ESR spectroscopy identifies inhibitory Cu 2þ sites in a DNA-modifying enzyme to reveal determinants of catalytic specificity Zhongyu Yang a,1,2 , Michael R. Kurpiewski b,1 , Ming Ji a,1 , Jacque E. Townsend b , Preeti Mehta b,3 , Linda Jen-Jacobson b,4 , and Sunil Saxena a,4 a Department of Chemistry; and b Department of Biological Sciences, University of Pittsburgh, Pittsburgh, PA 15260 Edited by Brian M. Hoffman, Northwestern University, Evanston, IL, and approved March 9, 2012 (received for review January 18, 2012) The relationship between DNA sequence recognition and catalytic specificity in a DNA-modifying enzyme was explored using para- magnetic Cu 2þ ions as probes for ESR spectroscopic and biochem- ical studies. Electron spin echo envelope modulation spectroscopy establishes that Cu 2þ coordinates to histidine residues in the EcoRI endonuclease homodimer bound to its specific DNA recognition site. The coordinated His residues were identified by a unique use of Cu 2þ -ion based long-range distance constraints. Double elec- tron-electron resonance data yield Cu 2þ -Cu 2þ and Cu 2þ -nitroxide distances that are uniquely consistent with one Cu 2þ bound to His114 in each subunit. Isothermal titration calorimetry confirms that two Cu 2þ ions bind per complex. Unexpectedly, Mg 2þ -cata- lyzed DNA cleavage by EcoRI is profoundly inhibited by Cu 2þ bind- ing at these hitherto unknown sites, 13 Å away from the Mg 2þ positions in the catalytic centers. Molecular dynamics simulations suggest a model for inhibition of catalysis, whereby the Cu 2þ ions alter critical protein-DNA interactions and water molecule positions in the catalytic sites. In the absence of Cu 2þ , the Mg 2þ -dependence of EcoRI catalysis shows positive cooperativity, which would en- hance EcoRI inactivation of foreign DNA by irreparable double- strand cuts, in preference to readily repaired single-strand nicks. Nonlinear Poisson-Boltzmann calculations suggest that this coop- erativity arises because the binding of Mg 2þ in one catalytic site makes the surface electrostatic potential in the distal catalytic site more negative, thus enhancing binding of the second Mg 2þ . Taken together, our results shed light on the structural and electrostatic factors that affect site-specific catalysis by this class of endonu- cleases. DEER ∣ ESEEM ∣ metalloenzyme catalysis ∣ protein-DNA recognition ∣ DNA distortion T he biochemical basis of specificity in the interaction of pro- teins with DNA sites is a major problem of modern molecular genetics. Studies of many protein-DNA complexes by crystallo- graphy have elucidated the intermolecular recognition contacts (1), but it is clear that point-to-point contacts cannot fully explain specificity. Solution biochemical and computational studies have shown that a comprehensive view of specificity determination must also include factors such as shape recognition (2), mutual accommodation of the macromolecules through DNA distortion or conformational selection (1, 3–6), and/or DNA-induced pro- tein folding (5, 7). Thermodynamic studies have revealed that specific protein-DNA association is often driven primarily by the favorable entropy increase provided by desolvation of the ap- posed complementary surfaces (8–10). For those DNA-binding proteins that are also DNA-modifying enzymes (nucleases, methylases, recombinases, repair enzymes, etc.) a key question is the relationship between DNA-binding specificity and catalytic specificity. One exemplary system for addressing specificity determination is the EcoRI endonuclease (3, 11), a 62 kDa homodimer that recognizes the DNA site 5′- GAATTC-3′ and binds as much as 90,000-fold better (3, 12) than at sites that differ by one base pair from the specific site. Full binding specificity is achieved in the absence of divalent metal (3, 12, 13). Like most phosphotransferases and all metallonucleases, EcoRI requires a divalent metal as an obligatory participant in catalysis. The fully symmetrical complex of EcoRI homodimer with its palindromic cognate DNA contains two catalytic centers, each formed from elements of both subunits. In each center, one Mg 2þ coordinates with a pair of carboxylate side chains and the scissile phosphate GpAATTC (14, 15). Catalytic rates for EcoRI vary according to the series Mg 2þ ≈ Mn 2þ > Co 2þ ≫ Zn 2þ ≫ Cd 2þ > Ni 2þ (16, 17). Ca 2þ cannot support catalysis, but can act as an inhibitor by competing with the physiological cofactor Mg 2þ (15). Based on studies with stereospecific phosphate analogues and molecular dynamics simulations, Kurpiewski et al. (15) proposed a model of EcoRI catalysis in which the phosphate at GApATTC (one step downstream from the scissile phosphate GpAATTC) precisely orients the attacking nucleophilic water through an in- termediate “relay” water molecule. This phosphate is uniquely positioned by the “kinked” DNA distortion in the fully correct recognition complex (15). The postulated role of sequence- dependent DNA distortion may account for the fact that catalytic rate depends on recognition of the correct DNA site: When Mg 2þ is added to prebound protein-DNA complexes, the cataly- tic rates for the chemical steps of double-strand DNA cleavage are 10 6 -fold higher for the correct GAATTC DNA sites than for sites that differ from GAATTC by as little as one base pair (3, 15). That is, only the fully correct recognition complex sup- ports the maximum catalytic rate. The multiplicative binding and catalytic specificities enable EcoRI to distinguish a single target site from the competing excess of near-correct DNA sites in vivo (3, 9, 12). Here we combine electron spin resonance (ESR) spectroscopy and biochemical studies to study the locations and functional con- sequences of paramagnetic Cu 2þ ions bound at hitherto unknown sites in the EcoRI endonuclease-DNA complex. Cu 2þ ions, which do not support DNA cleavage (16), have the potential to provide Author contributions: L.J.-J. and S.S. designed research; Z.Y., M.R.K., M.J., J.E.T., and P.M. performed research; Z.Y., M.R.K., M.J., J.E.T., L.J.-J., and S.S. analyzed data; and Z.Y., M.J., L.J.-J., and S.S. wrote the paper. The authors declare no conflict of interest. This article is a PNAS Direct Submission. 1 Z.Y., M.R.K., and M.J. contributed equally to this work. 2 Present address: Jules Stein Eye Institute and Department of Chemistry and Biochemistry, University of California, Los Angeles, CA 90095. 3 Present address: Department of Biochemistry, Stony Brook University, Stony Brook, NY 11794. 4 To whom correspondence may be addressed. E-mail: [email protected] or [email protected]. See Author Summary on page 6366 (volume 109, number 17). This article contains supporting information online at www.pnas.org/lookup/suppl/ doi:10.1073/pnas.1200733109/-/DCSupplemental. www.pnas.org/cgi/doi/10.1073/pnas.1200733109 PNAS ∣ Published online April 9, 2012 ∣ E993–E1000 BIOCHEMISTRY CHEMISTRY PNAS PLUS
Transcript
ESR spectroscopy identifies inhibitory Cu2þ
sites in a DNA-modifying enzyme to revealdeterminants of catalytic specificityZhongyu Yanga,1,2, Michael R. Kurpiewskib,1, Ming Jia,1, Jacque E. Townsendb, Preeti Mehtab,3,Linda Jen-Jacobsonb,4, and Sunil Saxenaa,4
aDepartment of Chemistry; and bDepartment of Biological Sciences, University of Pittsburgh, Pittsburgh, PA 15260
Edited by Brian M. Hoffman, Northwestern University, Evanston, IL, and approved March 9, 2012 (received for review January 18, 2012)
The relationship between DNA sequence recognition and catalyticspecificity in a DNA-modifying enzyme was explored using para-magnetic Cu2þ ions as probes for ESR spectroscopic and biochem-ical studies. Electron spin echo envelope modulation spectroscopyestablishes that Cu2þ coordinates to histidine residues in the EcoRIendonuclease homodimer bound to its specific DNA recognitionsite. The coordinated His residues were identified by a uniqueuse of Cu2þ-ion based long-range distance constraints. Double elec-tron-electron resonance data yield Cu2þ-Cu2þ and Cu2þ-nitroxidedistances that are uniquely consistent with one Cu2þ bound toHis114 in each subunit. Isothermal titration calorimetry confirmsthat two Cu2þ ions bind per complex. Unexpectedly, Mg2þ-cata-lyzed DNA cleavage by EcoRI is profoundly inhibited by Cu2þ bind-ing at these hitherto unknown sites, 13 Å away from the Mg2þ
positions in the catalytic centers. Molecular dynamics simulationssuggest a model for inhibition of catalysis, whereby the Cu2þ ionsalter critical protein-DNA interactions andwatermolecule positionsin the catalytic sites. In the absence of Cu2þ, the Mg2þ-dependenceof EcoRI catalysis shows positive cooperativity, which would en-hance EcoRI inactivation of foreign DNA by irreparable double-strand cuts, in preference to readily repaired single-strand nicks.Nonlinear Poisson-Boltzmann calculations suggest that this coop-erativity arises because the binding of Mg2þ in one catalytic sitemakes the surface electrostatic potential in the distal catalytic sitemore negative, thus enhancing binding of the second Mg2þ. Takentogether, our results shed light on the structural and electrostaticfactors that affect site-specific catalysis by this class of endonu-cleases.
