Extraction—Metals Processing THOMAS W. CHAPMAN Department of Chemical Engineering University of Wisconsin Madison, Wisconsin 8.1 INTRODUCTION Although electrolytes normally do not exhibit significant solubility in nonpolar solvents, many metal ions can react with a wide variety of organic compounds to form species that are soluble in organic solvents. Such solubility, which depends on a chemical reaction, provides a basis for separating and concentrating metals that are present as ions in aqueous solution. As with many separation processes, solvent extraction of metals was first developed as a tool of the analytical chemist. Chemical reagents and solvents are known for isolating virtually every metallic element of the Periodic Table.'^ In the 1940s and 1950s some of this basic analytical chemistry was used to develop continuous processes for separating nuclear 5 and rare-earth 6 elements. Subsequently, the availability of inexpensive or especially effective reagents led to the establishment of large-scale processes for extraction of copper, zinc, uranium, and other metals from hydrometallurgical leach liquors. 7 " 10 The flowsheet of two proposed processes, based on solvent extraction, for separating the constituents of deep-sea manganese nodules are shown in Figs. 8.1-1 and 8.1-2. 11 The solvent extraction of metals is similar in concept to ion exchange, which is treated in Chapter 13, and provides specific examples of separation by chemical complexation, discussed in Chapter 15. Tech- nological implementation is based on the concepts of ordinary solute extraction, presented in Chapter 7. The choice of solvent extraction over alternative separation schemes is a complex one, but it clearly depends on the availability of an effective extraction agent. A reagent that provides a very high distribution coefficient from dilute solution usually can be found, but reagent selectivity, stability, solubility, kinetics, and cost are all important factors. The ability to reverse the extraction reaction to recover the metal and regenerate the reagent is crucial. In favor of extraction is the relative ease of handling liquid-liquid systems, compared with the solid-liquid operations required in most of the alternative processes. An extensive literature has developed that offers many choices of chemical systems, and progress is being made in refining the engineering of metal extraction processes. Relevant publications appear in the analytical chemistry literature, the Journal of Inorganic and Nuclear Chemistry, Separation Science and Technology, metallurgical journals and conferences, Hydrometallurgy, and most recently in the standard chemical engineering periodicals, such as Industrial and Engineering Chemistry. Although publications in this field have been widely dispersed, a focal point has been the Proceedings of the triennial International Solvent Extraction Conference. The Handbook of Solvent Extraction 12 provides a number of useful chapters, and Ritcey and Ashbrook 13 discuss many practical aspects of metal extraction processes. 8.2 EXTRACTION CHEMISTRY AND REAGENTS The extraction of a metal ion from an aqueous solution into an organic solvent is accomplished by the chemical formation of an uncharged species that is soluble in the organic phase. Because metal salts usually CHAPTER O
Transcript
E x t r a c t i o n — M e t a l s P r o c e s s i n g
THOMAS W. CHAPMANDepartment of Chemical EngineeringUniversity of WisconsinMadison, Wisconsin
8.1 INTRODUCTION
Although electrolytes normally do not exhibit significant solubility in nonpolar solvents, many metal ionscan react with a wide variety of organic compounds to form species that are soluble in organic solvents.Such solubility, which depends on a chemical reaction, provides a basis for separating and concentratingmetals that are present as ions in aqueous solution.
As with many separation processes, solvent extraction of metals was first developed as a tool of theanalytical chemist. Chemical reagents and solvents are known for isolating virtually every metallic elementof the Periodic Table.'^ In the 1940s and 1950s some of this basic analytical chemistry was used to developcontinuous processes for separating nuclear5 and rare-earth6 elements. Subsequently, the availability ofinexpensive or especially effective reagents led to the establishment of large-scale processes for extractionof copper, zinc, uranium, and other metals from hydrometallurgical leach liquors.7"10 The flowsheet of twoproposed processes, based on solvent extraction, for separating the constituents of deep-sea manganesenodules are shown in Figs. 8.1-1 and 8.1-2.11
The solvent extraction of metals is similar in concept to ion exchange, which is treated in Chapter 13,and provides specific examples of separation by chemical complexation, discussed in Chapter 15. Tech-nological implementation is based on the concepts of ordinary solute extraction, presented in Chapter 7.The choice of solvent extraction over alternative separation schemes is a complex one, but it clearly dependson the availability of an effective extraction agent. A reagent that provides a very high distribution coefficientfrom dilute solution usually can be found, but reagent selectivity, stability, solubility, kinetics, and costare all important factors. The ability to reverse the extraction reaction to recover the metal and regeneratethe reagent is crucial. In favor of extraction is the relative ease of handling liquid-liquid systems, comparedwith the solid-liquid operations required in most of the alternative processes.
An extensive literature has developed that offers many choices of chemical systems, and progress isbeing made in refining the engineering of metal extraction processes. Relevant publications appear in theanalytical chemistry literature, the Journal of Inorganic and Nuclear Chemistry, Separation Science andTechnology, metallurgical journals and conferences, Hydrometallurgy, and most recently in the standardchemical engineering periodicals, such as Industrial and Engineering Chemistry. Although publications inthis field have been widely dispersed, a focal point has been the Proceedings of the triennial InternationalSolvent Extraction Conference. The Handbook of Solvent Extraction12 provides a number of useful chapters,and Ritcey and Ashbrook13 discuss many practical aspects of metal extraction processes.
8.2 EXTRACTION CHEMISTRY AND REAGENTS
The extraction of a metal ion from an aqueous solution into an organic solvent is accomplished by thechemical formation of an uncharged species that is soluble in the organic phase. Because metal salts usually
C H A P T E R O
Basic nickel carbonateFIGURE 8.1-1 Solvent extraction portion of the Kennecott copper-nickel carbonate process for deep-seamanganese nodules. Adapted from U.S. Patent 3,907,966 in Ref. 11.
are not soluble in organic solvents, the process requires the introduction of a reagent, or extractant, thatwill combine with the metal ion to form an organic-soluble species. For economic and environmentalreasons, the extractant chosen to separate a metal must be practically insoluble in water. Thus, the extractantsometimes can constitute the organic-phase solvent of the process. In most cases, however, an inert organiccarrier, or diluent, is used as the solvent of the organic phase. Additional organic components, calledmodifiers, also are employed. The proper use of diluents and modifiers can prevent formation of veryviscous or solid organic phases, can modify extraction kinetics as well as equilibria, can reduce the aqueoussolubility of the extractant, and allows flexibility in the choice of phase ratios in contacting equipment.
The extractants that are used to form organic-soluble metal species from aqueous metal salts usuallyare grouped into three classes according to the type of reaction that occurs. Solvating extractants competewith water in coordination bonds around a metal ion and carry neutral salts into the organic phase. Cation
Cu Extraction4 stages
40%LIX 64N Crude leach
liquor
NH3 Scrub2 stages
Ni Scrub2 stages
NH4HCO3
solution RaffinateNH3AX)2
solution
Cu Stripping? stages
Ni Extraction2 stages
Electrowinning
Cathodecopper
Ni Stripping3 stages
Ammoniumcarbonatesolution
NH3 Ammoniaremoval
Solid-liquidseparation
Return chloride Co chloride toelectrolyte electrolysis
FIGURE 8.1-2 Deepsea Ventures solvent extraction flowsheet for separation of metals in a manganesenodule leach liquor. From Ref. 11, with permission.
Aqueous feedMn, Fe, Ni, Co1 Cu
chlorides
Fe Extraction4 stages
15(20%) secondaryamine
15(20%) isodecanolkerosene
Fe Stripping3 stages FeCI3 solution to
pyrohydrolysisAcid pH 2pH adjust to 2
with 2 N NaOHMn, Ni, Co, Cu
• chlorides •(sulfates)
r~
Cu Extraction5 stages
2 iV NaOH added tomaintain pH2
10% LIX 64N20% isodecanol
napoleum
Return sulfateelectrolyte3 № n H +
CuSO4 solutionto electrolysis
Cu Stripping5 stages
pH adjust to 4.5with 2 N NaOH
Mn chloride(sulfate) Ni-Co Extraction
5 stages
Mn, Ni, Cochlorides •(sulfates)
10% Kelex 10020% isodecanol
napoleum
2 N NaOH added tomaintain pH 4.05
Co Stripping4 stages
Ni Stripping3 stages
20%HCI
Return chloride orsulfate electrolyte
adjusted to 3 N in H+
Ni chloride orsulfate to
electrolysisAcidic
Co chlorideCo Re-extraction
3 stages
10% TIOAkerosene
Co Restripping3 stages
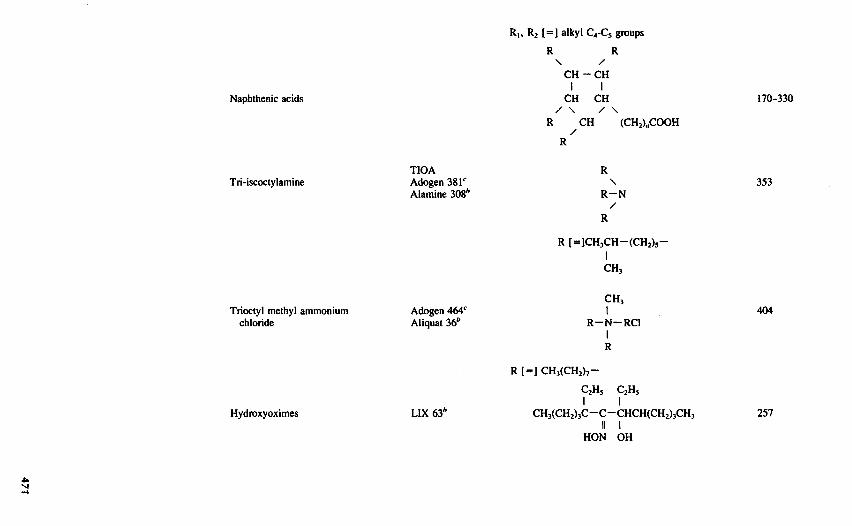
TABLE 8.2-1 Examples of Some Common Metal Extractants*
°Ritcey and Ashbrook1 and Flett et al. give more complete lists.*Henkel Corp.c Sherex Corp.
exchangers include organic acids as well as specific chelating extractants. Anton exchangers extract metalsthat form anionic complexes. Synergistic mixtures of extractants are sometimes found to be effective inpromoting a particular separation. The chemical structures of some common extractants are shown in Table8.2-1.
