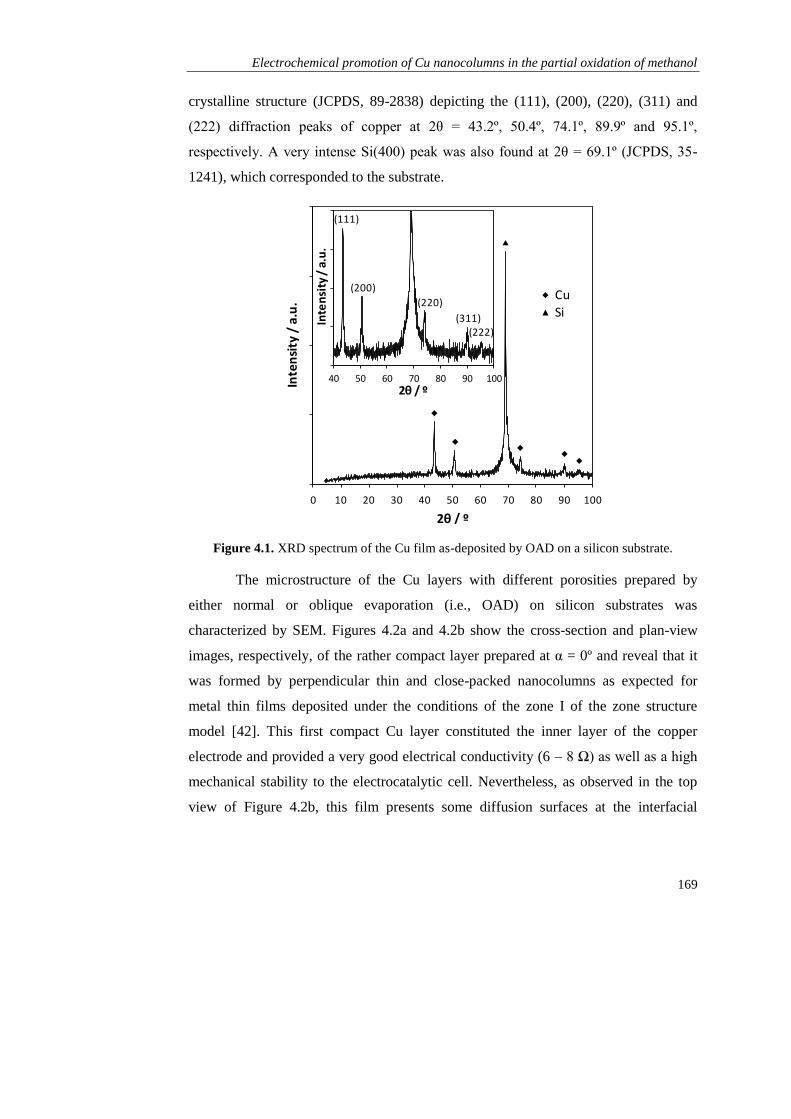
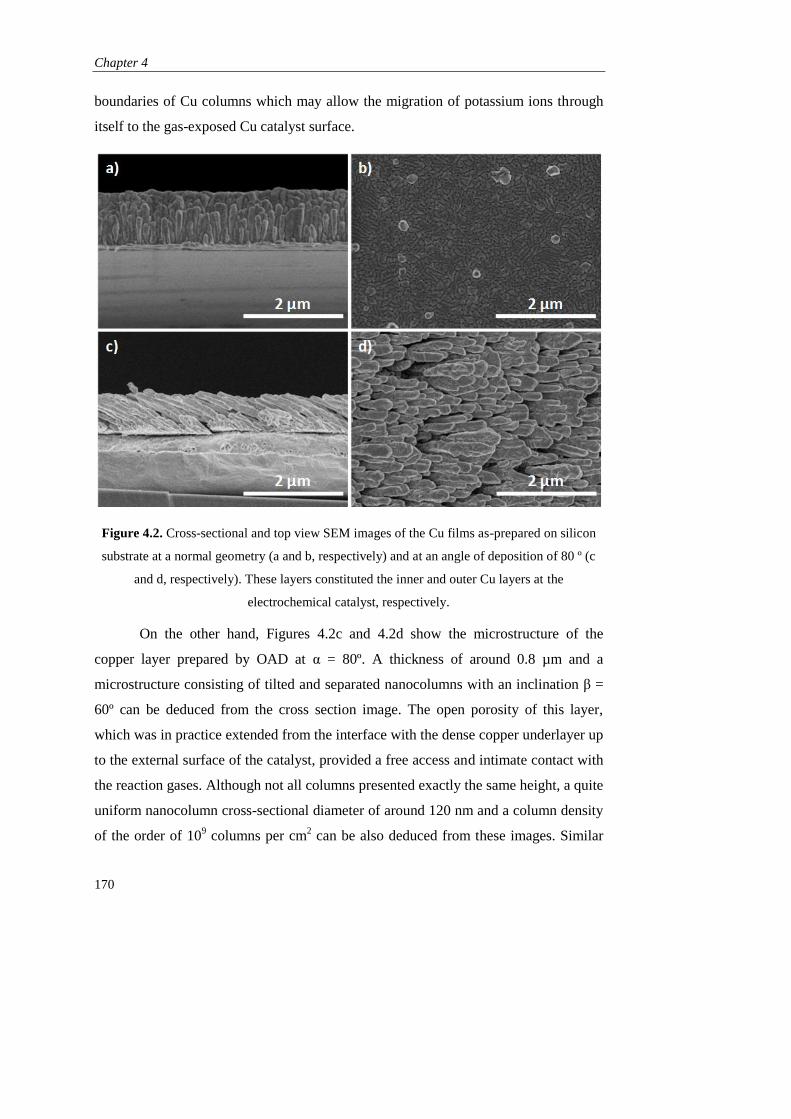
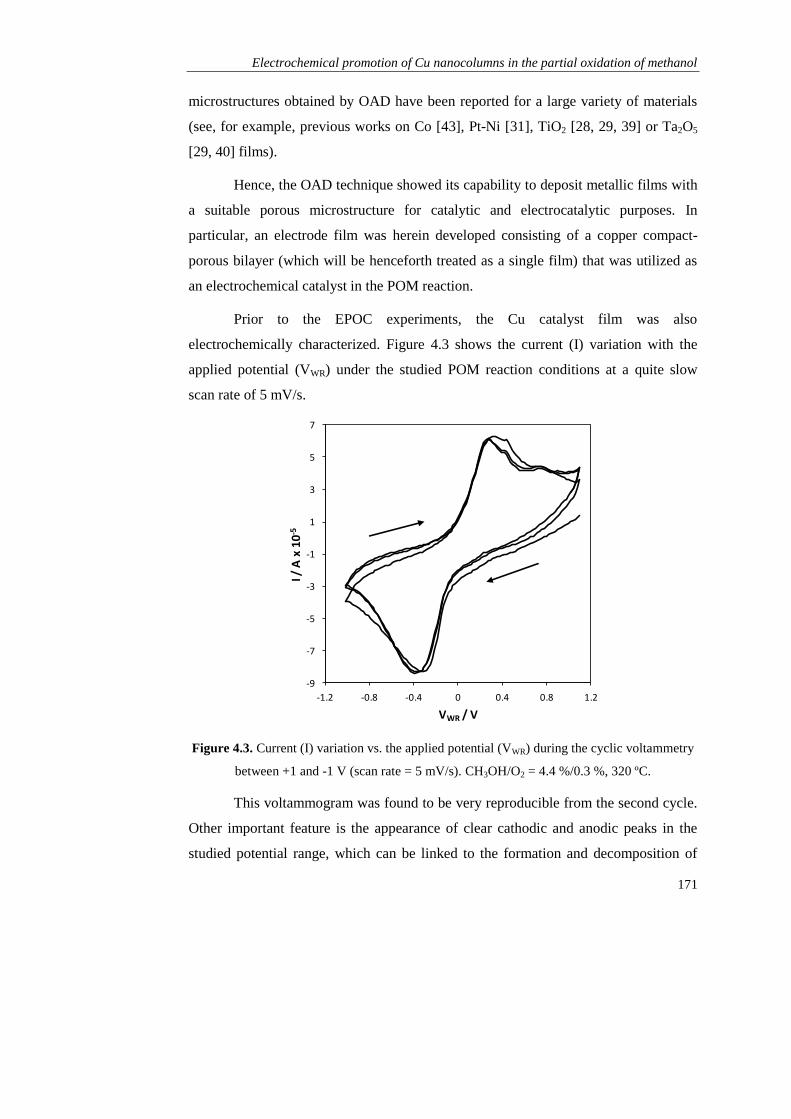
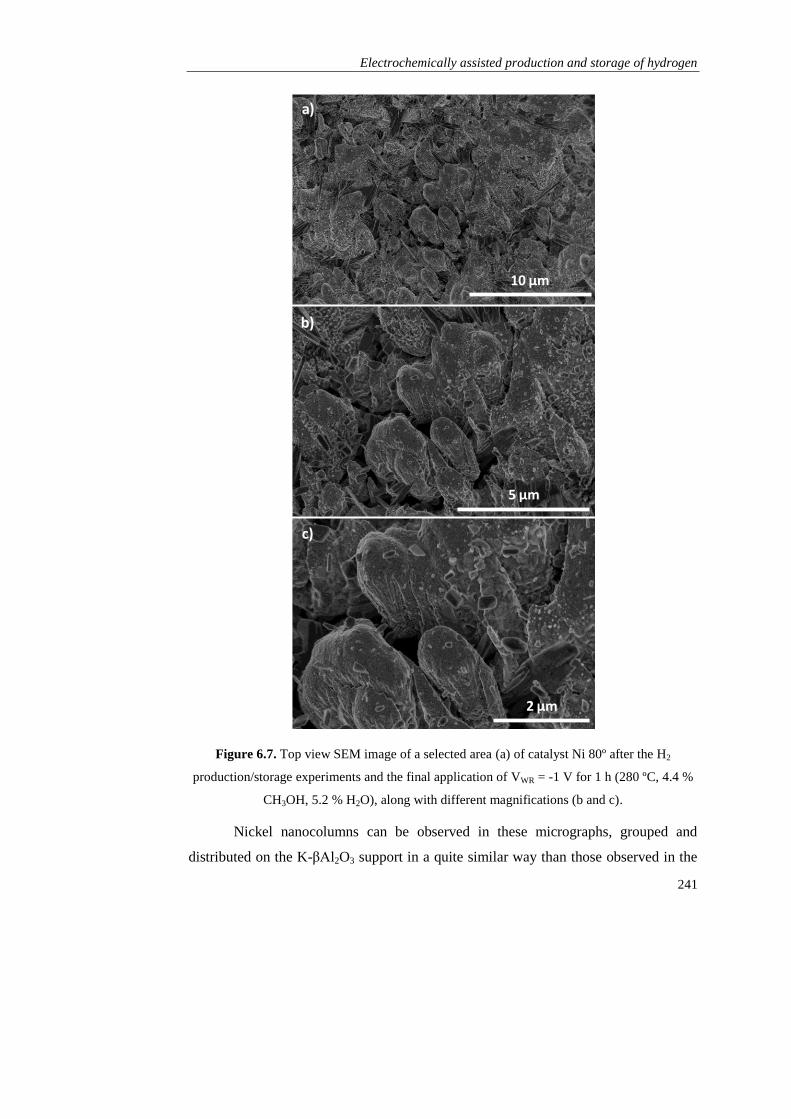
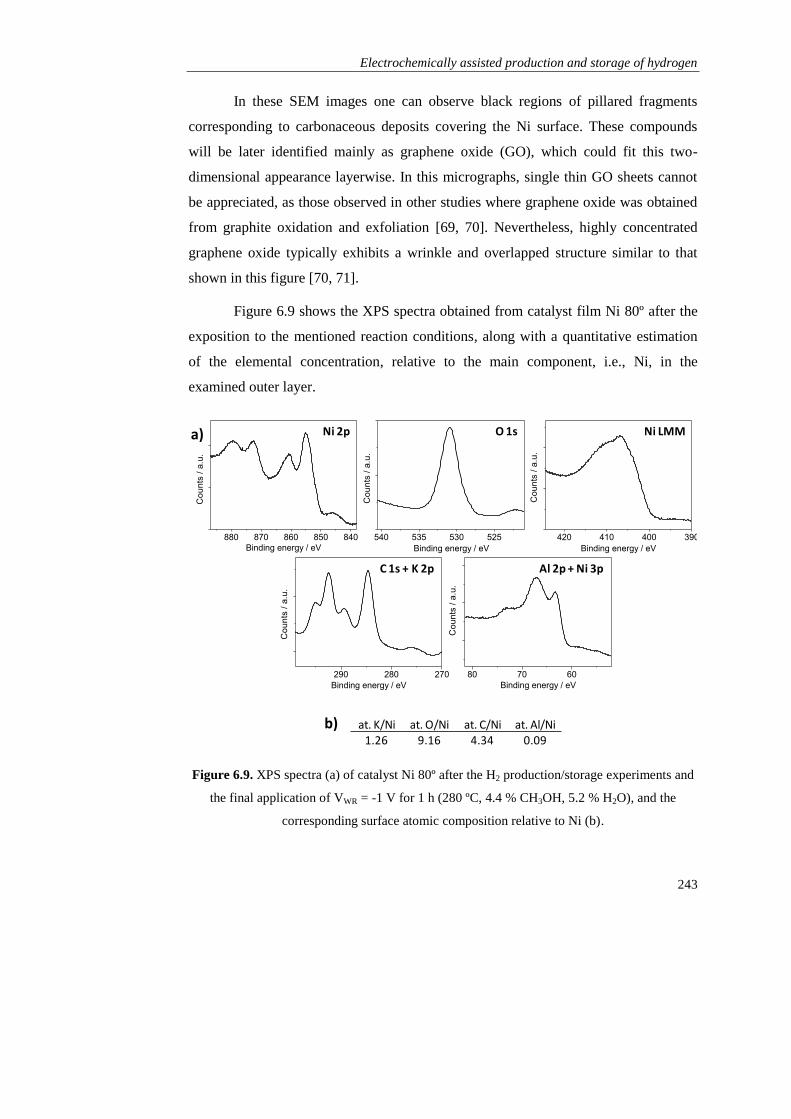
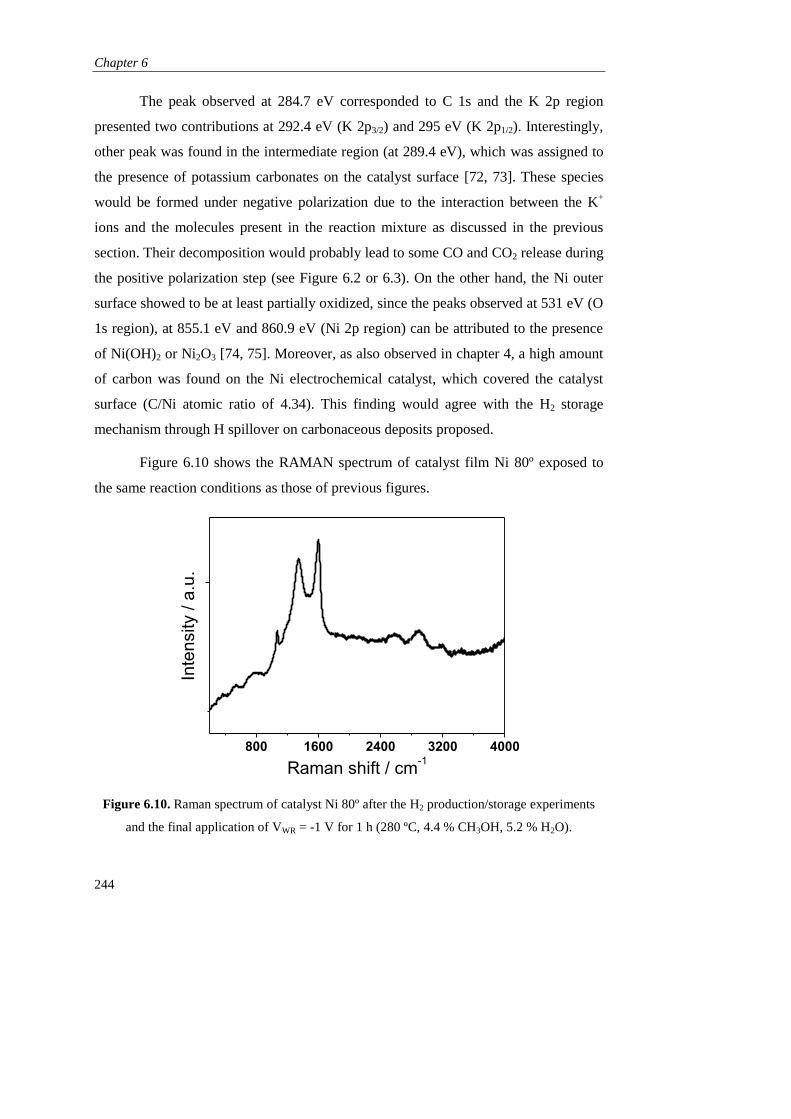
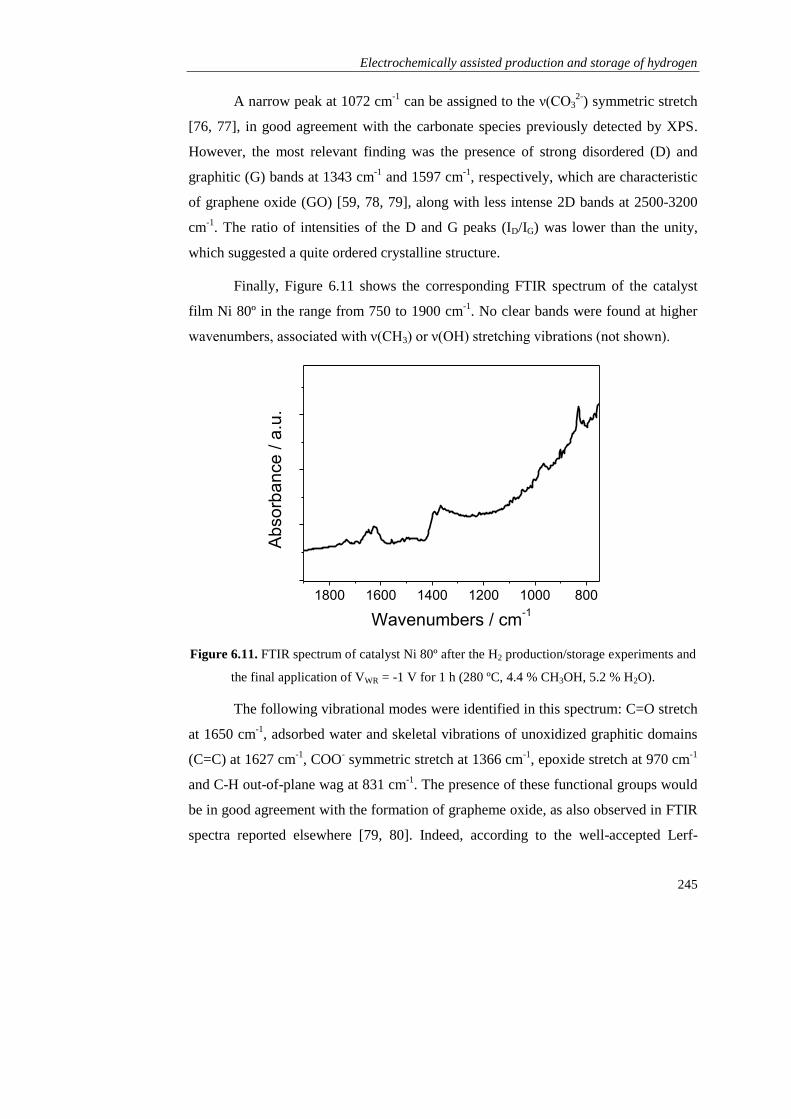
Page 1
UNIVERSIDAD DE CASTILLA-LA MANCHA
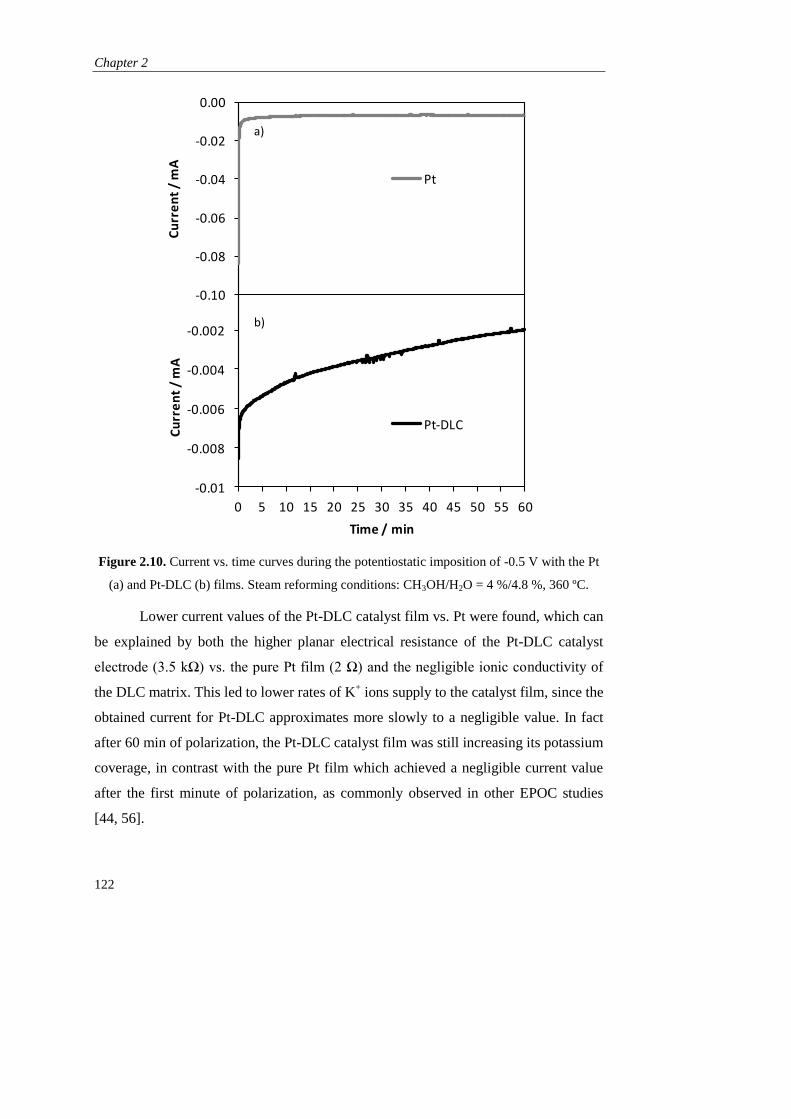
FACULTAD DE CIENCIAS Y TECNOLOGÍAS QUÍMICAS
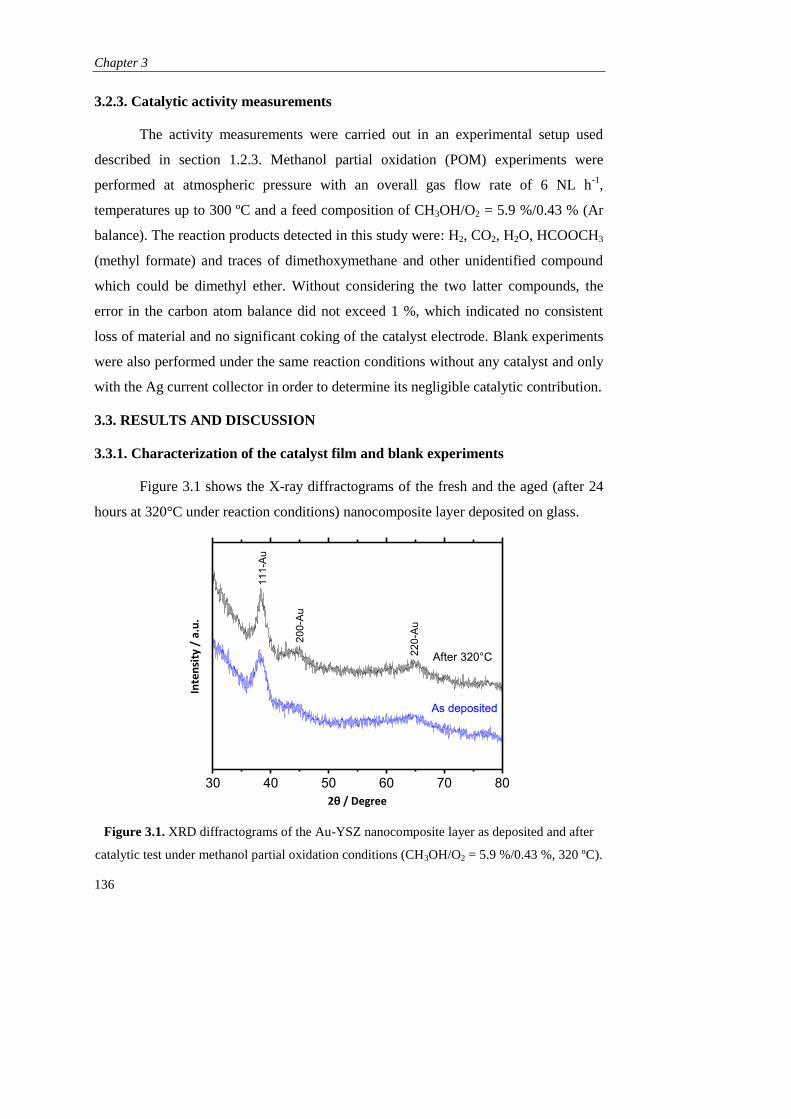
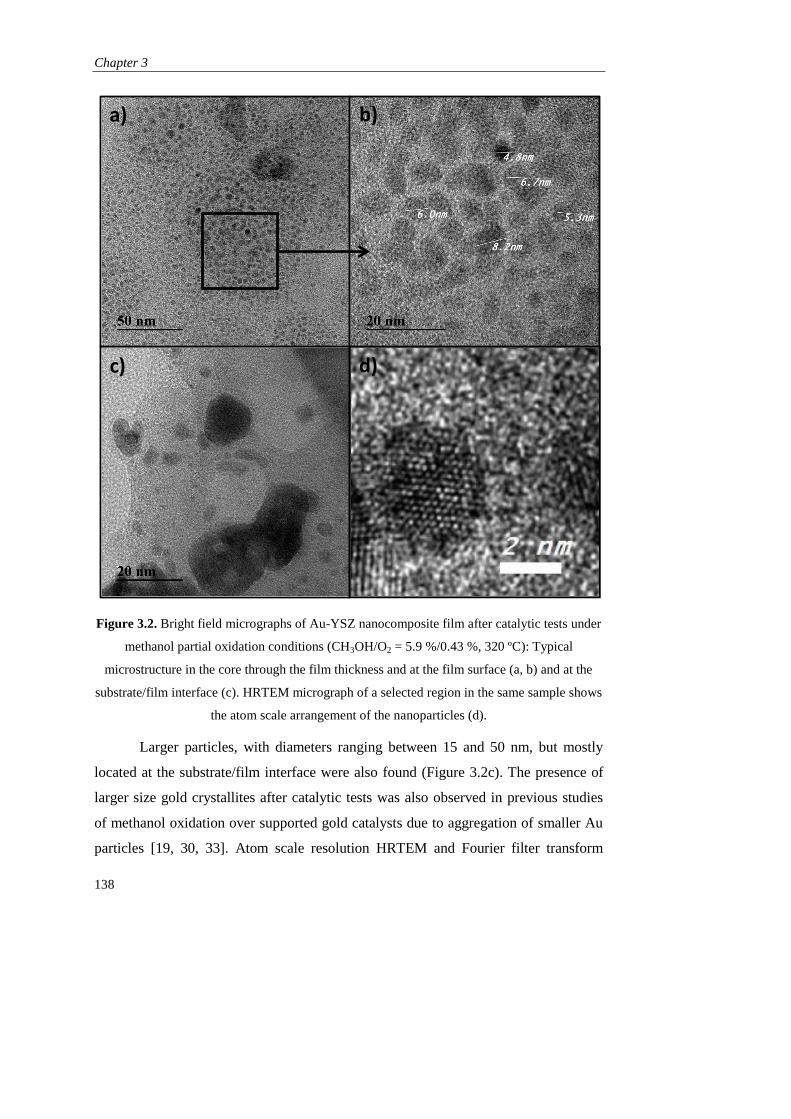
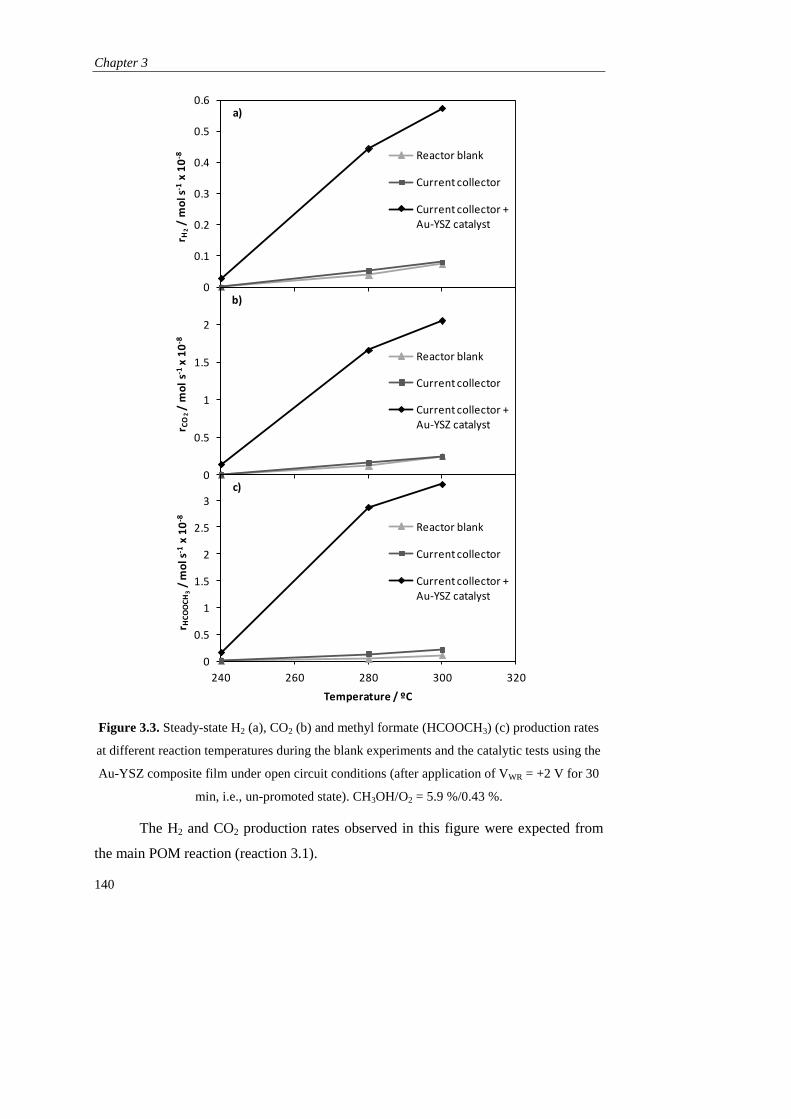
DEPARTAMENTO DE INGENIERÍA QUÍMICA
ELECTROCHEMICAL PROMOTION OF NOVEL
CATALYSTS WITH ALKALINE CONDUCTORS FOR
HYDROGEN PRODUCTION FROM METHANOL
Memoria que para optar al grado de Doctor en Ingeniería Química
(Doctorado Internacional) presenta
JESÚS GONZÁLEZ COBOS
Directores:
Dr. José Luis Valverde Palomino
Dr. Antonio de Lucas Consuegra
Ciudad Real, 2015
Page 3
Nomenclature
v
Acronyms
CAD Cathodic arc deposition
CE Counter electrode
CV Cyclic voltammetry
DLC Diamond-like carbon
EBE Electron beam evaporation
EDX Energy-dispersive X-ray spectroscopy
EELS Electron energy loss spectroscopy
EPOC Electrochemical promotion of catalysis
FCC Face-centered cubic structure
FTIR Fourier transform infrared spectroscopy
GC Gas chromatograph
GLAD Glancing angle deposition
GO Graphene oxide
HRTEM High-resolution transmission electron microscopy
JCPDS Joint committee on powder diffraction standards
LSV Linear sweep voltammetry
MD Methanol decomposition
MEPR Monolithic electrochemically promoted reactor
NASICON Sodium super ionic conductor
NEMCA Non faradaic electrochemical modification of catalytic activity
OAD Oblique angle deposition
OC Open circuit
PEMFC Proton exchange membrane fuel cell
POM Partial oxidation of methanol
PVD Physical vapor deposition
RE Reference electrode
SCY Strontia-ceria-ytterbia
SEM Scanning electron microscopy
SOFC Solid oxide fuel cell
SRM Steam reforming of methanol
STEM Scanning transmission electron microscopy
TOF Turnover frequency
tpb three-phase boundaries
TPS Temperature-programmed stabilization
XPS X-ray photoelectron spectroscopy
XRD X-ray diffraction
XRF X-ray fluorescence
Page 4
Nomenclature
vi
WE Working electrode
WGS Water-gas shift
YSZ Yttria-stabilized zirconia
Symbols
B Full width at half maximum (FWHM) of a XRD peak
D Metal dispersion
d Metal particle size
F Faraday constant (96485 C)
Fi Molar flow of i compound
I Current
i Current density
KW Half-width Scherrer constant
Mm Metal atomic weight
N Total surface area
NA Avogadro number (6.023 x 1023
)
NG Active surface area
n Charge of the ionic species (1 for K+)
Promotion index
r Catalytic reaction rate under promoted conditions
r0 Catalytic reaction rate under unpromoted conditions
re, rK+ Electrocatalytic reaction rate
t Time
VWR Catalyst potential
Catalyst potential under open circuit conditions
α In OAD, zenithal incident angle
β In OAD, zenithal tilt angle of deposited metal columns
γ Permanent enhancement ratio
θ Bragg angle
Promoter coverage
λ X-ray wavelength
ρ Rate enhancement ratio
ρm Metal density
σ Metal atomic surface
ϕ Work function
Λ Faradaic efficiency
Page 5
Table of contents
vii
Descripción del trabajo realizado……………………………………………….. 1
A. INTRODUCCIÓN....................................................................................... 2
A.1. El hidrógeno como vector energético………………………………... 2
A.2. El metanol como fuente de hidrógeno……………………………….. 8
A.3. La promoción electroquímica de la catálisis (EPOC)……………….. 10
A.4. Nuevas tendencias y perspectivas de la promoción electroquímica…. 18
A.5. Objetivo de la tesis doctoral…………………………………………. 25
B. MÉTODOS E INSTALACIÓN EXPERIMENTAL……………………… 27
B.1. Preparación de los catalizadores electroquímicos…………………… 27
B.2. Técnicas de caracterización………………………………………….. 29
B.3. Instalación experimental……………………………………………... 31
C. RESULTADOS OBTENIDOS…………………………………………… 33
D. CONCLUSIONES Y RECOMENDACIONES…………………………... 44
E. BIBLIOGRAFÍA………………………………………………………….. 47
Abstract……………. ……………………………………………………………... 55
Chapter 1. Electrochemical promotion of Pt for H2 production from
methanol partial oxidation and steam reforming: A better performance of
pysical vapor deposited catalyst films……………………………………………... 63
1.1. INTRODUCTION………………………………………………………... 64
1.2. EXPERIMENTAL……………………………………………………….. 67
1.2.1. Preparation of the electrochemical catalysts……………………... 67
1.2.2. Characterization measurements…………………………………... 69
1.2.3. Catalytic activity measurements………………………………….. 70
1.3. RESULTS AND DISCUSSION…………………………………………. 71
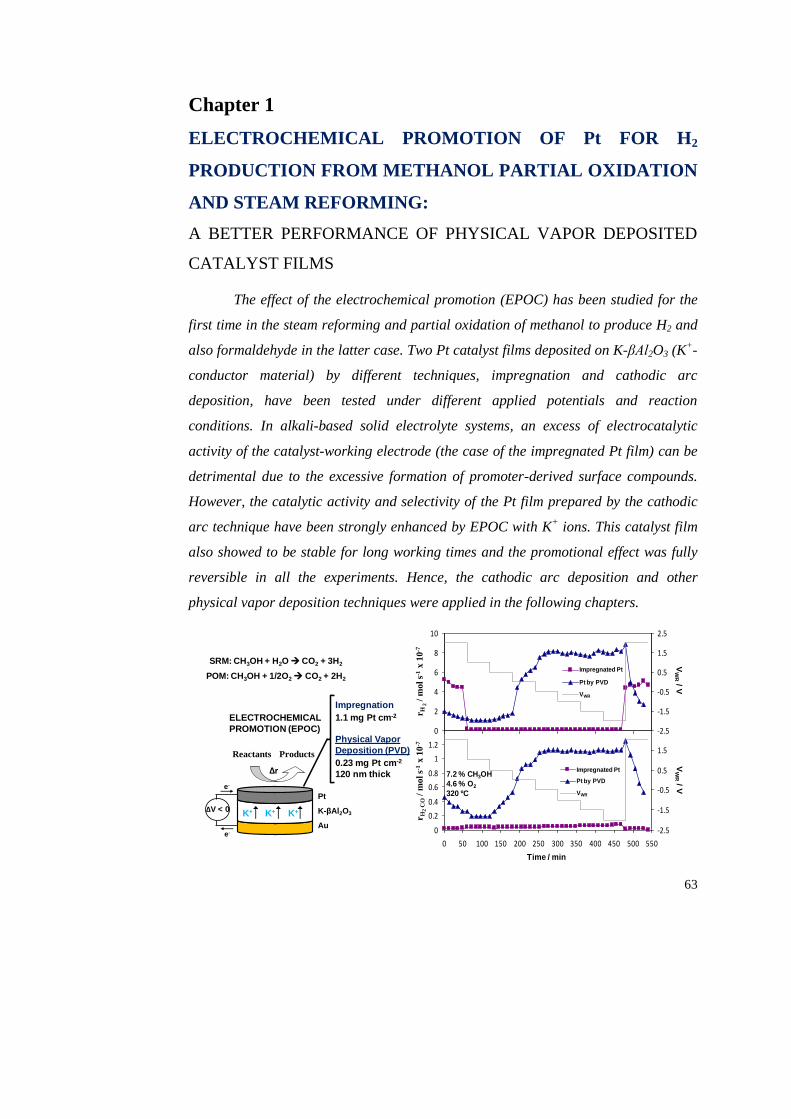
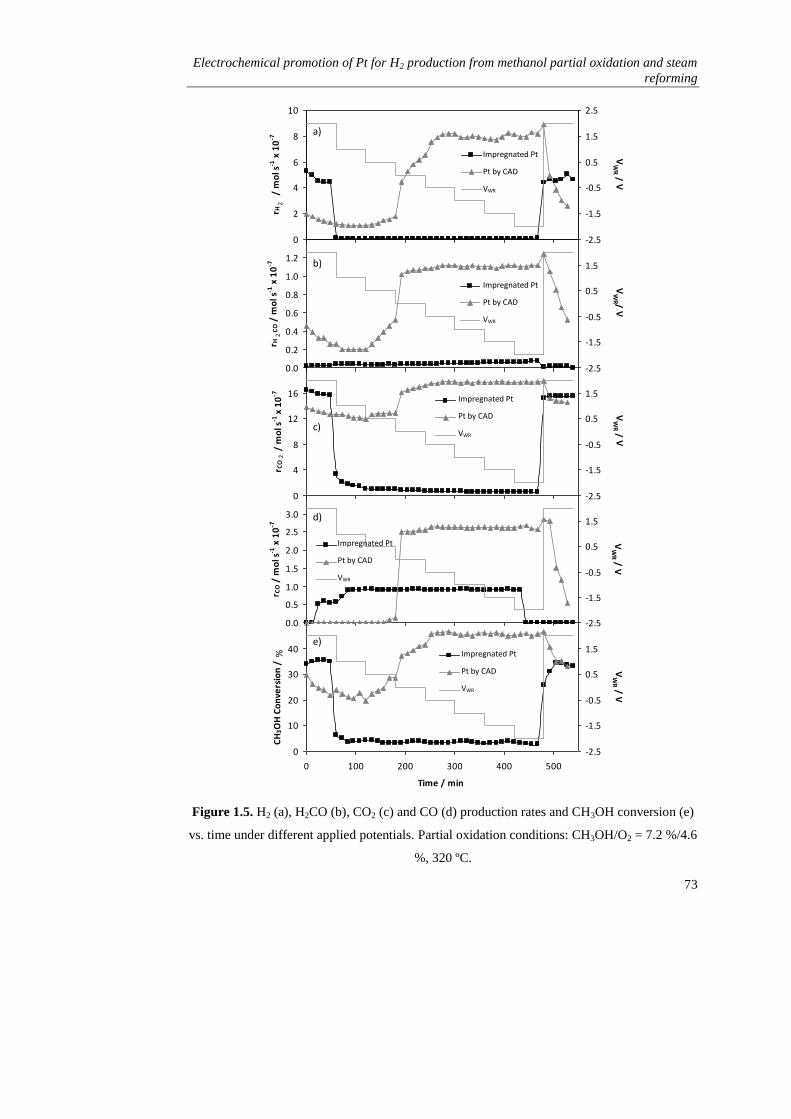
1.3.1. Partial oxidation of methanol…………………………………….. 71
i) Influence of the preparation technique………………………..... 71
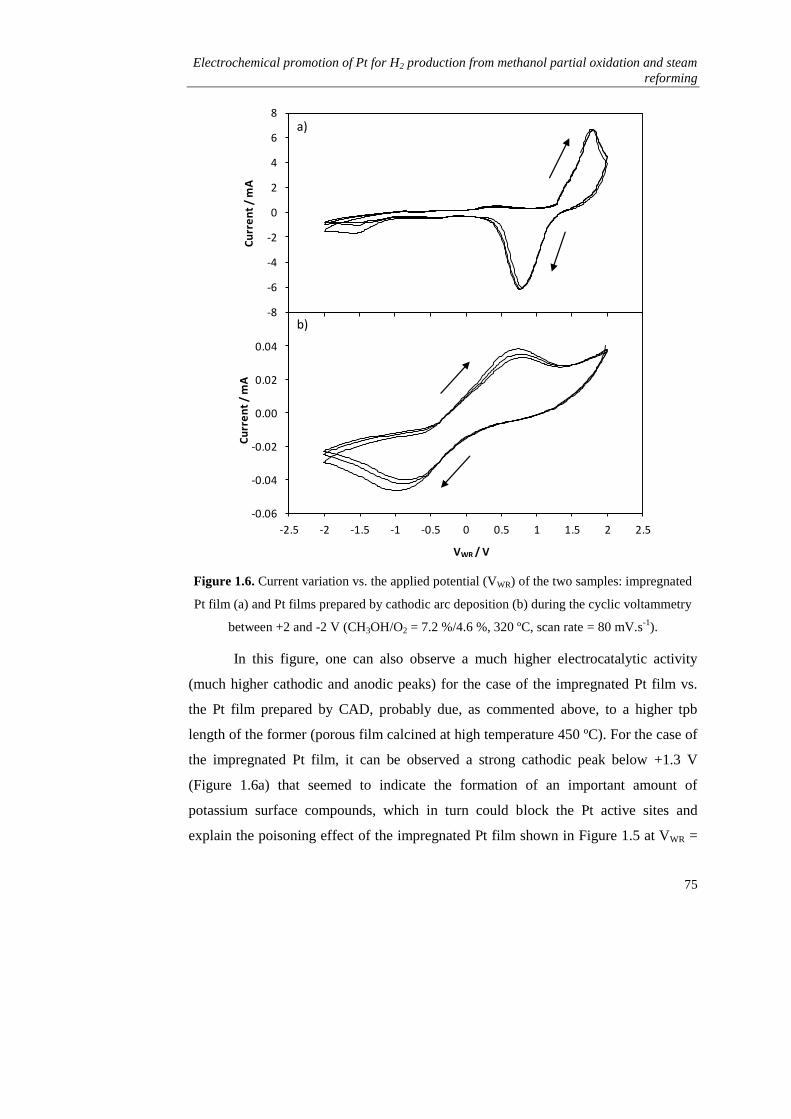
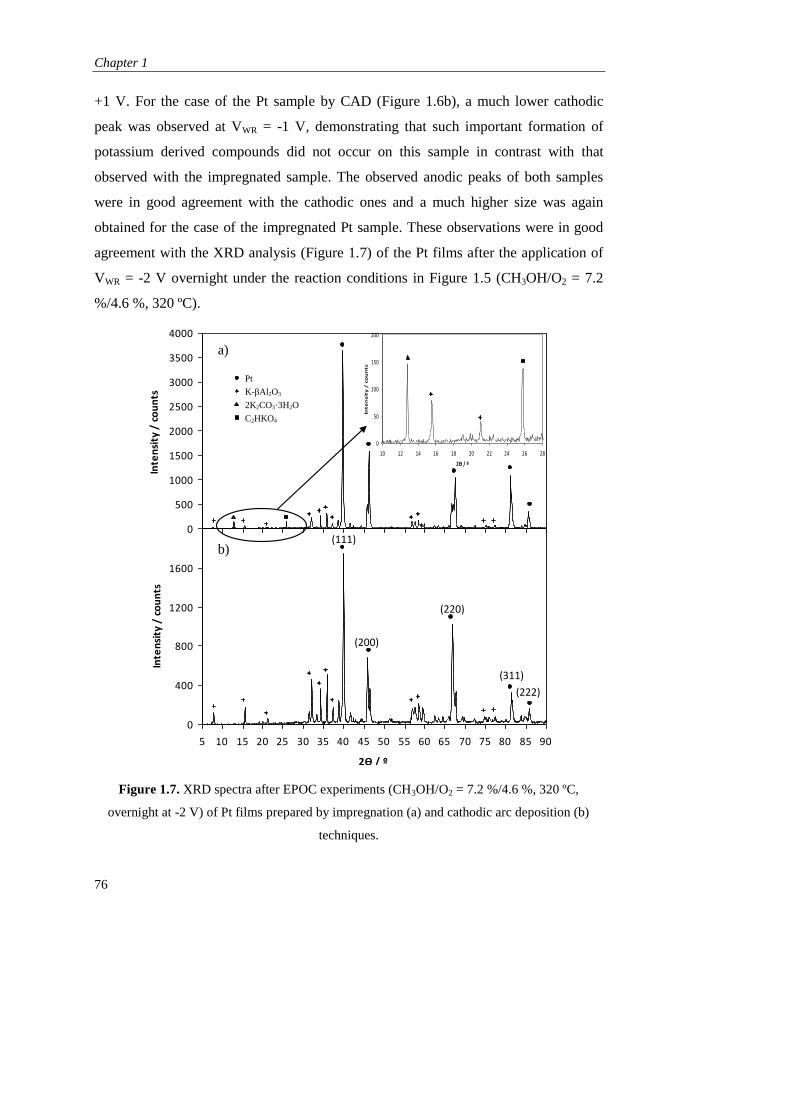
ii) Electrochemical promotion mechanism and parameters………. 79
iii) Influence of the reaction conditions……………………………. 83
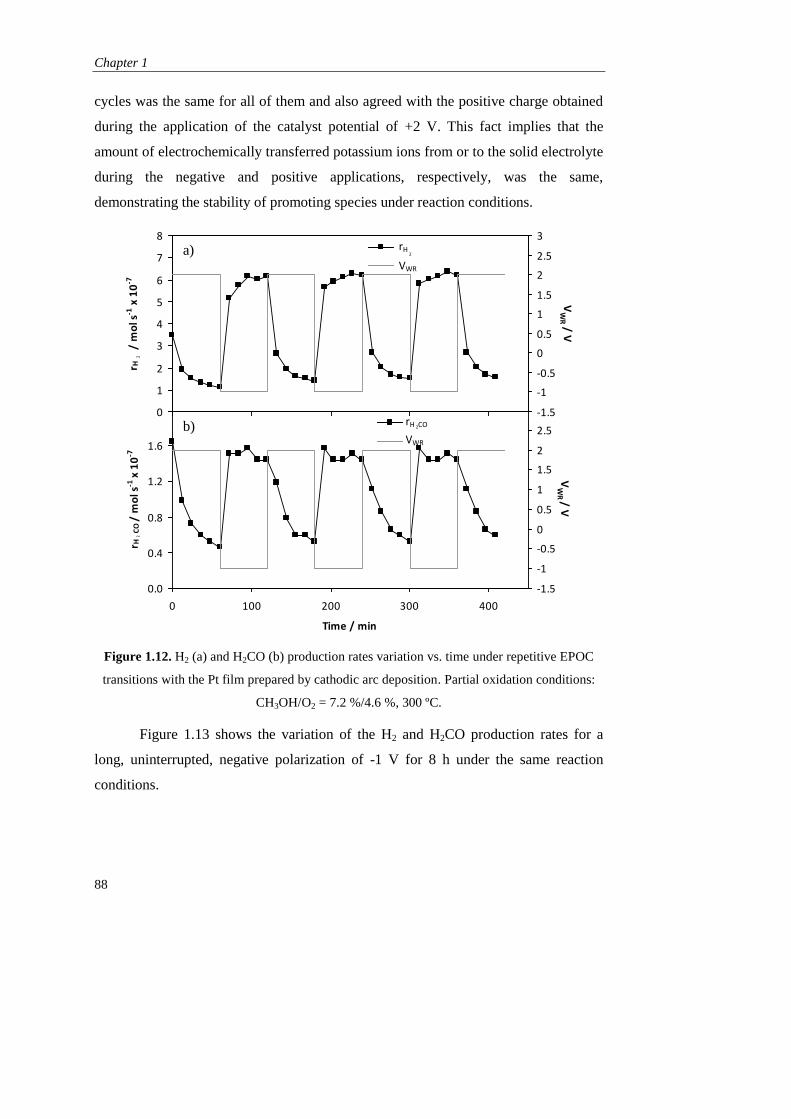
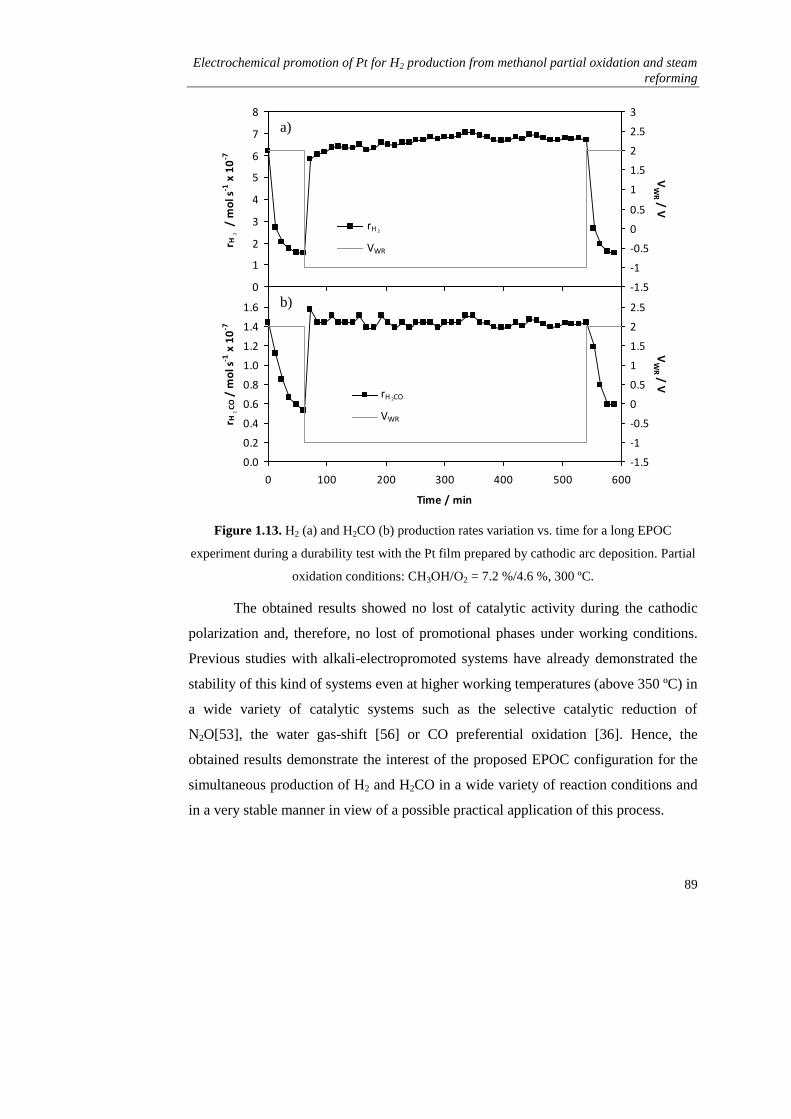
iv) Stability study…………………………………………………… 87
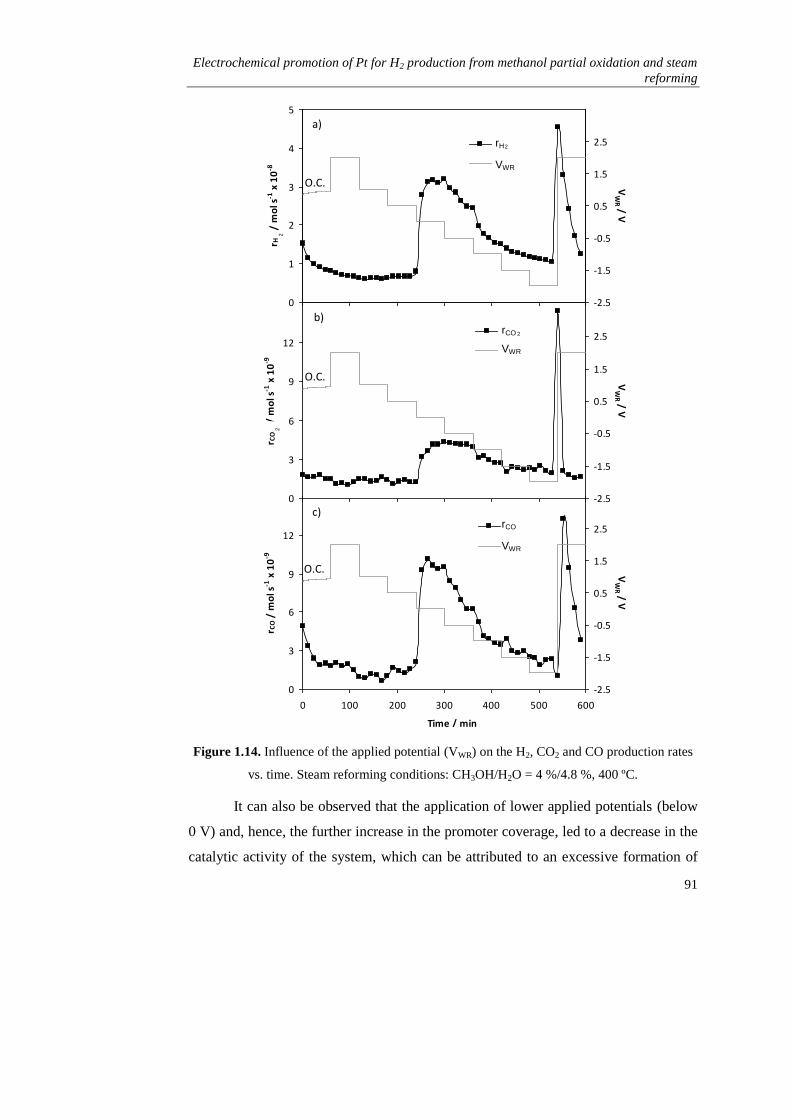
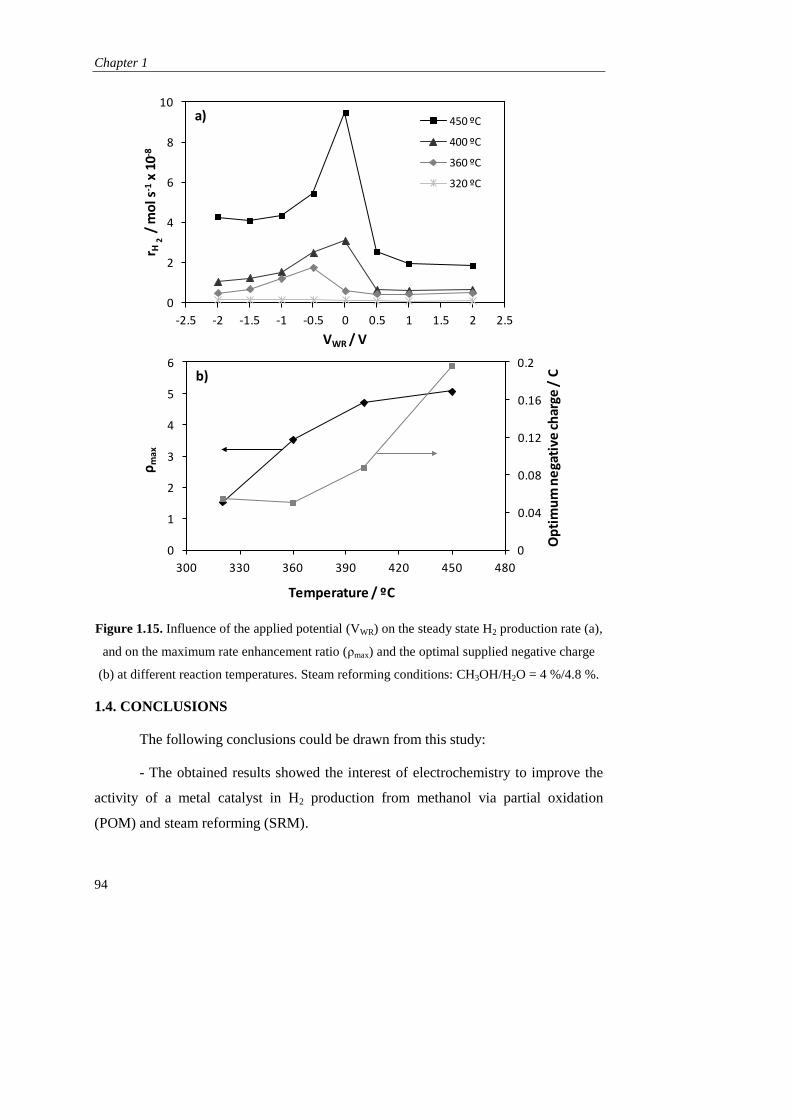
1.3.2. Steam reforming of methanol…………………………………….. 90
1.4. CONCLUSIONS…………………………………………………………. 94
1.5. REFERENCES…………………………………………………………… 95
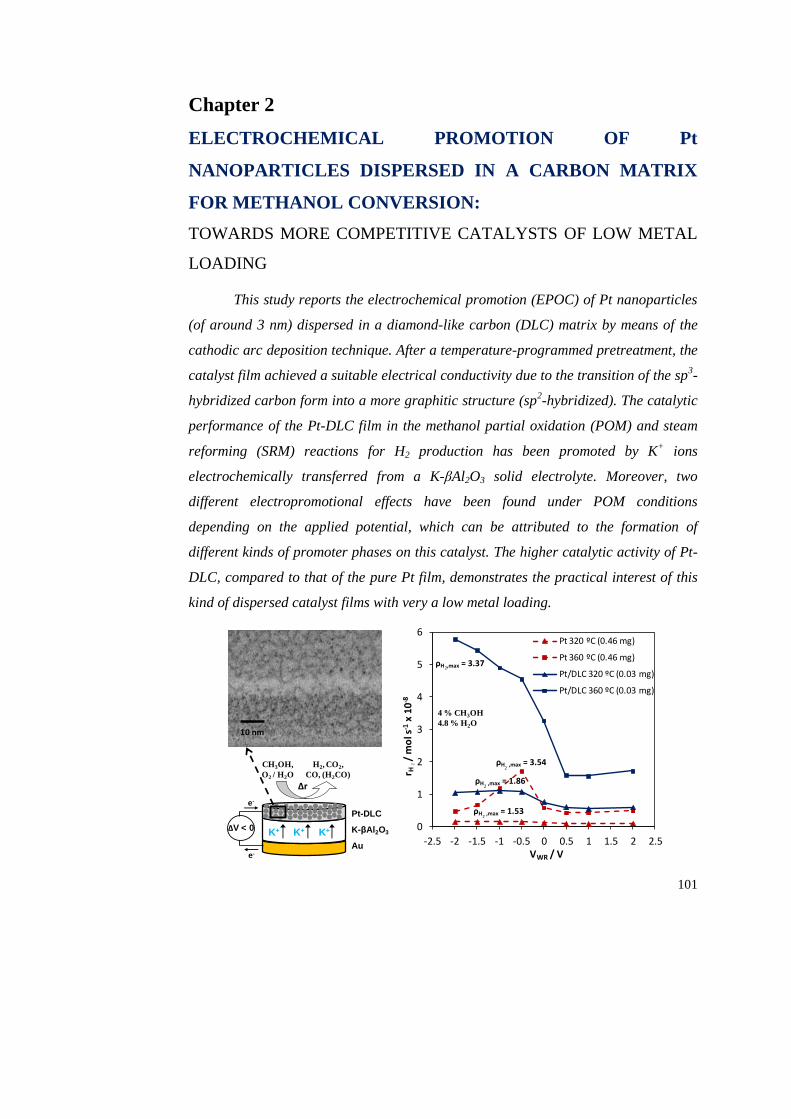
Chapter 2. Electrochemical promotion of Pt nanoparticles dispersed in a
carbon matrix for methanol conversion: Towards more competitive catalysts
of low metal loading.. ……………………………………………………………... 101
2.1. INTRODUCTION………………………………………………………... 102
2.2. EXPERIMENTAL………………………………………………………... 104
2.2.1. Preparation of the electrochemical catalyst………………………. 104
Page 6
Table of contents
viii
2.2.2. Characterization measurements…………………………………... 105
2.2.3. Catalytic activity measurements………………………………….. 106
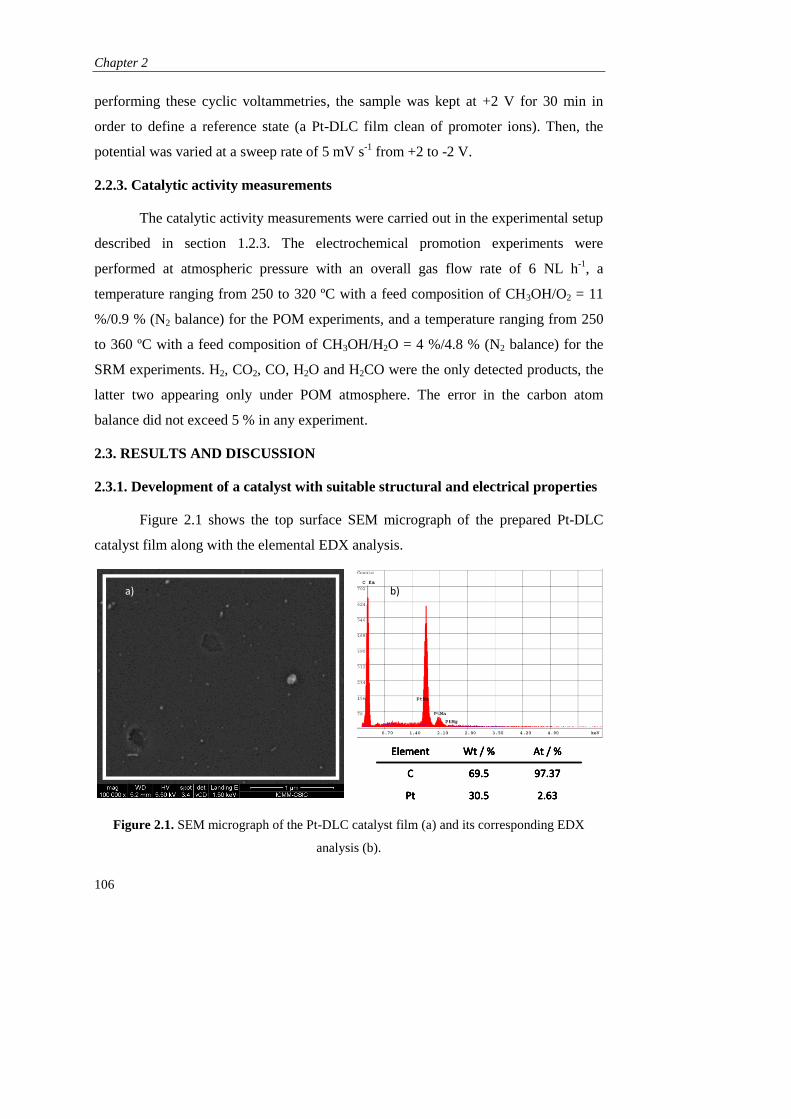
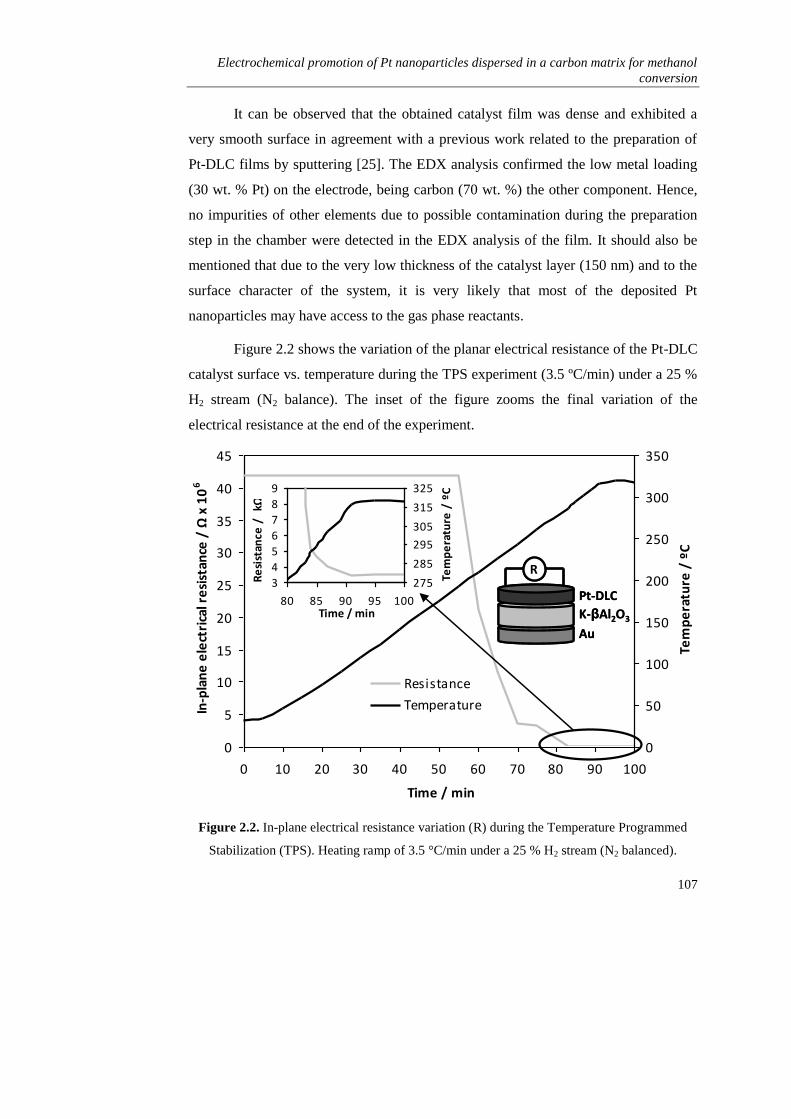
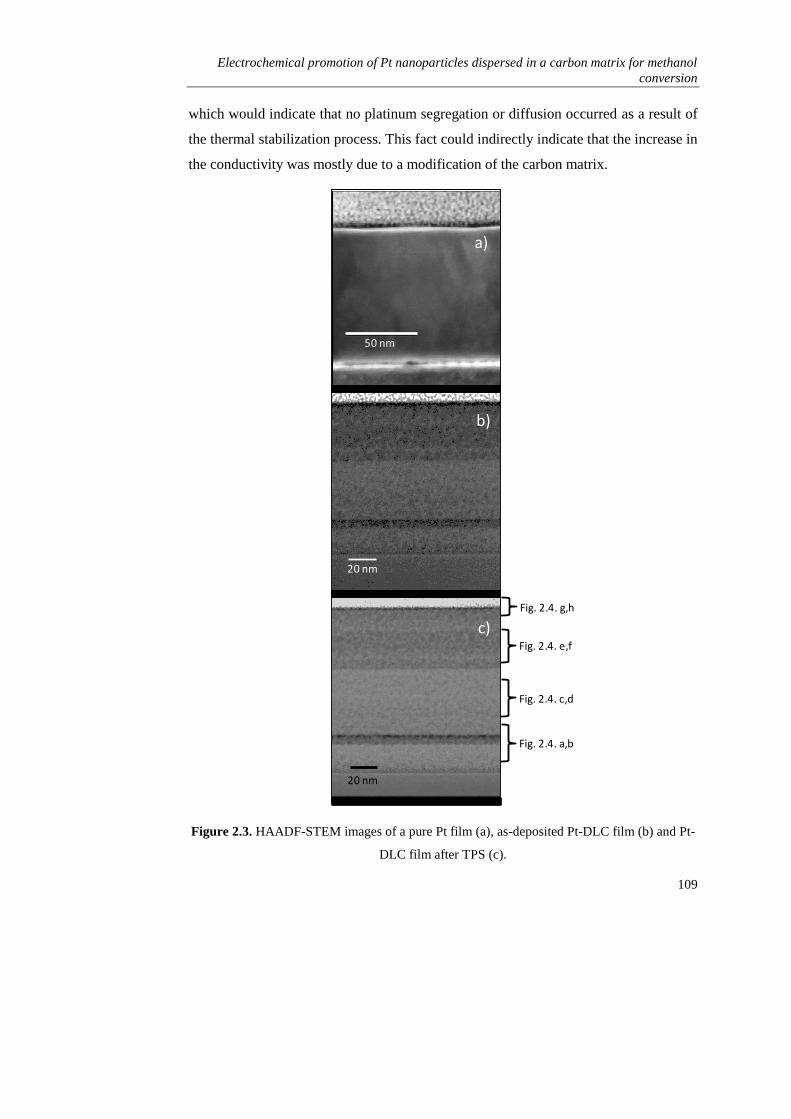
2.3. RESULTS AND DISCUSSION………………………………………... 106
2.3.1. Development of a catalyst with suitable structural and electrical
properties…………………………………………………………. 106
2.3.2. Electrochemical promotion experiments…………………………. 112
2.3.3. Comparison between Pt-DLC and Pt…………………………….. 120
2.4. CONCLUSIONS……………………………………………………….. 123
2.5. REFERENCES…………………………………………………………. 124
Chapter 3. Electrochemical activation of Au nanoparticles dispersed in
YSZ for methanol partial oxidation: Electrochemical promotion of a
non-conductive catalyst film..……………………………………………………... 131
3.1. INTRODUCTION………………………………………………………... 132
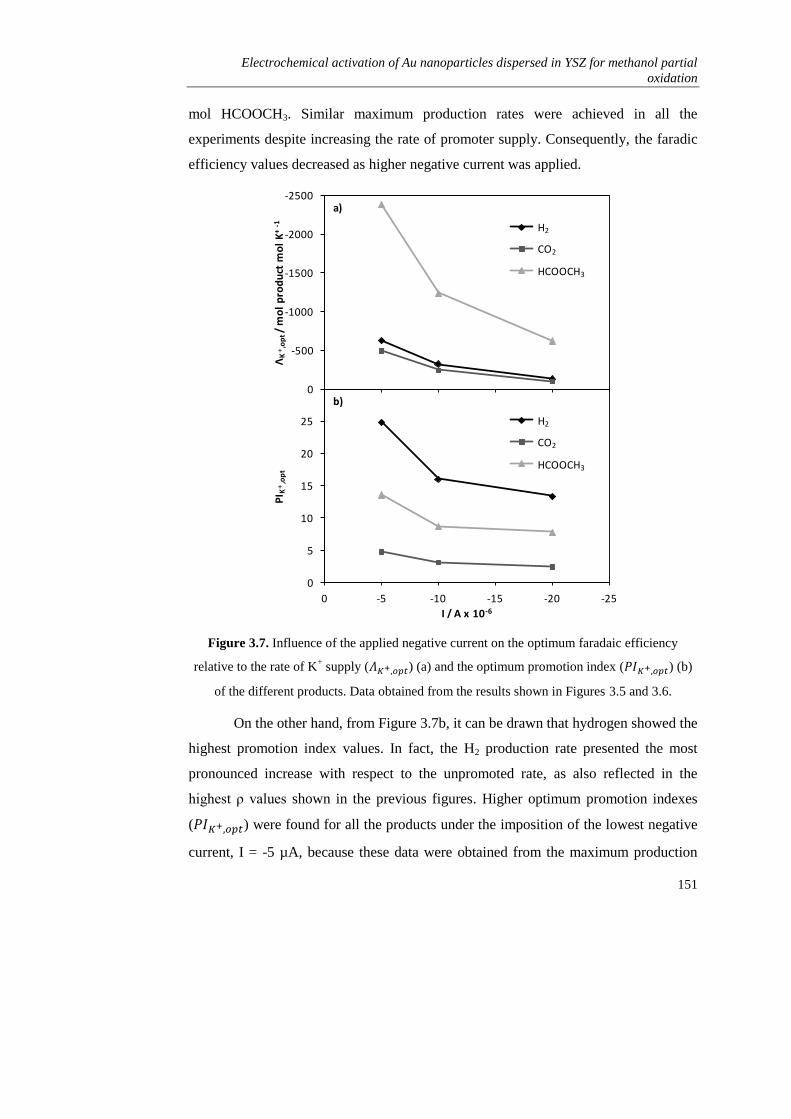
3.2. EXPERIMENTAL……………………………………………………….. 134
3.2.1. Preparation of the electrochemical catalyst………………………. 134
3.2.2. Characterization measurements…………………………………... 135
3.2.3. Catalytic activity measurements………………………………….. 136
3.3. RESULTS AND DISCUSSION…………………………………………. 136
3.3.1. Characterization of the catalyst film and blank experiments…….. 136
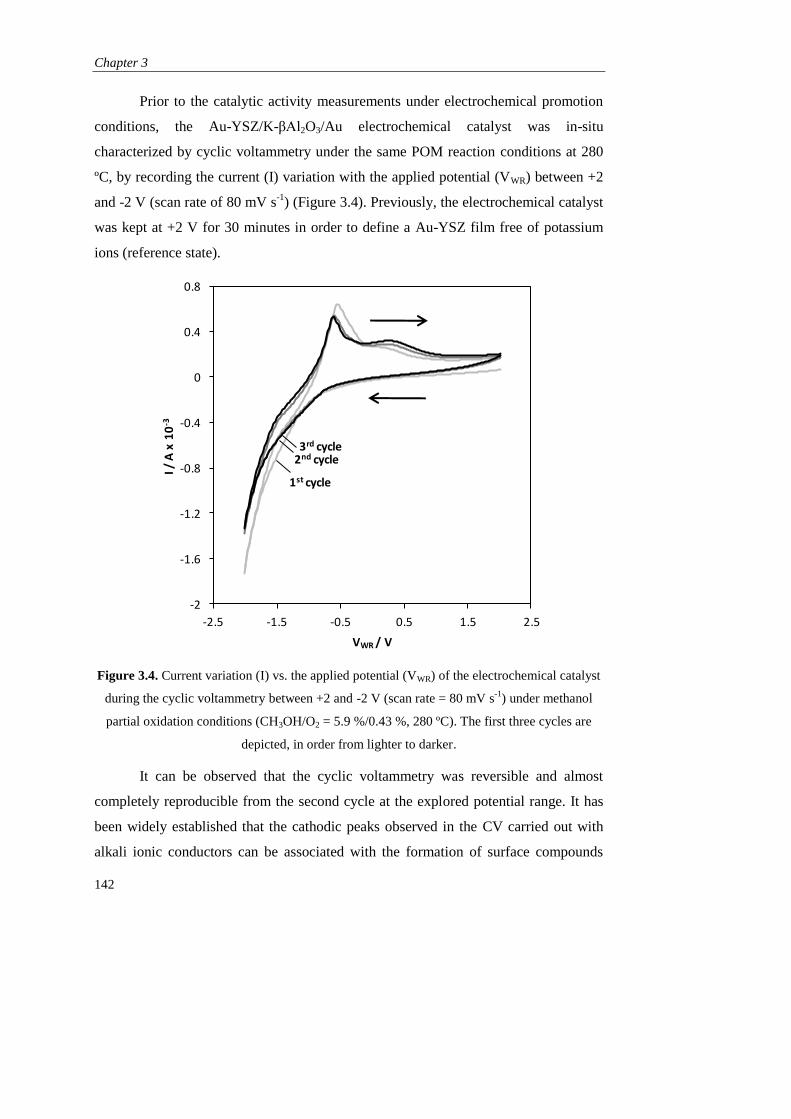
3.3.2. Electrochemical promotion via galvanostatic transitions………… 143
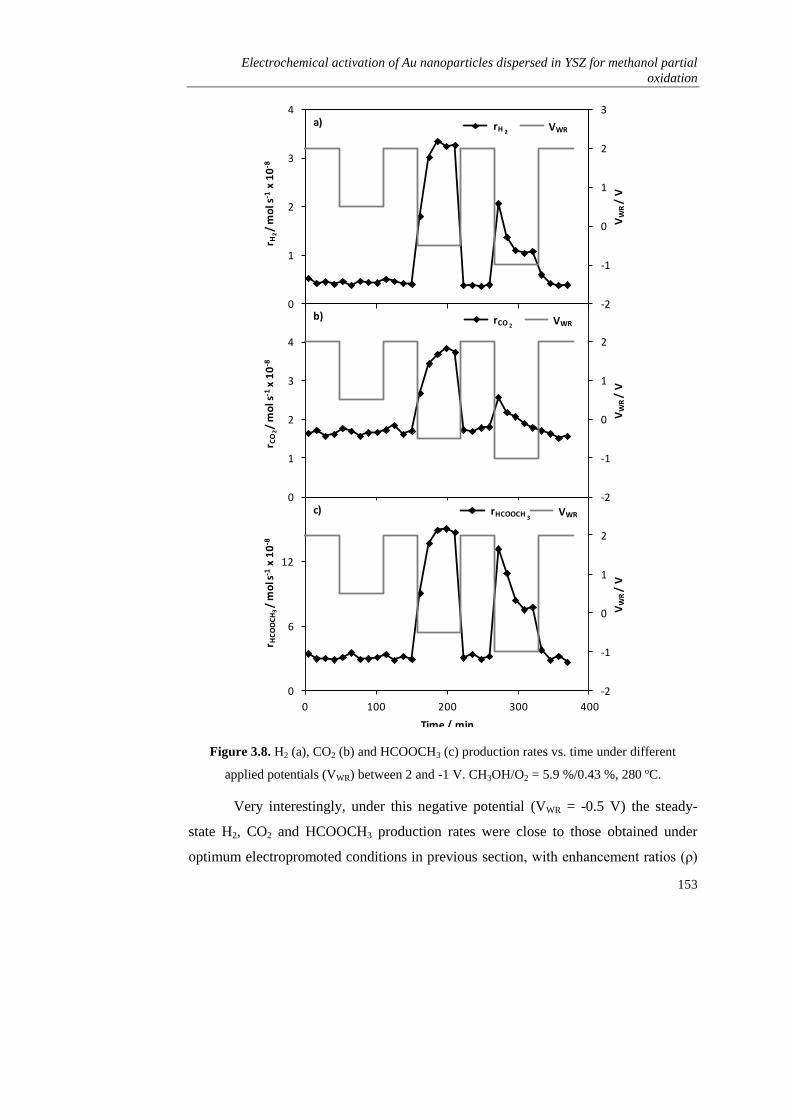
3.3.3. Electrochemical promotion via potentiostatic transitions………... 152
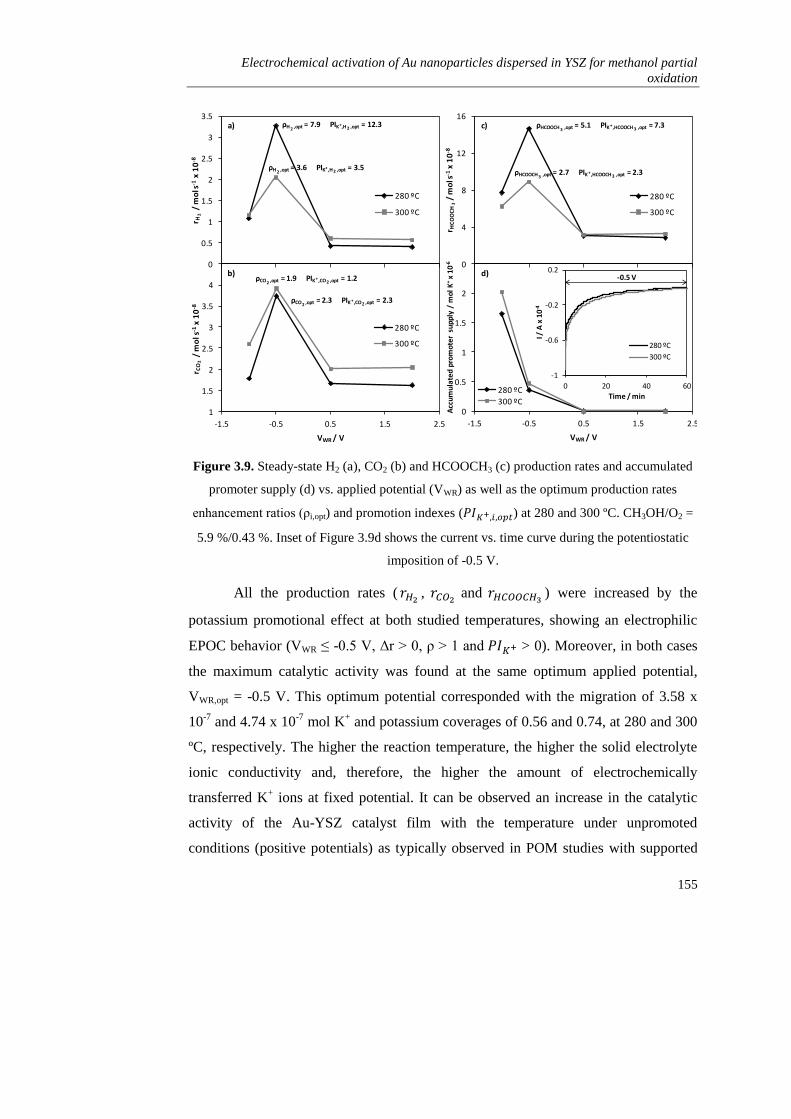
3.4. CONCLUSIONS…………………………………………………………. 156
3.5. REFERENCES…………………………………………………………… 157
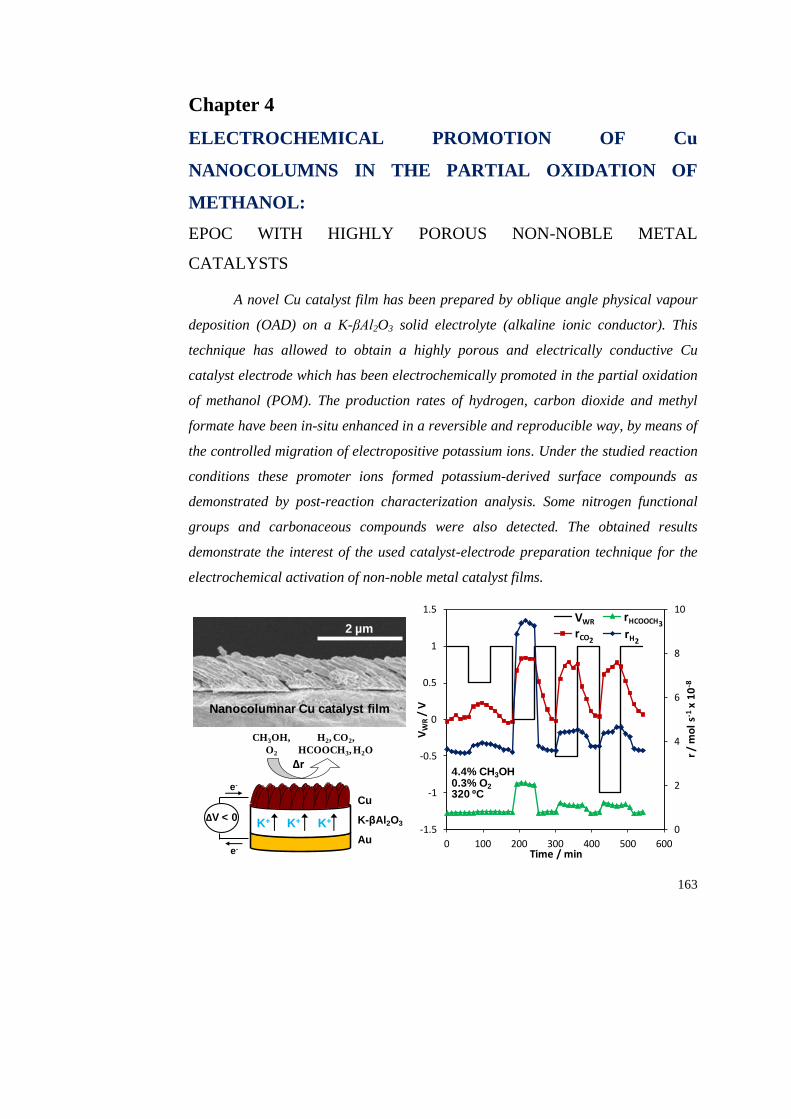
Chapter 4. Electrochemical promotion of Cu nanocolumns in the partial
oxidation of methanol: EPOC with highly porous non-noble metal catalysts…… 163
4.1. INTRODUCTION………………………………………………………... 164
4.2. EXPERIMENTAL……………………………………………………….. 166
4.2.1. Preparation of the electrochemical catalyst………………………. 166
4.2.2. Characterization measurements…………………………………... 167
4.2.3. Catalytic activity measurements………………………………….. 168
4.3. RESULTS AND DISCUSSION…………………………………………. 168
4.3.1. Preliminary characterization of the catalyst film………………… 168
4.3.2. Electrochemical promotion experiments…………………………. 172
4.3.3. Post-reaction characterization of the catalyst film……………….. 180
4.4. CONCLUSIONS…………………………………………………………. 185
4.5. REFERENCES…………………………………………………………… 185
Page 7
Table of contents
ix
Chapter 5. Electrochemical promotion of Ni in methanol conversion
reactions: Different applications of EPOC on a single catalytic system………….. 191
5.1. INTRODUCTION………………………………………………………... 192
5.2. EXPERIMENTAL……………………………………………………….. 194
5.2.1. Preparation of the electrochemical catalyst………………………. 194
5.2.2. Characterization measurements…………………………………... 195
5.2.3. Catalytic activity measurements………………………………….. 195
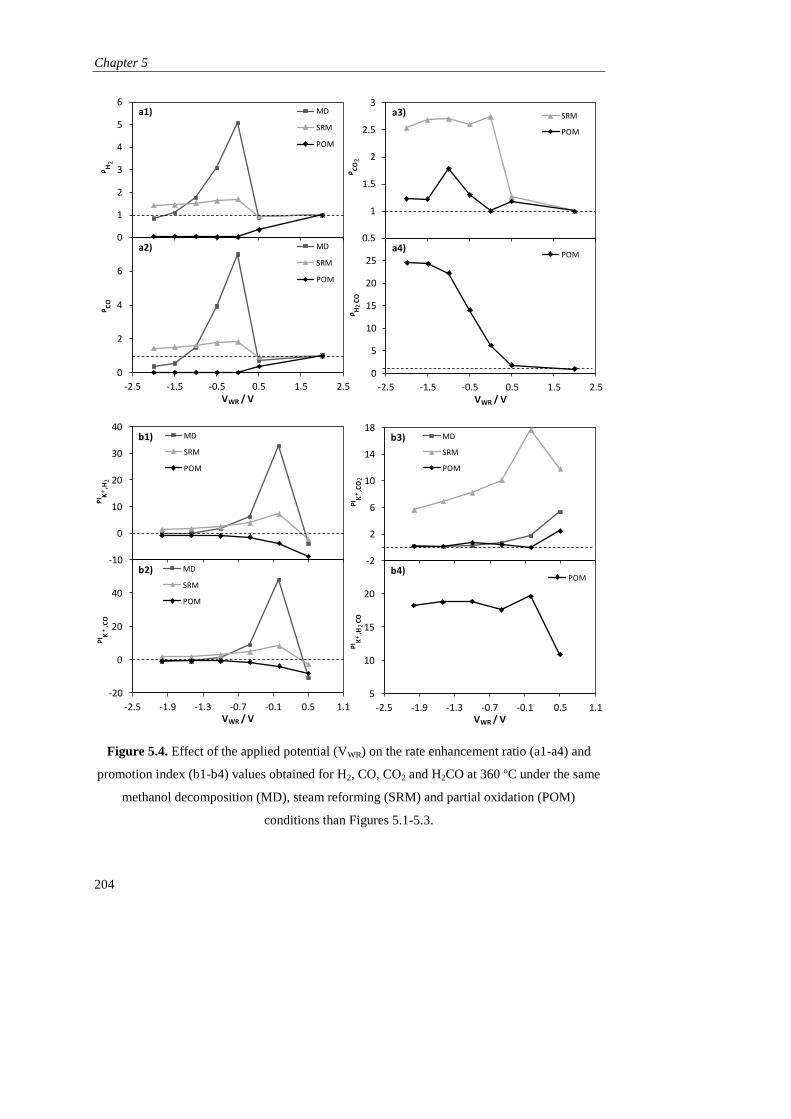
5.3. RESULTS AND DISCUSSION…………………………………………. 196
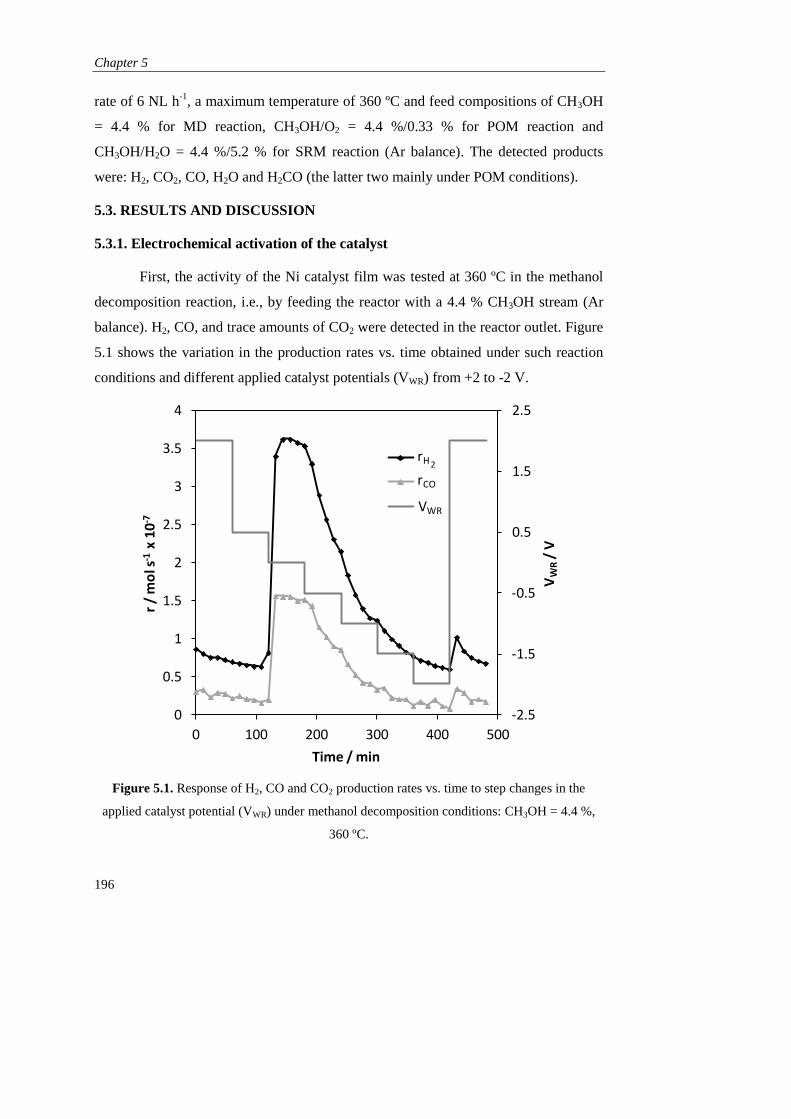
5.3.1. Electrochemical activation of the catalyst………………………... 196
5.3.2. Effect of the electrochemical promotion on the catalyst
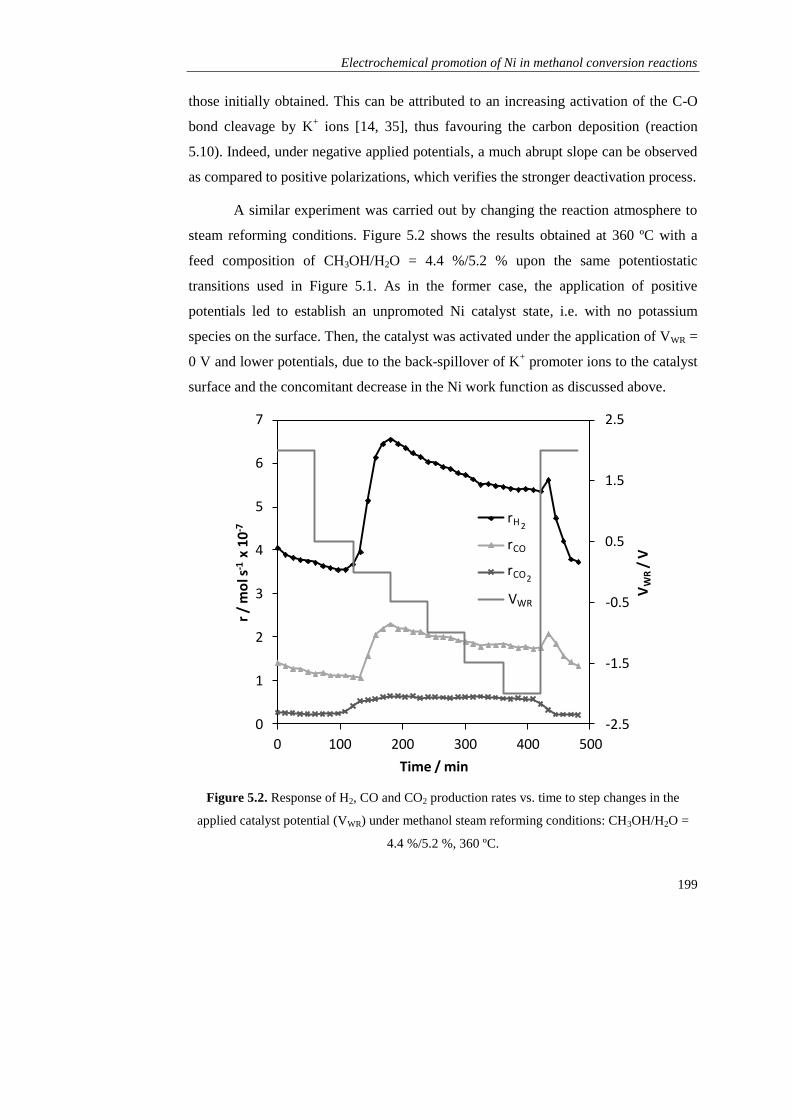
oxidation state……………………………………………………. 206
5.3.3. Control of the catalyst selectivity via electrochemical
promotion………………………………………………………… 210
5.4. CONCLUSIONS…………………………………………………………. 214
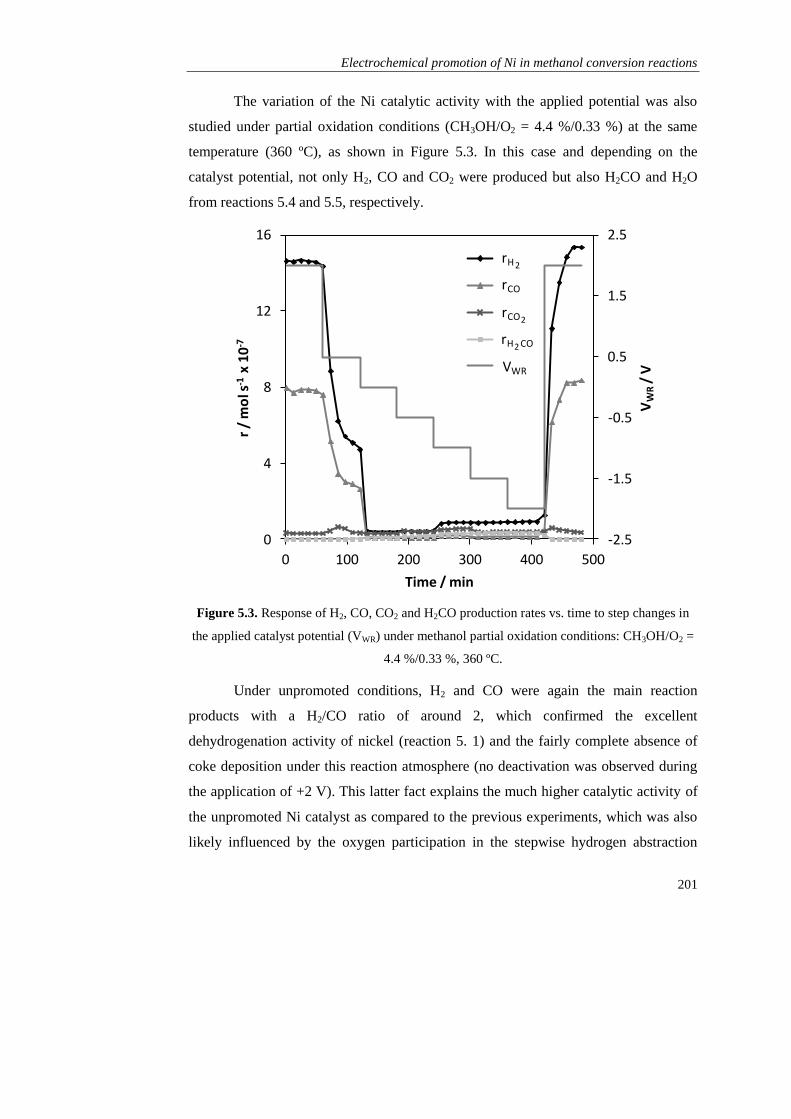
5.5. REFERENCES……………………………………………………………. 215
Chapter 6. Electrochemically assisted production and storage of hydrogen:
A novel contribution of alkaline electrochemical catalysts………………………... 221
6.1. INTRODUCTION………………………………………………………... 222
6.2. EXPERIMENTAL……………………………………………………….. 224
6.2.1. Preparation of the electrochemical catalyst………………………. 224
6.2.2. Characterization measurements…………………………………... 227
6.2.3. Catalytic activity measurements………………………………….. 228
6.3. RESULTS AND DISCUSSION………………………………………….. 228
6.3.1. Preliminary experiments of H2 production and storage………….. 228
6.3.2. Influence of the applied negative polarization and the reaction
atmosphere……………………………………………………….. 234
6.3.3. Investigation of possible surface compounds by catalyst
characterization…………………………………………………... 242
6.4. CONCLUSIONS………………………………………………………….. 246
6.5. REFERENCES……………………………………………………………. 247
Chapter 7. General conclusions and recommendations……………………….. 255
7.1. CONCLUSIONS…………………………………………………………. 256
7.2. RECOMMENDATIONS………………………………………………… 258
List of publications and conferences…………………………………………….. 261
Page 8
Table of contents
x
Page 9
1
DESCRIPCIÓN DEL TRABAJO REALIZADO
Este trabajo forma parte de un amplio programa de investigación sobre la
aplicación de sistemas electrocatalíticos en procesos de interés energético y
medioambiental que se está desarrollando durante los últimos años en el
Departamento de Ingeniería Química de la Universidad de Castilla-La Mancha
(UCLM).
