Dirección: Dirección: Biblioteca Central Dr. Luis F. Leloir, Facultad de Ciencias Exactas y Naturales, Universidad de Buenos Aires. Intendente Güiraldes 2160 - C1428EGA - Tel. (++54 +11) 4789-9293 Contacto: Contacto: [email protected]Tesis Doctoral Estudio de las vías visuales Estudio de las vías visuales superiores en el glaucoma superiores en el glaucoma experimental experimental Bordone, Melina Paula 2015-03-20 Este documento forma parte de la colección de tesis doctorales y de maestría de la Biblioteca Central Dr. Luis Federico Leloir, disponible en digital.bl.fcen.uba.ar. Su utilización debe ser acompañada por la cita bibliográfica con reconocimiento de la fuente. This document is part of the doctoral theses collection of the Central Library Dr. Luis Federico Leloir, available in digital.bl.fcen.uba.ar. It should be used accompanied by the corresponding citation acknowledging the source. Cita tipo APA: Bordone, Melina Paula. (2015-03-20). Estudio de las vías visuales superiores en el glaucoma experimental. Facultad de Ciencias Exactas y Naturales. Universidad de Buenos Aires. Cita tipo Chicago: Bordone, Melina Paula. "Estudio de las vías visuales superiores en el glaucoma experimental". Facultad de Ciencias Exactas y Naturales. Universidad de Buenos Aires. 2015-03-20.
Transcript
Di r ecci ó n:Di r ecci ó n: Biblioteca Central Dr. Luis F. Leloir, Facultad de Ciencias Exactas y Naturales, Universidad de Buenos Aires. Intendente Güiraldes 2160 - C1428EGA - Tel. (++54 +11) 4789-9293
Estudio de las vías visualesEstudio de las vías visualessuperiores en el glaucomasuperiores en el glaucoma
experimentalexperimental
Bordone, Melina Paula
2015-03-20
Este documento forma parte de la colección de tesis doctorales y de maestría de la BibliotecaCentral Dr. Luis Federico Leloir, disponible en digital.bl.fcen.uba.ar. Su utilización debe seracompañada por la cita bibliográfica con reconocimiento de la fuente.
This document is part of the doctoral theses collection of the Central Library Dr. Luis FedericoLeloir, available in digital.bl.fcen.uba.ar. It should be used accompanied by the correspondingcitation acknowledging the source.
Cita tipo APA:
Bordone, Melina Paula. (2015-03-20). Estudio de las vías visuales superiores en el glaucomaexperimental. Facultad de Ciencias Exactas y Naturales. Universidad de Buenos Aires.
Cita tipo Chicago:
Bordone, Melina Paula. "Estudio de las vías visuales superiores en el glaucoma experimental".Facultad de Ciencias Exactas y Naturales. Universidad de Buenos Aires. 2015-03-20.
UNIVERSIDAD DE BUENOS AIRES FACULTAD DE CIENCIAS EXACTAS Y NATURALES
Estudio de las vías visuales superiores en el glaucoma experimental
Tesis presentada para optar al título de Doctora de la Universidad de Buenos Aires en el área: CIENCIAS BIOLÓGICAS
Melina Paula Bordone
Directora de tesis: Prof. Dra. Ruth Estela Rosenstein Consejero de Estudios: Prof. Dr. Matías Pandolfi
Lugar de trabajo: Departamento de Bioquímica Humana, Laboratorio de Neuroquímica Retiniana y Oftalmología Experimental, Facultad de Medicina, UBA. CEFyBO/CONICET. Buenos Aires, 2015
Estudio de las vías visuales superiores en el glaucoma experimental
El glaucoma, una de las principales causas de ceguera irreversible, se caracteriza por una pérdida
progresiva de las funciones visuales, que se asocia a la muerte de células ganglionares retinianas
(CGRs) y atrofia de la cabeza del nervio óptico (NO). El principal factor de riesgo es el aumento de
la presión intraocular (PIO). Aunque el glaucoma fue concebido como una enfermedad limitada al
ojo, los axones de las CGRs son extraoculares, con componentes intraorbitales e intracraneales. En
este trabajo de Tesis se examinaron los efectos del glaucoma experimental agudo y crónico sobre el
sistema visual consciente (SV-C) y el sistema visual no formador de imagen (SV-NFI). El SV-C en
la rata, se compone de las CGRs clásicas y sus axones que proyectan al colículo superior (CS) y al
núcleo geniculado lateral (NGL). El SV-NFI se compone por las CGRs intrínsecamente fotosensibles
que expresan melanopsina (CGRsm) y proyectan a los núcleos supraquiasmáticos (NSQ), al núcleo
pretectal olivar (NPO) y al intergeniculado (IG), que participan en la sincronización del reloj
circadiano y el reflejo pupilar, entre otros. El modelo de glaucoma agudo consistió en un aumento de
la PIO a 70 mm de Hg durante 90 min (hipertensión ocular aguda, HOA), en tanto que el glaucoma
crónico se indujo a través de inyecciones intracamerales de condroitín sulfato (CSU), una vez por
semana, durante 15 semanas. A los 7 días post-HOA se observó una alteración significativa de la
función y estructura retinianas, con una pérdida significativa de CGRs, así como cambios astro- y
microgliales en el CS y una disminución en el transporte anterógrado desde la retina al CS y NGL,
pero no a los NSQ y al NPO. El número de CGRsm, los niveles de melanopsina y el reflejo pupilar
consensual permanecieron inalterados aún a las 4 semanas post-HOA. La administración de CSU por
6 semanas indujo alteraciones en la función retiniana y en la vía visual, una disminución en el
transporte anterógrado a todas las áreas de proyección y cambios gliales a nivel del NO, el CS y la
retina. En el CS, estas alteraciones incluyeron una marcada respuesta micro- y oligodendroglial y una
moderada respuesta astrocitaria, que se acompañaron de una disminución del contenido lipídico. A
las 15 semanas de glaucoma crónico, estas alteraciones fueron más marcadas e incluyeron
alteraciones en los axones, sin afectar el número de neuronas coliculares. La minociclina previno
algunas de las alteraciones inducidas por la hipertensión ocular crónica, como el déficit en el
transporte anterógrado desde la retina al CS. En cuanto al SV-NFI, el glaucoma crónico indujo una
caída significativa en el número de CGRsm y los niveles de melanopsina, así como en el transporte
desde la retina a los NSQ y el NPO, con una disminución en el reflejo pupilar consensual. En suma,
los resultados obtenidos en esta Tesis aportan datos de relevancia respecto a la participación de las
áreas visuales post-retinianas en el daño glaucomatoso.
Describir lo que significó la realización de esta Tesis inevitablemente me traslada a lo vivido durante mi
primer Maratón. Entrenar para un maratón no solo requiere preparación física sino también mental.
Motivación, confianza, dedicación, responsabilidad, tolerancia a la frustración y al dolor, paciencia,
perseverancia, compañerismo y entrega, son cualidades que se cultivan en las pistas y se trasladan a la
vida, y en este caso también, al quehacer científico. El científico, y sobre todo el que aspira a serlo,
motivado por la curiosidad y un eventual descubrimiento a veces se ve frustrado por experimentos que no
dan, técnicas que no salen e incluso aparatos que no andan, reactivos que no llegan y otras cuestiones que
superan la realidad para la que uno ha sido entrenado durante sus épocas de estudiante. Luego, la
paciencia, las charlas con compañeros de labo más experimentados o con amigos y familiares que tocan el
tema de oído y mucha perseverancia, permiten volverse a embarcar en la búsqueda de alguna nueva pista.
Aprender a aceptar la incertidumbre y confiar en que en algún momento todo va a llegar a buen puerto
resulta imprescindible para no bajar los brazos y volverlo a intentar. Tal como le ocurre al maratonista,
los primeros kilómetros casi no se sienten, pasada la primera mitad afloran un mar de dudas e incontables
dolores y sin embargo, uno sigue. Sólo cuando ya falta poco se toma conciencia del esfuerzo invertido y
el camino transitado. Es entonces cuando la balanza se inclina y brotan energías desde lo más profundo,
para tirar hasta el 40 y disfrutar del sprint final, esos 2,195 km llenos de emociones indescriptibles junto
con la imagen de todas aquellas personas que hicieron posible tremenda locura. Les agradezco:
A Ruth, por confiar en mí para lanzarme a la aventura, por transmitirme su amor por la retina, por
acompañarme en los primeros pasos y en los últimos, por sacar la brújula en los momentos clave,
por su rigurosidad y sus críticas para hacer de éste un trabajo más sólido y de mí una persona más
fuerte.
A Laura Pasquini, por enseñarme el mundo de los oligodendrocitos, la infinidad de inmunos y
anticuerpos, las horas en el confocal, el IMARIS, las charlas y los papers.
A Dami, por su amistad, sus consejos y su guía, su entusiasmo, su generosidad inmensa y su
tiempo, muchas veces hasta largas horas de la noche.
A Moni, por ser madre y compañera al mismo tiempo. ¡Trabajo difícil ese, si los hay! Por ser mi
ejemplo desde que tengo 6, por guiarme hasta acá y ¿después?, por la búsqueda de papers, la
puesta a punto de técnicas y la bibliografía de la Tesis. A vos Ma, gracias infinitas.
A Ine, por la paciencia y ayuda con las compras de reactivos, trámites burocráticos, las meriendas
todas y los descuentos, Jaja!
A Dani, por las charlas y los mates, las recetas vegetarianas y por localizar algo en el labo cada
vez que necesitaba.
A Pablito, por las inyecciones de CSU, sus chistes malos, su entusiasmo y su carisma, su amistad.
Agradecimientos
A los peques, Flor, Marquitos y Magui, por rejuvenecer al labo, la buena onda, el inmenso
compañerismo, los mates, las horas de compu, los VEPs, las charlas de biología y de la vida.
Chiquis, los quiero!
A Maga, por ofrecerme su hogar durante el doctorado y sus consejos para los formularios de las
becas.
A Diego, Eze, Nuria, Nico, con quienes compartí los últimos años de la Licenciatura y los
primeros del Doctorado, por enseñarme mucho de lo que sé de la “mesada” y dejarme “meter
mano” en sus experimentos.
A Charly, por procesar todos mis “nervios”, las horas y charlas en el MET y por su buena onda.
A Mariana, por ayudarme con el Red Oil, su compañía en las interminables cuantificaciones de
los procesos de la glía, las charlas de la vida y su amistad.
A los “Lemmings”, por los reactivos y la yerba prestada/robada, el microscopio, los almuerzos
compartidos, los festejos conjuntos. En especial a Memi y, también en este último tiempo, a
Silvia, por sus charlas y su amistad.
A mis padres, por darme la vida y por hacer de mí hoy una mujer independiente. A Mamá, por
estar en los momentos más difíciles, darme su garra, su sensibilidad y su valor y por su inmenso
amor. A Papá, por decir sí a todas mis locuras, inculcarme el amor por la naturaleza y la aventura,
y por los no sé cuántos miles de km de ruta para verme cruzar la Cordillera de los Andes a pie!
A mis hermanos, Aye y Ale porque son únicos, por el amor/odio, porque los veo y me veo y los
amo.
A Javi, por entregarme su corazón, hacer mi vida más hermosa y el mundo un lugar más feliz, por
su infinito compañerismo y generosidad, por su ternura y espontaneidad, por su música y por su
gran ayuda en toda esta última etapa.
A mis primos Lore y Victor y sus pimpollos, Celes y Tizi, por su apoyo incondicional, su amor y
sus abrazos.
A Jorge y a Pablito, por las intensas charlas y consejos y por su incondicional apoyo y su cariño.
A mi abuela, por ofrecerme su casa para internarme a estudiar y por bancarse mis nervios antes de
los exámenes.
Agradecimientos
A Laura Colman, por estos 5 años que llevamos juntas, por enseñarme a superar dificultades y
miedos, por animarme a animarme y perseguir mis sueños y por confiar en mí antes que yo
misma.
A “las chichis del Nacional” (Ceci, Flor, Naty, Meli, Lu y Andy) y a mis “compañeritas de la
vida” (Romi, Taty, Nadia, Fer, Lau y Adri), por cultivar todos estos años de amistad repletos de
los más hermosos recuerdos.
A mis amigos de la Facu (Ale, Vero, Belu y Camilo), por las innumerables catarsis doctorales y
científicas. Ya casi estamos, chicos! =)
A Eric Rosenberg, por quererme como un hermano, por las interminables charlas a la salida de los
entrenamientos, por las marchas, su confianza y su apoyo. HLVS!
A Pablito y a Pau, por enseñarme la otra cara del deporte, a correr con una sonrisa y a respirar
para desechar lo negativo, por su tiempo, su entrega, su sensibilidad y su amistad sincera.
A los TROTAS, por su compañerismo y su generosidad desinteresada, por contagiarme su
entusiasmo y energía, por los domingos a las 6 AM, por los fondos y decenas de km, por los
nervios de las largadas y el llanto de llegada, por demostrarme que no existen imposibles, por los
PISTA, PISTA, PISTA que pasaron y que están por venir!
A la Facultad de Medicina de la UBA, en especial al CEFyBO, por el lugar de trabajo brindado
para la realización de esta Tesis.
Al Sistema Nacional de Microscopía por haberme permitido la utilización de los equipos de
microscopía confocal (Dpto. de Fisiología de la Fac. de Medicina de la UBA) y de microscopía
electrónica (Instituto de Biología Celular y Neurociencia “Prof. E. De Robertis”).
Al IQUIFIB del Dpto. de Química Biológica de la Fac. de Farmacia y Bioquímica de la UBA, por
las innumerables horas en el crióstato y en el microscopio de fluorescencia.
Al Estado Nacional que financió mi educación primaria, secundaria y hasta mi formación
universitaria y de post-grado.
A mis padres, Mónica y Sergio.
Al NROE, porque esta Tesis es el
reflejo de un trabajo en equipo.
A Javi, por acompañarme desde
el 28 y hacer los últimos km
más felices y divertidos.
“The difficulty lies, not in the new ideas, but in escaping from the old ones…”
John Maynard Keynes (1883-1946)
“…Mi abuelo era ciego. Año tras año se sentó en la misma habitación ante un brasero de carbón, en el mismo rincón vuelto hacia el Este. Cada tanto volvía la cabeza hacia el Sur, pero nunca al Norte. Una vez que me di cuenta de ese hábito suyo de volver la cara sólo en una dirección, me sentí tremendamente perturbado (…) Eso me provocaba malestar. Me parecía misterioso. Al sur había lugares soleados y me pregunté si, aun siendo ciego, podría percibir esa dirección como algo un poco más luminoso…”
Historias en la palma de la mano, Yasunari Kawabata, 1971.
“The pressures on scientists today oppose truly creative thinking.”
Craig Loehle, 1990.
Los resultados presentados en este trabajo de Tesis forman parte de las siguientes publicaciones
- Effect of retinal ischemia on the non-image forming visual system.
Ma. Florencia González Fleitas 1, Melina P. Bordone1, Ruth E. Rosenstein, Damián Dorfman.
Chronobiology International 2014; 19:1-12. (versión electrónica previa a publicación) 1: ambos autores prestaron igual contribución al trabajo citado.
- Experimental glaucoma in rats induces early glial alterations in the superior colliculus
Melina P. Bordone, Laura A. Pasquini, Pablo H. Sande, Marcos Aranda, Ezequiel Salido, Ruth
Rosenstein. (manuscrito en redacción)
- Involvement of microglia in the early disruption of the retinal anterograde transport
induced by experimental glaucoma
Melina P. Bordone, Damián Dorfman, Pablo H. Sande, Laura A. Pasquini, Ma. Florencia
González Fleitas, Ruth Rosenstein. (manuscrito en redacción)
de un proceso patológico en animales de experimentación, aún con diferencias respecto al
humano, contribuye significativamente a la comprensión de un proceso en su contraparte
humana. A pesar de múltiples intentos, el desarrollo de modelos para el estudio del glaucoma
todavía no ha alcanzado consenso en un modelo que reproduzca completamente la enfermedad
en animales de laboratorio y sea reproducible. Dado el vínculo estrecho entre la hipertensión
ocular y el daño glaucomatoso, los modelos animales de glaucoma generalmente se desarrollan a
través de la inducción de hipertensión ocular o hacen uso de cepas de animales con aumento
espontáneo de la PIO (Pang y Clark 2007; Sappington y col., 2010). En este sentido, las
estrategias más frecuentemente utilizadas son: 1) la cauterización u oclusión de 2 ó 3 de las
venas epiesclerales (Shareef y col., 1995); 2) la inyección de solución salina hipertónica en las
venas acuosas (Morrison y col., 1997); 3) el bloqueo del flujo acuoso por fotocoagulación luego
de la inyección de tinta china en la cámara anterior (Ueda y col., 1998); 4) la inyección de
microesferas de látex en la cámara anterior del ojo que bloquea el drenaje del humor acuoso a
nivel del trabeculado (Weber y Zelenak, 2001) y 5) el uso de cepas de animales en los que la
patología se produce por alteraciones genéticas, de los cuales el más frecuentemente utilizado es
el ratón DBA/2J (Anderson y col., 2002). Estos ratones desarrollan una hipertensión ocular
espontánea, muy variable entre individuos, entre sexos e incluso entre ojos de un mismo animal,
que se hace evidente entre los 6 - 10 meses de vida. Este aumento de la PIO se debe a la
mutación recesiva de dos genes, Gpnmb y Tyrp-1 y tiene como consecuencia una atrofia del
estroma del iris y una dispersión del pigmento que produce la obstrucción de la vía de salida del
humor acuoso, provocando el aumento de la PIO que se asimila al glaucoma pigmentario en
humanos (Anderson y col., 2002). Ninguno de estos modelos es ideal y todos ellos tienen
algunas desventajas, como la necesidad del uso de equipos especializados y de técnicas
quirúrgicas complejas, o presentan una cinética de aumento de la PIO marcadamente diferente a
la de los humanos.
Introducción
43
Como ya se mencionó, los tipos predominantes de GAGs en el trabeculado son el AH y el
CSU. Trabajos pioneros de Barany y Scotchbrook (1954) demostraron que el tratamiento de ojos
de gato con hialuronidasa testicular bovina disminuye la resistencia al filtrado de humor acuoso
en el ángulo, a casi la mitad de su valor inicial. Desde entonces, se ha dedicado considerable
atención a los mucopolisacáridos sensibles a hialuronidasa en el sistema de salida del flujo
acuoso. En este sentido, se ha demostrado que la hialuronidasa testicular incrementa el flujo de
humor acuoso en ojos de perros (Van Buskirk y Brett, 1978) aunque la evidencia sugiere un
efecto menor en ojos humanos (Hayasaka y Sears, 1978). Asimismo, se demostró una
correlación entre la remoción del AH y la disminución en el flujo de humor acuoso en ojos de
conejo (Knepper y col., 1984), perro (Van Buskirk y Brett, 1978) y vaca (Barany y Scotchbrook,
1954), pero no en ojos de primates (Hubbard y col., 1997; Sawaguchi y col., 1992).
En la práctica oftalmológica, se dispone de evidencias considerables a partir del uso de
agentes viscoelásticos que avalan el vínculo entre los GAGs y la PIO. Estos agentes contienen
AH y CSU y se han convertido en una herramienta esencial en la cirugía del segmento anterior
para generar espacio y reducir el trauma y la pérdida de células endoteliales. Estos agentes son
responsables de causar o exacerbar en forma transitoria, pero a veces significativa, una elevación
post-quirúrgica de la PIO (Jurgens y col., 1997). Estos resultados sugieren que el AH y el CSU
podrían participar en la regulación del flujo de humor acuoso y que la acumulación de estos
GAGs en el trabeculado podría cumplir un rol significativo en el glaucoma crónico de ángulo
abierto. En trabajos previos de nuestro laboratorio hemos desarrollado un modelo experimental
de glaucoma a través de la administración semanal de AH en la cámara anterior del ojo. En este
sentido, hemos demostrado que la inyección aguda o crónica de AH aumenta significativamente
la PIO en el ojo de rata (Benozzi y col., 2002) y su administración en forma crónica reproduce
aspectos centrales del glaucoma crónico humano tanto a nivel funcional como histológico
(Moreno y col., 2005a). Sin embargo, el análisis bioquímico del perfil de GAGs de la red
Introducción
44
trabecular de pacientes con glaucoma demuestra una marcada disminución en el contenido de
AH y un aumento en el contenido de CSU (Knepper y col., 1996). Resultados similares se
demostraron en un modelo de glaucoma en conejos (Knepper y col., 1985). Estos resultados
sugieren que el CSU podría participar activamente en la regulación de la salida del humor acuoso
y que una acumulación de CSU en el trabeculado podría desempeñar un rol central en el
glaucoma crónico. De hecho, se demostró que el aumento en el contenido de CSU en el tejido
conectivo yuxtacanalicular de pacientes con glaucoma tiene mayor impacto sobre la resistencia a
la salida de humor acuoso que el aumento en los niveles de AH (Knepper y col., 2005). Estas
evidencias avalan la posibilidad de que un aumento en el contenido de CSU en la cámara anterior
del ojo podría provocar un aumento en la PIO, aspecto que se analizará en este trabajo de Tesis.
6.3. La vía visual en el glaucoma
El glaucoma es usualmente concebido como una enfermedad limitada al ojo; sin
embargo, los axones de las CGRs son extraoculares, con componentes intraorbitales e
intracraneales. En el sistema visual, como en otros sistemas cerebrales, la función y
supervivencia neuronal depende de las conexiones con otras neuronas. Se ha demostrado una
reducción marcada en la densidad de fibras retinianas procedentes de ojos con glaucoma
experimental en el NGLd y el CS (Drouyer y col., 2008). Sin embargo, la información disponible
respecto a cambios trans-sinápticos a nivel del NGL, el CS y la corteza visual primaria (CV1)
han sido hasta ahora sólo escasamente examinados. Estudios en modelos experimentales de
glaucoma en monos han demostrado una disminución significativa en el número, la densidad y el
tamaño de neuronas, así como en el volumen laminar del NGL ipsilateral al ojo glaucomatoso en
las capas retinorreceptivas del NGL que reciben aferencias del ojo glaucomatoso (Weber y col.,
2000; Yücel y col., 2001). En algunos casos, se ha observado un daño preferencial en la vía M
(Weber y col., 2000), en otros de la vía P (Yücel y col., 2001), en tanto que otros estudios
demostraron daños comparables en ambos sistemas (Vickers y col., 1997; Yücel y col., 2003).
Introducción
45
Esta contradicción se extiende también a los resultados obtenidos en un número pequeño de
autopsias humanas (Chaturvedi y col., 1993; Gupta y col., 2006).
