L’épidémiologie analytique (ou étiologique) vise à identifier des facteurs de risque, c'est‐à‐
dire des facteurs associés à un risque accru de développer une maladie, et à donner une
interprétation causale des relations observées. Cette discipline est particulièrement importante car
une bonne compréhension des relations qui existent entre facteurs de risque et AVC peut aider à
identifier de véritables causes et permet de définir et d’évaluer des stratégies de prévention. Il est
classique de séparer les facteurs de risque en facteurs modifiables, potentiellement modifiables, et
non modifiables, même si cette séparation est quelque peu subjective (tableau 1). Un facteur de
risque est considéré comme bien documenté lorsque l’association est prouvée par des études
épidémiologiques solides et lorsque des essais thérapeutiques randomisés montrent que la réduction
du facteur ou sa suppression diminue le risque d’AVC.1 Les effets des traitements sur la réduction du
risque d’AVC ne seront toutefois pas détaillés ici (voir chapitre Jean‐Louis Mas).
Certains paramètres physiologiques comme la pression artérielle (PA), le taux de cholestérol,
la glycémie, et le poids sont des variables continues. Comme nous le verrons, il existe une relation
continue entre le niveau de ces paramètres et le risque d’événement cérébrovasculaire. De plus, la
réduction relative de risque associée à une diminution du niveau de ces paramètres est souvent
indépendante du niveau initial, avec un bénéfice absolu d’autant plus important que le risque absolu
initial est élevé. Ceci signifie que, bien que permettant de définir des niveaux de risque différents, la
notion de seuil « pathologique » permettant de définir le facteur de risque est arbitraire pour ces
facteurs.2
La sténose carotide asymptomatique, la fibrillation auriculaire, et la drépanocytose qui sont
non seulement des facteurs de risque, mais aussi des causes bien documentées. Ils sont traités dans
des chapitres séparés de même que la migraine et les états d’hypercoagulabilité.
3
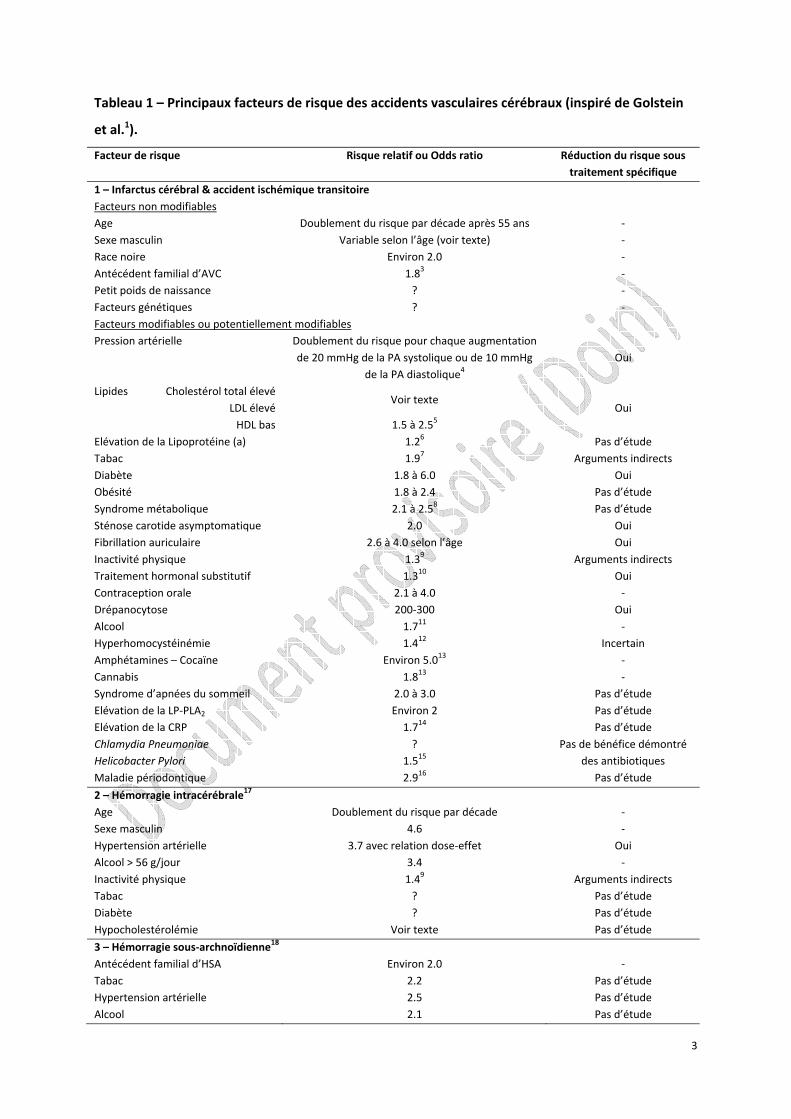
Tableau 1 – Principaux facteurs de risque des accidents vasculaires cérébraux (inspiré de Golstein
et al.1).
Facteur de risque Risque relatif ou Odds ratio Réduction du risque sous traitement spécifique
1 – Infarctus cérébral & accident ischémique transitoire Facteurs non modifiables Age Doublement du risque par décade après 55 ans ‐ Sexe masculin Variable selon l’âge (voir texte) ‐ Race noire Environ 2.0 ‐ Antécédent familial d’AVC 1.83 ‐ Petit poids de naissance ? ‐ Facteurs génétiques ? ‐ Facteurs modifiables ou potentiellement modifiables Pression artérielle Doublement du risque pour chaque augmentation
de 20 mmHg de la PA systolique ou de 10 mmHg de la PA diastolique4
Oui
Lipides Cholestérol total élevé Voir texte
Oui LDL élevé HDL bas 1.5 à 2.55 Elévation de la Lipoprotéine (a) 1.26 Pas d’étude Tabac 1.97 Arguments indirects Diabète 1.8 à 6.0 Oui Obésité 1.8 à 2.4 Pas d’étude Syndrome métabolique 2.1 à 2.58 Pas d’étude Sténose carotide asymptomatique 2.0 Oui Fibrillation auriculaire 2.6 à 4.0 selon l’âge Oui Inactivité physique 1.39 Arguments indirects Traitement hormonal substitutif 1.310 Oui Contraception orale 2.1 à 4.0 ‐ Drépanocytose 200‐300 Oui Alcool 1.711 ‐ Hyperhomocystéinémie 1.412 Incertain Amphétamines – Cocaïne Environ 5.013 ‐ Cannabis 1.813 ‐ Syndrome d’apnées du sommeil 2.0 à 3.0 Pas d’étude Elévation de la LP‐PLA2 Environ 2 Pas d’étude Elévation de la CRP 1.714 Pas d’étude Chlamydia Pneumoniae ? Pas de bénéfice démontré
des antibiotiques Helicobacter Pylori 1.515 Maladie périodontique 2.916 Pas d’étude
2 – Hémorragie intracérébrale17 Age Doublement du risque par décade ‐ Sexe masculin 4.6 ‐ Hypertension artérielle 3.7 avec relation dose‐effet Oui Alcool > 56 g/jour 3.4 ‐ Inactivité physique 1.49 Arguments indirects Tabac ? Pas d’étude Diabète ? Pas d’étude Hypocholestérolémie Voir texte Pas d’étude
3 – Hémorragie sous‐archnoïdienne18 Antécédent familial d’HSA Environ 2.0 ‐ Tabac 2.2 Pas d’étude Hypertension artérielle 2.5 Pas d’étude Alcool 2.1 Pas d’étude
4
Notions fondamentales L’épidémiologie est définie comme l’étude de la fréquence des maladies (épidémiologie
descriptive) et de ses déterminants (épidémiologie analytique ou étiologique). L’épidémiologie
analytique vise à rechercher des associations entre une exposition (facteur de risque) et une
maladie, et à en donner une interprétation causale. Pour bien interpréter les études
épidémiologiques, il est nécessaire de rappeler quelques notions fondamentales.
Définitions Le risque (R) est un indice qui quantifie la probabilité de devenir malade durant une période
de temps (∆t). C’est un nombre compris entre 0 et 1. On peut facilement le calculer dans une
population où il n’y a pas de censure, c’est‐à‐dire si tous les sujets ont été suivis pendant toute la
période. Le risque est alors égal au nombre de nouveaux cas enregistrés divisé par le nombre de
sujets au début de la période. Si il y a des censures (par exemple des perdus de vue), le calcul du
risque doit faire intervenir le taux d’incidence (TI). Cependant, le taux d’incidence n’est pas égal au
risque, car il donne la variation du risque par unité de temps et peut donc être supérieur à 1. On
montre que R(∆t)=1‐exp(‐TI∆t), ce qui peut se simplifier en R(∆t)≈TI∆t, si TI est petit.
Un facteur de risque est une caractéristique individuelle qui permet de partager la
population en catégories présentant des valeurs distinctes du risque. Cela n’implique pas
nécessairement que le facteur est la cause de la maladie. L’étude de l’association entre une
exposition (ou facteur de risque) et une maladie est une des étapes majeures de la recherche des
facteurs étiologiques de cette maladie. Elle utilise des indicateurs et des tests statistiques. Les
principaux indicateurs utilisés pour mesurer une association entre une exposition et une maladie
sont l’excès de risque, le risque relatif, et le rapport de cotes (odds ratios en anglais) (Tableau 2). Le
risque relatif, qui exprime le facteur par lequel le risque de maladie est multiplié en présence de
l’exposition, est aisément compréhensible. L’interprétation d’un rapport de cotes est moins
immédiate. Cependant, lorsque la maladie est rare, les 2 indices sont proches. Le rapport de cotes a
l’avantage de pouvoir être estimé dans tous les types d’études (en particulier les études cas‐témoins)
et d’avoir de meilleures propriétés mathématiques qui lui permettent d’être utilisé dans des modèles
logistiques multivariés. Dans les études de cohorte, c’est souvent le Hazard Ratio (HR) qui est
calculé. Il s’agit d’une approximation du risque relatif, obtenu par un modèle de survie multivarié
(modèle de Cox).
Le risque relatif et le rapport de cotes mesurent la force du lien entre un facteur de risque et
la maladie au niveau individuel, mais n’indiquent pas l’importance du facteur de risque au niveau de
la population. La fraction de risque attribuable (RA) mesure la proportion des cas attribuable à
5
l’exposition étudiée. Cependant, la notion de cas « attribuables » n’a de sens que si l’exposition est
un agent causal de la maladie. Cette fraction dépend à la fois de la valeur du risque relatif et de la
fréquence de l’exposition dans la population (Tableau 2).
