Accepted Manuscript EQCM and XPS analysis of 1,2,4-triazole and 3-amino-1,2,4-triazole as Copper Corrosion Inhibitors in Chloride Solution Matjaž Finšgar PII: S0010-938X(13)00393-4 DOI: http://dx.doi.org/10.1016/j.corsci.2013.08.026 Reference: CS 5522 To appear in: Corrosion Science Received Date: 12 April 2013 Accepted Date: 29 August 2013 Please cite this article as: M. Finšgar, EQCM and XPS analysis of 1,2,4-triazole and 3-amino-1,2,4-triazole as Copper Corrosion Inhibitors in Chloride Solution, Corrosion Science (2013), doi: http://dx.doi.org/10.1016/j.corsci. 2013.08.026 This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Transcript
Accepted Manuscript
EQCM and XPS analysis of 1,2,4-triazole and 3-amino-1,2,4-triazole as Copper
EQCM and XPS analysis of 1,2,4-triazole and 3-amino-1,2,4-triazole as Copper Corrosion
Inhibitors in Chloride Solution
Matjaž Finšgar∗
University of Maribor, Faculty of Chemistry and Chemical Engineering (UM FKKT), Smetanova ulica
17, 2000 Maribor, Slovenia
Abstract
In this study, the influence of the amino functional group in the 1,2,4-triazole at position C3 (the
3-amino-1,2,4-triazole compound), on the surface layer formation and surface chemistry of these two
Cu corrosion inhibitors is explored. Special attention is devoted to the orientation of these two
molecules and the way they bond to the Cu surface. With the aim of obtaining electrochemical quartz
crystal microbalance and X-ray photoelectron spectroscopy measurements, the article discusses why
differences in the corrosion inhibition effectiveness of these two molecules exist. Moreover, the
thicknesses of the inhibitor surface layers formed on the Cu are determined.
Keywords: A. copper, B. XPS, C. interfaces, C. neutral inhibition
∗ Corresponding author Address: University of Maribor, Faculty of Chemistry and Chemical Engineering (UM FKKT), Smetanova ulica 17, 2000 Maribor, Slovenia e-mail: [email protected]; phone: +386 2 2294 447
2
1 Introduction
Copper and its alloys are used in many environments and applications due to their desirable
properties. However, the corrosion resistance of copper-based materials strongly depends on their
environment. Corrosion cannot be completely prevented, but an effort is made to minimize it, which is
the ultimate goal of corrosion studies. One convenient approach to minimizing corrosion damage is to
use corrosion inhibitors, which are substances used in very low amounts that efficiently slow the rate
of corrosion.
The studies mentioned below show that 1,2,4-triazole compounds are particularly worth
investigating as they act as efficient corrosion inhibitors for various materials in diverse environments.
In this work, the surface layer formation and surface chemistry of 1,2,4-triazole (TRZ) and its
derivative, 3-amino-1,2,4-triazole (3-AT), adsorbed on Cu from 3 wt.% aqueous solution, were
investigated (the molecular structures are given in Fig. 1). Sometimes the acronyms TA and ATA are
used in the literature for 1,2,4-triazole and 3-amino-1,2,4-triazole, respectively, but not herein. Thus
far, TRZ action as a Cu corrosion inhibitor has been relatively unexamined in chloride media, most
likely because it is not very effective [1]. Moreover, corrosion tests performed for 0.1, 1.0, and 10.0
mM TRZ or 3-AT in 3 wt.% NaCl solutions showed that 3-AT was a more effective Cu corrosion
inhibitor than TRZ (these results are not shown herein, because that was not the aim of this work).
However, study of the influence of different functional groups (amino in the present case) for the
given compound (1,2,4-triazole in the present case) is especially important for an understanding of the
inhibitor action, which consequently provides an opportunity to design new potential inhibitor
compounds (for example, sometimes this is needed to reduce toxicity). Herein, the reason why 3-AT
(and not TRZ) is an effective Cu corrosion inhibitor will be discussed. In particular, in this work
electrochemical quartz crystal microbalance (EQCM) was employed to study the kinetics of the
4. Angle-resolved XPS measurements suggest that TRZ and 3-AT molecules are directed to the
Cu surface via N atoms.
