Evaluation of a setting reaction pathway in the novel composite TiHA–CSD bone cement by FT-Raman and FT-IR spectroscopy Czesława Paluszkiewicz, Joanna Czechowska ⇑ , Anna S ´ lósarczyk, Zofia Paszkiewicz AGH University of Science and Technology, al. A. Mickiewicza 30, 30-059 Krakow, Poland highlights " FT-Raman and FT-IR are feasible techniques for analyzing the pathway of cement setting process. " The close relationship between heat treatment of TiHA, composition and microstructure of final materials was stated. " Calcium sulfate was the main setting phase in cements composed with untreated or calcined at 800 °C TiHA. " a-TCP was the main setting phase in the case of cement consisted of TiHA sintered at 1250 °C. " The ATR–FTIR and SEM indicated on high bioactive potential of the obtained bone cements. article info Article history: Received 5 June 2012 Received in revised form 22 October 2012 Accepted 22 October 2012 Available online 2 November 2012 Keywords: Raman spectroscopy FT-IR Bone cement Titanium modified hydroxyapatite Calcium sulfate abstract The aim of this study was to determine a setting reaction pathway in a novel, surgically handy implant material, based on calcium sulfate hemihydrate (CSH) and titanium doped hydroxyapatite (TiHA). The previous studies confirmed superior biological properties of TiHA in comparison to the undoped hydroxy- apatite (HA) what makes it highly attractive for future medical applications. In this study the three types of titanium modified HA powders: untreated, calcined at 800 °C, sintered at 1250 °C and CSH were used to produce bone cements. The Fourier Transform-InfraRed (FT-IR) spectroscopy and Raman spectroscopy were applied to evaluate processes taking place during the setting of the studied materials. Our results undoubtedly confirmed that the reaction pathways and the phase compositions differed significantly for set cements and were dependent on the initial heat treatment of TiHA powder. Final materials were multiphase composites consisting of calcium sulfate dihydrate, bassanite, tricalcium phosphate, hydroxy- apatite and calcium titanate (perovskite). The FT-IR and Scanning Electron Microscopy (SEM) measure- ments performed after the incubation of the cement samples in the simulated body fluid (SBF), indicate on high bioactive potential of the obtained bone cements. Ó 2012 Elsevier B.V. All rights reserved. 1. Introduction Calcium phosphates (CaPs) occupy an important place in the hierarchy of bone grafting materials. The vast majority of CaP bioceramics is based on a hydroxyapatite (HA–Ca 10 (PO 4 ) 6 (OH) 2 ), tricalcium phosphates (a-TCP, b-TCP) and biphasic calcium phos- phate (BCP). Hydroxyapatite is a well-known synthetic biomate- rial, widely used for orthopedic and dental applications due to its similarity to the inorganic phase of human bone and teeth, high biocompatibility and bioactivity [1]. Biological and physicochemi- cal properties of hydroxyapatite can be improved by the substitu- tion with various ions, such as magnesium, carbonate, zinc, silicon and others. After the incorporation into the HA structure they may cause changes in properties of final materials i.e. crystallinity, sol- ubility and biological response [2,3]. Recently a few researches have been conducted to determinate the influence of doping hydroxyapatite with titanium. Promising results regarding en- hance of bioactive potential of these new titanium modified hydroxyapatite sintered materials have been received [4,5]. The bone cements (BCs) are a remarkable development in the field of bone substitutes, overcoming the disadvantages of prefab- ricated bioceramics, such as a lack of shapeability and difficulties in the delivery of preformed material to the defects. Cement type biomaterials are easy to handle because of their ability to be formed into any complex shape during the surgical procedure. Cal- cium phosphate bone cements (CPCs) are obtained by mixing for- mulations containing calcium phosphate powder ingredients with water or aqueous solutions, to produce self-setting pastes. The first calcium phosphate bone cement (CPC) composed of tetracalcium phosphate (TTCP–Ca 4 (PO 4 ) 2 O) and dicalcium phosphate anhydrous (DCPA–CaHPO 4 ) was introduced in 1986 by Brown and Chow [6]. 0022-2860/$ - see front matter Ó 2012 Elsevier B.V. All rights reserved. http://dx.doi.org/10.1016/j.molstruc.2012.10.044 ⇑ Corresponding author. Tel./fax: +48 12 617 2327. E-mail address: [email protected](J. Czechowska). Journal of Molecular Structure 1034 (2013) 289–295 Contents lists available at SciVerse ScienceDirect Journal of Molecular Structure journal homepage: www.elsevier.com/locate/molstruc
Transcript
Journal of Molecular Structure 1034 (2013) 289–295
Contents lists available at SciVerse ScienceDirect
Evaluation of a setting reaction pathway in the novel composite TiHA–CSDbone cement by FT-Raman and FT-IR spectroscopy
Czesława Paluszkiewicz, Joanna Czechowska ⇑, Anna Slósarczyk, Zofia PaszkiewiczAGH University of Science and Technology, al. A. Mickiewicza 30, 30-059 Krakow, Poland
h i g h l i g h t s
" FT-Raman and FT-IR are feasible techniques for analyzing the pathway of cement setting process." The close relationship between heat treatment of TiHA, composition and microstructure of final materials was stated." Calcium sulfate was the main setting phase in cements composed with untreated or calcined at 800 �C TiHA." a-TCP was the main setting phase in the case of cement consisted of TiHA sintered at 1250 �C." The ATR–FTIR and SEM indicated on high bioactive potential of the obtained bone cements.