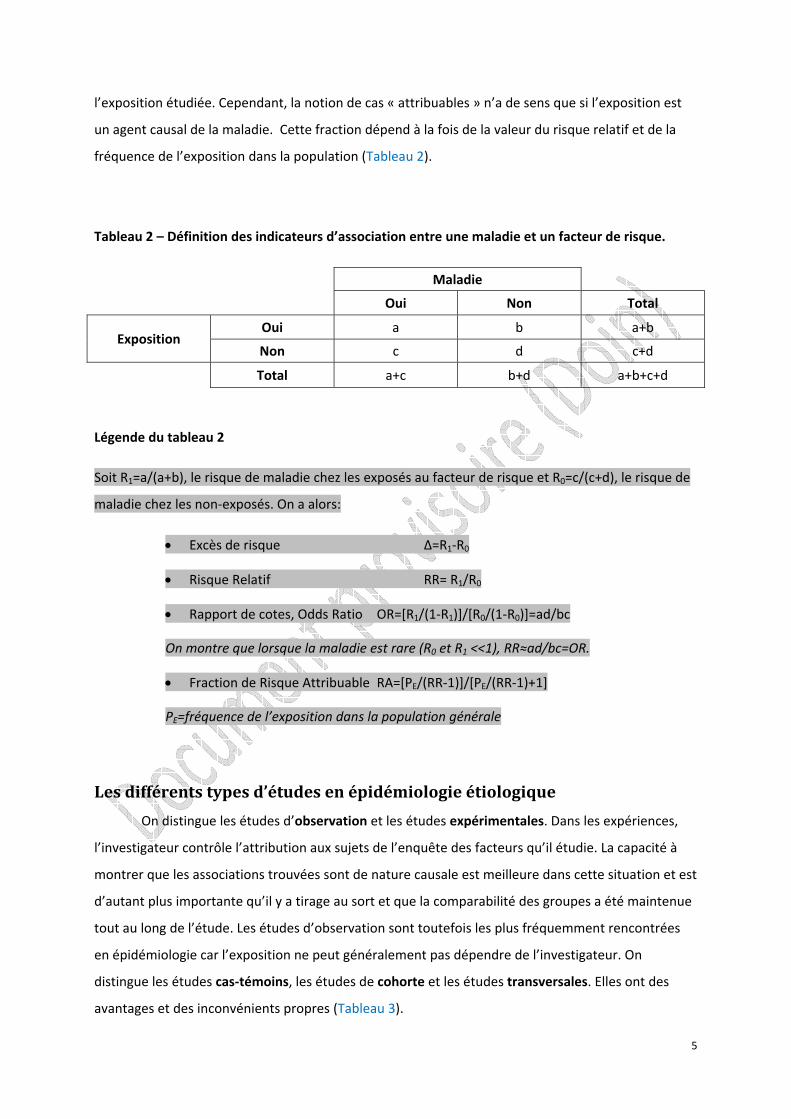
Tableau 2 – Définition des indicateurs d’association entre une maladie et un facteur de risque.
Maladie
Oui Non Total
Exposition Oui a b a+b
Non c d c+d
Total a+c b+d a+b+c+d
Légende du tableau 2
Soit R1=a/(a+b), le risque de maladie chez les exposés au facteur de risque et R0=c/(c+d), le risque de
maladie chez les non‐exposés. On a alors:
• Excès de risque ∆=R1‐R0
• Risque Relatif RR= R1/R0
• Rapport de cotes, Odds Ratio OR=[R1/(1‐R1)]/[R0/(1‐R0)]=ad/bc
On montre que lorsque la maladie est rare (R0 et R1 <<1), RR≈ad/bc=OR.
• Fraction de Risque Attribuable RA=[PE/(RR‐1)]/[PE/(RR‐1)+1]
PE=fréquence de l’exposition dans la population générale
Les différents types d’études en épidémiologie étiologique
On distingue les études d’observation et les études expérimentales. Dans les expériences,
l’investigateur contrôle l’attribution aux sujets de l’enquête des facteurs qu’il étudie. La capacité à
montrer que les associations trouvées sont de nature causale est meilleure dans cette situation et est
d’autant plus importante qu’il y a tirage au sort et que la comparabilité des groupes a été maintenue
tout au long de l’étude. Les études d’observation sont toutefois les plus fréquemment rencontrées
en épidémiologie car l’exposition ne peut généralement pas dépendre de l’investigateur. On
distingue les études cas‐témoins, les études de cohorte et les études transversales. Elles ont des
avantages et des inconvénients propres (Tableau 3).
6
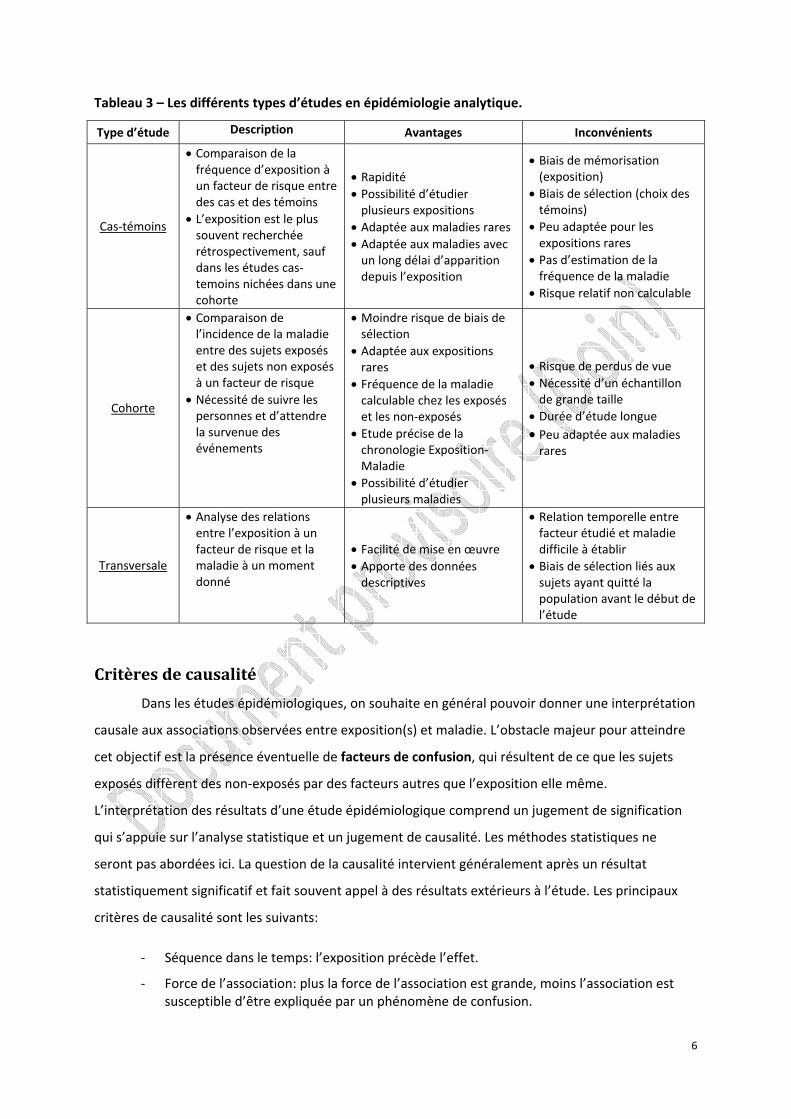
Tableau 3 – Les différents types d’études en épidémiologie analytique.
Type d’étude Description Avantages Inconvénients
Cas‐témoins
• Comparaison de la fréquence d’exposition à un facteur de risque entre des cas et des témoins
• L’exposition est le plus souvent recherchée rétrospectivement, sauf dans les études cas‐temoins nichées dans une cohorte
• Rapidité • Possibilité d’étudier plusieurs expositions
• Adaptée aux maladies rares • Adaptée aux maladies avec un long délai d’apparition depuis l’exposition
• Biais de mémorisation (exposition)
• Biais de sélection (choix des témoins)
• Peu adaptée pour les expositions rares
• Pas d’estimation de la fréquence de la maladie
• Risque relatif non calculable
Cohorte
• Comparaison de l’incidence de la maladie entre des sujets exposés et des sujets non exposés à un facteur de risque
• Nécessité de suivre les personnes et d’attendre la survenue des événements
• Moindre risque de biais de sélection
• Adaptée aux expositions rares
• Fréquence de la maladie calculable chez les exposés et les non‐exposés
• Etude précise de la chronologie Exposition‐Maladie
• Possibilité d’étudier plusieurs maladies
• Risque de perdus de vue • Nécessité d’un échantillon de grande taille
• Durée d’étude longue • Peu adaptée aux maladies rares
Transversale
• Analyse des relations entre l’exposition à un facteur de risque et la maladie à un moment donné
• Facilité de mise en œuvre • Apporte des données descriptives
• Relation temporelle entre facteur étudié et maladie difficile à établir
• Biais de sélection liés aux sujets ayant quitté la population avant le début de l’étude
Critères de causalité
Dans les études épidémiologiques, on souhaite en général pouvoir donner une interprétation
causale aux associations observées entre exposition(s) et maladie. L’obstacle majeur pour atteindre
cet objectif est la présence éventuelle de facteurs de confusion, qui résultent de ce que les sujets
exposés diffèrent des non‐exposés par des facteurs autres que l’exposition elle même.
L’interprétation des résultats d’une étude épidémiologique comprend un jugement de signification
qui s’appuie sur l’analyse statistique et un jugement de causalité. Les méthodes statistiques ne
seront pas abordées ici. La question de la causalité intervient généralement après un résultat
statistiquement significatif et fait souvent appel à des résultats extérieurs à l’étude. Les principaux
critères de causalité sont les suivants:
‐ Séquence dans le temps: l’exposition précède l’effet.
‐ Force de l’association: plus la force de l’association est grande, moins l’association est susceptible d’être expliquée par un phénomène de confusion.
7
‐ Spécificité de la cause et de l’effet: le facteur étudié est présent chez tous les malades (ou presque) et seulement chez eux, et que pour cette maladie.
‐ Relation de type « dose‐effet » entre l’exposition et la fréquence de la maladie.
‐ Prise en compte des facteurs de confusion et minimisation des biais de classement et de sélection.
‐ Constance de l’association et reproductibilité dans diverses situations.
‐ Plausibilité biologique et cohérence avec les connaissances générales.
‐ Parallélisme de distribution (dans l’espace et dans le temps) du facteur et de la maladie.
‐ En cas d’expérience randomisée, la suppression du facteur de risque fait diminuer ou disparaître le risque de maladie.
Facteurs de risque non modifiables des AVC Bien que non modifiables, ces facteurs sont importants pour identifier les personnes à haut
risque d’AVC. L’âge est le facteur de risque non modifiable le plus puissant, puisqu’on estime que le
risque d’AVC double chaque décennie après 55 ans.1, 19 A la différence de ce qui est observé dans la
maladie coronaire, le sexe masculin n’est pas un facteur de risque majeur d’infarctus cérébral ou
d’AIT. Le taux d’incidence des infarctus cérébraux (IC) est un peu plus élevé chez l’homme dans les
tranches d’âge de moins de 75 ans, mais la tendance s’inverse ensuite.19 Cette tendance n’a toutefois
pas été observée partout (voir chapitre épidémiologie descriptive). De plus, puisque les femmes ont
une plus grande espérance de vie, le nombre absolu d’IC et d’AIT est plus important chez les femmes
que chez les hommes. Le sexe masculin est associé à un risque accru d’hémorragie cérébrale (HC),17
alors que l’hémorragie sous‐arachnoïdienne (HSA) est plus fréquente chez la femme.20 Les sujets de
race noire ont un risque accru d’AVC. Aux USA, le risque d’AVC est 2 fois plus élevé chez les sujets de
race noire avec une incidence ajustée sur l’âge de 6.6 pour 1000 chez les hommes de race noire, de
3.6 chez les hommes de race blanche, de 4.9 chez les femmes de race noire, et 2.3 chez les femmes
de race blanche. De même, les hispano‐américains ont 2 fois plus de risque d’IC ou d’HC que les non
hispaniques.1 Il est cependant souvent difficile de séparer le rôle des facteurs raciaux de celui des
facteurs socio‐économiques et culturels, qui peuvent être à l’origine de différences dans la
prévalence des facteurs de risque.21
Des études conduites notamment en Angleterre et au Pays de Galles ont observé une
corrélation entre un petit poids de naissance et un risque accru de mortalité par AVC à l’âge adulte,
suggérant qu’il pourrait y avoir des phénomènes de « programmation » du risque vasculaire pendant
la vie fœtale.22 Cependant, les rôles respectifs des facteurs génétiques et environnementaux restent
indéterminés et le caractère causal de l’association non prouvé.