5. The thicknesses of the TRZ and 3-AT layers on the Cu surface formed after 1 h of immersion
are 1.6 ± 0.5 nm and 0.9 ± 0.3 nm, respectively, as determined by the Tougaard method.
Acknowledgements
The author would like to thank Dr. Janez Kovač, Dr. Miha Čekada, and Dr. Peter Panjan for their
valuable discussions.
20
5 References
[1] S. El Issami, L. Bazzi, M. Mihit, B. Hammouti, S. Kertit, E.A. Addi, R. Salghi, Triazolic compounds as corrosion inhibitors for copper in hydrochloric acid, Pigm. Resin. Technol., 36 (2007) 161-168. [2] P.G. Fox, P.A. Bradley, 1 : 2 : 4-triazole as a corrosion inhibitor for copper, Corrosion Science, 20 (1980) 643-649. [3] M.L. Zheludkevich, K.A. Yasakau, S.K. Poznyak, M.G.S. Ferreira, Triazole and thiazole derivatives as corrosion inhibitors for AA2024 aluminium alloy, Corrosion Science, 47 (2005) 3368-3383. [4] B. Trachli, M. Keddam, H. Takenouti, A. Srhiri, Protective effect of electropolymerized 3-amino 1,2,4-triazole towards corrosion of copper in 0.5 M NaCl, Corrosion Science, 44 (2002) 997-1008. [5] E.-S.M. Sherif, R.M. Erasmus, J.D. Comins, Effects of 3-amino-1,2,4-triazole on the inhibition of copper corrosion in acidic chloride solutions, Journal of Colloid and Interface Science, 311 (2007) 144-151. [6] E.-S.M. Sherif, R.M. Erasmus, J.D. Comins, Corrosion of copper in aerated synthetic sea water solutions and its inhibition by 3-amino-1,2,4-triazole, Journal of Colloid and Interface Science, 309 (2007) 470-477. [7] M. Finšgar, I. Milošev, B. Pihlar, Inhibition of copper corrosion studied by electrochemical and EQCN techniques, Acta Chim. Slov., 54 (2007) 591-597. [8] A. Kokalj, S. Peljhan, M. Finšgar, I. Milošev, What Determines the Inhibition Effectiveness of ATA, BTAH, and BTAOH Corrosion Inhibitors on Copper?, Journal of the American Chemical Society, 132 (2010) 16657-16668. [9] A. Lalitha, S. Ramesh, S. Rajeswari, Surface protection of copper in acid medium by azoles and surfactants, Electrochimica Acta, 51 (2005) 47-55. [10] W. Qafsaoui, C. Blanc, N. Pébère, H. Takenouti, A. Srhiri, G. Mankowski, Quantitative characterization of protective films grown on copper in the presence of different triazole derivative inhibitors, Electrochimica Acta, 47 (2002) 4339-4346. [11] W. Qafsaoui, C. Blanc, J. Roques, N. Pebere, A. Srhiri, C. Mijoule, G. Mankowski, Pitting corrosion of copper in sulphate solutions: inhibitive effect of different triazole derivative inhibitors, Journal of Applied Electrochemistry, 31 (2001) 223-231. [12] W. Qafsaoui, C. Blanc, N. Pebere, H. Takenouti, A. Srhiri, G. Mankowski, Quantitative characterization of protective films grown on copper in the presence of different triazole derivative inhibitors, Electrochimica Acta, 47 (2002) 