψ, repolarization and ADP lag phase.
52
Figure 5. Effect of exercise and DOX treatment on heart mitochondria ∆ψ fluctuations (A) maximal energization, (B) ADP-induced depolarization, (C) repolarization and (D) ADP phosphorylation lag phase. Data are mean±SEM for heart mitochondria (0.5 mg/mL protein) obtained from different mitochondrial preparations for each experimental group. Figure shows the average response of maximal mitochondrial membrane potential developed with glutamate (5 mM) plus malate (2.5 mM), the decrease in membrane potential after ADP addition (depolarization), the repolarization value after ADP phosphorylation, and the lag phase. Mitochondrial transmembrane potential was measured using a TPP+-selective electrode at 30ºC in a total volume of 1 mL. Reaction medium and other experimental details are provided in methods. Different letters are significantly different (p≤0.05). * Significant (p≤0.05) effects of Exercise (E), Treatment (T), or their interaction (E x T) are shown; Non Significant (NS, p>0.05)
5.4 Mitochondrial osmotic swelling during MPTP induction
The effects of both types of exercise training and DOX treatment on in vitro
susceptibility to Ca2+-induced MPTP opening were investigated. The addition of
Ca2+ on mitochondria suspension resulted in a decrease in absorbance with
three distinct phases. Initially, an increase in absorbance was observed, which
most likely results from the formation of opaque Ca2+ crystals inside
mitochondria (Andreyev, Fahy et al. 1998). Upon MPTP opening, a decrease of
53
absorbance with a slow followed by a fast kinetic rate is usually observed in
cardiac mitochondria. Incubation of mitochondrial suspension with cyclosporine
A, a specific MPTP inhibitor (Broekemeier, Dempsey et al. 1989), limits the
absorbance decrease after Ca2+ addition, which demonstrate the association
with MPTP opening.
Figure 6 shows different end-points measured from the recordings obtained,
namely (A) swelling amplitude (the difference between the initial and the final
absorbance value) and (B) the average swelling rate. The results demonstrate
that DOX treatment significantly increased susceptibility to Ca2+-induced MPTP
opening (DOX + SED vs. SAL + SED). Heart mitochondria isolated from
SAL+TM group, but not SAL+FW were less susceptible to Ca2+-induced MPTP
opening (SAL + TM vs. SAL + SED). Both types of exercise were able to
mitigate DOX-induced increased susceptibility to MPTP opening (DOX + TM
and DOX + FW vs. DOX + SED).
Figure 6. Effect of exercise and DOX treatment on heart mitochondria to Ca2+-induced MPTP (A) Swelling amplitude; (B) Average swelling rate. Data are mean ± SEM. The absorbance of mitochondrial suspension was followed at 540 nm. Mitochondria were incubated as described in methods. A 80 nmol of Ca2+ pulse (160 nmol/mg protein) was added to 0.5 mg of mitochondrial protein in order to attain the cyclosporin A-sensitive swelling, indicating that the decreased optical density corresponding to the increased swelling was due to MPTP opening. Different letters are significantly different (p≤0.05). * Significant (p≤0.05) effects of Exercise (E), Treatment (T), or their interaction (E x T) are shown; Non Significant (NS, p>0.05)
54
5.5 Oxidative stress markers
The next step was to ascertain whether exercise and DOX treatment-modulated
mitochondrial oxidative stress markers. According to the protective phenotype
seen in Fig.7 DOX treatment increased MDA levels and decreased –SH content
(SAL + SED vs. DOX + SED). Exercise, particularly TM decreased
mitochondrial MDA levels (SAL + TM vs. SAL + SED). Both types of exercise
were able to revert DOX-induced alterations in MDA level and –SH content
(DOX + SAL vs. DOX + TM and DOX + FW).
Figure 7. Heart mitochondrial (A) MDA and (B) reduced sulfhydryl contents. Data are means±SEM for heart mitochondria obtained from different mitochondrial preparations for each experimental group. Different letters are significantly different (p≤0.05). Significant (p<0.05) effects of Exercise (E), Treatment (T), or their interaction (E x T) are shown; Non Significant (NS, p> 0.05).
55
6. Discussion
The current study provided additional support to understand the effects of both
endurance treadmill training and voluntary wheel running activity performed
before, during and after sub-chronic DOX treatment schedule on cardiac
mitochondrial bioenergetics. Only male rats were used to avoid hormone-
dependent influence in drug-induced mitochondrial toxicity (Lagranha,
Deschamps et al. 2010). Rats were sub-chronically treated with DOX in an
attempt to mimic human’s treatment, a protocol previously used by others
(Pereira, Pereira et al. 2012, Santos, Moreno et al. 2002). Moreover, in the
present study 1st DOX injection was administrated 5 weeks after the beginning
of the exercise protocol with subsequent weekly injection until the end of
protocol. This set up can be understood with both a preconditioning (preventive)
and a therapeutic strategy against DOX treatment schedules. Two different
types of exercise were analyzed: voluntary free wheel run and treadmill run.
Considerable attention has been focused on the efficacy of exercise protocols
since it has been speculated that further stress induced by exercise could be
potentially detrimental, exacerbating the impairments induced by DOX (Emter
and Bowles 2008). In fact, patients undergoing chemotherapy experience
severe fatigue or exercise intolerance. Consequently, the intensity and duration
of exercise they are able to tolerate is likely to be severely limited (Emter and
Bowles 2008). For these reason, and because we wanted to analyze the
response on heart mitochondria of DOX treated rats at distinct intensity and
duration, both free wheel and run treadmill were performed.
The results on mitochondrial function obtained in the present study confirm at
least in part, the cardiac protection afforded by both endurance treadmill training
and voluntary free wheel running (for refs see Ascensao, Oliveira et al. 2012)
against DOX toxicity. Cardiac dysfunction and defective mitochondrial function
in DOX-treated animals has been studied previously (Ascensao, Lumini-Oliveira
et al. 2011, Ascensão, Magalhães et al. 2006, Chicco, Schneider et al. 2005,
Kavazis, Smuder et al. 2010, Sardao, Pereira et al. 2008, Wallace 2003). The
present study confirmed that sub-chronic DOX administration in heart
56
mitochondria results in: (i) worsening heart mitochondrial respiration; (ii)
decreased maximally developed ΔΨ, repolarization and increased
phosphorylative lag phase; (iii) decreased ability of heart mitochondria to
accumulate Ca2+ before MPTP induction and (iv) increased MDA levels and
decreased -SH content. Both types of exercise performed before and during
DOX treatment resulted in attenuation or complete prevention of the heart
mitochondrial impairments induced by DOX.
6.1 Heart mitochondrial oxygen consumption and transmembrane electric potential
Previous studies have shown that exercise attenuates DOX-induced cardiac
damage, diminishing the increased biochemical and morphological signs of
toxicity induced (Ascensao, Magalhaes et al. 2005, Ascensao, Oliveira et al.
2012, Chicco, Schneider et al. 2006, Hydock, Lien et al. 2008, Kanter, Hamlin et
al. 1985, Kavazis, Smuder et al. 2010); however, no data are available
concerning the cross-tolerance effect of both voluntary free wheel running and
endurance training on sub-chronic DOX treatment mitochondrial malfunction.
Present results demonstrate that sub-chronic treatment of DOX induces
impairments on mitochondrial respiration, and that 12 weeks of endurance
running training and voluntary free wheel running prevented the inhibition of
mitochondrial respiration. Alterations in mitochondrial oxidative phosphorylation
induced by DOX relies in several factors including the decreased aconitase
activity; increased ROS production and activity; and the decreased activity,
content or organization of the electron transport chain complexes or proteins of
the phosphorylation system (Ascensao, Lumini-Oliveira et al. 2011). Also, the
depressed activity of mitochondrial complexes I and II caused by DOX (Santos,
Moreno et al. 2002) could partially justify the diminished electron transport
through electron transport chain (ETC) in the SAL+DOX group. Thus, the
unaltered state 3 respiration observed in exercised groups suggest that, among
other possible effects, training probably prevented the inactivation of complex I
57
and II in DOX-treated heart mitochondria. The enhanced ETC functionality
could also be due to an up regulation of oxido-redutase activity or increased
availability of reduced equivalents formation, consequently increasing the
supply of electrons to the ETC (Nulton-Persson and Szweda 2001) or even to
enhanced capability of phosphorylative system due an upregulation of Krebs
cycle enzymes (Holloszy, Oscai et al. 1970). Furthermore, our experiments
reveal that mitochondria isolated from hearts of animals treated with DOX
exhibited impaired coupling (i.e., lower RCR). RCR is known as a respiratory
parameter associated with mitochondrial functionality and structural integrity
(Brand and Nicholls 2011). As exercise training prevent DOX-induced
uncoupled cardiac mitochondrial respiration, this might suggest that exercise
training enhance mitochondrial respiratory activity due increased
phosphorilative system functionality. Interestingly, regarding RCR, FW running
protocol was more effective at counteracting DOX-induced impairments, which
may be consequence of a slight decreased in sate 4 observed in DOX+FW
group. Concerning ADP/O, both TM and FW groups reverted the values of
DOX+SED group, which suggest that training prevented the heart mitochondrial
impairments in oxidative phosphorylation capacity in DOX rats.