8
Les facteurs familiaux pourraient en fait être classés en potentiellement modifiables. Un
antécédent familial paternel ou maternel d’AVC constitue un facteur de risque d’accident ischémique
cérébral , d’HC, ou d’HSA, multipliant par environ 2 le risque.3, 20, 23 Il existe des maladies
mendéliennes responsables de formes familiales d’AVC, mais ces maladies sont très rares et ne
permettent d’expliquer qu’une très faible partie des AVC. En l’absence de maladie génétique
familiale, l’association entre AVC et antécédent familial d’AVC pourrait être expliquée par l’hérédité
des facteurs de risque vasculaire (notamment hypertension artérielle, diabète, et fibrillation
auriculaire), ou d’une susceptibilité à développer un AVC en présence de ces facteurs; par des
facteurs environnementaux communs au sein d’une famille; et par des interactions entre facteurs
génétiques et environnementaux. Il pourrait par ailleurs exister des interactions complexes avec le
sexe. Il a ainsi été montré que les femmes ayant un IC ou un AIT avaient plus souvent un antécédent
maternel que paternel d’AVC, alors que chez les hommes, les antécédents maternels et paternels
avaient la même prévalence.24 Cette relation, difficilement attribuable à une transmission génétique
classique, pourrait être expliquée par des interactions complexes entre le sexe du probant et des
facteurs épigénétiques (modifications de l’expression des gènes qui ne résultent pas de modifications
de l’ADN) et/ou des facteurs non génétiques (environnement intra‐utérin ou exposition à des
facteurs environnementaux pendant la petite enfance).24
En dehors des rares maladies monogéniques, le rôle des facteurs génétiques est beaucoup
plus difficile à établir. Des généticiens ont fait l’hypothèse que l’AVC est un modèle de maladie
polygénique, dans lequel de nombreux gènes agiraient de façon synergique.25 Chacun de ses facteurs
génétiques n’aurait cependant qu’une influence faible individuellement. Ces facteurs génétiques
pourraient prédisposer aux facteurs de risque vasculaire, aux causes des AVC (athérosclérose,
cardiopathies…), à une susceptibilité à l’ischémie ou l’hémorragie, ou à la survenue d’un AVC en cas
d’exposition à un autre facteur de risque (interactions gène‐environnement) ou à un autre gène
(interaction gène‐gène). De très nombreuses études ont été conduites pour rechercher des
associations entre des polymorphismes génétiques et les AVC.26 Cependant, les relations mises en
évidence dans les études d’association étaient souvent faibles et surtout, les résultats ont rarement
pu être répliqués dans des populations indépendantes. Il s’agit majoritairement d’études cas‐
témoins portant généralement sur de petits effectifs, et il existe de nombreux biais de publications
car les études négatives sont rarement publiées. De plus, de nombreuses études n’ont pas pris en
compte l’hétérogénéité des AVC. Les techniques modernes de criblage complet du génome n’ont
pas, pour l’instant, permis de mieux identifier des polymorphismes associés de façon convaincante à
un risque accru d’AVC.25 Les différentes études dans les IC ont examiné les gènes des protéines
impliquées dans l’hémostase, le système rénine‐angiotensine, le métabolisme monoxyde d’azote, le
9
métabolisme de l’homocystéine, le métabolisme lipidique, et l’inflammation.26, 27 Dans les HC, des
études, peu nombreuses, ont montré une association avec des polymorphismes de gènes impliqués
dans l’hémostase et du gène de l’apoE.28 Les principaux gènes candidats incriminés dans la genèse
des anévrismes intracrâniens sont ceux impliqués dans les protéines de la matrice extracellulaire.20
Facteurs de risque modifiables ou potentiellement modifiables
Pression artérielle
Il existe une relation continue entre le niveau de pression artérielle (PA) systolique ou
diastolique et le risque d’IC et d’HC.4, 29 Une méta‐analyse a montré que cette relation, qui existe dès
115/75 mmHg, est loglinéaire: chaque augmentation de la PA systolique de 20 mmHg ou de la PA
diastolique de 10 mmHg est associée à un doublement du risque d’AVC, quel que soit l’âge (figure
1).4 Or, chez un sujet non hypertendu à 55 ans, le risque de développer une HTA durant le reste de sa
vie (définie par une PA ≥ 140/90 mmHg) est d’environ 90%.29 C’est dire que la fraction de risque
d’AVC attribuable à une PA élevée est très importante. Le Joint National Committee on Prevention,
Detection, Evaluation, and Treatment of High Blood pressure a ainsi redéfini les différents stades
d’HTA, avec une PA normale < 120/80 mmHg (tableau 4). En plus de l’élévation, la variabilité de la
PA, en particulier nocturne, pourrait aussi constituer un facteur de risque indépendant d’AVC.
Cependant, les mécanismes de cette association restent mal compris et la causalité de la relation non
démontrée.29 Il existe aussi une association entre PA élevée et risque d’HSA, l’hypertension artérielle
multipliant le risque par 3 environ, dans les études cas‐témoins, comme dans les études de cohorte.18
Le bénéfice d’abaisser la PA sur le risque d’IC ou d’HC a été très largement démontré, aussi bien en
prévention primaire qu’en prévention secondaire.
10
Figure 1 – Mortalité (en échelle logarithmique) par AVC dans chaque décennie d’âge en fonction de la PA systolique (A) ou diastolique (B) usuelle. (Lancet, 2002;360:1903–1913).
Tableau 4 – Classification des niveaux de pression artérielle (JNC7).29
PA Systolique/Diastolique (mmHg) Stade <120/80 Normal
Alors qu’il existe une relation très claire, continue, et indépendante de l’âge entre élévation
du cholestérol et risque d’infarctus du myocarde, les relations entre cholestérol et AVC sont
complexes et encore incomplètement comprises. Il existe en effet une contradiction apparente entre
les résultats des études épidémiologiques et ceux des essais thérapeutiques sur les statines.30, 31
Une méta‐analyse conduite sur données individuelles de 61 études épidémiologiques
(environ 90000 sujets) a montré qu’il existait une association entre cholestérol élevé et risque de
mort par AVC.31 Cependant, cette relation est faible et présente uniquement pour les IC et chez des
sujets d’âge moyen (40‐69 ans). Il existe une relation inverse avec le risque d’HC (figure 2). Cette
approche présente toutefois plusieurs limites. Tout d’abord, ces études ont considéré uniquement
les AVC mortels, ce qui a entraîné une surreprésentation des HC (majoritaires dans les quelques
études où ce renseignement était disponible). De plus, même si l’on considère uniquement les IC, il
est possible que les IC mortels soient moins fortement associés au cholestérol, en raison de causes
particulières. Enfin, l’hétérogénéité étiologique des IC n’a été prise en compte dans aucune de ces
études. D’autres études, dont les données individuelles n’ont pas été incluses dans la méta‐analyse
sus‐citée, étayent la relation entre cholestérol et IC.30, 32 Par exemple, l’étude Multiple Risk Factor
Interven
cholesté
femmes
associati
artères,
différent
du taux d
le niveau
une rédu
relation
L
d’IC ont
riche en
et risque
triglycér
contradi
métabol
une mét
Figure 2 A – Relatio
en échelle
B – Relatio
et cholesté
cholestéro
ntion Trial (M
érol et IC mo
< 55 ans con
ion semble p
mais peu d’é
tes populatio
de cholestér
u initial du LD
uction relativ
entre choles
Les quelques
pour l’instan
protéines, e
e d’IC,5 qui p
ides comme
ctoires.1 En f
ique. Une re
ta‐analyse.18
– Mortalité on entre AVC et
e logarithmique
on entre AVC (s
érol. Les résulta
ol.
MRFIT), condu
rtel et une re
nforte égalem
plus forte da
études ont e
ons à risque
rol (LDL en pa
DL cholestér
ve du risque
stérol et IC.
s études con
nt montré de
en particulier
ourrait être
facteur de r
fait, l’hypert
elation invers
par AVC en t cholestérol da
e.
séparé en infarc
ats sont présen
uite chez des
elation inver
ment la relat
ns les accide
examiné cette
d’événemen
articulier) ét
ol, une dimin
d’IC de 19%
sacrées spéc
es résultats c
r l’ApoA1. Il e
plus forte da
risque d’IC es
triglycéridém
se entre hyp
fonction duans différentes
ctus cérébral, h
ntés en réductio
s hommes, a
rse avec les H
tion entre ch
ents liés à l’at
e question. E
nts vasculaire
tait associé à
nution de 1 m
.34 Tous ces
cifiquement
contradictoir
existe une re
ans les accide
st incertain c
mie s’intègre
ercholestéro
taux de choclasses d’âge. L
hémorragie intr
on relative de r
a montré une
HC mortelles
holestérol et
théroscléros
Enfin, les ess
es ont clairem
à une réducti
mmol/L du L
arguments s
aux relation
res.1 Le HDL c
elation invers
ents liés à l’a
car les étude
très souvent
olémie et risq
olestérol usuLes risques rela
a‐cérébrale, hé
isque pour un a
e association
s.32 Une étud
infarctus cé
se ou dans la
sais thérapeu
ment montr
ion du risque
LDL cholestér
sont donc bie
s entre LDL c
cholestérol e
se entre taux
athéroscléro
es montrent d
t dans le cad
que d’HSA a
uel. (Lancet, 20atifs (hazard rat
émorragie méni
abaissement de
n entre éléva
de conduite c
rébral.33 Cet
a maladie des
utiques mené
é qu’un abai
e d’IC. Quel q
rol est assoc
en en faveur
cholestérol e
est une fract
x de HDL cho
se. Le rôle d
des résultats
dre d’un synd
été suggéré
007;370:1829–1tios ici) sont rep
ingée et AVC no
e 1 mmol/L du t
11
tion du
chez des
te
s petites
és dans
issement
que soit
iée à
d’une
et risque
tion très
olestérol
es
s
drome
e par
839). présentés
on classé)
taux de
12
La lipoprotéine a [Lp(a)] est un complexe lipide‐protéine qui ressemble aux particules LDL.
