Available online at www.pelagiaresearchlibrary.com Pelagia Research Library Der Pharmacia Sinica, 2011, 2 (5):169-181 ISSN: 0976-8688 CODEN (USA): PSHIBD 169 Pelagia Research Library Enhancement of Dissolution rate and stability study of Ofloxacin Solid dispersion Pintu K De 1* , Basudev Sahana 2 and Soumen Rakshit 2 1 Department of Pharmaceutics, Dr. B. C. Roy College of Pharmacy and Allied Health Sciences, Dr. Megnad Saha Sarani, Bidhannagar Durgapur, W.B. INDIA 2 Institute of Pharmacy, Jalpaiguri, Govt. of West Bengal, Jalpaiguri, W.B., India ______________________________________________________________________________ ABSTRACT In case of relatively insoluble drugs i.e., the drugs having solubility less than 1%, the rate of dissolution is usually the rate determining step in the overall absorption process to become bioavailable. However for relatively soluble drugs i.e., the drugs having intrinsic dissolution rate more than 1mg/cm 2 /min, the permeability of the drug may become the rate determining step. Poor solubility causing potential bio- absorption problems unless the dosage forms are specially designed. Solid dispersion is the dispersion of the drug in a biologically inert matrix. The objective of the present study was to enhance the dissolution rate of Ofloxacin by making a molecular dispersion of drug in the polymeric matrix of Polyethylene Glycol-6000. Solid dispersion of Ofloxacin was prepared following fusion and solvent evaporation techniques. The drug polymer interaction was studied by Differential Scanning Calorimetry (DSC) and Fourier Transform Infrared Spectroscopy (FTIR), which revealed the absence of major interactions in both the methods. Dissolution study of the solid dispersions shows the enhancement of dissolution rate of Ofloxacin. After 15 minutes of study only 65% of the pure drug is dissolved in the aqueous medium whereas at the same time 98% dissolution was observed with the formulation of solid dispersion in presence of a surfactant. Among the two methods; considering the absence of interaction and higher rate of dissolution the solvent evaporation technique is found most suitable to enhance the dissolution rate and consequently the bioavailability of the drug. The stability study of the formulations as per ICH guideline Q1A in stability chamber both at intermediate and accelerated conditions ascertained that the formulations are stable at wide range of storage conditions. Key words: Solid dispersion, Ofloxacin, FTIR, DSC, TGA. ______________________________________________________________________________ INTRODUCTION Ofloxacin is a synthetic fluoroquinolone broad spectrum antimicrobial agent for oral administration. It acts by inhibiting bacterial DNA gyrase enzyme which is required for DNA replication and thus causes bacterial lysis. The solubility characteristics of ofloxacin at room
Transcript
Available online at www.pelagiaresearchlibrary.com
Pelagia Research Library
Der Pharmacia Sinica, 2011, 2 (5):169-181
ISSN: 0976-8688 CODEN (USA): PSHIBD
169 Pelagia Research Library
Enhancement of Dissolution rate and stability study of Ofloxacin Solid dispersion
Pintu K De1*, Basudev Sahana2 and Soumen Rakshit2
1Department of Pharmaceutics, Dr. B. C. Roy College of Pharmacy and Allied Health Sciences,
Dr. Megnad Saha Sarani, Bidhannagar Durgapur, W.B. INDIA 2Institute of Pharmacy, Jalpaiguri, Govt. of West Bengal, Jalpaiguri, W.B., India
______________________________________________________________________________ ABSTRACT In case of relatively insoluble drugs i.e., the drugs having solubility less than 1%, the rate of dissolution is usually the rate determining step in the overall absorption process to become bioavailable. However for relatively soluble drugs i.e., the drugs having intrinsic dissolution rate more than 1mg/cm2/min, the permeability of the drug may become the rate determining step. Poor solubility causing potential bio-absorption problems unless the dosage forms are specially designed. Solid dispersion is the dispersion of the drug in a biologically inert matrix. The objective of the present study was to enhance the dissolution rate of Ofloxacin by making a molecular dispersion of drug in the polymeric matrix of Polyethylene Glycol-6000. Solid dispersion of Ofloxacin was prepared following fusion and solvent evaporation techniques. The drug polymer interaction was studied by Differential Scanning Calorimetry (DSC) and Fourier Transform Infrared Spectroscopy (FTIR), which revealed the absence of major interactions in both the methods. Dissolution study of the solid dispersions shows the enhancement of