Epidemiology of equine rhinitis B virus (ERBV) A report for the Rural Industries Research and Development Corporation by C.A. Hartley, W. Black, K.Dynon, J. Huang and M.J. Studdert March 2001 RIRDC Publication No 01/021 RIRDC Project No UM-40A
Transcript
Epidemiology of equine rhinitis B virus (ERBV)
A report for the Rural Industries Research and Development Corporation by C.A. Hartley, W. Black, K.Dynon, J. Huang and M.J. Studdert
March 2001 RIRDC Publication No 01/021 RIRDC Project No UM-40A
Professor Michael J Studdert Centre for Equine Virology School of Veterinary Sciences The University of Melbourne VIC 3010 Phone: +61-3-8344-7373 Fax: +61-3-8344-7374 Email: [email protected]
In submitting this report, the researcher has agreed to RIRDC publishing this material in its edited form. RIRDC Contact Details Rural Industries Research and Development Corporation Level 1, AMA House 42 Macquarie Street BARTON ACT 2600 PO Box 4776 KINGSTON ACT 2604 Phone: 02 6272 4539 Fax: 02 6272 5877 Email: [email protected]. Website: http://www.rirdc.gov.au Published in March 2001 Printed on environmentally friendly paper by Canprint
iii
Foreword Surveys by the RIRDC Equine Research and Development program to identify the research needs of the Australian horse industry identified a number of key strategies of importance to the Australian horse industry. A consistent and high priority need was found to be respiratory diseases caused by viruses and the need for improved diagnostic, treatment and preventative strategies for these diseases. Viral diseases causing respiratory diseases, abortion and central nervous system diseases are responsible for serious economic losses to the Australian horse industry. Respiratory diseases can require many expensive treatments, are responsible for poor performance and many lost training days. All of the viruses known to cause endemic viral diseases in Australia have been isolated and studied at the Centre for Equine Virology (CEV), The University of Melbourne. In collaboration with Racing Victoria and the RIRDC, the CEV has previously developed a set of rapid diagnostic tests for endemic virus diseases that impact on the horse industry in Australia (RIRDC project UM-23A). These tests were developed for viruses already known to cause disease in the Australian horse population, including the five equine herpesviruses (EHV), equine adenovirus (EAdV) types 1 and 2, equine rhinitis A virus (ERAV, previously known as equine rhinovirus type 1) and equine arteritis virus (EVA). A test for equine influenza virus was also developed although this virus is exotic to Australia. These viruses are either known to cause, or are suspected to cause respiratory disease in Australian horses. Several other viruses are known to cause respiratory disease in horses in other parts of the world, but their existence in Australia has not been investigated. One such virus, equine rhinitis B virus (ERBV previously known as equine rhinovirus type 2) which is known to cause significant respiratory disease in other countries, however its presence in Australia has notbeen established. The purpose of the work performed in this study was to determine the prevalence of ERBV in the Australian horse population, and to begin to develop methodologies for its diagnosis and control. This was a two-year project funded from industry revenue which is matched by funds provided by the Federal Government. This report, an addition to RIRDC’s diverse range of almost 800 research publications, forms part of our Equine R&D program, which aims to assist in developing the Australian horse industry and developing its research potential Most of our publications are available for viewing, downloading or purchasing online through our website: • downloads at www.rirdc.gov.au/reports/Index.htm • purchases at www.rirdc.gov.au/eshop Simon Hearn Managing Director Rural Industries Research and Development Corporation
iv
Acknowledgments We thank Professors W.H. McCollum and P.J. Timoney, University of Kentucky USA, Dr E. Dubovi, Cornell University USA and Dr M Weiss, University of Berne, Switzerland for the gifts of ERBV virus isolates, and Dr James Gilkerson, Macquarie University, New South Wales for the serum samples. This work is also funded by Racing Victoria and a Special Virology Fund.
Abbreviations bp base pairs cDNA complementary deoxyribonucleic acid CEV Centre for Equine Virology DNA deoxyribonucleic acid EAdV1 equine adenovirus 1 EAdV2 equine adenovirus 2 EAV equine arteritis virus EHV1 equine herpesvirus 1 (equine abortion virus) EHV2 equine herpesvirus 2 (equine gammaherpesvirus) EHV3 equine herpesvirus 3 (equine coital exanthema virus) EHV4 equine herpesvirus 4 (equine rhinopneumonitis virus) EHV5 equine herpesvirus 5 (equine gammaherpesvirus) EIV equine influenza virus ELISA enzyme linked immunosorbent assay ERAV equine rhinitis A virus (previously known as equine rhinovirus type 1) ERBV equine rhinitis B virus (previously known as equine rhinovirus type 2) FMDV foot-and-mouth-disease virus kD kilodaltons PCR polymerase chain reaction RNA ribonucleic acid RT-PCR reverse transcription-polymerase chain reaction SDS-PAGE sodium dodecyl sulphate-polyacrylamide gel electrophoresis TCID50 50% tissue culture infectious dose VLP virus-like particles VP1-4 virus proteins 1-4 (equine rhinitis virus structural proteins)
v
Contents
Foreword .................................................................................................................... iii Acknowledgments ...................................................................................................... iv Abbreviations.............................................................................................................. iv Executive Summary....................................................................................................vi 1. Introduction .......................................................................................................... 1 2. Objectives ............................................................................................................ 2 3. Methodology ........................................................................................................ 3 4. Results ................................................................................................................. 5
4.1 Seroepidemiology of ERBV infection in Australian horses ............................................5 4.2 Development of a diagnostic RT-PCR assay for the detection of ERBV in clinical samples. ........................................................................................................................7 4.3 Development of a diagnosticELISA to detect antibodies to ERBV. ...............................9 4.4 Production of recombinant baculovirus-expressed ERBV virus-like particles. ............11
6. References......................................................................................................... 15 List of Figures Figure 1 Comparison of ERAV and ERBV VN antibody titres of selected serums. .............5 Figure 2. Age distribution of ERBV VN antibody titres in equine serum samples. ................6 Figure 3 Age distribution of ERBV seropositive (VN titres ≥ 80) in equine serum samples .6 Figure 4 RT-PCR of ERBV isolates (1 - 13) from infected RK13 cell supernatants using ....