a r t i c l e i n f o
Article history:Received 5 June 2012Received in revised form 22 October 2012Accepted 22 October 2012Available online 2 November 2012
The aim of this study was to determine a setting reaction pathway in a novel, surgically handy implantmaterial, based on calcium sulfate hemihydrate (CSH) and titanium doped hydroxyapatite (TiHA). Theprevious studies confirmed superior biological properties of TiHA in comparison to the undoped hydroxy-apatite (HA) what makes it highly attractive for future medical applications. In this study the three typesof titanium modified HA powders: untreated, calcined at 800 �C, sintered at 1250 �C and CSH were usedto produce bone cements. The Fourier Transform-InfraRed (FT-IR) spectroscopy and Raman spectroscopywere applied to evaluate processes taking place during the setting of the studied materials. Our resultsundoubtedly confirmed that the reaction pathways and the phase compositions differed significantlyfor set cements and were dependent on the initial heat treatment of TiHA powder. Final materials weremultiphase composites consisting of calcium sulfate dihydrate, bassanite, tricalcium phosphate, hydroxy-apatite and calcium titanate (perovskite). The FT-IR and Scanning Electron Microscopy (SEM) measure-ments performed after the incubation of the cement samples in the simulated body fluid (SBF),indicate on high bioactive potential of the obtained bone cements.
� 2012 Elsevier B.V. All rights reserved.
1. Introduction
Calcium phosphates (CaPs) occupy an important place in thehierarchy of bone grafting materials. The vast majority of CaPbioceramics is based on a hydroxyapatite (HA–Ca10(PO4)6(OH)2),tricalcium phosphates (a-TCP, b-TCP) and biphasic calcium phos-phate (BCP). Hydroxyapatite is a well-known synthetic biomate-rial, widely used for orthopedic and dental applications due to itssimilarity to the inorganic phase of human bone and teeth, highbiocompatibility and bioactivity [1]. Biological and physicochemi-cal properties of hydroxyapatite can be improved by the substitu-tion with various ions, such as magnesium, carbonate, zinc, siliconand others. After the incorporation into the HA structure they maycause changes in properties of final materials i.e. crystallinity, sol-
ll rights reserved.
.
ubility and biological response [2,3]. Recently a few researcheshave been conducted to determinate the influence of dopinghydroxyapatite with titanium. Promising results regarding en-hance of bioactive potential of these new titanium modifiedhydroxyapatite sintered materials have been received [4,5].
The bone cements (BCs) are a remarkable development in thefield of bone substitutes, overcoming the disadvantages of prefab-ricated bioceramics, such as a lack of shapeability and difficultiesin the delivery of preformed material to the defects. Cement typebiomaterials are easy to handle because of their ability to beformed into any complex shape during the surgical procedure. Cal-cium phosphate bone cements (CPCs) are obtained by mixing for-mulations containing calcium phosphate powder ingredients withwater or aqueous solutions, to produce self-setting pastes. The firstcalcium phosphate bone cement (CPC) composed of tetracalciumphosphate (TTCP–Ca4(PO4)2O) and dicalcium phosphate anhydrous(DCPA–CaHPO4) was introduced in 1986 by Brown and Chow [6].
290 C. Paluszkiewicz et al. / Journal of Molecular Structure 1034 (2013) 289–295
Since that time a great number of bone cement compositions havebeen proposed [7–14].
Another well recognized, self-setting calcium salt is a hemihy-drate form of calcium sulfate (CSH–CaSO4�0.5H2O), known as a Plas-ter of Paris. Because of its biological safety and in vivo resorption thecalcium sulfate possess a long clinical history as a bone implantmaterial [15–18]. Calcium sulfate dihydrate (CSD–CaSO4�2H2O) –the product of the hydratation process of CSH is obtained in theform of a moldable paste with good handling properties, whichin several minutes sets giving a solid material. The specificmicrostructure, consisting of interlocking CSD crystals influenceproperties of final product [19–21]. The main disadvantage ofmaterials based on CSH is too rapid resumption rate. The solutionto this problem seems to be in composite type CPCs–CSH materials.
Thanks to their self-setting ability, excellent biocompatibilityand the capability of being replaced by new bone, calcium phos-phates and calcium sulfate cements possess a huge potential asbiomaterials for filling of bone defects and bone augmentation[13,22,23]. However obtaining synthetic self-setting material withall of the desirable properties such as a high osteoconductivity,appropriate mechanical properties, low price, easy handling, effi-cient drug delivery and optimal resorption rate is not trivial andstill remain a challenge. Furthermore there is a great need for abetter understanding of some processes taking place during settingreaction in complex cement compositions. Very little informationis actually available on the reaction mechanisms in the bone ce-ment systems consisted of three or more components. Accordingto our knowledge only a few studies regarding spectroscopic char-acterization of phase composition changes during the cement set-ting reaction were done. Fourier Transform Raman (FT-RS) andInfraRed (FT-IR) spectroscopies are well established, complemen-tary spectroscopic methods used for quantitative analysis of manykinds of materials [24–27]. Nowadays various conventional appli-cations of FT-RS spectroscopy are available including determina-tion of material composition [28–31]. Diversity of applicationsmakes FT-RS and FT-IR methods of a great interest as a potentiallyuseful for better understanding the physical and chemical pro-cesses taking place during the cement setting.