Los mecanismos involucrados en el daño neuronal del NGL, el CS y la CV1 en el
glaucoma son aún elusivos. Aunque es un hecho establecido que en el glaucoma las CGRs
mueren por apoptosis, no se han realizado estudios similares a nivel de las proyecciones
cerebrales. Sin embargo, se ha demostrado en situaciones experimentales en las que se produce
desconexión de vías aferentes que las neuronas desaferentadas se “encogen” y posteriormente
mueren por apoptosis (Guillery, 1973). Se ha sugerido que la atrofia neuronal inducida por la
hipertensión ocular podría representar tanto un estado de quiescencia o reducción en el consumo
de energía secundario a la pérdida del input retiniano (Zhang y col., 2009). Algunas evidencias
sugieren que estos cambios podrían ser reversibles, lo que permitiría inducir alguna forma de
intervención terapéutica para prevenir la muerte celular aún en estadíos en los que se observa
reducción del tamaño neuronal (Guillery, 1973; Matthews, 1964). Sin embargo, todos los
estudios dedicados a la búsqueda de nuevas estrategias terapéuticas para el glaucoma, no han
examinado si el beneficio terapéutico se extiende a las vías de procesamiento cortical de la
información visual.
Otro aspecto de interés es la elucidación del curso temporal del daño glaucomatoso en las
vías visuales superiores. Si bien en un modelo de glaucoma en monos se ha descripto una
relación lineal entre el grado de daño del NO y la atrofia de neuronas en el NGL (Yücel y col.,
2001; 2003), también se ha descripto una reducción del tamaño de neuronas del NGL en monos
con hipertensión ocular, sin pérdida evidente de fibras del NO. Esto podría sugerir que la atrofia
del NGL es un evento temprano, eventualmente relacionado en forma directa con la hipertensión
ocular o con cambios funcionales (pero no histológicos) retinianos. La degeneración de neuronas
del NGL y del CS podría ser un mecanismo activo de daño glaucomatoso sobre las CGRs y sus
axones al reducir su metabolismo y disminuir el suministro de factores tróficos a la retina (Gupta
Introducción
46
y Yücel, 2007; Pearson y Stoffler, 1992). Además, cambios en las neuronas del NGL pueden
provocar daños post-sinápticos (degeneración trans-sináptica) en la CV1, localizada en la parte
posterior del cerebro y organizada en columnas de dominancia ocular específica (Crawford y
col., 2001; Lam y col., 2000). Estudios de resonancia magnética demostraron alteraciones
significativas en la CV de pacientes con glaucoma, con una reducción en la densidad de la
sustancia gris en las zonas corticales de proyección visual (Boucard y col., 2009). Estos
resultados sugieren que el glaucoma induce una degeneración retinotópica específica en la CV.
En un modelo de glaucoma experimental en ratas se demostró una alteración del metabolismo de
colina y un aumento marginal en los niveles de glutamato en la CV1 contralateral al ojo
glaucomatoso (Chan y col., 2009). Asimismo, se han demostrado cambios metabólicos en la
CV1 en monos con glaucoma experimental (Lam y col., 2003).
Estos resultados, aunque aún son evidencias fragmentarias obtenidas en un número muy
reducido de muestras humanas y en diferentes modelos experimentales de glaucoma, sugieren
que el daño de las CGRs provoca cambios degenerativos en el NGL, el CS y la CV1. Sin
embargo, las evidencias resultan contradictorias en algunos aspectos, los mecanismos
involucrados en el proceso degenerativo y el curso temporal de este proceso no han sido
exhaustivamente examinados y no se dispone al presente de estrategias terapéuticas capaces de
prevenir, curar, o detener el daño cortical asociado a la pérdida visual glaucomatosa. Desde una
perspectiva clínica, un estudio detallado de la relación entre el déficit retiniano y los cambios
estructurales y funcionales corticales, podrían contribuir a una mayor comprensión de la
progresión en la disfunción visual glaucomatosa, así como al desarrollo de estrategias
terapéuticas diseñadas para la protección del daño cerebral asociado al glaucoma.
Introducción
47
7. La minociclina
La minociclina (MINO) es un análogo semi-sintético de la tetraciclina, de fácil absorción
por el tracto gastro-intestinal, con una vida media aproximada de 18 h (revisado por Chen y col.,
2011). La MINO es una molécula altamente liposoluble, por lo que puede atravesar fácilmente la
barrera hemato-encefálica y hemato-retiniana. En dosis terapéuticas, la MINO tiene buena
tolerancia para su uso en humanos (Chen y col., 2011; Garrido-Mesa y col., 2013). Además de
sus propiedades antimicrobianas, la MINO tiene un potente efecto antiinflamatorio
inmunomodulatorio y neuroprotector (Maier y col., 2007; Chen y col., 2011). El efecto anti-
inflamatorio de la MINO involucra la inhibición de la activación y la migración microglial,
estimulando su transformación al fenotipo en “reposo” tanto in vivo como in vitro (Garrido-Mesa
y col., 2013), así como la inhibición de la síntesis de óxido nítrico y citoquinas pro-inflamatorias
(Yang y col., 2007; Baptiste y col., 2005; Bosco y col., 2008). Asimismo, se ha demostrado que
la MINO induce cascadas de señales anti-apoptóticos, disminuye la toxicidad glutamatérgica (Du
y col., 2001; Chen y col., 2000; Levkovitch-Verbin y col., 2013; 2014; Sánchez Mejía y col.,
2001; Wang y col., 2004; Zhu y col., 2002), y actúa como scavenger de radicales libres (Kraus y
col., 2005, Wilkins y col., 2004).
Como ya se mencionó, luego de una lesión neuronal, la microglía se activa y libera
sustancias con alto poder deletéreo. De hecho, existe consenso en que la microglía constituye un
blanco potencialmente explotable con fines preventivos y terapéuticos en procesos
neuroinflamatorios y neurodegenerativos. Sobre la base de sus efectos sobre la reactividad
microglial, se han demostrado efectos beneficiosos de la MINO en diversos modelos de injuria
neuronal, como isquemia cerebral (Yrjänheikkii y col., 1998), daño en la médula espinal (Teng y
col., 2004), esclerosis múltiple (Zhang y col., 2008), esclerosis lateral amiotrófica (Kriz y col.,
2002; Zhu y col., 2002), enfermedad de Huntington (Chen y col., 2000; Wang y col., 2006),
enfermedad de Parkinson (Du y col., 2001), y en modelos de daño retiniano inducido por luz
Introducción
48
intensa (Zhang y col., 2004) y transección del NO (Baptiste y col., 2005; Levkovitch-Verbin y
col., 2006; 2011). De hecho, se han realizado estudios clínicos para analizar el efecto de la
MINO en el tratamiento de pacientes con esclerosis múltiple, con resultados alentadores (Zabad
y col, 2007; Zhang y col., 2008). Sin embargo, sólo existen escasos antecedentes sobre el uso de
MINO en el glaucoma (Bosco y col., 2008; Levkovitch-Verbin y col., 2006; 2014). Bosco y
colaboradores (2008) demostraron que el tratamiento de ratones DBA/2J con MINO aumenta la
fracción de células microgliales con fenotipo ramificado, reduce los niveles de Iba-1 y previene
la disminución del transporte axonal retrógrado (desde el CS a la retina). Por otra parte, en un
modelo de glaucoma experimental en ratas inducido por la fotocoagulación translimbal con láser,
se ha demostrado que la MINO aumenta significativamente la expresión del gen anti-apoptótico
Bcl-2, disminuye la expresión de IL-18 y reduce el número de células microgliales fagocíticas en
la retina (Levkovitch-Verbin y col., 2014). El conjunto de antecedentes mencionados, avala la
realización de estudios adicionales para la evaluación del efecto de la MINO con fines
terapéuticos en el glaucoma, aspecto que se analizará en el marco de este trabajo de Tesis.
Objetivos
49
OBJETIVOS
Sobre la base de las consideraciones mencionadas en la Introducción, los objetivos centrales de
este trabajo de Tesis fueron:
1. Analizar los efectos del glaucoma agudo experimental sobre la retina y las vías visuales
superiores.
2. Analizar los efectos del glaucoma agudo experimental sobre el sistema visual no
formador de imágenes.
3. Desarrollar un modelo de glaucoma crónico experimental en ratas.
4. Analizar los efectos del glaucoma crónico experimental sobre las vías visuales superiores.
5. Examinar el efecto de la minociclina sobre las alteraciones inducidas por el glaucoma
crónico a nivel de la retina, el nervio óptico y el colículo superior.
6. Analizar los efectos del glaucoma crónico experimental sobre el sistema visual no
formador de imágenes.
Materiales y Métodos
50
MATERIALES Y MÉTODOS
1. Animales
Se utilizaron ratas Wistar macho adultas (300 ± 50 g). Los animales se mantuvieron bajo
condiciones controladas de temperatura (21 ± 2°C) y humedad, bajo un fotoperíodo de 12 h de
luz (~ 150 lux en las jaulas) y 12 h de oscuridad (encendido de las luces a las 08.00 h), con agua
y alimento ad libitum. En todos los casos, los animales se anestesiaron con hidrocloruro de
ketamina (150 mg/kg) e hidrocloruro de xilacina (2 mg/kg), administradas intraperitonealmente.
En algunos casos, se aplicó anestesia local (hidrocloruro de proparacaína al 0,5%) en la córnea.
1.1. Inducción de glaucoma agudo. Bajo anestesia general, se canuló la cámara anterior de
cada ojo con una aguja 30G conectada a un reservorio presurizado, conteniendo solución Ringer
lactato. Mediante la regulación de la presión del reservorio, se modificó la presión intraocular
(PIO), que se elevó a 70 mm de Hg durante 90 minutos. De esta manera se indujo una
hipertensión ocular aguda (HOA), caracterizada por el bloqueo total del flujo sanguíneo
(observado mediante examinación del fondo de ojo) y pérdida de la respuesta
electrorretinográfica. Luego de completado el período de hipertensión ocular, se retiró la aguja
de la cámara anterior y se comprobó la inmediata reperfusión y recuperación del flujo sanguíneo.
En el caso del tratamiento simulado, se realizó el mismo procedimiento pero sin el aumento de la
PIO (Figura 13). Durante todo el procedimiento se mantuvo constante la temperatura corporal
mediante el uso de mantas térmicas. Se excluyeron de los experimentos aquellos animales que
sufrieron daño en el cristalino (desarrollo de cataratas) durante el procedimiento. Los animales
se sacrificaron al cabo de 1, 2 ó 4 semanas.
Materiales y Métodos
51
Figura 13. Fotografías de ojos canulados en la cámara anterior. Panel A: Se muestra un ojo canulado con
una aguja 30G durante el procedimiento simulado. Panel B: Se muestra un ojo con ausencia de rubidez
durante la HOA.
1.2. Inducción de glaucoma crónico. Las ratas se anestesiaron como ya se mencionó.
Mediante el uso de agujas 30G, se inyectaron 10 l de condroitín sulfato (CSU) (400 mg/ml) en
un ojo y el mismo volumen de solución fisiológica en el ojo contralateral. Para ello, los ojos se
enfocaron bajo un microscopio quirúrgico con luz coaxial (Colden, Argentina). Se movió la
aguja a través del limbo esclero-corneal hacia la cámara anterior con el bisel hacia abajo. Cuando
la punta del bisel alcanzó la cámara anterior, el líquido progresivamente incrementó la
profundidad de la cámara, separando la aguja del iris y evitando el contacto con el cristalino. Las
inyecciones se realizaron una vez/semana usando la fuerza necesaria para vaciar el contenido de
la jeringa de tuberculina. El sitio de inyección fue el limbo esclero-corneal, comenzando en la
hora 12 y cambiando semanalmente el sitio de la inyección de hora en hora para evitar la
formación de pannus y rotando el animal para lograr mejor acceso al limbo. Se excluyeron los
animales que desarrollaron cataratas o pthisis bulbi, cuya incidencia no superó el 5% del total de
animales utilizados. Además, casi todos los animales desarrollaron un edema corneal localizado
en el sitio de la inyección que no duró más de 24 h. Durante el período en que los animales
estuvieron anestesiados, se colocó gel sobre los ojos para evitar la sequedad corneal. No se
observaron diferencias en la incidencia de las complicaciones oculares entre los grupos
inyectados con vehículo o CSU. Como control del efecto de las inyecciones per se, se utilizó un
grupo de animales que se manipuló y anestesió en forma análoga, pero no se aplicaron
B A
Materiales y Métodos
52
inyecciones intracamerales. En algunos experimentos, se inyectó vehículo y el ojo contralateral
permaneció intacto. Los animales se sacrificaron a las 3, 6, 10 ó 15 semanas de tratamiento.
1.2.1. Tratamiento con minocilina. Un grupo de animales con glaucoma crónico unilateral al
cabo de cuatro semanas, se dividió al azar en dos subgrupos: uno de ellos recibió
intraperitonealmente una solución de 30 mg/kg de hidrocloruro de minociclina (Sigma, EE.UU.)
en solución salina, una vez por día durante 2 semanas y el otro grupo, fue tratado en forma
análoga con el mismo volumen de vehículo (solución salina). La dosis, vía y esquema de
administración seleccionados se basó en trabajos de otros autores (Guasti y col., 2009). Los
animales se sacrificaron luego de dos semanas de tratamiento.
En todos los procedimientos se respetaron estrictamente las Guías para el Cuidado y Uso
de Animales de Laboratorio de la Association for Research in Vision and Ophthalmology
(ARVO).
2. Determinación de la presión intraocular
La PIO se determinó mediante un TonoPen XL (Figura 14) (Mentor, Norwell, MA,
EE.UU.) en ratas conscientes, según la técnica descripta por Moore y col. (1995). Los animales
se envolvieron suavemente con una toalla, sin provocar estasis venosa ni efectos valsalva, y
mientras un operador lo mantenía firme, otro realizaba la determinación. Se obtuvieron cinco
lecturas de cada ojo, omitiendo las que se registraban cuando el aparato no estaba en contacto
firme con la córnea. La variación entre lecturas resultó menor al 10%. Los promedios de estas
lecturas se registraron como la PIO de un ojo y un día determinados. La PIO de cada animal se
promedió con las del resto de los animales del mismo grupo y el valor promedio resultante se
registró como la PIO media ± error estándar (EE). No se observaron diferencias significativas
entre el ojo derecho y el ojo izquierdo en animales intactos o inyectados con vehículo. Las
mediciones de la PIO se realizaron a la misma hora cada día o semana (entre las 11.00 h y las
12.00 h), para evitar las variaciones diarias en este parámetro.
Materiales y Métodos
53
Figura 14. Determinación de la PIO en ratas.
3. Función de la retina y la vía visual
3.1. Electrorretinografía escotópica. Los animales se adaptaron a oscuridad por 6 h y luego se
anestesiaron, como ya se describió. Se utilizó hidrocloruro de fenilefrina al 2,5% y tropicamida
al 1% (Laboratorios Alcon, Argentina) para dilatar la pupila y se administró solución salina
(Laboratorios Alcon, Argentina) en forma continua sobre la córnea, para mantener un registro
estable y prevenir una queratopatía. Los animales se colocaron a 20 cm de la fuente luminosa.
Todos los registros se realizaron dentro de los 20 min luego de la inducción de la anestesia y se
mantuvo constante la temperatura corporal usando una manta térmica. Se usó un electrodo de
referencia en la oreja (ipsilateral), un electrodo de tierra en la porción media de la cola y un
electrodo de oro en el centro de la córnea. Para la correcta implantación de los electrodos se
utilizó una luz roja de 15 W, que no afectó la adaptación de los animales a la oscuridad. Los
electrorretinogramas (ERGs) de ambos ojos se registraron simultáneamente y en completa
oscuridad. Para ello, se aplicaron 20 pulsos de luz blanca sin atenuación (5 ms, 0,2 Hz)
generados por un estimulador fótico de diodos aplicados con una intensidad máxima de 9 cd
s/m2. Los registros se amplificaron utilizando un equipo Akonic BIO-PC (Akonic, Argentina).
La señal se registró con una ganancia de 100 V, sin atenuación, con filtros de ruido de baja (1,5
Hz) y de alta (1000 Hz) frecuencia para optimizar el registro. Se determinó la amplitud (en V)
de la onda a, como la diferencia en amplitud entre la línea isoeléctrica y el pico negativo, y la
Materiales y Métodos
54
amplitud de la onda b, como la diferencia entre el pico de la onda a y el pico positivo de la onda
b (Figura 15A). Se determinaron las latencias de cada onda. La respuesta electrorretinográfica se
obtuvo como el promedio de los valores de cada corrida. Se realizaron tres estímulos, con un
intervalo de 5 min.
3.2. Potenciales oscilatorios. Los potenciales oscilatorios (POs) se registraron utilizando el
mismo estimulador fótico (5 ms, 0,2 Hz), con filtros de baja (100 Hz) y alta frecuencia (300 Hz).
La amplitud de los POs se calculó como el punto de máxima amplitud de cada pico en relación
a la línea de base (Figura 15B). El resultado obtenido de la suma de los tres picos de los POs se
utilizó para el análisis estadístico.
Figura15. Determinación de la función retiniana. Panel A: Se muestra un registro de ERG escotópico
característico y el método utilizado para evaluar la amplitud y latencia de las ondas a y b. Panel B: Se
muestra un registro de los POs, y se señalan los picos P1, P2 y P3.
3.3. Potenciales visuales evocados (VEPs). Para el registro de los VEPs se colocaron
quirúrgicamente dos electrodos de acero inoxidable 4 mm lateral a la cisura inter-hemisférica y
5,6 mm por detrás de bregma (electrodo activo). Se colocaron electrodos de referencia en
posición 2 mm lateral a la línea media y 2 mm antes de bregma, y un electrodo de tierra en la
cola. Ambos electrodos se aislaron y fijaron con acrílico dental, y la piel se suturó con nylon 5/0.
Los VEPs se registraron 5 días después de la implantación de los electrodos de la siguiente
Materiales y Métodos
55
manera: luego de 6 h de adaptación a la oscuridad, las ratas se anestesiaron, las pupilas se
dilataron y la córnea se irrigó de forma intermitente como ya se describió, bajo luz roja tenue.
Cada ojo se registró individualmente, para lo cual se ocluyó el ojo contralateral, y se registró un
promedio de 70 estímulos. Los ojos se estimularon con luz blanca sin atenuar (1 Hz) con un
estimulador fótico (diodos emisores de luz), fijado a la máxima intensidad de luz y los registros
se amplificaron, filtraron (0,5 Hz filtro de baja frecuencia, 100 Hz filtro de alta frecuencia) y
promediaron. Se evaluaron las amplitudes entre el pico P1 y la deflexión N2 (componente P1-
N2) y la deflexión N2 y el pico P2 (componente N2-P2), y desde el inicio de los registros se
determinaron las latencias de P1, N2 y P2 (Figura 16).
Figura 16. Determinación de la función de la vía visual. Se muestra un registro de VEPs y el método
utilizado para evaluar la amplitud de cada componente (P1-N2 y N2-P2) y latencias de P1, N2 y P2.
4. Evaluación del reflejo pupilar
Los animales se adaptaron a la oscuridad por 1 h. Se aplicó un pulso de luz blanca de
baja intensidad (120 lux) en un ojo y se registró la contracción de la pupila en el ojo
contralateral. Luego se re-adaptaron los animales a oscuridad por 1 h y se evaluó el reflejo
pupilar consensual frente a un estímulo de luz blanca de alta intensidad (1200 lux). Los registros
se realizaron bajo luz infrarroja con una cámara de video digital (Sony DCR-SR60, Japón). Se
determinó el diámetro de la pupila antes y 30 s después de aplicar el pulso de luz. La velocidad
de muestreo fue de 30 imágenes/s. Se adquirieron las imágenes a partir de la grabación de video
Materiales y Métodos
56
digital utilizando el programa OSS Video Decompiler Software (One Stop Soft, EE.UU.). El
porcentaje de contracción de la pupila se calculó como el porcentaje del área de la pupila a los
30 s del inicio del pulso (estado estacionario) en relación con el tamaño de la pupila dilatada.
Los datos obtenidos de 10 animales por grupo se promediaron y la media se utilizó como el
valor representativo de cada grupo.
5. Procesamiento histológico
Bajo anestesia profunda, los animales se perfundieron con ~ 250 ml de solución
fisiológica, seguida de ~ 300 ml de solución fijadora (paraformaldehído al 4% en buffer fosfato
(PBS) 0,1 M, pH 7,4). Se disecó cuidadosamente el globo ocular con la porción intraorbital del
nervio óptico (NO) proximal y el cerebro. Las muestras se post-fijaron en la misma solución
fijadora durante toda la noche. Los ojos se procesaron como se describirá posteriormente, y se
obtuvieron cortes transversales en parafina de la retina para el análisis morfométrico (espesor
total y de las capas retinianas y número de CGRs) y estudios de inmunomarcación y cortes
transversales del NO para inmunofluorescencia. Los cerebros se cortaron transversalmente (~ 2
mm por encima del quiasma óptico) y se procesaron para la obtención de cortes por congelación
(cortes coronales) de los distintos núcleos de proyección retiniana.
5.1. Secciones semifinas. Bajo anestesia profunda, se expuso el corazón de los animales y se
inyectó intracardiacamente 0,5 ml de heparina (como anticoagulante) y 0,5 ml de nitroprusiato
de sodio al 2,4% (como vasodilatador). Los animales se perfundieron con ~ 200 ml de solución
fisiológica seguida de ~ 300 ml de glutaraldehído al 2% y paraformaldehído al 4% en PBS. Se
disecó cuidadosamente el globo ocular y la porción proximal del NO y el cerebro a la altura del
colículo superior (CS) y se procesaron para la obtención de muestras semifinas. Luego de varios
lavados con PBS, las muestras se post-fijaron durante 1 h en OsO4 al 1% en el mismo buffer.
Posteriormente, se realizaron varios lavados, las muestras se deshidrataron mediante series
crecientes de etanol, se aclararon en acetona y se incluyeron en araldita. Se obtuvieron secciones
Materiales y Métodos
57
transversales semifinas (0,5 m) del NO y parasagitales del CS mediante el uso de navajas de
vidrio y un ultramicrótomo Ultracut E (Reichert-Jung, Austria), que se colorearon con azul de
toluidina-bórax (Bancroft y Stevens, 1990).
5.2. Obtención de cortes en parafina. Luego de varios lavados, las muestras se deshidrataron
(serie creciente de alcohol: 70%, 96%, 100%), se aclararon en acetato de n-butilo y se
embebieron en parafina. Se obtuvieron secciones a lo largo del eje sagital del ojo, manteniendo
la membrana nictitante para facilitar su posterior orientación y secciones transversales del NO
proximal. Los cortes se utilizaron para el análisis histológico y/o morfométrico,
inmunohistoquímica, inmunofluorescencia y análisis de TUNEL (terminal deoxynucleotidyl
transferase dUTP nick end labeling). Todos los cortes se montaron sobre portaobjetos con carga
electrostática (Superfrost Plus Esco, EE.UU.).
