Page 1
MEl'AIOLITES OF ASPERGILLUS VEBSICOLOR
by
Paul N. Chen
Dissertation subnitted to the Graduate Faculty of the
Virginia Polytechnic Institute and State University
in partial fulfillment of the requirements for the degree of
APPROVED,
OOaI'OR OF PHILOSOPHY
in
Chemistry
Dr. D.G.I. Kingston, Chairman
Dr. H.M. Bell
Dr. J.R. Vercellotti
June, 1977 Blacksburg, Virginia
fir. H.D. Smith, Jr.
ifr. J.F. Wolfe
Page 2
ACKNOWLEIX;EMENTS
I would like to express my gratitude to my research advisor,
Dr. David G.I. Kingston, who helped me with ideas and encouragement
during the research project. His special concern and understanding
made the job easier and thoroughly gratifying. I also extend thanks
to Dr. John R. Vercellotti and Miss Sue Ellen Jolly, who provided the
fermentation metabolites and gave valuable criticism during the
performance of this research. The author is indebted to the U.S. Food
and Drug Administration for financial support in this project. (Grant
Number 223-74-2146). Finally, I wish to express appreciation to my
wife, Ruth, who stood by throughout.
ii
Page 3
TABLE OF CONTENTS
AC".t<:NOWLEDGEI1ENTS • • • • • • • • . . . . . . . • • • • • • • • • • ii
TABLE OF CONTENTS • • • • • • • • • • • • • • • • • • • • • • • • iii
LIST OF TABLES •
LIST OF FIGURES
• • • • • • •
• • • • • • •
• • • • • • • • • • • • • • • • • • vi
• • • • • • • • • • • • • • • • • • viii
LIST OF SCHEME3 • • • • • • • • • • • • • • • • • • • • • • • • • ix
LIST OF CHARI'S.
LIST OF SPECTRUM
• • • • • • • I I I I I I I I I I I I I I I I I I
• • • • • • • • • • • • • • • • • • • • • • • • •
INTRODUCTION •••••
RE3ULTS AND DISCUSSION
• •
• •
I I I I • • • • • • • •
• • • • • • • • • • • •
• • • • • • • •
• • • • • • • •
X
xi
1
10
I. Growth of Aspergillus species ••••••••••••••• 10
I. l. Growth of Aspergillus versicolor NRIU, 5213, H 1004 and H 1214 • • • • • • • • • • • • • • • • • • • • • • • 10
I.2o Growth of Asnergillus parasiticus, Yellow Mutant • • • • 10
II. Extraction and Purification of PiQllents from AsJ2er~illus versicolor • • • • • • • • • . • • 0 • • • • • • • • • • • • 11
II.l. Extraction and Purification of Pii:;ments • • • • • • • • 11
II.2. Isolation of Metabolites from ~o versicolor (NRIU, 5213) • • • • • • • • • • • • • • • • • • • • • • 14
II.3. Isolation of Metabolites from !}_. versicolor (M 1214) . • • • • • • • • • • • • • • • . • • • • • • . 16
II.4. Isolation of Metabolites from!• versicolor (M 1004) • • • • • • • • • • • • • • • • • • • • • • • • 17
II.5. Isolation of Metabolites from A. parasiticus (Yellow Mutant) -
• • • • • • • • • • • • • • • • • • • • 18
iii
Page 4
III, Analytical Methodology ••••• , • • • • • • • •
III,l.
III,2.
Development of Thin-layer Chromatography (TLC) System • • • • • • • , • , , • • , , • • , • •
High-performance Liquid Chromatography (HPLC) Analysis of Metabolites of!• versicolor, • •
IV. Structure Elucidation of Isolated!• versicolor
• • • •
• • • •
• • • •
Pigments , , • • • • • , • • , , , • , , , • , • , , , , ,
v. Preparation and Reduction of Sterigmatocystin-hemiacetal • • · • • • • • • • • • • , • • • • • • • • • • •
VI. lJC-NMR Studies of Sodium Borohydride Reduced Derivatives
~
18
18
19
28
J9
of Sterigmatocystin-hemiacetal. • • • • • • • • • • • • • 64
VII. Chemical Modification of Partially Reduced Sterigmato-cystin-hemiacetal • • • • • • • • • • • • • • • • • • • • 85
VIII. Preparation and Reduction of Versicolorin A-hemiacetal. • 90
IX. B:losynthesis of Aflatoxin B_i_ ••••••••••••••• 106
EXPERIMENT AL • • • • • • • • • • • • • • • • • • • • • • • • • • • I.
II.
III,
IV.
v.
VI.
General Information • • • • • • • • • • • • • • • • • • • I.l. Thin-layer Chromatography (TLC) • • • • • • • • • • • I.2. High-performance Liquid Chromatography (HPLC)
Extraction and Fractionation of Netabolites of A. versicolor (NRRL 521J) •••••••••••• 7, Extraction and Fractionation of Netabolites of!• versicolor (M 1214) • • • • • • • • • • • • • • •
Extraction and Fractionation of Metabolites of!• versicolor (M 1004) •••••••••••••••
Isolation of Versicolorin A from A. parasiticus
• • • •
• • • •
• • • •
• • • •
(Yellow Mutant) •••••••• 7 . ..... . • • • • • Structure Elucidation of Isolated Pigments •••••• • •
116
116
116
117
118
122
129
lJJ
1J7 VII. Synthesis and Reduction of Hemiacetals of Sterigmatocystin
and Versicolorin A • • • • • • • • • • • • • • • • • • • • 14 J
iv
Page 5
~
REFERENCE:> • • • • • • • • • • • • • • • • • • • • • • • • • • • • 1.54
VITA • • • • • • • • • • • • • • • • • • • • • • • • • • • • • • • 159
AISTRAcr
V
Page 6
LIST OF TABLES
Table
1. Versicolori.ns Isolated from Aspergillus versicolor. • • • • J
2. Avermutins Isolated from Aspergillus versicolor • • • • • • 4
J. Hydroxylated Anthraquinones Isolated from Aspergillus versicolor • • • • • • • • • • • • • • • • • • • • • • • • • 5
4. Averufin and Niduru:fin Isolated from Aspergillus 6 versicolor ••••••••••••• • • • • • • • • • • • •
5. Sterigmatocystins Isolated from Aspergillus versicolor. • • 7
6. Niscellaneous Metabolites Isolated from Aspergillus versicolor • • • • • • • • • • • • • • • • • a . • • • • • • • 8
7. Separation of Aspergillus versicolor metabolites in various TLC systems • • • • • • • • • • • • • • • • • • • • 20
8. Silica Packings for Liquid-Solid Chromatography •••••• 22
9.
10.
11.
12.
lJ.
14.
Retention Volumes and Capacity Factors of Aspcrgillus versicolor Netabolites ••••••• a ••••••• • • • • lJC-Chemical Shifts of Sterigmatocystin and Dihydro-steri.gmatocystin •••••••••••••••••••• • • lJc-Chemical Shifts of Steri.gmatodiol (XII) • • • • • • • • lJC-Chemical Shifts of Iso-dihydrosteri.gmatocystin (XIII). • lJc-Chemical Shifts of Partially Reduced Sterigmatocystin-hemiacetal (XI) • • • • • • I • • • • . • • • . • . • • • . Separation of Derivatives of Sterigmatocystin in Various TLC Systems • • • • • • • • • • • • • • • • • • • • • • • •
25
68
72
75
79
91
15. Radioactivity Incorporation of Aflatoxin B:i_ Derived from Various Labeled Precursors ••••••• o • o ••••••• 109
16.
17.
18.
Column Chromatography of Pigments from Aspergillus versicolor (NRRL 5213) •••••••••••••• • • • •• 120
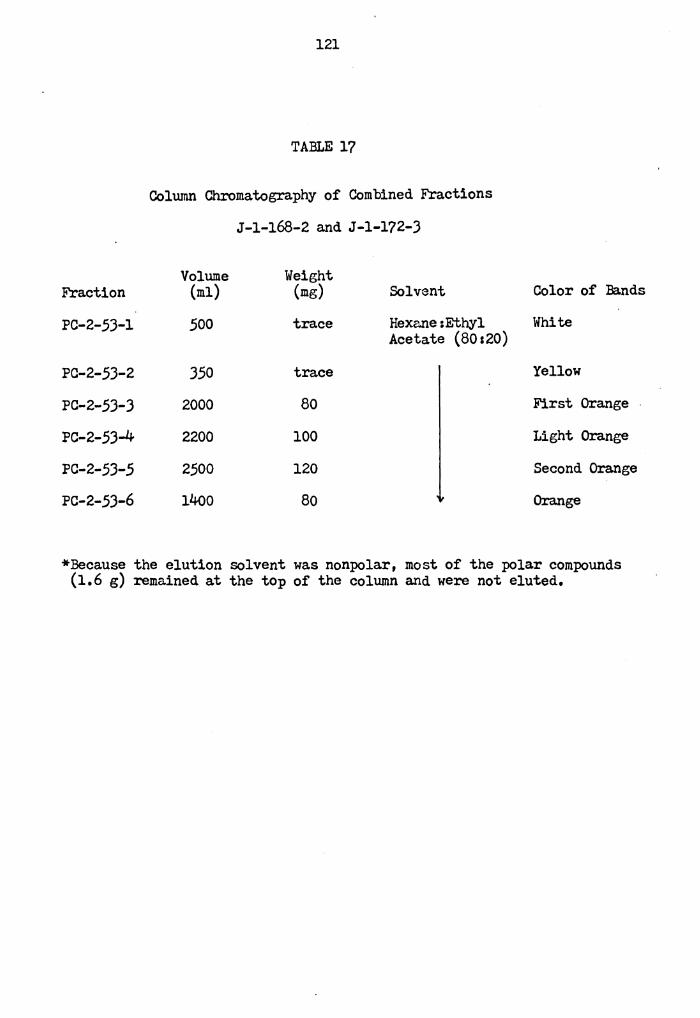
Column Chromatography of Combined Fractions J-1-168-2 and J-1-172-3 e e O e O e e O I e e o e e e o e e e e e
Column Chromatography of Fraction J-1-90 •• • • • • • •
vi
• •
• •
121
123
Page 7
Table
19. Colwnn Chromatography of Fractions J-1-90-1 & 2. • • • • • 124
20.
21 •
22.
23.
24.
25.
Column Chromatography of Fractions J-1-90-5,6 and Similar Fractions •••••••••••••••••
Column Chromatography of Metabolites of Aspergillus . versicolor (M 1214) ••••••••••••••••
Column Chromatography of Metabolites of Aspergillus versicolor (M 1214) ••••••••••••••••
• • • •
• • • •
• • • •
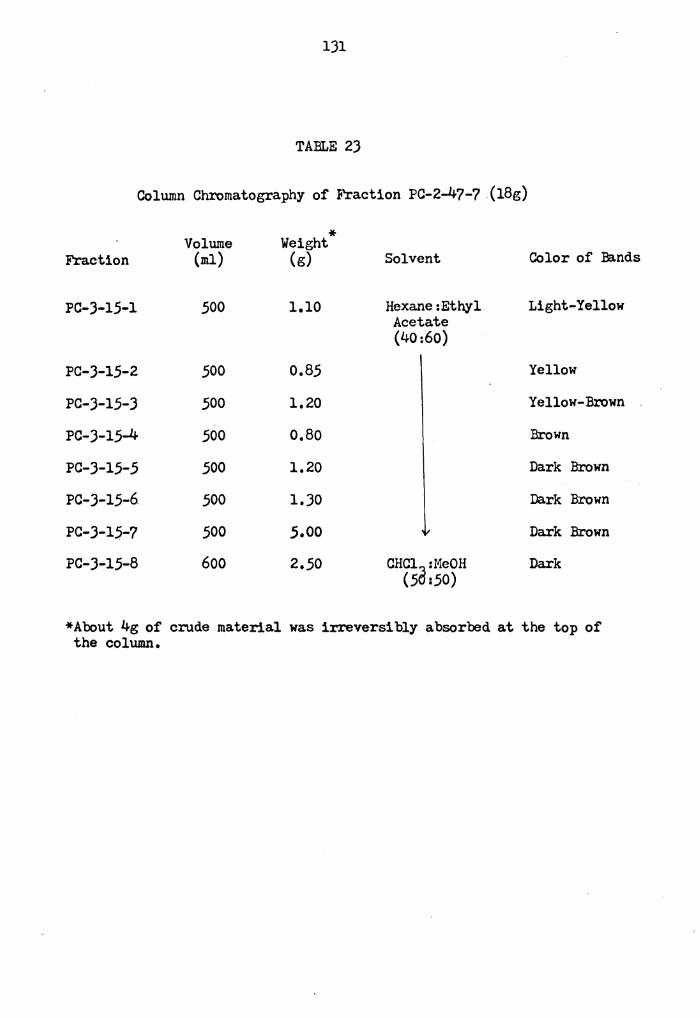
Column Chromatography of Fraction PC-2-47-7 •••
Colwnn Chromatography of Fraction J-1-82-1 & 2 • • • • •
• • • • • •
Colwnn Chromatography of Metabolites of Yellow.Mutant. • •
vii
125
128
130
131
134 136
Page 8
1.
2.
J.
4.
LIST OF FIGURES
HPLC Separation of averufin (AV), avermutin (AM), vericolorin A (VA), versicolorin C (VC) on Pora.sil.
HPLC Separation of sterigmatocystin (ST), demethyl-sterigmatocystin (DMST), 5-methoxysterigmatocystin (MeOST), avermutin (AM), averufin (AV), versicolorin A (VA), and versicolorin C (VC) on Partisil 10-PAC.
• • •
• • • Natural-abundance proton-noise-decoupled lJC-NMR spectrum of sterigmatodiol (XII) •••••••• • e I e I
Natural-abundance proton-noise-decoupled lJC-N11R spectrum of iso-dihydrosterigmatocystin (XIII) •
13 . Natural-abundance proton-noise-decoupled C-NI1R spectrum of partially reduced sterigmatocystin-hemiacetal (XI), ••••••••••••••••
viii
• • • • •
• • • • •
26
70
77
78
Page 9
LIST OF SCHEMES
Scheme
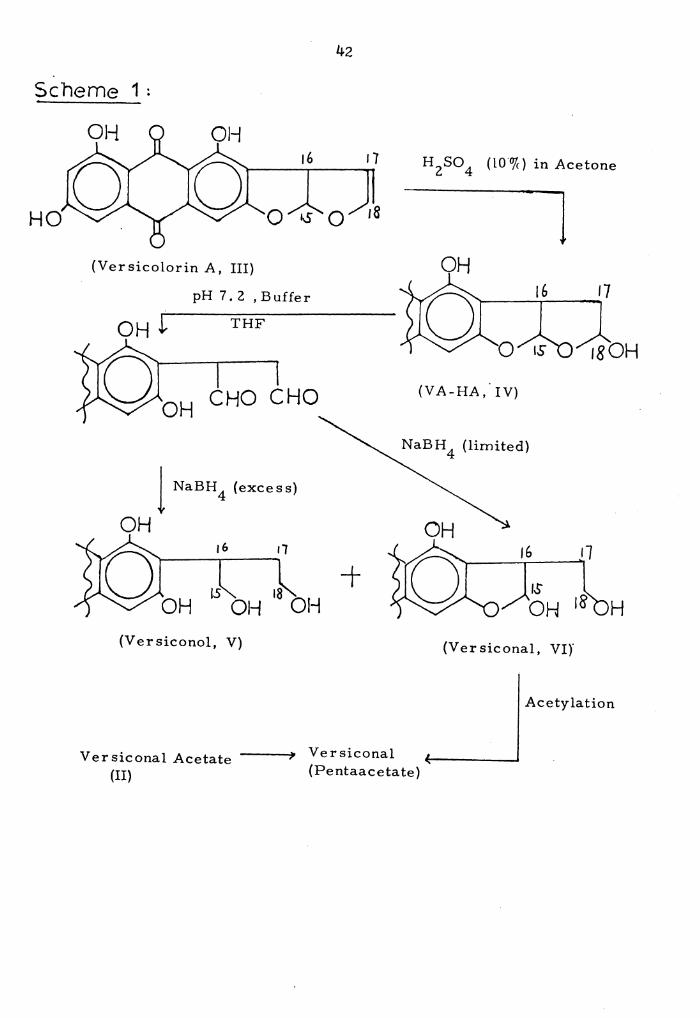
1. Preparation of Versiconal Penta-acetate. • • • • • • • • • 42
2. Tautomeric Structures of Sterigmatocystin-Hemiacetal (VIII) • 0 •••••• I I ••• I I I I I I I I I I •• I 44
J. Sodium Borohydride Reduction of Sterigmatocystin-Hemiacetal (VIII) , • , •• , , , , ••• , •• , I e e I •
4, lJC-Chemical Shifts of Dihydro-stericmatocystin (XIV), Sterigmatodiol (XII), Iso-dihydrosterigmatocystin (XIII), , 82
5. Preparation and Reduction of Sterigmatocystin-Hemiacetal (VIII) • I I • • • I I I I I I • • • I • • • • • I • • e I
6. Preparation and Reduction of Versicolorin A-Hemiacetal (VI). • • • • • • • • • • • • • • • • • • • • • • • • e O I
Acid Cyclization of Partially Reduced Versicolorin A-Hemiacetal (VI) • • • • • • • • • • • • • • • • , , • • e I
8, Biosynthesis of Versicolorin A (III) • • • • • • • • • • •
88
94
100
111
9. A Modified Bio synthetic Pathway of AflatoXin 1\ . . , • . . 112
ix
Page 10
LIST OF CHARI'S
Chart
1. Isolation of Metabolites from Aspergillus versicolor ( NRRL 5213) • • • • • • • • • • • • • • • • • • • • • • • • 14
2. Isolation of Metabolites from Aspergillus versicolor · (M 1214) • • • • I I • I I I • • I I I I • • • • I • I I • 16
J. Isolation of Metabolites from Aspergillus versicolor (I11004) • I I • • • • • • • I • • • • • • • • • • • • • • 17
4. Liquid-Liquid Partition of Crude .Myc·elium from Aspergillus versicolor (NRRL 5213) •••••••••• • • ••• • • • 118
5. Isolation of Metabolites from Aspergillus versicolor (NnRL 5213) • • • • • • • • • • • • • • • • ... • • • • • • 126
6. Isolation of Metabolites from Aspergillus versicolor (NRRL 5213) • • • • • • • • • • • • • • • • • • • • • • • • 127
7, Isolation of Metabolites from Asnergillus versicolor (N 1214) • • I • • • • • • I • • • • • • • I • • • • • • • 132
8. Isolation of Metabolites from Asner~illus versicolor (M 1004) • • • • • • • • • • • • • • • • • • • • • • • • • 135
X
Page 11
LIST OF SPEGrRA
Spectrum
1.
2.
• • • • 1H-m1R spectrum of sterigmatocystin-ethoxyacetal •
1H-NMR spectrum of isodihydrosterigmatocystin • • • • • •
J; 1H-NHR spectrum of dihydrosterigmatocystin • • •
4. 1H-Double resonance NMR-spectrum of isodihydro-sterigmatocystin o •••••••••••••••
Xi
• • • • •
• • • • •
47
60
61
62
Page 12
INTRODUCTION
For the past 15 years, there has been a keen interest in the
mycotoxins derived from the polyketides 1 because of their complex
structure and biological activities. One of the most important
discoveries in this area in recent years has been that the common
molds, Aspergillus flavus and ~spergillus parasiticus produce large
quantities of highly toxic and hepatocarcinogenic compounds known as
aflatoxins, such as aflatoxin Bi and G1•
0
Aflatoxin Bi Aflatoxin G1
The related fungus Aspergillus versicolor produces a number of
metabolites which share certain common structural features with the
aflatoxins. These compounds, the ster-lgmatocystins and versicolorins,
were isolated and their structure elucidated before their carcinogenic
activity had been recogniGed. The marked similarity in structure
between the sterigmatocystins,.the versicolorins, and the aflatoxins
makes it important to obtain sufficient quantities of the versicolorins
so that their toxicity and carcinogenicity can be established, In
1
Page 13
2
addition, the presence or absence of other potentially toxic metabolites
in Aspergillus versicolor should also be established, so that the total
hazard to human health from this fungus can be estimated. For these
reasons a study of the metabolites of Aspergillus versicolor was
initiated. The majority of the metabolites from Aspergillus versicolor
contain a hydroxylated anthraquinone or xanthone ring system, although
several non-quinonoid metabolites are also produced by this mould. The
following tables list all metabolites which have been isolated to date
from different strains of Aspergillus versicolor.
