Electron transfer catalysis with monolayer protected Au 25 clusters† Sabrina Antonello, * Mahdi Hesari, Federico Polo and Flavio Maran * Received 1st May 2012, Accepted 4th June 2012 DOI: 10.1039/c2nr31066j Au 25 L 18 (L ¼ S(CH 2 ) 2 Ph) clusters were prepared and characterized. The resulting monodisperse clusters were reacted with bis(pentafluorobenzoyl) peroxide in dichloromethane to form Au 25 L 18 + quantitatively. The kinetics and thermodynamics of the corresponding electron transfer (ET) reactions were characterized via electrochemistry and thermochemical calculations. Au 25 L 18 + was used in homogeneous redox catalysis experiments with a series of sym-substituted benzoyl peroxides, including the above peroxide, bis(para-cyanobenzoyl) peroxide, dibenzoyl peroxide, and bis(para- methoxybenzoyl) peroxide. Peroxide dissociative ET was catalyzed using both the Au 25 L 18 /Au 25 L 18 and the Au 25 L 18 + /Au 25 L 18 redox couples as redox mediators. Simulation of the CV curves led to determination of the ET rate constant (k ET ) values for concerted dissociative ET to the peroxides. The ET free energy DG could be estimated for all donor–acceptor combinations, leading to observation of a nice activation–driving force (log k ET vs. DG ) relationship. Comparison with the k ET obtained using a ferrocene-type donor with a formal potential similar to that of Au 25 L 18 /Au 25 L 18 showed that the presence of the capping monolayer affects the ET rate rather significantly, which is attributed to the intrinsic nonadiabaticity of peroxide acceptors. Introduction When gold nanoparticles are sufficiently small, electronic-band energetics leads to quantum confinement effects, which are conve- niently studied by electrochemistry. 1,2 Quantized double-layer charging behavior of phenylethanethiolate or hexanethiolate coated Au 140 (core diameter, 1.6 nm) 3–5 and Au 225 clusters (2.0 nm) 6 has been evidenced by sensitive transient electrochemical techniques such as differential pulse voltammetry (DPV), square wave vol- tammetry, and cyclic voltammetry (CV). For these monolayer- protected clusters (MPCs), stepwise charging of the metal core is detected as an approximately regular sequence of redox peaks. When the core size decreases, the separation between the formal potential (E ) values of such redox couples increases. When the core radius is made progressively smaller, a transition between quantized- double layer charging behavior and molecule-like regime eventually takes place, 2 as detected through the observation of a sizeable separation between the first oxidation and first reduction steps. This E separation, which is related to the energy gap between the highest occupied and lowest unoccupied molecular orbitals (HOMO– LUMO), 7–11 becomes evident 2 for clusters smaller than 1.5 nm, such as Au 75 , 12 Au 38 , 4,13 Au 25 , 8,9,11,14 Au 13 , 15 Au 11 , 16 and Au 9 . 17 Within the few small gold nanoclusters so far characterized and displaying distinct molecule-like redox and optical behav- iors, Au 25 L 18 (1 nm) occupies a special position. 18 In its native diamagnetic form, such as when synthesized in the presence of tetra-n-octylammonium, 19,20 Au 25 L 18 bears a negative charge. 21 Au 25 L 18 tends to undergo aerobic oxidation to form the neutral state Au 25 L 18 0 . 22 The structures of both anionic and neutral forms were unraveled in 2008. 22–24 Although the structure of [n-Oct 4 N + ][Au 25 L 18 ] shows some distortion, both redox states share the same general features, i.e., the presence of a core composed of a Au 13 icosahedron capped by six Au 2 L 3 staple-like elements. The presence of these Au atoms, stellated on 12 faces of the Au 13 core, causes the ligands to be of two types: whereas in 6 ligands each sulfur atom is connected to two stellated Au atoms, in the remaining 12 ligands the sulfur atom is connected to one stellated Au atom and one core Au atom. Provided the sample is truly monodisperse, 1 H and 13 C NMR spectroscopy quantita- tively shows that the corresponding resonances of the two ligand families are not equivalent. 19 Whereas basic electrochemical information (such as E values and E differences) has been reported for some molecule-like clusters and under different environment conditions (solvents, electrolytes, and temperature), much less is known about their actual electron-transfer (ET) properties, such as intrinsic barriers, activation parameters, and ET rate constants. The ET properties of Au 25 L 18 clusters (L ¼ phenylethanethiolate) have been studied in more detail, albeit in former papers they were erroneously assigned to Au 38 L 24 0 instead of Au 25 L 18 . 2 ET characterization of the native cluster, Au 25 L 18 , showed that Au 25 behaves in all regards as a Department of Chemistry, University of Padova, via Marzolo 1, 35131 Padova, Italy. E-mail: fl[email protected]; sabrina.antonello@unipd. it; Tel: +39 049 827 5147 † This article was submitted as part of a Themed Issue on metallic clusters. Other papers on this topic can be found in issue 14 of vol. 4 (2012). This issue can be found from the Nanoscale homepage [http://www.rsc.org/nanoscale]. This journal is ª The Royal Society of Chemistry 2012 Nanoscale, 2012, 4, 5333–5342 | 5333 Dynamic Article Links C < Nanoscale Cite this: Nanoscale, 2012, 4, 5333 www.rsc.org/nanoscale PAPER Published on 07 June 2012. Downloaded by University of Leeds on 13/08/2013 02:55:05. View Article Online / Journal Homepage / Table of Contents for this issue
Transcript
Dynamic Article LinksC<Nanoscale
Cite this: Nanoscale, 2012, 4, 5333
www.rsc.org/nanoscale PAPER
Publ
ishe
d on
07
June
201
2. D
ownl
oade
d by
Uni
vers
ity o
f L
eeds
on
13/0
8/20
13 0
2:55
:05.