The biochemical basis of specificity in the interaction of pro-teins with DNA sites is a major problem of modern molecular
genetics. Studies of many protein-DNA complexes by crystallo-graphy have elucidated the intermolecular recognition contacts(1), but it is clear that point-to-point contacts cannot fully explainspecificity. Solution biochemical and computational studies haveshown that a comprehensive view of specificity determinationmust also include factors such as shape recognition (2), mutualaccommodation of the macromolecules through DNA distortionor conformational selection (1, 3–6), and/or DNA-induced pro-tein folding (5, 7). Thermodynamic studies have revealed thatspecific protein-DNA association is often driven primarily by thefavorable entropy increase provided by desolvation of the ap-posed complementary surfaces (8–10).
For those DNA-binding proteins that are also DNA-modifyingenzymes (nucleases, methylases, recombinases, repair enzymes,etc.) a key question is the relationship between DNA-bindingspecificity and catalytic specificity. One exemplary system foraddressing specificity determination is the EcoRI endonuclease(3, 11), a 62 kDa homodimer that recognizes the DNA site 5′-GAATTC-3′ and binds as much as 90,000-fold better (3, 12) than
at sites that differ by one base pair from the specific site. Fullbinding specificity is achieved in the absence of divalent metal(3, 12, 13).
Like most phosphotransferases and all metallonucleases,EcoRI requires a divalent metal as an obligatory participant incatalysis. The fully symmetrical complex of EcoRI homodimerwith its palindromic cognate DNA contains two catalytic centers,each formed from elements of both subunits. In each center, oneMg2þ coordinates with a pair of carboxylate side chains and thescissile phosphate GpAATTC (14, 15). Catalytic rates for EcoRIvary according to the series Mg2þ ≈Mn2þ > Co2þ ≫ Zn2þ ≫Cd2þ > Ni2þ (16, 17). Ca2þ cannot support catalysis, but canact as an inhibitor by competing with the physiological cofactorMg2þ (15).
Based on studies with stereospecific phosphate analogues andmolecular dynamics simulations, Kurpiewski et al. (15) proposeda model of EcoRI catalysis in which the phosphate at GApATTC(one step downstream from the scissile phosphate GpAATTC)precisely orients the attacking nucleophilic water through an in-termediate “relay” water molecule. This phosphate is uniquelypositioned by the “kinked” DNA distortion in the fully correctrecognition complex (15). The postulated role of sequence-dependent DNA distortion may account for the fact that catalyticrate depends on recognition of the correct DNA site: WhenMg2þ is added to prebound protein-DNA complexes, the cataly-tic rates for the chemical steps of double-strand DNA cleavageare 106-fold higher for the correct GAATTC DNA sites thanfor sites that differ from GAATTC by as little as one base pair(3, 15). That is, only the fully correct recognition complex sup-ports the maximum catalytic rate. The multiplicative bindingand catalytic specificities enable EcoRI to distinguish a singletarget site from the competing excess of near-correct DNA sitesin vivo (3, 9, 12).
Here we combine electron spin resonance (ESR) spectroscopyand biochemical studies to study the locations and functional con-sequences of paramagnetic Cu2þ ions bound at hitherto unknownsites in the EcoRI endonuclease-DNA complex. Cu2þ ions, whichdo not support DNA cleavage (16), have the potential to provide
Author contributions: L.J.-J. and S.S. designed research; Z.Y., M.R.K., M.J., J.E.T., andP.M. performed research; Z.Y., M.R.K., M.J., J.E.T., L.J.-J., and S.S. analyzed data; and Z.Y.,M.J., L.J.-J., and S.S. wrote the paper.
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.1Z.Y., M.R.K., and M.J. contributed equally to this work.2Present address: Jules Stein Eye Institute and Department of Chemistry and Biochemistry,University of California, Los Angeles, CA 90095.
3Present address: Department of Biochemistry, Stony Brook University, Stony Brook,NY 11794.
fixed points of reference for ESR studies of the structure anddynamics of these complexes and other phosphotransferases.We used continuous wave (CW) ESR and electron spin echoenvelope modulation (ESEEM) to probe the interaction of theCu2þ electron spin with surrounding nuclei. These experimentsestablished that Cu2þ ions are coordinated to EcoRI histidines.Isothermal titration calorimetry (ITC) determined a stoichiome-try of two Cu2þ bound per EcoRI dimer-DNA complex. We iden-tified His114 (one in each monomer) as the site of Cu2þ bindingby double electron-electron resonance (DEER)-ESR measure-ments of both Cu2þ-Cu2þ and Cu2þ-nitroxide distances. We con-firmed this identification by showing that the His114Tyr mutationabolished the effect of Cu2þ on both DNA binding and DNAcleavage.
Unexpectedly, we found that Cu2þ inhibits the chemical stepof DNA cleavage, despite binding at sites 13 Å away from those atwhich Mg2þ binds. Molecular dynamics simulations suggest thatupon Cu2þ binding at His114, protein side chains and watermolecules that are important in catalysis are displaced and thecrucial position of the 3′-neighboring DNA phosphate is altered.This model differs from any previously proposed for metal inhi-bition of a metallonuclease. We also show that there is positivecooperativity in Mg2þ association with the EcoRI-DNA complexto promote catalysis, and we propose an electrostatic basis for thisinteraction between catalytic sites. Positive cooperativity betweencatalytic sites may help ensure that EcoRI fulfills its biologicalfunction of inactivating foreign DNA by irreparable double-strand DNA cuts, in preference to single-strand nicks that arereadily repaired by DNA ligase.
Results and DiscussionESEEM Shows Cu2þ Is Coordinated to a Histidine. The signal fromCu2þ binding to the EcoRI-DNA complex is readily detected inthe CW-ESR spectrum (Fig. 1A). There are two distinct spectralcomponents, and the best-fit simulated spectrum (black dotted
lines, Fig. 1A) is obtained when the ratio of these two componentsis approximately 1∶1. The magnetic g‖ and A‖ tensor values arewithin the range found for type-II Cu2þ complexes (18), indicat-ing that both Cu2þ components have four equatorial ligands. Thevalues of the magnetic g‖ and A‖ tensors of the first componentare consistent with either one, two, or three nitrogen ligands, withoxygens providing the remaining ligands (18). The second com-ponent has a smaller A‖ value, which can be attributed to distor-tion in the planarity of the four equatorial ligands (19). Evidencefrom distance measurements combined with experimental deter-mination of stoichiometry (below) shows that a single Cu2þ ionbinds at the same location in each subunit; thus, the two compo-nents do not arise from heterogeneity in the locations of boundCu2þ ions. We further address the origin of two components be-low in the context of ITC, DEER, and molecular dynamics (MD)results.
To further characterize the Cu2þ coordination, we obtained athree-pulse ESEEM spectrum at a magnetic field that corre-sponds to the g⊥ region of the Cu2þ ESR spectrum, so that bothCu2þ components contribute to the ESEEM signal. The ESEEMspectrum (Fig. 1B) has two sharp peaks (approximately 0.60 MHzand approximately 1.60 MHz) and one broad shoulder peak (ap-proximately 1.09 MHz). The sum of the two lower frequencies isclose to the highest one, indicating that they are mainly due to thenuclear quadrupole interactions of a weakly coupled 14N (20–22).Both the quadrupole parameters derived from these frequencies(e2Qq∕h approximately 1.73 MHz and η approximately 0.69) (22)and the three peaks in the ESEEM spectrum are typical ofremote, noncoordinated 14N from a histidine imidazole ringbound to Cu2þ (20, 21, 23–27). Broad peaks at approximately4.0 MHz are also resolved (Fig. 1B). These peaks may arise fromthe double quantum transition of 14N (21). Hyperfine sublevelcorrelation experiments (Fig. S1) show characteristic cross-peaksthat support the assignment that all the peaks at approximately4.0 MHz in the ESEEM spectrum are from the double quantumtransition. The peak at 14.3 MHz in the ESEEM spectrum isfrom weakly coupled protons around the Cu2þ electron spin.These protons may come from either the solvent or the macro-molecules.