4339-4346. [13] Z. Mountassir, A. Srhiri, Electrochemical behaviour of Cu–40Zn in 3% NaCl solution polluted by sulphides: Effect of aminotriazole, Corrosion Science, 49 (2007) 1350-1361. [14] K. Rahmouni, H. Takenouti, N. Hajjaji, A. Srhiri, L. Robbiola, Protection of ancient and historic bronzes by triazole derivatives, Electrochimica Acta, 54 (2009) 5206-5215. [15] M. Elbakri, R. Touir, M. Ebn Touhami, A. Srhiri, M. Benmessaoud, Electrosynthesis of adherent poly(3-amino-1,2,4-triazole) films on brass prepared in nonaqueous solvents, Corrosion Science, 50 (2008) 1538-1545. [16] B.D. Mert, M.E. Mert, G. Kardaş, B. Yazıcı, Experimental and theoretical studies on electrochemical synthesis of poly(3-amino-1,2,4-triazole), Applied Surface Science, 258 (2012) 9668-9674. [17] Z. Khiati, A.A. Othman, M. Sanchez-Moreno, M.C. Bernard, S. Joiret, E.M.M. Sutter, V. Vivier, Corrosion inhibition of copper in neutral chloride media by a novel derivative of 1,2,4-triazole, Corrosion Science, 53 (2011) 3092-3099. [18] E.-S.M. Sherif, A.M. El Shamy, M.M. Ramla, A.O.H. El Nazhawy, 5-(Phenyl)-4H-1,2,4-triazole-3-thiol as a corrosion inhibitor for copper in 3.5% NaCl solutions, Materials Chemistry and Physics, 102 (2007) 231-239.
21
[19] Y.-C. Pan, Y. Wen, R. Zhang, Y.-Y. Wang, Z.-R. Zhang, H.-F. Yang, Electrochemical and SERS spectroscopic investigations of 4-methyl-4H-1,2,4-triazole-3-thiol monolayers self-assembled on copper surface, Applied Surface Science, 258 (2012) 3956-3961. [20] K. Rahmouni, N. Hajjaji, M. Keddam, A. Srhiri, H. Takenouti, The inhibiting effect of 3-methyl 1,2,4-triazole 5-thione on corrosion of copper in 3% NaCl in presence of sulphide, Electrochimica Acta, 52 (2007) 7519-7528. [21] Sudheer, M.A. Quraishi, Electrochemical and theoretical investigation of triazole derivatives on corrosion inhibition behavior of copper in hydrochloric acid medium, Corrosion Science, 70 (2013) 161-169. [22] E.-S.M. Sherif, R.M. Erasmus, J.D. Comins, Corrosion of copper in aerated acidic pickling solutions and its inhibition by 3-amino-1,2,4-triazole-5-thiol, Journal of Colloid and Interface Science, 306 (2007) 96-104. [23] M.M. Antonijević, S.M. Milić, M.B. Petrović, Films formed on copper surface in chloride media in the presence of azoles, Corrosion Science, 51 (2009) 1228-1237. [24] S. Ramesh, S. Rajeswari, Evaluation of inhibitors and biocide on the corrosion control of copper in neutral aqueous environment, Corrosion Science, 47 (2005) 151-169. [25] S. Ramesh, S. Rajeswari, S. Maruthamuthu, Corrosion inhibition of copper by new triazole phosphonate derivatives, Applied Surface Science, 229 (2004) 214-225. [26] F.M. Al-Kharafi, F.H. Al-Hajjar, A. Katrib, 3-phenyl-1,2,4-triazol-5-one as a corrosion inhibitor