Page 14
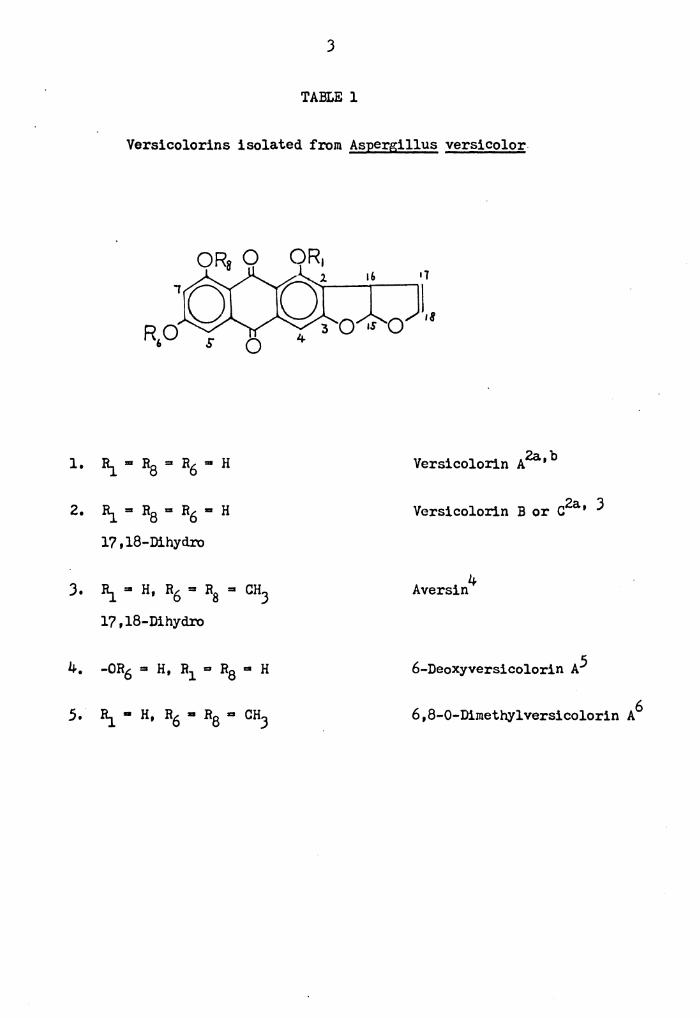
J
TABLE l
Versicolorins isolated from Aspergillus versicolor
$ 0
1. I\ ... Ra = R6 "' H 2a b Versicolorin A '
2. I\= R8 = R6 "' H 2a Versicolorin B or C ' J
17,18-Dihyd.ro
J. I\ = H, R6 = R8 = CHJ 4 Aversin
17,18-Dihydro
4. -OR6 = H, R1 = R8 "' H 6-Deoxyversicolorin A5
5. I\ • H, R6 • RB ... CHJ 6,8-0-Dimethylversicolorin A 6
Page 15
4
TABLE 2
Avermutins isolated from Aspergillus versicolor
• Avermutin?a,b 1. li_ = RJ = R6 • Ra = RJ .. H
R.5 • CHJ
2. ' li_ .. R.3 .. R6 = R.3 • H a-O-Methylavermutin?a,b
Ra • R.5 • CHJ
J. li_ =- RJ = Rj = H 6,8-Di-O-Methylaver-• R6 •Ra= RS• CH.'.3 mutin?a,b
4. 1i. • RJ • R6 = Ra .. R; • H a Versicorufin • R.3 =- OAc
Page 16
5
TABLE J
Hydroxylated Anthraquinones isolated from
Aspergillus versicolor
• 0 2. Bi .. OH, R2 .. H, 1 ,2 -Dihydro
J. Ri_ 111 OCHJ' R/ .. H, 1' ,2'=Dihydro
0
Norsolorinic Acidlla,b
Averythrin9
AverantinlOa,b
l,J,6,8-Tetrahydroxy-2-
(1'-methoxyhexyl)-Anthra-
quinone?a
OH
OH OH
12 Versiconol
Page 17
1. Rj_6 "" H
2. Rj_6 ... OH
6
TABLE 4
Averufin and Nidurufin isolated from
Aspergillus versicolor
, R,,
Averuf1nl3a,b
Nidurufin 7a
Page 18
7
TABLE 5
Sterigmatocystins isolated from Aspergillus versicolor
1. R18 ... CHJ' R19 = R20 = H 14a b Sterigmatocystin '
2. 8is = CHJ' 8i 9 = H, R20 = OCHJ 6-Methoxysterigmatocyst1n 15
J. 8is = 8i9 = R20 = H Demethylsterigmatocystin5
4. 8is .,. CHJ' R19 = R20 = H Dihydrosterigmatocyst1n 16
16,17-Dihydro
.5. 1\8 = 11_9 "" R20 • H Dihydrodemethylsterigmato- 16
16,17-Dihydro cystin
Page 19
8
TABLE 6
Miscellaneous Metabolites isolated from Aspergillus versicolor
OHO OH
Sterigmatin 17
OH rAYCH3
~OH OH
Versicolin 19
Versio1 21
18 Versimide
20 Aspercolorin
Page 20
I 2. R = Hc,_-,,c( CH.3) 2
' , '-o·
9
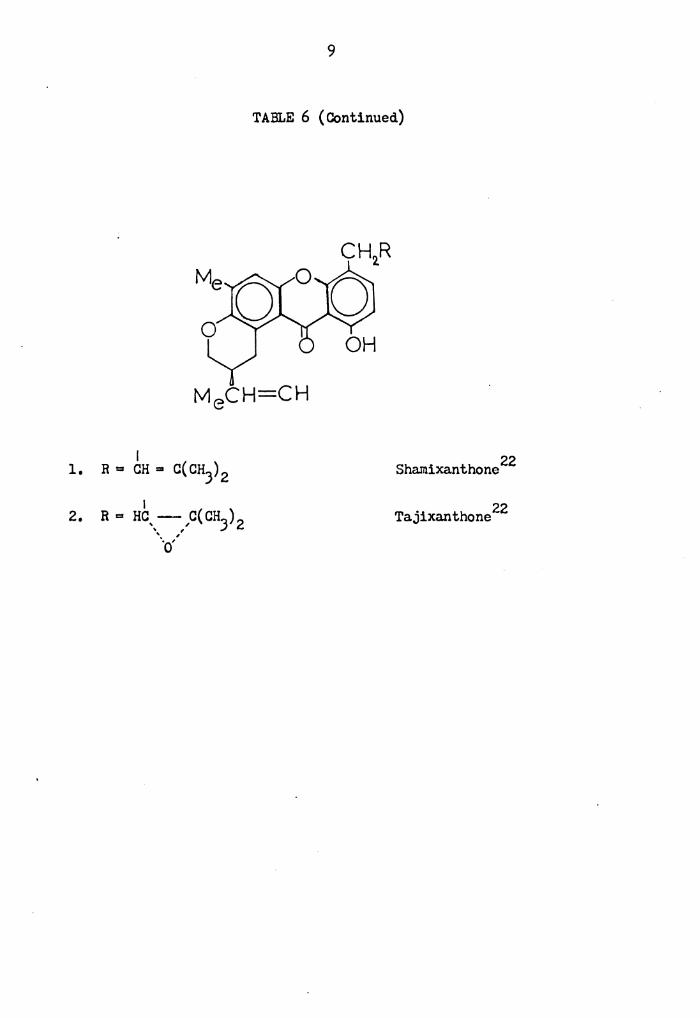
TABLE 6 ( Continued)
22 Shamixanthone
Tajixanthone 22
Page 21
RESULTS AND DISCUSSION
I, Growth of Asnersillus Species*
I,l, Growth of Aspergillus versicolor NRRL 521J, M 1004 and M 1214
Two of the strains M 1004 and M 1214, were made available from
the U.S. Food and Drug Administration, The third, NRRL 521J, came
from the U.S. Department of Agriculture. Slants of these strains
were maintained on Czapek-Dox agar (5% glucose per liter with sodium
nitrate (Jg), magnesium sulfate (0,5 g), potassium chloride (0,5 g),
and ferrous chloride (0,01 g)). Inoculation was carried out by using
sterile water with Tween-20 (0,5%) to make uniform suspension of spores,
Before inoculation, the medium, usually steamed rice, was auto-
claved at 110° for 15 min. The strain NRRL 521J grew best in still 0 culture at J5 for 2-J weeks, whereas strains M 1004 and M 1214 gave
better results in still culture at room temperature.
I,2, Growth of ~gillus parasiticus, Yellow Mutant**
(Obtained from Dr. J.C. Bennett, Tulane University, New Orleans, LA)
Cultures of this organism were best carried out on potato-
dextrose-agar, Both shake flasks and still culture of their yellow
mutant produced anthraquinone pigments in a modified Ad.ye and Mateles
*The growth research was done by Dr. J.R. Vercellotti and Miss Sue Ellen Jolly in Dapartment of Biochemistry & Nutrition, VPI & SU, Blacksburg, Virginia.
**There appears to be some question as to whether this mutant is in fact Aspergillus parasiticus; some mycologists (as Dr. P. Mislevic, FDA, Bureau of Foods, Washington, D.C.) believe that it may be a strain of Aspergillus versicolor,
10
Page 22
11
medium23 developed by Dr. J.C. Bennett. Still culture.needed 2-J
weeks for growth, whereas shake culture took 6-8 days. The "yellow
mutant'' did produce good amounts of versicolorin A in the modified
Adye and Mateles liquid medium,
II. Extraction and Purification of Pigments from Aspergillus
versicolor,
II.l. Extraction and Purification of Pigments
Most extractions were performed initially with a chloroform:
acetonesmethanol mixture. A sequential clean up procedure was devised 24 based on the method of Stack and Rodricks, because large quantities
of oils were obtained during the initial extractions and these made
the chromatographic separation of the pigments difficult. The clean
up procedure involved initial removal of the oils by extraction of an
aqueous acetonitrile solution of the pigments with hexane; the pigments
were then recovered from the acetonitrlle by dilution with water and
extraction with chloroform, The defatted pigments were then purified
by column chromatography and preparative thin-layer chromatography
(PI'LC),
In this work, preparative TLC was usually used when the sample
size ranged from 10 to 200 mg. For sample sizes exceeding 0.2 g,
column chromatography was the method of choice. Based on the separation
mechanism, generally there are four different types of thin-layer and
column chromatography. These are adsorption chromatography, partition
chromatography, ion-exchange chromatography and size exclusion chroma-
tography. The metatolites of Aspergillus versicolor of interest were
Page 23
12
mainly either neutral (xanthones) or weakly acidic (hydroxylated
anthraquinones) compounds, with molecular weight )JO to 400. Because
of the small molecular weight differences of these non-ionic metabolites,
the use of ionic and size exclusion chromatographic techniques for the
isolation of the metabolites were not considered.
Partition chromatography usually requires ten times more sorbent,
such as cellulose, than adsorption chromatography for the separation
of the same amount of sample. From the economical point of view,
adsorption chromatography is a better choice than partition chromato-
graphy. Also the purity of the isolated metabolites· should be checked
by TLC and high-performance liquid chromatography (HPLC) at the final
stage, so it was desirable to use the same type of packing materials
in all the column chromatographic, thin-layer chromatographic and
high-performance liquid chromatographic separations. The common parti-
tion chromatographic sorbent used in TLC and column chromatography is
cellulose, but this sorbent is not commercially available in HPLC.
TLC on cellulose has tested in the early stage of this work, but it
did not give satisfactory separation of the metabolites of interest.
For these reasons, partition column chromatography and TLC were not
used in this research.
The only mode left to separate the Aspergillus versicolor
metaboli tcs is thus adsorption chromatography. Three adsorption
chromatographic sorbents have been used in general separations; these
are alumina, silica and polyamide. Alumina is usually basic, which
would ionize and irreversibly adsorb the weakly acidic hydroxylated
Page 24
1)
anthraquinone pigments of interest, so it was not considered. Poly-
amide columns have been proved very useful in the separation of phenolic
metabolites such as flavones, 25 but after several early tests it was
observed that this sorbent could only be used with aqueous methanolic
solvent systems. Since these systems tend to decompose the materials
of interest by reaction with the dihydrofUran double bond in the pre-
sence of traces of acid, the use of polyamide was discontinued early
in, this study.
Because silica sorbent has a better efficiency, higher capacity
and lower reactivity (less sample decomposition) than alumina, and
because most of the literature references to the isolation of the
anthraquinone metabolites have cited the use of silica gel column or
thin-layer chromatogra.phy2,J•26 it was finally chosen for this research.
Three different kinds of silica were used by us, namely (1) the neutral
(pH 6,8) E. Merck Silica Gel 60 for analytical TLC and column chroma-
tography; (ii) the neutral E. Merck Silica Gel 60 PF-2.54 for prepara-
tive TLC; (iii) the acidic (pH 4.6) Mallinckrodt silica AR cc-4 for
column chromatography. With both the neutral silica gels some
irreversible adsorption of pigments on the column was observed,
presumably due to ionization of the acidic anthraquinones in the
solvent systems used. For the acidic silica sorbent, however, another
problem presented itself in that the furofuran ring of versicolorin A
might be decomposed by the acidic nature of this column packing. After
a study of the various factors involved, both the neutral and acidic
silica packings were used in the purification of pigments, although
neither was entirely satisfactory.
Page 25
14
The crude defatted pigments were usually first purified by column
chromatography on a silica gel sorbent using chlorofonn and chlorofonn-
met_hanol eluents. The eluted fractions containing the desired pigments
were then rechromatographed on a silica gel column with various solvent
systems. After much experimentation, it was found that a hexane:ethyl
acetate eluent gave the most efficient separation of the desired com-
ponents, and this system was used in most of the separations described.
II. 2. Isolation of Meta.boli tes from A. versicolor (NRRL .521))
De-0-methylsterigmatocystin (DM-ST), Sterigmato_cystin (ST),
Averufin (AV), Versicolorin A (VA) and Versicolorin C (VC) were isolated
from this strain on cultured rice, as shown in Chart l.
Hexane (Oil)
Chart l
~. versicolor NRRL 521) I
Extract mycelium with CHc13/MeOH I
Defat by extraction of aqueous CHJCN solution with Hexane I
CHJCN & 4% aq. KCl (90:10) I
Extlct with CHClJ
Pigments I ·--- - ,
I Silica AR CC-4 Column
(CHClJ/MeOH) s.G. 60 Column ( CHCl:/MeOH)
I * ~ I
Page 26
15
t A-3 A-4 A-5 A-6 A-7 A-1 A-2
I S.G. 60 Colwnn
(Hexane/Ethyl Acetate)
I Combined with similar frac-
I I I I I I
tions I s.G. 60 Column (Hexane/Ethyl Acet1ate)
B-1 B-2 B-3 B-4 B-5
B-1 I
I I I I I C-1 C-2 C-3 C-4 C-5
B-2 B-3
S.G. PI'LC (Benzene/Ethyl Acetate) I
DM-ST
I I I I C-1 C-2 C-3 C-4
I Recrystallize from Acetone
l VA
I
~
-..h.
'""' r - - -,- - - i" - L - T - - - i - - - l I I I I I I I I I I 1
D-1 D-2 D-3 D-4 D-5 D-6 I I
Combine with similar frac-tions
I ,s.G. 60 Co-1lwnn ; ( Hexane/Ethyl ,Acetate)
r - -,- J-, - - r - - 1- - , I I I I I I
E-1 E-2 E-J E-4 E-5 E-6
I B-4 B-5 I
Recrystalize from Acetone
I C-5
I ST
r - - --- r------,- - ------,------- - ,------- --, I I I I I I
E-1 E-2 E-3 E-4 E-5 E-6 I I I I s.G. Pl'LC s.G. PI'LC
(Benzene/Ethyl (Benzene/Ethyl Acetate) Acetate)
I I I t
AV VC
Page 27
16
II.J. Isolation of Metabolites from!• versicolor (M 1214)
Dechlorogrlseofulvin, Griseofulvin and J,8-Dihydro:xy-6-metho:xy-1-
methylxanthone were isolated from strain M 1214 grown on solid rice
still culture, as shown on Chart 2.
Chart 2
!• versicolor M 1214 I
~ract with CHClJ/MeOH
Defat by extraction of aq. CH3CN solution with Hexane
Hexane C~CN & 4% aq. IKCl (90:10)· (Oil)
F-1
Extract with CHClJ
[email protected] I
S.G. 60 Colwnn (Hexane/Ethyl Acetate)
F-2 F-J F-4 I
F-5 F-6 F-7 F-8
Recrystallize from Acetone
I 3,8-Dihydroxy-6-methoxy-l-methylxanthone
1 I I
I Recrystallize from Acetone
I Dechlorogriseo-fulvin
S.G. 60 Column
(Hexane/Ethyl Acetate) I t I I I
G-1 G-2 G-J G-4 G-5 G-6 G-7 I Recrystallize
from Acetone I
Griseofulvin
Page 28
17
II.4. Isolation of :Metaooli tes from A. versicolor (M 1004)
6-Methoxysterigmatocystin (:MeO-ST), Aversin and 6,8-Di-0-
methylnidurufin were isolated from this strain grown on solid rice
still culture, as shown on Chart J.
Chart J
!• versicolor M 1004
I Extract with CHc13/MeOH)
Pigments r
Silic~ AR CC-4 Column (CHCl:3/MeOH)
H-1 I
S.G. 60 Column
(Hexane/Ethyl Acetate)
I I I-1 I-2 I-J I-4 I-5 I-6 I-7
I S.G. Pl'LC (Benzene/Ethyl Acetate)
Aversin
MeO-ST
H-2 I
S.G. 60 PF 2.54 Column
(Hexane/Ethyl Acetate)
I J-1 J-2 J-J J-4
I 6,8-Di-0-methyl nidurufin
Page 29
18
II.5. Isolation of Versicolorin A from A. parasiticus (Yellow
Mutant)
A mutant strain of!• parasiticus, named "Yellow Mutant" has been
shown to produce versicolorin A as the major metabolite. Since large
amounts of versicolorin A were required for biological testing, this
organism was used as the major source of the required material. The
mold was grown on modified Adye & Matele' s liquid medium. 23 The crude
extract was subjected to chromatography on Silica gel 60, with elution
by hexane/ethyl acetate. The major fraction containing versicolorin A
was collected, and the versicolorin A was collected, and the versi-
colorin A purified by recr.ystalization from acetone.
III. Analytical Methodology:
III.l. Development of Thin-layer Chromatography (TLC) system
Previous methods for the analytical detection of sterigmatocystins
by TLC techniques have been published, 24 a, 27 but little was known
about the separation of the anthraquinone pigments. Previous methods
for their separations have relied heavily on column chromatography
with silica gel as the sorbent. 2
Small reference samples of sterigmatocystion, 6-methoxy-
sterigmatocystin and of the versicolorins A,B and C were made available
by the Food and Drug Administration, while samples of averufin and
avermutin were obtained from England,* and a sample of versiconol
from Japan.** It thus became possible to develop TLC systems to
*J,S,E, Holker, Robert Robinson Lab., Univ. of Liverpool, Liverpool, Eng,
** Y. Hatsuda, Faculty of Agriculture, Tottori Univ,, Tottori, Japan.
Page 30
19
separate these materials, and hence to monitor the fermentation,
extraction and isolation procedures used. The results with approximate
Rf values are listed in Table 7. It may be seen from Table 7 that conditions have been developed
to separate every compound of interest except the pair averufin/
avermutin; these compounds have been separated in the past by multiple
development TLc.28 It may also be noted that one TLC system, hexane/
ethyl acetate (70:30), gives efficient separation of the pigments of
interest into three groupss versiconol (Rf 0.01), the versicolorins
(Rf 0.22) and averufin/avemutin (Rf 0.38). Several solvent systems
in addition to those listed in Table 7 have been tested, but none of
them gave superior separations to these listed. Recently, it has been
shown that the system chloroform/acetone/acetic acid (97:211), will
separate averufin/avermutin: 29 this system was not used in the work
that is described, however.