3A (A) and 3D (B) region primer pairs. ..................................................................8 Figure 5 RT-PCR of ERBV isolates (14 – 23) from infected Vero cell culture supernatants
using 3A (A) and 3D (B) region primer pairs. ...............................................8 Figure 6 Electron micrograph (A) and Coomassie Brilliant Blue stained SDS-PAGE (B) of
purified ERBV.1436/71 particles..........................................................................10 Figure 7 ELISA to detect ERBV antibodies in equine sera ................................................10
vi
Executive Summary The RIRDC Equine Research and Development program continues to identify a range of key strategies for the development of the Australian horse industry. A consistent high priority was the need to improve the respiratory health of horses through improved diagnostic, treatment and preventative strategies. It is suggested that 50% of all calls veterinarians make to horses in racing are for viral respiratory disease. These diseases are responsible for many expensive treatments, poor performance and many lost training days. Diagnosis of endemic equine virus diseases is based on isolation of viruses from clinical samples linked to electron microscopy, and a range of blood tests that measure the immune response of an infected horse to an invading virus. These techniques such as virus isolation are relatively slow, since the virus must be allowed time to replicate in cell culture in order to be detected. This can take as little as overnight for some viruses, but up to two weeks for other viruses. Indeed, some viruses cannot be grown in cell culture and therefore cannot be detected using this method. Since its discovery, polymerase chain reaction (PCR) technology has been introduced into many areas of medical and veterinary research. The enormous potential of PCR in diagnostic technology results from the ability of PCR to specifically detect minute amounts of target DNA. PCR involves repeated cycles of enzymatic synthesis of specific DNA sequences to result in an exponential accumulation of a specific DNA product, where the number of target DNA copies doubles every cycle. Thus 20 cycles of PCR yields about a million-fold amplification of the original target DNA. The PCR offers several advantages over conventional techniques for the identification of unknown pathogens. It is highly sensitive and has been shown to be over 1000 times more sensitive than virus isolation in cell culture. Once established in the diagnostic laboratory, it is extremely rapid with testing completed within 8 hours. As the techniques are simple, many samples can be processed simultaneously. All of the viruses known to cause endemic viral respiratory diseases in Australia have been isolated and studied at the Centre for Equine Virology (CEV). With funding from Racing Victoria and RIRDC (UM-23A) we have previously developed of a suite of rapid and sensitive PCRs for the diagnosis of equine herpesvirus 1 (EHV1, equine abortion virus), EHV4 (equine rhinopneumonitis virus), EHV3 (equine coital exanthema virus) the equine gammaherpesviruses EHV2 and EHV5, equine adenovirus 1 (EAdV1), EAdV2, equine arteritis virus (EVA) and equine rhinitis A virus (ERAV). Despite being recognised as a cause of acute respiratory disease in horse overseas, no study to confirm the presence of ERBV in Australia has ever been performed. In this report we now confirm the presence of this virus in Australia indirectly by detecting ERBV-specific antibody in serum from Australian horses which have been collected since 1995 and was found to be the most prevalent in horses between 2 and <3 years of age. Recognising the causative agents of respiratory disease is the first stage in developing methods for its diagnosis and control. ERBV has not been isolated in Australia, which is probably due to the difficulties associated with primary cultivation of the virus from clinical material. The development of an ERBV specific RT-PCR will not only circumvent the difficulties associated with virus culture, but will also significantly reduce the amount of time required to obtain and report a diagnosis once a sample has been received. One difficulty encountered in the design of these primers results from the variability of the RNA genome of these viruses. We have obtained over 23 different ERBV isolates from the USA and Switzerland and have examined the molecular variation amongst within their genomes. A set of primers able to detect all ERBV isolates has now been designed. Once we have demonstrated the ability of these primers to detect all 23 available ERBV isolates, we will begin screening nasopharyngeal swabs taken from horses suffering acute respiratory disease.
vii
The suite of diagnostic PCRs for respiratory viruses, including ERBV, will then be applied to these samples in attempts to diagnose the cause of the illness. Detection of ERBV RT-PCR positive swabs would provide direct confirmation of the presence of ERBV in Australia, complementing the sero-epidemiological evidence for the prevalence of this virus in the Australian horse population. Progress has also been made in the development of recombinant DNA technology to engineer antigens that could be used in a blood test and vaccine for ERBV. ERBV grows only to low levels in cell culture, therefore obtaining sufficient quantities of native virus particles for the production of a blood test or vaccine would be difficult. Therefore DNA encoding the structural (capsid) proteins and some processing enzymes of ERBV were engineered into a vector to facilitate expression of fully processed ERBV capsid proteins in an insect cell system. This system has been shown to enable extremely high levels of expression of recombinant proteins, which are structurally very similar to their authentic counterparts. The expression cassettes for ERBV have now been prepared and trials will soon begin to investigate the suitability of these recombinant proteins for use in the blood test and vaccine.
1
1. Introduction Equine rhinitis viruses are members of the Picornaviridae family and have been shown to cause acute respiratory disease in horses. Three serotypes of equine rhinitis viruses have been identified. The RNA genome of equine rhinitis A virus (ERAV) has been completely sequenced at the Centre for Equine Virology and the virus was shown to be most closely related to foot-and-mouth-disease virus (6). In a previous RIRDC funded program we designed PCR primers to amplify the genome of ERAV from clinical samples to provide rapid diagnosis of infection. Results from this study showed that ERAV is a significant cause of respiratory disease in Australian horses and that it had been underestimated as a pathogen because of difficulties in isolating the virus (7). An ELISA to detect antibodies in equine serum in horses for the diagnosis of ERAV infection is now under development. Sequence of the genome of ERBV has also been determined (13); ERBV is most closely related to the encephalomyocarditis virus (EMCV), but has been assigned as the sole members of a new genus Erbovirus in the family Picornaviridae. In a study of 92 horses in Canada (3), ERBV was identified as a significant cause of viral respiratory disease. ERBV was the causative agent of acute respiratory disease in 21% of the horses. The virus was also isolated from 22% of horses with respiratory disease that did not have a significant rise in antibody levels to ERBV suggesting that the virus can establish persistent infections which may serve as a source of new infections in susceptible populations. Although it is believed that ERBV is present in Australian horses, no study to confirm its presence has ever been performed. The purpose of this study is to investigate the prevalence of ERBV in Australian horses and to develop diagnostic technologies to diagnose ERBV infection based on PCR and enzyme linked immunosorbent assay (ELISA).
2
2. Objectives a. To investigate the prevalence of ERBV infection in Australian horses. b. To develop diagnostic tests for ERBV based on PCR and ELISA. c. To investigate the potential of an ERBV vaccine based on recombinant baculovirus
expressed virus-like particles.