Strategy for Identifying the Cu2þ Binding Histidine Residue.There arefive His residues in each subunit of EcoRI, at positions 31, 114,147, 162, and 225. We employed a four-stage strategy to identifythe specific His residues that bind Cu2þ in the EcoRI-DNAcomplex. First, we used DEER-ESR to measure the distance be-tween Cu2þ ions, and compared this to inter-His distances in thecocrystal structure. The single distance obtained narrowed thefield of candidate binding sites. Second, we made a Ser180Cysmutant and nitroxide spin-labeled the Cys residues, and then usedDEER-ESR to measure Cu2þ-nitroxide distances. The datayielded two Cu2þ-nitroxide distances (intersubunit and intrasubu-nit), from which triangulation unambiguously identified His114as the Cu2þ-binding residue. Third, the binding site was confirmedby showing that a His114Tyr mutation completely abolishes theability of Cu2þ to stimulate protein-DNA binding and to inhibitDNA cleavage. Fourth, we showed by ITC a stoichiometry oftwo Cu2þ ions bound per EcoRI dimer.
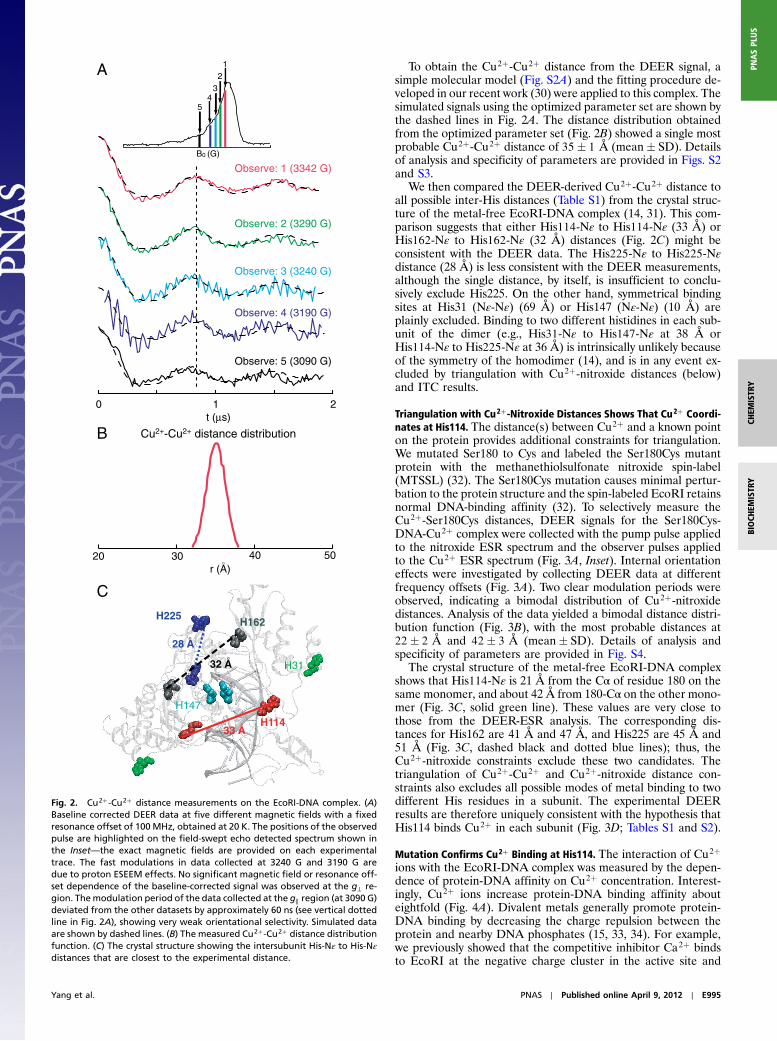
Cu2þ-Cu2þ Distance Excludes Some Candidate Binding Sites. To nar-row the set of candidate Cu2þ binding sites, we used the DEERexperiment to measure the distance between the two Cu2þ ions(one in each EcoRI subunit). DEER data were collected at fivemagnetic fields and a fixed resonance offset (Fig. 2 A), and alsowith multiple resonance offsets (Fig. S2B). The field and/orresonance offset variation of the DEER signal provides informa-tion about Cu2þ-Cu2þ distance as well as the relative orientationof the two Cu2þ centers (28–30). The data showed only very weakorientational effects, as described in the legend in Fig. 2.
A
B
Cu2+
B (G)
N
NH
2,200 2,600 3,000 3,400 3,800
0 4 8 12 16ν (MHz)
H1{
NQI Transitions
DQ Transitions
Fig. 1. ESR data on the Cu2þ bound EcoRI-DNA complex. (A) The CW-ESRspectrum at 80 K (gray solid line) shows two components. The second com-ponent is clear in the magnified section of the spectrum, which is shown inthe Inset. The simulated spectrum is shown by the dotted line. The best-fitsimulation was obtained with the following parameters: component 1:g‖ ¼ 2.289, A‖ ¼ 163 G, g⊥ ¼ 2.06, A⊥ ¼ 20 G and component 2: g‖ ¼ 2.228,A‖ ¼ 143 G, g⊥ ¼ 2.06, A⊥ ¼ 20 G. The relative ratio of the two componentsis approximately 1∶1. (B) The three-pulse ESEEM spectrum at 20 K obtained ata magnetic field of 3369 G. The peaks between 0–2 MHz, indicated by thetrident, are assigned to the imidazole 14N from a histidine residue. The broadpeaks at approximately 4.0 MHz are assigned to the double quantum transi-tion. The peak at approximately 14.3 MHz is assigned to the proton ESEEMpeak. (Inset) The Cu2þ coordination derived from the ESEEM results.
E994 ∣ www.pnas.org/cgi/doi/10.1073/pnas.1200733109 Yang et al.
To obtain the Cu2þ-Cu2þ distance from the DEER signal, asimple molecular model (Fig. S2A) and the fitting procedure de-veloped in our recent work (30) were applied to this complex. Thesimulated signals using the optimized parameter set are shown bythe dashed lines in Fig. 2A. The distance distribution obtainedfrom the optimized parameter set (Fig. 2B) showed a single mostprobable Cu2þ-Cu2þ distance of 35� 1 Å (mean� SD). Detailsof analysis and specificity of parameters are provided in Figs. S2and S3.
We then compared the DEER-derived Cu2þ-Cu2þ distance toall possible inter-His distances (Table S1) from the crystal struc-ture of the metal-free EcoRI-DNA complex (14, 31). This com-parison suggests that either His114-Nε to His114-Nε (33 Å) orHis162-Nε to His162-Nε (32 Å) distances (Fig. 2C) might beconsistent with the DEER data. The His225-Nε to His225-Nεdistance (28 Å) is less consistent with the DEER measurements,although the single distance, by itself, is insufficient to conclu-sively exclude His225. On the other hand, symmetrical bindingsites at His31 (Nε-Nε) (69 Å) or His147 (Nε-Nε) (10 Å) areplainly excluded. Binding to two different histidines in each sub-unit of the dimer (e.g., His31-Nε to His147-Nε at 38 Å orHis114-Nε to His225-Nε at 36 Å) is intrinsically unlikely becauseof the symmetry of the homodimer (14), and is in any event ex-cluded by triangulation with Cu2þ-nitroxide distances (below)and ITC results.
Triangulation with Cu2þ-Nitroxide Distances Shows That Cu2þ Coordi-nates at His114. The distance(s) between Cu2þ and a known pointon the protein provides additional constraints for triangulation.We mutated Ser180 to Cys and labeled the Ser180Cys mutantprotein with the methanethiolsulfonate nitroxide spin-label(MTSSL) (32). The Ser180Cys mutation causes minimal pertur-bation to the protein structure and the spin-labeled EcoRI retainsnormal DNA-binding affinity (32). To selectively measure theCu2þ-Ser180Cys distances, DEER signals for the Ser180Cys-DNA-Cu2þ complex were collected with the pump pulse appliedto the nitroxide ESR spectrum and the observer pulses appliedto the Cu2þ ESR spectrum (Fig. 3A, Inset). Internal orientationeffects were investigated by collecting DEER data at differentfrequency offsets (Fig. 3A). Two clear modulation periods wereobserved, indicating a bimodal distribution of Cu2þ-nitroxidedistances. Analysis of the data yielded a bimodal distance distri-bution function (Fig. 3B), with the most probable distances at22� 2 Å and 42� 3 Å (mean� SD). Details of analysis andspecificity of parameters are provided in Fig. S4.