Because mitochondrial complexes rely on enzymatic machinery (Bernstein,
Bucher et al. 1978), they can become prone to impairments induced by ROS,
resulting in accumulation of products of protein oxidation. DOX-induced
impairments in oxidative damage markers, such as MDA level and -SH content
were consistent with alterations in mitochondrial respiratory function, which can
suggest that exercise may counteract DOX-induced impairments in redox
homeostasis. One possible justification for these alterations is that exercise
induced up-regulation of mitochondrial defenses including HSPs or SOD
contributing to the up-regulation of mitochondrial tolerance against DOX effects
(Ascensao, Ferreira et al. 2007). The up-regulation of other antioxidants such
as GSH and CAT has also been described with exercise and DOX (Ascensao,
Magalhaes et al. 2005, Kavazis, Smuder et al. 2010).
58
The complementary study of Δψ is indispensable for a complete analysis of
mitochondrial function being that it reflects the basic energetic relation to
cellular homeostasis maintenance. In fact, the electrochemical gradient due the
pumping of protons through the inner membrane (Murphy and Brand 1988) is
indispensable to ADP phosphorylation (Stock, Leslie et al. 1999). Moreover,
when cytosolic concentration of Ca2+ increases, mitochondria act as Ca2+
buffers due its ability to uptake and accumulate Ca2+ (Gunter, Yule et al. 2004).
It has been suggested that intramitochondrial Ca2+ concentration, whose flow is
directed in accordance with the protomotriz gradient, has a controlling function
in metabolic rate of oxidative energy production through the activation of Ca2+-
sensitive dehydrogenases, F0F1ATPase and ANT (Glancy, Willis et al. 2013).
Our results showed that DOX decreased maximal Δψ, repolarization and
increased the lag phase. The lag phase represents the time elapsed to
phosphorylate ADP. In the present study, exercise led to increased maximal
Δψ, repolarization and decreased time to restore membrane potential after
addition and consequent ADP phosphorylation in the DOX+FW and DOX+TM
groups. One possible explanation for the observed protective effect of exercise
may be associated with the preservation of mitochondrial complex activity,
namely complex I and V in exercised groups (Ascensao, Lumini-Oliveira et al.
2011). It is however important to note that the Δψ values above −200 mV in all
experimental groups do not seem to compromise ATP synthase flow or the
transport of ions and metabolites. In fact, the range of the Δψ is −120 to −220
mV. For instance, regarding the driving force for ATP generation, it has been
shown that the kinetics of the ATP synthase follow a sigmoid pattern in
response to Δψ, reaching saturation at approximately − 100 mV (Kaim and
Dimroth 1999).
6.2. Mitochondrial osmotic swelling during MPTP induction
In addition to their role in energy supply, mitochondria are also considered
determinant players in the establishment of cytosolic Ca2+ homeostasis,
59
uptaking and accumulation of Ca2+ in the matrix, in a process that is favored by
the electrochemical gradient formed across the inner mitochondrial membrane
(Gustafsson and Gottlieb 2008). However, mitochondria have a finite capability
to accumulate Ca2+ before undergoing Ca2+-dependent MPTP opening, and
thereafter to the release of pro-apoptotic proteins, which in turn results in
apoptosis (Ascensao, Lumini-Oliveira et al. 2011). In this regard, the study of
exercise in the context of MPTP modulation may assume an important clinical
relevance. Also, it has been described that, among others, a characteristic of
MPTP after Δψ loss, is the increased osmotic swelling amplitude induced by
Ca2+ in vitro (Gunter, Yule et al. 2004). Furthermore, endogenous Ca2+ levels in
matrix are greatly higher in oxidative tissue, limiting heart ability to uptake Ca2+
before MPTP induction (Picard, Csukly et al. 2008).
In the present study, only TM exercise per se was able to increase the
mitochondrial capability to accumulate Ca2+ after MPTP induction. However,
both protocols afforded protection against DOX impairments. Briefly, our results
showed that DOX per se decreased the mitochondrial Ca2+ tolerance
(SED+SAL vs. SED+DOX) and both exercise protocols counteract DOX-
induced impairments, possibly activating some defense mechanisms that might
contribute to prevent the increased DOX-induced MPTP opening susceptibility
(Marcil, Bourduas et al. 2006).
As MPTP is known to be formed/regulated by several proteins including ANT,
hexokinase VDAC, phosphate carrier or Cyp D (Ascensao, Lumini-Oliveira et al.
2011, Crompton 1999, Halestrap and Brenner 2003), it is possible that exercise
may positively modulate the expression and activity of those proteins. Also,
given the refereed close relationship between increased mitochondrial oxidative
stress and the susceptibility to MPTP induction (Kowaltowski, Castilho et al.
2001), it is possible that the up-regulation and modulation of some mechanisms
involving stress chaperones, as HSPs antioxidants or other defense systems
(Ascensao, Ferreira et al. 2007), as well as the decreased heart mitochondrial
free radical production found in rats undergoing regular exercise (Judge, Jang
et al. 2005) may contribute to these protective effects. Moreover, the possible
60
increased functionality of the phosphorylative system in general, and the ETC in
particular, induced by both exercise protocols may have some implications in
Ca2+ uptake capacity. However, to better understand this phenomenon, further
studies need to be addressed.
6.3 Oxidative stress markers
Prevailing hypotheses suggest that myocardial oxidative stress is a primary
event in DOX-induced cardiotoxicity and it is believed to initiate several of the
deleterious cellular events reported following DOX treatment (Zucchi and
Danesi 2003). In fact, at present the principal mechanism of DOX-induced
cardiotoxicity is believed to be increased mitochondrial oxidant production
leading to protease activation and induction of apoptosis (Ascensão, Magalhães
et al. 2006, Ascensao, Magalhaes et al. 2005, Chicco, Hydock et al. 2006,
Chicco, Schneider et al. 2005). Accordingly, oxidative injury of fatty acids at
subcellular level measured by increased levels of lipid peroxidation products
has been frequently reported following DOX exposure (for refs see Chicco,
Schneider et al. 2005).
The present results show that TM, but not FW per se was able to decrease
MDA level and increase -SH groups. In accordance to previous reports, DOX
induced a significant decrease in -SH, indicating increased disulfide linkages
from both proteins and GSH. As polyunsaturated fatty acids are considered
highly susceptible to ROS attack, the increased oxidative stress caused by DOX
led to peroxidative modification of lipid membranes affecting membrane integrity
and permeability, which leads to decoupled mitochondria, altering normal
mitochondrial respiratory function.
Myocardial antioxidant enzymes defend the heart against the damaging effects
of ROS and have been hypothesized to play an important role in exercise-
induced resistance to oxidative stress (Powers, Lennon et al. 2002) and in the
attenuation of DOX cardiotoxicity (Singal, Iliskovic et al. 1997). In particular,
61
some studies suggested that the presence of myocardial SOD might be
important for the prevention of DOX cardiotoxicity (Ascensao, Lumini-Oliveira et
al. 2011, Sarvazyan, Askari et al. 1995, Yen, Oberley et al. 1996). This is
reasonable, as SOD dismutates superoxide into H2O2, thereby providing the
first line of defense against DOX-induced oxidative stress. Furthermore,
increasing evidence suggest that myocardial HSP72 induction plays a pivotal
role in exercise-induced cardioprotection against oxidative stress (Powers,
Lennon et al. 2002, Powers, Locke et al. 2001, Taylor and Starnes 2003).
6.4 Meaning for exercise-induced cardioprotection in aging
Considering the present results in the context of exercise-induced
cardioprotection in advanced age, they can be interpreted as preliminary.