3
3. Methodology Serum. Equine serums used in this study came from the equine serum bank that is maintained and stored at –20°C at the CEV. These serums were submitted to the CEV from all over Australia for routine serologic testing. Serums used in this study were submitted during and after 1995. Additional serums were obtained from samples collected by Dr James Gilkerson, Macquarie University, New South Wales. Viruses. Viruses used in this study were obtained from Professor W.H. McCollum, University of Kentucky USA, Dr E. Dubovi, Cornell University USA and Dr M Weiss, University of Berne, Switzerland. ERBV.1436/71 was used in all virus neutralisation (VN) assays as the prototype strain since it was the first ERBV isolated and the genome of this virus has been almost completely sequenced (13). ERBV has not been isolated in Australia and therefore there are no known Australian ERBV strains. Virus Neutralisation (VN) assays. Infection with ERBV was determined using conventional virus neutralisation tests carried out in a microtitre plates using RK-13 cells (11). Serums were heat-inactivated for 30 minutes at 56°C prior to use. Plates were read after 3 days incubation in a humidified atmosphere of 5% CO2. The VN titre is the reciprocal of the final dilution of serum that inhibited cytopathic effect (cpe) by 100 TCID50 of virus. Reverse transcriptase - PCR (RT-PCR). Viral RNA from the supernatant from ERBV-infected cells was extracted using the QIAamp viral RNA mini kit (Qiagen) according to manufacturer’s instructions. Copy DNA (cDNA) copies of the viral RNA was obtained by RT at 42°C using Superscript reverse transcriptase (Life Technologies) according to manufacturer’s instructions with the exception that 100U of enzyme is used. One microlitre of the cDNA made in this reaction is then used as template in the subsequent PCR. PCR was performed in a 25µL reaction volume containing 0.5 units of Taq DNA polymerase (Promega), 0.2 mM (each) deoxynucleotide triphosphate (dNTP) in 1 x thermophilic reaction buffer (50 mM KCl, 10 mM Tris-HCl pH 8.6, 0.1% Triton X-100) with MgCl2 and primer concentrations at 1.5 mM and 2 µM respectively. Amplification was carried out in a thermal cycler (Hybaid) where conditions varied according to the primers used. In general, reactions began with a 5 mins denaturation at 95°C, followed by 40 cycles of 30 sec denaturation at 95°C, 30 sec annealing at 60°C and 30 sec extension at 72°C. A final incubation of 5 mins at 72°C was then performed. Products were visualised on ethidium bromide stained agarose gels using a transillumination UV light box. DNA sequencing. In order to determine the DNA sequence of PCR products obtained from different virus isolates, PCR products were extracted from agarose gel slices using the Qiagen gel extraction kit. Purified products were then cloned into the pGEM-T vector (Promega) and sequenced with the SP6 promoter sequencing primer in a Big Dye Terminator reaction (Applied Biosystems). Reactions were separated and read by the Australian Genome Research Facility, Walter and Elisa Hall Institute for Medical Research. Chromatograms were then analysed using Applied Biosystem’s EditView 1.0.1 software package. Sequence alignments were performed using ClustalX (12).
4
Viral protein analysis by polyacrylamide gel electrophoresis (PAGE) Samples were analysed on polyacylamide gels in the presence of 0.1% sodium dodecyl sulphate (SDS-PAGE) according to the method of Laemmli (5) under reducing conditions. Proteins were then either visualised by staining with 0.01% Coomassie Brilliant Blue. ELISA Wells of a 96 well polystyrene microtitre tray (MaxiSorp, Nunc) were coated with purified ERBV at 10 µg/mL diluted in 0.05M carbonate-bicarbonate buffer (pH 9.6) overnight at 4°C. Plates were incubated with 100µL volumes per well at all steps and were washed four times between all steps with phosphate buffered saline (PBS, pH 7.5) containing 0.05% (v/v) Tween 20 (PBST). Unoccupied sites were blocked by incubation for 2 hours with 10 mg/mL bovine serum albumin (BSA) in PBS. Dilutions of serum sample were then prepared in 5 mg/mL BSA in PBST (BSA5PBST) and added to wells for 2 hours, prior to incubation with horseradish peroxidase-conjugated, affinity purified goat anti-horse immunoglobulin G (IgG) (Kirkegaard and Perry Laboratories Inc) diluted 1/1000 in BSA5PBST. Wells were developed with a 3,3’,5,5’-tetramethyl benzidine (TMB) substrate (Sigma), and the reactions stopped after 6 minutes by the addition of 50 µL 1M HCl. The absorbance at 450 nm was read in each well with a Titertek Multiskan MC3. Baculovirus expression systems Recombinant baculoviruses have become popular vectors for the expression of heterologous proteins, including those from viruses, fungi, plants and animals. Expression of foreign genes is usually driven by the polyhedrin promoter of the Autographa californica nuclear polyhedrosis virus (AcNPV). The recombinant proteins are often expressed at high levels in cultured insect cells or infected larvae and are often functionally similar to their authentic counterparts. A rapid method to generate recombinant baculoviruses, designated as Bac-to-Bac baculovirus expression system (Life Technologies), has been developed which is based on site-specific transposition of an expression cassette into a baculovirus shuttle vector (bacmid). The recombinant bacmid replicates in E. coli as a large plasmid and remains infectious when introduced into insect cells. Transfection of bacmid DNA containing the foreign DNA insert into SF9 (Spodoptera frugiperda) cells then results in the production of the recombinant baculovirus.