The crystal structure of the metal-free EcoRI-DNA complexshows that His114-Nε is 21 Å from the Cα of residue 180 on thesame monomer, and about 42 Å from 180-Cα on the other mono-mer (Fig. 3C, solid green line). These values are very close tothose from the DEER-ESR analysis. The corresponding dis-tances for His162 are 41 Å and 47 Å, and His225 are 45 Å and51 Å (Fig. 3C, dashed black and dotted blue lines); thus, theCu2þ-nitroxide constraints exclude these two candidates. Thetriangulation of Cu2þ-Cu2þ and Cu2þ-nitroxide distance con-straints also excludes all possible modes of metal binding to twodifferent His residues in a subunit. The experimental DEERresults are therefore uniquely consistent with the hypothesis thatHis114 binds Cu2þ in each subunit (Fig. 3D; Tables S1 and S2).
Mutation Confirms Cu2þ Binding at His114. The interaction of Cu2þions with the EcoRI-DNA complex was measured by the depen-dence of protein-DNA affinity on Cu2þ concentration. Interest-ingly, Cu2þ ions increase protein-DNA binding affinity abouteightfold (Fig. 4A). Divalent metals generally promote protein-DNA binding by decreasing the charge repulsion between theprotein and nearby DNA phosphates (15, 33, 34). For example,we previously showed that the competitive inhibitor Ca2þ bindsto EcoRI at the negative charge cluster in the active site and
210t (µs)
B0 (G)
Observe: 1 (3342 G)
Observe: 2 (3290 G)
Observe: 3 (3240 G)
Observe: 4 (3190 G)
A
B
Observe: 5 (3090 G)
20 30 40 50r (A)
O
12
34
5
C
Cu2+-Cu2+ distance distribution
28 AO
H162
32 AO
H147
H11433 A
O
H31
H225
Fig. 2. Cu2þ-Cu2þ distance measurements on the EcoRI-DNA complex. (A)Baseline corrected DEER data at five different magnetic fields with a fixedresonance offset of 100 MHz, obtained at 20 K. The positions of the observedpulse are highlighted on the field-swept echo detected spectrum shown inthe Inset—the exact magnetic fields are provided on each experimentaltrace. The fast modulations in data collected at 3240 G and 3190 G aredue to proton ESEEM effects. No significant magnetic field or resonance off-set dependence of the baseline-corrected signal was observed at the g⊥ re-gion. Themodulation period of the data collected at the g‖ region (at 3090 G)deviated from the other datasets by approximately 60 ns (see vertical dottedline in Fig. 2A), showing very weak orientational selectivity. Simulated dataare shown by dashed lines. (B) The measured Cu2þ-Cu2þ distance distributionfunction. (C) The crystal structure showing the intersubunit His-Nε to His-Nεdistances that are closest to the experimental distance.
Yang et al. PNAS ∣ Published online April 9, 2012 ∣ E995
thereby increases the equilibrium association constant (KA) asmuch as 380-fold (15). Cu2þ evidently has a weaker effect thanCa2þ because the Cu2þ (coordinated at His114) lies at greaterdistance (10 Å) from the nearest DNA phosphate GApATTC.
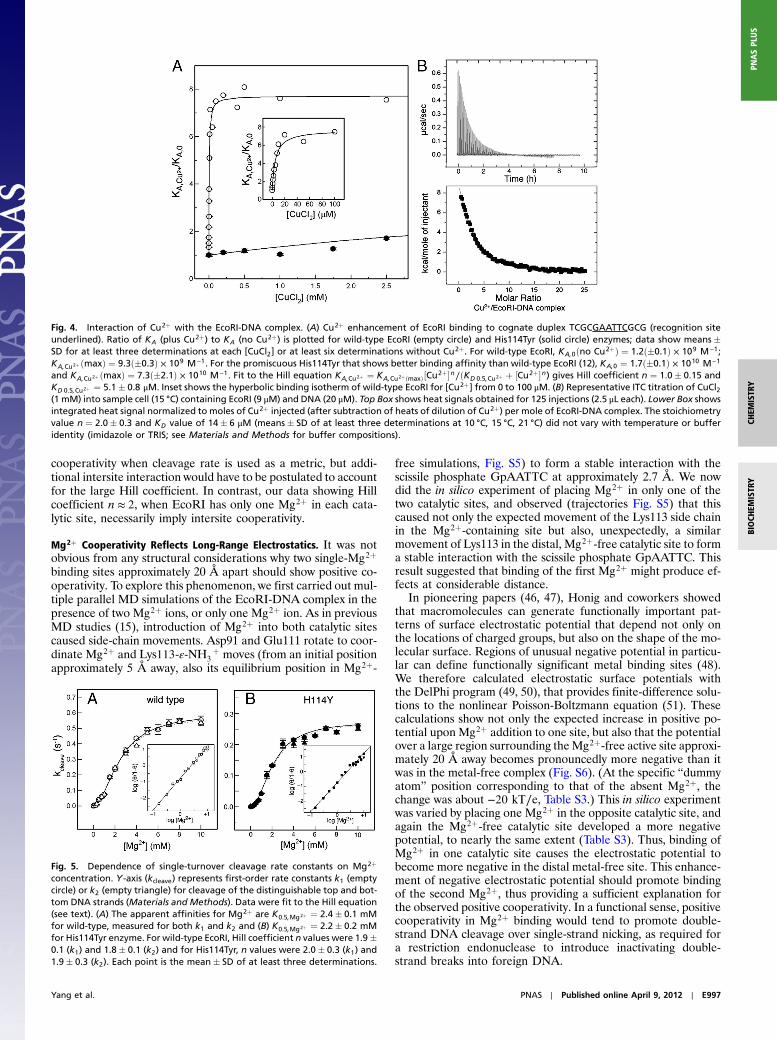
When the putative Cu2þ binding residue is removed in His114-Tyr mutant EcoRI, the stimulation by Cu2þ is almost completelyabolished (Fig. 4A). The apparent affinity for Cu2þ is reducedby 1,600-fold by the mutation; compare apparent KD;Cu2þ ¼5.1� 0.8 μM for wild-type EcoRI to KD;Cu2þ approximately8 mM for the His114Tyr mutant. These observations confirm thatCu2þ ion binds at His114, a site distinct from that whereMg2þ binds.
Direct Determination of Cu2þ Binding Stoichiometry by ITC. The ESRdata above give a single Cu2þ-Cu2þ distance in the complex,suggesting one Cu2þ is bound to each EcoRI subunit. Detectionof only two Cu2þ-nitroxide distances (one intersubunit and oneintrasubunit) is also indicative of only one Cu2þ bound per sub-unit. To confirm this inference, we determined stoichiometry di-rectly by calorimetric measurement of the heat of binding. Cu2þwas titrated into a solution of the wild-type EcoRI-DNA complex(Fig. 4B). The parameters obtained were KD;Cu2þ ¼ 14� 6 μM,and a stoichiometry of 2.0� 0.3 Cu2þ bound per EcoRI-DNAcomplex. The ITC results thus confirm that each subunit ofthe EcoRI dimer coordinates one Cu2þ ion.
Cooperative Mg2þ Interactions in the Two Catalytic Sites. To providea sound basis for understanding Cu2þ effects on catalysis (seebelow), we carefully reinvestigated the interaction of Mg2þ withthe EcoRI-DNA complex. In contrast to some metallonucleasesthat use two Mg2þ ions per catalytic site, EcoRI is generally con-sidered (14, 17, 35) to use only a single Mg2þ in each catalytic site.Each Mg2þ is coordinated to Glu111, Asp91, and GpAATTC(see last section of Results and Discussion). On the basis of co-crystal structures (14) and MD simulations (15), one phosphoryloxygen (O1P) at the scissile GpAA is coordinated by Mg2þ whilethe other phosphoryl oxygen (O2P) hydrogen bonds to positivelycharged side chains of Lys113 and Arg145; these side chains thusmimic both the position and the functional role of the secondmetal ion proposed for many two-metal-ion phosphotransferasereactions, namely to stabilize the phosphorane transition stateand/or the oxyanion leaving group (36–40).
To determine effects on catalysis independent of bindingeffects (3), we preformed EcoRI-DNA complexes in the absenceof Mg2þ under conditions of molar excess of enzyme to DNA,and then initiated reaction by the addition of Mg2þ at variousconcentrations. The resulting single-turnover first-order rate con-stants, measured separately for each DNA strand, thus representa composite of Mg2þ binding to the EcoRI-DNA complex (12)and the chemical step of catalysis. The rate constants for wild-type EcoRI cleavage of the two DNA strands show sigmoidaldependence on [Mg2þ] (Fig. 5 A); the best fits of the kinetic datato the Hill equation kcleave ¼ kmax½Mg2þ�n∕ðKMg2þ þ ½Mg2þ�nÞyield values of n ¼ 1.9� 0.1 and n ¼ 1.8� 0.1 for cleavage ofeach of the two DNA strands. The slopes of log-log Hill plots(Inset, Fig. 5A) for the kinetic data give similar values. The ob-served value of n ¼ 1.8–1.9, given that EcoRI uses one metal ionper catalytic center (11, 14, 17, 41), implies interaction betweenthe single-Mg2þ binding sites of the protein homodimer.