III.2. High-performance liquid chromatoir,ra.phy (HPLC) analysis of
metabolites of Aspergillus versicolor
Modern HPLC is one of the most powerful tools now available for
separating and anaiyzing complex mixtures of chemical compounds.
Like column chromatography, there are four separation mode~ in HPLC.
Liquid-Solid (Adsorption) Chromatography
Liquid-Liquid (Partition) Chromatography
Ion-Exchange Chromatography
Size Exclusion Chromatography
Page 31
Tah l<..• 7 • Supar:i.th>11 or !• vcrsicolor metabolites in various TLC systems. 11
--.
Sterig-Solvent System Plnte matocystin Averutin
-CHCJ 3 : llcOH 99:1 S"G-E 95 GG
c,,u6:HC00Et: IICOOII 75:24:l SG-E 85 GO
Call l1 : Ac-OE t 70: :lO SG-E 95 38
CHCI3 :CH3COCH2CH-(Cll3)2 8) :20 !;G-E --- GO
c0n6 : AcO!I: ~IcOH 05:5:5 !iG-E --- 85
CIIC13 : Cll 3cocn3 : 97:2:1 :,G-E --- 50
CUC13:cH3COCII3 : c6n14 85: 15:20 SG-E --- 50
c6n6 :Dioxane: AcOII 90:25:4 SG-E --- 80
!Ic0II PA-B --- 50
a. SG-E Silica gl.•1; Enstmnn chromagram plates PA-D Polymnide:- plates; supplied by Baker.
Rt X 100
Vcrsicolorin Vcr:;icolorin Avermutin A C
GG 52 50
60 55 52
3S 22 22 .
60 --- ---
85 --- ---
50 --- ---
50 --- ---
80 --- ---50 --- ---
-
\'crsiconol
1
2
1
~ ------------
------
Page 32
21
For the same reasons as in column chromatography, ion-exchange
and size exclusion chromatography were not considered. For reversed-
phase (partition) HPLC, the reverse phase sorbents have been widely
used in the separation of fused-ring aromatics,JO but because of the
nat~ of the non-polar reverse phase packing, the separations require
aqueous or aqueous-alcoholic solvent system. Since the anthraquinone
pigments are quite acidic, it was necessary to add a trace of acid to
the solvent to suppress ionization of the pigment and prevent irrever-
sible adsorption of the pigments. These conditions (aqueous-alcoholic
acid) are just those sufficient to convert the furofuran double bond
(as in versicolorin A) to its hemiacetal derivatives, and thus nullify
the purpose of the experiment. For this reason, non-polar reverse
phase (as Partisil ODS) sorbent was not used in our HPLC separations.
The remaining mode of nonnal phase (adsorption) HPLC is thus the
only possible mode for the separation of Aspergillus versicolor
metabolites. Silica and alumina are the two most popular general
purpose sorbent, and for the same reasons as described in the previous
section, only silica sorbent was used in the separation of these
metabolites. A polar bonded phase silica column, Partisil 10 PAC,
was also used and proved to give successful separation of Aspergillus
·· versicolor metabolites. Silica sorbents are usually described in
terms of the type of particle (porous versus pellicular), and particle
size. In Table 8, the kind of silica sorbents used in this work are
summarized.
Page 33
TABLE 8
Silica Packings for Liquid-Solid Chromatography
Type Commercial Name Company Particle Size Surface Area Shape* (Alm) (m2/g)
Pellicu- Corasil II Waters 37-50 14 s lar
Porous Porasil A Waters 37-75 350-500 s Porasis B 37-75 125-250 s ~
Porous .U.-Porasil Waters 10 350 s
Porous Partisil 10 PAC Whatman 10 I
Bonded Polar Eonded
Phase Phase
* S: Spherical; I, Irregular
Page 34
23
Corasil II is a pellicular silica packing material designed for
analytical scale separations. It consists of a solid, spherical,
glass core surrounded by two layers of porous silica, The pellicular
nature of Corasil permits fast mass transfer and rapid equilibration.
Corasil, therefore, is a good choice of packing material for the
development of an analytical separation.Jl
Porasil is a fully porous silica packing material, designed
primarily for preparative-scale separations. When cost and loadability
are of major importance - more important than efficiency and speed -
Porasil is the silica packing material of choice. Porasil A and B
differ only in their surface area.
ll-Porasil is a small particle (-10 .am), fully porous silica
packing material, designed for difficult analytical or preparative
separations. The small particle size and high surface area of
.U-Porasil combine to give extremely high efficiency (rapid, well-
resolved separations) and good loadability,3 2
Partisil 10 PAC is a microparticle with a polar lx>nded phase,
It is manufactured by chemically and permanently l:x>nding a cyano type
moiety to a substrate of Partisil 10, a 10 am silica gel of structured-
irregular shape, High stability Si-0-Si bonding is formed between the
stationary phase and the cyano-type moiety, and this offers exceptional
stability of the material, Partisil 10 PAC is highly polar and hence
is very effective in separating polar compounds,
The silica sorbents used initially were of the porous variety
(Porasil A and B), but to date no solvent system tested was successful
Page 35
24
in giving adequate resolution of the test mixtures studied on these
materials, The pellicular sorbent, Corasil II was also briefly
investigated, it behaved similarly to the Porasil A and Band no
good separation was obtained,
~uccessful liquid chromatographic separations were performed on
a pre-packed .tl-Porasil column (4 mm I.D. x JO cm L, Waters), and a
pre-packed Pa.rtisil 10 PAC column (4.6 mm I.D. x 25 cm L, Whatman).JJ
Of several solvent mixture-packing combinations investigated, three
systems proved effective at separating the mixtures tested, These
systems are summarized in Table 9, and Figure 1 and 2 show typical
separations for these systems. The use of small amounts of acetic
acid in the solvent was found to be helpful in suppressing tailing of
the anthraquinones, presumably because it suppressed the ionization
of these strongly acidic quinones,
Slight changes in the capacity ratios for the compounds studied
were noted as the columns became older; the values reported are for
columns that had been in use for approximately 200 hours, The flow
was 2 ml/min, and the sample injector and columns were at room
temperature, It is particularly noteworthy that the Partisil 10 PAC
column is capable of separating averufin from avermutin, in addition
to separating the three sterigmatocystins. The silica gel micro-
particle column (-lt-Porasil) gave good separations of averufin and the
versicolorins, but did not separate averufin from avermutin, In
addition, the sterigmatocystin peak overlapped the versicolorin A
peak on this column, The bonded phase cyano-type packing (PAC) thus
Page 36
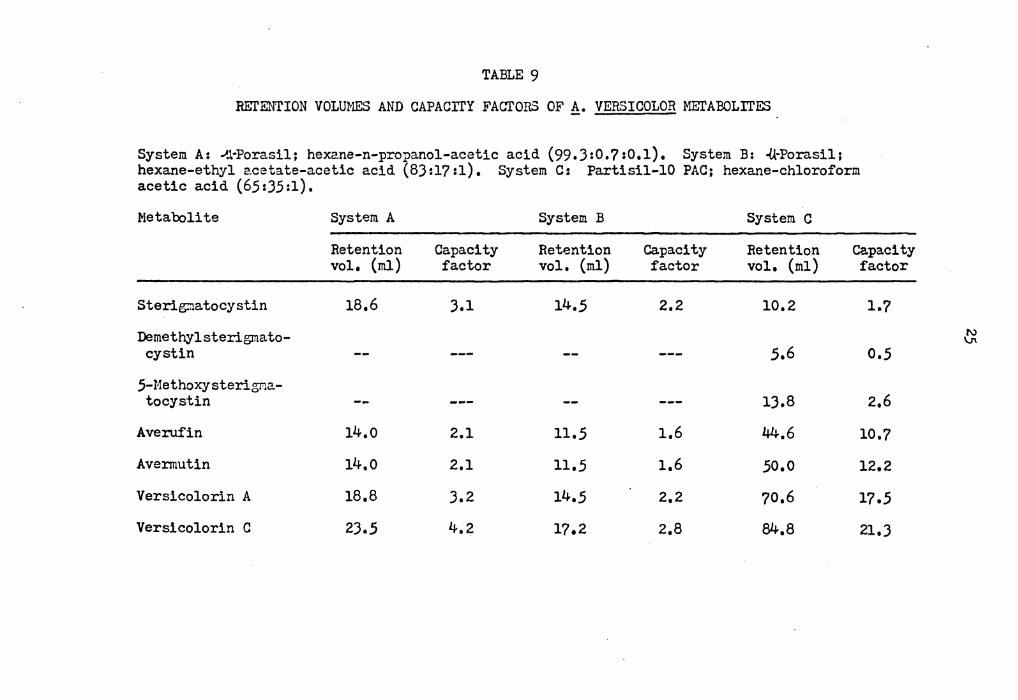
TABLE 9
REl'ENTION VOLUMES AND CAPACITY FACTORS OF!!_. VERSICOLOR METABOLITES
System A: ~.1-Porasil; hexane-n-propanol-acetic acid (99.3:0.?:0.1), System B: ·(kPorasil; hexane-ethyl 2.cetate-acetic acid (83:l? :1), System C: Partisil-10 PAC; hexane-chloroform acetic acid (65:35:1).
Metabolite
Sterigr:iatocystin
Demethylsterigmato-cystin
5-MethoX'Jsterigma-tocystin
Averuf'in
Avennutin
Versicolorin A
Versicolorin C
System A
Retention vol, {ml)
18,6
14.o
14.o
18,8
23,5
Capacity factor
3,1
2.1
2.1
3,2
4,2
System B
Retention vol, {ml)
14,5
11,5
11,5
14,5
17.2
Capacity factor
2,2
1.6
1,6
2,2
2,8
System C
Retention vol, (ml)
10,2
5,6
13.8
44.6
50,0
70,6
84,8
Capacity factor
l.?
0,5
2,6
10,?
12.2
17,5
21,3
~
Page 37
26
AV, AM
VA
vc
• 1r.1p
0 5 10 ;5 20 Ti:-nc Cm!n>
Fig .1. HPLC separation of averufin (AV), avermutin (AM), ver sicolorin A (VA), ver sicolorin C (VC) on.U.--Porasil. Solvent system, A (see Table 9); flow rate, 2.0 ml/min. imp.= impurity.
Page 38
ST Mc0ST
.. -.OM.ST
AV
AM .VA vc
0 10 20 t.:o -~o 1im0 (~.!n)
Fig. 2. HPLC separation of sterigmatocystin (ST), demethyl-sterigmatocystin (DMST), 5-methoxysterigmatocystin (MeOST), avermutin (AM), averufin (AV), versicolorin A (VA), and ver-sicolorin C (VC) on Partisil-10 PAC. Solvent system, C (see Table 9); flow rate, 2. 0 ml /min.
Page 39
28
seems to be the packing material of choice for the separation of
materials such as hyd.roxylated anthraquinones and xanthones; this
conclusion has been confirmed by another recent study.34
Independently of this work, Y. Hatsuda33b and coworkers investi-
gated .HPLC separations of the sterigmatocystins and versicolorins and
reported that these xanthone and anthraquinone derivatives could be
well sepa.ra.ted on a microsilica column (Zorba.x Sil, DuPont).
IV. Structure Elucidation of Isolated A. Versicolor Pigments
The metabolites isolated from!• versicolor were identified by
comparing their spectroscopic properties, melting points, and TLC or
HPLC data. with those of authentic samples or literature data.
1. Identification of PC-2-46-4 (Sterigmatocystin)
'" \ '1
The light yellow crystalline material showed mp 244°c (lit.
460) 14 2 C. It gave the same TLC Rf value and HPLC capacity factor as
those of an authentic sample. Its spectroscopic properties were
identical with those of the literature data.
Page 40
29
2. Identification of PC-2-73-23 (6-Methoxysterigmatocystin)*
The yellow crystals showed mp 218-220° C (lit, 223°). 15 Its TLC
and HPLC data were identical to those of an authentic sample. The
spectroscopic data were consistent with those of literature data,
3, Identification of PC-2-5-5-3 (Averufin)
'"
HO 0
The orange c:rystals had mp 280-282° C (dee.) (lit. 283-289°C,
dec,),l3a,b The compound gave an identical TLC and HPLC data as those
of an authentic sample. The spectroscopic data were consistent with
the assigned structure.
*Originally described in the literature as 5-methoJ5Ysterigmatocystin, but later corrected to 6-methoxysteigmatocystin,15
Page 41
JO
4. Identification of PC-2-53-5 (Versicolorin C)
OHO OH
0
The orange crystals showed mp >J00°C (lit. mp >J10°c).2a,Ja,b
On TLC and HPLC, it gave an Rf value and HPLC capacity factor identical
to these of authentic versicolorin c. Its spectroscopic data were
consistent with those of literature data.
5. Identification of PC-2-40-3 (Versicolorin A)
OH
0
The orange crystals showed mp 281° (dee.) (lit. 289°c, dec.).2a,b
The compound gave identical Rf value and capacity factor on TLC and
HPLC respectively as an authentic sample,
Page 42
31
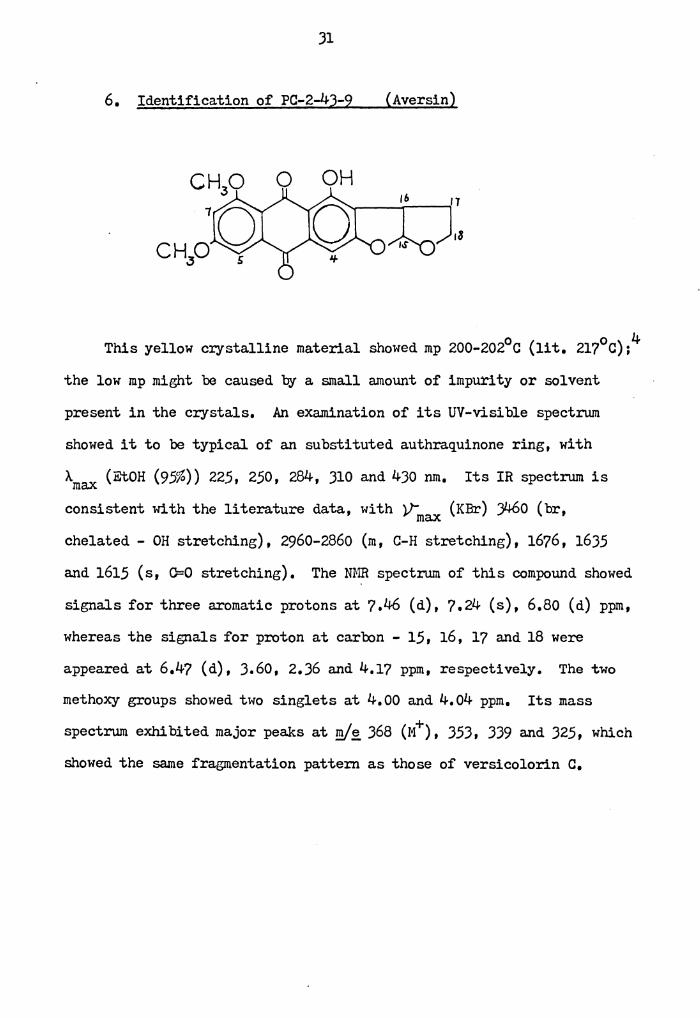
6. Identification of PC-2-43-9 (Aversin)
0 OH
This yellow crystalline material showed mp 200-202°c (lit. 217°c);4
the low mp might be caused by a small amount of impurity or solvent
present in the crystals, An examination of its UV-visible spectrum
showed it to be typical of an substituted authraquinone ring, with
Amax (EtOH (95%)) 225, 250, 284, 310 and 430 nm, Its IR spectrum is
consistent with the literature data, with V: (KBr) ;460 (br, max chelated - OH stretching), 2960-2860 (m, C-H stretching), 1676, 1635
and 1615 (s, O=O stretching). The mm spectrum of this compound showed
signals for three aromatic protons at 7,46 (d), 7.24 (s), 6,80 (d) ppm,
whereas the signals for proton at carbon - 15, 16, 17 and 18 were
appeared at 6.47 (d), 3,60, 2,36 and 4,1? ppm, respectively. The two
methoxy groups showed two singlets at 4,00 and 4.04 ppm. Its mass
spectrum exhibited major peaks at '!Y~ 368 (M+), 353, 339 a.nd 325, which
showed the same fragmentation pattern as those of versicolorin c.
Page 43
.32
OH OH
+· H (Aversin, Mt ,m/,g_ 368)
OH OH l
(m/,g_ 325) (m/,g_ 339)
OH OH
H3 +·
OH OH l H' 2.
"'O. (m/e JJ9) +
(m/,g_ 353)
Page 44
JJ
7. Identification of PC-2-43-9 (6 18-Di-O-Methylnidurufin)
OH
This material showed UV-visible absorption indicating the presence
of a hydro:xylated authraquinone ring system similar to that in 6.8-6 .
Di-0-methylversicolorin A, while its IR spectrum showed absorptions
indicating the presence of oonded and free quinone carbonyls. Its NMR
spectrum absorptions consistent with those expected for a dimethylated
derivative of nidurufin.?a Thus a )-proton singlet at 1.62 ppm is
characteristic of the deshielded ketal methyl group and appears at
about this position in nidurufin (1.58 ppm)?a and tri-0-methylaverufin
(1.64 ppm)l)a and the aromatic proton signals are consistent with
those recorded for other compounds with this substitution pattern.
In addition, the 1-proton doublet at 5.28 is almost identical to that
observed in nidurufin (5.17 ppm),?a conclusively indicating the presence
of only one proton on the 2' caroon adjacent to the deshielded proton
in the 11 position.
Confirmation of the nature of the 2' substituent and of the side
chain is provided by the mass spectrum of the new compound, which
showed prominent peaks at m/~ 412 (M+), 394 (M-18), )14 (M-98) and 99.
Page 45
The peaks at (M-18) and (M-98) occur as prominent peaks only in
niduruf'in and averufin among versions related authraquinones, 7a and
only a dimethyl nidurufin would have a molecular weight of 412,
The methoxy groups are assigned to the 6~ and 8- positions on the
basis .of the chemical shifts observed for the aromatic protons on
acetylation of dimethylnidurutin, The proton in the 4-position showed
an acylation shift of almost 0,4 ppm (from 7,28 to 7,64 ppm) while the
protons in the .5- and ?-positions showed essentially unchanged chemical
shifts, This evidence confi:cms the presence of a free hydroxyl group
in the 1-position and the absence of such a group in.the 6- and 8-
positions, and enables us to identify the new compound as 6,8-di-0-
methylnidurufin,
8, Identifica.tion of PC-2-4?-3
meth.yl-xanthone)
OH 0
(3,8-dihydroxy-6-methoxy-l-
OH
The white crystals showed mp 2.51-25J0 c (lit, 25J-255°c).J5 Its
UV spectrum showed AEtOH (95%) at 24), 267 and JlO run, indicating the max
presence of a substituted xanthone ring. The IR absorption is consis-
tent with the assigned structure, with yKBr at )290 ( s, -OH stretching) ,
16.50 (S, O=O stretching), 1)00-100 (m, C-0-C stretching), Its NMR
Page 46
35
spectrum showed the presence of four aromatic protons at 6 6.84 (2H),
6.50 and 6.J6 ppm. In addition, it showed two singlet at 4.01 (-OcH3)
and 2.85 (-cH3) ppm. The mass spectrum showed a molecular ion at Z'/2
and an M-29 peak at 24J.
OH
0 CH3
l + (M , ml£.. 272)
H 0 0
l
l
(m/~243)
Page 47
)6
9. Identification of PC-2-47-6 (Dechlorogriseofulvin)
The white c:rystals had mp 18o0 c (lit. 179°c).3 6a The compound's
UV spectrum showed AEtOH (95%) 237 (calcd. 245), 292 (calcd. 285) and ma.x
J25 run, indicating the presence of two unconjugated chromophone.