5.3. Cortes por congelación. Las muestras se sumergieron en soluciones crecientes de sacarosa
(10%, 20% y 30%), y se armó un taco con solución criopreservante (Cryoblock, O.C.T,
EE.UU.). En un crióstato CM 1850 (Leica, Alemania) se realizaron secciones coronales seriadas
de 30 m a nivel de los NSQ, que se montaron en portaobjetos con carga electrostática. A nivel
del NGL, el NPO y el CS, se realizaron cortes coronales seriados y alternados de 40 m, de los
cuales el primero se descartó, el segundo se montó en portaobjetos cargados y el tercero se crio-
preservó en PBS y glicerol (1:1) a -20° C, para la realización de estudios de inmunomarcación
en cortes flotantes de cerebro para aquellos antígenos que requirieron una mayor penetración de
los anticuerpos. Para obtener cortes de retinas por congelación se realizaron dos pasos
adicionales: se extrajo la córnea y el cristalino antes del pasaje a sacarosa al 10% y previo al
armado del taco con solución criopreservante, se adsorbió con un papel de filtro el remanente de
sacarosa al 30% junto con el vítreo. Luego, los ojos se seccionaron sagitalmente para obtener
retinas en corte transversal junto con la cabeza del NO y la porción proximal del NO en corte
longitudinal de 15 m de espesor.
Materiales y Métodos
58
6. Microscopía electrónica
6.1. Análisis ultraestructural del nervio óptico. Se evaluó la ultraestructura de secciones
transversales de la región proximal del NO. Se realizaron secciones ultrafinas (70 nm) mediante
el uso de navajas de vidrio y un ultramicrótomo Ultracut E (Reichert-Jung, Austria). Las
secciones se montaron sobre grillas de 200 Mesh y se tiñeron con acetato de uranilo (2% en
etanol al 70%) y con citrato de plomo de Reynolds. Las secciones se analizaron en un
microscopio electrónico de transmisión Zeiss 109T (Zeiss, Alemania), equipado con una cámara
digital Gatan ES1000W, en el Laboratorio Nacional de Investigación y Servicio de Microscopía
Electrónica (LANAIS-MIE, CONICET-UBA, Buenos Aires, Argentina).
6.2. Análisis ultraestructural del colículo superior. Se utilizó un slicer de cerebro para ratas
de 200 - 300 g (# catálogo: NP 62-0047, Harvard Apparatus, EE.UU.) para obtener secciones
coronales de cerebro a la altura del CS de 1 mm de espesor. Se disecaron ambos CS tomando la
capa SGI como borde ventral y se orientaron las muestras mediante una muesca realizada en la
porción medial del CS. Posteriormente, se realizaron secciones ultrafinas parasagitales de la
porción lateral del CS como se muestra en la Figura 17, a fin de obtener cortes transversales de
los axones de las CGRs que ingresan a este nivel desde el tracto óptico (TO). A continuación, el
procedimiento utilizado fue esencialmente similar al que se describió para el NO.
Figura 17. Procesamiento del CS para microscopía electrónica. La figura de la izquierda esquematiza la
entrada de las fibras del tracto óptico (TO) al CS. La imagen central muestra un esquema del CS en corte
coronal y el plano de corte parasagital. La imagen de la derecha representa el plano parasagital con la
porción del CS lateral seccionada, indicada en verde. CSS: CS superficial, SO: Stratum opticum, D:
dorsal, L: lateral, C: caudal, y R: rostral.
Materiales y Métodos
59
7. Análisis de células TUNEL(+)
Para la detección de células apoptóticas, se utilizó el kit de detección in situ ApopTag
(S7110, Chemicon-Millipore, EE.UU.), según las especificaciones del fabricante. En cortes de
retina (a la altura de la cabeza del NO) se cuantificó el número de células TUNEL(+) por
sección. Los resultados obtenidos de 4 secciones separadas se promediaron y el valor promedio
de 5 animales se consideró representativo de cada grupo.
8. Análisis de inmunomarcación y detección con isolectina Griffonia simplicifolia I
Se utilizaron los siguientes anticuerpos primarios:
Anticuerpo Antígeno Especie y tipo Dilución Empresa
Brn3a Factor de transcripción neuronal Cabra, policlonal 1:500 Santa Cruz,
EE.UU.
ED1 (CD68) Glicoproteína de superficie y de vesículas fagocíticas (microglía activada, macrófagos, neutrófilos y basófilos).
Ratón, monoclonal 1:200 Abcam, EE.UU.
GFAP Proteína glial fibrilar ácida (astrocitos y células de Müller reactivas).
Ratón, monoclonal 1:1200 Sigma, EE.UU.
(conjugado-Cy3)
Conejo, monoclonal
1:200 Cell Signaling,
EE.UU.
Iba-1 Proteína adaptadora de unión a calcio ionizado- 1 (microglía y macrófagos).
Cabra, policlonal 1:500 Abcam, EE.UU.
MBP Proteína básica de mielina (oligodendrocitos mielinizantes maduros).
Conejo, policlonal 1:400 *
Melanopsina Fotopigmento de las CGRsm. Conejo, policlonal 1:750 Affinity
Bioreagent, EE.UU.
Nestina
Filamento intermedio característico de progenitores neurales durante el desarrollo. En el adulto, puede re-expresarse en condiciones de daño del SNC.
Pollo 1:150 *
Neuromics, EE.UU.
NeuN Proteína nuclear específica de neuronas. Ratón, monoclonal
IgG 1:200
Millipore, EE.UU.
pNF-H (SMI-310)
Cadena pesada de neurofilamento extensivamente fosforilado (axones maduros).
Ratón, monoclonal 1:1000 Abcam, EE.UU.
NG2
Proteoglicano condroitín sulfato - 4 (polidendrocitos, entre las que se encuentran células precursoras de oligodendrocitos y oligodendrocitos premielinizantes).
Conejo 1:200 *
O1 (GalC) Marcador de oligodendrocitos – 1 o Galactocererebrósido (oligodendrocitos mielinizantes).
Ratón, policlonal IgM
1:50 *
VgluT2 Transportador vesicular de glutamato - 2 característico de terminales axónicas de CGRs.
Cobayo, policlonal 1:4000 Millipore, EE.UU.
Materiales y Métodos
60
Se utilizaron los siguientes anticuerpos secundarios:
Anticuerpo 2rio Especie Dilución Empresa Conjugado
Anti-cabra Burro 1:500 Life Technologies, EE.UU. Alexa Fluor 568
Anti-cobayo Cabra 1/500 Life Technologies, EE.UU. Alexa Fluor 568
plexiforme externa; CNE: capa nuclear externa; SE: segmento externo de los fotorreceptores.
A continuación, se analizó el número de CGRs a través de la inmunorreactividad para
Brn3a (un marcador específico de este tipo celular) en retinas de ojos sometidos a un
procedimiento simulado o a HOA. La HOA indujo una disminución en el número de células
Brn3a(+), como se muestran en la Figura 26. Luego de 2 semanas de la HOA, se observó un
perfil similar y no se observaron diferencias en este parámetro entre ojos intactos y ojos
sometidos a un procedimiento simulado (datos no mostrados).
Figura 26. Efecto de la HOA sobre el número de CGRs. Se muestran imágenes representativas de la
inmunomarcación para Brn3a (rojo) y las capas nucleares de la retina coloreadas con DAPI (azul). Al
cabo de 7 días, la HOA provocó una disminución evidente en el número de células Brn3a(+), respecto al
grupo sometido al tratamiento simulado. Barra de escala: 20 m.
Resultados
75
Estos resultados fueron confirmados mediante el ensayo de TUNEL. Como se muestra en
la Figura 27, en la CCG de ojos sometidos a HOA, se observó un aumento significativo en el
número de células TUNEL(+).
Figura 27. Efecto de la HOA sobre la apoptosis de las CGRs a los 7 días post-tratamiento. En el panel
izquierdo se muestran imágenes representativas. En el panel derecho se muestra la cuantificación del
número de células TUNEL(+) (verde). La HOA indujo un aumento significativo de este parámetro. Se
muestran las medias ± EE (5 retinas/grupo), ** p < 0,01 vs. grupo sham, test de Student.
A continuación, se determinó el estado funcional de los axones de las CGRs a través de la
evaluación del transporte anterógrado de la subunidad de la toxina colérica (CTB) desde la
retina hacia su principal blanco sináptico en la vía visual consciente en la rata, el colículo
superior (CS). A los 7 días, la HOA indujo una marcada reducción en la marca de CTB en el área
retinorreceptiva del CS contralateral, en comparación con el CS contralateral a ojos con
tratamiento simulado (Figura 28). No se observaron diferencias en el transporte de CTB al CS
con aferencias de ojos con tratamiento simulado, respecto de ojos intactos.
Resultados
76
Figura 28. Efecto de la HOA sobre el transporte anterógrado de CTB al CS a los 7 días post-tratamiento.
Se muestra la reconstrucción en sentido rostro-caudal del CS a partir de secciones coronales de cerebro. A
la izquierda se muestra el transporte de CTB de un animal que recibió tratamiento simulado (sham)
unilateral y a la derecha el transporte de CTB de un animal con tratamiento simulado en un ojo e HOA en
el ojo contralateral. La HOA indujo una marcada reducción del transporte anterógrado respecto del
tratamiento simulado. El tratamiento simulado no tuvo efecto per se sobre este parámetro. Barra de
escala: 3 mm.
En la siguiente serie de experimentos, se examinó el estado de la microglía y macroglía
en el CS a los 7 días y 4 semanas post-HOA. Como se muestra en la Figura 29, a los 7 días se
observó una marcada reactividad microglial (evaluada por la inmunorreactividad para Iba-1).
Estas alteraciones se mantuvieron sin cambios luego de 4 semanas post-HOA.
Resultados
77
Figura 29. Estudio de la inmunorreactividad para Iba-1 en el CS a los 7 días y 4 semanas post-
tratamiento. En ambos períodos se observó una respuesta microglial manifiesta en el CS contrateral al ojo
sometido a HOA.
Asimismo, se observó un aumento en la inmunorreactividad para GFAP en el CS a los 7
días, que fue más marcada luego de 4 semanas post-HOA (Figura 30).
Figura 30. Análisis de la inmunorreactividad para GFAP en el CS. A los 7 días post-HOA, se observó
una moderada respuesta astroglial en las capas superficiales y en la SGI del CS contrateral al ojo
sometido a HOA. Estas alteraciones fueron más marcadas luego de 4 semanas post-HOA.
En conjunto, estos resultados indican que la HOA provocó una alteración funcional
retiniana, junto con cambios estructurales marcados que incluyeron una reducción significativa
en el número de CGRs, así como alteraciones en el transporte anterógrado al CS y en la
Resultados
78
estructura glial del área retinoceptiva y adyacente intermedia del CS. Al menos en la ventana
temporal analizada (7 días post-HOA), este conjunto de alteraciones ocurrieron en forma
simultánea. Por lo tanto, a través de este modelo no fue posible disecar el curso temporal de las
alteraciones visuales a nivel de la retina, el NO y del CS. Sin embargo, se consideró de interés
examinar la vulnerabilidad de las CGRs melanopsina(+) (CGRsm) en particular y del sistema
visual no formador de imagen en general, en este modelo experimental. Los resultados obtenidos
se describen a continuación.
1.2. Sistema visual no formador de imagen
Con el fin de analizar el efecto del glaucoma agudo sobre el sistema visual no formador
de imagen, se seleccionó un período de 4 semanas post-HOA, de manera de maximizar los
cambios en el sistema visual y evaluar las posibles consecuencias sobre este sistema en
particular. Como se muestra en la Figura 31, a las 4 semanas post-HOA se confirmó una
alteración electrorretinográfica altamente significativa en cuanto a la amplitud de la onda a, la
onda b y los POs. En la misma Figura se muestran registros representativos de los grupos
experimentales.
Figura 31. Estudio funcional a las 4 semanas post-HOA o tratamiento simulado. Panel A: Se muestran
las medias ± EE de las amplitudes de las ondas a, b y de los POs del ERG escotópico (5 animales/grupo),
Resultados
79
La HOA indujo una caída significativa en todos los parámetros analizados a las 4 semanas post-
tratamiento. **p < 0,01 vs. grupo sham, test de Student. Panel B: Se muestran registros representativos
para cada grupo experimental.
En este mismo período, la estructura retiniana se encontró marcadamente alterada, con
cambios en las distintas capas retinianas y en el número de células en la CCG (Figura 32).
Figura 32. Estudio de la histología retiniana a las 4 semanas post-HOA o tratamiento simulado. En el
panel superior se muestran imágenes representativas de retinas de ambos grupos experimentales en
sección transversal (hematoxilina-eosina). Barras de escala: 100 y 50 m (detalles). En el panel inferior
se muestra el espesor total de la retina y el número de células en la CCG. La HOA indujo una marcada
desorganización de la estructura retiniana y una reducción significativa del espesor total y del número de
células en la CCG, con respecto del grupo sham. Se muestran las medias ± EE (5 retinas / grupo), ** p <
0,01 vs. grupo sham, test de Student.
Con el objeto de determinar el número total de CGRsm, el estudio inmunohistoquímico
para Brn3a y melanopsina se realizó en retinas en montaje plano, como se muestra en la Figura
33. Los resultados obtenidos indican que concomitantemente con una caída significativa en el
número de células Brn3a(+), el número de células melanopsina(+) no se modificó
significativamente.
Resultados
80
Figura 33. Estudio de la inmunorreactividad para Brn3a y melanopsina a las 4 semanas post-HOA o
tratamiento simulado. Panel A: Se muestran imágenes representativas de CGRsm en retinas en montaje
plano. Barra de escala: 1000 m. Panel B: a la izquierda se muestra un esquema de la retina y de los
cuadrantes analizados. A la derecha, se muestran imágenes representativas de la inmunorreactividad para
Brn3a (rojo) o melanopsina (verde) para ambos grupos experimentales. Barra de escala: 50m. Panel C:
Se muestra el área retiniana total y el número de células Brn3a(+) y melanopsina(+). Se observó una
disminución significativa del número de células Brn3a(+), en tanto que el número de células
melanopsina(+) permaneció inalterado. Se muestran las medias ± EE (5 retinas/grupo), ** p < 0,01 vs.
grupo sham, test de Student.
Estos resultados se confirmaron por un análisis de Western blot (Figura 34), que indicó
una caída significativa en los niveles retinianos de Brn3a en retinas de ojos que fueron sometidos
a HOA, en tanto que los niveles retinianos de melanopsina no se modificaron significativamente.
Resultados
81
Figura 34. Determinación de los niveles proteicos de Brn3a y melanopsina retinianos a las 4 semanas
post-HOA o tratamiento simulado. Panel A: Se muestran geles representativos. Panel B: Se muestran los
niveles de Brn3a y melanopsina relativos a los niveles de -actina. La HOA indujo una reducción
significativa en los niveles de Brn3a, en tanto que los niveles de melanopsina no se modificaron. Se
muestran las medias ± EE (5 retinas/grupo), ** p < 0,01 vs. grupo con tratamiento simulado (sham), test
de Student.
Con el objeto de evaluar las proyecciones de la retina a áreas centrales del sistema visual
no formador de imagen, se examinó el transporte anterógrado de CTB a los NSQ y el NPO.
Como se muestra en la Figura 35, el transporte de CTB al NPO y los NSQ no se modificó en
forma evidente, en tanto que la marca de CTB disminuyó significativamente en el área
retinorreceptiva del CS y del NGL contralateral a ojos sometidos a HOA.
Figura 35. Efecto de la HOA sobre el transporte anterógrado a las 4 semanas post-tratamiento. En el
panel izquierdo se muestran imágenes representativas del CS, los NGL, NPO y NSQ de un animal que fue
inyectado con CTB-Alexa Fluor 488 (verde) en el ojo con tratamiento simulado y CTB-Alexa Fluor 568
Resultados
82
(roja) en el ojo con HOA. En el panel derecho se muestra un esquema de los núcleos analizados. La HOA
indujo una disminución en la marca de CTB en el CS y los NGL, pero no en los NPO y los NSQ.
Finalmente, se analizó el reflejo pupilar consensual en animales en los que un ojo fue
sometido a HOA o tratamiento simulado y el ojo contralateral permaneció intacto. Como se
muestra en la Figura 36, el reflejo pupilar consensual inducido por 120 lux disminuyó
significativamente en los animales sometidos a HOA, en tanto que cuando el estímulo luminoso
fue de 1200 lux, no se observaron diferencias significativas entre los grupos experimentales.
Figura 36. Efecto de la HOA sobre el reflejo pupilar consensual a las 4 semanas post-tratamiento. Panel
A: Se representa el porcentaje de contracción pupilar en respuesta a un estímulo luminoso de 120 ó 1200
lux. Se observó una reducción significativa en la contracción pupilar frente a un estímulo de 120 (pero no
de 1200) lux. Se muestran las medias ± EE (5 ojos/ grupo), ** p < 0,01 vs. grupo sham, test de Student.
Panel B: Se muestran fotos representativas de la contracción pupilar máxima para ambos grupos
experimentales.
2. Modelo experimental de glaucoma crónico.
2.1. Sistema visual formador de imagen
Como se mencionó en la Introducción, en trabajos previos de nuestro laboratorio
demostramos que la administración semanal de ácido hialurónico (AH) en la cámara anterior del
ojo reproduce aspectos centrales del glaucoma humano. Sin embargo, la utilización de este
modelo debió ser abandonada porque la empresa fabricante (Sigma®) interrumpió en forma
definitiva la producción de este tipo particular de AH (catálogo # H1751). No hemos logrado
Resultados
83
reproducir los resultados originales obtenidos con el tipo de AH con el que se realizaron
experimentos previos, que dieron origen a diversas publicaciones del laboratorio (Belforte y col.,
2007, 2010; Benozzi y col., 2002; Moreno y col., 2004, 2005a, 2005b, 2008;), incluso con
diferentes AH de ésta u otras empresas. Por lo tanto, resultó necesario desarrollar un modelo de
glaucoma experimental alternativo. Para ello, considerando los antecedentes mencionados en la
Introducción sobre la relevancia de los GAGs como barrera al pasaje de humor acuoso, así como
el hecho de que el condroitín sulfato (CSU) junto con el AH son los principales GAGs de la red
trabecular, se examinó el efecto de la administración intracameral de CSU sobre la PIO, la
función y la histología retiniana. Los resultados obtenidos se describen a continuación.
En una primera serie de experimentos, se inyectaron animales con diferentes
concentraciones de CSU en un ojo y vehículo en el ojo contralateral y se determinó la PIO en
ambos ojos de cada animal 24 h después de la inyección. En la Figura 37 se muestran los valores
de PIO de ojos inyectados con vehículo o CSU (10, 20 ó 40%). Se observó un aumento leve pero
no significativo de la PIO en los ojos inyectados con 10 ó 20% de CSU, en tanto que la
inyección con CSU al 40% indujo un aumento significativo de este parámetro.
Figura 37. Efecto de una única inyección intracameral de CSU sobre la PIO. Los ojos se inyectaron con
vehículo o CSU (10, 20 ó 40%), 24 h antes de la determinación de la PIO. Se observó un aumento
significativo de este parámetro en los ojos inyectados con 40 (pero no con 10 ó 20) % de CSU. Se
Resultados
84
representan las medias ± EE (n = 10 ojos / grupo), ** p < 0,01 vs. ojos inyectados con vehículo, test de
Dunnett.
Con el fin de analizar el curso temporal del efecto del CSU sobre la PIO, se realizó una
única inyección de CSU al 40% en un ojo y vehículo en el ojo contralateral y se determinó la
PIO en ambos ojos antes de la inyección (día 0) y diariamente a partir de 24 h post-inyección,
como se muestra en la Figura 38. La inyección de CSU indujo un aumento de casi al doble de la
PIO que duró 7 días, mientras que en el 8vo día post-inyección, la PIO en los ojos inyectados con
CSU alcanzó valores similares al control.
Figura 38. Determinaciones de la PIO en ojos tratados con una única inyección intracameral de vehículo
(círculos blancos) o CSU (círculos negros). El CSU aumentó significativamente la PIO hasta el día 7
post-inyección. Se representan las medias ± EE (n = 10 ojos/grupo), ** p < 0,01 vs. ojos inyectados con
vehículo en los mismos intervalos post-inyección, test de Student.
Con el objeto de inducir una hipertensión ocular persistente, los animales se inyectaron
una vez por semana con CSU (en un ojo) y vehículo (en el ojo contralateral). Se determinó
semanalmente la PIO en ambos ojos, antes de una nueva inyección, como se muestra en la
Figura 39. La PIO de los ojos tratados con CSU fue significativamente mayor que la de los ojos
inyectados con vehículo a lo largo de todo el estudio (15 semanas). No se observaron diferencias
significativas en la PIO de los ojos inyectados con vehículo entre los intervalos analizados, ni
entre ojos intactos o inyectados con vehículo (datos no mostrados).
Resultados
85
Figura 39. Efecto de la administración crónica de CSU sobre la PIO. Se realizaron inyecciones
semanales de vehículo (círculos blancos) o CSU (círculos negros), después de la determinación de la
PIO. El CSU indujo una hipertensión ocular persistente a lo largo de todo el estudio. Se representan las
medias ± EE (n = 15 ojos/grupo), ** p < 0,01 vs. ojos inyectados con vehículo, test de Student.
A continuación, se evaluó el efecto de la hipertensión ocular inducida por CSU sobre la
función de la retina y la vía visual. Para ello, se registraron ERGs escotópicos a las 3, 6, 10 y 15
semanas de tratamiento con vehículo o CSU. En la Figura 40 se muestran las amplitudes de la
onda a, la onda b y los POs en ratas no inyectadas o inyectadas semanalmente con vehículo en un
ojo y CSU en el ojo contralateral. No se observaron diferencias significativas en estos
parámetros entre el grupo de animales con ojos intactos y el grupo tratado con vehículo. A las 3
semanas, la administración de CSU no indujo cambios significativos en ninguno de los
parámetros analizados (datos no mostrados), en tanto que a las 6 semanas de hipertensión ocular
crónica se observó una leve (pero significativa) reducción de la amplitud de la onda a, la onda b
y los POs. La disfunción retiniana fue más evidente luego de 10 semanas de hipertensión ocular
y continuó su progresión a las 15 semanas de tratamiento. Esta disminución fue
significativamente mayor a las 10 y 15 semanas de tratamiento con CSU, respecto del grupo
tratado durante 6 semanas.
Resultados
86
Figura 40. Efecto de la hipertensión ocular crónica sobre el ERG escotópico. Se representa la amplitud
de la onda a, b y POs del ERG de animales con ojos intactos o inyectados semanalmente con CSU en un
ojo y vehículo en el ojo contralateral durante 6, 10 ó 15 semanas. El tratamiento con CSU indujo una
disminución significativa de los tres parámetros analizados en todos los intervalos. A las 10 y 15 semanas
de hipertensión ocular, el efecto del CSU sobre la amplitud de la onda a y los POs fue significativamente
mayor que a las 6. A las 15 semanas, la caída en la amplitud de la onda b fue significativamente mayor
que a las 6 y 10 semanas de tratamiento con CSU. Se representan las medias ± EE (n = 15 ojos/grupo), *
p < 0,05 y ** p < 0,01 vs. ojos inyectados con vehículo, a: p < 0,05 y b: p < 0,01 vs. ojos con 6 semanas
de tratamiento con CSU y # p < 0,05 vs. ojos con 10 semanas de tratamiento con CSU, test de Tukey.