5
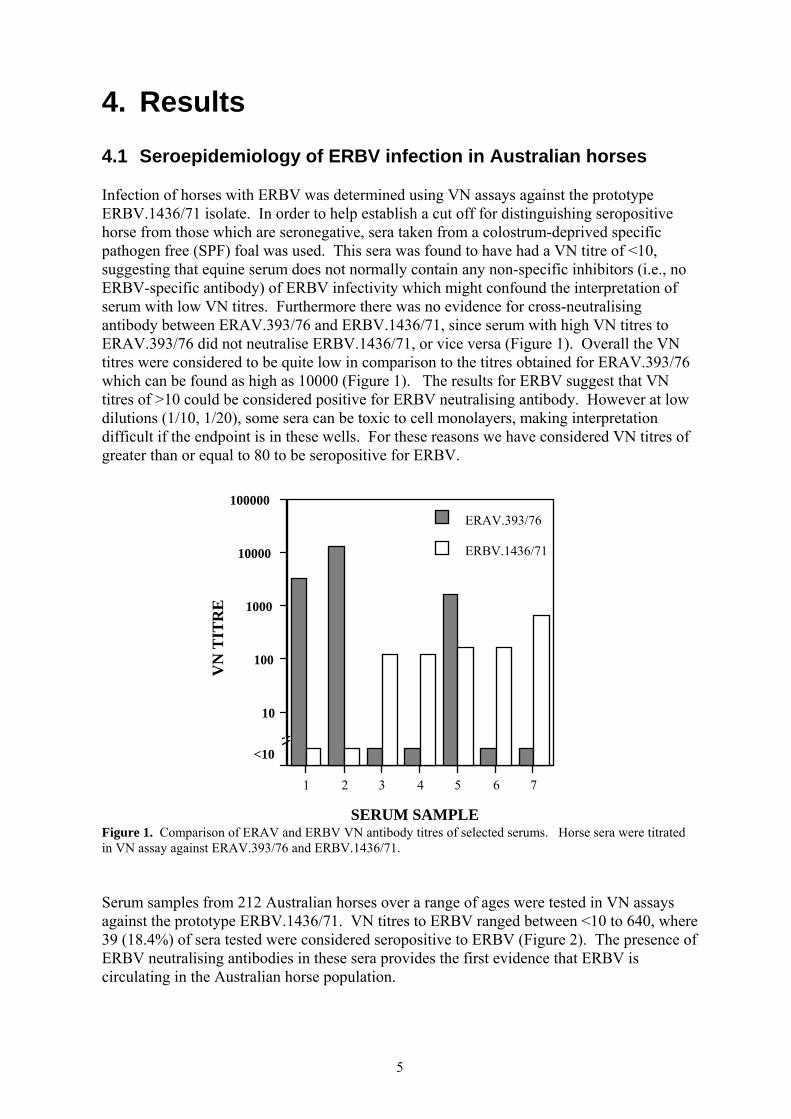
4. Results 4.1 Seroepidemiology of ERBV infection in Australian horses Infection of horses with ERBV was determined using VN assays against the prototype ERBV.1436/71 isolate. In order to help establish a cut off for distinguishing seropositive horse from those which are seronegative, sera taken from a colostrum-deprived specific pathogen free (SPF) foal was used. This sera was found to have had a VN titre of <10, suggesting that equine serum does not normally contain any non-specific inhibitors (i.e., no ERBV-specific antibody) of ERBV infectivity which might confound the interpretation of serum with low VN titres. Furthermore there was no evidence for cross-neutralising antibody between ERAV.393/76 and ERBV.1436/71, since serum with high VN titres to ERAV.393/76 did not neutralise ERBV.1436/71, or vice versa (Figure 1). Overall the VN titres were considered to be quite low in comparison to the titres obtained for ERAV.393/76 which can be found as high as 10000 (Figure 1). The results for ERBV suggest that VN titres of >10 could be considered positive for ERBV neutralising antibody. However at low dilutions (1/10, 1/20), some sera can be toxic to cell monolayers, making interpretation difficult if the endpoint is in these wells. For these reasons we have considered VN titres of greater than or equal to 80 to be seropositive for ERBV.
Figure 1. Comparison of ERAV and ERBV VN antibody titres of selected serums. Horse sera were titrated in VN assay against ERAV.393/76 and ERBV.1436/71. Serum samples from 212 Australian horses over a range of ages were tested in VN assays against the prototype ERBV.1436/71. VN titres to ERBV ranged between <10 to 640, where 39 (18.4%) of sera tested were considered seropositive to ERBV (Figure 2). The presence of ERBV neutralising antibodies in these sera provides the first evidence that ERBV is circulating in the Australian horse population.
VN
TIT
RE
1 2 3 4 5 6 7
SERUM SAMPLE
ERBV.1436/71
ERAV.393/76
10
100
1000
10000
100000
<10
6
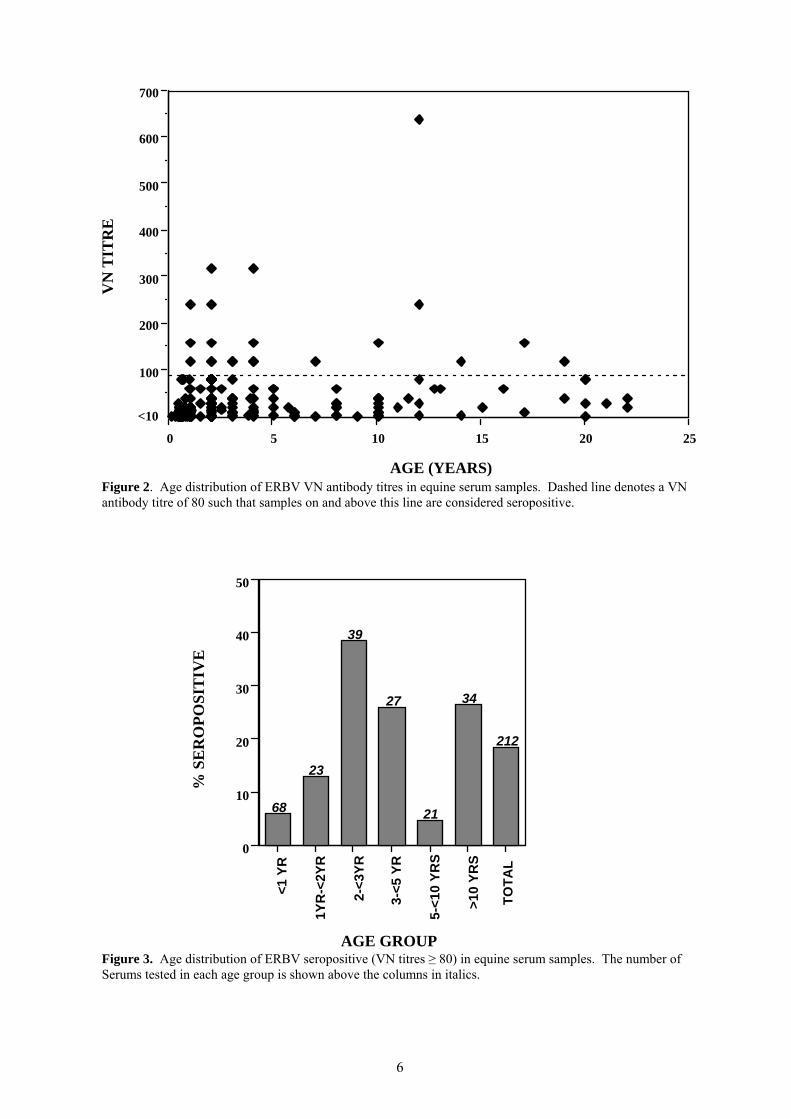
Figure 2. Age distribution of ERBV VN antibody titres in equine serum samples. Dashed line denotes a VN antibody titre of 80 such that samples on and above this line are considered seropositive.