Indeed, it can be carefully speculated that the observed protective phenotype
caused by both chronic models of exercise against DOX can also be observed
in aged rats. In fact, Quindry et al. (2005) reported that aged rats submitted to
exercise training ameliorate cardiac hemodynamic response with significant
improvements in the apoptotic levels and signaling caused by IR injury.
Furthermore, the authors observed that trained old rats increased MnSOD
activity, which can be interpreted as a sign of cardiac mitochondrial adaptations
induced by chronic exercise in old rats compared to their young counterparts.
Similar results were found by Starnes et al. (2003), which suggest that, although
observing cardioprotective protein phenotype alterations with age, exercise can
enhance cardioprotection regardless of elderly. Furthermore, physical exercise
has the ability to positively modulate some gene expression associated with
improved heart function in aged rats. In fact, heart is known for its ability to
produce energy from fatty acids because of its important β-oxidation equipment,
which capacity is reduced with age (Starnes, Beyer et al. 1983). Confirming the
potential beneficial effects of physical exercise on cardiac metabolism in elderly,
Iemitsu et al. (2002) reported that exercise training improved the aging-induced
decreased expression of peroxisome proliferator-activated receptor, which
regulates genes related to fatty acid metabolism in the heart. Giving those
62
alterations reported in aged hearts, it is possible to speculate that the results of
the present work could also be observed in aged rats; affording protection and
mitigating the deleterious consequences associated with sub-chronic DOX
treatment schedules.
63
7. Conclusion
In summary, the data from the present work provide additional support about
the effect of two types of physical exercise (treadmill endurance training and
free-wheel voluntary physical activity) against heart mitochondrial dysfunction
induced by sub-chronic treatment of Doxorubicin (DOX). Our results showed us
that:
• Regarding mitochondrial respiratory function both types of exercise
reverted the effects induced by DOX on state 3, RCR and ADP/O.
Interestingly, free wheel voluntary physical activity was more efficient at
counteracting DOX-induced defects on RCR;
• Both types of exercise were able to counteract DOX-induced
impairments in mitochondrial transmembrane endpoints. Importantly, free
wheel voluntary physical activity was also more efficient at normalizing
DOX-induced increases in lag phase;
• Regarding mitochondrial osmotic swelling during MPTP induction, both
exercise protocols reverted DOX-induced impairments. In fact, both
types of exercise mitigated DOX-induced increases in swelling amplitude
and average swelling rate. However, once again free wheel voluntary
physical activity was more efficient at counteracting Ca2+-induced MPTP
induction
• Exercise protocols were able to revert DOX-induced increases in MDA
content and decrease in sulfhydryl groups.
The mechanisms by which treadmill endurance training and free-wheel
voluntary physical activity seems to confer additional protection against DOX
remain elusive and further studies need to be addressed in order to
comprehend the role of the different systems, such as those related to
mitochondria, in this process.
65
8. References
Adhihetty, P. J., V. Ljubicic and D. A. Hood (2007). Effect of chronic contractile activity on SS and IMF mitochondrial apoptotic susceptibility in skeletal muscle. Am J Physiol Endocrinol Metab,292(3): E748-‐755.
Andreyev, A. Y., B. Fahy and G. Fiskum (1998). Cytochrome c release from brain mitochondria is independent of the mitochondrial permeability transition. FEBS Lett,439(3): 373-‐376.
Anversa, P., B. Hiler, R. Ricci, G. Guideri and G. Olivetti (1986). Myocyte cell loss and myocyte hypertrophy in the aging rat heart. J Am Coll Cardiol,8(6): 1441-‐1448.
Ascensão, A. (2003). Exercício e Stress Oxidativo Cardíaco. Rev Port Cardiol,22(5).
Ascensão, A. (2011). Mitochondria as a target for exercise-‐induces cardioprotection. Current Drug Targets,12(6): 860-‐871.
Ascensao, A., R. Ferreira and J. Magalhaes (2007). Exercise-‐induced cardioprotection-‐-‐biochemical, morphological and functional evidence in whole tissue and isolated mitochondria. International Journal of Cardiology,117(1): 16-‐30.
Ascensao, A., R. Ferreira and J. Magalhaes (2007). Exercise-‐induced cardioprotection-‐-‐biochemical, morphological and functional evidence in whole tissue and isolated mitochondria. Int J Cardiol,117(1): 16-‐30.
Ascensao, A., R. Ferreira, P. J. Oliveira and J. Magalhaes (2006). Effects of endurance training and acute Doxorubicin treatment on rat heart mitochondrial alterations induced by in vitro anoxia-‐reoxygenation. Cardiovasc Toxicol,6(3-‐4): 159-‐172.
Ascensao, A., J. Lumini-‐Oliveira, N. G. Machado, R. M. Ferreira, I. O. Goncalves, A. C. Moreira, F. Marques, V. A. Sardao, P. J. Oliveira and J. Magalhaes (2010). Acute exercise protects against calcium-‐induced cardiac mitochondrial permeability transition pore opening in doxorubicin-‐treated rats. Clin Sci (Lond),120(1): 37-‐49.
Ascensao, A., J. Lumini-‐Oliveira, N. G. Machado, R. M. Ferreira, I. O. Goncalves, A. C. Moreira, F. Marques, V. A. Sardao, P. J. Oliveira and J. Magalhaes (2011). Acute exercise protects against calcium-‐induced cardiac mitochondrial permeability transition pore opening in doxorubicin-‐treated rats. Clin Sci (Lond),120(1): 37-‐49.
Ascensao, A., J. Lumini-‐Oliveira, P. J. Oliveira and J. Magalhaes (2011). Mitochondria as a target for exercise-‐induced cardioprotection. Curr Drug Targets,12(6): 860-‐871.
Ascensao, A., J. Lumini-‐Oliveira, P. J. Oliveira and J. Magalhaes (2011). Mitochondria as a Target for Exercise-‐Induced Cardioprotection. Current Drug Targets,12(6): 860-‐871.
Ascensão, A., J. Magalhães, J. Soares, R. Ferreira, M. Neuparth, F. Marques and J. Duarte (2006). Endurance exercise training attenuates morphological signs of cardiac muscle damage induced by doxorubicin in male mice.
Ascensao, A., J. Magalhaes, J. Soares, R. Ferreira, M. Neuparth, F. Marques, J. Oliveira and J. Duarte (2005). Endurance training attenuates doxorubicin-‐induced cardiac oxidative damage in mice. Int J Cardiol,100(3): 451-‐460.
Ascensao, A., J. Magalhaes, J. M. Soares, R. Ferreira, M. J. Neuparth, F. Marques, P. J. Oliveira and J. A. Duarte (2005). Moderate endurance training prevents doxorubicin-‐induced in vivo mitochondriopathy and reduces the development of cardiac apoptosis. Am J Physiol Heart Circ Physiol,289(2): H722-‐731.
Ascensao, A., J. Magalhaes, J. M. Soares, R. Ferreira, M. J. Neuparth, F. Marques, P. J. Oliveira and J. A. Duarte (2006). Endurance training limits the functional alterations of rat heart mitochondria submitted to in vitro anoxia-‐reoxygenation. Int J Cardiol,109(2): 169-‐178.
66
Ascensao, A., P. J. Oliveira and J. Magalhaes (2012). Exercise as a beneficial adjunct therapy during Doxorubicin treatment-‐-‐role of mitochondria in cardioprotection. Int J Cardiol,156(1): 4-‐10.
Aydin, C., E. Ince, S. Koparan, I. T. Cangul, M. Naziroglu and F. Ak (2007). Protective effects of long term dietary restriction on swimming exercise-‐induced oxidative stress in the liver, heart and kidney of rat. Cell Biochem Funct,25(2): 129-‐137.
Bagchi, M., D. Bagchi, E. B. Patterson, L. Tang and S. J. Stohs (1996). Age-‐related changes in lipid peroxidation and antioxidant defense in Fischer 344 rats. Ann N Y Acad Sci,793: 449-‐452.
Beckman, K. B. and B. N. Ames (1998). The free radical theory of aging matures. Physiol Rev,78(2): 547-‐581.
Bejma, J., P. Ramires and L. L. Ji (2000). Free radical generation and oxidative stress with ageing and exercise: differential effects in the myocardium and liver. Acta Physiol Scand,169(4): 343-‐351.
Bernstein, J. D., J. R. Bucher and R. Penniall (1978). Origin of mitochondrial enzymes. V. The polypeptide character and the biosynthesis of rat liver cytochrome c oxidase polypeptides by mitochondria. J Bioenerg Biomembr,10(1-‐2): 59-‐74.