8.2-1 Solvating Extractants
Extraction of metals by solvating extractants occurs formally by the reaction
(8.2-1)
where MXn is an uncharged salt coming out of the aqueous phase and S is the extractant. Organic-phasespecies are denoted by an overline. Inorganic acid can also be extracted by a reaction of the form
(8.2-2)
The most common solvating extractants are alkyl phosphate esters, such as tributyl phosphate (TBP)and trioctyl phosphine oxide (TOPO), but thiophosphate esters, phosphine sulfides, ketones, alcohols, andesters also have been used.
The common features of these solvating extractants is that they contain both electron-rich oxygen orsulfur atoms that can coordinate metal cations and multiple alkyl chains that make them hydrophobic. It isthe electron-rich atoms that can bind protons to extract acids. Generally, those reagents possessing a carbon-bound oxygen tend to extract water with the metal salt, whereas the phosphorus-based reagents do not.
Because the bonds formed in the solvation-extraction reactions are not highly specific, the stoichiometriccoefficients p and qy as well as the amount of coextracted water, are not definite and may vary withextraction conditions. Increased polarity of the functional group in the extractant, as is obtained in elimi-nating the ester linkages in trioctyl phosphate to obtain TOPO, increases the strength of the solvation, butit also tends to increase the aqueous solubility of the reagent. Solvating extractants can be used undiluted,but usually they are mixed with an inexpensive diluent such as kerosene.
The solvation extraction of a metal, according to the form of reaction (8.2-1), depends on the formationin the aqueous phase of an uncharged ion pair or complex MXn to some significant level of activity.Therefore, the extent of the extraction reaction will depend strongly on the aqueous solution chemistry ascomplexation, association, and hydrolysis affect the distribution of metal species. Similarly, the reversalof reaction (8.2-1) to strip the metal from the organic phase usually depends on altering the aqueous-phasechemistry. This may be accomplished by stripping with an aqueous phase low in the concentration of theligand X or by using a competing aqueous-phase complexing agent. Temperature may have some effect onthe distribution equilibrium, and precipitation or redox reactions might be used to reverse the extractionreaction.
Table 8.2-2 summarizes some specific applications of solvating extractants. More extensive reviewsare given in Refs. 1-5.
8.2-2 Liquid Cation Exchangers
Solvent extraction of metals can be accomplished by use of organic-phase extractants that function as ionexchangers. For metal cations extractants possessing an acidic functionality are appropriate. There are twotypes of cationic, or acidic, ion-exchange extractant available. The same functional groups that are usedin cation-exchange resins, namely, carboxylic, phosphoric, and sulfonic acids, can be incorporated in asoluble organic molecule to obtain a liquid ion exchanger. Structures of moderate molecular weight (around300) are used such that the extractant is soluble in a diluent like kerosene but exhibits a low aqueoussolubility. Common examples of these acidic extractants include di-2-ethylhexyl phosphoric acid (DEHPA)and alkyl monocarboxylic acids with 10-20 carbons in various branched (e.g., Versatic acids) or cyclic(e.g., naphthenic acid) structures.
The other type of cation-exchange extractant available is a class of chelating extractants. Chelatingextractants contain two functional groups that form bidentate complexes with metal ions. Although thereare many chelating reagents used in analytical chemistry to achieve highly selective complexing of a widevariety of metals, only a few have been developed for large-scale commercial use. The notable exampleis the family of LIX reagents developed especially for copper extraction by General Mills Inc., now HenkelCorp., which are hydroxyoximes and possess an acidic hydrogen. Similar reagents based on the oximegroup have been developed by several other chemical companies. Other acidic chelating extractants, whichact as cation exchangers, include other hydroxyoximes, such as SME259 of Shell Chemical Company andthe P5000 reagents of Acorga Limited, and substituted 8-hydroxyquinolines, such as the Kelex reagents ofSherex Chemical Company.
With both simple and chelating acidic extractants the metal extraction reaction generally follows theform
TABLE 8.2-2 Hydrometallurgical Applications of Some Common Solvating Extractants
Extractant Application
Tributyl phosphate (TBP) U extraction from nitric acidZr separation from Hf in nitric acidFeCl3 separation from Cu and Co in HCl solution
Methyl isobutyl ketone Extraction of Nb and Ta from HF solutions, followed by selective(MIBK) stripping
Trioctyl phosphine oxide Extraction of U from phosphoric acid(TOPO)
(8.2-3)
such that the reaction is strongly pH dependent and stripping occurs into strong acid.Table 8.2-3 summarizes the applications of some acidic extractants. Ritcey and Ashbrook6 provide a
historical account of the evolution of the commercial chelating extractants.
8.2-3 Liquid Anion Exchangers
In some electrolytic solutions, especially those strong in halides, many metal cations are complexed byanions to the extent that they exist primarily as neutral ion pairs or anionic species. The formation ofanionic complexes may retard metal extraction by solvation or cation exchange, but it can be exploited byuse of anion-exchanging extractants.
The most important liquid anion exchangers for commercial use are quaternary ammonium salts andprimary, secondary, and tertiary amines, which become anion exchangers by combining with mineral acidsto form alkyl ammonium salts. In the former case, such a metal extraction reaction might be written
(8.2-4)
where m is the charge number of the metal cation M +m and X " is a univalent anion. Because aqueouscomplexes of various coordination numbers n coexist in an equilibrium distribution, the overall extractionreaction may be viewed as either anion exchange, with n > m, or an association reaction with n — m. Ineither case, the stability of the organic-soluble species depends on the tendency of the metal ion to formcomplexes with the anion.
With other amines, such as tri-isooctylamine (TIOA), an overall metal extraction reaction might bewritten
(8.2-5)
where HX and MXm both combine with the amine in an apparent addition reaction. Alternatively, thisreaction could be written, and can occur, in two separate steps,
TABLE 8.2-3 Hydrometallurgical Applications of Some Common Cation-Exchanging Extractants
Extractant Applications
Versatic 911 (carboxylic Separation of rare earths in nitrate solutionacid)
Separation of Co from Ni in sulfate solutionNaphthenic acids Extraction of Cu, Ni, and Co
(carboxylic acids)Di-2-ethylhexyl Extraction of rare earths, V, Be, Al, Co, Ni, Zn
phosphoric acid(DEHPA)
U extraction from sulfuric acidLIX 64N Selective extraction of Cu from acidic solutions
Extraction of Cu and Ni from alkaline ammonia solutionKelex 100 Selective extraction of Cu from acidic solutions
Extraction of Cu, Ni, Co, and Zn from alkaline ammonia solution
(8.2-6)
amine salt formation, followed by
(8.2-7)
an addition reaction between the ion pair and the amine salt. If attention were focused on an anionic metalcomplex MX™"" rather than the uncharged ion pair MXm, an alternative to reaction (8.2-7) would be ananion-exchange reaction analogous to reaction (8.2-4),
(8.2-8)
Different amine extraction systems exhibit different stoichiometries from that shown here, and a numberof different organic species may form in any one system, but the common feature of such extractions isthat they depend on combining metal cations, anions, and amine into uncharged organic-soluble molecules.The extraction may be reversed by suppressing the activity of any of the product's constituents. For example,a metal extracted with a tertiary amine can usually be stripped by an aqueous phase that has either asufficiently high pH to reverse the amine salt formation or sufficiently low coordinating-anion concentrationto suppress formation of the complex.
For quantitative process calculations it is important to know what chemical species form in a givenextraction system. The stoichiometry of the overall reactions affects the process material balance. Also,the number of different species formed determines the number of degrees of freedom required to specifythe thermodynamic state of a system. On the other hand, the detailed mechanism of a reaction, for example,whether amine extraction occurs by addition [reaction (8.2-5)] or ion exchange [reaction (8.2-7)], is of nodirect concern unless slow reaction kinetics become an issue.
Table 8.2-4 summarizes applications of amines for metal extraction. The viability of amine extractiondepends directly on the identity of the anions present in the aqueous phase. Amine extraction has beendiscussed by Schmidt.7
8.2-4 Diluents
Although the solvent extraction of a metal occurs by reaction with an extractant that is essentially insolublein water, it is seldom practical to use pure extractant as the solvent phase. The pure extractants are usuallyvery viscous and may have specific gravities near 1 so that both mixing and separating phases may bedifficult. They are intrinsically surface active and tend to form emulsions with water. To avoid theseproblems and to provide flexibility with respect to phase ratios and the hydraulics of liquid-liquid contacting,one uses a diluent, which is often the major component in the organic phase.
Inexpensive hydrocarbon solvents are chosen as diluents in large-scale processes. Table 8.2-5 lists someexamples. These solvents are chosen for their low solubilities in water and high flash points as well assome less obvious chemical properties.
The diluent in a solvent extraction system does not serve simply as an inert carrier for the extractantand its metal compounds. It has been found repeatedly that the choice of diluent can affect significantlythe performance of various extractants, presumably through both chemical and physical interactions withthe solute species. For example, Murray and Bouboulis8 reported the effects of systematically varying thearomatic content of the diluent on a copper extraction process, and Akiba and Freiser9 demonstrated solventeffects on system chemistry. Ritcey and Ashbrook10 review the various solvent properties that influenceextractant performance.
From the combination of a hydrocarbon solvent as a diluent with extracted metal species, which oftencontain some water molecules or other polar groups, there is a tendency in many systems for a third phase
TABLE 8.2-4 Hydrometallurgical Applications of Some Common Anion-Exchanging Extractants
Extractant Applications
Tertiary amines Separation of Co and Cu from Ni in acidic chloride solutions(TIOA)
Extraction of Zn and Hg from acidic chloride solutionsExtraction of U, V, W, and Mo from acidic sulfate solutionsExtraction of Re from acidic nitrate solution
Quaternary amines Extraction of Mo from carbonate solutionExtraction of Na2CrO4
Extraction of V from caustic solution
TABLE 8.2-5 Some Commercial Solvents Used as Diluents in Metal Extraction Processes
a Union Oil Co."Chevron Oil.'Shell.''Esso or Exxon.'Kerr McGee.