The aim of this study was to investigate the setting reactionpathway of the novel, composite type bone cements on the basisof titanium doped hydroxyapatite (TiHA) and CSH using FT-IRand FT-Raman spectroscopic methods together with XRD measure-ments. Bioactivity of obtained materials was also evaluatedin vitro.
2. Materials and methods
Titanium doped hydroxyapatite (TiHA) and calcium sulfatehemihydrate (CSH, Acros Organics) were used to produce the ini-tial cement mixtures (powder batches). Hydroxyapatite dopedwith titanium was synthesized by a wet method using chemicalgrade reactants: CaO (POCH, Poland), 85% solution of H3PO4 (POCH,Poland) and 15% solution of TiCl3 in 10% HCl (MERCK, Germany),introducing 2.0 wt.% of Ti in proportion to HA. During the precipi-tation process, pH was kept constant at 11 by addition of ammoniasolution. Three types of Ti doped HA powders were used: initial-untreated (TiHA), calcined at 800 �C (TiHA/c) and sintered at1250 �C (TiHA/s). All of them were ground in the rotary vibratingball mill to the grain size below 0.06 mm and sieved.
Three different powder batches: A, B and C were prepared bymixing the 60 wt.% of CSH with 40 wt.% of either TiHA, TiHA/c orTiHA/s powders respectively. Mixing was performed using mixermill (Retsch MM400, 5 Hz/5 min). In the case of cement A distilledwater and for cements B and C-1.0 wt.% Na2HPO4 solution wereapplied as liquid phases.
The setting times of the varied formulations and commercialbone cement (HydroSet™, Stryker�), used as a reference, weredetermined with Gillmore apparatus according with ASTM C266-04 specification [32]. Briefly, the self–setting cement samples wereprepared by mixing powder batches with appropriate amount of li-quid phases, for 1 min to obtain moldable paste. Afterward, ob-tained pastes were put into the mold and initial (I) and final (F)setting times were determined. All experiments were performedat 23 ± 2 �C.
FT-IR and FT-Raman spectroscopy methods were used for thecharacterization of obtained powders and the hardened cementbodies. FTIR investigations of materials were conducted on a Bio-Rad FTS 6000 spectrometer with the FT-Raman attachment (Nd-YAG laser excitation line of 1064 nm) in the wavenumber rangeof 3800–200 cm�1. The transmission technique was used and thesamples were prepared as standard KBr pellets. To determinatesurface phase composition before and after 28 days of cement sam-ples incubation in SBF, Attenuated Total Reflectance – FourierTransform Spectroscopy (ATR–FTIR) technique was applied. ATR–FTIR spectra were collected using a Bio-Rad Excalibur spectrometerwith a attenuated total reflectance spectroscopy sampling acces-sory (diamond crystal, Pike attachment).
Phase composition of the samples was determined using X-raydiffraction analysis using X’Pert Pro diffractometer (Panalytical)with CuKa1 radiation, Ni-filter and the standard Bragg–Brentanogeometry. The diffraction patterns were collected in the 2h anglerange of 10–90� with formal step of 0.002�. Phase quantificationwas made according to the Rietveld method, which based on thesimulation of the diffraction profile from structures of the phasecomponents in the material and is well known and established inliterature [4,5,8,33,34]. The crystalline phases were identified bymeans of the JCPDS cards: PDF 01-074-9761 for hydroxyapatite,PDF 01-075-0437 for perovskite, PDF 01-081-1849 for bassaniteand PDF 00-006-0047 for calcium sulfate dihydrate.
Microstructure of the cements after setting and hardening wasexamined on fractured samples with Scanning Electron Micros-copy (SEM, Nova NanoSem 200) with the attachment of the EnergyDispersive Spectrometer (EDS), used for determination of chemicalcompositions in microareas. All samples were previously sputteredwith carbon to avoid charging effect.
The bioactive potential of cement B and C was tested in vitro inthe simulated body fluid (SBF). The cement cylindrical samples,8 mm in diameter and 2 mm in thickness, were incubated for4 weeks in the SBF, in sterilized bottles, at 37 ± 0.1 �C. Simulatedbody fluid solution was made according to the recipe by Kokuboand Takadama [35]. After immersion, all samples were taken out,gently washed with distilled water and dried below 40 �C for24 h in an air atmosphere.
