Swiss Institute of Bioinformatics Torsten Schwede Biozentrum - Universität Basel Swiss Institute of Bioinformatics Klingelbergstr 50-70 CH - 4056 Basel, Switzerland Tel: +41-61 267 15 81 EMBnet course: Introduction to Protein Structure Bioinformatics Homology Modeling I Basel, September 30, 2004 [ PDB: http://www.pdb.org ] Growth of the Protein Data Bank PDB
Transcript
1
Swiss Institute of Bioinformatics
Torsten SchwedeBiozentrum - Universität Basel Swiss Institute of BioinformaticsKlingelbergstr 50-70 CH - 4056 Basel, Switzerland Tel: +41-61 267 15 81
EMBnet course: Introduction to Protein Structure Bioinformatics
Homology Modeling IBasel, September 30, 2004
[ PDB: http://www.pdb.org ]
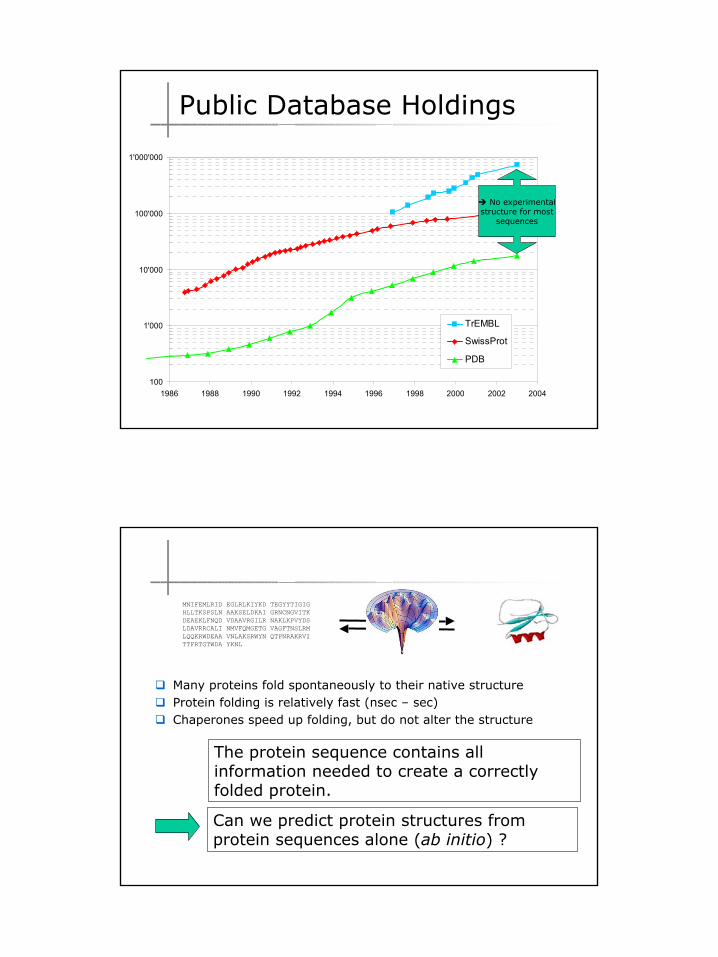
Growth of the Protein Data Bank PDB
2
100
1'000
10'000
100'000
1'000'000
1986 1988 1990 1992 1994 1996 1998 2000 2002 2004
TrEMBL
SwissProt
PDB
No experimentalstructure for most
sequences
Public Database Holdings
The protein sequence contains all information needed to create a correctly folded protein.
Can we predict protein structures from protein sequences alone (ab initio) ?