Positive cooperativity for Mg2þ interaction has also beenobserved for restriction endonuclease PvuII, which binds two me-tal ions in each of its two catalytic sites (42, 43); Hill coefficientsof n ¼ 3.6 to 4 were reported (44, 45). Although the precise roleof the second metal ion is in general controversial for manymetallonucleases (17, 35, 39, 40), for PvuII the first Mg2þ boundin a catalytic site is both necessary and sufficient for catalysis (45).The second Mg2þ acts as a powerful accelerant for DNA clea-vage. This accelerant function, in itself, would produce intrasite
C
210t (µs)
B0 (G)
A
B
Pump: B, Observe: 2, ∆ν = 100 MHz
A
B
Pump: A, Observe: 1, ∆ν = 266 MHz
Pump: A, Observe: 3, ∆ν = 448 MHz
Pump: A, Observe: 4, ∆ν = 548 MHz
20 30 40 50r (A)
10 60O
1
23
4
Cu2+-S180C distance distribution
41 AO47 A
O
21 AO
42 AO
H114
51 AO
45 AO
H162
H225
S180
S180
22 AO
42 AO
S180C
S180CH114H114
35 AO
Cu2+
Cu2+
D
Fig. 3. Cu2þ-nitroxide distance measurements on the MTSSL-Ser180Cys-EcoRI-DNA-ðCu2þÞ2 complex. (A) Baseline corrected DEER data at four differ-ent resonance offsets, 266 MHz, 100 MHz, 448 MHz, and 548 MHz (from Topto Bottom), obtained at 20 K. The positions of the pump and observer pulsesare indicated on the field-swept echo detected spectrum shown on the inset.Simulated data are shown by dashed lines. (B) The measured Cu2þ-nitroxidedistance distribution function. (C) The intersubunit and intrasubunit dis-tances between candidate His-Nε and residue 180-Cα. (D) Summary of trian-gulation results, showing Cu2þ-Cu2þ distance (red) and Cu2þ-nitroxidedistances (green).
E996 ∣ www.pnas.org/cgi/doi/10.1073/pnas.1200733109 Yang et al.
cooperativity when cleavage rate is used as a metric, but addi-tional intersite interaction would have to be postulated to accountfor the large Hill coefficient. In contrast, our data showing Hillcoefficient n ≈ 2, when EcoRI has only one Mg2þ in each cata-lytic site, necessarily imply intersite cooperativity.
Mg2þ Cooperativity Reflects Long-Range Electrostatics. It was notobvious from any structural considerations why two single-Mg2þbinding sites approximately 20 Å apart should show positive co-operativity. To explore this phenomenon, we first carried out mul-tiple parallel MD simulations of the EcoRI-DNA complex in thepresence of two Mg2þ ions, or only one Mg2þ ion. As in previousMD studies (15), introduction of Mg2þ into both catalytic sitescaused side-chain movements. Asp91 and Glu111 rotate to coor-dinate Mg2þ and Lys113-ε-NH3
þ moves (from an initial positionapproximately 5 Å away, also its equilibrium position in Mg2þ-
free simulations, Fig. S5) to form a stable interaction with thescissile phosphate GpAATTC at approximately 2.7 Å. We nowdid the in silico experiment of placing Mg2þ in only one of thetwo catalytic sites, and observed (trajectories Fig. S5) that thiscaused not only the expected movement of the Lys113 side chainin the Mg2þ-containing site but also, unexpectedly, a similarmovement of Lys113 in the distal, Mg2þ-free catalytic site to forma stable interaction with the scissile phosphate GpAATTC. Thisresult suggested that binding of the first Mg2þ might produce ef-fects at considerable distance.
In pioneering papers (46, 47), Honig and coworkers showedthat macromolecules can generate functionally important pat-terns of surface electrostatic potential that depend not only onthe locations of charged groups, but also on the shape of the mo-lecular surface. Regions of unusual negative potential in particu-lar can define functionally significant metal binding sites (48).We therefore calculated electrostatic surface potentials withthe DelPhi program (49, 50), that provides finite-difference solu-tions to the nonlinear Poisson-Boltzmann equation (51). Thesecalculations show not only the expected increase in positive po-tential uponMg2þ addition to one site, but also that the potentialover a large region surrounding theMg2þ-free active site approxi-mately 20 Å away becomes pronouncedly more negative than itwas in the metal-free complex (Fig. S6). (At the specific “dummyatom” position corresponding to that of the absent Mg2þ, thechange was about −20 kT∕e, Table S3.) This in silico experimentwas varied by placing one Mg2þ in the opposite catalytic site, andagain the Mg2þ-free catalytic site developed a more negativepotential, to nearly the same extent (Table S3). Thus, binding ofMg2þ in one catalytic site causes the electrostatic potential tobecome more negative in the distal metal-free site. This enhance-ment of negative electrostatic potential should promote bindingof the second Mg2þ, thus providing a sufficient explanation forthe observed positive cooperativity. In a functional sense, positivecooperativity in Mg2þ binding would tend to promote double-strand DNA cleavage over single-strand nicking, as required fora restriction endonuclease to introduce inactivating double-strand breaks into foreign DNA.
Fig. 4. Interaction of Cu2þ with the EcoRI-DNA complex. (A) Cu2þ enhancement of EcoRI binding to cognate duplex TCGCGAATTCGCG (recognition siteunderlined). Ratio of KA (plus Cu2þ) to KA (no Cu2þ) is plotted for wild-type EcoRI (empty circle) and His114Tyr (solid circle) enzymes; data show means�SD for at least three determinations at each [CuCl2] or at least six determinations without Cu2þ. For wild-type EcoRI, KA;0ðno Cu2þÞ ¼ 1.2ð�0.1Þ × 109 M−1;KA;Cu2þ ðmaxÞ ¼ 9.3ð�0.3Þ × 109 M−1. For the promiscuous His114Tyr that shows better binding affinity than wild-type EcoRI (12), KA;0 ¼ 1.7ð�0.1Þ × 1010 M−1
and KA;Cu2þ ðmaxÞ ¼ 7.3ð�2.1Þ × 1010 M−1. Fit to the Hill equation KA;Cu2þ ¼ KA;Cu2þðmaxÞ½Cu2þ�n∕ðKD 0.5;Cu2þ þ ½Cu2þ�n) gives Hill coefficient n ¼ 1.0� 0.15 andKD 0.5;Cu2þ ¼ 5.1� 0.8 μM. Inset shows the hyperbolic binding isotherm of wild-type EcoRI for [Cu2þ] from 0 to 100 μM. (B) Representative ITC titration of CuCl2(1 mM) into sample cell (15 °C) containing EcoRI (9 μM) and DNA (20 μM). Top Box shows heat signals obtained for 125 injections (2.5 μL each). Lower Box showsintegrated heat signal normalized to moles of Cu2þ injected (after subtraction of heats of dilution of Cu2þ) per mole of EcoRI-DNA complex. The stoichiometryvalue n ¼ 2.0� 0.3 and KD value of 14� 6 μM (means� SD of at least three determinations at 10 °C, 15 °C, 21 °C) did not vary with temperature or bufferidentity (imidazole or TRIS; see Materials and Methods for buffer compositions).
Fig. 5. Dependence of single-turnover cleavage rate constants on Mg2þ
concentration. Y-axis (kcleave) represents first-order rate constants k1 (emptycircle) or k2 (empty triangle) for cleavage of the distinguishable top and bot-tom DNA strands (Materials and Methods). Data were fit to the Hill equation(see text). (A) The apparent affinities for Mg2þ are K0.5;Mg2þ ¼ 2.4� 0.1 mMfor wild-type, measured for both k1 and k2 and (B) K0.5;Mg2þ ¼ 2.2� 0.2 mMfor His114Tyr enzyme. For wild-type EcoRI, Hill coefficient n values were 1.9�0.1 (k1) and 1.8� 0.1 (k2) and for His114Tyr, n values were 2.0� 0.3 (k1) and1.9� 0.3 (k2). Each point is the mean� SD of at least three determinations.