for copper, Corrosion Science, 26 (1986) 257-264. [27] M.M. El-Naggar, Bis-aminoazoles corrosion inhibitors for copper in 4.0 M HNO3 solutions, Corrosion Science, 42 (2000) 773-784. [28] M. Finšgar, I. Milošev, Corrosion study of copper in the presence of benzotriazole and its hydroxy derivative, Materials and Corrosion-Werkstoffe Und Korrosion, 62 (2011) 956-966. [29] M. Finšgar, S. Peljhan, A. Kokalj, J. Kovač, I. Milošev, Determination of the Cu2O Thickness on BTAH-Inhibited Copper by Reconstruction of Auger Electron Spectra, Journal of the Electrochemical Society, 157 (2010) C295-C301. [30] M. Finšgar, A. Lesar, A. Kokalj, I. Milošev, A comparative electrochemical and quantum chemical calculation study of BTAH and BTAOH as copper corrosion inhibitors in near neutral chloride solution, Electrochimica Acta, 53 (2008) 8287-8297. [31] M. Finšgar, I. Milošev, Inhibition of copper corrosion by 1,2,3-benzotriazole: A review, Corrosion Science, 52 (2010) 2737-2749. [32] M. Finšgar, 2-Mercaptobenzimidazole as a Copper Corrosion Inhibitor: Part I. Long-term Immersion, 3D-Profilometry, and Electrochemistry, Corrosion Science, 72 (2013) 82-89. [33] M. Finšgar, J. Kovač, I. Milošev, Surface Analysis of 1-Hydroxybenzotriazole and Benzotriazole Adsorbed on Cu by X-Ray Photoelectron Spectroscopy, Journal of the Electrochemical Society, 157 (2010) C52-C60. [34] Y.I. Kuznetsov, L.P. Kazansky, Physicochemical aspects of metal protection by azoles as corrosion inhibitors, Russian Chemical Reviews, 77 (2008) 219. [35] M. Finšgar, 2-Mercaptobenzimidazole as a Copper Corrosion Inhibitor: Part II. Surface Analysis Using X-ray Photoelectron Spectroscopy, Corrosion Science, 72 (2013) 90-98. [36] M. Finšgar, I. Milošev, Corrosion behaviour of stainless steels in aqueous solutions of methanesulfonic acid, Corrosion Science, 52 (2010) 2430-2438. [37] M. Finšgar, Galvanic series of different stainless steels and copper- and aluminium-based materials in acid solutions, Corrosion Science, 68 (2013) 51-56. [38] D.A. Shirley, High-Resolution X-Ray Photoemission Spectrum of the Valence Bands of Gold, Physical Review B, 5 (1972) 4709–4714. [39] V.K. Kaushik, Identification of oxidation states of copper in mixed oxides and chlorides using ESCA, Spectrochimica Acta Part B: Atomic Spectroscopy, 44 (1989) 581-587. [40] S. Tougaard, Quantification of Nano-structures by Electron Spectroscopy in Surface Analysis by Auger and X-Ray Photoelectron spectroscopy, in: D. Briggs, J.T. Grant (Eds.) Surface Spectra, IM Publications, Manchester, UK, 2003, pp. 295-343. [41] S. Tougaard, Surface nanostructure determination by x-ray photoemission spectroscopy peak shape analysis, in, AVS, Mineapolis, Minnesota (USA), 1996, pp. 1415-1423.
