Page 1
doi.org/10.26434/chemrxiv.12380330.v2
Efficient Reduction of Electron-Deficient Alkenes Enabled by aPhotoinduced Hydrogen Atom TransferXacobe Cambeiro, Natalia A. Larionova, Jun Miyatake Ondozabal
Submitted date: 01/06/2020 • Posted date: 02/06/2020Licence: CC BY-NC-ND 4.0Citation information: Cambeiro, Xacobe; Larionova, Natalia A.; Miyatake Ondozabal, Jun (2020): EfficientReduction of Electron-Deficient Alkenes Enabled by a Photoinduced Hydrogen Atom Transfer. ChemRxiv.Preprint. https://doi.org/10.26434/chemrxiv.12380330.v2
Direct hydrogen atom transfer from a photoredox-generated Hantzsch ester radical cation to electron-deficientalkenes has enabled thedevelopment of an efficient formal hydrogenation under mild, operationally simple conditions. The HAT-drivenmechanism, key to circumventthe problems associated with the low electron affinity of alkenes, is supported by experimental andcomputational studies. The reaction is appliedto a variety of cinnamate derivatives and related structures, irrespective of the presence of electron-donatingor electron-withdrawingsubstituents in the aromatic ring and with good functional group compatibility.
File list (2)
download fileview on ChemRxivHAT reduction ChemRxiv v2.pdf (523.61 KiB)
download fileview on ChemRxivHAT reduction QMUL - ESI.pdf (7.07 MiB)
Page 2
Efficient reduction of electron-deficient alkenes enabled by a photoinduced hydrogen atom transfer
Natalia A. Larionova, Jun Miyatake Ondozabal and Xacobe C. Cambeiro*
Department of Chemistry, School of Biological and Chemical Sciences, Queen Mary University of London. Mile End Rd, London E1 4NS (UK). * [email protected]
Direct hydrogen atom transfer from a photoredox-generated Hantzsch ester radical cation to electron-deficient alkenes has enabled the
development of an efficient formal hydrogenation under mild, operationally simple conditions. The HAT-driven mechanism, key to circumvent
the problems associated with the low electron affinity of alkenes, is supported by experimental and computational studies. The reaction is applied
to a variety of cinnamate derivatives and related structures, irrespective of the presence of electron-donating or electron-withdrawing
substituents in the aromatic ring and with good functional group compatibility.
Introduction
The hydrogenation of C-C multiple bonds and related reductive
transformations are among the most important processes in
chemical industry.1 In particular, transfer hydrogenation,
which avoids the use of hydrogen gas, is of much practical
interest in both industrial and laboratory settings, and the
development of new methods and strategies is of continued
importance.2
The recent fast development of photocatalysis has resulted in
the discovery of a wide variety of reductive, oxidative and
redox-neutral transformations.3 Photocatalytic reduction
methods have been explored in a number of instances, and
actually some of the pioneering research in photocatalysis
dealt with the formal hydrogenation of alkenes using a
dihydropyridine (N-benzyl 1,4-dihydronicotinamide, BNAH) as
the reductant.4 However, this early work was handicapped by
a very narrow substrate scope ―the reaction only worked on
extremely electron-deficient alkenes bearing at least two
electron-withdrawing groups (Scheme 1A).5 This is attributed
to the low electron affinity of alkenes, which results in direct
electron transfer reductions being difficult. For other
functional groups, similar limitations have been overcome by
activating the substrate with Lewis or Brønsted acid additives
or co-catalysts, which make it more electron-deficient.6
However, to the best of our knowledge, such strategies have
not been successfully applied to the formal hydrogenation of
alkenes.7 On a different approach, reduction could be forced
by using catalytic systems with increased reductive power, for
example by the exploitation of two-photon excited states8 or
combining electro- and photocatalysis.9 Nevertheless, the
possible reduction of other functional groups under these very
strongly reducing conditions limits the scope of such
strategies. One recent report has described the application of
a two-photon strategy for the formal hydrogenation of 1,2-
diarylethylenes by sequential electron and proton transfer
reactions (Scheme 1A).10
Finally, an alternative strategy to achieve the reduction of
alkenes circumventing the issue of their low electron affinity
would be to exploit hydrogen atom transfer (HAT) reactions.11
Thus, a catalytic system capable of promoting the transference
of a H atom to an alkene would provide an easy route for their
reduction without requiring highly reducing potentials
(Scheme 1B). HAT is frequently invoked in light-promoted
hydrofunctionalisation,12 always in a termination step where
an organic radical abstracts a H atom from radical cations of
dihydropyridines, such as Hantzsch ester (HE), or other
donors.13,14 However, methodologies using HAT into a stable,
closed-shell compound have not been reported to date. Herein
we report an efficient and operationally simple reduction of
cinnamate derivatives under mild conditions, initiated by HAT
from a photoredox-generated Hantzsch ester radical cation.
Scheme 1. Strategies for the photoreduction of alkenes promoted by visible light
Results and discussion
We started our investigation by studying the reduction of
methyl cinnamate (1a) as a model substrate with combinations
of known photocatalysts and potential hydrogen atom
donors13 (summary in Table 1, entries 1-5). Gratifyingly, we
found that [Ir(ppy)2(dtbbpy)]PF6 (Ir1) was indeed capable of
promoting the reaction with either iPr2NEt, 1,4-
dihydronicotinamide (BNAH) or Hantzsch ester (HE) as the
Page 3
reductant and H atom donor, with HE providing quantitative
yield (entry 1). Other similar photocatalysts such as Ru(bpy)3Cl2
and Ir(ppy)3 did not provide virtually any conversion to 2a.15
Application of these conditions to the more electron-rich
substrate 1b resulted initially in a significant erosion of the
yield (entry 6). Nonetheless, re-optimisation of the reductant
to substrate ratio (entry 7) and concentration (entry 8) brought
the yield back to satisfactory levels (91%, entry 8). As expected,
control experiments in the absence of the Ir photocatalyst
(entry 9) or in the dark (entry 10) showed that both are
essential for the reaction to proceed. It is worth noting that the
reduced form of Ir1 is not predicted to be a strong enough
reductant to perform a SET to 1a (E½(Ir1) = ‒1.51 V (MeCN),3d
E½(1a) = -1.87 V, see below for further discussion).
Table 1. Summary of reaction optimisation.a
Entry Substrate Conditions Yield (%)b
1 1a 50 mM, Ir1 2.5 mol%, HE (1 equiv) 100
2 Ru(bpy)3Cl2 as the catalyst 1
3 Ir(ppy)3 as the catalyst 2
4 EtNiPr2 as the reductant 36
5 BNAH as the reductant 42
6 1b As entry 1 64
7 HE (2 equiv) 70
8 12.5 mM, HE (2 equiv) 91
9 1b No photocatalystc 1
10 Darkd 0
Ir1: [Ir(ppy)2(dtbbpy)]PF6. a Reactions run with substrate 1 (0.1 mmol), reductant
(see table) and catalyst (2.5 mol%) at RT in MeOH, following general procedure B
(see ESI). b Yield determined by 1H NMR using an internal standard. c Reaction
performed at 50 mM concentration of 1b. d Reaction performed at 25 mM
concentration of 1b.
Then, we set out to explore the substrate scope and limitations of
our method (Table 2). The photoreduction worked to high yields
with a wide variety of cinnamate derivatives bearing different
substituents on the aryl ring (2a-u). Electron-donating MeO group
was tolerated at all three possible positions (2b-d), as well as
electron-withdrawing CF3 (2e-g). Likewise, no steric influence on
reaction yield was apparent from substitution at the ortho- position
(2k-m). The reaction was compatible with halogen substituents (2h-
j, 2l-2n), although a lower yield for I (2n) may suggest a competing
reduction of the C‒I bond. In the case of Br, para-substituted
compound (2h) was only obtained in moderate yield, in contrast
with the good results for the ortho- and meta- analogues (60% and
97%, respectively, for 2j and 2i).
Table 2. Substrate scope and limitations.a
a Unless stated otherwise, reactions were run with substrate 1 (0.2 mmol,
0.025 M), HE (2 equiv) and catalyst Ir1 (2.5 mol%) following general procedure B
(see ESI). Yields are of isolated product unless otherwise noted. b NMR yield in
this case was significantly higher (89%). c Yield determined by 1H NMR using an
internal standard. d The ethyl ester was used in this case instead of methyl. e
Starting from the corresponding ethyleneglycol acetal. f MeCN was used as the
solvent.
The reaction tolerated unprotected alcohols (2p), carboxylic acids
(2r) and amines, even when containing free NH bonds (2t and 2u),
although phenol and aniline provided decreased yields (2o and 2s).
Aldehyde-substituted product 2q could be obtained by performing
the reaction with the corresponding ethyleneglycol acetal, which
was deprotected in situ during work-up.
Replacing the aryl group in the cinnamate ester structure for
heterocycles such as pyridyl or furyl resulted in low yields of product
(2v-w) and the reaction did not proceed at all in the absence of an
aromatic group (2x).
Page 4
Scheme 2. Mechanistic elucidation of the HAT-based photoreduction. a Experiments performed at 0.01 mM concentration of Ir1 in a quartz cuvette. b Conditions as in Table 2. c Reduction
potentials are in MeCN vs. saturated calomel electrode (SCE). 1a, [1aHα]• and [1aHβ]• are calculated by DFT (M06-2X/631+G(d,p), see ESI for details). d Relative energies respect of
independent starting materials (M06-2X/6-31+G(d,p), see ESI for details).
Moving away from the methyl cinnamate structure,
cinnamonitrile (2y) and coumarin (2aa) were both efficiently
reduced under our reaction conditions Finally, our method
worked well for the reduction of a tetrasubstituted alkene
(2ab), highlighting its robustness with respect to steric
hindrance.
To probe the mechanism of the reaction we decided to
perform a series of additional experiments, summarised in
Scheme 2. Firstly, Stern-Volmer quenching experiments
(Scheme 2A) showed fluorescence quenching by HE to
significantly outcompete quenching by substrate 1a (quencher
rate coefficient kq(HEH)/kq(1a) ≈ 13).16 This confirmed our
expectation that the reaction likely starts with a reductive
quenching of the excited catalyst [Ir1]* by HE to give the
reduced form of the catalyst [Ir1]‒ together with formation of
the Hantzsch ester radical cation [HE]•+.17
Then, we explored the use of deuterium labelling (Scheme 2B)
to assess the possibility of the reduction proceeding through a
sequence of electron and proton transfer events, as previously
proposed for related alkene photoreductions.4,5,10 The
photoreduction of 1a using MeOD instead of MeOH led to
formation of product 2a with 9% incorporation of deuterium
at the β-position and 44% at α (out of an expected maximum
of 50%). Conversely, the same reaction in MeOH with 4,4-d2-
HE resulted in 2a with 25% deuteration exclusively at α. These
results are in clear contrast with those previously reported for
reductions based on consecutive electron and proton
transfer.18 The extent of deuterium incorporation in these
experiments combined with the marked regioselectivity
strongly supports a mechanism where the α-H would be
incorporated by a HAT from a HE derivative, while the β-H
would come from the solvent. The incomplete deuteration
may be attributed to adventitious H2O, and some extent of H/D
exchange between the reagents and solvent during the
reaction.
With these data in hand, we contemplated two distinct
possible mechanisms –either: (a) The reaction is initiated by
SET to give, after protonation, an intermediate radical species,
which is in turn transformed into the final product 2 by HAT
from [HE]•+; or (b) the reaction is initiated by HAT from [HE]•+
to 1a, giving an intermediate radical species, which is then
transformed into 2a by consecutive SET and H+ transfer.
Interestingly, as mentioned above, while the role of [HE]•+ as a
H atom donor is frequently proposed in the literature, its
involvement is always mentioned in termination steps,
reacting with another radical intermediate (i.e. type a
mechanisms).12 Mechanisms of the type b where [HE]•+
transfers a H atom to a ground state, closed shell species are
absent from the literature. However, comparison of the
reduction potentials suggests that the reduced catalyst [Ir1]‒ is
Page 5
not a strong enough reductant to engage in a direct SET with
cinnamate derivatives, which would be a requirement of type
a mechanisms (reduction potentials in MeCN referenced to
SCE: E½(Ir1) = ‒1.54 V,3d E½(1a) = ‒1.87 V,19 Scheme 2C). To assess
the feasibility of type b mechanism, we resourced to DFT
modelling of the reaction (M06-2X/6-31+G(d,p), see ESI for
details).20 Pleasingly, we could find transition states for the
HAT step from [HE]•+ to either position α or β of 1a, both of
which are predicted to be readily accessible energetically (ΔG‡
= 14.9 and 19.0 kcal/mol for α and β, respectively, Scheme 2D).
Moreover, computed reduction potentials of the resulting
radicals to their corresponding carbanions were consistent
with their easy reduction by [Ir1]‒ (E½(1aHα) = ‒1.43 V, Scheme
2C).21,22
All this evidence, thus, provides support for a mechanism as
depicted in Scheme 2E, where excitation of Ir1 by visible light
enables it to oxidise HE to its radical cation [HE]•+.
Subsequently, [HE]•+ transfers a H atom to the alkene 1a giving
place to a benzylic radical [1aHα]•, which is finally further
reduced by SET from [Ir1]‒ and protonated to give the reduced
product 2a.
Conclusions
In conclusion, we have developed a simple method for the
photoreduction of olefins which enables the use of moderately
electron-deficient, synthetically meaningful substrates with
good functional group compatibility. Our mechanistic
investigations support a hydrogen atom transfer to the
substrate as the key step which enables the reduction to
proceed without requiring the generation of a very highly
reducing medium. We believe this unprecedented mode of
substrate activation offers new opportunities for photoredox
transformations by enabling the generation of radical
intermediates that otherwise are not easily accessible.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We gratefully acknowledge Queen Mary University of London
for partially funding this research. NAL thanks the European
Commission for an MSCA fellowship (Project LIONCAT). JMO
thanks QMUL for a PhD scholarship. This research utilised
Queen Mary's Apocrita HPC facility, supported by QMUL
Research-IT. http://doi.org/10.5281/zenodo.438045.