Elle aurait une action pro‐athérogène, en favorisant le dépôt du cholestérol dans la paroi artérielle,
et prothrombotique, en raison d’une homologie de séquence avec le plasminogène.35 Les
importantes variations inter‐individuelles de la concentration de Lp(a) sont expliquées par des
polymorphismes dans le gène de l’apoprotéine (a). Alors qu’il existe une association assez forte entre
taux élevé de Lp(a) et maladie coronaire,36 la relation avec les accidents ischémiques cérébraux
semble plus faible (RR=1.22 ; IC95%: 1.04‐1.43) et surtout démontrée dans des études cas‐témoins.6
Le taux de Lp(a) peut être réduit d’environ 25% par l’acide nicotinique, mais il n’y pas d’étude
randomisée ayant démontré une réduction du risque par ce traitement.1
L’enzyme « lipoprotein‐associated phospholipase A2 » (Lp‐PLA2), sécrétée par les
macrophages, est liée au LDL circulant. Elle semble impliquée dans l’oxydation des particules LDL et
un taux élevé de Lp‐PLA2 pourrait être un marqueur de l’instabilité des plaques d’athérosclérose. Une
association entre taux élevé de Lp‐PLA2 et événement coronaire a été observée dans plusieurs
cohortes.37 Dans l’ensemble, les quelques études consacrées aux IC montrent une association entre
un taux élevé de Lp‐PLA2 et IC. Chez les femmes, une étude a suggéré que cette association ne serait
présente qu’en l’absence de THS.38 Le gemfibrozil diminue légèrement le taux de Lp‐PLA2, mais l’effet
de ce traitement sur le risque cérébrovasculaire est inconnu.
Diabète et hyperglycémie
Plusieurs études épidémiologiques montrent que la prévalence mondiale du diabète
augmente. Ainsi, on estime que la prévalence du diabète dans le monde sera de 6,3% en 2025 contre
5,1% en 2003 et celle de l’intolérance au glucose de 9,0% contre 8,2%. Le diabète de type 2 est un
facteur de risque bien établi d’IC, multipliant le risque par un facteur allant de 1.8 à 6.0 selon les
études.39 La relation entre diabète et IC semble plus forte chez la femme que chez l’homme, sans que
cette différence soit clairement expliquée.39 Chez les patients diabétiques (type I ou II), il existe une
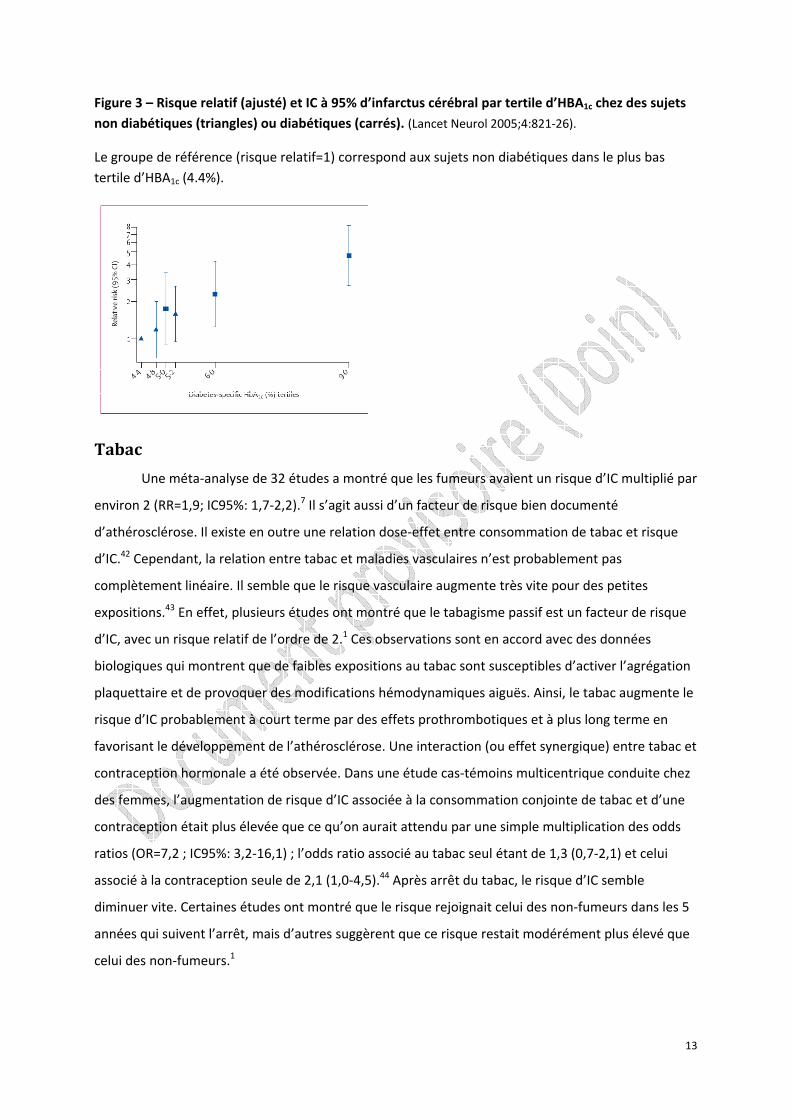
relation loglinéaire entre le niveau d’hémoglobine glyquée (HBA1c) et le risque d’IC.40 Le risque d’IC
semble aussi augmenté chez sujets ayant une hyperglycémie sans diabète avéré. Dans l’étude ARIC
(Atherosclerosis Risk in Communities), il a été observé une relation continue entre le taux d’HBA1c et
le risque d’IC que le sujet soit diabétique ou non (Figure 3).40
Bien que le bénéfice du bon contrôle glycémique chez les patients diabétiques sur la
réduction du risque d’événements macrovasculaires a été longtemps discuté, une méta‐analyse a
montré que ce bénéfice existait, en particulier chez les diabétiques de type I.41
13
Figure 3 – Risque relatif (ajusté) et IC à 95% d’infarctus cérébral par tertile d’HBA1c chez des sujets non diabétiques (triangles) ou diabétiques (carrés). (Lancet Neurol 2005;4:821‐26).
Le groupe de référence (risque relatif=1) correspond aux sujets non diabétiques dans le plus bas tertile d’HBA1c (4.4%).
Tabac
Une méta‐analyse de 32 études a montré que les fumeurs avaient un risque d’IC multiplié par
environ 2 (RR=1,9; IC95%: 1,7‐2,2).7 Il s’agit aussi d’un facteur de risque bien documenté
d’athérosclérose. Il existe en outre une relation dose‐effet entre consommation de tabac et risque
d’IC.42 Cependant, la relation entre tabac et maladies vasculaires n’est probablement pas
complètement linéaire. Il semble que le risque vasculaire augmente très vite pour des petites
expositions.43 En effet, plusieurs études ont montré que le tabagisme passif est un facteur de risque
d’IC, avec un risque relatif de l’ordre de 2.1 Ces observations sont en accord avec des données
biologiques qui montrent que de faibles expositions au tabac sont susceptibles d’activer l’agrégation
plaquettaire et de provoquer des modifications hémodynamiques aiguës. Ainsi, le tabac augmente le
risque d’IC probablement à court terme par des effets prothrombotiques et à plus long terme en
favorisant le développement de l’athérosclérose. Une interaction (ou effet synergique) entre tabac et
contraception hormonale a été observée. Dans une étude cas‐témoins multicentrique conduite chez
des femmes, l’augmentation de risque d’IC associée à la consommation conjointe de tabac et d’une
contraception était plus élevée que ce qu’on aurait attendu par une simple multiplication des odds
ratios (OR=7,2 ; IC95%: 3,2‐16,1) ; l’odds ratio associé au tabac seul étant de 1,3 (0,7‐2,1) et celui
associé à la contraception seule de 2,1 (1,0‐4,5).44 Après arrêt du tabac, le risque d’IC semble
diminuer vite. Certaines études ont montré que le risque rejoignait celui des non‐fumeurs dans les 5
années qui suivent l’arrêt, mais d’autres suggèrent que ce risque restait modérément plus élevé que
celui des non‐fumeurs.1
14
Une relation entre tabagisme et HC a été inconstamment observée et était de faible
amplitude lorsqu’elle était présente. Une méta‐analyse a également montré des résultats
discordants avec un RR allant de 1.06 (0.89‐1.26) à 1.36 (1.07‐1.73).17 Il a été suggéré que les
discordances entre les études pourraient venir de la méthode de mesure du tabagisme, avec une
relation plus constante dans les études qui ont quantifié le nombre de paquets‐années.45 Si une
association entre tabagisme et HC existe, elle est donc probablement faible.
Le tabagisme est aussi un facteur de risque majeur d’HSA, multipliant le risque par 2 à 3, avec
une relation dose‐effet.7, 18 Il constitue ainsi le principal facteur de risque modifiable de cette
maladie.
Alcool
Une méta‐analyse de 35 études observationnelles a montré qu’une consommation régulière
d’alcool >60 g/J est associé a un risque accru d’IC (RR=1.7 ; IC95%: 1.3‐2.2) et d’AVC hémorragique
(incluant ici HC et HSA) (RR=2.2 ; IC95%: 1.5‐3.2) par rapport à des sujets abstinents.11 Il existe une
relation dose‐effet pour des consommations élevées, notamment pour les HC.17 Comme dans la
maladie coronaire, une consommation régulière modérée d’alcool (12‐24 g/J) est associée à une
diminution du risque d’IC (RR=0,7 ; IC95%: 0,6‐0,9).11 La relation en J entre alcool et IC semble
indépendante du type d’alcool, même si l’effet protecteur d’une consommation modérée semble
moindre avec la bière.46 Cette relation a une certaine plausibilité biologique car l’alcool à petites
doses diminue l’agrégation plaquettaire, augmente le taux de HDL cholestérol, et diminue le taux de
fibrinogène plasmatique. Il n’existe cependant pas d’étude d’intervention démontrant cette relation
et une consommation modérée d’alcool n’est pas associée à une diminution du risque d’HC.
Obésité
L’obésité est classiquement définie par index de masse corporel (IMC) (poids divisé par la
taille au carré) ≥ 30 kg/m2 et le surpoids par un IMC compris entre 25 et 29,9 kg/m2. L’obésité
abdominale est mesurée par le rapport tour de taille (ou périmètre abdominal) sur tour de hanches
ou par le tour de taille seul. L’obésité abdominale est définie par un tour de taille > 102 cm chez les
hommes et > 88 cm chez les femmes. La prévalence de l’obésité (quelle que soit la définition) ne
cesse d’augmenter dans les pays développés.1 Aux Etats‐Unis, elle est passée de 12% en 1991 à
19,8% en 2000 et 20,9% en 2001, affectant particulièrement les populations noires et hispaniques.
Les obèses ont une pression artérielle, un cholestérol plasmatique, un taux de triglycérides, une
15
glycémie, et un hématocrite plus élevés que les sujets non obèses,47 ce qui peut rendre difficile
l’interprétation des études si les analyses n’ont pas été ajustées sur ces facteurs.