View Article Online / Journal Homepage / Table of Contents for this issue
Electron transfer catalysis with monolayer protected Au25 clusters†
Sabrina Antonello,* Mahdi Hesari, Federico Polo and Flavio Maran*
Received 1st May 2012, Accepted 4th June 2012
DOI: 10.1039/c2nr31066j
Au25L18 (L ¼ S(CH2)2Ph) clusters were prepared and characterized. The resulting monodisperse
clusters were reacted with bis(pentafluorobenzoyl) peroxide in dichloromethane to form Au25L18+
quantitatively. The kinetics and thermodynamics of the corresponding electron transfer (ET) reactions
were characterized via electrochemistry and thermochemical calculations. Au25L18+ was used in
homogeneous redox catalysis experiments with a series of sym-substituted benzoyl peroxides, including
the above peroxide, bis(para-cyanobenzoyl) peroxide, dibenzoyl peroxide, and bis(para-
methoxybenzoyl) peroxide. Peroxide dissociative ET was catalyzed using both the Au25L18/Au25L18�
and the Au25L18+/Au25L18 redox couples as redox mediators. Simulation of the CV curves led to
determination of the ET rate constant (kET) values for concerted dissociative ET to the peroxides. The
ET free energy DG� could be estimated for all donor–acceptor combinations, leading to observation of
a nice activation–driving force (log kET vs. DG�) relationship. Comparison with the kET obtained using
a ferrocene-type donor with a formal potential similar to that of Au25L18/Au25L18� showed that the
presence of the capping monolayer affects the ET rate rather significantly, which is attributed to the
intrinsic nonadiabaticity of peroxide acceptors.
Introduction
When gold nanoparticles are sufficiently small, electronic-band
energetics leads to quantum confinement effects, which are conve-
niently studied by electrochemistry.1,2 Quantized double-layer
charging behavior of phenylethanethiolate or hexanethiolate coated
Au140 (core diameter, 1.6 nm)3–5 and Au225 clusters (2.0 nm)6 has
been evidenced by sensitive transient electrochemical techniques
such as differential pulse voltammetry (DPV), square wave vol-
tammetry, and cyclic voltammetry (CV). For these monolayer-
protected clusters (MPCs), stepwise charging of the metal core is
detected as an approximately regular sequence of redox peaks.
When the core size decreases, the separation between the formal
potential (E�) values of such redox couples increases. When the core
radius is made progressively smaller, a transition between quantized-
double layer charging behavior and molecule-like regime eventually
takes place,2 as detected through the observation of a sizeable
separation between the first oxidation and first reduction steps. This
E� separation, which is related to the energy gap between the highestoccupied and lowest unoccupied molecular orbitals (HOMO–
LUMO),7–11 becomes evident2 for clusters smaller than 1.5 nm, such
as Au75,12 Au38,
4,13 Au25,8,9,11,14 Au13,
15 Au11,16 and Au9.
17
Department of Chemistry, University of Padova, via Marzolo 1, 35131Padova, Italy. E-mail: [email protected]; [email protected]; Tel: +39 049 827 5147
† This article was submitted as part of a Themed Issue on metallicclusters. Other papers on this topic can be found in issue 14 of vol. 4(2012). This issue can be found from the Nanoscale homepage[http://www.rsc.org/nanoscale].
This journal is ª The Royal Society of Chemistry 2012