dissolution rate of Ofloxacin. After 15 minutes of study only 65% of the pure drug is dissolved in the aqueous medium whereas at the same time 98% dissolution was observed with the formulation of solid dispersion in presence of a surfactant. Among the two methods; considering the absence of interaction and higher rate of dissolution the solvent evaporation technique is found most suitable to enhance the dissolution rate and consequently the bioavailability of the drug. The stability study of the formulations as per ICH guideline Q1A in stability chamber both at intermediate and accelerated conditions ascertained that the formulations are stable at wide range of storage conditions. Key words: Solid dispersion, Ofloxacin, FTIR, DSC, TGA. ______________________________________________________________________________
INTRODUCTION Ofloxacin is a synthetic fluoroquinolone broad spectrum antimicrobial agent for oral administration. It acts by inhibiting bacterial DNA gyrase enzyme which is required for DNA replication and thus causes bacterial lysis. The solubility characteristics of ofloxacin at room
Pintu K De et al Der Pharmacia Sinica, 2011, 2(5):169-181 ______________________________________________________________________________
170 Pelagia Research Library
temperature, as defined by USP is sparingly to slightly soluble in aqueous solutions with pH 7 (solubility falls to 4 mg/mL). The rate and extent of absorption of an orally administered drug are dependent upon two independent processes, its dissolution characteristics in an aqueous medium and its permeability characteristics across the gastric or intestinal mucosa. Ideally for optimum absorption a drug should have sufficient aqueous solubility to dissolve in the fluids at absorption site and lipid solubility high enough to facilitate partitioning in the lipoidal membrane and into the systemic circulation. Solubility values are useful in a candidates early development are those in distilled water, 0.9% Nacl, 0.01M Hcl, 0,1M Hcl and 0.1M NaoH, all at room temperature as well as at pH 7.4 buffer at 37°C. Drug’s having limited solubility (<1% means 10mg/ml) over the pH range 1-7 at 37°C, often exhibit potential bioabsorption problem unless dosage forms are specially designed. If the intrinsic dissolution rate was greater than 1mg/cm2/min then absorption was unimpeded. Dissolution rates less than 0.1mg/cm2/min were likely to give dissolution rate limited absorption. This tenfold difference in dissolution rate translates to a lower limit for solubility of 1mg/ml. Solubility less than 1mg/ml indicates the potential bioabsorption problems unless the dosage forms are specially designed. Solubility profiles are not predictors of biological performance but do provide rationale for more extensive in-vivo studies and formulation development prior to drug evaluation in humans.
Up to 40 per cent of new chemical entities discovered by the pharmaceutical industry today are poorly soluble or lipophilic compounds. . The various techniques are available for enhancement of solubility. Solid dispersion is one of the most promising approaches for solubility enhancement. The solid dispersion approach to reduce particle size and therefore increase the dissolution rate and absorption of drugs was first recognized in 1961[1]. It improves dissolution rate by reducing particle size, higher porosity, drug is in amorphous state, improving wettability and hence improves bioavailability of poorly water soluble drugs. The term “solid dispersions” refers to the dispersion of one or more active ingredients in an inert carrier in a solid state, frequently prepared by the melting (fusion) method, solvent method, or fusion solvent-method [2]. Novel additional preparation techniques have included rapid precipitation by freeze drying [3], often in the presence of amorphous hydrophilic polymers and also using methods such as melt extrusion [4]. The most commonly used hydrophilic carriers for solid dispersions include polyvinylpyrrolidone [5,6] polyethylene glycols [7], Plasdone-S630 [8]. Many times surfactants
Pintu K De et al Der Pharmacia Sinica, 2011, 2(5):169-181 ______________________________________________________________________________
171 Pelagia Research Library
may also used in the formation of solid dispersion. Surfactants like Tween-80, Docusate sodium, Myrj-52, Pluronic-F68 and Sodium Lauryl Sulphate used [8]. The solubility of etoposide [9], Felodipine [10], itraconazole [11], aceclofenac [12], valdecoxib [13], celecoxib [14], Ketoprofen [15] can be improved by solid dispersion using suitable hydrophilic carriers.