0
The IR spectrum showed strong absorption bands at VKBr 1650 and 1620 ma.x
(O=O, stretching), 1600 (O=C, stretching) cm-1• Its NMR spectrum
showed two aromatic proton signals and a vinylic proton signal at
6.21, 6.0) and 5.52 ppm, respectively. In addition, three methoxy
groups (J.91-J.65), one methylene group (J.25-2.20), one methine group
(2.88) and one methyl group (1.0) protons were consistent with the
assigned structure. The mass spectrum gave a strong molecular ion
peak at ml~ 318, and important fragment peaks at ml~ 287, Z'/6, 250,
181, 138 and 69. This fragmentation pattern was similar to that
reported for griseofulvin.J 6b
10. Identification of PC-3-15-5 (Griseofulvin)
Page 48
37
The white crystals had mp 215-216°0 (lit. 220-221°0).37 The UV
spectrum of the material showed AEtOH (95%) at 250, 287 and 325 nm. max The IR spectrum showed strong absorption bands at ',-KBr 1660 and 1620 Ymax
(O=O, stretching), 1590 (O=C, stretching) cm-1• ItsNMR spectrum
showed one aromatic proton signal and one vinylic proton signal at
6.,56 and ,5.64 ppm, respectively. In addition, signals for three
methoxy groups (4.14, 4.04 and 3.77), one methylene group and one
methine group (2.90-2.4o), and one methyl group (0.93) were consistent
with the assigned structure. The mass spectrum showed predominant
peaks at fill~ 352 (M+), 321, 310, 284, 21.5 and 1J8.J6a.
Page 49
J8
(m Is;, 215)
I 0
CH3 0 Cl
CH=CO
Cm/~ 3l0)
Cm/~284)
Page 50
J9
V, Preparation and Reduction of Sterigmatocystin-hemiacetal
The highly toxic and carcinogenic properties of the a.flatoxins and
the omniprescence of the fungi producing these toxins in food and feed
crops have encouraged much basic and applied research,J8 Although
detection, prevention and elimination of a.flatoXin contaminator have
. had priority, investigations of the basic metab:>lic functions of the
causative organisms and of the effectiveness of various natural and
artificial. stimuli have not been neglected,
Rao and HarienJ9 investigated the insecticide dichlorvos as an
inhibitor of a.flatoxin production when applied to rice, corn, wheat
and peanuts, They found that dichlorvos, at 5 and 10.ag/ml, reduced
a.flatoXin production by an average of 62 and 59%, respectively, At
20 JJ.g/ml, a.flatoxin, production was reduced to levels too low for
analysis by ultraViolet (UV) spectrophotometry, 40 Cole, et al, have
confi:rmed that, in general, dichlorvos significantly reduced aflatoxin
production by toxin-producing strains of Aspergillus flavus and
!, parasi ticus, Cole also observed that reduction in yield of
a.flatoxins was accompanied by the appearance of a previously unidenti-
fied orange pigment, Spectral analyses of the pigment and of its
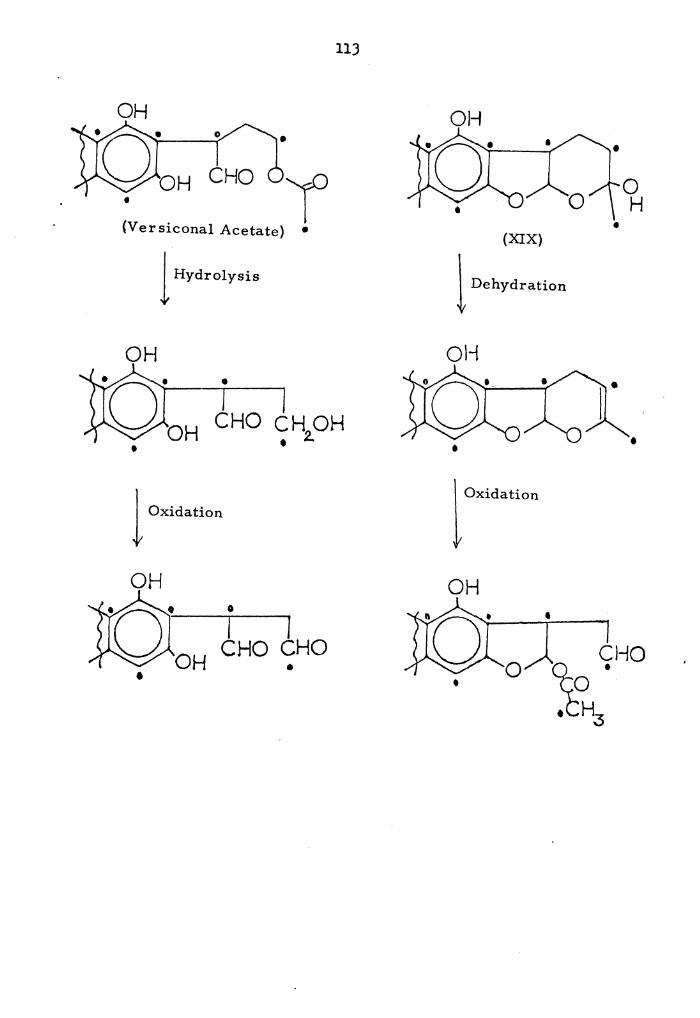
0 OAc 16 17
0
Page 51
40
methylated and acetylated derivatives indicated the compound to be
versiconal acetate, and the tentative structure (I-A) was proposed for
this pigment, although it was recognized that other structures were
also possible,
~ consideration of the spectral data in the original paper, 40
however, led us to the conclusion tha.t the structure (II-A) was a more
probable structure than (I-A).
0 OH 11 ~------
(II-A)
H2.0Ac ,&
The reasoning behind this structural assignment is that the "C-18" pro-
tons give a triplet in the PMR spectrum of versiconal acetate at
4,08 ppm; the predicted value for the "C-18" protons of structure
(I-A) would be a.t 3,60 ppm and those of structure (II-A) around 4.1
ppm. Secondly, no signal was seen for a free aldehyde proton, implied
by structure (I-A).
Versiconal acetate is a potentially important intermediate in the
biosynthetic pathway leading from acetate through averufin to versi-
colorin A and hence to sterigmatocystin and aflatoxin 1\; this pathway
is discussed in section IX of the results and discussion part of this
dissertation, For this reason, and because of the uncertainty
surrounding the structure of "versiconal acetate," it was decided to
Page 52
41
attempt to confirm its structure by synthesis. The proposed scheme
is shown below in Scheme l; since there would be some difficulty in
preparing a monoacetate such as versiconal acetate, the intention was
to prepare a penta-acetate and compare it with the corresponding
compound already prepared by Dr. Cole.
Because versicolorln A, the starting point for the synthesis of
versiconal acetate, was available only in limited quantities, it was
decided to carry out a detailed investigation of the synthetic scheme
using the more readily available material sterigmatocystin (VII). The
use of this compound had the added advantage that the products would
be expected to be less polar than those from the versicolorin reaction,
and thus more easily handled.
Conversion of sterigmatocystin to the hemiacetal (VIII) was
carried out under the conditions described by Pohland for the prepara-B 42 2a•
IS 16
0
(Sterigmatocystin, VII)
IS 16
(ST-HA, VIII)
Page 53
42
sc·heme 1;
HO
H 2SO 4 (lO"o/c) in Acetone
l (Ver sicolorin A, III)
pH 7 , 2 , Buffer
THF
CHO CHO (VA-HA,. IV)
NaBH 4 (limited)
1 NaBH 4 (excess)
OH OH 16 r1
0 ~ 18 OH OH OH
+ H
(Versiconol, V) (Ver siconal, VI)'
Acetylation
Versiconal Acetate ---(II)
Versiconal (Pentaacetate)
Page 54
4)
The hemiacetal (VIII) was obtained in 40% yield on treatment of
sterigrnatocystin with 10% sulfuric acid in acetone at 60°. The
isolated material showed UV-visible absorption indicating the
presence of a hydroxylated xanthone ring similar to that of sterigma-
matocystin, while its IR spectrum showed absorptions indicating the
presence of a bonded xanthone carbonyl group. Its mm spectrum in
(DMSO-d6) showed absorptions consistent with those expected for a
hydrated derivative of sterigmatocystin, although the spectrum was
poorly resolved and difficult to assign precisely. Thus a 1-proton
triplet at 7.51, two 1-proton doublets at 6.82 and 6~62, together
with a 1-proton singlet at 6.45 were consistent with these aromatic
protons. Three 1-proton multiplets at 6.50-6.)2, 5.56-5.)8 and
4.18-4.oo were assigned to protons at carbons-14, 17 and 15, respec-
tively. A )-proton singlet at J.84 was due to the aromatic methoxy
group, and the two protons of carbon-16 showed a multiplet signal at
2.20 ppm. In addition, a very weak signal at 9.46 ppm assignable to
an aldehyde proton was observed. It's mass spectrum showed prominent
peaks at m/~ 342 Ot) )24 (M-18), JlJ (M-29), )06 (M-)6), 296 (M-46)·,
295 (M-47).
The various possible tautomeric structures for sterigmatocystin
hemiacetal (VIII) are shown in Scheme 2. Because only a very weak
absorption for an aldehyde proton was observed in the N:rIR spectrum
of the hemiacetal, the three tautomers VIII-B, C and D, were excluded
as major contributions to the equilibrium. The remaining two structures
VIII-A and VIII-E could be differentiated by the NMR spectrum. The
Page 55
44
Scheme 2:
IS 16 ' n OJO IS- 16 (VIII-A)
CHO
(VIII-B)
,.r 16
CHO CHO
(VIII-C)
CHO OH
(VIII-D) (VIII-E)
Page 56
45
proton signals at carlx>ns-15 and 16 were complex, due to multiple-
coupling, while carlx>n-17 in structure VIII-A or carlx>n-14 in
structure VIII-E is an epimeric carlx>n so its associated proton also
showed a complex resonance pattern. The only signal that can be used
to differentiate the tautomers is the signal at 6.53 ppm, which can
be assigned to H-14 in structure VIII-A or H-17 in VIIl-E. The FT-NMR
spectrwn of compound VIII suggested that the proton on carbon-14
resonates as a 1-p:roton doublet at 6 • .53 ppm. This confims that the
hemiacetal VIII exists largely in NMR solution (CDC13) and presumably
in other sol vents also, predominantly as the tautomer VIII-A, since
only this structure would give the observed coupling. Small amounts
of other tautomers may also be present, however, and it is significant
that the chelated hydxoxyl proton in the )-position appears as a
three-line signal, suggesting three significant contributors to the
structure of the hemiacetal.
In the preparation of one batch of sterigmatocystin-hemiacetal,
recycled ethyl acetate was used to extract the hemiacetal from the
acidic acetone solution. The crude product in this case showed two
major components on TLC, and these were separated by preparative TLC
( Benzene/Ethyl Acetate 70: JO) • The more polar compound was the
expected hemiacetal, while as the less polar compound was identified
as a derivative of the hemiacetal. It showed UV-visible absorption
similar to that of sterigmatocystin-hemiacetal (VIII-A), while its IR
spectrum showed the disappearance of the free-OH stretching at about -1 3300 cm observed in the hemiacetal. Its NMR spectrum in cn013 showed
Page 57
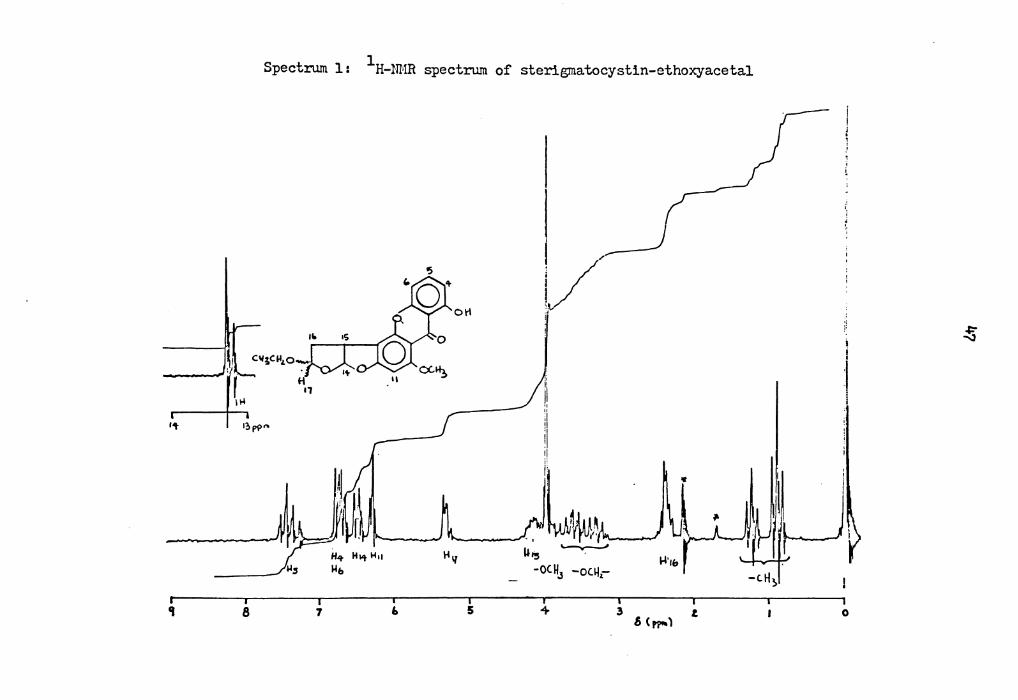
46
absorptions at o ppm; 1).28 and 1).20 (lH, 2s, chelated-OH), 6.84-6.64
(2H, m, -H4 and H6), 7.46 (lH, t, -H5), 6.JO (lH, distorted s, -8i1),
6.50 (1H, m, -~ 4), 5.)4 (1H, m, -~ 7), 4.18 (lH, m, -~ 5), 4.o (JH, s,
-OCHJ)' J.84 and J.16 (2H, 2q), 2.40 (2H, m, -~ 6), 1.14 and 0.89 (JH,
2t). _(Spectrum 1)
The chelated hydroxyl proton in the )-position of this hemiacetal
derivative appears as a two-line signal in a ratio of 2:1, suggesting
two significant contributors (i.e., two epimers) to the structure of
this compound. The two quartets at J.84 and J.16 ppm together with two
triplets at 1.14 and 0.89 ppm, indicated the existence of a pair of
epimeric ethoxyl groups in this hemiacetal derivative. Based on the
NMR spectrum, the structure of the derivative was proposed to be a
mixture of two epimeric sterigmatocystin-ethoxyacetals IX-A and IX-A'
(211). They are presumably formed through the reaction of sterigmato-
cystin-hemiacetal VIII-A with trace amounts of ethanol in the recycled
ethyl acetate. The triplet methyl signal at 6 1.14 ppm was assigned to
the methyl group at carbon-20 of structure IX-A, because models indi-
cate that the C-20 methyl group in this isomer is far away from the
aromatic ring and thus would have a shift similar to that of the methyl
group of ethanol (6 1.16 ppm). The methyl group of carbon-20 of
structure IX-A' on the other hand, is oriented towards the aromatic
ring, and it was thus assigned the chemical shift of 0.89 ppm. This
assignment was made by comparing the relative position of the benzene
ring and the methyl group, and by applying the diagram of Johnson and 42b Bovey for the long-range shielding associated with the benzene ring.
Page 58
1 Spectrum 1: H-NMR spectrum of sterigrnatocystin-ethoxyacetal
~
J 'L c11;Cll'-o
"4
.... 1?,p?"'
er 8
H .,
7 6
1-111
5
l,/-1
i:
I! :1
:1
' i:L ~. ~ ., ~'·to; ,
' "' U15 . -oC.H3 -oc.~,-
4 3
..i·,&
.s (~) t
i i
:I l
0
~
Page 59
II
(IX-A)
HO 0 0 0
II
(IX-A)
48
lb
IS 1b
(IX-A')
11
'•oc~CH;, 1.9 ~o
(IX-A')
Page 60
49
Just as in the case of sterigmatocystin-hemiacetal {VIII),
various isomeric structures for sterigmatocystin-ethoxyacetal IX
are possible. These are shown below as the structures IX-A - IX-E.
0 H
(IX-A)
CHO
(IX-D)
lb
IS-
HO 0 IL/-OCH CH 2. 3
(IX-B)
0
(IX-E)
Because no aldehyde proton was observed in the NMR spectrum of the
ethoxyacetal, the two isomers IX-B and D, were excluded as possible
structures. The remaining two structures IX-A and IX-E could be
differentiated by the NMR spectrum.
Page 61
.50
In the following section, we obtained the NMR spectrum of iso-
dihydrosterigmatocystin (XIII) and dihydrosterigmatocystin (XIV). The
chemical shift of protons at carbon-14 of structure XIII is at 4.19 ppm,
whereas the protons at carbon-17 of structure XIV were at 4.1.5 ppm. This
suggested that these protons in those two structures are almost equivalent.
(XIII) H
(4.1.9 ppm) H
0
(S, 80 ppm)
(XIV)
,s 16
14 11 H 0 t4 0 14.IS"rr.:!)
~ 6.'tO ppm.)
By analogy to this, the chemical shift of proton at carbon-14 of structure
IX-E of sterigmatocystin-ethoxyacetal should have a comparable value to
that of the proton at carbon-17 of structure IX-A.
ll ( 6.3S c.c&ccA..)
(IX-E) l6.S-O obsd.J
(IX-A)
<S,30)
it O 110CH2C~ l6.9S c.,tfoA) t 6,SO obsd)
Page 62
.51
We also know that the chemical shift of the a-protons of tetrahydro-
furan is at '.3.60 ppm, whereas the corresponding protons of dihydro-
sterigmatocystin (XIV) resonate at 4.15 ppm. This 0.55 downfield shift
is caused by the replacement of the a'-proton of tetrahydrofuran with
a.n al~oxyl group.
0 H
(XIV)
~I ~ o;O~ 0 l3,60 fP'm)
(THF)
By applying this 0.55 ppm downfield shift, and comparing the
chemical shifts of the proton nt carbon-17 of structure XIII and of
the proton at carbon-14 of structure XIV, we nere able to calculate
the chemical shifts of these corresponding protons of tautomeric
structures IX-A and Eat 6.95 ~nd 6.:35 ppm, respectively. A signal
at 6.50 ppm of sterlgmatocystin-ethoxyacetal, IX, was t'1us assigned
to the proton of a bridgo carbon which has two alkoxyl substituents.
Another signal at .5 • .30 ppm was assigned to tho proton of a carbon
which has an ethoxy group bonded to it. After assigning these aliphatic
proton signals of fiterignatocystin-ethoxyacetal, the two tautomeric
structure IX-A and IX-E could then be differentiated by the double
Page 63
52
resonance decoupled NMR spectru;.1. Thus irradiation at 4. 2 ppm caused
the resonance at 6.5 ppm to collapse to a singlet, while irradiation
at 6.5 ppm clearly simplified the complex signal at 4.2 ppm. Irradiation
at 5.J ppm also simplified the adsorption at 2.4 ppm. From these
results, it is clear that structure IX-A is the structure of sterigma-
tocystin-ethoxyacetal. This also supports the previous conclusion that
sterigmatocystin-hemiacetal exists predominantly as the structure VIII-A.
The mass spectrum of the ethoxyacetal IX-A showed prominent peaks at
!/!. 370 (M+), 342, )41, 297, and 286.
0 0
(IX-A, fvl+, m/.!! 370)
0
(m/~ 341)
0 +•
0 ~OH H
Cm.I~ 342)
Page 64
0
53
~ ~OEt
(IX-A, M+, m/~ 370)
+ H
(m/~ 297)
0 +· OEt
This fragmentation pattern is consistent with the assigned structure
IX-A, as outlined in the scheme above.
The redu:tion of sterigm~tocystin-hemiacetal, VIII-A, with sodium
borohydride could in principle yield some of or all of the reduction
products shown in Scheme J. Although as we have shown, the hemiacetal
exists in the ring closed fonn VIII-A in org<'.nic sol vents, in aqueous
solution at p:1 7.2 the fonns VIII-B, VIII-C and VIII-D would also be
expected to contribate to the equilibrium (for convenience, ionic forms
of e.g., VIII-Care not shown).