Figure 3. Age distribution of ERBV seropositive (VN titres ≥ 80) in equine serum samples. The number of Serums tested in each age group is shown above the columns in italics.
0
10
20
30
40
50
% S
ER
OPO
SIT
IVE
<1 Y
R
1YR
-<2Y
R
2-<3
YR
3-<5
YR
5-<1
0 YR
S
>10
YRS
TOTA
L
AGE GROUP
68
23
39
27
21
34
212
100
200
300
400
500
600
700V
N T
ITR
E
0 5 10 15 20 25
AGE (YEARS)
<10
7
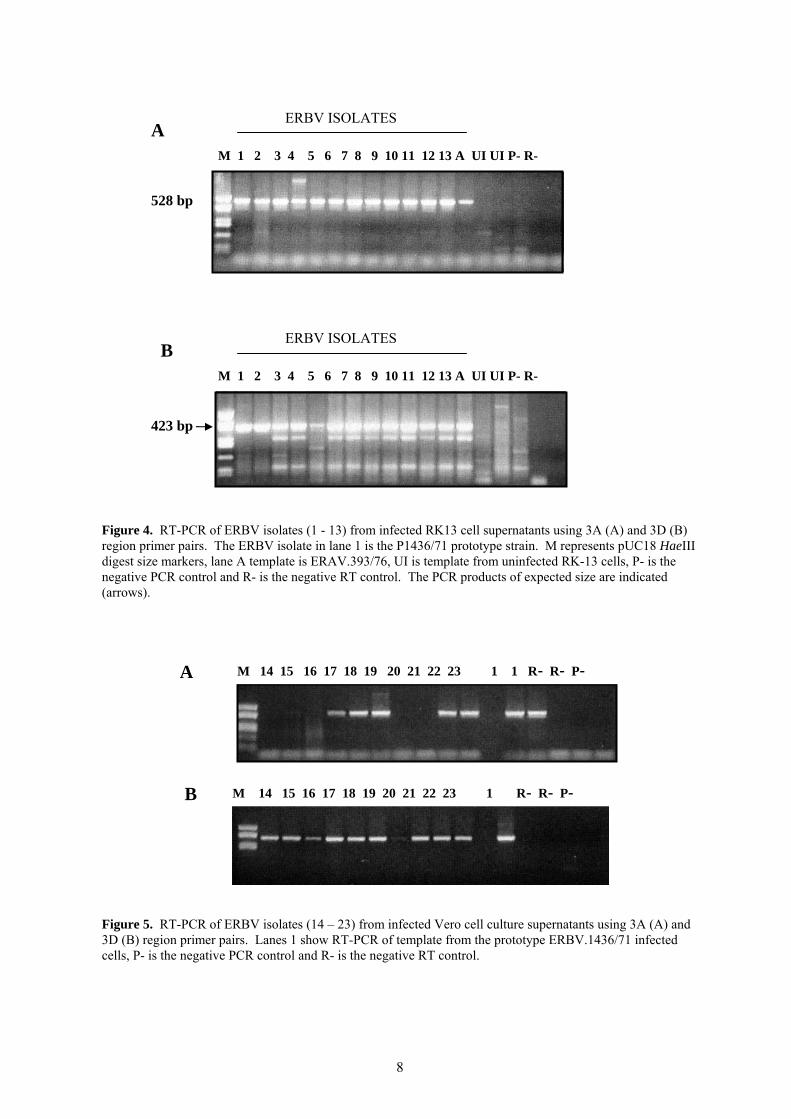
Seropositive samples are shown to cluster mainly in samples from horses in the 1 to 5 year old age range (Figure 2). When further divided into age groups, it can be seen that foals less than 1 year are generally seronegative to ERBV (Figure 3). Only 4 foals (5.9%) in this age group had low VN titres (VN = 80) in this group. Highest prevalence (38.5%) was found in the 2 to <3 year old age group, and this gradually decreased in older horses. A sharp reduction in prevalence was found in horses 5 to <10 years. The significance of this drop is not understood, however only 21 sera have been tested in this group. The inclusion of further samples in this age range is therefore required. 4.2 Development of a diagnostic PCR assay for the detection of ERBV in clinical samples. The development of a diagnostic PCR would provide a highly sensitive technique for the detection of ERBV infection. Equine rhinitis viruses have proven to be particularly difficult to culture from nasal swabs and other clinical samples and as a result have probably been under estimated in their significance as equine pathogens. Indeed despite our findings based on serology that ERBV infects Australian horses, ERBV has not been isolated in Australia. The development of an ERBV specific RT-PCR would provide a highly sensitive diagnostic tool, which has been shown for other viruses to be as much as 1000 times more sensitive than virus isolation by cell culture (8). PCR provides a rapid method of diagnosis where many samples can be processed simultaneously and results reported within 8 hours of receiving the samples. Oligonucleotide primers were designed based on the genome sequence of the prototype strain of ERBV (13). Primers were designed to the 3’ end of the VP3 coding region and the 3’ end of the VP1 coding region. RNA was prepared from Vero cells infected with 13 different ERBV isolates. An RT-PCR was performed and products were visualised after electrophoresis in an ethidium bromide stained agarose gels. A product of the correct size (1061 base pairs) was obtained from the prototype strain, to which these primers were designed. No products were observed from any of the other strains, suggesting the sequence in the region to which the primers bind is highly variable (data not shown). In order to overcome the difficulties associated with genomic variability between ERBV isolates, new primers were designed to more conserved regions of the ERBV genome. Primers designed to both the 3A and the 3D regions of the genome, amplified products of the correct size from the prototype strain ERBV.1436/71 as well as 12 other ERBV isolates (Figure 4). These reactions did not amplify any product from ERAV, suggesting that these primers are specific for ERBV isolates only (Figure 4). The conditions for the RT-PCR using the 3A region primers were optimised to obtain an assay with maximum sensitivity for detecting the prototype virus strain. Conditions tested included MgCl2 concentration, annealing temperature, primer concentration, reverse transcriptase and Taq polymerase concentrations, cycle number and extension time. Using these optimised conditions, the 3A directed ERBV RT-PCR was found to be extremely sensitive and able to detect 500 TCID50’s of virus in a nasal swab which was ‘spiked’ with ERBV.1436/71.