MATERIALS AND METHODS
Materials Ofloxacin was received as a gift sample from Cadila Health Care Ltd.(Ahmedabad, India); PEG-6000 (Hydroxyl number 16-23, Molecular weight 5000-7000, Melting range 60-63° C), was purchased from Merck, Mumbai, India. Chloroform was procured from Ranbaxy Laboratories Limited, SAS Nagar, India. Other materials used in this study like Tween-80 are of analytical grade. Preparation of Ofloxacin Solid Dispersions Two commonly used methods have been followed for preparation of ofloxacin solid dispersion. 1) Hot Melt Method and 2) solvent evaporation method. Hot Melt Method Ofloxacin and PEG-6000 mixture of different composition (10 & 20% drug load) were melted in crucible in a muffle furnace at 257°c and rapidly cooled in a refrigerator. The solid residue was powdered and sifted through Sieve no.100 and stored in desiccators prior to use. Solvent Evaporation Method The drug, Ofloxacin and the polymer, Polyethylene glycol 6000 (PEG 6000) have been dissolved together in a common solvent i.e. chloroform. After evaporation of solvent the formulation were kept in desiccators containing activated silica gel and dried to constant weight. The dried product was powdered and sifted through Sieve no.100 and stored in desiccators prior to use for further evaluation.
Table2: Different formulations of Ofloxacin solid dispersion
FORMULATION CODE COMPOSITION METHOD
