Ó 2012 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Ethylene Oxide SIEGFRIED REBSDAT, Winh€ oring, Federal Republic of Germany DIETER MAYER, Hoechst Aktiengesellschaft, Frankfurt, Federal Republic of Germany 1. Introduction......................... 547 2. Physical Properties ................... 547 3. Chemical Properties .................. 549 4. Production .......................... 553 4.1. Catalysts ........................... 553 4.2. Mechanism of Catalysis ................ 554 4.3. Technology.......................... 556 5. Environmental Protection and Ecology .... 560 6. Quality Specifications.................. 561 7. Analysis ............................ 561 8. Handling, Storage, and Transportation .... 561 9. Uses ............................... 564 10. Economic Aspects .................... 565 11. Toxicology and Occupational Health ...... 565 12. Acknowledgement .................... 568 References .......................... 568 1. Introduction Ethylene oxide (oxirane) [75-21-8], M r 44.05, is the simplest cyclic ether. It is a colorless gas or liquid and has a sweet, etheric odor. The ethylene oxide molecule with its short C–C bond and strained angles is shown in Figure 1 [6]. Theo- retical calculations of the structure are in good agreement with experimental results [7]. Ethylene oxide is very reactive because its highly strained ring can be opened easily, and is thus one of the most versatile chemical inter- mediates. Because of its reactivity and toxicity it is also a hazardous compound that has been involved in a number of serious incidents. A good understanding of its properties is a neces- sary prerequisite for its safe handling. Ethylene oxide was first described in 1859 by WURTZ [8], who prepared it by eliminating hydrochloric acid from ethylenechlorohydrin, using potassium hy- droxide solution. Industrial production by the chlorohydrin process began in 1914 and was based on WURTZ’s discovery. Since then the production and importance of ethylene oxide have steadily grown. In 1931, LEFORT [9] discovered the direct catalytic oxidation of ethylene [74-85-1], which gradually superseded the chlorohydrin process. Currently, ethylene oxide is produced by direct oxidation of ethylene with air or oxygen; annual worldwide production capacity is ca. 15 10 6 t, making it an important industrial chemical. Eth- ylene oxide itself is used as a disinfectant, steril- izing agent, and fumigant. Its most important derivative is ethylene glycol [107-21-1], which is used in antifreeze (car radiators) (! Anti- freezes) and for the manufacture of polyester fibers (! Fibers, 5. Polyester Fibers; ! Polye- sters). Other ethylene oxide derivatives, amines and poly(ethylene glycols), are used in surfac- tants, solvents, etc. There is some endogenous formation from ethylene in humans [10], and it has been discov- ered in an interstellar source [11]. As atmospheric pollutant it is present in natu- ral gas, cigarette smoke, and diesel exhaust. Though ethylene oxide in nature is rare its bio- logical action may have a fundamental impact on DNA and RNA evolution of whole biological systems [12]. 2. Physical Properties Some physical properties of ethylene oxide are summarized in Table 1. In the pressure range 0 – 101.3 kPa and at 40 and 60 C, the solubility of ethylene oxide in water obeys Henry’s law. The Henry constants for these temperatures are 2.875 and 1.448, DOI: 10.1002/14356007.a10_117
Transcript
� 2012 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim
Article No : a10_117
Ethylene Oxide
SIEGFRIED REBSDAT, Winh€oring, Federal Republic of Germany
DIETER MAYER, Hoechst Aktiengesellschaft, Frankfurt, Federal Republic of Germany
Ethylene oxide (oxirane) [75-21-8], Mr 44.05, isthe simplest cyclic ether. It is a colorless gas orliquid and has a sweet, etheric odor. The ethyleneoxide molecule with its short C–C bond andstrained angles is shown in Figure 1 [6]. Theo-retical calculations of the structure are in goodagreement with experimental results [7].
Ethylene oxide is very reactive because itshighly strained ring can be opened easily, and isthus one of the most versatile chemical inter-mediates. Because of its reactivity and toxicity itis also a hazardous compound that has beeninvolved in a number of serious incidents. Agood understanding of its properties is a neces-sary prerequisite for its safe handling. Ethyleneoxide was first described in 1859 by WURTZ [8],who prepared it by eliminating hydrochloric acidfrom ethylenechlorohydrin, using potassium hy-droxide solution. Industrial production by thechlorohydrin process began in 1914 and wasbased on WURTZ’s discovery. Since then theproduction and importance of ethylene oxidehave steadily grown.
In 1931, LEFORT [9] discovered the directcatalytic oxidation of ethylene [74-85-1], whichgradually superseded the chlorohydrin process.Currently, ethylene oxide is produced by directoxidation of ethylene with air or oxygen; annual
worldwide production capacity is ca. 15 � 106 t,making it an important industrial chemical. Eth-ylene oxide itself is used as a disinfectant, steril-izing agent, and fumigant. Its most importantderivative is ethylene glycol [107-21-1], whichis used in antifreeze (car radiators) (! Anti-freezes) and for the manufacture of polyesterfibers (! Fibers, 5. Polyester Fibers; ! Polye-sters). Other ethylene oxide derivatives, aminesand poly(ethylene glycols), are used in surfac-tants, solvents, etc.
There is some endogenous formation fromethylene in humans [10], and it has been discov-ered in an interstellar source [11].
As atmospheric pollutant it is present in natu-ral gas, cigarette smoke, and diesel exhaust.Though ethylene oxide in nature is rare its bio-logical action may have a fundamental impact onDNA and RNA evolution of whole biologicalsystems [12].
2. Physical Properties
Some physical properties of ethylene oxide aresummarized in Table 1.
In the pressure range 0 – 101.3 kPa and at 40and 60 �C, the solubility of ethylene oxide inwater obeys Henry’s law. The Henry constantsfor these temperatures are 2.875 and 1.448,
DOI: 10.1002/14356007.a10_117
respectively. However, at 20 �C a more thanproportional increase in solubility occurs withthe partial pressure [15].
Liquid ethylene oxide and water arecompletely miscible in all proportions. Table 2shows some of the properties of aqueous ethyleneoxide solutions. Of particular note are the rela-tively high melting points, which are due toclathrate formation [17]. Clathrates consist oforganic molecules that are enclosed in a cage
structure. In this case, the cage is an ice latticewhich is composed of unit cells, each containing46 water molecules, and two types of cavities: sixlarger ones (14-sided, tetradecahedra) and twosmaller ones (12-sided, dodecahedra). If the clath-rates are made to crystallize from increasinglyconcentrated aqueous solutions of ethylene oxide,their density and ethylene oxide content alsoincrease. Ethylene oxide first fills the six tetra-decahedral cavities of the water lattice, followedby 20 – 40% of the dodecahedral cavities [18].
The temperatures for the formation of ice andhydrate from mixtures of ethylene oxide andwater for concentrations of 0 to 97.89 wt%ethylene oxide gave a highest hydrate formationtemperature of 11.08 �C at an ethylene oxideconcentration of 26.15 wt%. The hydrate corre-sponds to a composition of 6.91 molecules ofwater per molecule ethylene oxide. The unit cellconsists of 46 water molecules and 6.66 ethyleneoxide molecules. The eutectic point of the mix-ture has been determined to lie at 2.107 �C and4.75 wt% ethylene oxide (Fig. 2) [19].
There are also mixed clathrates, in which thesmall cages are occupied by ethylene oxide andthe big cages by molecules with an higher spacerequirement, e.g., tetrahydrofuran [20].
The solubilities of gases in ethylene oxidevary, increasing in the order nitrogen, argon,methane, ethane. Earlier data [21] have beenrevised [22]. The Henry constants for these gasesin ethylene oxide at different temperatures aregiven in Table 3.
Table 4 shows other temperature-dependentphysical properties of gaseous and liquid ethyl-ene oxide.
Figure 1. The ethylene oxide molecule
Table 1. Physical properties of ethylene oxide [5], [13], [14]
mp at 101.3 kPa �112.5 �Cbp at 101.3 kPa 10.8 �CCritical temperature 195.8 �CCritical pressure 7.2 MPa
Critical density 314 kg/m3
Refractive index, n7D 1.3597
Explosive limits in air at 101.3 kPa
lower 2.6 vol%
upper 100.0 vol%
Electrical conductivity 4 � 10�6 S/m
Dielectric constant
at �1 �C (liquid) 13.9
at 15 �C (vapor) 1.01
Heat of combustion at 25 �C,101.3 kPa 29.648 kJ/kg
Entropy of the vapor at 101.3 kPa
10.5 �C 5.439 kJ kg�1 K�1
25.0 �C 5.495 kJ kg�1 K�1
Ignition temperature in air at
101.3 kPa 429 �CIgnition energy in air at
101.3 kPa and 25 �C 0.087 mJ
Minimum ignition energy of the gas at
220 kPa and 100 �C 64 mJ
Decomposition temperature of
the vapor at 101.3 kPa 571 �CHeat of polymerization 2091 kJ/kg
Heat of fusion 117.86 kJ/kg
Heat of decomposition of the vapor 1901 kJ/kg
Coefficient of cubic expansion
at 22 �C 0.00161
at 55 �C 0.00170
Heat of solution in water at 25 �C 142.57 kJ/kg
Table 2. Physical properties of aqueous ethylene oxide solutions [3],
[5], [16]
Ethylene oxide
content, wt % mp, �C bp, �CDensity at
10 �C, g/L Flash point, �C
0 0.0 100 0.9991
0.5 41.5
1 �0.4 31
2 3
3 �1.3
5 �1.6 58 0.9988 �2
10 5.6 42.5 0.9980
20 10.4 32 0.9945 �21
30 11.08 (max) 27 0.9882 �28
40 10.4 21 0.9792 �35
60 7.8 16 0.9534 �45
80 3.7 13 0.9194 �53
100 �112.5 10.4 0.8826 �57
548 Ethylene Oxide Vol. 13
3. Chemical Properties
Ethylene oxide is a very reactive, versatile com-pound. Its reactions proceed mainly via ringopening and are highly exothermic. Explosivedecomposition of the ethylene oxide vapor mayoccur at higher temperatures if heat dissipation isinadequate. Only the most important types of the
large number of possible reactions are brieflydiscussed here.More detailed information can befound in [25–29].