Source: Reference 2.
to form when the extraction proceeds. This presents serious problems in fluid handling and is avoided bythe addition of modifiers. The most common modifiers are alcohols, phenols, and TBP, which act ascosolvents by virtue of their polar groups. Modifier concentrations are usually 5-10% by volume of theorganic phase, a level comparable to that of the extractant in many cases. Like the diluent, the modifiercan also interact with the solute species and affect the extraction reaction in various ways.10
8.2-5 Competing and Supporting Reactions
In the outline of common extraction mechanisms given above, the phenomenon of metal extraction isviewed as a chemical reaction occurring across a liquid-liquid phase boundary. It is clear, however, thatmore than one reaction occurs in almost all extraction systems. In the aqueous solutions the metal ionsoften interact with anions or complexing agents or water (by hydrolysis) to form a variety of species. Suchreactions may promote the extraction of the metal, as in the case of anionic complexes with amines, orthey may hinder it and promote stripping, as with precipitation of hydroxides or sulfides or with ammoniacomplexing that competes with cation exchange. In the organic phase there are parallel interactions, suchas association of extractant molecules into dimers and even polymers or micelles. Between phases otherreactions may occur simultaneously. Water, acids, or other metals may be extracted by interaction withthe extractant, and multiple species of organic-soluble metal may also form. Introduction of modifiers aswell as diluents increases the number of possible interactions and the complexity of these systems.
In spite of the large number of experimental studies of various extraction systems reported in theliterature, the range of variables available in these highly interacting, multicomponent systems leaves manyregions unexplored. Therefore, the development of a solvent-extraction process requires extensive experi-mentation. The general behavior of many available reagents is known, but the effects of interactions in aspecific system must be determined experimentally.
It must also be recognized that one is seldom working with pure chemicals in these extraction systems.Not only are the aqueous feed streams usually complex mixtures, but the commercial extractants, diluents,and modifiers are themselves mixtures and contain unknown impurities. Therefore, each system of interestmust be characterized by experimental investigation.
To attain a quantitative understanding of a particular metal extraction system, one must gather as muchinformation as possible about the chemical species forming in the system. Molecular weights and states ofaggregation should be determined for both extractant and extracted metal species. Maximum metal loadingshould be measured as an indication of the extraction-reaction stoichiometry. Degree of water coextractionshould be determined, and the tendency of other species such as acids to extract should be examined.Although extensive analytical work is required, such information is needed to construct meaningful materialbalances as well as to formulate equilibrium and rate analyses.
8.3 PHASEEQUILIBRIA
Often the equilibrium extent of extraction of a metal is described in terms of the phase distribution coef-ficient,
__ (concentration of metal in organic phase)(concentration of metal in aqueous phase)
Much of the published information about phase equilibria is presented in terms of D. Although the distri-bution coefficient depends on many variables, because of the multicomponent and reactive nature of thesesystems, many studies report D for a trace amount of metal in fixed compositions of aqueous and organicsolutions, usually at ambient temperature. Such data are independent of metal concentration.
Early reagent-screening experiments are summarized by Marcus and Kertes1 in a form such as shownin Figs. 8.3-1 and 8.3-2. Figure 8.3-1 presents low-concentration distribution coefficient values for variousmetals in a 5% solution of TOPO in toluene for several concentrations of hydrochloric acid in the aqueousphase. Figure 8.3-2 shows a similar presentation of distribution coefficients from sulfuric acid solutionsinto undiluted TBP.
As an example of the use of such information, suppose that one wished to separate zinc and copperpresent in an acidic chloride solution. Figure 8.3-1 indicates that the distribution coefficients of the twometals with 5% TOPO in toluene are on the order of 100 and 0.01, respectively, for an aqueous phasecontaining 1.0 M HCl. Therefore, this extractant should be effective in separating zinc from copper if theaqueous chloride level is not too high. Furthermore, the distribution coefficient of zinc falls to 0.01 in 12M HCl so that a concentrated hydrochloric acid solution would be a candidate for accomplishing thestripping of zinc from the loaded organic phase. This information suggests that some experimental workwith TOPO and the zinc-copper solution would be justified to determine the phase equilibria more com-pletely.
Trace-level distribution coefficients are often plotted as functions of acid or anion concentration for agiven extractant and presented as an overlay of the Periodic Table to indicate the efficacy and selectivityof the reagent. Figures 8.3-3 and 8.3-4 summarize extraction of metals by some tertiary and quaternaryamines from hydrochloric acid solutions. The ordinate of each graph is log D, which ranges from 10~2 to104, and the abscissa is HCl concentration, ranging from 0 to 12 Af. The high distribution coefficients offerric chloride with amines shown in Figs. 8.3-3 and 8.3-4 indicate the basis for using amine extraction asthe first step of the process shown in Fig. 8.1-2. Numerous charts of this form are available for variousextraction systems.2"4
Another way to present tracer-level metal equilibrium data is to plot the fraction of the metal extractedinto the organic phase as a function of some composition variable, often pH. Implicit in such a presentationis a phase volume ratio of unity. Figure 8.3-5 shows the percent extraction of several metal cations bynaphthenic acid as a function of pH. Such data indicate the potential for separating certain metals with aparticular reagent; they also show the possibility of stripping from the loaded organic by pH control. Forexample, Fig. 8.3-5 suggests that cupric and ferric ions can both be extracted by naphthenic acid if the pHof the aqueous phase at equilibrium is held above 4 by buffering or addition of base. On the other hand,only ferric ion should be extracted if the pH is adjusted to 2.5. With regard to stripping, addition of acidto lower the pH to 1.0 should strip all metal from the organic phase. Alternatively, the curve for copperin ammonium nitrate suggests that copper can be stripped at high pH in the presence of ammonia.
Although representative values of the distribution coefficients of various metals with different extractantsare useful in guiding the selection of a reagent for a particular separation process, the information providedby low-concentration D values is incomplete. For example, Fig. 8.3-3 indicates that the distribution coef-ficient of mercuric ion between hydrochloric acid and a solution of tri-isooctylamine in xylene(0.14 M) ison the order of 100. Figure 8.3-6 shows the distribution coefficient of mercuric ion from 4.63 M NaClsolution at pH = 1 and indicates that the value can be considerably higher and that it varies with the levelof mercury in solution. At high concentrations of metal, extractant is consumed by the extraction reaction,and the distribution coefficient falls as the finite stoichiometric loading of the organic phase is approached.
A further complication in the equilibrium is shown in Fig. 8.3-7, which shows the mercury distributioncoefficient as a function of amine concentration for a fixed level of mercury. At low amine concentrationsthe distribution coefficient increases with amine level in the way that is expected for reaction (8.2-7) or(8.2-8), but above a certain amine level the distribution coefficient becomes constant. This behavior indi-cates that some separate phenomenon, such as limited solubility or association of the amine-hydrochloridesalt, keeps its activity constant with increasing concentration. This is a common experimental observationin such systems.
Although distribution coefficients in metal extraction systems are highly variable, they are thermody-namic quantities that can be measured and correlated as functions of the state of the system. Because ofthe multicomponent nature of metal extraction systems, one must take care to identify a proper set ofindependent variables in constructing a correlation of phase equilibrium data. Another consideration intreating equilibrium extraction data is that one desires a systematic method for treating the effects of
FIGURE 8.3-1 Trace-level distribution coefficients of metal ions into a 5% solution of TOPO in tolueneas functions of aqueous HCl concentration. From Ref. 1, with permission.
additional species, such as other metals, modifiers, and so on, on the equilibrium distribution of a particularmetal. A chemical-reaction model of extraction equilibria provides an appropriate framework for correlatingdata because the large, but reversible, distribution coefficients of most interesting systems are the result ofreversible chemical reactions.
For a cation-exchanging extractant HR, which extracts a divalent metal ion M2 + according to reaction(8.2-3), a chemical-reaction model of the phase equilibrium would be the mass-action equilibrium expres-sion
(8.3-1)
where the quantities in brackets are activities and the equilibrium constant K for a specific metal-extractantsystem depends on temperature and the identity of the diluent. If activities were identical to analytical
FIGURE 8.3-2 Trace-level distribution coefficients of metal ions into undiluted TBP as functions ofaqueous sulfuric acid concentration. From Ref. 1, with permission.
concentrations, knowledge of A" in Eq. (8.3-1) would allow calculation of the equilibrium metal distribution,or D = (MR2V(M2+), for any given experimental conditions. For example, for the case of a trace amountof metal, equal phase volumes, and an extractant concentration fixed at 0.1 Af, Eq. (8.3-1) with constantK yields the metal distribution curves shown in Fig. 8.3-8, which may be compared with Fig. 8.3-5. Avalue of K = 1.0 represents an extraction equilibrium that provides complete extraction at pH > 2 andstripping at pH < 0.
To account for nonidealities in solution one can use activity coefficients or simply replace activitieswith species concentrations in Eq. (8.3-1) to obtain an empirical mass-action equilibrium quotient that isconcentration dependent. Bauer and Chapman5 treated data for copper extraction by Kelex 100 by a chemicalmodel, both with and without explicit activity coefficient terms, and were successful in extrapolating fromresults with 5% Kelex to obtain an accurate copper distribution curve for 20% Kelex. Hoh and Bautista6
used a similar approach to correlate equilibrium data for copper with LIX 65N and with Kelex 100. Theconcentration-based equilibrium constant at 25 0C was found to be on the order of 0.03 for LIX 65N in
FIGURE 8.3-3 Trace-level distribution coefficients of metal ions into solutions of tertiary amines asfunctions of aqueous HCl concentration: —, 0.14 M TIOA in xylene or kerosene; • • • , 0.1 M Alamine336 in diethylbenzene; <, D < 10~2 at 0.5-12 M HCl. From Ref. 17, with permission.