F-1 10% drug in polymer Fusion F-2 20% drug in polymer Fusion F-3 Drug: polymer=5:5 in chloroform Solvent evaporation F-4 Drug: polymer: surfactant (tween 80) =5:5:1.in chloroform. Solvent evaporation
Drug-polymer compatibility study The physicochemical interactions between Ofloxacin and the polymer (PEG 6000) used in the formulation of solid dispersion were studied using Fourier transform infrared spectroscopy (FTIR). The infrared spectra were recorded in the FTIR (Perkin Elmer) instrument in the wave length region between 4400 and 600cm-1 by KBr pellet method. The spectra obtained for pure drug, polymer and different solid dispersion formulations were compared.
Pintu K De et al Der Pharmacia Sinica, 2011, 2(5):169-181 ______________________________________________________________________________
172 Pelagia Research Library
Pintu K De et al Der Pharmacia Sinica, 2011, 2(5):169-181 ______________________________________________________________________________
173 Pelagia Research Library
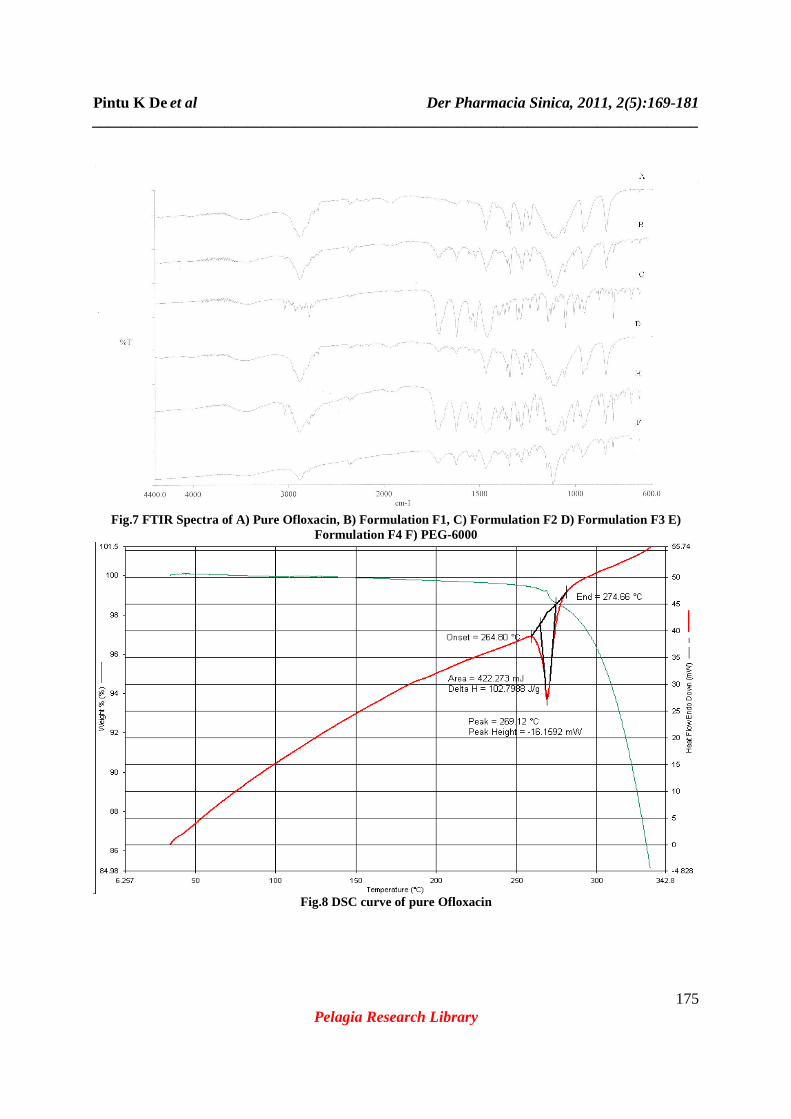
Fig.4 FTIR Spectra of the pure drug, Ofloxacin.
Pintu K De et al Der Pharmacia Sinica, 2011, 2(5):169-181 ______________________________________________________________________________
174 Pelagia Research Library
Fig.5 FTIR Spectra of Formulation F1 prepared by fusion method containing 10% drug.
Fig.6 FTIR Spectra of Formulation F3 prepared by solvent evaporation method, drug to polymer ratio 5:5.
Pintu K De et al Der Pharmacia Sinica, 2011, 2(5):169-181 ______________________________________________________________________________