Notes and references
1 J. A. Kent, T. V. Bommaraju and S. D. Barnicki, Eds., Handbook of Industrial Chemistry and Biotechnology, Springer International Publishing, 2017.
2 (a) D. Wang and D. Astruc, Chem. Rev., 2015, 115, 6621–6686; (b) L. Lloyd, in Fundamental and Applied Catalysis, eds. M. Twigg and M. Spencer, Springer US, 2011, pp. 73–117.
3 (a) R. C. McAtee, E. J. McClain and C. R. J. Stephenson, Trends Chem., 2019, 1, 111–125; (b) L. Marzo, S. K. Pagire, O. Reiser and B. König, Angew. Chem. Int. Ed., 2018, 57, 10034–10072; (c) C. R. J. Stephenson, T. P. Yoon and D. W. C. MacMillan, Eds., Visible Light Photocatalysis in Organic Chemistry, Wiley-VCH, Weinheim, 2018; (d) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363.
4 (a) C. Pac, Y. Miyauchi, O. Ishitani, M. Ihama, M. Yasuda and H. Sakurai, J. Org. Chem., 1984, 49, 26–34; C. Pac, M. Ihama, M. Yasuda, Y. Miyauchi and H. Sakurai, J. Am. Chem. Soc., 1981, 103, 6495–6497.
5 Similarly, a few cases of formal hydrogenation of alkylidene malonates have been reported: (a) R. C. Betori and K. A. Scheidt, ACS Catal., 2019, 9, 10350–10357. (b) R. C. Betori, B. R. McDonald and K. A. Scheidt, Chem. Sci., 2019, 10, 3353–3359; (c) B. R. McDonald and K. A. Scheidt, Org. Lett., 2018, 20, 6877–6881.
6 Selected papers: (a) E. Speckmeier, P. J. W. Fuchs and K. Zeitler, Chem. Sci., 2018, 9, 7096–7103; (b) T. Rossolini, J. A. Leitch, R. Grainger and D. J. Dixon, Org. Lett., 2018, 20, 6794–6798; (c) K. N. Lee, Z. Lei and M. Y. Ngai, J. Am. Chem. Soc., 2017, 139, 5003–5006; (d) M. Nakajima, E. Fava, S. Loescher, Z. Jiang and M. Rueping, Angew. Chem. Int. Ed., 2015, 54, 8828–8832; (e) L. J. Rono, H. G. Yayla, D. Y. Wang, M. F. Armstrong and R. R. Knowles, J. Am. Chem. Soc., 2013, 135, 17735–17738.
7 Y. Nakano, M. J. Black, A. J. Meichan, B. A. Sandoval, M. M. Chung, K. F. Biegasiewicz, T. Zhu and T. K. Hyster, Angew. Chem. Int. Ed., 2020, DOI: 10.1002/anie.202003125.
8 For a review on multi-photon excitation, see: (a) F. Glaser, C. Kerzig and O. S. Wenger, Angew. Chem. Int. Ed., 2020, 2–21. For selected recent papers, see: (b) M. Giedyk, R. Narobe, S. Weiß, D. Touraud, W. Kunz and B. König, Nat. Catal., 2020, 3, 40–47; (c) T. U. Connell, C. L. Fraser, M. L. Czyz, Z. M. Smith, D. J. Hayne, E. H. Doeven, J. Agugiaro, D. J. D. Wilson, J. L. Adcock, A. D. Scully, D. E. Gómez, N. W. Barnett, A. Polyzos and P. S. Francis, J. Am. Chem. Soc., 2019, 141, 17646–17658; (d) C. Kerzig and O. S. Wenger, Chem. Sci., 2019, 10, 11023–11029.
9 (a) N. G. W. Cowper, C. P. Chernowsky, O. P. Williams and Z. K. Wickens, J. Am. Chem. Soc., 2020, 142, 2093–2099; (b) H. Kim, H. Kim, T. H. Lambert and S. Lin, J. Am. Chem. Soc., 2020, 142, 2087–2092.
10 (a) T. Horngren, M. S. Taylor, M. Czyz and A. Polyzos, 2020, ChemRxiv: 10.26434/chemrxiv.12235928.v1. For a related pioneering report, see: (b) D. R. Arnold and A. J. Maroulis, J. Am. Chem. Soc., 1977, 99, 7355–7356.
11 (a) L. Capaldo, L. L. Quadri and D. Ravelli, Green Chem. 2020, DOI: 10.1039/D0GC01035A; (b) L. Capaldo and D. Ravelli, Eur. J. Org. Chem., 2017, 2056–2071;
12 Selected examples: (a) K. A. Margrey and D. A. Nicewicz, Acc. Chem. Res., 2016, 49, 1997-2006; (b) D. A. Nicewicz and D. S. Hamilton, Synlett, 2014, 25, 1191–1196. For selected papers, see: (c) J. Dong, X. Wang, Z. Wang, H. Song, Y. Liu and Q. Wang, Chem. Commun., 2019, 55, 11707–11710; (d) C. P. Seath, D. B. Vogt, Z. Xu, A. J. Boyington and N. T. Jui, J. Am. Chem. Soc., 2018, 140, 15525–15534; (e) S. Sumino, M. Uno, T. Fukuyama, I. Ryu, M. Matsuura, A. Yamamoto and Y. Kishikawa, J. Org. Chem., 2017, 82, 5469–5474; (f) C. Wang, K. Harms and E. Meggers, Angew. Chem. Int. Ed., 2016, 55, 13495–13498.
Page 6
13 (a) P. Z. Wang, J. R. Chen and W. J. Xiao, Org. Biomol. Chem., 2019, 17, 6936–6951; (b) J. Hu, J. Wang, T. H. Nguyen and N. Zheng, Beilstein J. Org. Chem., 2013, 9, 1977–2001; (c) Y. Nakano, K. F. Biegasiewicz and T. K. Hyster, Curr. Opin. Chem. Biol., 2019, 49, 16–24.
14 HE has also been applied as a hydride donor in organocatalytic transfer hydrogenation reactions: (a) C. Zheng and S. L. You, Chem. Soc. Rev., 2012, 41, 2498–2518; (b) S. G. Ouellet, A. M. Walji and D. W. C. Macmillan, Acc. Chem. Res., 2007, 40, 1327–1339.
15 E/Z isomerisation of 1a was observed as a significant side-reaction, particularly with Ir(ppy)3. For related isomerisations, see: ; (a) J. B. Metternich and R. Gilmour, J. Am. Chem. Soc., 2015, 137, 11254–11257; (b) K. Singh, S. J. Staig and J. D. Weaver, J. Am. Chem. Soc., 2014, 136, 5275–5278.
16 The degree of quenching by 1a was very small, being difficult to differentiate from experimental error. It is reasonable to assume that at least some quenching takes place, which was consistent with the observation of Z-1a in small amounts by 1H NMR at early stages of the reaction (see ESI for details).
17 This has been frequently proposed in related transformations: (a) A. Trowbridge, D. Reich and M. J. Gaunt, Nature, 2018, 561, 522–527; (b) L. Qi and Y. Chen, Angew. Chem. Int. Ed., 2016, 55, 13312–13315.
18 For example in ref. 4(a), photoreduction of dimethyl fumarate by BNAH catalysed by Ru1 in MeOD gave 59% d2- and 37% d-dimethyl succinate, with only 4% non-deuterated product, while reaction with 4,4-d2-BNAH in MeOH gave 6% mono- and 94% non-deuterated product. Similar numbers were reported for dimethyl maleate.
19 Calculated value. Compare to ‒1.98 V experimental in MeOH, see: T. Shiragami, H. Ankyu, S. Fukami, C. Pac, S. Yanagida, H. Mori and H. Fujita, J. Chem. Soc. Faraday Trans., 1992, 88, 1055–1061.
20 Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215-241.
21 Calculated reduction potential for the isomeric radical [1aHβ]•, obtained from H addition on the β position, was E½(1aHβ) = -0.72 V.
22 Method for calculating reduction potentials: H. G. Roth, N. A. Romero and D. A. Nicewicz, Synlett, 2016, 27, 714-723.
Page 7
download fileview on ChemRxivHAT reduction ChemRxiv v2.pdf (523.61 KiB)
Page 8
1
Efficient reduction of electron-deficient alkenes through
photoinduced hydrogen atom transfer
Supporting Information
Natalia A. Larionova, Jun Miyatake Ondozabal and Xacobe C. Cambeiro*
School of Biological and Chemical Sciences, Queen Mary University of London,
E1 4NS, London, United Kingdom. E-mail: [email protected]
Table of content
General Considerations .......................................................................................................... 2
Synthesis of Starting Material ................................................................................................ 4
Photoreduction and Characterization ................................................................................... 11
NMR Yields ......................................................................................................................... 18
Mechanistic Studies ............................................................................................................. 26
Computational Details ......................................................................................................... 28
References ............................................................................................................................ 37
Spectra.................................................................................................................................. 40
Page 9
2
General Considerations
All experiments were performed under inert atmosphere in dry microwave vials.
Anhydrous solvents were purchased or collected from a MBRAUN SPS-800 solvent
purification system. Commercially available starting materials and photocatalysts were used as
purchased.
The photocatalytic reactions were performed in a tin container with aluminium foil and
strips of LED lights wrapped around the inner walls. The reaction tubes were held in place
using a plastic rack that sat at the top of the reactor. A thermometer was placed in a tube filled
with either water or paraffin oil at the centre of the reactor to monitor the temperature. The
reactor temperature was kept at room temperature by passing a stream of compressed air. The
top of the reactor was covered with a lid fashioned with cardboard/foil and holes were made
where appropriate (Figure S1).
Figure S1. Photographs of the reactor used for photocatalytic reactions.
LED strips, connectors, power supply and wireless IR remote were purchased from
superbrightleds.com. Specifications of the LED strips: Outdoor RGBW LED strip lights –
weatherproof 12V LED tape light with white and multicolour LEDs – 380 Lumens/ft. – natural
white (WFLS-RGBNW60X3-WHT).
NMR data was obtained from a Bruker Avance III 400 spectrometer. All spectra are
recorded at 303K and reported in ppm unless otherwise stated. Chemical shifts ( values) are
reported in ppm. Coupling constants are reported in Hz and multiplicities abbreviated as
follows: s (singlet); d (doublet); t (triplet); q (quartet); br (broad); m (multiplet); dd (doublet of
doublets); ddd (doublet of doublet of doublets); dt (doublet of triplets); td (triplet of doublets);
app (apparent).
GC-MS analyses were performed using Agilent Technologies GC6890N/MS5973. An
Agilent J&W HP-5MS capillary column of 30 m x 0.32 mm, film thickness 0.25 µm was used.
The oven temperature was programmed to start at 40 ℃ (hold for 1 minute) and increase at
Page 10
3
20 ℃ min-1 to 300 ℃ (hold for 5 minutes). The carrier gas was helium at a flow rate of 2.0 mL
min-1. Data was processed using MSD ChemStation Data Analysis software.
High-resolution mass spectra were obtained using an Acquity H Class UPLC
instrument interfacing though an electrospray ionization (ESI) LockSpray source to a High
Definition Mass Spectrometer Synapt G2Si, both from Waters Corporation. The sample was
separated by reverse phase using a Waters BEH C18 column (2.1x 50 mm, 1.7 µm). The mobile
phases were water + 0.1% formic acid (A), and acetonitrile + 0.1% formic acid (B) using a
gradient of 2% B to 98% B in 8 minutes. The column temperature was 60 ℃. Data were
acquired and analysed using MassLynx v4.1.
IR spectra were recorded on a Perkin Elmer Spectrum 65 FT-IR spectrometer or an
Agilent Technologies Cary 630 FT-IR and are reported in frequency of absorption at the peak
maximum (cm–1). Data were acquired and analysed using Perkin Elmer Spectrum
v10.00.00.0018.
Page 11
4
Synthesis of Starting Material
General Procedure A for the Synthesis of Methyl Cinnamate Derivatives1
Monomethyl malonate was synthesised from commercially available starting materials
following reported methodology.2 To a solution of malonic acid monomethyl ester (0.89 g; 7.5
mmol) in pyridine (2 mL) containing a catalytic amount of piperidine (0.045 mL) was added
5.0 mmol of the aldehyde derivative. The reaction mixture was refluxed until TLC analysis
indicated total consumption of the aromatic aldehyde (2-6 hours). Then, after cooling at room
temperature, water (ca. 10 mL) was added, the pH was adjusted to 7.0 with 20% aq. HCl and
the aqueous phase was extracted with ethyl ether (3*20 mL). The organic extracts were joined,
dried over anhydrous sodium sulphate and concentrated under reduced pressure.