3. Results and discussion
The initial heat treatment of the TiHA powders influenced theirbehavior during the preparation of cement pastes. The powdermixtures based on the untreated (TiHA) and calcined titaniumdoped hydroxyapatite (TiHA/c) needed higher liquid to powder ra-tio (0.58 and 0.54 ml/g) than the sintered one (TiHA/s) (0.44 ml/g)to produce a putty-like consistency (Table 1). On the basis of prac-tical experience with clinicians Khairoun et al. [36] formed require-ments regarding setting times of bone cements. It is believed thatinitial setting time (TI) of cement paste cannot be shorter than3 min, whereas for orthopedic applications should be near to8 min. Final setting time (TF) should not be longer than 15 min.Taking into account these requirements distilled water was the li-quid phase of choice in the case of cement A, with the initial andfinal setting times equal 4 and 9 min, respectively. When distilled
Table 1The initial powder and liquid components of the investigated cements.
Cement Solid phase-P Liquid phase-L L/P (ml/g)
A 40 wt.% TiHA Distilled water 0.5460 wt.% CSH
B 40 wt.% TiHA/c 1.0 wt.% Na2HPO4 0.5860 wt.% CSH
C 40 wt.% TiHA/s 1.0 wt.% Na2HPO4 0.4460 wt.% CSH
Table 2Wavenumbers of Raman bands in calcium sulfate dihydrate (CSD) and hemihydrate(CSH).
C. Paluszkiewicz et al. / Journal of Molecular Structure 1034 (2013) 289–295 291
water was used for B and C powder mixtures the final setting timesof obtained pastes (2 min and 34 min, respectively) were not ade-quate for their potential medical applications. Therefore 1.0 wt.%solution of Na2HPO4 was used as a liquid component to producecements B and C. In such a case obtained initial and final settingtimes were improved and were equal 6 and 9 min for cement B,and 7 and 21 min for cement C. For commercial bone cement (Hyd-roSet™, Stryker�) used as the reference material setting timeswere as follows: TI = 8 min and TF = 17 min.
In the calcium sulfate based cements the hydration reaction isbelieved to be crucial for the process of setting. The structuralchanges occurring during the phase transition of calcium sulfatecan be easily noticed on its spectrum (Fig. 1).
In the case of calcium sulfate dihydrate a downward shift of m1
(SO4) Raman symmetric stretching mode to 1007 cm�1 in compar-ison to calcium sulfate hemihydrate – 1015 cm�1 can be noticed.The bending modes m2 (SO4) of CSD became stronger and wereshifted from 428 cm�1 to 412 cm�1 and from 490 cm�1 to493 cm�1. The same tendency was observed in the case of m4
(SO4) mode, where characteristic for CSH Raman bands at 625
Fig. 1. Raman spectra of: (a) CSH, (b) CSD and FT-IR spectra of (c) CSH, and (d) CSD.Spectral regions – (a) 3800–200 cm�1, (b) 1040–400 cm�1.
Fig. 2. Raman spectra of: (a) TiHA/c powder, (b) set and hardened cement B. FT-IRspectra of (c) TiHA/c powder, and (d) set and hardened cement B. Spectral regions –(a) 3800–200 cm�1, (b) 1400–400 cm�1.
and 667 cm�1 were downward shifted to 617 and 671 cm�1 inthe case of CSD. According to infrared spectra (Fig. 1) the bands:m1 (H2O) around 3400 cm�1 and m2 (H2O) – 3600 cm�1 were verystrong for calcium sulfate dihydrate, while for CSH bands assignedto vibrations of water molecules were significantly weaker. Similarchanges were observed in the OH bending modes at around 1680and 1620 cm�1 characteristic for calcium sulfate dihydrate, withthe disappearance of higher energy vibrations in the calcium sul-fate hemihydrate phase. Band assignments for calcium sulfatedihydrate and hemihydrate are presented in Table 2.
The Raman spectrum of cement B (Fig. 2) revealed the presenceof bands corresponding to calcium sulfate dihydrate, namely: 413,494 cm�1 (m2), 617 and 671 cm�1 (m4), 1134 cm�1 (m3). and a veryintensive one at 1007 cm�1 (m1). The lack of bands characteristicfor CSH indicated that all amount of calcium sulfate hemihydratecompleted the hydration reaction and formed CSD. The bands at961 cm�1 (m1) and weaker at 1075 and 1050 cm�1 (m3) attributedto stretching modes of the PO4 group in the hydroxyapatite
Fig. 3. Raman spectra of: (a) TiHA/s powder, (b) set and hardened cement C. FT-IRspectra of (c) TiHA/s powder, and (d) set and hardened cement C. Spectral regions –(a) 3800–200 cm�1, (b) 1400–400 cm�1.
Fig. 4. XRD patterns of the set and hardened: (a) cement A, (b) cement B and (c)cement C.
Table 3Positions of the chosen characteristic peaks of the phases present in the set andhardened cement bodies.
Phase 2h <hkl> I/I100
HA (PDF 01-074-9761) 25.8 002 4031.8 121 10032.9 300 60
292 C. Paluszkiewicz et al. / Journal of Molecular Structure 1034 (2013) 289–295
structure were found. The Raman spectra of cement A was analog-ical and exhibited the same phase composition as cement B.