Decomposition. Gaseous ethylene oxidestarts to decompose at ca. 400 �C to formmainlyCO, CH4, as well as C2H6, C2H4, H2, C, andCH3CHO. The first step in the decomposition ispresumed to be the isomerization of ethyleneoxide to acetaldehyde [30]. Once the decompo-sition reaction has been initiated (ignitionsource), it can be propagated through the gasphase and, under certain conditions, may beexplosive (see Chap. 8).
Addition to Compounds with a LabileHydrogen Atom. Ethylene oxide reacts withcompounds containing a labile hydrogen atomto form a product containing a hydroxyethylgroup:xs
Figure 2. Hydrate-formation temperature as a function ofethylene oxide concentration
Table 3. Solubility of gases in ethylene oxide (Henry constants in
MPa) [22], [23]
Temperature, �C Solubility in ethylene oxide
Nitrogen Argon Methane Ethane
0 284 169 62.1 8.5
25 221 144 62.2 11.0
50 184 129 62.3 13.0
Table 4. Physical properties of ethylene oxide at various temperatures [3], [24]
Examples of XH are: HOH, H2NH, HRNH,R2NH, RCOOH, RCONH2, HSH, RSH, ROH,N�CH, andB2H6 (R ¼ alkyl, aryl). The reactionis accelerated by acids and bases. During acidcatalysis, the ethylene oxide is first protonated toform an epoxonium ion (1), which is in equilib-rium with the corresponding hydroxycarbeniumion (2). The anionX� can then react with 1 or 2 inan SN2 or SN1 reaction, respectively. In an alka-line medium, the SN2 mechanism is favored. Adetailed discussion of reaction mechanisms canbe found in [27], [31].
The protonation of ethylene oxide by variousH-active compounds in tetrachloromethane canbe detected by IR spectroscopy. Broadening andshift of the IR bands in the region of 530 to740 cm�1 depend on the type of H-active com-pound (OH, NH, CH) [32]. Analogous protonat-ed intermediates have been postulated in gas-phase SN2 reactions of ethylene oxide on thebasis of theoretical and experimental results [33].
All common acids and Lewis acids as well aszeolites, ion exchangers [34], and aluminumoxide are effective catalysts. Solid polymericacids (cation exchangers) are especially recom-mended for the reaction of ethylene oxide withmethanol [35].
As the end product of the above reactioncontains at least one hydroxyl group, it may reactsuccessively with further ethylene oxide mole-cules:
The molecular mass of the resulting polymersdepends on the ratio of the reactants, the catalystused, and the reaction conditions [1], [28].
A byproduct of the acid-catalyzed polymeri-zation is dioxane, which may even be the soleproduct with a large excess of ethylene oxide. Anexplanation for this behavior is offered by theassumption that two different reaction mechan-
isms are effective. The first is the active chain end(ACE) mechanism, which leads to dioxane andpoly(ethylene glycol).
And the second is the activated monomer(AM) mechanism, which leads exclusively topoly(ethylene glycol).
One of the conditions which favor the ACMmechanism and therefore the formation of diox-ane is an increasing excess of ethylene oxide[36].
A large variety of reactions occur betweenethylene oxide and compounds containing labilehydrogen atoms; therefore, such compounds areoften used to produce derivatives.
Commercially, themost important of this typeof reaction is the hydrolysis of ethylene oxide toethylene glycol. About 60% of the total ethyleneoxide production is converted into ethylene gly-col in this way (! Ethylene Glycol). However,this reaction, which is often referred to as ethox-ylation, is also used to produce the bulk of all theother commercially important ethylene oxidederivatives. The ethoxylation products of alkyl
550 Ethylene Oxide Vol. 13
phenols, ammonia, fatty alcohols, fatty amines,and fatty acids have a variety of uses [28].Although polymer formation according to theabove equation is more likely to be an unwantedside reaction, the production of poly(ethyleneglycols) of widely varying molecular mass bythis route is of some importance [37]. Undesiredpolymerization may be catalyzed by rust in rustycontainers [38].
Reaction with Water. The formation ofhydrates is discussed in Chapter 2. Hydrolysisis the most important reaction of ethylene oxide:about 60% of the total ethylene oxide productionis transformed into ethylene glycol. The reactionof ethylene oxide with water in the absence ofcatalysts at 20 �C is relatively slow, as shown bythe half-life of 20 d. The dependence of the half-life of a 3% solution of ethylene oxide in wateron pH is shown in Figure 3. The dependence ischaracterized by a bell-shaped curve with arelatively broad plateau from pH 5 to 11 and asteep slope on both sides. A strong accelerationoccurs only below pH 4 and above pH 12. By-products of the hydrolysis are diethylene glycol,triethylene glycol, and higher molecular masspoly(ethylene glycols). The percentage of ethyl-ene oxide converted to these products dependsalmost solely on the ratio of water to ethylene
oxide in the startingmaterial and onlymarginallyon pH and temperature.
Reaction with DNA, RNA and Proteins.The major product of reaction with DNA is 7-(2-hydroxyethyl) guanine. Further reaction pro-ducts are (2-hydroxyethyl) guanine and 3-(2-hydroxyethyl) adenine [12].
In men exposed to ethylene oxide, adductswith hemoglobin can be found and its level maybe evaluated by the concentration of (2-hydro-xyethyl) valine [39].
Addition to Double Bonds. Ethylene oxidecan add to compounds with double bonds, e.g.,carbon dioxide [40], to form cyclic products:
Quaternary ammonium compounds are suit-able catalysts for this reaction, which then pro-ceeds at 200 �C and 8 MPa [41]. Use of porphinecomplexes of aluminum in solvents such aschloroform allows the reaction to take place atnormal pressure and at room temperature [42].Hydrolysis of ethylene carbonate (1,3-dioxolan-2-one) [96-49-1] yields pure ethylene glycol[43]. Therefore, the reaction is of interest for theselective production of ethylene glycol fromethylene oxide, i.e., for avoiding the partly un-wanted formation of poly(ethylene glycols) [44].
Ethylene oxide also adds to other double bondsystems, e.g., to R2C¼O [45], SC¼S, O2S¼O,RN¼CO, and OS¼O.
Catalytic Isomerization to Acetaldehyde.Aluminum oxide (Al2O3) [46], phosphoric acidand phosphates [47], and under certain condi-tions silver [48] catalyze the isomerization ofethylene oxide to acetaldehyde.
Reduction to Ethanol. Reduction of ethyl-ene oxide to ethanol is catalyzed by Ni, Cu, and
Figure 3. Half-life of 3 wt% ethylene oxide in water as afunction of pH at 8 and 20 �C
Vol. 13 Ethylene Oxide 551
Cr on Al2O3 [49].
Reaction with Grignard Reagents.Grignard reagents react with ethylene oxide toproduce compounds with a primary hydroxylgroup [29], [50].
Reaction with Lithium Acetylide. Acety-lene is treated with lithium in liquid ammonia toform the lithium salt, which reacts with ethyleneoxide at �33 �C to give 3-butyne-1-ol in 80%yield [51].
Reaction with Synthesis Gas. Ethylene ox-ide is carbonylatedwith CO/H2 in the presence ofrhodium catalysts in tetraethylene glycol dimeth-yl ether to give 1,3-propanediol in good yields[52].
Oligomerization to Crown Ethers. Ethyl-ene oxide oligomerizes to form cyclic polyethers(crown ethers) in the presence of a fluorinatedLewis acid catalyst (! Crown Ethers).
Synthesis is successful when groups that sat-urate potential terminal hydroxyl groups areabsent [53]. Suitable catalysts are the BF�4 ,PF�6 , or SbF
�6 salts of metal cations. The reaction
can be directed to produce a particular oligomerby choosing an appropriatemetal cation. The sizeof the cation determines the size of the crownether ring by acting as a template [54]. Forexample, with CsBF4, only the cyclic hexamer(n ¼ 3), 1,4,7,10,13,16-hexaoxacyclooctade-cane (18-crown-6) [17455-13-9], is produced;if Cu(BF4)2 is used, 90% of the product consistsof the pentamer (n ¼ 2), 1,4,7,10,13-penta-oxacyclopentadecane (15-crown-6) [33100-27-5], and with Ca(BF4)2, a mixture of the tetramer(50%, n ¼ 1), 1,4,7,10-tetraoxacyclodecane(12-crown-4) [294-93-9], and the pentamer,1,4,7,10,13-pentaoxacyclopentadecane (18-crown-6) [17455-13-9], is produced.
Reaction with Dimethyl Ether. Ethyleneoxide reacts with dimethyl ether to producepoly(ethylene glycol) dimethyl ethers (! Di-methyl Ether); this reaction is used for the indus-trial production of the lower molecular masshomologues, which are widely used as solvents[55].
Reaction with Bromotrimethylsilane.Ethylene oxide adds to bromotrimethylsilane[2857-97-8] in a highly exothermic reaction withexcellent yields [56].
Disproportionation in the presence of ironoxides (especially g-Fe2O3 and g-FeOOH ) hasbeen described as the cause of an explosion (seeChap. 8) [57].