Within the few small gold nanoclusters so far characterized
and displaying distinct molecule-like redox and optical behav-
iors, Au25L18 (1 nm) occupies a special position.18 In its native
diamagnetic form, such as when synthesized in the presence of
tetra-n-octylammonium,19,20 Au25L18 bears a negative charge.21
Au25L18� tends to undergo aerobic oxidation to form the neutral
state Au25L180.22 The structures of both anionic and neutral
forms were unraveled in 2008.22–24 Although the structure of
[n-Oct4N+][Au25L18
�] shows some distortion, both redox states
share the same general features, i.e., the presence of a core
composed of a Au13 icosahedron capped by six Au2L3 staple-like
elements. The presence of these Au atoms, stellated on 12 faces of
the Au13 core, causes the ligands to be of two types: whereas in 6
ligands each sulfur atom is connected to two stellated Au atoms,
in the remaining 12 ligands the sulfur atom is connected to one
stellated Au atom and one core Au atom. Provided the sample is
truly monodisperse, 1H and 13C NMR spectroscopy quantita-
tively shows that the corresponding resonances of the two ligand
families are not equivalent.19
Whereas basic electrochemical information (such as E� values
and E� differences) has been reported for some molecule-like
clusters and under different environment conditions (solvents,
electrolytes, and temperature), much less is known about their
actual electron-transfer (ET) properties, such as intrinsic barriers,
activation parameters, and ET rate constants. The ET properties of
Au25L18 clusters (L ¼ phenylethanethiolate) have been studied in
more detail, albeit in former papers they were erroneously assigned
to Au38L240 instead of Au25L18
�.2ET characterization of the native
cluster, Au25L18�, showed that Au25 behaves in all regards as a
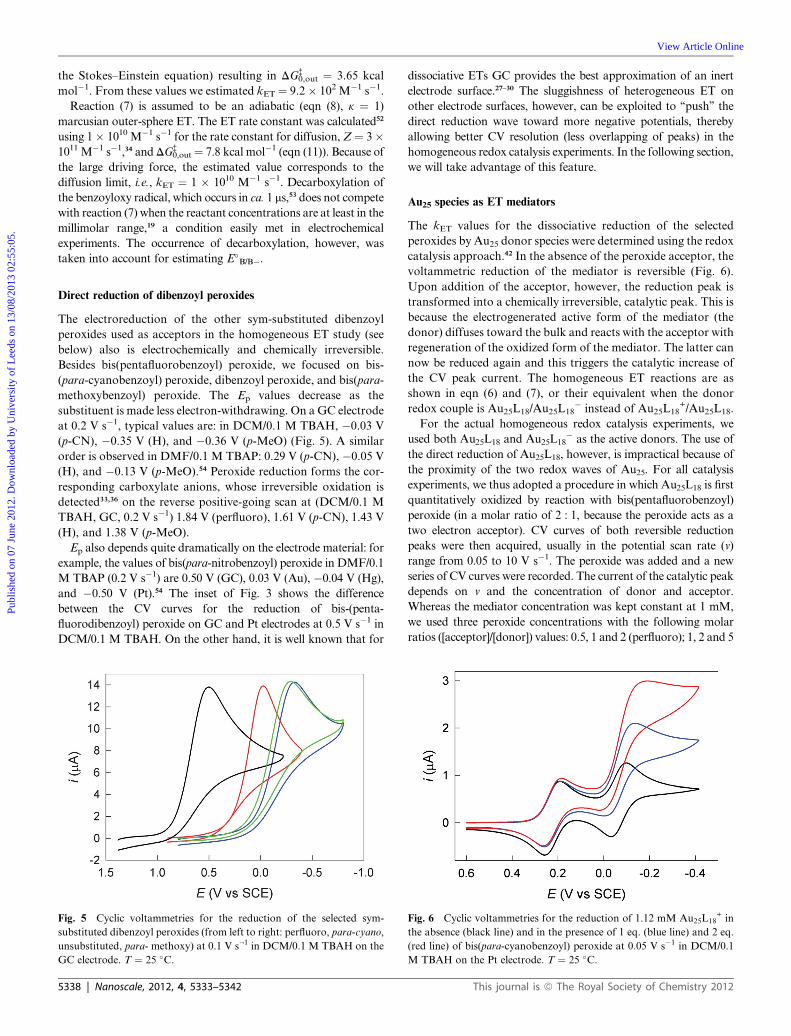
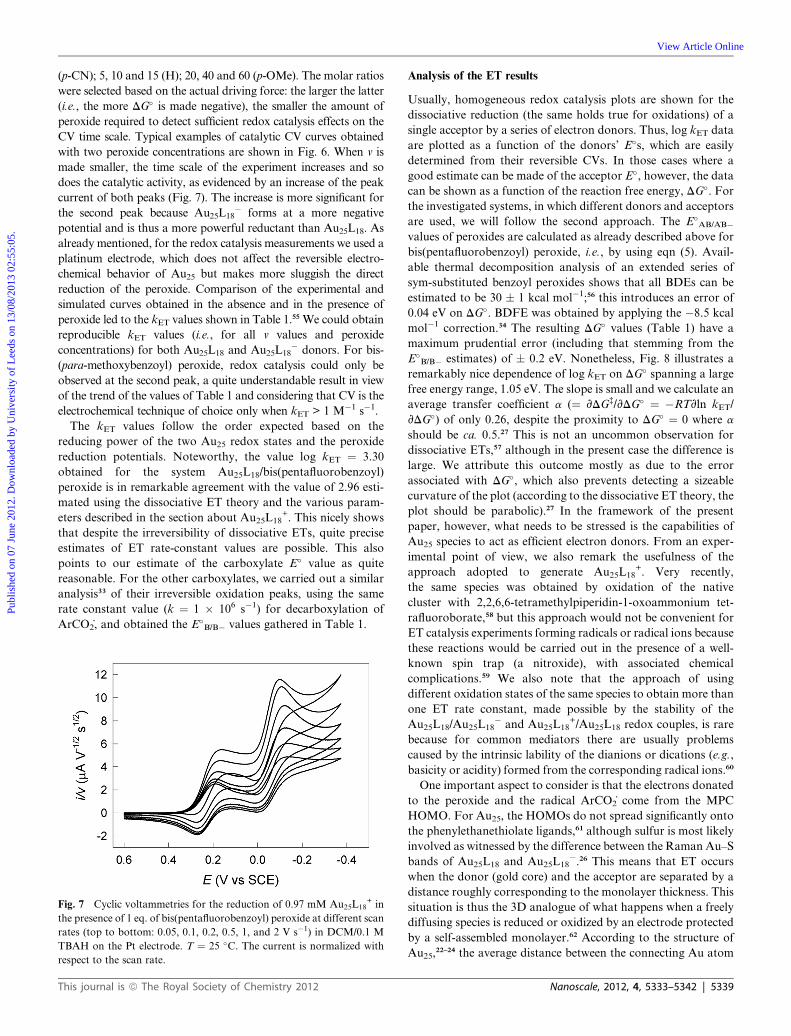
(p-CN); 5, 10 and 15 (H); 20, 40 and 60 (p-OMe). The molar ratios
were selected based on the actual driving force: the larger the latter
(i.e., the more DG� is made negative), the smaller the amount of
peroxide required to detect sufficient redox catalysis effects on the
CV time scale. Typical examples of catalytic CV curves obtained
with two peroxide concentrations are shown in Fig. 6. When v is
made smaller, the time scale of the experiment increases and so
does the catalytic activity, as evidenced by an increase of the peak
current of both peaks (Fig. 7). The increase is more significant for
the second peak because Au25L18� forms at a more negative
potential and is thus a more powerful reductant than Au25L18. As
already mentioned, for the redox catalysis measurements we used a
platinum electrode, which does not affect the reversible electro-
chemical behavior of Au25 but makes more sluggish the direct
reduction of the peroxide. Comparison of the experimental and
simulated curves obtained in the absence and in the presence of
peroxide led to the kET values shown in Table 1.55 We could obtain
reproducible kET values (i.e., for all v values and peroxide
concentrations) for both Au25L18 and Au25L18� donors. For bis-
(para-methoxybenzoyl) peroxide, redox catalysis could only be
observed at the second peak, a quite understandable result in view
of the trend of the values of Table 1 and considering that CV is the
electrochemical technique of choice only when kET > 1 M�1 s�1.