8
Figure 4. RT-PCR of ERBV isolates (1 - 13) from infected RK13 cell supernatants using 3A (A) and 3D (B) region primer pairs. The ERBV isolate in lane 1 is the P1436/71 prototype strain. M represents pUC18 HaeIII digest size markers, lane A template is ERAV.393/76, UI is template from uninfected RK-13 cells, P- is the negative PCR control and R- is the negative RT control. The PCR products of expected size are indicated (arrows). Figure 5. RT-PCR of ERBV isolates (14 – 23) from infected Vero cell culture supernatants using 3A (A) and 3D (B) region primer pairs. Lanes 1 show RT-PCR of template from the prototype ERBV.1436/71 infected cells, P- is the negative PCR control and R- is the negative RT control.
B
M 1 2 3 4 5 6 7 8 9 10 11 12 13 A UI UI P- R-
528 bp
ERBV ISOLATES A
M 1 2 3 4 5 6 7 8 9 10 11 12 13 A UI UI P- R-
423 bp
ERBV ISOLATES
B M 14 15 16 17 18 19 20 21 22 23 1 R- R- P-
A M 14 15 16 17 18 19 20 21 22 23 1 1 R- R- P-
9
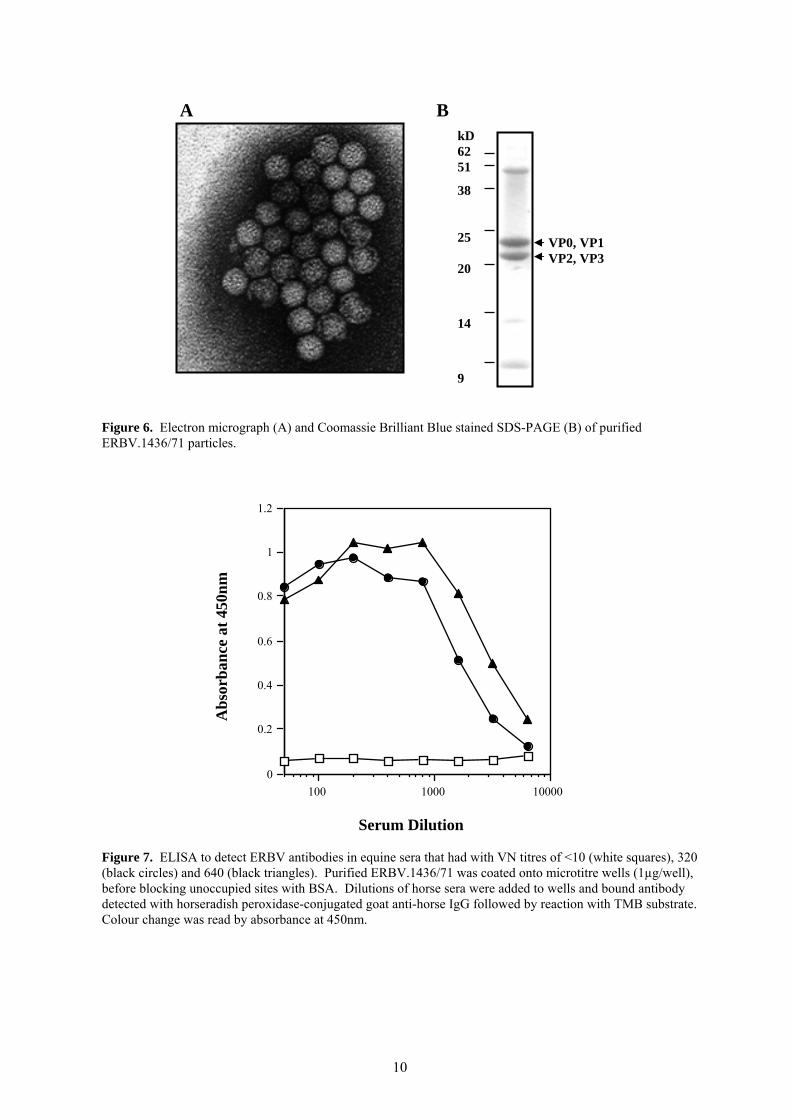
To further investigate the ability to detect all ERBV isolates, the RT-PCR was tested on a further 10 isolates of ERBV obtained from Dr M Weiss, University of Berne, Switzerland. The 3A region primers amplified a product from 6 of the 10 Swiss ERBV isolates. The 3D primer pair amplified a product all 10 of these isolates; amplification of one of these isolates (lane 20, Figure 5) by the 3D primer pair did not appear to be particularly sensitive. Therefore while the 3D region primers are able to amplify all strains, their efficiency requires improvement. The most efficient way to design primers capable of recognising all ERBV viruses is to obtain nucleotide sequence data for more than one ERBV isolate. Only ERBV.1436/71 sequence is currently known (13) and it is this sequence to which the 3A and 3D primer pairs were designed. Sequence analysis of the 3D regions of several other ERBV isolates would enable the design of a primer pair capable of efficiently detecting all ERBV strains. Six different ERBV isolates were chosen for sequence analysis based on the country of isolation and the efficiency with which the 3A and 3D primer pairs amplified a product. While nucleotide sequence in the other regions is well conserved (92% identity between the most divergent strains), there is some nucleotide changes interspersed throughout the sequenced region. The prototype strain P1436/71 appears to be the most divergent such that primers designed using this sequence would be the least likely to amplify all isolates. This information has been used to design a new set of primers to the 3D region. The ability of these primers to amplify each of the different ERBV isolates is currently being evaluated. 4.3 Development of a diagnostic ELISA to detect antibodies to ERBV. Traditional methods of determining antibody titres by virus neutralisation assays are labour intensive and time consuming, therefore we also plan to improve techniques available for ERBV serology by developing an ELISA to provide more rapid diagnosis. The basic design of most ELISA’s for the detection of viral antibody entails a virus (or part thereof) coated solid phase (usually a plastic microtitre well) which is able to capture the virus specific antibody from the serum or plasma sample. To investigate the potential of this type of ELISA for ERBV, purified virus was needed. A purification method for of ERAV has been developed at the CEV that involves high-speed centrifugation of virus from infected cell supernatant, followed by purification over a continuous sucrose density gradient. Approximately 100-200 µg of virus can be routinely obtained from 700 mL of ERAV-infected cell culture supernatant. When this method was used to purify ERBV from 700 mL of ERBV.1436/71-infected cell culture supernatant only small amounts (<50 µg) of virus was obtained. Since ERBV is distantly related to EMCV methods for the purification of EMCV were therefore used to purify ERBV. A method was adapted from that of Rueckert (9) which precipitates virus particles from infected cell supernatant using PEG6000, followed by DNase I and trypsin treatment and high speed sedimentation through a sucrose cushion. High yields (300-400µg) of virus were obtained with this method which was of reasonable purity when examined by electron microscopy and Coomassie brilliant blue stained SDS-PAGE (Figure 6).