The infrared spectrum (Fig. 2) showed the existence of bandscharacteristic for PO4 group stretching vibrations at 1045 cm�1
(m3) and bending vibrations at 572 cm�1 (m4) for all examinedmaterials. The presence of a strong doublet near 602 and669 cm�1 was attributed to the SO4 bending vibrations (m4). How-ever the coincidence of PO4 or SO4 bending vibration bands can benoticed at 602 cm�1. The strongest bands were assigned to SO4
stretching vibrations (m3) and were observed as a doublet near1045 and 1142 cm�1. Absorption features in the 2080–2400 cm�1
region present in the spectrum of cement B were referred to thefirst order overtones and combinations of the m3 absorptions ofSO4. Both cements, A and B, were biphasic and composed withthe calcium sulfate dihydrate and titanium doped hydroxyapatite.The spectrum of cement C (Fig. 3), based on the sintered at 1250 �CTiHA powder (TiHA/s), significantly differed from the others.
As can be seen in Fig. 3 only a small intensity of Raman bandscorresponding to calcium sulfate dihydrate, could be noticed. Fur-thermore, characteristic for not fully hydrated calcium sulfate, veryintensive Raman band at 1011 cm�1, weaker at 428, 486 cm�1, 630,667 cm�1 and bands situated around 1130, 1170 cm�1 were ob-served. The band assigned to the symmetric stretching mode ofthe PO4 group (m1) was also present at around 960 cm�1. FT-IRstudy of the cement C revealed that the OH stretching modes ob-served at 3555 and 3610 cm�1 were analogical for those in calciumsulfate hemihydrate. Described bands were also notably weaker forthe cement C than bands at 3407 and 3545 cm�1 for the material B.Furthermore, in the case of the cement C only one band at1621 cm�1 originating from OH bending mode and the disappear-
ance of higher energy vibration (present in the cement B at1686 cm�1) was noticed. These spectral features can indicate onthe existence of not fully hydrated calcium sulfate phase in theexamined material. On the other hand, the presence of very weakband at 3408 cm�1 could be the evidence for the existence ofCSD phase simultaneously. The bands around 1520–1400 cm�1
can be assigned to –CO3 groups in titanium doped hydroxyapatite.The quantitative Rietveld analysis of XRD patterns confirmed
that TiHA and TiHA/c consisted of monophasic hydroxyapatite
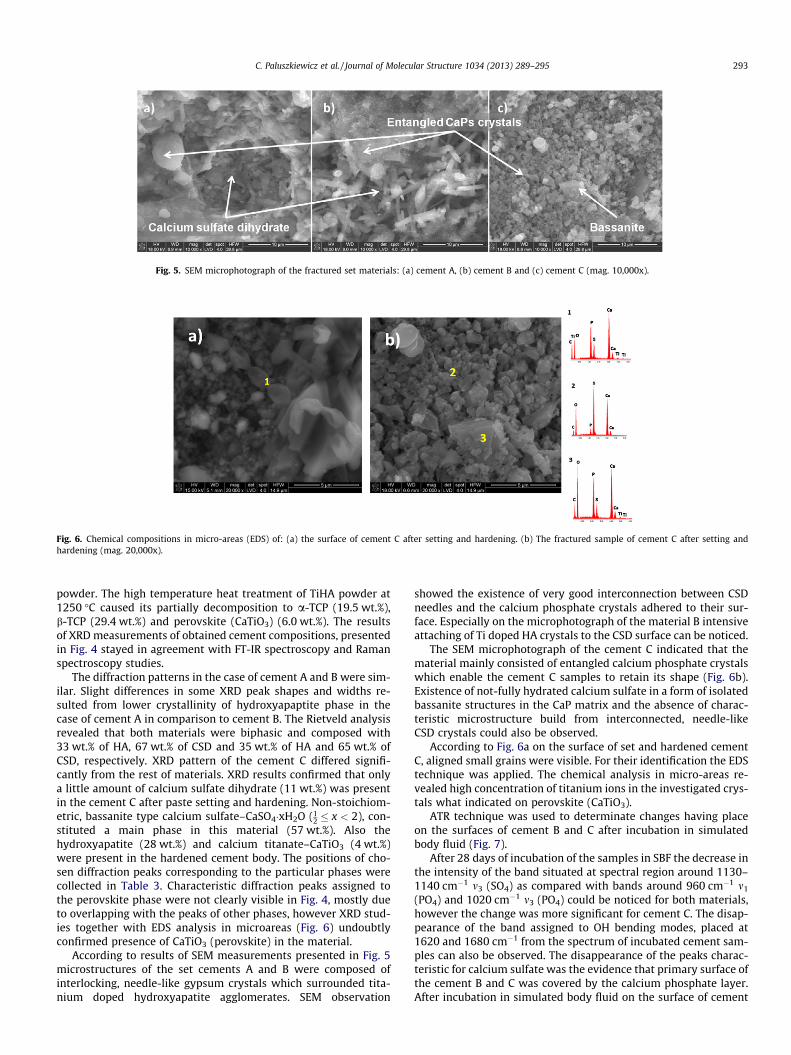
Fig. 5. SEM microphotograph of the fractured set materials: (a) cement A, (b) cement B and (c) cement C (mag. 10,000x).