4 C2H4O!3 C2 H4þ2 CO2þ2 H2O
5 C2H4O!4 C2H4þ2 CO2þH2þH2O
6 C2H4O!5 C2H4þ2 CO2þ2 H2O
552 Ethylene Oxide Vol. 13
4. Production
As mentioned in the introduction, ethylene oxidewas produced formerly by the chlorohydrin pro-cess (! Chlorohydrins, Chap. 4.). However, thismethod is no longer used on an industrial scale; adescription is given in [58]. Although the selec-tivity of this process (80 %) was satisfactory,practically all of the chlorine that was used waslost as calcium chloride and unwanted chlorine-containing byproducts were generated. This notonly was inefficient, but also caused pollutionproblems so that this method has now beenreplaced by the direct oxidation process.
WURTZ, who had discovered ethylene oxide,attempted as early as 1863 to produce it by directoxidation of ethylene with oxygen, but did notsucceed [59]. Many other unsuccessful attemptswere made [60] before LEFORT made the crucialdiscovery in 1931 that the formation of ethyleneoxide from ethylene and oxygen was catalyzedby metallic silver [9]. Since the catalyst plays acentral role in the production process, it is dis-cussed first.
4.1. Catalysts
To date no other metal has been found that cancompete with silver in the catalysis of the directoxidation of ethylene to ethylene oxide. Howev-er, the silver catalysts have been substantiallyimproved since their discovery by LEFORT [9].
Only supported catalysts are used, the silverbeing deposited on a porous support material inconcentrations of 7 – 20%. The supportmaterialis of critical importance [61]; currently, prefer-ence is given to ultrapure (over 99%) aluminumoxide [57828-03-2] fired at a high temperaturethat has a defined pore structure (pore diameter0.5 – 50 mm) and low specific surface area (<2m2/g). Although supports with higher specificsurface areas are very active, their selectivity islow, presumably because ethylene oxide candiffuse only slowly out of the smaller pores and,therefore, can be further oxidized [62]. Supportswith chemical activity towards ethylene or eth-ylene oxide are unsuitable. All support contain-ing hydroxyl groups catalyze the isomerizationof ethylene oxide to acetaldehyde. As total oxi-dation of acetaldehyde is very fast on silver,catalysts consisting of silver and carriers like
g-alumina, SiO2, MgO, SiC, TiO2, Y2O3, andZrO2 give only poor selectivities or form onlyCO2 and H2O [63]. Silane treatment of supportscontaining hydroxyl groups improves the perfor-mance in ethylene oxide formation, and thisconfirms the detrimental effect of hydroxylgroups, which result in isomerization of ethyleneoxide to acetaldehyde [64].
The impregnationmethods used to deposit thesilver on the support are being constantly im-proved. Complexes of silver salts with aminocompounds are used which decompose to giveevenly and finely distributed silver particles witha diameter of 0.1 – 1 mm [65]. Figure 4 shows ascanning electronmicrograph (SEM) of the innersurface of a silver catalyst particle.
In addition, 100 – 500 mg/kg of promoterssuch as salts or other compounds of alkali andalkaline earth metals are added to the catalyst,significantly improving the selectivity. Amongthe alkali metal salts, those of cesium are espe-cially effective [66]. Promoters are often com-bined. An especially effective combination con-sists of rhenium, sulfur, tungsten, molybdenumand results in outstanding selectivities of up to90%, albeit at higher temperature and reducedcatalyst life [67].
Addition of chlorine compounds to the reac-tion gases improves selectivity [68]. These in-hibitors (e.g., 1,2-dichloroethane, vinyl chloride[69], ethyl chloride) [70] suppress the combus-tion of ethylene to carbon dioxide andwater; theyensure that the silver surface is covered evenlywith a supply of chlorine [71].
The advances made in silver-based catalystssince LEFORT’s original discovery (i.e., optimizedsupport materials, silver distribution, and use ofpromoters and inhibitors) have improved selec-tivity from 50 to about 90%. Silver has main-tained its position as the only known metal thatcan catalyze the oxidation of ethylene to ethyleneoxide with a commercially viable selectivity.
The silver catalysts used in ethylene oxideproduction are not suited for the production ofepoxides from olefins with allylic hydrogenatoms (e.g., propene); total oxidation is the pre-vailing reaction. Nonallylic olefins such as buta-diene are reported to form epoxides in reasonableyields [72].
Aging of the Catalyst. Modern silver-basedcatalysts have an initial selectivity of 80 – 90%;a maximum selectivity of 90% is commerciallyfeasible [44]. The disadvantages associated withhighly selective catalysts are that they age rela-tively quickly and less heat is produced. As thecatalyst is used, its selectivity and activity grad-ually deteriorate [73–75] due to
1. Abrasion, dust formation, and blocking ofpores.
2. Accumulation of detrimental impurities intro-duced with the reaction gases (e.g., sulfurfrom ethylene or methane).
3. Changes in the silver particles, which enlarge,form agglomerates, and become unevenlydistributed. Thus, the installed silver surface,which is about 20 km2 for 100 t/a ethyleneoxide capacity, drops to about 50% of itsinitial value by sintering during two years onstream. To compensate this reduction in silversurface the reaction temperature (reactionvelocity) has to be increased accordingly tomaintain the production rate.
Generally applicable methods of regeneratingsuch catalysts are unknown; the only method ofany importance is the use ofmethanolic solutionsof cesium salts [76]. If regeneration is impossi-
ble, it becomes economically necessary to re-place the catalyst when selectivity has fallen to acritical level or becomes technically necessarybecause the reaction temperature has reached themaximumdesign value. The lifetime of amoderncatalyst is two to five years, depending on thetype of catalyst (high-selectivity catalysts haveconsiderably shorter life times), the rate of eth-ylene oxide production, and the purity of thereaction gases (sulfur is extremely poisonous).From the spent catalysts silver is recovered inhigh purity and with only small losses, while theused supports are waste.
4.2. Mechanism of Catalysis
Two reactions take place simultaneously at thesilver surface. In addition to ethylene oxideformation (partial oxidation, Eq. 1), completecombustion (total oxidation, Eq. 2) to CO2 andwater also takes place. Small amounts of acetal-dehyde and formaldehyde are also formed [77].The reactions given by Equations (1) and (2) areexothermic, their enthalpies being �106.7 and�1323 kJ/mol, respectively at 250 �C and1.5 MPa [44].
Although many attempts have been made todiscover the mechanism responsible for silver’sunique action, opinions remain divided [78].Evaluation of the various studies is difficultbecause often they were carried out under differ-ent conditions that were not always suitable forindustrial purposes. It is agreed that silver canadsorb oxygen in a number of ways and that thisphenomenon is the basis of its unequalled effi-ciency in catalyzing the oxidation of ethylene toethylene oxide. The following adsorption formsof oxygen are of critical importance:
Opinions differ as to the role of these differenttypes of adsorbed oxygen; two conflicting me-chanisms have been proposed.
Mechanism 1. Only molecular oxygen re-actswith ethylene to formethylene oxide,whereasatomic oxygen only reacts to form carbon dioxideandwater[79].Chlorine(fromtheinhibitor)blocksthe adsorption of atomic oxygen on the silversurface so that, in the ideal case, the optimallyinhibited silver surface only adsorbs molecularoxygen (Eq. 3). The adsorbed molecular oxygenthen reacts with ethylene to form ethylene oxide,leaving behind one oxygen atom after desorption(Eq. 4). This atomic oxygen then causes the com-bustion of ethylene to CO2 and H2O (Eq. 5).
Since six oxygen atoms are needed for thecomplete oxidation of one ethylene molecule, sixethylene oxide molecules must be formed beforeone ethylene molecule can be completely oxi-dized. Therefore, if inhibition of atomic oxygenadsorption is optimal, the maximum selectivity is6/7�100 ¼ 85.7%. This mechanism plausiblyexplains the way in which the inhibitor acts. Fora long time themolecular oxygenmechanismwasstrongly supported by the fact that efforts to im-prove the catalysts and the process seemed to leadto selectivities approaching the limit of 85.7%predicted by the mechanism, but not exceeding it.
A variant of this mechanism is based on theassumption that molecular oxygen can causeboth ethylene oxide formation and total oxidationvia a common intermediate [80], [81]:
The effect of the inhibitor is interpreted differ-ently here. Reaction 2 requires a greater spacethan reaction 1. The inhibitor favors reaction 1 byreducing the available space on the surface of thecatalyst. On an optimally inhibited surface, reac-tion 2 is totally suppressed, so that total oxidationtakes place only via reaction 3. This results againin a maximum selectivity of 85.7%.
Mechanism 2. In this mechanism, atomicoxygen, possibly together with the subsurfaceoxygen, is thought to be responsible for both totaland partial oxidation. The contribution of themolecular oxygen, if any, is indirect [82], [83].The environment of the adsorbed oxygen atomdetermines whether the reaction with ethyleneleads to ethylene oxide or CO2 and H2O [84],[85]. Chlorine (inhibitor), promoters, and sub-surface oxygen in the proximity of the reactingoxygen atom are thought to influence the latter tofavor partial oxidation, e.g., by reducing thenegative charge of the adsorbed oxygen [86].Unlike mechanism 1, the theoretical upper limitof selectivity of 6/7 does not apply formechanism 2.
Since with modern catalysts (containing Cs,W, Re, and S in promoting quantities on thesilver) maximum selectivities of 90% are feasi-ble even on an industrial scale, mechanism 1 canat least not exclusively be valid with these cat-alysts. Also, it has been shown that atomic oxy-gen, formed on the silver surface by decomposi-tion of N2O, can produce ethylene oxide, thoughwith low selectivities [87], [88]. For the epoxi-dation of nobornene and styrene it has beenshown under ultrahigh-vacuum conditions thatthe atomic oxygen species on the silver is respon-sible for the epoxidation [89], [90]. Furthermore,theoretical calculations seem to favor mecha-nism 2 and can explain the action of inhibitorand promotor [91], [92].