Dans l’ensemble, les études montrent une association entre obésité et IC chez l’homme et
chez la femme.48, 49 La majorité des études suggère que le risque d’IC augmente de façon continue
avec l’IMC. Dans une étude Coréenne ayant porté sur 234863 sujets, le risque d’IC augmentait de
façon linéaire avec l’IMC (RR=1,06 ; IC95%: 1,04‐1,07 pour 1 kg/m2).50 Les données sur la relation
entre obésité abdominale et IC sont plus limitées. Des études ont suggéré que le tour de taille
pourrait être un facteur de risque plus puissant que l’IMC.1 Cependant, une étude finlandaise a
suggéré que cette relation ne serait présente que chez l’homme.48 A ce jour, il n’existe pas d’étude
d’intervention ayant évalué le bénéfice d’une perte de poids sur le risque d’IC. Cependant, il est
clairement montré qu’une diminution de poids est associée à une réduction de la PA.1
Une relation entre poids et HC a été inconstamment observée. Cependant, cette relation ne
semble pas linéaire, les sujets très maigres ayant aussi un risque accru d’HC, suggérant une relation
en J.48, 50
Syndrome métabolique
Le syndrome métabolique est une entité clinique et biologique définie par l’association de
plusieurs facteurs de risque: obésité abdominale, dyslipidémie, insulino‐résistance et hypertension
artérielle. Ces paramètres sont influencés à des degrés divers par des facteurs environnementaux,
comme l’alimentation et l’activité physique, et des facteurs génétiques qui en favorisent l’expression
clinique. L’insulino‐résistance et l’obésité abdominale sont vraisemblablement au centre du
processus physiopathologique, mais la cause de ce syndrome reste inconnue. Il existe actuellement
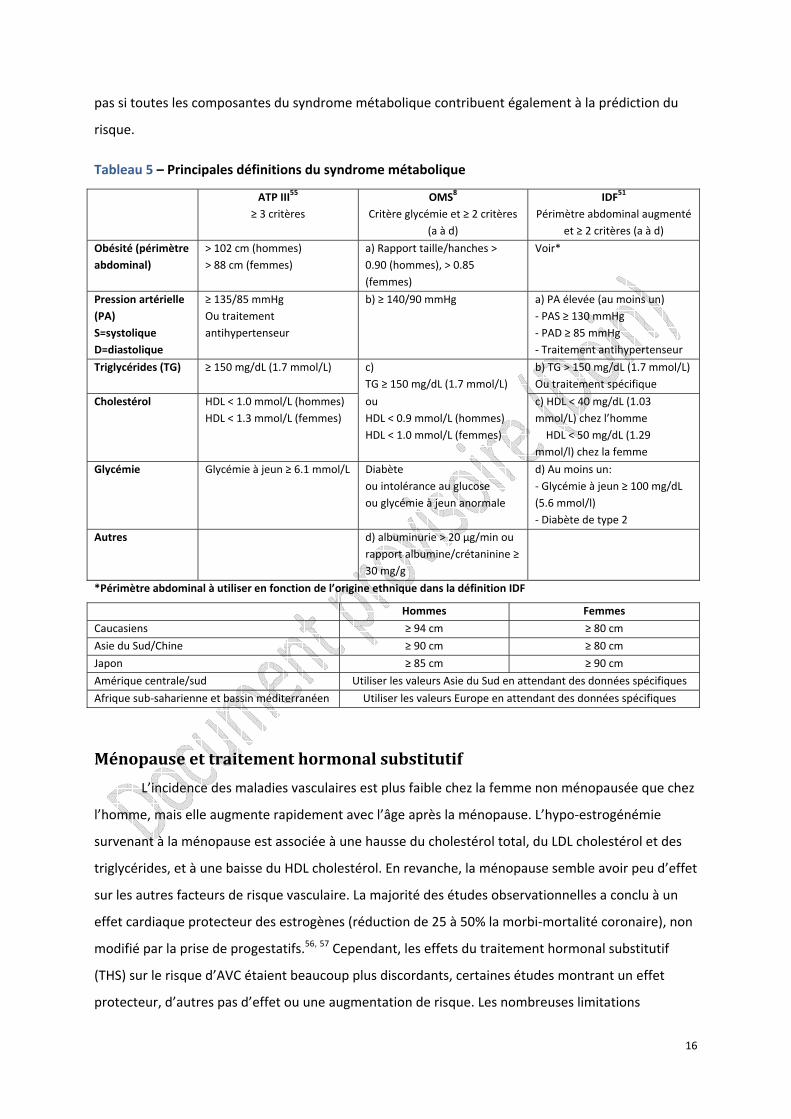
plusieurs définitions du syndrome métabolique, les principales étant celle de l’OMS, celle de l’Institut
National de Recherche en Santé Américain (NCEP), et celle de la Fédération Internationale du
Diabète (IDF) (tableau 5).51, 52 Le syndrome métabolique est un facteur de risque de diabète de type 2
et d’événements cardiovasculaires, incluant les IC. Cependant, la question reste ouverte de savoir si
le syndrome métabolique est authentiquement un syndrome, justifiant le regroupement de ces
différents facteurs de risque dans une même entité, ou si le syndrome métabolique n’est finalement
qu’une appellation qui n’apporte rien à la liste des différents facteurs de risque le constituant.53, 54 De
plus, l’existence de multiples définitions dont le rationnel n’est pas toujours clair et les résultats de
plusieurs études montrant le faible apport du syndrome dans la prédiction du risque cardiovasculaire
par rapport au score de Framingham ont remis en cause l’intérêt de ce syndrome.53 Enfin, on ne sait
16
pas si toutes les composantes du syndrome métabolique contribuent également à la prédiction du
risque.
Tableau 5 – Principales définitions du syndrome métabolique
ATP III55 ≥ 3 critères
OMS8 Critère glycémie et ≥ 2 critères
(a à d)
IDF51 Périmètre abdominal augmenté
et ≥ 2 critères (a à d)
Obésité (périmètre abdominal)
> 102 cm (hommes) > 88 cm (femmes)
a) Rapport taille/hanches > 0.90 (hommes), > 0.85 (femmes)
c) HDL < 40 mg/dL (1.03 mmol/L) chez l’homme HDL < 50 mg/dL (1.29 mmol/l) chez la femme
Glycémie Glycémie à jeun ≥ 6.1 mmol/L Diabète ou intolérance au glucose ou glycémie à jeun anormale
d) Au moins un: ‐ Glycémie à jeun ≥ 100 mg/dL (5.6 mmol/l) ‐ Diabète de type 2
Autres d) albuminurie > 20 µg/min ou rapport albumine/crétaninine ≥ 30 mg/g
*Périmètre abdominal à utiliser en fonction de l’origine ethnique dans la définition IDF
Hommes Femmes
Caucasiens ≥ 94 cm ≥ 80 cm
Asie du Sud/Chine ≥ 90 cm ≥ 80 cm
Japon ≥ 85 cm ≥ 90 cm
Amérique centrale/sud Utiliser les valeurs Asie du Sud en attendant des données spécifiques
Afrique sub‐saharienne et bassin méditerranéen Utiliser les valeurs Europe en attendant des données spécifiques
Ménopause et traitement hormonal substitutif
L’incidence des maladies vasculaires est plus faible chez la femme non ménopausée que chez
l’homme, mais elle augmente rapidement avec l’âge après la ménopause. L’hypo‐estrogénémie
survenant à la ménopause est associée à une hausse du cholestérol total, du LDL cholestérol et des
triglycérides, et à une baisse du HDL cholestérol. En revanche, la ménopause semble avoir peu d’effet
sur les autres facteurs de risque vasculaire. La majorité des études observationnelles a conclu à un
effet cardiaque protecteur des estrogènes (réduction de 25 à 50% la morbi‐mortalité coronaire), non
modifié par la prise de progestatifs.56, 57 Cependant, les effets du traitement hormonal substitutif
(THS) sur le risque d’AVC étaient beaucoup plus discordants, certaines études montrant un effet
protecteur, d’autres pas d’effet ou une augmentation de risque. Les nombreuses limitations
17
méthodologiques de ces études (absence de prise en compte de l’hétérogénéité des AVC, différences
dans la définition de l’exposition, absence de prise en compte des facteurs de confusion…) incitaient
toutefois à la prudence dans l’interprétation des résultats. Les études spécifiquement dédiées aux IC
avaient aussi montré des résultats discordants, avec 2 études de grande envergure (Framingham et la
Nurses’ Health Study) ayant montré que le THS augmentait le risque d’IC.
Les grands essais randomisés récents ont complètement remis en cause l’hypothèse que le
THS pourrait avoir un effet protecteur. Ils ont unanimement montré que THS augmentait le risque de
premier IC ou de récidive d’IC.57 Une méta‐analyse de 16 études randomisées montre que le THS
augmente de 29% (IC95% : 6 ‐56) le risque d’IC, alors que le risque d’AVC hémorragique et d’AIT n’est
pas modifié.10 Le THS serait aussi plus fortement associé aux accidents sévères.10 Dans l’étude WHI,
alors que le risque d’événements coronaires était plus élevé dans la première année de traitement
par THS, le risque d’IC n’apparaissait qu’après la première année.58 Cette étude a aussi montré que
l’augmentation de risque était indépendante de l’âge, des autres facteurs de risque vasculaire, des
traitements, de l’utilisation de progestatifs, et n’était pas expliqué par l’élévation de la PA observée
dans le groupe traité. Le THS estroprogestatif ou par estrogènes seuls est donc associé à un risque
accru d’IC. Ceci incite à ne pas prescrire de THS et à l’arrêter chez les femmes à haut risque
vasculaire.
Une relation inverse entre THS et HSA a été observée dans les études cas‐témoins.18
Alimentation
Il existe maintenant un bon nombre d’études observationnelles montrant que le risque
d’AVC est influencé par des facteurs alimentaires.59 La majorité des études prospectives montre
qu’une consommation importante de fruits et de légumes est associée une diminution du risque d’IC,
avec une relation dose‐effet. Dans l’analyse combinée de la Nurses’ Health Study et de la Health
Professionals’ Follow‐up Study, la consommation d’une part supplémentaire de fruit et de légume
par jour était associée à une diminution relative du risque d’IC de 3 à 5%.59 L’effet protecteur
semblait plus important pour les agrumes, les jus de fruits et les légumes crucifères ou verts. Les
quelques études qui ont examiné l’effet des fruits et légumes sur le risque HC ont montré des
résultats semblables à ceux observés pour les IC.59 Un régime riche en fruits et légumes et pauvre en
graisses, notamment saturées diminue la PA.1 Une consommation régulière de poisson (≥1 fois par
mois) est associée à une réduction de 40 à 50% du risque d’IC chez les hommes,60 et chez la femme.61
La relation avec les HC est incertaine.
18
Plusieurs études de cohorte ont montré une relation entre risque d’AVC et consommation
élevée de sodium ou consommation faible de potassium.62, 63 Il a aussi été observé une association
entre consommation de magnésium (notamment contenu dans les céréales complètes) et diminution
du risque d’IC chez les hommes.64 Cependant, ces études présentent des limites en raison des
difficultés de mesure de la consommation d’électrolytes. Les effets de la consommation de sodium,
de potassium, ou de magnésium semblent être, au moins en partie, expliqués par des effets sur la
pression artérielle. Il reste à démontrer que le risque d’IC peut être modifié par une modification de
la consommation de ces électrolytes.1 Il faut aussi souligner que les facteurs alimentaires sont
souvent très liés à d’autres éléments du style de vie, et qu’il est parfois difficile d’individualiser les
effets propres de ces différentes composantes du style de vie.
Activité physique
Une méta‐analyse de 31 études observationnelles a montré qu’une activité physique
régulière (dans un cadre professionnel ou de loisir) était associée à une réduction du risque d’IC
(RR=0,78 ; IC95%: 0,71‐0,85) et d’HC (RR=0,74 ; IC95%: 0,57‐0,96).9 Une relation dose‐effet est
généralement observée dans les études.9 Les effets bénéfiques d’une activité physique régulière
pourraient être notamment expliqués par une réduction de la PA, une perte de poids, et une
amélioration de la régulation de la glycémie.