175 Pelagia Research Library
Fig.7 FTIR Spectra of A) Pure Ofloxacin, B) Formulation F1, C) Formulation F2 D) Formulation F3 E)
Formulation F4 F) PEG-6000
Fig.8 DSC curve of pure Ofloxacin
Pintu K De et al Der Pharmacia Sinica, 2011, 2(5):169-181 ______________________________________________________________________________
176 Pelagia Research Library
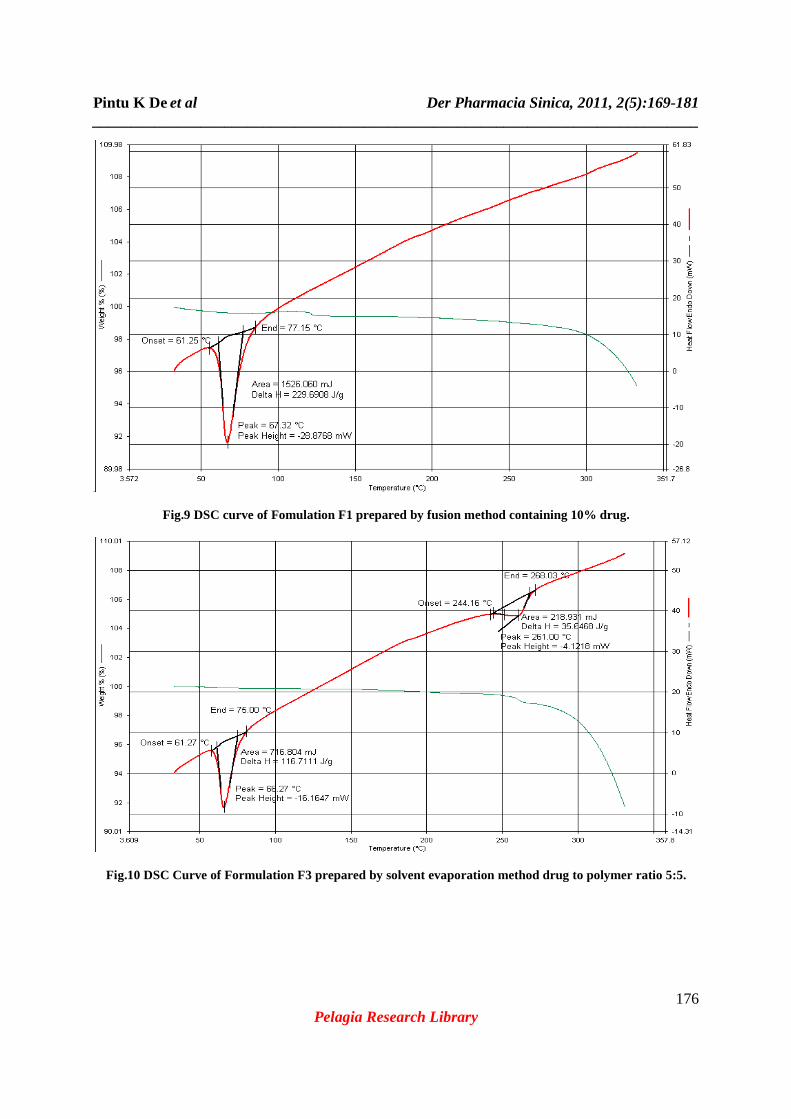
Fig.9 DSC curve of Fomulation F1 prepared by fusion method containing 10% drug.
Fig.10 DSC Curve of Formulation F3 prepared by solvent evaporation method drug to polymer ratio 5:5.
Pintu K De et al Der Pharmacia Sinica, 2011, 2(5):169-181 ______________________________________________________________________________
177 Pelagia Research Library
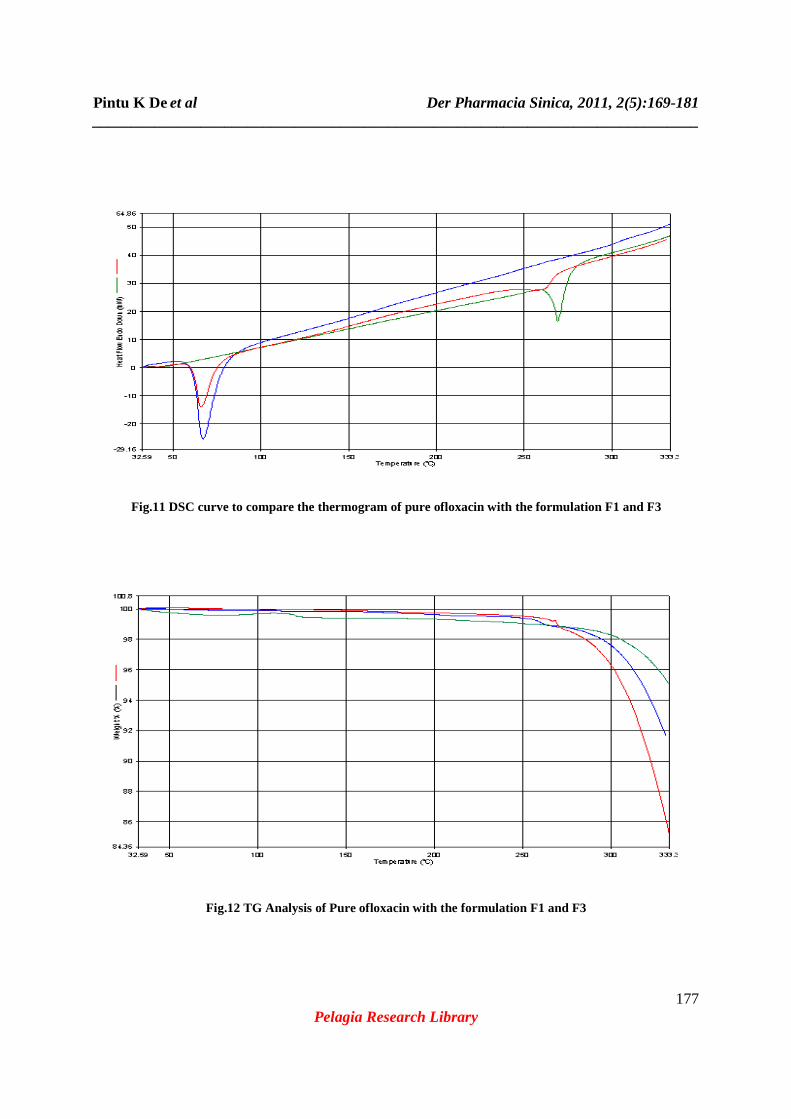
Fig.11 DSC curve to compare the thermogram of pure ofloxacin with the formulation F1 and F3
Fig.12 TG Analysis of Pure ofloxacin with the formulation F1 and F3
Pintu K De et al Der Pharmacia Sinica, 2011, 2(5):169-181 ______________________________________________________________________________
178 Pelagia Research Library
Thermal Analysis A differential scanning calorimeter (DSC) [DSC60, SHIMADZU Corporation, Japan] is used under nitrogen gas flow of 60mL/min, at a heating rate of 5°c/min. the samples were sealed in an aluminum pan. A sample of 2-3mg was accurately weight was subjected to DSC run over the temperature range 