Many proteins fold spontaneously to their native structureProtein folding is relatively fast (nsec – sec)Chaperones speed up folding, but do not alter the structure
3. Experimental conditions (ligands and cofactors)
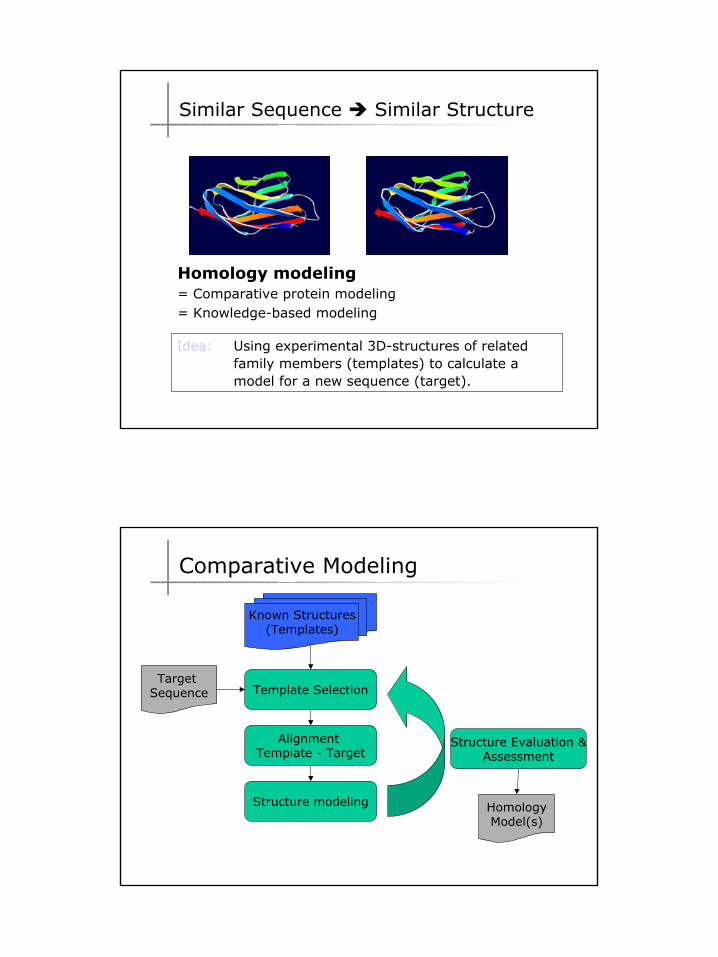
Comparative Modeling
10
Known Structures(Templates)
Target Sequence Template Selection
Alignment Template - Target
Structure modeling
Structure Evaluation &Assessment
HomologyModel(s)
• Multiple sequence alignment for pairs > 40% identity
or• Use structural alignment of
templates to guide sequence alignment of target
or• Use separate profiles for
template and targets
Comparative Modeling
Known Structures(Templates)
Target Sequence Template Selection
Alignment Template - Target
Structure modeling
Structure Evaluation &Assessment
HomologyModel(s)
• Errors in template selection or alignment result in bad models
iterative cycles of alignment, modeling and evaluation
Built many models, choose best.
Comparative Modeling
11
Known Structures(Templates)
Target Sequence Template Selection
Alignment Template - Target
Structure modeling
Structure Evaluation &Assessment
HomologyModel(s)
I. Manual Model building
II. Template based fragment assembly
– Composer (Sybyl, Tripos)– SWISS-MODEL
III. Satisfaction of spatial restraints– Modeller (Insight II, MSI)– CPH-Models
Comparative Modeling
[ http://www.expasy.org/spdbv/ ]
I. Manual Modeling
12
II. Template based fragment assembly
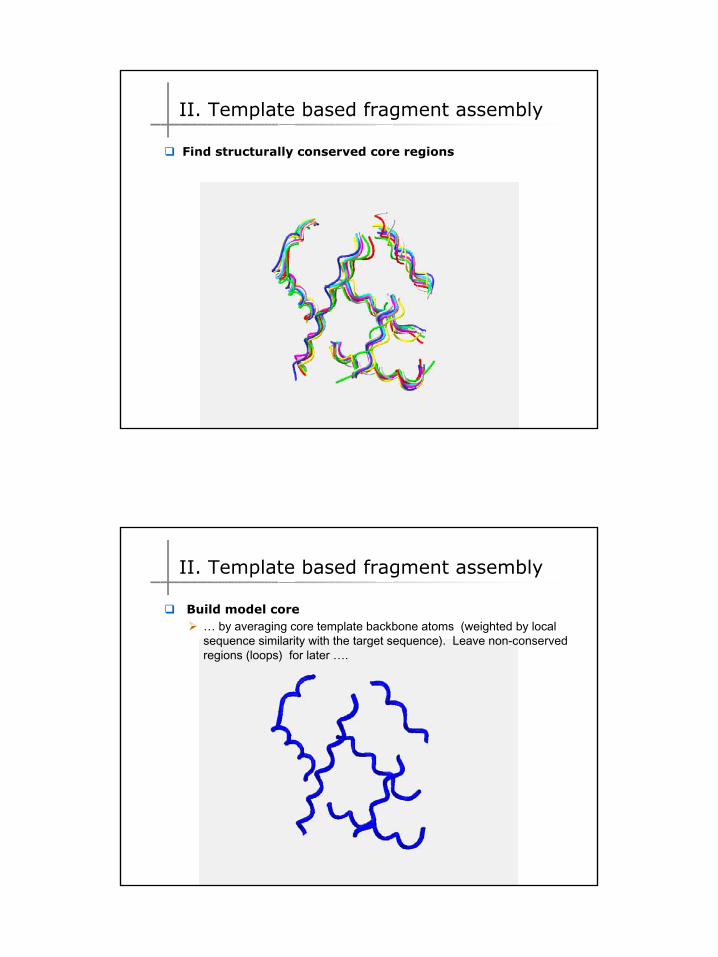
Find structurally conserved core regions
II. Template based fragment assembly
Build model core… by averaging core template backbone atoms (weighted by local sequence similarity with the target sequence). Leave non-conserved regions (loops) for later ….
13
II. Template based fragment assembly
Loop (insertion) modelingUse the “spare part” algorithm to find compatible fragments in a Loop-Database, or “ab-initio” rebuilding (e.g. Monte Carlo, MD, GA, etc.) to build missing loops.
II. Template based fragment assembly
Side Chain placementFind the most probable side chain conformation, using
Only a small fraction of all possible side chain conformations is observed in experimental structures
Rotamer libraries provide an ensemble of likely conformations