Reduction of VIII-B would be expected to yield the products, X,
while reduction of VIII-C could yield the partially reduced compounds
Page 65
54
Scherne 3 :
,s , , OH
1l (VIII-A) 1l (VIn-D)
1& ~
~ HO HO CHO OH 3 (VIII-B)
NaBH4 l (VIII-C)
\i,l) ,s If>
HO H
H (XII)
Page 66
OH
H HO (X-B)
H
l+.8) l~.:q,rrn) -----,o-)6 Q Q U.8J
H t.t.6>
(X-C)
.5.5
(+.8) l~,.1. ppm)
H i'f.
,~.B> (XI-C)
Page 67
56
X or XI or the fully reduced compound, XII. Reduction of VIII-D would
yield only the reduced compounds XI. The product Xis, of course, the
compound analogous to the desired "versiconal acetate" and it was
expected that this compound would constitute at least 5o% of the
partially reduced material,
In the e,ent, reduction of the hemiacetal VIII in tetrahydrofuran
and sodium phosphate buffer (0.05 M, pH 7,2) with sodium borohydride
yielded only two products isolable by Pl'LC. The more polar of the two
materials wa~ identified as the totally reduced compound, XII. It
showed UV-viEtble absorption indicating the presence· of a hydroxylated
xanthone ring structure, while its IR spectrum was consistent with the
assigned structure, showing )Y !! 3400 (br), 1645 (s), 1605 (s), etc.
Its NMR spectrum in DMSO-d6 showed a 1-proton triplet, two 1-~roton
doublets and a 1-proton singlet at 7.68, 6.99, 6,77 .:;nd 6.45, respective-
ly, assigned to the aromatic protc·ns. A 3-proton singlet and a 2-proton
triplet at 3,90 and 3,36 were assigned to the protons of the aromatic
methoxy group and carbon-17, respectively. The protons at ca.rbons-14
and 15 overlapped with water in the sol•rent and formed a broad signal
around 3,68. A 2-proton multiplet at 2.04 was assigned to the protons
at carbon-16. Its mass spectrum gave p:~ominent peaks at m/! 346 (M+),
315 (M-31), 297 (M-49), 285 (M-61) and 271 (M-75).
The less polar product was identified as the desired partially
reduced sterig:aatoc;rstin-hemiacetal. Its UV-visible absorption indi-
cated the presence of a substituted xanthone ring structure, while its
IR spectrum showed absorptions consistent with its functional groups.
Its mass spectrum showed prominent peaks at '!!Y! 344 (M+), 326 (M-18),
Page 68
57
313 (M-31), 235 (M-59) and 283 (M-31). Its NMR spectrum in DMSO-d6
showed absorptions consistent with those expected for a partially
reduced derivative of sterigmatocystin-hemiacetal. Thus a 1-proton
singlet at lJ.15 was characteristic of the chelated aromatic hydroxy
proton. In addition, a 1-proton triplet, two 1-proton doublets and
a 1-proton singlet at 7.54, 6.84, 6.6) and 6.Jo were due to the four
aromatic protons at carbons - 5,6,4 and 11, respectively. Two 1-proton
multiplets and a )-proton singlet at 5.64-5.42, 5.20-4.78 and J.8) ppm
were assignable to an acetal proton, a deshielded benzylic proton, and
the aromatic methoxy group. A 2-proton signal at J.80 ppm was partially
overlapped with the methoxy proton signal, while a two proton signal was
observed as a multiplet at 2.20 ppm.
This information, an<l specifically the absence of any signal due
to an aldehyda group, indicates that the i3olated material does not have
the open-chain structure X-B or XI-A. It <loes not, however, distinguish
satisfactorily between the two basic struc·~ures X and XI, since both of
these could be rationalized as giving rise to the observed NMR spectrum.
(The possible assignments are shown in Scheme J). Spin decoupling
experiments did, however, permit a choice to be made between structures
X and XI. II.Ta.diation at 2.2 ppm caused trc signal at 5.6 ppm to
simplify significantly: the signal did not collapse to a singlet,
presumably because the compound isolated mc:,t probably consisted of a
mixture epimeric at the hemiacetal carbon. Irradiation at J.8 ppm
caused the signal at 4.8 ppm to simplify, a:id the reverse was also true,
with irradiation at 4.8 ppm causing noticerble changes in the signal
Page 69
at J.8 ppm. This evidence conclusively proves that the material
isolated does not have the general structure X, as hoped and expected,
but instead has the structure XI.
Further proof of this assignment was derived from the conversion
of th~ partially reduced compound to its anhydro-fom. Treatment of
compound XI with dilute acid yielded a compound named isodihydrosterig-
matocystin, XIII. The compound was shown to be different from dihydro-
sterigmatocystin, XIV (prepared by hydrogenation of sterigmatocystin
over palladium/charcoal) in physical properties (TLC Rf' value, IR
spectrum.)
0 H
0
(ST, VII)
0
Hydrogenation
(Di hydro ste rigmatocystin) (XIV)
Page 70
59
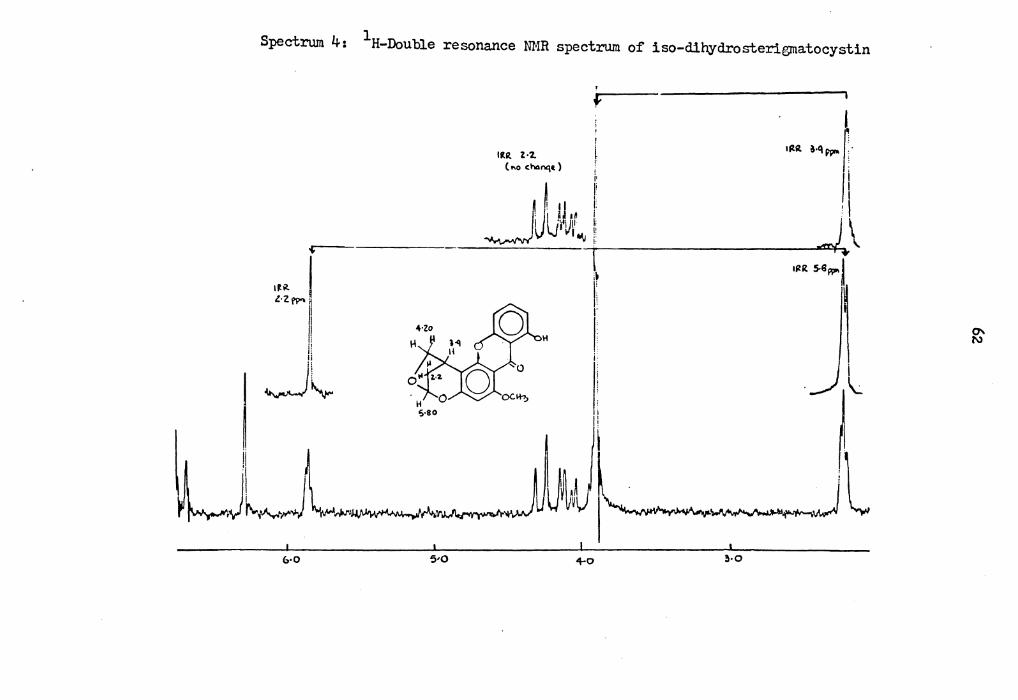
It showed UV-visible absorption indicating the presence of a substi-
tuted xanthone ring structure. Its NMR spectrum (Spectrum 2) in cnc13 showed absorptions different from those of dihydrosterigmatocystin.
(Spectrum J). Thus a 1-proton singlet at 12.96 ppm was due to the
chelated aromatic hydroxy proton. One 1-proton triplet, two 1-proton
doublets and a 1-proton singlet at ?.J6, 6.69, 6.62 and 6.20 ppm were
assigned to the aromatic protons. In addition, a 1-proton doublet
and one 2-proton multiplet at 5.79 a.nd 4.19 ppm were assigned to protons
at carbon-17 and 14. The aromatic methoxy protons showed a singlet at
J.90. A 1-proton multiplet and a 2-proton multiplet at J.88 and 2.20
ppm were assigned to protons at carbon-15 and 16. The doublet reson-
ance spin decoupled spectrum confi:rmed the as~igned structure. Irra-
diation at 2.20 ppm caused th3 sign.'.1.ls at 5.79 ppm to collapse to a
singlet. Irradiation at J.88 ppm cc.used the signal at 2.20 ppm to
simplify and irradiation at 5.8 ppm caused noticeable changes in the
signal at 2.20 ppm. Its mass spectr,.un showed prominent peaks at fill~ .326 (M+), .308 (M-18)~ 297 (M-29), 283 (M-4J) and 265 (M-61).
The evidence to date indicates ~hat the partially reduced sterigma-
tocystin-hemiacetal has structure XI, but does not distinguish between
the possible tautomers XI-B and XI-C. It was hoped through the
preparation of the acetyl derivative of XI-Band XI-C that it would
be possible to differentiate these two isomers.
Page 71
60
·--I ' \ I \ ' •• j~--
:
.. C\l
Page 72
Spectrum 3: 1H-ID1R spectrum· of dihydrosterigmatocystin
I , , ,;
dl 'o·°" ;- 11, I~
11
~ i ' I
I l .
111 lil
JU\. ,__,._ ___ ~17 I H •S w ...
1 8 7 ' 5 ) ., t .5 C Pf"I)
0
Page 73
1 Spectrum 4: H-Double resonance NMR spectrum of iso-dihydrosterigmatocystin
f I ........ r /1 , I
.J.\ IIZR S-8W" l
\ ~
lltR. l•'Z. ( f\O ~h4nq~ ) i
U 1:
I J i . · I •ft i' _J ~l'Jr ... 1!
i. 14
i
V~11
-r -~l
&•O 5•0 +o 3•0
Page 74
6J
(Ac)2 0, Pyridine
(XI-B)
OAc (XI-B ')
(XI-C)
(Ac) 2 0, Pyridine 1
0 (XI-C ')
Page 75
64
If it were in the XI-C form, the acetyl derivative would not
significantly change the chemical shift of protons at carbon-14. If
it were in form XI-B, the acetyl derivative would cause a 0.5 ppm
downfield shift of protons at carbon-14'. However, it did not prove
possible to prepare an acetyl derivative in adequate quantity, given
the small amounts of material available; in the one attempt to prepare
such a derivative, a complex mixture of products was obtained.
VI. lJC-NMR Studies of Sodium B'Jrohydride Redur.ed Derivatives of
Sterigmatocystin-Hemiacetal
The evidence presented so far has established th~ structure of
partially reduced sterigmatocystin hemiacetal as eith~r form XI-B or
XI-C. Because attempted aeetylation of this compound gave a mixture of
products, and because of the difficulties inherent in using a chemical
method to distinguish between tautomeric st.ructures, it was decided to
attempt to distinguish between the two structures on the basis of
lJC-NMR spectroscopy.
lJC-NMR has a much greater potential than 1H-NMR for structure
studies on organic, polymeric and biological mo:i.ecules, because the
chemical shift range of 13c-NMR covers 600 ppm, whereas for 1H-NMR it
only has a range of 20 ppm. Thus it is not unu.sual tha.t lJC-NMR gives
much better resolution and the capability of assigning the resonance of
each individual carbon atom in a complex molecule. In our case, 1H-NMR
showed clearly the signals of the aromatic protons, but the signals due
to the upfield aliphatic protons could not be used to differentiate the
tautomeric structures XI-Band XI-C, because of overlapping signals,
Page 76
65
complex coupling patterns, and essentially equivalent predicted spectra
for the two tautomers,
In 1H-NJR, three types of information can in principle be obtained
about the nature of the protons in the molecule, These are their chem-
ical. shift, their multiplicity (including the approximate coupling
constants), and their peak area. In lJC-NMR, chemical shifts are the
most important and useful parameter. Quantitative coupling constants
can also be obtained by applying modern techniques, such as off-resonance
and gated decoupling, but peak area measureme:1ts usually do not give
adequate information, This is because the nuclear Overhauser effect
causes the loss of the direct relationship be·;ween peak area and the
number of carl:x:m atoms giving rise to that peak.
It has been experimentally found that subs ti tuent effects on the
chemical. shifts of carbon atoms are usutlly ,.dditive; thus predictions
of carbon chemical shifts by comparison with model compounds are usually
successful, Assignments of chemical. shifts can be made using different
techniques, such as off-resonance decoupling, gated decoupling, specific
labeling, chemical shift reagents, etc. The::;e techniques 4J will be
briefly discussed in the following sections,
Off-resonance decouplin~
This technique will tell the degr)e of protonation of each indivi-
dual carbon atom in a molecule. Thus, a methyl carbon will give a
quartet signal, a methylene carbon will show a triplet signal and a
methine carbon will appear as a doublet.. .The observation of this
effect is possible because this technique involves the use of either
Page 77
66
an insufficient 1H decoupling power or a decoupling power which was not
set exactly at the resonance frequency of the protons coupled to the
carbon of interest, thus allowing some residual coupling between the
carbon a.tom and its directly bonded proton(s). Generally, "off-
resonance" spectra can be obtained in a.bout the same time as the
"no.ise-decoupled" spectra.
Gated-decouplin~
These spectra are obtained by automatically turning off the proton
decoupler beforn the pulse and on again after a short period. The
result of this technique is that the proton and carbon coupling will
be retained. Usually, remote (two bond) l3c- 1H couplings are observed
as well as the directly bonded l3c- 1H coupling.
Specific Labelin~
The most common specific labeling technique is that of deuterium
labeling. Deuterium has a spin of 1, and would produce a l3c- 2H
multiplet when it is coupled with a carbon e.tom. The signal of a
l3c- 2H coupled carbon atom is less intense c.s compared with a 13c-1H
coupled carbon atom, because the spin-lattice relaxation time is greatly
increased in the l3c- 2H coupled carbon. Generall~·, the resonance peak
of a carbon would decrease or disappe,.1r wher.. this specific carbon atom
has been deutera.ted. The technique of lJC-la.belling has also been
widely used. In this technique one or a few carbons of interest are
enriched with 13c-atom by a synthetic process. The enriched carbon
a.tom gives a. strong peak a.s compared with the natural abundance lJC-NMR
spectrum at its corresponding chemical shift.
Page 78
Chemical Shift Rea.gents
The Lanthanide chemical shift reagents that are currently being
used to simplify 1H-NMR spectra can also be used in 13c-N~m studies.
It is possible to aid l)C-NMR spectrum assignments with pseudo-contact
shift.measurements made with these reagents. An exa.mple of the appli-44 cation of this technique is the work on Steyn and co-workers, who
have employed Eu(FOD)3 chemical shift reagents to assign some aromatic
lJC resonances in aflatoxin Bi_ and sterigmatocystin.
Selective Proton Decoupling
This is a single-frequency on resonance decoupling technique. It ·
includes irradiation at a resonance frequency of ths specific proton(s),
with the result that the carbon atom directly bonded to the irradiated
protons collapses into a sharp singlet, while other non-coupled carbons
remain as multiplets.
The assignments of the l)C-NMR spectra of sterigmatocystin and
dihydrosterigmatocystin have been reported previously. 44,45 Their
reported chemical shifts together with the chemical shifts we o bta:ined
are listed in Table 10.
In order to distinguish between the two possible structures XI-B
and XI-C for the partially reduced sterigmatocystin-hemiacetal, it is
necessary to bs able to pkedict the chemical shifts of key carbon atoms
in the two structures. With this in mind, the l)C-NM.R spectra of some
model compounds were obtained.
The natural abundance l)C-NMR spectrum of sterigmatodiol, XII, is
shown in Figure J. The l)C-NMR data derived from proton-noise-decoupled
Page 79
68
TABLE 10
lJC-Chemical Sh1~s* of Sterigmatocystin and Dihydrosterigmatocystin
s
(VII)
Carbon VIIa XIVa VIIb XIVb XIVc
1 180,9 181,0 180.9 180,8 180,0
2 1Q8,8 108.8 108.8 108,7 108,0
J 162.1 162.1 1.54. 7 151~.6 161.1
4 111.0 111.0 106.4 10.5,5 110,2
.5 135,4 135,3 135,4 13.5,2 135,6 6 10.5. 7 105.6 111.0 110.7 105.8
7 1.54,7 1.54. 7 162.1 161.9 1.54,3
8 1.53,7 1.54.) 15). 7 1.54.6 15),6
9 106,4 10.5.J 106.4 106.7 105.4
10 164.J 16.5.9 164.J 16.5.7 16.5,6
Page 80
TABLE 10 (continued)
Carbon VIIa XIVa VIIb XIVb XIVc
11 90.4 89.7 90.4 89.6 90.0
12 163.0 163.2 163.0 163.1 162.8
lJ 105.7 105.6 105.7 105.1 105.8
14 llJ.l llJ.4 llJ.1 113.1 llJ.J
15 47.9 44.2 47.9 44.2 4J.2
16 102.4 Jl.4 105.7 Jl.4 Jo.5
17 145.1 67.7 145.1 67.6. 67.0
18 56.6 56.6 56.6 56.6 56.J
a. 44 Assigned by Steyn, et al. in cnc13 b. Assigned by Cox, et ai. 45 in CDClJ
c. This woI:k in d~-DMSO (The ass:tgnments of aromatic carbons were based on struc ure VIIa-steyn's work).
*Chemical shifts in 6 ppm downfield from TMS.
Page 81
..-, s
Figu1'8 J.
r-,
DMIO
M .. I
17
II
u • • !16
Natural.-abmdance proton-noise-decoupled lJC-NMR spectrum of sterigmatodiol (XII).
~ 0
Page 82
71
(p.n.d.), off-resonance decoupled and gated decoupled spectra are given
in Table ll. The aromatic carbon signals have been assigned by com-
paring their chemical shi~s with those of the corresponding carbons
of sterigmatocystin, VII, (Assigned by Steyn and coworkers). The
carbo~ signals at carbon - 14,15,16,17 and 18, have been assigned by
correlating the residual splittings in off-resonance and gated decoupled
spectra with the known proton chemical shifts. The ma.gnit~des of the
observed directly bonded and long range lJc- 1H coupling constants
(Table 11) support these assignments.
The two signals at JJ.2 and J4,9 could be assigned to carbons-16
and 15, respectively. In the off-resonance decoupled spectrum, the
signal at JJ.2 was split into a triplet, indicating that it derived
from carbon-16, and the doublet sicnal at 34,9 w,1s assigned to carbon-15,
Signals due to carbons-14 and 17 both showad triplet patterns in the
off-resonance spectrum, so their differentiation and assignment was
based on the gated decoupled spectrum. Th3 assigned signals of carbon-14
(o 59,9 ppm, triplet of doublets) a~d carcJn-17 (6J.5 ppm, triplet of
triplets) are consistent with the predicted lon~ range lJC- 1H coupling
for these two cartons, The methoxy carbon atom showed a quartet signal
at 55.8 ppm in both the off-resonance and gated decoupled spectrum. It
is known that the resonance of an aromatic carbon bearing a hydroxyl
group occurs r.bout 5 ppm upfield of the slI!le carbon bearing an alkoxyl
group, 46 and it is thus :possible to calculate ani assign the chemical
shifts of carcon atom-10 and 12 based on l3c substituent effects of 46 substituted benzenes. The carbon-10 signal was assigned at 159.7 ppm
(Calcd, 159,3), and carbon-12 at 16J.6 (Calcd, 16J.4),
Page 83
72
TABLE 11
lJC-Chemical Shifts of Sterigmatodiol, XII
IS 16
H
(XII)
a. J13 directly bonded 13c- H coupling C- H lJ
J13~ long range C- H coupli1g C- H
b. "*", "+","•"may be interchanged
c. Capital letters refer to the pattern resulting f5om directly bonded p:rotons and small letters to long range C- H coupling.
d. T: triplet; D: doublet; Q, quartet
Carbon Atom No. 8 ppm(TMS) J lJC- H (Hz)
J ~ 13c- H (Hz)
1 180.?