The kET values follow the order expected based on the
reducing power of the two Au25 redox states and the peroxide
reduction potentials. Noteworthy, the value log kET ¼ 3.30
obtained for the system Au25L18/bis(pentafluorobenzoyl)
peroxide is in remarkable agreement with the value of 2.96 esti-
mated using the dissociative ET theory and the various param-
eters described in the section about Au25L18+. This nicely shows
that despite the irreversibility of dissociative ETs, quite precise
estimates of ET rate-constant values are possible. This also
points to our estimate of the carboxylate E� value as quite
reasonable. For the other carboxylates, we carried out a similar
analysis33 of their irreversible oxidation peaks, using the same
rate constant value (k ¼ 1 � 106 s�1) for decarboxylation of
ArCO2_, and obtained the E�B_/B� values gathered in Table 1.
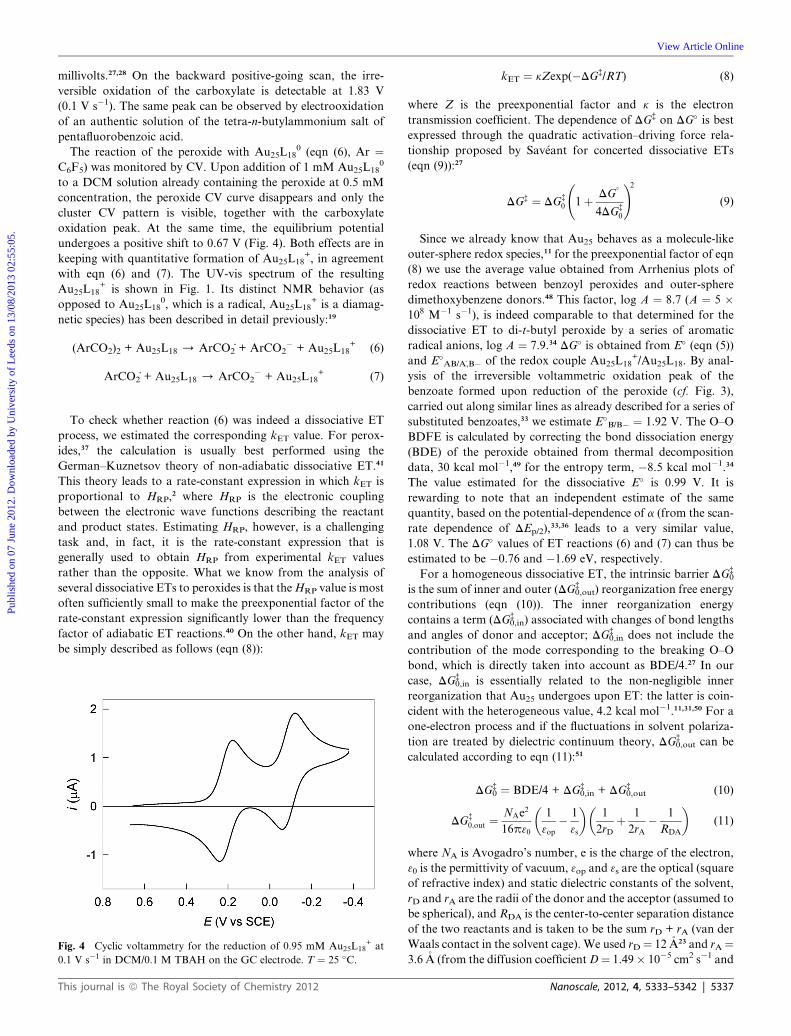
Fig. 7 Cyclic voltammetries for the reduction of 0.97 mM Au25L18+ in
the presence of 1 eq. of bis(pentafluorobenzoyl) peroxide at different scan
rates (top to bottom: 0.05, 0.1, 0.2, 0.5, 1, and 2 V s�1) in DCM/0.1 M
TBAH on the Pt electrode. T ¼ 25 �C. The current is normalized with
respect to the scan rate.