En particular, esta Tesis Doctoral tiene como objetivo el estudio del fenómeno
de la promoción electroquímica de la catálisis en la producción de hidrógeno a partir
de metanol empleando conductores alcalinos. Este trabajo ha sido financiado por el
Ministerio de Ciencia e Innovación a través del proyecto del plan Nacional CTQ
2010-16179/PQ, y por el Ministerio de Economía y Competitividad a través del
proyecto CTQ 2013-45030-R.
Esta tesis doctoral se ha realizado en colaboración con el Instituto de Ciencia
de Materiales de Madrid (CSIC), el Institut Jean Lamour de Université de Lorraine
(Francia) y el Instituto de Ciencia de Materiales de Sevilla (CSIC).
Page 10
Descripción del trabajo realizado
2
A. INTRODUCCIÓN
A.1. El hidrógeno como vector energético
El consumo masivo de combustibles fósiles como el petróleo, asociado al
fuerte desarrollo tecnológico de las últimas décadas, ha dado lugar a una alarma
permanente sobre el irremediable agotamiento de las reservas de estos recursos
naturales y el grave deterioro del medioambiente que está ocasionando. Por ello la
humanidad se enfrenta actualmente a un triple desafío: satisfacer las necesidades
energéticas de la población, buscar alternativas al agotamiento de los combustibles
fósiles, y abordar seriamente la amenaza que supone el sobrecalentamiento del planeta
debido a la emisión de gases de efecto invernadero como el CO2 [1].
En este sentido, el hidrógeno se está postulando como una de las alternativas
energéticas más prometedoras, llegándose a hablar incluso de una futura “economía
del hidrógeno” [2]. Uno de los principales motivos es su gran densidad energética
gravimétrica en comparación con el resto de combustibles (Tabla A.1).
Tabla A.1. Densidad energética de diferentes combustibles (adaptado de Dutta y col. [3]).
Combustible Densidad energética (kWh kg-1
)
Hidrógeno 33,3
Gas natural licuado 15,1
Propano 13,8
Gasolina de aviación 13,0
Gasolina de automoción 12,9
Gasoil de automoción 12,7
Etanol 8,2
Carbón 7,5
Metanol 5,5
Madera seca 4,5
Además, el H2 obtenido de forma renovable es considerado mundialmente
como un vector energético limpio, puesto que el único subproducto derivado de su
combustión con oxígeno es vapor de agua [4].
Page 11
Introducción
3
A.1.1. Aplicaciones del hidrógeno
i) Materia prima en la industria química.
En torno a la mitad del hidrógeno consumido anualmente en el mundo (sobre 5
x 1010
kg H2 [4]) se emplea en la elaboración de fertilizantes basados en amoníaco por
medio del proceso Haber-Bosh (reacción A.1), que típicamente tiene lugar a elevadas
presiones (200 bar) y temperaturas (400-500 ºC) con catalizadores de Fe o Ru [5].
N2 + 3H2 → 2NH3 (A.1)
Otra gran parte del H2 se consume en la industria petroquímica,
particularmente en el refinado del petróleo (proceso de hidrocraqueo catalítico) y para
reducir la cantidad de aromáticos y sobre todo de azufre en la gasolina y el gasóleo
(hidrodesulfuración). El hidrógeno también se utiliza en la elaboración de agua
oxigenada y junto con CO (gas de síntesis) en la obtención de metanol [6] y otros
muchos combustibles (proceso Fischer-Tropsch) [7].
ii) Generación de energía.
Existen principalmente dos vías para la obtención de energía a partir del H2.
Por un lado, el empleo de motores de combustión interna o turbinas de gas permite
transformar la energía química del hidrógeno en energía térmica y esta en energía
mecánica. Por otro lado, el hidrógeno se puede oxidar electroquímicamente en una
celda de combustible, en cuyo caso la energía química se transforma directamente en
energía eléctrica, evitando la limitación del ciclo de Carnot. Por ejemplo, un coche que
necesitara unos 24 kg de gasolina para recorrer 400 km, consumiría en su lugar
aproximadamente 8 kg H2 de hidrógeno en un motor de combustión interna, o 4 kg H2
en el caso de un coche eléctrico con pila de combustible [4].
iii) Otras aplicaciones.
El hidrógeno también se emplea para refrigerar motores y generadores
eléctricos y como gas protector en el método de soldadura por arco eléctrico. Debido a
su baja densidad (0,0899 kg m-3
), en el pasado incluso se utilizaba como gas de
elevación en globos aerostáticos y dirigibles.
Page 12
Descripción del trabajo realizado
4
A.1.2. Tecnologías de producción de hidrógeno
El hidrógeno es el elemento más abundante del planeta, pero menos del 1 % se
encuentra en forma de gas molecular (H2) y es necesario obtenerlo a partir de agua,
hidrocarburos u otra materia orgánica por medio de muy diversos procesos. A
continuación se mencionan algunos de los más importantes.
i) Obtención de H2 a partir de agua.
Existen básicamente tres maneras de provocar la ruptura de la molécula de
agua: termólisis (por acción de la temperatura), fotoelectrólisis (por acción de la luz
solar) y electrólisis (por acción de la electricidad). Este último proceso es de los más
estudiados y se suele llevar a cabo en electrolizadores alcalinos, electrolizadores de
membranas de intercambio de protones (tipo PEM) o de óxido sólido (tipo SOEC) [8,
9].
ii) Producción de H2 mediante procesos biológicos.
El H2 se puede producir a partir de la conversión de la biomasa mediante los
procesos de gasificación, pirolisis, o hidrólisis [10]. También se puede obtener a partir
de la fotólisis directa del agua empleando microalgas verdes o cianobacterias y
procesos fermentativos, entre otros [3, 10].
iii) Procesos de conversión de hidrocarburos y compuestos oxigenados.
Actualmente la principal vía de producción de hidrógeno a nivel industrial es
el reformado de metano con vapor de agua, si bien es cierto que el gas de síntesis (CO
+ H2) obtenido se suele utilizar en las refinerías para su consumo propio. Existen tres
procesos tradicionales de producción de H2 a partir de hidrocarburos:
Reformado con vapor de agua (“Steam Reforming”, SR):
CnHm + nH2O → nCO + (n + m/2)H2 (A.2)
Esta es una reacción altamente endotérmica que se suele llevar a cabo a
temperaturas superiores 500 ºC y 1-25 atm. Los catalizadores empleados se basan en
Ni, Cu, o metales nobles como Pt o Rh [11]. Los primeros son más baratos pero son
Page 13
Introducción
5
más propensos a la desactivación debido a la sinterización térmica de las partículas
metálicas o a la deposición de carbón [12]. Para evitar estos efectos sobre los
catalizadores, estos se suelen combinar formando aleaciones como Ni-Cr o Ni-Pt, o
soportar sobre una gran variedad de óxidos metálicos como SiO2, Al2O3, CeO2 o ZrO2
[11, 13]. Otra forma de modificar estos catalizadores consiste en la adición de
promotores alcalinos o alcalino térreos [14, 15].
Oxidación parcial (“Partial Oxidation”, PO):
CnHm + n/2O2 → nCO + m/2H2 (A.3)
Esta reacción se puede llevar a cabo en ausencia de quemadores externos para
el mantenimiento de la temperatura e incluso sin catalizador (a 1300-1500 ºC), aunque
este se suele utilizar para disminuir la deposición de carbón y la temperatura de trabajo
[11]. Por otra parte, el control de esta es complicado debido a la exotermicidad de la
reacción y a la formación de puntos calientes [16], que puede dar lugar a la
sinterización localizada de la fase activa y por tanto a la desactivación del catalizador.
Se suelen utilizar catalizadores similares a los del proceso de SR.
Reformado autotérmico (“Autothermal Reforming”, AR):
CnHm + n/2H2O + n/4O2 → nCO + (n/2 + m/2)H2 (A.4)
Esta reacción consiste en una combinación de las dos anteriores, y es muy
interesante desde el punto de vista del aprovechamiento energético ya que utiliza la
exotermicidad de la reacción de oxidación parcial para aportar el calor necesario a la
reacción de reformado con vapor de agua [17].
En los últimos años también se ha intensificado el estudio de algunas variantes
de estos procesos como el reformado seco de metano (con CO2) [18]. Además, los
procesos mencionados anteriormente suelen estar acompañados de otros sistemas de
reacción donde se llevan a cabo la oxidación de CO (reacción A.5) y/o la reacción de
“Water-Gas Shift” (WGS, reacción A.6) con objeto de purificar el H2 obtenido.
CO + 1/2O2 → CO2 (A.5)
CO + H2O → CO2 + H2 (A.6)
Page 14
Descripción del trabajo realizado
6
Ambas reacciones son exotérmicas y esta última se suele llevar a cabo en dos
etapas consecutivas: la primera de ellas con catalizadores de Fe3O4/Cr2O3 a elevada
temperatura (300-530 ºC), y la segunda con catalizadores de Cu/ZnO/Al2O3 a
temperaturas inferiores (200-250 ºC).
Las ventajas de utilizar hidrocarburos como el CH4 como materia prima para la
obtención de H2 son su gran disponibilidad, bajo coste y amplia red de distribución.
Sin embargo, son recursos no renovables y presentan los inconvenientes asociados a
su estado gaseoso. En este sentido destaca el empleo de compuestos oxigenados
líquidos en condiciones ambientales, como el metanol y otros alcoholes. Además, al
igual que el agua, estos se pueden utilizar directamente en las celdas de combustible
[13]. Por otro lado, se puede afirmar que los procesos de obtención de hidrógeno aquí
mencionados son respetuosos con el medio ambiente, siempre y cuando en los
procesos de electrólisis se obtenga la electricidad a partir de energías renovables como
la eólica o la solar, y en los procesos de conversión catalítica se utilicen materias
primas de origen renovable como los bioalcoholes (biometanol, bioglicerol) [3].
A.1.3. Tecnologías de almacenamiento de hidrógeno
Una de las principales desventajas del hidrógeno está asociada a la dificultad
de su transporte, manipulación y almacenamiento en condiciones seguras. Presenta un
elevado calor de combustión (142 kJ g-1
), un amplio rango de inflamabilidad (4 – 75
% en aire) y una temperatura de autoignición de 560 ºC. Sin embargo, se puede
considerar seguro en recintos abiertos debido a su enorme difusividad en el aire (0,61
cm2 s
-1) y a su carácter inocuo. Por otro lado, el desarrollo de sistemas eficientes de
almacenamiento de H2 resulta de capital importancia si se obtiene por medio de
energías renovables dadas las fluctuaciones de disponibilidad que estas suponen.
Además, este aspecto es de particular importancia debido a su naturaleza gaseosa,
principalmente para su aplicación en automoción. Por ejemplo, en el caso del coche
eléctrico comentado anteriormente, 4 kg de hidrógeno almacenados en condiciones
ambientales ocuparían un volumen de unos 45 m3 [4]. En este sentido, por ejemplo, el
Departamento de Energía de EEUU (DOE) ha establecido como objetivo para el año
2015 el desarrollo de sistemas con una capacidad de almacenamiento de H2 del 9 % en
Page 15
Introducción
7
peso (incluyendo válvulas, sistemas de calentamiento, enfriamiento, aislamiento, etc.)
[19].
Los sistemas de almacenamiento de hidrógeno se pueden dividir
principalmente en tres categorías:
i) Sistemas de almacenamiento convencionales.
El H2 se suele comprimir a 200 bar en cilindros de acero convencional, o hasta
450 bar si se emplean materiales reforzados con fibra de carbono y recubrimientos
inertes especiales. Los principales inconvenientes de estos sistemas son la necesidad
de un sistema de control de la presión y el riesgo inherente al proceso de compresión.
Por otro lado, el hidrógeno licuado (por debajo de -250 ºC) presenta una mayor
densidad que el comprimido, pero requiere infraestructuras adicionales para
aprovechar las cantidades de hidrógeno que se pierden por evaporación [4, 20].
ii) Almacenamiento de hidrógeno por adsorción física.
El H2 se puede adsorber en forma molecular sobre la superficie de una gran
variedad de sólidos porosos: nanotubos y nanofibras de carbono, grafito, carbones
activados, estructuras organometálicas (MOFs), zeolitas, etc. [4, 20, 21]. Estos
sistemas presentan una rápida cinética de adsorción/desorción y una gran
reversibilidad. Sin embargo, las débiles interacciones entre el H2 y los adsorbentes dan
lugar a una baja capacidad de almacenamiento de H2 (1-5 % en peso de H2). Esta
depende del área superficial del adsorbente y de las condiciones de operación,
requiriendo elevadas presiones (hasta 100 bar) y temperaturas criogénicas (-196 ºC).
iii) Almacenamiento de hidrógeno por adsorción química.
El otro mecanismo principal de captura de H2 es su disociación y la formación
de determinados compuestos como nitruros e hidruros de metales o de aleaciones
metálicas [4, 20, 21]. Estos compuestos sólidos son muy fáciles de manipular y en
estos casos la máxima cantidad de hidrógeno admitida es superior a la obtenida por
adsorción física. Por ejemplo, el MgH2 contiene idealmente un 7,6 % H2 en peso, y
otros hidruros interrmetálicos como Li3Be2H7 o BaReH9 en torno al 9 % en peso. Sin
Page 16
Descripción del trabajo realizado
8
embargo, la formación de estos compuestos es lenta y su descomposición requiere
temperaturas superiores a 300 ºC. Los alanatos y borohidruros (hidruros complejos de
Al y B, respectivamente) admiten cantidades de H2 incluso mayores. Por ejemplo, el
LiBH4 contiene un 18,3 % en peso de H2, pero se descompone a temperaturas de hasta
600 ºC y presenta cierta irreversibilidad en el proceso de captura de hidrógeno [4].
A.2. El metanol como fuente de hidrógeno
El CH3OH es un producto fundamental en la industria química mundial que se
utiliza en la obtención de formaldehido, MTBE, ácido acético, metil metacrilato,
formato de metilo, etc. También está cobrando una enorme importancia como fuente
de H2, e incluso se puede utilizar directamente para generar energía mediante celdas
de combustible (“Direct Methanol Fuel Cells”, DMFC) [13]. De hecho, al igual que
sucede con el H2, se habla de una posible “economía del metanol” [22].
En esta tesis se ha escogido el metanol como fuente de H2 debido a sus
numerosas ventajas. Tiene una elevada relación H/C (4/1), la misma que el metano,
pero su naturaleza líquida en condiciones ambientales facilita mucho su transporte,
almacenamiento y manipulación. El reformado de metanol se puede llevar a cabo a
menores temperaturas (150 – 350 ºC) que muchos otros combustibles porque no
contiene enlaces C-C, no requiere procesos de desulfuración y produce una cantidad
de CO inferior al metano [13]. No es de los combustibles más tóxicos (lo es más que
el diésel y el etanol, pero menos que la gasolina, por ejemplo), ya que es fácilmente
metabolizado por el cuerpo humano y por los microorganismos presentes en el medio
ambiente. Además, el CH3OH se puede producir de forma sostenible a partir de
fuentes biológicas [23], e incluso en procesos de revalorización de CO2, vía
hidrogenación [24] o conversión fotocatalítica [25].
A.2.1. Reacciones de conversión catalítica de metanol
Las principales reacciones de producción de hidrógeno a partir de metanol son
las de descomposición (MD, reacción A.7), reformado con vapor de agua (SRM,
reacción A.8) y oxidación parcial (POM, reacción A.9).
CH3OH → 2H2 + CO (A.7)
Page 17
Introducción
9
CH3OH + H2O → 3H2 + CO2 (A.8)
CH3OH + 1/2O2 → 2H2 + CO2 (A.9)
El SRM suele tener lugar a temperaturas de 250-300 ºC y presenta la mayor
proporción de hidrógeno por átomo de carbono (H/C = 3). La oxidación parcial de
metanol obtiene mayores conversiones a menores temperaturas (a partir de 120 ºC), y
por tanto se puede llevar a cabo en reactores de menor tamaño [13]. Además,
dependiendo del catalizador empleado, se pueden obtener otros subproductos a partir
de metanol, algunos de gran valor añadido como el formaldehído (H2CO) o el formato
de metilo (HCOOCH3), por medio de mecanismos como el mostrado en la Figura A.1.
Figura A.1. Esquema de reacción de la oxidación de metanol (adaptado de Tatibouët [26]).
A.2.2. Catalizadores empleados para la producción de H2
Los catalizadores utilizados en las reacciones A.7, A.8 y A.9, así como en la
síntesis de metanol (reacción inversa de A.8) y water-gas shift (reacción A.6), se
pueden clasificar en dos categorías: catalizadores de Cu o de metales del grupo VIII.
i) Catalizadores basados en Cu.
El cobre es un metal muy económico y presenta una excelente actividad en
todas estas reacciones. Por otro lado, es pirofórico cuando se expone al aire y es
susceptible de desactivarse por la deposición de carbono sobre su superficie y,
principalmente, por la sinterización de las partículas metálicas. De hecho, entre los
metales más utilizados, solo la plata es menos estable que el Cu en este sentido según
Page 18
Descripción del trabajo realizado
10
la clasificación de Hughes: Ag < Cu < Au < Pd < Fe < Ni < Co < Pt < Rh < Ru < Ir <
Os < Re [12]. Por estos motivos se suelen emplear soportes óxidos como ZnO, Al2O3,
CeO2, SiO2, TiO2 o ZrO2, que actúan como promotores estructurales (mejorando la
dispersión del metal) y/o electrónicos (modificando sus propiedades catalíticas) [11,
13, 27, 28]. También se puede promocionar el Cu adicionando sales o hidróxidos de
metales alcalinos, cuyo efecto aumenta con el radio atómico (Cs > Rb > K > Na > Li)
[29-31].
ii) Catalizadores basados en metales del grupo VIII.
Aunque estos no presentan tantos problemas de estabilidad como el Cu (el Pt y
el Pd principalmente), algunos como el Ni son mucho menos selectivos a la reacción
de reformado con vapor de agua (reacción A.8) que a la descomposición de metanol
(reacción A.7) [13]. Este metal presenta además una gran tendencia a desactivarse por
la formación de depósitos de carbono derivados de la disociación de CO o CH4 [11].
Al igual que el Cu, los metales de este grupo se suelen soportar sobre óxidos metálicos
[11, 13, 28] y también se pueden promocionar con compuestos alcalinos [32-34].
A.3. La promoción electroquímica de la catálisis (EPOC)
El fenómeno de la promoción electroquímica (“Electrochemical Promotion of
Catalysis”, EPOC) se basa en el uso de la electroquímica para activar y sintonizar un
catalizador heterogéneo en una reacción espontánea, de un modo que parece obviar la
ley de Faraday [35].
A.3.1. Origen y mecanismo de la promoción electroquímica
En 1981 Vayenas [36] observó por primera vez que la actividad y selectividad
de un catalizador metálico depositado sobre un electrolito sólido (conductor iónico) se
pueden modificar electroquímicamente durante el propio transcurso de una reacción
química. Esta modificación puede llevarse a cabo de modo controlado y reversible,
mediante la aplicación de una diferencia de potencial o intensidad eléctrica entre esta
película metálica (catalizador-electrodo de trabajo) y un segundo electrodo
(contraelectrodo) depositado sobre el lado opuesto del electrolito. Así, si sobre el
electrodo de trabajo tiene lugar una reacción catalítica (Figura A.2), la aplicación de
Page 19
Introducción
11
una corriente eléctrica, (I, del orden de µA) puede ocasionar un incremento en la
velocidad de reacción (Δr), que puede superar en varios órdenes de magnitud al valor
previsto por la ley de Faraday (re). Por ello este fenómeno se conoce también como
“modificación electroquímica no faradaica de la actividad catalítica” (NEMCA).
Figura A.2. Representación del efecto EPOC empleando K/β-Al2O3 como electrolito sólido.
Las especies iónicas promotoras se generan en la región llamada tpb (three-
phase boundaries), que consiste en la interfase entre el electrolito sólido, el catalizador
y la fase gas. Estas especies son acompañadas por los correspondientes iones de
compensación de carga, formando dipolos neutros que se distribuyen a lo largo de la
superficie metálica y que constituyen la llamada doble capa efectiva (Figura A.3). Esta
modifica la densidad electrónica del catalizador y por tanto su función de trabajo (Δϕ)
que se relaciona con la variación del potencial (ΔVWR) según la ecuación A.10 [37]:
Δϕ = eΔVWR (A.10)
De este modo se varía la capacidad de enlace del catalizador con cada uno de
los reactivos y por tanto la velocidad de la reacción. Para confirmar esta teoría se han
empleado diversas técnicas de caracterización tanto catalíticas (TPD, medida de la
función de trabajo), como electroquímicas (voltametría cíclica, espectroscopía de
impedancia) y de análisis de superficies (XPS, UPS, PEEM, STM), poniendo de
manifiesto que este fenómeno es análogo a la promoción química convencional en
re I < 0
Δr
Δr >> re
re = I/nF
K+
K+ K
+
K+K+
Page 20
Descripción del trabajo realizado
12
catálisis heterogénea [35, 38]. Sin embargo, mientras en la promoción química los
promotores se adicionan al catalizador durante su preparación, en la promoción
electroquímica estos son iones que migran desde el electrolito sólido hacia el metal, de
modo que se puede controlar su movimiento por medio de la corriente aplicada.
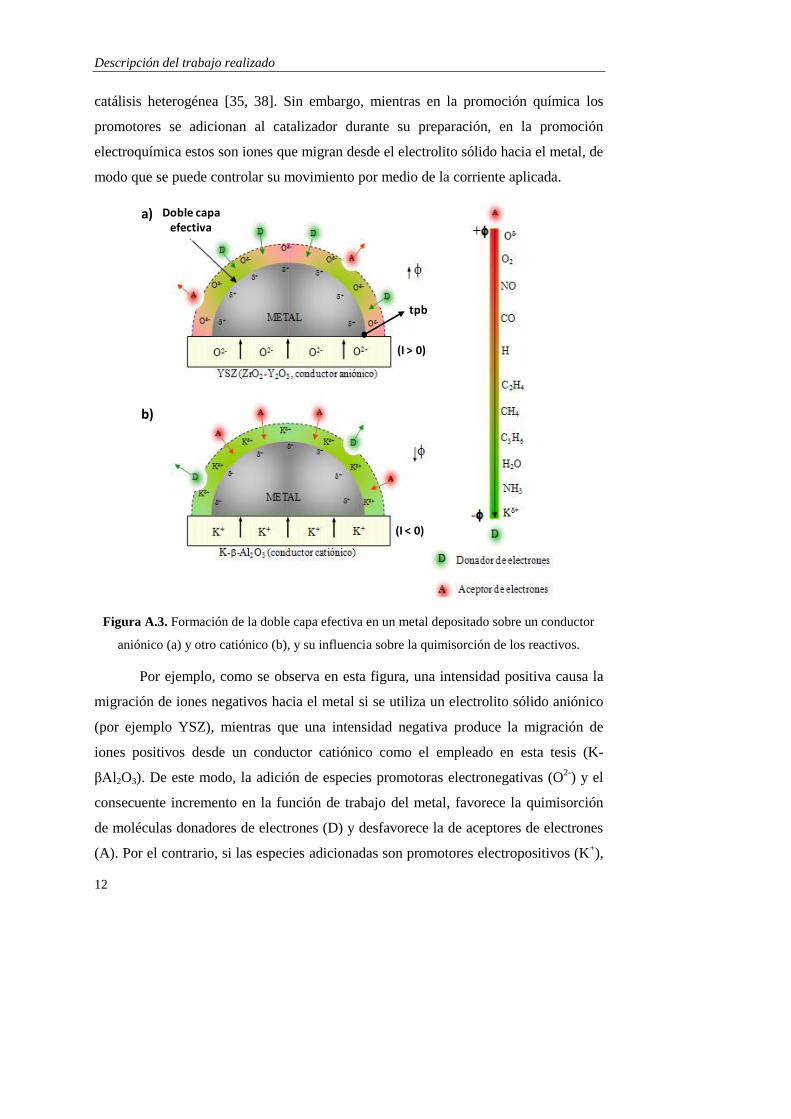
Figura A.3. Formación de la doble capa efectiva en un metal depositado sobre un conductor
aniónico (a) y otro catiónico (b), y su influencia sobre la quimisorción de los reactivos.
Por ejemplo, como se observa en esta figura, una intensidad positiva causa la
migración de iones negativos hacia el metal si se utiliza un electrolito sólido aniónico
(por ejemplo YSZ), mientras que una intensidad negativa produce la migración de
iones positivos desde un conductor catiónico como el empleado en esta tesis (K-
βAl2O3). De este modo, la adición de especies promotoras electronegativas (O2-
) y el
consecuente incremento en la función de trabajo del metal, favorece la quimisorción
de moléculas donadores de electrones (D) y desfavorece la de aceptores de electrones
(A). Por el contrario, si las especies adicionadas son promotores electropositivos (K+),
Doble capaefectiva
tpb
(I > 0)
(I < 0)
a)
b)
Page 21
Introducción
13
la función de trabajo disminuye (Figura A.3b) y se favorece la quimisorción de los
aceptores de electrones frente a los donadores. Por tanto, dependiendo de las
condiciones de reacción, del catalizador empleado y del carácter electronegativo o
electropositivo de los distintos adsorbatos, una determinada polarización tendrá un
efecto positivo o negativo sobre la cinética global de la reacción. Se pueden distinguir
cuatro tipos de reacciones basadas en la promoción electroquímica [39]:
Reacciones electrofóbicas: muestran un incremento de la velocidad de
reacción para valores positivos del potencial. Este tipo de comportamiento tiene lugar
cuando la cinética es de orden positivo respecto al donador de electrones y de orden
cero o negativo respecto al aceptor de electrones; es decir, el donador de electrones es
el que se encuentra más débilmente adsorbido sobre el catalizador.
Reacciones electrofílicas: muestran un incremento de la velocidad de reacción
para valores negativos del potencial. Este tipo de comportamiento tiene lugar cuando
la cinética es de orden positivo respecto al aceptor de electrones y de orden cero o
negativo respecto al donador de electrones; es decir, el aceptor de electrones es el que
se encuentra más débilmente adsorbido sobre el catalizador.
Reacciones tipo volcán: presentan un máximo en la velocidad de reacción
respecto al potencial. Este tipo de comportamiento tiene lugar cuando tanto el donador
como el aceptor de electrones se encuentran fuertemente adsorbidos sobre el
catalizador (orden cero o negativo de ambos).
Reacciones tipo volcán invertido: presentan un mínimo en la velocidad de
reacción respecto al potencial. Este tipo de comportamiento tiene lugar cuando tanto el
donador como el aceptor de electrones se encuentran débilmente adsorbidos sobre el
catalizador (orden positivo de ambos).
Durante las tres últimas décadas el fenómeno EPOC se ha demostrado en más
de 80 sistemas catalíticos diferentes de interés industrial y medioambiental, y no
parece estar limitado a ningún tipo de reacción catalítica heterogénea, catalizador
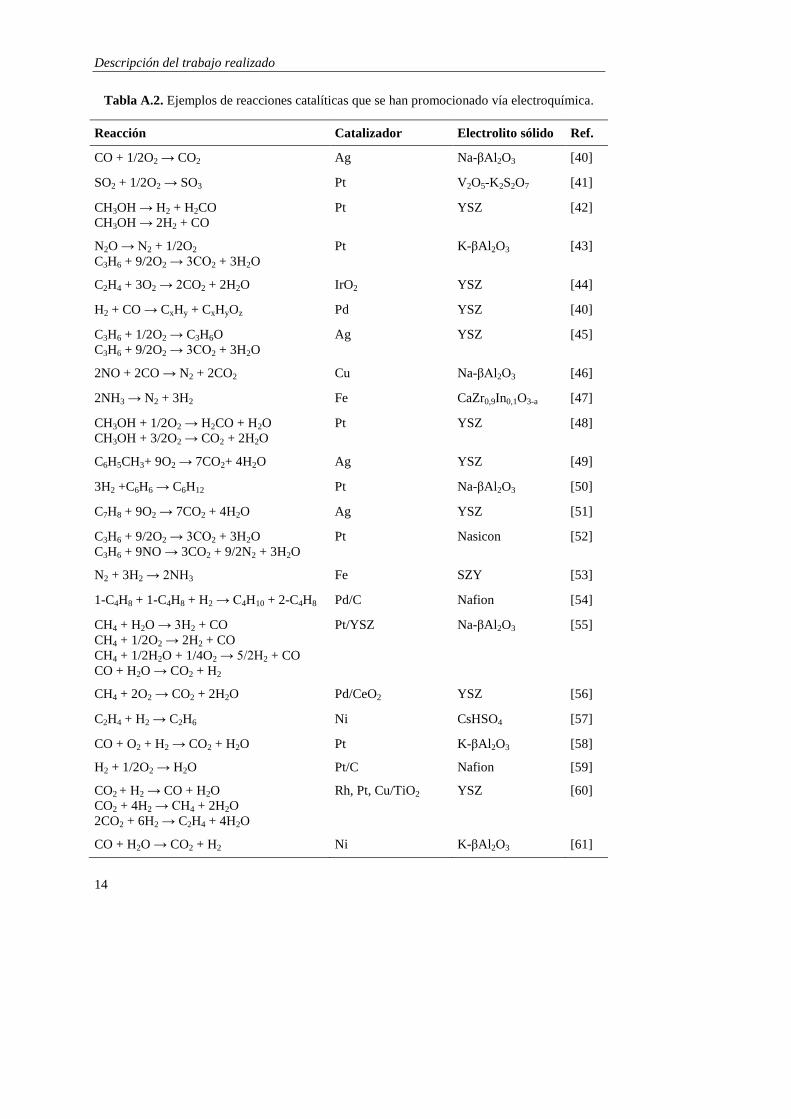
metálico o electrolito sólido [35, 38]. A modo de ejemplo, en la Tabla A.2 se muestran
algunos de los sistemas catalíticos en los que se ha aplicado este fenómeno.
Page 22
Descripción del trabajo realizado
14
Tabla A.2. Ejemplos de reacciones catalíticas que se han promocionado vía electroquímica.
Reacción Catalizador Electrolito sólido Ref.
CO + 1/2O2 → CO2 Ag Na-βAl2O3 [40]
SO2 + 1/2O2 → SO3 Pt V2O5-K2S2O7 [41]
CH3OH → H2 + H2CO
CH3OH → 2H2 + CO
Pt YSZ [42]
N2O → N2 + 1/2O2
C3H6 + 9/2O2 → 3CO2 + 3H2O
Pt K-βAl2O3 [43]
C2H4 + 3O2 → 2CO2 + 2H2O IrO2 YSZ [44]
H2 + CO → CxHy + CxHyOz Pd YSZ [40]
C3H6 + 1/2O2 → C3H6O
C3H6 + 9/2O2 → 3CO2 + 3H2O
Ag YSZ [45]
2NO + 2CO → N2 + 2CO2 Cu Na-βAl2O3 [46]
2NH3 → N2 + 3H2 Fe CaZr0,9In0,1O3-a [47]
CH3OH + 1/2O2 → H2CO + H2O
CH3OH + 3/2O2 → CO2 + 2H2O
Pt YSZ [48]
C6H5CH3+ 9O2 → 7CO2+ 4H2O Ag YSZ [49]
3H2 +C6H6 → C6H12 Pt Na-βAl2O3 [50]
C7H8 + 9O2 → 7CO2 + 4H2O Ag YSZ [51]
C3H6 + 9/2O2 → 3CO2 + 3H2O
C3H6 + 9NO → 3CO2 + 9/2N2 + 3H2O
Pt Nasicon [52]
N2 + 3H2 → 2NH3 Fe SZY [53]
1-C4H8 + 1-C4H8 + H2 → C4H10 + 2-C4H8 Pd/C Nafion [54]
CH4 + H2O → 3H2 + CO
CH4 + 1/2O2 → 2H2 + CO
CH4 + 1/2H2O + 1/4O2 → 5/2H2 + CO
CO + H2O → CO2 + H2
Pt/YSZ Na-βAl2O3 [55]
CH4 + 2O2 → CO2 + 2H2O Pd/CeO2 YSZ [56]
C2H4 + H2 → C2H6 Ni CsHSO4 [57]
CO + O2 + H2 → CO2 + H2O Pt K-βAl2O3 [58]
H2 + 1/2O2 → H2O Pt/C Nafion [59]
CO2 + H2 → CO + H2O
CO2 + 4H2 → CH4 + 2H2O
2CO2 + 6H2 → C2H4 + 4H2O
Rh, Pt, Cu/TiO2 YSZ [60]
CO + H2O → CO2 + H2 Ni K-βAl2O3 [61]
Page 23
Introducción
15
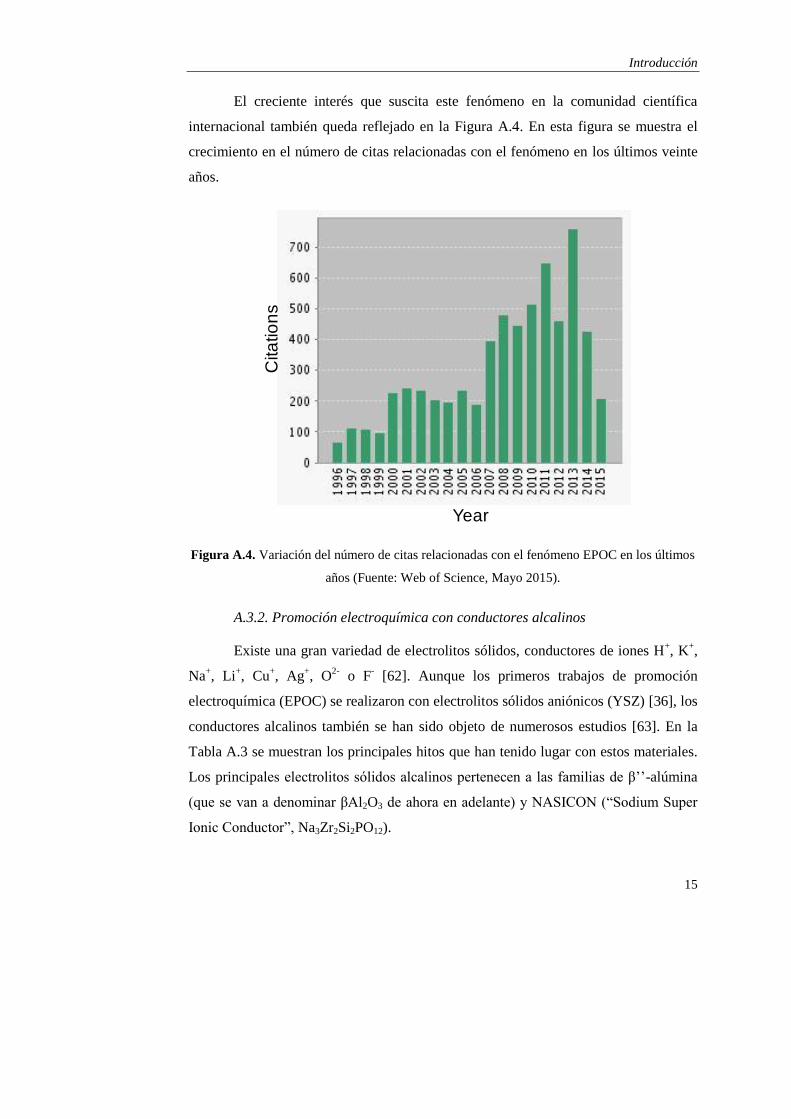
El creciente interés que suscita este fenómeno en la comunidad científica
internacional también queda reflejado en la Figura A.4. En esta figura se muestra el
crecimiento en el número de citas relacionadas con el fenómeno en los últimos veinte
años.
Figura A.4. Variación del número de citas relacionadas con el fenómeno EPOC en los últimos
años (Fuente: Web of Science, Mayo 2015).
A.3.2. Promoción electroquímica con conductores alcalinos
Existe una gran variedad de electrolitos sólidos, conductores de iones H+, K
+,
Na+, Li
+, Cu
+, Ag
+, O
2- o F
- [62]. Aunque los primeros trabajos de promoción
electroquímica (EPOC) se realizaron con electrolitos sólidos aniónicos (YSZ) [36], los
conductores alcalinos también se han sido objeto de numerosos estudios [63]. En la
Tabla A.3 se muestran los principales hitos que han tenido lugar con estos materiales.
Los principales electrolitos sólidos alcalinos pertenecen a las familias de β’’-alúmina
(que se van a denominar βAl2O3 de ahora en adelante) y NASICON (“Sodium Super
Ionic Conductor”, Na3Zr2Si2PO12).
Year
Cita
tio
ns
Page 24
Descripción del trabajo realizado
16
Tabla A.3. Algunas de las contribuciones más relevantes en la historia del EPOC con
conductores alcalinos (adaptado de de Lucas-Consuegra [63]).
Año Contribución Referencia
1991 Primer estudio EPOC usando un conductor alcalino (Pt/Na-βAl2O3) [64]
1995 Confirmación del mecanismo de backspillover de los iones Na+
mediante XPS
[65]
1996 Demostración la promoción de Pt con Na+ mediante caracterización
STM
[66]
1997 Primer estudio de EPOC usando un conductor de K+
(Fe/K2YZr(PO4)3)
[47]
1998 Simulación Monte Carlo de la promoción con Na+ de la reacción de
CO + NO sobre Pt
[67]
1999 Primera demostración de la promoción controlada in-situ de un metal
base (Cu) con Na+ en la reacción de CO + NO
[46]
2001 Reglas de la promoción electroquímica [68]
2001 Aplicación de la espectroscopía electrónia y EPOC a la reacción de
CO + NO sobre Rh. Mecanismo de la promoción alcalina
[69]
2003 SCR de NO con propeno usando Pt/NASICON bajo condiciones de
mezcla pobre en combustible
[52]
2005 Promoción electroquímica de Rh con K+ en la síntesis de Fischer-
Tropsch a 14 bar
[70]
2007 Promoción electroquímica de Pt/K-βAl2O3 a baja temperatura (200
ºC): Efecto permanente y estudio FTIR-EDX de las fases promotoras
[71]
2008 Desarrollo de un tubo de Pt/K-βAl2O3 para el
almacenamiento/reducción de NOx asistido electroquímicamente
[72]
2010 Regeneración in-situ de un catalizador de Pt de la deposición de
carbono mediante Na-βAl2O3
[55]
2013 Estudio a escala bancada de la promoción electroquímica de la
hidrogenación de CO2 con Cu/K-βAl2O3
[73]
Vayenas realizó el primer estudio de EPOC con electrolito alcalino en 1991
[64]. Desde entonces, tanto el Na-βAl2O3 como el NASICON (ambos conductores de
Na+) se han empleado en numerosos procesos catalíticos como la oxidación de etileno,
de CO, de propano o de propileno, la reducción de NO y la hidrogenación de benceno
o de CO2 [63]. Por otro lado, el primer estudio EPOC con un conductor de K+
(K2YZr(PO4)3) data de 1997 [47], y no es de extrañar que estuviera aplicado a la
descomposición de NH3 con un catalizador de Fe, ya que esta reacción y la de síntesis
Page 25
Introducción
17
de amoniaco son ejemplos clásicos de procesos químicos industriales promocionados
con potasio [74]. Más adelante, de Lucas-Consuegra introdujo el empleo de K-βAl2O3
para la promoción electroquímica de catalizadores de Pt en la oxidación de CO y de
propileno, así como en la reducción de NOx y N2O [63]. También caben destacar en
los últimos años los estudios de EPOC con este electrolito sólido en la reacción de
hidrogenación de CO2 [73, 75].
En esta tesis se ha seleccionado K-βAl2O3 como conductor debido a su
adecuación a las condiciones de reacción empleadas en esta investigación (puede
operar a partir de 150 ºC) y a los estudios previos en bibliografía que demuestran el
efecto promotor de los metales alcalinos, y en especial del potasio (favorecido por su
mayor radio atómico en comparación con el Na, por ejemplo) en las reacciones de
descomposición de metanol [29], reformado de metanol con vapor de agua [32, 33] y
oxidación parcial de metanol [31]. En todos estos estudios, el efecto electrónico del
potasio causó una mejora de la actividad catalítica atribuida al debilitamiento del
enlace C-H en los intermedios de reacción. Sin embargo, como se ha señalado
anteriormente, en este tipo de promoción alcalina (clásica) el potasio se adiciona en
forma de sal o de hidróxido durante la etapa de preparación del catalizador. En
cambio, en los estudios de EPOC el potasio se suministra al catalizador en el propio
transcurso de la reacción por vía electroquímica mediante la aplicación de una
corriente o potencial eléctricos. Esto le confiere a este tipo de sistemas una serie de
ventajas adicionales que se enumeran en la Tabla A.4.
Algunas de estas ventajas son particularmente importantes, como la capacidad
de optimizar in-situ la cantidad de promotor bajo diferentes condiciones de reacción, y
de sintonizar el catalizador para maximizar su actividad y su selectividad hacia el
producto deseado en todo momento. También resulta de especial interés la posibilidad
de prevenir la desactivación del catalizador e incluso de regenerarlo. Estas
aplicaciones del EPOC se han estudiado previamente en diferentes sistemas de
reacción, como se recoge en distintos trabajos de revisión [38, 63]. Sin embargo, en
esta tesis se han estudiado por primera vez en procesos de producción de hidrógeno a
partir de metanol. Además, algunos aspectos como el efecto de los iones promotores
Page 26
Descripción del trabajo realizado
18
sobre el estado de oxidación del metal se han estudiado por primera vez con un
electrolito alcalino en el presente trabajo de investigación.
Tabla A.4. Principales ventajas de la promoción electroquímica alcalina frente a la promoción
química clásica, y sus aplicaciones.
Ventaja Aplicación
Control in-situ de la cantidad de promotor
sobre la superficie del catalizador (mediante
la aplicación de una corriente o potencial
eléctricos)
Posibilidad de estudiar de forma rápida y
sencilla el efecto de un determinado promotor
sobre un sistema catalítico, e interpretarlo en
base a las reglas EPOC. No es necesario
preparar varios catalizadores con distintas
cantidades de promotor
Adición de la cantidad de promotor óptima
bajo diferentes condiciones de reacción
Optimización in-situ de la actividad y
selectividad de un catalizador. Utilidad en
procesos no estacionarios, como en la catálisis
de automoción
Utilización de la celda de electrolito sólido
como un sensor
Posibilidad de anticipar las condiciones
óptimas que maximizan una velocidad de
reacción
Modificación in-situ del estado de oxidación
del metal
Control de la fase activa del catalizador sin
modificar las condiciones de reacción
Control y atenuación y la adsorción
competitiva de unas moléculas frente a otras
Prevención del envenenamiento de la
superficie del catalizador
Promoción in-situ de reacciones de
eliminación de especies adsorbidas (como
carbón depositado)
Regeneración de un catalizador sin necesidad
de cambiar las condiciones de reacción
Almacenamiento de compuestos sobre la
superficie del catalizador mediante la
formación de especies derivadas alcalinas
Gran utilidad en procesos cíclicos como la
captura/reducción de NOx y el
almacenamiento de gases de interés como
CO2 o H2
A.4. Nuevas tendencias y perspectivas de la promoción electroquímica
A.4.1. Desarrollo de reactores más eficientes y catalizadores dispersos y más
competitivos
A pesar de las múltiples posibilidades que ofrecen los sistemas
electrocatalíticos empleados en los estudios de promoción electroquímica, debido a
sus particulares características presentan también ciertas limitaciones que disminuyen
su aplicabilidad. Por un lado, el reactor que se emplea en este tipo de estudios debe
Page 27
Introducción
19
tener un diseño especial que le permita conectar el catalizador electroquímico con el
sistema de polarización, y generalmente no está optimizado. Por otro lado, los
catalizadores empleados en estudios de promoción electroquímica suelen presentar
una baja dispersión de las partículas y una elevada carga metálica. Por todo ello, con
vistas a una posible etapa de comercialización futura del EPOC, resulta esencial
dedicar mayores esfuerzos al diseño de reactores más compactos y eficientes, así
como a minimizar el coste de los materiales [76].
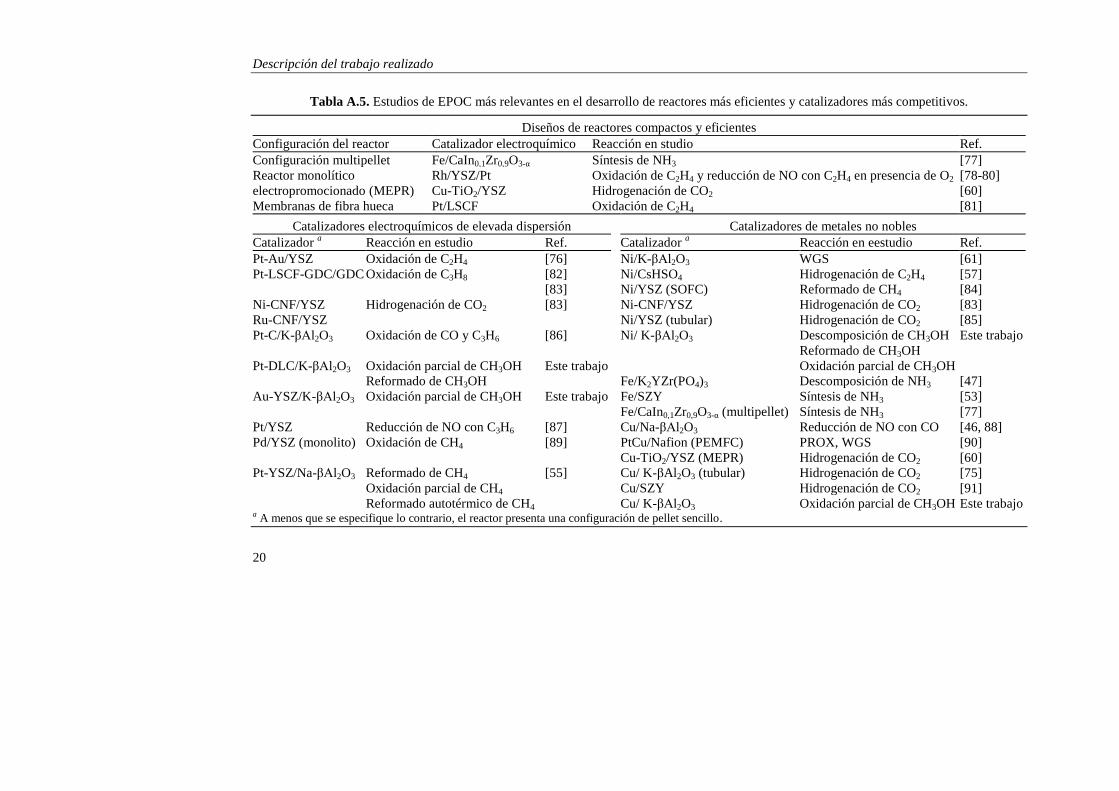
En la tabla A.5 se recogen los estudios de promoción electroquímica más
relevantes realizados durante los últimos años en ambos sentidos, algunos de los
cuales ya han sido mencionados anteriormente. Se incluyen también los catalizadores
desarrollados durante la presente investigación.
Una de las primeras configuraciones para la posible aplicación práctica del
fenómeno EPOC fue desarrollada por Yiokari y col., [77] para la síntesis de amoniaco
con catalizadores de Fe comerciales. Ésta consistía en un conjunto de 24 pellets de
CaZr0,9In0,1O3-α (conductor de H+) conectados eléctricamente en paralelo. Además, este
fue también uno de los primeros estudios de promoción electroquímica realizado a
elevadas presiones (50 atm). Otro gran avance en este sentido fue el desarrollo del
reactor monolítico promocionado electroquímicamente (“Monolithic
Electrochemically Promoted Reactor”, MEPR) [78]. Este reactor fue desarrollado en
2004 tras la consolidación de la técnica de pulverización catódica (“sputtering”) como
método de deposición de películas metálicas delgadas. Este reactor permite trabajar a
una mayor escala y se puede considerar un híbrido entre uno monolítico y otro de tipo
celda de combustible de óxido sólido. De este modo se han desarrollado varias
configuraciones de MEPR basadas en 22 unidades de Rh/YSZ/Pt o Cu-TiO2/YSZ/Au
conectadas en serie para la hidrogenación de CO2 a CO y CH4 [60] y para la reducción
catalítica selectiva de NO con etileno [78-80].
Page 28
Descripción del trabajo realizado
20
Tabla A.5. Estudios de EPOC más relevantes en el desarrollo de reactores más eficientes y catalizadores más competitivos.
Diseños de reactores compactos y eficientes
Configuración del reactor Catalizador electroquímico Reacción en studio Ref.
Configuración multipellet Fe/CaIn0,1Zr0,9O3-α Síntesis de NH3 [77]
Reactor monolítico Rh/YSZ/Pt Oxidación de C2H4 y reducción de NO con C2H4 en presencia de O2 [78-80]
electropromocionado (MEPR) Cu-TiO2/YSZ Hidrogenación de CO2 [60]
Membranas de fibra hueca Pt/LSCF Oxidación de C2H4 [81]
Catalizadores electroquímicos de elevada dispersión Catalizadores de metales no nobles
Catalizador a Reacción en estudio Ref. Catalizador
a Reacción en eestudio Ref.
Pt-Au/YSZ Oxidación de C2H4 [76] Ni/K-βAl2O3 WGS [61]
Pt-LSCF-GDC/GDC Oxidación de C3H8 [82] Ni/CsHSO4 Hidrogenación de C2H4 [57]
[83] Ni/YSZ (SOFC) Reformado de CH4 [84]
Ni-CNF/YSZ Hidrogenación de CO2 [83] Ni-CNF/YSZ Hidrogenación de CO2 [83]
Ru-CNF/YSZ Ni/YSZ (tubular) Hidrogenación de CO2 [85]
Pt-C/K-βAl2O3 Oxidación de CO y C3H6 [86] Ni/ K-βAl2O3 Descomposición de CH3OH Este trabajo
Reformado de CH3OH
Pt-DLC/K-βAl2O3 Oxidación parcial de CH3OH Este trabajo Oxidación parcial de CH3OH
Reformado de CH3OH Fe/K2YZr(PO4)3 Descomposición de NH3 [47]
Au-YSZ/K-βAl2O3 Oxidación parcial de CH3OH Este trabajo Fe/SZY Síntesis de NH3 [53]
Fe/CaIn0,1Zr0,9O3-α (multipellet) Síntesis de NH3 [77]
Pt/YSZ Reducción de NO con C3H6 [87] Cu/Na-βAl2O3 Reducción de NO con CO [46, 88]
Pd/YSZ (monolito) Oxidación de CH4 [89] PtCu/Nafion (PEMFC) PROX, WGS [90]
Cu-TiO2/YSZ (MEPR) Hidrogenación de CO2 [60]
Pt-YSZ/Na-βAl2O3 Reformado de CH4 [55] Cu/ K-βAl2O3 (tubular) Hidrogenación de CO2 [75]
Oxidación parcial de CH4 Cu/SZY Hidrogenación de CO2 [91]
Reformado autotérmico de CH4 Cu/ K-βAl2O3 Oxidación parcial de CH3OH Este trabajo a A menos que se especifique lo contrario, el reactor presenta una configuración de pellet sencillo.
Page 29
Introducción
21
En cuanto al desarrollo de catalizadores dispersos, se pueden encontrar
estudios en los que se emplea un material adicional que puede actuar simultáneamente
como soporte de las partículas del catalizador y como material conductor eléctrico, por
ejemplo, oro [76], carbón [54], nanofibras de carbono [83] o materiales conductores
mixtos (de iones y electrones) [81, 82]. Por otro lado, en la Tabla A.5 se muestran
diversos trabajos de promoción electroquímica, en los que se emplearon metales como
Ni [57, 61, 83, 85, 92], Fe [47, 53, 77] o Cu [46, 60, 75, 90, 91], de menor coste
económico que los típicos metales nobles empleados en estudios de EPOC como Pt,
Rh o Pd. En este sentido uno de los objetivos principales de esta Tesis ha sido el
desarrollo de catalizadores más competitivos que los empleados tradicionalmente para
su activación electroquímica en procesos de producción de H2 a partir de metanol. Así,
en los dos primeros capítulos se emplearon catalizadores electroquímicos de Pt de bajo
contenido metálico preparados mediante una técnica de deposición física de vapor
(PVD). En el segundo caso las partículas de Pt estaban, además, dispersas en una
matriz de carbono, presentando diámetros de unos 3 nm. En la misma línea, en el
capítulo 3 se desarrolló una película compuesta de nanopartículas de Au inmersas en
una matriz de zirconia estabilizada con itria (YSZ), que además de dispersar las
nanopartículas metálicas contribuía a evitar su sinterización. Por último, a partir del
capítulo 4 se emplearon catalizadores de metales no nobles (Cu y Ni) preparados
mediante técnicas de PVD que igualmente consiguieron reducir el contenido metálico
por unidad de área o aumentar el área superficial de estos electrodos.
A.4.2. Aplicación de la promoción electroquímica en la producción de H2
Al margen de la necesaria mejora y optimización de los sistemas
electrocatalíticos, otra posible estrategia para impulsar la aplicación práctica de la
promoción electroquímica es explorar nuevos procesos catalíticos en los ámbitos
energético o medioambiental donde este fenómeno pueda resultar interesante. Uno de
estos sistemas podría ser la producción y almacenamiento de H2 que tiene un gran
protagonismo en la actualidad ya que se postula como el posible vector energético del
futuro [2, 3]. Sin embargo, a excepción de los trabajos de Pitselis y col. de
descomposicón de NH3 [47] y de Yentekakis y col. de reformado de metano con vapor
Page 30
Descripción del trabajo realizado
22
de agua en una celda de combustible de óxido sólido [92], solo el presente grupo de
investigación ha llevado a cabo estudios EPOC enfocados a reacciones de producción
de hidrógeno: water-gas shift [61] y conversión de metano mediante diversas vías [55,
93].
Como se mencionó en la sección A.2, la producción de hidrógeno a partir de
metanol es especialmente interesante debido fundamentalmente a su elevado
contenido en H2, su naturaleza líquida y su posible obtención por medios sostenibles.
En cuanto a los procesos de conversión de metanol, hasta el inicio de la presente tesis
solo había publicados 4 estudios de promoción electroquímica, en los que se empleó
Ag [42, 94] o Pt [48, 95] como catalizador-electrodo de trabajo, YSZ como electrolito
sólido en todos los casos, y elevadas temperaturas de trabajo de hasta 500 ºC. Por
tanto, en esta tesis se ha aplicado por primera vez el fenómeno de la promoción
electroquímica a reacciones de conversión de metanol para la producción de
hidrógeno, utilizando para ello un electrolito sólido alcalino (K-βAl2O3) y
temperaturas de operación generalmente inferiores a los estudios anteriores. En la
Tabla A.6 se resumen todos los estudios de EPOC realizados hasta la fecha en los que
se han estudiado reacciones de conversión de metanol (incluyendo los de la presente
tesis).
Las principales reacciones catalíticas que tuvieron lugar en los estudios con
YSZ fueron la descomposición del metanol a CO (reacción A.7) y H2CO (reacción
A.11), la reacción de metanación (reacción A.12) [42], y la oxidación de metanol por
medio de las reacción A.13 y A.14 [48, 94, 95], obteniendo principalmente
formaldehído como producto interés.
CH3OH → H2CO + H2 (A.11)
CH3OH + H2 → CH4 + H2O (A.12)
CH3OH + 1/2O2 → H2CO + H2O (A.13)
CH3OH + 3/2O2 → CO2 + 2H2O (A.14)
Page 31
Introducción
23
Tabla A.6. Estudios de promoción electroquímica aplicada a reacciones de conversión de metanol.
Reacción Catalizador Electrolito sólido Método de preparación Principales productos Efecto EPOC Ref.
MD Ag YSZ Impregnación CO, H2CO, CH4, H2, H2O Electrofílico [42]
Ni K-βAl2O3 CADa H2, CO, C Electrofílico Este trabajo
SRM Pt K-βAl2O3 CADa H2, CO, CO2 Electrofílico Este trabajo
Pt-DLC K-βAl2O3 CADa H2, CO, CO2 Electrofílico Este trabajo
Ni K-βAl2O3 CADa H2, CO, CO2 Electrofílico Este trabajo
Ni K-βAl2O3 OADa H2, CO, CO2, C Electrofílico Este trabajo
POM Pt YSZ Pasta organometálica CO2, H2CO, H2O Volcán invertido [48]
Pt YSZ Pasta organometálica CO2, H2CO, H2O, H2 Electrofóbico [95]
Ag YSZ Pasta organometálica CO2, H2CO, H2O Electrofílico [94]
Pt K-βAl2O3 Impregnación H2, CO, CO2, H2CO, H2O Electrofóbicob Este trabajo
Pt K-βAl2O3 CADa H2, CO, CO2, H2CO, H2O Electrofílico Este trabajo
Pt-DLC K-βAl2O3 CADa H2, CO, CO2, H2CO, H2O Electrofílico Este trabajo
Au-YSZ K-βAl2O3 Pulverización catódica H2, CO, CO2, HCOOCH3, H2O Electrofílico Este trabajo
Cu K-βAl2O3 OADa H2, CO, CO2, HCOOCH3, H2O Electrofílico Este trabajo
Ni K-βAl2O3 CADa H2, CO, CO2, H2CO, H2O Electrofóbico
b Este trabajo
aCAD = Deposición por arco catódico; OAD = Deposición en ángulo oblicuo. bEfecto aparente, afectado por variaciones en el estado de oxidación del catalizador o por el bloqueo de centros activos por compuestos de potasio
Page 32
Descripción del trabajo realizado
24
Así pues, esta tesis doctoral se ha enfocado a la conversión de metanol para la
producción de H2 y CO2 a través de las reacciones A.8 (SRM) y A.9 (POM), aunque
en general también se ha observado una elevada formación de CO y de otros
compuestos como formaldehído o formato de metilo, dependiendo del catalizador.
A.4.3. Aplicación de la promoción electroquímica en la captura de compuestos
En los últimos años se ha dado a conocer una nueva aplicación de los sistemas
electrocatalíticos alcalinos, que consiste en la posibilidad de almacenar
electroquímicamente compuestos sobre la superficie de una película catalítica porosa.
De Lucas-Consuegra y col. demostraron por primera vez este concepto hace siete años
[72]. Para ello depositó una película de catalizador de Pt sobre un tubo de K-βAl2O3
con el cual estudió la captura/reducción de NOx. Así pues, en una primera etapa del
proceso llevada a cabo a potenciales negativos, los iones K+ desplazados desde el
electrolito sólido hasta el catalizador no solo promocionaron la reacción de oxidación
de NO, sino también almacenaron NOx sobre la superficie del catalizador en forma de
nitratos de potasio. Además, otro estudio en la misma línea de investigación [96]
demostró la posibilidad de regenerar electroquímicamente la superficie de este tipo de
catalizadores para su empleo bajo una composición de gas pobre.
Más recientemente, Ruiz y col. aplicaron este fenómeno de captura asistida por
vía electroquímica a la eliminación de CO2 [97]. Este estudio demostró que se puede
almacenar CO2 sobre la superficie de un catalizador metálico bajo determinadas
condiciones de reacción y en presencia de iones K+ (dando lugar a la formación de
carbonatos de potasio). Adicionalmente, las especies capturadas pueden ser liberadas
al invertir la polarización regenerando el catalizador y mostrando la completa
reversibilidad de esta tecnología de captura. Por tanto, se consideró interesante
explorar también la posible utilización de estos catalizadores electroquímicos
catiónicos en sistemas acoplados de producción y almacenamiento de H2.