40-350°c. The temperature was calibrated using pure indium with a melting point of 156.6°c. an empty pan was used as a reference. Stability Study The stability study of the formulations was done according to ICH guideline Q1A in stability chamber. The stability study was performed at intermediate and accelerated conditions in closed containers at specific storage conditions (Table 3). In both cases samples were analyzed for drug content at 0, 3 and 6 months to find out the effect of temperature and humidity on product stability.
Table3 Different stability study condition according to the ICH guideline
Table 4 Drug content analysis after stability study of intermediate (IM) and accelerated (AC) conditions
Formulation 0 months 3 months 6 months
IM AC IM AC IM AC F1 98.6 98.6 98.4 98.5 98.2 98.4 F2 97.5 97.5 97.5 97.3 97.3 97.04 F3 99.3 99.3 99.0 99.1 99.05 98.6 F4 99.7 99.7 99.6 99.4 99.01 97.9
Dissolution Study of Ofloxacin Solid Dispersions: 100 mg equivalent weight of pure ofloxacin was taken for each formulation and dissolution study was done in 6 stages USP Apparatus II (rotating paddle type). 900ml of distilled water was taken as dissolution medium. The temperature was maintained at 37±0.5°c. stirring speed was adjusted to 75 rpm. 5ml sample was withdrawn at 05 minutes interval and same volume of distilled water was added to it. Then all the samples are measured in U.V Spectrophotometer (Shimadzu UV-1700) at 280nm against appropriate blank.