Yang et al. PNAS ∣ Published online April 9, 2012 ∣ E997
Cu2þ Inhibits Mg2þ-Dependent Catalysis. To examine the catalyticconsequences of Cu2þ binding, we performed single-turnover as-says to measure rate constants for the chemical step. Significantlywe found that Cu2þ is a powerful inhibitor of DNA cleavage bywild-type EcoRI (Fig. 6). This inhibition is unexpected becauseCu2þ does not bind directly at the catalytic site. Because this sin-gle-turnover assay uses preformed EcoRI-DNA complexes withmolar excess of enzyme over DNA, Cu2þ must affect the chemi-cal step rather than protein-DNA association or product release.Note that the apparent Cu2þ affinity as an inhibitor of catalysis(K0.5;Cu2þ ≈ 70 μM) is weaker than the affinity in the absence ofMg2þ from calorimetry (14 μM, Fig. 4B) or the apparent affinityfrom equilibrium binding stimulation (5 μM, Fig. 4A), also in theabsence of Mg2þ. This difference likely reflects Mg2þ-Cu2þrepulsion in the cleavage assays, at an inter-ion distance of ap-proximately 13 Å (see Fig. 7A below). This difference in Cu2þ
affinities is also responsible for the sigmoid shape of the Cu2þ
inhibition curve.
Strikingly, for the His114Tyr mutant EcoRI, Cu2þ (even at200 μM) does not inhibit the Mg2þ-catalyzed cleavage reaction,whereas 100 μM Cu2þ completely inhibits catalysis by wild-type EcoRI (Fig. 6). As a control, we showed that the His114Tyrmutation does not affect affinity for Mg2þ or the cooperativityof Mg2þ action (Fig. 5B). The insensitivity of His114Tyr proteinto Cu2þ inhibition thus confirms our inference from ESR and thebinding assays (see above) that His114 is the coordinating sitefor Cu2þ.
It is interesting that the effective Cu2þ concentrations forinhibition of wild-type enzyme are approximately equivalent tonormal Cu2þ concentrations in vivo (52), suggesting that coordi-nation to histidine sites in proteins could have a biologically sig-nificant role in Cu2þ toxicity.
How Does Cu2þ Inhibit EcoRI Catalysis? In the wild-type EcoRI-DNA complex, His114 donates a hydrogen bond to the phos-phate at GApATTC. In the proposed catalytic mechanism (15)in the absence of Cu2þ, this phosphate serves to position a watermolecule (WC in Fig. 7A), that in turn hydrogen bonds to theMg2þ-bound water that carries out nucleophilic attack atGpAATTC. In addition, studies with chiral phosphate analogs(15) implied that DNA distortion in the complex is requiredfor catalysis and that the precise positioning of the phosphoryloxygen at GApA that participates in the “water relay” is impor-tant for the rate of cleavage. This preorganized arrangement isthus an example of what Bruice and Benkovic (53) termed a“near-attack conformer.”
To investigate how Cu2þ inhibits catalysis, we carried outMD simulations of the EcoRI-DNA-ðMg2þÞ2 complex, with orwithout Cu2þ (see below). Coordination of the Cu2þ ions atHis114-Nε yields a stabilized mean intersubunit Cu2þ-Cu2þ dis-tance of 38 Å, close to the 35 Å from ESR-DEER; both ESRand MD distributions have distribution widths at half-height ap-proximately 2–3 Å.
Detailed MD simulations with explicit solvent show that whenone Cu2þ is inserted into the EcoRI•DNA•ðMg2þÞ2 complex atHis114-Nε (see above), His114 assumes a position in which Cu2þis coordinated by His114-Nε, Glu170 and Glu245; this requiresrotations of the Glu170 and Glu245 side chains (Fig. 7A). Thesimulations suggest that transient rotation of the Glu170 side
Fig. 6. Inhibition of EcoRI cleavage by Cu2þ. See procedure in Materialsand Methods. Ratio of first-order cleavage rate constant kcleaveðþCu2þÞ∕kcleaveðno Cu2þÞ as a function of [CuCl2]. Wild-type EcoRI cleavage reactionswere performed at Mg2þ concentrations 3 mM (upside down triangle), 4 mM(empty triangle), 6 mM (empty square), and 8 mM (empty circle); His114Tyrreaction was at 8 mM (solid circle). Plots for k1 (cleavage in top strand) andk2 (cleavage in bottom strand) were precisely superimposable. For clarity,only k1 data are shown. Each point represents the mean of at least threedeterminations. Error bars are too small to be seen at this scale.
Fig. 7. Simulation of Cu2þ-induced structural changes in the catalytic site of the EcoRI-DNA complex. Superimposed images of EcoRI•DNA•ðMg2þÞ2 complex(protein side chains green; DNA cyan) and the EcoRI•DNA•ðMg2þÞ2•ðCu2þÞ1 complex (protein side chains orange; DNA magenta). Structures from MD snap-shots (see Materials and Methods) were matched at the DNA nucleotides CGA using Chimera software (59). Hydrogen bonding distances (yellow dashed linesand numbers) are given in Ångstroms. (A) In the complex without Cu2þ, a water molecule (WA) coordinated to Mg2þ carries out nucleophilic attack (whitearrow) at the scissile phosphate GpAATTC. This water is hydrogen-bonded to the stable “relay water” (WC ), which is precisely positioned by H-bonding to theadjacent phosphate GApATTC, while His114-Nδ H-bonds to the phosphate GApATTC. In the Mg2þ þ Cu2þ complex, His114 and GApATTC assume completelydifferent positions and there is no stable equivalent of WC (see text). (B) Rotated view of the region, showing how Arg145-guanidino in the Mg2þ þ Cu2þ
complex makes H-bonds (double-headed arrows) to GpAATTC and GApAATTC (thus bridging both phosphates), while retaining an H-bond to recognize Ade-N7. Within <2 ps after the start of the simulation for the EcoRI•DNA•ðMg2þÞ2 complex, stable (�0.1 Å) coordination distances to Mg2þ were achieved asfollows: GpA-O1P, 1.85 Å; Asp91-OD1/OD2, 1.9 Å, Glu111-OE2, 1.9 Å, WA, 1.95 Å. An additional coordination fluctuates between Ala112-carbonyl(3.0� 0.5 Å) and a transiently visiting water.
E998 ∣ www.pnas.org/cgi/doi/10.1073/pnas.1200733109 Yang et al.
chain allows both carboxylate oxygens to coordinate alternately tothe Cu2þ ion. This rotation can create distortions in the planarityof the equatorial ligands and account for the second spectral com-ponent (with small A‖ value) that we observed in CW-ESR.
After these side chain adjustments, His114 is approximately4 Å from its normal position. (No comparable repositioning oc-curs in the Cu2þ-free catalytic site, suggesting that the Cu2þ bind-ing sites in each subunit have no mutual influence at a range of36 Å. Similar movement occurs in both catalytic sites when Cu2þions are inserted in both.) Lacking interaction with His114-Nδ,the GApA phosphate rotates to where it cannot support thewater network (Fig. 7A). The GApA is stabilized in this abnormalposition by a newly formed interaction with Arg145 (Fig. 7B).Arg145 remains in position to donate hydrogen bonds to the scis-sile GpAA (15), and simultaneously to N7 of the inner adenine(Fig. 7B). Arg145 was proposed (15) to polarize the scissile phos-phate and/or to stabilize the phosphorane intermediate in thetransition state; this function may be compromised by the brid-ging of one Arg145 guanidino nitrogen between the GpAA andGApA phosphates (double arrows, Fig. 7B) upon addition ofCu2þ. Crucially, the water relay (water C, Fig. 7A) from theGApA to the nucleophilic water no longer exists in stable form.In the absence of Cu2þ, this position has 98–100% occupancy(in various parallel simulations) with average residence timesfor the waters in the range 300 to 500 ps, but in the Cu2þ-boundsite it is only 20–40% occupied (in various parallel simulations) bywaters with short residence times (<100 ps). The overall pictureis that the “near-attack conformer” essential for catalysis no long-er exists in the Cu2þ complex.
Our findings therefore strengthen the view that coupling be-tween recognition and catalytic specificity for EcoRI derives from(i) A role of DNA distortion in both recognition and catalysis; (ii)the use of functional groups (e.g., Arg145, Fig. 7B) for dual rolesin sequence recognition and the catalytic mechanism; (iii) andcommunication between the two active sites. It will be intriguingto learn if additional principles have been evolved by other site-specific DNA-modifying enzymes.
Materials and MethodsEnzyme Expression and Purification. The EcoRI endonuclease gene was ex-pressed as a fusion with maltose-binding protein and cleaved after purifica-tion to produce the complete EcoRI protein with no extra amino acids. Detailsare in SI Text.