En la Figura 41 se muestran registros electrorretinográficos representativos de ojos
inyectados con CSU o vehículo durante 6, 10 ó 15 semanas. El tratamiento con CSU no afectó
significativamente las latencias de las ondas a y b y de los POs en ninguno de los intervalos
analizados (datos no mostrados).
Resultados
87
Figura 41. Efecto de la hipertensión ocular crónica sobre el ERG escotópico. Se muestran registros
representativos de ojos de animales inyectados con vehículo (línea gris llena) o CSU (línea punteada)
durante 6, 10 ó 15 semanas.
Con el fin de evaluar la actividad de toda la vía visual (desde los fotorreceptores a la
corteza visual) se registraron potenciales visuales evocados (VEPs), a las 3, 6 y 15 semanas de
hipertensión ocular. Se observó una disminución significativa en las amplitudes de los
componentes P1-N2 y N2-P2 en los ojos inyectados con CSU durante 6 ó 15 semanas en
comparación con los ojos inyectados con vehículo (Figura 42). No se observaron cambios en los
VEPs a las 3 semanas de hipertensión ocular (datos no mostrados). Los VEPs en ojos intactos no
difirieron significativamente de los registrados en ojos inyectados con vehículo durante 3 (datos
no mostrados), 6 ó 15 semanas (Figura 42). No se observaron diferencias significativas en las
latencias de los componentes de los VEPs entre los grupos experimentales (datos no mostrados).
Resultados
88
Figura 42. Efecto de la hipertensión ocular sobre la amplitud de los VEPs. Panel A: A las 6 y 15 semanas
de tratamiento con CSU, las amplitudes promedio de los componentes P1-N2 y N2-P2 de los VEPs
disminuyeron significativamente. Se representan las medias ± EE (n = 8 animales/grupo), * p < 0,05 y **
p < 0,01 vs. ojos inyectados con vehículo, test de Student. Panel B: Se muestran registros representativos
de todos los grupos experimentales: ojos intactos (control, línea negra), ojos inyectados con vehículo
(línea gris llena) o con CSU (línea punteada). En el registro control se señalan los componentes P1, P2 y
N2.
En la Figura 43 se muestran imágenes representativas de retinas en sección transversal de
un ojo tratado con vehículo o con CSU por 10 semanas. Aunque el espesor total de la retina y de
la CPI, la CNI y la CNE no difirió entre los grupos experimentales, se observó una disminución
significativa en el número de células en la CCG en los ojos inyectados con CSU durante 10
semanas (Figura 43, Tabla 2).
A B
Resultados
89
Figura 43. Efecto de la hipertensión ocular crónica sobre la morfología retiniana. Se muestran secciones
transversales de retinas de un ojo inyectado con solución salina (A) y un ojo inyectado con CSU durante 10
semanas (B). Nótese la disminución del número de células en la CCG en el ojo inyectado con CSU, en tanto
que la estructura general de la retina fue similar en ambos grupos. Hematoxilina-eosina.
A las 3 ó 6 semanas de tratamiento con vehículo o CSU no se observaron alteraciones en
la morfología de la retina y a las 15 semanas los resultados fueron esencialmente similares a los
observados a las 10 semanas de tratamiento (datos no mostrados).
10 semanas de tratamiento vehículo CSU
Nº de células en la CCG/100 m 9,2 ± 0,5 5,9 ± 0,7** Espesor total (µm) 132,0 ± 7.0 129,0 ± 6,8 Espesor de la CPI (µm) 32,7 ± 4,6 31,6 ± 4,2 Espesor de la CNI (µm) 25 ± 2,8 23,3 ± 2,3 Espesor de la CNE (µm) 37,1 ± 3.0 35,5 ± 4,7
Tabla 2. Número de células en la CCG y espesor total de la retina y de sus capas. Se representan las
medias ± EE, ** p 0,01 (n = 5 retinas/grupo) vs. grupo inyectado con vehículo, test de Student.
Con el objeto de analizar el efecto de la hipertensión ocular crónica específicamente
sobre las CGRs, se evaluó la inmunorreactividad para Brn3a en cortes transversales de retina.
Como se muestra en la Figura 44, el número de células Brn3a(+) no se modificó
significativamente luego de 6 semanas de hipertensión ocular, en tanto que a las 10 semanas de
glaucoma experimental, se observó una reducción significativa en este parámetro, que fue
similar a la observada luego de 15 semanas de hipertensión ocular.
Resultados
90
Figura 44. Efecto de la hipertensión ocular crónica sobre el número de células Brn3a(+). En el panel
izquierdo se muestran imágenes representativas de retinas de animales con 6, 10 ó 15 semanas de
tratamiento con vehículo o CSU y en el panel derecho, la cuantificación del número de células Brn3a(+).
Este parámetro disminuyó significativamente a las 10 y 15 (pero no a las 6) semanas de tratamiento con
CSU. Se representan las medias ± EE (n = 5 retinas/grupo), ** p < 0,01 vs. ojos inyectados con vehículo,
a: p < 0,01 y b: p < 0,05 vs. CSU durante 6 semanas, test de Tukey.
Por una limitación en la disponibilidad de animales, en los experimentos cuyos resultados
se describirán a continuación se evaluaron dos intervalos de hipertensión ocular crónica: una
etapa temprana, es decir a las 6 semanas de tratamiento (en forma previa a la caída en el número
de CGRs) y una etapa avanzada de glaucoma experimental, es decir a las 15 semanas de
tratamiento con CSU (a posteriori de la caída en este parámetro).
En la siguiente serie de experimentos, se examinó el transporte axonal anterógrado desde
la retina al CS y al NGL, a través de la inyección intravítrea de CTB. En una primera instancia,
se examinaron las retinas de ojos inyectados con CTB para corroborar la incorporación de la
toxina en las CGRs de los grupos experimentales. En ambos intervalos, se observó
cualitativamente que las CGRs mantuvieron la capacidad de captación de la toxina (Figura 45).
Resultados
91
Figura 45. Incorporación de CTB en las CGRs de animales inyectados con vehículo o CSU por 6 ó 15
semanas. En secciones transversales de retina se observó marca para CTB, particularmente en los cuerpos
y los axones de las CGRs (detalle). Barras de escala: 50 m.
Sin embargo, se observó una disminución en el transporte de CTB ya a las 6 semanas de
hipertensión ocular en la porción del NO proximal a la retina (Figura 46).
Figura 46. Transporte de CTB en animales inyectados con vehículo o CSU por 6 semanas. En el panel
izquierdo se muestran cortes longitudinales representativos de 4 NOs/grupo y en el derecho se representa
el % de marca de CTB en función de la distancia a la retina. En el NO de ojos inyectados con vehículo, se
observó acumulación de la toxina en la porción más cercana a la retina (0 - 400 m, región amielínica),
que decayó progresivamente al alcanzar la zona de transición (400 - 600 m) y la región mielinizada del
NO. En el NO de ojos inyectados con CSU por 6 semanas se observó un perfil de decaimiento similar de
CTB en función de la distancia a la retina, aunque la marca fue notablemente menor en toda su extensión.
En cuanto a los núcleos cerebrales, el transporte de CTB disminuyó en el CS contralateral
a ojos hipertensos ya a partir de las 6 semanas de tratamiento con CSU. A las 15 semanas de
tratamiento, se observó ausencia casi completa de marca de CTB en el CS, como se muestra en
Resultados
92
intacto
la Figura 47. Resultados similares se obtuvieron en cuanto al transporte de CTB al NGL, como
se muestra en la Figura 48.
Figura 47. Transporte anterógrado de CTB en animales con ojos intactos o inyectados semanalmente con
vehículo en un ojo y CSU en el ojo contralateral durante 6 ó 15 semanas. Microfotografías representativas
de 5 animales/grupo que muestran el patrón de transporte de CTB hacia las capas superficiales (SZ y
SGS) del CS en cortes coronales. En el CS contralateral al ojo inyectado con vehículo (Veh), no se
observaron signos de alteración con respecto a los controles no inyectados, en tanto que en el CS
contralateral a ojos inyectados con CSU, se observó una marcada reducción de las áreas marcadas con
CTB, tanto a las 6 como a las 15 semanas de tratamiento. Nótese la representación del disco óptico en el
CS, indicada por las flechas blancas.
Resultados
93
Figura 48. Transporte anterógrado de CTB al NGL. Microfotografías representativas de 5
animales/grupo del patrón de transporte de CTB al intergeniculado (IG), NGL dorsal (NGLd) y ventral
(NGLv) en cortes coronales. En el NGL contralateral a ojos inyectados con CSU, se observó una marcada
reducción en la marca de CTB, tanto a las 6 como a las 15 semanas de tratamiento.
A continuación se evaluó el estado de los terminales presinápticos de las CGRs, a través
de la inmunodetección de VgluT-2 en el CS. Este parámetro no se modificó en forma evidente ni
a las 6 ni a las 15 semanas de hipertensión ocular, como se muestran en la Figura 49.
Figura 49. Análisis de la inmunorreactividad para VgluT-2 en el CS de animales inyectados con vehículo
en un ojo y CSU en el ojo contralateral. En el panel izquierdo se muestran imágenes representativas de la
inmunorreactividad para VgluT-2. En el panel derecho se muestra la cuantificación del % de área
inmunopositiva para VgluT-2, que no difirió significativamente entre los grupos experimentales en
ninguno de los intervalos estudiados. Se representan las medias ± EE (n = 5 CS/grupo).
Resultados
94
2.1.1. Efecto del glaucoma crónico experimental a nivel del nervio óptico
En la siguiente serie de experimentos se analizó la morfología y ultraestructura del NO de
ojos inyectados con vehículo o CSU. En cortes semifinos de NO proximal, se cuantificó el área
transversal y el número total de axones y se determinó la distribución del número de axones en
función del área axonal. El glaucoma experimental indujo una disminución moderada pero
significativa del número total de axones a las 6 semanas, que progresó a las 15 semanas de
hipertensión ocular, mientras que el área del NO disminuyó significativamente sólo a las 15
semanas (Figura 50).
Figura 50. Efecto del glaucoma experimental sobre el área transversal del NO y el número de axones a
las 6 ó 15 semanas de tratamiento. En el panel superior se muestran imágenes representativas de cortes
semifinos transversales de la porción proximal del NO (azul de toluidina). En el panel inferior se muestra
la cuantificación del área transversal del NO y del número de axones. El CSU indujo una reducción
significativa del número de axones tanto a las 6 como a las 15 semanas de tratamiento, en tanto que el
área del NO disminuyó significativamente sólo a las 15 semanas. Se representan las medias ± EE (n = 5
nervios/grupo), * p < 0,01 y ** p < 0,01 vs. NOs de ojos inyectados con vehículo, a: p < 0,01 vs. NOs de
ojos inyectados con CSU por 6 semanas, test de Tukey.
Resultados
95
Con el fin de determinar la susceptibilidad diferencial a los efectos deletéreos del
glaucoma en términos del tamaño de los axones, se analizó la distribución de los axones en
función del área axonal (Figura 51). A las 6 semanas, sólo los axones de mayor área (> 1,5 m2)
fueron afectados significativamente por la hipertensión ocular crónica, en tanto que a las 15
semanas se redujo significativamente el número de axones de todos los rangos de área evaluados.
Figura 51. Distribución del número de axones en función del área axonal. A las 6 semanas de
hipertensión ocular, se redujo significativamente el número de axones grandes (entre 1,5 - 2,5 m2 y > 2,5
m2), en tanto que a las 15 semanas disminuyó significativamente el número de axones de todos los
tamaños analizados. Se representan las medias ± EE (n = 5 nervios/grupo), * p < 0,01 y ** p < 0,01 vs.
NOs de ojos inyectados con vehículo, test de Student.
En la Figura 52 se muestran microfotografías representativas de NOs de ojos control o
con 6 ó 15 semanas de hipertensión ocular. Los axones del NO de los ojos inyectados con
vehículo por 6 ó 15 semanas presentaron una forma uniforme (redondeada/oval) y un tamaño
variable, con niveles moderados de glía perivascular y del neuropilo. En el NO de los ojos
tratados con CSU durante 6 semanas, se observaron algunos axones degenerados (áreas densas
de mielina), axones con distensiones y distorsiones que se apartaron de la morfología
característica de axones normales, así como signos de gliosis, evidenciados por un aumento en la
distancia entre los haces de axones del nervio. A las 15 semanas de hipertensión ocular, los
Resultados
96
efectos fueron más marcados y se evidenciaron por una pérdida global de la uniformidad e
integridad axonal (menor número total de axones, mayor número de axones degenerados) y
claros signos de gliosis reactiva (Figura 52).
Figura 52. Efecto de la hipertensión ocular crónica sobre la estructura del NO. Se muestran
microfotografías representativas de cortes transversales del NO proximal, luego de 6 ó 15 semanas de
tratamiento con CSU o vehículo (azul de toluidina). Nótese la distribución homogénea de los axones en
todo el campo, con contenido glial moderado, que se corresponde con un nervio sano. Luego de 6
semanas de hipertensión ocular, se observó una menor densidad de axones, algunas fibras en
degeneración y procesos gliales más numerosos y de mayor tamaño. A las 15 semanas, el efecto del
glaucoma fue más marcado, con evidentes signos de gliosis reactiva. En estos nervios, la mayoría de los
axones presentaron degeneración axonal, siendo escasos los axones con apariencia normal. Imágenes
representativas de 5 NOs/grupo. Barra de escala: 25 m.
A nivel ultraestructural, se observó que los NOs de ojos inyectados con vehículo por 6 ó
15 semanas presentaron una estructura conservada, en la que haces de axones se encontraron
delimitados por finos procesos astrocitarios de citoplasma electrón-lúcido. A mayor aumento, las
Resultados
97
vainas de mielina presentaron una apariencia normal, compacta y muy electrón-densas, rodeando
el axoplasma sin alteraciones evidentes, con fibras del citoesqueleto (microtúbulos y
neurofilamentos) y organelas (principalmente mitocondrias). A las 6 semanas de hipertensión
ocular, la arquitectura global del NO fue similar a la de los NOs de ojos inyectados con vehículo,
con fibras en apariencia normales a bajo aumento. A mayor magnificación, entre fibras normales
se encontraron algunos axones con mielina fragmentada, axones con áreas de mielina
distorsionada y vacuolizada, en las que las lamelas de las vainas se observaron descompactadas.
En baja proporción, se observaron axones completamente degenerados sin axoplasma, en los que
la mielina formó una masa electrón-densa y redondeada. A las 15 semanas de hipertensión
ocular, las alteraciones ultraestructurales fueron más marcadas: el NO presentó una estructura
desorganizada, gruesos procesos astrocitarios intercalados entre los haces de fibras nerviosas con
distinto grado de degeneración y fue frecuente distinguir células microgliales en las
proximidades de axones degenerados. Las células microgliales se reconocieron por poseer
núcleos con forma arriñonada o con una leve escotadura, heterocromatina perinuclear y
citoplasma moderadamente electrón-denso, con vacuolas, lisosomas y áreas de debris, y se
distinguieron de los oligodendrocitos, con núcleos de forma circular u ovoide de gran tamaño
con escaso citoplasma perinuclear levemente electrón-denso, y de los astrocitos, con núcleos
ovoides eucromáticos (Figura 53 y 54). En este estadío, prevalecieron las fibras con mielina
descompactada y axones con degeneración densa de dos tipos: acúmulos de mielina condensada
(como se observó a las 6 semanas) y otros en los que el axoplasma aparentemente fue
reemplazado por un material granular indefinido y electrón-denso. En menor proporción, se
identificaron axones con mielina fragmentada con exposición del axoplasma al medio
extracelular y axones con degeneración laxa (watery degeneration), en el que el contenido
axoplásmico (citoesqueleto y organelas) fue reemplazado por un material amorfo y en apariencia
laxo y claro.
Resultados
98
Figura 53. Análisis ultraestructural del NO proximal. Se muestras imágenes representativas a distintos
aumentos de los NOs de ojos inyectados con vehículo o CSU durante 6 ó 15 semanas. A las 6 semanas de
hipertensión ocular, la estructura general del NO fue similar a la de los NOs correspondientes a ojos
inyectados con vehículo. Sin embargo, intercalados entre axones con apariencia normal, se observaron
axones con áreas de mielina descompactada (flechas negras), axones con ruptura de las vainas (flechas
blancas) y algunos axones completamente degenerados (estrellas blancas). A las 15 semanas, se
observaron procesos astrocitarios (A) más gruesos, así como células microgliales (Mi) y axones con
distinto grado de degeneración. Además, se observaron axones cuyo axoplasma fue reemplazado por una
masa homogénea granular y electrón-densa (asteriscos blancos) y minoritariamente, algunas fibras con
contenido axoplásmico alterado, ausencia del citoesqueleto y organelas (estrellas negras). Ol:
oligodendrocito. Imágenes representativas de 4 NOs/grupo. Barras de escala: 6 m, 1,5 m y 400 nm
para la primera, segunda y tercera fila, respectivamente.
Resultados
99
A B C
Figura 54. Detalles al MET de células microgliales en NOs de animales con 15 semanas de hipertensión
ocular. Paneles A y B: Se muestran dos células microgliales en diferentes estados de activación, con
axones degenerados en su interior (estrellas blancas y negras), localizados en las proximidades de axones
con degeneración densa (asteriscos blancos) y laxa (Ad). Nótese la diferencia de contrastes entre el
citoplasma microglial (electrón-denso) y el neuropilo (electrón-lúcido), que facilitó la identificación de
los límites celulares. Panel C: Se muestra un detalle del área señalada en (B) en la que se observan dos
axones degenerados (estrellas negras) en el interior de citoplasma microglial, delimitado por su membrana
plasmática (mp). Nótese en B-C la presencia de múltiples vacuolas (v) y otras organelas electrón-densas
(posiblemente lisosomas), indicativas de un estado microglial fagocítico. f: filamentos intermedios de
astrocitos; Nu: núcleos. Barras de escala: 1 m (A-B) y 0,4 m (C).
Los resultados de la distribución de frecuencias de las líneas intraperiódicas (Li) de los
NOs de animales con 6 ó 15 semanas de tratamiento con vehículo o CSU, se muestran en la
Figura 55. Debido a la presencia de axones degenerados en los animales con hipertensión ocular,
sólo fue posible analizar en estos grupos los axones normales remanentes y los axones con
degeneración leve, que intercalaron focos de mielina descompactada y mielina compacta, en los
que las Li fueron claramente identificadas. El glaucoma experimental a las 15 (pero no a las 6)
semanas indujo un corrimiento de la distribución de frecuencias hacia la izquierda, es decir,
hacia axones con vainas de mielina más delgadas.
Resultados
100
Figura 55. Efecto del glaucoma experimental sobre la distribución de frecuencias del número de líneas
intraperiódicas de los axones del NO proximal. En el panel superior se muestran fotografías
representativas utilizadas para la cuantificación manual del número de líneas intraperiódicas (Li) de las
vainas de mielina (VM) de axones normales a levemente degenerados (con algunos focos de mielina
descompactada en los que aún pueden contarse las Li). En el panel inferior se muestran histogramas
representativos de la distribución de frecuencias del número de Li de los axones de los NOs en animales
con 6 ó 15 semanas de CSU en un ojo y vehículo en el ojo contralateral. La hipertensión ocular por 15
(pero no 6) semanas indujo el corrimiento de la distribución de frecuencias hacia la izquierda. Al:
ser posible proveer nuevos enfoques terapéuticos para la protección de estas células, así como de
las CGRs en su conjunto, particularmente en el glaucoma crónico.
Como ya se mencionó, el “escenario” fue notablemente diferente en el glaucoma crónico
experimental con respecto al glaucoma agudo, en el que se observó una afectación significativa
de las CGRsm y sus axones. Como se muestra en este trabajo de Tesis, el glaucoma crónico
podría provocar alteraciones no sólo visuales, sino también en funciones visuales no formadoras
de imagen. Trastornos del sistema circadiano pueden provocar falta de concentración, menor
rendimiento, disminución de habilidades cognitivas, coordinación psicomotora pobre y dolores
de cabeza, entre muchos otros. Existen diversas estrategias terapéuticas para restablecer el
equilibrio circadiano. Por lo tanto, a través de la identificación de los trastornos circadianos en el
glaucoma crónico, estos resultados podrían contribuir a mejorar la calidad de vida de los
pacientes con esta enfermedad ocular.
4. Consecuencias conceptuales de los resultados obtenidos y perspectivas futuras
El objetivo central de este trabajo fue examinar las consecuencias del glaucoma sobre el
sistema visual en general, más allá de los reconocidos efectos a nivel retiniano (específicamente
la pérdida de CGRs) y las comparativamente menos exploradas consecuencias a nivel del NO.
Para ello, en una primera instancia se utilizó un modelo experimental que remeda la situación
clínica del glaucoma agudo humano, que como ya se mencionó, no resultó una herramienta
adecuada para la descripción de la secuencia temporal de eventos deletéreos en la retina, el NO y
el CS por su rápida evolución. En cambio, en el modelo de glaucoma crónico utilizado en este
trabajo, que tiene ventajas comparativas con otros modelos de glaucoma crónico de ángulo
abierto, se obtuvieron indicios de significación respecto al efecto de la hipertensión ocular sobre
estructuras visuales post-retinianas. En este sentido, en el curso de esta Tesis se han demostrado
alteraciones axo-gliales significativas en la retina, el NO y el CS, algunas de las cuales incluso
precedieron a la muerte de las CGRs, que es considerado el signo patognomónico de la
Discusión
176
enfermedad. Es muy probable que una lesión del SNC no consista en cambios independientes en
las diferentes clases de células que lo componen, sino más bien, es plausible que las
interacciones entre las neuronas, los axones y las células gliales también se alteren en detrimento
de las demandas fisiológicas y las funciones del sistema nervioso. En este sentido, aunque aún
no estamos en condiciones de establecer si el daño glaucomatoso ocurre inicialmente en los
somas de las CGRs, sus axones o en los componentes gliales de la retina, el NO o el CS, nuestros
resultados podrían sugerir que la comunicación multidireccional entre todos estos “actores del
sistema visual” puede ser negativamente afectada por la enfermedad. Esta concepción más
holística del glaucoma podría tener consecuencias de transferencia clínica, de manera tal que las
terapias de nueva generación deberían tener en cuenta las alteraciones en las distintas estaciones
de relevo del procesado visual al momento de elegir un tratamiento exitoso. A partir de estos
resultados (a pesar de estar en una etapa experimental), resulta evidente que un tratamiento será
efectivo si y sólo si permite evitar o al menos reducir la progresión del daño glaucomatoso a
nivel de la vía visual en su conjunto. También desde esta nueva perspectiva, es posible predecir
que una terapéutica anti-glaucomatosa eficiente debería involucrar una estrategia compleja, dada
la multiplicidad de los eventos deletéreos y de los componentes celulares afectados. Como ya se
mencionó, al presente, la principal estrategia terapéutica para el glaucoma está dirigida a
disminuir farmacológica o quirúrgicamente la PIO. Los resultados obtenidos en esta Tesis
podrían proveer una interpretación racional a la evidencia de que el éxito del tratamiento (en
términos de protección de la función visual) sea por ahora muy limitado, como lo sustenta el
hecho de que el glaucoma sigue siendo una de las principales causas de ceguera irreversible, a
pesar de los constantes esfuerzos invertidos en mejorar las estrategias para reducir la
hipertensión ocular.