Page 33
Introducción
25
A.5. Objetivo de la tesis doctoral
El objetivo principal de esta tesis doctoral es estudiar el fenómeno de
promoción electroquímica de la catálisis en reacciones de producción de hidrógeno a
partir de metanol empleando conductores alcalinos. Para ello se ha colaborado con
distintos grupos de investigación especializados en la preparación de películas
delgadas que puedan actuar como catalizador-electrodo de trabajo. De este modo se
pretende contribuir al campo de la producción catalítica de H2 mediante una
aproximación novedosa, que prácticamente no ha sido estudiada hasta la fecha. Con
este fin se ha desarrollado un programa de trabajo que se puede dividir en las
siguientes etapas:
- Realizar una revisión bibliográfica y poner en marcha una instalación
experimental donde llevar a cabo los experimentos de promoción electroquímica
(equipos de análisis, polarización, control de flujo, reactor electroquímico, etc).
- Estudiar la promoción electroquímica aplicada a la oxidación parcial de
metanol y al reformado de metanol con vapor de agua, utilizando catalizadores de Pt
depositados sobre K-βAl2O3 (conductor de K+) mediante una técnica de deposición
física de vapor (PVD) y otras más tradicionales (impregnación).
- Desarrollar películas delgadas de catalizador que sean más competitivas que
las comúnmente empleadas en estudios de promoción electroquímica, mediante la
aplicación de una técnica PVD (deposición por arco catódico, “sputtering”, deposición
en ángulo oblicuo). Para ello se ha procurado disminuir la carga metálica y aumentar
la dispersión por medio de una matriz (DLC, YSZ) que contenga nanopartículas del
metal, así como obtener películas de metales no nobles (Cu, Ni) que presenten cierta
estabilidad.
- Adecuar el catalizador empleado en cada estudio para poder llevar a cabo la
promoción electroquímica, mediante la deposición de una película de catalizador que
presente al mismo tiempo una buena conductividad eléctrica y adecuadas propiedades
catalíticas, y si es necesario, mediante la realización de algún proceso de
acondicionamiento del catalizador o el empleo de un colector de corriente.
Page 34
Descripción del trabajo realizado
26
- Evaluar el efecto los iones K+ sobre cada catalizador en las reacciones de
producción de H2 a partir de metanol y discutir los resultados en base a la teoría de la
promoción química y con el apoyo de diversas técnicas de caracterización.
- Estudiar la posibilidad de promocionar mediante iones K+ la producción y
almacenamiento simultáneos de H2 en un único sistema catalítico, y su liberación bajo
las mismas condiciones de operación, todo ello de forma controlada en base a
variaciones en la polarización del catalizador.
Page 35
Métodos e instalación experimental
27
B. MÉTODOS E INSTALACIÓN EXPERIMENTAL
B.1. Preparación de los catalizadores electroquímicos
B.1.1. Deposición de pasta organometálica
Este método consiste en la deposición de una pasta orgánica del metal y su
posterior calcinación a elevada tempertaura (800 ºC) para lograr la descomposición del
componente orgánico y una adecuada adherencia entre el electrodo y el electrolito
sólido. Esta técnica es muy rápida y sencilla, y se ha utilizado en todos los capítulos
para la deposición del contraelectrodo (CE) y electrodo de referencia (RE) de Au
(inertes).
B.1.2. Impregnación
Esta técnica se ha utilizado en la preparación de uno de los dos catalizadores
de Pt empleados en el capítulo 1. Consiste en la descomposición térmica de una
solución de H2PtCl6 0,1 M en una mezcla agua-2-propanol, por medio de sucesivas
etapas de impregnación (de varios µL de esta disolución), secado y calentamiento a
450 ºC. A diferencia del método de deposición de una pasta organometálica, este
permite establecer la cantidad exacta de metal depositado. Sin embargo, ninguno de
estos dos métodos permite controlar la microestructura de la película de catalizador.
B.1.3. Deposición física de vapor (PVD)
Este es un grupo de técnicas de preparación de películas delgadas, que se
basan en el calentamiento en vacío de una placa del material a depositar (“target”)
hasta lograr una elevada presión de vapor y su posterior condensación sobre un
sustrato frío (K-βAl2O3). Las principales diferencias entre las distintas técnicas PVD
residen en la naturaleza de la fuente del vapor (sólida en estos casos) y en el método
usado para producirlo. Así pues, en esta tesis se han utilizado tres métodos de
deposición física de vapor diferentes:
i) Deposición por arco catódico (CAD).
Esta técnica utiliza un arco eléctrico para vaporizar el material del cátodo que
posteriormente se va a condensar. Los electrodos de Pt, Pt-DLC y Ni utilizados en los
Page 36
Descripción del trabajo realizado
28
capítulos 1, 2 y 5, respectivamente, se han preparado mediante esta método. Esta
técnica se ha llevado a cabo en colaboración con el Dr. José Luis Endrino del
Instituto de Ciencia de Materiales de Madrid (CSIC).
ii) Pulverización catódica (comúnmente conocida como “sputtering”).
El catalizador de Au-YSZ del capítulo 3 se ha preparado por este método. En
este caso, el material sólido objetivo (Au, Zr e Y) se evapora mediante un bombardeo
con iones energéticos. Esta técnica se ha llevado a cabo en colaboración con el Dr.
David Horwat del Institut Jean Lamour de Université de Lorraine (Francia).
iii) Evaporación por bombardeo electrónico (EBE).
Esta técnica se ha utilizado para depositar películas de Cu y Ni en los capítulos
4 y 6, respectivamente. En concreto se ha aplicado una modificación de este método
denominada “deposición en ángulo oblicuo” o “deposición en ángulo rasante” (OAD o
GLAD, respectivamente), que se basa en colocar el sustrato (K-βAl2O3) formando un
ángulo rasante respecto de la dirección de propagación del vapor que se va a depositar.
De este modo, cuando el material (Cu o Ni) incide sobre el sustrato se produce el
mecanismo de deposición con efecto sombra o “shadowing”, que favorece el
crecimiento de la película de forma columnar. Controlando el ángulo de incidencia del
material y la velocidad de giro de la muestra se consiguen todo tipo de morfologías de
la película. Una de las propiedades que hace más atractiva esta técnica de deposición
es el aumento de la superficie interna y porosidad de la lámina delgada respecto a una
capa compacta del mismo material, lo que puede ser de gran interés desde el punto de
vista electrocatalítico. Esta técnica se ha llevado a cabo en colaboración con el Dr.
Agustín González-Elipe y el Dr. Víctor Joaquín Rico del Instituto de Ciencia de
Materiales de Sevilla (CSIC – Universidad de Sevilla).
Las condiciones concretas de deposición de cada película de catalizador se
detallan en los diferentes capítulos.
Page 37
Métodos e instalación experimental
29
B.2. Técnicas de caracterización
B.2.1. Microscopia electrónica de barrido (SEM)
Esta técnica se ha empleado en la mayoría de los capítulos de la Tesis. Permite
la observación y análisis de la película del catalizador de forma sencilla, aportando
información sobre la textura, tamaño y forma de las partículas. Además, si se
acompaña de una espectroscopía de energía dispersiva (EDX) permite estimar la
composición química superficial de la lámina y de posibles especies depositadas.
B.2.2. Microscopía electrónica de transmisión (TEM)
Este tipo de microscopía también ofrece información estructural y
morfológica de la muestra del catalizador, y se suele utilizar cuando se requiere una
resolución mayor que con la microscopía SEM, como es el caso de los catalizadores
dispersos Pt-DLC y Au-YSZ en los capítulos 2 y 3, respectivamente. Concretamente
se ha empleado una variante denominada STEM (“scanning transmission electron
microscopy”) que es especialmente adecuada para la obtención de imágenes de
resolución atómica donde el contraste está directamente relacionado con el número
atómico, y para el acoplamiento de técnicas espectroscópicas como EDX o EELS.
B.2.3. Espectroscopía electrónica de pérdidas de energías (EELS)
Esta técnica permite medir la composición atómica de una muestra, enlaces
químicos y propiedades electrónicas de las bandas de valencia y de conducción. Con
respecto al EDX esta técnica es generalmente más adecuada para muestras con
números atómicos relativamente bajos, como el carbono. En concreto se ha utilizado
en el capítulo 2 para identificar la estructura de la matriz carbonosa donde se
encuentran dispersas las nanopartículas de Pt.
B.2.4. Espectroscopía de infrarrojos por transformada de Fourier (FTIR)
Esta técnica aporta información acerca de los modos y tipos de vibraciones
moleculares, y permite de este modo identificar grupos funcionales de moléculas
presentes en la muestra. Se ha utilizado en el capítulo 6 para determinar los tipos de
compuestos carbonosos depositados sobre el Ni bajo promoción electroquímica.
Page 38
Descripción del trabajo realizado
30
B.2.5. Espectroscopía Raman
Esta técnica es complementaria a la espectroscopía infrarroja en el estudio de
las frecuencias vibracionales de las moléculas, de modo que algunas bandas pueden
observarse solo en el espectro Raman y otras en el infrarrojo. También se utilizó en el
capítulo 6 para la identificación de los compuestos carbonosos adsorbidos.
B.2.6. Espectroscopía de fotoelectrones emitidos por rayos X (XPS)
Esta técnica se suele utilizar para estimar la estequiometria, estado químico y
estructura electrónica de los elementos presentes sobre la superficie del material. Se ha
realizado en los capítulos 4 y 6.
B.2.7. Difracción de rayos X (XRD)
Esta técnica se ha llevado a cabo prácticamente en todos los capítulos, ya que
permite analizar la estructura cristalina del catalizador y obtener un tamaño
aproximado de partícula por el método de Scherrer.
B.2.8. Variación de resistencia eléctrica superficial a temperatura
programada
Esta es una técnica muy sencilla que se ha llevado a cabo con el catalizador Pt-
DLC en el capítulo 2 para obtener una medida aproximada, in-situ, de la variación de
la resistencia eléctrica de la superficie del catalizador durante un proceso realizado a
temperatura programada y así detectar posibles cambios estructurales.
B.2.9. Voltamperometría cíclica (CV)
Esta es una técnica de caracterización electroquímica que se ha utilizado en
gran parte de los capítulos para evaluar los procesos electrocatalíticos asociados a los
iones K+ que tienen lugar sobre la superficie del catalizador y determinar el rango de
potencial adecuado para llevar a cabo los experimentos de promoción electroquímica
de modo reversible y reproducible.
Las condiciones de caracterización y las características de los equipos se
detallan en cada capítulo.
Page 39
Métodos e instalación experimental
31
B.3. Instalación experimental
Para llevar a cabo los experimentos de promoción electroquímica se preparó y
puso en marcha la instalación experimental que se muestra en la Figura B.1.
Figura B.1. Instalación experimental utilizada en los estudios de promoción electroquímica.
Esta instalación consta de cuatro partes diferenciadas:
i) Sistema de alimentación: Está constituido por cuatro líneas de flujo de gases,
análogas e independientes que pueden suministrar aire (fuente de O2 en la reacción de
POM), H2 (agente reductor) y N2 o Ar, según el capítulo (gas portador de la mezcla de
reacción). Una de estas líneas de gas inerte es saturada a temperatura controlada en
metanol y la otra en agua (en los experimentos de SRM).
ii) Sistema de reacción: Es un reactor de electrolito sólido de cámara sencilla,
donde se localiza el catalizador electroquímico, de modo que los tres electrodos
(trabajo, contraelectrodo, referencia) se encuentran bajo la misma mezcla de reacción.
Page 40
Descripción del trabajo realizado
32
iii) Sistema de polarización: Consiste en un potenciostato/galvanostato que
permite aplicar potenciales o corrientes eléctricas sobre el catalizador electroquímico.
iv) Sistema de análisis de los productos de reacción: Consiste en un
cromatógrafo de gases situado en linea a la salida del reactor.
Esta instalación se describe detalladamente en el primer capítulo (sección
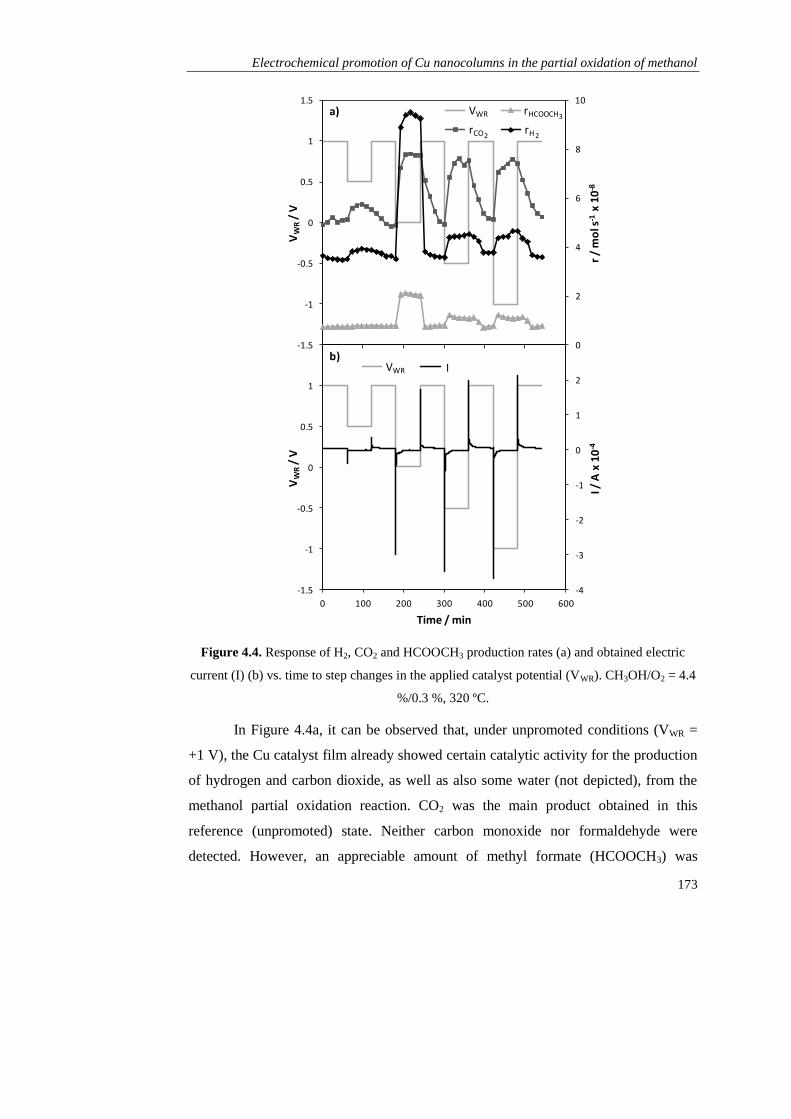
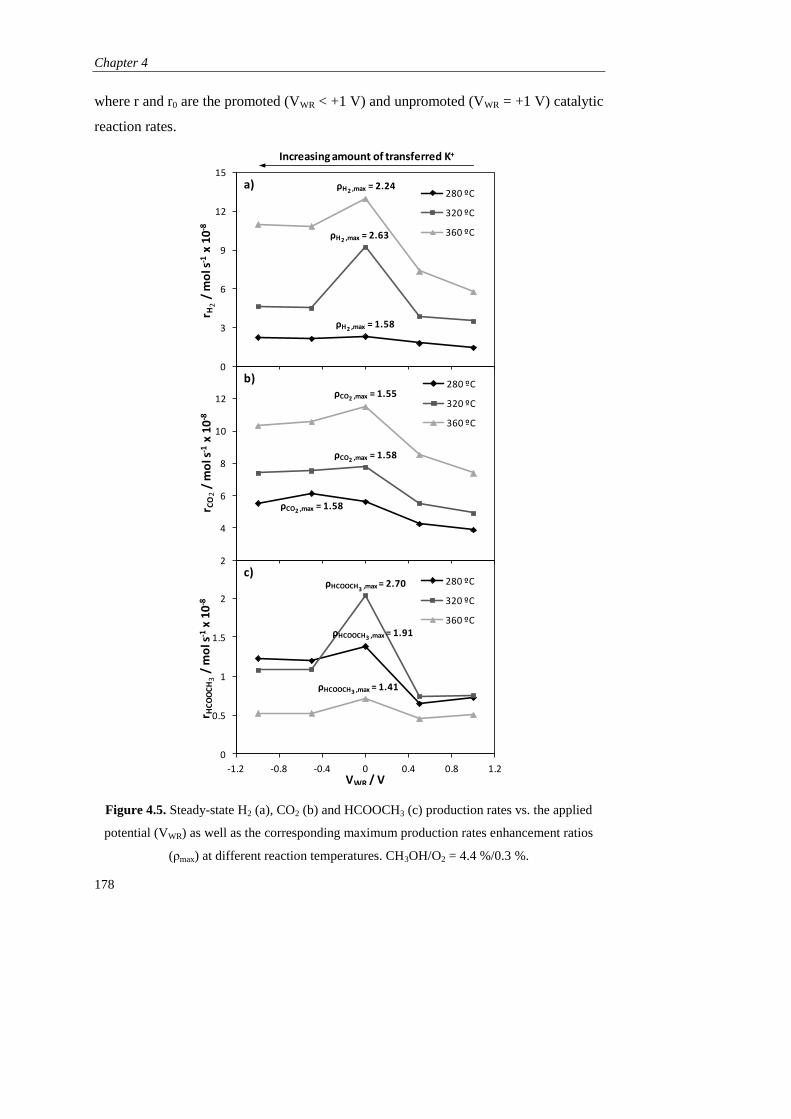
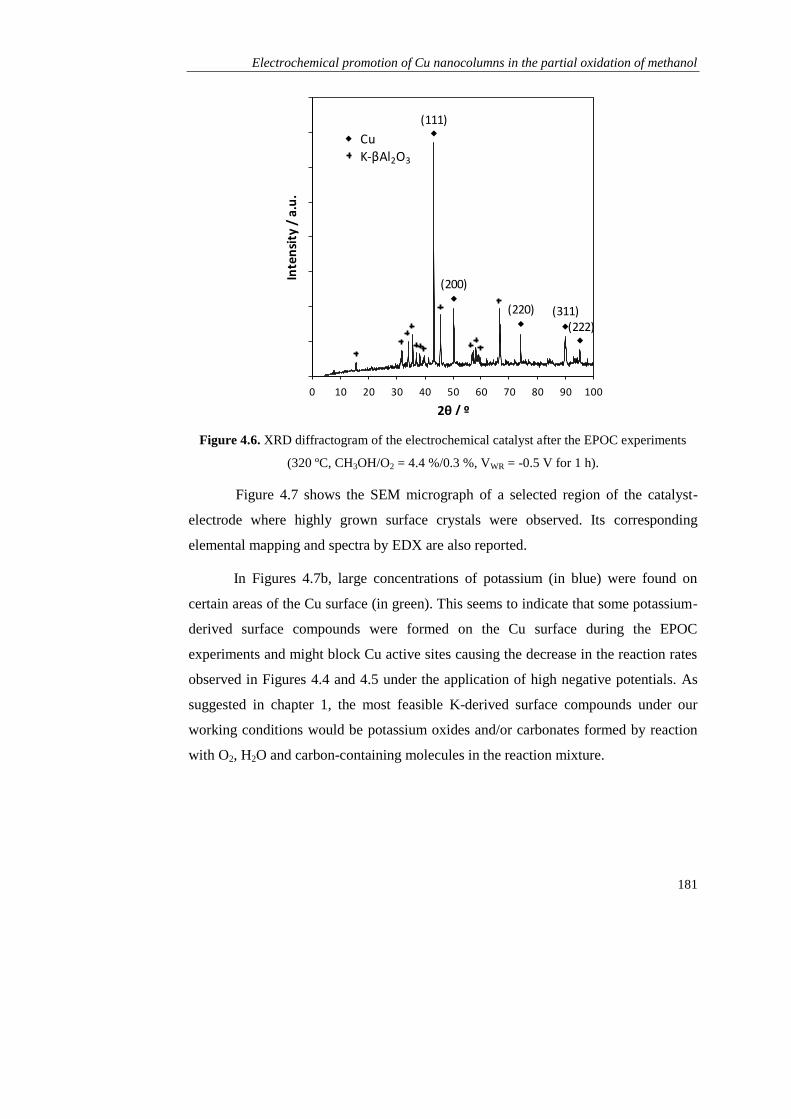
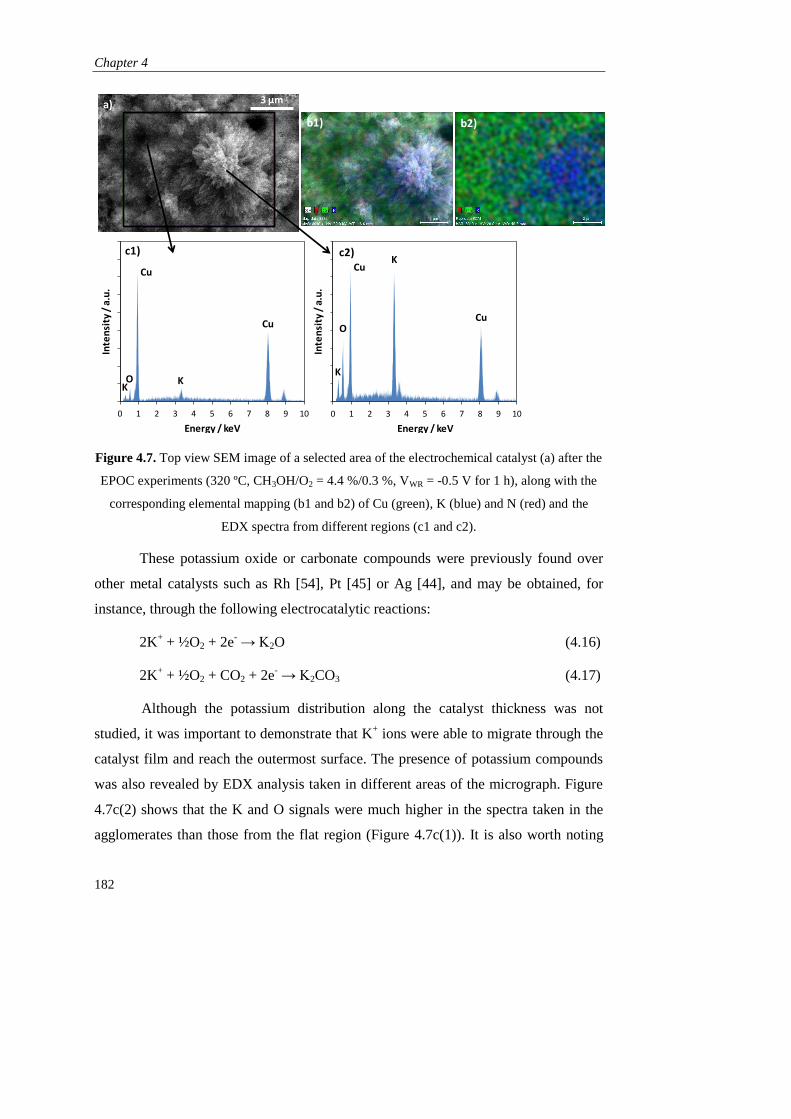
1.2.3).
Page 41
Resultados obtenidos
33
C. RESULTADOS OBTENIDOS
Como se ha mencionado, esta tesis está dirigida al estudio de la promoción
electroquímica de la catálisis (EPOC) aplicada a procesos de producción de hidrógeno
empleando metanol como materia prima. Para ello se seleccionó K-βAl2O3 como
soporte del catalizador y electrolito sólido (fuente de iones promotores K+) en todos
los estudios.
En el Capítulo 1 se realizaron los primeros estudios de promoción
electroquímica en las reacciones de oxidación parcial de metanol (POM, CH3OH +
1/2O2 → 2H2 + CO2) y de reformado de metanol con vapor de agua (SRM, CH3OH +
H2O → 3H2 + CO2), empleando como catalizador metálico de partida Pt, en base a sus
buenos resultados obtenidos en bibliografía tanto en las reacciones de POM y SRM
como en otros estudios de promoción electroquímica. Se prepararon dos tipos de
películas de Pt mediantes técnicas diferentes: una mediante impregnación de una
disolución precursora de este metal (dando lugar a un contenido final de 1,1 mg Pt
cm-2
), y otra en colaboración con el Instituto de Ciencia de Materiales de Madrid
(CSIC), mediante deposición por arco catódico (CAD), que es un tipo de técnica de
deposición física de vapor (PVD). Esta permitió obtener una película densa con un
espesor de 120 nm y un contenido metálico de solo 0,23 mg Pt cm-2
.
En primer lugar se compararon los dos catalizadores de Pt/K-βAl2O3/Au en la
reacción de oxidación parcial de metanol (CH3OH/O2 = 7,2 %/4,6 %, 320 ºC) bajo
condiciones de promoción electroquímica, es decir, bajo la aplicación de diferentes
potenciales eléctricos (entre +2 y -2 V). En ambos casos, al igual que en el resto de
experimentos de este capítulo y de toda la tesis, se aplicó un potencial positivo (VWR =
+2 V) al principio y al final del experimento para asegurar la retirada de todos los
iones K+ de la película del catalizador y por tanto para definir un estado no
promocionado o de referencia. A continuación, la disminución del potencial causó la
migración de iones K+ desde el electrolito sólido hasta el catalizador y su distribución
a lo largo de la superficie (lo que se conoce como “back-spillover”), dando lugar a la
formación de una doble capa efectiva que puede modificar la quimisorción de los
distintos reactivos sobre el Pt, tal y como se explicó en la sección A.3.1.
Page 42
Descripción del trabajo realizado
34
En el caso del Pt depositado por arco catódico (CAD), el suministro de iones
K+ causó un incremento de las velocidades de producción del H2 y CO2 (productos de
la oxidación parcial de metanol) así como del CO, derivado de la descomposición del
metanol (MD, CH3OH → 2H2 + CO) y del formaldehído, obtenido también como
subproducto (CH3OH + 1/2O2 → H2CO + H2O). Este efecto promotor del potasio se
puede explicar en base a una disminución de la función de trabajo del catalizador, que
fortalecería la quimisorción sobre el Pt del aceptor de electrones (O2) y debilitaría la
del donador de electrones (CH3OH). De este modo el efecto EPOC obtenido fue de
tipo electrofílico y consiguió aumentar la producción de hidrógeno y formaldehído 6 y
4 veces, respectivamente, bajo condiciones óptimas de promoción electroquímica.
Por otro lado, en el caso del Pt preparado por impregnación, el descenso en el
potencial dio lugar a una disminución brusca de la actividad catalítica del Pt, que se
atribuyó a la mayor porosidad de este electrodo con respecto al preparado por CAD, y
a la formación de una excesiva cantidad de compuestos superficiales derivados del
potasio (tipo carbonatos) que pudieron bloquear centros activos del Pt. Esta hipótesis
quedó demostrada mediante diferentes técnicas de caracterización (voltametría cíclica,
XRD, SEM-EDX), reflejando que el comportamiento EPOC de un catalizador
electroquímico catiónico no solo depende de su capacidad de quimisorción de las
distintas moléculas de reactivo sino también de su actividad electrocatalítica y su
facilidad para formar compuestos superficiales derivados del promotor.
El catalizador de Pt preparado por CAD fue, por tanto, seleccionado para la
realización de los siguientes experimentos, en los que se estudió la influencia tanto de
la temperatura como de la composicón del alimento. Se comprobó que el efecto de la
promoción electroquímica no solo mejoró la actividad catalítica del platino, sino que
también incrementó su selectividad hacia el mecanismo de la oxidación parcial frente
a la oxidación total. Esto permitió obtener simultáneamente dos productos de elevado
interés como son el H2 y el H2CO en una sola etapa de reacción. Además, se realizaron
ensayos de reproducibilidad y durabilidad que demostraron la reversibilidad y
estabilidad del efecto promocional obtenido con este tipo de catalizadores
electroquímicos. Por último se evaluó la actividad catalítica de este catalizador en
Page 43
Resultados obtenidos
35
condiciones de reformado de metanol con vapor de agua (CH3OH/H2O = 4 %/4,8 %),
y aunque fue mucho menor que en las condiciones de oxidación parcial, también se
promocionó electroquímicamente con los iones K+, activando en este caso la
quimisorción del agua (aceptor de electrones) frente al metanol (donador de
electrones). Por tanto se demostró la utilidad de los métodos de deposición física de
vapor (PVD) en la obtención de películas delgadas para su uso en estudios de
promoción electroquímica con conductores alcalinos, y por tanto los catalizadores
empleados en el resto de capítulos se prepararon mediante alguna técnica PVD.
En el Capítulo 2 se llevó a cabo una variante de la técnica de CAD empleada
en el capítulo anterior para obtener un catalizador de Pt con mejores propiedades. Esta
consistió en depositar conjuntamente dos materiales diferentes. En este caso, la
película de catalizador resultante estaba formada por nanopartículas de Pt de unos 3
nm de diámetro dispersas en una matriz de carbono tipo diamante (“diamond-like
carbón”, DLC). Con un espesor (del orden de 100 nm) similar a la película de Pt puro
preparada por el mismo método (CAD), la película de Pt-DLC contenía una carga
metálica menor (solo 0,014 mg Pt cm-2
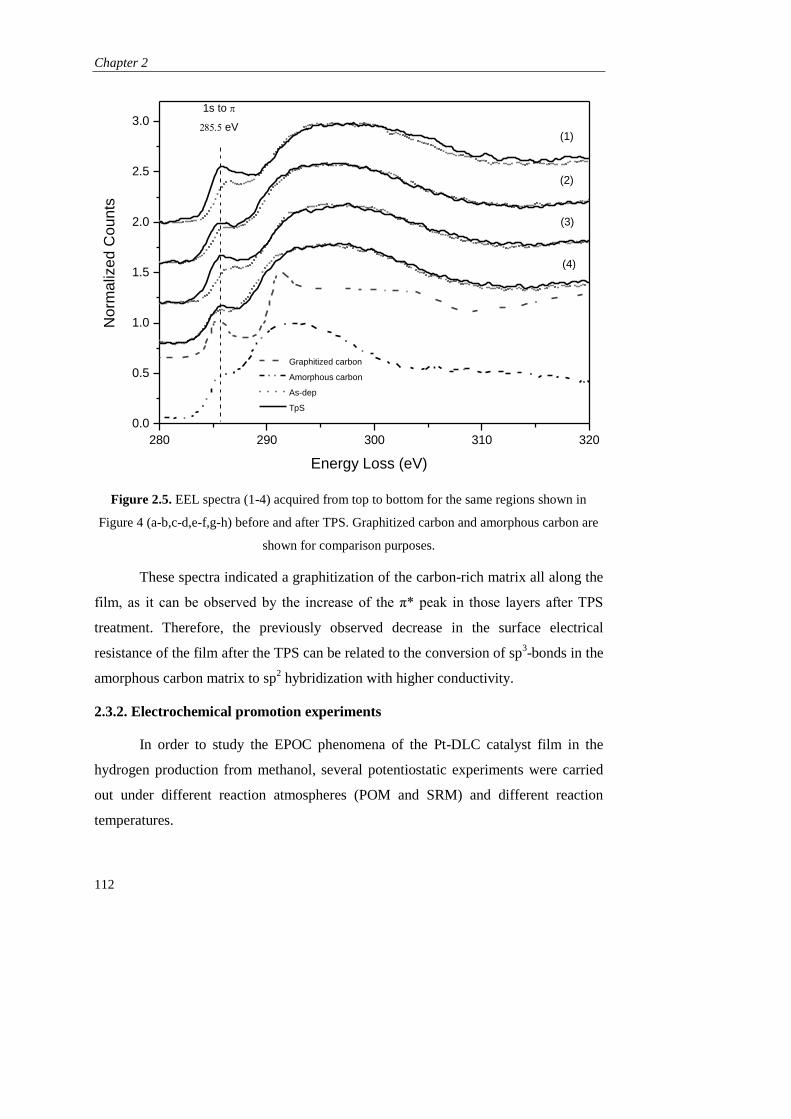
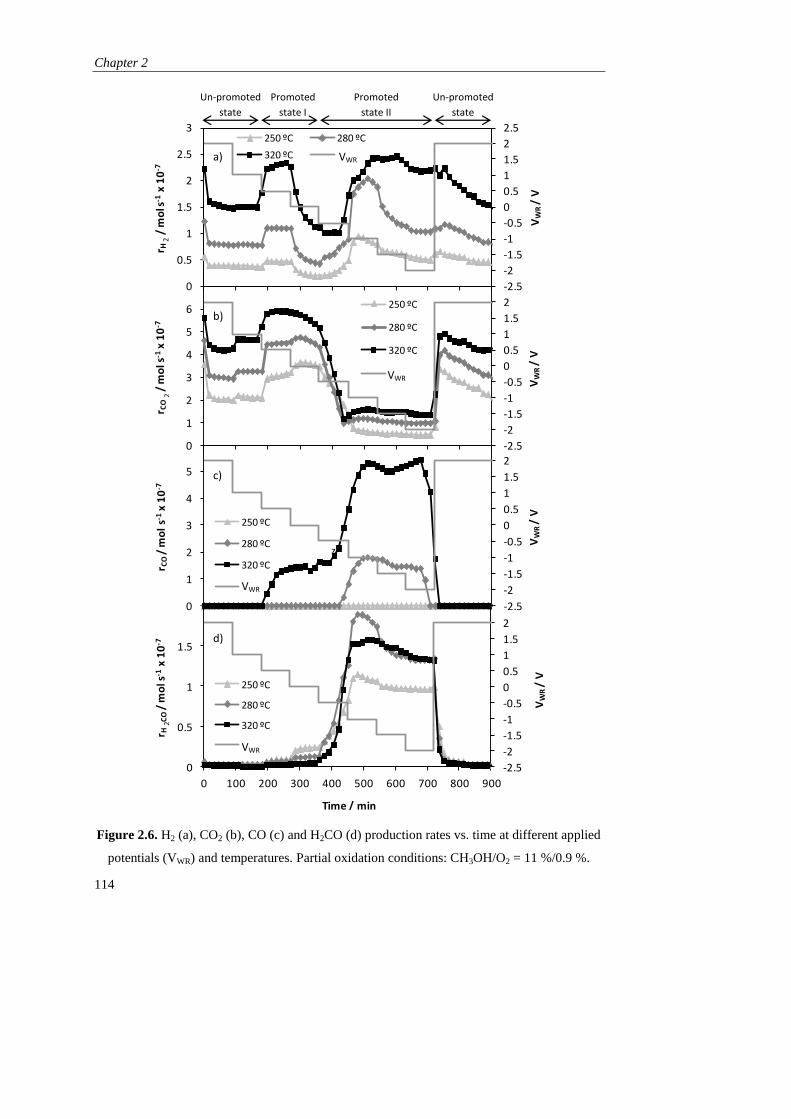
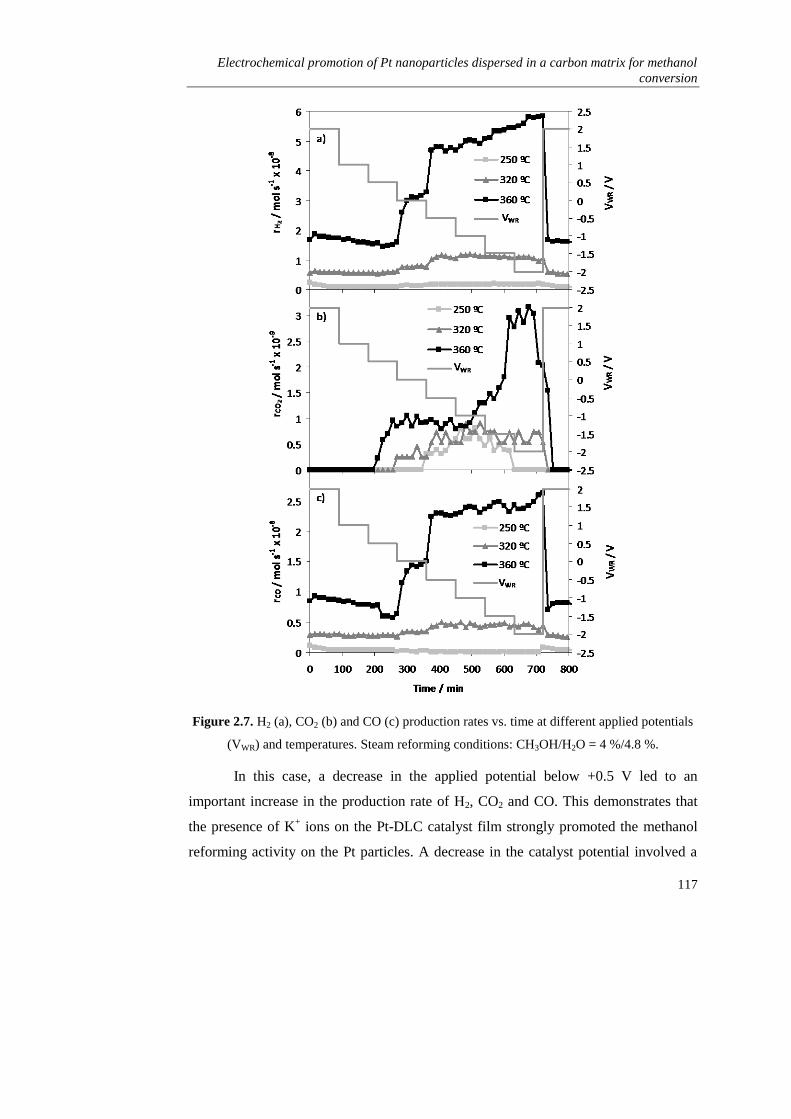
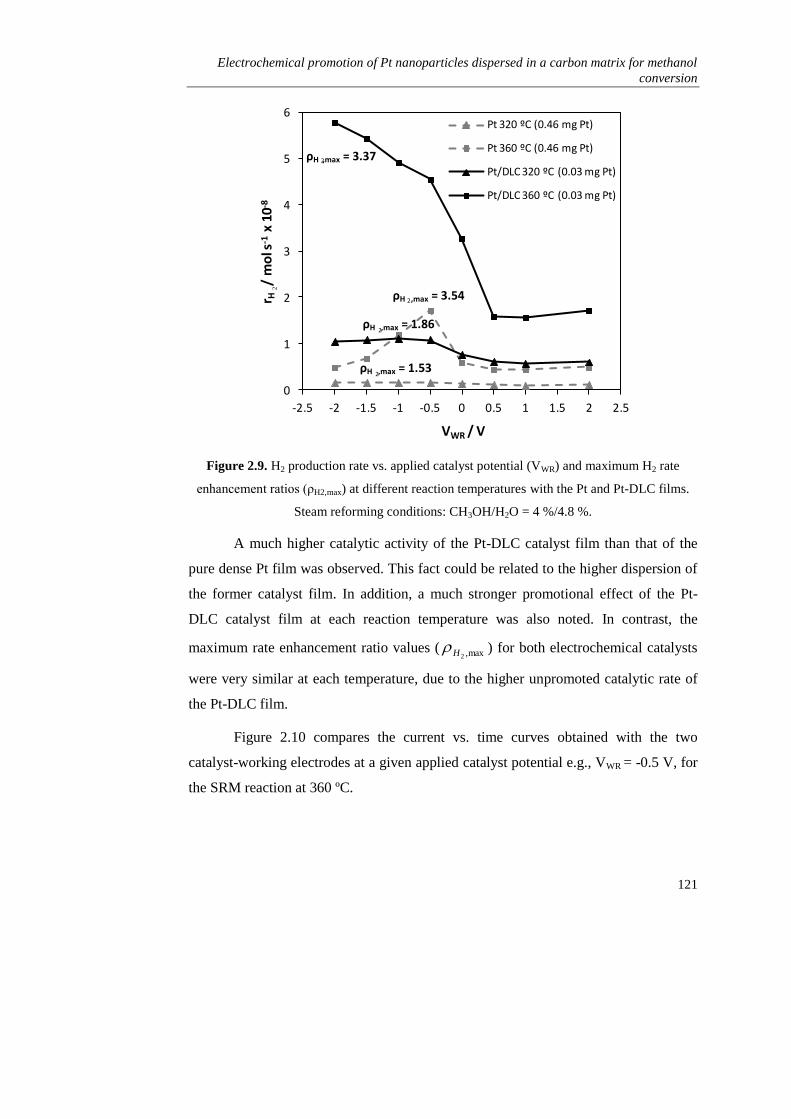
). Como principal inconveniente, este
catalizador no presentaba conductividad eléctrica tras su deposición. Sin embargo, tras
someter el catalizador Pt-DLC/K-βAl2O3/Au a un experimento inicial a temperatura
programada en atmósfera de hidrógeno, su resistencia eléctrica superficial (medida in-
situ) descendió hasta un valor de 3,5 kΩ, que permitió su polarización en los
posteriores experimentos electrocatalíticos. Esta disminución de la resistencia eléctrica
tuvo lugar bruscamente en torno a 200 ºC, lo cual se atribuyó a una transición de la
matriz de carbono (con una hibridación mayoritaria sp3) hacia una forma más similar a
la del grafito (una mayor hibridación sp2), como se verificó mediante EELS. Además,
se comprobó la estabilidad de las nanopartículas de Pt durante este proceso mediante
microscopía STEM.
La película de catalizador Pt-DLC consiguió ser promocionada mediante la
migración electroquímica de iones alcalinos K+ de forma reversible tanto en la
oxidación parcial de metanol (CH3OH/O2 = 11 %/0,9 %), obteniendo H2, CO, CO2 y
H2CO y H2O como productos de reacción, como en el reformado de metanol con
Page 44
Descripción del trabajo realizado
36
vapor de agua (CH3OH/H2O = 4 %/4,8 %), produciendo en este caso H2, CO y CO2.
Al igual que en el capítulo anterior, el efecto de promoción de tipo electrofílico
obtenido en ambos casos al aplicar un potencial lo suficientemente negativo se
atribuyó a una mayor quimisorción del aceptor de electrones (O2 o H2O) frente al
donador de electrones (CH3OH). Sin embargo, en este estudio se observó un primer
incremento de la actividad catalítica, bajo condiciones de oxidación parcial, al
disminuir el potencial desde +2 V hasta tan solo +0,5 V. Este efecto sugirió la
formación de una nueva fase de promotor sobre las nanopartículas de Pt, y pareció
confirmarse mediante voltamperometría cíclica. Durante este procedimiento se
observaron varios picos catódicos y anódicos en atmósfera de POM, mientras que solo
un único pico en cada barrido en atmósfera de SRM, lo cual pareció indicar que
dependiendo de las condiciones de reacción y del potencial aplicado los iones K+
pueden formar diferentes fases promotoras sobre el catalizador.
Por último, comparando en condiciones de SRM los dos catalizadores
preparados por arco catódico (Pt y Pt-DLC), se pudo comprobar que las
nanopartículas dispersas de Pt presentaron una mayor actividad catalítica y un
incremento de esta (vía EPOC) de una magnitud similar al obtenido con el catalizador
denso de Pt. Este estudio abre, por tanto, una nueva posibilidad para la aplicación de la
promoción electroquímica en catalizadores dispersos de muy bajo contenido metálico
y soportados sobre un material no conductor iónico.
En la misma línea de empleo de películas de catalizadores dispersos, en el
Capítulo 3 se desarrolló un catalizador Au-YSZ/K-βAl2O3/Au que se basaba en
nanopartículas de Au soportadas en una matriz de zirconia estabilizada con itria
(YSZ), preparado en esta ocasión mediante pulverización catódica (“sputtering”). Este
electrodo fue depositado en colaboración con el Institut Jean Lamour de Université de
Lorraine (Francia). Concretamente se depositó mediante el “co-sputtering” simultáneo
de Au y Y-Zr en atmósfera de Ar y O2. La película de Au-YSZ se caracterizó
mediante diferentes técnicas (XRD, STEM, voltamperometría cíclica) y se evaluó su
actividad en la oxidación parcial catalítica de metanol (CH3OH/O2 = 5,9 %/0,43 %). A
diferencia del contraelectrodo y el electrodo de referencia de Au depositados en todos
Page 45
Resultados obtenidos
37
los capítulos mediante la calcinación a elevada temperatura de una pasta orgánica de
este metal, el catalizador-electrodo de trabajo formado por nanopartículas de Au
dispersas en YSZ mostró cierta actividad catalítica y, en particular, una selectividad
muy elevada hacia formato de metilo (HCOOCH3).
Debido a que el Au-YSZ no presentaba conductividad eléctrica, se utilizó una
malla de Ag como colector eléctrico y se consiguió polarizar y promocionar
electroquímicamente el catalizador con iones alcalinos. Se llevó a cabo una serie de
experimentos galvanostáticos en los cuales, tras limpiar la superficie del catalizador de
iones K+ por medio de una polarización positiva (VWR = +2 V), se aplicó una
intensidad de corriente negativa de unos pocos µA, que suponía la migración
electroquímica de iones K+ desde el electrolito sólido hacia la película de catalizador
con un flujo constante determinado por la ley de Faraday, r = I/nF. Se realizaron
distintos experimentos en los que se varió esta intensidad negativa, y también otros en
los que la promoción electroquímica se llevó a cabo de modo potenciostático
(aplicando potenciales eléctricos constantes) como en los capítulos anteriores. En
estos últimos experimentos también se podía controlar la cantidad de promotor
suministrada, si bien en estos casos el flujo de K+ no era constante y estaba
determinado por el valor de intensidad obtenido en cada momento. Así pues, en todos
los experimentos realizados en las mismas condiciones de reacción se obtuvo la
misma cantidad óptima de promotor y las mismas velocidades máximas de producción
de los distintos compuestos, demostrando que la magnitud del efecto de promoción
electroquímica con electrolitos sólidos alcalinos no depende de la intensidad eléctrica
aplicada (es decir, del flujo de promotor) ni del modo de operación (potenciostático o
galvanostático), sino de la cantidad total de promotor adicionada.
Además, al interrumpir la corriente eléctrica en estos experimentos
(condiciones de circuito abierto), se observó un mantenimiento de las distintas
velocidades de producción obtenidas durante la polarización previa negativa,
reflejando una gran estabilidad de las especies promotoras en las condiciones de
reacción estudiadas y por tanto un efecto EPOC permanente. Solo aplicando de nuevo
un potencial positivo (VWR = +2 V) al final de cada experimento se conseguía eliminar
Page 46
Descripción del trabajo realizado
38
estas especies del catalizador y restablecer la actividad catalítica inicial (no
promocionada). En definitiva, no solo se consiguió promocionar electroquímicamente
una película que no era conductora eléctrica, sino que además una primera
polarización podía ser suficiente para mantener la modificación de la actividad
catalítica durante un cierto periodo de tiempo. Por otro lado, se comprobó la
estabilidad de la película de Au-YSZ a lo largo de todos estos experimentos,
demostrando la enorme importancia del soporte de YSZ para dispersar nanopartículas
de metal y evitar su sinterización.
En el Capítulo 4 se aplicó por primera vez el fenómeno de la promoción
electroquímica a la reacción de oxidación parcial de metanol, empleando un metal no
noble (Cu) como catalizador. En esta ocasión, la película de catalizador-electrodo de
trabajo se depositó sobre el soporte de K-βAl2O3 en colaboración con el Instituto de
Ciencia de Materiales de Sevilla (CSIC - Universidad de Sevilla), mediante una
técnica de deposición física de vapor muy innovadora denominada “Deposición en
ángulo oblicuo” o “Deposición en ángulo rasante” (OAD o GLAD). Esta se basa en
hacer incidir el flujo evaporado del material a depositar (Cu) con un determinado
ángulo (α) sobre el sustrato (K-βAl2O3). De este modo, durante el crecimiento de la
película se produce un “efecto sombra” que causa la formación de nanocolumnas
inclinadas del metal y por tanto la obtención de una elevada porosidad y área
superficial. El ángulo de deposición α se varió durante el proceso de deposición, de
modo que en una primera etapa (α = 0º) se forzó el crecimiento de una película con
una buena conductividad eléctrica formada por nanocolumnas Cu perpendiculares al
electrolito sólido con una estructura compacta, y en una segunda etapa (α = 80º) se
formó una película mucho más porosa (en torno al 50 %) encima de la anterior,
mediante la deposición de nanocolumnas inclinadas de Cu. El objetivo de este
procedimiento fue conseguir un electrodo de trabajo de Cu que en su conjunto
presentara buenas propiedades tanto catalíticas como electrocatalíticas.
Mediante la realización de voltamperometrías cíclicas con el catalizador de
Cu/K-βAl2O3/Au se observó que un rango de potencial entre +1 y -1 era suficiente
para formar y descomponer las especies promotoras derivadas del potasio de forma
Page 47
Resultados obtenidos
39
reversible y reproducible. Por tanto se realizaron transiciones potenciostáticas entre
estos dos valores en los experimentos de promoción electroquímica. La película de
cobre demostró ser activa en la oxidación parcial de metanol (CH3OH/O2 = 4,4 %/0,3
%) obteniendo H2 y CO2, además de H2O y HCOOCH3, y la velocidad de producción
de todos estos productos se vio incrementada al disminuir el potencial aplicado y
dopar electroquímicamente la superficie del catalizador con iones K+. Al igual que en
los capítulos anteriores, este efecto promocional electrofílico se explicó en base a un
fortalecimiento de la quimisorción de oxígeno que favorecería la deshidrogenación del
metanol y de las especies metóxido intermedias (la cual se suele considerar la etapa
limitante de este tipo de reacciones).
Tras los experimentos de promoción electroquímica el catalizador se sometió a
un acondicionamiento bajo condiciones de POM y una polarización negativa para
establecer un estado promocionado, y se caracterizó mediante diferentes técnicas ex-
situ (XRD, SEM-EDX, XPS). De este modo se confirmó que bajo condiciones de
promoción electroquímica se formaron especies superficiales derivadas de los iones
K+, probablemente carbonatos u óxidos. También se detectó la presencia de cierta
cantidad de carbono superficial (aunque no se observó una apreciable desactivación
durante los ensayos catalíticos) y de compuestos de nitrógeno que probablemente se
formaron bajo el efecto promocional del potasio a partir del N2 e H2 presentes en la
artmósfera de reacción.
Cabe destacar que a pesar de presentar esta película de Cu una actividad
catalítica menor que los catalizadores de Pt de capítulos anteriores, se obtuvo un
efecto promocional de magnitud similar. Esto, por un lado, puso de manifiesto el gran
interés que puede tener en este tipo de estudios la técnica de deposición en ángulo
oblicuo para la obtención de películas porosas, y por otro lado, motivó a continuar
estudiando el fenómeno EPOC con catalizadores no nobles.
En el Capítulo 5 se preparó un catalizador de Ni/K-βAl2O3/Au por arco
catódico de modo similar a la película fina de Pt del capítulo 1 y se aplicó el fenómeno
de la promoción electroquímica con iones K+ bajo diferentes condiciones de reacción:
descomposición de metanol (MD, CH3OH = 4,4 %), reformado de metanol con vapor
Page 48
Descripción del trabajo realizado
40
de agua (SRM, CH3OH/H2O = 4,4 %/5,2 %) y oxidación parcial de metanol (POM,
CH3OH/O2 = 4,4 %/0,33 %). El efecto de los iones K+, unido a las características
particulares del Ni (una gran tendencia a la deposición de carbón y a la oxidación), dio
lugar a comportamientos catalíticos muy diferentes en las tres atmósferas de reacción
y posibilitó, en definitiva, el estudio de tres aplicaciones fundamentales del fenómeno
EPOC: la activación del catalizador, la modificación de la selectividad catalítica y la
oxidación parcial del catalizador.
Bajo condiciones de MD, la promoción del catalizador de Ni mediante la
migración electroquímica de iones K+ por medio de transiciones potenciostáticas
favoreció la deshidrogenación del metanol y de las especies intermedias para obtener
H2 y CO. En estas condiciones también tuvo lugar una fuerte desactivación por
deposición de carbono que además se acrecentó por efecto de los iones K+ a
potenciales demasiado negativos. Por otro lado, bajo condiciones de SRM la actividad
catalítica del Ni fue mayor y se obtuvo también CO2, aunque los principales productos
siguieron siendo H2 y CO. El suministro de iones K+ dio lugar a un incremento en la
velocidad de producción de los tres compuestos, lo cual se atribuyó a la activación del
agua como en los capítulos 1 y 2 (con catalizadores de Pt). Del mismo modo, este
efecto propició además una atenuación de la desactivación del Ni por deposición de
carbono bajo esta atmósfera de reacción.
Bajo condiciones de POM el Ni no promocionado (VWR = +2 V) presentó una
producción de H2 y CO superior a los casos anteriores. Sin embargo, la disminución
en el potencial aplicado y el fortalecimiento de la quimisorción del oxígeno en este
caso causó un descenso brusco de la actividad del Ni y de la selectividad hacia H2 y
CO, al mismo tiempo que aumentó ligeramente la selectividad hacia CO2, agua y
formaldehído. Este efecto negativo de los iones K+ parecía estar en contra del orden
positivo de la reacción, observado en estado no promocionado, con respecto a la
concentración de oxígeno (para valores bajos como el empleado en los experimentos
de EPOC). Finalmente se atribuyó a un incremento de la formación de NiO derivado
de una mayor adsorción de oxígeno de acuerdo con los resultados obtenidos mediante
XRD. Cabe señalar, no obstante, que el efecto promocional observado en todos los
Page 49
Resultados obtenidos
41
casos se llevó a cabo de una forma controlada (variando tan solo el potencial aplicado)
y totalmente reversible, en base a una completa reproducibilidad de la actividad
catalítica del Ni en cada imposición del estado de referencia (VWR = +2 V). De este
modo se demostró la posibilidad de controlar la oxidación parcial del catalizador por
vía electroquímica, y con ello la modificación de su selectividad hacia la formación de
distintas especies.
Por último, en el Capítulo 6 se ha estudiado la posibilidad de aplicar el
fenómeno de la promoción electroquímica conjuntamente para la producción y el
almacenamiento de hidrógeno. La idea fue desarrollar un catalizador electroquímico
alcalino que presentara un rendimiento adecuado en la producción de hidrógeno
mediante reformado de metanol con vapor de agua (SRM), y que fuese capaz de
almacenarlo y liberarlo de forma controlada, operando bajo diferentes polarizaciones y
condiciones suaves de reacción. Para ello se seleccionó Ni como fase activa debido a
la buena actividad catalítica en la reacción SRM mostrada en el capitulo anterior. Se
prepararon diferentes películas de Ni sobre K-βAl2O3 mediante el método de
deposición en ángulo oblicuo (OAD) utilizado en el capítulo 4), variando el ángulo de
incidencia (α) del Ni sobre la K-βAl2O3: En un caso (catalizador Ni 0º) el flujo de
evaporación de Ni incidió perpendicularmente sobre el electrolito sólido (α = 0º)
dando lugar a nanocolumnas compactas y verticales de Ni, y en otro caso el
catalizador (Ni 80º) se depositó con un ángulo de inclinación de α = 80º , obteniendo a
una película formada por nanocolumnas de Ni inclinadas con una porosidad del 35 %.
Se llevó a cabo una serie de experimentos que constaban de las siguientes
cuatro etapas: 1) Aplicación de un potencial positivo (VWR = +2 V) bajo condiciones
de SRM (CH3OH/H2O = 4,4 %/5,2 %) para asegurar la ausencia de iones K+ sobre el
catalizador y establecer un estado de referencia; 2) Aplicación de una polarización
negativa bajo condiciones de SRM para trasladar iones K+ a la superficie de Ni y
promocionar la producción y captura de compuestos; 3) Purga del reactor con gas
inerte bajo circuito abierto; y 4) Aplicación de una polarización positiva en atmósfera
inerte para la retirada de los iones K+ del catalizador y la liberación de los compuestos.
Page 50
Descripción del trabajo realizado
42
Unos experimentos preliminares realizados con el catalizador Ni 0º ya
reflejaron una liberación de H2 junto con CO y CO2 (en cantidades mucho menores)
durante la última etapa. También permitió optimizar el intervalo de los análisis y el
procedimiento de polarización durante esta etapa. Posteriormente se estudió la
influencia de la corriente negativa aplicada durante la etapa de almacenamiento, de la
microestructura del catalizador y de la atmósfera de reacción en la capacidad de
almacenamiento de H2 del catalizador y en la velocidad de liberación.
En todos los casos los catalizadores de Ni mostraron una actividad mucho
mayor en la producción de H2 y CO (MD, CH3OH → 2H2 + CO) frente a la
producción CO2 (SRM, CH3OH + H2O → 3H2 + CO2). También se observó cierta
desactivación de los catalizadores atribuida a la deposición de especies carbonosas
sobre la superficie. Esta podía estar además favorecida bajo polarización negativa por
la presencia de iones K+ y su efecto promocional en la disociación de CO.
En una primera etapa se plantearon los distintos posibles mecanismos de
captura de H2. Posteriormente, se fueron analizando y descartando algunos de ellos en
base a los resultados obtenidos y a diversas técnicas de caracterización realizadas. La
gran cantidad liberada de H2 observada en todos los casos en comparación con el CO,
el CO2 y los iones K+ transferidos electroquímicamente sugirió que el principal
mecanismo de almacenamiento no podía ser la formación directa de compuestos de
potasio: KH, KOH o bicarbonatos de potasio, ni la acumulación de intermedios de
reacción tipo metóxido sobre la superficie del catalizador. De este modo el
almacenamiento de H2 se atribuyó principalmente a la quimisorción de átomos de
hidrógeno procedentes de la conversión catalítica del metanol sobre centros activos
del niquel, y su posible acumulación posterior, a través de un proceso de spillover,
sobre otros adsorbentes como los depósitos carbonosos formados durante la reacción.
De hecho, la hipótesis de una captura de H2 ligada en gran medida a estos compuestos
carbonosos cobró una especial relevancia a la vista de los resultados obtenidos con el
Ni 80º, donde los moles de H2 liberados superaron en un orden de magnitud a la
cantidad de Ni presente en el catalizador, reflejando el papel fundamental de la
porosidad y área superficial del electrodo.
Page 51
Resultados obtenidos
43
El Ni 80º presentó una mayor actividad catalítica y una capacidad de
almacenamiento de H2 muy superior al Ni 0º (19 vs 0,5 g H2 x 100 g Ni-1
), lo cual se
atribuyó a su mayor porosidad y facilidad para formar depósitos carbonosos. Esto
mostró la gran influencia de la microestructura del catalizador sobre el proceso de
producción/captura de H2, y por tanto la enorme utilidad del método de deposición en
ángulo oblicuo (OAD) en el diseño de este tipo de catalizadores. Por otro lado, el
aumento de la intensidad negativa en la etapa de almacenamiento causó un incremento
de la cantidad de H2 capturado (hasta cierto valor de saturación), atribuido a un doble
efecto promotor de los iones K+: en la formación de compuestos carbonosos sobre el
Ni, y en la adsorción de hidrógeno sobre estos compuestos.
En línea con el mecanismo de almacenamiento de H2 propuesto, unos
experimentos adicionales donde se emplearon diferentes atmósferas de reacción en la
etapa de polarización negativa demostraron el papel clave de los compuestos
carbonosos, ya que el hidrogeno capturado a partir de una corriente de H2 (en lugar de
metanol) fue despreciable. Por otro lado, la presencia de agua no supuso ninguna
diferencia en la capacidad de captura de H2, aunque sí aceleró su liberación.
Por último, el catalizador Ni/K-βAl2O3/Au se caracterizó mediante diferentes
técnicas ex-situ (SEM-EDX, XPS, FTIR, Raman) y se identificó óxido de grafeno
como principal compuesto carbonoso localizado sobre la superficie del catalizador.
Por tanto, el catalizador electroquímico de Ni desarrollado en este trabajo permite
producir H2 y óxido de grafeno a partir de metanol, almacenar el H2 con un elevado
rendimiento por unidad de área y cantidad de metal, y liberarlo con una elevada
pureza bajo las mismas condiciones de reacción (280 ºC); todo ello controlado vía
electroquímica, mediante un soporte conductor de iones K+.
Page 52
Descripción del trabajo realizado
44
D. CONCLUSIONES Y RECOMENDACIONES
De los resultados obtenidos en la presente investigación se pueden obtener las
siguientes conclusiones:
- Una película catalítica de Pt con un espesor 120 nm se ha preparado
mediante el método de deposición por arco catódico (CAD) y se ha promocionado
electroquímicamente con iones K+ en las reacciones de oxidación parcial de metanol
(POM) y reformado de metanol con vapor de agua (SRM). En cambio, otro
catalizador de Pt preparado por impregnación presentó un exceso de actividad
electrocatalítica y el bloqueo de centros activos de Pt por compuestos derivados del
potasio. Por tanto, los demás catalizadores empleados en esta tesis se han preparado
mediante el método de CAD u otra técnica de deposición física de vapor (PVD), como
“sputtering” o OAD.
- La promoción electroquímica de la catálisis (EPOC) permite mejorar in-situ
la actividad del catalizador de Pt preparado por CAD y su selectividad hacia la
oxidación parcial de metanol (frente a la oxidación total), mediante la migración
controlada de iones K+ desde un electrolito sólido. Además, el efecto EPOC observado
con este catalizador y con todos los demás empleados en la tesis demostró ser
reversible al retirar los iones K+ del catalizador (bajo un potencial positivo).
- Se ha desarrollado una película de catalizador formada por nanopartículas de
Pt (~3 nm) dispersas en una matriz de carbono (Pt-DLC), preparada por CAD. Se ha
podido promocionar electroquímicamente tras un proceso de grafitización previo y ha
mostrado diferentes efectos promocionales del K+ en función del potencial aplicado y
de la atmósfera de reacción (POM o SRM). Esta película ha presentado una mayor
actividad catalítica y un efecto EPOC similar que el catalizador de Pt puro, incluso
con peores propiedades eléctricas y una menor carga metálica (solo 0,014 mg cm-2
).
- Se ha conseguido promocionar electroquímicamente mediante un colector de
corriente una película catalítica no conductora eléctrica, compuesta por nanopartículas
de Au dispersas en zirconia estabilizada con itria (Au-YSZ) mediante “co-sputtering”,
para la oxidación selectiva de metanol. Se ha optimizado la cantidad de iones
Page 53
Conclusiones y recomendaciones
45
promotores K+ suministrada a este catalizador, demostrando además que el efecto
EPOC obtenido es permanente (hasta una nueva polarización positiva) y que su
magnitud no depende de la velocidad del suministro de los iones ni del modo de
operación (potenciostático o galvanostático) sino exclusivamente de la cantidad total
suministrada.
- Es posible promocionar electroquímicamente películas de metales no nobles
como Cu en la reacción de POM con razones de incremento de la actividad catalítica
comparables a las obtenidas con el catalizador de Pt. Este catalizador de Cu se ha
preparado mediante una novedosa modificación del método de PVD que consiste en la
deposición en ángulo oblicuo (OAD) de nanocolumnas del metal, obteniendo películas
conductoras con una elevada porosidad muy útiles para aplicaciones catalíticas o
electrocatalíticas.
- Se ha demostrado la posible aplicación de la promoción electroquímica con
iones K+ para activar un catalizador metálico, modificar su selectividad, y controlar
parcialmente su estado de oxidación, empleando una misma película de Ni (praparada
por CAD). En atmósfera de metanol y de metanol+agua el suministro de iones K+
ocasiona la activación del mecanismo de deshidrogenación del metanol, y en este
último caso también una atenuación de la desactivación del catalizador por deposición
de carbono. Sin embargo, en atmósfera de metanol+oxígeno el fortalecimiento de la
quimisorción del oxígeno por efecto del K+ deriva en la formación de NiO, y
consecuentemente en la disminución de la selectividad hacia H2 y CO y el aumento de
la selectividad hacia CO2, H2O y H2CO.
- Se ha desarrollado un catalizador poroso de Ni depositado en ángulo oblicuo
que permite producir simultáneamente H2 y óxido de grafeno a partir de metanol, y
capturar parte del H2 sobre el Ni y el óxido de grafeno (que también actúa como
adsorbente) bajo la acción de iones K+. Además, este H2 también se puede liberar de
forma controlada vía electroquímica bajo las mismas condiciones de reacción (280
ºC), obteniendo valores de almacenamiento reversible de H2 muy elevados (19 g H2 x
100 g Ni-1
). Estos resultados abren la puerta a una nueva aplicación del fenómeno
EPOC en el campo de la producción y el almacenamiento de hidrógeno.
Page 54
Descripción del trabajo realizado
46
Con objeto de completar los resultados obtenidos y continuar esta
investigación, se recomienda:
- Realizar estudios de promoción electroquímica de las reacciones de
conversión de metanol con otros tipos de electrolitos sólidos, catiónicos (Na-βAl2O3,
NASICON, SCY), aniónicos (YSZ) o conductores mixtos (tipo perovskita), y
comparar los resultados con los obtenidos en esta tesis con K-βAl2O3.
- Estudiar la promoción electroquímica de reacciones de producción de
hidrógeno empleando otras materias primas como etanol o bioalcoholes (mezclas
reales).
- Aplicar algunas de las técnicas de deposición de películas delgadas expuestas
en esta tesis (evaporación por bombardeo electrónico, pulverización catódica) para la
preparación de catalizadores soportados de Cu o Ni similares a los empleados en
catálisis convencional (Ni/Al2O3, Cu/Al2O3, Ni/CeO2, Cu/CeO2), u otros tipos de
catalizadores, como aleaciones (NiPt, NiCu) o configuraciones multicapa.
- Investigar otras técnicas para la preparación películas metálicas con una
elevada dispersión, como por ejemplo algún modo de prensar el catalizador en forma
de polvo sobre el electrolito sólido.
- Realizar estudios de promoción electroquímica de reacciones de conversión
de metanol en reactores de doble cámara, bien en configuración de pellet, o bien en
celda de combustible.
- Escalar los experimentos de promoción electroquímica de producción y/o
almacenamiento de hidrógeno, empleando configuraciones de mayor área de
electrodo, como las tubulares o monolíticas, que permitan obtener mayores valores de
conversión de metanol y/o de capacidad de almacenamiento de hidrógeno.
Page 55
Bibliografía
47
E. BIBLIOGRAFÍA
[1] J. Rifkin, La economía del hidrógeno. La creacción de la red energética mundial y la
redistribución del poder en la tierra, Paidos Iberica, Barcelona, 2002.
[2] J.N. Armor, Catalysis and the hydrogen economy, Catalysis Letters, 101 (2005) 131-135.
[3] S. Dutta, A review on production, storage of hydrogen and its utilization as an energy
resource, Journal of Industrial and Engineering Chemistry, 20 (2014) 1148-1156.
[4] L. Schlapbach, A. Züttel, Hydrogen-storage materials for mobile applications, Nature, 414
(2001) 353-358.
[5] J.R. Jennings, Catalytic Ammonia Synthesis: Fundamentals and Practice, Plenum Press,
New York, 1991.
[6] J.P. Lange, Methanol synthesis: A short review of technology improvements, Catalysis
Today, 64 (2001) 3-8.
[7] H. Jahangiri, J. Bennett, P. Mahjoubi, K. Wilson, S. Gu, A review of advanced catalyst
development for Fischer-Tropsch synthesis of hydrocarbons from biomass derived syn-gas,
Catalysis Science and Technology, 4 (2014) 2210-2229.
[8] M. Carmo, D.L. Fritz, J. Mergel, D. Stolten, A comprehensive review on PEM water
electrolysis, International Journal of Hydrogen Energy, 38 (2013) 4901-4934.
[9] M.A. Laguna-Bercero, Recent advances in high temperature electrolysis using solid oxide
fuel cells: A review, Journal of Power Sources, 203 (2012) 4-16.
[10] J.D. Holladay, J. Hu, D.L. King, Y. Wang, An overview of hydrogen production
technologies, Catalysis Today, 139 (2009) 244-260.
[11] R.M. Navarro, M.A. Peña, J.L.G. Fierro, Hydrogen production reactions from carbon
feedstocks: Fossil fuels and biomass, Chemical Reviews, 107 (2007) 3952-3991.
[12] R. Hughes, Deactivation of catalysts, Academic Press, New York, 1984.
[13] D.R. Palo, R.A. Dagle, J.D. Holladay, Methanol steam reforming for hydrogen
production, Chemical Reviews, 107 (2007) 3992-4021.
[14] P.O. Graf, B.L. Mojet, L. Lefferts, Influence of potassium on the competition between
methane and ethane in steam reforming over Pt supported on yttrium-stabilized zirconia,
Applied Catalysis A: General, 346 (2008) 90-95.
[15] F. Frusteri, S. Freni, V. Chiodo, L. Spadaro, G. Bonura, S. Cavallaro, Potassium improved
stability of Ni/MgO in the steam reforming of ethanol for the production of hydrogen for
MCFC, Journal of Power Sources, 132 (2004) 139-144.
Page 56
Descripción del trabajo realizado
48
[16] C. Song, Fuel processing for low-temperature and high-temperature fuel cells:
Challenges, and opportunities for sustainable development in the 21st century, Catalysis
Today, 77 (2002) 17-49.
[17] G. Nahar, V. Dupont, Recent advances in hydrogen production via autothermal reforming
process (ATR): A review of patents and research articles, Recent Patents on Chemical
Engineering, 6 (2013) 8-42.
[18] M. Usman, W.M.A. Wan Daud, H.F. Abbas, Dry reforming of methane: Influence of
process parameters - A review, Renewable and Sustainable Energy Reviews, 45 (2015) 710-
744.
[19] United States Department of Energy (DOE),
http://energy.gov/sites/prod/files/2014/03/f11/targets_onboard_hydro_storage_explanation.pdf,
[20] A.F. Dalebrook, W. Gan, M. Grasemann, S. Moret, G. Laurenczy, Hydrogen storage:
Beyond conventional methods, Chemical Communications, 49 (2013) 8735-8751.
[21] A.W.C. Van Den Berg, C.O. Areán, Materials for hydrogen storage: Current research
trends and perspectives, Chemical Communications, (2008) 668-681.
[22] S. Tibdewal, U. Saxena, A.V.P. Gurumoorthy, Hydrogen economy vs. Methanol economy,
International Journal of Chemical Sciences, 12 (2014) 1478-1486.
[23] M. Paleologou, T. Radiotis, L. Kouisni, N. Jemaa, T. Mahmood, T. Browne, D. Singbeil,
New and emerging biorefinery technologies and products for the canadian forest industry, J-
FOR, 1 (2011) 6-14.
[24] E.L. Fornero, P.B. Sanguineti, D.L. Chiavassa, A.L. Bonivardi, M.A. Baltanás,
Performance of ternary Cu-Ga2O3-ZrO2 catalysts in the synthesis of methanol using CO2-rich
gas mixtures, Catalysis Today, 213 (2013) 163-170.
[25] C.A. Trudewind, A. Schreiber, D. Haumann, Photocatalytic methanol and methane
production using captured CO2 from coal-fired power plants. Part i - A Life Cycle Assessment,
Journal of Cleaner Production, 70 (2014) 27-37.
[26] J.M. Tatibouët, Methanol oxidation as a catalytic surface probe, Applied Catalysis A:
General, 148 (1997) 213-252.
[27] S.T. Yong, C.W. Ooi, S.P. Chai, X.S. Wu, Review of methanol reforming-Cu-based
catalysts, surface reaction mechanisms, and reaction schemes, International Journal of
Hydrogen Energy, 38 (2013) 9541-9552.
[28] S. Sá, H. Silva, L. Brandão, J.M. Sousa, A. Mendes, Catalysts for methanol steam
reforming-A review, Applied Catalysis B: Environmental, 99 (2010) 43-57.
Page 57
Bibliografía
49
[29] A. Guerrero-Ruiz, I. Rodriguez-Ramos, J.L.G. Fierro, Dehydrogenation of methanol to
methyl formate over supported copper catalysts, Applied Catalysis, 72 (1991) 119-137.
[30] K. Klier, Preparation of bifunctonal catallysts, Catalysis Today, 15 (1992) 361-382.
[31] L. Alejo, R. Lago, M.A. Peña, J.L.G. Fierro, Partial oxidation of methanol to produce
hydrogen over Cu-Zn-based catalysts, Applied Catalysis A: General, 162 (1997) 281-297.
[32] M. Kusche, F. Enzenberger, S. Bajus, H. Niedermeyer, A. Bösmann, A. Kaftan, M.
Laurin, J. Libuda, P. Wasserscheid, Enhanced activity and selectivity in catalytic methanol
steam reforming by basic alkali metal salt coatings, Angewandte Chemie - International
Edition, 52 (2013) 5028-5032.
[33] H.N. Evin, G. Jacobs, J. Ruiz-Martinez, U.M. Graham, A. Dozier, G. Thomas, B.H.
Davis, Low temperature water-gas shift/methanol steam reforming: Alkali doping to facilitate
the scission of formate and methoxy C-H bonds over Pt/ceria catalyst, Catalysis Letters, 122
(2008) 9-19.
[34] J.M. Pigos, C.J. Brooks, G. Jacobs, B.H. Davis, Low temperature water-gas shift: The
effect of alkali doping on the Csingle bondH bond of formate over Pt/ZrO2 catalysts,
Applied Catalysis A: General, 328 (2007) 14-26.
[35] C.G. Vayenas, S. Bebelis, C. Pliangos, S. Brosda, D. Tsiplakides, Electrochemical
Activation of Catalysis: Promotion, Electrochemical Promotion and Metal-Support
Interactions, Kluwer Academic Publishers/Plenum Press, New York, 2001.
[36] M. Stoukides, C.G. Vayenas, The effect of electrochemical oxygen pumping on the rate
and selectivity of ethylene oxidation on polycrystalline silver, Journal of Catalysis, 70 (1981)
137-146.
[37] C.G. Vayenas, S. Bebelis, S. Ladas, Dependence of catalytic rates on catalyst work
function, Nature, 343 (1990) 625-627.
[38] P. Vernoux, L. Lizarraga, M.N. Tsampas, F.M. Sapountzi, A. De Lucas-Consuegra, J.L.
Valverde, S. Souentie, C.G. Vayenas, D. Tsiplakides, S. Balomenou, E.A. Baranova, Ionically
conducting ceramics as active catalyst supports, Chemical Reviews, 113 (2013) 8192-8260.
[39] C. Vayenas, S. Brosda, Electrochemical promotion: Experiment, rules and mathematical
modeling, Solid State Ionics, 154-155 (2002) 243-250.
[40] C.G. Vayenas, M.M. Jaksic, S. Bebelis, S. Neophytides, Modern Aspects of
Electrochemistry, Plenum, New York, 1996.
[41] I.M. Petrushina, V.A. Bandur, F. Cappeln, N.J. Bjerrum, Electrochemical promotion of
sulfur dioxide catalytic oxidation, Journal of the Electrochemical Society, 147 (2000) 3010-
3010.
Page 58
Descripción del trabajo realizado
50
[42] S. Neophytides, C.G. Vayenas, Non-faradaic electrochemical modification of catalytic
activity. 2. The case of methanol dehydrogenation and decomposition on Ag, Journal of
Catalysis, 118 (1989) 147-163.
[43] A. de Lucas-Consuegra, F. Dorado, C. Jiménez-Borja, J.L. Valverde, Influence of the
reaction conditions on the electrochemical promotion by potassium for the selective catalytic
reduction of N2O by C3H6 on platinum, Applied Catalysis B: Environmental, 78 (2008) 222-
231.
[44] E. Varkaraki, J. Nicole, E. Plattner, C. Comninellis, C.G. Vayenas, Electrochemical
promotion of IrO2 catalyst for the gas phase combustion of ethylene, Journal of Applied
Electrochemistry, 25 (1995) 978-981.
[45] M. Stoukides, C.G. Vayenas, ELECTROCATALYTIC RATE ENHANCEMENT OF
PROPYLENE EPOXIDATION ON POROUS SILVER ELECTRODES USING A ZIRCONIA
OXYGEN PUMP, 131 (1984) 839-845 jesoan.
[46] F.J. Williams, A. Palermo, M.S. Tikhov, R.M. Lambert, First Demonstration of in Situ
Electrochemical Control of a Base Metal Catalyst: Spectroscopic and Kinetic Study of the CO
+ NO Reaction over Na-Promoted Cu, Journal of Physical Chemistry B, 103 (1999) 9960-
9966.
[47] G.E. Pitselis, P.D. Petrolekas, C.G. Vayenas, Electrochemical promotion of ammonia
decomposition over Fe catalyst films interfaced with K+- & H+- conductors, Ionics, 3 (1997)
110-116.
[48] C.G. Vayenas, S. Neophytides, Non-faradaic electrochemical modification of catalytic
activity III. The case of methanol oxidation on Pt, Journal of Catalysis, 127 (1991) 645-664.
[49] N. Li, F. Gaillard, A. Boréave, Electrochemical promotion of Ag catalyst for the low
temperature combustion of toluene, Catalysis Communications, 9 (2008) 1439-1442.
[50] C.A. Cavalca, G.L. Haller, Solid electrolytes as active catalyst supports: Electrochemical
modification of benzene hydrogenation activity on Pt/β″(Na)Al2O3, Journal of Catalysis, 177
(1998) 389-395.
[51] F. Gaillard, N. Li, Electrochemical promotion of toluene combustion on an inexpensive
metallic catalyst, Catalysis Today, 146 (2009) 345-350.
[52] P. Vernoux, F. Gaillard, C. Lopez, E. Siebert, Coupling catalysis to electrochemistry: A
solution to selective reduction of nitrogen oxides in lean-burn engine exhausts?, Journal of
Catalysis, 217 (2003) 203-208.
Page 59
Bibliografía
51
[53] M. Ouzounidou, A. Skodra, C. Kokkofitis, M. Stoukides, Catalytic and electrocatalytic
synthesis of NH3 in a H+ conducting cell by using an industrial Fe catalyst, Solid State Ionics,
178 (2007) 153-159.
[54] L. Ploense, M. Salazar, B. Gurau, E.S. Smotkin, Proton spillover promoted isomerization
of n-butylenes on Pd-black cathodes/nafion 117 [8], Journal of the American Chemical
Society, 119 (1997) 11550-11551.
[55] A. De Lucas-Consuegra, A. Caravaca, P.J. Martínez, J.L. Endrino, F. Dorado, J.L.
Valverde, Development of a new electrochemical catalyst with an electrochemically assisted
regeneration ability for H2 production at low temperatures, Journal of Catalysis, 274 (2010)
251-258.
[56] C. Jiménez-Borja, F. Dorado, A. de Lucas-Consuegra, J.M. García-Vargas, J.L. Valverde,
Complete oxidation of methane on Pd/YSZ and Pd/CeO2/YSZ by electrochemical promotion,
Catalysis Today, 146 (2009) 326-329.
[57] T.I. Politova, V.A. Sobyanin, V.D. Belyaev, Ethylene hydrogenation in electrochemical
cell with solid proton-conducting electrolyte, Reaction Kinetics & Catalysis Letters, 41 (1990)
321-326.
[58] A. de Lucas-Consuegra, A. Princivalle, A. Caravaca, F. Dorado, C. Guizard, J.L.
Valverde, P. Vernoux, Preferential CO oxidation in hydrogen-rich stream over an
electrochemically promoted Pt catalyst, Applied Catalysis B: Environmental, 94 (2010) 281-
287.
[59] D. Tsiplakides, S.G. Neophytides, O. Enea, M. Jaksic, C.G. Vayenas, Nonfaradaic
electrochemical modification of the catalytic activity of Pt-black electrodes deposited on nafion
117 solid polymer electrolytes, Journal of the Electrochemical Society, 144 (1997) 2072-2078.