FIGURE 8.3-4 Trace-level distribution coefficients of metal ions into solutions of quaternary ammoniumsalts as functions of aqueous HCl concentration: • • • , 0.1 Af Aliquat 336 in diethylbenzene + 3% tride-canol; —, 0.1 M Hyamine 1622 in 1,2-dichloroethane; , 0.2 M tetra-fl-hexylammonium iodide inhexone; <, D < 10"2 at 0.2-5 M HCl. From Ref. 17, with permission.
EQUILIBRIUM pMFIGURE 8.3-5 Fractional amount of extraction of trace levels of some metal cations from aqueous solutioninto organic solutions of naphthenic acid as a function of pH. From Ref. 18, with permission.
toluene,6 5 for Kelex 100 in toluene,6 and 90 for Kelex 100 in xylene5. From trace-level measurementsAkiba and Freiser7 found the equilibrium constant for copper extraction by LIX 65N to range from 10"° 2
to 1013 in various solvents.Virtually no metal extraction system involves only one reaction. In aqueous metal salt solutions there
occur homogeneous association and complexation reactions that make true species concentrations differfrom the analytical concentrations that are measured and that enter into material balances. In sulfate solutionsbisulfate formation occurs at low pH, and metal-sulfate ion pairing may take place. In chloride and ammoniasolutions metal complexation is common. Dimerization and other association reactions often occur in theorganic phases. And two-phase extraction reactions of acids or other species may take place simultaneouslywith metal extraction or stripping. The equilibria of these reactions must be modeled concurrently with the
PERC
ENT
EXTR
AC
TIO
NDI
STRI
BUTIO
N CO
EFFIC
IENT
S,
CONCENTRATION IN THE ORGANIC PHASE (̂ g Hg/mL)
FIGURE 8.3-6 Distribution coefficient of mercuric ion from 4.63 M NaCl solution at 22°C and pH = 1as a function of organic-phase mercury concentration. The organic phase is TIOA in xylene at 0.1 M and0.5 M as indicated. From Ref. 13, with permission.
TOTAL AMINE CONCENTRATION, 5> (mol/L)FIGURE 8.3-7 The effect of amine concentration on the distribution coefficient shown in Fig. 8.3-6. FromRef. 13, with permission.
metal-extraction reaction if the true reactive species concentrations are affected by them. If such reactionsare overlooked in an equilibrium model, system behavior will appear to be highly nonideal, and extremevalues of empirical activity coefficients will be required.
The relatively large values found for the extraction equilibrium constant of copper with Kelex 100 (5and 90) indicate that stripping of copper from this reagent should be difficult. It is found, however, thatcopper does strip readily into sulfuric acid solutions because Kelex 100 reacts with sulfuric acid in preferenceto copper. Fitting the extraction of sulfuric acid by Kelex 100 by a chemical-reaction equilibrium constant,
(8.3-2)
at 500C, and solving the two equilibria simultaneously describes adequately the equilibrium distribution ofcopper under stripping conditions.5 This is shown in Fig. 8.3-9.
The acidic copper-chelating extractants also can extract other metals from alkaline ammonia solutions,which are used in some hydrometallurgical processes to exclude iron. DeRuiter8 modeled the extraction ofcopper(II) by LIX 64N by considering the following reversible reactions: complexation of aqueous copperby four and by five ammonia ligands, the ion-exchange extraction of the free copper cation, hydrolysis ofammonia to ammonium, and extraction of ammonia by both physical solubility and by chemical associationwith LIX. Not only does this chemical-reaction model fit the copper-extraction equilibrium, but the equi-librium constant for extraction of free copper cations, found to be 0.02, is in good agreement with thatdetermined in acidic sulfate solutions.
DIS
TRIB
UTI
ON
C
OE
FFIC
IEN
T,
EQUILIBRIUM pHFIGURE 8.3-8 Fractional amount of extraction of trace levels of metal cations from aqueous solution intoorganic solutions of an acidic extractant as a function of pH. Computed from Eq. (8.3-1) for several valuesof the equilibrium constant K.
Nickel is extracted from alkaline ammonia solutions by LIX 64N, but its equilibrium constant is fiveorders of magnitude smaller than that of copper.8 Also, it forms ammonia complexes more extensivelythan copper, which enhances the selectivity of LIX 64N for copper over nickel. Under conditions ofsimultaneous extraction the parameters of the chemical-reaction model for each individual metal are ableto predict the two-metal equilibria provided that the equilibrium distribution of species in the aqueous phaseis taken into account.
It appears that solutions of the common chelating agents behave ideally in the sense that the organic-phase species in these systems usually exhibit activities that are proportional to their concentrations. Un-fortunately, such behavior is the exception rather than the rule. Figure 8.3-7 illustrated nonideal behaviorof TIOA in the extraction of mercuric chloride. Organic-phase nonideality is commonly encountered withsolvating extractants and most cation exchangers as well as amines. As with the aqueous phase most ofthe nonideality can be accounted for by identifying association reactions in the organic phase and modelingthe chemical equilibrium among species in solution.
An example of nonideal organic-phase behavior arises with di-2-ethylhexyl phosphoric acid (DEHPA)in the extraction of metal cations. Equilibrium data of Troyer9 for extraction of copper in the presence ofnickel from sulfate solution into xylene solutions of DEHPA are shown in Fig. 8.3-10. Although theorganic-phase copper concentration would be expected to rise in proportion to that in the aqueous phase,
PE
RC
EN
T
EX
TRA
CTE
D
OR
GA
NIC
CO
PPER
CO
NC
ENTR
ATI
ON
(k
g/m
3)
OATA15% KELEX 10020% NONYL PHENOLIN ESCAIO 100
H2SO4 IN AQUEOUS PHASE (kg/ms)
FIGURE 8.3-9 Comparison of chemical-equilibrium extraction models with stripping data for copper inKelex 100. Curve A follows Eq. (8.3-1). Curve B is obtained by solving Eq. (8.3-1) simultaneously withEq. (8.3-2). From Ref. 5, with permission.
INITIAL CuSO4 CONCENTRATION (mole/liter)FIGURE 8.3-10 Predicted and observed organic copper concentrations in equilibrium with O.1 M DEHPAin xylene for different amounts of base added and several copper-to-nickel feed ratios. Curves are predictedby the model for the indicated initial NaOH concentrations. From Ref. 8.4-10, with permission.
the data indicate that the former is rather insensitive to the latter. Detailed data analysis indicates that theactivity of free extractant is nearly independent of its analytical concentration, and this is caused byreversible polymerization of DEHPA. The equilibrium data can be modeled by accounting for the followingreactions: complete dimerization of DEHPA, formation of an organic metal species of the formMR2 • 2HR, metal hydrolysis to form a monohydroxy complex, formation of a metal-sulfate ion pair,formation of bisulfate, extraction of sodium to form NaR * 3HR, and association of the extractant dimersto form a hierarchy of oligomers and polymers. The equilibrium of each of these reactions is governed bya mass-action equilibrium expression. Many aqueous-phase stability constants are reported in the litera-ture,10 and the sodium-extraction equilibrium constant can be measured separately. Thus, metal-extractionequilibrium data can be fit with only two additional parameters, the metal-extraction-reaction equilibriumconstant K and a parameter characterizing the degree of polymerization. The latter parameter must relatethe concentration of the reactive extractant species to the total analytical concentration of extractant insolution.
The curves in Fig. 8.3-9 have been computed from such a chemical-reaction model.9 For an extractionreaction of the form
(8.3-3)
the equilibrium constant is found to be 0.148 M"1 for copper and 1.65 X 10~4 Af"1 for nickel. Thesevalues indicate that copper is extracted at a lower pH than is nickel, starting at 2 or 3 compared with 3 or4. If enough base is used, the extractant can be loaded with either metal, but when both metals are present,and the amount of DEHPA or base is limited, the extractant is quite selective for copper. The individualand simultaneous extractions of both metals as well as stripping are described adequately by the chemical-reaction model.
Amine-extraction equilibria can also be modeled by chemical-reaction equilibrium constants. Figure8.3-3 indicates that cations such as iron(III), zinc, cobalt(II) and copper(II) exhibit high distribution coef-ficients with chloride solutions, whereas nickel, iron(II), and manganese are not extracted to any greatextent. The basis for the differences in distribution coefficients lies mainly in the tendency for the formergroup of cations to form chloride complexes. Stability constants for these complexes are available in theliterature,11 and they can be used to develop quantitative phase-equilibrium models.
For TIOA with hydrochloric acid the concentration-based equilibrium constant for salt formation12
according to reaction (8.2-6) is 1.51 x 104 M~2, and the equilibrium constant for amine-hydrochloridesalt dimerization13 is 8.0 M~x. Combination of these parameters and the ion-complex stability constantswith experimental metal-distribution data allows determination of the equilibrium constants for reactions(8.2-5) or (8.2-7). This completes the description of the amine-metal extraction-phase equilibria. Forcobalt(II) in acidic sodium chloride solutions the equilibrium constant14 for reaction (8.2-7) with TIOA is2.0 x 104 M~2, and that for copper(II) is 370 Af"2. The corresponding value for zinc15 is 7.5 x 10* Af"2
In spite of these relative values, the order of selectivity of TIOA for extraction of the metals is Zn > Cu> Co because of the relative extent of chloride complex formation. For the same reason, zinc stripping isdifficult in this system, and copper has a tendency to be reduced to cuprous, which also complexes andextracts extensively.
phos« ratio* I:I
FEED RATIO
Unfortunately, few of the published studies of extraction equilibria have provided complete quantitativemodels that are useful for extrapolation of data or for predicting multiple metal distribution equilibria fromsingle metal data. The chemical-reaction equilibrium formulation provides a framework for constructingsuch models. One of the drawbacks of purely empirical correlations of distribution coefficients is that pHhas often been chosen as an independent variable. Such a choice is suggested by the form of Figs. 8.3-5and 8.3-8. Although pH is readily measured and controlled on a laboratory scale, it is really a dependentvariable, which is determined by mass balances and simultaneous reaction equilibria. An appropriate phase-equilibrium model should be able to predict equilibrium pH, at least within a moderate activity coefficientcorrection, concurrently with other species concentrations.