Desde una perspectiva terapéutica y considerando que la activación microglial fue un
aspecto consistente en todas las áreas visuales evaluadas, se analizó el efecto de la minociclina.
Discusión
177
El efecto de la minociclina sobre la activación microglial (un evento central en las enfermedades
neurodegenerativas en general y eventualmente también en el glaucoma), así como su efecto
beneficioso en distintos modelos de enfermedad neurológica y su buena tolerancia para su uso en
humanos, acredita la potencialidad terapéutica de la minociclina para su consideración en el
tratamiento del glaucoma. En este sentido, si bien aún quedan diversos aspectos por analizar, los
resultados obtenidos avalan el uso de minociclina, (eventualmente en conjunto con hipotensores
oculares y compuestos neuroprotectores) para el tratamiento de esta enfermedad, pero además,
estos resultados aportan evidencias sobre la participación microglial en el daño glaucomatoso,
análogamente a lo descripto en otros procesos neurodegenerativos.
Otro de los aspectos analizados en esta Tesis fue el estudio del sistema visual no
formador de imagen en ambas formas de glaucoma experimental. También en este sentido,
quedan aún muchos interrogantes por contestar, como las causas de la resistencia de las CGRsm
al glaucoma agudo y de su susceptibilidad al glaucoma crónico, así como los mecanismos
involucrados en la protección y el daño de estas células, respectivamente. Sin embargo, estos
resultados avalan la importancia de incluir al sistema visual no formador de imagen en el marco
del estudio de enfermedades oftalmológicas en general y del glaucoma en particular. El papel
central de la retina en el sistema generador de ritmos circadianos y las consecuencias
considerables de las alteraciones circadianas sobre la salud humana, acreditan la importancia de
evaluar este sistema y desarrollar estrategias terapéuticas para el tratamiento frente a un mal
funcionamiento o pérdida de las CGRsm. En suma, si bien en el intento de incorporar nuevos
conceptos sobre la neuropatía glaucomatosa, esta Tesis muy probablemente haya generado más
preguntas que respuestas (como corresponde a cualquier trabajo científico), los hallazgos de este
trabajo generan genuinas expectativas de que las nuevas respuestas que surjan de los nuevos
interrogantes contribuyan en forma significativa a mejorar la calidad de vida de los pacientes con
glaucoma.
Bibliografía
178
BIBLIOGRAFÍA
Aarum J, Sandberg K, Haeberlein SL, Persson MA. Migration and differentiation of neural precursor cells can be directed by microglia. Proc Natl Acad Sci USA. 2003; 100(26):15983-8.
Abrahamson EE, Moore RY. Suprachiasmatic nucleus in the mouse: retinal innervation, intrinsic organization and efferent projections. Brain Res. 2001; 916(1-2):172-91.
Acott TS, Kelley MJ. Extracellular matrix in the trabecular meshwork. Exp Eye Res. 2008; 86(4):543-61.
Aihara M, Lindsey JD, Weinreb RN. Aqueous humor dynamics in mice. Invest Ophthalmol Vis Sci. 2003; 44(12):5168-73.
Aloisi F, Penna G, Cerase J, Menendez Iglesias B, Adorini L. IL-12 production by central nervous system microglia is inhibited by astrocytes. J Immunol. 1997; 159:1604-1612.
Anderson DR, Hendrickson A. Effect of intraocular pressure on rapid axoplasmic transport in monkey optic nerve. Invest Ophthalmol. 1974; 13(10):771-83.
Anderson MG, Smith RS, Hawes NL, Zabaleta A, Chang B, Wiggs JL, John SW. Mutations in genes encoding melanosomal proteins cause pigmentary glaucoma in DBA/2J mice. Nat Genet. 2002; 30:81-5.
Anthony DC, Pitossi FJ. Special issue commentary: the changing face of inflammation in the brain. Mol Cell Neurosci. 2013; 53:1-5.
Back SA, Riddle A, McClure MM. Maturation-dependent vulnerability of perinatal white matter in premature birth. Stroke. 2007; 38(2 Suppl):724-30.
Baker GE. Prechiasmatic reordering of fibre diameter classes in the retinofugal pathway of the ferret. Eur J Neurosci. 1990; 2:24-33.
Bancroft JD, Stevens A. Theory and practice of histological techniques. 3º edición: Edinburgh: Churchill-Livingstone; 1990.
Baptiste DC, Powell KJ, Jollimore CA, Hamilton C, LeVatte TL, Archibald ML, Chauhan BC, Robertson GS, Kelly ME. Effects of minocycline and tetracycline on retinal ganglion cell survival after axotomy. Neuroscience 2005; 134(2):575-82.
Barany EH, Scotchbrook S. Influence of testicular hyaluronidase on the resistance to flow through the angle of the anterior chamber. Acta Physiol Scand. 1954; 30:240-8.
Baumann N, Pham-Dinh D. Biology of oligodendrocyte and myelin in the mammalian central nervous system. Physiol Rev. 2001; 81(2):871-927.
Bayer AU, Danias J, Brodie S, et al. Electroretinographic abnormalities in a rat glaucoma model with chronic elevated intraocular pressure. Exp Eye Res. 2001a; 72:667-77.
Bayer AU, Neuhardt T, May AC, et al. Retinal morphology and ERG response in the DBA/2NNia mouse model of angle-closure glaucoma. Invest Ophthalmol Vis Sci. 2001b; 42:1258-65.
Beaulé C, Robinson B, Lamont EW, Amir S. Melanopsin in the circadian timing system. J Mol Neurosci. 2003; 21(1):73-89.
Belenky MA, Smeraski CA, Provencio I, Sollars PJ, Pickard GE.Melanopsin retinal ganglion cells receive bipolar and amacrine cell synapses. J Comp Neurol. 2003; 460(3):380-93.
Belforte N, Moreno MC, Cymeryng C, Bordone M, Keller Sarmiento MI, Rosenstein RE. Effect of ocular hypertension on retinal nitridergic pathway activity. Invest Ophthalmol Vis Sci. 2007; 48(5):2127-33.
Belforte NA, Moreno MC, de Zavalía N, Sande PH, Chianelli MS, Keller Sarmiento MI, Rosenstein RE. Melatonin: a novel neuroprotectant for the treatment of glaucoma. J Pineal Res. 2010; 48(4):353-64.
Bell-Pedersen D, Cassone VM, Earnest DJ, Golden SS, Hardin PE, Thomas TL, Zoran MJ. Circadian rhythms from multiple oscillators: lessons from diverse organisms. Nat. Rev. Genet. 2005; 6(7):544-56.
Benozzi J, Nahum LP, Campanelli JL, Rosenstein RE. Effect of hyaluronic acid on intraocular pressure in rats. Invest Ophthalmol Vis Sci. 2002; 43(7):2196-200.
Berson DM, Dunn FA, Takao M. Phototransduction by retinal ganglion cells that set the circadian clock. Science 2002; 295(5557):1070-3.
Betmouni, S, Perry VH, Gordon JL. Evidence for an early inflammatory response in the central nervous system of mice with scrapie. Neuroscience 1996; 74, 1–5.
Biber K, Neumann H, Inoue K, Boddeke HW. Neuronal ‘On’ and ‘Off’ signals control microglia. Trends Neurosci. 2007; 30:596-602.
Bill A. Uveoscleral drainage of aqueous humor: physiology and pharmacology. Prog Clin Biol Res. 1989; 312:417-27.
Binder DK, Steinhäuser C. Functional changes in astroglial cells in epilepsy. Glia 2006; 54(5):358-68.
Bizzozero OA, Bixler HA, Davis JD, Espinosa A, Messier AM. Chemical deacylation reduces the adhesive properties of proteolipid protein and leads to decompaction of the myelin sheath. J Neurochem. 2001;76(4):1129-41.
Bongarzone ER, Pasquini JM, Soto EF. Oxidative damage to proteins and lipids of CNS myelin produced by in vitro generated reactive oxygen species. J Neurosci Res 1995; 41(2):213-21.
Bosco A, Inman DM, Steele MR, Wu G, Soto I, Marsh-Armstrong N, Hubbard WC, Calkins DJ, Horner PJ, Vetter ML. Reduced retina microglial activation and improved optic nerve integrity with minocycline treatment in the DBA/2J mouse model of glaucoma. Invest Ophthalmol Vis Sci. 2008; 49(4):1437-46.
Bosco A, Steele MR, Vetter ML. Early microglia activation in a mouse model of chronic glaucoma. J Comp Neurol. 2011; 519: 599-620.
Boucard CC, Hernowo AT, Maguire RP, Jansonius NM, Roerdink JB, Hooymans JM, Cornelissen FW. Changes in cortical grey matter density associated with long-standing retinal visual field defects. Brain. 2009;132(Pt 7):1898-906.
Bouhenni RA, Dunmire J, Sewell A, Edward DP. Animal models of glaucoma. J Biomed Biotechnol. 2012; 2012:692609.
Brainard GC, Hanifin JP, Greeson JM, Byrne B, Glickman G, Gerner E, Rollag MD. Action spectrum for melatonin regulation in humans: evidence for a novel circadian photoreceptor. J Neurosci. 2001; 21: 6405-12.
Bringmann A, Pannicke T, Grosche J, Francke M, Wiedemann P, Skatchkov SN, Osborne NN, Reichenbach A. Müller cells in the healthy and diseased retina. Prog Retin Eye Res. 2006; 25(4):397-424.
Bron AJ, Tripathi RC, Tripathi BJ. Wolff’s anatomy of the eye and orbit. Chapman & Hall Medical, London, 1997, p. 736.
Broom K, Anthony D, Lowe J, Griffin J, Scott H, Blamire A, Styles P, Perry V, Sibson N, MRI and MRS alterations in the preclinical phase of murine prion disease: association with neuropathological and behavioural changes. Neurobiol. Dis. 2007;26: 707-717.
Bu J, Akhtar N, Nishiyama A. Transient expression of the NG2 proteoglycan by a subpopulation of activated macrophages in an excitotoxic hippocampal lesion. Glia 2001; 34(4):296-310.
Buckingham BP, Inman DM, Lambert W, Oglesby E, Calkins DJ, Steele MR, Vetter ML, Marsh-Armstrong N, Horner PJ. Progressive ganglion cell degeneration precedes neuronal loss in a mouse model of glaucoma. J Neurosci. 2008; 28:2735-44.
Bui BV, Edmunds B, Cioffi GA, Fortune B. The gradient of retinal functional changes during acute intraocular pressure elevation. Invest Ophthalmol Vis Sci. 2005; 46(1):202-13.
Buller C, Johnson DH, Tschumper RC. Human trabecular meshwork phagocytosis. Observations in an organ culture system. Invest Ophthalmol Vis Sci. 1990; 31(10):2156-63.
Burgoyne CF, Downs JC, Bellezza AJ, Suh JK, Hart RT. The optic nerve head as a biomechanical structure: a new paradigm for understanding the role of IOP-related stress and strain in the pathophysiology of glaucomatous optic nerve head damage. Prog Retin Eye Res. 2005; 24(1):39-73.
Busch SA, Horn KP, Cuascut FX, Hawthorne AL, Bai L, Miller RH, Silver J. Adult NG2+ cells are permissive to neurite outgrowth and stabilize sensory axons during macrophage-induced axonal dieback after spinal cord injury. J. Neurosci. 2010; 30(1):255-65.
Butt AM, Ibrahim M, Ruge FM, Berry M. Biochemical subtypes of oligodendrocyte in the anterior medullary velum of the rat as revealed by the monoclonal antibody Rip. Glia. 1995; 14(3):185-97.
Calkins DJ. Critical pathogenic events underlying progression of neurodegeneration in glaucoma. Prog Retin Eye Res. 2012; 31(6):702-19.
Carbonell AL, Boya J, Calvo JL, Marin JF. Ultrastructural study of the neuroglial and macrophagic reaction in Wallerian degeneration of the adult rat optic nerve. Histol Histopathol. 1991; 6(4):443-51.
Carden WB, Datskovskaia A, Guido W, Godwin DW, Bickford ME. Development of the cholinergic, nitrergic, and GABAergic innervation of the cat dorsal lateral geniculate nucleus. J Comp Neurol. 2000; 418(1):65-80.
Cernea P. The resistance to drainage of the aqueous humor. Oftalmología 1993; 37(4):289-98.
Chan KC, So KF, Wu EX. Proton magnetic resonance spectroscopy revealed choline reduction in the visual cortex in an experimental model of chronic glaucoma. Exp Eye Res. 2009; 88(1):65-70.
Chaturvedi N, Hedley-Whyte ET, Dreyer EB. Lateral geniculate nucleus in glaucoma. Am J Ophthalmol. 1993; 116(2):182-8.
Chauhan BC, Pan J, Archibald ML, LeVatte TL, Kelly ME, Tremblay F. Effect of intraocular pressure on optic disc topography, electroretinography, and axonal loss in a chronic pressure-induced rat model of optic nerve damage. Invest Ophthalmol Vis Sci. 2002; 43:2969-76.
Chen M, Ona VO, Li M, Ferrante RJ, Fink KB, Zhu S, Bian J, Guo L, Farrell LA, Hersch SM, Hobbs W, Vonsattel JP, Cha JH, Friedlander RM. Minocycline inhibits caspase-1 and caspase-3 expression and delays mortality in a transgenic mouse model of Huntington disease. Nat Med. 2000; 6(7):797-801.
Chen X, Ma X, Jiang Y, Pi R, Liu Y, Ma L. The prospects of minocycline in multiple sclerosis. J Neuroimmunol. 2011;235(1-2):1-8.
Chidlow G, Ebneter A, Wood JP, Casson RJ. The optic nerve head is the site of axonal transport disruption, axonal cytoskeleton damage and putative axonal regeneration failure in a rat model of glaucoma. Acta Neuropathol. 2011; 121(6):737-51.
Choong YF, Devarajan N, Pickering A, Pickering S, Austin MW. Initial management of ocular hypertension and primary open-angle glaucoma: an evaluation of the royal college of ophthalmologists' guidelines. Eye 2003; 17(6):685-9.
Colwell CS. Rhythmic coupling among cells in the suprachiasmatic nucleus. J Neurobiol. 2000; 43(4):379-88.
Crawford ML, Harwerth RS, Smith EL 3rd, Mills S, Ewing B. Experimental glaucoma in primates: changes in cytochrome oxidase blobs in V1 cortex. Invest Ophthalmol Vis Sci. 2001; 42(2):358-64.
Crish SD, Calkins DJ. Neurodegeneration in glaucoma: progression and calcium-dependent intracellular mechanisms. Neuroscience 2011; 176:1-11.
Crish SD, Dapper JD, MacNamee SE, Balaram P, Sidorova TN, Lambert WS, Calkins DJ. Failure of axonal transport induces a spatially coincident increase in astrocyte BDNF prior to synapse loss in a central target. Neuroscience 2013; 229:55-70.
Crish SD, Sappington RM, Inman DM, Horner PJ, Calkins DJ. Distal axonopathy with structural persistence in glaucomatous neurodegeneration. Proc Natl Acad Sci USA. 2010;107(11):5196-201.
Dai C, Khaw PT, Yin ZQ, Li D, Raisman G, Li Y. Structural basis of glaucoma: the fortified astrocytes of the optic nerve head are the target of raised intraocular pressure. Glia. 2012; 60(1):13-28.
Daubs JG, Crick RP. Effect of refractive error on the risk of ocular hypertension and open angle glaucoma. Trans Ophthalmol Soc UK. 1981; 101(1):121-6.
Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, Jung S, Littman DR, Dustin ML, Gan WB. ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci. 2005; 8:752-758.
De Keyser J, Mostert JP, Koch MW. Dysfunctional astrocytes as key players in the pathogenesis of central nervous system disorders. J Neurol Sci. 2008; 267(1-2):3-16.
Defaux A, Zurich MG, Braissant O, Honegger P, Monnet-Tschudi F. Effects of the PPAR-beta agonist GW501516 in an in vitro model of brain inflammation and antibody-induced demyelination. J. Neuroinflammation 2009; 6:15.
Dengler-Crish CM, Smith MA, Inman DM, Wilson GN, Young JW, Crish SD. Anterograde transport blockade precedes deficits in retrograde transport in the visual projection of the DBA/2J mouse model of glaucoma. Front Neurosci. 2014; 8:290.
DeParis S, Caprara C, Grimm C. Intrinsically photosensitive retinal ganglion cells are resistant to N-methyl-D-aspartic acid excitotoxicity. Mol Vis. 2012; 18:2814-27.
Dorfman D, Fernandez DC, Chianelli M, Miranda M, Aranda ML, Rosenstein RE. Post-ischemic environmental enrichment protects the retina from ischemic damage in adult rats.Exp Neurol. 2013;240:146-56.
Dräger UC, Hubel DH. Responses to visual stimulation and relationship between visual, auditory, and somatosensory inputs in mouse superior colliculus. J Neurophysiol. 1975; 38(3):690-713.
Drouyer E, Dkhissi-Benyahya O, Chiquet C, WoldeMussie E, Ruiz G, Wheeler LA, Denis P, Cooper HM. Glaucoma alters the circadian timing system. PLoS One. 2008; 3(12):e3931.
Du Y, Ma Z, Lin S, Dodel RC, Gao F, Bales KR, Triarhou LC, Chernet E, Perry KW, Nelson DL, Luecke S, Phebus LA, Bymaster FP, Paul SM. Minocycline prevents nigrostriatal dopaminergic neurodegeneration in the MPTP model of Parkinson's disease. Proc Natl Acad Sci U S A. 2001;98(25):14669-74.
Duan Y, Sahley CL, Muller KJ. ATP and NO dually control migration of microglia to nerve lesions. Dev Neurobiol. 2009; 69(1):60-72.
Duguay D, Cermakian N. The crosstalk between physiology and circadian clock proteins. Chronobiol. Int. 2009; 26(8):1479-513.
Edmunds B, Thompson JR, Salmon JF, Wormald RP. The National Survey of Trabeculectomy. I. Sample and methods. Eye 1999; 13 (Pt 4):524-30.
Eng LF, Ghirnikar RS, Lee YL. Glial fibrillary acidic protein: GFAP-thirty-one years (1969-2000). Neurochem. Res. 2000; 25:1439-51.
Esquiva G, Lax P, Cuenca N. Impairment of intrinsically photosensitive retinal ganglion cells associated with late stages of retinal degeneration. Invest Ophthalmol Vis Sci. 2013; 54:4605-18.
Fan W, Li X, Wang W, Mo JS, Kaplan H, Cooper NG. Early Involvement of Immune/Inflammatory Response Genes in Retinal Degeneration in DBA/2J Mice. Ophthalmol. Eye Dis. 2010; 1:23-41.
Faulkner JR, Herrmann JE, Woo MJ, Tansey KE, Doan NB, Sofroniew MV. Reactive astrocytes protect tissue and preserve function after spinal cord injury. J. Neurosci. 2004; 24(9):2143-55.
Fei PF, Yue BY, Tso MO. Effects of chronic intracameral injections of chondroitin sulfate on cat eyes. Graefes Arch Clin Exp Ophthalmol. 1984; 222:1-8.
Felts PA, Woolston AM, Fernando HB, Asquith S, Gregson NA, Mizzi OJ, Smith KJ. Inflammation and primary demyelination induced by the intraspinal injection of lipopolysaccharide. Brain 2005; 128(Pt 7):1649-66.
Fernandez DC, Bordone MP, Chianelli MS, Rosenstein RE. Retinal neuroprotection against ischemia-reperfusion damage induced by postconditioning. Invest Ophthalmol Vis Sci. 2009; 50(8):3922-30.
Fernandez DC, Sande PH, de Zavalía N, Belforte N, Dorfman D, Casiraghi LP, Golombek D, Rosenstein RE. Effect of experimental diabetic retinopathy on the non-image-forming visual system. Chronobiol Int. 2013; 30(4):583-97.
Fortune B, Choe TE, Reynaud J, Hardin C, Cull GA, Burgoyne CF, Wang L. Deformation of the rodent optic nerve head and peripapillary structures during acute intraocular pressure elevation. Invest Ophthalmol Vis Sci. 2011; 52(9):6651-61.
Fournier AE, Beer J, Arregui CO, Essagian C, Aguayo AJ, McKerracher L. Brain-derived neurotrophic factor modulates GAP-43 but not T alpha1 expression in injured retinal ganglion cells of adult rats. J Neurosci Res. 1997; 15; 47(6):561-72.
Freddo TF. A contemporary concept of the blood-aqueous barrier. Prog Retin Eye Res. 2013; 32:181-95.
Freedman MS, Lucas RJ, Soni B, von Schantz M, Munoz M, David-Gray Z, Foster R. Regulation of mammalian circadian behavior by non-rod, non-cone, ocular photoreceptors. Science 1999; 284: 502-504.
Fröjdman K, Pelliniemi LJ, Lendahl U, Virtanen I, Eriksson JE. The intermediate filament protein nestin occurs transiently in differentiating testis of rat and mouse. Differentiation 1997; 61(4):243-9.
Fuchshofer R, Birke M, Welge-Lussen U, Kook D, Lütjen-Drecoll E. Transforming growth factor-beta 2 modulated extracellular matrix component expression in cultured human optic nerve head astrocytes. Invest Ophthalmol Vis Sci. 2005; 46(2):568-78.
Funk RH. The vessel architecture of the pars plana in the cynomolgus monkey, rat and rabbit eye. A scanning electron microscopic study of plastic corrosion casts. Ophthalmic Res. 1993; 25(6):337-48.
Garden GA, Möller T. Microglia biology in health and disease. J Neuroimmune Pharmacol. 2006; 1(2):127-37.
Garrido-Mesa N, Zarzuelo A, Gálvez J. Minocycline: far beyond an antibiotic. Br J Pharmacol. 2013;169(2):337-52.
Geneser F. Histología, 3ra Ed. Panamericana, 2006.
Gerometta RM, Malgor LA, Vilalta E, Leiva J, Candia OA. Cl- concentrations of bovine, porcine and ovine aqueous humor are higher than in plasma. Exp Eye Res.2005; 80(3):307-12.
Gibson EM, Purger D, Mount CW, Goldstein AK, Lin GL, Wood LS, Inema I, Miller SE, Bieri G, Zuchero JB, Barres BA, Woo PJ, Vogel H, Monje M. Neuronal activity promotes oligodendrogenesis and adaptive myelination in the mammalian brain. Science 2014; 344(6183):1252304.
Glovinsky Y, Quigley HA, Dunkelberger GR. Retinal ganglion cell loss is size dependent in experimental glaucoma. Invest Ophthalmol Vis Sci. 1991; 32:484-91.
Goldman D. Müller glial cell reprogramming and retina regeneration. Nat. Rev. Neurosci. 2014; 15(7):431-42.