Contraception orale
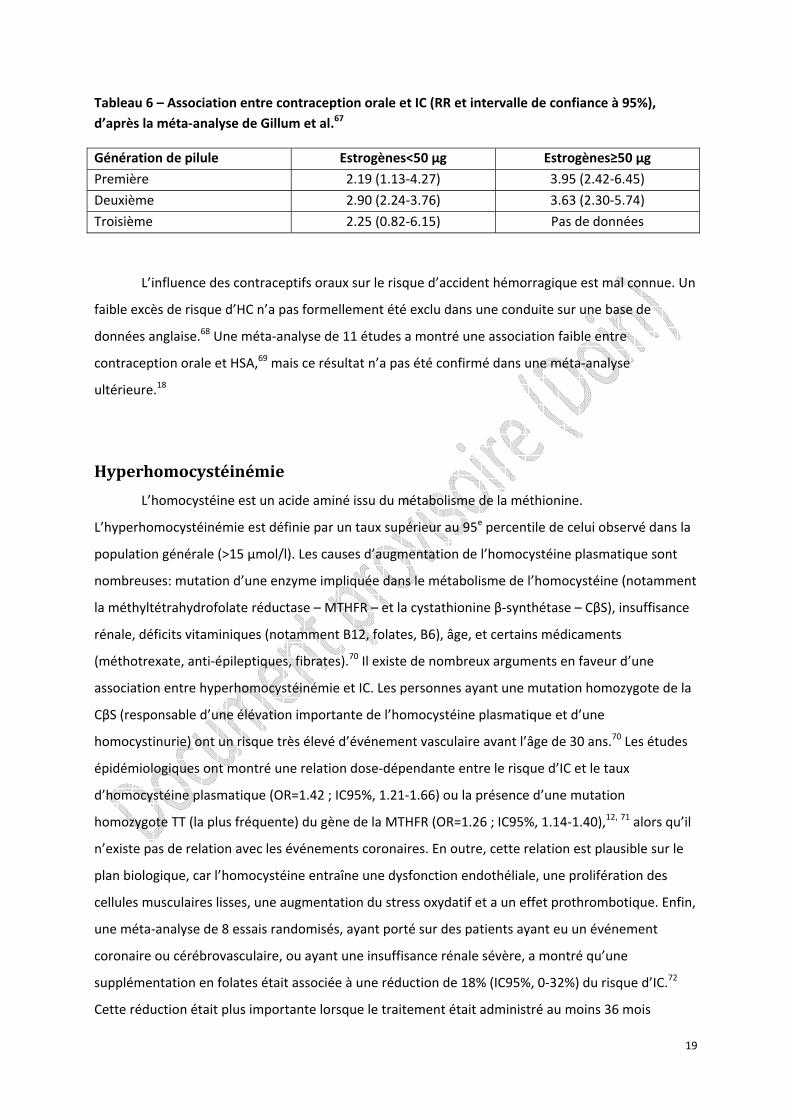
Les relations entre contraception orale et IC ont été largement étudiées.65 Deux méta‐
analyses ont montré que la contraception orale était associée à un risque accru d’IC.65 L’association
existe quelle que soit la génération de pilule et le dosage en estrogènes même si elle est plus faible
avec les pilules microdosées (< 50 µg d’estrogènes) (tableau 6). Cependant, compte tenu de la très
faible incidence de la maladie dans la tranche d’âge des femmes qui utilisent les contraceptifs oraux,
le risque attribuable aux contraceptifs et l’excès de risque absolu sont très faibles. L’absence de
différence entre les générations de pilules et les doses d’estrogènes a été aussi observée dans une
étude publiée après ces méta‐analyses.66 La force de l’association semble augmenter avec l’âge de la
femme, en présence d’un autre facteur de risque vasculaire, ou d’une anomalie congénitale d’une
protéine de la coagulation.44, 66 Les mécanismes d’action possibles des estrogènes sont une
augmentation de l’inflammation systémique (élévation du taux de CRP par exemple), des troubles de
la vasoréactivité, et une hypercoagulabilité.65
19
Tableau 6 – Association entre contraception orale et IC (RR et intervalle de confiance à 95%), d’après la méta‐analyse de Gillum et al.67
Génération de pilule Estrogènes<50 µg Estrogènes≥50 µg
Première 2.19 (1.13‐4.27) 3.95 (2.42‐6.45)
Deuxième 2.90 (2.24‐3.76) 3.63 (2.30‐5.74)
Troisième 2.25 (0.82‐6.15) Pas de données
L’influence des contraceptifs oraux sur le risque d’accident hémorragique est mal connue. Un
faible excès de risque d’HC n’a pas formellement été exclu dans une conduite sur une base de
données anglaise.68 Une méta‐analyse de 11 études a montré une association faible entre
contraception orale et HSA,69 mais ce résultat n’a pas été confirmé dans une méta‐analyse
ultérieure.18
Hyperhomocystéinémie
L’homocystéine est un acide aminé issu du métabolisme de la méthionine.
L’hyperhomocystéinémie est définie par un taux supérieur au 95e percentile de celui observé dans la
population générale (>15 µmol/l). Les causes d’augmentation de l’homocystéine plasmatique sont
nombreuses: mutation d’une enzyme impliquée dans le métabolisme de l’homocystéine (notamment
la méthyltétrahydrofolate réductase – MTHFR – et la cystathionine β‐synthétase – CβS), insuffisance
rénale, déficits vitaminiques (notamment B12, folates, B6), âge, et certains médicaments
(méthotrexate, anti‐épileptiques, fibrates).70 Il existe de nombreux arguments en faveur d’une
association entre hyperhomocystéinémie et IC. Les personnes ayant une mutation homozygote de la
CβS (responsable d’une élévation importante de l’homocystéine plasmatique et d’une
homocystinurie) ont un risque très élevé d’événement vasculaire avant l’âge de 30 ans.70 Les études
épidémiologiques ont montré une relation dose‐dépendante entre le risque d’IC et le taux
d’homocystéine plasmatique (OR=1.42 ; IC95%, 1.21‐1.66) ou la présence d’une mutation
homozygote TT (la plus fréquente) du gène de la MTHFR (OR=1.26 ; IC95%, 1.14‐1.40),12, 71 alors qu’il
n’existe pas de relation avec les événements coronaires. En outre, cette relation est plausible sur le
plan biologique, car l’homocystéine entraîne une dysfonction endothéliale, une prolifération des
cellules musculaires lisses, une augmentation du stress oxydatif et a un effet prothrombotique. Enfin,
une méta‐analyse de 8 essais randomisés, ayant porté sur des patients ayant eu un événement
coronaire ou cérébrovasculaire, ou ayant une insuffisance rénale sévère, a montré qu’une
supplémentation en folates était associée à une réduction de 18% (IC95%, 0‐32%) du risque d’IC.72
Cette réduction était plus importante lorsque le traitement était administré au moins 36 mois
20
(réduction de risque, 29%) et lorsqu’une diminution du taux d’homocystéine d’au moins 20% était
observée (réduction de risque, 23%), suggérant une possible relation dose‐effet.72 La réduction était
plus faible dans les études menées dans les pays où les céréales sont supplémentées en acides
foliques. Cependant, les intervalles de confiance des réductions de risque observées dans cette
méta‐analyse étaient larges et ce résultat contraste avec l’absence d’effet des folates sur le risque
d’événement coronaire.73 D’autres essais sont en cours.
Syndrome d’apnées obstructive du sommeil
Le syndrome d’apnées obstructives du sommeil (SAS) est défini sur une polysomnographie
par un index d’apnées‐hypopnées nocturnes augmenté et une somnolence diurne.74 Les relations
entre SAS et AVC restent controversées, notamment parce que les 2 affections surviennent sur le
même terrain, et qu’il est donc difficile de bien prendre en compte tous les facteurs de confusion
dans les analyses. Il existe une association entre SAS et élévation de la PA nocturne et diurne, avec
une relation plus forte chez les sujets présentant les index d’apnées les plus élevés. La causalité de la
relation entre SAS et HTA est attestée par la réduction de PA observée sous ventilation nocturne en
pression positive. Une association entre ronflement (défini par l’interrogatoire) et AVC a été
observée dans plusieurs études cas‐témoins ou de cohorte. Bien que peu nombreuses et plus
récentes, les études de cohorte ayant recherché le SAS par une polysomnographie ont toutes mis en
évidence une association (risque relatif entre 2 et 3) entre SAS et IC/AIT ou de décès, IDM et IC/AIT
combinés.75, 76 Les mécanismes physiopathologiques par lesquels le SAS pourrait augmenter le risque
d’IC/AIT sont probablement multiples et restent mal connus. En plus d’une élévation de la PA, le SAS
aurait des effets délétères sur l’hémodynamique et l’oxygénation cérébrale favoriserait la survenue
de troubles du rythme cardiaque, l’athérogenèse et la thrombose.74 La ventilation nocturne en
pression positive améliore l’index d’apnées, l’hypoxémie nocturne, la somnolence diurne, la qualité
de vie, et entraîne une réduction de la PA. Mais, il n’existe pas d’étude randomisée ayant évalué le
bénéfice de ce traitement sur le risque vasculaire.
Inflammation
Il existe de nombreux arguments biologiques et épidémiologiques montrant que les
processus inflammatoires jouent un rôle majeur dans le développement de l’athérosclérose et dans
la survenue de ses manifestations cliniques.1 Chaque étape du processus physiopathologique de
l’athérosclérose peut être considérée comme une réaction inflammatoire à une agression de la paroi
21
vasculaire. L’inflammation favoriserait aussi la survenue des complications athérothrombotiques
aiguës de l’athérosclérose. En effet, d’une part, les cellules de l’inflammation produisent des
molécules protéolytiques (en particulier les métalloprotéinases) dégradant la chape fibreuse qui,
devenant fine, est susceptible de se rompre. D’autre part, les macrophages produisent aussi des
facteurs procoagulants favorisant la thrombose.
Les relations entre protéine C‐réactive (CRP) et athérothrombose restent encore mal
comprises. Il existe une association entre élévation du taux de CRP et risque d’événement coronaire
ou cérébrovasculaire.77 Cependant, on ne sait pas si l’élévation de la CRP est un facteur de risque
d’athérosclérose ou simplement un marqueur d’une activité inflammatoire plus importante de la
maladie athérosclérose. De plus, les seuils de CRP associés à un risque accru d’événement au cours
de la maladie coronaire ne sont peut être pas applicables à l’accident ischémique cérébral. Il est donc
impossible actuellement d’identifier les personnes à risque d’accident ischémique cérébral sur la
base du taux de CRP. Enfin, bien que les statines diminuent le taux de CRP, il n’est pas démontré que
cet effet est indépendant d’un effet sur la maladie athéroscléreuse elle‐même et que cette
diminution est associée à une réduction du risque d’événement vasculaire. Ainsi, l’utilité de mesurer
la CRP pour estimer le risque d’accident ischémique cérébral reste à démontrer.77
L’élévation du fibrinogène plasmatique est un facteur de risque indépendant d’IC avec une
relation dose‐effet mais qui semble s’atténuer avec l’âge.78 Cependant, la causalité de la relation
reste non démontrée.