Workers at Oak Ridge National Laboratory have announced the establishment of a Separations ScienceData Base, which should be useful in seeking equilibrium data for specific extraction systems.16
8.4 EXTRACTION KINETICS
Because the metal-extraction separation process involves chemical reactions, rates may be slow comparedwith ordinary liquid extraction. Although slow kinetics have important process design implications, moreattention has been focused on mechanistic interpretations than on quantitative phenomenonological char-acterization. Cox and Flett1 have reviewed some chemical aspects of extraction kinetics.
One issue in the study of extraction kinetics is the question of whether the metal-extraction reaction,or its rate-determining step, occurs heterogeneously or homogeneously in the aqueous phase.2 For practicalpurposes, the locale of the reaction is usually irrelevant because the kinetics of most systems can be fit bya heterogeneous model. In practical extraction systems, the aqueous solubility of the extractant is necessarilyvery low; organic-to-aqueous distribution coefficients of some common extractants have been determinedto be 103 and higher.3 A small homogeneous reaction rate constant combined with very low extractantconcentrations in the aqueous phase would provide an extraction rate so slow as to make the processuninteresting. On the other hand, if the homogeneous rate constant is large enough to provide finiteextraction rates, the Hatta number4 for the reaction between metal ion and aqueous extractant will normallybe large. The Hatta number is a dimensionless ratio of the homogeneous reaction rate constant to the filmmass transfer coefficient. According to the theory of mass transfer with homogeneous reaction, a Hattanumber greater than 3 causes the reaction to go to completion within the mass transfer boundary layer.This condition requires only that the pseudo-first-order rate constant be greater than about 10 s ~ \ whereasreported values of rate constants in "slow" extraction systems have generally been found to be muchlarger.5
Furthermore, for a large ratio of bulk metal ion concentration to the interfacial aqueous extractantconcentration, the reaction zone is located very close to the liquid-liquid interface, and the extraction rateper unit area of interface becomes independent of the aqueous-film mass transfer coefficient as well as thesystem volume. Under these conditions a truly heterogeneous reaction and a homogeneous process areindistinguishable in the sense that the rate of each will be a definite function of reactant concentrations inthe vicinity of the interface. In either case, the functional form of the kinetics and its parameters must bedetermined experimentally.
The critical question regarding metal-extraction (or stripping) kinetics is whether the chemical reactionsthat accomplish the phase transfer of the metal are fast or slow relative to the prevailing mass transfer rates.Mass transfer rates may be characterized conveniently in terms of a two-film model, with film mass transfercoefficients being typically on the order of 10"2 cm/s in liquid-liquid systems. (This value corresponds toan effective stagnant-film thickness of 10~3 cm; a homogeneous reaction zone might lie within 10~5 cmof the interface for a bulk metal concentration 100 times the aqueous extractant solubility.) Within thiscontext maximum, or mass-transfer-limited, extraction rates, obtained when the extraction reaction main-tains equilibrium at the interface, can be computed.
For a simple cation-exchange extraction, such as reaction (8.2-3), concentration profiles near the in-terface are represented by Fig. 8.4-1. The fluxes at the interface of all four species are related by thereaction stoichiometry. The interfacial concentrations depend on the bulk-solution concentrations, the in-terfacial metal flux, and the respective mass transfer resistances. Requiring the interfacial concentrationsto be in equilibrium according to Eq. (8.3-1) yields the following equation6 for the metal-extraction rate:
(8.4-1)
in which the interfacial metal flux Nm is an implicit function of bulk concentrations, film mass transfercoefficients kh and the equilibrium constant. Figure 8.4-2 is a plot of Eq. (8.4-1) for several combinationsof bulk-phase concentrations; the ordinate is the ratio of the metal flux to its value for (M2+) = 0 at theinterface and the abscissa represents deviation from equilibrium of the bulk concentrations. Figure 8.4-2demonstrates that the extraction rate is affected by transport of ail reactive species because all transportrates must be considered in estimating interfacial concentrations.
FIGURE 8.4-1 Concentration profiles near the interface during metal extraction by an acidic extractant.
If slow heterogeneous kinetics affect the metal-extraction rate, an appropriate kinetics model should beexpressed in terms of interfacial species concentrations, which are obtained from bulk values by correctingfor mass transfer resistances. Figure 8.4-3 combines an empirical kinetics model7 for copper extraction byKelex 100 with a two-film mass transfer model to indicate the relative importance of mass transfer andkinetics in different regions of composition. For this system, the reaction kinetics become fast at higherpH and high extractant and metal concentrations. As the flux approaches the mass-transfer-limited flux,given by Eq. (8.4-1), the interfacial concentrations approach the corresponding equilibrium values. Atlower pH and low extractant level, the kinetics are considerably slower. Under these conditions the inter-facial concentrations maintain their bulk values.
ORGANICPHASE
AQUEOUSPHASE
FIGURE 8.4-2 Dimensionless extraction flux as a function of distance of bulk-phase concentrations fromequilibrium as computed from Eq. (8.4-1). Curves 1 and 2 represent different bulk concentration ratios.Curve P-B represents a pseudobinary calculation where gradients in H+ and HR arc neglected. From Ref.6, with permission.
CC|I*2 (MOLESAJTER)FIGURE 8.4-3 The rate of extraction of copper from sulfate solution by Kelex 100 in xylene. Theexperimental flux is presented relative to the mass-transfer-limited metal flux for kM2* = 10"2 cm/s. FromRef. 6, with permission.
There is considerable confusion in the literature about extraction kinetics, and little information directlyapplicable to process calculations is available. Part of the problem is that many rate studies have been doneunder conditions that were not well characterized with respect to mass transfer effects or interfacial areaso that quantitative heterogeneous rate models cannot be derived. Another problem is irreproducibilitycaused by unknown impurity levels in extractant solutions. Kinetics are much more sensitive to impurities,additives, and changes in diluent than are equilibrium properties. Finally, it is clear that interfacial phe-nomena have a strong, and variable, effect on extraction kinetics. Most extractants are surface-active agentsas are many modifiers and impurities. Interactions with the surface can affect extraction kinetics directlyby blocking sites for heterogeneous reaction steps or providing an interfacial transport resistance, or indi-rectly by affecting hydrodynamic conditions. It is difficult to control these effects to obtain reproducibleand meaningful data.
A pragmatic approach to extraction kinetics is to measure extraction rates under mass transfer conditionsthat are similar to those expected in processing equipment and that are characterized well enough to allowestimation of interfacial concentrations. With such measurements one can determine whether the kineticsof a particular system are fast or slow compared to mass transfer, and if they are slow, the rate can becorrelated with interfacial conditions. Several methods are used to measure extraction kinetics; these includemeasurements with (1) well-stirred vessels, (2) single drops or jets, and (3) the Lewis cell.
Rate measurements with well-stirred vessels may be continuous or batch. An example of the latter issimply a stirred flask. The former include the AKUFVE apparatus8 and bench-scale models of mixer-settlercontactors.9 Because the interfacial area and mass transfer conditions in these systems are not known, theygenerally do not yield intrinsic heterogeneous rate data for metal-extraction systems. Nevertheless, theyare useful for screening relative rates and identifying cases where slow kinetics may be a problem.
The advantage of single-drop and liquid-jet experiments is that the interfacial area is known. Also, thehydrodynamics may be simple enough to allow calculation of film mass transfer coefficients. With risingor falling drops, however, adsorption of surfactants can alter the hydrodynamics and possibly interfere withthe extraction as well. A growing-drop experiment,10 analogous to the dropping mercury electrode ofpolarography, offers the advantages of continuous generation of fresh interface as well as known masstransfer conditions. Laminar-jet measurements11 offer similar advantages except that surfactants tend tobuild up on the downstream end of the interface and make the surface rigid.
The Lewis cell12 is a stirred cell with a planar interface between the two liquid phases. Although thehydrodynamics are complex and surfactants can build up on the interface, it is possible to calibrate themass transfer coefficients empirically. Also, it appears that sufficiently intense stirring can disrupt stagnantsurface layers.13
Although not a great amount of systematic kinetics data are yet available for metal-extraction systems,a few cases have been studied in detail. It is well established that the acidic chelating extractants generallyreact so slowly compared to ordinary mass transfer rates that special precautions must be taken in processdesign to provide adequate contacting time. According to the commercial importance of the copper-LIXextraction systems, much of the kinetics work to date has been aimed at this system.14"16 All the methods
FIGURE 8.4-4 Schematic drawing of the species profiles near the interface during the extraction of copperfrom ammonia solution by an acidic extractant.
listed above have been used, but differences in experimental conditions and mechanistic interpretationsmake quantitative comparison of the results difficult. With alkaline solutions containing ammonia theextraction of copper by LIX 64N becomes mass transfer limited, but extraction of nickel is controlled partlyby slow kinetics.17
From other experiments in which the effects of mass transfer have been analyzed, it appears that thefollowing systems are mass transfer limited: uranyl nitrate extraction by TBP,18 copper extraction bysodium-loaded DEHPA,19 and extraction of zinc and copper(II) chlorides by TIOA.20 Zinc extraction bydithizone in carbon tetrachloride is mass transfer limited at high zinc concentrations but kinetically controlledat low zinc levels.21 Ferric ion extractions are reputed to be slow because of its sluggish ligand-exchangekinetics.22 Extraction of ferric chloride by TIOA, for example, is controlled by a slow heterogeneousreaction.23
In mass-transfer-controlled systems in which extensive complexing or association takes place in thebulk phases, a proper mass transfer model must account for transport of all species. Otherwise, the transportmodel will not be consistent with a chemical model of phase equilibrium. For example, Fig. 8.4-4 indicatesschematically the species concentration profiles established during the extraction of copper from ammonia-ammonium sulfate solution by a chelating agent such as LIX. In most such cases the reversible homogeneousreactions, like copper complexation by ammonia, will be fast and locally equilibrated. The method ofOIander24 can be applied in this case to compute individual species profiles and concentrations at theinterface for use in an equilibrium or rate equation. This has been done in the rate analyses of several ofthe chloride and ammonia systems cited above.25
Stripping rates are as important for process viability as extraction rates, but less attention has been paidto their determination. In principle, stripping rates should be related through microscopic reversibility toextraction rates, but far from equilibrium the complete kinetics model is difficult to discern.