Oligodeoxynucleotide Substrates. Oligodeoxynucleotides (Integrated DNATechnologies) were purified as described previously (12). The self-comple-mentary TCGCGAATTCGCG (12 bp with dangling T) was used for ESRand equilibrium binding experiments (see below). For cleavage rate assays,duplex deoxyoligonucleotide, (23 base-pairs plus dangling end base), was an-nealed from purified single-stranded 24 nt top (5′-GATGGGTGCAGAATTCTG-CAGGTA-3′) and bottom (5′-CTACCTGCAGAATTCTGCACCCAT-3′) sequencesas described by Sapienza, et al. (12). For both equilibrium binding and clea-vage experiments, the DNA was 5′-end labeled with 32P-ATP using T4 poly-nucleotide kinase as described (54). The substrate was designed such thatcleavage in the top and bottom strands produces 5′-32P-labeled 11 nt and10 nt products, respectively.
Preparation of Cu2þ-Wt EcoRI-DNA and Cu2þ-S180C-MTSSL-DNA ESR Samples. Asolution of wild-type EcoRI (5 μM) in the presence of fivefold molar excessof TCGCGAATTCGCG was exchanged into 30 mM N-ethylmorpholine(NEM) buffer containing 0.3MNH4Cl, 10% dioxane, 30% deuterated glycerol(d8), 65% D2O (pH 8.0) and concentrated. The final concentrations of EcoRIand 13mer DNA were 380 μM and 1.5 mM, respectively.
Details of nitroxide spin-labeling are in SI Text. The final concentrationsof Ser180Cys protein and 13mer DNA were 180 μM and 1.8 mM respectively.For both Cu2þ-EcoRI-DNA and Cu2þ-MTSSL-S180C-DNA samples, isotopicallyenriched 63CuCl2 (Cambridge Isotope Labs, Inc) was added at a 4∶1 molarratio (Cu2þ: protein dimer). The samples were stored at −80 °C and flash fro-zen to 20 K before each DEER ESR experiment.
ESR Experiments. All of the pulsed ESR experiments were performed on aBruker Elexsys 580 spectrometer at 20 K. CW-ESR and ESEEM experimentswere performed with a MS3 resonator. DEER experiments were performedwith a MD5 resonator. Details of pulse sequences, data acquisition, and ana-lysis are in SI Text.
Equilibrium Binding Studies. Equilibrium association constants (KA) ofSer180Cys protein and MTSSL-Ser180Cys protein to DNA were determinedas described (32). Experiments assessing Cu2þ enhancement of binding usedthe same double-strand deoxyoligonucleotide TCGCGAATTCGCG as that inthe ESR experiments. Equilibrium association constants (KA) for wild-typeEcoRI and His114Tyr (�Cu2þ) were measured by the nitrocellulose filter bind-ing assay as described (34, 55). For assays in the absence of Cu2þ, EDTA (1mM)was included in the binding buffer (30 mMNEM, 10% glycerol, 10% dioxane,100 μM dithiothreitol, 100 μg∕mL bovine serum albumin) plus 0.3 M KCl;pH 8.0, 22 °C.
Cleavage Kinetics. Single-turnover cleavage rates (3, 12) using 32P-labeled 23base-pair DNA (see above) were determined as a function of Mg2þ concen-tration by preforming EcoRI-DNA complexes (fivefold molar excess of EcoRIover DNA) and initiating the reaction by addition of Mg2þ. Inhibition by Cu2þ
was determined by two different protocols (SI Text) that gave identicalresults.
Isothermal Titration Calorimetry. ITC experiments were performed by re-peated injections of CuCl2 into a solution of EcoRI-DNA complex until com-plete saturation, where there was no further change in the heat of reaction.Observed heats of reaction were corrected for the heat of dilution. Details ofprocedure and data analysis are in SI Text.
Molecular Dynamics Simulations.MD simulations using the combined all-atomCHARMM22/CMAP and CHARMM27 force fields (56–58) were performed onEcoRI-TCGCGAATTCGCG complexes with (i) no addedmetal ion, (ii) one activesite (either A or B) with Mg2þ, (iii) both active sites A and B with Mg2þ, (iv)one subunit (either A or B) with Cu2þ, (v) both subunits with Cu2þ, (vi) bothactive sites A and B filled withMg2þ, one Cu2þ added to one subunit (either Aor B), (vii) both active sites A and B filled with Mg2þ, and a Cu2þ ion added toboth subunits. The starting point for all simulations was a highly refined,high-resolution (1.85 Å) crystal structure model (14) based on PDB ID code1CKQ. For each case (with or without Mg2þ and/or Cu2þ), multiple parallelproduction runs (ranging from 4 ns to 10 ns) were performed. Details are in SIText. Molecular graphics images were produced using the UCSF Chimerapackage (59) from the Resource for Biocomputing, Visualization, and Infor-matics at the University of California, San Francisco.
Electrostatic Potential Calculations. Electrostatic potentials were calculatedand mapped to molecular surfaces using an updated version (v.5) of theDelPhi program which uses finite-difference methods to solve the nonlinearPoisson-Boltzmann equation (FDPB method) (49, 50). Input coordinate filesfor the DelPhi calculations were generated from snapshots from multipleparallel molecular dynamics simulations (see above) of the EcoRI-DNA com-plex in the absence of Mg2þ or with oneMg2þ added to either active site A orB. Details are given in SI Text. Electrostatic potential maps of the molecularsurfaces of the EcoRI-DNA complexes were visualized using PyMOL (60).
ACKNOWLEDGMENTS. This work was supported by a National Science Founda-tion (MCB 0842956) grant to S. S. and L. J-J.; a National Institutes of HealthMERIT (5R37-GM029207) grant to L. J-J.; and the Center for Simulation andModeling at the University of Pittsburgh.
1. Garvie CW, Wolberger C (2001) Recognition of specific DNA sequences. Mol Cell
8:937–946.
2. Rohs R, et al. (2009) The role of DNA shape in protein-DNA recognition. Nature
461:1248–1253.
3. Lesser DR, Kurpiewski MR, Jen-Jacobson L (1990) The energetic basis of specificity in-
the Eco RI endonuclease—DNA interaction. Science 250:776–786.
4. Olson WK, Gorin AA, Lu X-J, Hock LM, Zhurkin VB (1998) DNA sequence-dependent
deformability deduced from protein-DNA crystal complexes. Proc Natl Acad Sci USA
95:11163–11168.
5. Jen-Jacobson L, Engler LE, Jacobson LA (2000) Structural and thermodynamic strate-
gies for site-specific DNA binding proteins. Structure 8:1015–1023.
6. Rohs R, et al. (2010) Origins of specificity in protein-DNA recognition. Annu Rev Bio-
chem 79:233–269.
Yang et al. PNAS ∣ Published online April 9, 2012 ∣ E999
7. Spolar RS, Record MT, Jr (1994) Coupling of local folding to site-specific binding ofproteins to DNA. Science 263:777–784.
8. Ha J-H, Spolar RS, Record MT, Jr (1989) Role of the hydrophobic effect in stability ofsite-specific protein-DNA complexes. J Mol Biol 209:801–816.
9. Jen-Jacobson L (1997) Protein-DNA recognition complexes: conservation of structureand binding energy in the transition state. Biopolymers 44:153–180.
10. Jen-Jacobson L, Jacobson LA (2008) Protein-Nucleic Acid Interactions: Structural Biol-ogy, eds PA Rice and CC Correll (Royal Society of Chemistry Publishing, Cambridge),pp 13–46.
11. Pingoud A, Fuxreiter M, Pingoud V, WendeW (2005) Type II restriction endonucleases:structure and mechanism. Cell Mol Life Sci 62:685–707.
12. Sapienza PJ, dela Torre CA, McCoy WH, IV, Jana SV, Jen-Jacobson L (2005) Thermo-dynamic and kinetic basis for the relaxed DNA sequence specificity of “promiscuous”mutant EcoRI endonucleases. J Mol Biol 348:307–324.
13. Thielking V, Alves J, Fliess A, Maass G, Pingoud A (1990) Accuracy of the EcoRI restric-tion endonuclease: Binding and cleavage studies with oligodeoxynucleotide sub-strates containing degenerate recognition sequences. Biochemistry 29:4682–4691.
14. Grigorescu A, Horvath M, Wilkosz P, Chandrasekhar K, Rosenberg J (2004) RestrictionEndonucleases (Springer-Verlag, Heidelberg), pp 137–177.
15. Kurpiewski MR, et al. (2004) Mechanisms of coupling between DNA recognitionspecificity and catalysis in EcoRI endonuclease. Structure 12:1775–1788.