Fig. 6. Chemical compositions in micro-areas (EDS) of: (a) the surface of cement C after setting and hardening. (b) The fractured sample of cement C after setting andhardening (mag. 20,000x).
C. Paluszkiewicz et al. / Journal of Molecular Structure 1034 (2013) 289–295 293
powder. The high temperature heat treatment of TiHA powder at1250 �C caused its partially decomposition to a-TCP (19.5 wt.%),b-TCP (29.4 wt.%) and perovskite (CaTiO3) (6.0 wt.%). The resultsof XRD measurements of obtained cement compositions, presentedin Fig. 4 stayed in agreement with FT-IR spectroscopy and Ramanspectroscopy studies.
The diffraction patterns in the case of cement A and B were sim-ilar. Slight differences in some XRD peak shapes and widths re-sulted from lower crystallinity of hydroxyapaptite phase in thecase of cement A in comparison to cement B. The Rietveld analysisrevealed that both materials were biphasic and composed with33 wt.% of HA, 67 wt.% of CSD and 35 wt.% of HA and 65 wt.% ofCSD, respectively. XRD pattern of the cement C differed signifi-cantly from the rest of materials. XRD results confirmed that onlya little amount of calcium sulfate dihydrate (11 wt.%) was presentin the cement C after paste setting and hardening. Non-stoichiom-etric, bassanite type calcium sulfate–CaSO4�xH2O (1
2 � x < 2), con-stituted a main phase in this material (57 wt.%). Also thehydroxyapatite (28 wt.%) and calcium titanate–CaTiO3 (4 wt.%)were present in the hardened cement body. The positions of cho-sen diffraction peaks corresponding to the particular phases werecollected in Table 3. Characteristic diffraction peaks assigned tothe perovskite phase were not clearly visible in Fig. 4, mostly dueto overlapping with the peaks of other phases, however XRD stud-ies together with EDS analysis in microareas (Fig. 6) undoubtlyconfirmed presence of CaTiO3 (perovskite) in the material.
According to results of SEM measurements presented in Fig. 5microstructures of the set cements A and B were composed ofinterlocking, needle-like gypsum crystals which surrounded tita-nium doped hydroxyapatite agglomerates. SEM observation
showed the existence of very good interconnection between CSDneedles and the calcium phosphate crystals adhered to their sur-face. Especially on the microphotograph of the material B intensiveattaching of Ti doped HA crystals to the CSD surface can be noticed.
The SEM microphotograph of the cement C indicated that thematerial mainly consisted of entangled calcium phosphate crystalswhich enable the cement C samples to retain its shape (Fig. 6b).Existence of not-fully hydrated calcium sulfate in a form of isolatedbassanite structures in the CaP matrix and the absence of charac-teristic microstructure build from interconnected, needle-likeCSD crystals could also be observed.
According to Fig. 6a on the surface of set and hardened cementC, aligned small grains were visible. For their identification the EDStechnique was applied. The chemical analysis in micro-areas re-vealed high concentration of titanium ions in the investigated crys-tals what indicated on perovskite (CaTiO3).
ATR technique was used to determinate changes having placeon the surfaces of cement B and C after incubation in simulatedbody fluid (Fig. 7).
After 28 days of incubation of the samples in SBF the decrease inthe intensity of the band situated at spectral region around 1130–1140 cm�1 m3 (SO4) as compared with bands around 960 cm�1 m1
(PO4) and 1020 cm�1 m3 (PO4) could be noticed for both materials,however the change was more significant for cement C. The disap-pearance of the band assigned to OH bending modes, placed at1620 and 1680 cm�1 from the spectrum of incubated cement sam-ples can also be observed. The disappearance of the peaks charac-teristic for calcium sulfate was the evidence that primary surface ofthe cement B and C was covered by the calcium phosphate layer.After incubation in simulated body fluid on the surface of cement
Fig. 7. ATR spectra of the surface of examined bone cements before and after28 days of incubation in simulated body fluid. Spectral regions – (a) 3800–550 cm�1, (b) 1800–550 cm�1.
294 C. Paluszkiewicz et al. / Journal of Molecular Structure 1034 (2013) 289–295
C the existence of bands at 872 cm�1 and 1420–1450 cm�1, corre-sponding to carbonated hydroxyapatite (type B) was clearly visible.