Gooley JJ, Lu J, Chou TC, Scammell TE, Saper CB. Melanopsin in cells of origin of the retinohypothalamic tract. Nat Neurosci. 2001;4(12):1165.
Gooley JJ, Lu J, Fischer D, Saper CB. A broad role for melanopsin in nonvisual photoreception. J Neurosci. 2003; 23:7093-106.
Gordon MO, Beiser JA, Brandt JD, Heuer DK, Higginbotham EJ, Johnson CA, Keltner JL, Miller JP, Parrish RK 2nd, Wilson MR, Kass MA. The Ocular Hypertension Treatment Study: baseline factors that predict the onset of primary open-angle glaucoma. Arch Ophthalmol. 2002; 120(6):714-20; discussion 829-30.
Graeber MB. Changing face of microglia. Science 2010; 330(6005):783-8.
Greenwood K, Butt AM. Evidence that perinatal and adult NG2-glia are not conventional oligodendrocyte progenitors and do not depend on axons for their survival. Mol. Cell Neurosci. 2003; 23(4):544-58..
Grippo TM, Hood DC, Kanadani FN, Ezon I, Greenstein VC, Liebmann JM, Ritch R. A comparison between multifocal and conventional VEP latency changes secondary to glaucomatous damage. Invest Ophthalmol Vis Sci. 2006; 47:5331-5336.
Grosche J, Grimm D, Clemens N, Reichenbach A.Retinal light damage vs. normal aging of rats: altered morphology, intermediate filament expression, and nuclear organization of Müller (glial) cells. J Hirnforsch. 1997;38(4):459-70.
Grozdanic SD, Betts DM, Sakaguchi DS, Kwon YH, Kardon RH, Sonea IM. Temporary elevation of the intraocular pressure by cauterization of vortex and episcleral veins in rats causes functional deficits in the retina and optic nerve. Exp Eye Res. 2003; 77(1):27-33.
Grozdanic SD, Matic M, Sakaguchi DS, Kardon RH. Evaluation of retinal status using chromatic pupil light reflex activity in healthy and diseased canine eyes. Invest Ophthalmol Vis Sci. 2007; 48:5178-83.
Guasti L, Richardson D, Jhaveri M, Eldeeb K, Barrett D, Elphick MR, Alexander SP, Kendall D, Michael GJ, Chapman V. Minocycline treatment inhibits microglial activation and alters spinal levels of endocannabinoids in a rat model of neuropathic pain. Mol. Pain. 2009; 5:35.
Guillery C. Quantitative studies of transneuronal atrophy in the dorsal lateral geniculate nucleus of cats and kittens. J Comp Neurol. 1973; 149(4):423-38.
Guillery RW, Walsh C. Changing glial organization relates to changing fiber order in the developing optic nerve of ferrets. J Comp Neurol. 1987; 265:203-217.
Güldner FH. Numbers of neurons and astroglial cells in the suprachiasmatic nucleus of male and female rats. Exp Brain Res. 1983; 50(2-3):373-6.
Guo L, Moss SE, Alexander RA, Ali RR, Fitzke FW, Cordeiro MF. Retinal ganglion cell apoptosis in glaucoma is related to intraocular pressure and IOP-induced effects on extracellular matrix. Invest Ophthalmol Vis Sci. 2005; 46(1):175-82.
Gupta N, Yücel YH. What changes can we expect in the brain of glaucoma patients? Surv Ophthalmol. 2007; 52 Suppl 2:S122-6.
Gupta N, Ang LC, Noël de Tilly L, Bidaisee L, Yücel YH. Human glaucoma and neural degeneration in intracranial optic nerve, lateral geniculate nucleus, and visual cortex. Br J Ophthalmol. 2006; 90(6):674-8.
Hailer NP, Grampp A, Nitsch R: Proliferation of microglia and astrocytes in the dentate gyrus following entorhinal cortex lesion: a quantitative bromodeoxyuridine-labelling study. Eur J Neurosci. 1999; 11:3359-3364.
Hanisch UK, Kettenmann H. Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat Neurosci. 2007, 10:1387-1394.
Hanisch UK. Microglia as a source and target of cytokines. Glia 2002; 40(2):140-55.
Hankins MW, Lucas RJ. The primary visual pathway in humans is regulated according to long-term light exposure through the action of a nonclassical photopigment. Curr Biol. 2002; 12: 191-198.
Hannibal J, Hindersson P, Ostergaard J, Georg B, Heegaard S, Larsen PJ, Fahrenkrug J. Melanopsin is expressed in PACAP-containing retinal ganglion cells of the human retinohypothalamic tract. Invest Ophthalmol Vis Sci. 2004; 45(11):4202-9.
Hannibal J. Roles of PACAP-containing retinal ganglion cells in circadian timing. Int Rev Cytol. 2006; 251:1-39.
Harooni M, Freilich JM, Abelson M, Refojo M. Efficacy of hyaluronidase in reducing increases in intraocular pressure related to the use of viscoelastic substances. Arch Ophthalmol. 2000; 118:445-6.
Hattar S, Kumar M, Park A, Tong P, Tung J, Yau KW, Berson DM. Central projections of melanopsin-expressing retinal ganglion cells in the mouse. J Comp Neurol. 2006; 497:326-49.
Hattar S, Lucas RJ, Mrosovsky N, Thompson S, Douglas RH, Hankins MW, Lem J, Biel M, Hofmann F, Foster RG, Yau KW. Melanopsin and rod-cone photoreceptive systems account for all major accessory visual functions in mice. Nature 2003; 424(6944):76-81.
Hayasaka S, Sears ML. Distribution of acid phosphatase, glucuronidase, and lysosomal hyaluronidase in the anterior chamber of the rabbit eye. Invest Ophthalmol Vis Sci.1978; 17:982-7.
Haynes SE, Hollopeter G, Yang G, Kurpius D, Dailey ME, Gan WB, Julius D: The P2Y12 receptor regulates microglial activation by extracellular nucleotides. Nat Neurosci. 2006, 9:1512-1519.
Heijl A, Leske MC, Bengtsson B, Hyman L, Bengtsson B, Hussein M; Early Manifest Glaucoma Trial Group. Reduction of intraocular pressure and glaucoma progression: results from the Early Manifest Glaucoma Trial. Arch Ophthalmol. 2002; 120(10):1268-79.
Henkind P, Hansen RI, Szalay J. In: Physiology of the Human Eye and Visual System. (RE Records, ed.), Harper, Hagerstown, Maryland. 1979.
Hernandes MS, Britto LR. Inflammatory responses in the rat superior colliculus after eye enucleation. Brain Res Bull. 2014; 101:1-6.
Hernandez MR. The optic nerve head in glaucoma: role of astrocytes in tissue remodeling. Prog Retin Eye Res. 2000; 19(3):297-321.
Herrmann JE, Imura T, Song B, Qi J, Ao Y, Nguyen TK, Korsak RA, Takeda K, Akira S, Sofroniew MV. STAT3 is a critical regulator of astrogliosis and scar formation after spinal cord injury. J. Neurosci. 2008; 28(28):7231-43.
Hill RA, Nishiyama A. NG2 cells (polydendrocytes): listeners to the neural network with diverse properties. Glia 2014; 62(8):1195-210.
Hill RA, Patel KD, Medved J, Reiss AM, Nishiyama A. NG2 cells in white matter but not gray matter proliferate in response to PDGF. J. Neurosci. 2013; 33(36):14558-66.
Hoffman RM. The potential of nestin-expressing hair follicle stem cells in regenerative medicine. Expert Opin. Biol. Ther. 2007; 7(3):289-91.
Holländer H, Makarov F, Stefani FH, Stone J. Evidence of constriction of optic nerve axons at the lamina cribrosa in the normotensive eye in humans and other mammals. Ophthalmic Res. 1995; 27(5):296-309.
Horky LL, Galimi F, Gage FH, Horner PJ. Fate of endogenous stem/progenitor cells following spinal cord injury. J. Comp. Neurol. 2006; 498(4):525-38.
Horn KP, Busch SA, Hawthorne AL, van Rooijen N, Silver J. Another barrier to regeneration in the CNS: activated macrophages induce extensive retraction of dystrophic axons through direct physical interactions. J. Neurosci. 2008; 28(38):9330-41.
Hotta CT, Gardner MJ, Hubbard KE, Baek SJ, Dalchau N, Suhita D, Dodd AN, Webb AA. Modulation of environmental responses of plants by circadian clocks. Plant Cell Environ. 2007; 30(3):333-49.
Howell GR, Libby RT, Jakobs TC, Smith RS, Phalan FC, Barter JW, Barbay JM, Marchant JK, Mahesh N, Porciatti V, Whitmore AV, Masland RH, John SW. Axons of retinal ganglion cells are insulted in the optic nerve early in DBA/2J glaucoma. J. Cell Biol. 2007; 179:1523-1537.
Hubbard WC, Johnson M, Gong H, Gabelt BT, Peterson JA, Sawhney R, Freddo T, Kaufman PL. Intraocular pressure and outflow facility are unchanged following acute and chronic intracameral chondroitinase ABC and hyaluronidase in monkeys. Exp Eye Res. 1997; 65:177-90
Hubel DH. Eye, brain and vision. Scientific American Library, WH Freeman (ed.), serie # 22, New York, 1988.
Hughes EG, Kang SH, Fukaya M, Bergles DE. Oligodendrocyte progenitors balance growth with self-repulsion to achieve homeostasis in the adult brain. Nat. Neurosci. 2013;16(6):668-76.
Bibliografía
188
Hur J, Lee P, Kim MJ, Kim Y, Cho YW. Ischemia-activated microglia induces neuronal injury via activation of gp91phox NADPH oxidase. Biochem Biophys Res Commun. 2010; 391(3):1526-30.
Imamura K, Hishikawa N, Sawada M, Nagatsu T, Yoshida M, Hashizume Y. Distribution of major histocompatibility complex class II-positive microglia and cytokine profile of Parkinson’s disease brains. Acta Neuropathol. 2003; 106:518-26.
Imamura K, Onoe H, Shimazawa M, Nozaki S, Wada Y, Kato K, Nakajima H, Mizuma H, Onoe K, Taniguchi T, Sasaoka M, Hara H, Tanaka S, Araie M, Watanabe Y. Molecular imaging reveals unique degenerative changes in experimental glaucoma. Neuroreport. 2009; 20(2):139-44.
Inman DM, Horner PJ. Reactive nonproliferative gliosis predominates in a chronic mouse model of glaucoma. Glia 2007; 55(9):942-53.
Isenmann S, Cellerino A, Gravel C, Bähr M. Excess target-derived brain-derived neurotrophic factor preserves the transient uncrossed retinal projection to the superior colliculus. Mol. Cell. Neurosci. 1999; 14(1):52-65.
Ito Y, Shimazawa M, Chen YN, Tsuruma K, Yamashima T, Araie M, Hara H. Morphological changes in the visual pathway induced by experimental glaucoma in Japanese monkeys. Exp. Eye Res. 2009; 89(2):246-55.
Ito Y, Shimazawa M, Inokuchi Y, Fukumitsu H, Furukawa S, Araie M, Hara H. Degenerative alterations in the visual pathway after NMDA-induced retinal damage in mice. Brain Res. 2008; 1212:89-101.
Jain V, Ravindran E, Dhingra NK. Differential expression of Brn3 transcription factors in intrinsically photosensitive retinal ganglion cells in mouse. J Comp Neurol. 2012; 520(4):742-55.
Jakobs TC, Libby RT, Ben Y, John SW, Masland RH. Retinal ganglion cell degeneration is topological but not cell type specific in DBA/2J mice. J. Cell Biol. 2005;171(2):313-25.
Jones LL, Yamaguchi Y, Stallcup WB, Tuszynski MH. NG2 is a major chondroitin sulfate proteoglycan produced after spinal cord injury and is expressed by macrophages and oligodendrocyte progenitors. J. Neurosci. 2002;22(7):2792-803.
Jurgens I, Matheu A, Castilla M. Ocular hypertension after cataract surgery: a comparison of three surgical techniques and two viscoelastics. Ophthalmic Surg Lasers 1997; 28:30-6.
Kaback MB, Podos SM, Harbin TS, Jr, Mandell A, Becker B. The effects of dipivalyl epinephrine on the eye. Am J Ophthalmol. 1976; 81(6):768–772.
Kalloniatis M, Tomisich G. Amino acid neurochemistry of the vertebrate retina. Prog Retin Eye Res. 1999; 18:811-66.
Kandel E. y col. Principles of Neural Science. Mc Graw Hill Companies, USA, 4th Ed, 2000, Part V (27): 523-548.
Bibliografía
189
Kanski JJ. Clinical Ophthalmology, 4d edition, Elsevier Health Sciences. 2005.
Karlstetter M, Scholz R, Rutar M, Wong WT, Provis JM, Langmann T. Retinal microglia: Just bystander or target for therapy? Prog Retin Eye Res. 2014 Dec 2.
Keller KE, Bradley JM, Vranka JA, Acott TS. Segmental versican expression in the trabecular meshwork and involvement in outflow facility. Invest Ophthalmol Vis Sci. 2011; 52(8):5049-57.
Kierdorf K, Prinz M. Factors regulating microglia activation. Front Cell Neurosci. 2013; 7:44.
Kigerl KA, Gensel JC, Ankeny DP, Alexander JK, Donnelly DJ, Popovich PG. Identification of two distinct macrophage subsets with divergent effects causing either neurotoxicity or regeneration in the injured mouse spinal cord. J. Neurosci. 2009; 29(43):13435-44.
Kim SU, de Vellis J. Microglia in health and disease. J Neurosci Res. 2005; 81(3):302-13.
Klein BE, Klein R, Jensen SC. Open-angle glaucoma and older-onset diabetes. The Beaver Dam Eye Study. Ophthalmology 1994; 101(7):1173-7.
Klein D, Moore RY, Reppert SM. (Eds.). Suprachiasmatic Nucleus: The Mind’s Clock. Oxford University Press, Oxford. 1991.
Klerman EB, Gershengorn HB, Duffy JF, Kronauer RE. Comparisons of the variability of three markers of the human circadian pacemaker. J Biol Rhythms. 2002; 17(2):181-93.
Knepper PA, Collins JA, Frederick R. Effects of dexamethasone, progesterone, and testosterone on IOP and GAGs in the rabbit eye. Invest Ophthalmol Vis Sci.1985; 26:1093-1100.
Knepper PA, Fadel JR, Miller AM, Goossens W, Choi J, Nolan MJ, Whitmer S. Reconstitution of trabecular meshwork GAGs: influence of hyaluronic acid and chondroitin sulfate on flow rates. J Glaucoma. 2005; 14:230-238.
Knepper PA, Farbman AI, Telser AG. Exogenous hyaluronidases and degradation of hyaluronic acid in the rabbit eye. Invest Ophthalmol Vis Sci. 1984; 25(3):286-93.
Knepper PA, Goossens W, Hvizd M, Palmberg PF. Glycosaminoglycans of the human trabecular meshwork in primary open-angle glaucoma. Invest Ophthalmol Vis Sci. 1996; 37:1360-7.
Kohn AN, Moss AP, Podos SM. Relative afferent pupillary defects in glaucoma without characteristic field loss. Arch Ophthalmol. 1979; 97(2):294–296.
Kohno H, Sakai T, Kitahara K. Induction of nestin, Ki-67, and cyclin D1 expression in Müller cells after laser injury in adult rat retina. Graefes Arch. Clin. Exp. Ophthalmol. 2006; 244(1):90-5.
Kolker AE, Hetherington J Jr. Becker-Shaffer's diagnosis and therapy of the glaucomas. 5th ed. St Louis, CV Mosby, 1981.
Kong YX, Crowston JG, Vingrys AJ, Trounce IA, Bui VB. Functional changes in the retina during and after acute intraocular pressure elevation in mice. Invest Ophthalmol Vis Sci. 2009; 50(12):5732-40.
Kraus RL, Pasieczny R, Lariosa-Willingham K, Turner MS, Jiang A, Trauger JW. Antioxidant properties of minocycline: neuroprotection in an oxidative stress assay and direct radical-scavenging activity. J Neurochem. 2005;94(3):819-27.
Kreutzberg GW. Microglia: a sensor for pathological events in the CNS. Trends Neurosci. 1996; 19:312-318.
Kriz J, Nguyen MD, Julien JP. Minocycline slows disease progression in a mouse model of amyotrophic lateral sclerosis. Neurobiol Dis. 2002; 10(3):268-78.
Krout KE, Kawano J, Mettenleiter TC, Loewy AD. CNS inputs to the suprachiasmatic nucleus of the rat. Neuroscience 2002; 110(1):73-92.
Krstić, RV. Human microscopic anatomy. Springer-Verlag. 1997.
Kuchibhotla KV, Lattarulo CR, Hyman BT, Bacskai BJ. Synchronous hyperactivity and intercellular calcium waves in astrocytes in Alzheimer mice. Science 2009; 323(5918):1211-5.
Kursula P. Structural properties of proteins specific to the myelin sheath. Amino Acids 2008; 34(2):175-85.
La Morgia C, Ross-Cisneros FN, Sadun AA, Hannibal J, Munarini A, Mantovani V, Barboni P, Cantalupo G, Tozer KR, Sancisi E, Salomao SR, Moraes MN, Moraes-Filho MN, Heegaard S, Milea D, Kjer P, Montagna P, Carelli V. Melanopsin retinal ganglion cells are resistant to neurodegeneration in mitochondrial optic neuropathies. Brain 2010; 133:2426-38.
Lafuente MP, Villegas-Pérez MP, Sellés-Navarro I, Mayor-Torroglosa S, Miralles de Imperial J, Vidal-Sanz M. Retinal ganglion cell death after acute retinal ischemia is an ongoing process whose severity and duration depends on the duration of the insult. Neuroscience 2002; 109(1):157-68.
Lam DY, Kaufman PL, Gabelt BT, Matsubara JA. Cortical consequences of elevated intraocular pressure in primate model of glaucoma. Invest Ophthalmol Vis Sci. 2000; 41(4)S831. Abstract 4413.
Lam DY, Kaufman PL, Gabelt BT, To EC, Matsubara JA. Neurochemical correlates of cortical plasticity after unilateral elevated intraocular pressure in a primate model of glaucoma. Invest Ophthalmol Vis Sci. 2003; 44(6):2573-81.
Lam D, Jim J, To E, Rasmussen C, Kaufman PL, Matsubara J. Astrocyte and microglial activation in the lateral geniculate nucleus and visual cortex of glaucomatous and optic nerve transected primates. Mol. Vis. 2009; 15:2217-29.
LaVail MM. Rod outer segment disk shedding in rat retina: relationship to cyclic lighting. Science. 1976; 194(4269):1071-4.
Lendahl U, Zimmerman LB, McKay RD. CNS stem cells express a new class of intermediate filament protein. Cell 1990; 60(4):585-95.
Levine JM, Enquist LW, Card JP. Reactions of oligodendrocyte precursor cells to alpha herpesvirus infection of the central nervous system. Glia 1998; 23(4):316-28.
Levkovitch-Verbin H, Kalev-Landoy M, Habot-Wilner Z, Melamed S. Minocycline delays death of retinal ganglion cells in experimental glaucoma and after optic nerve transection. Arch Ophthalmol. 2006; 124(4):520-6.
Levkovitch-Verbin H, Makarovsky D, Vander S. Comparison between axonal and retinal ganglion cell gene expression in various optic nerve injuries including glaucoma. Mol Vis. 2013; 19:2526-41.
Levkovitch-Verbin H, Martin KR, Quigley HA, Baumrind LA, Pease ME, Valenta D. Measurement of amino acid levels in the vitreous humor of rats after chronic intraocular pressure elevation or optic nerve transection. J Glaucoma 2002; 11:396-405.
Levkovitch-Verbin H, Spierer O, Vander S, Dardik R. Similarities and differences between primary and secondary degeneration of the optic nerve and the effect of minocycline. Graefes Arch Clin Exp Ophthalmol. 2011; 249(6):849-57.
Levkovitch-Verbin H, Waserzoog Y, Vander S, Makarovsky D, Piven I. Minocycline upregulates pro-survival genes and downregulates pro-apoptotic genes in experimental glaucoma. Graefes Arch. Clin. Exp. Ophthalmol. 2014; 252(5):761-72.
Li RS, Chen BY, Tay DK, Chan HH, Pu ML, So KF. Melanopsin-expressing retinal ganglion cells are more injury-resistant in a chronic ocular hypertension model. Invest Ophthalmol Vis Sci. 2006; 47:2951-8.
Li SY, Yau SY, Chen BY, Tay DK, Lee VW, Pu ML, Chan HH, So KF. Enhanced survival of melanopsin-expressing retinal ganglion cells after injury is associated with the PI3 K/Akt pathway. Cell Mol Neurobiol. 2008; 28:1095-107.
Libby RT, Anderson MG, Pang IH, Robinson ZH, Savinova OV, Cosma IM, Snow A, Wilson LA, Smith RS, Clark AF, John SW. Inherited glaucoma in DBA/2J mice: pertinent disease features for studying the neurodegeneration. Vis Neurosci. 2005; 22(5):637-48.
Libby RT, Li Y, Savinova OV, Barter J, Smith RS, Nickells RW, John SW. Susceptibility to neurodegeneration in a glaucoma is modified by Bax gene dosage. PLoS Genet. 2005; 1(1):17-26.
Liu KM, Shen CL. Ultrastructural sequence of myelin breakdown during Wallerian degeneration in the rat optic nerve. Cell Tissue Res. 1985; 242(2):245-56.
Liu M, Guo L, Salt TE, Cordeiro MF. Dendritic changes in rat visual pathway associated with experimental ocular hypertension. Curr Eye Res. 2014; 39(9):953-63.
Liu W, Tang Y, Feng J. Cross talk between activation of microglia and astrocytes in pathological conditions in the central nervous system. Life Sci. 2011; 89:141-6.
Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951; 193:265-75.
Lucas RJ, Douglas RH, Foster RG. Characterization of an ocular photopigment capable of driving pupillary constriction in mice. Nat Neurosci. 2001;4(6):621-6.
Lucas RJ, Hattar S, Takao M, Berson DM, Foster RG, Yau KW. Diminished pupillary light reflex at high irradiances in melanopsin-knockout mice. Science 2003; 299: 245-247.
Luna G, Lewis GP, Banna CD, Skalli O, Fisher SK. Expression profiles of nestin and synemin in reactive astrocytes and Müller cells following retinal injury: a comparison with glial fibrillar acidic protein and vimentin. Mol. Vis. 2010; 16:2511-23.
Lund RD, Land PW, Boles J. Normal and abnormal uncrossed retinotectal pathways in rats: an HRP study in adults. J. Comp. Neurol. 1980; 189(4):711-20.
Lund RD. Uncrossed visual pathways of hooded and albino rats. Science 1965; 149:1506-1507.
Luo XG, Chen SD. The changing phenotype of microglia from homeostasis to disease. Transl Neurodegener. 2012; 1(1):9.
Lytle JM, Chittajallu R, Wrathall JR, Gallo V. NG2 cell response in the CNP-EGFP mouse after contusive spinal cord injury. Glia 2009; 57(3):270-85.