This journal is ª The Royal Society of Chemistry 2012
Analysis of the ET results
Usually, homogeneous redox catalysis plots are shown for the
dissociative reduction (the same holds true for oxidations) of a
single acceptor by a series of electron donors. Thus, log kET data
are plotted as a function of the donors’ E�s, which are easily
determined from their reversible CVs. In those cases where a
good estimate can be made of the acceptor E�, however, the datacan be shown as a function of the reaction free energy, DG�. Forthe investigated systems, in which different donors and acceptors
are used, we will follow the second approach. The E�AB/A_B�
values of peroxides are calculated as already described above for
bis(pentafluorobenzoyl) peroxide, i.e., by using eqn (5). Avail-
able thermal decomposition analysis of an extended series of
sym-substituted benzoyl peroxides shows that all BDEs can be
estimated to be 30 � 1 kcal mol�1;56 this introduces an error of
0.04 eV on DG�. BDFE was obtained by applying the �8.5 kcal
mol�1 correction.34 The resulting DG� values (Table 1) have a
maximum prudential error (including that stemming from the
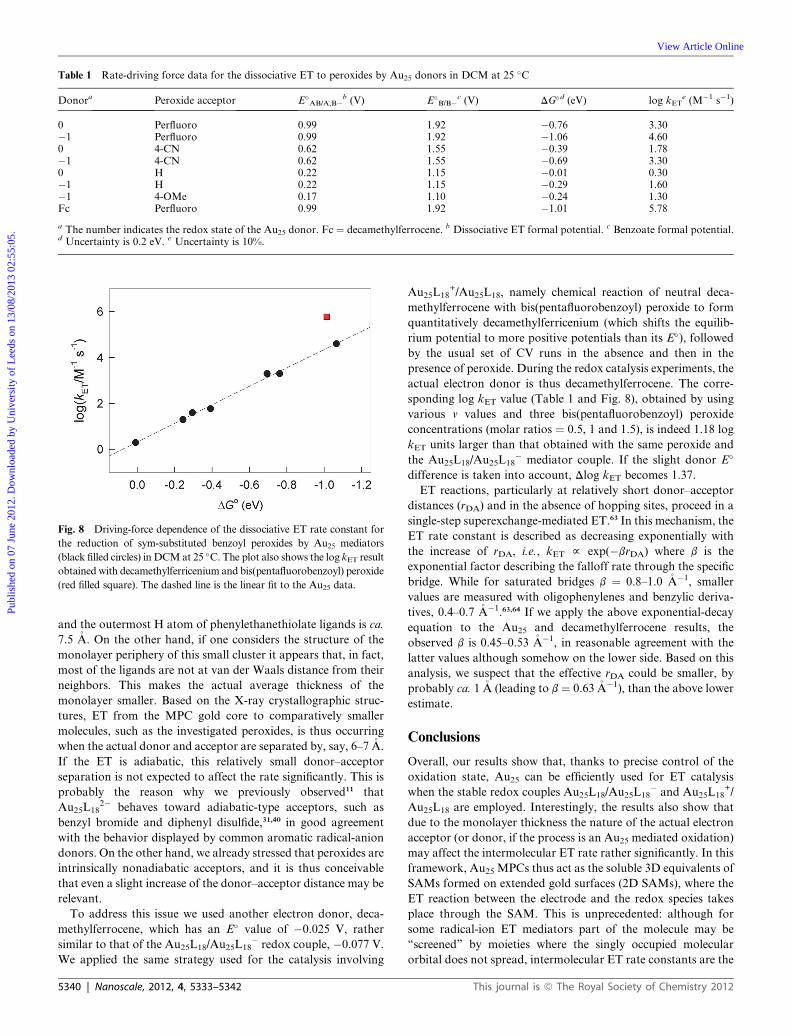
E�B_/B� estimates) of � 0.2 eV. Nonetheless, Fig. 8 illustrates a
remarkably nice dependence of log kET on DG� spanning a large
free energy range, 1.05 eV. The slope is small and we calculate an
average transfer coefficient a (¼ vDG‡/vDG� ¼ �RTvln kET/
vDG�) of only 0.26, despite the proximity to DG� ¼ 0 where a
should be ca. 0.5.27 This is not an uncommon observation for
dissociative ETs,57 although in the present case the difference is
large. We attribute this outcome mostly as due to the error
associated with DG�, which also prevents detecting a sizeable
curvature of the plot (according to the dissociative ET theory, the
plot should be parabolic).27 In the framework of the present
paper, however, what needs to be stressed is the capabilities of
Au25 species to act as efficient electron donors. From an exper-
imental point of view, we also remark the usefulness of the
approach adopted to generate Au25L18+. Very recently,
the same species was obtained by oxidation of the native
cluster with 2,2,6,6-tetramethylpiperidin-1-oxoammonium tet-
rafluoroborate,58 but this approach would not be convenient for
ET catalysis experiments forming radicals or radical ions because
these reactions would be carried out in the presence of a well-
known spin trap (a nitroxide), with associated chemical
complications.59 We also note that the approach of using
different oxidation states of the same species to obtain more than
one ET rate constant, made possible by the stability of the
Au25L18/Au25L18� and Au25L18
+/Au25L18 redox couples, is rare
because for common mediators there are usually problems
caused by the intrinsic lability of the dianions or dications (e.g.,
basicity or acidity) formed from the corresponding radical ions.60
One important aspect to consider is that the electrons donated
to the peroxide and the radical ArCO2_ come from the MPC
HOMO. For Au25, the HOMOs do not spread significantly onto
the phenylethanethiolate ligands,61 although sulfur is most likely
involved as witnessed by the difference between the Raman Au–S
bands of Au25L18 and Au25L18�.26 This means that ET occurs
when the donor (gold core) and the acceptor are separated by a
distance roughly corresponding to the monolayer thickness. This
situation is thus the 3D analogue of what happens when a freely
diffusing species is reduced or oxidized by an electrode protected
by a self-assembled monolayer.62 According to the structure of
Au25,22–24 the average distance between the connecting Au atom
a The number indicates the redox state of the Au25 donor. Fc ¼ decamethylferrocene. b Dissociative ET formal potential. c Benzoate formal potential.d Uncertainty is 0.2 eV. e Uncertainty is 10%.
Fig. 8 Driving-force dependence of the dissociative ET rate constant for
the reduction of sym-substituted benzoyl peroxides by Au25 mediators
(black filled circles) in DCMat 25 �C. The plot also shows the log kET resultobtained with decamethylferricenium and bis(pentafluorobenzoyl) peroxide
(red filled square). The dashed line is the linear fit to the Au25 data.
Publ
ishe
d on
07
June
201
2. D
ownl
oade
d by
Uni
vers
ity o
f L
eeds
on
13/0
8/20
13 0
2:55
:05.
View Article Online
and the outermost H atom of phenylethanethiolate ligands is ca.