[60] E.I. Papaioannou, S. Souentie, A. Hammad, C.G. Vayenas, Electrochemical promotion of
the CO2 hydrogenation reaction using thin Rh, Pt and Cu films in a monolithic reactor at
atmospheric pressure, Catalysis Today, 146 (2009) 336-344.
[61] A. De Lucas-Consuegra, A. Caravaca, J. González-Cobos, J.L. Valverde, F. Dorado,
Electrochemical activation of a non noble metal catalyst for the water-gas shift reaction,
Catalysis Communications, 15 (2011) 6-9.
[62] P.J. Gellings, H.J.M. Bouwmeester, The CRC Handbook of Solid State Electrochemistry,
CRC Press1997.
[63] A. de Lucas-Consuegra, New trends of Alkali Promotion in Heterogeneous Catalysis:
Electrochemical Promotion with Alkaline Ionic Conductors, Catalysis Surveys from Asia,
2015, DOI:10.1007/s10563-014-9179-6.
Page 60
Descripción del trabajo realizado
52
[64] C.G. Vayenas, S. Bebelis, M. Despotopoulou, Non-faradaic electrochemical modification
of catalytic activity 4. The use of β″-Al2O3 as the solid electrolyte, Journal of Catalysis, 128
(1991) 415-435.
[65] I.R. Harkness, R.M. Lambert, Electrochemical Promotion of the No + Ethylene Reaction
over Platinum, Journal of Catalysis, 152 (1995) 211-214.
[66] M. Makri, G.G. Vayenas, S. Bebelis, K.H. Besocke, C. Cavalca, Atomic resolution STM
imaging of electrochemically controlled reversible promoter dosing of catalysts, Surface
Science, 369 (1996) 351-359.
[67] F.J. Williams, C.M. Aldao, A. Palermo, R.M. Lambert, A Monte Carlo simulation of the
NO + CO reaction on Na-promoted platinum, Surface Science, 412-413 (1998) 174-183.
[68] C.G. Vayenas, S. Brosda, C. Pliangos, Rules and mathematical modeling of
electrochemical and chemical promotion: 1. Reaction classification and promotional rules,
Journal of Catalysis, 203 (2001) 329-350.
[69] F.J. Williams, A. Palermo, M.S. Tikhov, R.M. Lambert, Mechanism of alkali promotion in
heterogeneous catalysis under realistic conditions: Application of electron spectroscopy and
electrochemical promotion to the reduction of NO by CO and by propene over rhodium,
Surface Science, 482-485 (2001) 177-182.
[70] A.J. Urquhart, F.J. Williams, R.M. Lambert, Electrochemical promotion by potassium of
Rh-catalysed fischer-tropsch synthesis at high pressure, Catalysis Letters, 103 (2005) 137-141.
[71] A. de Lucas-Consuegra, F. Dorado, J.L. Valverde, R. Karoum, P. Vernoux, Low-
temperature propene combustion over Pt/K-βAl2O3 electrochemical catalyst:
Characterization, catalytic activity measurements, and investigation of the NEMCA effect,
Journal of Catalysis, 251 (2007) 474-484.
[72] A. de Lucas-Consuegra, Á. Caravaca, P. Sánchez, F. Dorado, J.L. Valverde, A new
improvement of catalysis by solid-state electrochemistry: An electrochemically assisted NOx
storage/reduction catalyst, Journal of Catalysis, 259 (2008) 54-65.
[73] E. Ruiz, D. Cillero, P.J. Martínez, Á. Morales, G.S. Vicente, G. De Diego, J.M. Sánchez,
Bench scale study of electrochemically promoted catalytic CO2 hydrogenation to renewable
fuels, Catalysis Today, 210 (2013) 55-66.
[74] B. Lin, K. Wei, X. Ma, J. Lin, J. Ni, Study of potassium promoter effect for Ru/AC
catalysts for ammonia synthesis, Catalysis Science and Technology, 3 (2013) 1367-1374.
[75] E. Ruiz, D. Cillero, P.J. Martínez, Á. Morales, G.S. Vicente, G. De Diego, J.M. Sánchez,
Electrochemical synthesis of fuels by CO2 hydrogenation on Cu in a potassium ion conducting
membrane reactor at bench scale, Catalysis Today, 236 (2014) 108-120.
Page 61
Bibliografía
53
[76] M. Marwood, C.G. Vayenas, Electrochemical promotion of a dispersed platinum catalyst,
Journal of Catalysis, 178 (1998) 429-440.
[77] C.G. Yiokari, G.E. Pitselis, D.G. Polydoros, A.D. Katsaounis, C.G. Vayenas, High-
pressure electrochemical promotion of ammonia synthesis over an industrial iron catalyst,
Journal of Physical Chemistry A, 104 (2000) 10600-10602.
[78] S. Balomenou, D. Tsiplakides, A. Katsaounis, S. Thiemann-Handler, B. Cramer, G. Foti,
C. Comninellis, C.G. Vayenas, Novel monolithic electrochemically promoted catalytic reactor
for environmentally important reactions, Applied Catalysis B: Environmental, 52 (2004) 181-
196.
[79] S.P. Balomenou, D. Tsiplakides, C.G. Vayenas, S. Poulston, V. Houel, P. Collier, A.G.
Konstandopoulos, C. Agrafiotis, Electrochemical promotion in a monolith electrochemical
plate reactor applied to simulated and real automotive pollution control, Topics in Catalysis,
44 (2007) 481-486.
[80] S. Souentie, A. Hammad, S. Brosda, G. Foti, C.G. Vayenas, Electrochemical promotion of
NO reduction by C2H4 in 10% O2 using a monolithic electropromoted reactor with Rh/YSZ/Pt
elements, Journal of Applied Electrochemistry, 38 (2008) 1159-1170.
[81] D. Poulidi, M.E. Rivas, B. Zydorczak, Z. Wu, K. Li, I.S. Metcalfe, Electrochemical
promotion of a Pt catalyst supported on La 0.6Sr 0.4Co 0.2Fe 0.8O 3 - δ hollow fibre
membranes, Solid State Ionics, 225 (2012) 382-385.
[82] A. Kambolis, L. Lizarraga, M.N. Tsampas, L. Burel, M. Rieu, J.P. Viricelle, P. Vernoux,
Electrochemical promotion of catalysis with highly dispersed Pt nanoparticles,
Electrochemistry Communications, 19 (2012) 5-8.
[83] V. Jiménez, C. Jiménez-Borja, P. Sánchez, A. Romero, E.I. Papaioannou, D. Theleritis, S.
Souentie, S. Brosda, J.L. Valverde, Electrochemical promotion of the co2 hydrogenation
reaction on composite ni or ru impregnated carbon nanofiber catalyst-electrodes deposited on
YSZ, Applied Catalysis B: Environmental, 107 (2011) 210-220.
[84] A.C. Kaloyannis, C.A. Pliangos, D.T. Tsiplakides, I.V. Yentekakis, S.G. Neophytides, S.
Bebelis, C.G. Vayenas, Electrochemical promotion of catalyst surfaces deposited on ionic and
mixed conductors, Ionics, 1 (1995) 414-420.
[85] E. Ruiz, D. Cillero, P.J. Martínez, Á. Morales, G.S. Vicente, G. De Diego, J.M. Sánchez,
Bench-scale study of electrochemically assisted catalytic CO2 hydrogenation to hydrocarbon
fuels on Pt, Ni and Pd films deposited on YSZ, Journal of CO2 Utilization, 8 (2014) 1-20.
[86] A. de Lucas-Consuegra, A. Princivalle, A. Caravaca, F. Dorado, A. Marouf, C. Guizard,
J.L. Valverde, P. Vernoux, Preparation and characterization of a low particle size Pt/C
Page 62
Descripción del trabajo realizado
54
catalyst electrode for the simultaneous electrochemical promotion of CO and C3H6 oxidation,
Applied Catalysis A: General, 365 (2009) 274-280.
[87] A. Lintanf, E. Djurado, P. Vernoux, Pt/YSZ electrochemical catalysts prepared by
electrostatic spray deposition for selective catalytic reduction of NO by C3H6, Solid State
Ionics, 178 (2008) 1998-2008.
[88] R.M. Lambert, A. Palermo, F.J. Williams, M.S. Tikhov, Electrochemical promotion of
catalytic reactions using alkali ion conductors, Solid State Ionics, 136-137 (2000) 677-685.
[89] V. Roche, R. Revel, P. Vernoux, Electrochemical promotion of YSZ monolith honeycomb
for deep oxidation of methane, Catalysis Communications, 11 (2010) 1076-1080.
[90] F.M. Sapountzi, M.N. Tsampas, C.G. Vayenas, Methanol reformate treatment in a PEM
fuel cell-reactor, Catalysis Today, 127 (2007) 295-303.
[91] G. Karagiannakis, S. Zisekas, M. Stoukides, Hydrogenation of carbon dioxide on copper
in a H+ conducting membrane-reactor, Solid State Ionics, 162-163 (2003) 313-318.
[92] I.V. Yentekakis, Y. Jiang, S. Neophytides, S. Bebelis, C.G. Vayenas, Catalysis,
electrocatalysis and electrochemical promotion of the steam reforming of methane over Ni film
and Ni-YSZ cermet anodes, Ionics, 1 (1995) 491-498.
[93] A. Caravaca, A. De Lucas-Consuegra, C. Molina-Mora, J.L. Valverde, F. Dorado,
Enhanced H2 formation by electrochemical promotion in a single chamber steam electrolysis
cell, Applied Catalysis B: Environmental, 106 (2011) 54-62.
[94] J.K. Hong, I.H. Oh, S.A. Hong, W.Y. Lee, Electrochemical oxidation of methanol over a
silver electrode deposited on yttria-stabilized zirconia electrolyte, Journal of Catalysis, 163
(1996) 95-105.
[95] C.A. Cavalca, G. Larsen, C.G. Vayenas, G.L. Haller, Electrochemical modification of
CH3OH oxidation selectivity and activity on a Pt single-pellet catalytic reactor, Journal of
Physical Chemistry, 97 (1993) 6115-6119.
[96] A. de Lucas-Consuegra, A. Caravaca, M.J. Martín de Vidales, F. Dorado, S. Balomenou,
D. Tsiplakides, P. Vernoux, J.L. Valverde, An electrochemically assisted NOx
storage/reduction catalyst operating under fixed lean burn conditions, Catalysis
Communications, 11 (2009) 247-251.
[97] E. Ruiz, D. Cillero, Á. Morales, G.S. Vicente, G. De Diego, P.J. Martínez, J.M. Sánchez,
Bench scale study of electrochemically promoted CO2 capture on Pt/K-βAl2O3,
Electrochimica Acta, 112 (2013) 967-975.
Page 63
55
ABSTRACT
The present work is part of a wider research program dealing with the
application of electrocatalytic systems in environmental and energy processes. It is
being conducted for the last years in the Laboratory of Catalysis and Materials at the
Department of Chemical Engineering at the University of Castilla-La Mancha
(UCLM).
In particular, this doctoral thesis aims at the study of the phenomenon of
electrochemical promotion of catalysis in hydrogen production processes from
methanol by using alkaline conductors. The present work has been financially
supported by the Spanish Government through the research projects CTQ 2010-
16179/PQ and CTQ 2013-45030-R.
This thesis has been carried out in collaboration with the Institute of Materials
Science of Madrid (CSIC), the Institute Jean Lamour at University of Lorraine
(France) and the Institute of Materials Science of Seville (CSIC-University of Seville).
Page 64
Abstract
56
Hydrogen is a very important feedstock in the chemical industry and a
promising energy carrier with main application in internal combustion engines and
fuel cell technology as an alternative to the massive consumption of fossil fuels. H2
presents a high gravimetric energy density and can be considered as a clean synthetic
fuel depending on the sustainability of the energy and raw material employed for its
production. Hydrogen is currently obtained mainly via methane steam reforming.
However, the use of liquid hydrogen carriers such as methanol is acquiring increasing
interest.
H2 production from methanol is typically carried out through its
decomposition (MD, CH3OH → 2H2 + CO), steam reforming (SRM, CH3OH + H2O
→ 3H2 + CO2) or partial oxidation (POM, CH3OH + 1/2O2 → 2H2 + CO2), by using
catalysts based on Cu or Group VIII metals (Pt, Pd, Ni). A high catalytic activity, a
low CO selectivity and a good durability of the catalyst are the main targets. Other
valuable byproducts such as formaldehyde or methyl formate can also be obtained
from these processes. On the other hand, the low volumetric energy density of gaseous
H2 makes the development of efficient hydrogen storage systems to be of paramount
importance.
The coupling of catalysis and electrochemistry has been widely investigated
for the last years for H2 production through electrolysis processes. In this thesis, the
electrochemistry has been used to activate and tune different catalysts for H2
production from methanol through the Electrochemical Promotion Of Catalysis
(EPOC). This phenomenon is based on the modification of the chemisorption
properties of a metal catalyst by the electrochemical migration of promoter ions from
a solid electrolyte support (via application of an electric current or potential). Hence,
while in classical chemical promotion a specific amount of a promoter is added during
the preparation step of the catalyst, in the case of the electrochemical promotion,
promoter ions are electrochemically pumped between the metal catalyst and the solid
electrolyte in a controlled way during the reaction step. Then, the electrochemical
promotion presents several additional advantages, such as the possibility of optimizing
Page 65
Abstract
57
the promoter coverage on the catalyst surface at different reaction conditions and the
in-situ enhancement of the catalytic activity and selectivity.
In this sense, previous studies have reported the interest of the phenomenon of
electrochemical promotion on the methanol decomposition and partial oxidation
reactions over Pt and Ag catalysts, by using yttria-stabilized zirconia (YSZ) as the
solid electrolyte, which is an O2-
conductor. However, these works were only focused
on formaldehyde production (not H2) and were carried out at relatively high reaction
temperatures. With respect to the application of the EPOC phenomenon to the
trapping and storage of surface compounds, previous works have demonstrated the
interest of using cationic electrochemical catalysts for NOx storage-reduction process
(NSR) and CO2 capture.
On the other hand, although a large number of studies have demonstrated the
electrochemical promotion mechanism and this has been applied in a wide variety of
catalytic reactions of industrial and environmental interest, a further technological
progress is necessary for its practical application. Nowadays, some of the main
challenges of EPOC are the development of compact and efficient reactors, and more
competitive catalysts with a higher dispersion (composed of metal nanoparticles) or
based in non noble metals such as Ni or Cu.
Hence, in view of the mentioned above, this doctoral thesis aims to the study
of the phenomenon of electrochemical promotion of catalysis (EPOC) in H2
production processes from methanol by using alkaline conductors. Furthermore, the
possibility of applying the EPOC in the field of H2 storage was also investigated.
Firstly, the effect of electrochemical promotion was studied in the H2
production via steam reforming and partial oxidation of methanol, being formaldehyde
simultaneously obtained in the latter case. For this purpose, two kinds of Pt catalyst
films were deposited on K-βAl2O3 (K+-conductor material) by different techniques:
impregnation and cathodic arc deposition (CAD). Both electrochemical catalysts were
compared under electrochemical promotion conditions for the methanol partial
oxidation reaction. It was found that in alkali-based solid electrolyte cells, an excess of
Page 66
Abstract
58
electrocatalytic activity of the catalyst-working electrode (the case of the impregnated
Pt film) can be detrimental for the electropromotional effect due to the excessive
formation of promoter-derived surface compounds, which may block catalytic active
sites. On the other hand, the catalytic activity and selectivity of the thin Pt film of low
metal loading prepared by cathodic arc deposition were strongly enhanced by EPOC,
i.e., by the electrochemical transfer of K+ ions to the catalyst under negative potentials.
In this way, the Pt catalyst film prepared by this technique allowed to produce both H2
and H2CO at mild reaction conditions in a single reaction step. This electrochemical
catalyst presented much less catalytic activity in the steam reforming of methanol,
although it was also electrochemically promoted under these reaction conditions. The
K+ promotional effect was attributed to the decrease in the catalyst work function and,
hence, the strengthening of the Pt chemical bond with the electron acceptor (O2 or
H2O) against that with the electron donor (CH3OH). Furthermore, the Pt catalyst film
showed to be stable for long working times and the EPOC effect was found to be fully
reversible in all the experiments, given the good reproducibility observed in the
catalytic activity under every positive polarization (unpromoted or reference state). On
the basis of the obtained results, the catalyst films used in the following studies were
prepared by cathodic arc deposition or by other kind of physical vapor deposition
(PVD) technique, such as sputtering or oblique angle deposition (OAD).
Then, in other study, the phenomenon of electrochemical promotion (EPOC)
was applied on a catalyst film composed of Pt nanoparticles (of around 3 nm)
dispersed in a diamond-like carbon (DLC) matrix which was prepared by the cathodic
arc deposition technique. In first place, a temperature-programmed pretreatment was
carried out in order to achieve a suitable electrical conductivity for the electrochemical
promotion experiments. The decrease in the surface electrical resistance was due to
the transition of the sp3-hybridized carbon form into a more graphitic structure (sp
2-
hybridized) as confirmed by EELS. Moreover, the stability of the Pt nanoparticles was
verified by STEM. The catalytic performance of the Pt-DLC film in the methanol
partial oxidation (POM) and steam reforming (SRM) reactions for H2 production was
promoted by K+ ions electrochemically transferred from a K-βAl2O3 solid electrolyte.
Page 67
Abstract
59
Hence, it was demonstrated that the EPOC phenomenon may be applied to catalysts
based on metal nanoparticles dispersed in an electronic non-ionic conductor support.
Moreover, two different electropromotional effects were found under POM conditions
depending on the applied potential, which were attributed to the formation of different
kinds of promoter phases on this catalyst. The higher catalytic activity of Pt-DLC,
compared to that of the pure Pt film, demonstrated the practical interest of this kind of
dispersed catalyst films with lower metal loading.
Other novel electrochemical catalyst based on Au nanoparticles dispersed in a
Yttria-Stabilized Zirconia (YSZ) matrix was deposited by reactive co-sputtering of
Zirconium-Yttrium and Au targets on a K-βAl2O3 solid electrolyte. The Au-YSZ film
was electrically non-conductive and a silver current collector was used to polarize the
catalyst film that would be used in the electrochemical promotion experiments. The
Au nanoparticles showed to be active in the partial oxidation of methanol with a high
selectivity toward methyl formate production. This configuration allowed to decrease
the amount of metal used in the solid electrolyte cell, and to activate a highly
dispersed Au catalyst via electrochemical promotion (EPOC), by in-situ controlling
and optimizing the supplied amount of K+ ions. A number of experiments confirmed
that the observed electropromotional effect did depend on neither the rate of K+ supply
nor the operation mode (galvanostatic or potentiostatic). It only depended on the
achieved final promoter coverage. The stability of the Au nanoparticles under the
explored conditions was also confirmed under EPOC reaction conditions.
Then, a novel Cu catalyst film was prepared by oblique angle physical vapour
deposition (OAD) on a K-βAl2O3 solid electrolyte. This technique allowed to obtain a
highly porous and electrically conductive Cu catalyst electrode composed of metal
nanocolumns, which was electrochemically promoted in the partial oxidation of
methanol (POM). The production rates of hydrogen, carbon dioxide and methyl
formate were in-situ enhanced in a reversible and reproducible way, by means of the
controlled migration of electropositive potassium ions. Moreover, the enhancement
ratios were comparable to those obtained with the Pt-DLC catalyst. Under the studied
reaction conditions, these promoter ions also formed potassium-derived surface
Page 68
Abstract
60
compounds as demonstrated by post-reaction characterization analysis. Some nitrogen
functional groups and carbonaceous compounds were also detected. The obtained
results demonstrate the interest of the used catalyst-electrode preparation technique for
the electrochemical activation of non-noble metal catalyst films with high gas-exposed
surface area.
Then, three possible applications of the phenomenon of electrochemical
promotion of catalysis (EPOC) were demonstrated with a Ni catalyst for methanol
conversion processes: activation of the catalyst, modification of the catalytic
selectivity and partial oxidation of the catalyst. The Ni catalyst film was prepared by
cathodic arc deposition and electrochemically promoted by K+, upon negative
polarization, in the methanol decomposition (MD) and steam reforming (SRM)
reactions, showing an electrophilic EPOC behavior in both cases. In the presence of
water, the K+ ions promotional effect also attenuated the Ni deactivation by carbon
deposition. On the other hand, under methanol partial oxidation conditions (POM), the
K+ ions caused a sharp decrease in the catalytic selectivity toward H2 and CO while
the production rates of CO2 and H2CO slightly enhanced, due to an increase in the Ni
oxidation state by the alkali-induced O2 activation. All the potassium-derived effects
were fully reversible between the negative and positive polarizations, which showed
different interesting possibilities of the EPOC phenomena in heterogeneous catalysis.
Finally, the possibility of applying the EPOC phenomenon with alkaline
conductors in the fields of H2 production and storage was also explored. In this case, a
porous Ni catalyst film was deposited on K-βAl2O3 by the oblique angle deposition
technique. Under steam reforming conditions and negative polarization, this
electrocatalytic system allowed to produce and store H2 with a very high yield per
amount of metal (up to 19 g H2 x 100 g Ni-1
) due to the promotional effect of K+ ions.
Moreover, it was possible to release the stored, highly pure, H2 in a controlled way
under positive polarization, which represents a new application of the EPOC
phenomenon of great interest. The influence of the catalyst microstructure, the applied
negative current and the reaction atmosphere was studied. A H2 storage mechanism
was proposed on the basis of the obtained results. Although some K+-derived surface
Page 69
Abstract
61
compounds such as bicarbonates were detected by post-reaction characterization of the
Ni catalyst surface, the very high observed H2 storage capacity was mainly attributed
to the chemisorption of H atoms on both Ni active sites and carbonaceous surface
compounds through a spillover process. These carbonaceous species were
simultaneously formed under K+-promoted reaction conditions and showed to be
mainly composed of graphene oxide.
Page 71
63
Chapter 1
ELECTROCHEMICAL PROMOTION OF Pt FOR H2
PRODUCTION FROM METHANOL PARTIAL OXIDATION
AND STEAM REFORMING:
A BETTER PERFORMANCE OF PHYSICAL VAPOR DEPOSITED
CATALYST FILMS
The effect of the electrochemical promotion (EPOC) has been studied for the
first time in the steam reforming and partial oxidation of methanol to produce H2 and
also formaldehyde in the latter case. Two Pt catalyst films deposited on K-βAl2O3 (K+-
conductor material) by different techniques, impregnation and cathodic arc
deposition, have been tested under different applied potentials and reaction
conditions. In alkali-based solid electrolyte systems, an excess of electrocatalytic
activity of the catalyst-working electrode (the case of the impregnated Pt film) can be
detrimental due to the excessive formation of promoter-derived surface compounds.
However, the catalytic activity and selectivity of the Pt film prepared by the cathodic
arc technique have been strongly enhanced by EPOC with K+ ions. This catalyst film
also showed to be stable for long working times and the promotional effect was fully
reversible in all the experiments. Hence, the cathodic arc deposition and other
physical vapor deposition techniques were applied in the following chapters.
ΔV < 0
Pt
K+ K+ K+
e-
e-
K-βAl2O3
Au
Reactants Products
SRM: CH3OH + H2O CO2 + 3H2
POM: CH3OH + 1/2O2 CO2 + 2H2
Δr
ELECTROCHEMICAL
PROMOTION (EPOC)
1.1 mg Pt cm-2
0.23 mg Pt cm-2
120 nm thick
Impregnation
Physical Vapor
Deposition (PVD)
0
0.2
0.4
0.6
0.8
1
1.2
0 50 100 150 200 250 300 350 400 450 500 550
Tiempo / min
r H C
O /
mo
l s-1
x 1
0-7
-2.5
-1.5
-0.5
0.5
1.5
VW
R / V
0
2
4
6
8
10
r H
/
mo
l s-1
x 1
0-7
-2.5
-1.5
-0.5
0.5
1.5
2.5
VW
R / V
Pt impregnado
Pt por CAD
VWR
Pt impregnado
Pt por CAD
VWR
22
7.2 % CH3OH
4.6 % O2
320 ºC
VW
R/ V
VW
R/ V
Impregnated Pt
Pt by PVD
VWR
Impregnated Pt
Pt by PVD
VWR
Time / min
rH
CO
/ m
ol s-1
x 1
0-7
rH
/ m
ol
s-1x
10
-72
2
Page 72
Chapter 1
64
1.1. INTRODUCTION
Hydrogen is attracting increasing interest as a future clean energy carrier with
main application in fuel cell technology. It can be obtained by many different
processes and from a wide variety of raw materials (water, biomass or hydrocarbons),
the steam reforming of methane currently being the most used in industry [1-3].
However, the use of liquid hydrogen carriers at atmospheric pressure and room
temperature, such as methanol, is acquiring relevance due to the facilities arising from
its transport, storage and handling. Other advantages of methanol lie in its high H/C
ratio (4:1) and the absence of C-C bonds which allows its conversion at lower
temperatures (150-350 ºC) than most other fuels [4, 5]. Moreover, CH3OH can be
produced not only from fossil fuels (natural gas, coal, oil shale, tar sands, etc.), but
also from agricultural products and municipal waste, wood and varied biomass such as
landfill gas, sugar beets, driftwood or rice straw. It can even be obtained from
chemical recycling of carbon dioxide [6]. H2 production from methanol is mainly
carried out through the steam reforming (SRM, reaction 1.1) and partial oxidation
(POM, reaction 1.2).
CH3OH + H2O → 3H2 + CO2 (1.1)
CH3OH + 1/2O2 → 2H2 + CO2 (1.2)
The first reaction is endothermic and produces a higher H2/C ratio, while the
second one is exothermic and typically leads to higher reaction rates at lower
temperatures, also requiring smaller reactors [7]. This latter reaction can even be run
in an autothermal mode where the heat necessary for the reaction comes from the
reaction itself. The catalysts typically used in both processes, steam reforming and
partial oxidation of methanol, are similar as those employed in the methanol synthesis
(reverse reaction 1.1) and water-gas shift (WGS) reactions and have been reviewed by
several authors [4, 5, 8]. They can be divided into two main categories: copper-based
and group 8-10 metal-based catalysts. In particular, Pt-based catalysts have shown
very good activity for hydrogen production by both SRM [9-12] and POM [13-17].
Page 73
Electrochemical promotion of Pt for H2 production from methanol partial oxidation and steam
reforming
65
Moreover, regarding the partial oxidation of methanol, it is also possible to obtain
formaldehyde (H2CO), through the following reaction:
CH3OH + 1/2O2 → H2CO + H2O (1.3)
H2CO is also of great industrial interest, as a precursor to urea formaldehyde
resin, melamine resin, phenol formaldehyde resin and 1,4-butanediol, among others
[18]. However, Pt catalysts may also lead to high deep oxidation rates, which would
decrease the selectivity to H2 and formaldehyde production. Then, it is important to
enhance the partial oxidation mechanism vs. the deep oxidation one for the production
of both kinds of products. Then, the performance of metal catalysts employed in the
SRM and POM reactions has shown to be promoted by different dopants such as Al
[19], Zn [20] or alkali metals [21-23].
In this sense, the coupling of catalysis and electrochemistry has also emerged
in the last 30 years through the Electrochemical Promotion Of Catalysis (EPOC). This
phenomenon, also known as Non-faradaic Electrochemical Modification of Catalytic
Activity (NEMCA effect), opens a new way to promote the catalytic
activity/selectivity of a metal catalyst under working reaction conditions [24, 25]. It is
based on the modification of the catalytic properties of a metal catalyst by the
electrochemical pumping of promoter ions from an electro-active catalyst support,
which is a solid electrolyte material [26]. In the case of an alkaline solid electrolyte (as
in the present thesis, K-βAl2O3), the application of a cathodic polarization (negative
current) between a catalyst-working electrode, which is deposited on one side of the
electrolyte, and an inert counter electrode located at the opposite side (Figure 1.1) lead
to the migration of promoter (K+) ions to the catalyst film (back-spillover). Once
located over the catalyst surface, as in chemical (classical) promotion, these ionic
species modify the chemisorption properties of the catalyst and hence the reaction rate
[27]. This phenomenon has been demonstrated by means of several characterization
techniques and has been applied in more than 80 catalytic systems with several
important technological possibilities [28-31].
Page 74
Chapter 1
66
Figure 1.1. Scheme of the electrochemical cell used in the electrochemical promotion studies
with an alkaline ionic conductor solid electrolyte.
For instance, previous works have demonstrated the interest of EPOC in
enhancing the selectivity of several catalytic processes such as the selective catalytic
reduction of NOx [32, 33], CO2 hydrogenation [34], Fischer–Tropsch synthesis [35] or
CO preferential oxidation [36]. On the other hand, other studies have also reported the
application of the EPOC on the methanol decomposition and dehydrogenation on Ag
[37] and the methanol partial oxidation on Pt [38, 39] and Ag [40] by using yttria-
stabilized zirconia (YSZ) as the solid electrolyte, which is a O2-
conductor. These four
previous works were only focused on formaldehyde production (not H2 production)
and were carried out at relatively high reaction temperatures (up to 900 K). Here, a
main difference with respect to the present research lies, where all the electrochemical
promotion studies have been performed by means of akali conductor materials.
In the present work, it is proposed, for the first time, the electrochemical
promotion of the production of H2 via partial oxidation and steam reforming of
methanol, and also the simultaneous production of formaldehyde in the former case.
The effect of the catalyst film preparation technique, the reaction conditions and the
durability of the system for long working times have been investigated on a Pt/K-
βAl2O3 electrochemical catalyst. Under POM conditions, the novel proposed EPOC
Catalyst – Working
electrode
Counter
electrode
(Au)
Reference
electrode
(Au)
I Solid electrolite
(K-βAl2O3)
VWR
ΔrReactants Products
K+ K+ K+ Back-spilloverCathodic
polarization
I < 0
ELECTROCHEMICAL PROMOTION (EPOC)
Page 75
Electrochemical promotion of Pt for H2 production from methanol partial oxidation and steam
reforming
67
configuration may have significant importance since it would allow to produce in a
single reaction step both kinds of products: H2 and H2CO, which could be easily
separated for their further use in different applications.
1.2. EXPERIMENTAL
1.2.1. Preparation of the electrochemical catalysts
The electrochemical catalyst consisted of a continuous, thin Pt film (geometric
area of 2.01 cm2) deposited on a side of a 19-mm-diameter, 1-mm-thick K-βAl2O3
(Ionotec) disc, which was used as a cationic solid electrolyte (i.e. as source of K+
promoter ions). The electrochemical catalyst configuration used in this chapter and in
the rest of the thesis can be observed in Figure 1.2.
Figure 1.2. Scheme of the Pt/K-βAl2O3/Au electrochemical catalyst.
In first place, Au counter (CE) and reference (RE) electrodes were deposited
on the other side of the electrolyte. These electrodes were deposited by applying thin
coatings of gold paste (Gwent Electronic Materials C1991025D2), followed by
calcination at 800 ºC for 2 h (heating ramp of 5 ºC min-1
). Au was chosen because of
its inertness for the methanol partial oxidation reaction (checked via blank
experiments), which makes it an adequate pseudo-reference electrode when using the
Catalyst – Working electrode (Pt)
Solid electrolyte (K-βAl2O3)
Counter electrode (Au)
Reference electrode (Au)
Page 76
Chapter 1
68
single pellet cell design. Then, the active Pt catalyst film (WE), which also behaved as
a working electrode, was deposited on the other side of the electrolyte by two different
preparation techniques:
- In the first case, the Pt film was prepared by an impregnation method
described in detail elsewhere [41], consisting of successive steps of deposition and
thermal decomposition (450 ºC for 1 h, heating ramp of 5 ºC min-1
) of a H2PtCl6
precursor solution over a mixture of 1/1 H2O/2-propanol with a metal concentration of
0.1 M. The final Pt loading was 1.09 mg Pt cm-2
.
- In the second one, the Pt film was prepared by the pulse cathodic arc
technique (denoted as CAD; Cathodic Arc Deposition) [42]. The individual pulses
were 1 ms long and had a current of approximately 300 A. A total of 2800 pulses were
used for the deposition of the Pt layer. The substrate holder was at ground during the
deposition and was rotated at 2 r.p.m. This electrode preparation technique allowed
the preparation of a thin, highly dense, Pt film with 122 nm of thickness with high
adhesion to the substrate. The final Pt loading was 0.23 mg Pt cm-2
in this case.
This catalyst preparation method was carried out in collaboration with Dr.
José Luis Endrino from the Institute of Materials Science of Madrid (CSIC).
The resulting Pt/K-βAl2O3/Au electrochemical catalysts were placed into a
single chamber solid electrolyte cell reactor. A scheme of this reactor is shown in
Figure 1.3. It was made of a quartz tube with appropriate feed-through, where all the
electrodes were exposed to the same gas atmosphere.
The three electrodes (working, counter and reference) were connected to an
Autolab PGSTAT320-N potentiostat-galvanostat (Metrohm Autolab) by using gold
wires. Then, in the electrochemical promotion experiments, a catalyst potential (VWR)
was applied according to the procedure generally used in conventional three-
electrodes electrochemical cells [32, 33, 36].
Page 77
Electrochemical promotion of Pt for H2 production from methanol partial oxidation and steam
reforming
69
Figure 1.3. Scheme of the single chamber solid electrolyte cell reactor.
Prior to the catalytic activity measurements, both catalyst films were subjected
to a 25 % H2 stream (N2 balance) while heating to 450 ºC (ramp of 5 ºC min-1
), to
ensure they were in a fully reduced state, and were kept at this temperature under
POM conditions (CH3OH/O2 = 7.2 %/4.6 %) overnight for their stabilization.
1.2.2. Characterization measurements
Cyclic voltammetry (CV) measurements were performed under reaction
conditions with the potentiostat–galvanostat in order to investigate the formation and
decomposition of promoter species. The samples were prior to the measurement kept
at +2 V for 30 min in order to define a reference state (Pt films clean of promoter
ions). Then, the potential was varied at a scan rate of 80 mV s-1
from +2 to -2 V.
Reference electrode (R)
Counter electrode (C)
Working electrode (W)
Thermocouple
InletOutlet
Cooling coil
Metal lid
Quartz tubes
Perforated alumina
tube
Gold wires
Reference electrode (R)
Counter electrode (C)
Working electrode (W)
Thermocouple
InletOutlet
Cooling coil
Metal lid
Quartz tubes
Perforated alumina
tube
Gold wires
Pt, W
Page 78
Chapter 1
70
The two different Pt electrode films were characterized, after the EPOC
experiments, by X-ray diffraction (XRD) with a Philips PW 1710 instrument using Ni-
filtered Cu Ka radiation. Before the XRD analysis, the electrochemical catalysts were
kept under POM reaction conditions (CH3OH/O2 = 7.2 %/4.6 %) at 320 ºC overnight
under a potential VWR = -2 V in order to favour the formation of the promoter species.
The diffractograms were then compared with the JCPDS-ICDD references for
identification purposes of the promoter phases. The catalyst films were also
characterized after the electrochemical promotion experiments via scanning electron
microscopy (SEM) and EDX analysis using a FEI Nova NANOSEM 230 instrument.
1.2.3. Catalytic activity measurements
The catalytic activity measurements were carried out in the experimental setup
shown in Figure 1.4.
Figure 1.4. Scheme of the experimental set-up employed in the electrochemical promotion
experiments.
N2 / Ar
H2
Air / O2
N2 / Ar
FlowmeterTermometer
Mass
flowmeters
Potentiostat/
Galvanostat
Gas chromatograph
Reactor
Furnace
FIC
FIC
FIC
FIC
TIC
TIC
To vent
H2O
saturator
CH3OH
saturator
TIC
Page 79
Electrochemical promotion of Pt for H2 production from methanol partial oxidation and steam
reforming
71
The reaction gases (Praxair, Inc.) were certified standards (99.999 % purity) of
air (source of the O2 used in the methanol partial oxidation experiments), H2
(reductant agent) and Ar (carrier gas). The gas flow rates were controlled by a set of
mass flowmeters (Bronkhorst EL-FLOW). Methanol (Panreac, 99.8 % purity) and
water (distilled and deionized, used in the methanol steam reforming experiments)
were fed by sparging Ar through thermostated saturators. All lines placed downstream
from the saturators were heated above 100 ºC to prevent condensation.
The electrochemical promotion experiments were performed at atmospheric
pressure with an overall gas flow rate of 6 NL h-1
, a temperature ranging from 250 to
320 ºC with a feed composition of CH3OH/O2 = 7.2 %/0 - 8.9 % (N2 balance) for the
POM experiments, and a temperature ranging from 320 to 450 ºC with a feed
composition of CH3OH/H2O = 4 %/4.8 % (N2 balance) for the SRM experiments.
Reactant and product gases were on-line analyzed by using a double channel gas
chromatograph (Bruker 450-GC) equipped with Hayesep and Q-Molsieve 13X
consecutive columns and a CP-Wax 52 CB column, along with thermal conductivity
and flame ionization detectors, respectively. The detected products were: H2, CO2,
CO, H2O and formaldehyde (H2CO), the latter two only under POM reaction
conditions. The error in the carbon atom balance did not exceed 5 % in any
experiment, which indicated no consistent loss of material, no significant formation of
other oxygenated species and/or coking of the catalyst-electrodes. Unless otherwise
stated, the error remained below this value in the following chapters.
1.3. RESULTS AND DISCUSSION
1.3.1. Partial oxidation of methanol
i) Influence of the preparation technique.
In order to study the influence of the Pt film preparation technique, a
potentiostatic experiment was carried out with both catalysts: the impregnated Pt film
and the Pt prepared by Cathodic Arc Deposition (CAD), under methanol partial
oxidation (POM) conditions. Figure 1.5 shows the variation of the different product
Page 80
Chapter 1
72
reaction rates: H2, H2CO, CO2 and CO, as well as the overall CH3OH conversion vs.
time under different applied potentials (VWR), each for 60 min, under the following
POM reaction conditions: CH3OH/O2 = 7.2 %/4.6 % (N2 balance), 320 ºC.
Initially, the catalyst was in Open Circuit (OC) conditions, i.e., no electric
current or potential was being applied. Then, at the beginning and at the end of the
experiment, a catalyst potential VWR = +2 V was applied in order to remove all the K+
ions that could be located on the Pt surface. In this way, a clean (unpromoted) Pt
catalyst film was defined, and hence a reference state [36, 41]. Then, a significant
difference in the activity of the two electrochemical catalysts with varying the applied
potential can be observed in this figure. For the impregnated Pt film, a decrease in the
catalyst potential from +2 V led to a sharp reduction in the catalytic activity of the
system, while in the case of CAD Pt film, a decrease in the catalyst potential from +2
V led to a strong increase in the activity of the system (increasing the production rate
of all the products as well as the CH3OH conversion). Then, according to previous
studies of electrochemical promotion [27], one can apparently classify the case of the
impregnated Pt film as “electrophobic EPOC behaviour” and the case of the CAD Pt
film as “electrophilic EPOC behaviour”.
A decrease in the catalyst potential involved a decrease in the catalyst work
function due to the migration of positively charged potassium species, which would
weaken the Pt chemical bond with electron-donor adsorbates and would strengthen
those with electron acceptor ones. According to the previous work reported by
Vayenas and Neophytides on methanol partial oxidation [38], CH3OH can be
identified as the donor molecule and O2 as the acceptor one. In principle one can
explain the different observed EPOC behavior in Figure 1.5 according to the different
reaction orders of the two reaction molecules: acceptor (O2) and donor (CH3OH) for
the two catalyst films. Hence, the preparation method used for the two Pt films: i.e.,
impregnation of a Pt precursor solution or that based on the pulse cathodic arc
technique, seems to have a key role.
Page 81
Electrochemical promotion of Pt for H2 production from methanol partial oxidation and steam
reforming
73
Figure 1.5. H2 (a), H2CO (b), CO2 (c) and CO (d) production rates and CH3OH conversion (e)
vs. time under different applied potentials. Partial oxidation conditions: CH3OH/O2 = 7.2 %/4.6
%, 320 ºC.
0
4
8
12
16
r CO
/
mo
l s-1
x 1
0-7
-2.5
-1.5
-0.5
0.5
1.5
VW
R / V
0
10
20
30
40
0 100 200 300 400 500
Time / min
CH
3O
H C
on
vers
ion
/
-2.5
-1.5
-0.5
0.5
1.5
VW
R / V
0.0
0.2
0.4
0.6
0.8
1.0
1.2
r H
CO
/ m
ol s
-1 x
10
-7
-2.5
-1.5
-0.5
0.5
1.5
VW
R / V
0.0
0.5
1.0
1.5
2.0
2.5
3.0
r CO
/ m
ol s
-1 x
10
-7
-2.5
-1.5
-0.5
0.5
1.5
VW
R / V
0
2
4
6
8
10
r H
/ m
ol s
-1 x
10
-7
-2.5
-1.5
-0.5
0.5
1.5
2.5
VW
R / V
2
2
2
%
a)
b)
c)
d)
e)
Impregnated Pt
Pt by CAD
VWR
Impregnated Pt
Pt by CAD
VWR
Impregnated Pt
Pt by CAD
VWR
Impregnated Pt
Pt by CAD
VWR
Impregnated Pt
Pt by CAD
VWR
Page 82
Chapter 1
74
Previous studies have demonstrated [43] that the method of catalyst
preparation, affects the catalytic behavior due to a difference in the morphology
(surface roughness) and dispersion of the Pt particles. In addition, a work of Mutoro et
al. [44] also demonstrated important differences in the magnitude of the EPOC effect
in dense and porous Pt films prepared by two different techniques: application of Pt
metal pastes and pulse cathodic arc deposition. The higher EPOC effect observed with
the porous Pt film deposited on YSZ vs. the dense pulse layer deposited Pt film was
attributed to the higher tpb (three phase boundaries) length of the former, which
facilitated the formation of oxygen spillover promoting species. Then, very likely the
different morphology and tpb length of the two Pt catalyst films may be responsible of
the observed different EPOC behavior in Figure 1.5. In order to prove this hypothesis,
additional measurements (characterization techniques) were carried out.
Figure 1.6 shows the current variation vs. the applied potential (VWR) of the
two samples: impregnated Pt film (a) and Pt by CAD (b) during the cyclic
voltammetry (CV) between +2 and -2 V carried out under the same reaction
conditions of Figure 1.5 (CH3OH/O2 = 7.2 %/4.6 %, 320 ºC). As reported in previous
studies with alkali electro-promoted systems [45-47], the cathodic scan of the cyclic
voltammetry (negative potentials) could be linked to the migration of ions from the
solid electrolyte to the catalyst-working electrode surface with the consequent
formation of promoter-derived surface compounds after interaction with the
chemisorbed species. On the other hand, the anodic scan of the cyclic voltammetry
(positive potentials) could be linked to the decomposition of the previously formed
species and the migration of the ions back to the solid electrolyte. Thus, taking into
account that the gold counter electrode is constituted by big particles of negligible
activity, all the electrochemical processes displayed on the CV could be mainly
attributed to the formation and decomposition of surface compounds on the Pt
catalyst-working electrode [47].
Page 83
Electrochemical promotion of Pt for H2 production from methanol partial oxidation and steam
reforming
75
Figure 1.6. Current variation vs. the applied potential (VWR) of the two samples: impregnated
Pt film (a) and Pt films prepared by cathodic arc deposition (b) during the cyclic voltammetry
between +2 and -2 V (CH3OH/O2 = 7.2 %/4.6 %, 320 ºC, scan rate = 80 mV.s-1
).
In this figure, one can also observe a much higher electrocatalytic activity
(much higher cathodic and anodic peaks) for the case of the impregnated Pt film vs.
the Pt film prepared by CAD, probably due, as commented above, to a higher tpb
length of the former (porous film calcined at high temperature 450 ºC). For the case of
the impregnated Pt film, it can be observed a strong cathodic peak below +1.3 V
(Figure 1.6a) that seemed to indicate the formation of an important amount of
potassium surface compounds, which in turn could block the Pt active sites and
explain the poisoning effect of the impregnated Pt film shown in Figure 1.5 at VWR =
-8
-6
-4
-2
0
2
4
6
8
Cu
rre
nt
/ m
A
-0.06
-0.04
-0.02
0.00
0.02
0.04
-2.5 -2 -1.5 -1 -0.5 0 0.5 1 1.5 2 2.5
Cu
rre
nt
/ m
A
VWR / V
a)
b)
Page 84
Chapter 1
76
+1 V. For the case of the Pt sample by CAD (Figure 1.6b), a much lower cathodic
peak was observed at VWR = -1 V, demonstrating that such important formation of
potassium derived compounds did not occur on this sample in contrast with that
observed with the impregnated sample. The observed anodic peaks of both samples
were in good agreement with the cathodic ones and a much higher size was again
obtained for the case of the impregnated Pt sample. These observations were in good
agreement with the XRD analysis (Figure 1.7) of the Pt films after the application of
VWR = -2 V overnight under the reaction conditions in Figure 1.5 (CH3OH/O2 = 7.2
%/4.6 %, 320 ºC).
Figure 1.7. XRD spectra after EPOC experiments (CH3OH/O2 = 7.2 %/4.6 %, 320 ºC,
overnight at -2 V) of Pt films prepared by impregnation (a) and cathodic arc deposition (b)
techniques.
0
400
800
1200
1600
5 10 15 20 25 30 35 40 45 50 55 60 65 70 75 80 85 90
2Ѳ / º
Inte
nsi
ty /
co
un
ts
0
500
1000
1500
2000
2500
3000
3500
4000
Inte
nsi
ty /
co
un
ts
a)
b)
0
50
100
150
200
10 12 14 16 18 20 22 24 26 28
2Ѳ / º
Inte
nsit
y /
co
un
ts
(111)
(200)
(220)
(311)
(222)
Pt
K-βAl2O3
2K2CO3·3H2O
C2HKO4
Page 85
Electrochemical promotion of Pt for H2 production from methanol partial oxidation and steam
reforming
77
On both samples, peaks at 2θ = 39.6º, 47.4º, 67.1º, 81.2º and 83.6º
corresponded to the reflections (111), (200), (220), (311) and (222), respectively, and
were consistent with a Pt face-centered cubic (FCC) structure (JCPDS, 04-0802). The
βAl2O3 phase corresponding to the solid electrolyte can also be clearly identified
(JCPDS, 02-0921). It can be observed that the ratio of the Pt peaks vs. solid electrolyte
peaks was much higher in the impregnated sample, which is indicative of a much
higher thickness of the impregnated Pt vs. the CAD film. However, the most
interesting part of the figure comes from the occurrence of two new diffraction peaks
in the impregnated sample (inset of Figure 1.7a) that were assigned to potassium
carbonate hydrate oxide: 2K2CO3·3H2O (JCPDS, 01-1014) and potassium hydrogen
carbonate oxide: C2HKO4 (JCPDS, 11-0286). These results would support the
formation of an excess of promoter phases under the negative potential application in
the impregnated Pt film due to its higher electrocatalytic activity confirmed by the
cyclic voltammetry in Figure 1.6, which resulted in the apparent electrophobic EPOC
behaviour shown in Figure 1.5.
Figure 1.8 shows the SEM micrographs along with its corresponding EDX
analysis (in the marked regions) of the impregnated Pt film at various levels of
magnification (after the application of VWR = -2 V overnight under the reaction
conditions considered in Figure 1.5). Two different regions are observed in all the
micrographs. According to the EDX analysis, the lighter region corresponds to Pt
while the darker region corresponds to a potassium-derived carbonaceous surface
compound (combination of carbon, potassium and oxygen) in good agreement with
the XRD analysis (Figure 1.7a). In fact, the elemental analysis shown in Figure 1.8c is
in quite accordance with the two compounds detected by the XRD analysis:
2K2CO3·3H2O and C2HKO4. On the other hand, the SEM analysis of the Pt film by
CAD after the EPOC experiments (not shown) just showed the lighter region
corresponding to Pt with a more dense structure.
Page 86
Chapter 1
78
Figure 1.8. Top surface SEM micrographs of the Pt film prepared by impregnation after EPOC
experiments (CH3OH/O2 = 7.2 %/4.6 %, 320 ºC, overnight at -2 V) at 50 µm (a), 20 µm (b)
and 10 µm (c) along with the corresponding EDX analysis of the marked region.
0
10
20
30
40
50
At
(%)
C O Pt K
0
10
20
30
40
50
At
(%)
C O Pt K
0
10
20
30
40
50
60
At
(%)
C O Pt K
Page 87
Electrochemical promotion of Pt for H2 production from methanol partial oxidation and steam
reforming
79
Hence, the impregnated Pt film showed a important ability to form and store
potassium surface compounds, which clearly blocked an important area of it. Very
likely, it led to a decrease in the Pt number of active sites under EPOC conditions (K+
pumping conditions in Figure 1.5 at VWR < +2V), which explaines the observed
electrophobic EPOC behaviour of the impregnated sample in Figure 1.5. Then,
according to these observations, it is interesting to note that the EPOC phenomena
with alkali ionic conductors does not only depend on the kinetics order of both
acceptor and donor molecules but also on the electrocatalytic activity of the catalyst
film to form and store surface compounds between the promoter ions and the
chemisorbed species. Thus, with cationic solid electrolyte systems such as the Pt/K-
βAl2O3 used here, an excess of electrocatalytic activity of the catalyst-electrode film
could be detrimental for an electropromotional effect, in contrast to the O2-
conductors
[27, 44].
ii) Electrochemical promotion mechanism and parameters.
In order to quantify the promotional effect of both catalyst films, Figure 1.9
shows the values of the electrochemical promotion parameters [27] calculated for H2
and H2CO production: ρ (reaction rate enhancement ratio) and PIK+ (promotion index)
vs. the potassium coverage (θK+) achieved during the potentiostatic transitions of
Figure 1.5 (correlated with VWR) and calculated from the integration of the current vs.
time curves via the Faraday’s law (equation 1.4):
θ =
(1.4)
where I is the current, t is the time, n is the potassium ion charge, i.e., +1, F is the
Faraday constant and NG is the number of Pt active sites. In this case, NG was
estimated from the total number of Pt sites in the sample and the dispersion values
obtained from the Pt particle size estimated by the Scherrer equation [48] from the
XRD measurements. The promotional parameters where calculated as follows:
ρ =
(1.5)
Page 88
Chapter 1
80
θ (1.6)
where r and r0 are the promoted (VWR < +2 V) and unpromoted (VWR = +2 V) catalytic
reaction rates, respectively, and ∆r = r – r0 is the K+-induced change in the catalytic
reaction rate.
Figure 1.9. Variation of the promotional parameters: rate enhancement ratio (ρ) and promotion
index (PIK+) for H2 (a) and H2CO (b) vs. potassium coverage on the catalyst film (θK+) and
catalyst potential (VWR). Partial oxidation conditions: CH3OH/O2 = 7.2 %/4.6 %, 320 ºC.
0
1
2
3
4
5
6
7
ρH
-20
-10
0
10
20
30
40
PIK
+,H
0
1
2
3
4
0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1
θK+ / mol K+ x mol active Pt-1
ρH
C
O
-20
-10
0
10
20
PIK
+,H
CO
a)
b)
Impregnated Pt
Pt by CAD
Impregnated Pt
Pt by CAD
2
2
2
2
2 1 0 -1 -2 Pt by CAD
2 1 0 -1 -2 Impregnated Pt
VWR / V
Page 89
Electrochemical promotion of Pt for H2 production from methanol partial oxidation and steam
reforming
81
Firstly, it can be observed that much lower potassium coverage was obtained
in the Pt film prepared by the CAD technique as compared to that of the impregnated
Pt film. In the latter case, a high potassium coverage (θK+ = 0.4) was almost obtained
upon decreasing the catalyst potential (VWR) just to +1 V. This was not the case for the
CAD-Pt film, where much lower potassium coverage was obtained under all the
different applied potentials, i.e., the highest potassium coverage of 0.3 was attained in
the last polarization of -2 V. These results, which are in good agreement with the
previous characterization measurements, support the poisoning behaviour of the
impregnated Pt film observed in Figure 1.5 when decreasing the catalyst potential to
+1 V and lower potentials. It has already been reported in several alkali
electropromoted [41, 49] and alkali promoter catalytic systems [50] that an excess of
promoter may lead to a poisoning catalytic behaviour due to the blocking of active
sites.
Hence, these results seem to indicate that such poisoning regime is easy to
achieve for the case of the impregnated film, i.e., a high potassium coverage around
θK+ = 0.4 was already obtained in the first polarization of +1V in Figure 1.5. As can be
observed in this figure, regarding the Pt impregnated film, a negative effect of
polarization was obtained for the case of H2 production (ρ < 1 and PIK+ < 0) while a
slight increase in the reaction rate was observed for the case of H2CO (up to ρ = 2.7
and PIK+ = 1.8) with a decrease of the potential from +2 V. However, in this latter case
and according to the results of Figure 1.5, a negligible production rate of H2CO was
observed due to the previously mentioned suppression of methanol reaction rate (the
conversion decreased below 5 %).
Regarding the CAD-Pt film, the effect of electrochemical pumping of
potassium ions led to a strong increase in both H2 and H2CO production rates (ρ > 1
and PIK+ > 0). In this case, the rate enhancement ratio (solid line) for both products
reflects an electrophilic EPOC behaviour. The promotion index (dashed line) had an
optimum value at a potassium coverage θK+ = 0.1. An increase in the potassium
coverage above this value (VWR < -0.5 V in Figure 1.5) did not further improve the
Page 90
Chapter 1
82
catalytic activity of the system. This fact could be explained considering that the
promotional effect of the K+ ions began to be balanced with the negative effect of
blocking of Pt active sites at higher promoter coverage (θK+ > 0.1). Under optimal
potential conditions, the H2 production rate was increased more than 6 times leading to
a promotion index higher than 34, although for the case of H2CO that effect was less
pronounced (ρ~4 and PIK+~25). Hence, it is clear that the presence of K+ ions on the
CAD-Pt film led to a strong promotional effect in the partial oxidation mechanism,
enhancing in a very pronounced way the production rates of both H2 and H2CO. One
could explain such strong promotional effect attending to the mechanism of methanol
partial oxidation reaction on Pt catalysts. Previous spectroscopic studies [51]
demonstrated that methanol molecule is adsorbed on Pt as a methoxy specie by
dissociating H atom from the OH group according to reaction (8):
CH3OH → CH3O(a) + H(a) (1.7)
Then, the methoxy intermediate is converted to adsorbed formaldehyde by the
abstraction of a hydrogen atom by surface oxygen:
CH3O(a) + O(a) → H2CO(a) + OH(a) (1.8)
According to this mechanism, the observed promotional effect upon negative
polarization (in the presence of an electropositive promoter) can be attributed to an
increase in metal-oxygen (electron acceptor molecule) bonds strength, which would
enhance the formaldehyde production rate through the reaction step 1.8. Thus, more
active sites would be available for methoxy species chemisorption also increasing the
H2 production. Very likely, part of the produced H2CO can be further decomposed and
oxidized with stronger chemisorbed O(a) species leading to the production of CO and
CO2, respectively. It would also explain the observed lower promotional effect (lower
promotion index and enhancement ratio) of H2CO vs. H2 in Figure 1.9.
In short, all these results clearly demonstrate the superior performance of the
CAD-Pt film vs. the impregnated Pt film for the simultaneous production of H2 and
H2CO at the explored reaction conditions and potential range (VWR = +2, -2 V). Hence,
Page 91
Electrochemical promotion of Pt for H2 production from methanol partial oxidation and steam
reforming
83
the Pt film by CAD was selected for further investigation in the reaction conditions of
the methanol partial oxidation reaction.
iii) Influence of the reaction conditions.
Figure 1.10 shows the steady state variation (after 1 h of polarization at each
potential) vs. the applied potential (VWR) of different reaction parameters: H2, H2CO
and CO2 selectivity as well CH3OH conversion at different temperatures (250-320 ºC)
for the following composition CH3OH/O2 = 7.2 %/4.6 %. These parameters were
calculated through the following equations:
(1.9)
(1.10)
(1.11)
(1.12)
where Fi,in and Fi,out are the molar flow rate of the i species at the inlet and at the outlet
of the reactor, respectively.
In good agreement with the previously obtained results, it can be observed for
the CAD-Pt film that a decrease in the catalyst potential and, hence, an increase in the
potassium coverage, increased the CH3OH partial oxidation activity favouring the H2
and H2CO selectivity (vs. CO2 selectivity) as well as the CH3OH conversion at all
explored reaction temperatures.
Page 92
Chapter 1
84
Figure 1.10. H2 (a), H2CO (b), CO2 (c) selectivities and CH3OH conversion (d) vs. applied
catalyst potential (VWR) at different reaction temperatures with the Pt film prepared by cathodic
arc deposition. Partial oxidation conditions: CH3OH/O2 = 7.2 %/4.6 %.
20
25
30
35
40
45
-2.5 -2 -1.5 -1 -0.5 0 0.5 1 1.5 2 2.5
VWR / V
CH
3O
H C
on
vers
ion
/ %
250ºC280ºC320ºC
0
3
6
9
12
15
18
21
H2 S
ele
ctiv
ity
/ %
250ºC280ºC320ºC
75
80
85
90
95
CO
2 S
ele
ctiv
ity
/ %
250ºC280ºC320ºC
0
2
4
6
8
10
H2C
O S
ele
ctiv
ity
/ %
250ºC280ºC320ºC
a)
b)
c)
d)
Page 93
Electrochemical promotion of Pt for H2 production from methanol partial oxidation and steam
reforming
85
On the other hand, an increase in the temperature enhanced the methanol
partial oxidation activity (toward H2 production, Figure 1.10a) under both:
unpromoted (VWR = +2V) and electro-promoted (VWR < +2V) conditions. A similar
increase in the POM activity with the reaction temperature has been reported in
previous works where Pt supported catalysts were used [13, 17]. However, in the
explored temperature range the CO2 selectivity (Figure 1.10c) was nearly 100 %
under unpromoted conditions (VWR = +2 V) and it was not greatly influenced by
temperature, in agreement with a study of McCabe and McCready [52]. Under
unpromoted conditions (+2 V), the methanol-derived carbon products (CO and
H2CO) were easily oxidized to CO2. Nevertheless, under electro-promoted conditions
the CO2 selectivity decreased and the production of less oxidized carbon products,
such as H2CO increased due to the previously mentioned enhancement in the partial
oxidation mechanism under the effect of K+.
It can also be observed that the promotional effect on the H2 production rate
(H2 selectivity) seemed to increase with the reaction temperature, which can be
attributed to an increase in the solid electrolyte ionic conductivity and, therefore, in
the amount of electrochemically transferred potassium ions at fixed potential. At
higher temperatures, the electrochemical activation occurred at higher potentials
(closer to +2 V). An increase in the promotional effect at higher temperatures was
also observed in previous EPOC studies with cationic solid electrolytes [30, 53].
However, in those studies the promotional effect usually increased up to an optimum
temperature was reached, which it was not the case in this study. On the other hand,
for the case of formaldehyde, an increase in the reaction temperature led to a decrease
in the H2CO selectivity (Figure 1.10b) probably due to the further oxidation of the
produced H2CO to CO2 [52].
Figure 1.11 shows the evolution of the production rates of H2, H2CO and CO2
as well as the CH3OH conversion with the O2/CH3OH ratio in the feed composition for
both: unpromoted (VWR = +2 V) and electropromoted (VWR < +2 V) conditions at a
fixed reaction temperature of 300 ºC.
Page 94
Chapter 1
86
Figure 1.11. H2 (a), H2CO (b), CO2 (c) production rates and CH3OH conversion (d) variation
vs. O2/CH3OH ratio in the feed stream at 300 ºC for the unpromoted (+2 V) and electro-
promoted (-0.5 V) Pt film prepared by cathodic arc deposition.
0
1
2
3
4
5
6
7
8
r H
/ m
ol s
-1 x
10
-7
2 V
-0.5 V
0.0
0.4
0.8
1.2
1.6
r H
CO /
mo
l s-1
x 1
0-7
2 V
-0.5 V
0
10
20
30
40
50
60
0 0.25 0.5 0.75 1 1.25
O2/CH3OH ratio
CH
3O
H C
on
vers
ion
/ % 2 V
-0.5 V
0.0
0.5
1.0
1.5
2.0
2.5
3.0
r CO
/
mo
l s-1
x 1
0-6 2 V
-0.5 V
2
2
2
a)
b)
c)
d)
Page 95
Electrochemical promotion of Pt for H2 production from methanol partial oxidation and steam
reforming
87
At negative polarization, a similar promotional effect on the methanol partial
oxidation activity was again found under all explored O2/CH3OH ratios. In this figure,
an optimum O2/CH3OH ratio can be observed for the partial oxidation mechanism vs.
the total oxidation, which has been typically observed in previous investigations using
Pt catalysts [17, 52]. At a low O2 concentration range, O2/CH3OH < 0.5, the partial
oxidation of methanol exhibited a positive order with respect to the O2 concentration.
In fact, the Pt catalytic activity was negligible in the absence of oxygen (methanol
decomposition conditions). A further increase in the O2 concentration enhanced the
total oxidation process and favoured the subsequent oxidation of the H2 and H2CO
obtained. In the case of O2/CH3OH > 0.5, a further increase in the O2 concentration
also led to a stronger poisoning effect on the partial oxidation mechanism under
electropromoted (VWR = -0.5 V) conditions vs. unpromoted conditions (VWR = +2 V)
due to the previously mentioned higher O2 coverage of the potassium-modified Pt
surface. For the case of the overall methanol reaction rate (partial oxidation and deep
oxidation), the methanol conversion curve exhibited positive order dependence with
respect to the oxygen concentration in the feed for the overall studied range. Hence, in
this case the increase in the O2 coverage under electropromoted conditions enhanced
the methanol reaction rate for the overall O2/CH3OH ratio, in good agreement with the
rules of the chemical and electrochemical promotion in heterogeneous catalysis [27,
54, 55].
iv) Stability study.
Figure 1.12 shows the variation in the production rate of H2 and H2CO vs. time
on stream at different repetitive applied potentials, at a temperature of 300 ºC and with
a feed composition of CH3OH/O2 = 7.2 %/4.6 %.
It can be observed a quite reproducible behaviour of both reaction rates during
the three cycles of positive and negative potentials. This completely reversible
NEMCA effect would demonstrate the stability of the solid electrolyte cell (working
electrode and solid electrolyte) under reaction conditions. The obtained negative
charged (not shown in the figure) upon the application of -1 V during the different
Page 96
Chapter 1
88
cycles was the same for all of them and also agreed with the positive charge obtained
during the application of the catalyst potential of +2 V. This fact implies that the
amount of electrochemically transferred potassium ions from or to the solid electrolyte
during the negative and positive applications, respectively, was the same,
demonstrating the stability of promoting species under reaction conditions.
Figure 1.12. H2 (a) and H2CO (b) production rates variation vs. time under repetitive EPOC
transitions with the Pt film prepared by cathodic arc deposition. Partial oxidation conditions:
CH3OH/O2 = 7.2 %/4.6 %, 300 ºC.
Figure 1.13 shows the variation of the H2 and H2CO production rates for a
long, uninterrupted, negative polarization of -1 V for 8 h under the same reaction
conditions.
0
1
2
3
4
5
6
7
8
r H
/ m
ol s
-1 x
10
-7
-1.5
-1
-0.5
0
0.5
1
1.5
2
2.5
3V
WR / V
0.0
0.4
0.8
1.2
1.6
0 100 200 300 400
Time / min
r H
CO
/ m
ol s
-1 x
10
-7
-1.5
-1
-0.5
0
0.5
1
1.5
2
2.5
VW
R / V
2
2
2
2
rH
VWR
rH CO
VWR
a)
b)
Page 97
Electrochemical promotion of Pt for H2 production from methanol partial oxidation and steam
reforming
89
Figure 1.13. H2 (a) and H2CO (b) production rates variation vs. time for a long EPOC
experiment during a durability test with the Pt film prepared by cathodic arc deposition. Partial
oxidation conditions: CH3OH/O2 = 7.2 %/4.6 %, 300 ºC.
The obtained results showed no lost of catalytic activity during the cathodic
polarization and, therefore, no lost of promotional phases under working conditions.
Previous studies with alkali-electropromoted systems have already demonstrated the
stability of this kind of systems even at higher working temperatures (above 350 ºC) in
a wide variety of catalytic systems such as the selective catalytic reduction of
N2O[53], the water gas-shift [56] or CO preferential oxidation [36]. Hence, the
obtained results demonstrate the interest of the proposed EPOC configuration for the
simultaneous production of H2 and H2CO in a wide variety of reaction conditions and
in a very stable manner in view of a possible practical application of this process.
0
1
2
3
4
5
6
7
8
r H
/ m
ol s
-1 x
10
-7
-1.5
-1
-0.5
0
0.5
1
1.5
2
2.5
3
VW
R / V
0.0
0.2
0.4
0.6
0.8
1.0
1.2
1.4
1.6
0 100 200 300 400 500 600
Time / min
r H
CO
/ m
ol s
-1 x
10
-7
-1.5
-1
-0.5
0
0.5
1
1.5
2
2.5
VW
R / V
2
2
2
2
rH
VWR
rH CO
VWR
a)
b)
Page 98
Chapter 1
90
1.3.2. Steam reforming of methanol
The Pt catalyst film prepared by CAD was also tested in the methanol steam
reforming (SRM) reaction with a feed composition of CH3OH/H2O = 4 %/4.8 % (N2
balance). H2, CO and CO2 were produced but neither H2CO nor other possible
methanol-derived compounds (such as CH4) were obtained under this reaction
atmosphere.
Figure 1.14 shows the variation of the different production rates vs. time under
open circuit and different applied potentials at 400 ºC. As in the previous experiments
under POM conditions, the application of a potential of +2 V (un-promoted state)
allowed to define a Pt catalyst film free of promoting ions and hence a reference state.
Potassium ions that probably migrated thermally to the Pt film were removed, leading
to the observed slight decrease in the catalytic activity during the first polarization.
The subsequent application of lower potentials increased the amount of
electrochemically transferred positive K+ ions to the Pt catalyst-working electrode.
Hence, the application of a catalyst potential of 0 V strongly enhanced the H2, CO and
CO2 production rates through the promotion of the methanol reforming reaction by K+
ions.
It should be noted that in these experiments the methanol conversion was kept
below 5 %, even in the promoted state. Unlike the previous POM results, the low
conversions obtained under the studied steam reforming conditions were more similar
to those typically found with this kind of electrocatalytic systems due to the low
surface area of the active catalyst film and the reactant bypass to the catalyst surface.
However, under optimum applied potential conditions (VWR = 0 V) the H2 production
rate was still increased in 5 times with respect to the un-promoted catalytic rate (VWR
= +2 V).
Page 99
Electrochemical promotion of Pt for H2 production from methanol partial oxidation and steam
reforming
91
Figure 1.14. Influence of the applied potential (VWR) on the H2, CO2 and CO production rates
vs. time. Steam reforming conditions: CH3OH/H2O = 4 %/4.8 %, 400 ºC.
It can also be observed that the application of lower applied potentials (below
0 V) and, hence, the further increase in the promoter coverage, led to a decrease in the
catalytic activity of the system, which can be attributed to an excessive formation of
VWR
2
0
1
2
3
4
5
r H
/ m
ol s
-1 x
10
-8
-2.5
-1.5
-0.5
0.5
1.5
2.5
VW
R / V
0
3
6
9
12
r CO
/
mo
l s-1
x 1
0-9
-2.5
-1.5
-0.5
0.5
1.5
2.5
VW
R / V
0
3
6
9
12
0 100 200 300 400 500 600
Time / min
r CO
/ m
ol s
-1 x
10
-9
-2.5
-1.5
-0.5
0.5
1.5
2.5
VW
R / V
rH 2
VWR
rCO 2
rCO
VWR
2
a)
b)
c)
O.C.
O.C.
O.C.
Page 100
Chapter 1
92
promoter-derived compounds [45] which may block catalytic active sites. Anyway,
these promoting species were completely removed when the potential was again fixed
at +2 V (unpromoted state), demonstrating the reversibility of the EPOC phenomena
in terms of catalytic activity. On the way of reaching the initial unpromoted values, a
strong increase in the H2, CO and CO2 production rates was firstly observed, which
was attributed to the progressive removal and decomposition of potassium-derived
carbonaceous products formed during the previous applied potentials. As reported in
previous works, potassium ions may form carbonates or bicarbonates species under
H2-, H2O-, CO- and CO2-containing atmospheres [35, 57]. This shows the ability of
this kind of systems to partially store reaction products [58], e.g., H2, which may be
also of significant interest for other applications.
Regarding the strong promotional effect previously mentioned (below 0 V),
one can rationalize the K+-induced variation in the catalytic activity on the basis of the
electrode work function modification [27, 59]. As mentioned in the previous section
under POM reaction conditions, a decrease in the catalyst potential involves a
decrease in the catalyst work function due the migration of positively charged
potassium species, thus weakening the Pt chemical bonds with the electron-donor
adsorbates (i.e., CH3OH), and strengthening those with the electron acceptor
adsorbates (i.e., H2O in this case, instead of O2). A similar promotional effect in the
presence of electro-positive ions has also been observed due to the activation of H2O
molecules in the steam reforming of methane [60] and water gas shift reaction[56].
In particular, previous studies on the mechanism of methanol reforming on Pt
[10] stated that the reaction proceeds via: (1) dissociative adsorption of methanol to
produce methoxy species and adsorbed hydrogen; (2) oxidative decomposition of the
methoxy groups by adsorbed water to liberate two hydrogen molecules and generate
surface formate; (3) water-assisted decomposition of formate to hydrogen and
unidentate carbonate; and (4) decomposition of carbonate to carbon dioxide. Then,
very likely, the water activation in the presence of K+ ions would favour the
decomposition of carbonaceous species (methoxy and formate species) increasing the
activity of the system. This explanation is also in good agreement with the observed
Page 101
Electrochemical promotion of Pt for H2 production from methanol partial oxidation and steam
reforming
93
increase in the catalytic rate under unpromoted SRM conditions with respect to the
methanol decomposition reaction rate, i.e., in the absence of water (see, for instance,
O2/CH3OH ratio = 0, in Figure 1.11). Under methanol decomposition conditions a
negligible activity of the system was obtained, which demonstrates the positive effect
of water activation for the decomposition of the reaction intermediates and hence for
the methanol reforming catalytic process.
Figure 1.15 shows, under the same feed composition and different reaction
temperatures, the steady state variation of the H2 production rate vs. the applied
catalyst potential, and the variation of the optimum supplied negative charge which
maximized the H2 production rate in each case, along with the associated (maximum)
rate enhancement ratios, ρmax. The latter parameter was calculated by equation 1.5 and
the supplied charge was directly obtained from the integration of the current vs. time
curves.
As the reaction temperature raised, both higher promotional effects and
optimum supplied negative charges were observed. Thus, an increase of the
temperature not only favoured the methanol reforming kinetics [10] but also the
electrochemically assisted water activation. Above 400 ºC, the maximum rate
enhancement ratio reached a plateau at around 5, which was associated to the increase
in the unpromoted reaction rate (r0). These results demonstrate the interest of the
EPOC phenomena to promote to the optimal grade the performance of a metal catalyst
at varying reaction conditions (e.g., different temperatures).
Finally it is also interesting to note that the cathodic arc deposition method
allowed to prepare an electrochemical catalyst based on a very thin Pt layer (120 nm),
which has shown stable catalytic activity values for long experiments (10 h) under
POM and SRM conditions. Hence, this seems to be an appropriate method for the
application of thin metallic films for catalytic and electrocatalytic applications. Either
CAD or other kind of physical vapour deposition technique will be henceforth used
for the preparation of electrochemical catalysts.
Page 102
Chapter 1
94
Figure 1.15. Influence of the applied potential (VWR) on the steady state H2 production rate (a),
and on the maximum rate enhancement ratio (ρmax) and the optimal supplied negative charge
(b) at different reaction temperatures. Steam reforming conditions: CH3OH/H2O = 4 %/4.8 %.
1.4. CONCLUSIONS
The following conclusions could be drawn from this study:
- The obtained results showed the interest of electrochemistry to improve the
activity of a metal catalyst in H2 production from methanol via partial oxidation
(POM) and steam reforming (SRM).
0
2
4
6
8
10
-2.5 -2 -1.5 -1 -0.5 0 0.5 1 1.5 2 2.5
r H
/
mo
l s-1
x 10
-8
VWR / V
450 ºC
400 ºC
360 ºC
320 ºC
0
0.04
0.08
0.12
0.16
0.2
0
1
2
3
4
5
6
300 330 360 390 420 450 480O
pti
mu
m n
ega
tive
ch
arge
/ C
ρm
ax
Temperature / ºC
2
a)
b)
Page 103
Electrochemical promotion of Pt for H2 production from methanol partial oxidation and steam
reforming
95
- In contrast to the metal catalyst preparation method based on the
impregnation of a precursor solution, the cathodic arc deposition allowed to obtain a
very thin catalyst film (around 120 nm-thick) with a low metal loading (0.23 mg Pt
cm-2
). This kind of physical vapour deposition techniques is very useful for the
deposition of the catalysts employed in EPOC studies.
- The selectivity of the methanol partial oxidation reaction for H2 and H2CO
production can be greatly enhanced vs. the total combustion mechanism by
electrochemical promotion with K+ ions on a dense Pt film prepared by cathodic arc
deposition.
- The presence of electropositive K+ ions increased the metal bond strength
with the electron acceptor molecule, i.e., O2 in POM and H2O in SRM, favouring the
conversion of methoxy intermediates to hydrogen, and also to formaldehyde (in the
former case).
- This kind of electrochemically promoted catalysts may be of great interest
due to its ability of simultaneously producing H2 and H2CO with high yield in a single
reaction step. Its stability has also been verified for long working times.
- The EPOC behaviour of an alkali-based electrochemical catalyst (Pt/K-
βAl2O3) not only depends on the reaction conditions and the kinetic order of the
reactant molecules but also on the electrocatalytic activity of the catalyst-electrode
film (which is related to the deposition method) and its ability to form and store
promoter derived surface compounds, which may block active sites. This was not the
case of the cathodic arc deposited Pt. Hence, this technique or similar physical vapour
deposition methods were selected for subsequent experiments and chapters.
1.5. REFERENCES
[1] S. Dutta, A review on production, storage of hydrogen and its utilization as an energy
resource, Journal of Industrial and Engineering Chemistry, 20 (2014) 1148-1156.
[2] J.N. Armor, Catalysis and the hydrogen economy, Catalysis Letters, 101 (2005) 131-135.
Page 104
Chapter 1
96
[3] J.D. Holladay, J. Hu, D.L. King, Y. Wang, An overview of hydrogen production
technologies, Catalysis Today, 139 (2009) 244-260.
[4] D.R. Palo, R.A. Dagle, J.D. Holladay, Methanol steam reforming for hydrogen production,
Chemical Reviews, 107 (2007) 3992-4021.
[5] R.M. Navarro, M.A. Peña, J.L.G. Fierro, Hydrogen production reactions from carbon
feedstocks: Fossil fuels and biomass, Chemical Reviews, 107 (2007) 3952-3991.
[6] E.L. Fornero, D.L. Chiavassa, A.L. Bonivardi, M.A. Baltanás, CO2 capture via catalytic
hydrogenation to methanol: Thermodynamic limit vs. 'kinetic limit, Catalysis Today, 172
(2011) 158-165.
[7] C. Cao, K.L. Hohn, Study of reaction intermediates of methanol decomposition and
catalytic partial oxidation on Pt/Al2O3, Applied Catalysis A: General, 354 (2009) 26-32.
[8] S. Sá, H. Silva, L. Brandão, J.M. Sousa, A. Mendes, Catalysts for methanol steam
reforming-A review, Applied Catalysis B: Environmental, 99 (2010) 43-57.
[9] G. Jacobs, B.H. Davis, In situ DRIFTS investigation of the steam reforming of methanol
over Pt/ceria, Applied Catalysis A: General, 285 (2005) 43-49.
[10] P. Tolmacsov, A. Gazsi, F. Solymosi, Decomposition and reforming of methanol on Pt
metals supported by carbon Norit, Applied Catalysis A: General, 362 (2009) 58-61.
[11] S.H. Ahn, O.J. Kwon, I. Choi, J.J. Kim, Synergetic effect of combined use of Cu-ZnO-
Al2O3 and Pt-Al2O3 for the steam reforming of methanol, Catalysis Communications, 10
(2009) 2018-2022.
[12] G. Kolb, S. Keller, S. Pecov, H. Pennemann, R. Zapf, Development of micro-structured
catalytic wall reactors for hydrogen production by methanol steam reforming over novel
Pt/In2O 3/Al2O3 catalysts, Chemical Engineering Transactions, 2011, pp. 133-138,
DOI:10.3303/cet1124023.
[13] A. Wan, C.t. Yeh, Ignition of methanol partial oxidation over supported platinum catalyst,
Catalysis Today, 129 (2007) 293-296.
[14] M.P. Zum Mallen, L.D. Schmidt, Oxidation of methanol over polycrystalline Rh and Pt:
Rates, OH desorption, and model, Journal of Catalysis, 161 (1996) 230-246.
[15] N. Kizhakevariam, E.M. Stuve, Promotion and poisoning of the reaction of methanol on
clean and modified platinum (100), Surface Science, 286 (1993) 246-260.
[16] B.E. Traxel, K.L. Hohn, Partial oxidation of methanol at millisecond contact times,
Applied Catalysis A: General, 244 (2003) 129-140.
[17] R. Ubago-Pérez, F. Carrasco-Marín, C. Moreno-Castilla, Methanol partial oxidation on
carbon-supported Pt and Pd catalysts, Catalysis Today, 123 (2007) 158-163.
Page 105
Electrochemical promotion of Pt for H2 production from methanol partial oxidation and steam
reforming
97
[18] G. Reuss, W. Disteldorf, A.O. Gamer, A. Hilt, Formaldehyde, Ullmann's Encyclopedia of
Industrial Chemistry, Wiley-VCH, Weinheim, 2002.
[19] G.C. Behera, K. Parida, Selective gas phase oxidation of methanol to formaldehyde over
aluminum promoted vanadium phosphate, Chemical Engineering Journal, 180 (2012) 270-276.
[20] N. Iwasa, T. Mayanagi, W. Nomura, M. Arai, N. Takezawa, Effect of Zn addition to
supported Pd catalysts in the steam reforming of methanol, Applied Catalysis A: General, 248
(2003) 153-160.
[21] M. Kusche, F. Enzenberger, S. Bajus, H. Niedermeyer, A. Bösmann, A. Kaftan, M.
Laurin, J. Libuda, P. Wasserscheid, Enhanced activity and selectivity in catalytic methanol
steam reforming by basic alkali metal salt coatings, Angewandte Chemie - International
Edition, 52 (2013) 5028-5032.
[22] H.N. Evin, G. Jacobs, J. Ruiz-Martinez, U.M. Graham, A. Dozier, G. Thomas, B.H.
Davis, Low temperature water-gas shift/methanol steam reforming: Alkali doping to facilitate
the scission of formate and methoxy C-H bonds over Pt/ceria catalyst, Catalysis Letters, 122
(2008) 9-19.
[23] L. Alejo, R. Lago, M.A. Peña, J.L.G. Fierro, Partial oxidation of methanol to produce
hydrogen over Cu-Zn-based catalysts, Applied Catalysis A: General, 162 (1997) 281-297.
[24] M. Stoukides, C.G. Vayenas, The effect of electrochemical oxygen pumping on the rate
and selectivity of ethylene oxidation on polycrystalline silver, Journal of Catalysis, 70 (1981)
137-146.