16. Woodhead JL, Bhave N, Malcolm ADB (1981) Cation dependence of restriction endo-nuclease EcoRI activity. Eur J Biochem 115:293–296.
17. Vipond IB, Baldwin GS, Halford SE (1995) Divalent metal ions at the active sites of theEcoRV and EcoRI restriction endonucleases. Biochemistry 34:697–704.
18. Peisach J, Blumberg WE (1974) Structural implications derived from the analysis ofElectron Paramagnetic Resonance spectra of natural and artificial copper proteins.Arch Biochem Biophys 165:691–708.
19. Bertini I, Canti G, Grassi R, Scozzafava A (1980) Effects of planar and tetrahedral dis-tortions on the ESR parameters of bis(salicylaldiminato)copper(II) complexes. InorgChem 19:2198–2200.
20. Mims WB, Peisach J (1978) The nuclear modulation effect in electron spin echoes forcomplexes of Cu2þ and imidazole with 14N and 15N. J Chem Phys 69:4921–4930.
21. Flanagan HL, Singel DJ (1987) Analysis of 14N ESEEM patterns of randomly orientedsolids. J Chem Phys 87:5606–5616.
22. Singh V, Zhu Z, Davidson VL, McCracken J (2000) Characterization of the tryptophanTryptophyl-Semiquinone catalytic intermediate of methylamine dehydrogenase byelectron spin-echo envelope modulation spectroscopy. J Am Chem Soc 122:931–938.
23. McCracken J, Peisach J, Dooley DM (1987) Cu(II) coordination chemistry of amineoxidases. Pulsed EPR studies of histidine imidazole, water, and exogenous ligandcoordination. J Am Chem Soc 109:4064–4072.
24. McCracken J, et al. (1988) Electron spin-echo studies of the copper binding site inphenylalanine hydroxylase from Chromobacterium violaceum. J Am Chem Soc110:1069–1074.
25. Jiang F, McCracken J, Peisach J (1990) Nuclear quadrupole interactions in copper(II)-diethylenetriamine-substituted imidazole complexes and in copper(II) proteins. JAm Chem Soc 112:9035–9044.
26. Burns CS, et al. (2002) Molecular features of the copper binding sites in the Octarepeatdomain of the prion protein. Biochemistry 41:3991–4001.
27. Huffman DL, et al. (2002) Spectroscopy of Cu(II)-PcoC and the multicopper oxidasefunction of PcoA, two essential components of Escherichia coli pco copper resistanceOperon. Biochemistry 41:10046–10055.
28. Yang Z, Becker J, Saxena S (2007) On Cu(II)-Cu(II) distance measurements using pulsedelectron electron double resonance. J Magn Reson 188:337–343.
29. Bode BE, Plackmeyer J, Prisner TF, Schiemann O (2008) PELDOR measurements on aNitroxide-labeled Cu(II) Porphyrin: Orientation selection, spin-density distribution,and conformational flexibility. J Phys Chem A 112:5064–5073.
30. Yang Z, Kise D, Saxena S (2010) An approach towards the measurement of nanometerrange distances based on Cu2þ ions and ESR. J Phys Chem B 114:6165–6174.
31. Kim Y, Grable JC, Love R, Greene PJ, Rosenberg JM (1990) Refinementof Eco RI endo-nuclease crystal structure: a revised protein chain tracing. Science 249:1307–1309.
32. Stone KM, et al. (2008) Electron spin resonance shows common structural features fordifferent classes of EcoRI-DNA complexes. Angew Chem Int Ed 47:10192–10194.
33. Engler LE, et al. (2001) The Energetics of the interaction of BamHI Endonuclease withits recognition site GGATCC. J Mol Biol 307:619–636.
34. Hancock SP, Hiller DA, Perona JJ, Jen-Jacobson L (2011) The energetic contribution ofinduced electrostatic asymmetry to DNA bending by a site-specific protein. J Mol Biol406:285–312.
35. Pingoud V, et al. (2009) On the divalent metal ion dependence of DNA cleavage byrestriction endonucleases of the EcoRI family. J Mol Biol 393:140–160.
36. Steitz TA, Steitz JA (1993) A general two-metal-ion mechanism for catalytic RNA. ProcNatl Acad Sci USA 90:6498–6502.
37. Steitz TA (1998) A mechanism for all polymerases. Nature 391:231–232.38. Baldwin GS, Sessions RB, Erskine SG, Halford SE (1999) DNA cleavage by the EcoRV
restriction endonuclease: Roles of divalent metal ions in specificity and catalysis. JMol Biol 288:87–103.
39. Horton NC, Perona JJ (2004) DNA Cleavage by EcoRV endonuclease: Two metal ions inthree metal ion binding sites. Biochemistry 43:6841–6857.
40. Yang W, Lee JY, Nowotny M (2006) Making and breaking nucleic acids: two-Mg2þ-ioncatalysis and substrate specificity. Mol Cell 22:5–13.
41. Dupureur CM (2010) One is enough: Insights into the two-metal ion nuclease mechan-ism from global analysis and computational studies. Metallomics 2:609–620.
42. Horton JR, Cheng X (2000) PvuII endonuclease contains two calcium ions in active sites.J Mol Biol 300:1049–1056.
43. Dupureur CM, Conlan LH (2000) A catalytically deficient active site variant of PvuIIendonuclease binds Mg(II) ions. Biochemistry 39:10921–10927.
44. Conlan LH, Dupureur CM (2002) Dissecting the metal ion dependence of DNA bindingby PvuII endonuclease. Biochemistry 41:1335–1342.
45. Xie F, Qureshi SH, Papadakos GA, Dupureur CM (2008) One- and two-metal ion cat-alysis: global single-turnover kinetic analysis of the PvuII endonuclease mechanism.Biochemistry 47:12540–12550.
46. Honig B, Nicholls A (1995) Classical electrostatics in biology and chemistry. Science268:1144–1149.
47. Klapper I, Hagstrom R, Fine R, Sharp K, Honig B (1986) Focusing of electric fields in theactive site of Cu-Zn superoxide dismutase: effects of ionic strength and amino-acidmodification. Proteins 1:47–59.
48. Chin K, Sharp KA, Honig B, Pyle AM (1999) Calculating the electrostatic properties ofRNA provides new insights into molecular interactions and function. Nat Struct Biol6:1055–1061.
49. Rocchia W, Alexov E, Honig B (2001) Extending the applicability of the nonlinear Pois-son–Boltzmann equation: multiple dielectric constants and multivalent ions. J PhysChem B 105:6507–6514.
50. Rocchia W, et al. (2002) Rapid grid-based construction of the molecular surface andthe use of induced surface charge to calculate reaction field energies: applications tothe molecular systems and geometric objects. J Comput Chem 23:128–137.
51. Honig B, Sharp K, Yang AS (1993) Macroscopic models of aqueous solutions: Biologicaland chemical applications. J Phys Chem 97:1101–1109.
52. Brewer GJ (2007) Iron and copper toxicity in diseases of aging, particularly athero-sclerosis and Alzheimer’s disease. Exp Biol Med 232:323–335.
53. Bruice TC, Benkovic SJ (2000) Chemical basis for enzyme catalysis. Biochemistry39:6267–6274.
54. Lesser DR, et al. (1992) Stereoselective interaction with Chiral Phosphorothioates atthe Central DNA kink of the EcoRI endonuclease-GAATTC complex. J Biol Chem267:24810–24818.
55. Connolly BA, et al. (2001) Assay of restriction endonucleases using oligonucleotides.Methods in Molecular Biology 148:465–490.
56. MacKerell AD, Jr, Feig M, Brooks CL (2004) Extending the treatment of backboneenergetics in protein force fields: Limitations of gas-phase quantum mechanics in re-producing protein conformational distributions in molecular dynamics simulations. JComput Chem 25:1400–1415.
57. Foloppe N, MacKerell AD, Jr (2000) All-atom empirical force field for nucleic acids: I.Parameter optimization based on small molecule and condensed phase macromole-cular target data. J Comput Chem 21:86–104.
58. MacKerell AD, Jr, Banavali NK (2000) All-atom empirical force field for nucleic acids: II.Application to molecular dynamics simulations of DNA and RNA in solution. J ComputChem 21:105–120.
59. Pettersen EF, et al. (2004) UCSF Chimera—a visualization system for exploratoryresearch and analysis. J Comput Chem 25:1605–1612.
60. DeLanoWLT (2002) The PyMOLMolecular Graphics System (DeLano Scientific, San Car-los, CA).
E1000 ∣ www.pnas.org/cgi/doi/10.1073/pnas.1200733109 Yang et al.