(E)-methyl p-methoxycinnamate (1b)
Compound 1b was prepared following general procedure A. The
crude product was purified by column chromatography (silica gel,
n-hexane:EtOAc, 9:1) to afford a white solid, 48% yield. 1H NMR
(400 MHz, CDCl3) δ 7.65 (d, J = 16.0 Hz, 1H), 7.48 (d, J = 8.8 Hz, 2H), 6.91 (d, J = 8.8 Hz,
2H), 6.31 (d, J = 16.0 Hz, 1H), 3.84 (s, 3H), 3.79 (s, 3H). 13C NMR (101 MHz, CDCl3) δ
167.92, 161.55, 144.68, 129.87, 127.29, 115.43, 114.48, 55.53, 51.72. The analytic data are in
agreement with the literature.3
(E)-methyl m-methoxycinnamate (1c)
Compound 1c was prepared following general procedure A. The crude
product was purified by column chromatography (silica gel, n-
hexane:EtOAc, 9:1) to afford a colourless oil, 65% yield. 1H NMR (400
MHz, CDCl3) δ 7.66 (d, J = 16.0 Hz, 1H), 7.30 (t, J = 7.9 Hz, 1H), 7.12
(d, J = 7.6 Hz, 1H), 7.05-7.04 (m, 1H), 6.94 (dd, J = 8.1, 2.3 Hz, 1H), 6.43 (d, J = 16.0 Hz,
1H), 3.83 (s, 3H), 3.81 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 167.44, 160.0, 144.87, 135.84,
129.97, 120.84, 118.19, 116.22, 113.08, 55.36, 51.78. The analytic data are in agreement with
the literature.4
Page 12
5
(E)-methyl o-methoxycinnamate (1d)
Compound 1d was prepared following general procedure A. The crude
product was purified by column chromatography (silica gel, n-
hexane:EtOAc, 9:1) to afford a colourless oil, 88% yield. 1H NMR
(400 MHz, CDCl3) δ 8.00 (d, J = 16.2 Hz, 1H), 7.51 (dd, J = 7.7, 1.7
Hz, 1H), 7.35 (ddd, J = 8.4, 7.5, 1.7 Hz, 1H), 6.98 – 6.94 (m, 1H), 6.92 (d, J = 8.3 Hz, 1H),
6.53 (d, J = 16.2 Hz, 1H), 3.89 (s, 3H), 3.80 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 13C NMR
(101 MHz, CDCl3) δ 13C NMR (101 MHz, CDCl3) δ 167.87, 158.33, 140.23, 131.50, 128.85,
123.32, 120.68, 118.26, 111.14, 55.41, 51.52. The analytic data are in agreement with the
literature.5
(E)-methyl p-trifluoromethyl cinnamate (1e)
Compound 1e was prepared following general procedure A. The
crude product was purified by column chromatography (silica gel,
n-hexane:EtOAc, 9:1) to afford a white solid, 25% yield. 1H NMR
(400 MHz, CDCl3) δ 7.70 (d, J = 16.1 Hz, 1H), 7.66 – 7.61 (m, 4H), 6.51 (d, J = 16.0 Hz, 1H),
3.83 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 166.98, 143.13, 137.91, 132.99 (q, J = 32.5 Hz),
128.33, 126.02 (q, J = 3.8 Hz), 123.96 (q, J = 272.4 Hz), 120.53, 52.06. 19F NMR (377 MHz,
CDCl3) δ -62.90. The analytic data are in agreement with the literature.6
(E)-methyl m-trifluoromethyl cinnamate (1f)
Compound 1f was prepared following general procedure A. The crude
product was purified by column chromatography (silica gel, n-
hexane:EtOAc, 9:1) to afford a white solid, 41% yield. 1H NMR (400
MHz, CDCl3) δ 7.75 (s, 1H), 7.72-7.68 (m, 2H), 7.63 (d, J = 7.7 Hz,
1H), 7.51 (t, J = 7.8 Hz, 1H), 6.50 (d, J = 16.1 Hz, 1H), 3.82 (s, 3H). 13C NMR (101 MHz,
CDCl3) 166.98, 143.14, 135.32, 131.5 (q, J = 32.8 Hz),131.18, 129.58, 126.79 (q, J = 3.7 Hz),
124.73 (q, J = 3.8 Hz), 123.91 (q, J = 273.0 Hz), 119.93, 51.99. 19F NMR (377 MHz, CDCl3)
δ -62.96. The analytic data are in agreement with the literature.5,7
Page 13
6
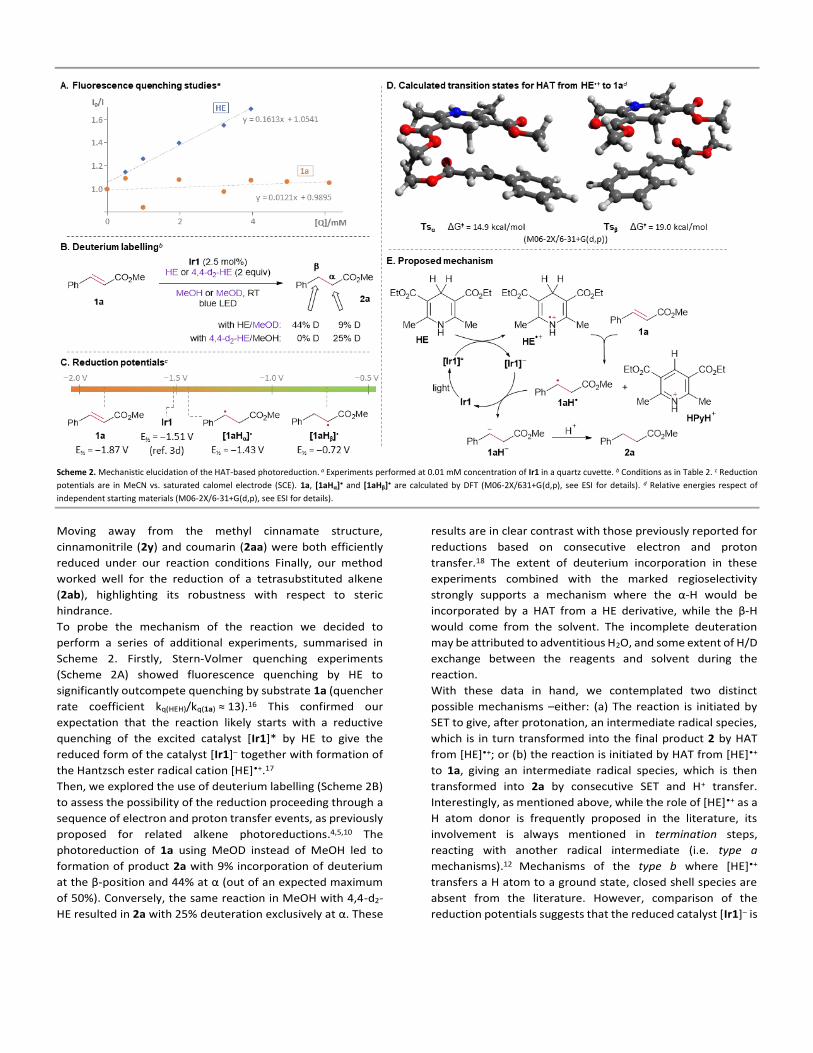
(E)-methyl o-trifluoromethyl cinnamate (1g)
Compound 1g was prepared following general procedure A. The crude
product was purified by column chromatography (silica gel, n-
hexane:EtOAc, 9:1) to afford a colourless oil, 33% yield. 1H NMR (400
MHz, CDCl3) δ 8.06 (dq, J = 15.8, 2.2 Hz, 1H), 7.71-7.69 (m, 2H), 7.57 (t, J = 7.6 Hz, 1H),
7.48 (t, J = 7.6 Hz, 1H), 6.41 (d, J = 15.8 Hz, 1H), 3.83 (s, 3H). 13C NMR (101 MHz, CDCl3)
δ 166.66, 140.47 (d, J = 2.2 Hz), 133.53 (d, J = 1.8 Hz), 132.24, 129.73, 129.17 (q, J = 30.4
Hz), 128.05, 126.32 (q, J = 5.6 Hz), 124.05 (q, J = 273 Hz), 122.34, 52.07. The analytic data
are in agreement with the literature.8 19F NMR (377 MHz, CDCl3) δ -58.98.
(E)-methyl o-bromomethyl cinnamate (1j)
Compound 1j was prepared following general procedure A. The crude
product was purified by column chromatography (silica gel, n-
hexane:EtOAc, 9:1) to afford a yellow oil, 83% yield. 1H NMR (400
MHz, CDCl3) δ 8.05 (d, J = 15.9 Hz, 1H), 7.62-7.58 (m, 2H), 7.34-7.30 (m, 1H), 7.24 – 7.20
(td, J = 7.7, 1.6 Hz, 1H), 6.39 (d, J = 15.9 Hz, 1H), 3.82 (s, 3H). 13C NMR (101 MHz, CDCl3)
δ 166.71, 143.07, 134.39, 133.39, 131.21, 127.73, 127.71, 125.29, 120.63, 51.81. The analytic
data are in agreement with the literature.9
(E)-methyl o-methylcinnamate (1k)
Compound 1k was prepared following general procedure A. The crude
product was purified by column chromatography (silica gel, n-
hexane:EtOAc, 9:1) to afford a colourless oil, 90% yield. 1H NMR (400
MHz, CDCl3) δ 7.98 (d, J = 15.9 Hz, 1H), 7.56 – 7.54 (m, 1H), 7.30 – 7.19 (m, 3H), 6.36 (d,
J = 15.9 Hz, 1H), 3.81 (s, 3H), 2.44 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 167.43, 142.51,
137.63, 133.36, 130.79, 130.04, 126.40, 126.35, 118.84, 51.64, 19.75. The analytic data are in
agreement with the literature.10
(E)-methyl o-fluoromethyl cinnamate (1l)
Compound 1l was prepared following general procedure A. The crude
product was purified by column chromatography (silica gel, n-
hexane:EtOAc, 9:1) to afford a colourless oil, 89% yield. 1H NMR (400
MHz, CDCl3) δ 7.82 (d, J = 16.2 Hz, 1H), 7.53 (td, J = 7.6, 1.7 Hz, 1H), 7.38 – 7.33 (m, 1H),
7.16 (td, J = 7.6, 0.9 Hz, 1H), 7.10 (ddd, J = 10.6, 8.3, 1.0 Hz, 1H), 6.54 (d, J = 16.3 Hz, 1H),
Page 14
7
3.82 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 167.25, 161.40 (d, J = 253.8 Hz), 137.5 (d, J =
3.2 Hz), 131.76 (d, J = 8.8 Hz), 129.10 (d, J = 2.8 Hz), 124.49 (d, J = 3.6 Hz), 122.50 (d, J =
11.9 Hz), 120.44 (d, J = 6.7 Hz), 116.23 (d, J = 22.3 Hz), 51.79. 19F NMR (377 MHz, CDCl3)
δ -114.42. The analytic data are in agreement with the literature.5,11
(E)-methyl o-chloromethyl cinnamate (1m)
Compound 1m was prepared following general procedure A. The crude
product was purified by column chromatography (silica gel, n-
hexane:EtOAc, 9:1) to afford a colourless oil, 67% yield. 1H NMR (400
MHz, CDCl3) δ 8.10 (d, J = 16.0 Hz, 1H), 7.62 (dd, J = 7.3, 2.2 Hz, 1H), 7.43-7.41 (m, 1H),
7.33 – 7.27 (m, 2H), 6.44 (d, J = 16.0 Hz, 1H), 3.83 (s, 3H). 13C NMR (101 MHz, CDCl3) δ
166.72, 140.40, 134.81, 132.51, 130.98, 130.05, 127.52, 127.01, 120.36, 51.68. The analytic
data are in agreement with the literature.5
(E)-methyl o-iodomethyl cinnamate (1n)
Compound 1n was prepared following general procedure A. The crude
product was purified by column chromatography (silica gel, n-
hexane:EtOAc, 9:1) to afford a yellow oil, 68% yield. 1H NMR (400
MHz, CDCl3) δ 7.90 (d*, J = 15.8 Hz, 1H), 7.90 (dd*, J = 8.0, 1.1 Hz, 1H), 7.56 (dd, J = 7.8,
1.6 Hz, 1H), 7.38-7.34 (m, 1H), 7.05 (td, J = 7.7, 1.6 Hz, 1H), 6.32 (d, J = 15.8 Hz, 1H), 3.83
(s, 3H). *Signals partially overlap. 13C NMR (101 MHz, CDCl3) δ 166.62, 147.85, 140.01,
137.73, 131.26, 128.58, 127.37, 120.81, 101.22, 51.88. The analytic data are in agreement with
the literature.12
(E)-methyl p-hydroxycinnamate (1o)
Compound 1o was prepared following general procedure A. The
crude product was purified by column chromatography (silica gel,
n-hexane:EtOAc, 9:1) to afford a white solid, 79% yield. 1H NMR
(400 MHz, CDCl3) δ 7.64 (d, J = 16.0 Hz, 1H), 7.44 (d, J = 8.6 Hz, 2H), 6.84 (d, J = 8.6 Hz,
2H), 6.31 (d, J = 16.0 Hz, 1H), 5.06-5.02 (m, 1H), 3.80 (s, 3H). 13C NMR (101 MHz, CDCl3)
δ 168.01, 157.69, 144.66, 130.12, 127.52, 116.02, 115.51, 51.80. The analytic data are in
agreement with the literature.13,14
Page 15
8
(E)-methyl p-(hydroxymethyl)cinnamate (1p)
Compound 1p was prepared according to the procedure reported
in the literature from commercially available methyl 4-
formylcinnamate.15 The crude product was purified by column
chromatography (silica gel, n-hexane:EtOAc, 3:1) and recrystallized from benzene to afford a
white solid, 78% yield. 1H NMR (400 MHz, CDCl3) δ 7.69 (d, J = 16.0 Hz, 1H), 7.52 (d, J =
8.1 Hz, 2H), 7.39 (d, J = 8.1 Hz, 2H), 6.44 (d, J = 16.0 Hz, 1H), 4.72 (d, J = 5.8 Hz, 2H), 3.81
(s, 3H). OH peak was not observed. 13C NMR (101 MHz, CDCl3) δ 167.59, 144.61, 143.35,
133.88, 128.44, 127.43, 117.90, 65.00, 51.87. The analytic data are in agreement with the
literature.16
(E)-methyl p-carboxylcinnamate (1q)
Compound 1q was prepared following general procedure A. The
crude product was purified by column chromatography (silica
gel, n-hexane:EtOAc, 3:1) to afford a white solid, 35% yield. 1H
NMR (400 MHz, CDCl3) δ 8.12 (d, J = 8.4 Hz, 2H), 7.73 (d, J =
16.1 Hz, 1H), 7.62 (d, J = 8.4 Hz, 2H), 6.55 (d, J = 16.1 Hz, 1H), 3.83 (s, 3H). 13C NMR (101
MHz, DMSO-d6) δ 166.77, 166.38, 143.24, 138.06, 132.08, 129.70, 128.43, 120.14, 51.62.