Intermediates of the total oxidation arethought to include acetaldehyde, acetic acid,formic acid, and oxalic acid [84]. Desorption ofintermediates from the silver surface producesacetaldehyde when a helium stream is used;ethanol and acetic acid are produced when ahydrogen stream is used [93].
Formate, acetate, and oxalate were detectedon a silver surface by 13C NMR spectroscopyafter contacting the silver first with oxygen andsubsequently with ethylene [94].
Vol. 13 Ethylene Oxide 555
Kinetics. Three reactions are usually con-sidered in kinetic models:
1. Partial oxidation2. Total oxidation3. Consecutive oxidation
The consecutive combustion of ethylene ox-ide (3) is negligible only if temperatures are nottoo high and if the reaction takes place underchemical control. To avoid diffusion control lowsurface area catalyst supports with high porediameters are required. The surface reactionleading to ethylene oxide is considered to be ofthe Langmuir – Hinshelwood type, that is, oxy-gen and ethylene are both adsorbed on the silversurface before reaction. Ethylene is not adsorbedon a clean, oxygen-free silver surface; this onlyoccurs after preadsorption of oxygen. The reac-tion of ethylene with oxygen on the silver surfaceis also thought to be the rate-determining step inthe absence of diffusion control. Kinetic data andmodels are available in the literature [95], [96],but these are usually restricted to very specificreaction conditions and catalysts, far from thecomplex situation of the industrial production.Ref. [97] describes kinetic studies with an indus-trial catalyst in different laboratory reactors toclarify the influence of the reaction products butdoes not take into account the influence of in-hibitors, which is crucial to achieve high selec-tivities on an industrial scale.
4.3. Technology
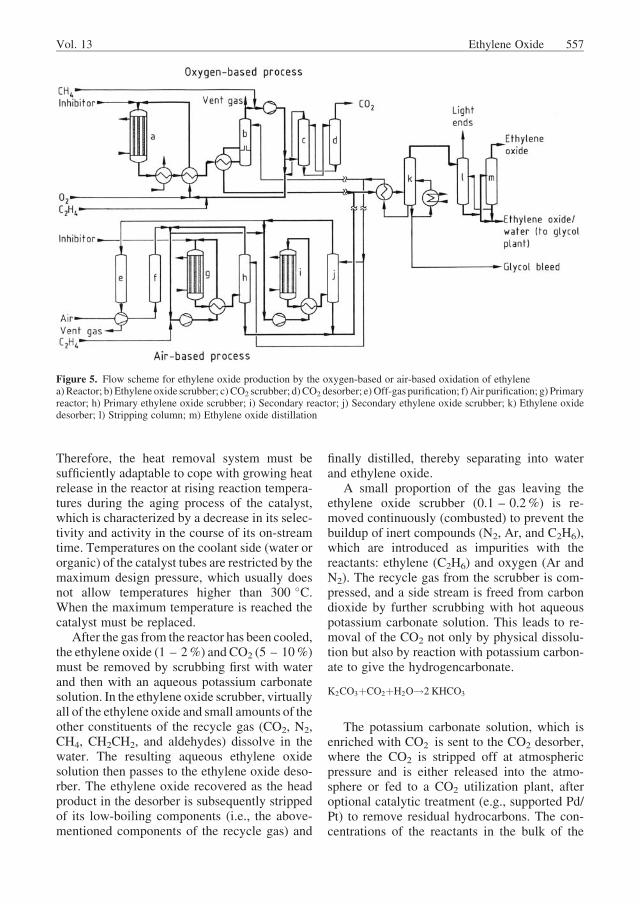
An overview of the beginnings of commercialethylene oxide production can be found in [1].Production technologies for ethylene oxideplants based on the direct oxidation process arelicensed by Shell, Scientific Design (SD), UCC,Japan Catalytic, Snam Progetti, and H€uls. Due toimproved catalysts and production technology,large plants with capacities of up to 400 000 t/acan now be built.
The technologies are very similar, but differ-ences exist, depending on whether air or pureoxygen is used for oxidation [44]. Shell plantsuse only pure oxygen, while Scientific Designand UCC have developed air-based oxidationplants as well. Figure 5 shows a simplifiedscheme for both the air- and oxygen-based pro-cesses. They both employ a recycle gas stream,which is continuously circulated through thereactors by compressors. The reactors consistof large bundles of several thousand tubes thatare 6 – 13 m long and have an internal diameterof 20 – 50 mm. The catalyst is packed in thetubes in the form of spheres or rings with adiameter of 3 – 10 mm; the initial selectivity ofmodern catalysts is ca. 80 – 90%, depending onthe type of catalysts used. The catalysts areeither very active, operating at an initial tem-perature of about 200 �C but with relatively lowselectivities of 80 – 84%, or they are highlyselective but then need an initial temperaturethat is ca. 40 �C higher. Ethylene is converted at200 – 300 �C and 1 – 3 MPa to produce ethyl-ene oxide, CO2, H2O, and heat, as well as tracesof acetaldehyde and formaldehyde; these pro-ducts must be removed or separated from therecycle gas stream. The recycle gas is thenreloaded with oxygen and ethylene and returnedto the reactor.
Oxygen-Based Oxidation Process. Atpresent, ethylene oxide is produced mainly bythe oxygen-based process. The reactor tubesfilled with the catalyst are surrounded by a cool-ant (water or a high-boiling hydrocarbon) thatremoves the reaction heat and permits tempera-ture control. Heat is extracted either by pumpingor by evaporating the coolant. If organic heattransfer media are used, the extracted energy isused to generate steam in secondary cycles,which is then employed for heating. The reactionheat also heats the recycle gas during its passagethrough the reactor. After leaving the reactor, thegas is likewise cooled by means of steam gener-ation and/or used directly to heat the reactor inletgases. Because of the different enthalpies of thepartial and total oxidation reactions (see Section4.1), the total available heat depends on theselectivity for the former (S) and amounts to47 250 � (434�S) kJ/kg ethylene.
Consequently, the quantity of energy releasedincreases rapidly with decreasing selectivity.
556 Ethylene Oxide Vol. 13
Therefore, the heat removal system must besufficiently adaptable to cope with growing heatrelease in the reactor at rising reaction tempera-tures during the aging process of the catalyst,which is characterized by a decrease in its selec-tivity and activity in the course of its on-streamtime. Temperatures on the coolant side (water ororganic) of the catalyst tubes are restricted by themaximum design pressure, which usually doesnot allow temperatures higher than 300 �C.When the maximum temperature is reached thecatalyst must be replaced.
After the gas from the reactor has been cooled,the ethylene oxide (1 – 2%) and CO2 (5 – 10%)must be removed by scrubbing first with waterand then with an aqueous potassium carbonatesolution. In the ethylene oxide scrubber, virtuallyall of the ethylene oxide and small amounts of theother constituents of the recycle gas (CO2, N2,CH4, CH2CH2, and aldehydes) dissolve in thewater. The resulting aqueous ethylene oxidesolution then passes to the ethylene oxide deso-rber. The ethylene oxide recovered as the headproduct in the desorber is subsequently strippedof its low-boiling components (i.e., the above-mentioned components of the recycle gas) and
finally distilled, thereby separating into waterand ethylene oxide.
A small proportion of the gas leaving theethylene oxide scrubber (0.1 – 0.2%) is re-moved continuously (combusted) to prevent thebuildup of inert compounds (N2, Ar, and C2H6),which are introduced as impurities with thereactants: ethylene (C2H6) and oxygen (Ar andN2). The recycle gas from the scrubber is com-pressed, and a side stream is freed from carbondioxide by further scrubbing with hot aqueouspotassium carbonate solution. This leads to re-moval of the CO2 not only by physical dissolu-tion but also by reaction with potassium carbon-ate to give the hydrogencarbonate.
K2CO3þCO2þH2O!2 KHCO3
The potassium carbonate solution, which isenriched with CO2 is sent to the CO2 desorber,where the CO2 is stripped off at atmosphericpressure and is either released into the atmo-sphere or fed to a CO2 utilization plant, afteroptional catalytic treatment (e.g., supported Pd/Pt) to remove residual hydrocarbons. The con-centrations of the reactants in the bulk of the
Figure 5. Flow scheme for ethylene oxide production by the oxygen-based or air-based oxidation of ethylenea) Reactor; b) Ethylene oxide scrubber; c) CO2 scrubber; d)CO2 desorber; e) Off-gas purification; f) Air purification; g) Primaryreactor; h) Primary ethylene oxide scrubber; i) Secondary reactor; j) Secondary ethylene oxide scrubber; k) Ethylene oxidedesorber; l) Stripping column; m) Ethylene oxide distillation
Vol. 13 Ethylene Oxide 557
recycle gas, which is free from ethylene oxideand has a reduced CO2 content, are restored totheir starting levels by separate addition of oxy-gen, ethylene, inhibitor (1,2-dichloroethane, eth-yl chloride or vinyl chloride), and if necessary, adiluent (CH4). The gas is then returned to thereactor.
The oxygen that is used must be extremelypure (> 99%) and is obtained by air separation.Nevertheless, a purge streamof recycle gas is stillnecessary due to the presence of traces of N2 andAr. Oxygen is added in a special mixing devicethat ensures rapid homogenization with the recy-cle gas. This is necessary because the explosivelimit is locally exceeded at the mixing point.
The ethylene is also usually very pure(> 99.5%) and must be free from the strongcatalyst poisons sulfur and acetylene. Methane,used as a diluent, also must be free from sulfurcompounds.