The propensity of rotamers depends on the backbone geometry:
g+
trans
g-
p(g+ | phi)
p(t | phi)
p(g- | phi)
p(g+ | psi)
p(t | psi)
p(g- | psi)
Phe,Tyr, His
Backbone-dependent rotamer libraries
15
II. Template based fragment assembly
Energy minimization
modeling method will produce unfavorable contacts and bonds
Energy minimization is used to
• regularize local bond and angle geometry
• Relax close contacts and geometric strain
extensive energy minimization will move coordinates away from real structure ⇒ keep it to a minimum
SWISS-MODEL is using GROMOS 96 force field for a steepest descent
III. Satisfaction of Spatial restraints
Alignment of target sequence with templates
Extraction of spatial restraints from templates
Modeling by satisfaction of spatial restraints
M
A
T
EA
F
TS
G
Q
16
Some features of a protein structure:
R resolution of X-ray experimentr amino acid residue typeΦ, Ψ main chain anglest secondary structure classM main chain conformation classΧ i,, ci side chain dihedral angle classa residue solvent accessibilitys residue neighborhood differenced Ca - Ca distance∆d difference between two Ca - Ca distances
III. Satisfaction of Spatial restraints
Feature properties can be associated with
a protein (e.g. X-ray resolution)
residues (e.g. solvent accessibility)
pairs of residues (e.g. Ca - Ca distance)
other features (e.g. main chain classes)
How can we derive modeling restraints from this data?A restraint is defined as probability density function (pdf) p(x):
∫=<≤1
2
)()21(x
x
dxxpxxxp1)( =∫ dxxp
with
0)( >xp
III. Satisfaction of Spatial restraints
17
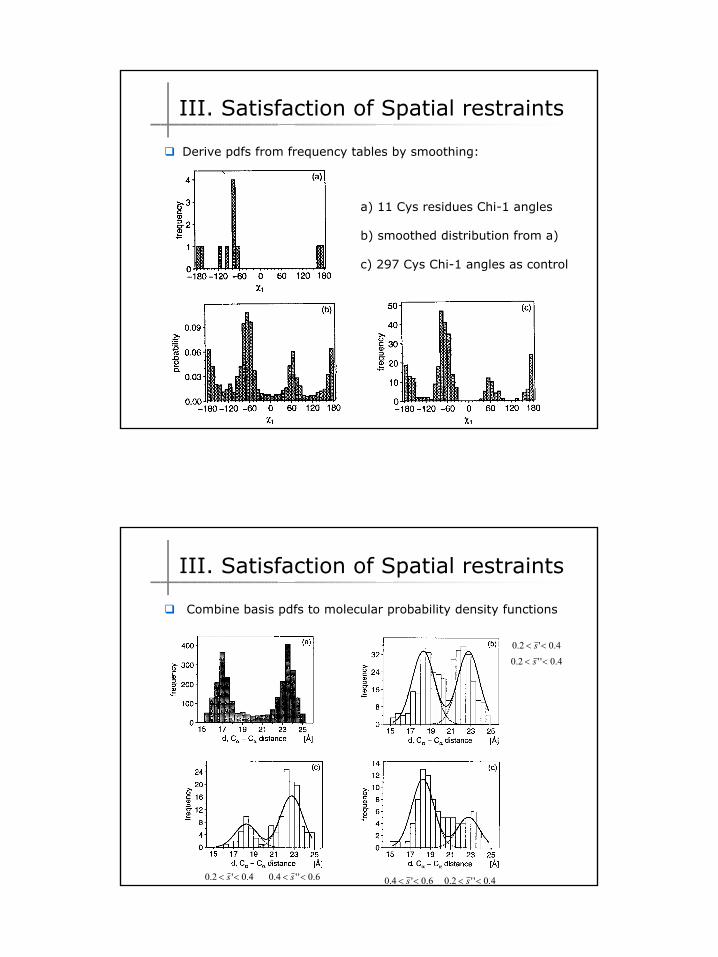
a) 11 Cys residues Chi-1 angles
b) smoothed distribution from a)
c) 297 Cys Chi-1 angles as control
III. Satisfaction of Spatial restraints
Derive pdfs from frequency tables by smoothing:
4.0'2.0 << s4.0''2.0 << s
4.0'2.0 << s 6.0''4.0 << s 4.0''2.0 << s6.0'4.0 << s
III. Satisfaction of Spatial restraints
Combine basis pdfs to molecular probability density functions
18
Satisfaction of spatial restraints
Find the protein model with the highest probability
Variable target function:
Start with a linear conformation model or a model close to
the template conformation
At first, use only local restraints