The analytic data are in agreement with the literature.17,18
(E)-methyl p-(1,3-dioxolan-2-yl)cinnamate (1r)
Compound 1r was prepared following general procedure A from
4-(1,3-dioxolan-2-yl)benzaldehyde synthesised according to the
literature.19 The crude product was purified by column
chromatography (silica gel, n-hexane:EtOAc, 5:1) to afford a
white solid, 57% yield. 1H NMR (400 MHz, CDCl3) δ 7.70 (d, J = 16.0 Hz, 1H), 7.54 (d, J =
8.3 Hz, 2H), 7.50 (d, J = 8.3 Hz, 2H), 6.45 (d, J = 16.0 Hz, 1H), 5.83 (s, 1H), 4.14 – 4.03 (m,
4H), 3.81 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 167.46, 144.45, 140.22, 135.35, 128.22,
127.14, 118.51, 103.32, 65.51, 51.87. HRMS (ESI) m/z calc. for C13H14O4, [MH+]: 235.0970,
found: 235.1001. IR (neat): 2950, 2887, 1706, 1171, 823 cm-1.
Page 16
9
(E)-methyl p-(n-butyl)aminomethylcinnamate (1t)
Compound 1t was prepared according to the procedure
reported in the literature (a yellow oil, 81% yield).20 1H
NMR (400 MHz, CDCl3) δ 7.65 (d, J = 16.1 Hz, 1H),
7.44 (d, J = 8.0 Hz, 2H), 7.30 (d, J = 8.0 Hz, 2H), 6.38 (d, J = 16.1 Hz, 1H), 3.75 (s, 5H), 2.58
(t, J = 7.1 Hz, 2H), 1.62 (br s, 1H, NH), 1.49 – 1.42 (m, 2H), 1.36 – 1.27 (m, 2H), 0.87 (t, J =
7.3 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 167.48, 144.68, 143.25, 133.04, 128.56, 128.15,
117.27, 53.69, 51.62, 49.19, 32.22, 20.46, 14.01. HRMS (ESI) m/z calc. for C15H21NO2,
[MH+]: 248.1651, found: 248.1637. IR (neat): 2957, 2930, 1717, 1637, 1166 cm-1.
(E)-methyl p-dimethylaminomethylcinnamate (1u)
Compound 1u was prepared according to the procedure reported
in the literature (a yellow oil, 65% yield).21 1H NMR (400 MHz,
CDCl3) δ 7.68 (d, J = 16.0 Hz, 1H), 7.48 (d, J = 8.0 Hz, 2H),
7.33 (d, J = 8.0 Hz, 2H), 6.42 (d, J = 15.9 Hz, 1H), 3.80 (s, 3H), 3.42 (s, 2H), 2.24 (s, 6H). 13C
NMR (101 MHz, CDCl3) δ 167.50, 144.70, 141.60, 133.29, 129.55, 128.09, 117.42, 64.02,
51.66, 45.42. HRMS (ESI) m/z calc. for C13H17NO2, [MH+]: 220.1338, found: 220.1360. IR
(neat): 2950, 2819, 2770, 1718, 1636, 1165 cm-1.
(E)-methyl 3-(pyridin-2-yl)acrylate (1v)
Compound 1v was prepared following general procedure A. The crude
product was purified by column chromatography (silica gel, n-
hexane:EtOAc, 9:1) to afford a colourless oil, 66% yield. 1H NMR (400
MHz, CDCl3) δ 8.66-8.64 (m, 1H), 7.73 – 7.67 (m, 2H), 7.42 (d, J = 7.8 Hz, 1H), 7.28-7.25
(m, 1H), 6.93 (d, J = 15.7 Hz, 1H), 3.82 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 167.37,
153.06, 150.32, 143.70, 136.89, 124.39, 124.37, 122.12, 51.99. The analytic data are in
agreement with the literature.22
(E)-methyl 3-(thiophen-2-yl)acrylate (1w)
Compound 1w was prepared following general procedure A. The crude
product was purified by column chromatography (silica gel, n-
hexane:EtOAc, 9:1) to afford a yellow oil, 81% yield. 1H NMR (400
MHz, CDCl3) δ 7.50 – 7.46 (m, 1H), 7.43 (d, J = 15.7 Hz, 1H), 6.61 (d, J = 3.4 Hz, 1H), 6.47-
6.46 (m, 1H), 6.32 (d, J = 15.8 Hz, 1H), 3.79 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 167.64,
Page 17
10
151.02, 144.88, 131.34, 115.58, 114.92, 112.40, 51.79. The analytic data are in agreement with
the literature.3
(E)-2-Styrylpyridine (1z)
Compound 1z was prepared according to the procedure reported in the
literature.23 The crude product was purified by column chromatography
(silica gel, n-hexane:EtOAc, 95:5) to afford an off-white solid, 31% yield.
1H NMR (400 MHz, CDCl3) δ 8.62-8.60 (m, 1H), 7.68 – 7.62 (m, 2H), 7.60 – 7.57 (m, 2H),
7.40 – 7.36 (m, 3H), 7.32 – 7.28 (m, 1H), 7.18 (d*, J = 16 Hz, 1H), 7.16-7.13 (m*, 1H). *Signals
partially overlap. 13C NMR (101 MHz, CDCl3) δ 155.80, 149.84, 136.82, 136.67, 132.88,
128.87, 128.48, 128.12, 127.26, 122.24, 122.20. The analytic data are in agreement with the
literature.24
Page 18
11
Photoreduction and Characterization
General procedure B for photocatalytic reduction of alkenes
Substrate (0.2 mmol), Hantzsch ester (2 eq., 0.4 mmol) and [Ir(dtbbpy)(ppy)2]PF6 (2.5 mol%)
were weighted out into a microwave vial equipped with a magnetic stirrer. Dry methanol (8
mL) was added and the vial was quickly capped. The mixture was degassed with argon for 10
minutes and the septum was covered with parafilm. The vial was placed in a photoreactor and
irradiated under blue light for 16 hours. Thereafter, the mixture was transferred to a round
bottom flask, the vial washed with Et2O and the solvent was evaporated.
Work-up A: 5 mL of HCl in Et2O (1.0-1.7M) was added to the residue and filtered through a
packed layer of silica. The solution was neutralized with a saturated NaHCO3 solution and the
extracted organic layer washed with water. The organic phase was dried over MgSO4, filtered
and concentrated in vacuo.
Work-up B: 0.5 mL of HCl in Et2O (1.0-1.7M) was added to the residue and filtered through a
packed layer of silica. The solution was neutralized with NaHCO3 powder, filtered and
concentrated in vacuo.
Method for crude NMR Analysis: An internal standard, either 1,3,5-trimethoxybenzene (0.33
eq.) or 1-bromo-4-nitrobenzene (1 eq.) was dissolved with CDCl3 in a volumetric flask. 1 mL
of the stock solution was added to the crude mixture and the resulting sample was analysed by
NMR. Spectra were referenced to 1,3,5-trimethoxybenzene peak at 6.09 ppm as the residual
CDCl3 was hard to identify unless otherwise stated.25
Methyl 3-phenylpropanoate (2a)
Synthesised according to general procedure B and the crude mixture
obtained after the first evaporation was directly purified by flash column
chromatography (n-hexane/ethyl acetate, 98:2). The desired product was
obtained as a colourless oil (87% yield). 1H NMR (400 MHz, CDCl3) δ 7.32 – 7.28 (m, 2H),
7.23-7.20 (m, 3H), 3.68 (s, 3H), 2.98-2.94 (m, 2H), 2.66-2.62 (m, 2H). 13C NMR (101 MHz,
CDCl3) δ 173.46, 140.64, 128.63, 128.39, 126.39, 51.73, 35.83, 31.07. The observed results
are in accordance with the literature values.26
Page 19
12
Methyl 3-(4-methoxyphenyl)propionate (2b)
Synthesised according to general procedure B/work-up B and the
resulting oil was purified by flash column chromatography (n-
hexane/ethyl acetate, 95:5). The desired product was obtained as
a pale-yellow oil (61% yield). 1H NMR (400 MHz, CDCl3) δ 7.12 (d, J = 8.7 Hz, 2H), 6.83
(d, J = 8.7 Hz, 2H), 3.78 (s, 3H), 3.67 (s, 3H), 2.91-2.88 (m, 2H), 2.62-2.58 (m, 2H).
13C NMR (101 MHz, CDCl3) δ 173.55, 158.23, 132.73, 129.36, 114.06, 55.39, 51.71, 36.15,
30.24. The observed results are in accordance with the literature values.27
Methyl 3-(3-methoxyphenyl)propanoate (2c)
Synthesised according to general procedure B/work-up A and the
resulting oil was purified by flash column chromatography (n-
hexane/ethyl acetate, 95:5). The desired product was obtained as a pale-
yellow oil (85% yield). 1H NMR (400 MHz, CDCl3) δ 7.23-7.18 (m,
1H), 6.80-6.74 (m, 3H), 3.79 (s, 3H), 3.68 (s, 3H), 2.95-2.91 (m, 2H), 2.65-2.61 (m, 2H). 13C
NMR (101 MHz, CDCl3) δ 173.46, 159.87, 142.28, 129.64, 120.75, 114.20, 111.74, 55.29,
51.78, 35.76, 31.13. The observed results are in accordance with the literature values.28
Methyl 3-(2-methoxyphenyl)propanoate (2d)
Synthesised according to general procedure B/work-up A and the
desired product was obtained as a pale-yellow oil (98% yield). 1H NMR
(400 MHz, CDCl3) δ 7.20 (td, J = 7.8, 1.6 Hz, 1H), 7.15 (dd, J = 7.4,
1.5 Hz, 1H), 6.90-6.84 (m, 2H), 3.83 (s, 3H), 3.67 (s, 3H), 2.97-2.93 (m, 2H), 2.64-2.60 (m,
2H). 13C NMR (101 MHz, CDCl3) δ 173.95, 157.60, 130.03, 128.94, 127.72, 120.55, 110.33,
55.29, 51.61, 34.15, 26.25. The observed results are in accordance with the literature values.29
MS(EI) m/z 194.00 [M+].
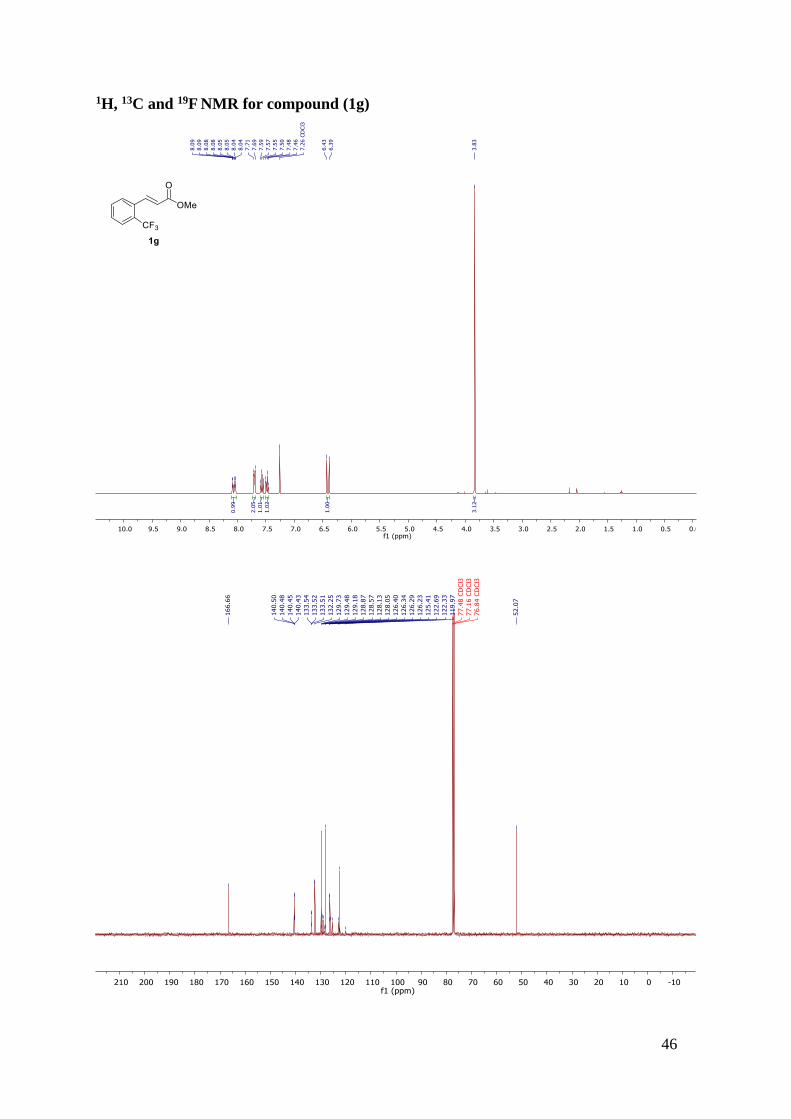
Methyl 3-(4-(trifluoromethyl)phenyl)propanoate (2e)
Synthesised according to general procedure B/work-up B and the
resulting oil was purified by flash column chromatography (n-
hexane/ethyl acetate, 95:5). The desired product was obtained as a
colourless oil (71% yield). 1H NMR (400 MHz, CDCl3) δ 7.54 (d, J = 8.0 Hz, 2H), 7.31 (d, J
= 8.0 Hz, 2H), 3.67 (s, 3H), 3.01 (t, J = 7.7 Hz, 2H), 2.65 (m, 2H). 13C NMR (101 MHz,
CDCl3) δ 173.01, 144.73 (d, J = 1.3 Hz), 128.85 (q, J = 32.4 Hz), 128.79, 125.59 (q, J = 3.8
Page 20
13
Hz), 124.39 (q, J = 271.9 Hz), 51.86, 35.31, 30.79. The observed results are in accordance with
the literature values.30 19F NMR (377 MHz, CDCl3) δ -62.43.