Spectroscopic and XRD studies revealed that set materials differin phase composition. Cement A and B consisted of calcium sulfatedihydrate and hydroxyapatite. In the case of cement C Raman andinfrared measurements clearly indicated on the structural changesin the calcium sulfate hemihydrate, which were the consequenceof its partially hydratation and creation of mainly bassanite phase– CaSO4�xH2O (1
2 � x < 2). Obtained results showed that only a lim-ited part of the CSH had been transformed into calcium sulfatedihydrate and the spectral features can be interpreted as coexistingphases: CSD and bassanite. The presence of the non-totallyhydrated calcium sulfate can origin from the existence of solublecalcium phosphates (a-TCP) in the initial powder composition.Phosphate ions are well known as retarders of calcium sulfate set-ting reaction. The hydratation process of CSH, leading to crystalli-zation of the calcium sulfate dihydrate, has been the subject ofmany investigations [6,28,37,38]. The reaction of CSH with waterpossess three time region: an induction period, an acceleratoryperiod and a period involving a very slow reaction – the comple-tion of hydratation [19]. The induction period starts immediatelyafter the CSH powder is mixed with water solution. The calciumsulfate hemihydrate dissolves and the solution becomes supersat-urated with respect to calcium and sulfate ions, what lead to pre-cipitation of CSD crystals. If the cement mixture composed of twosetting phases, a competitive chemical processes take place whatcan led to the blockage of setting reaction. As a consequence ofpresence of TCP phase, chemical reaction between the calciumand phosphate ions occurred and the physical barrier composedwith calcium phosphate precipitated crystals was formed on the
calcium sulfate grains. A surface blockage of hydration processled to presence of non-totally hydrated, bassanite type calcium sul-fate in the set material. Similar process was observed by Nilssonet al. [23] in the a-TCP–CSH based cement compositions. Further-more a-TCP present in powder composition reacted with watermolecules, forming a crystalline hydroxyapatite structure, accord-ing to the Eq. (1).
This competitive process impeded the reaction between waterand the inner calcium sulfate particles and completion of CSHhydration process was not possible. Considering that only a smallamount of CSD phase has been found in the material, a-tricalciumphosphate seemed to be a main setting phase in cement C. TheSEM microphotography of the fractured cement body confirmedthat after setting the entanglement of calcium phosphate crystalshad delivered the required cohesion. The calcium sulfate crystals,mainly in the form of blocks and plates, were embedded in the cal-cium phosphate matrix and after in vivo implantation may permitgradual porosity in the material, when resorbed. Fernandez et al.[10] observed similar process in biphasic bone cement with im-proved osteointegration, composed of a-TCP and CSD, in whicha-TCP during setting process transformed into calcium deficientapatite whereas porosity appeared during passive dissolution ofcalcium sulfate dihydrate. In the case of the rest materials (ce-ments A and B) a well interconnected calcium sulfate dihydratecrystal matrix was created. When sintered titanium dopedhydroxyapatite (TiHA/s) is used as a one of the initial components,in the set final material, beside calcium sulfate and hydroxyapatite,also another secondary phase – the perovskite was present. Theseconstituent came from the decomposition of titanium dopedhydroxyapatite during the sintering process and the existence ofCaTiO3 grains in the cement C was confirmed by SEM observations.
The differences in ATR–FTIR spectrum measured before andafter 28 days of incubation in SBF clearly indicated on formationof the apatitic layer on the cement samples and indicate on highbioactive potential of obtained materials. The more significantspectral changes in the case of the cement C can suggest that thick-er apatitic layer was created on its surface and indicate on thehigher bioactive potential in comparison to cement B. The presenceof resorbable TCP phase and perovskite in the material resulted inbetter in vitro bioactivity as revealed by hence apatite layer forma-tion. The presence of CaTiO3 in the material can positively impact abiological response to obtained cement. Webster et al. [39] in hisresearches regarding coating titanium with hydroxyapatite layerconfirmed good influence of perovskite phase on the osteoblastadhesion compared to the pure HA and titanium.
4. Conclusions
FT-Raman and FT-IR spectroscopy allowed, together with XRDstudies to determinate the composition of setting in situ compositetype bone cements. Above complementary techniques were provedto be suitable for analyzing the pathway of cement setting processand determination of the phase composition. The new bonecements based on calcium sulfate and titanium doped hydroxyap-atite consisted of: calcium sulfate dihydrate, hydroxyapatite,a-tricalcium phosphate and perovskite, depending on the heattreatment of initial TiHA powder. The received results revealedthat the calcium sulfate was the main setting phase in cementscomposed with untreated or calcined at 800 �C titanium dopedhydroxyapatite. In the case of cement consisted of TiHA sinteredat 1250 �C – a-TCP remained to be the main setting phase, creating
C. Paluszkiewicz et al. / Journal of Molecular Structure 1034 (2013) 289–295 295
calcium phosphate matrix. The existence of bassanite phase in theset material provided evidence that calcium sulfate hemihydratehydration process can be retarded or even prevented to be com-pleted by the presence of a-tricalcium phosphate phase.
The usefulness of infrared spectroscopy for evaluation of bioac-tive potential of implant materials in vitro was also confirmed. Theresults of ATR–FTIR measurements confirmed that incubation ofthe cement samples in the simulated body fluid led to apatite layerformation on the surfaces of the hardened bodies. The highestbioactive potential possessed cement consisted of sintered TiHA.
Obtained results helped to gain a better understanding of pro-cesses having place during setting of complex multiphase cementmaterials.
Acknowledgment
This work has been supported by the Project No UDA-POIG.01.03.01-00-005/09.
[4] J. Huang, S.M. Best, W. Bonfield, T. Buckland, Acta Biomater. 6 (2010) 241–249.[5] A. Slósarczyk, A. Zima, Z. Paszkiewicz, J. Szczepaniak, A.H. De Aza, A.