Natural gas is used as methane source andusually requires cleaning steps to attain the re-quired purity. Sulfur compounds, which poisonthe catalyst irreversibly, are removed by adsorp-tion beds. Higher hydrocarbons, which affect thereactor performance, mainly by removing thechlorine inhibitor from the silver surface, areremoved by distillation or with molecular sieves.
Air-Based Oxidation Process. The air-based process is similar to the oxygen process,but some differences exist. Air introduces a largeamount of nitrogen into the recycle gas, whichmeans that a large amount of purge gas must bevented to maintain a constant nitrogen concen-tration in the recycle stream. The quantity of gasthat is vented removes sufficient CO2 to makeCO2 scrubbing unnecessary. However, the off-gas leaving the primary reactor still contains somuch ethylene that itmust be further converted ina subsequent secondary or purge reactor before itcan be vented into the atmosphere.
The reaction conditions cannot be tailored tothe needs of ethylene oxide formation as opti-mally as in the oxygen-based process. The con-version of ethylene is higher than in the oxygen-based process, especially in the secondary reac-tors, so as to obtain an acceptable level of ethyl-ene loss in the purge gas. Since selectivity isinversely related to ethylene conversion, it fol-lows that the air-based process has a lowerselectivity.
The conditions and gas compositions used inthe oxygen- and air-based processes are listed inTable 5.
Reactor. Since oxidation of ethylene ishighly exothermic, reaction in a fluidized bedwould appear to be appropriate. However, at-tempts to develop such a process on a commer-cial basis have not produced any advantages forselectivity [98], [99] and have led to problemsdue to abrasion and sintering. As a result, allethylene oxide plants currently employ fixed-bedtubular reactors. The diameter of the tubes mustnot be excessive so as to ensure sufficient heattransfer to the heat transfer medium. A tubediameter of 20 – 40 mm is common. The diam-eter of the catalyst particles has an upper limitdetermined by the need for satisfactory gas mix-ing and a lower limit resulting from an increasingdrop in pressure.A diameter of 3 – 8 mm is used.
Cooling. Each of the two possible heat ex-traction systems – circulation or evaporation ofthe coolant – has its advantages and disadvan-tages. Coolant circulation requires a largeamount of liquid (pumping power), but allowsa defined temperature increase in the direction ofgas flow. Ethylene and oxygen concentrations inthe cycle gas decrease on passage through thereactor; this leads to lower conversion, which canbe compensated by controlled temperature in-crease toward the reactor outlet.
In evaporative cooling, an organic coolant(e.g., isododecane) or water is used. The reactiontemperature is controlled very effectively by a
Table 5.Operating parameters used in the air-based and oxygen-based
production of ethylene oxide
Parameter
Air-based
process
Oxygen-based
process
C2H4 concentration, vol% 2 – 10 15 – 40
O2 concentration, vol% 4 – 8 5 – 9
CO2 concentration, vol% 5 – 10 5 – 15
C2H6 concentration, vol% 0 – 1 0 – 2
Ar concentration, vol% 5 – 15
CH4 concentration, vol% 1 – 60
Temperature, �C 220 – 277 220 – 275
Pressure, MPa 1 – 3 1 – 2.2
GHSV*, h�1 2000 – 4500 2000 – 4000
Pressure drop, kPa, 40 – 200
C2H4 conversion, % 20 – 65 7 – 15
Selectivity, % 80 80 – 90
*GHSV ¼ gas hourly space velocity.
558 Ethylene Oxide Vol. 13
control valve that keeps the pressure of theboiling coolant at the appropriate pressure. Thewhole temperature range from 190 to 290 �C,required by the aging catalyst, can be covered byevaporative cooling. Organic coolants with aboiling point of about 190 �C at atmosphericpressure are used. The hydrostatic height of thecoolant in the reactor results in an unavoidabletemperature increase towards the bottom of thereactor. Since evaporation extracts heat moreefficiently than circulation, evaporative coolingrequires a smaller volume of circulating coolant.The axial temperature differences can be mini-mized more easily. Large amounts of liquidcoolant leave the reactor with the coolant vapor.The liquid is recovered in a separator and re-turned to the reactor. The coolant present in thereactor and in the separator constitutes a safetyreserve during uncontrolled increases in ethyleneconversion in the reactor (‘‘runaway’’).
The flammability of organic coolants is a cleardisadvantage compared to water, especially atthe necessary high temperatures. However, waterresults in much higher pressures on the coolantside and hence higher apparatus costs.
When organic coolants are used, the coolantvapors are condensed in heat exchangers produc-ing steam, after which the liquefied coolant isreturned to the reactor. The steam serves to heatdistillation columns in the plant. If water is usedas coolant the steam leaving the reactors can beused directly. The higher the selectivity of thecatalyst, the more steam must be imported intothe plant.
Improved catalysts with high activities andselectivities operate at a lower temperature (ca.220 �C) and allow smaller reactor volumes. Thishas encouraged the use of water-cooled reactors;however, organic heat transfer media (e.g., Dow-therm) are still widely employed. A detaileddiscussion of heat transfer with organic heattransfer media in terms of optimum selectivityand safety can be found in [100].
Ethylene Conversion. The selectivity ofethylene oxide formation depends on ethyleneconversion. Selectivity decreases more or lesslinearly with increasing ethylene conversion[101]. Therefore, the highest selectivities areachieved with minimum conversions, but theresulting ethylene oxide concentrations are thentoo low for commercial purposes. Thus, ethylene
conversion is chosen to achieve ethylene oxideconcentrations of 1 – 3 vol% at the reactoroutlet.
Byproducts. Besides large quantities ofCO2 produced by total oxidation of ethylene,ethylene glycols and small amounts of acetalde-hyde and formaldehyde are formed and must beprocessed in ethylene oxide plants. The acetal-dehyde stems from isomerization of ethyleneoxide, catalyzed by the catalyst support and byrust on apparatus walls. Formaldehyde is pro-duced by oxidation of ethylene oxide with andwithout participation of the wall surface [102].Ethylene glycols are unavoidably producedwhenethylene oxide is scrubbed from the recycle gaswith water and is subsequently stripped from theaqueous solution by heating. Monoethylene gly-col [107-21-1], diethylene glycol [111-46-6],triethylene glycol [112-27-6], and higher poly(ethylene glycols) are formed when ethyleneoxide comes into contact with water [103] (!Ethylene Glycol). Some of the cycle water mustbe bled off continuously and the ethylene glycolremoved to prevent it from accumulating (seeFig. 5). This glycol is of inferior quality to thatsynthesized by ethylene oxide hydrolysis. Spe-cial methods for purifying the resulting glycolshave been described, e.g., treatment with ionexchangers and activated charcoal [104].
Materials. Since ethylene oxide is noncor-rosive, the reactors and the sections of the plantthat convey ethylene oxide are usually made ofmild steel. To reduce the formation of acetalde-hyde by isomerization of ethylene oxide on rust,stainless steel is increasingly being used for thereactors and the ensuing cycle gas lines, wheretemperatures are highest. The sealing materialsfor valves, flanges, and pumps must be chosencarefully [105].Manymaterials used in chemicalindustry for O-rings, packings, and gaskets arenot resistant to the influence of ethylene oxideand have been the cause of severe incidents[106]. Thematerials in the systemused to removethe carbon dioxide must also be carefully select-ed because of possible CO2 corrosion [107].
If rust is present in steel tubes or containers,polymer formation [38], increased viscosity, andbrown discoloration are to be expected.
Current plant design is predominantly tailoredto the oxygen-based process because this is nor-
Vol. 13 Ethylene Oxide 559
mally more economical than the air-based pro-cess. In exceptional cases, however, the air-basedprocess may be preferred depending on localfactors (e.g., the availability of oxygen) [108].
Possible Developments. Alternative routesfrom ethylene to ethylene oxide are being stud-ied, e.g., Tl(III)-catalyzed oxidation in solution[108], electrochemical oxidation [109], and en-zymatic oxidation [110]. However, these pro-cesses appear to be far from industrial applica-tion. About half of the ethylene oxide produced isconverted into ethylene glycol. Alternative syn-theses for ethylene glycol based on carbon mon-oxide, formaldehyde, and ethylene are beingdeveloped [111–114] (! Ethylene Glycol);these are more promising than those for ethyleneoxide. With increasing raw material prices, theappearance of an economical alternative is fore-seeable. Naturally, such an alternative wouldhave an adverse effect on ethylene oxide capaci-ties, but the economics of ethylene oxide produc-tion could be improved by
1. Selective production of monoethylene glycolfrom ethylene oxide, without the formation ofhigher ethylene glycol homologues, e.g., viaglycol carbonate [44]
2. Reducing energy consumption3. Improving the selectivity, capacity, life-span,
and activity of the catalysts
5. Environmental Protection andEcology
Ethylene oxide plants using the direct oxidationprocess represent a clear environmental im-provement over the chlorohydrin process. Envi-ronmental and other important aspects of ethyl-ene oxide production are discussed in EPA stud-ies [115]. Both oxygen- and air-based plants emittwo main off-gas streams: recycle vent gas andCO2. In the oxygen-based process, ca. 70%of therecycle vent gas consists of hydrocarbons (eth-ylene andmethane), which can be combusted (bytorch or in a steam generator). The correspondingoff-gas in the air-based process is produced inconsiderably greater quantities, but contains onlyup to ca. 2.3%hydrocarbons. It can be purified bycatalytic oxidation. The CO2-rich vent gas in theoxygen-based process contains ca. 0.2% hydro-
carbons. This concentration can be reduced sub-stantially by depressurizing the CO2-rich absor-bent in two steps. The gas from the first step isrich in hydrocarbons and is recycled, only the gasfrom the second step is vented [116]. A catalytictreatment (e.g., supported Pd/Pt) may be requiredby environmental regulations to remove hydro-carbons before the CO2 is released to the atmo-sphere but may also be necessary to ensure thepurity of the CO2 for further use. The CO2-richvent gas stream from air-based plants is producedin much smaller quantities and contains ca. 5%hydrocarbons that can be removed by combus-tion. Ethylene oxide can be removed from wastegases, such as are generated during sterilization,by combustion with oxygen [117] or by an acidscrub [118], [119], [121]. Biodegradation of theethylene glycol generated during ethylene oxidehydrolysis presents no difficulties. If ethyleneoxide occurs as a mixture with fluorohydrocar-bons, recovery by drying and subsequent com-pression is recommended [120]. Methods forhandling ethylene oxide reaction vessels withoutthe release of vent gas have been described [122].