Berthiaume, J. M., P. J. Oliveira, M. W. Fariss and K. B. Wallace (2005). Dietary vitamin E decreases doxorubicin-‐induced oxidative stress without preventing mitochondrial dysfunction. Cardiovasc Toxicol,5(3): 257-‐267.
Berthiaume, J. M. and K. B. Wallace (2007). Adriamycin-‐induced oxidative mitochondrial cardiotoxicity. Cell Biol Toxicol,23(1): 15-‐25.
Betik, A. C. and R. T. Hepple (2008). Determinants of VO2 max decline with aging: an integrated perspective. Appl Physiol Nutr Metab,33(1): 130-‐140.
Bhattacharya, S. K., J. H. Thakar, P. L. Johnson and D. R. Shanklin (1991). Isolation of skeletal muscle mitochondria from hamsters using an ionic medium containing ethylenediaminetetraacetic acid and nagarse. Anal Biochem,192(2): 344-‐349.
Bossy-‐Wetzel, E., M. J. Barsoum, A. Godzik, R. Schwarzenbacher and S. A. Lipton (2003). Mitochondrial fission in apoptosis, neurodegeneration and aging. Curr Opin Cell Biol,15(6): 706-‐716.
Bowles, D. K., R. P. Farrar and J. W. Starnes (1992). Exercise training improves cardiac function after ischemia in the isolated, working rat heart. Am J Physiol,263(3 Pt 2): H804-‐809.
Brand, M. D. and D. G. Nicholls (2011). Assessing mitochondrial dysfunction in cells. Biochem J,435(2): 297-‐312.
Bratic, I. and A. Trifunovic (2010). Mitochondrial energy metabolism and ageing. Biochim Biophys Acta,1797(6-‐7): 961-‐967.
Braunwald, E. and M. R. Bristow (2000). Congestive heart failure: fifty years of progress. Circulation,102(20 Suppl 4): IV14-‐23.
Broekemeier, K. M., M. E. Dempsey and D. R. Pfeiffer (1989). Cyclosporin A is a potent inhibitor of the inner membrane permeability transition in liver mitochondria. J Biol Chem,264(14): 7826-‐7830.
Buege, J. A. and S. D. Aust (1978). Microsomal lipid peroxidation. Methods Enzymol,52: 302-‐310.
Carvalho, C., R. X. Santos, S. Cardoso, S. Correia, P. J. Oliveira, M. S. Santos and P. I. Moreira (2009). Doxorubicin: the good, the bad and the ugly effect. Curr Med Chem,16(25): 3267-‐3285.
Chaudhary, K. R., H. El-‐Sikhry and J. M. Seubert (2011). Mitochondria and the aging heart. J Geriatr Cardiol,8(3): 159-‐167.
Chen, L. and A. A. Knowlton (2011). Mitochondrial dynamics in heart failure. Congest Heart Fail,17(6): 257-‐261.
67
Chicco, A. J., D. S. Hydock, C. M. Schneider and R. Hayward (2006). Low-‐intensity exercise training during doxorubicin treatment protects against cardiotoxicity. J Appl Physiol,100(2): 519-‐527.
Chicco, A. J., C. M. Schneider and R. Hayward (2005). Voluntary exercise protects against acute doxorubicin cardiotoxicity in the isolated perfused rat heart. Am J Physiol Regul Integr Comp Physiol,289(2): R424-‐R431.
Chicco, A. J., C. M. Schneider and R. Hayward (2006). Exercise training attenuates acute doxorubicin-‐induced cardiac dysfunction. J Cardiovasc Pharmacol,47(2): 182-‐189.
Chiong, M., Z. V. Wang, Z. Pedrozo, D. J. Cao, R. Troncoso, M. Ibacache, A. Criollo, A. Nemchenko, J. A. Hill and S. Lavandero (2011). Cardiomyocyte death: mechanisms and translational implications. Cell Death Dis,2: e244.
Condorelli, G., C. Morisco, G. Stassi, A. Notte, F. Farina, G. Sgaramella, A. de Rienzo, R. Roncarati, B. Trimarco and G. Lembo (1999). Increased cardiomyocyte apoptosis and changes in proapoptotic and antiapoptotic genes bax and bcl-‐2 during left ventricular adaptations to chronic pressure overload in the rat. Circulation,99(23): 3071-‐3078.
Coore, H. G., R. M. Denton, B. R. Martin and P. J. Randle (1971). Regulation of adipose tissue pyruvate dehydrogenase by insulin and other hormones. Biochem J,125(1): 115-‐127.
Crompton, M. (1999). The mitochondrial permeability transition pore and its role in cell death. Biochem J,341 ( Pt 2): 233-‐249.
Dai, D. F. and P. S. Rabinovitch (2009). Cardiac aging in mice and humans: the role of mitochondrial oxidative stress. Trends Cardiovasc Med,19(7): 213-‐220.
Dai, D. F., P. S. Rabinovitch and Z. Ungvari (2012). Mitochondria and cardiovascular aging. Circ Res,110(8): 1109-‐1124.
Dai, D. F., L. F. Santana, M. Vermulst, D. M. Tomazela, M. J. Emond, M. J. MacCoss, K. Gollahon, G. M. Martin, L. A. Loeb, W. C. Ladiges and P. S. Rabinovitch (2009). Overexpression of catalase targeted to mitochondria attenuates murine cardiac aging. Circulation,119(21): 2789-‐2797.
Desler, C., M. L. Marcker, K. K. Singh and L. J. Rasmussen (2011). The importance of mitochondrial DNA in aging and cancer. J Aging Res,2011: 407-‐536.
Emter, C. A. and D. K. Bowles (2008). Curing the cure: utilizing exercise to limit cardiotoxicity. Med Sci Sports Exerc,40(5): 806-‐807.
Estabrook, R. W. (1967). Mitochondrial respiratory control and the polarographic measurement of ADP/O ratios. Meth. Enzymol.,10: 41-‐57.
Fannin, S. W., E. J. Lesnefsky, T. J. Slabe, M. O. Hassan and C. L. Hoppel (1999). Aging selectively decreases oxidative capacity in rat heart interfibrillar mitochondria. Arch Biochem Biophys,372(2): 399-‐407.
Figueiredo, P. A., M. P. Mota, H. J. Appell and J. A. Duarte (2008). The role of mitochondria in aging of skeletal muscle. Biogerontology,9(2): 67-‐84.
Fortune, J. M. and N. Osheroff (2000). Topoisomerase II as a target for anticancer drugs: when enzymes stop being nice. Prog Nucleic Acid Res Mol Biol,64: 221-‐253.
French, J. P., K. L. Hamilton, J. C. Quindry, Y. Lee, P. A. Upchurch and S. K. Powers (2008). Exercise-‐induced protection against myocardial apoptosis and necrosis: MnSOD, calcium-‐handling proteins, and calpain. FASEB J,22(8): 2862-‐2871.
Gates, P. E., H. Tanaka, J. Graves and D. R. Seals (2003). Left ventricular structure and diastolic function with human ageing. Relation to habitual exercise and arterial stiffness. Eur Heart J,24(24): 2213-‐2220.
Geng, T., P. Li, M. Okutsu, X. Yin, J. Kwek, M. Zhang and Z. Yan (2010). PGC-‐1alpha plays a functional role in exercise-‐induced mitochondrial biogenesis and angiogenesis but not fiber-‐type transformation in mouse skeletal muscle. Am J Physiol Cell Physiol,298(3): C572-‐579.
68
Glancy, B., W. T. Willis, D. J. Chess and R. S. Balaban (2013). Effect of calcium on the oxidative phosphorylation cascade in skeletal muscle mitochondria. Biochemistry,52(16): 2793-‐2809.
Golbidi, S. and I. Laher (2011). Molecular mechanisms in exercise-‐induced cardioprotection. Cardiol Res Pract,2011: 972807.
Golbidi, S. and I. Laher (2012). Exercise and the cardiovascular system. Cardiol Res Pract,2012: 210852.
Goldspink, D. F., J. G. Burniston and L. B. Tan (2003). Cardiomyocyte death and the ageing and failing heart. Exp Physiol,88(3): 447-‐458.
Gornall, A. G., C. J. Bardawill and M. M. David (1949). Determination of serum proteins by means of the biuret reaction. J Biol Chem,177(2): 751-‐766.
Green, D. R. and J. C. Reed (1998). Mitochondria and apoptosis. Science,281(5381): 1309-‐1312.