2 1oa.5•
J 161.2+
4 • 110.0
5 1J5.9
Page 84
73
TABLE 11 (continued)
Carbon Atom No. 6 ppm(THS) J lJC- H (Hz)
J ts lJC- H (Hz)
• 6 106.1 * 7 157.2 * a 1.54.5
9 107.9 •
10 159.7 11 95.7 12 16).6+
13 103.7 •
14 59.9 (T,d) 1)6.4 ).5 15 )4.9 (D)
16 JJ.2 (T)
17 63.5 (T,t) 139.04 4.4 18 55.8 (Q) 147.8
Page 85
74
The natural-abundance lJC-NMR data of iso-dihydrosterigmatocystin
XIII, derived from the proton-noise-decoupling and gated decoupling
techniques are given in Table 12, and its ),n,d, spectrum is shown in
Figure 4. The assigrunents of its aromatic carbons were used on 44 comparison with those of dihydrosterigmatocystin, XIV, The proton-
bearing aliphatic carbon signals of isodihydrosterigmatocystin have
been assigned by correlating the l3c- 1H splitting in the gated decoupled
spectra with the predicted l3c- 1H coupling pattern, The magnitudes of
the observed directly bonded lJc- 1n coupling constants support these
assignments. Because of ring-strain, the quart.et signal of carbon-14
was shifted downfield to 78,7 ppm. The assignments of carbon-17 (99,J
ppm, doublet of doublets) and carlx>n-16 (Jl.5 ppm, triplet) are based
on the results of their lJC-H coupling pattern and comparisons of their
chemi'cal shift with those of model compounds, The doublet signal of
carbon-1.5 (28,7 ppm) was partially overlapped with the triplet signal
of carbon-16 in the gat.ed decoupled spectrum,
Because of the complexity associa. ted -..ri th the purification of
partially reduced sterigmatocystin-hemiacetal and its consequently
limited supply, only its proton-noise-decoupled lJC-NI1R spectrt. 1.m could
be obtained (Figure 5), The assignments of its aroma.tic and methoxy
carbon signals were based on compairson with these of model compounds,
such as sterigmatocystin, dihydroxtyterigma.tocyst.in, sterigma.tc·diol and
isodihydrosterigmatocystin. It should be noted t.hat although there is
some uncertainty in the exact value of the chemical shift for carbon-10,
nevertheless this must lie in the range 160-161.1 ppm. (see Table lJ).
The signals at JQ.2 and J2 • .5, which were of lowe:: intensity then the
Page 86
75
TABLE 12
lJC- Chemical Shifts of Isodihyd:ro-sterigmatocystin (XIII)
(XIII)
"C" Atom No. 6 ppm J lJC-H (Hz)
J (3 lJC-H
l 180.l
2 1oa.2•
J 158.l+
4 109.8 •
5 1)5.J
6 10.5. 7 11
7 * 1.54.l
8 * 152.8
9 107.a •
10 160.9+
Page 87
76
TABLE 12 (continued)
"C" Atom No. 6 J ppm 13c-H (Hz)
11 95.7 (D) 163.a
12 160.2+
lo4.J • 13
14 78.7 (T) 154.4
15 28.7 (D) 169.7
16 Jl.5 (T) 112.2
17 99.J (D,d) 169.7
18 55.9
a. J directly bonded lJC-H coupling 13c-H
J S long range 13c-H coupling 13C-H
b. "*'' , "+" , "•" may be interchanged
J s 13C-H
8.2
c. Capital lette::cs refer to the pa.tterr. resulting f*om directly bonded protons and snall letters to long range lJc-H coupling
d. Da doublet; T: tripl,}t
Page 88
5 (l_ • .,.._rtp,aaocyellal
10
1'7 u
14
II -
200 ''° 100 ~
Dta0 15
"
Figure 4. Natural abmdance proton-noise-decoupled lJC-NMR spectrum of iso-dihydroster1g,natocys1n (XIII).
~
Page 89
200
(Partially Reduc" •rlpnatocylltl ..
Hemlacetal)
.0 5
II
l! 14
15
150 100 50 0 6(Pfln\)
Figure 5. Natural abmdance proton-noise-decoupled l3C-NMR spectrum of partia.lly reduced sterigmatocystin-hemiacetal (XI).
--.J a>
Page 90
79
TABLE lJ
lJC-Chemical Shifts of Partially Reduced Sterigmatocystin-hemiacetal
(XI-B or XI-C)
11 OH
(XI-B)
Carbon Atom No. 6 (ppm)
1 180.2
2 110.f
J 162.7+
4 • llJ,8
5 1J5,9
6 106.1 •
7 * 1.54,3
8 l.5J,6 * 9 108.0 •
10 161.J+
Page 91
80
TABLE 13 (continued)
Carbon Atom No.
11
12
13
14
15
16
17 18
Note: "+", "*", "•" may be interchanged.
6 (ppm)
• 105.1 (or 104.4)
62.7
)2.5 30.7
Not Observed
95.9 56.2
Page 92
81
other signals, were assigned to carbon-15; the observation of two signals
is consistent with the existence of XI-B or XI-C in two epimeric foms.
For the two signals at 56.2 and 62.7, the methoxy carbon atom was
assigned as the signal at 56.2, with the same 6 (ppm) value as the
model ·compounds, and the signal at 62. 7 was thus assigned to carbon
atom-14. In both dihydro and isodihydrosterigmatocystin, signals of
the acetal carbon were downfield from those of carbon atom-11, and so
the two signals at 93.2 and 95.9 were assigned to carbon atoms 11 and
17, respectively. The signal due to carbon atom-16 was presumably
concealed under the solvent resonances, and could not be observed.
In order to differentiate the tautomers XI-Band XI-C by 13c-NMR, a careful comparison of the lJC-chemical shifts of these aliphatic
carbons with these of model structures is reg_uired as descrit:ed in
Scheme 4.
The carbon atom-14 of structure XI-Chan similar neighboring atoms
and ring strain as that of carbon atom-17 of structure XIV. The only
difference is that atom-17 of structure XIV is bonded to aliphatic
carbon-16, whereas the atom-14 of structure XI-C is lx>nded to benzylic
carbon atom-15. From a comparison of the chemical shifts of carbon
atoms-17 and ll:· of structure XIV, it appears that a J.6 ppm upfield
shift occurs wten the directly bonded aliphatic carbon atom (carbon
atom-16) was replaced by a benzylic carbon atom (carbon atom-15). So
the chemical shift of carbon atom-14 of structure XI-C could be
calculated by correcting the shift of carbon atom-17 of structure XIV
with the J.6 ppm upfield shift caused by replacing the directly bonded
Page 93
82
Scheme ·4:
1b
0 \4- 0 '1 <.113.3) (61,0J
(XIV)
(XIII)
(XII)
I\
(XI-B)
15 I(,
0
(XI-C)
14-
Page 94
8J
aliphatic carbon a.tom with a. benzylic carbon atom. The chemical shift
of carbon atom-14 of structure XI-C thus calculated was 67,0-J,6 =
6J.4 ppm.
The carbon atom-14 of structure XI-B was almost eqw.v&lent to the
carbon a.tom-14 of structure XII, so no correction was needed, A signal
of 59,9 ppm due to carbon a.tom-14 should be observed, if the compound
existed.as structure XI-B. The observed signal of carbon atom-14 was
at 62.7 ppm, which agrees better with the value calculated for structure
XI-C than that calculated for structure XI-B,
The structure suggested for the partially reduced sterigmatocystin
hemiaceta.l could also be confirmed through a consideration of the
chemical shift of the aroma.tic carbon atom-10. It is known from
previous studies that the resonance of an aromatic carbon bearing a
hydroxyl group occurs about 5 ppm upfield of the same carbon bearing 46 an alkoxyl group, and it is even pos3ibl.e to calculate the chemical
shift of carbon atom-10 based on lJc substit.uent effec-~s of substituted
benzenes, 46 When this is done the values obtained are 161,7 ppm for
structure XI-B. These calculated values aro further supported by
values from the model compounds XIV and XII I for the former compound
the alkoxylated C-10 has a chemical shift of 165,6 ppn, while for the
latter the bydroxyla.ted C-10 is assigned a chemical shift of 159,7 ppm,
It should be noted that isodihydrosterigma.tocystin (XIII) is not a. good
model for the shift of C-10 because ring strain in the bridged bicyclic
system could cause steric compression effects of unknown ma.gni.tude at
C-10; on the other hand; the ring strain in compounds XIV and XI-B
would be expected to be comparable, On this basis, the observed
Page 95
84
chemical shift of C-10 in structure XI of 160-161 ppm agrees well with
the value predicted for structure XI-C.
(XIV)
l l OH OH
(Xll)
(Xl-C)
Page 96
85
From the predicted and observed chemical shifts of carbons-10 and
14 of partially reduced sterigmatocystin-hemiacetal, structure XI-C is
a better choice than XI-Bas the actual structure.
Based on lJC NMR spectra, it is suggested that the structure of
partially reduced sterigmatocystin hemiacetal exists in the tautomeric
form, XI-C. It is desirable, however, that we confirm this suggestion
through another pathway, such as chemical modification of the structure
of the compound.
VII. Chemical Modification of Partially Reduced Sterigrnatocystin-
Hemiacetal (XI-C).
From TLC results, we knew that at least. two acetyl derivatives
were formed from the acetylation of partiar.y reduced st.erigmatocystin
hemiacetal. Because of the complex product3 from acetylation and the
difficulty associated with their separation, an alternate method of
confirming the tautomeric structure XI-C, w3.s sou.;ht. It was important
to select a procedure which, as far as possible , would be expected to
proceed without disturbing any possible tautomeric equilibrium between
the two forms. This requirement excluded a simple test such as the UV
spectrum of compound XI in base, since it could 1::3 argued that form
XI-B might undergo tautomerization to yield a free phenolic hydroxyl
group, which uould then display a characteristic bathochromic shift
in base.
Methyla.tion with diazometha.ne was an attractive alternative, since
it is known that diazomethane will only methylate acidic hydroxyl groups,
Page 97
86
such as the phenolic group of tautomeric XI-C, and does so under
extremely mild conditions. The tautomer XI-B would not be methylated
under the reaction conditions,
OH 1 (XI-C)
0 OH
(XV, isolated)
No Reaction OH
(XI-B)
Reaction of compound XI with aldohol-free diazomethane 47 in ether
yielded a product which was identified as the methyl derivative, XV,
on the basis of its spectroscopic properties; the use of alcohol-free
diazomethane was necessary because the alcohol might react with the
hemiacetal to form an acetal derivative. The isolated product, XV,
showed UV-visible absorption similar to that of sterigmatocystin,
indicating the presence of a substituted xanthone ring, while its IR
Page 98
87
spectrum showed absorptions consistent with those predicted for its
functional groups. Its NMR spectrum showed the presence of a xanthone
ring and two methoxy groups. Thus, a 1-proton broad singlet at lJ.02
ppm was due to the chelated aromatic hydroxyl proton. One 1-proton
triplet, two 1-proton doublets and one 1-proton singlet at 7.51, 6.82,
6.64 and 6.50 ppm, respectively, were assigned to the four aroma.tic
protons. One 1-proton doublet at 5.64 was assigned to the proton at
ca.rbon-17. In addition, two )-proton singlets were overlapped at J.98,
and were assigned to two aromatic methoxy groups. The signals of protons
at ca.rbon-14 and 15 were overlapped with the methoxy protons at 3.52
4.40. One 2-proton multiplet at 2.30 was assigned to protons at carbon-
16. Its mass spectrum showed prominent peaks at 358 (M+), 340 (M-18),
328 (M-30), 3~7 (M-31), 299 (M-59) 285 (M-73) and 272 (M-86). The
observation of a prominent {M-H20) peak confirmed that methylation had
not occurred at the hemiacetal hydroxyl at structure XI-B, since then
a peak at (M-32) and none at (~1-18) would be expected.
The successful methylation of partially reduced sterigmatocystin-13 · hemiacetal with diazomethane, together with the C-NMR results, confirms
that the compound possesses the structure XI-C. The preparation and
reduction of sterigm.atocystin-hemiacetal can be rationalized a.s shown
in Scheme 5.
The sterigmatocystin-hemiacetal (VIII-A) in phosphate buffer (pH 7.2)
and THF solution could exist in several tautomeric forms (i.e., VIII-A
to VIII-E), but the structure VIII-Dis probably the most stable one
because the six-membered ring dihydropyran hemiacetal form would be
expected to be less strained than the five-membered ring dihydrofuran
Page 99
Scheme 5:
(ST, VII)
15"
H HO (VIII-C)
CHO
H
88
H
(ST-HA, VIII-A)
pH 7.2 (buffer)
OH
Page 100
89
(XII)
(XIII)
,s- 16
H (XI-A)
r
H
H HO
OH
Page 101
90
hemiacetal. structure VIII-B. The hemiacetal structure VIII-Dis al.so
more stable than the open chain structure VIII-D; it is well-known that
six-membered ring hemiacetals such as the pyranose sugars are more
stable than the corresponding open-chain forms.
Reduction of hemiacetal. VIII-D with a limited a.mount of sodium
borohydride would thus give a reduced hemiacetal with structure XI-B,
which has a six-membered ring structure fused with a benzene ring, and
is therefore slightly strained. In order to release the ring strain,
structure XI-C is formed through the open chain fo:rm XI-A. The structure
XI-C will have greater entropy than structure XI-B, because its furan
ring is free to rotate, and it lacks any strain due to incorporation of
a double bond in the ring. The hcmiacetal with structure XI-C is thus
the most stable form of the partially reduced sterigmatocystin-
hemiacetal.s formed by reduction of the most stable reducible tautomer
(VIII-D) of the sterigmatocystin hemiacetal. In buffer solution with
excess sodium borohydride the open chain tautomer XI-A would be further
reduced to form ster1gmatodiol (XII).
Three TLC solvent systems have been developed for the rapid
differentiation of these derivatives. The results with approximate
Rf values are listed in Table 14.
VIII. Preparation and Reduction of VersicoJ_orin A-hemiacetal
Having established the structures of the reduction products of the
hemiacetal. of sterigmatocystin, it is now possible to discuss the
corresponding reduction products of versicolorin A. The structural
evidence in this case is less extensive than in the case of the
Page 102
TABLE 14
Separation of Derivatives of Sterigmatocystin in Various TLC Systems. (*On E.M. S.G. G.F. 2.54 plate)
Partially reduced Solvent Sterigma- Sterigmatocystin Sterigmatocystin Sterigmatocystiri System tocystin -hemiacetal -ethoxyacetal -hemiacetal
Hexane:Ethyl 48 21 47 5 Acetate(50:50)
Benzene:Ethyl Acetate :1·!athanol 65 44 62 29 '° (50:50:2)
....
Benzene:Ethyl Acetate:Acetone 72 64 69 52 :Methanol (JO:JO:JO:J)
Page 103
Solvent System
Hexane:Ethyl · Acetate (50 :50)
Benzene:Ethyl Acetate :I·lathanol (50:50:2)
Benzene:Et.hyl Acetate:Acetone :Methanol (30:JO:J0:3)
Sterigmato-diol
l
1
15
TABLE 14 (continued)
Dihydro-Sterigmatocystin
43
61
70
Iso-dihydro-Sterigmatocystin
37
56
69
Methylated Partially reduced Sterigmatocystin-hemiacetal
6
15
48 '° I\)
Page 104
93
sterigmatocystin products, 'because of the more limited supply of starting
materials, and hence analogy to the fozmer case will 'be a useful tool
in assigning structures to the various products.
A simple scheme for the preparation and reduction of versicolorin
A-hemiacetal, analogous to the sterigmatocystin-hemiacetal, is shown
in Scheme 6.
Versicolorin A-hemiacetal, IV, was prepared by refluxing versi-
colorln A with lo% H2so4 in acetone solution. It showed UV-visible
absorption indicating the presence of a hydroxylated anthraquinone ring
structure similar to that of versicolorin A, while its IR spectrum showed
absorptions indicating the presence of bonded and free quinone carbonyls.
Its FT-NMR spectrum in d6-acetone showed absorptions consistent with
those expected for a hydrated derivative of versicolorin A. Thus two
1-proton doublets at 7.18 and 6.61 ppm together with a 1-proton singlet
at 7.05 ppm were assigned to the three aromatic proton signals. A
1-proton doublet at 6.49 ppm was due to the proton on carbon-15. In
addition, two 1-proton multiplets at 5.73 - 5.53 and 4.27 - 4.01 ppm
were assigned to protons at carbon-18 and 16. The two proton signal
at carbon-17 was overlapped by the solvont and thus could not 'be
identified. The mass spectrum of this new compound showed prominent
peaks at m/~ 356 (M+), 355, J48 (M-18, small), 328 (H-28), 310 (M-46),
309 (M-47), JOO (M-56), 299 (M-57) and 285 (M-71).
Reduction of versicolorin A-hemiacetal, with a limited amount of
sodium borohydrlde in the presence of THF and a pH 7. 2 sodium phosphate
buffer yielded only two major new comJX>nents, as judged by TLC of the
reaction mixture; these products were separated by preparative TLC. The
Page 105
* 94 Scheme 6:
HO
(Ver sicolorin A, III)
OH OH lb q Buffe;-7'
(THF)
CHO CHO
(IV-A)
OH
\NaBH l (limit!d)
I b 11
OH ,s OH CHO
1l
OH lb
OH 0 H
(PR-VA-HA, VI-F)
(V A.,HA, IV) NaBH 4
( excess)
OH lb t1
IS' 0-1 18 OH
(Versiconol, V)
* The elucidation of these structures will be discussed in the following section.
Page 106
95
product with the lower Rf value had the same TLC behavior and mp as
those of authentic versiconol, a.nd its mass spectrum showed major pea.ks
·at y~ J40 (M-20), Jl2, Jll a.nd 297.
The pea.k at ml~ J40 is a.n unusual peak which is due to the loss of
20 mass units, this loss from the molecular ion is presumed to occur in
the steps shown below a.nd leads to the stable molecular ion of versi-
colorln c. On ionization by chemical ionization, the compound showed
a quasi molecular ion at ~ ~ J61, together with the (M-20) pea.k at
ml~ J41. 0 OH H
+ (M , m./i!. 360; not observed)
01-1
OH
~ H w (m/.e.. 340)
OH
>
OH
OH
-H 0 2
H
Page 107
OH
0
OH +
0
~
)
OH
0 t-
(m/.~ 297)
OH
0
l CH 2 +
cm/~ 311)
The NMR spectrum (d6-acetone) showed absorptions at o (ppm) 7.24 (2H,
s, br) and 6.64 (d, J 4Hz), which were assigned to the three aromatic
protons. A 3-proton multiplet at 4.18-J.76 was assigned to the three
protons at carbon-15 and 16, whe:reas a 2-proton triplet at J.40 was
due to the two protons at carbon-18. The expected two-proton signal at
carbon-17 was partially overlapped with the sol vent r.nd thus could not
be identified. These data indicated that the compound should most
reasonably have the structure v. A compound with structure V has been
isolated and named versiconol. A comparison of the TLC behavior and /
IR spectra of the synthetic and naturally occuring versiconol showed
that the two compounds were very similar; small differences observed
Page 108
97
in the IR spectra could be attributed to the existance of more than one
stereoisomer in the synthetic material. This reaction constitutes the
first synthesis of versiconol from versicolorin A. Although the yield
in this particular reaction was low (approximately 10% based on versi-
colorin A), this was largely due to the deliberate use of a limited
amount of borohydride in the reduction step, and there is reason to
believe that this reaction could become a high yield procedure, if it
became necessary to prepare versiconol in quantity.
The second product was beU.eved to be the hemiacetal formed by
cylization of the hydroxy aldehyde produced when only one of the two
masked aldehyde groups of "IV-A was reduced. One possible group of
structures of this partially reduced compound, PR VA-HA, would be
structure VI-A to VI-D, and a second possible group of structures
would be structures VI-E to VI-H.