10
Figure 6. Electron micrograph (A) and Coomassie Brilliant Blue stained SDS-PAGE (B) of purified ERBV.1436/71 particles.
Figure 7. ELISA to detect ERBV antibodies in equine sera that had with VN titres of <10 (white squares), 320 (black circles) and 640 (black triangles). Purified ERBV.1436/71 was coated onto microtitre wells (1µg/well), before blocking unoccupied sites with BSA. Dilutions of horse sera were added to wells and bound antibody detected with horseradish peroxidase-conjugated goat anti-horse IgG followed by reaction with TMB substrate. Colour change was read by absorbance at 450nm.
0
0.2
0.4
0.6
0.8
1
1.2
Abs
orba
nce
at 4
50nm
100 1000 10000
Serum Dilution
VP0, VP1 VP2, VP3
kD 62 51
38 25 20
14
9
A B
11
To investigate the suitability of this purified virus as an antigen for ELISA, purified ERBV was used to coat wells of a polystyrene microtitre tray, and an ELISA performed as describe in the Methodology. As can be seen in Figure 7 the SPF foal serum showed no reactivity by ELISA, whereas the sera with VN titres of 320 and 640 showed strong reactivity to the ERBV coated wells. These results suggest that this ELISA format can provide a sensitive technique for the detection of anti-ERBV antibodies in equine serum. Further investigations of this ELISA are required, including the preparation of a monospecific hyperimmune serum prepared in rabbits to purified ERBV.1436/71. Preparation of this serum has begun (July 2000) and rabbits have received their first inoculation of 30 µg of purified, UV inactivated ERBV.1436/71. 4.4 Production of recombinant baculovirus-expressed ERBV virus-like particles. To develop an ELISA for use on a large scale would require the production of large quantities of viral antigen. Purification of large quantities ERBV using the method described above would be a costly and time-consuming process and alternative methods of antigen production are therefore required. ELISA antigens can be produced in large quantities by engineering either bacteria or insect cells to produce the required protein. Expression of picornavirus capsid proteins, such as FMDV and poliovirus, in baculovirus (and E.coli) results in the formation of virus-like particles (VLPs). VLPs have been shown to be indistinguishable from cell culture grown virus when viewed by electron microscopy, but do not contain nucleic acid and are therefore not infectious. Studies using FMDV have demonstrated that VLPs are highly immunogenic and have similar antigenic structure to whole virions (1). Production of ERBV VLPs requires the expression of the structural proteins (P1) of the virus capsid as well as the viral protease 3C, which processes the P1 precursor polyprotein into the individual capsid proteins VP0, VP1 and VP3. The main strategy for making ERBV VLPs will be to produce a recombinant baculovirus that has been engineered to express the DNA from ERBV that encodes both P1-2A and 3C. Primer pairs were designed to amplify DNA encoding both the entire P1-2A and 3C regions from ERBV.1436/71. The P1 forward primer incorporates an EcoRI site, an ATG start codon and the myristoylation signal sequence. The myristoylation of P1 protein has been shown to be critical for correct assembly of virus particles (2). The P1 reverse primer contains a NotI site. The 3C forward primer incorporates a NotI site and ATG start codon and the reverse primer contains a XhoI site. An RT-PCR was performed using each primer pair on ERBV.1436/71 RNA using Pfu turbo DNA polymerase in the PCR step to ensure the highest possible fidelity of the DNA copies. Products were of the expected sizes (2713 bp for P1-2A, and 1154 bp for 3C) and after being A-tailed were ligated into pGEM-T vector (Promega). P12A was then cut from the pGEM-T vector using the EcoRI and NotI restriction sites which were designed into the primers, and this fragment was then cloned into the EcoRI and NotI site of the pFastBac plasmid to form mP1-2A.pFastBac. Similarly 3C was cloned into pFastBac using NotI and XhoI restriction sites (3C.pFastBac). Finally 3C was cloned into the remaining NotI and XhoI site of the mP12A.pFastBac to create the plasmid mP12A.3C.pFastBac. Each of the three plasmids were analysed by restriction digests to ensure inserts were of the correct size and orientation. Sequence analysis has also confirmed inserts are in the correct reading frame. These plasmids will now be used in transpositions with DH10Bac cells to create the recombinant bacmid DNA, and then transfected into SF9 cells to produce recombinant baculovirus
12
particles. The recombinant baculoviruses should then enable the production of ERBV VLPs by these insect cells. Insect cell supernatant and lysate will then be examined for picornavirus-like particles by electron microscopy and protein bands corresponding in size to VP0, VP1 and VP3 which react to the rabbit anti-ERBV.1436/71 antiserum by western blot. The presence of these bands and not the complete P1 polyprotein (97 kD) would suggest that expressed protease 3C has functioned to process P1 into the individual capsid proteins. The antigenicity and immunogenicity of these VLPs will require further investigation to determine their usefulness as antigens for an ERBV diagnostic ELISA and as a vaccine.
13
5. Discussion This study provides the first evidence of the presence of ERBV in the Australian equine population, which was found to be most prevalent in horses between two and three years of age. This finding adds another virus to the already extensive list of equine respiratory viruses that are endemic in Australia. Virus diseases, particularly those causing abortion and respiratory disease are responsible for serious losses to the horse industry in Australia and worldwide. Identification of the agents responsible for causing respiratory diseases is the first stage in the development and implementation of methods for their diagnosis and control. The most recent study of the association of ERBV with respiratory illness in horses was performed in Ontario, Canada where nasopharyngeal swabs and paired serum samples were taken from 92 horses suffering acute respiratory disease (3). Swabs were then tested by virus culture isolation and serums tested for a rise in antibody titre to a range of bacterial and viral pathogens (e.g., equine influenza type A, ERAV, ERBV, EHV1, EHV4, EAV, EAdV, mycoplasmas and bacteria). ERBV was identified as a significant cause of respiratory disease and was the causative agent of acute respiratory disease in 21% of the horses. The virus was also isolated from 22% of horses with respiratory disease that did not have a significant rise in antibody levels to ERBV suggesting that the virus can establish persistent infections which may serve as a source of new infections in susceptible populations. Evidence for the existence of ERBV in Australia was obtained indirectly through measurement of VN titre in serum. ERBV has not been isolated in Australia, which is likely due to difficulties in primary cultivation of the virus from clinical samples. An alternative to direct isolation of virus from nasopharyngeal swabs to diagnose ERBV infection would be RT-PCR. After characterising regions of sequence variability amongst ERBV isolates we have redesigned a set of primers that will be used in a diagnostic RT-PCR. The capacity of the primers to amplify the available ERBV isolates requires further validation. In a previously funded RIRDC project, we have developed a suite of diagnostic PCRs for endemic equine respiratory diseases. The finding that ERBV is present in Australia would indicate that an ERBV RT-PCR should be added to the suite to diagnose causes of viral respiratory disease from clinical samples such as nasopharyngeal swabs. Identification of ERBV RT-PCR positive nasopharyngeal swabs from horses with respiratory disease would provide more direct evidence of the presence of ERBV in Australia as well as direct efforts to isolate ERBV from these samples, and these studies are on going. As noted, VN antibody titres to ERBV were much lower than generally expected for a picornavirus infection, particularly when compared to those found to ERAV. The reason for lower titres is not known but could simply be due to an inability of the virus to induce high levels of neutralising antibodies, or be the result of a short half-life of circulating ERBV-neutralising antibody. The VN method and cell type used in the assay may also contribute to low titres. It is also notable that there was a strong correlation between VN antibody titres and ELISA antibody levels. Low VN antibody levels have been recognised by others (4, 10). In comparison, titration of serum by ELISA on whole-purified virus particles showed a high titre antibody response to ERBV. Since neutralising antibodies comprise only a fraction of the total antibody response to a virus, an ELISA may provide a more sensitive means of detecting ERBV antibodies from equine serum. The improved sensitivity in combination with more rapid turn around time makes the development of a diagnostic ELISA an attractive alternative to VN.