Guarente, L. (2008). Mitochondria-‐-‐a nexus for aging, calorie restriction, and sirtuins? Cell,132(2): 171-‐176.
Gunduz, F., U. K. Senturk, O. Kuru, B. Aktekin and M. R. Aktekin (2004). The effect of one year's swimming exercise on oxidant stress and antioxidant capacity in aged rats. Physiol Res,53(2): 171-‐176.
Gunter, T. E., D. I. Yule, K. K. Gunter, R. A. Eliseev and J. D. Salter (2004). Calcium and mitochondria. FEBS Lett,567(1): 96-‐102.
Gustafsson, A. B. and R. A. Gottlieb (2008). Heart mitochondria: gates of life and death. Cardiovasc Res,77(2): 334-‐343.
Gustafsson, A. B. and R. A. Gottlieb (2009). Autophagy in ischemic heart disease. Circ Res,104(2): 150-‐158.
Halestrap, A. P. and C. Brenner (2003). The adenine nucleotide translocase: a central component of the mitochondrial permeability transition pore and key player in cell death. Curr Med Chem,10(16): 1507-‐1525.
Hambrecht, R., E. Fiehn, C. Weigl, S. Gielen, C. Hamann, R. Kaiser, J. Yu, V. Adams, J. Niebauer and G. Schuler (1998). Regular physical exercise corrects endothelial dysfunction and improves exercise capacity in patients with chronic heart failure. Circulation,98(24): 2709-‐2715.
Hamilton, K. L., J. L. Staib, T. Phillips, A. Hess, S. L. Lennon and S. K. Powers (2003). Exercise, antioxidants, and HSP72: protection against myocardial ischemia/reperfusion. Free Radic Biol Med,34(7): 800-‐809.
Harman, D. (1956). Aging: a theory based on free radical and radiation chemistry. J Gerontol(11): 298-‐300 Harris, E. D. (1992). Regulation of antioxidant enzymes. FASEB J,6(9): 2675-‐2683.
Harris, M. B. and J. W. Starnes (2001). Effects of body temperature during exercise training on myocardial adaptations. Am J Physiol Heart Circ Physiol,280(5): H2271-‐2280.
Haunstetter, A. and S. Izumo (1998). Apoptosis: basic mechanisms and implications for cardiovascular disease. Circ Res,82(11): 1111-‐1129.
Hayward, R., C. Y. Lien, B. T. Jensen, D. S. Hydock and C. M. Schneider (2012). Exercise training mitigates anthracycline-‐induced chronic cardiotoxicity in a juvenile rat model. Pediatric blood & cancer,59(1): 149-‐154.
Hayward, R., C. Y. Lien, B. T. Jensen, D. S. Hydock and C. M. Schneider (2012). Exercise training mitigates anthracycline-‐induced chronic cardiotoxicity in a juvenile rat model. Pediatr Blood Cancer,59(1): 149-‐154.
Harman, D. (1972). The biologic clock: the mitochondria? J Am Geriatr Soc(20): 145-‐7 Holloszy, J. O., L. B. Oscai, I. J. Don and P. A. Mole (1970). Mitochondrial citric acid cycle and related enzymes: adaptive response to exercise. Biochem Biophys Res Commun,40(6): 1368-‐1373.
Honda, H. M., P. Korge and J. N. Weiss (2005). Mitochondria and ischemia/reperfusion injury. Ann N Y Acad Sci,1047: 248-‐258.
69
Hong, H. and P. Johnson (1995). Antioxidant enzyme activities and lipid peroxidation levels in exercised and hypertensive rat tissues. Int J Biochem Cell Biol,27(9): 923-‐931.
Hood, D. A., A. Balaban, M. K. Connor, E. E. Craig, M. L. Nishio, M. Rezvani and M. Takahashi (1994). Mitochondrial biogenesis in striated muscle. Can J Appl Physiol,19(1): 12-‐48.
Hoppins, S., F. Edlich, M. M. Cleland, S. Banerjee, J. M. McCaffery, R. J. Youle and J. Nunnari (2011). The soluble form of Bax regulates mitochondrial fusion via MFN2 homotypic complexes. Mol Cell,41(2): 150-‐160.
Hotta, H. and S. Uchida (2010). Aging of the autonomic nervous system and possible improvements in autonomic activity using somatic afferent stimulation. Geriatrics & Gerontology International,10: S127-‐S136.
Hu, M. L. (1990). Measurement of protein thiol groups and GSH in plasma. Methods in Enzymology. Parker, L. San Diego, CA: Academic: 380-‐385.
Huang, C., X. Zhang, J. M. Ramil, S. Rikka, L. Kim, Y. Lee, N. A. Gude, P. A. Thistlethwaite, M. A. Sussman, R. A. Gottlieb and A. B. Gustafsson (2010). Juvenile exposure to anthracyclines impairs cardiac progenitor cell function and vascularization resulting in greater susceptibility to stress-‐induced myocardial injury in adult mice. Circulation,121(5): 675-‐683.
Hydock, D. S., C. Y. Lien, B. T. Jensen, T. L. Parry, C. M. Schneider and R. Hayward (2012). Rehabilitative exercise in a rat model of doxorubicin cardiotoxicity. Experimental biology and medicine,237(12): 1483-‐1492.
Hydock, D. S., C. Y. Lien, C. M. Schneider and R. Hayward (2008). Exercise preconditioning protects against doxorubicin-‐induced cardiac dysfunction. Med Sci Sports Exerc,40(5): 808-‐817.
Iemitsu, M., T. Miyauchi, S. Maeda, T. Tanabe, M. Takanashi, Y. Irukayama-‐Tomobe, S. Sakai, H. Ohmori, M. Matsuda and I. Yamaguchi (2002). Aging-‐induced decrease in the PPAR-‐alpha level in hearts is improved by exercise training. American Journal of Physiology-‐Heart and Circulatory Physiology,283(5): H1750-‐H1760.
Ji, L. L. (1993). Antioxidant enzyme response to exercise and aging. Med Sci Sports Exerc,25(2): 225-‐231.
Ji, L. L., D. Dillon and E. Wu (1991). Myocardial aging: antioxidant enzyme systems and related biochemical properties. Am J Physiol,261(2 Pt 2): R386-‐392.
Ji, L. L., C. Leeuwenburgh, S. Leichtweis, M. Gore, R. Fiebig, J. Hollander and J. Bejma (1998). Oxidative stress and aging. Role of exercise and its influences on antioxidant systems. Ann N Y Acad Sci,854: 102-‐117.
Judge, S., Y. M. Jang, A. Smith, T. Hagen and C. Leeuwenburgh (2005). Age-‐associated increases in oxidative stress and antioxidant enzyme activities in cardiac interfibrillar mitochondria: implications for the mitochondrial theory of aging. FASEB J,19(3): 419-‐421.
Judge, S., Y. M. Jang, A. Smith, C. Selman, T. Phillips, J. R. Speakman, T. Hagen and C. Leeuwenburgh (2005). Exercise by lifelong voluntary wheel running reduces subsarcolemmal and interfibrillar mitochondrial hydrogen peroxide production in the heart. Am J Physiol Regul Integr Comp Physiol,289(6): R1564-‐1572.
Judge, S. and C. Leeuwenburgh (2007). Cardiac mitochondrial bioenergetics, oxidative stress, and aging. Am J Physiol Cell Physiol,292(6): C1983-‐1992.
Jung, K. and R. Reszka (2001). Mitochondria as subcellular targets for clinically useful anthracyclines. Adv Drug Deliv Rev,49(1-‐2): 87-‐105.
Kaim, G. and P. Dimroth (1999). ATP synthesis by F-‐type ATP synthase is obligatorily dependent on the transmembrane voltage. EMBO J,18(15): 4118-‐4127.
Kanter, M. M., R. L. Hamlin, D. V. Unverferth, H. W. Davis and A. J. Merola (1985). Effect of exercise training on antioxidant enzymes and cardiotoxicity of doxorubicin. J Appl Physiol,59(4): 1298-‐1303.
70
Kappagoda, T. and E. A. Amsterdam (2012). Exercise and heart failure in the elderly. Heart Fail Rev,17(4-‐5): 635-‐662.
Kavazis, A. N. (2009). Exercise preconditioning of the myocardium. Sports Med,39(11): 923-‐935.
Kavazis, A. N., S. Alvarez, E. Talbert, Y. Lee and S. K. Powers (2009). Exercise training induces a cardioprotective phenotype and alterations in cardiac subsarcolemmal and intermyofibrillar mitochondrial proteins. Am J Physiol Heart Circ Physiol,297(1): H144-‐152.