22
[42] V. Brusic, M.A. Frisch, B.N. Eldridge, F.P. Novak, F.B. Kaufman, B.M. Rush, G.S. Frankel, Copper Corrosion With and Without Inhibitors, Journal of The Electrochemical Society, 138 (1991) 2253-2259. [43] J.-H. Chen, Z.-C. Lin, S. Chen, L.-H. Nie, S.-Z. Yao, An XPS and BAW sensor study of the structure and real-time growth behaviour of a complex surface film on copper in sodium chloride solutions (pH = 9), containing a low concentration of benzotriazole, Electrochimica Acta, 43 (1998) 265-274. [44] P.E. Laibinis, G.M. Whitesides, D.L. Allara, Y.T. Tao, A.N. Parikh, R.G. Nuzzo, Comparison of the structures and wetting properties of self-assembled monolayers of n-alkanethiols on the coinage metal surfaces, copper, silver, and gold, Journal of the American Chemical Society, 113 (1991) 7152-7167. [45] M. Finšgar, S. Fassbender, S. Hirth, I. Milošev, Electrochemical and XPS study of polyethyleneimines of different molecular sizes as corrosion inhibitors for AISI 430 stainless steel in near-neutral chloride media, Materials Chemistry and Physics, 116 (2009) 198-206. [46] M. Finšgar, S. Fassbender, F. Nicolini, I. Milošev, Polyethyleneimine as a corrosion inhibitor for ASTM 420 stainless steel in near-neutral saline media, Corrosion Science, 51 (2009) 525-533. [47] A.V. Naumkin, A. Kraut-Vass, S.W. Gaarenstroom, C.J. Powell, NIST Standard Reference Database 20, Version 4.1 (web version), (http://srdata.nist.gov/xps/), in, 2003. [48] T. Hashemi, C.A. Hogarth, The mechanism of corrosion inhibition of copper in NaCl solution by benzotriazole studied by electron spectroscopy, Electrochimica Acta, 33 (1988) 1123-1127. [49] C.C. Chusuei, M.A. Brookshier, D.W. Goodman, Correlation of Relative X-ray Photoelectron Spectroscopy Shake-up Intensity with CuO Particle Size, Langmuir, 15 (1999) 2806-2808. [50] M.C. Biesinger, L.W.M. Lau, A.R. Gerson, R.S.C. Smart, Resolving surface chemical states in XPS analysis of first row transition metals, oxides and hydroxides: Sc, Ti, V, Cu and Zn, Applied Surface Science, 257 (2010) 887-898. [51] G. Xue, X.-Y. Huang, J. Dong, J. Zhang, The formation of an effective anti-corrosion film on copper surfaces from 2-mercaptobenzimidazole solution, Journal of Electroanalytical Chemistry and Interfacial Electrochemistry, 310 (1991) 139-148. [52] I. Platzman, R. Brener, H. Haick, R. Tannenbaum, Oxidation of Polycrystalline Copper Thin Films at Ambient Conditions, The Journal of Physical Chemistry C, 112 (2008) 1101-1108. [53] D.-D. Li, J.-L. Tian, W. Gu, X. Liu, S.-P. Yan, A novel 1,2,4-triazole-based copper(II) complex: Synthesis, characterization, magnetic property and nuclease activity, Journal of Inorganic Biochemistry, 104 (2010) 171-179. [54] T. Asaji, H. Sakai, D. Nakamura, Magnetic phase transitions in dibromo(4H-1,2,4-triazole)copper(II) and related copper(II) complexes as studied by nitrogen-14 nuclear quadrupole resonance and magnetic susceptibility measurements, Inorganic Chemistry, 22 (1983) 202-206. [55] G.V. Romanenko, Z.A. Savelieva, N.V. Podberezskaya, S.V. Larionov, Structure of the Cu(II) chloride complex with 4-amino-l,2,4-triazole Cu(C2H4N4)Cl2, J Struct Chem, 38 (1997) 171-176. [56] S.M. Ansar, R. Haputhanthri, B. Edmonds, D. Liu, L. Yu, A. Sygula, D. Zhang, Determination of the Binding Affinity, Packing, and Conformation of Thiolate and Thione Ligands on Gold Nanoparticles, The Journal of Physical Chemistry C, 115 (2010) 653-660. [57] A. Kokalj, N. Kovačević, S. Peljhan, M. Finšgar, A. Lesar, I. Milošev, Triazole, Benzotriazole, and Naphthotriazole as Copper Corrosion Inhibitors: I. Molecular Electronic and Adsorption Properties, Chemphyschem, 12 (2011) 3547-3555. [58] D. Chadwick, T. Hashemi, Electron spectroscopy of corrosion inhibitors: Surface films formed by 2-mercaptobenzothiazole and 2-mercaptobenzimidazole on copper, Surface Science, 89 (1979) 649-659. [59] G. Fonder, F. Laffineur, J. Delhalle, Z. Mekhalif, Alkanethiol-oxidized copper interface: The critical influence of concentration, Journal of Colloid and Interface Science, 326 (2008) 333-338. [60] C. Duret-Thual, D. Costa, W.P. Yang, P. Marcus, The role of thiosulfates in the pitting corrosion of Fe-17Cr alloys in neutral chloride solution: Electrochemical and XPS study, Corrosion Science, 39 (1997) 913-933.