RESULTS AND DISCUSSION
The IR spectra of pure ofloxacin (Fig.4) showed the presence of principal peaks responsible for different functional groups. The wave number 3044 cm-1 is due to stretching vibration of hydroxyl group and intermolecular hydrogen bond; 1714 cm-1 due to stretching vibrations of C=O group of acids; 1622 cm-1 due to N-H bending vibration; 1550 cm-1 due to the presence of alkyl groups; 1407 cm-1 due to stretching vibration of methylene group in benzoxazine; 1351 cm-1 due to hydroxyl bending vibration; 1200 cm-1 and 1240 cm-1 due to C-O-C stretching vibration; 1011 cm-1 due to C-F stretching. The presence of all these peaks gives confirmation about the purity of the drug. The FTIR spectra of the formulation F1 which was prepared by fusion method containing 10% drug in the polymer PEG-6000 showing the absence of the certain
Pintu K De et al Der Pharmacia Sinica, 2011, 2(5):169-181 ______________________________________________________________________________
179 Pelagia Research Library
principal peaks (Fig.5) like 1550, 1407, 1011cm-1.Whereas almost all the peaks were present in FTIR spectra of formulation F3 (Fig6) which was prepared by solvent evaporation method containing equal ratio of drug and polymer. Fig.7 shows the FTIR spectra of PEG-6000(A); Formulation F2 (B); pure drug, Ofloxacin (C); Formulation F1 (D); Formulation F3 (E); Formulation F4 (F) all together in the same scale for better understanding of appearance, disappearance or shifting of IR peaks. From the above observation it can be concluded that there are no interaction in the process of formulation by solvent diffusion method although a minor interaction was observed in fusion method. Fusion is one of the effective method for preparing solid dispersion, but when the drug is melted alone or in combination of the polymer PEG-6000, the color of the formulation gradually become yellowish brown from white, this may be due to physical degradation at the temperature above 150°c [16]. Whereas the color of the solidified mass prepared by solvent evaporation technique (F3) does not change. Thermal analytical techniques like Thermal Gravimetric Analysis (TGA) and DSC are potentially very useful in determining the stability and the presence of very low level of impurity. Also through DSC curve we can detect the different crystalline form of the same drug and the ratio of each polymorph. In DSC the difference in energy input (∆H) required to maintain the sample temperature is measured with respect to a thermally inert reference substance. ∆H is plotted against the temperature (T). In TGA the change in the experimental mass is measured as the temperature is increased at a predetermined rate. Percentage weight remaining of the compound is plotted against temperature (T). Comparison of TGA curve (Fig.12) shows almost same rate of change in the mass of the experimental formulations (F1 and F3) like pure Ofloxacin. Hence it can be ascertained that both the formulations contain the intact drug in unchanged form. The DSC curve of pure ofloxacin shown in fig.8 exhibited a sharp endothermic peak at 269.12°c corresponding to its melting [16]. The DSC curve of solid dispersion F1 (Fig.9) prepared by fusion method showed the absence of the endothermic peak corresponding to the melting of ofloxacin but it shows a sharp endothermic peak at67.32°c which is corresponding to the melting of PEG-6000. The DSC curve of solid dispersion F3 (Fig.10) prepared by solvent evaporation method showed the presence of both the endothermic peaks at 261°c and at 66.27°c which are corresponding to the melting of ofloxacin and PEG-6000 respectively. Comparisons of endothermic peaks of DSC curve (Fig.11) of pure ofloxacin along with the formulations F-1 and F-3 shows the absence of interaction and presence of drug in unchanged form. All the formulations (F1 to F4) were kept at intermediate and accelerated conditions (Table3) in stability chamber in the closed container. A portion of the sample was taken out at 0, 3 and 6 months and interval and tested them for drug content. The result (Table 4) shows no significant changes in appearance and also in drug content. The highest percentage of degradation was observed in formulation F-4 after 6 months of storing in accelerated condition is found 1.89%. Hence it can be said that the formulations are stable at wide variation in storage condition for long time. The in vitro drug release studies were performed in using 6stage dissolution test apparatus USP type II (rotating paddle type). The release profile for all the five formulations were fitted to zero
Pintu K De et al Der Pharmacia Sinica, 2011, 2(5):169-181 ______________________________________________________________________________
180 Pelagia Research Library
order kinetics (cumulative % drug release vs. time plot) in Fig.1, first order kinetics (log% remaining to be released vs. time plot) in Fig.2 and in Higuchi model (cumulative % release vs. square root of time plot) depicted in Fig.3. The results of release profile indicate that only 65% of drug dissolved after 15 minutes of dissolution study with pure drug. But the dissolution study of all solid dispersions of ofloxacin shows higher dissolution than the pure drug after a definite time which indicates in enhancement of dissolution rate. If we compare the dissolution of solid dispersions by fusion and solvent evaporation method; solvent evaporation method gives higher dissolution (98%) rate as compared to fusion method (90%). In the fusion method as we increase the drug load or decrease the polymer proportion the dissolution rate is further low as in F-2 (90%) as compared to F-1 (93%). The solid dispersions prepared by solvent evaporation method and containing Tween-80 (F-4) as surfactant gives highest dissolution (98%) as compared to dispersions without surfactant F-3 (96%). The drug release data was fit to different kinetic model to understand the release mechanism. The linearity was found more comparatively in Higuchi plot than zero or first order plot, and release mechanism may follow diffusion from solid dispersion.