Ethylene oxide is toxic tomicroorganisms andfish [123]. The LC50 value for fish (Pimephalespromelas) is 84 mg/L (exposure time 96 h).However, in free-flowing waters the ethyleneoxide concentration decreases continually (after4 h at 25 �C in moving water, the concentrationdrops by ca. 95%) due to a combination ofevaporation, hydrolysis (half-life at 25 �C ca.14 d), and biodegradation. The ethylene glycolproduced as a result of ethylene oxide hydrolysisis considerably less toxic to aquatic organisms(LC50 > 10 000 mg/L) and is readily biode-gradable. Effluents (e.g., from ethylene oxideproduction plants [115]) containing ethylene ox-ide are therefore treated biologically after theethylene oxide has been converted into ethyleneglycol.
Degradation of ethylene oxide in air is rela-tively slow and thought to proceed mainly by aslow reaction with hydroxyl radicals. Disregard-ing other removal mechanisms, this results in alifetime of 330 d. The reaction with ozone is tooslow to represent a significant removal pathway[124]. Another report indicates a lifetime of100 – 200 d [125]
According to TA Luft [126], vent gas streamsmust not contain more than 25 g/h or 5 mg/m3
ethylene oxide.
560 Ethylene Oxide Vol. 13
6. Quality Specifications
Ethylene oxide is a major industrial product thatis obtained with a consistently high purity irre-spective of the production process used. Typicalspecifications are given in Table 6.
7. Analysis
An overview of analytical methods for ethyleneoxide can be found in [127]. An establishedmethod for determining ethylene oxide is basedon its reaction with MgCl2 [128]. In view of thelow TRK value (5 mg/m3), accurate methods areneeded to determine traces of ethylene oxide inair.
An adsorption chromatography method isreported to be suitable for concentrations ex-ceeding 0.15 ppm [129]. The NIOSH method1607 [134] with a working range of 0.04 –1.7 ppm (0.09 – 3 mg/m3) is based on adsorp-tion with charcoal, derivatization with HBr togive 2-bromoethanol, and gas chromatography.The CS2 used in the NIOSH method can bereplaced by N,N-dimethylacetamide [130].Methods using adsorption on charcoal are sus-ceptible to the influence of temperature andhumidity [131], [132].
A field comparison of four methods of moni-toring ethylene oxide by portable devices – twopassive monitors, one sorbent tube (OSHA),and one GC method (NIOSH) – showed theirviability for measuring the workplace concen-tration for short-term and full-shift exposure[133].
Exposition to ethylene oxide can be moni-tored by the detection of (2-hydroxyethyl) valinein hemoglobin, the method resulting in accuratedata of time-integrated exposure [39].
8. Handling, Storage, andTransportation
Ethylene oxide has repeatedly caused seriousexplosions, fires, and accidents [106], [135],[136]. It is an extremely hazardous substancebecause it can explode and is both highly flam-mable and extremely reactive (exothermic reac-tions). Aqueous solutions containing > 4 wt%ethylene oxide are flammable; flash points aregiven in Table 2. Furthermore, ethylene oxide istoxic and poses a danger both to health and to theenvironment. The inherent hazards of the productmust be known; all prescribed safety measuresand legal requirements must be observed. Per-sonnel handling ethylene oxide must receiveappropriate training.
Safety precautions for handling ethylene ox-ide and its dangerous properties are described indetail in the data sheets provided by the manu-facturers (e.g., [106], [137]) and elsewhere (e.g.,[105], [138–141]). Therefore, only the most im-portant points are discussed here.
Explosion and Fire Control. Pure ethyleneoxide vapor or ethylene oxide vapor mixed withair or inert gases can decompose explosively.Explosiveness depends on pressure, temperature,concentration, the type, form, and energy of theignition source, and the type of container. Highpressurecanbegeneratedonexplosionof ethyleneoxide; therefore, for safe handling the exact ex-plosive limitsmust be known. Pure ethylene oxidevapor at 101.3 kPa decomposes when passedthrough a heated platinum coil [142]. The temper-ature at which decomposition starts (decomposi-tion temperature) was calculated to be 571 �C.Calculations were based on the temperature at thetube exit and the thermodynamic data for ethyleneoxideand its decompositionproducts.The thermaldecomposition of ethylene oxide and ethyleneoxide – nitrogen mixtures has been investigated[143]. If ethylene oxide vapor is introduced into apreheated vessel at 101.3 kPa, explosive decom-position takes place at ca. 500 �C [143]. Thedecomposition temperature is reduced by an in-crease in the pressure (e.g., ca. 450 �C at 1 MPa)but is increased by the addition of nitrogen. Ex-plosive thermal decomposition is still possiblebelow atmospheric pressure, but the necessarytemperature then increases above 500 �C.
Table 6. Typical specifications of ethylene oxide
Appearance clear, colorless
bp at 101.3 kPa 10.8 �CWater content* 50 mg/kg
CO2 content* 10 mg/kg
Aldehyde content* 50 mg/kg
Ethylene oxide content (min.)* 99.5%
*As determined by gas chromatography.
Vol. 13 Ethylene Oxide 561
The ignition properties of ethylene oxide andits mixtures with air and inert gases have beenstudied by several authors. Pure ethylene oxidevapor can ignite even at a pressure as low as48 kPa (electric spark) or 20.2 kPa (mercuryfulminate) [144]. The maximum theoretical ex-plosive pressure is ca. 10 times the inital pres-sure, but can increase to 20 times the initialpressure if liquid ethylene oxide is present. Thisphenomenon occurs because liquid ethylene ox-ide evaporates and participates in the decompo-sition reactions that take place in the vapor phase[145]. Mixtures of ethylene oxide with N2, CO2,methane, and air do not ignite over certain con-centration ranges; the upper explosive limit isalways 100% ethylene oxide, since pure ethyl-ene oxide also decomposes if ignited. In thepresence of oxygen (air), combustion and de-composition take place simultaneously and theignition temperature (or energy) is reduced. Theignition temperature of ethylene oxide in air at101.3 kPa is 429 �C [146]. As the ethylene oxideconcentration increases, the proportion de-stroyed by decomposition also increases [21],[147]. The minimum value cited for the lowerexplosive limit of ethylene oxide – air mixturesis 2.6% [148]; the explosive range of ethyleneoxide – air mixtures is accordingly 2.6 – 100%.Figures for an upper explosive limit of ca. 80%at101.3 kPa are probably due to differences in theapparatus and methods used [149–151].
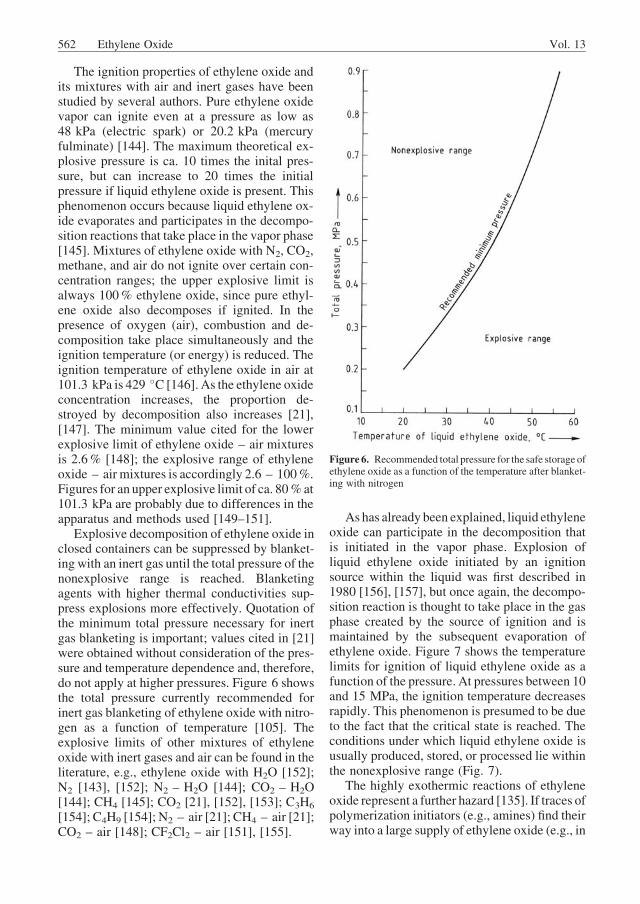
Explosive decomposition of ethylene oxide inclosed containers can be suppressed by blanket-ing with an inert gas until the total pressure of thenonexplosive range is reached. Blanketingagents with higher thermal conductivities sup-press explosions more effectively. Quotation ofthe minimum total pressure necessary for inertgas blanketing is important; values cited in [21]were obtained without consideration of the pres-sure and temperature dependence and, therefore,do not apply at higher pressures. Figure 6 showsthe total pressure currently recommended forinert gas blanketing of ethylene oxide with nitro-gen as a function of temperature [105]. Theexplosive limits of other mixtures of ethyleneoxide with inert gases and air can be found in theliterature, e.g., ethylene oxide with H2O [152];N2 [143], [152]; N2 – H2O [144]; CO2 – H2O[144]; CH4 [145]; CO2 [21], [152], [153]; C3H6
[154]; C4H9 [154]; N2 – air [21]; CH4 – air [21];CO2 – air [148]; CF2Cl2 – air [151], [155].