Chróscicka, Ceram. Mater. 62 (3) (2010) 369–375.[6] W.E. Brown, L.C. Chow, A new calcium phosphate, water-setting cement, in:
P.W. Brown (Ed.), Cements Research Progress, American Ceramic Society,Westerville, OH, 1986, pp. 352–379.
[7] S. Takagi, L.C. Chow, K. Ishikawa, Biomaterials 19 (1998) 1593–1599.[8] S. Pina, S.M. Olhero, S. Gheduzzi, A.W. Miles, J.M.F. Ferreira, Acta Biomater. 5
(4) (2009) 1233–1240.[9] C. Durucan, P.W. Brown, J. Mater. Sci. 37 (2002) 963–969.
[10] E. Fernandez, M.D. Vlada, M.M. Gel, J. Lopez, R. Torres, J.V. Cauich, M. Bohner,Biomaterials 26 (2005) 3395–3404.
[11] M. Bohner, H.P. Merkle, P. Van Landuyt, G. Trophardy, J. Lemaitre, J. Mater. Sci.:Mater. Med. 11 (2000) 111–116.
[12] E. Fernandez, F.J. Gil, M.P. Ginebra, F.C.M. Driessens, J.A. Planell, J. Mater. Sci.:Mater. Med. 10 (1999) 223–230.
[13] M.D. Vlad, E.V. Sindilar, M.L. Marinoso, I. Poeata, R. Torres, J. López, M. Barracó,E. Fernández, Acta Biomater. 6 (2010) 607–616.
[14] S. Del Valle, N. Mino, F. Munoz, A. Gonzalez, J.A. Planell, M.P. Ginebra, J. Mater.Sci.: Mater. Med. 18 (2007) 353–361.
[15] M. Nilsson, L. Wielanek, J.S. Wang, K.E. Tanner, L. Lingren, J. Mater. Sci.: Mater.Med. 14 (2003) 399–404.
[16] S.L. Bahn, Oral Surg. Oral Med. Oral Pathol. 21 (1966) 672–681.[17] L.F. Peltier, Clin. Orthop. 21 (1961) 1–29.[18] R. Strocchi, G. Orsini, G. Iezzi, A. Scarano, C. Rubini, G. Pecora, A. Piattelli, J. Oral
Implantol. 28 (2002) 273–278.[19] N.B. Singh, B. Middendorf, Prog. Cryst. Grow. Charact. 53 (2007) 57–77.[20] E. Finot, E. Lesniewska, J. Goudnnet, J.C. Mutin, Appl. Surf. Sci. 161 (2000) 316–
322.[21] P. Wang, E.J. Lee, C.S. Park, B.H. Yoon, D.S. Shin, H.E. Kim, Y.H. Koh, S.H. Park, J.
Am. Ceram. Soc. 91 (6) (2008) 2039–2042.[22] J.L. Ricci, M.J. Weiner, S. Mamidwar, H. Alexander, Calcium sulfate, in: T.
Kokubo (Ed.), Bioceramics and their Clinical Applications, WoodheadPublishing Limited, Cambridge England, 2008 (Chapter 14).
[23] M. Nilsson, E. Fernández, S. Sarda, L. Lidgren, J.A. Planell, J. Biomed. Mater. Res.61 (4) (2002) 600–607.
[24] Y. Liu, A. Wang, J. Freeman, Raman, MIR and NMR spectroscopic study oncalcium sulfates; gipsum, bassanite, and anhydrite, in: Proc. 40th Lunar nadPlanetary Science Conference, The Woodlands, Texas, 23–27 March 2009.
[25] B.A. Zapol, F.F. Alksnis, J. Appl. Spectrosc. 27 (4) (1977) 1346–1351.[26] A. Rapacz-Kmita, A. Slósarczyk, Z. Paszkiewicz, C. Paluszkiewicz, J. MolStruct.
704 (2004) 333–340.[27] A. Slósarczyk, Z. Paszkiewicz, C. Paluszkiewicz, J. Mol. Struct. 744–747 (2005)
657–661.[28] L.P. Sarma, P.S.R. Prasad, N. Ravikumar, J. Raman Spectrosc. 29 (1998) 851–
414–420.[32] ASTM C266–04. ASTM Annual Book of standards. Standard Test Method for
Time Setting of Hydraulic-Cement paste by Gillmore Needles. WestConshohocken PA, USA. pp. 19428–2959.
[33] H.M. Rietveld, Acta Crystallogr 22 (1967) 151–1152.[34] H.M. Rietveld, J. Appl. Crystallogr 2 (1969) 65–71.[35] T. Kokubo, H. Takadama, Biomaterials 27 (2006) 2907–2915.[36] I. Khairoun, M.G. Boltong, F.C.M. Driessens, J.A. Planell, J. Mater. Sci.: Mater.
Med. 9 (1998) 667–671.[37] B. Guan, Q. Ye, J. Zhang, W. Lou, Z. Wu, Cem. Concr. Res. 40 (2010) 253–259.[38] Q. Ye, B. Guan, W. Lou, L. Yang, B. Kong, Powder Technol. 207 (2011) 208–214.[39] T.J. Webster, C. Ergun, R.H. Doremus, W.A. Lanford, J. Biomed. Mater. Res., A