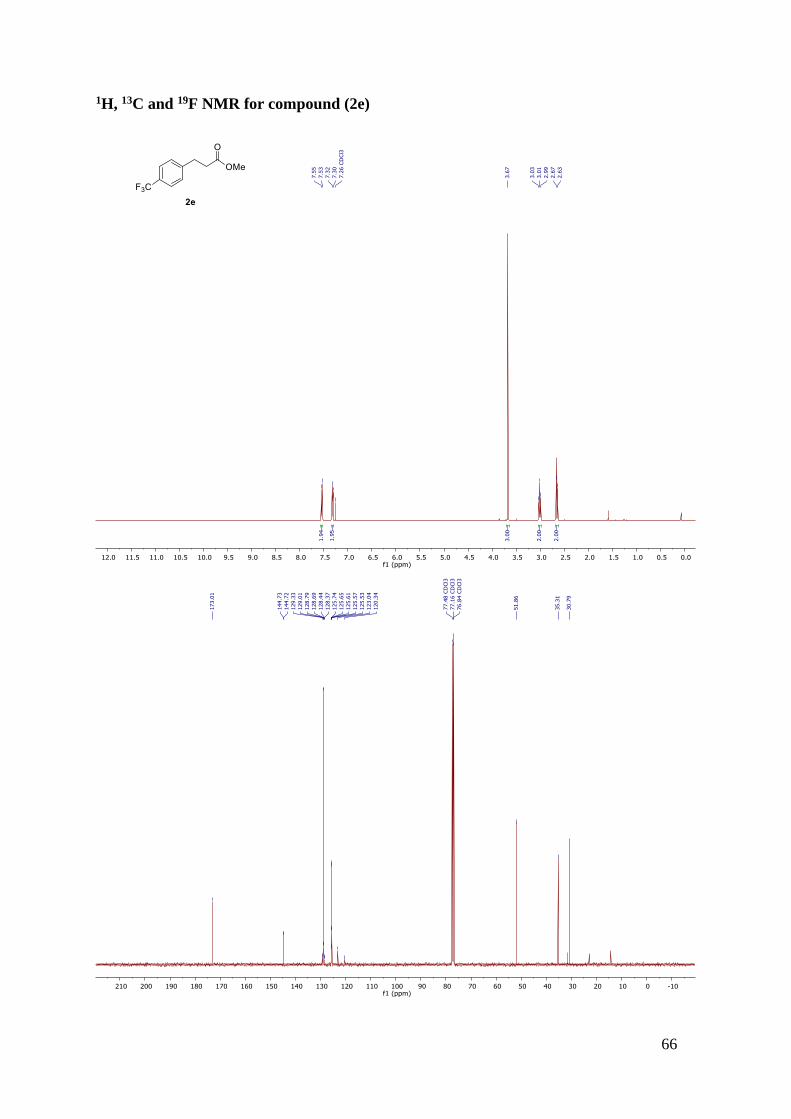
Methyl 3-(3-(trifluoromethyl)phenyl)propanoate (2f)
Synthesised according to general procedure B/work-up A and the
resulting oil was purified by flash column chromatography (n-
hexane/ethyl acetate, 95:5). The desired product was obtained as a pale-
yellow oil (86% yield). 1H NMR (400 MHz, CDCl3) δ 7.48-7.39 (m,
4H), 3.68 (s, 3H), 3.01 (t, J = 7.7 Hz, 2H), 2.68-2.64 (m, 2H). 13C NMR (101 MHz, CDCl3) δ
172.82, 141.53, 131.90 (d, J = 1.1 Hz), 130.99 (d, J = 32.0 Hz), 129.09, 125.20 (q, J = 3.8 Hz),
124.29 (q, J = 272.2 Hz), 123.37 (q, J = 3.8 Hz), 51.86, 35.48, 30.82. 19F NMR (377 MHz,
CDCl3) δ -62.64.The observed results are in accordance with the literature values.31
Methyl 3-(3-(trifluoromethyl)phenyl)propanoate (2g)
Synthesised according to general procedure B/work-up A and the
resulting oil was purified by flash column chromatography (n-
hexane/ethyl acetate, 95:5). The desired product was obtained as a
yellow oil (68% yield). 1H NMR (400 MHz, CDCl3) δ 7.63 (d, J = 7.9
Hz, 1H, HAr), 7.47 (t, J = 7.3 Hz, 1H, HAr), 7.36-7.30 (m, 2H, HAr), 3.70 (s, 3H, OMe), 3.16-
3.12 (m, 2H, CH2Ar), 2.65-2.61 (m, 2H, CH2CO). 13C NMR (101 MHz, CDCl3) δ 173.11
(C10), 139.37 (d, J = 1.5 Hz, C7), 132.11 (C5), 131.09 (C6), 128.74 (q*, J = 29.8 Hz, C2), 126.64
(C4), 126.27 (q, J = 5.8 Hz, C3), 124.67 (q*, J = 273.6 Hz, C1), 51.87 (C11), 35.77 (d, J = 1.0
Hz, C9), 27.96 (d, J = 1.8 Hz, C8). *Only the two central peaks of the quartet were visible. 19F
NMR (377 MHz, CDCl3) δ -59.82. MS (EI) m/z 233.05 [MH+], 232.05 [M+]. IR (neat): 1738,
1312, 1151, 1107 cm-1.
Methyl 3-(3-bromophenyl)propionate (2i)
Synthesised according to general procedure B and the crude mixture
obtained after the first evaporation was directly purified by flash column
chromatography (n-hexane/ethyl acetate, 95:5). The desired product was
obtained as a colourless oil (97% yield). 1H NMR (400 MHz, CDCl3) δ
7.35-7.33 (m, 2H), 7.18-7.11 (m, 2H), 3.68 (s, 3H), 2.92 (t, J = 7.7 Hz, 2H), 2.64-2.60 (m, 2H).
13C NMR (101 MHz, CDCl3) δ 173.09, 142.96, 131.55, 130.21, 129.60, 127.12, 122.67, 51.85,
35.49, 30.66. The observed results are in accordance with the literature values.32
Page 21
14
Methyl 3-(2-bromophenyl)propanoate (2j)
Synthesised according to general procedure B/work-up A and the
desired product was obtained as a yellow oil (68% yield). 1H NMR (400
MHz, CDCl3) δ 7.55-7.53 (m, 1H), 7.28 – 7.22 (m, 2H), 7.09 (ddd, J =
8.0, 6.5, 2.6 Hz, 1H), 3.69 (s, 3H), 3.10-3.06 (m, 2H), 2.69-2.65 (m, 2H). 13C NMR (101 MHz,
CDCl3) δ 173.18, 139.86, 133.04, 130.57, 128.23, 127.71, 124.48, 51.80, 34.04, 31.56. The
observed results are in accordance with the literature values.32
Methyl 3-(o-tolyl)propanoate (2k)
Synthesised according to general procedure B/work-up B and the
desired product was obtained as a pale-yellow oil (91% yield). 1H NMR
(400 MHz, CDCl3) δ 7.14 (s, 4H), 3.69 (s, 3H), 2.97-2.93 (m, 2H), 2.62-
2.58 (m, 2H), 2.33 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 173.60, 138.74, 136.10, 130.44,
128.62, 126.57, 126.27, 51.77, 34.56, 28.47, 19.35. The observed results are in accordance with
the literature values.33
Methyl 3-(2-fluorophenyl)propanoate (2l)
Synthesised according to general procedure B/work-up A and the
desired product was obtained as a pale-yellow oil (95% yield). 1H NMR
(400 MHz, CDCl3) δ 7.23 – 7.16 (m, 2H), 7.07 – 6.98 (m, 2H), 3.67 (s,
3H), 2.98 (t, J = 7.7 Hz, 2H), 2.66-2.62 (m, 2H). 13C NMR (101 MHz, CDCl3) δ 173.26,
161.30 (d, J = 245.2 Hz), 130.72 (d, J = 4.8 Hz), 128.24 (d, J = 8.1 Hz), 127.44 (d, J = 15.6
Hz), 124.19 (d, J = 3.6 Hz), 115.43 (d, J = 21.9 Hz), 51.76, 34.33 (d, J = 1.4 Hz), 24.73 (d, J =
2.7 Hz). The observed results are in accordance with the literature values.27 19F NMR (377
MHz, CDCl3) δ -118.59.
Methyl 3-(2-chlorophenyl)propanoate (2m)
Synthesised according to general procedure B/work-up A and the
desired product was obtained as a yellow oil (85% yield). 1H NMR
(400 MHz, CDCl3) δ 7.35-7.33 (m, 1H), 7.26 – 7.24 (m, 1H), 7.21 –
7.13 (m, 2H), 3.68 (s, 3H), 3.09-3.05 (m, 2H), 2.67-2.64 (m, 2H). The
observed results are in accordance with the literature values.34 13C NMR (101 MHz, CDCl3)
δ 173.27 (C9), 138.17 (C6), 134.10 (C1), 130.59 (C2), 129.71 (C3), 128.00 (C4), 127.05 (C5),
Page 22
15
51.79 (C10), 33.90 (C8), 29.09 (C7). MS (EI) m/z 198.00 [M+]. IR (neat): 2951, 1736, 1436,
1158, 1053, 751 cm-1.
Methyl 3-(4-(hydroxymethyl)phenyl)propanoate (2p)
Synthesised according to general procedure B/work-up B and the
desired product was obtained as yellow solids (81% yield).
1H NMR (400 MHz, CDCl3) δ 7.29 (d, J = 8.1 Hz, 2H), 7.19 (d,
J = 8.0 Hz, 2H), 4.65 (s, 2H), 3.67 (s, 3H), 2.97-2.93 (m, 2H), 2.64-2.60 (m, 2H). OH peak was
not observed. 13C NMR (101 MHz, CDCl3) δ 173.45, 140.05, 139.05, 128.57, 127.39, 65.17,
51.75, 35.77, 30.71.The observed results are in accordance with the literature values.35
4-(3-methoxy-3-oxopropyl)benzoic acid (2q)
Synthesised according to general procedure B and the crude
mixture obtained after the first evaporation was purified by flash
column chromatography DCM:EtOAc:AcOH, 95:4:1) and
concentrated. The residue was dissolved in EtOAc and washed
with brine. The organic phase was dried over MgSO4, filtered and concentrated in vacuo. The
desired product was obtained as a white solid (56% yield). 1H NMR (400 MHz, CDCl3) δ 8.04
(d, J = 8.3 Hz, 2H), 7.31 (d, J = 8.4 Hz, 2H), 3.68 (s, 3H), 3.03 (t, J = 7.7 Hz, 2H), 2.69-2.65
(m, 2H). 13C NMR (101 MHz, CDCl3) δ 173.09, 171.56, 147.08, 130.67, 128.65, 127.55,
51.90, 35.25, 31.09. The observed results are in accordance with the literature values.36
Methyl 3-(4-formylphenyl)propanoate (2r)
Synthesised according to general procedure B/work-up B using 1r
as the starting material and the desired product was obtained as a
pale-yellow oil (83% yield). 1H NMR (400 MHz, CDCl3) δ 9.98
(s, 1H), 7.81 (d, J = 8.2 Hz, 2H), 7.37 (d, J = 8.0 Hz, 2H), 3.68 (s,
3H), 3.04 (t, J = 7.7 Hz, 2H), 2.70-2.66 (m, 2H). 13C NMR (101 MHz, CDCl3) δ 192.01,
172.95, 147.91, 134.99, 130.18, 129.13, 51.87, 35.11, 31.12. The observed results are in
accordance with the literature values.37
Page 23
16
Methyl 3-(4-((butylamino)methyl)phenyl)propanoate (2t)
Synthesised according to general procedure B and the
crude mixture obtained after the first evaporation was
directly purified by flash column chromatography (n-
hexane/acetone, 9:1). The desired product was
obtained as a yellow oil (73% yield). 1H NMR (400 MHz, CDCl3) δ 7.23 (d, J = 8.0 Hz, 2H,
H7), 7.14 (d, J = 8.1 Hz, 2H, H8), 3.74 (s, 2H, H5), 3.66 (s, 3H, H13), 2.95-2.91 (m, 2H, H10),
2.63-2.59 (m, 4H, H4 and H11), 1.67 (br. s, 1H, NH), 1.53-1.45 (m, 2H, H3), 1.38-1.29 (m, 2H,
H2), 0.90 (t, J = 7.3 Hz, 3H, H1). 13C NMR (101 MHz, CDCl3) δ 173.45 (C12), 139.21 (C9),
138.54 (C6), 128.42 (C7), 128.39 (C8), 53.84 (C13), 51.69 (C4), 49.28 (C5), 35.83 (C11), 32.28
(C3), 30.70 (C10), 20.58 (C2), 14.11 (C1). HRMS (ESI) m/z calc. for C15H23NO2, [MH+]:
250.1807; found: 250.1831. IR (neat): 2958, 2930, 1738, 1438, 1155 cm-1.
Methyl 3-(4-((dimethylamino)methyl)phenyl)propanoate (2u)
Synthesised according to general procedure B and the crude
mixture obtained after the first evaporation was directly
purified by flash column chromatography (n-hexane/acetone,
9:1). The desired product was obtained as a yellow oil (67%
yield). 1H NMR (400 MHz, CDCl3) δ 7.21 (d, J = 8.1 Hz, 2H, H4), 7.14 (d, J = 8.1 Hz, 2H,
H5), 3.66 (s, 3H, H10), 3.38 (s, 2H, H2), 2.95-2.91 (m, 2H, H7), 2.64-2.60 (m, 2H, H8), 2.22 (s,
6H, H1). 13C NMR (101 MHz, CDCl3) δ 173.47 (C9), 139.4 (C6), 136.85 (C3), 129.41 (C4),
128.25 (C5), 64.13 (C2), 51.70 (C10), 45.41 (C1), 35.83 (C8), 30.72 (C7). HRMS (ESI) m/z calc.
for C13H19NO2, [MH+]: 222.1494; found: 222.1488. IR (neat): 2950, 2770, 1738, 1148, 1021
cm-1.