14
Since it would prove extremely labour intensive and costly to produce an ELISA based on whole purified ERBV particles, we have begun work to engineer high level production of ERBV VLPs from recombinant baculovirus infected insect cells. SF9 cells are grown in suspension culture, and this system is amenable to upgrading to large scale production. Our experience with production of ERAV VLPs has shown that this method of expressing viral protein provides significantly more viral antigen than traditional virus culture and purification. The viability of the ERBV VLPs as alternative ELISA and vaccine antigens will depend on their antigenicity and immunogenicity when compared to whole ERBV virions. VLPs described for other picornaviruses such as FMDV have shown they are highly immunogenic and have similar antigenic structure to whole virions (1). Similar studies for ERBV VLPs are on going. 5.1 Implications Viral respiratory diseases of horses are a major cause of economic loss to the industry not only in Australia but also throughout the world. Viruses are also recognised causes of serious losses from abortion, perinatal foal mortalities and central nervous system disease. We have identified of the presence of ERBV in Australia, which is a recognised cause of acute respiratory illness. Identification of viruses that cause respiratory disease is the first stage in development of methods for their diagnosis and control. Both a diagnostic RT-PCR to detect the presence of this virus in clinical samples, and an ELISA to detect ERBV antibodies are being developed to this end. The most efficient means of preventing and controlling virus disease is by vaccination, and preliminary work has begun to develop an ERBV vaccine. 5.2 Recommendations The ERBV diagnostic PCR will be incorporated into the respiratory virus PCR panel which is available at the CEV for routine diagnosis and incorporates PCRs for EHV1, EHV4, EAdV1, EAdV2, EHV2, EHV3, EHV5 and ERAV. We are now refining the technology whereby instead of each virus specific PCR being conducted separately, a single 'multiplex' reaction mixture could be used, for example, for all respiratory viruses, for viruses that cause abortion, or diahorrea. These multiplex reactions are part of ongoing work. The development of both the diagnostic ELISA and the ERBV is contingent upon the production of ERBV VLPs and this work is in progress.
15
6. References 1. Abrams, C. C., and G. J. Belsham. 1994. The antigenicity of foot-and-mouth disease virus P1-
2A polyproteins and empty capsids produced in vaccinia virus and baculovirus expression systems. Abstracts F6, 8th Meeting of the European Study Group on the Molecular Biology of Picornaviruses, Korpilampi.
2. Abrams, C. C., A. M. King, and G. J. Belsham. 1995. Assembly of foot-and-mouth disease
virus empty capsids synthesized by a vaccinia virus expression system. J Gen Virol. 76:3089-98. 3. Carman, S., S. Rosendal, L. Huber, C. Gyles, S. McKee, R. A. Willoughby, E. Dubovi, J.
Thorsen, and D. Lein. 1997. Infectious agents in acute respiratory disease in horses in Ontario. J Vet Diagn Invest. 9:17-23.
4. Holmes, D. F., M. J. Kemen, and L. Coggins. 1978. Equine rhinovirus infection - serologic
evidence of infection in selected horse populations. In J. T. Bryans and H. Gerber (ed.), Equine Infectious diseases. Karger, Basel.
5. Laemmli, U. K. 1970. Cleavage of structural proteins during the assembly of the head of
bacteriophage T4. Nature. 227:680-5. 6. Li, F., G. F. Browning, M. J. Studdert, and B. S. Crabb. 1996. Equine rhinovirus 1 is more
closely related to foot-and-mouth disease virus than to other picornaviruses. Proc Natl Acad Sci U S A. 93:990-5.
7. Li, F., H. E. Drummer, N. Ficorilli, M. J. Studdert, and B. S. Crabb. 1997. Identification of
noncytopathic equine rhinovirus 1 as a cause of acute febrile respiratory disease in horses. J Clin Microbiol. 35:937-43.
8. Reubel, G. H., B. S. Crabb, and M. J. Studdert. 1995. Diagnosis of equine gammaherpesvirus 2
and 5 infections by polymerase chain reaction. Arch Virol. 140:1049-60. 9. Rueckert, R. R., and M. A. Pallansch. 1981. Preparation and characterization of
encephalomyocarditis (EMC) virus. Methods Enzymol. 78:315-25. 10. Steck, F., B. Hofer, B. Schaeren, J. Nicholet, and H. Gerber. 1978. Equine rhinoviruses: new
serotypes. In J. T. Bryans and H. Gerber (ed.), Equine Infectious Diseases, vol. IV. Karger, Basel. 11. Studdert, M. J., and L. J. Gleeson. 1978. Isolation and characterisation of an equine rhinovirus.
Zentralbl Veterinarmed [B]. 25:225-37. 12. Thompson, J. D., T. J. Gibson, F. Plewniak, F. Jeanmougin, and D. G. Higgins. 1997. The
CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 25:4876-82.
13. Wutz, G., H. Auer, N. Nowotny, B. Grosse, T. Skern, and E. Kuechler. 1996. Equine
rhinovirus serotypes 1 and 2: relationship to each other and to aphthoviruses and cardioviruses. J Gen Virol. 77:1719-30.