The isolated material showed UV-visible absorption indicating the
presence of a hydroxylated anthraquinone ring system similar to versi-
colorin A-hemiacetal, while its IR spectrum showed absorptions indicating
the presence of bonded and free quinone carbonyls. Its FI'-NMR spectrum
in d6-acetone showed absorptions consistent with those expected for
structures of the general type VI. Thus two 1-proton doublets and a
1-proton singlet at 7.22, 6.62 and 7.10 were assigned to the aromatic
protons. In addition two 1-proton multiplets at 5,88-5,62 and J.90 were
assigned to the protons at carbons 15 and 16. Unfortunately, the sig-
nals due to the protons on carbons 17 and 18 were concealed under
solvent and water peaks, and this made it impossible to assign the
Page 109
(VI-A)
\ OH 16
(VI-B)
4-
1l (VI-E)
(VI-G)
98
lb 11
CHO '80H '
1l 17
18
H CHO
H
'
-..:.. ~
OH
OH
(VI-H)
16 11
180 H
(VI-C)
11
CH2oH 18
0 (VI-F)
Page 110
99
exact structure of partially reduced versicolorin A-hemiacetal. How-
ever, the absence of any absorption assignable to an aldehyde proton
did indicate that structures VI-A and VI-E could be excluded from
consideration.
The mass spectrum of the partially reduced product showed prominent
peaks at m/~ 356 (M+), )40 (M-18), 318 (~1-46), Jll and 297. The ions
giving rise to these peaks are characteristic of alcohols and quinones
in general, and thus do not offer any additional basis for distinguish-
ing between the various structures.
An alternate method for differentiating the various structures
was sought. Acid cyclization was an attractive procedure, since it
was expected that different structure would give different products,
as described in Scheme 7.
If the partially reduced versicoloi"i.n A-hemiacetal has structure
VI-B or VI-C, it would give versicolorin C, XVI, after acid hydrolysis.
Structures VI-D, F, G, and H, on the other hand, would give XVII-A
(ortho-VC), XVII-B (iso-VC), or XVII-C (iso-ortho-VC), respectively.
Since either versicolorin C or its isomers is expected to be less polar
than partially reduced versicolorin A-hemiacetal, it ought to be more
easily handled and its NMR spectrum should also be clearer and more
easily interpreted.
Acid treatment of partially reduced verslcolorin A-hemiacetal,
VI, yielded one major product only. After recrystallization of the
crude product, an orange compound was isolated, which showed a different
IR spectrum from that of authentic versicolorin c. The isolated mat-
erial had an HPLC capacity factor of lJ.4 (versicolorin C, 14.5) in two
Page 111
·scheme 7:
OH 0 OH
(VI-B)
OH
(VI-C)
(VI-D)
OH H
100
18 OH
OH
_J HCl
HCl
(Versicolorin C) . (XVI)
11
OH (ortho-VC, XVII-A)
Page 112
101
OH 16
OH 0 (VI-F)
HCl
(VI-G)
OH
HCl
CH20H 18
(VI-H)
'l HCl
,s OH
OH 16 18
0 (iso-VC, XVII-B)
0
~ 0
C XVII-C) (iso-ortho- V '
Page 113
102
linear connected Partisil 10 PAC columns with hexane:chloroform:acetic
acid (65:J5:1.0) as the elution solvent. Its mass spectrum showed
prominent peaks at m/~ :,40 (M+), Jl2 (~1-28), Jll (M-29) and 297 · (M-4J),
whereas versicolorin C showed peaks at m/~ (M+), J25 (M-15), Jll (M-29)
and 297 (M-4J). It had the same UV-visible absorption as versicolorin
c. The IDIR spectrum in DMSO-d6 showed two broad 1-proton singlets at
12.0 and 12.4 ppm which were assigned to the two chelated phenolic
protons at aromatic carbons-1 and 8. In addition, two 1-proton doublets
and one 1-proton singlet at 7.02, 6.50 and 6.91, respectively, were
assigned to the three aromatic protons. A 1-proton multiplet, one
2-proton multiplet and one 1-proton multiplet at 5.98, 4.14 and J.72
ppm were assigned to the protons at carbon-15, 18 and 16, respectively.
The two protons at carbon-17 showed a multiplet at 2.20 ppm. Because
of the observation of the two chelated phenolic protons, the structures
XVII-A and XVII-C could not be the correct structure of the acid
cyclization product, since these have only one chelated hydroxyl proton
each. This indicated that structures VI-D and VI-H could be excluded
as the structure of the partially reduced versicolorin A-hemiacetal.
Also the acid cyclization product showed different IR and mass spectra,
and HPLC capacity factor from those of versicolorin c. This suggested
that the cyclization product had the structure XVII-B, and hence the
partially reduced versicolorin A-hemiacetal must have had either
structure VI-F or VI-G.
The cyclization product, named isoversicolorin C (iso-VC), was
conclusively proved to have structure XVII-B by double resonance NMR.
Page 114
103
(XVII-B)
Irradiation at 2.20 ppm caused the signals at J.72 and 5.98 ppm to
simplify, while irradiation at J.72 ppm simplified the signal at 4.14
ppm. These results indicate that the aliphatic carl:xm atoms are linked
as in structure XVII-B rather than as in versicolorin c. Because of the limited quantity of the partially reduced versi-
colorin A-hemiacetal (VI-For VI-G) that was available, it was not
possible to distinguish between the two possible structures of this
compound. However, based on analogy to the sterigmatocystin case, it
is proposed that the hemiacetal has the structure VI-F; this structure
should be more stable than VI-G for the same reasons that XI-C is more
stable than XI-B.
The structure VI-Fis, of course, a different structure from that
of the parent structure of versiconal acetate, I, which was isolated
and tentatively assigned the structure I-A by Cole,40a After acid
hydrolysis of versiconal acetate, I, Cole isolated versicolorin C as
the only product.
Page 115
104
I CHO
H+/H 20
l---OH
-HOAc (Versiconal Acetate, I-A)
OH OH
(
0 OH (Versicolorin C, XVI)
From scheme 7, we knew structure VI-B would also give versicolorin
C as the acid cyclization product. This indicated there are several
possible structures for Dr. Cole's versiconal acetate, as described
below.
16 lb ---HO ---
0 (I-A) (I-B)
OAc OH H+
0 0
(I-C) (Versicolorin C, XVI)
Page 116
105
4oa The mm data in the original paper, however, suggests that
either structure II-A or II-Bis a more plausible structure than I-A,
I-Band I-C. Thus the protons on carbon-18 gave a triplet in the
l
OH
0 (II-A)
Ac
OH
(II_;B)
H-NMR spectrum of versiconal acetate at 4.08 ppm; the predicted value
for the protons on carbon-18 of structures I-A, I-B and I-C would be
at J.80, J.60 ppm, and those of structures II-A and II-B around 4.1
ppm. Secondly, no signal was seen for a free aldehyde proton, required
by structure I-A.
The structural assignment of versiconal acetate as either I or II
rests heaVily on its conversion to versicolorin C, and it was therefore
important to establish beyond doubt that the product obtained by Cole
was in fact versicolorin C and not an isomer of it. We thus obtained
from Cole a sample of his versicolorin C produced from versiconal
acetate, and compared it with authentic versicolorin C and with iso-
versicolorin c. The material supplied by Cole proved to be identical
in its HPLC retention time to versicolorin C, and different from
isoversicolor:l.n c. This result thus serves to confirm the assignment
of structure I or II to versiconal acetate, and as discussed above,
Page 117
106
the spectroscopic evidence indicates that versiconal acetate has
structure II, A recent paper confirms this finding, and adds the new
information that versiconal acetate exists as an approximately 60:40
mixture of forms II-A and II-B, 40b
IX, Biosynthesis of Aflato:cin ~
The aflatoxins are highly toxic and carcinogenic metabolites
produced by the common fungi Aspergillus versicolor andAspergillus
parasiticus on almost all food-stuffs. These compounds ha.ve become a
serious threat to food safety and public health, and .intensive studies
on their occurrence, chemistry and biological effects have been carried·
out during the pa.st decade,
Several theoretical and experimental studies have been employed
by different people in the elucidation of the biosynthetic pathway of
the aflatoxins. The polyketide theory has been widely accepted as the
most plausible route for the synthesis of aflatoxins by the fungi, In
this theory, averufin (AV), versicolorin A (VA) and sterigma.tocystin (ST)
have all been proposed as biosynthetic intermediates. 48 It wa.s suggested
that the aflatoxins were formed through the following pathway ba.sed on
the polyketide hypothesis,
anthra.quinones acetate -polyketide --> AV J
Coumarins (AF) +-- Xanthones (ST)
By using labeled (1-*C) or (2-*C) acetate, Biollaz 480 and
coworkers observed that a decrease in the yield of aflatoxins was
Page 118
107
accompanied by the accwnulation of averufin; this indicated that
averufin is probably a precursor of the aflatoxins, Hsieh, et al49
have shown that averufin and sterigmatocystin could be converted to
aflatoxins, Versicolorin A was also found to be a precursor of 2b aflatoXins, These experimental studies have further confimed the
biosynthetic pathw.cy-pJ:Oposed alx>ve for the aflatoxins.
Cole, et al 40 observed that the production of aflatoxins was
inhibited by the insecticide, dichlo:rvos. The inhibition of aflatoxin
formation was accompanied by the pJ:Oduction of an orange compound,
which was named versiconal acetate and its structure was tentatively
assigned as preViously discussed, This compound may also be an inter-
mediate in the biosynthesis of aflatoxins,
As has been mentioned, Hsieh50 found a high conversion of 14c labeled averufin, versicolorin A and sterigmatocystin into aflatoxin
~· pJ:OVing that these compounds are truly involved in the ma.in pathway
of Af'latoxin ~ biosynthesis. (Table 15) For versiconal acetate,
however, the conversion was less than one-third that of averufin and
versicolorin A, and it m.cy-or may not be directly involved in the path-
way of aflatoxin biosynthesis,
The above experimental evidence thus supports the pathway of
aflatoxin ~ biosynthesis shown below.
Page 119
(Acetate)
OH HO
108
OA HO C
(Versiconal Acetate,
i Path "A"
I-A) ~
/ Path "B"
OH
(Polyketide)
(Averufin)
---)~ Sterigmatocystin
(Ver sicolorin A, III) Aflatoxin
The suggested overall pathway5l from versicolorin A to aflatoxins
through sterigmatocystin has been widely accepted. The conversion of
averufin to versicolorin A is a key step, as shown in Scheme 7, but
this conversion has not yet been clearly demonstrated. This conversion
may ''involve!' versiconal acetate as an intennediate (path "A"), or may
pass through path "B", in which versiconal acetate is on a "shunt" off
the main pathway.
Our isolation of a derivative of nidurufin, 6,8-Di-0-methylniduru-
fin52 from a culture of Aspergillus versicolor serves to emphasize the
Page 120
109
TABLE 15
Radioactivity Incorporation of Aflatoxin.B:i_ Derived
from Various Labeled Precursors50
Labeled Precursor
Averufin
Versicolorin A
Versiconal Acetate
Sterigmatocystin
Radioact. Incorp. , %
49.4 41.5
13.7 65.0
Page 121
110
importance of nidurufin (hydroxylated averufin) in the metabolism of
Aspergillus versicolor, and suggests that it may also play a part in
the biosynthesis of versicolorin A. An interesting feature of the
putative pathway leading through versicolorin A is that the C-C bond
joining the aromatic part of the molecule to the dihydrofuran ring is
formed between two car'bon atoms which both originate in the methyl
groups (C-2) of acetate. Since averufin itself can readily be
OH • •
C~COOH • 0 0
(Ver sicolorin A, III)
constructed from acetate units in the normal manner, see Scheme 8, 1t
follows that the rearrangement which places two C-2 carlx>ns of acetate
adjacent to each other most likely occurs during the conversion of
averufin to versicolorin A.
At least two hypothetical mechanisms have been proposed for these
rearrangements 51, and I wish to suggest a third, outlined in Scheme 9.
In Scheme 9, the key rearrangement step occurs through a pinacol-
type rearrangement of the open-chain form of nidurufin to form an
intermediate with structure (XVIll). This intermediate may pass
through either pathway "A" or pathway "B" to form versicolorin A.
Page 122
111
Scheme 8:
•
• OH 0
(Averufin) •
0 0 t
• 0 (Ver sicolorin A)
Page 123
· Scheme 9:
• CHCOOH ~~
. 3
OH
•
•
Pinacol Rearrangement
OH
(XVIII)
Path "A"
Baeyer- Villiger Rearrangement
112
• 0
•
0
Hydroxylatio{
• • (Averufin)
OH
• (Nidurufin)
OH
0
• (XIX) • Path "B"
• Cyclization
OH
Page 124
113
• I (Versiconal Acetate) • l Hydrolysis
OH
CHO CH2.0H • •
l Oxidation
OH 0
I l CHO CHO
• •
OH
(XIX) l Dehydration
OH
• l Oxidation
OH •
• 0 H
•
CHO 0 \;' •
Page 125
114
OH • CHO CHO
•
Cyclization
01-1 •
0 0
(Ver sicolorin A-hemiacetal) (VIII-A)
Dehydration
OH •
• 0 CHO •
C • Hydrolysis
OH 0
0 OH ~HO
OH •
• (Versicolorin A, III)
Page 126
115
For path "A", versiconal acetate could be fonned through a "Ba.eyer-
Villiger" type of rearrangement of intermediate XVIII. After hydrolysis,
oxidation and cyclization, versiconal acetate would be converted to
versicolorin A-hemiacetal, which would then be dehydrated to form
versicolorin A.
For path "B", the intermediate (XVIII) would cyclize to form
another intermediate (XIX), which then would be converted to versi-
colorin A-hemiacetal through dehydration, oxidation, hydrolysis and
cyclization. The resulting hemiacetal would then be dehydrated to
produce versicolorin A. While it is recognized that this mechanism
(Scheme 9) is speculative, it does offer an alternative to the other
mechanisms that have been proposed and it also has the advantage of
involving the known metabolites, nidurufin and versiconal acetate, on
the biosynthetic pathway.
Page 127
EXPERIMENTAL
I. General Information
Unless otherwise stated, ultraviolet spectra were obtained in 95%
ethanol on a Cary Model 14 UV-visible spectrometer. Infrared spectra
were taken as KBr pellets on a Beckman IR-20AX infrared spectrophoto-
meter. Proton and carbon magnetic resonance spectra were measured on
a JDJLCO JNM-PS-100 instrument with TMS as internal reference. Mass
spectra were obtained on a Varian-MAT 112 double focusing spectrometer
with an ioniza.tion voltage of 70ev. unless otherwise noted. Melting
points taken on microscope cover plates were determined with a Thermo-
lyne heating stage with microscope, and are uncorrected.
1. Thin-layer chromatography (TLC)
The thin-layer chromatographic plates used in this laboratory were
prepared in our laboratory from E. Nerck silica gel ~F-254 with a
thickness of approximately JOO microns for analytical purposes. Home-
made preparative TLC plates (20 x 20 cm) were made from E. Merck silica
gel PF-2.54 with a thickness of approximat~ly 1000 microns. In normal
analytical TLC development, the plate was developed by the solvent
system only once. In preparative TLC, the plates were multiply deve-
loped by the solvent system three ,to five times. The TLC plates was
air dried between runs.
Three TLC solvent systems have been routinely used in this research,
they are,
Solvent system A: Hexane:Ethyl Acetate 70130
Solvent system B: BenzenesEthyl Acetate 70:JO
Solvent system Cs Benzene:Acetic Acid 90s10
116
Page 128
117
Visualization of TLC plates was performed using a Chromatovus
CC-20 ultraviolet viewing apparatus.
2, High-Performance Liquid Chromatography (HPLC)
1. Instrumentation
The liquid chromatographic system was equipped with a 2.54 and
280 nm dual wavelength UV monitor (Pharmacia Model 110), a refractive
index monitor (Waters M402), a Waters M 6000 pump, and a Fisher recorder
(series 5000). Sample injection was through a low dead-volume Valeo
valve injector.
2. Reagents
(A) Mobile phase: Solvents used in HPLC were Burdick and
Jackson distilled in glass grade or A.c.s. reagent grade supplied by
J ,T, Baker.
(B) Stationary phase: Porasil A & B, Corasil II and reverse
phase (Porasil C-18) packings were obtained from Waters Associates, The
pre-packed ..(.{-porasil column was also obtained from Haters Associates,
and the pre-packed Partisil 10 PAC column was obtained from Whatman,
Inc, Both pre-packed columns were used as received uithout pretreatment
except for equilibration with the solvent systems used.
(c) Chromatographic conditions: The flow rate was set at 2 ml/
minute for all analytical separations, and the pressure_ varied between
500 and 2000 psi depending on the solvent and column in use. All
separations were carried out at room temperature, and the elution
profile was measured at either 2.54 or 280 run by the UV detector.
Page 129
118
II. Extraction and Fractionation of Metabolites of A, versicolor
(NRRL 5213) Aspcrgillus versicolor (NRRL 5213) was cultured on autoclaved
rice at J8°c for 17 days (still culture) or 7 days (shake culture). The
pigmented mycelial mass, together with the rice, was extracted with
chloroform-methanol mixture, A typical extraction was of 12 flasks
of 200 g rice each, and this was extracted with 5 liters of methanolic
chloroform. After vaporation of the solvent, the oily mixed pigments
(19 g) were then dissolved in acetonitrile saturated aqueous potassium
chloride, 96:4. This solution, 600 mls, was extracted with hexane,
4 x 200 ml, The hexane layer, which contained the lipid fraction of
the crude extract, and which did not contain any of the desired pigments,
was not investigated further, The acetonitrile-water layer was then
mixed with chloroform, and the pigment-containing chloroform layer was
separated from the aqueous layer. Evaporation of the chloroform yielded
the crude pigments (6.J g) as a dark brown solid residue.
Chart 4. Liquid-Liquid Partition of Crude Mycelium
!• versicolor (NRRL 5213)
Hexane (Lipids)·
I Extract with CHc13-MeOH
Crude Extract
I Dissolved in CH3CN-aq. KCl ( 4%) solution
Extract with Hexane
I CHCl
( Crude ~igments)
CH3CN-aq. layer !Extract with CHClJ
I aq, layer
Page 130
119
II.1. Isolation of Averufin & Versicolorin C
One total of 6.J g of oil free dried pigments obtained from 2.4 Kg
of rice was subjected to chromatography on a column of E. Merck Silica
Gel 60. (Column size: 5.J cm i.d. x 64 cm; packed with 600 g of S.G.
60). Elution with chloroform-methanol and combination of fractions on
the basis of TLC data yielded six fractions (J-1-168-1,2,J,4,5 and 6)
as described in Table 16.
Based on TLC results in both systems A and C, fraction J-1-168-2-h
(1 g) was combined with another similar fraction J-1-172-J (0.9 g) which
was isolated from another batch of crude pigment by the same procedure.
The combined fraction (2.0 g) was rechromatographed on a column (4.2
cm i.d. x 65 cm) packed with 250 g of E. Merck silica gel 60. Elution
with hexane:ethyl acetate (80:20) yielded six fractions as listed in
Table 17.
Fraction PC-2-53-J (80 mg) was further purified by preparative TLC
using solvent system B. After collection of the major fraction, the
orange solid obtained was recrystallized from acetone to yield 40 mg
of orange crystals, which were later identified as averufin,
Preparative TLC of fraction PC-2-53-5 in system B, yielded an
orange solid. After recrystallization from acetone, a compound
identified as versicolorin C (60 mg) was obtained,
II,2. Isolation of De-0-rnethyl-sterigmatocystin, Sterigmatocystin
and Versicolorin A
Another dried pigment fraction J-1-90 (7.2 g), which was obtained
after the acetonitrile-water-hexane defatting process, was loaded on a
Page 131
120
TABLE 16
Column Chromatography of Pigments from !• versicolor (NRRL 5213)
Volume Weight Fraction (ml) (g.) Solvent Color of Bands
J-1-168-1 600 0.12 CHClJ Yellow-Orange
J-1-168-2 200 1.10 CHClJ Orange
J-1-168-3 525 1.10 CHClJ Orange
J-1-168-4 525 1.13 CHClJ Red
J-1-168-.5 975 0.70 CHC13:MeOH Deep Red-Brown (70: o)
J-1-168-6 1000 0.50 CHC13:MeOH Dark-Brown (50: o)
Page 132
121
TABLE 17
Column Chromatography of Combined Fractions
J-1-168-2 and J-1-172-3
Volume Weight Fraction (ml) (mg) Solvent Color of Bands
PC-2-53-1 500 trace Hexane:Ethyl White Acetate (80,20)
PC-2-53-2 350 trace Yellow
PC-2-53-3 2000 80 First Orange
PC-2-53-4 2200 100 Light Orange
PC-2-53-5 2500 120 Second Orange
PC-2-53-6 1400 80 Orange
*Because the elution solvent was nonpolar, most of the polar compounds (1,6 g) remained at the top of the column and were not eluted.