3-Phenylpropanenitrile (2y)
Synthesised according to general procedure B/work-up B and the desired
product was obtained as a yellow oil (83% yield).
1H NMR (400 MHz, CDCl3) δ 7.36 – 7.21 (m, 5H), 2.96 (t, J = 7.4 Hz, 2H), 2.62 (t, J = 7.4
Hz, 2H). Spectrum referenced to diethyl ether (Et2O) peak at 1.21 ppm as the residual CDCl3
was hard to identify.38
13C NMR (101 MHz, CDCl3) δ 138.19, 129.04, 128.40, 127.40, 119.25, 31.75, 19.51.
The observed results are in accordance with the literature values.39
Page 24
17
Chroman-2-one (2aa)
Synthesised according to general procedure B using acetonitrile as the solvent
and the crude mixture obtained after the first evaporation was directly purified
by flash column chromatography (n-hexane/acetone, 95:5). The desired product was obtained
as a yellow oil (84% yield, NMR purity = 81%; 16% of Hantzsch pyridine, 4% of starting
material and 1% of Et2O). 1H NMR (400 MHz, CDCl3) δ 7.28-7.24 (m, 1H), 7.21-7.19 (m,
1H), 7.10 (td, J = 7.4, 1.1 Hz, 1H), 7.06-7.04 (m, 1H), 3.03-2.99 (m, 2H), 2.81-2.78 (m, 2H).
Spectrum referenced to diethyl ether (Et2O) peak at 1.21 ppm as the residual CDCl3 was hard
to identify.38 13C NMR (101 MHz, CDCl3) δ 168.64, 152.14, 128.39, 128.12, 124.50, 122.75,
117.07, 29.35, 23.84. The observed results are in accordance with the literature values.40
Ethyl 2-cyano-3,3-diphenylpropanoate (2ab)
Synthesised according to general procedure B/work-up A and the crude
mixture was purified by flash column chromatography (n-hexane/ethyl
acetate, 98:2). The desired product was obtained as a white solid (93%
yield). 1H NMR (400 MHz, CDCl3) δ 7.39 – 7.23 (m, 10H), 4.74 (d, J =
8.6 Hz, 1H), 4.25 (d, J = 8.6 Hz, 1H), 4.10 (qd, J = 7.1, 1.6 Hz, 2H), 1.09 (t, J = 7.1 Hz, 3H).
Spectrum referenced to ethyl acetate (EtOAc) peak at 1.26 ppm as the residual CDCl3 was hard
to identify.38 13C NMR (101 MHz, CDCl3) δ 165.11, 139.43, 138.85, 128.98, 128.95, 128.31,
127.93, 127.90, 127.76, 115.87, 62.95, 51.18, 43.65, 13.79. The observed results are in
accordance with the literature values.41
Page 25
18
NMR Yields
Ethyl 3-(4-bromophenyl)propionate (2h)
Synthesised according to general procedure B and the crude yield
was determined by 1H NMR (30% yield) using 1,3,5-
trimethoxybenzene as internal standard.
Crude 1H NMR
SM: starting material
IS: Internal standard
HP: Hantzsch pyridine
Page 26
19
Methyl 3-(2-iodophenyl)propanoate (2n)
Synthesised according to general procedure B and the crude yield was
determined by 1H NMR (46% yield) using 1,3,5-trimethoxybenzene as
internal standard.
Crude 1H NMR
SM: starting material
IS: Internal standard
HP: Hantzsch pyridine
Page 27
20
Methyl 3-(4-hydroxyphenyl)propanoate (2o)
Synthesised according to general procedure B and the crude yield
was determined by 1H NMR (36% yield) using 1,3,5-
trimethoxybenzene as internal standard.
Crude 1H NMR
SM: starting material
IS: Internal standard
HP: Hantzsch pyridine
Page 28
21
Methyl 3-(4-aminophenyl)propanoate (2s)
Synthesised according to general procedure B and the crude yield
was determined by 1H NMR (35% yield) using 1,3,5-
trimethoxybenzene as internal standard.
Crude 1H NMR
SM: starting material
IS: Internal standard
HP: Hantzsch pyridine
Page 29
22
Methyl 3-(pyridin-2-yl)propanoate (2v)
Synthesised according to general procedure B and the crude yield was
determined by 1H NMR (25% yield) using 1-bromo-4-nitrobenzene as
internal standard.
Crude 1H NMR
SM: starting material
IS: Internal standard
HE: Hantzsch ester
HP: Hantzsch pyridine
Page 30
23
2-Phenethylpyridine (2z)
Synthesised according to general procedure B and the crude yield was
determined by 1H NMR (44% yield) using 1-bromo-4-nitrobenzene as
internal standard. Spectra were referenced to Hantzsch ester peak at 2.20
ppm as the residual CDCl3 was hard to identify.42
Crude 1H NMR
SM: starting material
IS: Internal standard
HE: Hantzsch ester
HP: Hantzsch pyridine
Page 31
24
Methyl 3-(furan-2-yl)propanoate (2w)
Synthesised according to general procedure B and the crude yield was
obtained by 1H NMR (16% yield) using 1,3,5-trimethoxybenzene as
internal standard.
Crude 1H NMR
SM: starting material
IS: Internal standard
HE: Hantzsch ester
HP: Hantzsch pyridine
Page 32
25
Ethyl butyrate (2x)
Synthesised according to general procedure B and the crude yield was
obtained by 1H NMR (not detected) using 1-bromo-4-nitrobenzene as
internal standard.
Crude 1H NMR
SM: starting material
IS: Internal standard
HE: Hantzsch ester
HP: Hantzsch pyridine
Page 33
26
Mechanistic Studies
Deuterium Labelling Studies
Hydrogenation of methyl cinnamate was performed according to general procedure B with
non-deuterated/deuterated Hantzsch esters. The results of these studies are summarized in
Table S1:
Table S1. Deuterium Labelling Experiments. N HE solvent Yield, % D incorporation, %
E-SM Z-SM Product α-d β-d
1
MeOD 0 0 100 9 44
2
MeOH 0 0 100 25 0
4,4’-Dideuterio-2,6-dimethyl-3,5-dicarboethoxy-1,4-dihydropyridine (4,4’-d2-HE)
4,4’-d2-HE was prepared according to the procedure reported in the
literature.43 A mixture of ethyl acetoacetate (4.25 mL, 33.3 mmol),
d2-paraformaldehyde (0.5 g, 16.7 mmol) and ammonium acetate (1.93
g, 25 mmol) was heated at 70°C in a water bath for 10 min. and then allowed to cool down to
room temperature. Cold water (30 mL) was added and the resulting mixture was stirred for 10
minutes and filtered. The precipitate was recrystallised from EtOH (25 mL) to afford the pure
deuterated Hantzsch ester 4b as a pale yellow solid (0.94 g, 22%). 1H NMR (400 MHz,
CDCl3) δ 5.21 (br s, 1H), 4.16 (q, J = 7.1 Hz, 4H), 2.19 (s, 6H), 1.28 (t, J = 7.1 Hz, 6H). The
analytic data are in agreement with the literature.43
Page 34
27
Stern-Volmer quenching studies
Stern-Volmer studies were conducted with methyl cinnamate (1a) and Hantzsch ester.
Standard solutions of 1a, Hantzsch ester and [Ir(dtbbpy)(ppy)2]PF6 were prepared in DMF. The
samples contained 0.01 mM Ir photocatalyst solution and varying quencher concentrations
were prepared in 3 mL quartz cuvettes, equipped with Suba-Seal septa, and degassed with
argon for 10 minutes. The cuvettes were irradiated at 450 nm and the emission intensity was
measured at 575 nm (two times) using an Agilent Eclipse Fluorescence Spectrophotometer.
Data were acquired and analysed using Cary Eclipse Scan Application v1.2. The ratio of I0/I
was plotted as a function of the quencher concentration (I0 = emission intensity of the
photocatalyst in isolation at the specified wavelength; I = observed intensity as a function of
the quencher concentration). These results show that the photocatalyst is readily quenched only
by Hantzsch ester.
Table S2. Stern-Volmer quenching study of Hantzsch ester and methyl cinnamate (1a).
[Hantzsch ester],mM I (average) Io/I
0 436.741 1
0.5 380.4695 1.1479
1 346.2595 1.261311
2 313.057 1.395085
3.2 282.0585 1.548406
4 258.6745 1.688381
Figure S2. Stern-Volmer quenching study of Hantzsch ester and methyl cinnamate (1a).
y = 0.1613x + 1.0541R² = 0.9802
y = 0.0121x + 0.9895R² = 0.1056
0
0.2
0.4
0.6
0.8
1
1.2
1.4
1.6
1.8
0 1 2 3 4 5 6 7
I0 /I
[Q], mM
Hantzsch ester
methylcinnamate
[Methyl cinnamate],mM I (average) Io/I
0 440.3755 1
0.5 403.83 1.090497
1 478.6915 0.843612
2 444.1135 1.077858
3.2 454.9835 0.976109
4 424.1305 1.072744
5 397.7035 1.066449
6.1 378.15 1.051708
Page 35
28
Computational Details
Methods. All calculations were performed using Gaussian 09.d01.44 All geometry
optimisations were performed using the M06-2X functional45 with 6-31+G(d,p) basis set
and SMD solvation model for MeOH or MeCN (for reduction potentials).46 Harmonic
vibrational frequencies were calculated with the same method and used to confirm the
nature of stationary points (minima or transition states) and to determine Gibbs free
energies (at 25 °C). Absolute half reaction reduction potentials were determined from the
difference of Gibbs free energies between oxidised and reduced form of the compound of
study in MeCN, applying equation 1,47 with 𝑛𝑒 = 1 and ℱ = 23.061. Relative potentials
were calculated using the reference values for SHE (Eabs(SHE) = 4.281 V) and SCE
(ESHE→SCE = ‒0.141 V in MeCN) reported by Isse and Gennaro.48
𝐸½𝑜 = −
Δ𝐺𝑜
𝑛𝑒ℱ (1)
Geometries and energies of minima and transition states:
1a
MeOH MeCN E = -537.3398988 Hartree E = -537.3415877 Hartree
G = -537.199653 Hartree G = -537.201648 Hartree
C 3.681316 0.975162 -0.000143 C 3.680758 0.977176 -0.000252
C 2.313735 1.243123 -0.000037 C 2.312405 1.242569 -0.000089
C 1.378151 0.198183 0.000096 C 1.378202 0.196311 0.000082
C 1.842963 -1.127666 0.000152 C 1.845937 -1.128576 0.000107
C 3.207401 -1.393998 0.000053 C 3.211146 -1.392690 -0.000050
C 4.130948 -0.344260 -0.000100 C 4.133189 -0.341472 -0.000232
H 4.392897 1.795382 -0.000253 H 4.390613 1.798785 -0.000389
H 1.961017 2.271385 -0.000064 H 1.957546 2.270041 -0.000099
H 1.138050 -1.953796 0.000290 H 1.142069 -1.955555 0.000258
H 3.554835 -2.422786 0.000105 H 3.560155 -2.420858 -0.000023
H 5.195785 -0.557694 -0.000174 H 5.198288 -0.552951 -0.000352
C -0.048619 0.540794 0.000170 C -0.050173 0.536487 0.000229
C -1.078729 -0.319317 -0.000078 C -1.080246 -0.322655 0.000175
H -0.276572 1.606555 0.000428 H -0.282246 1.601326 0.000390
H -0.957949 -1.397803 -0.000369 H -0.958276 -1.401136 -0.000004
C -2.461209 0.189383 0.000029 C -2.465281 0.192678 0.000370
O -2.791547 1.365387 0.000257 O -2.788154 1.365526 0.000285
O -3.353376 -0.807690 -0.000193 O -3.356083 -0.808823 -0.000099
C -4.736348 -0.422810 -0.000150 C -4.735969 -0.421461 -0.000346
H -4.963898 0.160524 -0.894798 H -4.965404 0.163984 -0.893358
Page 36
29
H -5.298471 -1.354959 -0.000412 H -5.302808 -1.350982 -0.000731
H -4.963966 0.160059 0.894785 H -4.965849 0.163515 0.892859
[1a]•‒
MeOH MeCN E = -537.4353901 Hartree E = -537.4310672 Hartree
G = -537.29944 Hartree G = -537.295253 Hartree
C 3.720784 0.938386 -0.000093 C 3.720572 0.941570 -0.000082
C 2.370357 1.250340 -0.000052 C 2.370646 1.251864 -0.000054
C 1.365117 0.239841 -0.000007 C 1.362517 0.240139 -0.000021
C 1.825387 -1.109586 -0.000002 C 1.828538 -1.110958 -0.000015
C 3.181839 -1.409966 -0.000045 C 3.185479 -1.408341 -0.000044
C 4.150021 -0.398138 -0.000091 C 4.154328 -0.395817 -0.000079
H 4.453201 1.742252 -0.000128 H 4.451472 1.746958 -0.000108
H 2.058622 2.293035 -0.000056 H 2.058184 2.294508 -0.000058
H 1.105949 -1.923592 0.000034 H 1.110420 -1.926220 0.000013
H 3.492487 -2.452308 -0.000041 H 3.497259 -2.450527 -0.000038
H 5.207875 -0.641771 -0.000124 H 5.212385 -0.637802 -0.000101
C -0.015807 0.601671 0.000029 C -0.017215 0.595894 0.000003
H -0.235091 1.668823 0.000022 H -0.244627 1.661513 0.000005
C -1.108962 -0.279728 0.000070 C -1.110014 -0.284242 0.000018
H -0.957949 -1.397803 -0.000369 H -0.958276 -1.401136 -0.000004
C -2.461209 0.189383 0.000029 C -2.465281 0.192678 0.000370
O -2.791547 1.365387 0.000257 O -2.788154 1.365526 0.000285
O -3.353376 -0.807690 -0.000193 O -3.356083 -0.808823 -0.000099
C -4.736348 -0.422810 -0.000150 C -4.735969 -0.421461 -0.000346
H -4.963898 0.160524 -0.894798 H -4.965404 0.163984 -0.893358
H -5.298471 -1.354959 -0.000412 H -5.302808 -1.350982 -0.000731
H -4.963966 0.160059 0.894785 H -4.965849 0.163515 0.892859
[1aHα]•
MeOH MeCN E = -537.9108336 Hartree E = -537.9125912 Hartree
G = -537.763546 Hartree G = -537.765226 Hartree
C -3.722892 0.964886 -0.001894 C -3.720284 0.968842 -0.013061
C -2.371179 1.271295 -0.002525 C -2.367529 1.271221 -0.019111