23
[61] V.I. Nefedov, M.N. Firsov, I.S. Shaplygin, Electronic structures of MRhO2, MRh2O4, RhMO4 and Rh2MO6 on the basis of X-ray spectroscopy and ESCA data, Journal of Electron Spectroscopy and Related Phenomena, 26 (1982) 65-78. [62] D. Chadwick, T. Hashemi, Benzotriazole adsorption on copper studied by X-ray photoelectron spectroscopy, Journal of Electron Spectroscopy and Related Phenomena, 10 (1977) 79-83. [63] M.A. Fazal, A.S.M.A. Haseeb, H.H. Masjuki, Corrosion mechanism of copper in palm biodiesel, Corrosion Science, 67 (2013) 50-59. [64] D. Chadwick, T. Hashemi, Adsorbed corrosion inhibitors studied by electron spectroscopy: Benzotriazole on copper and copper alloys, Corrosion Science, 18 (1978) 39-51. [65] C.J. Powell, A. Jablonski, NIST Electron Inelastic-Mean-Free-Path Database – version 1.1, National Institute of Standards and Technology, in, 2000. [66] A. Abdureyim, K.K. Okudaira, Y. Harada, S. Masuda, M. Aoki, K. Seki, E. Ito, N. Ueno, Characterization of 4-mercaptohydrocynnamic acid self-assembled film on Au(111) by means of X-ray photoelectron spectroscopy, Journal of Electron Spectroscopy and Related Phenomena, 114-116 (2001) 371-374. [67] C.M. Whelan, M.R. Smyth, C.J. Barnes, HREELS, XPS, and Electrochemical Study of Benzenethiol Adsorption on Au(111), Langmuir, 15 (1998) 116-126. [68] P.E. Laibinis, C.D. Bain, G.M. Whitesides, Attenuation of photoelectrons in monolayers of n-alkanethiols adsorbed on copper, silver, and gold, The Journal of Physical Chemistry, 95 (1991) 7017-7021. [69] G. Lewis, P.G. Fox, The thickness of thin surface films determined by photo-electron spectroscopy, Corrosion Science, 18 (1978) 645-650. [70] R.F. Roberts, X-ray photoelectron spectroscopic characterization of copper oxide surfaces treated with benzotriazole, Journal of Electron Spectroscopy and Related Phenomena, 4 (1974) 273-291.
24
Table 1: Calculated intensity ratios at different take-off angles
θ IC 1s/ICu 2p IO 1s/ICu 2p IN 1s/ICu 2p IC 1s/IN 1s IO 1s/IN 1s IO 1s/IC 1s
TRZ
5° 1.36 0.26 0.31 4.34 0.82 0.19
20° 0.72 0.16 0.31 2.35 0.53 0.22
45° 0.46 0.16 0.23 1.96 0.67 0.34
90° 0.16 0.10 0.10 1.64 1.04 0.63
3-AT
5° 0.57 0.13 0.38 1.49 0.35 0.24
20° 0.50 0.17 0.40 1.26 0.43 0.34
45° 0.30 0.15 0.34 0.89 0.47 0.52
90° 0.25 0.12 0.31 0.81 0.40 0.49
25
Figure captions
Figure 1: The structures of TRZ and 3-AT molecules.
Figure 2: The mass change vs. time during exposure of a Cu-plated electrode in 3 wt.% NaCl solution
containing 10 mM TRZ or 3-AT.