7.5 �A. On the other hand, if one considers the structure of the
monolayer periphery of this small cluster it appears that, in fact,
most of the ligands are not at van der Waals distance from their
neighbors. This makes the actual average thickness of the
monolayer smaller. Based on the X-ray crystallographic struc-
tures, ET from the MPC gold core to comparatively smaller
molecules, such as the investigated peroxides, is thus occurring
when the actual donor and acceptor are separated by, say, 6–7 �A.
If the ET is adiabatic, this relatively small donor–acceptor
separation is not expected to affect the rate significantly. This is
probably the reason why we previously observed11 that
Au25L182� behaves toward adiabatic-type acceptors, such as
benzyl bromide and diphenyl disulfide,31,40 in good agreement
with the behavior displayed by common aromatic radical-anion
donors. On the other hand, we already stressed that peroxides are
intrinsically nonadiabatic acceptors, and it is thus conceivable
that even a slight increase of the donor–acceptor distance may be
relevant.
To address this issue we used another electron donor, deca-
methylferrocene, which has an E� value of �0.025 V, rather
similar to that of the Au25L18/Au25L18� redox couple, �0.077 V.
We applied the same strategy used for the catalysis involving
5340 | Nanoscale, 2012, 4, 5333–5342
Au25L18+/Au25L18, namely chemical reaction of neutral deca-
methylferrocene with bis(pentafluorobenzoyl) peroxide to form
quantitatively decamethylferricenium (which shifts the equilib-
rium potential to more positive potentials than its E�), followedby the usual set of CV runs in the absence and then in the
presence of peroxide. During the redox catalysis experiments, the
actual electron donor is thus decamethylferrocene. The corre-
sponding log kET value (Table 1 and Fig. 8), obtained by using
various v values and three bis(pentafluorobenzoyl) peroxide
concentrations (molar ratios ¼ 0.5, 1 and 1.5), is indeed 1.18 log
kET units larger than that obtained with the same peroxide and
the Au25L18/Au25L18� mediator couple. If the slight donor E�
difference is taken into account, Dlog kET becomes 1.37.
ET reactions, particularly at relatively short donor–acceptor
distances (rDA) and in the absence of hopping sites, proceed in a
single-step superexchange-mediated ET.63 In this mechanism, the
ET rate constant is described as decreasing exponentially with
the increase of rDA, i.e., kET f exp(�brDA) where b is the
exponential factor describing the falloff rate through the specific
bridge. While for saturated bridges b ¼ 0.8–1.0 �A�1, smaller
values are measured with oligophenylenes and benzylic deriva-
tives, 0.4–0.7 �A�1.63,64 If we apply the above exponential-decay
equation to the Au25 and decamethylferrocene results, the
observed b is 0.45–0.53 �A�1, in reasonable agreement with the
latter values although somehow on the lower side. Based on this
analysis, we suspect that the effective rDA could be smaller, by
probably ca. 1 �A (leading to b ¼ 0.63 �A�1), than the above lower
estimate.
Conclusions
Overall, our results show that, thanks to precise control of the
oxidation state, Au25 can be efficiently used for ET catalysis
when the stable redox couples Au25L18/Au25L18� and Au25L18
+/
Au25L18 are employed. Interestingly, the results also show that
due to the monolayer thickness the nature of the actual electron
acceptor (or donor, if the process is an Au25 mediated oxidation)
may affect the intermolecular ET rate rather significantly. In this
framework, Au25 MPCs thus act as the soluble 3D equivalents of
SAMs formed on extended gold surfaces (2D SAMs), where the
ET reaction between the electrode and the redox species takes
place through the SAM. This is unprecedented: although for
some radical-ion ET mediators part of the molecule may be
‘‘screened’’ by moieties where the singly occupied molecular
orbital does not spread, intermolecular ET rate constants are the
This journal is ª The Royal Society of Chemistry 2012
average of random D/A distance and orientation distributions in
the encounter complex and thus screening effects are undetect-
able, as opposed to what was found for intramolecular ETs.38,40
MPCs thus provide a unique opportunity to test the behavior of
fully screened ET mediators. It follows that if from one hand
Au25 can be efficiently used for ET catalysis, on the other hand
efficient prediction of the ET rate depends on the actual acceptor
type.
This work was financially supported by the Foundation
CARIPARO (Progetto di Eccellenza), the Italian Ministero
dell’Istruzione, dell’Universit�a e della Ricerca (PRIN grant
20098Z4M5E), and the University of Padova (PRAT grant
CPDA103389).
Notes and references
1 (a) A. C. Templeton, W. P. Wuelfing and R. W. Murray, Acc. Chem.Res., 2000, 33, 27–36; (b) J. Z. Zhang, Z. Wang, J. Liu, S. Chen andG. Liu, Self-Assembled Nanostructures, Kluwer Academic/PlenumPublisher, New York, 2003, pp. 271–307.
2 R. W. Murray, Chem. Rev., 2008, 108, 2688–2720.3 (a) J. F. Hicks, A. C. Templeton, S. Chen, K. M. Sheran, R. Jasti,R. W. Murray, J. Debord, T. G. Schaaff and R. L. Whetten, Anal.Chem., 1999, 71, 3703–3711; (b) J. F. Hicks, D. T. Miles andR. W. Murray, J. Am. Chem. Soc., 2002, 124, 13322–13328; (c)D. T. Miles, M. C. Leopold, J. F. Hicks and R. W. Murray,J. Electroanal. Chem., 2003, 554–555, 87–97; (d) R. Guo,D. Georganopoulou, S. W. Feldberg, R. L. Donkers andR. W. Murray, Anal. Chem., 2005, 77, 2662–2669.