Lytle JM, Vicini S, Wrathall JR. Phenotypic changes in NG2+ cells after spinal cord injury. J. Neurotrauma 2006; 23(12):1726-38.
Ma M, Ferguson TA, Schoch KM, Li J, Qian Y, Shofer FS, Saatman KE, Neumar RW. Calpains mediate axonal cytoskeleton disintegration during Wallerian degeneration. Neurobiol Dis. 2013; 56:34-46.
Macknight AD, McLaughlin CW, Peart D, Purves RD, Carre DA, Civan MM. Formation of the aqueous humor. Clin Exp Pharmacol Physiol. 2000; 27(1-2):100-6.
Maier K, Merkler D, Gerber J, Taheri N, Kuhnert AV, Williams SK, Neusch C, Bähr M, Diem R. Multiple neuroprotective mechanisms of minocycline in autoimmune CNS inflammation. Neurobiol Dis. 2007; 25(3):514-25.
Malone P, Miao H, Parker A, Juarez S, Hernandez MR. Pressure induces loss of gap junction communication and redistribution of connexin 43 in astrocytes. Glia 2007; 55:1085-98.
Masland RH. Neuronal diversity in the retina. Curr Opin Neurobiol. 2001b; 11(4):431-6.
Masland RH. The fundamental plan of the retina. Nat Neurosci. 2001a; 4(9):877-86.
Matthews MR. Further observations on transneuronal degeneration in the lateral geniculate nucleus of the macaque monkey. J Anat.1964; 98:255-63.
Matute C, Melone M, Vallejo-Illarramendi A, Conti F. Increased expression of the astrocytic glutamate transporter GLT-1 in the prefrontal cortex of schizophrenics. Glia 2005; 49(3):451-5.
May PJ. The mammalian superior colliculus: laminar structure and connections. Prog Brain Res. 2006; 151:321-78.
McGeer PL, Itagaki S, Tago H, McGeer EG. Reactive microglia in patients with senile dementia of the Alzheimer type are positive for the histocompatibility glycoprotein HLA-DR. Neurosci Lett. 1987; 79:195-200.
McLoon SC. Response of astrocytes in the visual system to Wallerian degeneration: an immunohistochemical analysis of laminin and glial fibrillary acidic protein (GFAP). Exp Neurol. 1986; 91:613-21.
McTigue DM, Wei P, Stokes BT. Proliferation of NG2-positive cells and altered oligodendrocyte numbers in the contused rat spinal cord. J. Neurosci. 2001; 21(10):3392-400.
Mehlem A, Hagberg CE, Muhl L, Eriksson U, Falkevall A. Imaging of neutral lipids by oil red O for analyzing the metabolic status in health and disease. Nat. Protoc. 2013;8(6):1149-54.
Melyan Z, Tarttelin EE, Bellingham J, Lucas RJ, Hankins MW. Addition of human melanopsin renders mammalian cells photoresponsive. Nature 2005; 433(7027):741-5.
Meyer DB. The avian eye and its adaptations. En: The Visual System in Vertebrates. Crescitelli F, ed. Springer- Verlag, Berlín, 1977; p. 549-611.
Minckler DS, Bunt AH, Johanson GW. Orthograde and retrograde axoplasmic transport during acute ocular hypertension in the monkey. Invest Ophthalmol Vis Sci. 1977; 16(5):426-41.
Minckler DS, Tso MO, Zimmerman LE. A light microscopic, autoradiographic study of axoplasmic transport in the optic nerve head during ocular hypotony, increased intraocular pressure, and papilledema. Am J Ophthalmol. 1976; 82(5):741-57.
Miron VE, Boyd A, Zhao JW, Yuen TJ, Ruckh JM, Shadrach JL, van Wijngaarden P, Wagers AJ, Williams A, Franklin RJ, ffrench-Constant C. M2 microglia and macrophages drive oligodendrocyte differentiation during CNS remyelination. Nat. Neurosci. 2013; 16(9):1211-8.
Moga MM, Moore RY. Organization of neural inputs to the suprachiasmatic nucleus in the rat. J Comp Neurol. 1997; 389(3):508-34.
Moreno MC, Campanelli J, Sande P, Sánez DA, Keller Sarmiento MI, Rosenstein RE. Retinal oxidative stress induced by high intraocular pressure. Free Radic Biol Med. 2004; 37(6):803-12.
Moreno MC, de Zavalía N, Sande P, Jaliffa CO, Fernandez DC, Keller Sarmiento MI, Rosenstein RE. Effect of ocular hypertension on retinal GABAergic activity. Neurochem Int. 2008; 52(4-5):675-82.
Moreno MC, Marcos HJ, Oscar Croxatto J, Sande PH, Campanelli J, Jaliffa CO, Benozzi J, Rosenstein RE. A new experimental model of glaucoma in rats through intracameral injections of hyaluronic acid. Exp Eye Res. 2005a; 81(1):71-80.
Moreno MC, Sande P, Marcos HA, de Zavalía N, Keller Sarmiento MI, Rosenstein RE. Effect of glaucoma on the retinal glutamate/glutamine cycle activity. FASEB J. 2005b; 19(9):1161-2.
Morigiwa K, Quan M, Murakami M, Yamashita M, Fukuda Y. P2 Purinoceptor expression and functional changes of hypoxia-activated cultured rat retinal microglia. Neurosci Lett. 2000; 282:153-6.
Morin LP, Blanchard JH, Provencio I. Retinal ganglion cell projections to the hamster suprachiasmatic nucleus, intergeniculate leaflet, and visual midbrain: bifurcation and melanopsin immunoreactivity. J Comp Neurol. 2003; 465(3):401-16.
Morrison JC, DeFrank MP, Van Buskirk EM. Comparative microvascular anatomy of mammalian ciliary processes. Invest Ophthalmol Vis Sci. 1987; 28(8):1325-40.
Morrison JC, Dorman-Pease ME, Dunkelberger GR, Quigley HA. Optic nerve head extracellular matrix in primary optic atrophy and experimental glaucoma. Arch Ophthalmol. 1990; 108(7):1020-4.
Morrison JC, Moore CG, Deppmeier LM, Gold BG, Meshul CK, Johnson EC. A rat model of chronic pressure-induced optic nerve damage. Exp Eye Res. 1997; 64(1):85-96.
Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008; 8(12):958-69.
Mrak RE, Griffinbc WS. The role of activated astrocytes and of the neurotrophic cytokine S100B in the pathogenesis of Alzheimer's disease. Neurobiol Aging 2001; 22(6):915-22.
Mrosovsky N, Hattar S. Impaired masking responses to light in melanopsin-knockout mice. Chronobiol Int. 2003; 20: 989-999.
Murta V, Pitossi FJ, Ferrari CC. CNS response to a second pro-inflammatory event depends on whether the primary demyelinating lesion is active or resolved. Brain Behav. Immun. 2012;26(7):1102-15.
Nadal-Nicolás FM, Jiménez-López M, Sobrado-Calvo P, Nieto-López L, Cánovas-Martínez I, Salinas-Navarro M, Vidal-Sanz M, Agudo M. Brn3a as a marker of retinal ganglion cells: qualitative and quantitative time course studies in naive and optic nerve-injured retinas. Invest Ophthalmol Vis Sci. 2009; 50(8):3860-8.
Nakazawa T, Nakazawa C, Matsubara A, Noda K, Hisatomi T, She H, Michaud N, Hafezi-Moghadam A, Miller JW, Benowitz LI. Tumor necrosis factor-alpha mediates oligodendrocyte death and delayed retinal ganglion cell loss in a mouse model of glaucoma. J Neurosci. 2006; 26(49):12633-41.
Napoli I, Neumann H. Protective effects of microglia in multiple sclerosis. Exp Neurol. 2010; 225:24-28.
Narciso MS, Hokoç JN, Martinez AM. Watery and dark axons in Wallerian degeneration of the opossum's optic nerve: different patterns of cytoskeletal breakdown? An Acad Bras Cienc. 2001; 73(2):231-43.
Nave KA. Myelination and support of axonal integrity by glia. Nature. 2010; 468(7321):244-52.
Newman GC, Hospod FE. Rhythm of suprachiasmatic nucleus 2-deoxyglucose uptake in vitro. Brain Res. 1986; 381(2):345-50.
Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 2005; 308(5726):1314-8.
Nishiyama A, Suzuki R, Zhu X. NG2 cells (polydendrocytes) in brain physiology and repair. Front. Neurosci. 2014;8:133.
Niven JE, Laughlin SB. Energy limitation as a selective pressure on the evolution of sensory systems. J Exp Biol. 2008; 211(Pt 11):1792-804.
O’Donnell SL, Frederick TJ, Krady JK, Vannucci SJ, Wood TL: IGF-I and microglia/macrophage proliferation in the ischemic mouse brain. Glia 2002, 39:85-97.
Okada S, Nakamura M, Katoh H, Miyao T, Shimazaki T, Ishii K, Yamane J, Yoshimura A, Iwamoto Y, Toyama Y, Okano H. Conditional ablation of Stat3 or Socs3 discloses a dual role for reactive astrocytes after spinal cord injury. Nat. Med. 2006; 12(7):829-34.
Osborne NN, Block F, Sontag KH. Reduction in ocular blood flow results in glial fibrillary acidic protein (GFAP) expression in rat Muller cells. Vis. Neurosci.1991; 7(6):637- 9.
Panda S, Nayak SK, Campo B, Walker JR, Hogenesch JB, Jegla T. Illumination of the melanopsin signaling pathway. Science 2005; 307(5709):600-4.
Panda S, Provencio I, Tu DC, Pires SS, Rollag MD, Castrucci AM, Pletcher MT, Sato TK, Wiltshire T, Andahazy M, Kay SA, Van Gelder RN, Hogenesch JB. Melanopsin is required for non-image-forming photic responses in blind mice. Science 2003; 301(5632):525-7.
Panda S, Sato TK, Castrucci AM, Rollag MD, DeGrip WJ, Hogenesch JB, Provencio I, Kay SA. Melanopsin (Opn4) requirement for normal light-induced circadian phase shifting. Science 2002; 298: 2213-6.
Pang IH, Clark AF. Rodent models for glaucoma retinopathy and optic neuropathy. J. Glaucoma. 2007; 16(5):483-505.
Papst N, Bopp M, Schnaudigel OE. Pattern electroretinogram and visually evoked cortical potentials in glaucoma. Graefes Arch Clin Exp Ophthalmol. 1984; 222:29-33.
Paxinos G, Watson C. The rat brain in stereotaxic coordinates. Amsterdam: Elsevier, 1997.
Payne SC, Bartlett CA, Harvey AR, Dunlop SA, Fitzgerald M. Myelin sheath decompaction, axon swelling, and functional loss during chronic secondary degeneration in rat optic nerve. Invest Ophthalmol Vis Sci. 2012; 53(10):6093-101.
Pearson HE, Stoffler DJ. Retinal ganglion cell degeneration following loss of postsynaptic target neurons in the dorsal lateral geniculate nucleus of the adult cat. Exp Neurol. 1992; 116(2):163-71.
Pease ME, McKinnon SJ, Quigley HA, Kerrigan-Baumrind LA, Zack DJ. Obstructed axonal transport of BDNF and its receptor TrkB in experimental glaucoma. Invest Ophthalmol Vis Sci. 2000; 41(3):764-74.
Peirson SN, Halford S, Foster RG. The evolution of irradiance detection: melanopsin and the non-visual opsins. Philos Trans R Soc Lond B Biol Sci. 2009; 364(1531):2849-65.
Pekny M, Wilhelmsson U, Bogestål YR, Pekna M. The role of astrocytes and complement system in neural plasticity. Int. Rev. Neurobiol. 2007; 82:95-111.
Perganta G, Barnard AR, Katti C, Vachtsevanos A, Douglas RH, MacLaren RE, Votruba M, Sekaran S. Non-image-forming light driven functions are preserved in a mouse model of autosomal dominant optic atrophy. PLoS One 2013; 8:e56350.
Perge JA, Koch K, Miller R, Sterling P, Balasubramanian V. How the optic nerve allocates space, energy capacity, and information. J Neurosci. 2009; 29(24):7917-28.
Perge JA, Niven JE, Mugnaini E, Balasubramanian V, Sterling P. Why do axons differ in caliber? J Neurosci. 2012; 32(2):626-38.
Perry VH, Gordon S. Macrophages and microglia in the nervous system. Trends Neurosci. 1988; 11(6):273-7.
Pinteaux-Jones F, Sevastou IG, Fry VA, Heales S, Baker D, Pocock JM. Myelin-induced microglial neurotoxicity can be controlled by microglial metabotropic glutamate receptors. J. Neurochem. 2008; 106(1):442-54.
Polazzi E, Contestabile A. Reciprocal interactions between microglia and neurons: from survival to neuropathology. Rev Neurosci. 2002; 13:221-42.
Prins M, Eriksson C1, Wierinckx A, Bol JG, Binnekade R, Tilders FJ, Van Dam AM. Interleukin-1 and interleukin-1 receptor antagonist appear in grey matter additionally to white matter lesions during experimental multiple sclerosis. PLoS One 2013;8(12):e83835.
Provencio I, Rollag MD, Castrucci AM. Photoreceptive net in the mammalian retina. This mesh of cells may explain how some blind mice can still tell day from night. Nature 2002; 415(6871):493.
Pyo H, Yang MS, Jou I, Joe EH. Wortmannin enhances lipopolysaccharide-induced inducible nitric oxide synthase expression in microglia in the presence of astrocytes in rats. Neurosci Lett. 2003; 346:141-144.
Qiu X, Kumbalasiri T, Carlson SM, Wong KY, Krishna V, Provencio I, Berson DM. Induction of photosensitivity by heterologous expression of melanopsin. Nature 2005; 433(7027):745-9.
Quigley HA, Addicks EM, Green WR, Maumenee AE.Optic nerve damage in human glaucoma. II. The site of injury and susceptibility to damage. Arch Ophthalmol. 1981; 99(4):635-49.
Quigley HA, Addicks EM. Chronic experimental glaucoma in primates. II. Effect of extended intraocular pressure elevation on optic nerve head and axonal transport. Invest Ophthalmol Vis Sci. 1980; 19(2):137-52.
Quigley HA, Enger C, Katz J, Sommer A, Scott R, Gilbert D. Risk factors for the development of glaucomatous visual field loss in ocular hypertension. Arch Ophthalmol. 1994; 112(5):644-9.
Quigley HA, McKinnon SJ, Zack DJ, Pease ME, Kerrigan-Baumrind LA, Kerrigan DF, Mitchell RS. Retrograde axonal transport of BDNF in retinal ganglion cells is blocked by acute IOP elevation in rats. Invest Ophthalmol Vis Sci. 2000; 41(11):3460-6.
Ralph M, Lehman M. Transplantation: a new tool in the analysis of the mammalian hipothalamic circadian pacemaker. Trends Neurosci. 1991; 14:362-366.
Rao K, Lund RD. Degeneration of optic axons induces the expression of major histocompatibility antigens. Brain Res. 1989; 488(1-2):332-5.
Reese BE. 'Hidden lamination' in the dorsal lateral geniculate nucleus: the functional organization of this thalamic region in the rat. Brain Res. 1988; 472(2):119-37.
Reichenbach A, Stolzenburg JU, Eberhardt W, Chao TI, Dettmer D, Hertz L. What do retinal müller (glial) cells do for their neuronal 'small siblings'? J Chem Neuroanat. 1993; 6(4):201-13.
Reichstein D, Ren L, Filippopoulos T, Mittag T, Danias J. Apoptotic retinal ganglion cell death in the DBA/2 mouse model of glaucoma. Exp Eye Res. 2007; 84(1):13-21.
Richler M, Werner EB, Thomas D. Risk factors for progression of visual field defects in medically treated patients with glaucoma. Can J Ophthalmol. 1982; 17(6):245-8.
Ringvold A, Anderssen E, Kjønniksen I. Distribution of ascorbate in the anterior bovine eye. Invest Ophthalmol Vis Sci. 2000; 41(1):20-3.
Robinson GA, Madison RD. Axotomized mouse retinal ganglion cells containing melanopsin show enhanced survival, but not enhanced axon regrowth into a peripheral nerve graft. Vision Res. 2004; 44:2667-74.
Rodger J, Symonds AC, Springbett J, Shen WY, Bartlett CA, Rakoczy PE, Beazley LD, Dunlop SA. Eph/ephrin expression in the adult rat visual system following localized retinal lesions: localized and transneuronal up-regulation in the retina and superior colliculus. Eur. J. Neurosci. 2005; 22(8):1840-52.
Rodieck, RW. Visual pigments: bleaching and regeneration. The vertebrate retina. Principles of structure and function. WH Freeman and Company, San Francisco. 1973; Cap.VIII.199-237.
Rosenbaum DM, Rosenbaum PS, Singh M, Gupta G, Gupta H, Li B, Roth S. Functional and morphologic comparison of two methods to produce transient retinal ischemia in the rat. J Neuroophthalmol. 2001; 21(1):62-8.
Rouach N, Calvo CF, Glowinski J, Giaume C. Brain macrophages inhibit gap junctional communication and downregulate connexin 43 expression in cultured astrocytes. Eur J Neurosci. 2002; 15:403-7.
Ruby NF, Brennan TJ, Xie X, Cao V, Franken P, Heller HC, O'Hara BF. Role of melanopsin in circadian responses to light. Science 2002; 298: 2211-3.
Saggu SK, Chotaliya HP, Blumbergs PC, Casson RJ. Wallerian-like axonal degeneration in the optic nerve after excitotoxic retinal insult: an ultrastructural study. BMC Neurosci. 2010; 11:97.
Sakamoto K, Liu C, Kasamatsu M, Pozdeyev NV, Iuvone PM, Tosini G. Dopamine regulates melanopsin mRNA expression in intrinsically photosensitive retinal ganglion cells.Eur J Neurosci. 2005; 22:3129-36.
Salido EM, Dorfman D, Bordone M, Chianelli MS, Sarmiento MI, Aranda M, Rosenstein RE. Ischemic conditioning protects the rat retina in an experimental model of early type 2 diabetes. Exp Neurol. 2013;240:1-8.
Salinas-Navarro M, Jiménez-López M, Valiente-Soriano FJ, Alarcón-Martínez L, Avilés-Trigueros M, Mayor S, Holmes T, Lund RD, Villegas-Pérez MP, Vidal-Sanz M. Retinal ganglion cell population in adult albino and pigmented mice: a computerized analysis of the entire population and its spatial distribution. Vision Res. 2009; 49(6):637-47.
Sanchez Mejia RO, Ona VO, Li M, Friedlander RM. Minocycline reduces traumatic brain injury-mediated caspase-1 activation, tissue damage, and neurological dysfunction. Neurosurgery 2001; 48(6):1393-9; discussion 1399-401.
Sappington RM, Carlson BJ, Crish SD, Calkins DJ. The microbead occlusion model: a paradigm for induced ocular hypertension in rats and mice. Invest Ophthalmol Vis Sci. 2010; 51(1):207-16.
Sasaoka M, Nakamura K, Shimazawa M, Ito Y, Araie M, Hara H. Changes in visual fields and lateral geniculate nucleus in monkey laser-induced high intraocular pressure model. Exp. Eye Res. 2008; 86(5):770-82.
Sawaguchi S, Yue BY, Yeh P, Tso MO. Effects of intracameral injection of chondroitinase ABC in vivo. Arch Ophthalmol. 1992; 110:110-7.
Schlamp CL, Li Y, Dietz JA, Janssen KT, Nickells RW. Progressive ganglion cell loss and optic nerve degeneration in DBA/2J mice is variable and asymmetric. BMC Neurosci. 2006; 7:66.
Schmidt TM, Do MT, Dacey D, Lucas R, Hattar S, Matynia A. Melanopsin-positive intrinsically photosensitive retinal ganglion cells: from form to function. J Neurosci. 2011; 31(45):16094-101.
Schmidt-Kastner R, Wietasch K, Weigel H, Eysel UT. Immunohistochemical staining for glial fibrillary acidic protein (GFAP) after deafferentation or ischemic infarction in rat visual system: features of reactive and damaged astrocytes. Int J Dev Neurosci. 1993;11(2):157-74.
Schnitzer J. Retinal astrocytes: their restriction to vascularized parts of the mammalian retina. Neurosci Lett. 1987; 78(1):29-34.
Schnitzer J. Astrocytes in the guinea pig, horse, and monkey retina: their occurrence coincides with the presence of blood vessels. Glia 1988; 1(1):74-89.
Sears ML. Regulation of aqueous flow by the adenylate cyclase receptor complex in the ciliary epithelium. Am J Ophthalmol. 1985; 100:194-8.
Sejersen T, Lendahl U. Transient expression of the intermediate filament nestin during skeletal muscle development. J. Cell Sci. 1993; 106 (Pt 4):1291-300.
Semo M, Lupi D, Peirson SN, Butler JN, Foster RG. Light-induced c-fos in melanopsin retinal ganglion cells of young and aged rodless/coneless (rd/rd cl) mice.Eur J Neurosci. 2003;18:3007-17.
Seung HS, Sümbül U. Neuronal cell types and connectivity: lessons from the retina. Neuron 2014;83(6):1262-72.
Shareef SR, Garcia-Valenzuela E, Salierno A, Walsh J, Sharma SC. Chronic ocular hypertension following episcleral venous occlusion in rats. Exp Eye Res. 1995. 61(3):379-82.
Shields MB. Normal-tension glaucoma: is it different from primary open-angle glaucoma? Curr Opin Ophthalmol. 2008; 19(2):85-8.
Shin DH, Beker B, Kolker AE. Family history in primary open angle glaucoma. Arch Ophtalmol. 1977; 95:598.
Sihota R, Goyal A, Kaur J, Gupta V, Nag TC. Scanning electron microscopy of the trabecular meshwork: understanding the pathogenesis of primary angle closure glaucoma. Indian J Ophthalmol. 2012; 60(3):183-8.
Siminoff R, Schwassmann HO, Kruger L. An electrophysiological study of the visual projection to the superior colliculus of the rat. J Comp Neurol. 1966; 127(4):435-44.
Simon C, Götz M, Dimou L. Progenitors in the adult cerebral cortex: cell cycle properties and regulation by physiological stimuli and injury. Glia 2011; 59(6):869-81.
Sommer A. Intraocular pressure and glaucoma. Am J Ophthalmol. 1989; 107(2):186–188.
Son JL, Soto I, Oglesby E, Lopez-Roca T, Pease ME, Quigley HA, Marsh-Armstrong N. Glaucomatous optic nerve injury involves early astrocyte reactivity and late oligodendrocyte loss. Glia 2010; 58(7):780-9.
Steele MR, Inman DM, Calkins DJ, Horner PJ, Vetter ML. Microarray analysis of retinal gene expression in the DBA/2J model of glaucoma. Invest. Ophthalmol. Vis. Sci. 2006; 47(3):977-85.
Stence N, Waite M, Dailey ME. Dynamics of microglial activation: a confocal time-lapse analysis in hippocampal slices. Glia. 2001; 33(3):256-66.