CONCLUSION Solid dispersion of Ofloxacin can be prepared with Polyethylene Glycol 6000 by both Fusion and Solvent Evaporation technique. Solid dispersion of Ofloxacin by both the above mentioned methods increases the dissolution rate of Ofloxacin. The stability of the drug, ofloxacin is not affected by any of the two methods and the formulations can be stored without any significant degradation for long tome. Among two methods followed, highest dissolution was found with the formulation prepared by solvent evaporation technique in presence of surfactant. Hence preparation of solid dispersion of Ofloxacin can be useful in enhancing the dissolution and improvement of the bioavailability of the drug. Acknowledgements The authors are highly indebted to Principal, Institute of Pharmacy Jalpaiguri, Govt. of West Bengal. Authors are also thankful to Cadila Health Care Ltd., Ahmedabad, India for providing us gift sample of pure drug, Ofloxacin.
REFERENCES [1] Sekiguchi K, Obi N, Chem. Pharm. Bull., 1961, 9, 866-872. [2] Chiou WL, Riegelman S, J. Pharm. Sci., 1971, 60, 1281-1302. [3] Emara LH, Badr RM, Elbary AA, Drug. Dev. Ind. Pharm., 2002, 28, 795-807. [4] Forster A, Hempenstall J, Rades T, J. Pharm. Pharmacol., 2001, 53, 303-315. [5] Ambike AA, Mahadik KR, Paradkar A, Int. J. Pharm., 2004, 282, 151-162. [6] Paradkar A, Ambike AA, Jadhav BK, Mahadik KR, Int. J. Pharm., 2004, 271, 281-286. [7] Doshi DH, Ravis WR, Betageri GV, Drug. Dev. Ind. Pharm., 1967, 23, 1167-1176. [8] Ghebremeskel AN , Vemavarapu C, Lodaya M, International Journal of Pharmaceutics, 2007, 328, 119–129. [9] Shah JC, Chen JR, Chow D, International Journal of Pharmaceutics, 1995, 113, 103-111. [10] Tapas AR, Kawtikwar PS, Sakarkar DM, Der Pharmacia Sinica, 2010, 1(1), 136-146.
Pintu K De et al Der Pharmacia Sinica, 2011, 2(5):169-181 ______________________________________________________________________________
181 Pelagia Research Library
[11] Jung JY, Yoo SD, Lee SH, Kim KH, Yoon DS, Lee KH, International Journal of Pharmaceutics, 1999, 187, 209–218. [12] Arora G, Malik K, Sharma J, Nagpal M, Singh I, Der Pharmacia Sinica, 2011, 2(2), 142-151. [13] Modi A, Tayade P, AAPS Pharm. Sci. Tech 2006; 7 (3) Article 68. [14] Gupta P, Kakumanu VK and Bansal AK, Pharmaceutical Research, 2004, 21, 1762-1769. [15] Nagar G, Luhadiya A, Agarwal S, Dubey PK, Der Pharmacia Sinica, 2011, 2(4), 67-73.. [16] Okonogi S, Puttipipatkhachorn, AAPS Pharm Sci Tech, 2006, 7(2), E1 to E6.