4 B. M. Quinn, P. Liljeroth, V. Ruiz, T. Laaksonen and K. Kontturi,J. Am. Chem. Soc., 2003, 125, 6644–6645.
5 A. H. Holm, M. Ceccato, R. L. Donkers, L. Fabris, G. Pace andF. Maran, Langmuir, 2006, 22, 10584–10589.
6 R. L. Wolfe and R. W. Murray, Anal. Chem., 2006, 76, 1167–1173.7 D. Lee, R. L. Donkers, J. M. DeSimone and R. W. Murray, J. Am.Chem. Soc., 2003, 125, 1182–1183.
8 D. Lee, R. L. Donkers, G. Wang, A. S. Harper and R. W. Murray,J. Am. Chem. Soc., 2004, 126, 6193–6199.
9 V. L. Jimenez, D. G. Georganopoulou, R. J. White, A. S. Harper,A. J. Mills, D. Lee and R. W. Murray, Langmuir, 2004, 20, 6864–6870.
10 R. Guo and R. W. Murray, J. Am. Chem. Soc., 2005, 127, 12140–12143.
11 S. Antonello, A. H. Holm, E. Instuli and F. Maran, J. Am. Chem.Soc., 2007, 129, 9836–9837.
12 (a) S. Chen, R. S. Ingram, M. J. Hostetler, J. J. Pietron,R. W. Murray, T. G. Schaaff, J. T. Khoury, M. M. Alvarez andR. L. Whetten, Science, 1998, 280, 2098–2101; (b)R. Balasubramanian, R. Guo, A. J. Mills and R. W. Murray,J. Am. Chem. Soc., 2005, 127, 8126–8132.
13 O. Toikkanen, V. Ruiz, G. R€onnholm, N. Kalkkinen, P. Liljeroth andB. M. Quinn, J. Am. Chem. Soc., 2008, 130, 11049–11055.
14 D. Garc�ıa-Raya, R. Madue~no, M. Bl�azquez and T. Pineda, J. Phys.Chem. C, 2009, 113, 8756–8761.
15 L. D. Menard, S. Gao, H. Xu, R. D. Twesten, A. S. Harper, Y. Song,G. Wang, A. D. Douglas, J. C. Yang, A. I. Frenkel, R. G. Nuzzo andR. W. Murray, J.Phys. Chem. B, 2006, 110, 12874–12883.
16 Y. Yang and S. Chen, Nano Lett., 2003, 3, 75–79.17 F. Wen, U. Englert, B. Gutrath and U. Simon, Eur. J. Inorg. Chem.,
2008, 106–111.18 J. F. Parker, C. A. Fields-Zinna and R. W. Murray, Acc. Chem. Res.,
2010, 43, 1289–1296.19 A. Venzo, S. Antonello, J. Gascon, I. Guryanov, R. D. Leapman,
N. V. Perera, A. Sousa, M. Zamuner, A. Zanella and F. Maran,Anal. Chem., 2011, 83, 6355–6362.
20 J. P. Parker, J. E. F. Weaver, F. McCallum, C. A. Fields-Zinna andR. W. Murray, Langmuir, 2010, 26, 13650–13654.
21 (a) Y. Negishi, K. Nobusada and T. Tsukuda, J. Am. Chem. Soc.,2005, 127, 5261–5270; (b) J. B. Tracy, G. Kalyuzhny, M. C. Crowe,R. Balasubramanian, J.-P. Choi and R. W. Murray, J. Am. Chem.Soc., 2007, 129, 6706–6707.
This journal is ª The Royal Society of Chemistry 2012
22 M. Zhu, W. T. Eckenhoff, T. Pintauer and R. Jin, J. Phys. Chem. C,2008, 112, 14221–14224.
23 M. W. Heaven, A. Dass, P. S. White, K. M. Holt and R. W. Murray,J. Am. Chem. Soc., 2008, 130, 3754–3755.
24 M. Zhu, C. M. Aikens, F. J. Hollander, G. C. Schatz and R. Jin,J. Am. Chem. Soc., 2008, 130, 5883–5885.
25 J.-P. Choi and R. W. Murray, J. Am. Chem. Soc., 2006, 128, 10496–10502.
26 J. F. Parker, J.-P. Choi, W. Wang and R. W. Murray, J. Phys. Chem.C, 2008, 112, 13976–13981.
27 J.-M. Sav�eant, J. Am. Chem. Soc., 1997, 109, 6788–6795.28 F. Maran, D. D. M. Wayner and M. S. Workentin, Adv. Phys. Org.
Chem., 2001, 36, 85–166.29 S. Antonello and F. Maran, Chem. Soc. Rev., 2005, 34, 418–428.30 A. Houmam, Chem. Rev., 2008, 108, 2180–2237.31 A. B. Meneses, S. Antonello, M. C. Ar�evalo, C. C. Gonz�alez,
J. Sharma, A. N. Wallette, M. S. Workentin and F. Maran, Chem.–Eur. J., 2007, 13, 7983–7995.
32 S. Antonello, M. Musumeci, D. D. M. Wayner and F. Maran, J. Am.Chem. Soc., 1997, 119, 9541–9549.
33 S. Antonello and F. Maran, J. Am. Chem. Soc., 1999, 121, 9668–9676.34 R. L. Donkers, F. Maran, D. D. M. Wayner and M. S. Workentin,
J. Am. Chem. Soc., 1999, 121, 7239–7248.35 (a) M. S. Workentin, F. Maran and D. D. M. Wayner, J. Am. Chem.
Soc., 1995, 117, 2120–2121; (b) S. Antonello, F. Formaggio,A. Moretto, C. Toniolo and F. Maran, J. Am. Chem. Soc., 2003,125, 2874–2875; (c) F. Polo, S. Antonello, F. Formaggio,C. Toniolo and F. Maran, J. Am. Chem. Soc., 2005, 127, 492–493.