An interesting aspect of kinetics is the question of how species interact when multiple extractionreactions occur. Most extractants are not perfectly selective and are capable of extracting several speciesfrom a multicomponent aqueous solution. Complete equilibrium models should indicate extraction selec-tivity for a given system at equilibrium, but selectivity may vary considerably in a nonequilibrium processbecause of rate differences. In fact, iron(III) can be extracted by some of the copper-chelating extractants,but their effective selectivity for copper is based on the very slow kinetics of iron extraction.
Even in mass-transfer-limited processes, excursions in selectivity can be observed at finite contact times.This is predicted by rate models as simple as Eq. (8.4-1) for two metals with different equilibrium constantvalues.626 The phenomenon involves initially fast coextraction followed by crowding out of the lesspreferred metal during competition for extractant. This has been observed during simultaneous extractionof copper and zinc chlorides by TIOA in a growing-drop experiment10'25 and in extraction of uranyl nitrateand nitric acid by TBP in a Lewis cell,27 as shown in Fig. 8.4-5.
AQUEOUS PHASE ORGANIC PHASEBulk Stagnant Stagnant BulkFluid Film Film Fluid
Time (min)FIGURE 8.4-5 Measured and calculated organic-phase concentrations of nitric acid ( x ) and uranium (O)during coextraction by TBP in a Lewis cell. Concentrations are expressed as a percentage of their equilib-rium values. From Ref. 28, with permission.
8.5 CONTACTING EQUIPMENT AND DESIGN CALCULATIONS
The processing equipment used to conduct solvent extraction of metals is the same as that used in conven-tional liquid-liquid extraction.1'2 The most common choices have been mixer-settlers, columns with agi-tated internals, and static mixers. Some advantages and disadvantages of several classes of equipment aresummarized in Table 8.5-1. Many of the practical aspects of equipment selection are discussed by Prattand Hanson3 and by Ritcey and Ashbrook.4
Because detailed models of the chemical effects on phase equilibrium and rates in metal-extractionsystems have not been available, standard chemical engineering design procedures have not been appliedvery extensively in equipment sizing and comparison. Although McCabe-Thiele-type calculations are oftensuggested for designing equilibrium-stage operations, the results are only approximately correct becausethe locus of the metal-distribution equilibrium curve changes from stage to stage according to changes inpH and free extractant concentration. The problem is analogous to that encountered with multicomponentdistillation or nonisothermal gas absorption in the sense that the state of the system cannot be representedon a two-dimensional plot. Multiple conservation and rate equations must be solved simultaneously tosimulate process paths.
If the chemistry of an extraction system is well characterized and the equilibria among species andbetween phases are modeled adequately, equilibrium-stage calculations can be done numerically by solvingsimultaneously the algebraic equations that describe the equilibria and mass balances in each stage. Suchan approach has been demonstrated,5 and Table 8.5-2 shows calculated and experimental interstage con-centrations during multistage extraction of copper by Kelex. In such calculations it becomes clear thatanalytical reagent concentrations in feed streams must be used as independent process variables and thatpH is a dependent variable along with other individual species concentrations.
Although equilibrium-stage calculations are useful for preliminary design of staged contacting, char-acterization of equipment efficiency for a particular application requires experimental study. With mixer-settlers it is common to vary experimentally the number of real stages used as well as the residence timein each stage. Scale-up then is based on these parameters as well as power input per unit volume andsuperficial velocity in the settlers.3 By this approach large-scale copper-LIX processes have been designedwith three mixers for extraction and two for stripping.6*7 The slow kinetics of the reaction are accommodatedby a 2 min residence time in the mixers. Differential contactors can be characterized similarly in terms oftransfer units if the equilibria and rates are sufficiently well known.8
An interesting comparative study of six different contactors is summarized in Fig. 8.5-1.9 An alkalinefeed stream containing copper, nickel, zinc, and ammonia was contacted with a Kelex 100 solution in eachof the pilot-scale contactors. At a pH above 7 the reaction kinetics of Kelex are probably fast, but it losessome of its intrinsic selectivity for copper because of the tendency of nickel and zinc to be extracted.Although the various contactors could be made to operate at comparable levels with respect to specificthroughput and copper-extraction efficiency, they exhibited quite different selectivities. Detailed liquid-liquid reactor models would help to identify and predict such variations in efficiency and selectivity.
With both staged equipment and differential contactors, availability of adequate phase-equilibrium modelsand rate expressions would allow application of existing correlations and simulation algorithms. For ex-ample, knowledge of metal-extraction kinetics in terms of interfacial species concentrations could be com-bined with correlations of film mass transfer coefficients in a particular type of equipment to obtain theinterfacial flux as a function of bulk concentrations. Correlations or separate measurements of interfacialarea and an estimate of dispersion characteristics would allow calculation of extraction performance as a
TABLE 8.5-1. Some Advantages and Disadvantages of Several Classes of Liquid-Liquid Contactors
Low holdup volumeShort holdup timeSmall space requirementSmall inventory of
solvent
High initial costHigh operating costHigh maintenanceLimited number of
stages in single unit,although some unitshave 20 stages
Good dispersionReasonable costMany stages possibleRelatively easy scale-up
Limited throughput withsmall gravitydifference
Cannot handleemulsifying systems
Cannot handle high flowratio
Will not always handleemulsifying systems,except perhaps pulsecolumn
Low initial costLow operating costSimplest construction
Limited throughput withsmall gravitydifference
Cannot handle wide flowratio
High headroomSometimes low efficiencyDifficult scale-up
Good contacting ofphases
Can handle wide rangeof flow ratios (withrecycle)
Low headroomHigh efficiencyMany stagesReliable scale-upLow costLow maintenanceLarge holdupHigh power costsHigh solvent inventoryLarge floor spaceInterstage pumping may
be necessary
Advantages
Disadvantages
Source: From Ref. 4, with permission.
TABLE 8.5-2. Comparison of Experimental and Calculated Stream Concentrations for Copper Extraction by Kelex 100 in a Three-StageCountercurrent Cascade of Mixer-Setters
FIG URE 8.5-1 Comparisons of the operating characteristics of several contacting devices for the separationof copper from nickel and zinc by extraction from ammonia (pH 8) solution with Kelex. (a) Comparisonof specific throughput at flooding, (b) Comparison of copper extraction efficiency, (c) Comparison of Cu-Zn selectivity, (d) Comparison of Cu-Ni selectivity. From Ref. 9, with permission.
Podbielniak 25°C
Copper extraction (%)
Mixer-settlers g j r J g S
Mixco column organic cont.
Mixco column aq. cont.
Pulse column 600C
Pulse column 25°C
Kenics 60° C—4 min
Kenics25°C—4min
Graesser
Podbielniak 500C
(a)
Ken
ics
mix
er
Pul
se s
ieve
pla
te c
olum
n
Mix
co c
olum
n
Mix
er-s
ettle
rs
Gra
esse
r
Tota
l flo
w (
gal/h
-ft2
)
i -3
i
(d)
FIGURE 8.5-1 (Continued)
Cu/N
i Rat
io in
load
ed s
olven
t /%
/•»«
*.••
• ̂ -
i .
Cu/Z
n Ra
tio in
load
ed so
lvent
Pulse
25°
Cco
lum
n 60
0C
Mixe
r-set
tlers
60°
C-2
stag
es
Keni
cs25
°Cm
ixer 6
0° C
—4m
in
Mixc
o aq
.co
nt c
olum
n or
g. c
ontin
uous
25°
C
Grae
sser
Pod.
25°C
500C
(c)
Pulse
colu
mn
||££
Mixe
r-set
tlers
60°C
~2 st
ages
Kenic
s m
ixer
£QOQ
Mixc
o or
g. c
ont.
25°C
colum
n aq
. con
t.
Grae
sser
25°
C
Podb
ielnia
k 25
°C60
0C
function of equipment size. A strategy for developing such contactor models has been outlined,10 and theapproach has been demonstrated with a reaction-controlled" and mass-transfer-controlled12 extractions ina Kenics static mixer.
8.6 PROCESSDESIGNANDENGINEERING
Process flowsheets for separations of numerous metals have been published.1"5 Synthesis and design ofsuch processes for a given feed stream require consideration of the following factors:
1. Choice of Extractant. For separation of a particular metal, an effective extractant must be chosenthat has the capability of providing high distribution coefficients and high organic-phase loadings. Thischoice depends on the chemistry of the metal and the composition of the aqueous feed solution. For example,a chelating extractant may be appropriate for extracting copper from a weakly acidic sulfate solution, butan amine may be required for a solution that has a high level of hydrochloric acid.
A practical extractant must also be subject to regeneration. The extraction must be reversible such thatstripping is possible with minimal consumption of reagents. Stripping should yield a purified and concen-trated form of the metal product.
With feed streams that contain several metals, selectivity may be a major concern. To obtain selectivityone may seek a different extractant for each metal that is highly specific by virtue of its equilibrium orkinetics properties, or one may use a single extractant for several metals and design the contacting processto achieve effective fractionation by exploiting quantitative rather than qualitative differences in metalchemistry. For example, in the process6 represented by Fig. 8.6-1 TBP is chosen to extract iron from achloride feed stream containing copper and cobalt. Then copper and cobalt chlorides are both extractedinto an amine solution. The separation of cobalt from copper is accomplished by controlling the water-to-organic phase ratios in two separate stripping circuits. Similar strategies are represented by the flowsheetsshown in Figs. 8.1-1 and 8.1-2. Process models for multimetal fractionations with a single extractant havebeen presented7 for an idealized process chemistry.