Stone J, Dreher Z. Relationship between astrocytes, ganglion cells and vasculature of the retina. J Comp Neurol. 1987; 255(1):35-49.
Streit WJ. Microglia as neuroprotective, immunocompetent cells of the CNS. Glia 2002; 40:133-139.
Sun D, Lye-Barthel M, Masland RH, Jakobs TC. The morphology and spatial arrangement of astrocytes in the optic nerve head of the mouse. J Comp Neurol. 2009; 516(1):1-19.
Tamm ER, Carassa RG, Albert DM, Gabelt BT, Patel S, Rasmussen CA, Kaufman PL. Viscocanalostomy in rhesus monkeys. Arch Ophthalmol. 2004; 122(12):1826-38.
Tanaka H, Ito Y, Nakamura S, Shimazawa M, Hara H. Involvement of brain-derived neurotrophic factor in time-dependent neurodegeneration in the murine superior colliculus after intravitreal injection of N-methyl-D-aspartate. Mol Vis. 2009; 15:662-9.
Teng YD, Choi H, Onario RC, Zhu S, Desilets FC, Lan S, Woodard EJ, Snyder EY, Eichler ME, Friedlander RM. Minocycline inhibits contusion-triggered mitochondrial cytochrome c release and mitigates functional deficits after spinal cord injury. Proc Natl Acad Sci U S A. 2004;101(9):3071-6.
Tezel G, Hernandez MR, Wax MB. In vitro evaluation of reactive astrocyte migration, a component of tissue remodeling in glaucomatous optic nerve head. Glia 2001; 34(3):178-89.
Thapan K, Arendt J, Skene DJ. An action spectrum for melatonin suppression: evidence for a novel non-rod, non-cone photoreceptor system in humans. J Physiol. 2001; 535: 261-7.
Tian B, Geiger B, Epstein DL, Kaufman PL Cytoskeletal involvement in the regulation of aqueous humor outflow. Invest Ophthalmol Vis Sci. 2000; 41(3):619-23..
Tian GF, Azmi H, Takano T, Xu Q, Peng W, Lin J, Oberheim N, Lou N, Wang X, Zielke HR, Kang J, Nedergaard M. An astrocytic basis of epilepsy. Nat Med. 2005; 11(9):973-81.
Tielsch JM, Sommer A, Katz J, Royall RM, Quigley HA, Javitt J. Racial variations in the prevalence of primary open-angle glaucoma. The Baltimore Eye Survey. JAMA 1991; 266(3):369-74.
Tokuda K, Zorumski CF, Izumi Y. Effects of ascorbic acid on UV light-mediated photoreceptor damage in isolated rat retina. Exp Eye Res. 2007; 84:537–543.
Towle VL, Moskowitz A, Sokol S, Schwartz B. The visual evoked potential in glaucoma and ocular hypertension: effects of check size, field size, and stimulation rate. Invest Ophthalmol Vis Sci. 1983; 24:175-83.
Tsiperson V, Li X, Schwartz GJ, Raine CS, Shafit-Zagardo B. GAS6 enhances repair following cuprizone-induced demyelination. PLoS One 2010; 5(12):e15748.
Ueda J, Sawaguchi S, Hanyu T, et al. Experimental glaucoma model in the rat induced by laser trabecular photocoagulation after an intracameral injection of India ink. Jpn J Ophthalmol. 1998; 42: 337-344.
Valamanesh F, Monnin J, Morand-Villeneuve N, Michel G, Zaher M, Miloudi S, Chemouni D, Jeanny JC, Versaux-Botteri C. Nestin expression in the retina of rats with inherited retinal degeneration. Exp. Eye Res. 2013; 110:26-34.
Van Buskirk EM, Brett J. The canine eye: in vitro dissolution of the barriers to aqueous outflow. Invest Ophthalmol Vis Sci.1978; 17:258-71.
Vickers JC, Hof PR, Schumer RA, Wang RF, Podos SM, Morrison JH. Magnocellular and parvocellular visual pathways are both affected in a macaque monkey model of glaucoma. Aust N Z J Ophthalmol. 1997; 25(3):239-43.
Vinet J, Weering HR, Heinrich A, Kälin RE, Wegner A, Brouwer N, Heppner FL, Rooijen Nv, Boddeke HW, Biber K. Neuroprotective function for ramified microglia in hippocampal excitotoxicity. J Neuroinflammation. 2012; 9:27.
Viswanathan S, Frishman LJ, Robson JG. The uniform field and pattern ERG in macaques with experimental glaucoma: removal of spiking activity. Invest Ophthalmol Vis Sci. 2000; 41:2797-810.
Vugler AA, Semo M, Joseph A, Jeffery G. Survival and remodeling of melanopsin cells during retinal dystrophy.Vis Neurosci. 2008; 25:125-38.
Wachtmeister L. Oscillatory potentials in the retina: what do they reveal. Prog Retin Eye Res. 1998; 17(4):485-521.
Waller A. Experiments on the section of the glossopharyngeal and hypoglossal nerves of the frog, and observations of the alterations produced thereby in the structure of their primituve fibres. Phil. Transact. Royal Soc. London 1850; 140: 423-9.
Walsh DT, Perry VH, Minghetti L. Cyclooxygenase-2 is highly expressed in microglial-like cells in a murine model of prion disease. Glia 2000; 29, 392–396.
Walsh DT, Betmouni S, Perry VH. Absence of detectable IL-1beta production in murine prion disease: a model of chronic neurodegeneration. J. Neuropathol. Exp. Neurol. 2001; 60, 173-182
Wang HZ, Lu QJ, Wang NL, Liu H, Zhang L, Zhan GL. Loss of melanopsin containing retinal ganglion cells in a rat glaucoma model. Chin Med J. (Engl). 2008; 121:1015-9.
Wang J, Wei Q, Wang CY, Hill WD, Hess DC, Dong Z. Minocycline up-regulates Bcl-2 and protects against cell death in mitochondria. J Biol Chem. 2004; 279(19):19948-54.
Wang KK, Larner SF, Robinson G, Hayes RL. Neuroprotection targets after traumatic brain injury. Curr Opin Neurol. 2006; 19(6):514-9.
Wang SS, Shultz JR, Burish MJ, Harrison KH, Hof PR, Towns LC, Wagers MW, Wyatt KD. Functional trade-offs in white matter axonal scaling. J Neurosci. 2008; 28(15):4047-56.
Wanner IB, Anderson MA, Song B, Levine J, Fernandez A, Gray-Thompson Z, Ao Y, Sofroniew MV. Glial scar borders are formed by newly proliferated, elongated astrocytes that interact to corral inflammatory and fibrotic cells via STAT3-dependent mechanisms after spinal cord injury. J. Neurosci. 2013; 33(31):12870-86.
Watanabe M, Toyama Y, Nishiyama A. Differentiation of proliferated NG2-positive glial progenitor cells in a remyelinating lesion. J. Neurosci. Res. 2002; 69(6):826-36.
Weber AJ, Chen H, Hubbard WC, Kaufman PL. Experimental glaucoma and cell size, density, and number in the primate lateral geniculate nucleus. Invest Ophthalmol Vis Sci. 2000; 41(6):1370-9.
Weber AJ, Zelenak D. Experimental glaucoma in the primate induced by latex microspheres. J Neurosci Methods 2001; 111:39-48.
Wee R, Van Gelder RN. Sleep disturbances in young subjects with visual dysfunction. Ophthalmology. 2004; 111: 297-302; discussion 302-293.
Weinreb RN, Khaw PT. Primary open-angle glaucoma. Lancet. 2004; 22;363(9422):1711-20.
Wen L, Xu J, Zhan T, Wang H, Huang X, Liu W, Yang X, Zhan R. The occurrence of diffuse axonal injury in the brain: associated with the accumulation and clearance of myelin debris. Neural Regen. Res. 2014; 9(21):1902-6.
Wennström M, Hellsten J, Tingström A. Electroconvulsive seizures induce proliferation of NG2-expressing glial cells in adult rat amygdala. Biol. Psychiatry 2004; 55(5):464-71.
Whitmore AV, Libby RT, John SW. Glaucoma: thinking in new ways-a role for autonomous axonal self-destruction and other compartmentalised processes? Prog Retin Eye Res. 2005; 24(6):639-62.
Wilkins A, Nikodemova M, Compston A, Duncan I. Minocycline attenuates nitric oxide-mediated neuronal and axonal destruction in vitro. Neuron Glia Biol. 2004;1(3):297-305.
Woldemussie E, Wijono M, Ruiz G. Müller cell response to laser-induced increase in intraocular pressure in rats. Glia. 2004; 47(2):109-19.
Wu KH, Madigan MC, Billson FA, Penfold PL. Differential expression of GFAP in early vs. late AMD: a quantitative analysis. Br J Ophthalmol. 2003; 87(9):1159-66.
Xue LP, Lu J, Cao Q, Hu S, Ding P, Ling EA. Müller glial cells express nestin coupled with glial fibrillary acidic protein in experimentally induced glaucoma in the rat retina. Neuroscience 2006a; 139(2):723-32.
Xue LP, Lu J, Cao Q, Kaur C, Ling EA. Nestin expression in Müller glial cells in postnatal rat retina and its upregulation following optic nerve transection. Neuroscience 2006b; 143(1):117-27.
Yamada H, Takano T, Ito Y, Matsuzuka F, Miya A, Kobayashi K, Yoshida H, Watanabe M, Iwatani Y, Miyauchi A. Expression of nestin mRNA is a differentiation marker in thyroid tumors. Cancer Lett. 2009; 280(1):61-4.
Yang LP, Zhu XA, Tso MO. Minocycline and sulforaphane inhibited lipopolysaccharide-mediated retinal microglial activation. Mol Vis. 2007; 13:1083-93.
Yang Q, Shen J, Guo W, Wen J, Wang Z, Yu D. Effect of acute intraocular pressure elevation on blood flow velocity and resistance in the rabbit ophthalmic artery. Vet Ophthalmol. 2011; 14(6):353-7.
Yi CX, Habegger KM, Chowen JA, Stern J, Tschöp MH. A role for astrocytes in the central control of metabolism. Neuroendocrinology. 2011; 93(3):143-9.
Yokoyama A, Yang L, Itoh S, Mori K, Tanaka J. Microglia, a potential source of neurons, astrocytes, and oligodendrocytes. Glia. 2004; 45(1):96-104.
Yoshimura T, Ebihara S. Spectral sensitivity of photoreceptors mediating phase-shifts of circadian rhythms in retinally degenerate CBA/J (rd/rd) and normal CBA/N (+/+)mice. J Comp Physiol [A]. 1996; 178: 797-802.
Young MJ, Lund RD. The retinal ganglion cells that drive the pupilloconstrictor response in rats. Brain Res. 1998; 787:191-202.
Yrjänheikki J, Keinänen R, Pellikka M, Hökfelt T, Koistinaho J. Tetracyclines inhibit microglial activation and are neuroprotective in global brain ischemia. Proc. Natl. Acad. Sci. U S A. 1998; 95(26):15769-74.
Yuan L, Neufeld AH. Tumor necrosis factor-alpha: a potentially neurodestructive cytokine produced by glia in the human glaucomatous optic nerve head. Glia 2000; 32(1):42-50.
Yücel YH, Zhang Q, Gupta N, Kaufman PL, Weinreb RN. Loss of neurons in magnocellular and parvocellular layers of the lateral geniculate nucleus in glaucoma. Arch. Ophthalmol. 2000; 118(3):378-84.
Yücel YH, Zhang Q, Weinreb RN, Kaufman PL, Gupta N. Atrophy of relay neurons in magno- and parvocellular layers in the lateral geniculate nucleus in experimental glaucoma. Invest Ophthalmol Vis Sci. 2001; 42(13):3216-22.
Yücel YH, Zhang Q, Weinreb RN, Kaufman PL, Gupta N. Effects of retinal ganglion cell loss on magno-, parvo-, koniocellular pathways in the lateral geniculate nucleus and visual cortex in glaucoma. Prog Retin Eye Res. 2003; 22(4):465-81.
Zabad RK, Metz LM, Todoruk TR, Zhang Y, Mitchell JR, Yeung M, Patry DG, Bell RB, Yong VW. The clinical response to minocycline in multiple sclerosis is accompanied by beneficial immune changes: a pilot study. Mult Scler. 2007;13(4):517-26.
Zai LJ, Wrathall JR. Cell proliferation and replacement following contusive spinal cord injury. Glia 2005; 50(3):247-57.
Zhang C, Lei B, Lam TT, Yang F, Sinha D, Tso MO. Neuroprotection of photoreceptors by minocycline in light-induced retinal degeneration. Invest Ophthalmol Vis Sci. 2004; 45(8):2753-9.
Zhang R, Chopp M, Zhang ZG. Oligodendrogenesis after cerebral ischemia. Front. Cell Neurosci. 2013; 7:201.
Zhang S, Wang H, Lu Q, Qing G, Wang N, Wang Y, Li S, Yang D, Yan F. Detection of early neuron degeneration and accompanying glial responses in the visual pathway in a rat model of acute intraocular hypertension. Brain Res. 2009; 1303:131-43.
Zhang Y, Metz LM, Yong VW, Bell RB, Yeung M, Patry DG, Mitchell JR. Pilot study of minocycline in relapsing-remitting multiple sclerosis. Can J Neurol Sci. 2008; 35(2):185-91.
Zhu S, Stavrovskaya IG, Drozda M, Kim BY, Ona V, Li M, Sarang S, Liu AS, Hartley DM, Wu DC, Gullans S, Ferrante RJ, Przedborski S, Kristal BS, Friedlander RM. Minocycline inhibits cytochrome c release and delays progression of amyotrophic lateral sclerosis in mice. Nature 2002; 417(6884):74-8.








































































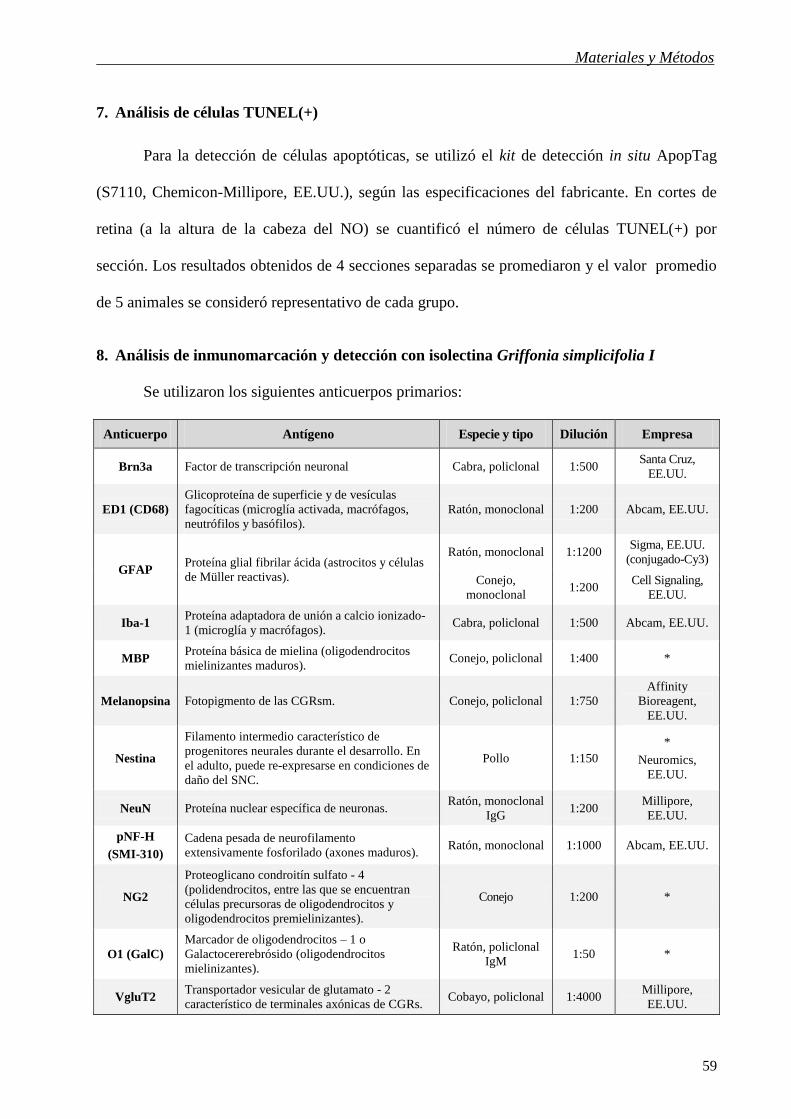


























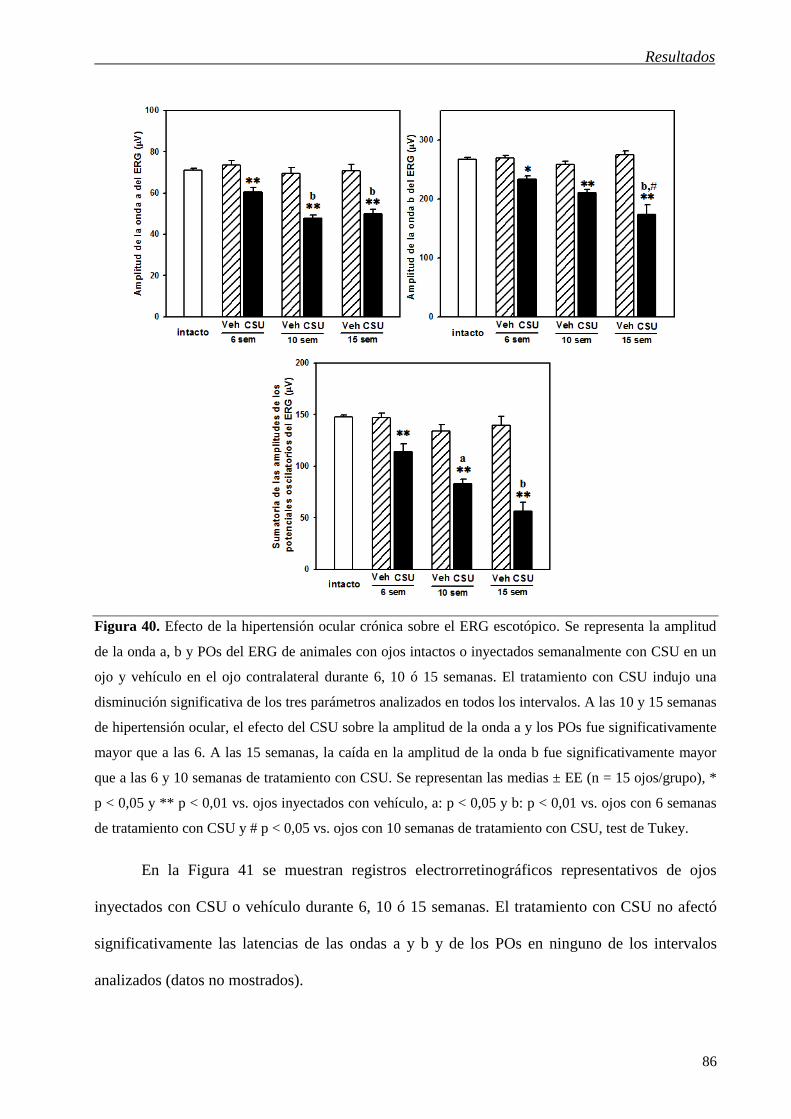




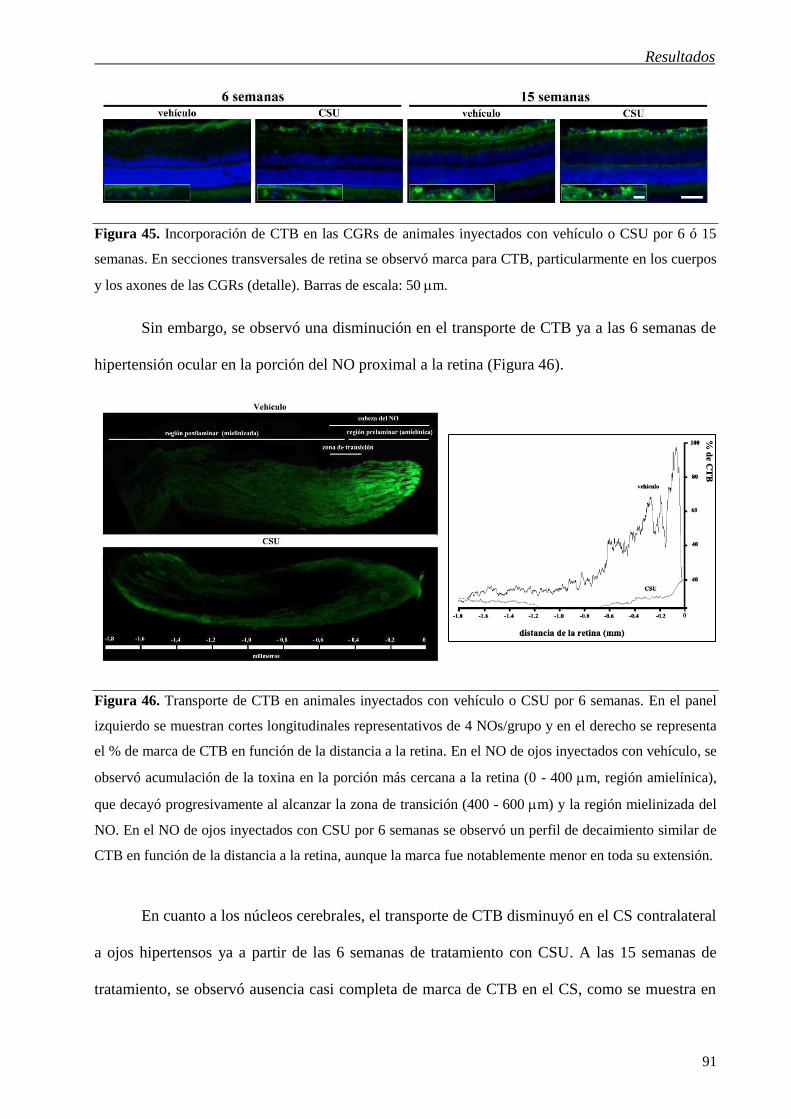





























































































































![Properties of the Nucleo-Olivary Pathway: An In Vivo …neurons excites the IO indirectly via nuclei located at the mesodiencephalic junction (MDJ) [7,8]. This olivo-cortico-nuclear](https://static.documents.pub/doc/80x56/5eaebd13fb48ac16bb08c20c/properties-of-the-nucleo-olivary-pathway-an-in-vivo-neurons-excites-the-io-indirectly.jpg)