Kavazis, A. N., J. M. McClung, D. A. Hood and S. K. Powers (2008). Exercise induces a cardiac mitochondrial phenotype that resists apoptotic stimuli. Am J Physiol Heart Circ Physiol,294(2): H928-‐935.
Kavazis, A. N., A. J. Smuder, K. Min, N. Tumer and S. K. Powers (2010). Short-‐term exercise training protects against doxorubicin-‐induced cardiac mitochondrial damage independent of HSP72. Am J Physiol Heart Circ Physiol,299(5): H1515-‐1524.
Kiyomiya, K., S. Matsuo and M. Kurebe (2001). Mechanism of specific nuclear transport of adriamycin: the mode of nuclear translocation of adriamycin-‐proteasome complex. Cancer Res,61(6): 2467-‐2471.
Kokkinos, P., J. Myers, C. Faselis, D. B. Panagiotakos, M. Doumas, A. Pittaras, A. Manolis, J. P. Kokkinos, P. Karasik, M. Greenberg, V. Papademetriou and R. Fletcher (2010). Exercise capacity and mortality in older men: a 20-‐year follow-‐up study. Circulation,122(8): 790-‐797.
Koltai, E., N. Hart, A. W. Taylor, S. Goto, J. K. Ngo, K. J. Davies and Z. Radak (2012). Age-‐associated declines in mitochondrial biogenesis and protein quality control factors are minimized by exercise training. Am J Physiol Regul Integr Comp Physiol,303(2): R127-‐134.
Kowaltowski, A. J., R. F. Castilho and A. E. Vercesi (2001). Mitochondrial permeability transition and oxidative stress. FEBS Lett,495(1-‐2): 12-‐15.
Kregel, K. C. and P. L. Moseley (1996). Differential effects of exercise and heat stress on liver HSP70 accumulation with aging. J Appl Physiol,80(2): 547-‐551.
Kroemer, G., L. Galluzzi and C. Brenner (2007). Mitochondrial membrane permeabilization in cell death. Physiol Rev,87(1): 99-‐163.
Lagranha, C. J., A. Deschamps, A. Aponte, C. Steenbergen and E. Murphy (2010). Sex differences in the phosphorylation of mitochondrial proteins result in reduced production of reactive oxygen species and cardioprotection in females. Circ Res,106(11): 1681-‐1691.
Lakatta, E. G. (2002). Age-‐associated cardiovascular changes in health: impact on cardiovascular disease in older persons. Heart Fail Rev,7(1): 29-‐49.
Landmesser, U. and H. Drexler (2005). The clinical significance of endothelial dysfunction. Curr Opin Cardiol,20(6): 547-‐551.
Lebrecht, D., B. Setzer, U. P. Ketelsen, J. Haberstroh and U. A. Walker (2003). Time-‐dependent and tissue-‐specific accumulation of mtDNA and respiratory chain defects in chronic doxorubicin cardiomyopathy. Circulation,108(19): 2423-‐2429.
Lee, H. C. and Y. H. Wei (2007). Oxidative stress, mitochondrial DNA mutation, and apoptosis in aging. Exp Biol Med (Maywood),232(5): 592-‐606.
Lee, S., S. Y. Jeong, W. C. Lim, S. Kim, Y. Y. Park, X. Sun, R. J. Youle and H. Cho (2007). Mitochondrial fission and fusion mediators, hFis1 and OPA1, modulate cellular senescence. J Biol Chem,282(31): 22977-‐22983.
Lehman, J. J., P. M. Barger, A. Kovacs, J. E. Saffitz, D. M. Medeiros and D. P. Kelly (2000). Peroxisome proliferator-‐activated receptor gamma coactivator-‐1 promotes cardiac mitochondrial biogenesis. J Clin Invest,106(7): 847-‐856.
Lenaz, G. (1998). Role of mitochondria in oxidative stress and ageing. Biochim Biophys Acta,1366(1-‐2): 53-‐67.
71
Lennon, S. L., J. C. Quindry, J. P. French, S. Kim, J. L. Mehta and S. K. Powers (2004). Exercise and myocardial tolerance to ischaemia-‐reperfusion. Acta Physiol Scand,182(2): 161-‐169.
Leung, F. P., L. M. Yung, I. Laher, X. Yao, Z. Y. Chen and Y. Huang (2008). Exercise, vascular wall and cardiovascular diseases: an update (Part 1). Sports Med,38(12): 1009-‐1024.
Li, L., C. Muhlfeld, B. Niemann, R. Pan, R. Li, D. Hilfiker-‐Kleiner, Y. Chen and S. Rohrbach (2011). Mitochondrial biogenesis and PGC-‐1alpha deacetylation by chronic treadmill exercise: differential response in cardiac and skeletal muscle. Basic Res Cardiol,106(6): 1221-‐1234.
Liesa, M., M. Palacin and A. Zorzano (2009). Mitochondrial dynamics in mammalian health and disease. Physiol Rev,89(3): 799-‐845.
Ljubicic, V., K. J. Menzies and D. A. Hood (2010). Mitochondrial dysfunction is associated with a pro-‐apoptotic cellular environment in senescent cardiac muscle. Mech Ageing Dev,131(2): 79-‐88.
Logue, S. E., A. B. Gustafsson, A. Samali and R. A. Gottlieb (2005). Ischemia/reperfusion injury at the intersection with cell death. J Mol Cell Cardiol,38(1): 21-‐33.
Lopez-‐Lluch, G., P. M. Irusta, P. Navas and R. de Cabo (2008). Mitochondrial biogenesis and healthy aging. Exp Gerontol,43(9): 813-‐819.
Lumini-‐Oliveira, J., J. Magalhaes, C. V. Pereira, A. C. Moreira, P. J. Oliveira and A. Ascensao (2011). Endurance training reverts heart mitochondrial dysfunction, permeability transition and apoptotic signaling in long-‐term severe hyperglycemia. Mitochondrion,11(1): 54-‐63.
Marcil, M., K. Bourduas, A. Ascah and Y. Burelle (2006). Exercise training induces respiratory substrate-‐specific decrease in Ca2+-‐induced permeability transition pore opening in heart mitochondria. Am J Physiol Heart Circ Physiol,290(4): H1549-‐1557.
Meng, R. R. Y. T. W. J. C. Q. (2007). Age-‐related changes in mitochondrial function and antioxidative enzyme activity in fischer 344 rats. Mechanisms of Ageing and Development(128): 286-‐92 Morley, J. E. and S. S. Reese (1989). Clinical implications of the aging heart. Am J Med,86(1): 77-‐86.
Moslehi, J., R. A. DePinho and E. Sahin (2012). Telomeres and mitochondria in the aging heart. Circ Res,110(9): 1226-‐1237.
Murphy, M. P. and M. D. Brand (1988). Membrane-‐potential-‐dependent changes in the stoichiometry of charge translocation by the mitochondrial electron transport chain. Eur J Biochem,173(3): 637-‐644.
Narula, J., N. Haider, R. Virmani, T. G. DiSalvo, F. D. Kolodgie, R. J. Hajjar, U. Schmidt, M. J. Semigran, G. W. Dec and B. A. Khaw (1996). Apoptosis in myocytes in end-‐stage heart failure. N Engl J Med,335(16): 1182-‐1189.
Navarro, A., C. Gomez, J. M. Lopez-‐Cepero and A. Boveris (2004). Beneficial effects of moderate exercise on mice aging: survival, behavior, oxidative stress, and mitochondrial electron transfer. Am J Physiol Regul Integr Comp Physiol,286(3): R505-‐511.
North, B. J. and D. A. Sinclair (2012). The intersection between aging and cardiovascular disease. Circ Res,110(8): 1097-‐1108.
Nulton-‐Persson, A. C. and L. I. Szweda (2001). Modulation of mitochondrial function by hydrogen peroxide. J Biol Chem,276(26): 23357-‐23361.
O'Brien, I. A., P. O'Hare and R. J. Corrall (1986). Heart rate variability in healthy subjects: effect of age and the derivation of normal ranges for tests of autonomic function. Br Heart J,55(4): 348-‐354.
Oliveira, P. J. and K. B. Wallace (2006). Depletion of adenine nucleotide translocator protein in heart mitochondria from doxorubicin-‐treated rats-‐-‐relevance for mitochondrial dysfunction. Toxicology,220(2-‐3): 160-‐168.