2. Choice of Diluent, Modifiers, and Extractant Concentration. The formulation and composition ofthe organic-phase solution play a major role in determining both the chemical and physical performancecharacteristics of an extraction system. The use of modifiers and mixed extractants alters both equilibriumand kinetics properties and can have a strong influence on interfacial phenomena. It is particularly importantto formulate a system that minimizes organic solubility in the aqueous phase, promotes phase disengage-ment, and prevents emulsion formation. Problems in any of these areas can quickly render a processuneconomical or infeasible.
3. Choice of Process Configuration. The sequence of processing steps and flowsheet configurationmust be selected concurrently with the choice of extraction chemistry. Some of the established heuristicsof separation process synthesis8 may be helpful here. For example, in multimetal fractionation it is probablybetter to try selective stripping from a single organic stream than to do multiple selective-extraction oper-
M/S = mixer-settlerFIGURE 8.6-1 Solvent extraction circuit of the Falconbridge Nikkelvert A/S matte leach plant for sepa-ration of iron, copper, and cobalt. From Ref. 6, with permission.
Feedsolution
Fe Loading Co-Cu Loading
Settler Settler TocrystallizationM/SM/SM/SM/SM/S
Fe Stripping Co-Cu Stripping
Wate
r
Wate
r
Wate
r
Fe-eluatestorage
TBPstorage
Co-eluatestorage
Cu-eluatestorage
TIOAstorage
M/S M/S M/S M/S M/S M/S M/S M/S M/S
ations. The former approach, if possible, would minimize handling of the total feed stream volume andpresumably reduce solvent losses and reagent consumption.
4. Choice of Contacting Equipment. Although each extraction and stripping operation in a metals-separation process is normally operated in a countercurrent mode, either stagewise or in differential contact,the large distribution coefficients provided by the chemical-reaction basis of the separations suggest that alarge number of stages, or transfer units, is not required. On the other hand, slow kinetics may restrictefficiency factors considerably so that high interfacial areas and residence times may be required. Theseconditions have favored the use of mixer-settlers. Where kinetics are fast, differential contactors may bepreferable because of smaller power requirements and solvent inventory requirements.
While efficiency is a factor in equipment selection, mechanical considerations often provide the deter-mining criteria. One must always be sure to minimize solvent losses, and concern about entrainment andemulsion formation can dictate the mode of operation if not the choice of contactor. For example, powerinput for mixing may be limited or the less viscous phase chosen to be continuous to ensure good phasedisengagement.
Many of these and other practical aspects of process design and operation, such as appropriate materialsof construction, prevention of * 1CiUd" formation, effluent treatment methods, and typical process costs,are discussed by Ritcey and Ashbrook.9
If any given extraction system can be characterized with respect to the dominant species appearing andreactions occurring, experimental equilibrium and rate data can be correlated in terms of the speciesconcentrations. Such information can be combined with material balances and available engineering cor-relations for liquid-liquid systems to generate process models useful for design, optimization, and control.
8.7 SUMMARY
Organic reagents are available that can extract metal ions into an organic solvent by virtue of liquid-liquidion exchange or association reactions. Such reactions are reversible and provide a basis for effective metal-separation processes. Large distribution coefficients are observed, but they vary with system composition.
Phase equilibria can be modeled in terms of equilibrium constants for the relevant reactions. Becauseof low mutual solubilities of the phases, extraction reactions appear to be heterogeneous. Some reactionsexhibit slow chemical kinetics, with the reaction step constituting a resistance to extraction in series withintraphase mass transfer.
Large-scale solvent extraction of metals is conducted in equipment that is similar to that used forconventional liquid extraction. Nonlinearities in the multicomponent, reactive phase equilibria and possiblyslow kinetics complicate design calculations, but existing correlations and methods for treating mass trans-fer, interfacial phenomena, and dispersion can be used if the effects of process chemistry are propertycharacterized.
An extensive technology and a large body of experience have developed from both laboratory studiesand commercial process operations. A variety of extractants, equipment, and processes is available forapplication to a wide range of metals-separation problems.
REFERENCES
Section 8.1
8.1-1 Y. Marcus and A. S. Kertes, Ion Exchange and Solvent Extraction of Metal Complexes, Wiley,
New York, 1969.8.1-2 H. Freiser, CHt. Rev. Anal. Chem., I9 47 (1970).8.1-3 J. Stary, The Solvent Extraction of Metal Chelates, Pergamon, Oxford, 1964.8.1-4 A. K. De, S. M. Khopkar, and R. A. Chalmers, Solvent Extraction of Metals, Van Nostrand,
New York, 1970.8.1-5 H. A. C. McKay, T. V. Healy, I. L. Jenkins, and A. Nyalor (Eds.), Solvent Extraction Chemistry
of Metals, MacMillan, London, 1965.8.1-6 J. Korkisch, Modern Methods for the Separation of Rarer Metal Ions, Pergamon, Oxford, 1969.8.1-7 J. F. C. Fisher and C. W. Notebaart, Commercial Processes for Copper, in T. C. Lo, M. H. I.
Baird, and C. Hanson (Eds.), Handbook of Solvent Extraction, Chap. 25.1, Wiley-Interscience,New York, 1983.
8.1-8 G. Thorsen, Commercial Processes for Cadmium and Zinc, in T. C. Lo, M. H. I. Baird, and C.Hanson (Eds.), Handbook of Solvent Extraction, Chap. 25.5, Wiley-Interscience, New York,1983.
8.1-9 P. J. D. Lloyd, Commercial Processes for Uranium from Ore, in T. C. Lo, M. H. L Baird, and
C. Hanson (Eds.), Handbook of Solvent Extraction, Chap. 25.11, Wiley-Interscience, New York,1983.
8.1-10 G. M. Ritcey and A. W. Ashbrook, Solvent Extraction, Principles and Applications to ProcessMetallurgy, Part II, Chaps. 4-6, Elsevier, Amsterdam, 1979.
8.1-11 A. J. Monhemius, The Extractive Metallurgy of Deep-Sea Manganese Nodules, in R. Burkin(Ed.), Topics in Non-ferrous Extractive Metallurgy, Blackwell Scientific Publications for the So-ciety of Chemical Industry, Oxford, 1980.
8.1-12 T. C. Lo, M. H. I. Baiixi, and C. Hanson (Eds.), Handbook of Solvent Extraction, Wiley-Interscience, New York, 1983.
8.1-13 G. M. Ritcey and A. W. Ashbrook, Solvent Extraction, Principles and Applications to ProcessMetallurgy, Parts I and II, Elsevier, Amsterdam, 1979.
Section 8.2
8.2-1 G. M. Ritcey and A. W. Ashbrook, Solvent Extraction, Principles and Applications to ProcessMetallurgy, Elsevier, Amsterdam, 1979.
8.2-2 D. S. Flett, J. Melling, and M, Cox, Commercial Solvent Systems for Inorganic Processes, in T.C. Lo, M. H. I. Baird, and C. Hanson (Eds.), Handbook of Solvent Extraction, Chap. 24, Wiley-Interscience, New York, 1983.
8.2-3 Proceedings of the International Solvent Extraction Conference, The Hague 1971, Society ofChemical Industry, London, 1971.
8.2-4 Proceedings of the International Solvent Extraction Conference, Lyons 1974, Society of ChemicalIndustry, London, 1974.
8.2-5 Proceedings of the International Solvent Extraction Conference, Toronto 1977, Canadian Instituteof Mining and Metallurgy, Montreal, 1979.
8.2-6 G. M. Ritcey and A. W. Ashbrook, Solvent Extraction, Principles and Applications to ProcessMetallurgy, Part I, Chap. 3, Elsevier, Amsterdam, 1979.
8.2-7 V. S. Schmidt, Amine Extraction, Israel Program for Scientific Translation, Keter Press, Jerusalem,1971.
8.2-8 K. J. Murray and C. J. Bouboulis, Eng. Mining J., 174, 74 (1973).8.2-9 K. Akiba and H. Freiser, Anal. Chim. Acta, 136, 329 (1982).8.2-10 G. M. Ritcey and A. W. Ashbrook, Solvent Extraction, Principles and Applications to Process
Metallurgy, Part I, Chap. 4, Elsevier, Amsterdam, 1979.
Section 8.3
8.3-1 Y. Marcus and A. S. Kertes, Ion Exchange and Solvent Extraction of Metal Complexes, Appendix
F, Wiley, New York, 1969.8.3-2 F. G. Seeley and D. J. Crouse, J. Chem. Eng. Data, 11, 424 (1966).8.3-3 F. G. Seeley and D. J. Crouse, J. Chem. Eng. Data, 16, 393 (1971).8.3-4 Japan Atomic Energy Research Institute, Data of Inorganic Solvent Extraction, JAERI1047 (1963),
JAERI 1062 (1964), JAERI 1106 (1966).8.3-5 G. L. Bauer and T. W. Chapman, Met. Trans., 7B, 519 (1976).8.3-6 Y. C. Hoh and R. G. Bautista, Met. Trans., 9B, 69 (1978).8.3-7 K. Akiba and H. Freiser, Anal. Chim. Acta, 136, 329 (1982).8.3-8 R. A. DeRuiter, "Copper and Nickel Extraction from Ammoniacal Solution by LIX 64N," M.S.
Thesis, University of Wisconsin, Madison, 1981.8.3-9 S. D. Troyer, "Liquid-Liquid Extraction of Copper and Nickel with Di-(2-Ethylhexyl) Phosphoric
Acid," M.S. Thesis, University of Wisconsin, Madison, 1975.8.3-10 L. G. Sillen and A. E. Martell, Stability Constants of Metal Ion Complexes, Special Publication
No. 17 (1962), and Supplement No. 1, Special Publication No. 25 (1971), The Chemical Society,Burlington House, London.
8.3-11 J. J. Christensen and R. M. Izatt, Handbook of Metal Ugand Heats, Marcel Dekker, New York,1983.
8.3-12 T. Kojima, H. Fukutomi, and H. Kakihana, Bull. Chem. Soc. Jpn., 42, 875 (1969).8.3-13 R. Caban and T. W. Chapman, AIChEJ., 18, 904 (1972).8.3-14 S. W. Tse, "Mass Transfer Rates in the Solvent Extraction of Metal Chlorides by Tri-isooctylam-ine," M.S. Thesis, University of Wisconsin, Madison, 1978.