[25] C.G. Vayenas, S. Bebelis, S. Ladas, Dependence of catalytic rates on catalyst work
function, Nature, 343 (1990) 625-627.
[26] S. Souentie, L. Lizarraga, E.I. Papaioannou, C.G. Vayenas, P. Vernoux, Permanent
electrochemical promotion of C3H8 oxidation over thin sputtered Pt films, Electrochemistry
Communications, 12 (2010) 1133-1135.
[27] C.G. Vayenas, S. Bebelis, C. Pliangos, S. Brosda, D. Tsiplakides, Electrochemical
Activation of Catalysis: Promotion, Electrochemical Promotion and Metal-Support
Interactions, Kluwer Academic Publishers/Plenum Press, New York, 2001.
[28] A. Katsaounis, Recent developments and trends in the electrochemical promotion of
catalysis (EPOC), Journal of Applied Electrochemistry, 40 (2010) 885-902.
[29] D. Tsiplakides, S. Balomenou, Electrochemical promoted catalysis: Towards practical
utilization, Chemical Industry and Chemical Engineering Quarterly, 14 (2008) 97-105.
Page 106
Chapter 1
98
[30] A. de Lucas-Consuegra, New trends of Alkali Promotion in Heterogeneous Catalysis:
Electrochemical Promotion with Alkaline Ionic Conductors, Catalysis Surveys from Asia,
2015, DOI:10.1007/s10563-014-9179-6.
[31] P. Vernoux, L. Lizarraga, M.N. Tsampas, F.M. Sapountzi, A. De Lucas-Consuegra, J.L.
Valverde, S. Souentie, C.G. Vayenas, D. Tsiplakides, S. Balomenou, E.A. Baranova, Ionically
conducting ceramics as active catalyst supports, Chemical Reviews, 113 (2013) 8192-8260.
[32] P. Vernoux, F. Gaillard, C. Lopez, E. Siebert, Coupling catalysis to electrochemistry: A
solution to selective reduction of nitrogen oxides in lean-burn engine exhausts?, Journal of
Catalysis, 217 (2003) 203-208.
[33] F.J. Williams, N. Macleod, M.S. Tikhov, R.M. Lambert, Electrochemical promotion of
bimetallic Rh-Ag/YSZ catalysts for the reduction of NO under lean burn conditions,
Electrochimica Acta, 47 (2002) 1259-1265.
[34] E.I. Papaioannou, S. Souentie, A. Hammad, C.G. Vayenas, Electrochemical promotion of
the CO2 hydrogenation reaction using thin Rh, Pt and Cu films in a monolithic reactor at
atmospheric pressure, Catalysis Today, 146 (2009) 336-344.
[35] A.J. Urquhart, F.J. Williams, R.M. Lambert, Electrochemical promotion by potassium of
Rh-catalysed fischer-tropsch synthesis at high pressure, Catalysis Letters, 103 (2005) 137-141.
[36] A. de Lucas-Consuegra, A. Princivalle, A. Caravaca, F. Dorado, C. Guizard, J.L.
Valverde, P. Vernoux, Preferential CO oxidation in hydrogen-rich stream over an
electrochemically promoted Pt catalyst, Applied Catalysis B: Environmental, 94 (2010) 281-
287.
[37] S. Neophytides, C.G. Vayenas, Non-faradaic electrochemical modification of catalytic
activity. 2. The case of methanol dehydrogenation and decomposition on Ag, Journal of
Catalysis, 118 (1989) 147-163.
[38] C.G. Vayenas, S. Neophytides, Non-faradaic electrochemical modification of catalytic
activity III. The case of methanol oxidation on Pt, Journal of Catalysis, 127 (1991) 645-664.
[39] C.A. Cavalca, G. Larsen, C.G. Vayenas, G.L. Haller, Electrochemical modification of
CH3OH oxidation selectivity and activity on a Pt single-pellet catalytic reactor, Journal of
Physical Chemistry, 97 (1993) 6115-6119.
[40] J.K. Hong, I.H. Oh, S.A. Hong, W.Y. Lee, Electrochemical oxidation of methanol over a
silver electrode deposited on yttria-stabilized zirconia electrolyte, Journal of Catalysis, 163
(1996) 95-105.
Page 107
Electrochemical promotion of Pt for H2 production from methanol partial oxidation and steam
reforming
99
[41] F. Dorado, A. de Lucas-Consuegra, P. Vernoux, J.L. Valverde, Electrochemical
promotion of platinum impregnated catalyst for the selective catalytic reduction of NO by
propene in presence of oxygen, Applied Catalysis B: Environmental, 73 (2007) 42-50.
[42] A. Anders, Cathodic Arcs: From Fractal Spots to Energetic Condensation. Springer Series
on Atomic, Optical, and Plasma Physics, Springer-Verlag, New York, 2008.
[43] E. Seker, E. Gulari, Improved N2 selectivity for platinum on alumina prepared by sol-gel
technique in the reduction of NOx by propene, Journal of Catalysis, 179 (1998) 339-342.
[44] E. Mutoro, C. Koutsodontis, B. Luerssen, S. Brosda, C.G. Vayenas, J. Janek,
Electrochemical promotion of Pt(111)/YSZ(111) and Pt-FeOx/YSZ(111) thin catalyst films:
Electrocatalytic, catalytic and morphological studies, Applied Catalysis B: Environmental,
100 (2010) 328-337.
[45] P. Vernoux, F. Gaillard, C. Lopez, E. Siebert, In-situ electrochemical control of the
catalytic activity of platinum for the propene oxidation, Solid State Ionics, 175 (2004) 609-613.
[46] N. Kotsionopoulos, S. Bebelis, In situ electrochemical modification of catalytic activity
for propane combustion of Pt/β-Al2O3 catalyst-electrodes, Topics in Catalysis, 44 (2007) 379-
389.
[47] N. Kotsionopoulos, S. Bebelis, Electrochemical characterization of the Pt/β″-Al 2O3
system under conditions of in situ electrochemical modification of catalytic activity for
propane combustion, Journal of Applied Electrochemistry, 40 (2010) 1883-1891.
[48] H.P. Klug, L.E. Alexander, X-Ray Diffraction Procedures for Polycrystalline and
Amorphous Materials, Wiley, New York, 1974.
[49] F.J. Williams, M.S. Tikhov, A. Palermo, N. Macleod, R.M. Lambert, Electrochemical
promotion of rhodium-catalyzed NO reduction by CO and by propene in the presence of
oxygen, Journal of Physical Chemistry B, 105 (2002) 2800-2808.
[50] I.V. Yentekakis, R.M. Lambert, M. Konsolakis, V. Kiousis, The effect of sodium on the
Pd-catalyzed reduction of NO by methane, Applied Catalysis B: Environmental, 18 (1998)
293-305.
[51] A. Peremans, F. Maseri, J. Darville, J.M. Gilles, Interaction of methanol with a
polycrystalline platinum surface studied by infrared reflection absorption spectroscopy,
Surface Science, 227 (1990) 73-78.
[52] R.W. McCabe, D.F. McCready, Kinetics and reaction pathways of methanol oxidation on
platinum, Journal of Physical Chemistry, 90 (1986) 1428-1435.
Page 108
Chapter 1
100
[53] A. de Lucas-Consuegra, F. Dorado, C. Jiménez-Borja, J.L. Valverde, Influence of the
reaction conditions on the electrochemical promotion by potassium for the selective catalytic
reduction of N2O by C3H6 on platinum, Applied Catalysis B: Environmental, 78 (2008) 222-
231.
[54] C. Vayenas, S. Brosda, Electrochemical promotion: Experiment, rules and mathematical
modeling, Solid State Ionics, 154-155 (2002) 243-250.
[55] S. Brosda, C.G. Vayenas, J. Wei, Rules of chemical promotion, Applied Catalysis B:
Environmental, 68 (2006) 109-124.
[56] A. De Lucas-Consuegra, A. Caravaca, J. González-Cobos, J.L. Valverde, F. Dorado,
Electrochemical activation of a non noble metal catalyst for the water-gas shift reaction,
Catalysis Communications, 15 (2011) 6-9.
[57] A. de Lucas-Consuegra, F. Dorado, J.L. Valverde, R. Karoum, P. Vernoux, Low-
temperature propene combustion over Pt/K-βAl2O3 electrochemical catalyst:
Characterization, catalytic activity measurements, and investigation of the NEMCA effect,
Journal of Catalysis, 251 (2007) 474-484.
[58] A. de Lucas-Consuegra, A. Caravaca, F. Dorado, J.L. Valverde, Pt/K-βAl2O3 solid
electrolyte cell as a "smart electrochemical catalyst" for the effective removal of NOx under
wet reaction conditions, Catalysis Today, 146 (2009) 330-335.
[59] A. Nakos, S. Souentie, A. Katsaounis, Electrochemical promotion of methane oxidation
on Rh/YSZ, Applied Catalysis B: Environmental, 101 (2010) 31-37.
[60] A. De Lucas-Consuegra, A. Caravaca, P.J. Martínez, J.L. Endrino, F. Dorado, J.L.
Valverde, Development of a new electrochemical catalyst with an electrochemically assisted
regeneration ability for H2 production at low temperatures, Journal of Catalysis, 274 (2010)
251-258.
Page 109
101
Chapter 2
ELECTROCHEMICAL PROMOTION OF Pt
NANOPARTICLES DISPERSED IN A CARBON MATRIX
FOR METHANOL CONVERSION:
TOWARDS MORE COMPETITIVE CATALYSTS OF LOW METAL
LOADING
This study reports the electrochemical promotion (EPOC) of Pt nanoparticles
(of around 3 nm) dispersed in a diamond-like carbon (DLC) matrix by means of the
cathodic arc deposition technique. After a temperature-programmed pretreatment, the
catalyst film achieved a suitable electrical conductivity due to the transition of the sp3-
hybridized carbon form into a more graphitic structure (sp2-hybridized). The catalytic
performance of the Pt-DLC film in the methanol partial oxidation (POM) and steam
reforming (SRM) reactions for H2 production has been promoted by K+ ions
electrochemically transferred from a K-βAl2O3 solid electrolyte. Moreover, two
different electropromotional effects have been found under POM conditions
depending on the applied potential, which can be attributed to the formation of
different kinds of promoter phases on this catalyst. The higher catalytic activity of Pt-
DLC, compared to that of the pure Pt film, demonstrates the practical interest of this
kind of dispersed catalyst films with very a low metal loading.
0
1
2
3
4
5
6
-2.5 -2 -1.5 -1 -0.5 0 0.5 1 1.5 2 2.5
r H
/ m
ol s
-1x
10
-8
VWR / V
Pt 320 ºC (0.46 mg)
Pt 360 ºC (0.46 mg)
Pt/DLC 320 ºC (0.03 mg)
Pt/DLC 360 ºC (0.03 mg)
2
2 ρH ,max = 3.37
ρH ,max = 3.54
2
ρH ,max = 1.86
2
ρH ,max = 1.53
2
ΔV < 0
Pt-DLC
K+ K+ K+
e-
e-
K-βAl2O3
Au
Δr
Catalyst film (Pt-DLC)
Dr
K+ ionsΔV Solid electrolyte (K-bAl2O3)
Counter/Reference (Au)
CH3OH
H2, CO,CO2, (H2CO)
10 nm
CH3OH/H2O = 4 %/4.8 %
-2.5
-2
-1.5
-1
-0.5
0
0.5
1
1.5
2
2.5
0
1
2
3
4
5
6
0 100 200 300 400 500 600 700 800
VW
R/
V
r H
/ m
ol s
-1x
10
-8
Time / min
250 ºC
320 ºC
360 ºC
VWR
2
-2.5
-2
-1.5
-1
-0.5
0
0.5
1
1.5
2
2.5
0
0.5
1
1.5
2
2.5
3
0 100 200 300 400 500 600 700 800
VW
R/
V
r H
/ m
ol s
-1cm
-2x
10
-8
Time / min
250 ºC
320 ºC
360 ºC
VWR
CH3OH/H2O = 4 %/4.8 %
2
CH3OH,
O2 / H2O
H2, CO2,
CO, (H2CO)
4 % CH3OH
4.8 % H2O
Page 110
Chapter 2
102
2.1. INTRODUCTION
As already pointed out, the present research is mainly focused on the
application of the phenomenon of Electrochemical Promotion Of Catalysis (EPOC)
[1] to H2 production catalytic processes by using CH3OH as the raw material. Indeed,
methanol is one of the most important feedstock in the chemical industry, and it is also
a very promising liquid energy carrier that can be used as a fuel in Direct Methanol
Fuel Cells (DMFC) [2, 3] or as a hydrogen source through the steam reforming (SRM)
and partial oxidation (POM) reactions [3-5]. As already mentioned, the main
advantages of using methanol for H2 production are the following: high energy
density, easy availability, and safe storage and transport conditions.
In the alkali electrochemical promotion experiments carried out in the first
chapter, Pt was selected as the active phase since this metal is one of the most
commonly employed catalysts in both SRM [6-9] and POM [10-14]. Hence, the EPOC
phenomenon showed to improve the catalytic behaviour of Pt by means of the
controlled electrochemical pumping of K+ promoter ions from a K-βAl2O3 solid
electrolyte (which also acted as catalyst support). Concretely, H2 production from
SRM and POM was enhanced up to 5 and 6 times through the electrochemical
promotion of a fairly dense Pt catalyst prepared by Cathodic Arc Deposition (CAD).
However, in view of a possible practical application of this kind of systems, novel
catalyst films with higher metal particle dispersions and with a more competitive cost
should be investigated [15, 16]. The use of conductive catalyst films of pure metal
implies high costs of these electro-catalytic configurations making it difficult to
compete with conventional supported catalysts (with dispersions typically above 10
%). This point is essential for a further commercialization step of the EPOC
phenomena for the H2 production and for other catalytic systems. In this sense, recent
efforts have been done in previous studies by using a second conductive material that
can simultaneously act as both catalyst particles support and electronic conductor
material: gold [16], graphite [17], carbon [18], carbon nanofibers [19] or mixed ionic
electronic composite materials [20, 21].
Page 111
Electrochemical promotion of Pt nanoparticles dispersed in a carbon matrix for methanol
conversion
103
In this work, we propose for the first time in literature the use of a novel Pt-
diamond like carbon (Pt-DLC) catalyst film prepared by the technique of cathodic arc
deposition. DLC is the denomination of an amorphous form of carbon in which the
bonding is composed of a mixture of diamond-type (sp3) and graphite-type (sp
2)
hybridization, exhibiting properties intermediate between them [22]. DLC presents
very unique characteristics to be used as catalyst particles support in an electrode film
such as: large potential window, high strength, thermal conductivity, low cost and
chemical inertness [23]. As expected for diamond, DLC is stable to high temperatures
(800 °C) and hence it could be used in a large number of catalytic processes.
Moreover, its properties can be further modified (improved) by the introduction of
metallic elements. In this way, the metal is dispersed on the diamond-like carbon. At
the same time, this addition should both decrease the internal stress and increase its
electrical conductivity [24, 25]. These kinds of composites (metal-DLC) are acquiring
great importance due to their potential applications, including microelectrodes in
electrochemical analysis and field emission properties [26, 27]. Pt-DLC films have
already been prepared for other applications in several previous studies by pulsed laser
deposition [24] and sputter deposition [25, 28-30]. However, to the best of our
knowledge there are no previous studies on the use of this kind of composites in
catalysis.
The aim of this study is to electrochemically promote a novel, thin, Pt-DLC
catalyst film of low metal loading where the Pt nanoparticles dispersed in the DLC
matrix could be electrochemically promoted. Thus, a Pt-DLC/K-βAl2O3
electrochemical catalyst has been prepared, characterized and tested for the H2
production via POM and SRM. The effect of the electrochemical pumping of
potassium ions on the active Pt-DLC catalyst film has been investigated under a wide
range of reaction conditions and explained according to the EPOC rules [1]. Finally,
the obtained EPOC behaviour of the Pt-DLC film has been compared with that shown
in the first chapter with a pure dense Pt film prepared by the same technique (cathodic
arc deposition) in order to evaluate the benefit of the Pt-DLC system proposed.
Page 112
Chapter 2
104
2.2. EXPERIMENTAL
2.2.1. Preparation of the electrochemical catalyst
The electrochemical catalyst consisted of a continuous, thin film (geometric
area of 2.01 cm2) composed of Pt nanoparticles dispersed in a diamond-like carbon
matrix (Pt-DLC), which was deposited on a side of a 19-mm-diameter, 1-mm-thick K-
βAl2O3 (Ionotec) disc. As in the first chapter, inert Au counter (CE) and reference
(RE) electrodes were firstly deposited on the other side of the electrolyte by the
application of coatings of gold paste (Gwent Electronic Materials C1991025D2)
followed by calcination at 800 ºC for 2 h (heating ramp of 5 ºC min-1
). Then, the active
Pt-DLC catalyst film, which also behaved as a working electrode (WE), was deposited
on the other side of the electrolyte by the filter cathodic arc deposition technique [31].
The deposition system consisted of two cathodes contained in a mini-gun source
designed to operate in pulsed mode [32]. The pulses used in the production of carbon
and platinum plasmas were 200 A and had a duration of 1 ms. Plasma produced by the
source was injected into a 90-degree filter contained in a cross-sectional chamber to
remove the majority of macroparticles which were created during the cathodic arc
process. During the deposition, the substrates were biased with negative 500 V pulses
that were 1 µs long and had a duty cycle of 10 %. The pulse repetition rate was set at 2
pulses per second and the number of pulses per deposition was 25000. During the
deposition, the substrate holder was rotated at a speed of 2 revolutions per minute. The
residual gas pressure was around 10-4
Pa. Hence, this technique allowed the
preparation of a thin (150 nm-thick), highly dense, Pt-DLC film which showed good
adhesion to the substrate. The final Pt loading was 0.014 mg Pt cm-2
.
This catalyst preparation method was carried out in collaboration with Dr.
José Luis Endrino from the Institute of Materials Science of Madrid (CSIC).
The resulting Pt-DLC/K-βAl2O3/Au electrochemical catalyst was placed into
the single chamber solid electrolyte cell reactor. Since the obtained catalyst film was
electrically nonconductive, it was subjected, prior to the catalytic activity
measurements, to a 25 % H2 stream (N2 balance) with an overall flow rate of 3.6 L h-1
Page 113
Electrochemical promotion of Pt nanoparticles dispersed in a carbon matrix for methanol
conversion
105
under temperature-programmed conditions (3.5 ºC min-1
) from room temperature to
320 ºC, in an effort to decrease its electrical resistance while keeping platinum in its
reduced state, as will be explained in the discussion section. All the electrodes were
connected to the potentiostat-galvanostat (PGSTAT320-N, Metrohom Autolab) as
reported in the first chapter, but in this case a second electric contact was also added to
the catalyst-working electrode and connected to a digital multimeter, in order to
monitor the in-plane electrical resistance of the Pt-DLC under working conditions.
2.2.2. Characterization measurements
The deposited Pt-DLC catalyst film was first characterized via scanning
electron microscopy (SEM) and EDX analysis using a FEI Nova NANOSEM 230
instrument, in order to investigate the morphology of the Pt-DLC film and identify and
approximately quantify the amount of the different elements (Pt and C). The
Temperature Programmed Stabilization process (TPS) prior to the catalytic
experiments was in-situ monitored by recording the variation of the in-plane electrical
resistance between the two contacts (1 cm separated) located on the Pt-DLC film vs.
temperature.
The cross-sectional properties of the Pt-DLC catalyst film were studied before
and after the H2 stabilization treatment by transmission electron microscopy (TEM).
Cross-section TEM lamellas of samples were fabricated in a FEI Helios 600 Nanolab
Dual-Beam system. The specimens were pre-thinned with an acceleration voltage of
30 kV of the ion column and a final thinning was performed at 5 kV to minimize any
possible preparation artifacts. TEM work was carried out using a probe corrected FEI
Titan 60-300. Scanning Transmission Electron Microscopy (STEM) imaging in bright
field (BF), and high-angle annular dark field (HAADF) modes combined with
Electron Energy Loss Spectroscopy (EELS) were also used to characterize the
morphology, chemical composition and electronic structure of the samples. Cyclic
voltammetry (CV) measurements were also in-situ performed with the potentiostat-
galvanostat under SRM and POM reaction conditions at a fixed temperature (360 ºC),
to investigate the formation and decomposition of promoter-derived species. Before
Page 114
Chapter 2
106
performing these cyclic voltammetries, the sample was kept at +2 V for 30 min in
order to define a reference state (a Pt-DLC film clean of promoter ions). Then, the
potential was varied at a sweep rate of 5 mV s-1
from +2 to -2 V.
2.2.3. Catalytic activity measurements
The catalytic activity measurements were carried out in the experimental setup
described in section 1.2.3. The electrochemical promotion experiments were
performed at atmospheric pressure with an overall gas flow rate of 6 NL h-1
, a
temperature ranging from 250 to 320 ºC with a feed composition of CH3OH/O2 = 11
%/0.9 % (N2 balance) for the POM experiments, and a temperature ranging from 250
to 360 ºC with a feed composition of CH3OH/H2O = 4 %/4.8 % (N2 balance) for the
SRM experiments. H2, CO2, CO, H2O and H2CO were the only detected products, the
latter two appearing only under POM atmosphere. The error in the carbon atom
balance did not exceed 5 % in any experiment.
2.3. RESULTS AND DISCUSSION
2.3.1. Development of a catalyst with suitable structural and electrical properties
Figure 2.1 shows the top surface SEM micrograph of the prepared Pt-DLC
catalyst film along with the elemental EDX analysis.
Figure 2.1. SEM micrograph of the Pt-DLC catalyst film (a) and its corresponding EDX
analysis (b).
2.6330.5Pt
97.3769.5C
At / %Wt / %Element
2.6330.5Pt
97.3769.5C
At / %Wt / %Element
2.6330.5Pt
97.3769.5C
At / %Wt / %Element
2.6330.5Pt
97.3769.5C
At / %Wt / %Element
a) b)
Page 115
Electrochemical promotion of Pt nanoparticles dispersed in a carbon matrix for methanol
conversion
107
It can be observed that the obtained catalyst film was dense and exhibited a
very smooth surface in agreement with a previous work related to the preparation of
Pt-DLC films by sputtering [25]. The EDX analysis confirmed the low metal loading
(30 wt. % Pt) on the electrode, being carbon (70 wt. %) the other component. Hence,
no impurities of other elements due to possible contamination during the preparation
step in the chamber were detected in the EDX analysis of the film. It should also be
mentioned that due to the very low thickness of the catalyst layer (150 nm) and to the
surface character of the system, it is very likely that most of the deposited Pt
nanoparticles may have access to the gas phase reactants.
Figure 2.2 shows the variation of the planar electrical resistance of the Pt-DLC
catalyst surface vs. temperature during the TPS experiment (3.5 ºC/min) under a 25 %
H2 stream (N2 balance). The inset of the figure zooms the final variation of the
electrical resistance at the end of the experiment.
Figure 2.2. In-plane electrical resistance variation (R) during the Temperature Programmed
Stabilization (TPS). Heating ramp of 3.5 °C/min under a 25 % H2 stream (N2 balanced).
0
5
10
15
20
25
30
35
40
45
0 10 20 30 40 50 60 70 80 90 100
Time / min
In-p
lan
e e
lect
rica
l re
sist
ance
/ Ω
x 1
06
0
50
100
150
200
250
300
350
Tem
pe
ratu
re /
ºC
Resistance
Temperature
3
4
5
6
7
8
9
80 85 90 95 100Time / min
Re
sist
ance
/
kΩ
275
285
295
305
315
325
Tem
pe
ratu
re /
ºC
K-βAl2O3
Pt-DLC
Au
R
K-βAl2O3
Pt-DLC
Au
R
Page 116
Chapter 2
108
It can be observed that at the beginning of the TPS experiment, the deposited
Pt-DLC catalyst film showed a high value of electrical resistance (42 x 106 Ω). It
seems to indicate that the as-prepared DLC film was on its most sp3-hybridized form
(similar to diamond). Above 200 ºC, the electrical resistance of the catalyst film
sharply decreased and finally stabilized at 3.5 kΩ at 315 ºC (zoom in Figure 2.2) due
to the transition toward a structure more similar to that of graphite (sp2-hybridized), as
will be confirmed below. These results are consistent with those obtained in previous
works [33-35] with DLC films prepared by filtered cathodic vacuum arc, in which a
transition temperature from a highly tetrahedral structure to that of a graphitic nature
took place abruptly around 200 ºC. The final achieved electrical resistance of the Pt-
DLC film (3.5 kΩ) was enough to electrochemically promote the active Pt particles as
will be also demonstrated later. This electrical resistance value was almost constant
during all the catalytic EPOC experiment, which also shows the stability of the
deposited Pt-DLC film on its most sp2-hybridized structure.
A microstructural characterization by HAADF-STEM in cross-sections of the
Pt and the Pt-DLC films, the latter both before and after the TPS treatment, is shown
in Figure 2.3. The pure Pt film employed in the first chapter (Figure 2.3a) shows a
highly dense crystalline platinum with a grain size of the order of the film thickness
(100 nm). The HAADF images of the composite Pt-DLC film before and after the
TPS (Figures 2.3b and 2.3c, respectively) indicate the formation of a nanocomposite
multilayer with different metal contents. Multilayer growth was due to the use of a
high energetic deposition technique such as a filter cathodic arc, where the high
kinetic energy of carbon and platinum ions sculpted layers with high and low metal
contents. HAADF images were characterized by presenting Z contrast, in which
brighter areas were composed by heavier atoms. On the other hand, darker regions
were composed by lighter materials. BF imaging contrast was reversed with respect to
HAADF. Therefore, the difference in contrast between the layers would correspond to
differences in the composition of the Pt-DLC thin film. The dark zones corresponded
to carbon-rich layers and the light ones to platinum-rich ones. It is interesting to note
that the same layered structure was maintained after the TPS treatment (Figure 2.3c),
Page 117
Electrochemical promotion of Pt nanoparticles dispersed in a carbon matrix for methanol
conversion
109
which would indicate that no platinum segregation or diffusion occurred as a result of
the thermal stabilization process. This fact could indirectly indicate that the increase in
the conductivity was mostly due to a modification of the carbon matrix.
Figure 2.3. HAADF-STEM images of a pure Pt film (a), as-deposited Pt-DLC film (b) and Pt-
DLC film after TPS (c).
a)
b)
c)Fig. 2.4. g,h
20 nm
20 nm
50 nm
Fig. 2.4. e,f
Fig. 2.4. c,d
Fig. 2.4. a,b
Page 118
Chapter 2
110
In order to gain in-depth structural characterization information of the
composite sample after TPS, Figure 2.4 shows both BF and HAADF images of the
sample taken in the regions indicated in Figure 2.3c.
Figure 2.4. BF-STEM images of bottom (a), middle (c,e), and top (g) of Pt-DLC film after
TPS, along with their corresponding Z-contrast HAADF-STEM images (b,d,f,h).
The morphology of the sample would correspond to that of a network of small
Pt nanoparticles of around 3 nm (bright in HAADF) surrounding carbon-rich areas
g)
e)
h)
a)
c) d)
f)
b)
10 nm
10 nm
10 nm
10 nm
f)
g)
e)
h)
a)
c) d)
f)
b)
10 nm
10 nm
10 nm
10 nm
f)
Page 119
Electrochemical promotion of Pt nanoparticles dispersed in a carbon matrix for methanol
conversion
111
(dark in HAADF). Assuming this Pt particle size, a number of Pt active sites of 5.74 x
10-8
mol has been estimated from the total number of Pt deposited in the sample (1.44
x 10-7
mol) and hence an approximate dispersion value of around 40 %. The size of the
carbon-rich areas varied along the growth direction of the film.
As shown in Figures 2.4a and 2.4b, a network of small Pt particles embedded
in a DLC matrix started to grow quickly on the top of a very thin amorphous carbon
layer which firstly nucleated on the substrate. However, after 30 nm, a thin 10 nm
platinum-poor layer was formed due to formation of stronger carbon bonds, which
slowed down the incorporation of larger amounts of platinum. It is worth noting, as
can be observed in Figure 2.4e and 2.4f (with higher magnification), that a similar
carbon rich-layer was formed again after some deposition time. The intermediate part
of the sample was mostly composed of a continuous network of extremely small
platinum grains, 1-3 nm, distributed in the hydrogen-free carbon matrix, which
appears to percolate (Figures 2.4c and 2.4d). TEM micrographs from the top of the
TPS-treated sample are shown in Figures 2.4g and 2.4h. It is particularly interesting to
note that the size of the platinum grains at the top of the samples was of the same
order of magnitude as that of platinum grains at the bottom, which would imply that
there was no agglomeration of platinum grains or metal surface segregation. This
underscores the usability of the cathodic arc deposition method in synthesizing
optimum active platinum sites for catalytic purposes.
Given that the TPS treatment did not result in any significant microstructural
change of the nanocomposite Pt-DLC, EEL spectra of carbon were collected in the
different areas shown in the micrographs of Figure 2.4. Figure 2.5 shows the C K-edge
spectra of the samples before and after TPS treatment (denoted as As-dep and TPS,
respectively) along the standard ones corresponding to amorphous carbon and Highly
Ordered Pyrolytic Graphite (HOPG carbon).
Page 120
Chapter 2
112
Figure 2.5. EEL spectra (1-4) acquired from top to bottom for the same regions shown in
Figure 4 (a-b,c-d,e-f,g-h) before and after TPS. Graphitized carbon and amorphous carbon are
shown for comparison purposes.
These spectra indicated a graphitization of the carbon-rich matrix all along the
film, as it can be observed by the increase of the π* peak in those layers after TPS
treatment. Therefore, the previously observed decrease in the surface electrical
resistance of the film after the TPS can be related to the conversion of sp3-bonds in the
amorphous carbon matrix to sp2 hybridization with higher conductivity.
2.3.2. Electrochemical promotion experiments
In order to study the EPOC phenomena of the Pt-DLC catalyst film in the
hydrogen production from methanol, several potentiostatic experiments were carried
out under different reaction atmospheres (POM and SRM) and different reaction
temperatures.
280 290 300 310 320
0.0
0.5
1.0
1.5
2.0
2.5
3.0
Graphitized carbon
Amorphous carbon
As-dep
TpS
(4)
(3)
(2)
Energy Loss (eV)
No
rma
lize
d C
ou
nts
1s to
eV(1)
Page 121
Electrochemical promotion of Pt nanoparticles dispersed in a carbon matrix for methanol
conversion
113
Figure 2.6 shows the variation of the different product formation rates (H2,
CO2, CO and H2CO) vs. time at different applied potentials and reaction temperatures
under POM conditions (CH3OH/O2 = 11 %/0.9 %, N2 balance). In agreement with
previous EPOC studies carried out with cationic electrochemical catalysts [36, 37], the
application of a high positive potential of 2 V (unpromoted state) at both the
beginning and the end of each experiment made it possible to define a clean catalyst-
working electrode film free of promoter ions and hence a reference state. The
subsequent decrease in the applied catalyst potential (VWR < +2 V) increased the
amount of positive K+ ions electrochemically transferred to the Pt catalyst-working
electrode film, according to the obtained negative currents (not shown here). It
allowed to obtain two different kinds of electrochemically promoted states: promoted
state I (VWR = +0.5 V) with an increase in the H2 and CO2 production rates (a slight
increase was also observed in CO production at 320 ºC) and promoted state II (VWR =
-0.5 V) with an increase in the H2, CO and H2CO production rates. At this point, it
should be mentioned that both promotional states were reproducible and appeared at
the three explored reaction temperatures. Moreover, in each promoted state,
reproducible optimum values of the product formation rates were attained, as typically
observed for alkali electropromotion reactions [38, 39], due to the occurrence of an
optimum promoter coverage on the catalyst surface (an excess of alkali promoter
coverage led to a poisoning effect).
The observed modifications of the catalytic activity vs. the applied potential
could be explained in terms of work function modifications as a consequence of the
back-spillover of electropositive ions from the solid electrolyte (K-βAl2O3) to the
catalyst-working electrode film (Pt-DLC) [40]. According to the current rules of
electrochemical promotion [1], the migration of K+ ions onto the catalyst-working
electrode (Pt-DLC surface) weakened the platinum chemical bond with the electron
donor adsorbates and strengthened that with electron acceptors. These donors and
acceptors have been identified as methanol and oxygen, respectively, under methanol
partial oxidation conditions [41, 42].
Page 122
Chapter 2
114
Figure 2.6. H2 (a), CO2 (b), CO (c) and H2CO (d) production rates vs. time at different applied
potentials (VWR) and temperatures. Partial oxidation conditions: CH3OH/O2 = 11 %/0.9 %.
-2.5
-2
-1.5
-1
-0.5
0
0.5
1
1.5
2
0
1
2
3
4
5
6
VW
R/
V
r CO
/
mo
l s-1
x 1
0-7
250 ºC
280 ºC
320 ºC
-2.5
-2
-1.5
-1
-0.5
0
0.5
1
1.5
2
0
1
2
3
4
5
VW
R/
V
r CO
/ m
ol
s-1x
10
-7
250 ºC
280 ºC
320 ºC
z
-2.5
-2
-1.5
-1
-0.5
0
0.5
1
1.5
2
2.5
0
0.5
1
1.5
2
2.5
3
VW
R/
V
r H
/ m
ol s
-1x
10
-7
250 ºC 280 ºC
320 ºC
-2.5
-2
-1.5
-1
-0.5
0
0.5
1
1.5
2
0
0.5
1
1.5
0 100 200 300 400 500 600 700 800 900
VW
R/
V
r H
CO
/ m
ol s
-1x
10
-7
Time / min
250 ºC
280 ºC
320 ºC
b)
c)
a)
d)
VWR
VWR
VWR
VWR
Un-promoted
state
Promoted
state I
Un-promoted
state
Promoted
state II
2
2
2
Page 123
Electrochemical promotion of Pt nanoparticles dispersed in a carbon matrix for methanol
conversion
115
As stated in the first chapter for a dense Pt film, the observed increase in H2,
CO and H2CO production rates (promoted state II) could be explained by the strong
promotional effect of K+
ions in the methanol partial oxidation mechanism, which was
in turn attributed to an increase in metal-oxygen bonds at a catalyst potential around
VWR < 0 V. Herein, this promotional effect could also be associated with the formation
of potassium carbonates and bicarbonates species on the catalyst film and allowed to
produce H2 and H2CO in a single reaction step. However, a new promotional effect
(promoted state I) appeared in the positive potential region (VWR = +0.5 V), which
increased the H2 and CO2 production rates. It should be noted that at this applied
potential, the overpotential (typically denoted as η in literature), was already negative
since the open circuit potential value (o
WRV ), was found in all experiments to be higher
than +0.5 V.
This new electro-promotional state, which was not reported before, could be
influenced by both the presence of Pt nanoparticles on the Pt-DLC catalyst film and
the formation of new promotional phases that may catalyze parallel processes. Hence,
we suggest that the formation of potassium oxides and superoxides species at VWR =
+0.5 V (promoted state I) may be also responsible for a parallel promotional effect on
the following reactions: water gas shift (with the H2O formed from the reaction
between H2 and O2) and CO oxidation reaction. It would lead to the observed increase
in the CO2 production rate instead of the CO one. This effect was probably hindered at
high temperature (320 ºC), where some CO was also produced at the promoted state I
(Figure 2.6c). In this case, it is probably that there was not enough O2 in the gas phase
to oxidize the higher amount of CO produced via the promotional effect on the partial
oxidation of methanol. Previous EPOC studies have already shown a promotional
effect of K+ ions on the water gas shift (WGS) [43] and CO oxidation [44] reactions.
In addition, the presence of this kind of K2O-derived promotional phases has been
reported in several studies with Pt/K-βAl2O3 systems [45, 46]. Previous works have
also demonstrated that the presence of smaller Pt particle sizes enhanced the formation
of K2O layers over platinum exposed to oxygen [47-49], which may explain the
Page 124
Chapter 2
116
presence of this new promotional state on Pt-DLC, not observed on the less dispersed
pure Pt film. It was also shown in other works that the addition of alkali oxides, like
K2O, to supported Pt catalysts may significantly enhance the CO oxidation and WGS
reactions [50].
Hence, under the studied reaction conditions, the application of different
potentials led to the occurrence of different kinds of promoter phases, which made it
possible to control and optimize the rate of the different possible catalytic reactions
and, hence, the formation of different products by tuning a heterogeneous catalyst. On
the other hand, it is interesting to note that both kinds of promoter phases can be
completely removed from the catalyst surface (returning the K+
ions to the solid
electrolyte) by the final application of VWR = +2 V, leading to a completely reversible
EPOC effect. This demonstrates the stability of the Pt-DLC catalyst film after the
long-term potentiostatic experiments. Regarding the influence of raising the
temperature, the production rates of H2, CO2 and CO increased, as expected from
POM kinetics [14]. However, in the case of formaldehyde (Figure 2.6d), the reaction
rate reached an optimum value at 280 ºC, due to its further oxidation to CO2 at higher
temperatures [51].
In order to quantify the magnitude of the observed EPOC effect, the rate
enhancement ratio, ρ, can be calculated at each different applied potential, through the
following equation:
ρ
(2.1)
where r and r0 are the promoted (VWR < +2 V) and unpromoted (VWR = +2 V) catalytic
reaction rates, respectively. Hence, in the case of H2 production, a maximum value of
this parameter, max,2H , of 2.5 was obtained at 280 ºC under the application of the
optimum potential, VWR = -1 V.
Figure 2.7 shows the variation of the different products reaction rates: H2, CO2
and CO vs. time at different applied potentials and temperatures under SRM
conditions (CH3OH/H2O = 4 %/4.8 %, N2 balance).
Page 125
Electrochemical promotion of Pt nanoparticles dispersed in a carbon matrix for methanol
conversion
117
Figure 2.7. H2 (a), CO2 (b) and CO (c) production rates vs. time at different applied potentials
(VWR) and temperatures. Steam reforming conditions: CH3OH/H2O = 4 %/4.8 %.
In this case, a decrease in the applied potential below +0.5 V led to an
important increase in the production rate of H2, CO2 and CO. This demonstrates that
the presence of K+ ions on the Pt-DLC catalyst film strongly promoted the methanol
reforming activity on the Pt particles. A decrease in the catalyst potential involved a
Page 126
Chapter 2
118
decrease in the catalyst work function due to the migration of positively charged
potassium ions which weakened the chemical bond between Pt and the electron-donor
adsorbates (i.e., CH3OH) and strengthened that between this metal and the electron
acceptor ones (i.e., H2O, in this case). According to the reaction mechanism reported
in previous studies [6], based on a dissociative pathway of methanol, the activation of
water molecules through electrochemical promotion (at negative potentials) would
favour the decomposition of the reaction intermediates and hence the methanol
reforming catalytic process. Moreover, it is clear that the SRM kinetics strongly
increased with the reaction temperature in agreement with previous SRM works using
Pt-based catalysts [7]. The H2 production enhancement was increased at higher
reaction temperatures whereas the maximum promotional effect, max,2H = 3.4, was
achieved at 360 ºC and VWR = -2 V.
The observed EPOC effect was reproducible at all reaction temperatures, since
the initial un-promoted values of all products reaction rates were reached again after
the final application of +2 V (after 15 h working at each temperature). This fact
demonstrates again the stability of the Pt-DLC catalyst film. The optimal promotional
rates for POM and SRM corresponded to the electrochemical supply of 1.38 x 10-6
and
5.14 x 10-7
mol K+, respectively, calculated from the integration of the current vs. time
curves. In addition, it should be noted that in both: POM (Figure 2.6) and SRM
(Figure 2.7) under optimum electropromoted conditions, the corresponding values of
turnover frequencies of methanol consumption (TOF) (8.45 and 0.51 s-1
, respectively)
were shown to be of the same order as those obtained with conventional catalysts such
as as Cu/ZnO- [52], Cu/CeO2- [53], PdZn- [54] and VO-based [55] catalysts under
similar reaction conditions.
However, unlike the experiments under POM conditions, a single promotional
effect was found (at VWR ≤ +0.5 V) for the SRM process. In the latter case, and
analogously to the SRM results obtained in the first chapter with a pure dense Pt
catalyst film, a unique kind of promotional phase would be formed on the Pt-DLC
catalyst surface, very likely potassium carbonates or bicarbonates species. This
Page 127
Electrochemical promotion of Pt nanoparticles dispersed in a carbon matrix for methanol
conversion
119
difference between both reaction atmospheres could be explained considering the
absence of O2 in the gas phase under SRM conditions, in contrast with POM where
both O2 and H2O were present on the gas phase. Thus, depending on both the reaction
atmosphere and the applied catalyst potential, several kinds of promoter phases, which
would affect the catalytic rates in different ways, could be formed via EPOC.
The different promotional effects, when the Pt-DLC catalyst was used for
POM and SRM processes, were characterized by cyclic voltammetry. Figure 2.8
shows the current variation vs. the applied potential of the catalyst film during a cyclic
voltammetry performed between +2 and -2 V (scan rate of 5 mV s-1
) at 360 ºC, with
two different feed compositions: CH3OH/O2 = 11 %/0.9 % (POM) and CH3OH/H2O =
4 %/4.8 % (SRM).
Figure 2.8. Current variation vs. the applied potential (VWR) of the Pt-DLC film during the
cyclic voltammetry between +2 and -2 V under POM (CH3OH/O2 = 11 %/0.9 %) and SRM
(CH3OH/O2 = 4 %/4.8 %) conditions. T = 360 ºC, scan rate = 5 mV s-1
.
-8
-6
-4
-2
0
2
4
6
8
-2.5 -2 -1.5 -1 -0.5 0 0.5 1 1.5 2 2.5
Cu
rre
nt
/ µ
A
VWR / V
SRM
POM
Page 128
Chapter 2
120
As already reported in previous EPOC studies with K-βAl2O3-based
electrolytes [46, 56], the presence of different kinds of potassium promoter species
(formation and decomposition) can be followed by this technique. The cathodic peaks
shown in this figure could be linked with the formation of promoter-derived
compounds caused by the migration of K+ ions toward the Pt-DLC layer. Otherwise,
the anodic peaks could be associated to the decomposition of these formed species and
the spillover of the promoter ions back to the solid electrolyte [57, 58]. Hence, the
occurrence of different cathodic peaks (at least two) and the other two well defined
anodic peaks under POM conditions revealed the formation and decomposition of
different kinds of promoter phases under this reaction atmosphere, which could
explain the presence of different promoted states in Figure 2.6. On the other hand,
under SRM conditions just one cathodic peak with the corresponding anodic one can
be clearly observed. It could be attributed to the formation and decomposition of a
unique kind of promoter phase under this reaction atmosphere, in agreement with the
observed promotional results in Figure 2.7.
It should also be mentioned that the maximum electrical power, calculated
from the integration of the current vs VWR curve during the cathodic scan, used for
activating the catalyst was very low, i.e., 3.78 x 10-6
and 3.85 x 10-6
W cm-2
under
POM and SRM conditions, respectively. Hence, this would preserve the operation life
of the electrochemical cell and lead to a very low electrical energy consumption, i.e.,
the amount of electrochemically transferred promoter ions is very low.
2.3.3. Comparison between Pt-DLC and Pt
Finally, the catalytic performance obtained under SRM conditions with the
novel Pt-DLC catalyst electrode film was compared with that obtained in the first
chapter with the pure Pt film prepared according to the procedure described in section
1.2.1. Both catalysts were synthesized by cathodic arc deposition and resulted in
similar geometric properties (2.01 cm2, thickness of 120-150 nm). Figure 2.9 shows
the steady-state H2 production rate under SRM conditions (CH3OH/H2O = 4 %/4.8 %,
N2 balance) vs. the applied potential for both electrochemical catalysts at two different
reaction temperatures (320 and 360 ºC).
Page 129
Electrochemical promotion of Pt nanoparticles dispersed in a carbon matrix for methanol
conversion
121
Figure 2.9. H2 production rate vs. applied catalyst potential (VWR) and maximum H2 rate
enhancement ratios (ρH2,max) at different reaction temperatures with the Pt and Pt-DLC films.
Steam reforming conditions: CH3OH/H2O = 4 %/4.8 %.
A much higher catalytic activity of the Pt-DLC catalyst film than that of the
pure dense Pt film was observed. This fact could be related to the higher dispersion of
the former catalyst film. In addition, a much stronger promotional effect of the Pt-
DLC catalyst film at each reaction temperature was also noted. In contrast, the
maximum rate enhancement ratio values ( max,2H ) for both electrochemical catalysts
were very similar at each temperature, due to the higher unpromoted catalytic rate of
the Pt-DLC film.
Figure 2.10 compares the current vs. time curves obtained with the two
catalyst-working electrodes at a given applied catalyst potential e.g., VWR = -0.5 V, for
the SRM reaction at 360 ºC.
0
1
2
3
4
5
6
-2.5 -2 -1.5 -1 -0.5 0 0.5 1 1.5 2 2.5
r H
/ m
ol s
-1x
10-8
VWR / V
Pt 320 ºC (0.46 mg Pt)
Pt 360 ºC (0.46 mg Pt)
Pt/DLC 320 ºC (0.03 mg Pt)
Pt/DLC 360 ºC (0.03 mg Pt)
2
2 ρH ,max = 3.37
ρH ,max = 3.54
2
ρH ,max = 1.86
2
ρH ,max = 1.53
2
Page 130
Chapter 2
122
Figure 2.10. Current vs. time curves during the potentiostatic imposition of -0.5 V with the Pt
(a) and Pt-DLC (b) films. Steam reforming conditions: CH3OH/H2O = 4 %/4.8 %, 360 ºC.
Lower current values of the Pt-DLC catalyst film vs. Pt were found, which can
be explained by both the higher planar electrical resistance of the Pt-DLC catalyst
electrode (3.5 kΩ) vs. the pure Pt film (2 Ω) and the negligible ionic conductivity of
the DLC matrix. This led to lower rates of K+ ions supply to the catalyst film, since the
obtained current for Pt-DLC approximates more slowly to a negligible value. In fact
after 60 min of polarization, the Pt-DLC catalyst film was still increasing its potassium
coverage, in contrast with the pure Pt film which achieved a negligible current value
after the first minute of polarization, as commonly observed in other EPOC studies
[44, 56].
a)
b)
-0.10
-0.08
-0.06
-0.04
-0.02
0.00
Cu
rre
nt
/ m
APt
-0.01
-0.008
-0.006
-0.004
-0.002
0 5 10 15 20 25 30 35 40 45 50 55 60
Cu
rre
nt
/ m
A
Time / min
Pt-DLC
Page 131
Electrochemical promotion of Pt nanoparticles dispersed in a carbon matrix for methanol
conversion
123
Thus, it seems clear that the higher electrical resistance and the absence of
ionic conductivity of the Pt-DLC catalyst film made the electrochemical supply of K+
ions to the Pt particles more difficult. However, the obtained EPOC catalytic results
showed higher electropromoted H2 production rates on the Pt-DLC catalyst film,
which presented much lower metal loading (0.014 mg Pt cm-2
) vs. the pure dense Pt
film (0.23 mg Pt cm-2
), thus demonstrating the interest of this novel kind of catalyst -
electrode materials. Finally it should also be noted that the electrical energy
consumption in EPOC systems is very low and is typically negligible vs. other
operational costs [1, 59]. This is because a low amount of electrical energy is required,
just to supply a small amount of promoter to the catalyst surface. For instance, the
overall supplied integral energy in the whole polarization at -0.5 V during 60 min
(Figure 2.10) was 3.11 x 10-3
J cm-2
for the Pt-DLC film and 6.32 x 10-3
J cm-2
for the
Pt film, which are a very low values.
2.4. CONCLUSIONS
The following conclusions could be drawn from this study:
- The technique of the cathodic arc deposition allowed to prepare a novel
catalyst film based on Pt nanoparticles (of around 3 nm) dispersed in a diamond-like
carbon (DLC) matrix with suitable properties for catalytic and electrocatalytic
purposes.
- The thermal treatment of the Pt-DLC catalyst film made it possible to
achieve a suitable surface electrical conductivity due to the transition of the sp3-
hybridized carbon form into a more graphitic structure (sp2-hybridized), as verified by
cross-section STEM and EELS characterization.
- The Pt-DLC catalyst film was successfully electrochemically promoted (by
potassium ions) for the hydrogen production from methanol via partial oxidation and
steam reforming reactions. All the observed electropromotional effects were
reproducible and reversible, the Pt-DLC catalyst film showing a good stability. Hence,
Page 132
Chapter 2
124
it was demonstrated the possibility of electrochemically promoting catalyst particles
dispersed on an electronic non-ionic conductor support.
- Two different electropromotional effects were observed under methanol
partial oxidation conditions, which were attributed to the formation of different kinds
of promoter phases depending on the applied potential.
- The catalytic activity of the Pt-DLC film was higher than that observed for
the pure Pt film prepared by the same technique due to the lower Pt particle size in the
former. This kind of catalyst films based on metal nanoparticles dispersed in a DLC
material of low metal loading open new possibilities for the further development of
the EPOC effect in view of its possible practical application.
2.5. REFERENCES
[1] C.G. Vayenas, S. Bebelis, C. Pliangos, S. Brosda, D. Tsiplakides, Electrochemical
Activation of Catalysis: Promotion, Electrochemical Promotion and Metal-Support
Interactions, Kluwer Academic Publishers/Plenum Press, New York, 2001.
[2] S. Sundarrajan, S.I. Allakhverdiev, S. Ramakrishna, Progress and perspectives in micro
direct methanol fuel cell, International Journal of Hydrogen Energy, 37 (2012) 8765-8786.
[3] D.R. Palo, R.A. Dagle, J.D. Holladay, Methanol steam reforming for hydrogen production,
Chemical Reviews, 107 (2007) 3992-4021.
[4] R.M. Navarro, M.A. Peña, J.L.G. Fierro, Hydrogen production reactions from carbon
feedstocks: Fossil fuels and biomass, Chemical Reviews, 107 (2007) 3952-3991.
[5] S. Sá, H. Silva, L. Brandão, J.M. Sousa, A. Mendes, Catalysts for methanol steam
reforming-A review, Applied Catalysis B: Environmental, 99 (2010) 43-57.
[6] G. Jacobs, B.H. Davis, In situ DRIFTS investigation of the steam reforming of methanol
over Pt/ceria, Applied Catalysis A: General, 285 (2005) 43-49.
[7] P. Tolmacsov, A. Gazsi, F. Solymosi, Decomposition and reforming of methanol on Pt
metals supported by carbon Norit, Applied Catalysis A: General, 362 (2009) 58-61.
[8] S.H. Ahn, O.J. Kwon, I. Choi, J.J. Kim, Synergetic effect of combined use of Cu-ZnO-
Al2O3 and Pt-Al2O3 for the steam reforming of methanol, Catalysis Communications, 10
(2009) 2018-2022.
[9] G. Kolb, S. Keller, S. Pecov, H. Pennemann, R. Zapf, Development of micro-structured
catalytic wall reactors for hydrogen production by methanol steam reforming over novel
Page 133
Electrochemical promotion of Pt nanoparticles dispersed in a carbon matrix for methanol
conversion
125
Pt/In2O 3/Al2O3 catalysts, Chemical Engineering Transactions, 2011, pp. 133-138,
DOI:10.3303/cet1124023.
[10] A. Peremans, F. Maseri, J. Darville, J.M. Gilles, Interaction of methanol with a
polycrystalline platinum surface studied by infrared reflection absorption spectroscopy,
Surface Science, 227 (1990) 73-78.
[11] M.P. Zum Mallen, L.D. Schmidt, Oxidation of methanol over polycrystalline Rh and Pt:
Rates, OH desorption, and model, Journal of Catalysis, 161 (1996) 230-246.
[12] B.E. Traxel, K.L. Hohn, Partial oxidation of methanol at millisecond contact times,
Applied Catalysis A: General, 244 (2003) 129-140.
[13] R. Ubago-Pérez, F. Carrasco-Marín, C. Moreno-Castilla, Methanol partial oxidation on
carbon-supported Pt and Pd catalysts, Catalysis Today, 123 (2007) 158-163.
[14] A. Wan, C.t. Yeh, Ignition of methanol partial oxidation over supported platinum catalyst,
Catalysis Today, 129 (2007) 293-296.
[15] C. Xia, M. Hugentobler, L. Yongdan, C. Comninellis, W. Harbich, Quantifying
electrochemical promotion of induced bipolar Pt particles supported on YSZ, Electrochemistry
Communications, 12 (2010) 1551-1554.
[16] M. Marwood, C.G. Vayenas, Electrochemical promotion of a dispersed platinum catalyst,
Journal of Catalysis, 178 (1998) 429-440.
[17] S.G. Neophytides, D. Tsiplakides, P. Stonehart, M.M. Jaksic, C.G. Vayenas,
Electrochemical enhancement of a catalytic reaction in aqueous solution, Nature, 370 (1994)
45-47.
[18] L. Ploense, M. Salazar, B. Gurau, E.S. Smotkin, Proton spillover promoted isomerization
of n-butylenes on Pd-black cathodes/nafion 117 [8], Journal of the American Chemical
Society, 119 (1997) 11550-11551.
[19] V. Jiménez, C. Jiménez-Borja, P. Sánchez, A. Romero, E.I. Papaioannou, D. Theleritis, S.
Souentie, S. Brosda, J.L. Valverde, Electrochemical promotion of the co2 hydrogenation
reaction on composite ni or ru impregnated carbon nanofiber catalyst-electrodes deposited on
YSZ, Applied Catalysis B: Environmental, 107 (2011) 210-220.
[20] D. Poulidi, M.E. Rivas, B. Zydorczak, Z. Wu, K. Li, I.S. Metcalfe, Electrochemical
promotion of a Pt catalyst supported on La 0.6Sr 0.4Co 0.2Fe 0.8O 3 - δ hollow fibre
membranes, Solid State Ionics, 225 (2012) 382-385.
Page 134
Chapter 2
126
[21] A. Kambolis, L. Lizarraga, M.N. Tsampas, L. Burel, M. Rieu, J.P. Viricelle, P. Vernoux,
Electrochemical promotion of catalysis with highly dispersed Pt nanoparticles,
Electrochemistry Communications, 19 (2012) 5-8.
[22] A. Grill, Diamond-like carbon: State of the art, Diamond and Related Materials, 8 (1999)
428-434.
[23] J. Robertson, AMORPHOUS CARBON, Advances in Physics, 35 (1986) 317-374.
[24] N. Menegazzo, C. Jin, R.J. Narayan, B. Mizaikoff, Compositional and electrochemical
characterization of noble metal-diamondlike carbon nanocomposite thin films, Langmuir, 23
(2007) 6812-6818.
[25] J.M. Ting, H. Lee, DLC composite thin films by sputter deposition, Diamond and Related
Materials, 11 (2002) 1119-1123.
[26] G.C. Fiaccabrino, X.M. Tang, N. Skinner, N.F. De Rooij, M. Koudelka-Hep,
Interdigitated microelectrode arrays based on sputtered carbon thin-films, Sensors and
Actuators, B: Chemical, 35 (1996) 247-254.
[27] G.Y. Chen, J.S. Chen, Z. Sun, Y.J. Li, S.P. Lau, B.K. Tay, J.W. Chai, Field emission
properties and surface structure of nickel containing amorphous carbon, Applied Surface
Science, 180 (2001) 185-190.
[28] H. Lee, J.M. Ting, Platinum-containing diamond-like carbon thin films, Journal of the
American Ceramic Society, 87 (2004) 2183-2186.
[29] Y.V. Pleskov, Y.E. Evstefeeva, A.M. Baranov, Threshold effect of admixtures of platinum
on the electrochemical activity of amorphous diamond-like carbon thin films, Diamond and
Related Materials, 11 (2002) 1518-1522.
[30] K.I. Schiffmann, M. Fryda, G. Goerigk, R. Lauer, P. Hinze, A. Bulack, Sizes and
distances of metal clusters in Au-, Pt-, W- and Fe-containing diamond-like carbon hard
coatings: a comparative study by small angle X-ray scattering, wide angle X-ray diffraction,
transmission electron microscopy and scanning tunneling microscopy, Thin Solid Films, 347
(1999) 60-71.
[31] A. Anders, Cathodic Arcs: From Fractal Spots to Energetic Condensation. Springer Series
on Atomic, Optical, and Plasma Physics, Springer-Verlag, New York, 2008.
[32] A. Anders, I.G. Brown, R.A. MacGill, M.R. Dickinson, 'Triggerless' triggering of vacuum
arcs, Journal of Physics D: Applied Physics, 31 (1998) 584-587.
[33] S.R.P. Silva, X. Shi, B.K. Tay, H.S. Tan, H.J. Scheibe, M. Chhowalla, W.I. Milne, The
structure of tetrahedral amorphous carbon thin films, Thin Solid Films, 290-291 (1996) 317-
322.
Page 135
Electrochemical promotion of Pt nanoparticles dispersed in a carbon matrix for methanol
conversion
127
[34] M. Chhowalla, J. Robertson, C.W. Chen, S.R.P. Silva, C.A. Davis, G.A.J. Amaratunga,
W.I. Milne, Influence of ion energy and substrate temperature on the optical and electronic
properties of tetrahedral amorphous carbon (ta-C) films, Journal of Applied Physics, 81
(1997) 139-145.
[35] J.L. Endrino, D. Horwat, R. Gago, J. Andersson, Y.S. Liu, J. Guo, A. Anders, Electronic
structure and conductivity of nanocomposite metal (Au, Ag, Cu, Mo)-containing amorphous
carbon films, Solid State Sciences, 11 (2009) 1742-1746.
[36] A. de Lucas-Consuegra, F. Dorado, J.L. Valverde, R. Karoum, P. Vernoux,
Electrochemical activation of Pt catalyst by potassium for low temperature CO deep oxidation,
Catalysis Communications, 9 (2008) 17-20.
[37] P. Vernoux, F. Gaillard, C. Lopez, E. Siebert, Coupling catalysis to electrochemistry: A
solution to selective reduction of nitrogen oxides in lean-burn engine exhausts?, Journal of
Catalysis, 217 (2003) 203-208.
[38] F.J. Williams, M.S. Tikhov, A. Palermo, N. Macleod, R.M. Lambert, Electrochemical
promotion of rhodium-catalyzed NO reduction by CO and by propene in the presence of
oxygen, Journal of Physical Chemistry B, 105 (2002) 2800-2808.
[39] F. Dorado, A. de Lucas-Consuegra, P. Vernoux, J.L. Valverde, Electrochemical
promotion of platinum impregnated catalyst for the selective catalytic reduction of NO by
propene in presence of oxygen, Applied Catalysis B: Environmental, 73 (2007) 42-50.
[40] C.G. Vayenas, S. Bebelis, S. Ladas, Dependence of catalytic rates on catalyst work
function, Nature, 343 (1990) 625-627.
[41] C.G. Vayenas, S. Neophytides, Non-faradaic electrochemical modification of catalytic
activity III. The case of methanol oxidation on Pt, Journal of Catalysis, 127 (1991) 645-664.
[42] C.A. Cavalca, G. Larsen, C.G. Vayenas, G.L. Haller, Electrochemical modification of
CH3OH oxidation selectivity and activity on a Pt single-pellet catalytic reactor, Journal of
Physical Chemistry, 97 (1993) 6115-6119.
[43] A. De Lucas-Consuegra, A. Caravaca, J. González-Cobos, J.L. Valverde, F. Dorado,
Electrochemical activation of a non noble metal catalyst for the water-gas shift reaction,
Catalysis Communications, 15 (2011) 6-9.
[44] A. de Lucas-Consuegra, A. Princivalle, A. Caravaca, F. Dorado, C. Guizard, J.L.
Valverde, P. Vernoux, Preferential CO oxidation in hydrogen-rich stream over an
electrochemically promoted Pt catalyst, Applied Catalysis B: Environmental, 94 (2010) 281-
287.
Page 136
Chapter 2
128
[45] A. Palermo, A. Husain, M.S. Tikhov, R.M. Lambert, Ag-catalysed epoxidation of propene
and ethene: An investigation using electrochemical promotion of the effects of alkali, NOx, and
chlorine, Journal of Catalysis, 207 (2002) 331-340.
[46] A. de Lucas-Consuegra, F. Dorado, J.L. Valverde, R. Karoum, P. Vernoux, Low-
temperature propene combustion over Pt/K-βAl2O3 electrochemical catalyst:
Characterization, catalytic activity measurements, and investigation of the NEMCA effect,
Journal of Catalysis, 251 (2007) 474-484.
[47] E.L. Garfunkel, G.A. Somorjai, Potassium and potassium oxide monolayers on the
platinum (111) and stepped (755) crystal surfaces: A LEED, AES, and TDS study, Surface
Science, 115 (1982) 441-454.
[48] J.H. Vleeming, B.F.M. Kuster, G.B. Marin, Effect of platinum particle size and catalyst
support on the platinum catalyzed selective oxidation of carbohydrates, Catalysis Letters, 46
(1997) 187-194.
[49] T. Frelink, W. Visscher, J.A.R. van Veen, On the role of Ru and Sn as promotors of
methanol electro-oxidation over Pt, Surface Science, 335 (1995) 353-360.
[50] C.H. Lee, Y.W. Chen, Effect of Basic Additives on Pt/Al2O3 for CO and Propylene
Oxidation under Oxygen-Deficient Conditions, Industrial and Engineering Chemistry
Research, 36 (1997) 1498-1506.
[51] R.W. McCabe, D.F. McCready, Kinetics and reaction pathways of methanol oxidation on
platinum, Journal of Physical Chemistry, 90 (1986) 1428-1435.
[52] L. Alejo, R. Lago, M.A. Peña, J.L.G. Fierro, Partial oxidation of methanol to produce
hydrogen over Cu-Zn-based catalysts, Applied Catalysis A: General, 162 (1997) 281-297.
[53] S.C. Yang, W.N. Su, S.D. Lin, J. Rick, B.J. Hwang, Preparation of highly dispersed
catalytic Cu from rod-like CuO-CeO 2 mixed metal oxides: Suitable for applications in high
performance methanol steam reforming, Catalysis Science and Technology, 2 (2012) 807-812.
[54] B. Halevi, E.J. Peterson, A. Roy, A. Delariva, E. Jeroro, F. Gao, Y. Wang, J.M. Vohs, B.
Kiefer, E. Kunkes, M. Hävecker, M. Behrens, R. Schlögl, A.K. Datye, Catalytic reactivity of
face centered cubic PdZn α for the steam reforming of methanol, Journal of Catalysis, 291
(2012) 44-54.
[55] G. Deo, I.E. Wachs, Reactivity of Supported Vanadium Oxide Catalysts: The Partial
Oxidation of Methanol, Journal of Catalysis, 146 (1994) 323-334.
[56] A. de Lucas-Consuegra, Á. Caravaca, P. Sánchez, F. Dorado, J.L. Valverde, A new
improvement of catalysis by solid-state electrochemistry: An electrochemically assisted NOx
storage/reduction catalyst, Journal of Catalysis, 259 (2008) 54-65.
Page 137
Electrochemical promotion of Pt nanoparticles dispersed in a carbon matrix for methanol
conversion
129
[57] P. Vernoux, F. Gaillard, C. Lopez, E. Siebert, In-situ electrochemical control of the
catalytic activity of platinum for the propene oxidation, Solid State Ionics, 175 (2004) 609-613.
[58] N. Kotsionopoulos, S. Bebelis, Electrochemical characterization of the Pt/β″-Al 2O3
system under conditions of in situ electrochemical modification of catalytic activity for
propane combustion, Journal of Applied Electrochemistry, 40 (2010) 1883-1891.
[59] P. Vernoux, L. Lizarraga, M.N. Tsampas, F.M. Sapountzi, A. De Lucas-Consuegra, J.L.
Valverde, S. Souentie, C.G. Vayenas, D. Tsiplakides, S. Balomenou, E.A. Baranova, Ionically
conducting ceramics as active catalyst supports, Chemical Reviews, 113 (2013) 8192-8260.
Page 139
131
Chapter 3
ELECTROCHEMICAL ACTIVATION OF Au
NANOPARTICLES DISPERSED IN YSZ FOR METHANOL
PARTIAL OXIDATION:
ELECTROCHEMICAL PROMOTION OF A NON-CONDUCTIVE
CATALYST FILM
An electrochemical catalyst based on Au nanoparticles dispersed in a Yttria-
Stabilized Zirconia (YSZ) matrix has been deposited by reactive co-sputtering of
Zirconium-Yttrium and Au targets on a K-βAl2O3 solid electrolyte. The Au/YSZ
catalyst film thus obtained has shown to be active in the partial oxidation of methanol
with a high selectivity toward methyl formate production. This configuration allowed
to decrease the amount of metal used in the solid electrolyte cell, and activate a highly
dispersed Au catalyst via electrochemical promotion (EPOC), by in-situ controlling
and optimizing the supplied amount of K+ ions. A number of experiments have
confirmed that the observed electropromotional effect did not depend on the rate of K+
supply or on the operation mode (galvanostatic or potentiostatic) and only depended
on the promoter coverage. The stability of the Au nanoparticles under the explored
conditions has also been verified.
-1
0
1
2
3
0
1
2
3
4
5
0 25 50 75 100 125 150 175
VW
R/
V
r H/
mo
l s-1
x 1
0-8
Time / min
0 2 41 3K+ supply / mol K+ x 10-7
VWR = 2 VVWR = 2 V I = 0 AI = -10 µA
ρH
2
0
3
6
9
12
2
ΔV < 0Au-YSZ
K+ K+ K+
e-
e-
K-βAl2O3
Au
Δr
CH3OH,
O2
H2, CO2,
HCOOCH3, H2O
5.9 % CH3OH
0.43 % O2
280 ºC
Optimum
Ag current
colector
Page 140
Chapter 3
132
3.1. INTRODUCTION
Alkali promoters play a key role in heterogeneous catalysis and are necessary
for the development of commercial catalysts for industrial applications [1, 2]. They are
commonly employed to modify the activity and selectivity of metal catalysts. In this
sense, the electrochemical promotion of catalysis (EPOC) is a straightforward
technique to study the effect of a certain electronic promoter on a reaction rate [3]. In
the previous chapters, Pt-based catalysts were observed to be promoted by alkali (K+)
ions in H2 production reactions from methanol via EPOC, i.e., through the
electrochemical pumping of these promoter ions from a solid electrolyte support (K-
βAl2O3). Hence, while the classical alkali promotion is carried out by adding a specific
amount of a promoter during the preparation step of the catalyst [1], in the case of the
electrochemical promotion, the electrically induced back-spillover of the promoter
species enables the in-situ control and enhancement of the performance of the catalyst
film during the reaction step [4].
In the present chapter, the alkali electrochemical promotion has been applied
on a gold-based catalyst for the methanol partial oxidation, which represented a great
challenge since Au is not among the most typical catalysts. In fact, counter and
reference electrodes made of gold have been selected for the EPOC experiments
herein performed given their proved inertness under the studied reaction conditions,
which is highly favoured by the deposition method employed in this case (calcination
of an Au organometallic paste at high temperature). Only a few works can be found in
literature related to electrochemical promotion with Au catalysts: in solid oxide fuel
cell configuration for the oxidation of CO [5] and CH4 [6] with O2-
promoter ions, in
the liquid-phase SO2 oxidation with molten V2O5-K2S2O7 catalyst [7] and in the
preferential oxidation of CO in a low temperature Proton Exchange Membrane (PEM)
fuel cell reactor promoted by H+ ions and O2 crossover through the membrane [8].
Certainly, until a few decades ago, gold has been generally considered
catalytically inactive due to its difficulty to both adsorb and dissociate molecular
oxygen [9] and to remain in a high dispersion state because of its low melting point as
compared to other noble metals such as Pt or Pd [10, 11]. However, several studies
Page 141
Electrochemical activation of Au nanoparticles dispersed in YSZ for methanol partial
oxidation
133
have shown good catalytic properties of unsupported nanoporous gold prepared by
leaching Ag from an Au-Ag alloy [12, 13], gold nanoparticles supported on highly
porous carbon materials [14] and, especially, on certain metal oxides (e.g. TiO2, CeO2,
Fe2O3, ZnO, or ZrO2) [10, 11, 15]. Numerous applications of gold-based catalysts in
the field of environmental catalysis have been recently reviewed [10]. In particular,
gold catalysts have demonstrated high activity in CO oxidation [11, 12, 16-19] even at
temperatures below 0 ºC [11, 12], which is of special interest in view of selective CO
sensing [16], CO removal from air at room temperature [11, 12], and preferential
oxidation of CO in H2 rich streams [11, 17]. Gold nanoparticles supported on oxide
supports have also been tested in other catalytic reactions such as CO2 hydrogenation
[20], NOx reduction [21], steam reforming of methanol [22] and ethanol [23], water
gas shift [22], and oxidation of methane [24], alkenes [25], alcohols both in liquid [26]
and in gas phase [19, 23, 27-33] and many other organic compounds [34].
In the case of the methanol partial oxidation processes [13, 19, 28-33], it
should be noted the complexity of the reaction pathways that may take place. A large
number of side reactions and intermediate species are generally involved, making
possible to obtain a wide variety of compounds. Along with the main oxidation
products, i.e., H2, CO2, CO and H2O, not only formaldehyde (H2CO) can be co-
produced [32, 33], as in the previous chapters, but also CH4 [29-32], dimethyl ether
(CH3OCH3) [33], dimethoxymethane (CH3O)2CH2 [33] or methyl formate HCOOCH3
[30-33]. In this sense, Au-based catalysts may acquire prominence against Pt or Pd,
due to the capability of Au to reach a very high selectivity in the methanol partial
oxidation toward some of these products of well-known interest in chemical
manufacturing, such as formaldehyde [19] and methyl formate [13]. The influence of
the gold particle size and the oxide support on the catalyst performance for the
methanol oxidation has been widely studied [19, 28-33] as well as the promoting
effect of a second active metallic component [28, 31, 32].
In this work, we report for the first time the electrochemical activation of Au
nanoparticles dispersed in a YSZ (Yttria-Stabilized Zirconia) matrix with
Page 142
Chapter 3
134
electropositive alkali ions. This novel catalyst film (Au-YSZ) has been deposited on a
K-βAl2O3 solid electrolyte by sputter deposition and electrochemically promoted by
controlling and optimizing the K+ promoter supply. The proposed configuration
allowed both to electrochemically activate highly dispersed Au nanoparticles and to
decrease the amount of metal used in the solid electrolyte cell. The Au-YSZ/K-βAl2O3
electrochemical catalyst has been characterized and tested under methanol partial
oxidation conditions (POM), also checking its reproducibility and stability.
3.2. EXPERIMENTAL
3.2.1. Preparation of the electrochemical catalyst
The electrochemical catalyst consisted of a thin continuous gold/yttria-
stabilized zirconia (Au-YSZ) film (geometric area of 3.14 cm2) composed of fine gold
nanoparticles dispersed in a YSZ matrix, which was deposited by magnetron
sputtering on a 20-mm-diameter, 1-mm-thick K-βAl2O3 (Ionotec) disc. Firstly, inert
Au counter (CE) and reference (RR) electrodes were deposited on one of the sides of
the solid electrolyte as in the previous chapters, i.e., by applying thin coatings of gold
paste (Gwent Electronic Materials C1991025D2) followed by calcination at 800 ºC for
2 h (heating ramp of 5 ºC/min). Then, the Au-YSZ nanocomposite catalyst film, which
also behaved as a working electrode (WE), was deposited by reactive co-sputtering of
Zr-Y and Au targets in an argon-oxygen gas mixture ensuring the complete oxidation
of the Zr and Y atoms in a nanocrystalline, nearly amorphous, YSZ matrix. The
deposition time was set such that the resulting Au-YSZ film thickness was around 170
nm, as measured by STEM imaging (not shown). The oxygen gas flow rate and the
current applied to the targets were fixed such that the volume fraction of gold particles
into the composite layer was 11 vol. %, which corresponded to a gold content of 7.5
at. % or a Au nanoparticles loading of 40 g Au cm-2
(2.03 x 10-7
mol Au cm-2
). More
details on the experimental procedure and principle of segregation of metallic gold
into the YSZ matrix during the formation of the film can be found in previous studies
[35]. For characterization purposes, the same layer was prepared on glass substrates.
Page 143
Electrochemical activation of Au nanoparticles dispersed in YSZ for methanol partial
oxidation
135
This catalyst preparation method was carried out in collaboration with Dr.
David Horwat from the Institute Jean Lamour at University of Lorraine (France).
The obtained Au-YSZ/K-βAl2O3/Au electrochemical catalyst was placed into a
single chamber solid electrolyte cell reactor and the three electrodes (working, counter
and reference) were connected to the potentiostat-galvanostat (PGSTAT320-N,
Metrohom Autolab). However, since the Au-YSZ nanocomposite was electrically
non-conductive from its preparation, a silver mesh (Fuel Cell Materials, AG-M40-
100) was used in this study which acted as current collector in order to carry out the
EPOC experiments. Moreover, the results reported here were obtained in both
operation modes: galvanostatic and potentiostatic, i.e. by setting the electrical current
(from 0 to -20 µA) or potential (from +2 to -2 V), respectively, according to the
procedure generally used in conventional three-electrodes electrochemical cells [3].
3.2.2. Characterization measurements
X-Ray Diffraction (XRD) was performed on the as-deposited Au-YSZ
nanocomposite layer with a AXS Brücker D8 advance diffractometer using a Cu
anode (Cu Kα radiation, λ = 1.54 Å). The thermal stability of this layer was verified
by the same technique after a long catalytic test of 12 h at 320 ºC under the studied
POM reaction conditions. Transmission Electron Microscopy (TEM) investigation and
High-Resolution TEM (HRTEM) imaging of the tested sample after all the catalytic
experiments were carried out with a JEOL ARM 200F TEM/STEM– Cold FEG (point
resolution: 0.19 nm) fitted with a GIF Quatum ER. Cyclic voltammetry (CV)
measurements were also performed prior to the EPOC experiments, in order to
evaluate the polarization of the solid electrolyte cell under POM reaction conditions.
Before the cyclic voltammetry experiment, the sample was kept at +2 V for 30 min in
order to define a reference state (Au-YSZ film clean of K+ promoter ions). Then, the
potential was cyclically varied between +2 and -2 V at a scan rate of 80 mV s-1
.
Page 144
Chapter 3
136
3.2.3. Catalytic activity measurements
The activity measurements were carried out in an experimental setup used
described in section 1.2.3. Methanol partial oxidation (POM) experiments were
performed at atmospheric pressure with an overall gas flow rate of 6 NL h-1
,
temperatures up to 300 ºC and a feed composition of CH3OH/O2 = 5.9 %/0.43 % (Ar
balance). The reaction products detected in this study were: H2, CO2, H2O, HCOOCH3
(methyl formate) and traces of dimethoxymethane and other unidentified compound
which could be dimethyl ether. Without considering the two latter compounds, the
error in the carbon atom balance did not exceed 1 %, which indicated no consistent
loss of material and no significant coking of the catalyst electrode. Blank experiments
were also performed under the same reaction conditions without any catalyst and only
with the Ag current collector in order to determine its negligible catalytic contribution.
3.3. RESULTS AND DISCUSSION
3.3.1. Characterization of the catalyst film and blank experiments
Figure 3.1 shows the X-ray diffractograms of the fresh and the aged (after 24
hours at 320°C under reaction conditions) nanocomposite layer deposited on glass.
Figure 3.1. XRD diffractograms of the Au-YSZ nanocomposite layer as deposited and after
catalytic test under methanol partial oxidation conditions (CH3OH/O2 = 5.9 %/0.43 %, 320 ºC).
30 40 50 60 70 80
22
0-A
u
20
0-A
u
After 320°C
Inte
nsity (
arb
. u
nits)
2 (Degree)
As deposited
11
1-A
u
Inte
nsi
ty /
a.u
.
2θ / Degree
Page 145
Electrochemical activation of Au nanoparticles dispersed in YSZ for methanol partial
oxidation
137
Both diffractogramms exhibit the characteristic diffraction peaks (111), (200)
and (220) at 2θ = 38.2º, 44.4º and 64.5º, respectively, corresponding to nanocrystalline
gold (JCPDS, 01-1172). Nevertheless, the diffracted intensity was slightly increased
after exposure to reaction conditions. The average size of the nanoparticles was
estimated from the width of the (111) diffraction peak using the Scherrer formula [36].
An average particle size of 3.3 nm was obtained in the fresh state, and slightly rose up
to 4.3 nm after exposure to reaction conditions, which indicated a minor diffusion of
the gold atoms and a good thermal stability of the nanocomposite layer. No diffraction
peaks corresponding to gold oxides (AuO or Au2O3) were detected. No diffraction
lines of Yttria-Stabilized Zirconia (YSZ) were identified in the X-ray diffractogramms
due to the ultrafine grained, nearly amorphous, nature of the YSZ matrix, which was
confirmed by HRTEM (not shown here). The XRD signal of YSZ could only be
detected after long time-annealing at 500°C, which is far above the temperatures
experienced by the samples in this study. Analyses of the films deposited on the K-
Al2O3 led to similar conclusions, therefore indicating a limited influence of the
substrate on the thermal stability of the nanocomposite layer.
Further details of the aged gold particles after exposure to reactions conditions
were obtained using TEM and HRTEM. Figure 3.2 shows bright field images of
selected zones in the Au-YSZ catalyst film. Figures 3.2a and 3.2b show extended and
local views of the typical distribution of gold nanoparticles in the core of the coatings
and at the surface exposed to the reaction mixture, respectively. The YSZ matrix
appears in clear while dark zones correspond to individual Au nanoparticles dispersed
in it. The particles were round shaped with a typical size distribution ranging between
2 and 10 nm. Nevertheless, it must be pointed out that thermally assisted diffusion
occurred upon exposure to the electron beam during experiments, which triggered a
slight increase of the particle diameter. This way, the size of the particles was slightly
over-estimated, being the measurements in accordance with the XRD data. This fact
would demonstrate the good stability of the gold nanoparticles after exposure to
reaction conditions.
Page 146
Chapter 3
138
Figure 3.2. Bright field micrographs of Au-YSZ nanocomposite film after catalytic tests under
methanol partial oxidation conditions (CH3OH/O2 = 5.9 %/0.43 %, 320 ºC): Typical
microstructure in the core through the film thickness and at the film surface (a, b) and at the
substrate/film interface (c). HRTEM micrograph of a selected region in the same sample shows
the atom scale arrangement of the nanoparticles (d).
Larger particles, with diameters ranging between 15 and 50 nm, but mostly
located at the substrate/film interface were also found (Figure 3.2c). The presence of
larger size gold crystallites after catalytic tests was also observed in previous studies
of methanol oxidation over supported gold catalysts due to aggregation of smaller Au
particles [19, 30, 33]. Atom scale resolution HRTEM and Fourier filter transform
c)
a) b)
d)
Page 147
Electrochemical activation of Au nanoparticles dispersed in YSZ for methanol partial
oxidation
139
(FFT) of the images (not shown) confirmed the presence of particles consisting of face
centered cubic (FCC) metallic assembly of gold atoms. For instance, the (111) atomic
arrangement of FCC gold was observed for particles in Figure 3.2d.
Figure 3.3 shows the steady-state variation at different reaction temperatures
of the production rates (hydrogen, carbon dioxide and methyl formate) obtained with
the Au-YSZ catalyst film as well as the blank results in the partial oxidation of
methanol (feed composition of CH3OH/O2 = 5.9 %/0.43 %, Ar balance). In the case of
the electrocatalytic system (current collector + Au-YSZ catalyst), the catalytic activity
was measured at each temperature under open circuit conditions, i.e. without applying
any electric current or potential, after the imposition of a catalyst potential (VWR) of +2
V for 30 minutes. In agreement with the previous chapters and other studies with K-
βAl2O3-based electrolytes [37, 38], the purpose of this preliminary positive
polarization was to remove from the Au-YSZ catalyst film the K+ ions that could have
thermally migrated from the solid electrolyte. This thereby defined a reference state
(unpromoted state).