72
Olivetti, G., R. Abbi, F. Quaini, J. Kajstura, W. Cheng, J. A. Nitahara, E. Quaini, C. Di Loreto, C. A. Beltrami, S. Krajewski, J. C. Reed and P. Anversa (1997). Apoptosis in the failing human heart. N Engl J Med,336(16): 1131-‐1141.
Oxenham, H. and N. Sharpe (2003). Cardiovascular aging and heart failure. Eur J Heart Fail,5(4): 427-‐434.
Pashkow, F. J. (2011). Oxidative Stress and Inflammation in Heart Disease: Do Antioxidants Have a Role in Treatment and/or Prevention? Int J Inflam,2011: 514623.
Pereira, G. C., S. P. Pereira, C. V. Pereira, J. A. Lumini, J. Magalhaes, A. Ascensao, M. S. Santos, A. J. Moreno and P. J. Oliveira (2012). Mitochondrionopathy phenotype in doxorubicin-‐treated Wistar rats depends on treatment protocol and is cardiac-‐specific. PLoS One,7(6): e38867.
Picard, M., K. Csukly, M. E. Robillard, R. Godin, A. Ascah, C. Bourcier-‐Lucas and Y. Burelle (2008). Resistance to Ca2+-‐induced opening of the permeability transition pore differs in mitochondria from glycolytic and oxidative muscles. Am J Physiol Regul Integr Comp Physiol,295(2): R659-‐668.
Pohjoismaki, J. L., T. Boettger, Z. Liu, S. Goffart, M. Szibor and T. Braun (2012). Oxidative stress during mitochondrial biogenesis compromises mtDNA integrity in growing hearts and induces a global DNA repair response. Nucleic Acids Res,40(14): 6595-‐6607.
Polla, B. S., S. Kantengwa, D. Francois, S. Salvioli, C. Franceschi, C. Marsac and A. Cossarizza (1996). Mitochondria are selective targets for the protective effects of heat shock against oxidative injury. Proc Natl Acad Sci U S A,93(13): 6458-‐6463.
Powers, S. K., H. A. Demirel, H. K. Vincent, J. S. Coombes, H. Naito, K. L. Hamilton, R. A. Shanely and J. Jessup (1998). Exercise training improves myocardial tolerance to in vivo ischemia-‐reperfusion in the rat. Am J Physiol,275(5 Pt 2): R1468-‐1477.
Powers, S. K., S. L. Lennon, J. Quindry and J. L. Mehta (2002). Exercise and cardioprotection. Curr Opin Cardiol,17(5): 495-‐502.
Powers, S. K., Locke and H. A. Demirel (2001). Exercise, heat shock proteins, and myocardial protection from I-‐R injury. Med Sci Sports Exerc,33(3): 386-‐392.
Powers, S. K., J. Quindry and K. Hamilton (2004). Aging, exercise, and cardioprotection. Ann N Y Acad Sci,1019: 462-‐470.
Powers, S. K., J. C. Quindry and A. N. Kavazis (2008). Exercise-‐induced cardioprotection against myocardial ischemia-‐reperfusion injury. Free Radic Biol Med,44(2): 193-‐201.
Pritsos, C. A. and J. Ma (2000). Basal and drug-‐induced antioxidant enzyme activities correlate with age-‐dependent doxorubicin oxidative toxicity. Chem Biol Interact,127(1): 1-‐11.
Quindry, J., J. French, K. Hamilton, Y. Lee, J. L. Mehta and S. Powers (2005). Exercise training provides cardioprotection against ischemia-‐reperfusion induced apoptosis in young and old animals. Exp Gerontol,40(5): 416-‐425.
Rabinovitch, P. (2012). Mitochondria and cardiovascular aging. American Heart Association, Inc.(110): 1109-‐24.
Rajendra Acharya, U., K. Paul Joseph, N. Kannathal, C. M. Lim and J. S. Suri (2006). Heart rate variability: a review. Med Biol Eng Comput,44(12): 1031-‐1051.
Rao, G., E. Xia and A. Richardson (1990). Effect of age on the expression of antioxidant enzymes in male Fischer F344 rats. Mech Ageing Dev,53(1): 49-‐60.
Reynolds, D. W. (2004). Understanding the aging cardiovascular system. Geriatrics and Gerontology International(4): 298–303.
73
Reznick, R. M., H. Zong, J. Li, K. Morino, I. K. Moore, H. J. Yu, Z. X. Liu, J. Dong, K. J. Mustard, S. A. Hawley, D. Befroy, M. Pypaert, D. G. Hardie, L. H. Young and G. I. Shulman (2007). Aging-‐associated reductions in AMP-‐activated protein kinase activity and mitochondrial biogenesis. Cell Metab,5(2): 151-‐156.
Rodgers, J. T., C. Lerin, W. Haas, S. P. Gygi, B. M. Spiegelman and P. Puigserver (2005). Nutrient control of glucose homeostasis through a complex of PGC-‐1alpha and SIRT1. Nature,434(7029): 113-‐118.
Santos, D. L., A. J. Moreno, R. L. Leino, M. K. Froberg and K. B. Wallace (2002). Carvedilol protects against doxorubicin-‐induced mitochondrial cardiomyopathy. Toxicol Appl Pharmacol,185(3): 218-‐227.
Sardao, V. A., P. J. Oliveira, J. Holy, C. R. Oliveira and K. B. Wallace (2009). Morphological alterations induced by doxorubicin on H9c2 myoblasts: nuclear, mitochondrial, and cytoskeletal targets. Cell Biol Toxicol,25(3): 227-‐243.
Sardao, V. A., S. L. Pereira and P. J. Oliveira (2008). Drug-‐induced mitochondrial dysfunction in cardiac and skeletal muscle injury. Expert Opin Drug Saf,7(2): 129-‐146.
Sarvazyan, N. A., A. Askari and W. H. Huang (1995). Effects of doxorubicin on cardiomyocytes with reduced level of superoxide dismutase. Life Sci,57(10): 1003-‐1010.
Searls, Y. M., I. V. Smirnova, B. R. Fegley and L. Stehno-‐Bittel (2004). Exercise attenuates diabetes-‐induced ultrastructural changes in rat cardiac tissue. Med Sci Sports Exerc,36(11): 1863-‐1870.
Seeley, R., T. Stephens and P. Tate (2005). Anatomy & Physiology, Martin J. Lange.
Sheu, S. (2009). Morphological Dynamics of Mitochondria-‐ A Special Emphasis on Cardiac Muscle Cells. J Mol Cell Cardiol,46(6).
Shioi, T. and Y. Inuzuka (2012). Aging as a substrate of heart failure. J Cardiol: 423-‐8. Singal, P. K., N. Iliskovic, T. Li and D. Kumar (1997). Adriamycin cardiomyopathy: pathophysiology and prevention. FASEB J,11(12): 931-‐936.
Souza, T. M. S. B. F. V. A. (2012). Reprodutibilidade do VO2Máx estimado na corrida pela frequência cardíaca e consumo de oxigênio de reserva. Rev. bras. educ. fís. esporte 26(1).
Spina, R. J., M. J. Turner and A. A. Ehsani (1998). Beta-‐adrenergic-‐mediated improvement in left ventricular function by exercise training in older men. Am J Physiol,274(2 Pt 2): H397-‐404.
Starnes, J. W., B. D. Barnes and M. E. Olsen (2007). Exercise training decreases rat heart mitochondria free radical generation but does not prevent Ca2+-‐induced dysfunction. J Appl Physiol,102(5): 1793-‐1798.
Starnes, J. W., R. E. Beyer and D. W. Edington (1983). Myocardial adaptations to endurance exercise in aged rats. Am J Physiol,245(4): H560-‐566.
Starnes, J. W., D. K. Bowles and K. S. Seiler (1997). Myocardial injury after hypoxia in immature, adult and aged rats. Aging (milano),9(4): 268-‐276.
Starnes, J. W. and R. P. Taylor (2007). Exercise-‐induced cardioprotection: endogenous mechanisms. Med Sci Sports Exerc,39(9): 1537-‐1543.
Starnes, J. W., R. P. Taylor and Y. Park (2003). Exercise improves postischemic function in aging hearts. Am J Physiol Heart Circ Physiol,285(1): H347-‐351.
Stock, D., A. G. Leslie and J. E. Walker (1999). Molecular architecture of the rotary motor in ATP synthase. Science,286(5445): 1700-‐1705.