C -1.386419 0.246613 -0.000835 C -1.385443 0.243708 -0.006966
C -1.837626 -1.100016 0.001475 C -1.840840 -1.101628 0.011161
C -3.194901 -1.394899 0.002069 C -3.199115 -1.392602 0.017428
C -4.147128 -0.370352 0.000402 C -4.148542 -0.365229 0.005422
H -4.455981 1.766658 -0.003177 H -4.450959 1.772615 -0.022698
Page 37
30
H -2.046329 2.308883 -0.004286 H -2.039248 2.307561 -0.033369
H -1.114816 -1.910848 0.002754 H -1.119618 -1.913746 0.020380
H -3.517000 -2.432455 0.003833 H -3.524272 -2.428997 0.031750
H -5.206465 -0.607998 0.000886 H -5.208452 -0.599685 0.010356
C -0.008980 0.587242 -0.001421 C -0.006804 0.579821 -0.012752
H 0.268216 1.636558 -0.003245 H 0.276252 1.627479 -0.025206
C 1.076444 -0.432082 0.000549 C 1.078589 -0.439552 -0.004218
H 1.013721 -1.100837 -0.870845 H 1.019272 -1.108374 -0.875373
C 2.460154 0.164054 0.001324 C 2.461671 0.165515 0.009614
O 2.723982 1.350986 0.006683 O 2.716806 1.349654 0.051946
O 3.398132 -0.784814 -0.003924 O 3.401469 -0.784480 -0.027192
C 4.762397 -0.332609 -0.002032 C 4.761505 -0.327170 -0.011950
H 5.368452 -1.236711 -0.006448 H 5.374035 -1.226519 -0.046117
H 4.960538 0.264492 -0.894360 H 4.959115 0.301128 -0.882938
H 4.960873 0.255952 0.895879 H 4.961882 0.234568 0.902772
H 1.012655 -1.097867 0.874263 H 1.006553 -1.104985 0.869003
[1aHβ]•
MeOH MeCN E = -537.9048103 Hartree E = -537.9056843 Hartree
G = -537.756348 Hartree G = -537.757848 Hartree
C 3.547436 -0.377408 -0.571412 C 3.669479 -0.436393 -0.435029
C 2.430244 -1.144125 -0.233473 C 2.495418 -1.158335 -0.208446
C 1.294570 -0.539615 0.310495 C 1.334531 -0.510243 0.220245
C 1.287833 0.847525 0.503257 C 1.364347 0.877062 0.412362
C 2.402278 1.613941 0.167968 C 2.534539 1.599719 0.187971
C 3.536304 1.002446 -0.371519 C 3.692166 0.943640 -0.237580
H 4.425945 -0.860279 -0.989921 H 4.564921 -0.954350 -0.766355
H 2.441729 -2.220205 -0.388968 H 2.480865 -2.234224 -0.364374
H 0.404579 1.324160 0.924080 H 0.463464 1.390120 0.743706
H 2.386882 2.688239 0.328226 H 2.544176 2.674438 0.345084
H 4.405478 1.598958 -0.632785 H 4.604493 1.505715 -0.413632
C 0.062629 -1.357991 0.669435 C 0.048311 -1.280441 0.469891
H 0.310372 -2.423094 0.608691 H 0.234935 -2.347047 0.292174
C -1.047918 -1.052697 -0.271831 C -1.043022 -0.812407 -0.425746
H -1.011540 -1.409720 -1.295467 H -0.933890 -0.891106 -1.502196
C -2.157681 -0.212486 0.116951 C -2.253444 -0.215307 0.100698
O -2.301985 0.295816 1.226660 O -2.497604 -0.051464 1.289042
O -3.045235 -0.034133 -0.878232 O -3.107448 0.150738 -0.875486
C -4.180579 0.785642 -0.570518 C -4.332040 0.748305 -0.437879
H -4.777443 0.811198 -1.480713 H -4.886696 0.982934 -1.345184
H -4.754075 0.345189 0.248203 H -4.898249 0.048905 0.181799
Page 38
31
H -3.857165 1.793064 -0.298988 H -4.130845 1.660053 0.129420
H -0.247691 -1.132356 1.694106 H -0.264467 -1.163241 1.512203
[1aHα]‒
MeOH MeCN E = -538.0202553 Hartree E = -538.0211752 Hartree
G = -537.873442 Hartree G = -537.875112 Hartree
C -3.752379 0.927041 -0.079233 C -3.755521 0.925048 -0.059442
C -2.416628 1.273225 -0.116253 C -2.420560 1.275919 -0.053089
C -1.353853 0.290991 -0.042889 C -1.354997 0.293744 -0.003569
C -1.819882 -1.075654 0.065866 C -1.819704 -1.077786 0.046373
C -3.172121 -1.394365 0.103377 C -3.171372 -1.400951 0.039477
C -4.173273 -0.415911 0.032270 C -4.174515 -0.422896 -0.014861
H -4.498545 1.718663 -0.138340 H -4.502934 1.716580 -0.099397
H -2.139446 2.323390 -0.202058 H -2.145000 2.329371 -0.088582
H -1.091797 -1.881918 0.116873 H -1.089588 -1.882666 0.088339
H -3.455094 -2.443417 0.186596 H -3.452625 -2.453011 0.077046
H -5.226008 -0.677914 0.059541 H -5.226591 -0.688323 -0.019958
C -0.009165 0.637765 -0.077129 C -0.011212 0.643069 -0.005042
H 0.282716 1.680043 -0.166428 H 0.285319 1.687350 -0.027977
C 1.068599 -0.402673 -0.009774 C 1.069948 -0.394576 0.043708
H 1.065329 -1.115326 -0.852292 H 1.029765 -1.115560 -0.791107
C 2.460532 0.160765 0.061834 C 2.465733 0.174234 0.037129
O 2.759748 1.312976 0.321493 O 2.770265 1.344326 0.139109
O 3.393349 -0.773695 -0.175979 O 3.389727 -0.795707 -0.087059
C 4.760645 -0.348855 -0.082034 C 4.755956 -0.368282 -0.076289
H 5.357306 -1.231870 -0.305339 H 5.350487 -1.274221 -0.186140
H 4.962100 0.439709 -0.810080 H 4.949621 0.313770 -0.907327
H 4.976201 0.010612 0.926545 H 4.994215 0.127966 0.867371
H 0.987611 -1.050186 0.884661 H 1.024873 -1.035347 0.944946
[1aHβ]‒
MeOH MeCN E = -538.0505524 Hartree E = -538.0404387 Hartree
G = -537.902141 Hartree G = -537.893932 Hartree
C 3.692713 -0.334255 0.265570 C 3.669572 -0.298006 0.300086
C 2.561809 -1.133831 0.076755 C 2.557896 -1.120857 0.092098
C 1.305841 -0.564444 -0.155627 C 1.296774 -0.577601 -0.172545
Page 39
32
C 1.208649 0.833718 -0.193590 C 1.174701 0.819090 -0.228321
C 2.334156 1.636049 -0.018032 C 2.280555 1.643929 -0.034321
C 3.583551 1.054320 0.215917 C 3.535427 1.087690 0.235569
H 4.657191 -0.798783 0.453007 H 4.637986 -0.742335 0.514171
H 2.656205 -2.217274 0.114374 H 2.671471 -2.202046 0.141742
H 0.235926 1.290683 -0.363580 H 0.193941 1.246272 -0.429112
H 2.238066 2.717813 -0.059263 H 2.166633 2.723428 -0.088373
H 4.460324 1.679179 0.360094 H 4.396452 1.730226 0.395570
C 0.074694 -1.428982 -0.385501 C 0.079310 -1.460519 -0.407916
H -0.174194 -1.374110 -1.454648 H -0.182586 -1.380891 -1.472558
C -1.140709 -1.050715 0.416737 C -1.142114 -1.118053 0.402265
H -1.215051 -1.373092 1.451246 H -1.220324 -1.461351 1.429779
C -2.115005 -0.232560 -0.089327 C -2.108576 -0.266501 -0.099860
O -2.205228 0.281756 -1.255040 O -2.165692 0.287382 -1.230253
O -3.139758 0.073237 0.825573 O -3.157460 -0.013054 0.804506
C -4.285124 0.725257 0.297961 C -4.186872 0.830550 0.328767
H -4.971003 0.853202 1.137748 H -4.905631 0.923670 1.146578
H -4.038295 1.706246 -0.119196 H -3.811161 1.824632 0.063450
H -4.770695 0.120599 -0.475744 H -4.690454 0.406857 -0.547089
H 0.357967 -2.471753 -0.193473 H 0.388848 -2.501420 -0.243115
HE
E = -783.60137 Hartree
G = -783.39199 Hartree
C -1.220169 1.594897 -0.000074
C -1.258591 0.234902 -0.000104
C 0.000000 -0.611607 -0.000417
C 1.258591 0.234902 -0.000106
C 1.220169 1.594897 -0.000061
H 0.000000 3.247637 0.000048
H -0.000002 -1.278285 0.871534
N 0.000000 2.235356 -0.000245
H 0.000003 -1.277701 -0.872829
C 2.398771 2.524861 0.000152
H 3.023170 2.357219 0.880383
H 3.023291 2.357456 -0.880042
H 2.060261 3.563011 0.000262
C -2.398771 2.524861 0.000126
H -3.023498 2.357197 -0.879867
H -3.022962 2.357477 0.880557
Page 40
33
H -2.060261 3.563011 -0.000138
C -2.541752 -0.462597 0.000072
O -3.662040 0.038398 0.000488
O -2.377625 -1.799685 -0.000241
C 2.541752 -0.462598 0.000029
O 3.662041 0.038397 0.000231
O 2.377625 -1.799685 -0.000054
C -3.576470 -2.584145 -0.000062
H -4.168034 -2.373727 -0.893809
H -3.245928 -3.621727 -0.000435
H -4.167461 -2.374186 0.894172
C 3.576469 -2.584146 0.000114
H 3.245927 -3.621728 0.000040
H 4.167886 -2.373937 -0.893780
H 4.167609 -2.373978 0.894201
HE•+
E = -783.3994389 Hartree
G = -783.192259 Hartree
C -1.226440 1.620027 -0.012816
C -1.250487 0.243379 0.006084
C 0.000000 -0.546856 0.002477
C 1.250486 0.243381 0.006147
C 1.226434 1.620030 -0.012734
H -0.000001 3.241389 -0.048316
H -0.000019 -1.251945 0.854389
N -0.000002 2.220645 -0.026473
H 0.000019 -1.237191 -0.862512
C 2.403124 2.541879 -0.022331
H 2.994157 2.404243 0.885932
H 3.047958 2.320503 -0.875143
H 2.072365 3.580048 -0.081068
C -2.403130 2.541876 -0.022500
H -3.047865 2.320553 -0.875400
H -2.994272 2.404197 0.885686
H -2.072366 3.580048 -0.081141
C -2.544109 -0.489913 0.023204
O -3.640539 0.032918 0.083675
Page 41
34
O -2.356316 -1.804280 -0.032940
C 2.544109 -0.489910 0.023288
O 3.640529 0.032908 0.084069
O 2.356327 -1.804262 -0.033234
C -3.546324 -2.613251 -0.021234
H -4.166448 -2.371549 -0.886463
H -3.195827 -3.641869 -0.074232
H -4.101883 -2.441141 0.902492
C 3.546335 -2.613234 -0.021600
H 3.195849 -3.641837 -0.074967
H 4.166582 -2.371266 -0.886666
H 4.101761 -2.441405 0.902258
TSα
E = -1320.739163 Hartree
G = -1320.368104 Hartree
C 4.999491 0.302824 1.040614
C 3.670474 0.133268 1.410592
C 2.666590 0.974348 0.886695
C 3.030576 1.983480 -0.031375
C 4.360097 2.153292 -0.389131
C 5.347060 1.317089 0.145793
H 5.763982 -0.350086 1.449886
H 3.390968 -0.650125 2.110680
H 2.274912 2.639974 -0.452098
H 4.634409 2.937214 -1.088202
H 6.385823 1.456556 -0.138493
C 1.310626 0.768714 1.329670
C 0.178085 1.399627 0.836454
H 1.162006 0.014697 2.101978
C -1.092507 1.244559 1.598826
O -1.291358 0.397853 2.450245
O -2.016238 2.108646 1.187043
C -3.289366 2.047022 1.856772
H -3.915538 2.786865 1.361797
H -3.718620 1.048727 1.755287
H -3.161048 2.295861 2.912251
H 0.243476 2.275244 0.194505
H -0.276465 0.463863 -0.220413
C -0.750958 -0.498365 -0.987083
Page 42
35
C -2.164905 -0.571085 -0.683437
C 0.079751 -1.614146 -0.587458
H -0.479647 0.010663 -1.912474
C -2.648890 -1.584625 0.119284
C -3.053899 0.519757 -1.152125
C -0.431310 -2.585632 0.248400
C 1.499339 -1.645772 -1.007604
N -1.755221 -2.513012 0.565249
C -4.067857 -1.799445 0.538268
O -4.257747 0.571534 -0.974780
O -2.366144 1.470837 -1.782371
C 0.311041 -3.744357 0.830634
O 2.312450 -2.493074 -0.681654
O 1.796873 -0.621953 -1.808308
H -2.121649 -3.249522 1.165684
H -4.440438 -0.936941 1.094604
H -4.702518 -1.919941 -0.342795
H -4.148398 -2.690946 1.162806
C -3.119124 2.618773 -2.205942
H 0.716184 -4.371372 0.033036
H 1.153021 -3.387597 1.428451
H -0.350066 -4.343014 1.459289
C 3.149538 -0.577025 -2.292173
H -3.572148 3.105720 -1.339327
H -2.396706 3.279160 -2.681706
H -3.892002 2.318970 -2.916139
H 3.216894 0.334752 -2.882721
H 3.848184 -0.549538 -1.453133
H 3.346125 -1.452660 -2.914944
TSβ
E = -1320.732105 Hartree
G = -1320.361618 Hartree
C -3.683682 -1.140080 -1.481453
C -2.319514 -0.869690 -1.592166
C -1.381152 -1.653002 -0.906029
C -1.826187 -2.717529 -0.107424
C -3.185301 -2.990051 -0.006620
C -4.117385 -2.199919 -0.688193
H -4.402025 -0.523037 -2.013081
H -1.973758 -0.048355 -2.216154
H -1.116779 -3.325456 0.446217
Page 43
36
H -3.523961 -3.814527 0.613778
H -5.178516 -2.412267 -0.595775
C 0.038990 -1.254959 -0.989302
C 1.123296 -2.051088 -0.622985
H 0.256748 -0.474981 -1.725047
H 1.023138 -2.963923 -0.046153
C 2.479510 -1.588189 -0.937233
O 2.740554 -0.541433 -1.515624
O 3.414524 -2.438315 -0.510422
C 4.774122 -2.090436 -0.821220
H 5.005261 -1.088181 -0.454112
H 5.387639 -2.836047 -0.318874
H 4.926681 -2.133511 -1.902106
H 0.068276 -0.302932 0.061017
C -0.023401 0.815480 0.813414
C -1.233332 1.458555 0.367088
C 1.232093 1.453446 0.498273
H -0.075173 0.256385 1.747308
C -1.156711 2.429956 -0.614875
C -2.543921 1.055204 0.935658
C 1.273468 2.427763 -0.474542
C 2.479078 0.941457 1.118524
N 0.083809 2.820979 -1.016467
C -2.304178 3.106143 -1.290668
O -3.592797 1.642589 0.741481
O -2.438221 -0.011095 1.724364
C 2.495633 3.097126 -1.010834
O 3.546259 1.524365 1.127566
O 2.285782 -0.253017 1.676512
H 0.127409 3.527300 -1.750055
H -3.047571 2.371039 -1.604881
H -2.792546 3.795049 -0.595766
H -1.954513 3.665900 -2.160129
C -3.637996 -0.418695 2.404875
H 2.898962 3.789655 -0.266661
H 3.267383 2.357311 -1.231448
H 2.254190 3.653179 -1.918222
C 3.409275 -0.829609 2.365388
H -4.438751 -0.599900 1.686512
H -3.375970 -1.336324 2.928819
H -3.936336 0.356530 3.114345
H 3.084011 -1.823668 2.666784
H 4.271866 -0.890459 1.700374
H 3.652649 -0.221695 3.239481
Page 44
37
References
1 N. M. Silva, J. L. M. Tributino, A. L. P. Miranda, E. J. Barreiro and C. A. M. Fraga,
Eur. J. Med. Chem., 2002, 37, 163–170.