As has already been explained, liquid ethyleneoxide can participate in the decomposition thatis initiated in the vapor phase. Explosion ofliquid ethylene oxide initiated by an ignitionsource within the liquid was first described in1980 [156], [157], but once again, the decompo-sition reaction is thought to take place in the gasphase created by the source of ignition and ismaintained by the subsequent evaporation ofethylene oxide. Figure 7 shows the temperaturelimits for ignition of liquid ethylene oxide as afunction of the pressure. At pressures between 10and 15 MPa, the ignition temperature decreasesrapidly. This phenomenon is presumed to be dueto the fact that the critical state is reached. Theconditions under which liquid ethylene oxide isusually produced, stored, or processed lie withinthe nonexplosive range (Fig. 7).
The highly exothermic reactions of ethyleneoxide represent a further hazard [135]. If traces ofpolymerization initiators (e.g., amines) find theirway into a large supply of ethylene oxide (e.g., in
Figure 6. Recommended total pressure for the safe storage ofethylene oxide as a function of the temperature after blanket-ing with nitrogen
562 Ethylene Oxide Vol. 13
a tank), polymerization starts slowly and thenaccelerates because of the resulting temperatureincrease. Polymerization proceeds quasi-adia-batically, leading to sudden and rapid increasesin temperature and pressure that can rupture thecontainer [158], [159],whichmay be followed byexplosive decomposition of the released ethyleneoxide vapor (see Fig. 8).
Although rust is only a mild catalyst forethylene oxide polymerization, its presence inheat exchangers or other hot equipment can leadto ignition of the ethylene oxide vapor by acombination of polymerization and dispropor-tionation to C2H4, CO2, H2, and H2O (explosionin the UCC plant, Seadrift, 1991). g-Fe2O3 and g-FeOOH are the active species in this reaction[160].
Insulation fires can occur if ethylene oxideleaks into insulating material such as asbestos,magnesium silicate, calcium silicate, andmineral
wool by a combination of effects and reactions.Like other organic compounds, ethylene oxidecan have considerably lower autoignition tem-peratures in insulation material than otherwise[161], [162]. Isomerization of ethylene oxide toacetaldehyde in the presence of rust results in anadditional decrease in the autoignition tempera-ture [141]. In the presence of water, ethyleneoxide may be hydrolyzed to ethylene glycol andsubsequently polymerize to poly(ethylene gly-cols) with evolution of heat. Eventually, theorganic material collected in the insulation (eth-ylene oxide, acetaldehyde, glycols) may be oxi-dized by air. The resulting hot spots on thesurface of the apparatus can trigger an explosivedecomposition of ethylene oxide vapor in thecolumn (explosion in the BP plant, Antwerp,1987 [163] and in the BASF plant, Antwerp,1989 [164]). To avoid these risks, special insu-lating material with a low specific surface andclosed cells (glass foam) is used, and specialmethods for insulation have been developed. Agap between the outer wall of the apparatus andthe insulation prevents accumulation of organicmaterial in the insulation and allows monitoringof organic compounds in the gap.
The accidents in ethylene oxide plants alsoinitiated a fundamental reevaluation of the dis-tillation section. The use of structured packinginstead of trays can prevent the propagation ofethylene oxide decomposition. A beginning de-composition in the column can be recognized intime to be quenched with water [165], [166].
Because of the above-mentioned hazards, thefollowing potentially dangerous situations mustbe prevented:
1. Leakage of liquid or gaseous ethylene oxide2. Entry of air, oxygen, or reactive impurities
into ethylene oxide containers3. Ignition sources in danger areas4. Overheating of ethylene oxide (chemical re-
action, fire, etc.)
Leaks or spills should be promptly diluted withsufficiently large volumes of water to reduce theethylene oxide concentration to less than 4 wt%.Mixtures of ethylene oxide and water will stillburn if the ethylene oxide concentration exceeds4 wt%. Fires should also be extinguished withlarge volumes of water, carbon dioxide extin-guishers can be used for small fires.
Figure 7. Explosive limits for pure liquid ethylene oxide as afunction of pressure and temperature
Figure 8. Runaway reactions of ethylene oxide (EO) [169]a) Dewar polymerizaation of EO (10 ml)/NaOH (0.02 g)/H2O [167]; b) Calculated adiabatic reaction of EO (40%) inH2O (initial temperature 27 �C) [168]; c) Polymerization ofEO/NMe3 (0.184%)
Vol. 13 Ethylene Oxide 563
Storage and Transportation. Ethylene ox-ide is transported in tank cars and containers andcan be conveyed by rail or by sea and, in manycountries, also by road. In Germany, conveyanceby road is possible with special permission andunder certain conditions (GGVS/ARD x 7).Hazard classifications for ethylene oxide follow:
Water hazard class WGK 2 (no. 235)
CFR 49 no. 172.101, flammable liquid
GGVS/GGVE/RID/ADR class 2, no. 4 ct
ADNR class 2, no. 8 AF
EMS 2–06
MFAG 365
GGVSee, IMDG Code class 2
UN no. 1040
JATA no. 722
Temperature class (DIN 57 165) 2
Explosion limit (DIN 57 165) IIB
Storage and transport containers for ethyleneoxide are usually made from steel, but steel issuitable only if special measures have been takento prevent rust formation [38]. Rust acts as amildpolymerization catalyst and is dispersed in theethylene oxide by polymerization starting onavailable surfaces. Polymer concentrations aslow as ca. 110 mg/kg produce an increase inviscosity, may cause brown discoloration, mayblock filters, valves etc., and form deposits on thecontainer walls. Therefore, steel containersshould be blasted before first use, the blastingresidues carefully removed, and the containerflushed with nitrogen. Stainless steel is increas-ingly being used to avoid these problems. Whenaqueous solutions of ethylene oxide are handled,precipitation of solid hydrates (cf. Table 2) maycause blockages.
In Europe, tank cars are tested at a pressure of1.6 MPa and they must be protected from the sunby a protective roof or insulation (GGVE). Eth-ylene oxide – nitrogen mixtures can be trans-ported at a maximum pressure of 1 MPa at 50 �C(GGVS/RID). The proportion of nitrogen in thevapor phase must be high enough to ensure thatexplosion cannot occur at or below this tempera-ture. In the United States railroad tanks are insu-lated and equipped with pressure-release valves.
Safetymeasures that should be observedwhenreactions with ethylene oxide are performed orwhen it is used for sterilization can be found in[105].
Personal Protective Equipment. To avoidskin contact with ethylene oxide, goggles as wellas protective clothing (gloves, boots, suits,aprons) have to be used. As ethylene oxidepermeates easily through most materials, thechoice of an adequate material is crucial. Butylrubber is normally a suitable material, but asquality and thickness play an important role, atest of the permeation resistance against ethyleneoxide is recommended before use.
Leather clothing and footwear represent anoften neglected risk when contacted with ethyl-ene oxide or its aqueous solution, because thesevere and slowly healing damage to the skinappears only after a induction period of manyhours. Therefore leather articles must be dis-carded at once when contaminated with ethyleneoxide. Other clothing contaminated with ethyl-ene oxide must be taken off immediately anddiscarded or decontaminated. Immediate flush-ing with copious water can avoid detrimentalaction on eyes and skin.
If ethylene oxide concentrations of 0.25 ppmin the air are reached, respiratory equipment mustbe used [170]. Gas masks are permitted only up tocertain exposure limits, and their duration of use isrestricted according to the ethylene oxide concen-tration [171]. Beyond a certain concentrationlevel, equipment has to be used which is indepen-dent of the ambient atmosphere (e.g., positive-pressure self-contained breathing apparatus)[172]. Also above ethylene oxide concentrationsof 0.25 ppm restrictions in the employment ofjuveniles and pregnantwomenare effective [170].
9. Uses
Ethylene oxide is an excellent disinfectant, ster-ilizing agent, and fumigant when it is used as anonexplosive mixture with N2, CO2, or dichlor-ofluoromethane. The gas penetrates into poresand through packaging or clothing. It can be used,for example, to sterilize surgical instruments inhospitals or to remove pests and microorganismsfrom spices, furs, etc.
However, most ethylene oxide is convertedinto other products. Figure 9 shows the percen-tages of ethylene oxide used in these derivativesin the United States. Although this distributionwas determined in 1978, it still applies today andis typical of the world market.
564 Ethylene Oxide Vol. 13
Products derived from ethylene oxide havemany different uses, only the most importantones are listed here:
Monoethylene Glycol[107-21-1] [173]: An-tifreeze for engines, production of poly(ethyleneterephthalate) (polyester fibers, foils, and bot-tles), and heat transfer liquids.
Diethylene Glycol[111-46-6] [174]: Polyur-ethanes, polyesters, softeners (cork, glue, casein,and paper), plasticizers, gas drying, solvents, anddeicing of aircraft and runways.
Triethylene Glycol[112-27-6] [175]: Lac-quers, solvents, plasticizers, gas drying, and hu-mectants (moisture-retaining agents).
Poly(ethylene Glycols) [176]: Cosmetics,ointments, pharmaceutical preparations, lubri-cants (finishing of textiles, ceramics), solvents(paints and drugs), and plasticizers (adhesivesand priinks).
Ethylene Glycol Ethers [177]: Brake fluids,detergents, solvents (paints and lacquers), andextractants for SO2, H2S, CO2, and mercaptansfrom natural gas and refinery gas.