D’autres marqueurs de l’inflammation (system CD40/CD40 ligand, interleukines), des
molécules d’adhésion cellulaire, (P‐selectine, ICAM‐1, CD40L) et des métalloprotéinases (gelatenases,
stromelysine) ont été récemment identifiés comme de possibles nouveaux facteurs de risque
d’accident ischémique cérébral, en particulier lié à l’athérosclérose. Mais, les relations entre ces
facteurs et les facteurs de risque traditionnels, ainsi que les mécanismes physiopathologiques sous‐
jacents restent à mieux préciser.1
Infections chroniques
L’implication d’agents infectieux bactériens ou viraux dans la genèse et le développement de
l’athérosclérose est suspectée depuis très longtemps et des agents infectieux ont été identifiés dans
des plaques d’athérosclérose coronaires ou carotides.79 Les mécanismes potentiels sous‐tendant
cette relation sont un effet pro‐inflammatoire, une réaction auto‐immune, un effet procoagulant, et
des interactions avec les facteurs de risque traditionnels. Cependant, les études ayant évalué
22
l’association entre maladie coronaire ou IC et des stigmates d’une infection par des agents bactériens
(Chlamydia pneumoniae et Helicobacter pylori) ou par des agents viraux (Cytomegalovirus et Herpes
Virus de type 1) ont montré des résultats contradictoires ou une association faible.15, 80 Les études
disponibles sont sujettes à des biais de sélection (études cas‐témoins majoritairement), des biais de
publications, et vraisemblablement à un défaut de prise en compte de facteurs de confusion. De plus,
il n’y a pas de preuve qu’un traitement anti‐infectieux réduise le risque d’événement vasculaire.
Ainsi, la causalité de la relation reste incertaine. La maladie périodontique est autre une source
d’infection bactérienne chronique. Une méta‐analyse a montré que la maladie périodontique était
associée à un risque accru d’événements vasculaires (RR=1.19 ; IC95%: 1.08‐1.32).16 Bien que l’effet
soit globalement de faible ampleur, le risque attribuable pourrait être très élevé compte tenu de la
forte prévalence de la maladie périodontique dans la population générale (jusqu’à 40% aux USA). De
plus, cette méta‐analyse a suggéré une association plus forte avec les AVC (RR=2.85).
Drogues
Nombreux cas isolés et des petites séries ont suggéré une relation entre l’utilisation
d’amphétamines, de cocaïne, ou d’héroïne et la survenue d’un IC ou d’une HC et quelques études
cas‐témoins ou transversales ont affirmé cette association.13 La relation entre amphétamines et HC
semble particulièrement forte (OR de l’ordre de 5).13 Les mécanismes possibles sont une élévation
brutale de la PA associée à une perte de l’autorégulation du débit sanguin cérébral, un vasospasme,
le développement d’une vascularite, une hyperagrégabilité plaquettaire, ou une embolie d’origine
cardiaque (infarctus du myocarde, trouble du rythme, cardiomyopathie).81 Une relation entre
cannabis et AVC a été suggérée dans des petites séries et évaluée dans une seule étude transversale
qui a montré une association entre cannabis et IC (OR=1.76 ; 1.15‐2.71) ou HC (OR=1.36 ; 0.90‐
2.06).13 Les mécanismes physiopathologiques supposés sont le vasospasme, l’hypotension artérielle,
et la vascularite.82
Facteurs précipitants
Si les facteurs de risque classiques permettent d’identifier les patients à risque de développer
un AVC, voire d’estimer un risque absolu moyen, il reste impossible de prédire à quel moment un
accident va survenir. L’identification de facteurs précipitants ou déclenchants, c'est‐à‐dire de facteurs
pouvant modifier le risque de façon très transitoire (quelques minutes à quelques semaines) est une
voie de recherche très vaste.83 La liste des principaux facteurs précipitants est donnée dans le
23
tableau 7. Les facteurs précipitants les mieux documentés sont les infections aiguës qui augmentent
le risque d’IC ou d’AIT dans la semaine qui suit84 et une consommation aiguë d’alcool (dès 40 g) qui
augmente le risque d’AVC (IC ou HC) dans les 24 heures qui suivent.
Tableau 7 – Principaux facteurs précipitants.
Prise aiguë d’alcool Infection aiguë (respiratoire, ORL, ou urinaire) Accès de colère Evénements de vie sévères Pollution atmosphérique Variation importante de la température extérieure Arrêt de l’aspirine
Prédiction du risque d’AVC L’estimation du risque d’AVC au niveau des individus peut s’avérer utile pour décider les
mesures de prévention à mettre œuvre chez un individu donné et orienter les politiques de
prévention au niveau des populations. Ce risque peut être estimé en utilisant l’équation d’un modèle
de risque ou d’un score qui en est dérivé, dont les composants sont des facteurs de risque. Alors qu’il
existe plusieurs méthodes relativement valides pour estimer le risque d’événement coronaire ou
d’événements vasculaires tous confondus, les modèles de prédiction d’IC sont peu nombreux et ne
sont pas toujours transposables dans des populations différentes de celle dans laquelle ils ont été
développés. Le plus connu est le score de Framingham qui prend en compte séparément chez
l’homme et chez la femme: l’âge, la pression artérielle ajustée sur le traitement, le diabète, le
tabagisme, les antécédents vasculaires (non cérébrovasculaires), la fibrillation auriculaire, et
l’hypertrophie ventriculaire gauche sur l’ECG.1 Il n’existe pas de score de risque pour le HIS ou les
HSA.
Facteurs de risque et soustype d’AVC Comme le montre le tableau 1, les facteurs de risque actuellement identifiés des HC et des
HSA sont moins nombreux. Certains sont communs avec les accidents ischémiques.
L’importance relative des différents facteurs de risque en fonction du sous‐type d’accident
ischémique a été peu évaluée. L’âge, l’élévation de la PA, le diabète et le tabagisme semblent être
impliqués dans tous les sous‐types. Les sujets de race noire sont plus à risque de développer une
maladie des petites artères, même après ajustement sur les facteurs de risque.85 Plusieurs études ont
24
suggéré que l’hypertension artérielle et le diabète étaient plus fréquemment observés chez les
patients ayant un infarctus lacunaire. Cependant, la majorité de ces études considérait
l’hypertension et/ou le diabète dans la définition de la maladie des petites artères. C’est d’ailleurs le
cas dans la classification étiologique TOAST.
Conclusion L’AVC est une maladie hétérogène dont les causes sont très nombreuses. Malgré les
importants progrès réalisés au cours des dernières années dans la compréhension des mécanismes
et des causes des AVC, une large proportion des AVC reste inexpliquée. L’épidémiologie analytique
fait partie de l’arsenal des outils de recherche pour identifier de potentielles nouvelles causes. Il faut
garder à l’esprit que cette recherche nécessite souvent des grandes populations et est en
conséquence longue et coûteuse. L’interprétation causale des résultats d’une étude épidémiologique
n’est pas toujours aisée et doit prendre en compte la signification statistique, l’amplitude du résultat,
la plausibilité biologique et surtout le caractère reproductible du résultat.
25
Références 1. Goldstein LB, Adams R, Alberts MJ et al. Primary prevention of ischemic stroke: a guideline
from the American Heart Association/American Stroke Association Stroke Council: cosponsored by the Atherosclerotic Peripheral Vascular Disease Interdisciplinary Working Group; Cardiovascular Nursing Council; Clinical Cardiology Council; Nutrition, Physical Activity, and Metabolism Council; and the Quality of Care and Outcomes Research Interdisciplinary Working Group: the American Academy of Neurology affirms the value of this guideline. Stroke 2006;37:1583‐633.
2. Law MR, Wald NJ. Risk factor thresholds: their existence under scrutiny. BMJ 2002;324:1570‐6.
3. Flossmann E, Schulz UG, Rothwell PM. Systematic review of methods and results of studies of the genetic epidemiology of ischemic stroke. Stroke 2004;35:212‐27.
4. Lewington S, Clarke R, Qizilbash N, Peto R, Collins R. Age‐specific relevance of usual blood pressure to vascular mortality: a meta‐analysis of individual data for one million adults in 61 prospective studies. Lancet 2002;360:1903‐13.
5. Sanossian N, Saver JL, Navab M, Ovbiagele B. High‐density lipoprotein cholesterol: an emerging target for stroke treatment. Stroke 2007;38:1104‐9.
6. Smolders B, Lemmens R, Thijs V. Lipoprotein (a) and stroke: a meta‐analysis of observational studies. Stroke 2007;38:1959‐66.
7. Shinton R, Beevers G. Meta‐analysis of relation between cigarette smoking and stroke. BMJ 1989;298:789‐94.
8. Arenillas JF, Moro MA, Davalos A. The metabolic syndrome and stroke: potential treatment approaches. Stroke 2007;38:2196‐203.
9. Wendel‐Vos GC, Schuit AJ, Feskens EJ et al. Physical activity and stroke. A meta‐analysis of observational data. Int J Epidemiol 2004;33:787‐98.
10. Bath PM, Gray LJ. Association between hormone replacement therapy and subsequent stroke: a meta‐analysis. BMJ 2005;330:342.
11. Reynolds K, Lewis B, Nolen JD, Kinney GL, Sathya B, He J. Alcohol consumption and risk of stroke: a meta‐analysis. JAMA 2003;289:579‐88.
12. Wald DS, Wald NJ, Morris JK, Law M. Folic acid, homocysteine, and cardiovascular disease: judging causality in the face of inconclusive trial evidence. BMJ 2006;333:1114‐7.
13. Westover AN, McBride S, Haley RW. Stroke in young adults who abuse amphetamines or cocaine: a population‐based study of hospitalized patients. Arch Gen Psychiatry 2007;64:495‐502.
14. Kuo HK, Yen CJ, Chang CH, Kuo CK, Chen JH, Sorond F. Relation of C‐reactive protein to stroke, cognitive disorders, and depression in the general population: systematic review and meta‐analysis. Lancet Neurol 2005;4:371‐80.
26
15. Cremonini F, Gabrielli M, Gasbarrini G, Pola P, Gasbarrini A. The relationship between chronic H. pylori infection, CagA seropositivity and stroke: meta‐analysis. Atherosclerosis 2004;173:253‐9.
16. Janket SJ, Baird AE, Chuang SK, Jones JA. Meta‐analysis of periodontal disease and risk of coronary heart disease and stroke. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 2003;95:559‐69.
17. Ariesen MJ, Claus SP, Rinkel GJ, Algra A. Risk factors for intracerebral hemorrhage in the general population: a systematic review. Stroke 2003;34:2060‐5.
18. Feigin VL, Rinkel GJ, Lawes CM et al. Risk factors for subarachnoid hemorrhage: an updated systematic review of epidemiological studies. Stroke 2005;36:2773‐80.
19. Rothwell PM, Coull AJ, Silver LE et al. Population‐based study of event‐rate, incidence, case fatality, and mortality for all acute vascular events in all arterial territories (Oxford Vascular Study). Lancet 2005;366:1773‐83.
20. Ruigrok YM, Rinkel GJ, Wijmenga C. Genetics of intracranial aneurysms. Lancet Neurol 2005;4:179‐89.
21. Cox AM, McKevitt C, Rudd AG, Wolfe CD. Socioeconomic status and stroke. Lancet Neurol 2006;5:181‐8.
22. Barker DJ, Lackland DT. Prenatal influences on stroke mortality in England and Wales. Stroke 2003;34:1598‐602.
23. Alberts MJ, McCarron MO, Hoffmann KL, Graffagnino C. Familial clustering of intracerebral hemorrhage: a prospective study in North Carolina. Neuroepidemiology 2002;21:18‐21.
24. Touzé E, Rothwell PM. Sex differences in heritability of ischemic stroke: a systematic review and meta‐analysis. Stroke 2008;39:16‐23.