36 S. Antonello and F. Maran, J. Am. Chem. Soc., 1997, 119, 12595–12600.
37 S. Antonello, F. Formaggio, A. Moretto, C. Toniolo and F. Maran,J. Am. Chem. Soc., 2001, 123, 9577–9584.
38 S. Antonello, M. Crisma, F. Formaggio, A. Moretto, F. Taddei,C. Toniolo and F. Maran, J. Am. Chem. Soc., 2002, 124, 11503–11513.
39 (a) R. L. Donkers and M. S. Workentin, Chem.–Eur. J., 2001, 7,4012–4020; (b) D. Magri and M. S. Workentin, Org. Biomol.Chem., 2003, 1, 3418–3429.
40 S. Antonello, A. Venzo and F. Maran, J. Electroanal. Chem., 2011,660, 234–242.
41 (a) E. D. German and A. M. Kuznetsov, J. Phys. Chem., 1994, 98,6120–6127; (b) E. D. German, A. M. Kuznetsov andV. A. Tikhomirov, J. Phys. Chem., 1995, 99, 9095–9101.
42 (a) C. P. Andrieux, C. Blocman, J. M. Dumas-Bouchiat, F. M’Hallaand J.-M. Sav�eant, J. Electroanal. Chem., 1980, 113, 19–40; (b)C. P. Andrieux and J.-M. Sav�eant, J. Electroanal. Chem., 1986, 205,43–58.
43 (a) T. B. Christensen and K. Daasbjerg, Acta Chem. Scand., 1997, 51,307–317; (b) S. Antonello, K. Daasbjerg, H. Jensen, F. Taddei andF. Maran, J. Am. Chem. Soc., 2003, 125, 14905–14916.
44 (a) H. Lund and K. Daasbjerg, Acta Chem. Scand., 1993, 47, 597–604;(b) Y. Huang and D. D. M. Wayner, J. Am. Chem. Soc., 1994, 116,2157–2158.
45 (a) C. G. Swain, W. H. Stockmayer and J. T. Clarke, J. Am. Chem.Soc., 1950, 72, 5426–5434; (b) C. A. Barson and R. A. Wisdom,Eur. Polym. J., 1972, 8, 1139–1144.
46 Y. Song, A. S. Harper and R. W. Murray, Langmuir, 2005, 21, 5492–5500.
47 R. S. Nicholson and I. Shain, Anal. Chem., 1964, 36, 706–723.48 C. Walling and C. X. Zhao, Tetrahedron, 1982, 38, 1105–1112.49 The BDE value was taken as the average between pertinent literature
values: (a) Ref. 45b; (b) C. X. Zhao, X. K. Jiang and J. Y. Zhang,J. Fluorine Chem., 1985, 27, 401–410.
50 Not explicitly reported in ref. 11 is that the inner reorganizationenergy corresponding to the Au25L18
+/Au25L180 redox couple is
slightly larger than that of the Au25L180/Au25L18
� couple, 3.7 kcalmol�1
51 R. A. Marcus, Faraday Discuss. Chem. Soc., 1982, 74, 7–15.52 R. A. Marcus and N. Sutin, Biochim. Biophys. Acta, 1985, 811, 265–
322.53 J. Chateauneuf, J. Lusztyk and K. U. Ingold, J. Am. Chem. Soc.,
1988, 110, 2886–2893.54 G. M. Rigodanza, Laurea in Chimica, M.Sc. thesis, Padova, 1995.55 Simulations took into account the E� values of the two redox steps of
Au25, eqn (1) (E� for the direct reduction of AB, with AB¼ peroxide),
eqn (4) (E� for the ArCO2_/ArCO2� redox couple), rate constant for
the decarboxylation of ArCO2_, reactions 6 and 7 and theirequivalent for Au25L18/Au25L18
� as the donor redox couple.56 A. T. Blomquist and A. J. Buselli, J. Am. Chem. Soc., 1951, 73, 3883–
3888.57 H. Lund, K. Daasbjerg, T. Lund and S. U. Pedersen,Acc. Chem. Res.,
1995, 28, 313–319.58 Z. Liu, M. Zhu, X. Meng, G. Xu and R. Jin, J. Phys. Chem. Lett.,
2011, 2, 2104–2109.59 L. Eberson, Adv. Phys. Org. Chem., 1998, 31, 91–141.60 Organic Electrochemistry, ed. H. Lund and M. M. Baizer, Marcel
Dekker, New York, 3rd edn, 1991.
5342 | Nanoscale, 2012, 4, 5333–5342
61 C. M. Aikens, J. Phys. Chem. Lett., 2010, 1, 2594–2599.62 (a) J. F. Smalley, H. O. Finklea, C. E. D. Chidsey, M. R. Linford,
S. E. Creager, J. P. Ferraris, K. Chalfant, T. Zawodzinsk,S. W. Feldberg and M. D. Newton, J. Am. Chem. Soc., 2003, 125,2004–2013; (b) J. J. Gooding, F. Mearns, W. Yang and J. Liu,Electroanalysis, 2003, 15, 81–96.
63 (a) M. D. Newton, Chem. Rev., 1991, 91, 767–792; (b) A. Nitzan,Annu. Rev. Phys. Chem., 2001, 52, 681–750; (c) M. N. Paddon-Row,Aust. J. Chem., 2003, 56, 729–748.
64 R. E. Holmlin, R. F. Ismagilov, R. Haag, V. Mujica, M. A. Ratner,M. A. Rampi and G. M. Whitesides, Angew. Chem., Int. Ed., 2001,40, 2316–2320.
This journal is ª The Royal Society of Chemistry 2012