Ethoxylation Products of fatty alcohols, fat-ty amines, alkyl phenols, cellulose, poly(propyl-ene glycol) [28]: Detergents and surfactants(nonionic), biodegradable detergents, emulsi-fiers, and dispersants.
Ethylene Carbonate[96-49-1]: Solvents.
10. Economic Aspects
In 1980, ca. 16% of world ethylene productionwas used to synthesize ethylene oxide; this usewas second only to polyethylene production(44%). World production of ethylene oxide in2000 was ca. 15 � 106 t/a. Ethylene oxide is animportant raw material for major consumergoods in virtually all industrialized countries.Table 7 gives an overview of worldwide ethyl-ene oxide capacities. Excess capacities werealready apparent in 1984 [178]
11. Toxicology and OccupationalHealth
The toxicological properties of ethylene oxideare mainly determined by its reactivity withnucleophilic groups such as carboxyl, amino,phenolic hydroxide, or sulfhydryl groups.
In humans, acute inhalative poisoning causesheadache, nausea, and vomiting within a fewminutes [179–181]. Local irritation results indyspnea [182]. Myocardial damage [183], exci-tation, numbness, and finally coma [179] follow.After dermal exposure, blisters are formed on theskin, and symptoms similar to those found afterinhalation appear due to skin absorption [184].Sensitization has been reported after repeateddermal contact [185]. Repeated inhalation ofethylene oxide leads to sensory-motor poly-neuropathy and impaired memory [186], [187].Cataracts may be formed after chronic exposure[188].
Oral Toxicity. The acute oral toxicity(LD50) of ethylene oxide in the rat is 330 mg/kg [189].
Inhalation. The4-hLC50value is 835 mL/m3
in the mouse and 1460 mL/m3 in the rat [190].Acute symptoms largely resemble those observed
Figure 9. Uses of ethylene oxide
Vol. 13 Ethylene Oxide 565
Table 7. World ethylene oxide capacities in 2000
Country Company Location Licensor* Capacity, 103 t/a
Australia ICI Botany SD 40
Canada 775
Dow Saskatchewan Dow 285
UCC Prentiss I UCC 220
Prentiss II UCC 270
W. Europe 2615
Belgium BASF Antwerp Shell, s 350
Ineos Antwerp Shell, s 350
Germany BASF Ludwigshafen Shell, s 215
Erd€olchemie Dormagen Shell, s 200
Clariant Gendorf Shell, s 200
RWE/DEA Marl H€uls, Shell, s 150
France BP Lavera Shell, s 200
Great Britain UCC Wilton Shell, s 300
Italy Enichem Priolo SD 30
Gela 40
Netherlands Dow Terneuzen Dow, s 150
Shell Moerdjik Shell, s 250
Sweden Akzo Nobel Stenungsund Nippon Sh./SD, s 80
Spain La Seda Tarragona Shell, s 100
E. Europe 950
Bulgaria Neftochim Burgas SD 80
Slovakia Slovnaft Bratislava Shell, s 40
Poland Petrochemia Plock Shell, s 60
Snam 30
Romania Romchim Brazi SD 35
Arpechim Pitesti SD 35
Ukrainia Nephtekhim Dzherzhinsk SD 200
Russia Techmashimp. Khazan Japan Catalytic 100
Nishnekamsk SD 200
Nishnekamsk SD 120
Salavat Neft Org. Salsvat 50
Asia 3845
China CNPC Liaoyang H€uls, 1 60
Fushun 50
Jilian 40
Jilin SD 80
Dushanzi 30
Sinopec Maoming 80
Yanshan 65
Yangzi 240
Jinshan 130
Tianjin 55
Zhejiang Chem. Pet Zhejiang 20
Beijing 50
India Glycols India Kashipur 20
Indian PC Kojali SD 20
Vadodana 5
Nagothane 45
SM Deyechem Ltd. Pune SD/Toyo 20
Reliance Ind. Hazira 265
NOCIL Bombay Shell, s 20
Indonesia O.T. Yasa Ganesha Pura Merak SD, s 185
Japan Nippon Shokubai Kawasaki Japan Catalytic 240
Mitsubishi PC Yokkaichi Shell, s 90
Kashima Shell, s 240
Mitsui PC Chiba Shell, s 120
Nisso Murazen Chiba SD, s 120
Nisso Yuka Yokkaichi Shell, s 85
Semppoku EO Daesan Osaka Shell, s 130
North Korea Puyangtang Japan Catalytic 10
566 Ethylene Oxide Vol. 13
in humans. Subchronic administration of ethyl-ene oxide (100 and 200mL/m3) to cats bymeansof inhalation for a period of 22 days producedanorexia, apathy, atoxia, and paralysis of thehind quarters [191]. Lethalities also occurred,postmortem examination revealed liver andkidney damage accompanied by hyperemia andperivascular bleeding in various organs (e.g.,the brain) [191]. Rodents that inhaled air con-taining ethylene oxide at a concentration of300 – 400 mL/m3 displayed additional local
irritation and severe, primary neurotoxicatrophy of the musculature in their hind quarters[190]. Hemotoxic effects have also beenobserved in rodents [192]. Monkeys exposed toinhalative concentrations of 204 mL/m3 for upto 226 days showed impaired reflexes, reducedsensitivity to pain, and neurotoxic muscularatrophy of their hind quarters [193].
Teratogenicity. In studies on rats exposedto maximum inhalative concentrations of 100
Table 7 (Continued)
Country Company Location Licensor* Capacity, 103 t/a
South Korea Honam PC Yeochon Shell, s 115
Yeochon Shell, s 100
Yeochon Shell, s 100
Hyundai PC Daesan SD, s 90
Daesan 160
Samsung Daesan SD, s 80
Singapur Ethylenegl. Sing. Merbau Shell, s 125
Taiwan Orient. UCC Koahsiung UCC, s 190
CMFC Koahsiung SD 40
Koahsiung SD 60
Nan-Ya Mailiao 240
Latin America 720
Brasil Oxiteno Camacari, Bahia SD, s 130
SD, s 105
Maua, Sao Paulo SD, s 45
Mexico Pemex La Cangrejera SD 110
Pajaritos SD 40
Morelos SD 220
Venezuela Pralca Santa Rita SD, s 70
Middle East 1370
Saudi Arabia Sharq Al Jubail Shell, s 360
Al Jubail Shell, s 360
Yanpet Yanbu SD, s 310
Kuwait Equate Shuiaba 270
Turkey Petkim Petro. Aliaga Shell, s 70
United States 4083
BASF Wyandotte Geismar, LA Shell, s 140
Geismar, LA Shell, s 160
Celanese Clear Lake, TX Shell, s 305
Dow Plaqemine, TX Dow, l 260
Eastman Longview, TX Shell, s 100
Huntsman Port Neches, TX SD, l 455
Sun Brandenburg, KY Shell, s 55
Marcus Hook, PA Shell, s 55
Equistar Bayport, TX Shell, s 340
Beaumont, TX SD 308
Shell Geismar, LA Shell, s 165
Geismar, LA Shell, s 240
Geismar, LA Shell, s 180
UCC Seadrift, TX UCC, l 420
Taft, LA UCC, s 350
Taft, LA UCC, l 310
Nan-Ya Point Comfort, TX SD, s 240
Total 14 396
* l ¼ air process; s ¼ oxygene process; SD ¼ Scientific Design.
Vol. 13 Ethylene Oxide 567
[195] or 150 mL/m3 [195], symptoms of intoler-ance were observed in the dams but no malfor-mations were observed in the fetuses. Ethyleneoxide does not produce teratogenic effects inrabbits [195].
Mutagenicity. Ethylene oxide is mutagenicdue to its alkylating properties. It alkylates thegerm cells of male rats [196] and producesmutations in Drosophila melanogaster [197],Neurospora crassa [198], and Salmonella typhi-murium (Ames test) [199], [200]. Dominant le-thal mutations and chromosome aberrations areinduced in male rats [201]. Increased numbers ofmicronuclei are found in the polynuclear ery-throcytes of rats [202] and mice [203], [204].Chromosomal defects are induced in culturedamniotic cells from humans [205]. Increasedexchange between sister chromatids is found inmonkeys and rats [206], [207]; the same effect isobserved in workers who have been exposed toethylene oxide (cumulative dose 0.5 – 50 g)[208–211].
Carcinogenicity. A large number of reportshave been published on the carcinogenic proper-ties of ethylene oxide in animals. Dilute ethyleneoxide applied to the skin of rats did not inducetumor formation [212]. Subcutaneous injectionof ethylene oxide in rats did not have any sys-temic carcinogenic effects [213], [214]. Howev-er, intragastric administration led to an increasein the incidence of epithelial carcinomas in thegastric mucosa [215]. An increased incidence ofbrain tumors andmononuclear cell leukemia wasfound in rats that had inhaled ethylene oxide atconcentrations of 10, 33, or 100 mL/m3 over aperiod of two years [216]. An increased inci-dence of peritoneal mesotheliomas was also ob-served in the animals exposed to concentrationsof 33 and 100 mL/m3. Results of human epide-miological studies on workers exposed to ethyl-ene oxide differ [217–220]. Ethylene oxide isclassified as a ‘‘putative human chemical carcin-ogen’’ [221].
Permissible Exposure Limits. Ethyleneoxide is classified as a class 2 carcinogen by theGerman MAK commission (TRK ¼ 1 mL/m3
¼ 2 mg/m3) and as a class A2 carcinogen bythe ACGIH (TLV-TWA ¼ 1 mL/m3 ¼ 2 mg/m3)[222].
12. Acknowledgement
With kind support from Clariant GmbH, WerkGendorf
References
General References1 G. O. Curme: Glycols, Reinhold Publ. Co., New York
1952, pp. 74 – 113.
2 S. A. Miller: Ethylene and its Industrial Derivatives,