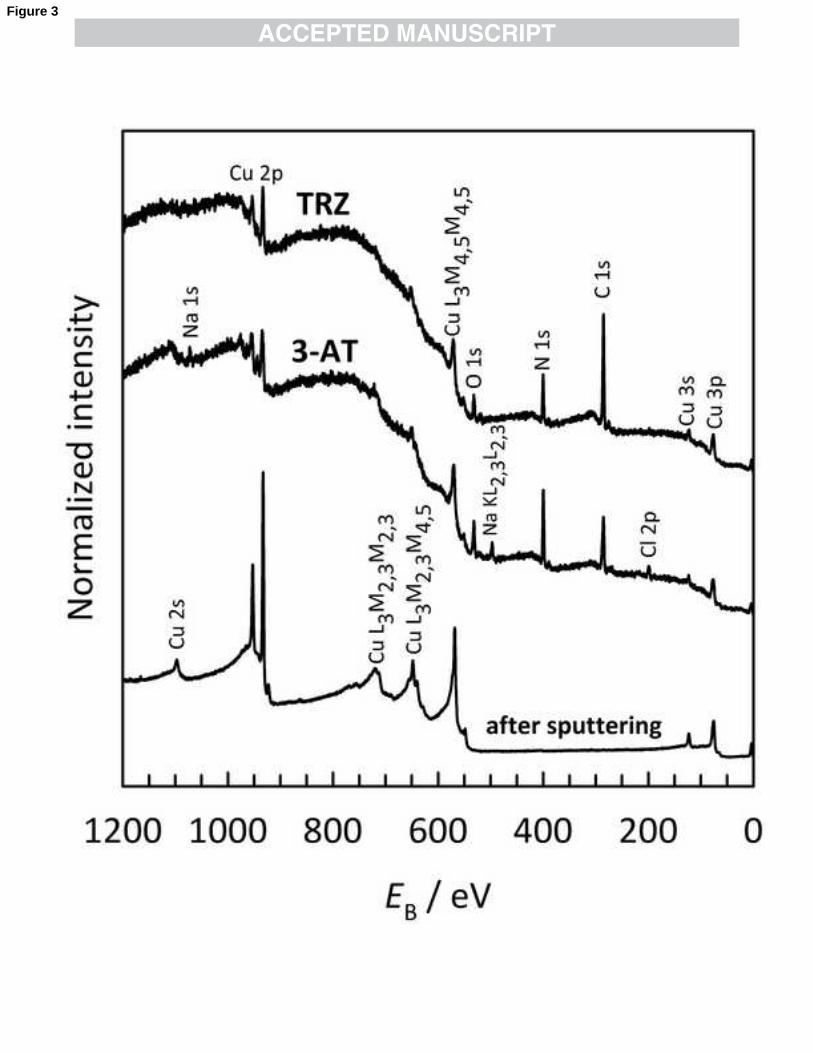
Figure 3: XPS survey spectra measured at θ = 45° of the 3-AT- and TRZ-treated (upper spectra) and
sputter-cleaned Cu (lower spectrum).
Figure 4: Cu 2p, Cu L3M4,5M4,5, N 1s, C 1s, and O 1s spectra analysed at different take-off angles. The
Cu sample was treated for 1 h in 3 wt.% NaCl solution containing 10 mM TRZ. The lowest curve
represents the sputter-cleaned Cu sample.
Figure 5: Cu 2p, Cu L3M4,5M4,5, N 1s, C 1s, and O 1s spectra analysed at different take-off angles. The
Cu sample was treated for 1 h in 3 wt.% NaCl solution containing 10 mM 3-AT. The lowest curve
represents the sputter-cleaned Cu sample.
Figure 6: The XPS measured spectra of the (a,b) TRZ- and (c,d) 3-AT-treated Cu samples and the
background corrected spectra for IMFP of 2 and 4 nm. Thicknesses of 1.6 ± 0.5 nm and 0.9 ± 0.3 nm
of the TRZ and 3-AT layers, respectively, were determined by the Tougaard method [40, 41].