Strait, J. B. and E. G. Lakatta (2012). Aging-‐Associated Cardiovascular Changes and Their Relationship to Heart Failure. Heart Failure Clinics,8(1): 143-‐+.
Strait, J. B. and E. G. Lakatta (2012). Aging-‐associated cardiovascular changes and their relationship to heart failure. Heart Fail Clin,8(1): 143-‐164.
74
Swain, S. M., F. S. Whaley and M. S. Ewer (2003). Congestive heart failure in patients treated with doxorubicin: a retrospective analysis of three trials. Cancer,97(11): 2869-‐2879.
T. Shioi, Y. I. (2012). Aging as a substrate of heart failure. Journal of Cardiology: 423-‐8. Taylor, A. H., N. T. Cable, G. Faulkner, M. Hillsdon, M. Narici and A. K. van Der Bij (2004). Physical activity and older adults: a review of health benefits and the effectiveness of interventions. Journal of Sports Sciences,22(8): 703-‐725.
Taylor, R. P. and J. W. Starnes (2003). Age, cell signalling and cardioprotection. Acta Physiologica Scandinavica,178(2): 107-‐116.
Tian, L., Q. Cai and H. Wei (1998). Alterations of antioxidant enzymes and oxidative damage to macromolecules in different organs of rats during aging. Free Radic Biol Med,24(9): 1477-‐1484.
van Acker, S. A., K. Kramer, E. E. Voest, J. A. Grimbergen, J. Zhang, W. J. van der Vijgh and A. Bast (1996). Doxorubicin-‐induced cardiotoxicity monitored by ECG in freely moving mice. A new model to test potential protectors. Cancer Chemother Pharmacol,38(1): 95-‐101.
Vertechy, M., M. B. Cooper, O. Ghirardi and M. T. Ramacci (1989). Antioxidant enzyme activities in heart and skeletal muscle of rats of different ages. Exp Gerontol,24(3): 211-‐218.
Wallace, D. C. (2010). Mitochondrial DNA mutations in disease and aging. Environ Mol Mutagen,51(5): 440-‐450.
Wallace, K. B. (2003). Doxorubicin-‐induced cardiac mitochondrionopathy. Pharmacol Toxicol,93(3): 105-‐115.
Wallace, K. B. (2007). Adriamycin-‐induced interference with cardiac mitochondrial calcium homeostasis. Cardiovasc Toxicol,7(2): 101-‐107.
Wei, J. Y. (2004). Understanding the aging cardiovascular system. Geriatrics & Gerontology International,4: S298-‐S303.
Weitzel, J. M., K. A. Iwen and H. J. Seitz (2003). Regulation of mitochondrial biogenesis by thyroid hormone. Exp Physiol,88(1): 121-‐128.
Wessells, R. J. and R. Bodmer (2007). Cardiac aging. Semin Cell Dev Biol,18(1): 111-‐116.
Whelan, R. S., V. Kaplinskiy and R. N. Kitsis (2010). Cell death in the pathogenesis of heart disease: mechanisms and significance. Annu Rev Physiol,72: 19-‐44.
Wonders, K. Y., D. S. Hydock, C. M. Schneider and R. Hayward (2008). Acute exercise protects against doxorubicin cardiotoxicity. Integr Cancer Ther,7(3): 147-‐154.
Wu, H., S. B. Kanatous, F. A. Thurmond, T. Gallardo, E. Isotani, R. Bassel-‐Duby and R. S. Williams (2002). Regulation of mitochondrial biogenesis in skeletal muscle by CaMK. Science,296(5566): 349-‐352.
Yen, H. C., T. D. Oberley, S. Vichitbandha, Y. S. Ho and D. K. St Clair (1996). The protective role of manganese superoxide dismutase against adriamycin-‐induced acute cardiac toxicity in transgenic mice. J Clin Invest,98(5): 1253-‐1260.
Yeragani, V. K., E. Sobolewski, J. Kay, V. C. Jampala and G. Igel (1997). Effect of age on long-‐term heart rate variability. Cardiovasc Res,35(1): 35-‐42.
Yilmaz, S., A. Atessahin, E. Sahna, I. Karahan and S. Ozer (2006). Protective effect of lycopene on adriamycin-‐induced cardiotoxicity and nephrotoxicity. Toxicology,218(2-‐3): 164-‐171.
Zhou, S., A. Starkov, M. K. Froberg, R. L. Leino and K. B. Wallace (2001). Cumulative and irreversible cardiac mitochondrial dysfunction induced by doxorubicin. Cancer Res,61(2): 771-‐777.
75
Zhu, X., L. Zuo, A. J. Cardounel, J. L. Zweier and G. He (2007). Characterization of in vivo tissue redox status, oxygenation, and formation of reactive oxygen species in postischemic myocardium. Antioxid Redox Signal,9(4): 447-‐455.
Zucchi, R. and R. Danesi (2003). Cardiac toxicity of antineoplastic anthracyclines. Curr Med Chem Anticancer Agents,3(2): 151-‐171.



























































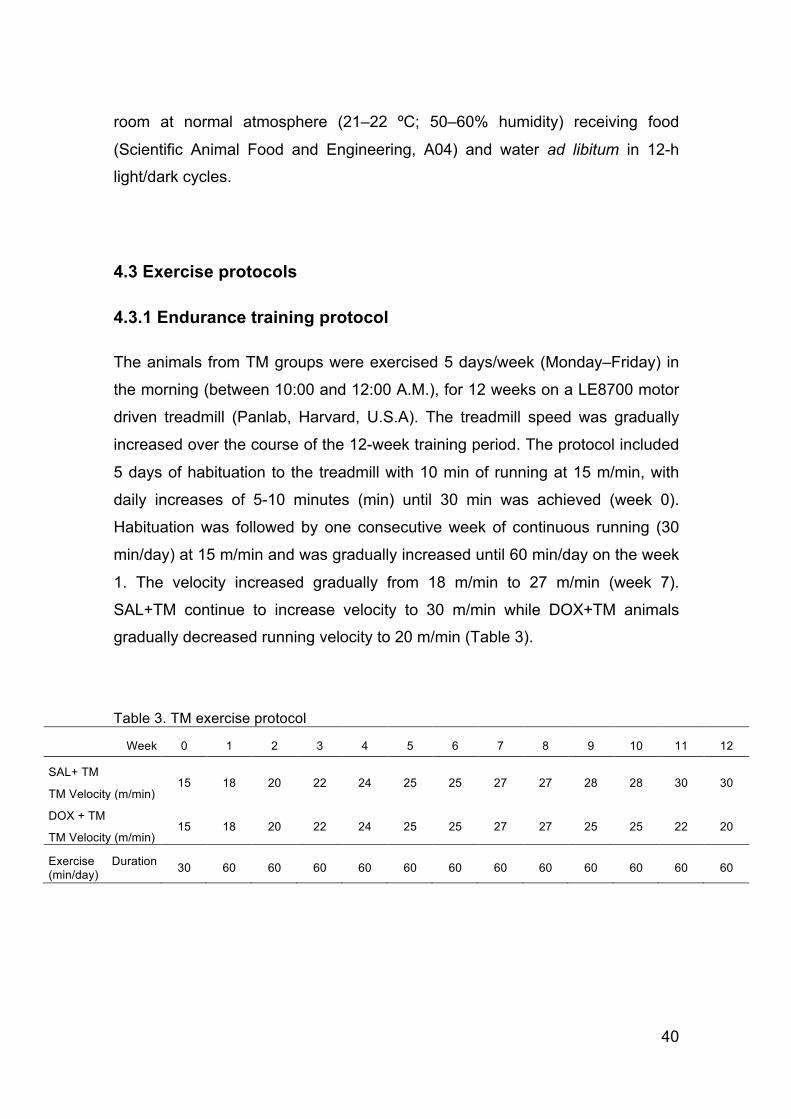



















































![Angel Carracedo [Modo de compatibilidad] - …biblioteca.climantica.org/resources/933/angel-carracedo.pdf · popularizada posteriormente como la “Eva mitocondrial”. ... Eva. Uniparental](https://static.documents.pub/doc/80x56/5bb06d2009d3f289598ba077/angel-carracedo-modo-de-compatibilidad-popularizada-posteriormente-como.jpg)