Both the reactor blank and the silver mesh showed a negligible activity under
the studied reaction conditions as compared to that of the electrochemical catalyst. On
the other hand, it was confirmed that the Au-YSZ catalyst film presented certain
catalytic activity in the methanol partial oxidation under unpromoted conditions.
While large Au particles and bulk gold are generally considered to be inert, catalysts
based on gold nanoparticles supported on different metal oxides have been reported by
several authors to be catalytically active in this reaction [19, 29-33]. In fact, the
smaller the Au particle size, the higher the methanol partial oxidation rate as well as
the H2 selectivity vs. CO or CH4 [30-32]. In particular, gold nanoparticles dispersed on
YSZ have already been tested in the CO oxidation reaction [18] but, to the best of our
knowledge, this is the first time that the catalytic activity of a Au-YSZ composite film
is studied in the methanol partial oxidation.
Page 148
Chapter 3
140
Figure 3.3. Steady-state H2 (a), CO2 (b) and methyl formate (HCOOCH3) (c) production rates
at different reaction temperatures during the blank experiments and the catalytic tests using the
Au-YSZ composite film under open circuit conditions (after application of VWR = +2 V for 30
min, i.e., un-promoted state). CH3OH/O2 = 5.9 %/0.43 %.
The H2 and CO2 production rates observed in this figure were expected from
the main POM reaction (reaction 3.1).
0
0.1
0.2
0.3
0.4
0.5
0.6
r H/
mo
l s-1
x 1
0-8 Reactor blank
Current collector
Current collector + Au-YSZ catalyst
a)
2
0
0.5
1
1.5
2
r CO
/ m
ol
s-1x
10
-8
Reactor blank
Current collector
Current collector + Au-YSZ catalyst
b)
2
0
0.5
1
1.5
2
2.5
3
240 260 280 300 320
r HC
OO
CH
/ m
ol s
-1x
10
-8
Temperature / ºC
Reactor blank
Current collector
Current collector + Au-YSZ catalyst
c)
3
Page 149
Electrochemical activation of Au nanoparticles dispersed in YSZ for methanol partial
oxidation
141
CH3OH + 0.5O2 → 2H2 + CO2 (3.1)
However, the most abundant product was methyl formate (HCOOCH3), with a
selectivity of 78 % (with respect to carbon), calculated as C C 3,out
C 2,out ,out , where C C 3,out and C 2,out are the molar flow rates
of methyl formate and carbon dioxide, respectively, at the outlet of the reactor. There
are many possible intermediate and side reactions involved in the methanol oxidation
as very recently summarized [33]. Methanol dissociation resulting in methoxy species
and hydroxyl group is generally considered the first step of the sequence. Then,
formaldehyde, formic acid and carbon dioxide may be produced, in this order, from
successive oxidation reactions, but also dimethyl ether, dimethoxymethane, methyl
formate and carbon monoxide may be obtained from the dehydrogenation of the
former compounds. Hence, the methyl formate could be formed from the following
consecutive steps [39]:
CH3OH + 0.5O2 → 2CO + H2O (3.2)
H2CO + 0.5O2 → HCOOH (3.3)
HCOOH + CH3 → C C 3 + H2O (3.4)
Under lack of surface oxygen, HCOOCH3 production on supported gold
catalysts has been postulated as a direct dehydrogenation of methanol (reaction 3.5)
[30, 31], and formaldehyde dimerization (reaction 3.6) has also be considered [40].
2CH3 ↔ C C 3 + 2H2 (3.5)
2H2C → C C 3 (3.6)
In any case, the weak adsorption of the methanol partial oxidation products on
Au, as compared to other noble metals, makes the gold-based catalysts suitable for the
low temperature selective oxidation of methanol to oxygenated compounds other than
CO2, such as HCOOCH3 [13]. Moreover, methyl formate is of interest in the chemical
industry as a solvent and a precursor in the synthesis of formic acid, formamide and
dimethylformamide, among other organic compounds [41].
Page 150
Chapter 3
142
Prior to the catalytic activity measurements under electrochemical promotion
conditions, the Au-YSZ/K-βAl2O3/Au electrochemical catalyst was in-situ
characterized by cyclic voltammetry under the same POM reaction conditions at 280
ºC, by recording the current (I) variation with the applied potential (VWR) between +2
and -2 V (scan rate of 80 mV s-1
) (Figure 3.4). Previously, the electrochemical catalyst
was kept at +2 V for 30 minutes in order to define a Au-YSZ film free of potassium
ions (reference state).
Figure 3.4. Current variation (I) vs. the applied potential (VWR) of the electrochemical catalyst
during the cyclic voltammetry between +2 and -2 V (scan rate = 80 mV s-1
) under methanol
partial oxidation conditions (CH3OH/O2 = 5.9 %/0.43 %, 280 ºC). The first three cycles are
depicted, in order from lighter to darker.
It can be observed that the cyclic voltammetry was reversible and almost
completely reproducible from the second cycle at the explored potential range. It has
been widely established that the cathodic peaks observed in the CV carried out with
alkali ionic conductors can be associated with the formation of surface compounds
-2
-1.6
-1.2
-0.8
-0.4
0
0.4
0.8
-2.5 -1.5 -0.5 0.5 1.5 2.5
I / A
x 1
0-3
VWR / V
1st cycle
2nd cycle3rd cycle
Page 151
Electrochemical activation of Au nanoparticles dispersed in YSZ for methanol partial
oxidation
143
derived from the interaction among the different reactants adsorbed on the catalyst-
working electrode and the alkali ions coming from the solid electrolyte [37, 38, 42,
43]. No electrocatalytic processes have been considered on the gold counter electrode
since it was constituted by large particles of negligible adsorption activity [37, 38, 42,
43]. In the present case, due to the migration of K+ ions to the Au-YSZ film and the
presence of O2, CO2 and H2O in the reaction atmosphere, K-derived surface
compounds such as potassium oxides/superoxides [37, 44] or carbonates/bicarbonates
[37, 45] may be formed. Then, the peaks observed during the anodic scan could be
linked to the decomposition of these promoter-derived species and the migration of the
K+ ions back to the solid electrolyte.
Hence two conclusions can be clearly obtained from this voltammogram:
- The applied potential of +2 V was high enough to electrochemically
decompose the promoter surface compounds, and this potential can be defined as a
reference un-promoted state for the further EPOC experiments.
- The reproducibility of the voltamogramm after the 2nd
cycle revealed a good
stability of the Au/YSZ catalyst during the polarizations under reaction conditions.
The reproducibility of the Au catalytic activity will be also confirmed in view of the
following experiments.
3.3.2. Electrochemical promotion via galvanostatic transitions
In order to study the EPOC phenomenon on the Au-YSZ catalyst film, several
closed and open circuit transitions were carried out under the following methanol
partial oxidation (POM) conditions: a feed composition of CH3OH/O2 = 5.9 %/0.43 %
(Ar balance) and a temperature of 280 ºC. The unpromoted catalyst was found to be
inactive below this temperature as shown on Figure 3.3.
Figure 3.5 shows the response vs. time in both the hydrogen, carbon dioxide
and methyl formate production rates and the catalyst potential (VWR) to the following
step changes: a positive potential (VWR = +2 V) for 32 min, a negative current (I = -5
Page 152
Chapter 3
144
µA) for 65 min, open circuit (I = 0) for 37 min and again a positive potential (VWR =
+2 V) for 37 min.
Figure 3.5. Variation of H2 (a), CO2 and HCOOCH3 (b) production rates as well as the
corresponding enhancement ratios (ρi) and catalyst potential (VWR) (a) vs. time upon different
potentiostatic and galvanostatic transitions. CH3OH/O2 = 5.9 %/0.43 %, 280 ºC.
Electrochemical promotion under the imposition of I = -5 µA.
In order to quantify the magnitude of the electropromotional effect, the rate
enhancement ratio of each compound (ρ) is also shown on the figure, calculated by the
following equation:
=
(3.7)
-1
0
1
2
3
0
1
2
3
4
5
VW
R/
V
r H/
mo
l s-1
x 1
0-8
0 1 20.5 1.5Promoter supply / mol K+ x 10-7
VWR = 2 VVWR = 2 V I = 0 AI = -5 µAρ
H2
0
3
6
9
12
2a)
0
6
12
0
1
2
3
4
0 25 50 75 100 125 150 175
r HC
OO
CH
/
mo
l s-1
x 1
0-8
r CO
/ m
ol
s-1x
10
-8
Time / min
3 ρH
CO
OC
H3
0
2
4
6
2
b)
2
1
0
ρC
O2
Page 153
Electrochemical activation of Au nanoparticles dispersed in YSZ for methanol partial
oxidation
145
where r and r0 are the promoted (VWR < +2 V) and unpromoted (VWR = +2 V) catalytic
reaction rates, respectively. Moreover, the inserted abscissa along the negative
polarization indicates the accumulated promoter supply, which varies linearly vs. time
in the case of galvanostatic experiments and is calculated from the following equation:
=
(3.8)
where n is the potassium ion charge in this case (+1), F is the Faraday constant (96485
C) and t is the polarization time.
The behaviour of the electrochemical catalyst has been evaluated upon the
different polarization regimes. Firstly, the application of a positive potential VWR = +2
V ensured the removal of K+ ions previously migrated to the catalyst surface by
thermal migration or as a consequence of the foregoing experiments [37, 38], and
resulted in a Au catalytic activity practically coincident with that observed at the same
temperature under open circuit conditions in Figure 3.3.
Then, at t = 32 min, a negative current I = -5 µA (current density, i = 1.6 µA
cm-2
) was applied between the Au-YSZ catalyst-working electrode and the Au counter
electrode, i.e. potassium promoter ions were electrochemically pumped from the solid
electrolyte to the Au/YSZ catalyst surface at a rate, = I/F = 5.18 x 10-11
mol K+ s
-1
according to the Faraday equation. The increase in the accumulated potassium supply
was found to enhance the overall methanol conversion (not shown) and the production
rates of hydrogen (Figure 3.5a), carbon dioxide and methyl formate (Figure 3.5b).
Selectivities toward H2 and HCOOCH3 were in turn enhanced vs. those toward CO2
and H2O. At the same time, the applied negative current caused a decrease in the
catalyst potential measured between the working and reference electrodes, VWR
(Figure 3.5a), as typically observed in studies with alkali solid electrolytes [38, 43,
46]. The trend of the catalytic activity instead depends on the EPOC behaviour of the
system. According to the theory of electrochemical promotion [3], the back-spillover
of potassium ions onto the Au-YSZ catalyst film under applied negative currents led
to the decrease in the catalyst potential and work function. It strengthened the Au
Page 154
Chapter 3
146
chemical bond with the electron acceptor adsorbates (oxygen) and weakened that with
the electron donors (methanol) [47]. In fact, Broqvist et al. [48] already reported the
interest of the alkali compounds as “oxygen attractors” for significantly improving the
activity of CO oxidation on a Au-based catalyst. It should be noted that the methanol
conversion obtained in this work did not exceed the 10 % probably due to the low
surface area of the active catalyst film and reactants bypass. However, very
interestingly, Au nanoparticles dispersed in a YSZ matrix were electrochemically
promoted by K+ ions under methanol partial oxidation conditions, and the obtained
EPOC effect was electrophilic as also observed in chapters 1 and 2 for Pt-based
catalysts.
Other studies have also successfully promoted metal catalysts dispersed on
YSZ by both alkali electropromotion [49] and alkali conventional promotion [50]. In
the present work, maximum production rates of 3.7 x 10-8
mol H2 s-1
, 4.3 x 10-8
mol
CO2 s-1
and 1.5 x 10-7
mol HCOOCH3 s-1
were achieved at the end of the negative
polarization, which corresponded to enhancement ratios (ρ) of 8.9, 2.5 and 5.3,
respectively, with respect to the production rates obtained with the unpromoted Au-
YSZ. Such an improvement in the catalytic activity was caused by an accumulated
promoter supply of 2.02 x 10-7
mol K+ (equation 3.8). Hence, the corresponding
optimum potassium coverage, , of 0.32 can be estimated by considering the total
number of deposited Au sites, N = 6.38 x 10-7
mol Au, by the following equation:
=
(3.9)
It is also interesting to note that upon current interruption (open circuit
conditions, at t = 97 min) a permanent EPOC phenomena was observed probably due
to the remaining of the promoter phases on the catalyst surface under open circuit
conditions, i.e., all the products reaction rates were kept at their electro-promoted
values. In addition, a very slight increase in the catalyst potential close to the open
circuit potential value typically observed in this work ( ≈ -0.4 V) was detected.
This kind of irreversible promotional effect (Permanent EPOC or Permanent
NEMCA) has been reported elsewhere for alkali-electropromoted systems [37, 38,
Page 155
Electrochemical activation of Au nanoparticles dispersed in YSZ for methanol partial
oxidation
147
46]. It has been mainly attributed to the high stability under reaction conditions of
certain promoter species formed on the catalyst surface, like potassium oxides or
superoxides [37, 38]. It is very important to highlight the practical application of the
observed irreversible EPOC behaviour: not only a very low current (a few µA) is
enough to enhance the catalytic performance of the gold nanoparticles by several
times, but also a first polarization step may be already sufficient to maintain this
catalytic modification for a certain time, with the consequent energy saving.
Finally, the imposition of a potential of +2 V again at t = 134 min restored the
initial production rates values within a few minutes, leading to a completely reversible
electropromotional effect. It was due to the decomposition of the promoter phases and
the electrochemical pumping of the K+ ions back to the solid electrolyte in good
agreement with the cyclic voltammetry. This reversible effect also sustains the
stability of the Au nanoparticles supported on the YSZ under EPOC conditions.
Hence, it is clear that the use of the YSZ support allowed to disperse the metal
nanoparticles, thus avoiding the metal sintering effect, very typical on this kind of
catalytic system.
Figures 3.6a and 3.6b show the results obtained in two additional experiments
under the same POM reaction conditions, which consisted of the same potentiostatic
and galvanostatic transitions than the previous one (Figure 3.5), but applying higher
currents during the negative polarization step: I = -10 µA (i = 3.2 µA cm-2
) in Figures
3.6a-1 and 3.6a-2, and -20 µA (i = 6.4 µA cm-2
) in Figures 3.6b-1 and 3.6b-2. The
application of a potential of +2 V at the beginning and the end of each experiment
defined again a Au-YSZ free of K-derived species (unpromoted state). In this way, all
the reaction rates remained almost at the same values upon every positive polarization,
confirming the complete reversibility of the promotional effect.
Page 156
Chapter 3
148
Figure 3.6. Variation of H2, CO2 and HCOOCH3 production rates as well as the corresponding
rate enhancement ratios (ρ) and catalyst potential (VWR) vs. time upon different potentiostatic
and galvanostatic transitions. CH3OH/O2 = 5.9 %/0.43 %, 280 ºC. Electrochemical promotion
under the imposition of I = -10 µA (a-1,a-2), and -20 µA (b-1,b-2).
As also observed in the first experiment during the negative polarization, the
migration of potassium ions to the catalyst surface at a rates, = I/F = 1.04 x 10-10
mol K+ s
-1 (Figures group 3.6a) and 2.07 x 10
-10 mol K s
-1 (Figures group 3.6b) sharply
enhanced the gold catalytic activity. However, it can be found under the polarizations
at -10 µA, and more clearly, at -20 µA, that all the production rates decreased from a
certain accumulated amount of supplied promoter, which was not achieved upon the
imposition of -5 µA. This poisoning effect could be attributed to the excessive
formation of alkali-derived surface compounds and the concomitant blocking of active
sites, as widely observed with alkali-based promoted systems [44, 50-52]. Moreover,
the catalyst potential measured after 65 min of negative polarization decreased from -
0.55 V (Figure 3.5a) to -0.60 V (Figure 3.6a-1) and -0.74 V (Figure 3.6b-1), due to the
increase in the maximum potassium coverage achieved in each experiment.
-1
0
1
2
3
0
1
2
3
4
VW
R/
V
r H/
mo
l s-1
x 1
0-8
0 4 82 6Promoter supply / mol K+ x 10-7
VWR = 2 VVWR = 2 V I = 0 AI = -20 µA
2
b-1)
ρH
2
0
4
6
8
10
2
0
6
12
0
1
2
3
0 25 50 75 100 125 150 175
r HC
OO
CH
/
mo
l s-1
x 1
0-8
r CO
/ m
ol
s-1x
10
-8
Time / min
3 ρH
CO
OC
H3
0
2
4
6
2
b-2)
2
1
0
ρC
O2
-1
0
1
2
3
0
1
2
3
4
5
VW
R/
V
r H/
mo
l s-1
x 1
0-8
0 2 41 3Promoter supply / mol K+ x 10-7
VWR = 2 VVWR = 2 V I = 0 AI = -10 µA
ρH
2
0
3
6
9
12
2
a-1)
0
6
12
0
1
2
3
4
0 25 50 75 100 125 150 175
r HC
OO
CH
/
mo
l s-1
x 1
0-8
r CO
/ m
ol
s-1x
10
-8
Time / min
3 ρH
CO
OC
H3
0
2
4
6
2
a-2)
2
1
0
ρC
O2
Page 157
Electrochemical activation of Au nanoparticles dispersed in YSZ for methanol partial
oxidation
149
A permanent EPOC was also observed in these experiments upon the
subsequent current interruption, although it was inevitably affected by the mentioned
poisoning effect. For instance, in the case of the hydrogen production, permanent rate
enhancement ratios ( ) of 8.7, 6.4 and 3.2 can be observed at the end of the open
circuit imposition in Figures 3.5a, 3.6a-1 and 3.6b-1, respectively. This parameter has
been calculated using the following equation:
=
(3.10)
where rper is the permanent catalytic production rate following the promoted state.
Nevertheless, the optimum amount of supplied K+ ions which maximized the catalytic
activity was found to be the same in each experiment, around 3 x 10-7
mol K+ (which
corresponded to a potassium coverage, , of about 0.5). In addition, the
maximum values of the reaction rates obtained were also practically the same, i.e., the
H2, CO2 and HCOOCH3 production rates were in all cases enhanced 9.2, 2.6 and 5.5
times, respectively, under optimum electrochemically promoted conditions. Then, it is
interesting to note that the applied current had no influence on the behaviour and
magnitude of the electropromotional effect, but only on the speed of the promotion
process. Finally, the stability of the Au/YSZ catalyst film in the different experiments
was demonstrated on the basis of the reproducible catalytic behaviour.
Besides the reaction rate enhancement ratio (ρ), other typical parameter used
in literature to describe the magnitude of the electrochemical promotion effect the
Faradaic efficiency (Λ). This parameter is calculated as follows:
Λ =
(3.11)
where Δr = r – r0 is the K+-induced change in the catalytic reaction rate and n is the
number of electrons involved in the corresponding electrocatalytic reaction. Under the
studied POM reaction conditions at 280 ºC, the increase in the oxygen catalytic
consumption rate (not shown) caused by the optimum promoter coverage was around
2.1 x 10-7
mol O s-1
, which was 4075 times larger than the rate of K+ supply ( =
Page 158
Chapter 3
150
I/F) to the catalyst film under the application of -5 µA ( = 5.18 x 10-11
mol K+ s
-1).
The corresponding Faradaic efficiency (Λ) would be -8150, i.e. by considering the
equivalent oxygen electrocatalytic reaction (n = 2 in equation 3.11). However, in the
present study the source of promoter ions is not any O2-
conductor material such as
YSZ, but K-βAl2O3 (a cationic conductor). Thus, the Faradaic efficiency must be
considered with caution since the alkali ions cannot interact with the gas reactants at
the same extent [53]. Hence, as stated in Chapter 1, the most significant parameters
from a catalytic point of view to characterize the alkali-based promoted systems are
the rate enhancement ratio (ρ, equation 3.7) and the promotion index ( ) [3], the
latter being defined according to the following equation:
=
θ (3.12)
Figure 3.7b depicts the promotion index ( ) for the maximum production
rates of the different compounds (H2, CO2 and HCOOCH3) obtained in the previous
figures under optimum promoted conditions, and Figure 3.7a shows the corresponding
faradaic efficiency values related to the rate of K+ promoter supply (n = 1 in equation
3.11). This latter parameter has been denoted as and has been expressed in units
of mol product/mol K+. In other words, in both Figure 3.7a and 3.7b, three points are
depicted which correspond with the maximum catalytic rates observed in the three
previous experiments (Figures 3.5, 3.6a and 3.6b).
Firstly, a clear EPOC effect can be observed in all the experiments ( > 0)
with an electrophilic behaviour ( < -1), since as previously discussed the
application of negative currents and the consequent back-spillover of K+ promoter ions
increased the Au catalytic activity (I < 0, Δr > 0). In Figure 3.7a, methyl formate
presented the highest values, being the most abundant product obtained under the
studied reaction conditions. This parameter represents the ratio between the mol of
product and the mol of K+ electrochemically supplied. In this sense, under the
application of I = -5 µA, each mol K+ migrated to the Au-YSZ catalyst film allowed
the simultaneous production of up to more than 600 mol H2, 500 mol CO2 and 2300
Page 159
Electrochemical activation of Au nanoparticles dispersed in YSZ for methanol partial
oxidation
151
mol HCOOCH3. Similar maximum production rates were achieved in all the
experiments despite increasing the rate of promoter supply. Consequently, the faradic
efficiency values decreased as higher negative current was applied.
Figure 3.7. Influence of the applied negative current on the optimum faradaic efficiency
relative to the rate of K+ supply ( ) (a) and the optimum promotion index ( ) (b)
of the different products. Data obtained from the results shown in Figures 3.5 and 3.6.
On the other hand, from Figure 3.7b, it can be drawn that hydrogen showed the
highest promotion index values. In fact, the H2 production rate presented the most
pronounced increase with respect to the unpromoted rate, as also reflected in the
highest ρ values shown in the previous figures. Higher optimum promotion indexes
( ) were found for all the products under the imposition of the lowest negative
current, I = -5 µA, because these data were obtained from the maximum production
0
5
10
15
20
25
-25-20-15-10-50
PI K
,o
pt
I / A x 10-6
+
H2
CO2
HCOOCH3
b)
-2500
-2000
-1500
-1000
-500
0
ΛK
,o
pt/
mo
l p
rod
uct
mo
l K
+ -
1+
H2
CO2
HCOOCH3
a)
Page 160
Chapter 3
152
rates observed at the end of this polarization step (Figure 3.5), which did not
correspond with the actual optimum electropromoted conditions. With such a low rate
of K+ supply in the first experiment (5.18 x 10
-11 mol K s
-1), the polarization time
should have been extended for another 35 min to achieve the optimum potassium
coverage (around 0.5). In this sense, the slight difference between the depicted
values obtained at I = -10 µA and -20 µA could be attributed to the non-
complete coincidence in the respective potassium coverages, which was likely due to
slight differences in the gas chromatograph analysis time. However, fixed promotion
index values of around 16, 3 and 9 for hydrogen, carbon dioxide and methyl formate,
respectively, could be considered, regardless the magnitude of applied negative
current, under optimum electrochemically promoted conditions ( ≈ 0.5).
Hence, in contrast to the parameter, it was demonstrated the independence of the
promotion index with respect to the rate of K+ ions supply, being possible to induce
the maximum improvement in the Au-YSZ catalytic activity under given reaction
conditions as long as a specific potassium coverage is reached.
3.3.3. Electrochemical promotion via potentiostatic transitions
Other kind of electrochemical promotion experiment under the same methanol
partial oxidation conditions consisted of successive potentiostatic transitions, similar
to the experiments carried out in the two first chapters. Figure 3.8 shows the dynamic
response in the H2 (a), CO2 (b) and HCOOCH3 (c) production rates to step changes in
the applied catalyst potential (VWR) at 280 ºC. A positive polarization at VWR = +2 V
before each different potential allowed starting from a reference (unpromoted) state in
all cases. The subsequent imposition of VWR = 0.5 V did not involve any modification
in the catalytic activity since it still remained above the open circuit potential value.
Then, the negative polarization at VWR = -0.5 V caused a sharp increase of all the
production rates as a result of the back-spillover of the promoter ions from the K-
βAl2O3 electrolyte to the Au-YSZ catalyst-working electrode (VWR ≤ -0.5 V, Δr > 0).
As noted in the previous galvanostatic experiments, the observed EPOC effect could
be explained in terms of the work function variation and the consequent change in
chemisorptive bond strengths on the catalyst surface [3].
Page 161
Electrochemical activation of Au nanoparticles dispersed in YSZ for methanol partial
oxidation
153
Figure 3.8. H2 (a), CO2 (b) and HCOOCH3 (c) production rates vs. time under different
applied potentials (VWR) between 2 and -1 V. CH3OH/O2 = 5.9 %/0.43 %, 280 ºC.
Very interestingly, under this negative potential (VWR = -0.5 V) the steady-
state H2, CO2 and HCOOCH3 production rates were close to those obtained under
optimum electropromoted conditions in previous section, with enhancement ratios (ρ)
-2
-1
0
1
2
3
0
1
2
3
4
VW
R/
V
r H/
mo
l s-1
x 1
0-8
a)2
rH VWR
2
-2
-1
0
1
2
0
1
2
3
4
VW
R/
V
r CO
/
mo
l s-1
x 1
0-8
2rCO VWR
b)
2
-2
-1
0
1
2
0
6
12
0 100 200 300 400
VW
R/
V
r HC
OO
CH
/ m
ol s
-1x
10
-8
Time / min
3rHCOOCH VWRc)
3
Page 162
Chapter 3
154
of around 8, 2 and 5, respectively. The associated amount of migrated promoter at the
end of this polarization was 3.58 x 10-7
mol K+, which was calculated from the
integration of the current vs. time curve (not shown here) via equation 3.8. It
corresponded to a potassium coverage equal to 0.56 (equation 3.9), which was
slightly higher than the optimum value found in previous experiments (around 0.5).
This fact could indicate some possible blocking of Au active sites by an excess of K-
derived surface compounds [44, 45] and explain the mildly lower catalytic activity
obtained under these promoted conditions compared to the maximum achieved in
Figures 3.6a and 3.6b. This poisoning effect was more clearly observed under the
application of VWR = -1 V. However, all the promoter species formed during the
negative polarizations were decomposed upon each successive imposition of VWR = +2
V, again proving, in good agreement with the Au-YSZ characterization results, the
reversibility and reproducibility of the EPOC phenomenon and the stability of the
YSZ-supported Au nanoparticles. Thus, in this experiment the electrochemical
catalyst definitely showed a similar qualitative and quantitative behaviour as that
observed in previous figures under applied negative currents.
Finally, the production rate of the different compounds was compared at
different temperatures with the same feed composition (CH3OH/O2 = 5.9 %/0.43 %)
under both unpromoted and promoted conditions. Figure 3.9 shows the variation of the
steady-state production rates and the accumulated promoter supply vs. the applied
catalyst potential at 280 and 300 ºC. The corresponding rate enhancement ratios (ρopt)
and promotion indexes ( ), calculated by equations 3.7 and 3.12 respectively,
are also indicated for the optimum applied potential in each case. Starting from the
reference positive polarization, VWR = +2 V (un-promoted state), the decrease in the
applied potential led to the electrochemical pumping of K+ ions from the solid
electrolyte to the Au-YSZ catalyst film, according to the obtained negative currents
(see inset of Figure 3.9d for the application of VWR = -0.5 V). As typically observed on
K-βAl2O3-based electrocatalytic systems under potentiostatic transitions [38], the
resultant negative currents were more pronounced at the beginning of the polarization
and then decreased to negligible residual values of the order of -1 µA.
Page 163
Electrochemical activation of Au nanoparticles dispersed in YSZ for methanol partial
oxidation
155
Figure 3.9. Steady-state H2 (a), CO2 (b) and HCOOCH3 (c) production rates and accumulated
promoter supply (d) vs. applied potential (VWR) as well as the optimum production rates
enhancement ratios (ρi,opt) and promotion indexes ( ) at 280 and 300 ºC. CH3OH/O2 =
5.9 %/0.43 %. Inset of Figure 3.9d shows the current vs. time curve during the potentiostatic
imposition of -0.5 V.
All the production rates ( ,
and ) were increased by the
potassium promotional effect at both studied temperatures, showing an electrophilic
EPOC behavior (VWR ≤ -0.5 V, Δr > 0, ρ > 1 and > 0). Moreover, in both cases
the maximum catalytic activity was found at the same optimum applied potential,
VWR,opt = -0.5 V. This optimum potential corresponded with the migration of 3.58 x
10-7
and 4.74 x 10-7
mol K+ and potassium coverages of 0.56 and 0.74, at 280 and 300
ºC, respectively. The higher the reaction temperature, the higher the solid electrolyte
ionic conductivity and, therefore, the higher the amount of electrochemically
transferred K+ ions at fixed potential. It can be observed an increase in the catalytic
activity of the Au-YSZ catalyst film with the temperature under unpromoted
conditions (positive potentials) as typically observed in POM studies with supported
0
0.5
1
1.5
2
-1.5 -0.5 0.5 1.5 2.5
Acc
um
ula
ted
pro
mo
ter
sup
ply
/ m
ol K
+x
10-6
VWR / V
280 ºC
300 ºC
d)
-1
-0.6
-0.2
0.2
0 20 40 60
I / A
x 1
0-4
Time / min
280 ºC
300 ºC
-0.5 V
0
0.5
1
1.5
2
2.5
3
3.5
r H
/ m
ol s
-1x
10
-8280 ºC
300 ºC2
a) ρH ,opt = 7.9 PIK ,H ,opt = 12.32 2
+
ρH ,opt = 3.6 PIK ,H ,opt = 3.52 2
+
0
4
8
12
16
r HC
OO
CH
/
mo
l s-1
x 1
0-8
280 ºC
300 ºC
3
c) ρHCOOCH ,opt = 5.1 PIK ,HCOOCH ,opt = 7.33 3
+
ρHCOOCH ,opt = 2.7 PIK ,HCOOCH ,opt = 2.33 3
+
1
1.5
2
2.5
3
3.5
4
-1.5 -0.5 0.5 1.5 2.5
r CO
/
mo
l s-1
x 1
0-8
VWR / V
280 ºC
300 ºC
2
b)ρCO ,opt = 1.9 PIK ,CO ,opt = 1.2
2 2+
ρCO ,opt = 2.3 PIK ,CO ,opt = 2.32 2
+
Page 164
Chapter 3
156
Au catalysts [19, 29-33]. However, under electrochemical promotion (VWR = -0.5 and
-1 V) the overall methanol conversion decreased at 300 ºC, very likely due to the
poisoning effect of the excessive promoter-derived species. In fact, the value of
potassium coverage, = 0.74, obtained at this temperature under the application of
-0.5 V was already far above the optimum value found in previous experiments
performed at 280 ºC ( ≈ 0.5). Furthermore, in good agreement with several
studies [13, 30, 31, 33], an increase in the temperature favored the formation of CO2
and hindered the HCOOCH3 selectivity due to the further oxidation of the latter.
As already remarked, the in-situ modification of the activity and selectivity of
a metallic catalysts by the controlled pumping of alkali ions (via EPOC) is of
paramount interest. This feature has been studied for different catalytic reactions such
as the epoxidation of alkenes [44], the Fischer-Tropsch synthesis [45], the selective
catalytic reduction of nitrogen oxides [51-53], and, in the first chapters, for H2 and
formaldehyde production from methanol partial oxidation. In the present work, gold
nanoparticles dispersed in a YSZ matrix were catalytically activated by K+ ions for the
production of hydrogen and methyl formate. Hence, two products of great interest can
be produced simultaneously and selectively by means of the electrochemical
promotion.
3.4. CONCLUSIONS
The following conclusions could be drawn from this study:
- An electrochemical catalyst based on Au nanoparticles dispersed in a YSZ
matrix was prepared by reactive co-sputtering of Zr-Y and Au targets. Similar to the
cathodic arc deposition in the previous chapter, this technique allowed depositing a
thin catalyst film (~170 nm) with very low particle size (~3 nm) and metal loading
(only 40 µg Au cm-2
).
- The obtained Au-YSZ catalyst showed to be active in the partial oxidation of
methanol with a very high selectivity toward methyl formate production, and it was
polarized by using a current collector, even without providing any electrical
Page 165
Electrochemical activation of Au nanoparticles dispersed in YSZ for methanol partial
oxidation
157
conductivity. Hence, the Au-YSZ was successfully promoted by electrochemical
pumping potassium ions under negative polarization.
- The stability of the Au/YSZ catalyst film was verified by different
characterization techniques, demonstrating the interest of the catalyst preparation
technique and the use of the YSZ support to disperse metal nanoparticles by avoiding
metal sintering.
- Under given reaction conditions, the magnitude of the electrochemical
promotion effect with alkali solid electrolyte did not depend on the applied current, i.e.
the rate of promoter supply, but on the promoter coverage. The H2, CO2 and
HCOOCH3 production rates were enhanced, regardless the operation mode
(galvanostatic or potentiostatic), more than 9, 2 and 5 times, respectively, at 280 ºC
upon an optimum potassium coverage of around 0.5.
- A permanent electropromoted effect was found upon negative current
interruption, showing the stability of the formed promoter species under the studied
reaction conditions, which may be of great interest for practical applications.
3.5. REFERENCES
[1] W.D. Mross, ALKALI DOPING IN HETEROGENEOUS CATALYSIS, Catalysis reviews, 25
(1983) 591-637.
[2] R.J. Farrauto, C.H. Bartholomew, Fundamentals of industrial catalytic processes, Chapman
& Hall, London, 1997.
[3] C.G. Vayenas, S. Bebelis, C. Pliangos, S. Brosda, D. Tsiplakides, Electrochemical
Activation of Catalysis: Promotion, Electrochemical Promotion and Metal-Support
Interactions, Kluwer Academic Publishers/Plenum Press, New York, 2001.
[4] A. de Lucas-Consuegra, New trends of Alkali Promotion in Heterogeneous Catalysis:
Electrochemical Promotion with Alkaline Ionic Conductors, Catalysis Surveys from Asia,
2015, DOI:10.1007/s10563-014-9179-6.
[5] O.A. Mar'ina, V.A. Sobyanin, The effect of electrochemical oxygen pumping on the rate of
CO oxidation on Au electrode-catalyst, Catalysis Letters, 13 (1992) 61-69.
Page 166
Chapter 3
158
[6] O.A. Mar'ina, V.A. Sobyanin, V.D. Belyaev, V.N. Parmon, The effect of electrochemical
oxygen pumping on catalytic properties of Ag and Au electrodes at gas-phase oxidation of
CH4, Catalysis Today, 13 (1992) 567-570.
[7] I.M. Petrushina, V.A. Bandur, F. Cappeln, N.J. Bjerrum, Electrochemical promotion of
sulfur dioxide catalytic oxidation, Journal of the Electrochemical Society, 147 (2000) 3010-
3010.
[8] F.M. Sapountzi, M.N. Tsampas, C.G. Vayenas, Electrochemical promotion of CO
conversion to CO2 in PEM fuel cell PROX reactor, Catalysis Today, 146 (2009) 319-325.
[9] X. Deng, B.K. Min, A. Guloy, C.M. Friend, Enhancement of O2 dissociation on Au(111) by
adsorbed oxygen: Implications for oxidation catalysis, Journal of the American Chemical
Society, 127 (2005) 9267-9270.
[10] G. Avgouropoulos, T. Tabakova, J.J. Spivey, Environmental Catalysis over Gold-Based
Materials, The Royal Society of Chemistry, Cambridge, 2013.
[11] M. Haruta, Gold as a novel catalyst in the 21st century: Preparation, working mechanism
and applications, Gold Bulletin, 37 (2004) 27-36.
[12] V. Zielasek, B. Jürgens, C. Schulz, J. Biener, M.M. Biener, A.V. Hamza, M. Bäumer,
Gold catalysts: Nanoporous gold foams, Angewandte Chemie - International Edition, 45
(2006) 8241-8244.
[13] A. Wittstock, V. Zielasek, J. Biener, C.M. Friend, M. Bäumer, Nanoporous gold catalysts
for selective gas-phase oxidative coupling of methanol at low temperature, Science, 327 (2010)
319-322.
[14] S. Gil, M. Marchena, C.M. Fernández, L. Sánchez-Silva, A. Romero, J.L. Valverde,
Catalytic oxidation of crude glycerol using catalysts based on Au supported on carbonaceous
materials, Applied Catalysis A: General, 450 (2013) 189-203.
[15] G.C. Bond, D.T. Thompson, Gold-catalysed oxidation of carbon monoxide, Gold Bulletin,
33 (2000) 41-50.
[16] T. Kobayashi, M. Haruta, S. Tsubota, H. Sano, B. Delmon, Thin films of supported gold
catalysts for CO detection, Sensors and Actuators: B. Chemical, 1 (1990) 222-225.
[17] S.S. Pansare, A. Sirijaruphan, J.G. Goodwin Jr, Au-catalyzed selective oxidation of CO: A
steady-state isotopic transient kinetic study, Journal of Catalysis, 234 (2005) 151-160.
[18] H.A.E. Dole, J.M. Kim, L. Lizarraga, P. Vernoux, E.A. Baranova, Catalytic CO oxidation
over Au nanoparticles supported on yttria-stabilized zirconia, ECS Transactions, 2012, pp.
265-274, DOI:10.1149/1.3701316.
Page 167
Electrochemical activation of Au nanoparticles dispersed in YSZ for methanol partial
oxidation
159
[19] A.N. Pestryakov, V.V. Lunin, N. Bogdanchikova, O.N. Temkin, E. Smolentseva, Active
states of gold in small and big metal particles in CO and methanol selective oxidation, Fuel,
110 (2013) 48-53.
[20] H. Sakurai, M. Haruta, Synergism in methanol synthesis from carbon dioxide over gold
catalysts supported on metal oxides, Catalysis Today, 29 (1996) 361-365.
[21] M.C. Kung, K.A. Bethke, J. Yan, J.H. Lee, H.H. Kung, Catalysts for lean NOx reduction:
Structure-property relationship, Applied Surface Science, 121-122 (1997) 261-266.
[22] N. Yi, R. Si, H. Saltsburg, M. Flytzani-Stephanopoulos, Active gold species on cerium
oxide nanoshapes for methanol steam reforming and the water gas shift reactions, Energy and
Environmental Science, 3 (2010) 831-837.
[23] A. Ciftci, D.A.J.M. Ligthart, P. Pastorino, E.J.M. Hensen, Nanostructured ceria supported
Pt and Au catalysts for the reactions of ethanol and formic acid, Applied Catalysis B:
Environmental, 130-131 (2013) 325-335.
[24] G. Walther, L. Cervera-Gontard, U.J. Quaade, S. Horch, Low temperature methane
oxidation on differently supported 2 nm Au nanoparticles, Gold Bulletin, 42 (2009) 13-19.
[25] M.D. Hughes, Y.J. Xu, P. Jenkins, P. McMorn, P. Landon, D.I. Enache, A.F. Carley, G.A.
Attard, G.J. Hutchings, F. King, E.H. Stitt, P. Johnston, K. Griffin, C.J. Kiely, Tunable gold
catalysts for selective hydrocarbon oxidation under mild conditions, Nature, 437 (2005) 1132-
1135.
[26] A. Abad, P. Concepción, A. Corma, H. García, A collaborative effect between gold and a
support induces the selective oxidation of alcohols, Angewandte Chemie - International
Edition, 44 (2005) 4066-4069.
[27] P.Y. Sheng, G.A. Bowmaker, H. Idriss, The Reactions of Ethanol over Au/CeO2, Applied
Catalysis A: General, 261 (2004) 171-181.
[28] H.C. Yang, F.W. Chang, L.S. Roselin, Hydrogen production by partial oxidation of
methanol over Au/CuO/ZnO catalysts, Journal of Molecular Catalysis A: Chemical, 276 (2007)
184-190.
[29] B.P.C. Hereijgers, T.M. Eggenhuisen, K.P. De Jong, H. Talsma, A.M.J. Van Der Eerden,
A.M. Beale, B.M. Weckhuysen, Understanding the promotion effect of lanthanum oxide on
gold-based catalysts in the partial oxidation of methanol by in situ XAFS and DSC studies,
Journal of Physical Chemistry C, 115 (2011) 15545-15554.
Page 168
Chapter 3
160
[30] F.W. Chang, S.C. Lai, L.S. Roselin, Hydrogen production by partial oxidation of
methanol over ZnO-promoted Au/Al2O3 catalysts, Journal of Molecular Catalysis A:
Chemical, 282 (2008) 129-135.
[31] W. Jianxin, L. Laitao, A comparative study of partial oxidation of methanol over zinc
oxide supported metallic catalysts, Catalysis Letters, 126 (2008) 325-332.
[32] T.C. Ou, F.W. Chang, L.S. Roselin, Production of hydrogen via partial oxidation of
methanol over bimetallic Au-Cu/TiO2 catalysts, Journal of Molecular Catalysis A: Chemical,
293 (2008) 8-16.
[33] K. Kähler, M.C. Holz, M. Rohe, A.C. Van Veen, M. Muhler, Methanol oxidation as probe
reaction for active sites in Au/ZnO and Au/TiO2 catalysts, Journal of Catalysis, 299 (2013)
162-170.
[34] S. Scirè, L.F. Liotta, Supported gold catalysts for the total oxidation of volatile organic
compounds, Applied Catalysis B: Environmental, 125 (2012) 222-246.
[35] R. Catrin, T. Gries, B. Raillard, F. Mücklich, S. Migot, D. Horwat, Influence of laser
interference patterning on microstructure and friction behavior of gold/yttria-stabilized
zirconia nanocomposite thin films, Journal of Materials Research, 27 (2012) 879-885.
[36] H.P. Klug, L.E. Alexander, X-Ray Diffraction Procedures for Polycrystalline and
Amorphous Materials, Wiley, New York, 1974.
[37] A. de Lucas-Consuegra, F. Dorado, J.L. Valverde, R. Karoum, P. Vernoux, Low-
temperature propene combustion over Pt/K-βAl2O3 electrochemical catalyst:
Characterization, catalytic activity measurements, and investigation of the NEMCA effect,
Journal of Catalysis, 251 (2007) 474-484.
[38] A. de Lucas-Consuegra, A. Princivalle, A. Caravaca, F. Dorado, C. Guizard, J.L.
Valverde, P. Vernoux, Preferential CO oxidation in hydrogen-rich stream over an
electrochemically promoted Pt catalyst, Applied Catalysis B: Environmental, 94 (2010) 281-
287.
[39] J.M. Tatibouët, Methanol oxidation as a catalytic surface probe, Applied Catalysis A:
General, 148 (1997) 213-252.
[40] N.W. Cant, S.P. Tonner, D.L. Trimm, M.S. Wainwright, Isotopic labeling studies of the
mechanism of dehydrogenation of methanol to methyl formate over copper-based catalysts,
Journal of Catalysis, 91 (1985) 197-207.
[41] J.S. Lee, J.C. Kim, Y.G. Kim, Methyl formate as a new building block in C1 chemistry,
Applied Catalysis, 57 (1990) 1-30.
Page 169
Electrochemical activation of Au nanoparticles dispersed in YSZ for methanol partial
oxidation
161
[42] N. Kotsionopoulos, S. Bebelis, Electrochemical characterization of the Pt/β″-Al 2O3
system under conditions of in situ electrochemical modification of catalytic activity for
propane combustion, Journal of Applied Electrochemistry, 40 (2010) 1883-1891.
[43] N. Kotsionopoulos, S. Bebelis, In situ electrochemical modification of catalytic activity
for propane combustion of Pt/β-Al2O3 catalyst-electrodes, Topics in Catalysis, 44 (2007) 379-
389.
[44] A. Palermo, A. Husain, M.S. Tikhov, R.M. Lambert, Ag-catalysed epoxidation of propene
and ethene: An investigation using electrochemical promotion of the effects of alkali, NOx, and
chlorine, Journal of Catalysis, 207 (2002) 331-340.
[45] A.J. Urquhart, J.M. Keel, F.J. Williams, R.M. Lambert, Electrochemical Promotion by
Potassium of Rhodium-Catalyzed Fischer-Tropsch Synthesis: XP Spectroscopy and Reaction
Studies, Journal of Physical Chemistry B, 107 (2003) 10591-10597.
[46] C.G. Vayenas, S. Bebelis, M. Despotopoulou, Non-faradaic electrochemical modification
of catalytic activity 4. The use of β″-Al2O3 as the solid electrolyte, Journal of Catalysis, 128
(1991) 415-435.
[47] C.G. Vayenas, S. Neophytides, Non-faradaic electrochemical modification of catalytic
activity III. The case of methanol oxidation on Pt, Journal of Catalysis, 127 (1991) 645-664.
[48] P. Broqvist, L.M. Molina, H. Grönbeck, B. Hammer, Promoting and poisoning effects of
Na and Cl coadsorption on CO oxidation over MgO-supported Au nanoparticles, Journal of
Catalysis, 227 (2004) 217-226.
[49] A. De Lucas-Consuegra, A. Caravaca, P.J. Martínez, J.L. Endrino, F. Dorado, J.L.
Valverde, Development of a new electrochemical catalyst with an electrochemically assisted
regeneration ability for H2 production at low temperatures, Journal of Catalysis, 274 (2010)
251-258.
[50] I.V. Yentekakis, R.M. Lambert, M.S. Tikhov, M. Konsolakis, V. Kiousis, Promotion by
sodium in emission control catalysis: A kinetic and spectroscopic study of the Pd-catalyzed
reduction of NO by propene, Journal of Catalysis, 176 (1998) 82-92.
[51] F. Dorado, A. de Lucas-Consuegra, C. Jiménez, J.L. Valverde, Influence of the reaction
temperature on the electrochemical promoted catalytic behaviour of platinum impregnated
catalysts for the reduction of nitrogen oxides under lean burn conditions, Applied Catalysis A:
General, 321 (2007) 86-92.
Page 170
Chapter 3
162
[52] I.V. Yentekakis, A. Palermo, N.C. Filkin, M.S. Tikhov, R.M. Lambert, In situ
electrochemical promotion by sodium of the platinum-catalyzed reduction of NO by propene,
Journal of Physical Chemistry B, 101 (1997) 3759-3768.
[53] P. Vernoux, F. Gaillard, C. Lopez, E. Siebert, Coupling catalysis to electrochemistry: A
solution to selective reduction of nitrogen oxides in lean-burn engine exhausts?, Journal of
Catalysis, 217 (2003) 203-208.
Page 171
163
Chapter 4
ELECTROCHEMICAL PROMOTION OF Cu
NANOCOLUMNS IN THE PARTIAL OXIDATION OF
METHANOL:
EPOC WITH HIGHLY POROUS NON-NOBLE METAL
CATALYSTS
A novel Cu catalyst film has been prepared by oblique angle physical vapour
deposition (OAD) on a K-βAl2O3 solid electrolyte (alkaline ionic conductor). This
technique has allowed to obtain a highly porous and electrically conductive Cu
catalyst electrode which has been electrochemically promoted in the partial oxidation
of methanol (POM). The production rates of hydrogen, carbon dioxide and methyl
formate have been in-situ enhanced in a reversible and reproducible way, by means of
the controlled migration of electropositive potassium ions. Under the studied reaction
conditions these promoter ions formed potassium-derived surface compounds as
demonstrated by post-reaction characterization analysis. Some nitrogen functional
groups and carbonaceous compounds were also detected. The obtained results
demonstrate the interest of the used catalyst-electrode preparation technique for the
electrochemical activation of non-noble metal catalyst films.
0
2
4
6
8
10
-1.5
-1
-0.5
0
0.5
1
1.5
0 100 200 300 400 500 600
r /
mo
l s-1
x 1
0-8
VW
R/
V
Time / min
VWR
rH2rCO2
rHCOOCH3
Δr
CH3OH,
O2
H2, CO2,
HCOOCH3, H2O
4.4% CH3OH0.3% O2
320 ºC
2 µm
Nanocolumnar Cu catalyst film
Cu
K+ K+ K+ K-βAl2O3
Au
ΔV < 0
e-
e-
Page 172
Chapter 4
164
4.1. INTRODUCTION
The phenomenon of Electrochemical Promotion of Catalysis (EPOC) or
NEMCA effect has been applied with a large variety of catalysts and solid electrolytes
(cationic, anionic or mixed conductors) and in a wide variety of catalytic reactions of
industrial and environmental interest [1]. However, as already pointed out, a further
technological progress is necessary for its practical application. Nowadays, some of
the main challenges of EPOC are the design of scaled-up reactors and the material cost
minimization. With respect to the former, increasingly more compact and efficient
reactors designs can be found in literature based, for example, on multi-pellet
configurations [2], hollow fibre membranes [3] or a sophisticated monolithic
electropromoted reactor (MEPR) [4-7]. Regarding the second outlook, several EPOC
studies have dealt with the improvement of catalytic materials and the development of
electrodes consisting of catalysts highly dispersed on gold [8], mixed ionic-electronic
conductors [9], different kinds of carbonaceous materials [10, 11] or yttria-stabilized
zirconia (YSZ) [12-14]. Other good examples have been reported in chapters 2 and 3
of this manuscript, where Pt and Au nanoparticles dispersed in carbon and YSZ
phases, respectively, were successfully electrochemically promoted for methanol
conversion with high selectivity toward H2 and methyl formate, respectively. A
complementary strategy on cost minimization would be the use of catalysts with a
higher extent of base metals. Some EPOC studies have already employed Ni- [10, 15-
18], Fe- [2, 19, 20] or Cu-based [7, 21-25] catalyst films in different reactions. In this
sense, the present chapter and the following have been focused on the electrochemical
promotion of non-noble metals for H2 production processes from CH3OH.
On the other hand, numerous properties of these metallic catalyst films such as
their porosity, surface area or metal dispersion, which are determining factors in the
performance of the catalytic reactions, strongly depend on the synthesis method.
Different techniques have been used in EPOC studies for the preparation of the active
catalyst film, some of which have been mentioned throughout the previous chapters.
Very common procedures for ease of application are, for instance, the deposition and
calcination of an organometallic paste [2, 14, 17, 19], the impregnation of a metal
Page 173
Electrochemical promotion of Cu nanocolumns in the partial oxidation of methanol
165
precursor solution [8, 9, 15] or the spray deposition in a carbon matrix [11]. Other less
conventional techniques which enable a better control of the microstructure of the film
include Physical Vapour Deposition (PVD) methods such as Pulsed Laser Deposition
(PLD) [26], sputtering [4, 6, 7, 21, 22] or Cathodic Arc Deposition (CAD), the two
latter already employed in chapters 3 and 2, respectively.
In the present work, a modification of the classical PVD evaporation technique
called Oblique or Glancing Angle Deposition (OAD or GLAD) for the preparation of
the film is proposed. By this method, the substrate is placed in an oblique or glancing
angle configuration with respect to the evaporated flux of deposition material to
enhance the shadowing effects during the film growth [27]. As a result, highly porous
films formed by tilted nanocolumns are obtained, which are characterized by a high
gas-exposed surface area and a controlled microstructure depending on the vapour
incident angle, the rotation speed of the substrate, and the temperature during the
deposition, among other factors. The open porosity of these films is very useful to
prepare host materials for devices with optical, photonic or magnetic applications [28,
29] or for the development of gas and liquid sensors [27]. They have also been used
for different energy related applications including photoelectrochemical cells [27],
solid-state hydrogen storage [30] and electrocatalysis for oxygen reduction in polymer
electrolyte membrane fuel cells [31] and for the 2-propanol oxidation in alkaline direct
alcohol fuel cells [32].
Hence, a novel Cu electrode prepared by OAD with both suitable catalytic and
electrical properties was employed in the partial oxidation of methanol (POM) by
electrochemical promotion. As already mentioned, this exothermic reaction is
especially interesting for on board H2 production due to the liquid nature of methanol,
its high H/C ratio and its availability from a wide variety of sources. The selection of a
Cu catalyst is justified by its lower cost in comparison with noble metals such as Pt or
Pd and by its proven excellent catalytic activity for the POM reaction [33-37]. For this
choice, we have also considered previous works showing the suitability of Cu-based
catalysts to study fundamental aspects of the EPOC phenomenon [38] and its previous
application in different catalytic reactions such as NO reduction by CO [21, 22]
Page 174
Chapter 4
166
preferential oxidation of CO [23] and CO2 hydrogenation [7, 24]. However, to the best
of our knowledge, this is the first time that this non-noble metal catalyst is
electrochemically promoted in a H2 production reaction.
4.2. EXPERIMENTAL
4.2.1. Preparation of the electrochemical catalyst
The electrochemical catalyst consisted of a continuous thin Cu layer
(geometric area of 2.01 cm2), which also behaved as working electrode (WE).
Similarly to the preceding chapters, a 19-mm-diameter, 1-mm-thick K-βAl2O3
(Ionotec) pellet was used as cationic solid electrolyte (i.e. as source of K+ promoter
ions), inert Au counter (CE) and reference (RE) electrodes were prepared from an
oganometalic paste (Fuel Cell Materials 233001). The active Cu catalyst-working
electrode film was deposited by evaporation of a Cu target (Goodfellow, 99.9999 %
purity) under vacuum conditions by bombardment with a high kinetic energy (< 5
keV) and intensity (150 mA) electron beam. The vapor was condensed onto the
surface of the substrate (K-βAl2O3) at room temperature in two steps by varying the
zenithal angle, α, formed between the perpendicular to the substrate surface and the
evaporation direction. Firstly, in order to provide a good electrical conductivity to the
catalyst, a fairly compact Cu layer was grown at α = 0º on the K-βAl2O3 pellet. The
microstructure of this compact film consisted of adjacent straight nanocolumns
perpendicular to the substrate (final inclination angle, β = 0º) and an approximate
thickness of 0.8 µm. Subsequently, a porous Cu layer with a tilted columnar
microstructure was deposited in a similar way as the former but fixing the zenithal
evaporation angle at α = 80º. This top layer presented a porosity of around 50 %
associated with large void spaces between separated nanocolumns tilted by an angle β
≈ 60º with respect to the perpendicular to the substrate. It was expected that the high
porosity of this outer layer favours the catalytic activity of the copper film. The total
thickness of the obtained Cu catalyst-working electrode was 1.6 µm approximately,
and the final metal loading was 1.08 mg Cu cm-2
(1.69 x 10-5
mol Cu cm-2
). Thus, this
novel preparation technique allowed to prepare a suitable copper electrode combining
both good electrical and catalytic properties, as shown below. Similar layers were also
Page 175
Electrochemical promotion of Cu nanocolumns in the partial oxidation of methanol
167
deposited at different evaporation angles on silicon supports for characterization
purposes. More details about the experimental setup employed in the thin catalyst film
preparation can be found elsewhere [39, 40].
This catalyst preparation method was carried out in collaboration with Dr.
Agustín R. González-Elipe and Dr. Víctor J. Rico from the Institute of Materials
Science of Seville (CSIC – University of Seville).
The resulting Cu/K-βAl2O3/Au electrochemical catalyst was placed into the
single chamber solid electrolyte cell reactor and, although the Cu catalyst film
presented good electrical conductivity after its deposition (surface electrical resistance
of around 8 Ω), prior to the catalytic activity measurements it was subjected to a 25 %
H2 stream (Ar balance) while heating to 280 ºC (ramp of 5 ºC/min) to ensure its
complete reduction. The electrochemical promotion (EPOC) experiments were carried
out in the potentiostatic operation mode, i.e., by varying the applied catalyst potential
measured between the working and reference electrodes (VWR) according to the
procedure generally used in conventional three-electrodes electrochemical cells [41].
4.2.2. Characterization measurements
The deposited porous Cu layer was firstly characterized by X-Ray Diffraction
(XRD) analysis using a Philips PW-1710 instrument with Cu Kα radiation (λ = 1.5404
Å). The surface and in-depth microstructure of the Cu films deposited on silicon
substrates were examined by plan-view and cross-sectional scanning electron
microscopy (SEM) analysis using a Hitachi S4800 field emission microscope operated
at 2 keV. Given the possibility of Cu oxidation under reaction conditions and thus
electrical conductivity loss, the in-plane surface electrical resistance was continuously
measured by connecting the catalyst film to an additional gold wire and a digital
multimeterv (as in chapter 2 during the temperature-programmed experiment). Cyclic
voltammetry (CV) was also carried out at a scan rate of 5 mV s-1
with the potentiostat-
galvanostat (PGSTAT320-N, Metrohom Autolab) before the EPOC experiments.
Moreover, the electrochemical catalyst was characterized after the
electrochemical promotion experiments. Previously, the Cu catalyst film was exposed
Page 176
Chapter 4
168
to the reaction mixture (CH3OH/O2 = 4.4 %/0.3 %) at 320 ºC while applying a
negative potential, VWR = -0.5 V in order to electrochemically supply K+ from the K-
βAl2O3 pellet to the Cu electrode. After 1 hour, the electrochemical catalyst was
cooled down to room temperature under reaction atmosphere. The applied potential
was interrupted (open circuit conditions) at 100 ºC. The aim of this procedure was to
“freeze out” the catalyst surface conditions pertaining to its electropromoted state.
Then the electrochemical catalyst was transferred to different characterization
equipments. In first place, the used catalyst film was characterized by XRD to
examine the final crystalline structure. SEM was performed along with Energy-
Dispersive X-ray spectroscopy (EDX) elemental mapping analysis by means of a
Bruker X-Flash Detector 4010 to identify the surface segregation of different
elements. X-ray photoelectron spectra (XPS) were also recorded with a PHOIBOS-
100 spectrometer with Delay Line Detector (DLD) from SPECS, which worked in the
constant pass energy mode fixed at 30 eV. Monochromatic Mg Kα radiation was used
as the excitation source and the binding energy (BE) scale of the spectra was
referenced to the C 1s of graphitic carbon taken at 284.6 eV.
4.2.3. Catalytic activity measurements
The catalytic activity measurements were carried out in the experimental setup
described in section 1.2.3. Methanol partial oxidation (POM) experiments were
performed at atmospheric pressure with an overall gas flow rate of 6 NL h-1
,
temperatures from 280 ºC to 360 ºC and a feed composition of CH3OH/O2 = 4.4 %/0.3
% (Ar balance). The detected products were: H2, CO2, H2O and HCOOCH3 (methyl
formate). The error in the carbon atom balance did not exceed 5 %, indicating no
consistent loss of material and no significant formation of other oxygenated species.
4.3. RESULTS AND DISCUSSION
4.3.1. Preliminary characterization of the catalyst film
Figure 4.1 shows the X-ray diffractogram of an oblique angle deposited copper
film on a silicon substrate at α = 80º. This diffractogram evidences that the as-
deposited Cu catalyst film exhibited a well-defined face-centered cubic (FCC)
Page 177
Electrochemical promotion of Cu nanocolumns in the partial oxidation of methanol
169
crystalline structure (JCPDS, 89-2838) depicting the (111), (200), (220), (311) and
(222) diffraction peaks of copper at 2θ = 43.2º, 50.4º, 74.1º, 89.9º and 95.1º,
respectively. A very intense Si(400) peak was also found at 2θ = 69.1º (JCPDS, 35-
1241), which corresponded to the substrate.
Figure 4.1. XRD spectrum of the Cu film as-deposited by OAD on a silicon substrate.
The microstructure of the Cu layers with different porosities prepared by
either normal or oblique evaporation (i.e., OAD) on silicon substrates was
characterized by SEM. Figures 4.2a and 4.2b show the cross-section and plan-view
images, respectively, of the rather compact layer prepared at α = 0º and reveal that it
was formed by perpendicular thin and close-packed nanocolumns as expected for
metal thin films deposited under the conditions of the zone I of the zone structure
model [42]. This first compact Cu layer constituted the inner layer of the copper
electrode and provided a very good electrical conductivity (6 – 8 Ω) as well as a high
mechanical stability to the electrocatalytic cell. Nevertheless, as observed in the top
view of Figure 4.2b, this film presents some diffusion surfaces at the interfacial
0 10 20 30 40 50 60 70 80 90 100
Inte
nsi
ty /
a.u
.
2θ / º
Cu
Si
40 50 60 70 80 90 100
Inte
nsi
ty /
a.u
.
2θ / º
(200)
(220)
(222)(311)
(111)
Page 178
Chapter 4
170
boundaries of Cu columns which may allow the migration of potassium ions through
itself to the gas-exposed Cu catalyst surface.
Figure 4.2. Cross-sectional and top view SEM images of the Cu films as-prepared on silicon
substrate at a normal geometry (a and b, respectively) and at an angle of deposition of 80 º (c
and d, respectively). These layers constituted the inner and outer Cu layers at the
electrochemical catalyst, respectively.
On the other hand, Figures 4.2c and 4.2d show the microstructure of the
copper layer prepared by OAD at α = 80º. A thickness of around 0.8 µm and a
microstructure consisting of tilted and separated nanocolumns with an inclination β =
60º can be deduced from the cross section image. The open porosity of this layer,
which was in practice extended from the interface with the dense copper underlayer up
to the external surface of the catalyst, provided a free access and intimate contact with
the reaction gases. Although not all columns presented exactly the same height, a quite
uniform nanocolumn cross-sectional diameter of around 120 nm and a column density
of the order of 109 columns per cm
2 can be also deduced from these images. Similar
Page 179
Electrochemical promotion of Cu nanocolumns in the partial oxidation of methanol
171
microstructures obtained by OAD have been reported for a large variety of materials
(see, for example, previous works on Co [43], Pt-Ni [31], TiO2 [28, 29, 39] or Ta2O5
[29, 40] films).
Hence, the OAD technique showed its capability to deposit metallic films with
a suitable porous microstructure for catalytic and electrocatalytic purposes. In
particular, an electrode film was herein developed consisting of a copper compact-
porous bilayer (which will be henceforth treated as a single film) that was utilized as
an electrochemical catalyst in the POM reaction.
Prior to the EPOC experiments, the Cu catalyst film was also
electrochemically characterized. Figure 4.3 shows the current (I) variation with the
applied potential (VWR) under the studied POM reaction conditions at a quite slow
scan rate of 5 mV/s.
Figure 4.3. Current (I) variation vs. the applied potential (VWR) during the cyclic voltammetry
between +1 and -1 V (scan rate = 5 mV/s). CH3OH/O2 = 4.4 %/0.3 %, 320 ºC.
This voltammogram was found to be very reproducible from the second cycle.
Other important feature is the appearance of clear cathodic and anodic peaks in the
studied potential range, which can be linked to the formation and decomposition of
-9
-7
-5
-3
-1
1
3
5
7
-1.2 -0.8 -0.4 0 0.4 0.8 1.2
I / A
x 1
0-5
VWR / V
Page 180
Chapter 4
172
promoter-derived surface compounds, respectively. The area of the two peaks from
their respective baselines showed to be quite similar, with a difference of near 10 %.
Moreover, this similarity in both cathodic and anodic operation was even higher by
considering the positive and negative I vs time curves obtained during the EPOC
experiments as will be observed below. This means that a positive potential of 1 V led
to the decomposition of practically all the promoter compounds previously formed on
the catalyst surface during the cathodic scan. One can find in literature a wide range of
positive unpromoted potentials employed in alkali-based electropromoted systems,
from +0.1 V [21] or +0.7 V [38] up to +3 V [11] or +4 V [24]. Hence, although in the
previous chapters the reference (un-promoted) state was set at VWR = +2 V, in the
present study a reference potential of +1 V was selected, since this was the lowest
possible value to avoid the deterioration of the catalyst film and/or the solid
electrolyte, while providing a completely reversible electrocatalytic behavior (Figure
4.3).
4.3.2. Electrochemical promotion experiments
The Cu/K-βAl2O3/Au solid electrolyte cell was tested in the methanol partial
oxidation reaction with a feed composition of CH3OH/O2 = 4.4 %/0.3 % (Ar balance)
under electrochemical promotion conditions.
Figure 4.4 shows the dynamic response of the reaction rate of the detected
products, i.e., hydrogen, carbon dioxide and methyl formate, as well as the current (I)
vs. time curves obtained upon the application of different catalyst potentials (VWR)
between +1 and -1 V at a temperature of 320 ºC. Each polarization step was performed
for 1 hour to reach a steady-state rate value. Similarly to the previous chapters and to
other EPOC studies based on K+-conductor materials [11, 15, 19, 44, 45], the
application of a positive potential (VWR = +1 V) at the beginning of the experiment led
to the removal of all the potassium promoter possibly located on the surface of the
catalyst/working electrode. Hence, this potential was defined as the reference state and
its imposition before each different polarization allowed to start from the unpromoted
state in all cases and to check the reversibility and reproducibility of the promotional
effect.
Page 181
Electrochemical promotion of Cu nanocolumns in the partial oxidation of methanol
173
Figure 4.4. Response of H2, CO2 and HCOOCH3 production rates (a) and obtained electric
current (I) (b) vs. time to step changes in the applied catalyst potential (VWR). CH3OH/O2 = 4.4
%/0.3 %, 320 ºC.
In Figure 4.4a, it can be observed that, under unpromoted conditions (VWR =
+1 V), the Cu catalyst film already showed certain catalytic activity for the production
of hydrogen and carbon dioxide, as well as also some water (not depicted), from the
methanol partial oxidation reaction. CO2 was the main product obtained in this
reference (unpromoted) state. Neither carbon monoxide nor formaldehyde were
detected. However, an appreciable amount of methyl formate (HCOOCH3) was
0
2
4
6
8
10
-1.5
-1
-0.5
0
0.5
1
1.5
r /
mo
l s-1
x 1
0-8
VW
R/
V
VWR
rH2rCO2
rHCOOCH3
-4
-3
-2
-1
0
1
2
-1.5
-1
-0.5
0
0.5
1
0 100 200 300 400 500 600I /
A x
10
-4
VW
R/
V
Time / min
VWR I
a)
b)
Page 182
Chapter 4
174
produced, as in chapter 3, although here the HCOOCH3 selectivity was appreciably
lower. As previously mentioned, this compound is a very interesting by-product with
many industrial applications; for example, as a precursor in the synthesis of formic
acid, formamide and dimethylformamide. The decrease in the applied potential to VWR
= +0.5 V caused only a slight increase in H2 and CO2 production rates. This potential
was close to the open circuit value ( ), which oscillated in the range of 0.3-0.5 V
throughout the entire chapter (under different reaction temperatures). Hence, this first
decrease in the applied potential did not constitute a clear negative overpotential, thus
not a significant transfer of potassium species.
However, under the application of VWR = 0 V, the reaction rate of all products
(H2, CO2 and HCOOCH3) sharply increased (electrochemically promoted state), being
H2 the main reaction product (9.27 x 10-8
mol H2 s-1
). It should be noted that, in this
case, the applied potential was already lower than the open circuit potential value,
leading to the observed negative currents (Figure 4.4b), i.e., under these conditions, K+
ions were electrochemically pumped from the K-βAl2O3 pellet to the Cu catalyst film.
An increase in the alkali concentration on the catalyst surface upon decreasing the
applied potential has been previously demonstrated, for instance, by Prof. Lambert and
co-workers by means of in-situ XP spectroscopy studies on Cu [21, 22, 38], Pt [46]
and Rh [47]. Hence, the observed modification of the Cu catalytic activity with the
applied potential can be attributed to a decrease in the catalyst work function as a
consequence of the migration of electropositive ions and to the subsequent
modification of the chemisorptive properties of the Cu during the reaction [38].
Regarding the reaction mechanism, it is widely accepted that the first reaction
step in the methanol oxidation processes is the formation of methoxy species from
methanol (reactions 4.1 and 4.2), which is favoured by the presence of surface oxygen
(reactions 4.3 and 4.4) through a dissociative adsorption mechanism [34-36].
CH3OH → CH3OH(a) (4.1)
CH3OH(a) → CH3O(a) + H(a) (4.2)
O2 → 2O(a) (4.3)
Page 183
Electrochemical promotion of Cu nanocolumns in the partial oxidation of methanol
175
CH3OH + O(a) → CH3O(a) + OH(a) (4.4)
Then, several consecutive reactions may take place leading to the production
of CO2 (reactions 4.5-4.7), H2 (reaction 4.8) and H2O (reaction 4.9) [36].
CH3O(a) + O(a) → H2CO(a) + OH(a) (4.5)
H2CO(a) + O(a) → HCOO(a) + H(a) (4.6)
HCOO(a) → H(a) + CO2 (4.7)
2H(a) → H2 (4.8)
H(a) + OH(a) → H2O (4.9)
On the other hand, the formation of methyl formate can be explained through
different reaction pathways and intermediate species reported elsewhere [34, 37, 48,
49] for the methanol oxidation over Cu catalysts. For instance, the following reaction
mechanism based on the occurrence of methoxy species could be proposed [49]:
CH3O(a) + O(a) → HCOO(a) + H2 (4.10)
CH3O(a) + HCOO(a) → HCOOCH3(a) + O(a) (4.11)
HCOOCH3(a) → HCOOCH3 (4.12)
According to the theory of the electrochemical promotion [41], under
electropromoted conditions (VWR ≤ 0 V in this work) the back-spillover of K+ ions
from the solid electrolyte onto the catalyst/working electrode would strengthen the
chemical bond between copper and electron-acceptor adsorbants, while the bonding
with the electron-donor ones should be weaken. Under POM conditions, these
molecules have been previously identified as O2 and CH3OH, respectively. Hence, the
enhancement of the Cu catalytic activity observed under negative polarization (Figure
4.4a) is in good agreement with POM studies [35] where a positive order of the
reaction rate was found with respect to the oxygen partial pressure (for low values, as
in this case). Likewise, an increase in the methanol reaction rate with O2 concentration
was found in previous EPOC studies with different electrochemical catalysts [50, 51],
and also in Figure 1.7 (chapter 1) until certain oxygen concentration was exceeded. A
Page 184
Chapter 4
176
beneficial effect of alkali addition upon cathodic polarization was also observed over
Cu catalysts in other studies on NO reduction by CO [21, 22] and, very recently, on
CO2 hydrogenation under certain reaction conditions [24]. In the present study, the
adsorbed oxygen would play an important role by favouring the O-H bond rupture in
reaction 4.4. Nevertheless, the rate-limiting step in this kind of processes is usually
considered the methoxy decomposition (reactions 4.5 and 4.10) [35, 36]. The
electrophilic EPOC effect could have also favoured the C-H bond cleavage in the
methanol and intermediates decomposition reactions [50, 51].
Figure 4.4a also shows that all the products reaction rates (H2, CO2 and
HCOOCH3) were restored to their initial values upon every positive polarization at
VWR = +1 V. This demonstrates the complete reversibility and reproducibility of the
electropromoted effect, as well as the high stability of the Cu electrode along the
whole experiment. Moreover, optimal promotional conditions were found at VWR = 0
V. A further decrease of the applied potential down to -0.5 and -1 V led again to an
electro-promotional effect. In this case, a less pronounced increase of all the reaction
rates with respect to those found under unpromoted conditions occurred, which can be
attributed to a detrimental effect of an excess of alkali promoter and the concomitant
formation of alkali-derived surface compounds that may partially block catalytic
active sites. This is a common effect observed both in EPOC [15, 19, 44, 45] and in
conventional alkali chemical promotion studies [52, 53].
Regarding the possible oxidation of copper during this experiment, the in-
plane surface electrical resistance was continuously measured leading to values lower
than 10 Ω throughout the entire study. As it will be later discussed, these low
resistance values would indicate that the catalyst/working electrode mainly remained
on its metallic state during the reaction. It should also be mentioned that the methanol
conversion obtained in this work (not depicted) did not exceed the 5 %, which was
probably influenced by the characteristics of the catalyst, in film form, and thus a still
low geometric area. Nevertheless, it is important to stress that both catalytic activity
and selectivity of the Cu film in the POM reaction can be in-situ improved under
polarization conditions and tuned depending on the applied potential (via
Page 185
Electrochemical promotion of Cu nanocolumns in the partial oxidation of methanol
177
electrochemical promotion). This feature of the EPOC phenomenon has been shown in
the previous chapters with alkali-promoted noble metals catalysts. In the present
study, the Cu-catalyzed H2 production was enhanced up to three times at 320 ºC, the
H2 selectivity being increased from 27.4 % (unpromoted conditions, VWR = +1 V) to
39.1 % (optimum electropromoted conditions, VWR = 0 V). These values were
calculated as H2,out CH3OH,in CH3OH,out , where Fi,in and Fi,out are the
molar flow rate of the i species at the inlet and at the outlet of the reactor, respectively.
Moreover, from the integration of the current vs. time curves (Figure 4.4b), the
transferred amount of promoter, and hence the promoter coverage (potassium to
copper ratio, θK+) can be estimated at each potential through the following equations:
=
(4.13)
=
(4.14)
where n is +1 in this case, F is the Faraday constant (96485 C), t is the polarization
time and N is the total amount of Cu deposited (3.4 x 10-5
mol Cu). θK+ was estimated
in this way due to the difficulty of calculating the Cu active sites in this
electrocatalytic system. The potassium coverage varied from 1 x 10-3
(at VWR = +0.5
V) to 8.5 x 10-3
(at VWR = -1 V). Hence, as in the previous chapters, the EPOC allowed
to determine the optimum promoter coverage (θK+,opt = 6.3 x 10-3
, obtained at VWR = 0
V) which led to the maximum reaction rate (9.27 x 10-8
mol H2 s-1
).
The influence of the reaction temperature on the copper catalytic activity was
studied at a fixed feed composition (CH3OH/O2 = 4.4 %/0.3 %) and different applied
potentials. Figure 4.5 shows the variation of the steady-state reaction rates vs. the
applied catalyst potential at different reaction temperatures between 280 and 360 ºC.
In order to evaluate the magnitude of the electropromotional effect, the maximum rate
enhancement ratios (ρmax) for the optimum applied potential are also included in the
figure. This parameter was calculated, as in previous chapters, as follows:
=
(4.15)
Page 186
Chapter 4
178
where r and r0 are the promoted (VWR < +1 V) and unpromoted (VWR = +1 V) catalytic
reaction rates.
Figure 4.5. Steady-state H2 (a), CO2 (b) and HCOOCH3 (c) production rates vs. the applied
potential (VWR) as well as the corresponding maximum production rates enhancement ratios
(ρmax) at different reaction temperatures. CH3OH/O2 = 4.4 %/0.3 %.
0
3
6
9
12
15
r H/
mo
l s-1
x 1
0-8280 ºC
320 ºC
360 ºC
2
ρH ,max = 1.582
ρH ,max = 2.632
ρH ,max = 2.242
2
4
6
8
10
12
r CO
/ m
ol s
-1x
10-8
280 ºC
320 ºC
360 ºC
2
ρCO ,max = 1.582
ρCO ,max = 1.582
ρCO ,max = 1.552
0
0.5
1
1.5
2
-1.2 -0.8 -0.4 0 0.4 0.8 1.2
r HC
OO
CH
/ m
ol s
-1x
10-8
VWR / V
280 ºC
320 ºC
360 ºC
3
ρHCOOCH ,max = 1.913
ρHCOOCH ,max = 2.703
ρHCOOCH ,max = 1.413
a)
b)
c)
Increasing amount of transferred K+
Page 187
Electrochemical promotion of Cu nanocolumns in the partial oxidation of methanol
179
Starting from the reference unpromoted state (VWR = +1 V) and according to
the previous considerations, the decrease in the applied potential caused the
electrochemical pumping of K+ ions from the solid electrolyte to the Cu catalyst film.
As a result, at the three studied temperatures, all the products reaction rates (H2, CO2,
HCOOCH3) increased because of the potassium promotional effect, evidencing an
electrophilic EPOC behavior (Δr > 0, ρ > 1, upon negative polarization) [41]. It should
be noted that at every reaction temperature, the same operational procedure was
carried out than in Figure 4.4, i.e., a potential of +1 V was applied at the end of every
experiment and before each different applied potential. In this way, both the
reversibility and the reproducibility of the electropromotional effect were verified at
all the reaction temperatures, as well as the stability of the Cu catalytic activity
throughout the entire study.
Under unpromoted conditions (VWR = +1 V), the Cu catalytic activity
increased with the reaction temperature in good agreement with the numerous POM
studies over conventional Cu supported catalysts (e.g., Cu/ZnO [35], Cu/ZnO/Al2O3
[37, 48] or Cu/ZrO2 [33]). As an exception, a decrease in the methyl formate
production rate was found at the highest temperature (360 ºC), which can be attributed
to its further oxidation to CO2 as reported in other works with Cu-based catalysts [37,
48, 49]. Meanwhile, under optimum electropromoted conditions (VWR = 0 V), an
increase in both the H2 and HCOOCH3 production rate enhancement ratios (ρ) was
found when increasing the temperature from 280 to 320 ºC, due to an increase in both
the copper catalytic activity and the solid electrolyte ionic conductivity at fixed
potential. The less pronounced EPOC effect found at 360 ºC should be likely related
with the higher unpromoted reaction rate (r0). In line with the results shown in Figure
4.4, a possible poisoning effect by potassium-derived surface compounds could also
take place under the application of the lowest applied potentials (VWR = -0.5 and -1 V).
The POM results obtained at different temperatures showed that the catalytic
performance of this novel Cu film prepared by OAD can be enhanced in a controlled
and reversible way via alkaline electrochemical promotion. Under optimal reaction
conditions (320 ºC, VWR = 0 V), the hydrogen and methyl formate production rates
Page 188
Chapter 4
180
increased by 2.63 and 2.70 times, respectively. It is clear that the catalytic activity of
this Cu catalyst is far below that of the Pt catalysts. However, it must be stressed that
the maximum enhancement of the H2 production rate obtained here ( )
was higher, for instance, than that previously obtained in chapter 2
with an electrochemical catalyst based on platinum nanoparticles (0.014 mg
Pt/cm2) and similar methanol partial oxidation conditions (320 ºC and O2/CH3OH ratio
= 0.08). These results show that, under the explored reaction conditions, a non-noble
metal catalyst such as Cu can be electrochemically activated in a similar or even more
pronounced way than a noble metal catalyst such as Pt. In this way, one can suggest
that EPOC phenomena could be used to increase the catalytic activity of a non-noble
metal up to the typically higher activity of a noble catalyst, for example, in H2
production processes.
4.3.3. Post-reaction characterization of the catalyst film
It should be remarked that, during the conditioning of the catalyst prior to the
post-reaction characterization (320 ºC, POM conditions, VWR = -0.5 V), the obtained
reaction rates were very similar to those obtained during the first experiment
performed at 320 ºC (Figure 4.4). This verifies the good reproducibility of Cu catalytic
activity even after heating up to 360 ºC (for the study of the temperature influence).
The XRD spectrum of the used catalyst film after the EPOC experiments is
shown in Figure 4.6. This diffractogram shows again the five diffraction peaks
corresponding to the typical face-centered cubic structure (JCPDS, 89-2838) of
metallic copper. An average Cu particle size (d) of 43 nm can be estimated from the
width of the main diffraction peak, Cu(111), by means of the Scherrer formula. Peaks
associated with the K-βAl2O3 pellet (JCPDS, 21-618) can also be observed, but no
diffraction peaks related to potassium compounds like carbonates or bicarbonates were
found. Neither peaks associated with copper oxides like CuO nor Cu2O were detected.
However, the minority presence of these compounds cannot be ruled out because
some overlapping may take place with the peaks of alumina, for instance, in the case
of the CuO peak at 2θ = 35.4º (JCPDS, 80-1917).
Page 189
Electrochemical promotion of Cu nanocolumns in the partial oxidation of methanol
181
Figure 4.6. XRD diffractogram of the electrochemical catalyst after the EPOC experiments
(320 ºC, CH3OH/O2 = 4.4 %/0.3 %, VWR = -0.5 V for 1 h).