25. Matarin M, Brown WM, Scholz S et al. A genome‐wide genotyping study in patients with ischaemic stroke: initial analysis and data release. Lancet Neurol 2007;6:414‐20.
26. Hassan A, Markus HS. Genetics and ischaemic stroke. Brain 2000;123:1784‐812.
27. Casas JP, Hingorani AD, Bautista LE, Sharma P. Meta‐analysis of Genetic Studies in Ischemic Stroke: Thirty‐two Genes Involving Approximately 18 000 Cases and 58 000 Controls. Arch Neurol 2004;61:1652‐61.
28. Haan J. Genetics of intrecerebral haemorrhage. In: Markus HS, editor. Stroke genetics.Oxford: Oxford University Press; 2003. p. 223‐41.
29. Chobanian AV, Bakris GL, Black HR et al. Seventh report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure. Hypertension 2003;42:1206‐52.
31. Lewington S, Whitlock G, Clarke R et al. Blood cholesterol and vascular mortality by age, sex, and blood pressure: a meta‐analysis of individual data from 61 prospective studies with 55,000 vascular deaths. Lancet 2007;370:1829‐39.
32. Iso H, Jacobs DR, Jr., Wentworth D, Neaton JD, Cohen JD. Serum cholesterol levels and six‐year mortality from stroke in 350,977 men screened for the multiple risk factor intervention trial. N Engl J Med 1989;320:904‐10.
33. Horenstein RB, Smith DE, Mosca L. Cholesterol predicts stroke mortality in the Women's Pooling Project. Stroke 2002;33:1863‐8.
34. Baigent C, Keech A, Kearney PM et al. Efficacy and safety of cholesterol‐lowering treatment: prospective meta‐analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet 2005;366:1267‐78.
35. Milionis HJ, Winder AF, Mikhailidis DP. Lipoprotein (a) and stroke. J Clin Pathol 2000;53:487‐96.
36. Danesh J, Collins R, Peto R. Lipoprotein(a) and coronary heart disease. Meta‐analysis of prospective studies. Circulation 2000;102:1082‐5.
37. Garza CA, Montori VM, McConnell JP, Somers VK, Kullo IJ, Lopez‐Jimenez F. Association between lipoprotein‐associated phospholipase A2 and cardiovascular disease: a systematic review. Mayo Clin Proc 2007;82:159‐65.
38. Wassertheil‐Smoller S, Kooperberg C, McGinn AP et al. Lipoprotein‐associated phospholipase A2, hormone use, and the risk of ischemic stroke in postmenopausal women. Hypertension 2008;51:1115‐22.
39. Almdal T, Scharling H, Jensen JS, Vestergaard H. The independent effect of type 2 diabetes mellitus on ischemic heart disease, stroke, and death: a population‐based study of 13,000 men and women with 20 years of follow‐up. Arch Intern Med 2004;164:1422‐6.
40. Selvin E, Coresh J, Shahar E, Zhang L, Steffes M, Sharrett AR. Glycaemia (haemoglobin A1c) and incident ischaemic stroke: the Atherosclerosis Risk in Communities (ARIC) Study. Lancet Neurol 2005;4:821‐6.
41. Stettler C, Allemann S, Juni P et al. Glycemic control and macrovascular disease in types 1 and 2 diabetes mellitus: Meta‐analysis of randomized trials. Am Heart J 2006;152:27‐38.
42. Robbins AS, Manson JE, Lee IM, Satterfield S, Hennekens CH. Cigarette smoking and stroke in a cohort of U.S. male physicians. Ann Intern Med 1994;120:458‐62.
43. Pechacek TF, Babb S. How acute and reversible are the cardiovascular risks of secondhand smoke? BMJ 2004;328:980‐3.
44. Ischaemic stroke and combined oral contraceptives: results of an international, multicentre, case‐control study. WHO Collaborative Study of Cardiovascular Disease and Steroid Hormone Contraception. Lancet 1996;348:498‐505.
45. Sturgeon JD, Folsom AR, Longstreth WT, Jr., Shahar E, Rosamond WD, Cushman M. Risk factors for intracerebral hemorrhage in a pooled prospective study. Stroke 2007;38:2718‐25.
28
46. Di CA, Rotondo S, Iacoviello L, Donati MB, De GG. Meta‐analysis of wine and beer consumption in relation to vascular risk. Circulation 2002;105:2836‐44.
47. Shaper AG, Wannamethee SG, Walker M. Body weight: implications for the prevention of coronary heart disease, stroke, and diabetes mellitus in a cohort study of middle aged men. BMJ 1997;314:1311‐7.
48. Hu G, Tuomilehto J, Silventoinen K, Sarti C, Mannisto S, Jousilahti P. Body mass index, waist circumference, and waist‐hip ratio on the risk of total and type‐specific stroke. Arch Intern Med 2007;167:1420‐7.
49. Kurth T, Gaziano JM, Rexrode KM et al. Prospective study of body mass index and risk of stroke in apparently healthy women. Circulation 2005;111:1992‐8.
50. Song YM, Sung J, Davey SG, Ebrahim S. Body mass index and ischemic and hemorrhagic stroke: a prospective study in Korean men. Stroke 2004;35:831‐6.
51. Alberti KG, Zimmet P, Shaw J. The metabolic syndrome‐‐a new worldwide definition. Lancet 2005;366:1059‐62.
53. Kahn R, Buse J, Ferrannini E, Stern M. The metabolic syndrome: time for a critical appraisal: joint statement from the American Diabetes Association and the European Association for the Study of Diabetes. Diabetes Care 2005;28:2289‐304.
54. Wang J, Ruotsalainen S, Moilanen L, Lepisto P, Laakso M, Kuusisto J. The metabolic syndrome predicts incident stroke: a 14‐year follow‐up study in elderly people in Finland. Stroke 2008;39:1078‐83.
55. Executive Summary of The Third Report of The National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, And Treatment of High Blood Cholesterol In Adults (Adult Treatment Panel III). JAMA 2001;285:2486‐97.
56. Nelson HD, Humphrey LL, Nygren P, Teutsch SM, Allan JD. Postmenopausal hormone replacement therapy: scientific review. JAMA 2002;288:872‐81.
57. Lamy C. [Postmenopausal hormone therapy and vascular risk]. Rev Neurol (Paris) 2007;163:851‐6.
58. Wassertheil‐Smoller S, Hendrix SL, Limacher M et al. Effect of estrogen plus progestin on stroke in postmenopausal women: the Women's Health Initiative: a randomized trial. JAMA 2003;289:2673‐84.
59. Johnsen SP. Intake of fruit and vegetables and risk of stroke: an overview. Curr Opin Clin Nutr Metab Care 2004;7:665‐70.
60. He K, Rimm EB, Merchant A et al. Fish consumption and risk of stroke in men. JAMA 2002;288:3130‐6.
61. Iso H, Rexrode KM, Stampfer MJ et al. Intake of fish and omega‐3 fatty acids and risk of stroke in women. JAMA 2001;285:304‐12.
29
62. Green DM, Ropper AH, Kronmal RA, Psaty BM, Burke GL. Serum potassium level and dietary potassium intake as risk factors for stroke. Neurology 2002;59:314‐20.
63. Nagata C, Takatsuka N, Shimizu N, Shimizu H. Sodium intake and risk of death from stroke in Japanese men and women. Stroke 2004;35:1543‐7.
64. Larsson SC, Virtanen MJ, Mars M et al. Magnesium, calcium, potassium, and sodium intakes and risk of stroke in male smokers. Arch Intern Med 2008;168:459‐65.
65. Bushnell CD. Oestrogen and stroke in women: assessment of risk. Lancet Neurol 2005;4:743‐51.
66. Kemmeren JM, Tanis BC, van den Bosch MA et al. Risk of Arterial Thrombosis in Relation to Oral Contraceptives (RATIO) study: oral contraceptives and the risk of ischemic stroke. Stroke 2002;33:1202‐8.
67. Gillum LA, Mamidipudi SK, Johnston SC. Ischemic stroke risk with oral contraceptives: A meta‐analysis. JAMA 2000;284:72‐8.
68. Jick SS, Myers MW, Jick H. Risk of idiopathic cerebral haemorrhage in women on oral contraceptives with differing progestagen components. Lancet 1999;354:302‐3.
69. Johnston SC, Colford JM, Jr., Gress DR. Oral contraceptives and the risk of subarachnoid hemorrhage: a meta‐analysis. Neurology 1998;51:411‐8.
70. Spence JD. Homocysteine‐lowering therapy: a role in stroke prevention? Lancet Neurol 2007;6:830‐8.
71. Casas JP, Bautista LE, Smeeth L, Sharma P, Hingorani AD. Homocysteine and stroke: evidence on a causal link from mendelian randomisation. Lancet 2005;365:224‐32.
72. Wang X, Qin X, Demirtas H et al. Efficacy of folic acid supplementation in stroke prevention: a meta‐analysis. Lancet 2007;369:1876‐82.
73. Bazzano LA, Reynolds K, Holder KN, He J. Effect of folic acid supplementation on risk of cardiovascular diseases: a meta‐analysis of randomized controlled trials. JAMA 2006;296:2720‐6.
74. Yaggi H, Mohsenin V. Obstructive sleep apnoea and stroke. Lancet Neurol 2004;3:333‐42.
75. Yaggi HK, Concato J, Kernan WN, Lichtman JH, Brass LM, Mohsenin V. Obstructive sleep apnea as a risk factor for stroke and death. N Engl J Med 2005;353:2034‐41.
76. Munoz R, Duran‐Cantolla J, Martinez‐Vila E et al. Severe sleep apnea and risk of ischemic stroke in the elderly. Stroke 2006;37:2317‐21.
77. Di Napoli M, Schwaninger M, Cappelli R et al. Evaluation of C‐reactive protein measurement for assessing the risk and prognosis in ischemic stroke: a statement for health care professionals from the CRP Pooling Project members. Stroke 2005;36:1316‐29.
78. Danesh J, Lewington S, Thompson SG et al. Plasma fibrinogen level and the risk of major cardiovascular diseases and nonvascular mortality: an individual participant meta‐analysis. JAMA 2005;294:1799‐809.
30
79. Lindsberg PJ, Grau AJ. Inflammation and infections as risk factors for ischemic stroke. Stroke 2003;34:2518‐32.
80. Danesh J. Coronary heart disease, Helicobacter pylori, dental disease, Chlamydia pneumoniae, and cytomegalovirus: meta‐analyses of prospective studies. Am Heart J 1999;138:S434‐S437.
81. Treadwell SD, Robinson TG. Cocaine use and stroke. Postgrad Med J 2007;83:389‐94.
82. Mateo I, Pinedo A, Gomez‐Beldarrain M, Basterretxea JM, Garcia‐Monco JC. Recurrent stroke associated with cannabis use. J Neurol Neurosurg Psychiatry 2005;76:435‐7.
83. Elkind MS. Why now? Moving from stroke risk factors to stroke triggers. Curr Opin Neurol 2007;20:51‐7.
84. Emsley HC, Hopkins SJ. Acute ischaemic stroke and infection: recent and emerging concepts. Lancet Neurol 2008;7:341‐53.
85. Ohira T, Shahar E, Chambless LE, Rosamond WD, Mosley TH, Jr., Folsom AR. Risk factors for ischemic stroke subtypes: the Atherosclerosis Risk in Communities study. Stroke 2006;37:2493‐8.