8.3-15 M. W. Thiel, "Separation of Copper and Zinc by Liquid-Liquid Extraction with Tri-isooctylam-ine," M.S. Thesis, University of Wisconsin, Madison, 1974.
8.3-16 W. J. McDowell, D. C. Michelson, B. A. Moyer, and C. F. Coleman, "A Data Base for SolventExtraction Chemistry," presented at International Solvent Extraction Conference, Denver, 1983.
8.3-17 C. F. Coleman, Amines as Extractants-Survey of the Descriptive and Fundamental ExtractionChemistry, Report ORNL-3516, Oak Ridge National Laboratory, Oak Ridge, TN, 1963. See alsoNucl. ScL Tech., 17, 274 (1963) and Prog. Nucl. Energy, Ser. Ill, Process Chemistry, Vol. 4,C. E. Stevenson, et al., eds., Pergamon, Oxford, pp. 233-285, 1970.
8.3-18 G. M. Ritcey and A. W. Ashbrook, Solvent Extraction, Principles and Applications to Process
Metallurgy, Part I, p. 112, Elsevier, Amsterdam, 1979.
Section 8.4
8.4-1 M. Cox and D. S. Flett, Metal Extractant Chemistry, in T. C. Lo, M. H. I. Baird, and C. Hanson
(Eds.), Handbook of Solvent Extraction, Chap. 2.2, Wiley-Interscience, New York, 1983.8.4-2 T. Kojima and T. Miyauchi, Ind. Eng. Chem. Fundam., 20, 14 (1981).8.4-3 K. Akiba and H. Freiser, Anal. Chim. Ada, 136, 329 (1982).8.4-4 G. F. Froment and K. B. Bischoff, Chemical Reactor Analysis and Design, p. 310, Wiley, New
York, 1979.8.4-5 T. Kojima and T. Miyauchi, Ind. Eng. Chem. Fundam., 21, 220 (1982).8.4-6 T. W. Chapman, R. Caban, and M. E. Tunison, AIChE Symp. Ser., 71 (152), 128 (1976).8.4-7 G. L. Bauer, "Solvent Extraction of Copper: Kinetics and Equilibrium Studies," Ph.D. Thesis,
University of Wisconsin, Madison, 1974.8.4-8 J. Rydberg, H. Reinhardt, and J. O. Liljensin, in J. A. Marinsky and Y. Marcus (Eds.), Ions
Exchange and Solvent Extraction, Vol. 3, p. I l l , Marcel Dekker, New York, 1973.8.4-9 M. J. Slater, G. M. Ritcey, and R. F. Pilgrim, in Proceedings of the International Solvent
Extraction Conference, Lyon, Vol. 1, p. 107, Society of Chemical Industry, London, 1974.8.4-10 T. W. Chapman, Solvent Extraction of Metals—Metal Transfer Rates and Contactor Design, in
R. G. Bautista (Ed.), Hydrometallurgical Process Fundamentals, Plenum, New York, 1984.8.4-11 R. W. Freeman and L. L. Tavlarides, Chem. Eng. ScL, 35, 559 (1980).8.4-12 J. B. Lewis, Chem. Eng. ScL, 3, 248 (1954).8.4-13 W. Nitsch and J. G. Kahni, Ger. Chem. Eng., 3, 96 (1980).8.4-14 D. S. Flett, D. N. Okuhara, and D. R. Spink, J. Inorg. Nucl. Chem., 35, 2471 (1973).8.4-15 R. L. Atwood, D. N. Thatcher, and J. D. Miller, Met. Trans., 6B, 465 (1975).8.4-16 R. J. Whewell, M. A. Hughes, and C. Hanson, J. Inorg. Nucl. Chem., 38, 2071 (1976).8.4-17 R. A. DeRuiter, "Copper and Nickel Extraction from Ammoniacal Solution by LIX64N," M.S.
Thesis, University of Wisconsin, Madison, 1981.8.4-18 W. Nitsch and U. Schuster, Separation ScL Tech., 18, 1509 (1983).8.4-19 C H . Hales, "The Effect of Bulk Concentrations on Liquid-Liquid Extraction of Copper with
Sodium-Loaded Di-(2-Ethylhexyl)Phosphoric Acid," M.S. Thesis, University of Wisconsin, Mad-ison, 1977.
8.4-20 S. W. Tse, "Mass Transfer Rates in the Solvent Extraction of Metal Chlorides by Tri-isooctylam-ine," M.S. Thesis, University of Wisconsin, Madison, 1978.
8.4-21 W. Nitsch and B. Kruis, J. Inorg. Nucl. Chem., 40, 857 (1978).8.4-22 J. W. Roddy, C. F. Coleman, and S. Arai, J. Inorg. Nucl. Chem., 33, 1099 (1971).8.4-23 J. C. S. Cassa and A. J. Monhemius, Kinetics of Extraction of Iron(III) from Chloride Solutions
by Trioctylamine, in R. G. Bautista (Ed.), Hydrometallurgical Process Fundamentals, p. 327,Plenum, New York, 1984,
8.4-24 D. R. Olander, AIChE J., 6, 233(1960).8.4-25 S. W. Tse, E. S. Vargas, and T. W. Chapman, Mass Transfer and Reaction Rates in the Solvent
Extraction of Metals, in Hydrometallurgical Recovery of Metals from Ores, Concentrates, andSecondary Sources, Chap. 19, SME-AIME, New York, 1981.
8.4-26 A. Altway, M. E. Tunison, and T. W. Chapman, Effect of Contactor Type on Metal Separationby Solvent Extraction, in Hydrometallurgical Recovery of Metals from Ores, Concentrates, andSecondary Sources, Chap. 25, SME-AIME, New York, 1981.
8.4-27 W. Nitsch, Faraday Discuss. Chem. Soc, 77, paper 77/8 (1984).8.4-28 W. Nitsch and A. Van Schoor, Chem. Eng. ScL, 38, 1947 (1983).
Section 8.5
8.5-1 R. E. Treybal, liquid Extraction, 2nd ed., McGraw-Hill, New York, 1963.8.5-2 G. S. Laddha and T. E. Degalessan, Transport Phenomena in Liquid Extraction, McGraw-Hill,
New York, 1978.8.5-3 H. P. C. Pratt and C. Hanson, Selection, Pilot Testing, and Scale-Up of Commercial Extractors,
in T. C. Lo, M. H. I. Baird, and C. Hanson (Eds.), Handbook of Solvent Extraction, Chap. 16,Wiley-Interscience, New York, 1983.
8.5-4 G. M. Ritcey and A. W. Ashbrook, Solvent Extraction, Principles and Applications to ProcessMetallurgy, Eisevier, Amsterdam, 1979.
8.5-5 T. W. Chapman, Equilibrium Stage Calculations for the Solvent Extraction of Metals, in Fun-damental Aspects of Hydrometallurgy, AIChE Symposium Series, Vol. 74, No. 173, p. 120,AIChE, New York, 1978.
8.5-6 G. M. Ritcey and A. W. Ashbrook, Solvent Extraction, Principles and Applications to ProcessMetallurgy, Part II, pp. 198-220, Eisevier, Amsterdam, 1979.
8.5-7 K. L. Power, in Proceedings of the International Solvent Extraction Conference, The Hague,1971, p. 1409, Society of Chemical Industry, London, 1971.
8.5-8 A. Altway, M. E. Tunison, and T. W. Chapman, Effect of Contactor Type on Metal Separationby Solvent Extraction, in Hydrometallurgical Recovery of Metals from Ores, Concentrates, andSecondary Sources, Chap. 25, SME-AIME, New York, 1981.
8.5-9 G. M. Ritcey and A. W. Ashbrook, Solvent Extraction, Principles and Applications to ProcessMetallurgy, Part II, p. 125, Eisevier, Amsterdam, 1979.
8.5-10 T. W. Chapman, Solvent Extraction of Metals—Metal Transfer Rates and Contactor Design, inR. G. Bautista (Ed.), Hydrometallurgical Processes Fundamentals, Plenum, New York, 1984.
8.5-11 M. E. Tunison and T. W. Chapman, Characterization of the Kenics Mixers as a Liquid-LiquidExtractive Reactor, in Fundamental Aspects of Hydrometallurgical Processes, AIChE SymposiumSeries, Vol. 74, No. 173, p. 112, AIChE, New York, 1978.
8.5-12 S. W. Tse, "Mass Transfer Rates in the Solvent Extraction of Metal Chlorides by Tri-isooctylam-ine," M.S. Thesis, University of Wisconsin, Madison, 1978.
Section 8.6
8.6-1 G. M. Ritcey and A. W. Ashbrook, Solvent Extraction, Principles and Applications to ProcessMetallurgy, Part II, Eisevier, Amsterdam, 1979.
8.6-2 T. C. Lo, M. H. I. Baird, and C. Hanson (Eds.), Handbook of Solvent Extraction, Wiley-Interscience, New York, 1983.
8.6-3 Proceedings of the International Solvent Extraction Conference, The Hague, 1971, Society ofChemical Industry, London, 1971.
8.6-4 Proceedings of the International Solvent Extraction Conference, Lyons 1974, Society of ChemicalIndustry, London, 1974.
8.6-5 Proceedings of the International Solvent Extraction Conference, Toronto 1977, Canadian Instituteof Mining and Metallurgy, Montreal, 1979.
8.6-6 P. G. Thomhill, E. Wigstol, and G. Van Veert, J. Metals, 23, 13 (1971).8.6-7 F. J. Brana-Mulero, "Studies on the Solvent Extraction of Metals: Process Synthesis Strategies
and Mathematical Consistency of the Models," Ph.D. Thesis, University of Wisconsin, Madison,1980.
8.6-8 N. Nishida, G. Stephanopoulos, and A. W. Westerberg, AlChE / . , 27, 320 (1981).8.6-9 G. M. Ritcey and A. W. Ashbrook, Solvent Extraction, Principles and Applications to Process