Figure 4.7 shows the SEM micrograph of a selected region of the catalyst-
electrode where highly grown surface crystals were observed. Its corresponding
elemental mapping and spectra by EDX are also reported.
In Figures 4.7b, large concentrations of potassium (in blue) were found on
certain areas of the Cu surface (in green). This seems to indicate that some potassium-
derived surface compounds were formed on the Cu surface during the EPOC
experiments and might block Cu active sites causing the decrease in the reaction rates
observed in Figures 4.4 and 4.5 under the application of high negative potentials. As
suggested in chapter 1, the most feasible K-derived surface compounds under our
working conditions would be potassium oxides and/or carbonates formed by reaction
with O2, H2O and carbon-containing molecules in the reaction mixture.
0 10 20 30 40 50 60 70 80 90 100
Inte
nsi
ty /
a.u
.
2θ / º
(111)
Cu
K-βAl2O3
(200)
(220)
(222)(311)
Page 190
Chapter 4
182
Figure 4.7. Top view SEM image of a selected area of the electrochemical catalyst (a) after the
EPOC experiments (320 ºC, CH3OH/O2 = 4.4 %/0.3 %, VWR = -0.5 V for 1 h), along with the
corresponding elemental mapping (b1 and b2) of Cu (green), K (blue) and N (red) and the
EDX spectra from different regions (c1 and c2).
These potassium oxide or carbonate compounds were previously found over
other metal catalysts such as Rh [54], Pt [45] or Ag [44], and may be obtained, for
instance, through the following electrocatalytic reactions:
2K+ + ½O2 + 2e
- → K2O (4.16)
2K+ + ½O2 + CO2 + 2e
- → K2CO3 (4.17)
Although the potassium distribution along the catalyst thickness was not
studied, it was important to demonstrate that K+ ions were able to migrate through the
catalyst film and reach the outermost surface. The presence of potassium compounds
was also revealed by EDX analysis taken in different areas of the micrograph. Figure
4.7c(2) shows that the K and O signals were much higher in the spectra taken in the
agglomerates than those from the flat region (Figure 4.7c(1)). It is also worth noting
0 1 2 3 4 5 6 7 8 9 10
Inte
nsi
ty /
a.u
.
Energy / keV
0 1 2 3 4 5 6 7 8 9 10
Inte
nsi
ty /
a.u
.
Energy / keV
3 µma)
b1) b2)
c2)c1)
Cu
OK
Cu
K
Cu
O
K
Cu
K
Page 191
Electrochemical promotion of Cu nanocolumns in the partial oxidation of methanol
183
that nitrogen (red points) was found uniformly distributed on the film surface. This
elemental nitrogen can only proceed from the fed air stream (source of the oxygen
reactant), and its presence on the catalyst surface needed to be confirmed by XPS
analysis.
Figure 4.8 shows all the obtained XPS spectra. In this way, the chemical state
and composition of the outer surface layers of the Cu catalyst were studied.
Figure 4.8. XPS spectra of the electrochemical catalyst after the EPOC experiments (320 ºC,
CH3OH/O2 = 4.4 %/0.3 %, VWR = -0.5 V for 1 h). Spectra taken at different binding energy
regions to show the presence of the different elements present on the surface of the sample (a).
The corresponding surface atomic composition expressed in percentages (b).
a)
b)
0
10
20
30
40
50
O C K Al N Cu Na
At
/ %
945 940 935 930 925
Counts
/ a
.u.
Binding energy (eV)
Cu 2p3/2
540 535 530 525
Binding energy (eV)
O 1s
405 400 395 390
Binding energy (eV)
N 1s
345 340 335 330
Counts
/ a
.u.
Binding energy (eV)
Cu LVV
300 295 290 285 280
Binding energy (eV)
C 1s + K 2p
85 80 75 70 65
Binding energy (eV)
Cu 3p + Al 2p
Page 192
Chapter 4
184
The Cu 2p photoemission (BE = 932.4 eV) and Cu LVV Auger (kinetic energy
915.14 eV) spectra obtained (Figure 4.8a) can be used to determine the Auger
parameter of copper (AP = BE photoemission peak + kinetic energy Auger peak)
which presented a value of 1847.54 eV, typical of Cu+ [55] (neither Cu
0 nor Cu
2+).
This finding would agree with some studies where the Cu active state in the methanol
partial oxidation reaction is considered Cu+ [48]. However, it should be noted that the
XPS characterization is only sensitive to a surface thickness of a few nanometers and
that, therefore, the Cu2O observed at the outermost catalyst layer could also be
attributed, at least in part, to the exposure of the sample to the air atmosphere during
its transfer from the reaction cell to the characterization equipment. An oxidation
limited to the outermost surface layers would be consistent with the very low surface
electrical resistance (below 10 ohms) and the XRD analysis of the utilized catalyst,
which would remain mainly as metallic copper under the studied reaction conditions.
On the other hand, the N 1s peak observed at 398 eV demonstrated the
presence of some reduced nitrogen on the catalyst surface, probably derived from a
certain K-promoted interaction between nitrogen from air and produced hydrogen. It is
well known that ammonia synthesis is typically promoted by potassium [53].
Furthermore, the ammonia synthesis reaction has been promoted by H+ ions in
previous EPOC studies with Fe catalysts under both high pressure [2] and atmospheric
pressure [20]. The C 1s signal in the spectrum of the used sample also shows the
presence of surface carbon mostly in the form of graphitic or hydrogenated carbon.
This indicates that a certain coking could have taken place at the highest reaction
temperatures. However, at least part of the O, C and K amounts observed in this figure
can be associated to promoter derived species as previously mentioned. The
aluminium also found in this XPS spectrum can be attributed to some uncovered
regions of the K-βAl2O3 electrolyte which might have appeared due to small scratches
caused by the contact with the gold wires. A quantitative estimation of the elemental
concentration in the examined outer layer of the electrochemical catalyst can be
observed in Figure 4.8b.
Page 193
Electrochemical promotion of Cu nanocolumns in the partial oxidation of methanol
185
4.4. CONCLUSIONS
The following conclusions can be drawn from this study:
- The Oblique Angle Deposition (OAD) technique is a straightforward method
to prepare a kind of novel Cu catalytic films with a tuneable microstructure.
Combining the physical vapour deposition at normal and oblique angles for the
fabrication of a bilayer film yielded a Cu catalyst film with both a high surface
electrical conductivity and a high porosity (around 50 %), very suitable for catalytic
and electrocatalytic applications.
- A non-noble catalyst was electrochemically activated in the partial oxidation
of methanol. The catalytic activity of the Cu electrode was enhanced by
electrochemically supplying of K+ ions in a controlled, reversible and reproducible
way. The observed promotional effect can be explained according to the rules of
chemical/electrochemical promotion.
- Under electrochemical promotion conditions, the formation of potassium-
derived surface compounds, likely potassium carbonates or oxides, was confirmed by
post-reaction EDX and XPS characterization.
- Adsorbed nitrogen functional groups seemed to be formed under the
potassium promotional effect due to the presence of N2 and H2 in the reaction
atmosphere. Some graphitic or hydrogenated carbon was also deposited during the
EPOC experiments.
4.5. REFERENCES
[1] P. Vernoux, L. Lizarraga, M.N. Tsampas, F.M. Sapountzi, A. De Lucas-Consuegra, J.L.
Valverde, S. Souentie, C.G. Vayenas, D. Tsiplakides, S. Balomenou, E.A. Baranova, Ionically
conducting ceramics as active catalyst supports, Chemical Reviews, 113 (2013) 8192-8260.
[2] C.G. Yiokari, G.E. Pitselis, D.G. Polydoros, A.D. Katsaounis, C.G. Vayenas, High-
pressure electrochemical promotion of ammonia synthesis over an industrial iron catalyst,
Journal of Physical Chemistry A, 104 (2000) 10600-10602.
Page 194
Chapter 4
186
[3] D. Poulidi, M.E. Rivas, B. Zydorczak, Z. Wu, K. Li, I.S. Metcalfe, Electrochemical
promotion of a Pt catalyst supported on La 0.6Sr 0.4Co 0.2Fe 0.8O 3 - δ hollow fibre
membranes, Solid State Ionics, 225 (2012) 382-385.
[4] S. Balomenou, D. Tsiplakides, A. Katsaounis, S. Thiemann-Handler, B. Cramer, G. Foti, C.
Comninellis, C.G. Vayenas, Novel monolithic electrochemically promoted catalytic reactor for
environmentally important reactions, Applied Catalysis B: Environmental, 52 (2004) 181-196.
[5] S.P. Balomenou, D. Tsiplakides, C.G. Vayenas, S. Poulston, V. Houel, P. Collier, A.G.
Konstandopoulos, C. Agrafiotis, Electrochemical promotion in a monolith electrochemical
plate reactor applied to simulated and real automotive pollution control, Topics in Catalysis,
44 (2007) 481-486.
[6] S. Souentie, A. Hammad, S. Brosda, G. Foti, C.G. Vayenas, Electrochemical promotion of
NO reduction by C2H4 in 10% O2 using a monolithic electropromoted reactor with Rh/YSZ/Pt
elements, Journal of Applied Electrochemistry, 38 (2008) 1159-1170.
[7] E.I. Papaioannou, S. Souentie, A. Hammad, C.G. Vayenas, Electrochemical promotion of
the CO2 hydrogenation reaction using thin Rh, Pt and Cu films in a monolithic reactor at
atmospheric pressure, Catalysis Today, 146 (2009) 336-344.
[8] M. Marwood, C.G. Vayenas, Electrochemical promotion of a dispersed platinum catalyst,
Journal of Catalysis, 178 (1998) 429-440.
[9] A. Kambolis, L. Lizarraga, M.N. Tsampas, L. Burel, M. Rieu, J.P. Viricelle, P. Vernoux,
Electrochemical promotion of catalysis with highly dispersed Pt nanoparticles,
Electrochemistry Communications, 19 (2012) 5-8.
[10] V. Jiménez, C. Jiménez-Borja, P. Sánchez, A. Romero, E.I. Papaioannou, D. Theleritis, S.
Souentie, S. Brosda, J.L. Valverde, Electrochemical promotion of the co2 hydrogenation
reaction on composite ni or ru impregnated carbon nanofiber catalyst-electrodes deposited on
YSZ, Applied Catalysis B: Environmental, 107 (2011) 210-220.
[11] A. de Lucas-Consuegra, A. Princivalle, A. Caravaca, F. Dorado, A. Marouf, C. Guizard,
J.L. Valverde, P. Vernoux, Preparation and characterization of a low particle size Pt/C
catalyst electrode for the simultaneous electrochemical promotion of CO and C3H6 oxidation,
Applied Catalysis A: General, 365 (2009) 274-280.
[12] A. Lintanf, E. Djurado, P. Vernoux, Pt/YSZ electrochemical catalysts prepared by
electrostatic spray deposition for selective catalytic reduction of NO by C3H6, Solid State
Ionics, 178 (2008) 1998-2008.
[13] V. Roche, R. Revel, P. Vernoux, Electrochemical promotion of YSZ monolith honeycomb
for deep oxidation of methane, Catalysis Communications, 11 (2010) 1076-1080.
Page 195
Electrochemical promotion of Cu nanocolumns in the partial oxidation of methanol
187
[14] A. De Lucas-Consuegra, A. Caravaca, P.J. Martínez, J.L. Endrino, F. Dorado, J.L.
Valverde, Development of a new electrochemical catalyst with an electrochemically assisted
regeneration ability for H2 production at low temperatures, Journal of Catalysis, 274 (2010)
251-258.
[15] A. De Lucas-Consuegra, A. Caravaca, J. González-Cobos, J.L. Valverde, F. Dorado,
Electrochemical activation of a non noble metal catalyst for the water-gas shift reaction,
Catalysis Communications, 15 (2011) 6-9.
[16] T.I. Politova, V.A. Sobyanin, V.D. Belyaev, Ethylene hydrogenation in electrochemical
cell with solid proton-conducting electrolyte, Reaction Kinetics & Catalysis Letters, 41 (1990)
321-326.
[17] I.V. Yentekakis, Y. Jiang, S. Neophytides, S. Bebelis, C.G. Vayenas, Catalysis,
electrocatalysis and electrochemical promotion of the steam reforming of methane over Ni film
and Ni-YSZ cermet anodes, Ionics, 1 (1995) 491-498.
[18] E. Ruiz, D. Cillero, P.J. Martínez, Á. Morales, G.S. Vicente, G. De Diego, J.M. Sánchez,
Bench-scale study of electrochemically assisted catalytic CO2 hydrogenation to hydrocarbon
fuels on Pt, Ni and Pd films deposited on YSZ, Journal of CO2 Utilization, 8 (2014) 1-20.
[19] G.E. Pitselis, P.D. Petrolekas, C.G. Vayenas, Electrochemical promotion of ammonia
decomposition over Fe catalyst films interfaced with K+- & H+- conductors, Ionics, 3 (1997)
110-116.
[20] M. Ouzounidou, A. Skodra, C. Kokkofitis, M. Stoukides, Catalytic and electrocatalytic
synthesis of NH3 in a H+ conducting cell by using an industrial Fe catalyst, Solid State Ionics,
178 (2007) 153-159.
[21] F.J. Williams, A. Palermo, M.S. Tikhov, R.M. Lambert, First Demonstration of in Situ
Electrochemical Control of a Base Metal Catalyst: Spectroscopic and Kinetic Study of the CO
+ NO Reaction over Na-Promoted Cu, Journal of Physical Chemistry B, 103 (1999) 9960-
9966.
[22] R.M. Lambert, F. Williams, A. Palermo, M.S. Tikhov, Modelling alkali promotion in
heterogeneous catalysis: in situ electrochemical control of catalytic reactions, Topics in
Catalysis, 13 (2000) 91-98.
[23] F.M. Sapountzi, M.N. Tsampas, C.G. Vayenas, Methanol reformate treatment in a PEM
fuel cell-reactor, Catalysis Today, 127 (2007) 295-303.
[24] E. Ruiz, D. Cillero, P.J. Martínez, Á. Morales, G.S. Vicente, G. De Diego, J.M. Sánchez,
Electrochemical synthesis of fuels by CO2 hydrogenation on Cu in a potassium ion conducting
membrane reactor at bench scale, Catalysis Today, 236 (2014) 108-120.
Page 196
Chapter 4
188
[25] G. Karagiannakis, S. Zisekas, M. Stoukides, Hydrogenation of carbon dioxide on copper
in a H+ conducting membrane-reactor, Solid State Ionics, 162-163 (2003) 313-318.
[26] E. Mutoro, C. Koutsodontis, B. Luerssen, S. Brosda, C.G. Vayenas, J. Janek,
Electrochemical promotion of Pt(111)/YSZ(111) and Pt-FeOx/YSZ(111) thin catalyst films:
Electrocatalytic, catalytic and morphological studies, Applied Catalysis B: Environmental,
100 (2010) 328-337.
[27] M. Matthew, M.T. Hawkeye, M.J.B. Taschuk, Glancing Angle Deposition of Thin Films:
Engineering the Nanoscale, Wiley2014.
[28] M. Oliveros, L. González-García, V. Mugnaini, F. Yubero, N. Roques, J. Veciana, A.R.
González-Elipe, C. Rovira, Novel guests for porous columnar thin films: The switchable
perchlorinated trityl radical derivatives, Langmuir, 27 (2011) 5098-5106.
[29] J.R. Sánchez-Valencia, A. Borrás, A. Barranco, V.J. Rico, J.P. Espinos, A.R. González-
Elipe, Preillumination of TiO2 and Ta2O5 photoactive thin films as a tool to tailor the
synthesis of composite materials, Langmuir, 24 (2008) 9460-9469.
[30] Y. He, J. Fan, Y. Zhao, The role of differently distributed vanadium nanocatalyst in the
hydrogen storage of magnesium nanostructures, International Journal of Hydrogen Energy, 35
(2010) 4162-4170.
[31] N.N. Kariuki, W.J. Khudhayer, T. Karabacak, D.J. Myers, GLAD Pt-Ni alloy nanorods for
oxygen reduction reaction, ACS Catalysis, 3 (2013) 3123-3132.
[32] S.A. Francis, R.T. Tucker, M.J. Brett, S.H. Bergens, Structural and activity comparison of
self-limiting versus traditional Pt electro-depositions on nanopillar Ni films, Journal of Power
Sources, 222 (2013) 533-541.
[33] H. Chen, A. Yin, X. Guo, W.L. Dai, K.N. Fan, Sodium hydroxide-sodium oxalate-assisted
co-precipitation of highly active and stable Cu/ZrO2 catalyst in the partial oxidation of
methanol to hydrogen, Catalysis Letters, 131 (2009) 632-642.
[34] S.T. Yong, C.W. Ooi, S.P. Chai, X.S. Wu, Review of methanol reforming-Cu-based
catalysts, surface reaction mechanisms, and reaction schemes, International Journal of
Hydrogen Energy, 38 (2013) 9541-9552.
[35] L.A. Espinosa, R.M. Lago, M.A. Peña, J.L.G. Fierro, Mechanistic aspects of hydrogen
production by partial oxidation of methanol over Cu/ZnO catalysts, Topics in Catalysis, 22
(2003) 245-251.
[36] S. Sakong, A. Gross, Total oxidation of methanol on Cu(110): A density functional theory
study, Journal of Physical Chemistry A, 111 (2007) 8814-8822.
Page 197
Electrochemical promotion of Cu nanocolumns in the partial oxidation of methanol
189
[37] R.M. Navarro, M.A. Peña, J.L.G. Fierro, Production of hydrogen by partial oxidation of
methanol over a Cu/ZnO/Al2O3 catalyst: Influence of the initial state of the catalyst on the
start-up behaviour of the reformer, Journal of Catalysis, 212 (2002) 112-118.
[38] F.J. Williams, A. Palermo, M.S. Tikhov, R.M. Lambert, The Origin of Electrochemical
Promotion in Heterogeneous Catalysis: Photoelectron Spectroscopy of Solid State
Electrochemical Cells, Journal of Physical Chemistry B, 104 (2000) 615-621.
[39] V. Rico, P. Romero, J.L. Hueso, J.P. Espinós, A.R. González-Elipe, Wetting angles and
photocatalytic activities of illuminated TiO2 thin films, Catalysis Today, 143 (2009) 347-354.
[40] V. Rico, A. Borrás, F. Yubero, J.P. Espinós, F. Frutos, A.R. González-Elipe, Wetting
angles on illuminated ta2o5 thin films with controlled nanostructure, Journal of Physical
Chemistry C, 113 (2009) 3775-3784.
[41] C.G. Vayenas, S. Bebelis, C. Pliangos, S. Brosda, D. Tsiplakides, Electrochemical
Activation of Catalysis: Promotion, Electrochemical Promotion and Metal-Support
Interactions, Kluwer Academic Publishers/Plenum Press, New York, 2001.
[42] J.A. Thornton, INFLUENCE OF APPARATUS GEOMETRY AND DEPOSITION
CONDITIONS ON THE STRUCTURE AND TOPOGRAPHY OF THICK SPUTTERED
COATINGS, J Vac Sci Technol, 11 (1974) 666-670.
[43] D.X. Ye, Y.P. Zhao, G.R. Yang, Y.G. Zhao, G.C. Wang, T.M. Lu, Manipulating the
column tilt angles of nanocolumnar films by glancing-angle deposition, Nanotechnology, 13
(2002) 615-618.
[44] A. Palermo, A. Husain, M.S. Tikhov, R.M. Lambert, Ag-catalysed epoxidation of propene
and ethene: An investigation using electrochemical promotion of the effects of alkali, NOx, and
chlorine, Journal of Catalysis, 207 (2002) 331-340.
[45] A. de Lucas-Consuegra, F. Dorado, J.L. Valverde, R. Karoum, P. Vernoux, Low-
temperature propene combustion over Pt/K-βAl2O3 electrochemical catalyst:
Characterization, catalytic activity measurements, and investigation of the NEMCA effect,
Journal of Catalysis, 251 (2007) 474-484.
[46] I.V. Yentekakis, A. Palermo, N.C. Filkin, M.S. Tikhov, R.M. Lambert, In situ
electrochemical promotion by sodium of the platinum-catalyzed reduction of NO by propene,
Journal of Physical Chemistry B, 101 (1997) 3759-3768.
[47] F.J. Williams, A. Palermo, M.S. Tikhov, R.M. Lambert, Electrochemical promotion by
sodium of the rhodium-catalyzed NO + CO reaction, Journal of Physical Chemistry B, 104
(2000) 11883-11890.
Page 198
Chapter 4
190
[48] Y. Choi, H.G. Stenger, Fuel cell grade hydrogen from methanol on a commercial
Cu/ZnO/Al2O3 catalyst, Applied Catalysis B: Environmental, 38 (2002) 259-269.
[49] V.A. Matyshak, O.N. Sil'Chenkova, I.T. Ismailov, V.F. Tret'Yakov, Properties of surface
compounds in methanol conversion on copper-containing catalysts based on CeO2 according
to in situ IR-spectroscopic data, Kinetics and Catalysis, 50 (2009) 784-792.
[50] C.G. Vayenas, S. Neophytides, Non-faradaic electrochemical modification of catalytic
activity III. The case of methanol oxidation on Pt, Journal of Catalysis, 127 (1991) 645-664.
[51] J.K. Hong, I.H. Oh, S.A. Hong, W.Y. Lee, Electrochemical oxidation of methanol over a
silver electrode deposited on yttria-stabilized zirconia electrolyte, Journal of Catalysis, 163
(1996) 95-105.
[52] I.V. Yentekakis, M. Konsolakis, R.M. Lambert, N. MacLeod, L. Nalbantian,
Extraordinarily effective promotion by sodium in emission control catalysis: NO reduction by
propene over Na-promoted Pt/γ-al2O3, Applied Catalysis B: Environmental, 22 (1999) 123-
133.
[53] B. Lin, K. Wei, X. Ma, J. Lin, J. Ni, Study of potassium promoter effect for Ru/AC
catalysts for ammonia synthesis, Catalysis Science and Technology, 3 (2013) 1367-1374.
[54] A.J. Urquhart, J.M. Keel, F.J. Williams, R.M. Lambert, Electrochemical Promotion by
Potassium of Rhodium-Catalyzed Fischer-Tropsch Synthesis: XP Spectroscopy and Reaction
Studies, Journal of Physical Chemistry B, 107 (2003) 10591-10597.
[55] J.P. Espinós, J. Morales, A. Barranco, A. Caballero, J.P. Holgado, A.R. González-Elipe,
Interface effects for Cu, CuO, and Cu2O deposited on SiO2 and ZrO2. XPS determination of
the valence state of copper in Cu/SiO2 and Cu/ZrO2 catalysts, Journal of Physical Chemistry
B, 106 (2002) 6921-6929.
Page 199
191
Chapter 5
ELECTROCHEMICAL PROMOTION OF Ni IN
METHANOL CONVERSION REACTIONS:
DIFFERENT APPLICATIONS OF EPOC ON A SINGLE
CATALYTIC SYSTEM
In this work, three possible applications of the phenomenon of electrochemical
promotion of catalysis (EPOC) are demonstrated for methanol conversion processes:
activation of the catalytic activity, modification of the catalytic selectivity and partial
oxidation of the catalyst. A Ni catalyst film prepared by cathodic arc deposition has
been electrochemically promoted by K+, upon negative polarization, in the methanol
decomposition (MD) and steam reforming (SRM) reactions, showing an electrophilic
EPOC behavior. On the other hand, under methanol partial oxidation conditions
(POM), the K+ ions caused the decrease in the catalytic selectivity toward H2 and CO
while favoured that toward CO2 and H2CO, due to an increase in the Ni oxidation
state by the alkali-induced O2 activation. All the potassium-derived effects have been
reversible between the negative and positive polarizations, which shows different
interesting possibilities of the EPOC phenomena in heterogeneous catalysis.
Methanoldecomposition
CHXO
Steam reformingof methanol
H2O CH3OH
Partial oxidation of methanol
O2 CH3OHA A AD D
ΔV < 0
Ni
K+ K+ K+
Au
e-
e-
ELECTOPROMOTEDSTATE
CH3OH CO + 2H2
CO(a) C(a) + O(a)
CH3OH CO + 2H2
CH3OH + H2O CO2 + 3H2
C(a) + H2O CO + H2
CH3OH CO + 2H2
CH3OH + 1/2O2 CO2 + 2H2
CH3OH + 3/2O2 CO2 + 2H2O
CH3OH + 1/2O2 H2CO + H2O
xNi + yO(a) NixOy
A D
A
D
Acceptor
Donor
Activation of thecatalyst
Partial oxidationof the catalyst
Modification of selectivity
Activation of thecatalyst
Modification of selectivity
Page 200
Chapter 5
192
5.1. INTRODUCTION
Based on the results presented in the previous chapters, one can appreciate
some of the main possibilities offered by the phenomenon of electrochemical
promotion (EPOC), in particular in H2 production processes from methanol. One can
also asseverate that the difference between electrochemical and chemical promotion is
operational and not functional, as Prof. Vayenas and co-workers stated in a seminal
book about electrochemical activation of catalysis through promotion, electrochemical
promotion and metal-support interactions (MSI) [1]. There is a very rich literature on
the similarities and differences between these three processes [1-5]. All of them are
based on the addition of promoting species to the catalytic active surface, which
modify its chemisorptive properties and hence its activity and/or its selectivity.
However, in the case of the EPOC phenomenon, these promoter ions are
electrochemically pumped between the metallic catalyst and a solid electrolyte
material (which acts as catalyst support) by means of the external imposition of an
electrical current or potential.
Then, the electrochemical promotion presents several additional advantages
against the classical promotion [3, 6]. One of the greatest is related to the first EPOC
studies with yttria-stabilized zirconia (YSZ) electrolytes, and lies on its capability to
continuously supply promoters on the catalyst surface, which is of special interest in
the case of short-lived effective promoters such as Oδ-
[1]. The electrochemical
promotion can be also carried out in a controlled, reversible way and, very
interestingly, in the course of the catalytic reaction, which has been pointed out
throughout the entire manuscript (especially in chapter 3). Thus, it is possible to
optimize the promoter coverage on the catalyst surface at different reaction conditions,
which is of great practical relevance in non-stationary processes like automotive
catalysis. This fact also enables a fast and direct evaluation of the effect of a specific
promoter on a catalytic system. Furthermore, the EPOC phenomenon is not only
aimed to enhance the catalytic activity and selectivity (as in all the previous chapters),
but also may affect the oxidation state of the catalyst [7-11] and can be focused to
other applications such as the prevention of poisoning effects and catalyst regeneration
Page 201
Electrochemical promotion of Ni in methanol conversion reactions
193
[7, 12]. A large number of examples on the different EPOC applications can be found
in exhaustive reviews [3, 5, 6].
In the case of the methanol conversion reactions, before this research started
up, only four studies [13-16] dealt with the application of the phenomenon of the
electrochemical promotion, specifically using YSZ (O2-
conductor) as solid electrolyte
and Pt [14, 15] or Ag [13, 16] catalyst films. The main catalytic reactions that took
place in these systems were the methanol decomposition to CO (reaction 5.1) and
H2CO (reactions 5.2) along with some methanation (reaction 5.3) [13], and the
methanol oxidation through reactions 5.4 and 5.5 [14-16], thus obtaining
formaldehyde as product of highest interest.
CH3OH → 2H2 + CO (5.1)
CH3OH → H2CO + H2 (5.2)
CH3OH + H2 → CH4 + H2O (5.3)
CH3OH + 1/2O2 → H2CO + H2O (5.4)
CH3OH + 3/2O2 → CO2 + 2H2O (5.5)
Herein, in chapters 1 and 2, the alkali electrochemical promotion was applied
on Pt-based catalysts focused on the production of H2 via methanol steam reforming
(SRM, reaction 5.6) and partial oxidation (POM, reaction 5.7), although formaldehyde
was also obtained through reaction (5.4).
CH3OH + H2O → 3H2 + CO2 (5.6)
CH3OH + 1/2O2 → 2H2 + CO2 (5.7)
CO was also produced in these chapters at large extent, which was not
surprising given the preference of Group VIII metals for the methanol decomposition
(MD, reaction 5.1). However, in chapters 3 and 4, where Au- and Cu-based catalysts
were employed, neither CO nor H2CO were detected. In these cases, besides methanol
partial oxidation (reaction 5.7), methyl formate production also took place, which is
typically postulated by some of the following reactions:
Page 202
Chapter 5
194
CH3OH + 1/2O2 → 1/2HCOOCH3 + H2O (5.8)
CH3OH → 1/2HCOOCH3 + H2 (5.9)
In the present study, the EPOC phenomenon has been applied in this kind of
reactions by using Ni as the active phase, which is indeed commonly employed
particularly in the decomposition and steam reforming of methanol [17-21]. Reactions
5.1, 5.2, 5.6 and 5.7 are of special interest from an energetic and environmental point
of view, as promising on-board H2 production processes from a sustainable liquid
source such as CH3OH. However, important drawbacks are typically found in this
kind of reactions with Group 8-11 metals, and especially with non noble ones such as
Cu or Ni, which are related to the thermal sintering, the deactivation by coke
deposition and undesirable changes in the oxidation state of the catalyst [22, 23].
Thus, these metals are usually supported on certain oxide supports such as CeO2,
ZrO2, Al2O3, ZnO, SiO2 or TiO2 [22-24], which may act as structural and/or electronic
promoters to improve the catalytic activity, decrease the CO selectivity and increase
the durability of the catalyst. The decomposition, steam reforming and partial
oxidation of methanol, as well as the water-gas shift and methanol synthesis reactions,
were also found to be promoted by alkali dopants, typically added as oxides or
hydroxides [25-29]. In the present work, the modifications of the activity and the
selectivity of a Ni catalyst via electrochemical promotion with K+ ions have been
studied under different reaction atmospheres (MD, SRM and POM). This way, three
different possibilities of the EPOC effect with Ni catalyst have been evaluated:
electrochemical activation of catalytic activity, alteration of the catalyst oxidation state
and electrochemical modification of the catalytic selectivity.
5.2. EXPERIMENTAL
5.2.1. Preparation of the electrochemical catalyst
The electrochemical catalyst consisted of a continuous thin Ni film (geometric
area of 2.84 cm2), which also behaved as a working electrode (WE), deposited on a
19-mm-diameter, 1-mm-thick K-βAl2O3 (Ionotec) pellet. Au counter (CE) and
reference (RE) electrodes were prepared by calcination of an organometallic paste
Page 203
Electrochemical promotion of Ni in methanol conversion reactions
195
(Gwent Electronic Materials). Then, the active Ni catalyst-working electrode was
deposited similarly to Pt in chapter 1, i.e., by the filter cathodic arc deposition (CAD)
technique [30]. This physical vapour deposition technique allowed the preparation of a
thin Ni film, 150 nm thick, with a high adhesion to the substrate and a good surface
electrical conductivity, which are necessary for the electrochemical promotion
(EPOC) studies. The final metal loading was 0.13 mg Ni cm-2
, and the obtained Ni/K-
βAl2O3/Au catalyst was placed into the single chamber solid electrolyte cell reactor.
This catalyst preparation method was carried out in collaboration with Dr.
José Luis Endrino from the Institute of Materials Science of Madrid (CSIC).
5.2.2. Characterization measurements
The Ni catalyst film was characterized by X-Ray Diffraction (XRD) analysis
using a Philips PW-1710 instrument with Cu Kα radiation (λ = 1.5404 Å) after
exposing to different reaction mixtures and applied potentials. To this end, the
following steps were carried out before each XRD analysis in order to keep the Ni
unpromoted/promoted states as unaltered as possible: 1) Every time the Ni catalyst
film was introduced into the reactor, a temperature programmed reduction (TPR) was
firstly performed under a 25 % H2 stream (Ar balance) from room temperature to 400
ºC (ramp of 5 ºC min-1
) in order to ensure the Ni fully reduced state. 2) A temperature
of 360 ºC was fixed and the catalyst was subjected for 1 hour to the reaction
atmosphere and the applied potential under study. 3) The reactor temperature was
decreased to 100 ºC by keeping constant the rest of reaction conditions. 4) The catalyst
was finally cooled down to room temperature under Ar atmosphere and open circuit
conditions. Then it was transferred to the characterization equipment under inert
conditions (N2 atmosphere).
5.2.3. Catalytic activity measurements
The catalytic activity measurements were performed in the experimental setup
described in section 1.2.3. Different methanol conversion reactions, i.e.,
decomposition (MD), partial oxidation (POM) and steam reforming (SRM) were
studied by carrying out experiments at atmospheric pressure with an overall gas flow
Page 204
Chapter 5
196
rate of 6 NL h-1
, a maximum temperature of 360 ºC and feed compositions of CH3OH
= 4.4 % for MD reaction, CH3OH/O2 = 4.4 %/0.33 % for POM reaction and
CH3OH/H2O = 4.4 %/5.2 % for SRM reaction (Ar balance). The detected products
were: H2, CO2, CO, H2O and H2CO (the latter two mainly under POM conditions).
5.3. RESULTS AND DISCUSSION
5.3.1. Electrochemical activation of the catalyst
First, the activity of the Ni catalyst film was tested at 360 ºC in the methanol
decomposition reaction, i.e., by feeding the reactor with a 4.4 % CH3OH stream (Ar
balance). H2, CO, and trace amounts of CO2 were detected in the reactor outlet. Figure
5.1 shows the variation in the production rates vs. time obtained under such reaction
conditions and different applied catalyst potentials (VWR) from +2 to -2 V.
Figure 5.1. Response of H2, CO and CO2 production rates vs. time to step changes in the
applied catalyst potential (VWR) under methanol decomposition conditions: CH3OH = 4.4 %,
360 ºC.
-2.5
-1.5
-0.5
0.5
1.5
2.5
0
0.5
1
1.5
2
2.5
3
3.5
4
0 100 200 300 400 500
VW
R/
V
r /
mo
l s-1
x 1
0-7
Time / min
rCO
VWR
rH2
Page 205
Electrochemical promotion of Ni in methanol conversion reactions
197
As discussed in previous chapters and typically stated at electrochemical
catalysts based on alkaline ionic conductors [6], such as K-βAl2O3, the application of a
positive current (I) or potential (VWR) led to the removal from the catalyst-working
electrode of the positive promoter ions (K+) that may have previously migrated
(thermally) or as a consequence of the foregoing polarizations. On the contrary, the
decrease in the applied current or potential below I < 0 or VWR < (open circuit
potential value), respectively, caused the migration of alkaline promoter ions from the
solid electrolyte to the catalyst surface (back-spillover). Hence, at the beginning and at
the end of each experiment, a catalyst potential of VWR = +2 V was imposed in order
to define a clean Ni catalyst film, free of promoter ions and thus a reference
(unpromoted) state. In this way, it was also possible to check the reversibility of the
electrochemical promotion effect. Under these unpromoted conditions (at +2 V and
still at +0.5 V) hydrogen and carbon monoxide were produced, through reaction 5.1, at
rates of around 7 x 10-8
mol H2 s-1
and 2 x 10-8
mol CO s-1
. No other reaction products
were detected. It can be observed, however, that these production rates were
progressively decreasing with time, and that the obtained H2 to CO ratio was far above
the stoichiometric value expected from reaction 5.1 (H2/CO = 2). This was likely due
to the deposition of carbonaceous species on the catalyst surface and the concomitant
poisoning effect. In fact, Ni is among the most susceptible metals to this kind of
deactivation derived from the following CO dissociation reaction [31, 32]:
CO(a) → C(a) + O(a) (5.10)
Then, a decrease in the applied potential to VWR = 0 caused a very sharp
increase in both H2 and CO production rates (electropromoted state), up to around 3.5
x 10-7
mol H2 s-1
and 1.5 x 10-7
mol CO s-1
, respectively, reaching a H2/CO ratio closer
to 2, i.e., the stoichiometric value from reaction 5.1. As in the previous EPOC study
on methanol decomposition with a Ag catalyst [13], this modification in the catalytic
activity upon decreasing the applied potential can be explained on the basis of a
decrease in the catalyst work function which led to strong modification in the catalyst
chemisorption properties. In the mentioned work, this effect was caused by the
removal of electronegative O2-
ions from the catalyst surface by means of an yttria-
Page 206
Chapter 5
198
stabilized zirconia (YSZ) solid electrolyte. In the present case, electropositive K+ ions
were electrochemically pumped from the K-βAl2O3 to the Ni catalyst at this applied
potential, according to the obtained negative currents (not shown here). Consequently,
in both cases, the lower catalyst work function would strengthen the chemical bond
between the metal and the electron acceptor adsorbate. This can be explained through
two approaches: the strengthening of the direct (electrostatic) attractive interactions
between the electron acceptor adsorbant and the electric field created by the
coadsorbed electropositive promoter, and the increase in the Fermi level with the
concomitant favouring of back-donation of binding electrons from the metal [33]. In
this point, it is important to analyze the role of the different adsorbates and
intermediate present in the reaction under study (reaction 5.1). For Group 10 metals
such as Ni, it is generally accepted that the MD reaction involves the adsorption of
CH3OH on the catalyst surface (reaction 5.11) and its successive dehydrogenation
(reactions 5.12-5.14), yielding CO (reaction 5.15) and H2 (reaction 5.16) [18].
CH3OH → CH3OH(a) (5.11)
CH3OH(a) → CH3O(a) + H(a) (5.12)
CH3O(a) → H2CO(a) + H(a) (5.13)
H2CO(a) → CO(a) + 2H(a) (5.14)
CO(a) → CO (5.15)
2H(a) → H2 (5.16)
Hydrogen abstraction from methoxy (reaction 5.13) is typically considered the
rate-limiting step [19, 34]. Hence, one could expect the methoxy group (CH3O) and
the successive intermediate species (CHxO, x = 0-2) to retain an electron acceptor
character versus the intramolecularly bonded H [8, 13]. This would explain the drastic
improvement in the methanol dehydrogenation rate, yielding H2 and CO, upon the
decrease in the applied potential and the consequent increase in the potassium
coverage. From Figure 5.1 it is also noteworthy that more negative potentials led to a
strong decrease in the Ni catalytic activity to H2 and CO production values close to
Page 207
Electrochemical promotion of Ni in methanol conversion reactions
199
those initially obtained. This can be attributed to an increasing activation of the C-O
bond cleavage by K+ ions [14, 35], thus favouring the carbon deposition (reaction
5.10). Indeed, under negative applied potentials, a much abrupt slope can be observed
as compared to positive polarizations, which verifies the stronger deactivation process.
A similar experiment was carried out by changing the reaction atmosphere to
steam reforming conditions. Figure 5.2 shows the results obtained at 360 ºC with a
feed composition of CH3OH/H2O = 4.4 %/5.2 % upon the same potentiostatic
transitions used in Figure 5.1. As in the former case, the application of positive
potentials led to establish an unpromoted Ni catalyst state, i.e. with no potassium
species on the surface. Then, the catalyst was activated under the application of VWR =
0 V and lower potentials, due to the back-spillover of K+ promoter ions to the catalyst
surface and the concomitant decrease in the Ni work function as discussed above.
Figure 5.2. Response of H2, CO and CO2 production rates vs. time to step changes in the
applied catalyst potential (VWR) under methanol steam reforming conditions: CH3OH/H2O =
4.4 %/5.2 %, 360 ºC.
-2.5
-1.5
-0.5
0.5
1.5
2.5
0
1
2
3
4
5
6
7
0 100 200 300 400 500
VW
R/
V
r /
mo
l s-1
x 10
-7
Time / min
rCO
VWR
rH2
rCO2
Page 208
Chapter 5
200
Under SRM conditions, H2 and CO were still the main obtained compounds
(likely from reaction 5.1) but an appreciable amount of CO2 was also produced
(reaction 5.6). The steam reforming of methanol can be globally considered as the sum
of the methanol decomposition and the water-gas shift reactions [23]. However,
unalloyed Group VIII metals in general, and Ni in particular, are especially active for
the methanol decomposition to CO rather than the steam reforming reaction [17, 20,
21]. A higher methanol conversion can be appreciated from this figure in comparison
with the methanol decomposition results (Figure 5.1), which is in good agreement
with other comparative studies [36]. This can be attributed to the key role of H2O in
assisting the decomposition of methoxy groups and subsequent intermediate species
involved in the MD reaction mechanism previously described [37]. Hence, one could
consider water as an electron acceptor adsorbate (vs. methanol) as also deduced from
other EPOC studies [12, 38]. It is then expected that a K+-derived decrease in the
catalyst work function upon negative polarizations favoured the water activation and
thus the production rate of H2, CO and CO2, in agreement with the SRM results
obtained with Pt catalysts in chapters 1 and 2.
On the other hand, although some coke deposition likely took place under
SRM conditions, a much lower catalyst deactivation was observed as compared to the
MD results. In fact, it is well known the beneficial effect of water on this kind of
catalytic systems through the performance of carbon gasification (reaction 5.17) [36,
39].
C(a) + H2O → CO + H2 (5.17)
The electrochemical activation of H2O by K+ ions would also favour this
reaction under negative applied potentials, as can be observed in Figure 5.2. The
deviation in the carbon atom balance obtained from the gaseous products decreased
approximately from 18 % at +2 V (unpromoted conditions) to 2 % at -0.5 V (promoted
conditions), which demonstrates that the H2O chemisorption enhancement via EPOC
attenuated the deactivation process.
Page 209
Electrochemical promotion of Ni in methanol conversion reactions
201
The variation of the Ni catalytic activity with the applied potential was also
studied under partial oxidation conditions (CH3OH/O2 = 4.4 %/0.33 %) at the same
temperature (360 ºC), as shown in Figure 5.3. In this case and depending on the
catalyst potential, not only H2, CO and CO2 were produced but also H2CO and H2O
from reactions 5.4 and 5.5, respectively.
Figure 5.3. Response of H2, CO, CO2 and H2CO production rates vs. time to step changes in
the applied catalyst potential (VWR) under methanol partial oxidation conditions: CH3OH/O2 =
4.4 %/0.33 %, 360 ºC.
Under unpromoted conditions, H2 and CO were again the main reaction
products with a H2/CO ratio of around 2, which confirmed the excellent
dehydrogenation activity of nickel (reaction 5. 1) and the fairly complete absence of
coke deposition under this reaction atmosphere (no deactivation was observed during
the application of +2 V). This latter fact explains the much higher catalytic activity of
the unpromoted Ni catalyst as compared to the previous experiments, which was also
likely influenced by the oxygen participation in the stepwise hydrogen abstraction
-2.5
-1.5
-0.5
0.5
1.5
2.5
0
4
8
12
16
0 100 200 300 400 500
VW
R/
V
r /
mo
l s-1
x 10
-7
Time / min
rCO
VWR
rH2
rCO2
rH CO2
Page 210
Chapter 5
202
from methanol (reaction 5.12) and methoxy species (reaction 5.13) [40, 41]. However,
the most interesting feature of this figure is the extremely sharp decrease in the Ni
catalytic activity observed upon decreasing the applied potential from VWR = +0.5 V,
which led to almost negligible H2 and CO production from reactions 5.1 and 5.7, while
some CO2, H2CO and H2O were obtained through reactions 5.4 and 5.5. One could
state that the back-spillover of K+ ions presented a strong negative effect in the
methanol partial oxidation for H2 production, as opposed to the enhancement effect
observed in the previous alkaline electrochemical promotion studies performed under
POM conditions with Pt-, Au- and Cu-based catalysts. Hence, in contrast with the two
previous experiments under MD and SRM conditions, a detrimental effect of negative
polarization was clearly observed for H2 and CO production. It could be attributed to
the presence of O2 in the gas phase which may lead to some modification in the Ni
catalyst oxidation state as will be discussed below. Anyway, it is clear that the
electrically-induced promotional (or poisoning) effects obtained under the different
reaction atmospheres (MD, SRM and POM) were completely reversible since, under
the final application of VWR = +2 V, all the reaction rates reached the same initial
values (Figures 5.1, 5.2 and 5.3, respectively).
The magnitude of the promotion effect obtained under the different methanol
conversion reactions was evaluated by means of two EPOC parameters commonly
used in this kind of studies: the reaction rate enhancement ratio (ρ) and the promotion
index (PIK+) defined by the following equations:
=
(5.18)
=
(5.19)
where r and r0 are the promoted (VWR < +2 V) and un-promoted (VWR = +2 V)
catalytic production rates, respectively, ∆r = r – r0 is the K+-induced change in the
catalytic reaction rate and K+ is the potassium coverage, which was calculated at each
applied catalyst potential (VWR) from the integration of the obtained current (I) vs.
time (t) curve through the Faraday’s law:
Page 211
Electrochemical promotion of Ni in methanol conversion reactions
203
=
(5.20)
where n is the potassium ion charge, i.e., +1, F is the Faraday constant (96485 C), and
NG is the active surface area, 2.34 x 10-7
mol Ni. This value was estimated from the
total amount of Ni deposited (2.28 x 10-6
mol Ni cm-2
) and an approximate catalyst
dispersion value (D) of 3.62 %, which was calculated from the following semi
empirical equation [42]:
ρ (5.21)
where Mm is the nickel atomic weight, ρm is the Ni density (in g m-3
), NA is the
Avogadro number, d is an average Ni particle diameter, 337 Å, as estimated from the
XRD measurements, and is the metal atomic surface, 5.38 x 10-20
m2 atom
-1, as
obtained for Ni(111) [43], since this was found to be the main Ni crystallographic
direction (as will be observed below).
Figure 5.4a depicts the steady-state H2, CO, CO2 and H2CO production rate
enhancement ratios (ρ) obtained in the three studied atmospheres (MD, POM and
SRM) upon the different applied catalysts potentials. Figure 5.4b shows the
corresponding promotional index values (PIK+,i).
As was discussed in previous chapters, the ρ parameter represents the factor by
which the production rate of one compound has increased, under certain reaction
conditions, at each applied potential with respect to its unpromoted value (i.e. at VWR
= +2 V). On the other hand, the PIK+ provides a measure of the K+-induced
improvement in the catalytic activity normalized by the promoter concentration
created on the Ni surface. Hence, the dashed lines in figures groups a and b represent
the neutral values of these two parameters (ρi = 1 and PIK+,i = 0, respectively), i.e., the
un-promoted state.
Page 212
Chapter 5
204
Figure 5.4. Effect of the applied potential (VWR) on the rate enhancement ratio (a1-a4) and
promotion index (b1-b4) values obtained for H2, CO, CO2 and H2CO at 360 ºC under the same
methanol decomposition (MD), steam reforming (SRM) and partial oxidation (POM)
conditions than Figures 5.1-5.3.
0
1
2
3
4
5
6ρ
HMD
SRM
POM
0
2
4
6
-2.5 -1.5 -0.5 0.5 1.5 2.5
ρC
O
VWR / V
MD
SRM
POM
0.5
1
1.5
2
2.5
3
ρC
O
SRM
POM
0
5
10
15
20
25
-2.5 -1.5 -0.5 0.5 1.5 2.5
ρH
C
O
VWR / V
POM
2 22
a1)
a2)
a3)
a4)
-2
2
6
10
14
18
PI K
,C
O
MD
SRM
POM
-10
0
10
20
30
40
PI K
,H
MD
SRM
POM
-20
0
20
40
-2.5 -1.9 -1.3 -0.7 -0.1 0.5 1.1
PI K
,C
O
VWR / V
MD
SRM
POM
5
10
15
20
-2.5 -1.9 -1.3 -0.7 -0.1 0.5 1.1
PI K
,H
C
O
VWR / V
POM
2 22
++
++
b1)
b2)
b3)
b4)
Page 213
Electrochemical promotion of Ni in methanol conversion reactions
205
It is clear that a decrease in the applied catalyst potential, under methanol
decomposition and steam reforming conditions, caused an enhancement in the
production of H2, CO and CO2. Thus, according to the rules of electrochemical
promotion [33], both MD (reaction 5.1) and SRM (reaction 5.6) are considered
electrophilic reactions under the studied conditions (upon ΔV < 0, ρ > 1 and PIK+ > 0).
The slight decrease in the H2 and CO production rates at VWR = +0.5 V (still
unpromoted state) and the progressive drop observed at very negative potentials can
be attributed to the concurrence of the CO dissociation on the catalyst surface
(reaction 5.10) as discussed above. Due to this deactivation under MD conditions, and
hence to the lower unpromoted reaction rates (vs. SRM), H2 and CO obtained via
methanol decomposition presented much higher ρ and PIK+ values, also in comparison
with the previous SRM studies with Pt and Pt-DLC catalyst films prepared by the
same deposition method.
On the other hand, it can also be observed the strong negative effect of K+ ions
on the H2 and CO production rates under POM conditions (upon ΔV < 0, ρ < 1 and
PIK+ < 0). However, CO2 and especially formaldehyde production showed favourable
EPOC parameters, which could not be well appreciated from Figure 5.3. When
reaction rates such as those of H2, CO and CO2 in the studied POM conditions barely
vary upon further decreasing the applied potential from a certain value (VWR = 0 V), it
is worth noting that the continuing rise in the K+ coverage makes the PIK+ values
approach zero. Hence, in such cases, this parameter actually provides useful
information only at low K+ coverages. Although the global partial oxidation reaction
presented apparent electrophobic behaviour, the Ni catalyst seemed then to be partially
activated for CO2 and H2CO production upon negative polarization. One can find in
literature other EPOC studies where Ni-based catalysts were activated in different
reactions such as water-gas shift [38], C2H4 hydrogenation [44], CH4 steam reforming
[45] or CO2 hydrogenation [35, 46]. In the present work, a Ni catalyst has been clearly
electrochemically promoted, for the first time, in the methanol decomposition and
steam reforming reactions for H2 production. However, given the unexpected sharp
drop in the Ni catalytic activity observed in the methanol partial oxidation under K+-
Page 214
Chapter 5
206
promoted conditions, as compared with the results previously obtained with Pt, Au
and Cu catalysts, additional experiments were carried out in this reaction atmosphere.
5.3.2. Effect of the electrochemical promotion on the catalyst oxidation state
Figure 5.5 shows the influence of the oxygen partial pressure ( ) on steady-
state reaction rates at the same temperature and feed methanol concentration than
those used in preceding experiments, upon the imposition of two different potentials:
+2 V (unpromoted state) and -1 V (electropromoted state). The dashed line depicts the
partial oxidation conditions studied in Figure 5.3.
In the overall studied range of feed oxygen concentration (0.33 – 1.53 % O2),
the same promotional behaviour was found as in the previous potentiostatic transition
experiment, i.e., H2 and CO production rates sharply decreased upon negative
polarization, while the formation of carbon dioxide and especially formaldehyde was
enhanced. Under unpromoted conditions, both CO2 and H2CO production showed a
positive order with respect to oxygen partial pressure, which agrees with the
experimental results previously discussed, i.e., the strengthening of the metal-oxygen
chemisorptive bond via K+ backspillover was derived in increasing both CO2 and
H2CO production rates, thus according to the rules of chemical and electrochemical
promotion [1]. In the same sense, one could then expect a zero to negative order of H2
and CO production with respect to oxygen partial pressure in view of the previous
EPOC experimental results. Nevertheless, it is striking that both formation rates were
favoured by increasing the oxygen partial pressure up to around 0.8 kPa. This fact
seemed then to disagree with the poisoning effect observed under negative
polarizations in H2 and CO production with a 0.33 % O2 feed composition (Figure
5.3).
Page 215
Electrochemical promotion of Ni in methanol conversion reactions
207
Figure 5.5. Effect of O2 partial pressure on H2 (a), CO (b), CO2 (c) and H2CO (d) production
under unpromoted (+2 V) and electropromoted (-1 V) state. CH3OH = 4.4 %, 360 ºC.
0
5
10
15
20
r H/
mo
l s-1
x 1
0-7
+2 V
-1 V
0
2
4
6
8r C
O/
mo
l s-1
x 1
0-7
+2 V
-1 V
0
0.5
1
1.5
2
r CO
/ m
ol s
-1x
10
-7
+2 V
-1 V
0
0.1
0.2
0.3
0 0.4 0.8 1.2 1.6
r HC
O/
mo
l s-1
x 1
0-7
PO / kPa
+2 V
-1 V
22
2
2
a)
b)
c)
d)
Page 216
Chapter 5
208
In order to clarify this behaviour, the Ni catalyst film was characterized by
XRD after exposing the catalyst to both steam reforming and partial oxidation
conditions (with two different feed oxygen concentrations in the latter case), under
both unpromoted (VWR = +2 V) and electropromoted (VWR = -1 V) conditions. The
obtained diffractograms are shown in Figure 5.6.
Figure 5.6. XRD diffractograms of the Ni catalyst film after exposure for 1 hour to the same
methanol partial oxidation (a-c) and steam reforming (d,e) conditions than Figures 5.2 and 5.3,
under unpromoted (+2 V) and electropromoted (-1 V) state. Figure 5.6f shows the pattern of β-
Al2O3 solid electrolyte.
In all cases, the three main diffraction peaks of metallic nickel, (111), (200)
and (220), can be clearly observed at 2 = 44.5º, 51.8º and 76.4º, respectively,
Inte
nsi
ty /
a.u
.
Ni/K-βAl2O3
after POM 0.33% O2
-1 V
b) NiNiONi2O3
NiO2
25 35 45 55 65 75
Inte
nsi
ty /
a.u
.
2 θ / º
Ni/K-βAl2O3
after POM 1.53% O2
+2 V
c) Ni
Inte
nsi
ty /
a.u
.
Ni/K-βAl2O3
after POM 0.33% O2
+2 V
a) Ni
Inte
nsi
ty /
a.u
.
Ni/K-βAl2O3
after SRM+2V
d) Ni
25 35 45 55 65 75
Inte
nsi
ty /
a.u
.
2 Ѳ / º
βAl2O3f)
Inte
nsi
ty /
a.u
.
Ni/K-βAl2O3
after SRM-1V
e) Ni
Page 217
Electrochemical promotion of Ni in methanol conversion reactions
209
exhibiting a face-centered cubic (FCC) crystalline structure (JCPDS, 87-0712). The
spectrum of the K-βAl2O3 solid electrolyte (Figure 5.6f) was also present in all the
diffractograms. This signal was even higher than that of Ni, probably due to the very
low thickness of the catalyst-working electrode (150 nm). No diffraction peaks
associated with nickel oxides were found under methanol steam reforming atmosphere
(Figures 5.6d and 5.6e) and neither under methanol partial oxidation conditions as
long as a positive potential was applied (Figures 5.6a and 5.6c), i.e., regardless of the
gas phase oxygen concentration in the overall range studied (see Figure 5.5).
However, very interestingly, upon negative polarization under POM atmosphere
(Figure 5.6b), the main diffraction peaks of NiO appeared at 2 = 37.26º, 43.29º,
62.89º and 79.43º (JCPDS, 78-0643). In addition, other peak at 2 = 39.13º and other
two peaks at 2 = 58.66º and 78.06º were also found, which could be attributed to the
presence of Ni2O3 (JCPDS, 14-0481) and NiO2 (JCPDS, 85-1977), respectively. From
this analysis, one cannot confirm the complete absence of oxidized Ni species under
other reaction conditions, but their presence was much more pronounced when the
catalyst film was subjected to POM atmosphere and negative applied potentials.
It becomes clear that the K+-promoted strengthening of the nickel-oxygen
chemisorptive bond under POM reaction conditions favoured the formation of
oxidized Ni species on the catalyst surface. Although this kind of effect is not very
commonly found in EPOC studies, some authors reported the oxidation or reduction
of Ru- [8], Rh- [9-11] and Pd-based [7] catalysts through the strengthening (upon
negative polarization) or weakening (upon positive polarization) of oxygen
chemisorption, respectively. While all the mentioned EPOC studies employed YSZ as
solid electrolyte, the oxidation state of a catalyst has been modified by alkali ions, for
the first time, in the present work. Then, this increase in the Ni oxidation state under
O2 atmosphere and negative polarization would explain the modification of the
reaction rates observed in Figure 5.3, since NiO is typically shown to be active in the
deep oxidation of methanol (CO2 and H2O production through reaction 5.5) instead of
the decomposition or partial oxidation reactions for CO and H2 production (reactions
5.1 and 5.7) [17, 47]. In addition it has been shown that NiO may present certain
Page 218
Chapter 5
210
selectivity toward formaldehyde production [48, 49]. NiO has already been detected in
previous studies with Ni catalysts under methanol partial oxidation conditions, for
instance, by XRD [17] or XPS [47] analysis. Thus, the most intriguing results obtained
in this work demonstrate that Ni was not significantly oxidized by increasing the feed
O2 concentration up to 1.53 % under VWR = +2 V (Figure 5.6c), but it was partially
oxidized under 0.33 % O2 by applying –1 V (Figure 5.6b). Moreover, in view of
Figure 5.3, the reversibility of the phenomenon is clear, i.e., the activity was again
recovered after the final application of +2 V and, very likely, the reduced state of the
Ni catalyst.
Hence, the results obtained in the present work with a very thin Ni film
prepared by the cathodic arc deposition technique show the ability of the EPOC
phenomenon for the in-situ, reversible, partial modification of the catalyst oxidation
state, by varying only the K+ coverage rather than the oxygen partial pressure.
Moreover, on the basis of the XRD spectra obtained after the catalyst
exposition to the different reaction atmospheres, an approximate Ni particle size was
calculated in each case from the width of the main diffraction peak, Ni(111), by means
of the Scherrer equation:
Å
(5.22)
where λ is the X-ray wavelength (in Å), B is the full width at half maximum (FWHM)
of the diffraction peak (in radians) corrected for instrumental broadening, is the
Bragg angle and KW is the half-width Scherrer constant which can be considered 0.9
for this kind of catalyst [50]. Ni particle sizes between 31.5 and 35 nm were estimated
in all cases, which also denotes the high stability of this Ni catalyst film under the
studied reaction conditions.
5.3.3. Control of the catalyst selectivity via electrochemical promotion
The variation in the catalyst selectivity with the applied potential and the
influence of the temperature were evaluated under POM conditions (CH3OH/O2 = 4.4
%/0.3 %), that was the reaction atmosphere which allowed to obtain a wider variety of
Page 219
Electrochemical promotion of Ni in methanol conversion reactions
211
compounds. Hence, Figure 5.7 shows the steady-state Ni selectivity toward H2, CO,
CO2 and H2CO under different applied catalyst potentials and reaction temperatures
between 280 and 360 ºC. As in previous chapters, the products selectivities were
calculated through the following equations:
(5.23)
(5.24)
(5.25)
(5.26)
where Fi,in and Fi,out are the molar flow rate of the i species at the inlet and outlet of the
reactor, respectively.
Figure 5.7. Steady-state H2 (a), CO (b), CO2 (c) and H2CO (d) selectivity vs. applied potential
(VWR) at different reaction temperatures. Methanol partial oxidation conditions: CH3OH/O2 =
4.4 %/0.33 %.
20
40
60
80
100
H2
Sele
ctiv
ity
/ %
280 ºC
320 ºC
360 ºC
0
20
40
60
80
-1.5 -0.5 0.5 1.5 2.5
CO
Sel
ecti
vity
/ %
VWR / V
280 ºC
320 ºC
360 ºC
0
15
30
45
60
75
CO
2Se
lect
ivit
y /
%
280 ºC
320 ºC
360 ºC
0
20
40
60
-1.5 -0.5 0.5 1.5 2.5
H2C
O S
elec
tivi
ty /
%
VWR / V
280 ºC
320 ºC
360 ºC
a)
b)
c)
d)
Increasing θK+ Increasing θK+
Page 220
Chapter 5
212
This figure clearly reflects the strong detrimental effect of the K+-induced
formation of NiO on the H2 and CO production from methanol under negative
potentials, as well as also the concomitant increase in the CO2 and H2CO selectivities.
As expected, under unpromoted conditions (VWR = +2 V), the methanol conversion
(not shown here) increased with the reaction temperature. The Ni activity in methanol
dehydrogenation for H2 and CO production (reaction 5.1) was also favoured by the
temperature increase, as typically reported [17, 20, 21], while formaldehyde selectivity
gradually decreased, probably due to its decomposition to CO and/or oxidation to CO2
[40, 48]. However, the most pronounced change in Ni catalytic performance was
derived from the K+ back-spillover and the likely increase in the NiO formation (as
discussed above). For instance, at 280 ºC, H2 and CO selectivity decreased from
around 80 % (under unpromoted conditions) to 20 and 0 % (under electropromoted
conditions), respectively, while formaldehyde selectivity increased from 0 % to 70 %.
As already pointed out, the possibility of modifying the selectivity of the catalyst
toward different products via in-situ alkali promotion is one of the main applications
of the EPOC phenomenon and it has been studied in other reactions such as CO2
hydrogenation [51], selective catalytic reduction of NOx [52, 53] or Fischer-Tropsch
synthesis [54]. In this sense, the large variety of products and intermediate species
involved in the methanol conversion processes, along with the ability of EPOC to
partially modify the catalyst oxidation state, offer a wide range of possibilities with
focus, for instance, on fine chemistry.
As a summary, Figure 5.8 shows the main catalytic reactions found to take
place under the different studied reaction atmospheres (MD, SRM and POM) and both
unpromoted (Figure 5.8a) and electropromoted (Figure 5.8b) conditions. In all cases,
as discussed in the first section, the decrease in the applied catalyst potential and thus
the increase in the potassium coverage on the Ni catalyst surface led to an
strengthening of the chemisorptive bond between the metal and the electron acceptor
adsorbates, which were methoxy and other CHXO–like intermediate species in the
methanol decomposition (MD), H2O in the methanol steam reforming (SRM) and O2
in the methanol partial oxidation (POM).
Page 221
Electrochemical promotion of Ni in methanol conversion reactions
213
Figure 5.8. Schematic representation of the main catalytic reactions taking place under positive
(a) and negative (b) polarization.
In the cases of MD and SRM, this K+ back-spillover caused the activation of
the methanol dehydrogenation mechanism. As a consequence, the Ni catalytic activity
for H2 and CO production via MD and also for H2 and CO2 production via SRM was
drastically enhanced showing a clear electrophilic behaviour, similarly to the previous
EPOC studies on methanol decomposition [13] and steam reforming (chapters 1 and
2) with noble metal catalysts. Under SRM atmosphere, the activation of water also
favoured the removal of carbonaceous species previously deposited on the Ni catalyst
MD
(4.4 % CH3OH)
CHXO
SRM
(4.4 % CH3OH, 5.2 % H2O)
H2O CH3OH
POM
(4.4 % CH3OH, 0.33 % O2)
O2 CH3OH
A
A
A
D
D
ΔV > 0
Ni (WE)
K+ K+ K+
Au (CE)
e-
e-
UNPROMOTED STATE
ΔV < 0
Ni (WE)
K+ K+ K+
Au (CE)
e-
e-
ELECTOPROMOTED STATE
CH3OH CO + 2H2
CO(a) C(a) + O(a)
CH3OH CO + 2H2
CO(a) C(a) + O(a)
CH3OH CO + 2H2
CH3OH + H2O CO2 + 3H2
C(a) + H2O CO + H2
CH3OH CO + 2H2
CO(a) C(a) + O(a)
CH3OH + H2O CO2 + 3H2
C(a) + H2O CO + H2
CH3OH CO + 2H2
CH3OH + 1/2O2 CO2 + 2H2
CH3OH + 3/2O2 CO2 + 2H2O
CH3OH CO + 2H2
CH3OH + 1/2O2 CO2 + 2H2
CH3OH + 3/2O2 CO2 + 2H2O
CH3OH + 1/2O2 H2CO + H2O
xNi + yO(a) NixOy
A D A D
A
D
Electron acceptor
Electron donor
a) b)
Page 222
Chapter 5
214
surface under positive applied potentials (decreasing the deactivation of the catalyst).
Then, this kind of electrocatalytic systems could also be useful to obtain highly pure
H2 or syngas depending on the catalyst promotion state. However, under the
application of negative potentials and the presence of oxygen in the reaction mixture
(POM conditions), H2 and CO production was sharply decreased while CO2 and H2CO
production rate was slightly increased. This was attributed to a K+-promoted increase
in the nickel oxidation state as confirmed by XRD analysis.
Similar promotional effects can be observed in related studies of classical
chemical promotion. For instance, the addition of different alkali compounds such as
Na, K or Cs already showed to promote the methanol decomposition [25] and steam
reforming [26] reactions by weakening the C-H bond in the methoxy and formate
intermediate species. Alkali and alkaline earth oxides have also been employed to
improve the catalyst resistance to carbon deposition via water activation [39].
Moreover, in the same sense, many metal oxides such as CeO2 or ZnO are commonly
used as catalyst supports to modify its activity and selectivity in methanol conversion
processes as well as to increase its stability, involving in many cases some transition
between the catalyst oxidation states [3]. It should be then remarked that, in the
present work, the possible potassium promotional effects have been in-situ studied for
a single catalytic system under different reaction atmospheres, by only varying the
applied electric potential. In this way, the promoter coverage was exhaustively
controlled and the consequent modifications of the Ni catalytic performance were
carried out in a fully reversible way, which shows a great versatility and practical
utility of the phenomenon of electrochemical promotion.
5.4. CONCLUSIONS
The following conclusions can be drawn from this study:
- The phenomenon of electrochemical promotion of catalysis (EPOC) was
applied in different methanol conversion reactions for H2 production by using a Ni
catalyst film prepared by the cathodic arc deposition technique. The catalytic activity
Page 223
Electrochemical promotion of Ni in methanol conversion reactions
215
and selectivity were in-situ modified by electrochemically pumping K+ ions from a
solid electrolyte support upon negative polarization.
- Under methanol decomposition conditions, the electrochemical promotion of
the Ni catalyst by K+ ions enhanced the dehydrogenation of methanol and intermediate
species to H2 and CO, although a strong deactivation by carbon deposition also took
place.
- Under methanol steam reforming conditions, the electrically-induced back-
spillover of K+ promoter ions favoured the H2, CO, and CO2 production, as well as
also the carbon gasification, via water activation.
- Under methanol partial oxidation conditions, the strengthening of oxygen
chemisorption caused a dramatic decrease in both the Ni catalytic activity and the
selectivity toward H2 and CO, probably due to an increase in the formation of surface
NiO, as observed in the XRD analysis. This shows the possibility of in-situ modifying
the catalyst oxidation state via electrochemical promotion with alkali ions.
- All the potassium-derived promotion effects were carried out in a controlled
way, by varying the applied electric potential, and showed to be fully reversible,
demonstrating the great interest of the EPOC phenomenon for many different practical
applications.
5.5. REFERENCES
[1] C.G. Vayenas, S. Bebelis, C. Pliangos, S. Brosda, D. Tsiplakides, Electrochemical
Activation of Catalysis: Promotion, Electrochemical Promotion and Metal-Support
Interactions, Kluwer Academic Publishers/Plenum Press, New York, 2001.
[2] A. Wieckowski, E.R. Savinova, C.G. Vayenas, Catalysis and Electrocatalysis at
Nanoparticles, Marcel Dekker, New York, 2003.
[3] P. Vernoux, L. Lizarraga, M.N. Tsampas, F.M. Sapountzi, A. De Lucas-Consuegra, J.L.
Valverde, S. Souentie, C.G. Vayenas, D. Tsiplakides, S. Balomenou, E.A. Baranova, Ionically
conducting ceramics as active catalyst supports, Chemical Reviews, 113 (2013) 8192-8260.
[4] C.G. Vayenas, Promotion, electrochemical promotion and metal-support interactions:
Their common features, Catalysis Letters, 143 (2013) 1085-1097.
Page 224
Chapter 5
216
[5] A. Katsaounis, Recent developments and trends in the electrochemical promotion of
catalysis (EPOC), Journal of Applied Electrochemistry, 40 (2010) 885-902.
[6] A. de Lucas-Consuegra, New trends of Alkali Promotion in Heterogeneous Catalysis:
Electrochemical Promotion with Alkaline Ionic Conductors, Catalysis Surveys from Asia,
2015, DOI:10.1007/s10563-014-9179-6.
[7] C. Jiménez-Borja, F. Dorado, A. De L.-Consuegra, J.M. G.-Vargas, J.L. Valverde,
Electrochemical promotion of CH4 combustion over a Pd/CeO 2-YSZ catalyst, Fuel Cells, 11
(2011) 131-139.
[8] D. Theleritis, S. Souentie, A. Siokou, A. Katsaounis, C.G. Vayenas, Hydrogenation of CO
2 over Ru/YSZ electropromoted catalysts, ACS Catalysis, 2 (2012) 770-780.
[9] E.A. Baranova, A. Thursfield, S. Brosda, G. Fóti, C. Comninellis, C.G. Vayenas,
Electrochemical promotion of ethylene oxidation over Rh catalyst thin films sputtered on YSZ
and TiO2/YSZ supports, Journal of the Electrochemical Society, 152 (2005) E40-E49.
[10] N. Kotsionopoulos, S. Bebelis, Electrochemical promotion of the oxidation of propane on
Pt/YSZ and Rh/YSZ catalyst-electrodes, Journal of Applied Electrochemistry, 35 (2005) 1253-
1264.
[11] A. Nakos, S. Souentie, A. Katsaounis, Electrochemical promotion of methane oxidation
on Rh/YSZ, Applied Catalysis B: Environmental, 101 (2010) 31-37.
[12] A. De Lucas-Consuegra, A. Caravaca, P.J. Martínez, J.L. Endrino, F. Dorado, J.L.
Valverde, Development of a new electrochemical catalyst with an electrochemically assisted
regeneration ability for H2 production at low temperatures, Journal of Catalysis, 274 (2010)
251-258.
[13] S. Neophytides, C.G. Vayenas, Non-faradaic electrochemical modification of catalytic
activity. 2. The case of methanol dehydrogenation and decomposition on Ag, Journal of
Catalysis, 118 (1989) 147-163.
[14] C.G. Vayenas, S. Neophytides, Non-faradaic electrochemical modification of catalytic
activity III. The case of methanol oxidation on Pt, Journal of Catalysis, 127 (1991) 645-664.
[15] C.A. Cavalca, G. Larsen, C.G. Vayenas, G.L. Haller, Electrochemical modification of
CH3OH oxidation selectivity and activity on a Pt single-pellet catalytic reactor, Journal of
Physical Chemistry, 97 (1993) 6115-6119.
[16] J.K. Hong, I.H. Oh, S.A. Hong, W.Y. Lee, Electrochemical oxidation of methanol over a
silver electrode deposited on yttria-stabilized zirconia electrolyte, Journal of Catalysis, 163
(1996) 95-105.
Page 225
Electrochemical promotion of Ni in methanol conversion reactions
217
[17] N. Iwasa, M. Yoshikawa, W. Nomura, M. Arai, Transformation of methanol in the
presence of steam and oxygen over ZnO-supported transition metal catalysts under stream
reforming conditions, Applied Catalysis A: General, 292 (2005) 215-222.
[18] M.S. Hegde, Electron spectroscopic studies of the adsorption and decomposition of
methanol on transition metals. A review, Proceedings of the Indian Academy of Sciences -
Chemical Sciences, 93 (1984) 373-387.
[19] G.C. Wang, Y.H. Zhou, Y. Morikawa, J. Nakamura, Z.S. Cai, X.Z. Zhao, Kinetic
mechanism of methanol decomposition on Ni(111) surface: A theoretical study, Journal of
Physical Chemistry B, 109 (2005) 12431-12442.
[20] B.S. Chen, J.L. Falconer, Alcohol Decomposition by Reverse Spillover, Journal of
Catalysis, 144 (1993) 214-226.
[21] N. Takezawa, N. Iwasa, Steam reforming and dehydrogenation of methanol: Difference in
the catalytic functions of copper and group VIII metals, Catalysis Today, 36 (1997) 45-56.
[22] R.M. Navarro, M.A. Peña, J.L.G. Fierro, Hydrogen production reactions from carbon
feedstocks: Fossil fuels and biomass, Chemical Reviews, 107 (2007) 3952-3991.
[23] D.R. Palo, R.A. Dagle, J.D. Holladay, Methanol steam reforming for hydrogen
production, Chemical Reviews, 107 (2007) 3992-4021.
[24] S. Sá, H. Silva, L. Brandão, J.M. Sousa, A. Mendes, Catalysts for methanol steam
reforming-A review, Applied Catalysis B: Environmental, 99 (2010) 43-57.
[25] A. Guerrero-Ruiz, I. Rodriguez-Ramos, J.L.G. Fierro, Dehydrogenation of methanol to
methyl formate over supported copper catalysts, Applied Catalysis, 72 (1991) 119-137.
[26] H.N. Evin, G. Jacobs, J. Ruiz-Martinez, U.M. Graham, A. Dozier, G. Thomas, B.H.
Davis, Low temperature water-gas shift/methanol steam reforming: Alkali doping to facilitate
the scission of formate and methoxy C-H bonds over Pt/ceria catalyst, Catalysis Letters, 122
(2008) 9-19.
[27] K. Klier, Preparation of bifunctonal catallysts, Catalysis Today, 15 (1992) 361-382.
[28] J.M. Pigos, C.J. Brooks, G. Jacobs, B.H. Davis, Low temperature water-gas shift: The
effect of alkali doping on the Csingle bondH bond of formate over Pt/ZrO2 catalysts,
Applied Catalysis A: General, 328 (2007) 14-26.
[29] L. Alejo, R. Lago, M.A. Peña, J.L.G. Fierro, Partial oxidation of methanol to produce
hydrogen over Cu-Zn-based catalysts, Applied Catalysis A: General, 162 (1997) 281-297.
[30] A. Anders, Cathodic Arcs: From Fractal Spots to Energetic Condensation. Springer Series
on Atomic, Optical, and Plasma Physics, Springer-Verlag, New York, 2008.
Page 226
Chapter 5
218
[31] P.B. Tøttrup, Kinetics of decomposition of carbon monoxide on a supported nickel
catalyst, Journal of Catalysis, 42 (1976) 29-36.
[32] Z. Murayama, I. Kojima, E. Miyazaki, I. Yasumori, Dissociation of carbon monoxide on
stepped nickel surfaces, Surface Science, 118 (1982) L281-L285.
[33] C.G. Vayenas, S. Brosda, Electron Donation-Backdonation and the Rules of Catalytic
Promotion, Top. Catal., 57 (2014) 1287-1301.
[34] Y. Liu, T. Hayakawa, T. Ishii, M. Kumagai, H. Yasuda, K. Suzuki, S. Hamakawa, K.
Murata, Methanol decomposition to synthesis gas at low temperature over palladium
supported on ceria-zirconia solid solutions, Applied Catalysis A: General, 210 (2001) 301-
314.
[35] E. Ruiz, D. Cillero, P.J. Martínez, Á. Morales, G.S. Vicente, G. De Diego, J.M. Sánchez,
Bench-scale study of electrochemically assisted catalytic CO2 hydrogenation to hydrocarbon
fuels on Pt, Ni and Pd films deposited on YSZ, Journal of CO2 Utilization, 8 (2014) 1-20.
[36] W. Cao, G. Chen, S. Li, Q. Yuan, Methanol-steam reforming over a ZnO-Cr2O3/CeO2-
ZrO2/Al2O3 catalyst, Chemical Engineering Journal, 119 (2006) 93-98.
[37] G. Jacobs, B.H. Davis, In situ DRIFTS investigation of the steam reforming of methanol
over Pt/ceria, Applied Catalysis A: General, 285 (2005) 43-49.
[38] A. De Lucas-Consuegra, A. Caravaca, J. González-Cobos, J.L. Valverde, F. Dorado,
Electrochemical activation of a non noble metal catalyst for the water-gas shift reaction,
Catalysis Communications, 15 (2011) 6-9.
[39] I. Alstrup, B.S. Clausen, C. Olsen, R.H.H. Smits, J.R. Rostrup-Nielsen, Promotion of
steam reforming catalysts, Studies in Surface Science and Catalysis, 1998, pp. 5-14,
[40] K. Kähler, M.C. Holz, M. Rohe, A.C. Van Veen, M. Muhler, Methanol oxidation as probe
reaction for active sites in Au/ZnO and Au/TiO2 catalysts, Journal of Catalysis, 299 (2013)
162-170.
[41] L.A. Espinosa, R.M. Lago, M.A. Peña, J.L.G. Fierro, Mechanistic aspects of hydrogen
production by partial oxidation of methanol over Cu/ZnO catalysts, Topics in Catalysis, 22
(2003) 245-251.
[42] J.R. Anderson, Structure of Metallic Catalysts, Academic Press1975.
[43] Y. Hoshino, S. Matsumoto, Y. Kido, Ultrathin Ni layers grown epitaxially on SiC(0001)
at room temperature, Physical Review B - Condensed Matter and Materials Physics, 69 (2004)
155303-155301-155303-155307.
Page 227
Electrochemical promotion of Ni in methanol conversion reactions
219
[44] T.I. Politova, V.A. Sobyanin, V.D. Belyaev, Ethylene hydrogenation in electrochemical
cell with solid proton-conducting electrolyte, Reaction Kinetics & Catalysis Letters, 41 (1990)
321-326.
[45] I.V. Yentekakis, Y. Jiang, S. Neophytides, S. Bebelis, C.G. Vayenas, Catalysis,
electrocatalysis and electrochemical promotion of the steam reforming of methane over Ni film
and Ni-YSZ cermet anodes, Ionics, 1 (1995) 491-498.
[46] V. Jiménez, C. Jiménez-Borja, P. Sánchez, A. Romero, E.I. Papaioannou, D. Theleritis, S.
Souentie, S. Brosda, J.L. Valverde, Electrochemical promotion of the co2 hydrogenation
reaction on composite ni or ru impregnated carbon nanofiber catalyst-electrodes deposited on
YSZ, Applied Catalysis B: Environmental, 107 (2011) 210-220.
[47] H. Iwai, T. Umeki, M. Yokomatsu, C. Egawa, Methanol partial oxidation on Cu-Zn thin
films grown on Ni(1 0 0) surface, Surface Science, 602 (2008) 2541-2546.
[48] S. Damyanova, M.L. Cubeiro, J.L.G. Fierro, Acid-redox properties of titania-supported
12-molybdophosphates for methanol oxidation, Journal of Molecular Catalysis A: Chemical,
142 (1999) 85-100.
[49] J. Estellé, J.E. Sueiras, Methanol oxidation on semiconducting oxides, Reaction Kinetics
& Catalysis Letters, 51 (1993) 119-124.
[50] J.I. Langford, A.J.C. Wilson, Scherrer after Sixty Years: A Survey and Some New Results
in the Determination of Crystallite Size, J. Appl. Cryst., 11 (1978) 102-113.
[51] E. Ruiz, D. Cillero, P.J. Martínez, Á. Morales, G.S. Vicente, G. De Diego, J.M. Sánchez,
Bench scale study of electrochemically promoted catalytic CO2 hydrogenation to renewable
fuels, Catalysis Today, 210 (2013) 55-66.
[52] F. Dorado, A. de Lucas-Consuegra, P. Vernoux, J.L. Valverde, Electrochemical
promotion of platinum impregnated catalyst for the selective catalytic reduction of NO by
propene in presence of oxygen, Applied Catalysis B: Environmental, 73 (2007) 42-50.
[53] A. Palermo, R.M. Lambert, I.R. Harkness, I.V. Yentekakis, O. Mar'ina, C.G. Vayenas,
Electrochemical promotion by Na of the platinum-catalyzed reaction between CO and NO,
Journal of Catalysis, 161 (1996) 471-479.
[54] A.J. Urquhart, F.J. Williams, R.M. Lambert, Electrochemical promotion by potassium of
Rh-catalysed fischer-tropsch synthesis at high pressure, Catalysis Letters, 103 (2005) 137-141.