minimize some steps using a conjugate gradient optimization
repeat with introducing more and more long range restraints
until all restraints are used
III. Satisfaction of Spatial restraints
EVA
Evaluation of Automatic protein structure prediction [ Burkhard Rost, Andrej Sali, http://cubic.bioc.columbia.edu/eva/ ]
CASPCommunity Wide Experiment on the Critical Assessment of Techniques for Protein Structure Prediction http://PredictionCenter.llnl.gov
Model Accuracy Evaluation
19
Evaluation of Automatic protein structure prediction
[ Burkhard Rost, Andrej Sali, http://cubic.bioc.columbia.edu/eva/ ]
Laskowski R A, MacArthur M W, Moss D S & Thornton J M (1993). PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Cryst., 26, 283-291. Morris A L, MacArthur M W, Hutchinson E G & Thornton J M (1992). Stereochemical quality of protein structure coordinates. Proteins, 12, 345-364.
PROCHECK
WHAT IF I check my structure?
Imagine ...• An everyday situation in a biocomputing lab: "Should they use the structure?" • An everyday situation in a crystallography lab: "Should they deposit the structure already?" In a WHAT_CHECK report, each reported fact has an assigned severity:
error:severe errors encountered during the analyses. Items marked as errors are considered severe problems requiring immediate attention.
warning:Either less severe problems or uncommon structural features. These still need special attention.
note:Statistical values, plots, or other verbose results of tests and analyses that have been performed.
WHAT IF: A molecular modeling and drug design program. G.Vriend, J. Mol. Graph. (1990) 8, 52-56. Errors in protein structures. R.W.W. Hooft, G. Vriend, C. Sander, E.E. Abola, Nature (1996) 381, 272-272.
WhatCheck / WhatIf
25
# 49 # Note: Summary report for users of a structureThis is an overall summary of the quality of the structure ascompared with current reliable structures. This summary is mostuseful for biologists seeking a good structure to use for modellingcalculations.
The second part of the table mostly gives an impression of how wellthe model conforms to common refinement constraint values. Thefirst part of the table shows a number of constraint-independentquality indicators.