2 S. Niwayama, H. Cho and C. Lin, Tetrahedron Lett., 2008, 49, 4434–4436.
3 T. Gatzenmeier, P. S. J. Kaib, J. B. Lingnau, R. Goddard and B. List, Angew. Chem.
Int. Ed., 2018, 57, 2464–2468.
4 C. M. Young, J. E. Taylor and A. D. Smith, Org. Biomol. Chem., 2019, 17, 4747–
4752.
5 C. E. Gregerson, K. N. Trentadue, E. J. T. Phipps, J. K. Kirsch, K. M. Reed, G. D.
Dyke, J. H. Jansen, C. B. Otteman, J. L. Stachowski and J. B. Johnson, Org.
Biomol. Chem., 2017, 15, 5944–5948.
6 K. Wilson, J. Murray, H. Sneddon, C. Jamieson and A. Watson, Synlett, 2018, 29,
2293–2297.
7 M.-H. Yang, D. L. Orsi and R. A. Altman, Angew. Chem. Int. Ed., 2015, 54, 2361–
2365.
8 R. J. Faggyas, M. Grace, L. Williams and A. Sutherland, J. Org. Chem., 2018, 83,
12595–12608.
9 F. Le Callonnec, E. Fouquet and F.-X. Felpin, Org. Lett., 2011, 13, 2646–2649.
10 M. E. Daub, H. Jung, B. J. Lee, J. Won, M.-H. Baik and T. P. Yoon, J. Am. Chem.
Soc., 2019, 141, 9543–9547.
11 J. Liu, K. Dong, R. Franke, H. Neumann, R. Jackstell and M. Beller, J. Am. Chem.
Soc., 2018, 140, 10282–10288.
12 T. Arif, C. Borie, A. Tintaru, J.-V. Naubron, N. Vanthuyne, M. P. Bertrand and M.
Nechab, Adv. Synth. Catal., 2015, 357, 3611–3616.
13 J. Avó, L. Cunha-Silva, J. C. Lima and A. J. Parola, Org. Lett., 2014, 16, 2582–
2585.
14 R. Ueda, T. Suzuki, K. Mino, H. Tsumoto, H. Nakagawa, M. Hasegawa, R. Sasaki,
T. Mizukami and N. Miyata, J. Am. Chem. Soc., 2009, 131, 17536–17537.
15 J. Takeo, S. Yamashita, K. Wada and Y. Chiba, US5955505A, 1999.
16 L. H. Riihimaki-Lampen, M. J. Vainio, M. Vahermo, L. L. Pohjala, J. M. S.
Heikura, K. H. Valkonen, V. T. Virtanen, J. T. Yli-Kauhaluoma and P. M. Vuorela,
J. Med. Chem., 2010, 53, 514–518.
17 T. Taira, T. Yanagimoto, K. Sakai, H. Sakai, A. Endo and T. Imura, Tetrahedron,
Page 45
38
2016, 72, 4117–4122.
18 M.-K. Zhu, J.-F. Zhao and T.-P. Loh, Org. Lett., 2011, 13, 6308–6311.
19 J. Isaad and A. El Achari, Dye. Pigment., 2013, 99, 878–886.
20 W. Pan, K. Hu, P. Bai, L. Yu, Q. Ma, T. Li, X. Zhang, C. Chen, K. Peng, W. Liu
and Z. Sang, Bioorg. Med. Chem. Lett., 2016, 26, 2539–2543.
21 S. Bhattacharyya, Synth. Commun., 2000, 30, 2001–2008.
22 L. Li, J. C. Stimac and L. M. Geary, Tetrahedron Lett., 2017, 58, 1379–1381.
23 E. Shirakawa, X. Zhang and T. Hayashi, Angew. Chem. Int. Ed., 2011, 50, 4671–
4674.
24 S. C. Söderman and A. L. Schwan, J. Org. Chem., 2012, 77, 10978–10984.
25 M. Kreis, A. Palmelund, L. Bunch and R. Madsen, Adv. Synth. Catal., 2006, 348,
2148–2154.
26 T. Ikeda, Z. Zhang and Y. Motoyama, Adv. Synth. Catal., 2019, 361, 673–677.
27 D. Leow, Y.-H. Chen, T.-H. Hung, Y. Su and Y.-Z. Lin, Eur. J. Org. Chem., 2014,
7347–7352.
28 D. Stubba, G. Lahm, M. Geffe, J. W. Runyon, A. J. Arduengo and T. Opatz, Angew.
Chem. Int. Ed., 2015, 54, 14187–14189.
29 P. H. Gehrtz, V. Hirschbeck and I. Fleischer, Chem. Commun., 2015, 51, 12574–
12577.
30 Q.-D. Wang, J.-M. Yang, D. Fang, J. Ren, B. Dong, B. Zhou and B.-B. Zeng,
Tetrahedron Lett., 2016, 57, 2587–2590.
31 K. Geoghegan, S. Kelleher and P. Evans, J. Org. Chem, 2011, 76, 2187–2194.
32 W. T. Jorgensen, D. W. Gulliver, T. A. Katte, E. L. Werry, T. A. Reekie, M.
Connor and M. Kassiou, Eur. J. Med. Chem., 2018, 143, 1644–1656.
33 T. O. Vieira, M. J. Green and H. Alper, Org. Lett., 2006, 8, 6143–6145.
34 H. Doušová, Z. Růžičková and P. Šimůnek, J. Heterocycl. Chem., 2018, 55, 670–
684.
35 J. Yin, S. Eller, M. Collot and P. H. Seeberger, Beilstein J. Org. Chem., 2012, 8,
2067–2071.
36 L. J. Oates, R. F. W. Jackson and M. H. Block, Org. Biomol. Chem., 2003, 140–
144.
37 H. Matsuhashi, S. Asai, K. Hirabayashi, Y. Hatanaka, A. Mori and T. Hiyama, Bull.
Chem. Soc. Jpn., 1997, 70, 437–444.
38 G. R. Fulmer, A. J. M. Miller, N. H. Sherden, H. E. Gottlieb, A. Nudelman, B. M.
Page 46
39
Stoltz, J. E. Bercaw and K. I. Goldberg, Organometallics, 2010, 29, 2176–2179.
39 J. M. Brunel, Tetrahedron, 2007, 63, 3899–3906.
40 H. K. Moon, G. H. Sung, B. R. Kim, J. K. Park, Y.-J. Yoon and H. J. Yoon, Adv.
Synth. Catal., 2016, 358, 1725–1730.
41 C. Hu, G. Hong, X. Qian, K. R. Kim, X. Zhu and L. Wang, Org. Biomol. Chem.,
2017, 15, 4984–4991.
42 T. E. Elton, G. E. Ball, M. Bhadbhade, L. D. Field and S. B. Colbran,
Organometallics, 2018, 37, 3972–3982.
43 M.-H. Larraufie, R. Pellet, L. Fensterbank, J.-P. Goddard, E. Lacôte, M. Malacria
and C. Ollivier, Angew. Chem. Int. Ed., 2011, 50, 4463–4466.
44 Gaussian 16, Revision A.03, M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E.
Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson,
H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R.
Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L.
Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B.
Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N.
Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J.
Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K.
Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J.
Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J.
Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi,
M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L.
Martin, K. Morokuma, O. Farkas, J. B. Foresman, and D. J. Fox, Gaussian, Inc.,
Wallingford CT, 2016.
45 Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215-241.
46 A. V. Marenich, C. J. Cramer, and D. G. Truhlar, J. Phys. Chem. B, 2009, 113,
6378-6396.
47 H. G. Roth, N. A. Romero and D. A. Nicewicz, Synlett, 2016, 27, 714-723.
48 A. A. Isse and A. Gennaro, J. Phys. Chem. B, 2020, 114, 7894.
Page 47
40
Spectra
1H and 13C NMR for compound (1b)
Page 48
41
1H and 13C NMR for compound (1c)
Page 49
42
1H and 13C NMR for compound (1d)
Page 50
43
1H, 13C and 19F NMR for compound (1e)
Page 51
44
1H, 13C and 19F NMR for compound (1f)
Page 53
46
1H, 13C and 19F NMR for compound (1g)
Page 54
47
1H and 13C NMR for compound (1j)
Page 55
48
1H and 13C NMR for compound (1k)
Page 56
49
1H, 13C and 19F NMR for compound (1l)
Page 58
51
1H and 13C NMR for compound (1m)
Page 59
52
1H and 13C NMR for compound (1n)
Page 60
53
1H and 13C NMR for compound (1o)
Page 61
54
1H and 13C NMR for compound (1p)
Page 62
55
1H and 13C NMR for compound (1q)
Page 63
56
1H and 13C NMR for compound (1r)
Page 64
57
1H and 13C NMR for compound (1t)
Page 65
58
1H and 13C NMR for compound (1u)
Page 66
59
1H and 13C NMR for compound (1v)
Page 67
60
1H and 13C NMR for compound (1w)
Page 68
61
1H and 13C NMR for compound (1z)
Page 69
62
1H and 13C NMR for compound (2a)
Page 70
63
1H and 13C NMR for compound (2b)
Page 71
64
1H and 13C NMR for compound (2c)
Page 72
65
1H and 13C NMR for compound (2d)
Page 73
66
1H, 13C and 19F NMR for compound (2e)
Page 74
67
1H, 13C and 19F NMR for compound (2f)
Page 76
69
1H, 13C and 19F NMR for compound (2g)
Page 77
70
1H and 13C NMR for compound (2i)
Page 78
71
1H and 13C NMR for compound (2j)
Page 79
72
1H and 13C NMR for compound (2k)
Page 80
73
1H and 13C NMR for compound (2l)
Page 82
75
1H and 13C NMR for compound (2m)
Page 83
76
1H and 13C NMR for compound (2p)
Page 84
77
1H and 13C NMR for compound (2q)
Page 85
78
1H and 13C NMR for compound (2r)
Page 86
79
1H and 13C NMR for compound (2t)
Page 87
80
1H and 13C NMR for compound (2u)
Page 88
81
1H and 13C NMR for compound (2y)
Page 89
82
1H and 13C NMR for compound (2aa)
NMR purity = 81%; 16% of Hantzsch pyridine, 4% of starting material and 1% of Et2O.
Page 90
83
1H and 13C NMR for compound (2ab)
Page 91
84
1H NMR for compound (4,4’-d2-HE)
Page 92
download fileview on ChemRxivHAT reduction QMUL - ESI.pdf (7.07 MiB)