Page 229
221
Chapter 6
ELECTROCHEMICALLY ASSISTED PRODUCTION AND
STORAGE OF HYDROGEN
A NOVEL CONTRIBUTION OF ALKALINE ELECTROCHEMICAL
CATALYSTS
In this last chapter, the possibility of applying the electrochemical promotion
(EPOC) with alkaline conductors in the fields of H2 production and storage was
investigated. For this purpose, Ni catalyst films were deposited on K-βAl2O3 by the
oblique angle deposition technique. Under methanol steam reforming conditions (280
ºC) and negative polarization, this electrocatalytic system allowed to produce and
store H2 with a very high yield per amount of metal (up to 19 g H2 x 100 g Ni-1
, on a
Ni catalyst film with a porosity of 35 %) due to the promotional effect of K+ ions.
Then, it was also possible to release the captured H2 under positive polarization. This
represents a new application of the EPOC of great interest for the in-situ production,
storage and release of H2 in a controlled way. The influence of the catalyst
microstructure, the applied negative current and the reaction atmosphere was studied,
and a H2 storage mechanism was proposed with the support of different
characterization techniques. It was mainly based on the spillover of H atoms on
grapheme oxide which was simultaneously formed under electropromoted conditions.
e-
Ni
K+ K+ K+ K-βAl2O3
Au
ΔV < 0
e-K+
K+K+
K+ K+
K+K+
K+
K+ K+ K+K+ K+
HH CO
K+
K+
K+
HH
H
HHH
H
K+
H
H
COH2
CH3OH
KOH
KH
K-C-O-H
K-C-O-H
GONi Ni
Ni
Graphene oxide (GO)
K+-assisted
H2 production
K+-assisted
GO formation
K+-assisted
H adsorption
on GO
K+
Page 230
Chapter 6
222
6.1 INTRODUCTION
As mentioned in previous chapters, hydrogen is a feedstock of paramount
importance in the chemical industry (around 5 x 1010
kg consumed per year worldwide
[1]). H2 is also a very promising energy carrier with application in internal combustion
engines and fuel cell technology. One of its main advantages is its high gravimetric
energy density, although its volumetric energy density is very low. It is the most
abundant element on Earth but less than 1 % is present as molecular hydrogen. In this
thesis, the electrochemical activation of different catalysts for its catalytic production
from methanol has been considered, but H2 can also be obtained from other fossil
fuels, water electrolysis or photoelectrolysis processes [2-4]. Hence, hydrogen can be
considered as a clean synthetic fuel if it is finally burnt with oxygen (in this case the
only exhaust gas is water). In addition, an improvement from the environmental point
of view would be attained if renewable energies are employed to split the water, or
sustainable sources such as bioalcohols are used in the catalytic conversion processes.
Moreover, despite its bad reputation, H2 is safe in open enclosures due to its high
volatility and non-toxicity. However, its storage remains a problem for stationary and
specially transportation applications.
Hydrogen is conventionally stored on its pure form either as a compressed gas
or as a cryogenic liquid. In the first case, H2 cylinders are typically filled at 200 bar (as
those employed in some sections of this manuscript), although pressures up to 450 bar
can be reached if carbon-fibre-reinforced composite materials are used. In the second
case, liquid hydrogen presents a higher density than the compressed one, but it
requires specialized infrastructure to avoid gaseous H2 losses [1, 5]. Hence, these
traditional storage methods suffer from difficult operational conditions and sizable
safety risks. Some chemical compounds like methanol, formic acid or ammonia can
also be themselves considered as H2 storage media, since these molecules can be
decomposed (dehydrogenated) likely in a catalytic process [5, 6]. This storage method
would be only reversible in the case that the dehydrogenated compound is collected
and re-hydrogenated, being totally subjected to the availability of the raw material.
Other method is, for example, the encapsulation of the H2 gas inside a guest (solid)
Page 231
Electrochemically assisted production and storage of hydrogen
223
structure, from which hydrogen can be released by a pressure and temperature swing
[5, 7]. However, most of the H2 storage systems can be divided into two main
categories: physisorption of molecular H2 and chemisorptions of atomic hydrogen.
Since H2 physisorption is a non-activated process, fast kinetics and
reversibility are expected. However, the interaction between H2 and the adsorbant
(dominated by weak van der Waals forces) usually leads to unspecific (non localized)
adsorption, thus, significant hydrogen storage is only possible under cryogenic
temperatures. H2 physisorption on all kinds of carbon materials (carbon nanotubes,
carbon fibers, graphite, graphite oxide, activated carbons, etc) as well as on metal-
organic frameworks (MOFs), covalent organic frameworks (COFs), different kinds of
porous polymers and zeolites has been widely studied [1, 5-8]. In all these studies, a
maximum H2 storage capacity of about 1.5 wt. % is generally achieved at room
temperature while values ranging from 5 to 6 wt. % can be obtained at -196 ºC. In
these processes, great efforts are being carried out to develop adsorbents with ultra
high surface area.
On the other hand, atomic hydrogen chemisorption is usually followed by
chemical compounds formation leading to H2 storage as metal hydrides or nitrides.
Many metals and alloys are capable of reversibly adsorbing large amounts of
hydrogen by this mechanism. Charging can be performed using molecular gas (which
needs to be dissociated before adsorption) or H atoms from an electrolyte (where the
main drawbacks are the covering of the adsorbent by water or H+ species and the harsh
acidic environment) [9]. For instance, alloys derived from LaNi5 or CoNi5 present
some promising properties such as fast and reversible sorption with small hysteresis,
good cycling life, pressure and temperature conditions inside the operational window
for hydrogen PEMFC (25-100 ºC, 1-10 atm) and good volumetric hydrogen density.
However, their gravimetric capacities are very low (e.g., 0.9 wt. % for LaNi5H6, 1.1
wt. % for CoNi5H4). A higher hydrogen storage capacity is obtained with other
hydrides such as Mg2NiH4 (3.6 wt. %), MgH2 (7.6 wt. %) or LiBH4 (18 wt. %), but in
these cases the hydride formation is much slower and high temperatures (up to 600 ºC)
may be required for its decomposition, leading to problems of storage reversibility [1,
Page 232
Chapter 6
224
5, 7]. In this kind of systems, the main strategies for the enhancement of the storage
performance consist of the decrease in the metal grain size, the increase in the defects
concentration, and the incorporation of additional materials on which H atoms may
spill over, thus increasing the global storage capacity [8, 10-13].
The aim of this chapter is to explore the possibility of applying the EPOC
phenomena in the field of H2 production and storage. Previous works have already
demonstrated the use of cationic electrochemical catalysts for NOx storage-reduction
process (NSR) [14, 15] and CO2 capture [16]. Thus, in the present work, a novel
electrocatalytic system is developed for the electrochemically assisted production and
storage of H2 from methanol-water streams. The idea is to prepare an electrochemical
catalyst of suitable performance for H2 production from methanol steam reforming
(SRM), which is capable of operating upon different electrochemical polarization for
the controllable H2 storage and release, under mild reaction conditions. For that
purpose, a porous Ni catalyst film was deposited on a K-βAl2O3 solid electrolyte. Ni
was selected as the active phase because of its good catalytic activity under methanol
steam reforming conditions (proven in the previous chapter). The Ni catalyst film was
prepared by an oblique angle deposition procedure, which is similar to that carried out
in chapter 4, in order to achieve a Ni catalyst film with suitable porosity and high
catalyst-electrode surface area. The influence of the applied electrical current, the
reaction atmosphere and the microstructure of the catalyst film on the H2 storage
capacity was studied. In addition, different ex-situ characterization techniques were
performed: SEM, XPS, Raman Spectroscopy and FTIR analysis in order to identify
the nature of the different H2 storage surface compounds formed under the different
polarization conditions.
6.2. EXPERIMENTAL
6.2.1. Preparation of the electrochemical catalyst
The electrochemical catalyst consisted of a continuous thin Ni film (geometric
area of 2.01 cm2), which also behaved as working electrode (WE). Similarly to the
previous chapters, a 19-mm-diameter, 1-mm-thick K-βAl2O3 (Ionotec) pellet was used
Page 233
Electrochemically assisted production and storage of hydrogen
225
as the cationic solid electrolyte (i.e. as source of K+ promoter ions), and inert Au
counter (CE) and reference (RE) electrodes were prepared from an oganometalic paste
(Fuel Cell Materials 233001). Two different active Ni catalyst films were deposited by
oblique angle deposition (OAD). In both cases, a Ni target pellets (Goodfellow,
99.9999 % purity) were evaporated under vacuum conditions by bombardment with a
high kinetic energy (< 5 keV) and intensity (150 mA) electron beam. Then, the vapour
was condensed onto the surface of the substrate (K-βAl2O3) at room temperature. Two
different zenithal angles, α, formed between the perpendicular to the substrate surface
and the evaporation direction, were used to prepare two different samples. The first
catalyst (called as Ni 0º) was grown at α = 0º and consisted of a fairly compact Ni film
with a thickness of around 1.3 µm, as obtained from the cross-sectional SEM image
(not shown). The second Ni catalyst (called as Ni 80º) was deposited at a zenithal
evaporation angle of α = 80º to enhance the shadowing effects during the film growth
(as in chapter 4) [17-19], leading to a 0.6 µm-thick film composed of tilted Ni
nanocolumns. The final metal loading was 1.09 mg Ni cm-2
in the case of Ni 0º and
0.36 mg Ni cm-2
in the case of Ni 80º, as determined by X-ray fluorescence (XRF) in a
Siemens SRS 3000 sequential spectrophotometer with a rhodium tube as the radiation
source. Hence, a relative film porosity of around 35 % was estimated in the latter case
on the basis of the thickness values obtained from cross-sectional micrographs (1.3
and 0.6 µm for the catalysts Ni 0º and Ni 80º, respectively). Similar films were also
prepared on (polished) silicon substrates for comparison purposes.
This catalyst preparation method was carried out in collaboration with Dr.
Agustín R. González-Elipe and Dr. Víctor J. Rico from the Institute of Materials
Science of Seville (CSIC – University of Seville).
Figure 6.1 shows the top-view SEM images of the nickel catalyst films as-
deposited by normal (Ni 0º) and oblique (Ni 80º) glancing angle deposition on silicon
substrate for a reference study (Figures 6.1a and 6.1b) and on the K-βAl2O3 pellet
(Figures 6.1c and 6.1d) which will be used for the subsequent electrochemical
experiments below.
Page 234
Chapter 6
226
Figure 6.1. Top view SEM images of the Ni films deposited at a normal geometry (Ni 0º) and
at a deposition angle of 80º (Ni 80º) on silicon (a and b) and K-βAl2O3 (c and d) substrates.
It can be observed that normal physical vapour deposition of Ni (i.e., at an
incident angle of α = 0º) led to fairly compact films composed of perpendicular, thin,
close-packed nanocolumns. On the other hand, the oblique angle deposited (OAD)
nickel at α = 80º resulted in separated groups of tilted nanocolumns, thus providing a
greater porosity (35 %), as can be observed in Figures 6.1b and 6.1d for the silicon
and K-βAl2O3 substrates, respectively. SEM images of the sample deposited on silicon
(Figures 6.1a and 6.1b) exhibited a very ordered and homogeneous distribution,
similar to oblique angle deposited films on silicon shown in previous studies based on
Co [20], Ti [21, 22], Pt-Ni [23], V-Mg [21, 22], TiO2 [18, 24] or Ta2O5 [19, 24]. The
Ni film prepared on the K-βAl2O3 pellet (Figures 6.1c and 6.1d) presented a much
more irregular microstructure, which was probably influence by the higher surface
roughness of this substrate and the different interaction between the Ni atoms on
silicon or K-βAl2O3 substrate. A more pronounced shadowing effect was also obtained
on K-βAl2O3 during the oblique angle deposition, giving rise to agglomerations of Ni
1 µm 1 µm
5 µm 5 µm
a) b)
c) d)
Ni 0º Ni 80º
Page 235
Electrochemically assisted production and storage of hydrogen
227
columns separated by large areas of alumina which were left in view (Figure 6.1d).
Anyway, as measured by XRF, the same amount of Ni was deposited on both kinds of
substrates. Thus, it is clear that the porosity, the morphology and the gas-exposed
metal surface area of the catalyst films can be severely modified by varying the
zenithal angle during their deposition.
Both obtained Ni catalyst-working electrodes (Ni 0º and Ni 80º) were
electrically conductive. The resulting Ni/K-βAl2O3/Au electrochemical catalysts were
placed into the single chamber solid electrolyte cell reactor and the three electrodes
(working, counter and reference) were connected to the potentiostat-galvanostat as
done in the previous chapters.
6.2.2. Characterization measurements
The surface microstructure of the Ni films deposited on silicon and K-βAl2O3
substrates was examined by scanning electron microscopy (SEM) analysis using a
Hitachi S4800 field emission microscope operated at 2 keV. In order to investigate the
composition of the catalyst surface during the H2 storage process and identify the
possible surface compounds on it, the catalyst film Ni 80º was characterized after the
catalytic activity measurements. Firstly, it was exposed to methanol steam reforming
conditions (CH3OH/H2O = 4.4 %/5.2 %, Ar balance, 6 NL h-1
) at 280 ºC while a
positive potential of VWR = +2 V was applied for 1 hour, in order to define a Ni
surface free of K+ ions and hence a cleaned (reference) catalyst state. A subsequent
negative potential of VWR = -1 V was applied to electrochemically transfer K+ ions
from the K-βAl2O3 solid electrolyte to the catalyst film, which favored the H2
production/storage process as will be explained below. After 1 hour under these
reaction conditions, the electrochemical catalyst was cooled down and the applied
potential was interrupted (open circuit conditions) at 100 ºC. Then, the catalyst was
transferred to different characterization equipments.
Besides SEM, X-ray photoelectron spectroscopy (XPS) was performed with a
PHOIBOS-100 spectrometer with Delay Line Detector (DLD) from SPECS, which
worked in the constant pass energy mode fixed at 30 eV. Monochromatic Mg Kα
Page 236
Chapter 6
228
radiation was used as excitation source and the binding energy (BE) scale of the
spectra referenced to the C 1s of graphitic carbon taken at 284.6 eV. The study of the
binding of the surface compounds was performed through specular reflectance FTIR
spectroscopy using a Jasco FT/IR-6200 spectrometer. All spectra were typically
obtained using 500 scans with a resolution of 4 cm-1
. To remove the background, the
signal obtained from a gold substrate was subtracted. Raman spectra were also
recorded with a HORIBA HR-800-UV microscope. For these measurements, a green
laser (532.14 nm) working at 600 lines per mm and a 100x objective were used.
6.2.3. Catalytic activity measurements
The catalytic activity measurements were carried out in the experimental setup
described in Chapter 1, section 1.2.3. A temperature of 280 ºC was fixed in the whole
study and the feeding stream was varied along the experiments. In general, the H2
production and storage was performed under methanol steam reforming (SRM)
conditions (CH3OH/H2O = 4.4 %/5.2 %, Ar balance, 6 NL h-1
), while the
electrochemical decomposition of the stored compounds was carried out in Ar stream.
In this way, released H2, CO and CO2 molecules were clearly detected in the outlet
stream. The different stages that comprised each experiment will be described in detail
in the next section.
6.3. RESULTS AND DISCUSSION
6.3.1. Preliminary experiments of H2 production and storage
Different systematic electrocatalytic experiments were carried out with both
kinds of catalyst films (Ni 0º and Ni 80º) in order to study the possibility of producing
and storing H2 at 280 ºC. Each experiment consisted of 4 consecutive steps, as
follows:
1) The Ni catalyst was subjected to methanol steam reforming (SRM)
conditions (CH3OH/H2O = 4.4 %/5.2 %, Ar balance, 6 NL h-1
) and to a positive
applied potential of VWR = 2 V for 1 hour in order to remove all the positive promoter
ions (K+) that may have remained on the catalyst/working electrode. In this way, a
reference cleaned Ni catalyst surface was established.
Page 237
Electrochemically assisted production and storage of hydrogen
229
2) A constant negative current, I, was applied for 45 min to electrochemically
transfer K+ ions at a constant rate, = I/F (F being the Faraday constant), from the
K-βAl2O3 solid electrolyte to the Ni catalyst. Once located on the Ni catalyst, these
potassium ions may likely migrate over the entire gas-exposed Ni catalyst-surface
and/or react with coadsorbed species [25]. Under the studied reaction conditions, as
analysed in previous chapters, the back-spillover of K+ ions could modify the catalytic
properties of Ni in the methanol steam reforming or in some side reaction due to the
promotional effect of the potassium ions. In addition, a purely electrocatalytic reaction
between potassium ions and adsorbed reactants or products would lead to the
formation of different surface compounds, which may contain hydrogen and be
decomposed by inverting the polarization [14-16, 26].
3) After adsorption, the reactor was purged with Ar and the system was kept
under open circuit conditions.
4) Finally, a positive potential was applied (still under Ar atmosphere) to
decompose the possible formed potassium-derived surface compounds and to return
the K+ ions back to the solid electrolyte. The experiment ended when gas compounds
stopped of being released from the catalyst surface.
Figure 6.2 shows the time variation of the outlet molar flow rates of the
different detected products, the catalyst potential (VWR) and the current (I) during one
of these experiments performed at 280 ºC with the catalyst Ni 0º. After one hour under
methanol steam reforming conditions and an applied catalyst potential of +2 V (not
depicted), the time (t) was set at 0 min and the unpromoted H2, CO and CO2
production rates reached the following values: 2.55 x 10-8
mol H2 s-1
, 5.8 x 10-9
mol
CO s-1
and 8 x 10-10
mol CO2 s-1
, respectively. According to these values, the methanol
decomposition reaction (MD, reaction 6.1) seemed to prevail over the steam reforming
reaction (SRM, reaction 6.2) as commonly found on supported Ni catalysts [27-29].
CH3OH 2H2 + CO (6.1)
CH3OH + H2O 3H2 + CO2 (6.2)
Page 238
Chapter 6
230
Figure 6.2. Time variation of H2, CO and CO2 molar flow rates (a), current (I) and catalyst
potential (VWR) (b) with catalyst film Ni 0º during the following steps at 280ºC: H2
production/storage, CH3OH/H2O = 4.4 %/5.2 %, I = -20 µA; Cleaning, 100 % Ar, OC; H2
release, 100 % Ar, VWR = +2 V.
Then, the imposition of a constant negative current of -20 µA under the same
reaction conditions led to the alkali ions supply to the catalyst film (at a rate of I/F =
2.1 x 10-10
mol K+ s
-1, according to Faraday’s law). After 45 min, a total number of 5.6
x 10-7
mol K+ was electrochemically migrated and the catalyst potential decreased to
around -0.7 V. During this step, the hydrogen obtained with respect to the CO and
CO2 widely exceeded the stoichiometric value according to the previous reactions, and
0
1
2
3
r /
mo
l s-1
x 10
-8
-1
0
1
2
-0.1
0.3
0.7
1.1
0 10 20 30 40 50 60 70
VW
R/
V
I / A
x 1
0-3
Time / min
-20 µA4.4 % CH3OH + 5.2 % H2O
+2 VAr
OCAr
rH2
rCO
rCO2
VWR
I
a)
b)
Page 239
Electrochemically assisted production and storage of hydrogen
231
both H2 and CO production rates decreased with time on stream to 1.92 x 10-8
mol H2
s-1
, 3.1 x 10-9
mol CO s-1
. This was attributed to the deactivation of the Ni catalyst as a
consequence of the occurrence of carbon deposits derived from the dehydrogenation
of methyl groups or, most likely, from CO dissociation [30, 31], which was probably
favoured by the K+ promotional effect. In fact, Ni-based catalysts are typically prone
to the formation of different forms of carbon, that may poison the catalytic activity,
from a wide variety of molecules such as: methane [32-34], ethylene [35], methanol
[36, 37] or other alcohols [38, 39]. At first glance one cannot appreciate any apparent
promotional effect of potassium on the Ni catalytic activity, probably hindered by the
previously mentioned poisoning effect. This kind of Ni catalyst films thus behaved
very differently from those previously prepared by CAD and electrochemically
promoted in the methanol steam reforming reaction in chapters 1, 2 and 5.
Once the steam reforming reaction was interrupted and all the lines were fully
cleaned by Ar, it can be observed that the final application of VWR = +2 V (which
caused the migration of the K+ ions back to the solid electrolyte according to the
obtained positive current), led to certain H2, CO and CO2 production. Due to the
absence of any possible gaseous molecules in this step (Ar atmosphere), these peaks
could only be attributed to the desorption of the previously stored surface species.
Hence, potassium ions very likely participated in the promotion/formation of such
storing phases under the previous methanol steam reforming conditions and the
negative current imposition. Moreover, the K+ ions not only presented a promotional
effect on the H2 capture mechanism, but also on its release. The subsequent removal of
potassium ions under Ar atmosphere by the positive polarization caused the
decomposition and the release of such stored phases leading to the observed presence
of H2, CO and CO2 in the gas phase. From the integration of each peak, total amounts
of 4.68 x 10-6
mol H2, 4.8 x 10-7
mol CO and 8 x 10-8
mol CO2 were released during
the last step of the experiment under anodic polarization.
The physisorption of H2 molecules on the Ni catalyst was fully dismissed
because of the employed operation conditions (1 atm, 280 ºC). Hence, one can suggest
different possibilities for explaining the above results through hydrogen chemisoprtion
Page 240
Chapter 6
232
mechanisms. Firstly, H atoms adsorbed on Ni active sites could lead to the formation
of nickel hydrides [1, 7]. On the other hand, the H adatoms could also migrate (via
spillover) from the Ni active sites and adsorb on the surface of a secondary material
[10, 11], e.g., carbonaceous surface species formed during the SRM reaction. Another
plausible explanation could be the H2 storage in the form of some methanol-derived
intermediate species on the Ni active sites, such as methoxy groups (CH3O). In fact,
potassium has shown to stabilize this kind of intermediate species on catalytically
active sites [40-42]. Moreover, one cannot rule out the electrocatalytic reaction
between K+ ions and the H2O, H2, CO2 and CO molecules present in the reaction
atmosphere to form potassium bicarbonates species [26, 43] as a source of stored H2
stored molecules. Finally, one could also suggest that K+ ions may react with H2 or
H2O adsorbed molecules to form potassium hydride or hydroxide [44, 45].
Looking at the ratio of the different desorbed molecules during the last step of
the experiment, the formation of either methoxy or bicarbonate species cannot be
considered as the main H2 storage mechanism because much higher CO and CO2
formation rates, according to stoichiometric factors, should then be observed during
the final positive polarization. Furthermore, the number of K+ ions transferred to the
solid electrolyte during the imposition of -20 µA for 45 min (5.6 x 10-7
mol K+) was
one order of magnitude lower than the amount of released H2 (4.68 x 10-6
mol H2) to
the gas phase. Hence, potassium hydride or hydroxide can also be diminished as the
main source of the H2 storage. Then, it seems to indicate that the main plausible
mechanism of H2 storage was the hydrogen chemisorption on either Ni actives sites or
other surface compounds (e.g., carbonaceous deposits).
Before going deeper into the mechanism of H2 storage, a second experiment
was carry out in order to confirm the reproducibility of this process (Figure 6.3). The
only modification in this experiment with respect to that reflected in Figure 6.2 was
the application of a linear sweep voltammetry (LSV) in the final release step. This
linear voltammetry was performed from -0.6 to +2 V at a scan rate of 1 mV s-1
under
Ar atmosphere (instead of the imposition of a fixed VWR = +2 V).
Page 241
Electrochemically assisted production and storage of hydrogen
233
Figure 6.3. Variation of H2, CO and CO2 molar flow rates (a), current (I) and catalyst potential
(VWR) (b) with catalyst film Ni 0º during the following steps at 280ºC: H2 production/storage,
CH3OH/H2O = 4.4 %/5.2 %, I = -20 µA; Cleaning, 100 % Ar, OC; H2 release, 100 % Ar, LSV
from -0.6 V to +2 V (1 mV s-1
).
It should be mentioned that almost the same catalytic activity and Ni
deactivation trend were observed during the imposition of -20 µA for 45 min in both
Figures 6.2 and 6.3. Very similar amounts of transferred K+ ions and released H2
molecules were also obtained during the LSV, which confirms the good
reproducibility of both catalytic and electrocatalytic activity of Ni under the studied
reaction conditions. The only difference with respect to the previous experiment was
0
1
2
3
r /
mo
l s-1
x 10
-8
-1
0
1
2
-3
-1
1
3
5
0 20 40 60 80 100
VW
R/
V
I / A
x 1
0-5
Time / min
-20 µA4.4 % CH3OH + 5.2 % H2O
From -0.6 V to +2 VAr
OCAr
rH2
rCO
rCO2
VWR
I
a)
b)
Page 242
Chapter 6
234
that, as the potential was linearly increased, the removal of K+ ions (according to the I
vs. time curve) and the H2 release took place in a gradual way. Hence, this operational
procedure was selected for further experiments since it allowed a better quantification
of the H2 release through the periodic analysis of the GC.
Moreover, the release peaks in both figures (6.2 and 6.3) seemed to behave in
a similar way than the positive current vs. time peaks (which were related to the
respective transfer of K+ ions back to the solid electrolyte). This means that the K
+
ions had a direct implication on both the H2 storage and its release.
6.3.2. Influence of the applied negative polarization and the reaction atmosphere
Different experiments were performed at 280 ºC with both kinds of Ni
catalysts, i.e., the normal (Ni 0º) and oblique (Ni 80º) angle deposited films, by
varying in each experiment the negative current applied during the H2 storage step.
Figures 6.4a and 6.4b show the time variation of the H2 molar flow rate and the
current (I) obtained for both catalysts during the release step (positive linear sweep
voltammetry under Ar stream) after imposition of three different negative currents
during the storage step (under a feed composition of CH3OH/H2O = 4.4 %/5.2 %).
It should be noted that under negative polarization and reaction conditions (not
shown) a higher catalytic activity and a more pronounced deactivation were observed
on the oblique angle deposited Ni if compared to those of catalyst Ni 0º, which is in
agreement with the higher porosity of the former (35 %). This different microstructure
was probably also a determining factor in the larger amount of released H2 observed
for the case of catalyst Ni 80º. From the integration of the H2 release peaks one can
calculate a maximum value of 6.99 x 10-5
mol H2 stored after 45 min at -30 µA for
catalyst Ni 80º (Figure 6.4b-1) vs. 5.3 x 10-6
mol H2 under analogous conditions for
catalyst Ni 0º (Figure 6.4a-1). Hence, one can observe an increase of one order of
magnitude in the H2 storage capacity of the more porous catalyst film (Ni 80 º). In
addition, the H2 release from the catalyst Ni 80º was extended in time (some hours). A
potential of VWR = +2 V was thus applied at the end of the linear sweep voltammetry
in this case to complete the depletion of H2.
Page 243
Electrochemically assisted production and storage of hydrogen
235
Figure 6.4. Variation of H2 molar flow rate and current (I) with catalyst films Ni 0º (a-1 and a-
2) and Ni 80º (b-1 and b-2) at 280 ºC during the H2 release step (100 % Ar, LSV from -0.6 V to
+2 V, 1 mV s-1
) after the imposition of different negative currents for 45 min during the H2
production/storage step (CH3OH/H2O = 4.4 %/5.2 %).
As in the previous figures (6.2 and 6.3), in all these experiments a much lower
amount of transferred K+ ions was obtained if compared to that of released H2, and
both CO and CO2 releases (not depicted) were again almost negligible. Apart from
certain possible formation of adsorbed methoxy species, potassium hydride, hydroxide
or bicarbonates, the main storage mechanism seemed to be the chemisorption of
atomic hydrogen on Ni and carbonaceous deposits. At this point, it is important to
remark that the H to Ni ratio obtained with catalyst Ni 80º was quite high (up to 11,
after 45 min at -30 µA), thus suggesting that the worthy H2 storage results were rather
due to the spillover of H atoms on carbon compounds formed under SRM conditions.
Furthermore, one can appreciate from this figure a very strong effect of the
transfer of K+ ions during the storage step (negative current supply) on the process
performance. The application of more negative currents during the H2
0
2
4
6
8
10
12
r H
/ m
ol s
-1x
10-9
-1
0
1
2
-1
2
5
8
0 10 20 30 40
VW
R/
V
I / A
x 1
0-5
Time / min
After -10 µA
After -20 µA
After -30 µA
I after -10 µA
I after -20 µA
I after -30 µA
a-1)
2
VWR
-1
0
1
2
-1
2
5
8
0 50 100 150 200 250
VW
R/
V
I / A
x 1
0-5
Time / min
0
3
6
9
12
15
18
21
r H
/ m
ol s
-1x
10-9
After -10 µA
After -20 µA
After -30 µA
I after -10 µA
I after -20 µA
I after -30 µA
b-1)
2
VWR
a-2) b-2)
Ni 0º Ni 80º
Page 244
Chapter 6
236
production/storage step caused a remarkable subsequent release of H2 up to certain
saturation value (similar hydrogen storage results were obtained after the application
of either -20 µA or -30 µA). As expected, higher positive current values were also
observed during the linear sweep voltammetries (Figures 6.4a-2 and 6.4b-2) after
higher negative polarizations. This information has been summarized in Figure 6.5. It
shows the results obtained with both Ni catalysts (Ni 0º and Ni 80º) as a function of
the negative current applied during the H2 production/storage step, in terms of total
hydrogen released (in units of g H2 x 100 g Ni-1
) during the subsequent LSV.
Figure 6.5. Influence of the negative current (I) applied for 45 min during the H2
production/storage step (CH3OH/H2O = 4.4 %/5.2 %) on the H2 storage capacity of catalyst
films Ni 0º and Ni 80º at 280 ºC, measured during the subsequent H2 release steps (100 % Ar,
LSV from -0.6 V to +2 V, 1 mV s-1
).
From both Figures 6.4 and 6.5, one can state that there is a clear enhancement
effect of K+ ions on the H2 storage capacity of the Ni catalysts. As discussed in the
previous chapter, the presence of alkali (K+) ions on Ni likely activated the CH3OH
adsorption and successive dehydrogenation toward H2 and CO (reactions 6.3-6.8).
CH3OH → CH3OH(a) (6.3)
CH3OH(a) → CH3O(a) + H(a) (6.4)
CH3O(a) → H2CO(a) + H(a) (6.5)
0
3
6
9
12
15
18
21
After -10 µA After -20 µA After -30 µA
Tota
l H
2re
leas
ed
/ g
H2
x 10
0 g
Ni-1
Ni 0º
Ni 80º
Page 245
Electrochemically assisted production and storage of hydrogen
237
H2CO(a) → CO(a) + 2H(a) (6.6)
2H(a) → H2 (6.7)
CO(a) → CO (6.8)
Then, potassium ions would also favor the subsequent dissociation of CO
molecules and hence the carbon deposition (reaction 6.9), as typically reported in
other EPOC [46, 47] and classical alkali promotion [48, 49] studies.
CO(a) → C(a) + O(a) (6.9)
K+ ions could also stabilize the hydrogen adsorbed on these carbon compounds
as observed in other studies where alkali ions were intercalated into graphitic layers
[50-52]. This would explain the similarity observed in all the experiments, especially
with catalyst Ni 0º, between the H2 release and the positive current peaks.
This way, the increase in the H2 storage capacity of the Ni catalyst films upon
increasing the applied negative current under SRM conditions could be attributed to a
triple promoting effect of potassium ions: 1) the promotion of the methanol
dehydrogenation reaction, 2) the increase in the formation of surface carbon
compounds which acted as hydrogen adsorbents and 3) the hydrogen chemisorption
and stabilization on the carbonaceous stored compounds. This storage mechanism
would also explain the slower H2 release rate observed in Figure 6.4b-1 after the
application of more negative currents. A reverse spillover of the H adatoms toward the
Ni active sites would be necessary before these H atoms can be recombined and
desorbed to the gas phase as H2 molecules [53]. This H back-spillover would be
slowed since the increase in the K+-assisted formation of carbonaceous compounds
would make the H adsorption to take place on less accessible sites.
In contrast to previous studies where NOx [14, 15] or CO2 [16] were captured
by using K+-conductor solid electrolytes through the formation of some nitrates or
carbonates, respectively, in the present study K+ ions should barely be bonded with the
stored compound (H2). Instead, these K+ ions could be involved in the formation of an
adsorbent carbonaceous material and the subsequent H2 adsorption.
Page 246
Chapter 6
238
Regarding the influence of the Ni catalyst microstructure on the H2 storage
capacity, Figure 6.5 depicts a great difference between both catalyst films. Whilst
catalyst Ni 0º stored/released up to 0.5 g H2 x 100 g Ni-1
, catalyst Ni 80º reached a
maximum H2 storage capacity of around 19 g H2 x 100 g Ni-1
. This increase was
attributed to the higher porosity of the latter (35 %) and its paramount role in the
formation of larger carbon deposits on which hydrogen could be adsorbed. The
potential interest of the oblique angle deposited catalysts for the H2 storage in form of
MgH2 was previously reported for V-decorated and V-doped Mg nanostructures [21,
22]. In these studies the H2 adsorption and desorption temperatures generally
decreased upon increasing the zenithal deposition angle [22], and the theoretical
hydrogen storage capacity for MgH2 (7.6 wt. %) was reached at around 280 ºC [21].
Herein, the deposition of a Ni catalyst film at an incident angle of α = 80º
allowed to obtain apparent H2 storage capacities (up to 19 wt. %) which were excellent
as compared to those shown, for instance, with Mg2NiH4 (3.6 wt. %), other hydrides
of Ni-containing alloys working at lower operation temperatures such as CoNi5H4 or
LaNi5H6 (less than 2 wt. %), and even the best-performing hydrides, i.e., the
borohydrides (around 18 wt. %), which typically require decomposition temperatures
up to 600 ºC [1, 5, 7]. However, this comparison must be carried out with caution. The
weight percentage values of stored H2 shown in Figure 6.5 were related to the amount
of Ni deposited, since this parameter was quantified by X-ray fluorescence
spectroscopy and remained, regardless the operation conditions, unalterable.
In order to clarify the mechanism of the adsorbed species responsible for the
hydrogen trapping, additional experiments were carried out with catalyst Ni 80º, in
which H2 was stored under the application of a potential of VWR = -1 V for 1 hour and
different feed compositions: methanol steam reforming conditions (4.4 % CH3OH, 5.2
% H2O), methanol decomposition conditions (only 4.4 % CH3OH, i.e., in absence of
water) and a 2.3 % H2 stream, all of them at 280 ºC and with an overall flow rate of 6
L h-1
(Ar balance). Figure 6.6 shows the variation in the released H2 flow rate and the
current during the last step of these experiments, consisting of a positive LSV from -1
to +2 V (scan rate = 1 mV s-1
) and maintenance of the latter potential.
Page 247
Electrochemically assisted production and storage of hydrogen
239
Figure 6.6. Variation of H2 molar flow rate (a) and current (I) (b) with catalyst film Ni 80º at
280 ºC during the H2 release step (100 % Ar, LSV from -1 V to +2 V, 1 mV s-1
) following the
H2 production/storage step (VWR = -1 V) under different atmospheres.
Regarding the LSV performed after the negative polarization under the 2.3 %
H2 stream, a small peak can be detected in Figure 6.6a that corresponded to 4 x 10-7
mol H2 released. This peak was quite synchronized with the positive current peak
(Figure 6.6b), which corresponded to an amount of transferred K+ ions of the same
order of magnitude (5 x 10-7
mol K+), thus suggesting that some H2 was stored during
the negative polarization step by interacting with the K+ ions. However, the H2 release
peak detected in this experiment was still negligible compared to those obtained after
the negative polarization step carried out in CH3OH or CH3OH+H2O atmospheres.
-1.5
-0.5
0.5
1.5
-1
2
5
0 50 100 150 200 250 300 350
VW
R/
V
I / A
x 1
0-5
Time / min
-1.5
-0.5
0.5
1.5
2.5
0
2
4
6
8
10
VW
R/
V
r H
/ m
ol s
-1x
10
-9
a)
b)
After H2
After CH3OHAfter CH3OH + H2O
VWR
After H2
After CH3OHAfter CH3OH + H2O
VWR
2
Page 248
Chapter 6
240
These findings are in good agreement with the proposed mechanism of H2 storage
based on a chemical adsorption of H atoms on carbon compounds, which were formed
during the SRM reaction under the effect of K+ supply. Furthermore, the total amount
of obtained H2 was very similar in both the absence and the presence of water (6.98 x
10-5
and 6.82 x 10-5
mol H2, respectively), the release process being faster in the later
case. Thus one could state that both the hydrogen and the carbonaceous adsorbent
were derived from the methanol itself, whereas water would facilitate the H spillover
process through the formation of surface hydroxyl groups [10, 53, 54].
The spillover-assisted hydrogen storage has been reported in literature for
many different carbon-based or metal oxide adsorbents which acted as catalyst support
[8, 10, 11, 13]. On the other hand, hydrogen is typically supplied either from an
aqueous solution (electrochemically, i.e., as H+) [9, 55-58] or from the gas phase (as
H2) [53, 59-68]. Group VIII metals such as Ni [67, 68], Pt [53, 59-63] or Pd [62-66]
are commonly employed catalysts in the latter case due to their good activity in
hydrogen dissociation. In the present work, both hydrogen and carbon-based adsorbent
were obtained from the very process of methanol conversion in a single reaction step
on Ni catalysts, which was assisted by the promotional effect of K+ ions. Hence, the
coupling of solid-state electrochemistry and catalysis, along with the employment of
this novel kind of oblique angle deposited catalyst films with high porosity, allowed to
obtain vey high yields of H2 storage per unit area and amount of metal. This would
represent an alternative to the H2 capture classically performed in metal hydrides with
high decomposition temperatures such as MgH2, alanates or borohydrides.
Summarizing, the proposed Ni/K-βAl2O3 electrocatalytic system shows the
potential application of EPOC for the in-situ controlled storage and release of
hydrogen under mild reaction conditions (280 ºC). The obtained highly pure hydrogen
could be then conditioned and used by either fuel cells or other catalytic processes.
The oblique angle deposited Ni catalyst film (Ni 80º), which showed a
superior performance for H2 production and storage, was characterized by several
techniques. Figure 6.7 shows SEM images of a selected area from catalyst Ni 80º,
after being exposed to the application of VWR = -1 V for 1 hour under SRM conditions.
Page 249
Electrochemically assisted production and storage of hydrogen
241
Figure 6.7. Top view SEM image of a selected area (a) of catalyst Ni 80º after the H2
production/storage experiments and the final application of VWR = -1 V for 1 h (280 ºC, 4.4 %
CH3OH, 5.2 % H2O), along with different magnifications (b and c).
Nickel nanocolumns can be observed in these micrographs, grouped and
distributed on the K-βAl2O3 support in a quite similar way than those observed in the
10 µm
a)
5 µm
b)
2 µm
c)
Page 250
Chapter 6
242
fresh sample (see Figure 6.1d). This seems to indicate that the Ni catalyst film did not
suffer significant modifications during the H2 production and storage experiments,
demonstrating the good stability of the oblique angle deposited (OAD) catalyst films.
Then, additional characterization measurements were carried out to identify
the deposited carbonaceous molecules and the storage process that takes place.
6.3.3. Investigation of possible surface compounds by catalyst characterization
Figure 6.8 shows other SEM micrographs of catalyst film Ni 80º after
exposure to methanol steam reforming conditions (CH3OH/H2O = 4.4 %/5.2 %) and
the application of VWR = -1 V.
Figure 6.8. Top view SEM images of selected areas of catalyst Ni 80º after the H2
production/storage experiments and the final application of VWR = -1 V for 1 h (280 ºC, 4.4 %
CH3OH, 5.2 % H2O).
5 µm
b)
10 µm
a)
Page 251
Electrochemically assisted production and storage of hydrogen
243
In these SEM images one can observe black regions of pillared fragments
corresponding to carbonaceous deposits covering the Ni surface. These compounds
will be later identified mainly as graphene oxide (GO), which could fit this two-
dimensional appearance layerwise. In this micrographs, single thin GO sheets cannot
be appreciated, as those observed in other studies where graphene oxide was obtained
from graphite oxidation and exfoliation [69, 70]. Nevertheless, highly concentrated
graphene oxide typically exhibits a wrinkle and overlapped structure similar to that
shown in this figure [70, 71].
Figure 6.9 shows the XPS spectra obtained from catalyst film Ni 80º after the
exposition to the mentioned reaction conditions, along with a quantitative estimation
of the elemental concentration, relative to the main component, i.e., Ni, in the
examined outer layer.
Figure 6.9. XPS spectra (a) of catalyst Ni 80º after the H2 production/storage experiments and
the final application of VWR = -1 V for 1 h (280 ºC, 4.4 % CH3OH, 5.2 % H2O), and the
corresponding surface atomic composition relative to Ni (b).
80 70 60
Co
un
ts / a
.u.
Binding energy / eV
880 870 860 850 840
Co
un
ts / a
.u.
Binding energy / eV
540 535 530 525
Co
un
ts / a
.u.
Binding energy / eV
420 410 400 390C
ou
nts
/ a
.u.
Binding energy / eV
290 280 270
Co
un
ts / a
.u.
Binding energy / eV
O 1sNi 2p
Al 2p + Ni 3p
Ni LMM
C 1s + K 2p
at. K/Ni at. O/Ni at. C/Ni at. Al/Ni1.26 9.16 4.34 0.09
a)
b)
Page 252
Chapter 6
244
The peak observed at 284.7 eV corresponded to C 1s and the K 2p region
presented two contributions at 292.4 eV (K 2p3/2) and 295 eV (K 2p1/2). Interestingly,
other peak was found in the intermediate region (at 289.4 eV), which was assigned to
the presence of potassium carbonates on the catalyst surface [72, 73]. These species
would be formed under negative polarization due to the interaction between the K+
ions and the molecules present in the reaction mixture as discussed in the previous
section. Their decomposition would probably lead to some CO and CO2 release during
the positive polarization step (see Figure 6.2 or 6.3). On the other hand, the Ni outer
surface showed to be at least partially oxidized, since the peaks observed at 531 eV (O
1s region), at 855.1 eV and 860.9 eV (Ni 2p region) can be attributed to the presence
of Ni(OH)2 or Ni2O3 [74, 75]. Moreover, as also observed in chapter 4, a high amount
of carbon was found on the Ni electrochemical catalyst, which covered the catalyst
surface (C/Ni atomic ratio of 4.34). This finding would agree with the H2 storage
mechanism through H spillover on carbonaceous deposits proposed.
Figure 6.10 shows the RAMAN spectrum of catalyst film Ni 80º exposed to
the same reaction conditions as those of previous figures.
Figure 6.10. Raman spectrum of catalyst Ni 80º after the H2 production/storage experiments
and the final application of VWR = -1 V for 1 h (280 ºC, 4.4 % CH3OH, 5.2 % H2O).
800 1600 2400 3200 4000
Inte
nsity /
a.u
.
Raman shift / cm-1
Page 253
Electrochemically assisted production and storage of hydrogen
245
A narrow peak at 1072 cm-1
can be assigned to the ν(CO32-
) symmetric stretch
[76, 77], in good agreement with the carbonate species previously detected by XPS.
However, the most relevant finding was the presence of strong disordered (D) and
graphitic (G) bands at 1343 cm-1
and 1597 cm-1
, respectively, which are characteristic
of graphene oxide (GO) [59, 78, 79], along with less intense 2D bands at 2500-3200
cm-1
. The ratio of intensities of the D and G peaks (ID/IG) was lower than the unity,
which suggested a quite ordered crystalline structure.
Finally, Figure 6.11 shows the corresponding FTIR spectrum of the catalyst
film Ni 80º in the range from 750 to 1900 cm-1
. No clear bands were found at higher
wavenumbers, associated with ν(CH3) or ν(OH) stretching vibrations (not shown).
Figure 6.11. FTIR spectrum of catalyst Ni 80º after the H2 production/storage experiments and
the final application of VWR = -1 V for 1 h (280 ºC, 4.4 % CH3OH, 5.2 % H2O).
The following vibrational modes were identified in this spectrum: C=O stretch
at 1650 cm-1
, adsorbed water and skeletal vibrations of unoxidized graphitic domains
(C=C) at 1627 cm-1
, COO- symmetric stretch at 1366 cm
-1, epoxide stretch at 970 cm
-1
and C-H out-of-plane wag at 831 cm-1
. The presence of these functional groups would
be in good agreement with the formation of grapheme oxide, as also observed in FTIR
spectra reported elsewhere [79, 80]. Indeed, according to the well-accepted Lerf-
1800 1600 1400 1200 1000 800
Ab
so
rba
nce
/ a
.u.
Wavenumbers / cm-1
Page 254
Chapter 6
246
Klinowski model of graphene oxide [81], this compound consists of aromatic units of
variable size which are separated from each other by aliphatic 6-membered rings,
where the basal plane contains atomic oxygen in the form of hydroxyl (C-OH) or
epoxide (C-O-C) groups. The edges of graphite and graphene oxide sheets and
oxidized carbons can also contain other oxygen species such as C-OH, carbonyl
(C=O) and carboxyl (O=C-OH) groups.
This way, the presence of graphene oxide on the catalyst surface was
confirmed by both Raman and infrared spectroscopy. Some concurrence of potassium-
derived species such as bicarbonates cannot be ruled out. However, according to the
analysis carried out, graphene oxide can be postulated as the main adsorbent involved
in the H2 storage mechanism discussed in the previous section. It is then supposed that
the hydroxyl and oxygen groups present on the graphene oxide favoured the spillover
of H atoms [13, 53, 54], which would specially adsorb at the edge sites of the layers
and in the vicinity of these functional groups [12, 13, 61, 82].
6.4. CONCLUSIONS
The following conclusions can be drawn from this study:
- The phenomenon of electrochemical promotion (EPOC) was for the first time
applied in the H2 production and storage processes, by using oblique angle deposited
Ni catalyst films and a K+-conductor support.
- H2 was simultaneously produced from methanol steam reforming (SRM) and
stored under the promotional effect of K+ ions (cathodic polarization). Then, hydrogen
was released with high purity upon removing the K+ ions from the catalyst film
(anodic polarization).
- A higher H2 release was obtained by increasing both the negative current
applied during the H2 production/storage step and the porosity of the Ni catalyst film
(which was related to the incident angle during the catalyst deposition). The presence
of methanol in the reaction atmosphere showed to be needed to obtain an appreciable
amount of released H2. On the other hand, in view of the amounts of transferred K+
ions, and those of released H2, CO and CO2 during the different experiments, the
Page 255
Electrochemically assisted production and storage of hydrogen
247
direct formation of potassium-derived compounds (hydride, hydroxide, bicarbonates)
or the stabilization of some reaction intermediate (like methoxy) were not considered
as the main sources of the observed released H2.
- Regarding the obtained catalytic results and different ex-situ characterization
measurements, a H2 storage mechanism was proposed based on the hydrogen
chemisorption on both Ni active sites and graphene oxide, which was deposited under
SRM electropromoted conditions, through a spillover process. Hence, a triple
promoting effect of K+ ions would take place: 1) promotion of the methanol
dehydrogenation, 2) promotion of the formation of graphene oxide, and 3) promotion
of the hydrogen chemisorption on graphene oxide.
- A very high H2 storage capacity per amount of metal was obtained (19 g H2 x
100 g Ni-1
with the oblique angle deposited Ni catalyst film), which shows a very
promising application of this kind of electrocatalytic systems and the EPOC
phenomenon.
6.5. REFERENCES
[1] L. Schlapbach, A. Züttel, Hydrogen-storage materials for mobile applications, Nature, 414
(2001) 353-358.
[2] S. Dutta, A review on production, storage of hydrogen and its utilization as an energy
resource, Journal of Industrial and Engineering Chemistry, 20 (2014) 1148-1156.
[3] J.N. Armor, Catalysis and the hydrogen economy, Catalysis Letters, 101 (2005) 131-135.
[4] J.D. Holladay, J. Hu, D.L. King, Y. Wang, An overview of hydrogen production
technologies, Catalysis Today, 139 (2009) 244-260.
[5] A.F. Dalebrook, W. Gan, M. Grasemann, S. Moret, G. Laurenczy, Hydrogen storage:
Beyond conventional methods, Chemical Communications, 49 (2013) 8735-8751.
[6] P. Makowski, A. Thomas, P. Kuhn, F. Goettmann, Organic materials for hydrogen storage
applications: From physisorption on organic solids to chemisorption in organic molecules,
Energy and Environmental Science, 2 (2009) 480-490.
[7] A.W.C. Van Den Berg, C.O. Areán, Materials for hydrogen storage: Current research
trends and perspectives, Chemical Communications, (2008) 668-681.
Page 256
Chapter 6
248
[8] L. Wang, R.T. Yang, Hydrogen storage on carbon-based adsorbents and storage at
ambient temperature by hydrogen spillover, Catalysis Reviews - Science and Engineering, 52
(2010) 411-461.
[9] S. Mukherjee, B. Ramalingam, S. Gangopadhyay, Hydrogen spillover at sub-2 nm Pt
nanoparticles by electrochemical hydrogen loading, Journal of Materials Chemistry A, 2
(2014) 3954-3960.
[10] W.C. Conner Jr, J.L. Falconer, Spillover in heterogeneous catalysis, Chemical Reviews,
95 (1995) 759-788.
[11] R. Prins, Hydrogen spillover. Facts and fiction, Chemical Reviews, 112 (2012) 2714-
2738.
[12] S. Gadipelli, Z.X. Guo, Graphene-based materials: Synthesis and gas sorption, storage
and separation, Progress in Materials Science, 69 (2015) 1-60.
[13] G.M. Psofogiannakis, G.E. Froudakis, Fundamental studies and perceptions on the
spillover mechanism for hydrogen storage, Chemical Communications, 47 (2011) 7933-7943.
[14] A. de Lucas-Consuegra, Á. Caravaca, P. Sánchez, F. Dorado, J.L. Valverde, A new
improvement of catalysis by solid-state electrochemistry: An electrochemically assisted NOx
storage/reduction catalyst, Journal of Catalysis, 259 (2008) 54-65.
[15] A. de Lucas-Consuegra, A. Caravaca, M.J. Martín de Vidales, F. Dorado, S. Balomenou,
D. Tsiplakides, P. Vernoux, J.L. Valverde, An electrochemically assisted NOx
storage/reduction catalyst operating under fixed lean burn conditions, Catalysis
Communications, 11 (2009) 247-251.
[16] E. Ruiz, D. Cillero, Á. Morales, G.S. Vicente, G. De Diego, P.J. Martínez, J.M. Sánchez,
Bench scale study of electrochemically promoted CO2 capture on Pt/K-βAl2O3,
Electrochimica Acta, 112 (2013) 967-975.
[17] M. Matthew, M.T. Hawkeye, M.J.B. Taschuk, Glancing Angle Deposition of Thin Films:
Engineering the Nanoscale, Wiley2014.
[18] V. Rico, P. Romero, J.L. Hueso, J.P. Espinós, A.R. González-Elipe, Wetting angles and
photocatalytic activities of illuminated TiO2 thin films, Catalysis Today, 143 (2009) 347-354.
[19] V. Rico, A. Borrás, F. Yubero, J.P. Espinós, F. Frutos, A.R. González-Elipe, Wetting
angles on illuminated ta2o5 thin films with controlled nanostructure, Journal of Physical
Chemistry C, 113 (2009) 3775-3784.
[20] D.X. Ye, Y.P. Zhao, G.R. Yang, Y.G. Zhao, G.C. Wang, T.M. Lu, Manipulating the
column tilt angles of nanocolumnar films by glancing-angle deposition, Nanotechnology, 13
(2002) 615-618.
Page 257
Electrochemically assisted production and storage of hydrogen
249
[21] Y. He, Y. Zhao, Hydrogen storage and cycling properties of a vanadium decorated Mg
nanoblade array on a Ti coated Si substrate, Nanotechnology, 20 (2009).
[22] Y. He, J. Fan, Y. Zhao, The role of differently distributed vanadium nanocatalyst in the
hydrogen storage of magnesium nanostructures, International Journal of Hydrogen Energy, 35
(2010) 4162-4170.
[23] N.N. Kariuki, W.J. Khudhayer, T. Karabacak, D.J. Myers, GLAD Pt-Ni alloy nanorods for
oxygen reduction reaction, ACS Catalysis, 3 (2013) 3123-3132.
[24] J.R. Sánchez-Valencia, A. Borrás, A. Barranco, V.J. Rico, J.P. Espinos, A.R. González-
Elipe, Preillumination of TiO2 and Ta2O5 photoactive thin films as a tool to tailor the
synthesis of composite materials, Langmuir, 24 (2008) 9460-9469.
[25] C.G. Vayenas, S. Bebelis, C. Pliangos, S. Brosda, D. Tsiplakides, Electrochemical
Activation of Catalysis: Promotion, Electrochemical Promotion and Metal-Support
Interactions, Kluwer Academic Publishers/Plenum Press, New York, 2001.
[26] A. de Lucas-Consuegra, F. Dorado, J.L. Valverde, R. Karoum, P. Vernoux, Low-
temperature propene combustion over Pt/K-βAl2O3 electrochemical catalyst:
Characterization, catalytic activity measurements, and investigation of the NEMCA effect,
Journal of Catalysis, 251 (2007) 474-484.
[27] N. Iwasa, M. Yoshikawa, W. Nomura, M. Arai, Transformation of methanol in the
presence of steam and oxygen over ZnO-supported transition metal catalysts under stream
reforming conditions, Applied Catalysis A: General, 292 (2005) 215-222.
[28] B.S. Chen, J.L. Falconer, Alcohol Decomposition by Reverse Spillover, Journal of
Catalysis, 144 (1993) 214-226.
[29] N. Takezawa, N. Iwasa, Steam reforming and dehydrogenation of methanol: Difference in
the catalytic functions of copper and group VIII metals, Catalysis Today, 36 (1997) 45-56.
[30] M.T. Tavares, I. Alstrup, C.A. Bernardo, J.R. Rostrup-Nielsen, Carbon formation and CO
methanation on silica-supported nickel and nickel-copper catalysts in CO + H2 mixtures,
Journal of Catalysis, 158 (1996) 402-410.
[31] Z. Murayama, I. Kojima, E. Miyazaki, I. Yasumori, Dissociation of carbon monoxide on
stepped nickel surfaces, Surface Science, 118 (1982) L281-L285.
[32] A. Serrano-Lotina, L. Daza, Long-term stability test of Ni-based catalyst in carbon dioxide
reforming of methane, Applied Catalysis A: General, 474 (2014) 107-113.
[33] J.L. Pinilla, I. Suelves, M.J. Lázaro, R. Moliner, J.M. Palacios, Parametric study of the
decomposition of methane using a NiCu/Al 2O3 catalyst in a fluidized bed reactor,
International Journal of Hydrogen Energy, 35 (2010) 9801-9809.
Page 258
Chapter 6
250
[34] H.F. Abbas, W.M.A. Wan Daud, Hydrogen production by methane decomposition: A
review, International Journal of Hydrogen Energy, 35 (2010) 1160-1190.
[35] V. Jiménez, A. Nieto-Márquez, J.A. Díaz, R. Romero, P. Sánchez, J.L. Valverde, A.
Romero, Pilot plant scale study of the influence of the operating conditions in the production
of carbon nanofibers, Industrial and Engineering Chemistry Research, 48 (2009) 8407-8417.
[36] P. Wei, W. Xia, J.Z. Li, H. Long, J. Chen, T. Li, M. Fan, Single-phase Ni3Sn alloy alkali-
leached for hydrogen production from methanol decomposition, Renewable Energy, 78 (2015)
357-363.
[37] J.H. Jang, Y. Xu, D.H. Chun, M. Demura, D.M. Wee, T. Hirano, Effects of steam addition
on the spontaneous activation in Ni3Al foil catalysts during methanol decomposition, Journal
of Molecular Catalysis A: Chemical, 307 (2009) 21-28.
[38] G. Wu, C. Zhang, S. Li, Z. Han, T. Wang, X. Ma, J. Gong, Hydrogen production via
glycerol steam reforming over Ni/Al 2O3: Influence of nickel precursors, ACS Sustainable
Chemistry and Engineering, 1 (2013) 1052-1062.
[39] R. Carrera Cerritos, R. Fuentes Ramírez, A.F. Aguilera Alvarado, J.M. Martínez Rosales,
T. Viveros García, I.R. Galindo Esquivel, Steam reforming of ethanol over Ni/Al2O3-La 2O3
catalysts synthesized by sol-gel, Industrial and Engineering Chemistry Research, 50 (2011)
2576-2584.
[40] J. Raskó, J. Bontovics, F. Solymosi, FTIR study of the interaction of methanol with clean
and potassium-doped Pd SiO2 catalysts, Journal of Catalysis, 146 (1994) 22-33.
[41] J.T. Miller, B.L. Meyers, F.S. Modica, G.S. Lane, M. Vaarkamp, D.C. Koningsberger,
Hydrogen Temperature-Programmed Desorption (H2 TPD) of Supported Platinum Catalysts,
Journal of Catalysis, 143 (1993) 395-408.
[42] F. Solymosi, A. Berkó, Z. Tóth, Adsorption and dissociation of CH3OH on clean and K-
promoted Pd(100) surfaces, Surface Science, 285 (1993) 197-208.
[43] P. Vernoux, F. Gaillard, C. Lopez, E. Siebert, In-situ electrochemical control of the
catalytic activity of platinum for the propene oxidation, Solid State Ionics, 175 (2004) 609-613.
[44] V.V. Simonyan, J.K. Johnson, Hydrogen storage in carbon nanotubes and graphitic
nanofibers, Journal of Alloys and Compounds, 330-332 (2002) 659-665.
[45] R.T. Yang, Hydrogen storage by alkali-doped carbon nanotubes-revisited, Carbon, 38
(2000) 623-626.
[46] C.G. Vayenas, S. Neophytides, Non-faradaic electrochemical modification of catalytic
activity III. The case of methanol oxidation on Pt, Journal of Catalysis, 127 (1991) 645-664.
Page 259
Electrochemically assisted production and storage of hydrogen
251
[47] E. Ruiz, D. Cillero, P.J. Martínez, Á. Morales, G.S. Vicente, G. De Diego, J.M. Sánchez,
Bench-scale study of electrochemically assisted catalytic CO2 hydrogenation to hydrocarbon
fuels on Pt, Ni and Pd films deposited on YSZ, Journal of CO2 Utilization, 8 (2014) 1-20.
[48] J.E. Crowell, G.A. Somorjai, The effect of potassium on the chemisorption of carbon
monoxide on the Rh(111) crystal face, Applied Surface Science, 19 (1984) 73-91.
[49] R.W. Joyner, R.A.v. Santen, Elementary Reaction Steps in Heterogeneous Catalysis,
Springer, Netherlands, 2012.
[50] H. Zabel, S.A. Solin, Graphite Intercalation Compounds II: Transport and Electronic
Properties, Springer-Verlag, Berlin Heidelberg, 2013.
[51] W.Q. Deng, X. Xu, W.A. Goddard, New alkali doped pillared carbon materials designed
to achieve practical reversible hydrogen storage for transportation, Physical Review Letters,
92 (2004) 166103-166101.
[52] M. Inagaki, O. Tanaike, Determining factors for the intercalation into carbon materials
from organic solutions, Carbon, 39 (2001) 1083-1090.
[53] W.J. Ambs, M.M. Mitchell Jr, Hydrogen spillover on platinum-alumina, effect of water,
Journal of Catalysis, 82 (1983) 226-229.
[54] Q. Li, A.D. Lueking, Effect of surface oxygen groups and water on hydrogen spillover in
pt-doped activated carbon, Journal of Physical Chemistry C, 115 (2011) 4273-4282.
[55] R. Krishna, E. Titus, L.C. Costa, J.C.J.M.D.S. Menezes, M.R.P. Correia, S. Pinto, J.
Ventura, J.P. Araújo, J.A.S. Cavaleiro, J.J.A. Gracio, Facile synthesis of hydrogenated reduced
graphene oxide via hydrogen spillover mechanism, Journal of Materials Chemistry, 22 (2012)
10457-10459.
[56] Z. Dongping, J. Velmurugan, M.V. Mirkin, Adsorption/desorption of hydrogen on Pt
nanoelectrodes: Evidence of surface diffusion and spillover, Journal of the American Chemical
Society, 131 (2009) 14756-14760.
[57] D.V. Esposito, I. Levin, T.P. Moffat, A.A. Talin, H2 evolution at Si-based metal-
insulator-semiconductor photoelectrodes enhanced by inversion channel charge collection and
H spillover, Nature Materials, 12 (2013) 562-568.
[58] S. Sata, M.I. Awad, M.S. El-Deab, T. Okajima, T. Ohsaka, Hydrogen spillover
phenomenon: Enhanced reversible hydrogen adsorption/desorption at Ta2O5-coated Pt
electrode in acidic media, Electrochimica Acta, 55 (2010) 3528-3536.
[59] V.H. Pham, T.T. Dang, K. Singh, S.H. Hur, E.W. Shin, J.S. Kim, M.A. Lee, S.H. Baeck,
J.S. Chung, A catalytic and efficient route for reduction of graphene oxide by hydrogen
spillover, Journal of Materials Chemistry A, 1 (2013) 1070-1077.
Page 260
Chapter 6
252
[60] F.H. Yang, A.J. Lachawiec Jr, R.T. Yang, Adsorption of spillover hydrogen atoms on
single-wall carbon nanotubes, Journal of Physical Chemistry B, 110 (2006) 6236-6244.
[61] P.C.H. Mitchell, A.J. Ramirez-Cuesta, S.F. Parker, J. Tomkinson, D. Thompsett,
Hydrogen spillover on carbon-supported metal catalysts studied by inelastic neutron
scattering. Surface vibrational states and hydrogen riding modes, Journal of Physical
Chemistry B, 107 (2003) 6838-6845.
[62] A.D. Lueking, R.T. Yang, Hydrogen spillover to enhance hydrogen storage - Study of the
effect of carbon physicochemical properties, Applied Catalysis A: General, 265 (2004) 259-
268.
[63] C.C. Huang, N.W. Pu, C.A. Wang, J.C. Huang, Y. Sung, M.D. Ger, Hydrogen storage in
graphene decorated with Pd and Pt nano-particles using an electroless deposition technique,
Separation and Purification Technology, 82 (2011) 210-215.
[64] A.J. Lachawiec Jr, G. Qi, R.T. Yang, Hydrogen storage in nanostructured carbons by
spillover: Bridge-building enhancement, Langmuir, 21 (2005) 11418-11424.
[65] E. Yoo, L. Gao, T. Komatsu, N. Yagai, K. Arai, T. Yamazaki, K. Matsuishi, T.
Matsumoto, J. Nakamura, Atomic hydrogen storage in carbon nanotubes promoted by metal
catalysts, Journal of Physical Chemistry B, 108 (2004) 18903-18907.
[66] L. Wang, F.H. Yang, R.T. Yang, M.A. Miller, Effect of surface oxygen groups in carbons
on hydrogen storage by spillover, Industrial and Engineering Chemistry Research, 48 (2009)
2920-2926.
[67] L. Zubizarreta, J.A. Menéndez, J.J. Pis, A. Arenillas, Improving hydrogen storage in Ni-
doped carbon nanospheres, International Journal of Hydrogen Energy, 34 (2009) 3070-3076.
[68] K.Y. Lin, W.T. Tsai, T.J. Yang, Effect of Ni nanoparticle distribution on hydrogen uptake
in carbon nanotubes, Journal of Power Sources, 196 (2011) 3389-3394.
[69] J. Kim, L.J. Cote, J. Huang, Two dimensional soft material: New faces of graphene oxide,
Accounts of Chemical Research, 45 (2012) 1356-1364.
[70] G. Sobon, J. Sotor, J. Jagiello, R. Kozinski, M. Zdrojek, M. Holdynski, P. Paletko, J.
Boguslawski, L. Lipinska, K.M. Abramski, Graphene Oxide vs. Reduced Graphene Oxide as
saturable absorbers for Er-doped passively mode-locked fiber laser, Optics Express, 20 (2012)
19463-19473.
[71] Y. Shao, J. Wang, M. Engelhard, C. Wang, Y. Lin, Facile and controllable
electrochemical reduction of graphene oxide and its applications, Journal of Materials
Chemistry, 20 (2010) 743-748.
Page 261
Electrochemically assisted production and storage of hydrogen
253
[72] A. Iordan, M.I. Zaki, C. Kappenstein, Interfacial chemistry in the preparation of catalytic
potassium-modified aluminas, Journal of the Chemical Society, Faraday Transactions, 89
(1993) 2527-2536.
[73] M.E. Gálvez, S. Ascaso, P. Stelmachowski, P. Legutko, A. Kotarba, R. Moliner, M.J.
Lázaro, Influence of the surface potassium species in Fe-K/Al2O3 catalysts on the soot
oxidation activity in the presence of NOx, Applied Catalysis B: Environmental, 152-153 (2014)
88-98.
[74] A.P. Grosvenor, M.C. Biesinger, R.S.C. Smart, N.S. McIntyre, New interpretations of
XPS spectra of nickel metal and oxides, Surface Science, 600 (2006) 1771-1779.
[75] K.S. Kim, N. Winograd, X-ray photoelectron spectroscopic studies of nickel-oxygen
surfaces using oxygen and argon ion-bombardment, Surface Science, 43 (1974) 625-643.
[76] Y. Morizet, M. Paris, F. Gaillard, B. Scaillet, Carbon dioxide in silica-undersaturated
melt. Part I: The effect of mixed alkalis (K and Na) on CO2 solubility and speciation,
Geochimica et Cosmochimica Acta, 141 (2014) 45-61.
[77] C. Xu, R. Reed, J.P. Gorski, Y. Wang, M.P. Walker, The distribution of carbonate in
enamel and its correlation with structure and mechanical properties, Journal of Materials
Science, 47 (2012) 8035-8043.
[78] S.C. Ray, S.K. Bhunia, A. Saha, N.R. Jana, Graphene oxide (GO)/reduced-GO and their
composite with conducting polymer nanostructure thin films for non-volatile memory device,
Microelectronic Engineering, 146 (2015) 48-52.
[79] C.Y. Ho, C.C. Liang, H.W. Wang, Investigation of low thermal reduction of graphene
oxide for dye-sensitized solar cell counter electrode, Colloids and Surfaces A:
Physicochemical and Engineering Aspects, 481 (2015) 222-228.
[80] C. Galande, A.D. Mohite, A.V. Naumov, W. Gao, L. Ci, A. Ajayan, H. Gao, A.
Srivastava, R. Bruce Weisman, P.M. Ajayan, Quasi-molecular fluorescence from graphene
oxide, Scientific Reports, 1 (2011).
[81] A. Lerf, H. He, M. Forster, J. Klinowski, Structure of graphite oxide revisited, Journal of
Physical Chemistry B, 102 (1998) 4477-4482.
[82] M. Kayanuma, U. Nagashima, H. Nishihara, T. Kyotani, H. Ogawa, Adsorption and
diffusion of atomic hydrogen on a curved surface of microporous carbon: A theoretical study,
Chemical Physics Letters, 495 (2010) 251-255.
Page 263
255
Chapter 7
GENERAL CONCLUSIONS AND RECOMMENDATIONS
This chapter lists the main conclusions obtained from the research carried out
in this doctoral thesis. In addition, some recommendations are suggested to be taken
into account in further studies.
Page 264
Chapter 7
256
7.1. CONCLUSIONS
The results obtained from the present research work support the following
main conclusions:
- A 120 nm-thick Pt catalyst film was prepared by the cathodic arc deposition
(CAD) technique on K-βAl2O3 (a K+ conductor material) and was electrochemically
promoted in the methanol partial oxidation (POM) and steam reforming (SRM)
reactions. On the other hand, a Pt catalyst film prepared by impregnation presented an
excess of electrocatalytic activity and a poisoning effect due to potassium-derived
compounds which blocked Pt active sites. Thus, the catalyst films henceforth used in
this thesis were prepared through cathodic arc deposition or a similar physical vapor
deposition (PVD), such as sputtering or oblique angle deposition (OAD).
- The electrochemical promotion of catalysis (EPOC) allowed in-situ
enhancing the catalytic activity of the Pt film prepared by CAD and its selectivity
towards methanol partial oxidation against total oxidation mechanism, through the
controlled migration of K+ ions (under negative polarization). Moreover, the EPOC
effect observed on this catalyst and the others showed to be reversible upon removal
of these ions from the catalyst (under positive polarization).
- A novel catalyst film composed of Pt nanoparticles (of around 3 nm)
dispersed in a carbon matrix (Pt-DLC) was developed by CAD. This film was
successfully electrochemically promoted after a graphitization process and different
K+-derived promotional effects were found depending on the applied potential and the
reaction atmosphere (POM or SRM). The Pt-DLC film showed a higher catalytic
activity and a similar enhancement ratio than the pure Pt catalyst, even with worse
electric properties and much lower amount of metal (only 0.014 mg cm-2
). These
results demonstrate the possibility of electrochemically promoting catalyst
nanoparticles dispersed in an electronic non-ionic conductor support.
- It was possible to electrochemically activate a non-conductive catalyst film
formed by Au nanoparticles dispersed in a YSZ matrix by the co-sputtering of Au, Y
and Zr targets. For this purpose a silver current collector was used on the Au-YSZ
Page 265
General conclusions and recommendations
257
catalyst film which was tested in the methanol partial oxidation reaction. The amount
of supplied K+ promoter ions was optimized and a permanent EPOC effect was found.
This study also demonstrated that the magnitude of the promotional effect obtained
with alkaline solid electrolytes does depend on neither the rate of promoter supply nor
the operational mode (either potentiostatic or galvanostatic) but on the final promoter
coverage achieved.
- A novel catalyst film configuration was developed to conduct
electrochemical promotion experiments, which consists of tilted nanocolumns of the
active metal grown on the support through an oblique angle deposition method. In this
way, a Cu catalyst film was prepared with a high porosity and gas-exposed surface
area, which results of great interest for catalytic and electrocatalytic applications. This
non noble metal catalyst was electrochemically promoted with K+ ions in the reaction
of partial oxidation of methanol and showed catalytic activity enhancement ratios of a
similar magnitude than those previously obtained with Pt catalysts.
- It was demonstrated the possible application of the EPOC phenomenon with
K+ ions for the activation of a metal catalyst, the modification of its catalytic
selectivity and the controlled variation of its oxidation state, by using a single Ni
catalyst film prepared by CAD. Under methanol decomposition and steam reforming
conditions, the K+ ions migration caused the activation of the methanol
dehydrogenation mechanism, and in the latter case, the K+ effect also attenuated the Ni
deactivation by carbon deposition. Under methanol partial oxidation conditions, the
K+-assisted strengthening of the oxygen chemisorption led to an increase in NiO
formation. As a consequence, the catalyst selectivity toward H2 and CO decreased
while that toward CO2, H2O and H2CO slightly increased.
- A novel porous Ni catalyst film prepared by the oblique angle deposition
technique with high porosity allowed to simultaneously produce H2 and graphene
oxide (GO) from methanol and to store part of the H2 on the GO (which acted as
adsorbent) under the promotional effect of K+ ions. Moreover, it was possible to
release the stored H2 in a controlled way, via electrochemical transfer of the promoter
ions under mild reaction conditions (280 ºC). The high obtained values of H2 storage
Page 266
Chapter 7
258
capacity (19 g H2 x 100 g Ni-1
) open a new interesting possibility for the practical
application of the EPOC phenomenon in the fields of hydrogen production and
storage.
7.2. RECOMMENDATIONS
The following proposals can be stated in order to complete and extend this
research work:
- To perform EPOC studies in the methanol conversion reactions by using
other kinds of solid electrolytes, either cationic (Na-βAl2O3, NASICON, SCY), or
anionic (YSZ) or mixed ionic electronic conductors (perovskite-like materials) and
comparing the results with those herein obtained with K-βAl2O3.
- To study the influence of the phenomenon of electrochemical promotion on
hydrogen production processes from other materials such as ethanol or bioalcohols
(real mixtures).
- To apply some of the thin film deposition techniques reported in this thesis
(OAD, sputtering) for the preparation of either supported Cu or Ni catalysts similar to
those employed in classical catalysis studies (Ni/Al2O3, Cu/Al2O3, Ni/CeO2, Cu/CeO2)
or other kinds of configurations such as bimetallic (NiPt, NiCu) or multilayered
catalysts.
- To investigate other techniques for the preparation of metal catalyst films
with high dispersion; for instance, by pressing some catalyst powder on the solid
electrolyte or by using some kind of binder, in such a way that a suitable adhesion to
the substrate is achieved.
- To develop electrochemical promotion studies of methanol conversion
reactions by using some kind of double chamber solid electrolyte membrane reactor,
in such a way that some product of interest (e.g. H2) is obtained separately, and/or
some reactant (e.g. O2) is supplied from one chamber to the other (where the reaction
takes place).
Page 267
General conclusions and recommendations
259
- To scale up the electrochemical promotion experiments for H2 production
and/or storage by using configurations with higher surface area, such as tubular or
monolithic catalysts, which would allow to obtain higher methanol conversion values
and/or higher H2 storage capacities.
Page 269
List of publications and conferences
261
The research results obtained in this doctoral thesis have led to the following
publications in international refereed journals:
1. Electrochemical activation of the catalytic methanol reforming reaction for H2
production. A. de Lucas-Consuegra, J. González-Cobos, Y. García-Rodríguez,
J.L. Endrino, J.L. Valverde. Electrochemistry Communications, 19 (2012) 55-
58.
2. Enhancing the catalytic activity and selectivity of the partial oxidation of methanol
by electrochemical promotion. A. de Lucas-Consuegra, J. González-Cobos, Y.
García-Rodríguez, A. Mosquera, J.L. Endrino, J.L. Valverde. Journal of
Catalysis, 293 (2012) 149-157.
3. Electrochemical promotion of Pt nanoparticles dispersed on a diamond-like
carbon matrix: A novel electrocatalytic system for H2 production. A. de Lucas-
Consuegra, J. González-Cobos, V. Carcelén, C. Magén, J.L. Endrino, J.L.
Valverde. Journal of Catalysis, 307 (2013) 18-26.
4. Electrochemical activation of Au nanoparticles for the selective partial oxidation
of metanol. J. González-Cobos, D. Horwat, J. Ghanbaja, J.L. Valverde, A. de
Lucas-Consuegra. Journal of Catalysis, 317 (2014) 293-302.
5. Electrochemical activation of an oblique angle deposited Cu catalyst film for H2
production. J. González-Cobos, V.J. Rico, A.R. González-Elipe, J.L. Valverde, A.
de Lucas-Consuegra. Catalysis Science and Technology, 5 (2015) 2203-2214.
6. Practical applications of the electrochemical promotion of catalysis in methanol
conversion processes. J. González-Cobos, J.L. Valverde, A. de Lucas-Consuegra.
Submitted to Topics in Catalysis.
Page 270
List of publications and conferences
262
In addition, results obtained in this doctoral thesis have been presented in
the following international and national conferences:
- Keynotes in international conferences:
1. Coupling catalysis and electrochemistry for H2 production from alcohols. A.
de Lucas-Consuegra, J.L. Endrino, J. González-Cobos, D. López, J.A. Díaz,
J.L. Valverde. ANQUE’s International Congress of Chemical Engineering
(ICCE). Sevilla (Spain), June 2012.
- Oral presentations in international conferences:
1. Electrochemical promotion of partial oxidation of methanol for the production
of H2 and H2CO. A. de Lucas-Consuegra, J. González-Cobos, Y. García-
Rodríguez, A. Caravaca, J.L. Endrino, J.L. Valverde. 7th International
Conference on Environmental Catalysis (ICEC). Lyon (France), September
2012.
2. H2 production from methanol by electrochemical promotion of metallic
catalysts using cationic solid electrolytes. J. González-Cobos, A. de Lucas-
Consuegra, A. Nieto-Prado, J.L. Endrino, J.L. Valverde. 11th International
Conference on Catalysis in Membrane Reactors (ICCMR). Porto (Portugal),
July 2013.
3. Electrochemical promotion of hydrogen production reactions with alkali ionic
conductors. A. de Lucas-Consuegra, A. Caravaca, J. González-Cobos, F.
Dorado, J.L. Valverde. International Workshop on Ionically Conducting
Ceramics for Catalysis (IC3). Lyon (France), September 2013.
4. Hydrogen production from methanol by electrochemical promotion of Pt
catalysts. J. González-Cobos, C. Jiménez-Borja, N. Gutiérrez, A. de Lucas-
Consuegra, J.L. Valverde. 2013 Energy and Environment Knowledge Week
(E2KW), Toledo (Spain), November 2013.
Page 271
List of publications and conferences
263
5. Electrochemical promotion of novel electrodes for the catalytic partial
oxidation of methanol. A. de Lucas-Consuegra, J. González-Cobos, D. López-
Pedrajas, D. Horwat, J. Ghanbaja, J.L. Valverde. 65th Annual Meeting of the
International Society of Electrochemistry (ISE). Lausanne (Switzerland),
August - September 2014.
6. Recent contributions of electrochemical promotion of catalysis for H2
production. A. de Lucas-Consuegra, J. González-Cobos, J.L. Valverde.
Accepted for oral presentation at the Hydrogen Power Theoretical and
Engineering Solutions International Symposium (HYPOTHESIS). Toledo
(Spain), September 2015.
7. Hydrogen production and storage by coupling of catalysis and
electrochemistry. J. González-Cobos, V.J. Rico, A.R. González-Elipe, J.L.
Valverde, A. de Lucas-Consuegra. Accepted for oral presentation at the 66th
Annual Meeting of the International Society of Electrochemistry (ISE). Taipei
(Taiwan), October 2015.
- Oral presentations in national conferences:
1. Promoción electroquímica para la producción de hidrógeno a partir de
alcoholes. J. González-Cobos. VI Jornadas de la Ciencia Joven. Ciudad Real
(Spain), May - June 2012.
- Posters in international conferences:
1. Electrochemical promotion of H2 production from methanol on novel metal
catalyst films. A. de Lucas-Consuegra, J. González-Cobos, V. Carcelén, C.
Magén, J.L. Endrino, J.L. Valverde. 11th European Congress on Catalysis
(EuropaCat). Lyon (France), September 2013.
2. Novel electrocatalytic systems for hydrogen production. J. González-Cobos,
N. Gutiérrez, C. Jiménez-Borja, A. de Lucas-Consuegra, J.L. Valverde. 64th
Annual Meeting of the International Society of Electrochemistry (ISE).
Santiago de Queretaro (Mexico), September 2013.
Page 272
List of publications and conferences
264
- Posters in national conferences:
1. Producción de hidrógeno a partir de metanol mediante promoción
electroquímica. J. González Cobos, J.L. Valverde, A. de Lucas Consuegra. II
Jornadas Doctorales de Castilla-La Mancha. Toledo (Spain), November 2012.
2. Promoción electroquímica de nuevos catalizadores nanoestructurados para la
producción de H2. J. González-Cobos, A. de Lucas-Consuegra, A. Nieto-
Prado, N. Gutiérrez, D. Horwat, F. Soldera, J.L. Valverde. Reunión de la
Sociedad Española de la Catálisis (SECAT). Sevilla (Spain), June 2013.
3. Oxidación parcial de metanol mediante promoción electroquímica de
nanopartículas de oro. J. González-Cobos, D. López-Pedrajas, D. Horwat,
J.L. Valverde, A. de Lucas-Consuegra. I Encuentro de Jóvenes Investigadores
de la SECAT, Málaga (Spain), June 2014.
4. Producción de hidrógeno mediante catalizadores dispersos promocionados
electroquímicamente. J. González-Cobos, J.L. Valverde, A. de Lucas-
Consuegra. IV Jornadas Doctorales de Castilla-La Mancha. Cuenca (Spain),
October 2014.