Page 133
122
column (5.7 cm i.d. x 70 cm) of Mallinckrodt Silica AR CC-4 sorbent
(700 g). Elution with chloroform-methanol yielded eight fractions as
described in Table 18.
Based on TLC data, fractions J-1-90-1 & 2 were combined and
purified by another column (5,3 cm i.d. x 64 cm) of E. Merck silica
gel 60 (400 g). The collected fractions are described in Table 19,
Fraction PC-2-46-1 (80 mg) was subjected to preparative TLC in
solvent system B. The major band yielded de-0-methylsterigmatocystin
(10 mg). Recrystallization of fraction PC-2-46-4, yielded sterigmato-
cystin (70 mg).
The fractions J-1-90-5 and 6 were combined with similar fractions
from two other extracts. The combined fraction (0,5 g) was chromato-
graphed on a 2.1 cm i.d. x 60 cm column packed with 100 g E. Merck
Silica gel 60. Elution with Hexane:E.Ae (70,30) yielded five fractions
after combination of similar fractions based on TLC data (Table 20).
Summary isolation trees of these isolated metabolites of
Aspergillus versicolor (NRRL 5213) are given in Charts 5 and 6.
III. Extraction and Fractionation of Metabolites of A. versicolor
(M 1214), Isolation of Griseofulvin, Dechlorogriseofulvin &
3.8-Dihydroxy-6-methoxy-l-methyl-xanthone
Extraction of the still culture of A. versicolor (M 1214) grown on
autoclaved dry rice (7.8 Kg) with chloroform yielded on oily extract
(62 g) which was loaded on a column (3.3 cm i.d. x 75 cm) of Mallinckrodt
Silica AR CC-4 sorbent (250 g) and eluted with chloroform to yield four
fractions which are described in Table 21. Fractions J-1-149-3 and 4
Page 134
123
TABLE 18
Column Chromatography of Fraction J-1-90
Volume Weight Fraction (ml) (g) Solvent Color of Bands
J-1-90-1 550 3.30 CHC13 Yellow
J-1-90-2 500 0.57 CHC13 Yellow-Orange
J-1-90-) 1000 0.13 CHC13 Orange
J-1-90-4 600 0.10 CHC13 Orange
J-1-90-5 700 0.30 CHC13 Deep Orange
J-1-90-6 1000 0.07 CHC13 Deep Orange
J-1-90-? 100 0.02 CHc131MeOH Orange Brown
J-1-90-8 300 3.10 (3:1) i Brown
Page 135
124
TABLE 19
Column Chromatography of Fractions J-1-90-1 & 2 (J.8 g)
Volume Weight Fraction (ml) (mg) Solvent Comment
PC-2-46-1 150 80 Hexane:Ethyl Collected Acetate by fraction (80,20) collector
l and conlined
PC-2-46-2 JOO 250 by TLC da.ta
PC-2-46-J 400 400
PC-2-46-4 JOO 140 Hexane:Ethyl Acetate (50:50)
PC-2-46-5 300 240 Ethyl Acetate
PC-2-46-6 JOO 170 Ethyl Acetate: MeOH (80:20)
PC-2-46-7 JOO 150 Methanol
*About 2.4 g of polar compounds were irreversibly absorbed on the top of the column.
Page 136
125
TABLE 20
Column Chromatography of Fractions J-1-90-5,6 and
Similar Fractions
Fraction
PC-2-40-1
PC-2-40-2
PC-2-40-J
PC-2-40-4
PC-2-40-5
Volume (ml)
1200
1500
1400
1000
1200
Weight (mg)
250
20
?O
40
50
Solvent
Hexane:Ethyl Acetate
(?O:JO)
Color of Bands
First Orange
Second Orange
Orange
Orange
Orange
*Recrystallization of fraction PC-2-40-J from yielded orange crystals (40 mg) which were later identified as versicolorin A.
Page 137
Aqueous layer
J-1-168-1 (0.12 g)
CHART 5: A Vsraisotor NRRL sz11 (2400 g. rice; atill cult uni
lcHCl 3 Ext. Crude ext.
Triturate With pet. ether
Insoluble portion J.\.167 (19 g.)
Diuolvcd in CH3CN
olution of lipids.
plus aqueous -I~ KCI (90110)
Ext. CH3CN-HzO with CH<;.13 pH=-1
Ext. with Hex.
CHCl 3 layer dried &. vap.
I EM S. G. column (CHCl.3 &. MeOH)
.z (1.10 g)
.3 (1.10 g)
-4 (I. 13 g)
-5 (0. 70 g)
Hex layer (Wax)
.l, (0. 50 g)
Yellow-orange band-. - -· •• - • • • - - - • - -- - - - - -Red-brown band- - - - - - - • - • Dark-brown band
PC- Z-53-1 (trace)
Combine with similar !rax. J-1-172-3 (0.9 g)
EM S.G. column (Hex /E. A. 80: ZO)
-2 -3 -4 (trace) (80 mg) (100 mg) rSG PTLC
(Ben/E. A.) 90:10 I Recrys.
AV (40 mg)
-5 . -b (120 mg) (80 mg)
EJi S.G. PTLC (Ben. /E. A.) 90:10
Jecrys.
I VC (t,0 mg)
..... S?-
Page 138
r-PC-7.-46-l -2
· CHART 6: A. V,•rsicc!l.!l.!:. NRRL 5213 (1200 g. rice; still culture)
I Ext. with CHCL 3
Crude Ext.
I Triturate with pel. ether
1 1
Insoluble portion l Solu. of lipic!sl J-1-90 (7. 2 g.)
Silicic acid column CHCl3 & MeOH
I I I J-l-90-1 (3.3g.)
-2 -3 -4 -5 -6 -7 -8 (0.57 g.) (0.13 g.) (0.1 g.) (0.30 g.) (0.07 g.) (0.02 g.) (3.1 g.)
Yellow band----------------- Red-orange band -----------Dark
EM S.G. column
(Hex/E.A.)I 80:20
-3 -4 -5 -6 -7
Combine with similar fractions from twu other ext.
EMS.G. column (HL'x/E.A.) 70: 30
(80 mg) (250 mg) (400 mg) (l-10 mg) (240 mg) (170 mg) (150 mg) PC-2-40-l (250 mg)
-2 (20 mg)
-3 -4 -5 (70 mg) (40 mg) (50 mg) I Recrys.
I S.G. PTLC (Ben/E. A.)
90:10
D.--nwthyl ST (LO mi;)
Recrys.
VA (40 mg) ST
(70 mg)
.... ~
Page 139
128
TABLE 21
Column Chromatography of Metabolites of A, vers1color (M1214)
Volume Weight Fraction (ml) (g) Solvent Color of Bands
J-1-149-1 450 9,38 CHClJ Yellow
J-1-149-2 J25 9.10 CHClJ Orange
J-1-149-J 600 9,45 MeOH Red-Brown
J-1-149-4 750 JO,lJ MeOH Dark-Brown
Page 140
129
were combined with the corresponding fractions from an extract of J.2
Kg rice to yield a total of 45 g of oily material. This was defatted
by a hexane-aqueous acetonitrile partitioning, and the crude aceto-
nitrile soluble material (35 g) was loaded onto a column (5 cm i.d. x
80 cm) of E. Merck S.G. 60 (100 g). The resulting separation is des-
cribed in Table 22.
Crystallization of the major component of fraction of PC-2-47-6
from acetone yielded a material identified later as dechlorogriseofulvin
(400 mg). Purification of fraction PC-2-47-7 on a second E. Merck S.G.
60 (400 g) column (4 cm i.d. x 65 cm) yielded griseofulvin (550 mg)
which was isolated after recrystallization of fraction PC-3-15-5 from
acetone.
The isolation tree of these metabolites from strain M 1214 is
desert bed in Chart 7.
IV. Extraction and Fractionation of Metabolites of A. versicolor
(M 1004). Isolation of 5-Methoxy-Sterigmatocystin, Aversin and
6,8-Di-0-Methylnidurufin
~ versicolor (M 1004) was cultured on autoclaved rice (4.3 Kg)
at J8°c for 17 ~s. The pigmented mycelial mass, together with the
rice, was extracted with chloroform-methanol mixture. The crude extract
(4,0 g) was subjected to chromatography on a column (4 cm i.d. x 65 cm)
of Mallinckrodt Silica AR CC-4 sorbent (400 g). Elution with chloroform
monitored by UV absorbance at 350 nm yielded three fractions: J-1-82-1
(350 ml, first yellow Band, 1.8 g), J-1-82-2 (2400 ml, pale yellow band,
O.J g) and J-1-82-J (lJOO ml CHClJ and then 600 ml MeOH, orange band,
Page 141
lJO
TABLE 22
Column Chromatography of Metabolites of!• verslcolor (M1214)
Volume Weight Fraction (ml) (g) Solvent Color of Bands
PC-2-47-1 500 0.75 Hexane:Ethyl Light-Yellow Acetate (70:JO)
PC-2-47-2 500 0.11 Hexane:Ethyl Yellow Acetate (50:50)
PC-2-47-J 400 1.00 l Yellow
PC-2-47-4 600 0.60 Yellow
PC-2-47-5 700 0.50 Hcxane:Ethyl Deep Yellow to Acetate Brown (25:75)
PC-2-47-6 1000 4.50 Ethyl Acetate: Brown }reOH (90:10)
PC-2-47-7 1900 18.00 Ethyl Acetate, Dark Brown MeOH (50150)
Page 142
lJl
TAm..E 2J
Column Chromatography of Fraction PC-2-47-7 (18g)
* Volume Weight Fraction (ml) (g) Solvent Color of Bands
PC-J-15-1 500 1.10 Hexane:Ethyl Light-Yellow Acetate (40:60)
PC-J-15-2 500 o.85 Yellow
PC-J-15-J 500 1.20 Yellow-Brown
PC-J-15-4 500 o.ao Brown
FC-J-15-5 500 1.20 Dark Brown
PC-J-15-6. 500 l,JO Dark Brown
PC-J-15-7 500 5.00 Dark Brown
PC-J-15-8 600 2,50 CHC1d:I1e0H Dark (5 :50)
*About 4g of crude material was irreversibly absorbed at the top of the column.
Page 143
• PC-Z-47-1 (0. 7 5 g.)
CHART 7:
f J-1-149-1
(9. 3 g.)
A. Versicolor M 1Zl4 (7. 8 g rice; still culture) I Ext. mycelium with CHC1 3 & MeOH
Oil Ext. (6Z g.)
Silicic acid column; CHCI & MeOH
-Z -3 -4 (9.lg.) (9.4g.) (30.lg.)
PC-Z-3Z-3 • Dissolve in CH3CNIHzO 90:10
plus aqueous 4 ~ KCl
Ext. with hex.
Ext. • CHCl3 layer
CH 3CN-l-lz0 layer
with CHCltl pH=4
Hex. layer (Wax)
' HzO layer I
PC-Z-3Z-3-a I EM S.G, column (Hex/E. A. =70:30)
-'z .'3 .'4 -s -b -t -'s (3.lg.) (0.06 g.) (1.0 g.) (0.60 g.) (0.5 g.) (4.5 g.) (18 g.)
rmy,. i""Y'· Xanthone Dechloro-
(60 mg) griseofulvin (400 mg)
EMS. G. column (Hex/E. A. =40:60)
PC~-15-1 (l.lg.)
-2 -3 -4 -5 -6 (0.85g.) (I.Zg.) (0.8 g.) (l.Z g.) (1. 3 g.)
recrys.
Griseofulvin (550 mg)
-7 (5 g.)
-8 (Z.5g.)
~ N
Page 144
lJJ
1.7 g). Chromatography of fraction J-1-82-J on a column of E. Merck
s.G. 60 PF 2.54 (column size: J cm i.d. x 58 cm, containing 100 g of
silica gel) and elution by ethyl acetate:hexane (1,4) with collection
of JO ml fractions yielded an orange material (20 mg) in fractions 22-29.
On recrystallization from acetone-hexane mixture, the crystals were
later identified as the new compound 6,8-Di-0-methylnidurufin.
Based on TLC, fractions J-1-82-1 and 2 were combined and loaded
on a E, Merck S.G. 60 (250 g) column (J.J cm i.d. x 75 cm) for further
purification as described on Table 24.
Crystallization of the major component of fraction PC-2-43-9 from
acetone yielded a material later identified as aversin (60 mg). Based
on TLC results, fractions PC-2-43-5,6 and 7 were combined and purified
by preparative TLC (hexane:ethyl acetate, J0:70). The isolated major
component was recrystallized from acetone to yield 5-methoxy-sterigmato-
cystin.
The isolation tree of these metabolites from strain M 1004 is
described in Chart 8.
v. Isolation of Versicolorin A from Aspergj_llus parasiticus (Yellow
Mutant) 2b
The mutant strain of A• parasiticus, designated 1-11-105 wh-1 was
grown in liquid culture in Adye and Mateles medium for 7 days. The
yellow mycelial mass from a total culture volume of 0.5 liters with
acetone or chloroform, and the crude extract (1.1 g) subjected to
chromatography on a column (J.2 cm i.d. x 57 cm) of E. Merck S.G. 60.
Page 145
134
TABLE 24
Column Chromatography of Fraction J-1-82-1 & 2 (2,lg)
* Volume Weight Fraction (ml) (mg) Solvent Color of Bands
PC-2-43-1 800 700 Hexane:Ethyl White Acetate (65:3.S)
PC-2-43-2 2.50 200 First Yellow
PC-2-43-3 300 200 Yellow
PC-2-43-4 300 100 Yellow
PC-2-43-.5 300 220 Yellow
PC-2-43-6 300 220 Second Yellow
PC-2-43-7 200 230 Yellow
PC-2-43-8 250 250 Yellow
PC-2-43-9 250 250 Yellow
*The collected weight was more than the loaded material, probably due to traces of solvent remaining in each fraction,
Page 146
PC-Z-43-1 (700 mg)
J-1-82-1
GHART 8: A. Versicolor M 1004
' -2 ' -3
(4. 3 kg rice; still culture) ixt mycelium with CHCl3 & McOH
Crude Ext. (7. 0 g.)
1 I I Silicic ~cid colu:nn;CHCl~ &
-4 -5 -6 -7 -8 I I i (1.1 g.)
MeOH
(4.J g.) I
EMS. G. column EMS.G. PF 254 column (Hex /E. A. =80:20) (Hex/E. A. =65:35) collected in 20 ml test tubes
-2 -3 -4 -5 -6 -7 -8 -9 (ZOO mg) (ZOO mg) (100 mg) (220 mg) (220 mg) (230 mg) (250 mg) (250 mg)
I I I I EMS. G. PTLC Req'."ys. (Ben/E.A.=90:10) I
I Aver sin Recrs. (60 mg)
ZZ--29 tube no. ~ Recrys.
I Dimethyl-nidurufin
(10 mg)
5-MeO-ST (50 mg)
~
Page 147
136
TABLE 25
Column Chromatography of Metabolites of Yellow Mutant
* Volume Weight Fraction (ml) (mg) Solvent Color of Bands
PC-3-40-1 250 trace Hexane:Ethyl Pale Yellow Acetate (70:JO)
PC-3-40-2 500 130 First Yellow
PC-J-40-3 JOO 20 Light Yellow
PC-J-40-4 600 45 Second Yellow
PC-3-40-5 250 17 Yellow
PC-J-40-6 200 38 Yellow
*About o. 7g of polar compound was absorbed by the pa.eking material and was not eluted in the solvent used.
Page 148
137
Elution with hexane:ethyl acetate (70:30) yielded six fractions as
described in Table 25.
Crystallization of the major component of fraction PC-J-40-2 from
acetone yielded an orange material which was later identified as
versicolorin A (60 mg).
VI. Structure Elucidation of Isolated Pigments
1. Identification of PC-2-46-4 (Sterigmatocystin)
The light yellow crystals showed mp 244°0 (lit. 246°c). 14a,b
The mixed mp determination with authentic sample showed no depression.
Following TLC and HPLC, it gave the same Rf value and capacity factor
as the reference sample. UV: AEtOH 23), 248 and 328 nm. IR: V-KBr max 2920 W, 1650 S, 1630 S, 1610 S, 1590 S, 1485 S, 1273 S, 1240 s, cm-1•
}1S: '!!}/~ (relative abundance) 324 (~, 100), 307 (64), 296 (83). NMR
(CDC13) o 3.94 (3H,s, -OCH3), 4.73 (lH, m, -8i_5), 5.40 (lH, t, -~6),
6.34 (lH, s, -8i_1) 6.43 (lH, t, J2Hz' -8i_7), 6.65 (lH, d, J8Hz' -8i4),
6.60 (lH, d, J 8Hz, -H4) 6.76 (lH, d, J8Hz, -H6) and 7.39 (lH, t,
J8Hz, -H5) ppm.
2. Identification of PC-2-46-1 (De-0-methylsterigmatocystin)
The light yellow crystals showed mp 251°0 (lit. 253-255°0).5
UV: A=H 233, 252 and 335 nm. IR: v-KBr 1663 S, 16JO S, 1605 S, 1475 S,
1288 S, 1230 S, 1155 S, 1120 S, 1095 S, 1060 S, 993 S, 932 S, 845, 820 S ~ _, ( + cm • MS: !!!J ~ relative abundance) 310 (M , 100) NMR ( cnc13) o 4.90
(lH, m, -8i5), 5.57 (1H, t, J 2Hz, -8i6), 6.55 (lH, s, -H11) 6.69 (lH,
t, J2Hz, -~ 7), 6.94 (1H, d, J8Hz, -H14), 7.10 (2H, t, J 8Hz, -H4 & H6)
7.71 (lH, t, J 8Hz, -H5) ppm.
Page 149
138
3. Identification of PC-2-73-23 (6-Methoxysterigmatocystin)
The yellow crystals showed m.p. 218-220°c (lit. 223°c) 14 and
a mixed m.p. determination with an authentic sample showed no depression,
Following TLC and HPLC, it gave identical Rf value and capacity factor
as those of authentic sample. UV: AEtOH 2JJ (E, 2,1 x 104),246 max (2.4 x 104) and 3)1 (1.1 x 104) nm. IR: ))-KBr 2975 w, 1650 s, 1625 s, 1610 S, 1585 S, 1484 S, 14)5, 1243 S, 1135 S, 1094, 1055, 973, 965, 820 S
-1 cm • This spectrum was superimposable with that of an authentic sample.
MSc ~~ (relative abundance) 3.54 (M+, 100) 339 (84), 325 (26), 295 (23).
NMR (CDCl.3) 6 3.92 & 3.98 (6H, d, 2-0CH3), 4,82 (1H, ·m, -~ 5) 5,47 (lH,_
t, J2Hz, -J\6) 6.)7 (lH, s, -8:i_1), 6.44 (lH, t, J2Hz, -11i7) 6.60 (lH, d,
J8Hz, -~ 4), 6,76 (1H, d, J8Hz, -H4), 7.10 (1H, d, J8Hz, -H5) ppm.
4. Identification of PC-2-53-J (Averufin)
The orange crystals showed m.p. 280°-282°0 (dee.) (lit, 283-
2890C, decl3a,b), Following TLC and HPLC, it gave identical Rf value
and capacity factor as those of authentic sample, UV: AEtOH 223 254 max ' '
· 260, 276, 294, )21 and 45) nm. IR: })KBr 3360 S, 2920 M, 1660 M, 1610 S,
1585 S, 1555 S, 1460 S, 1390 S, 1315 S, 1230 S, 1155 S, 1120 M, 1105 M,
1090 M, 1020 M, 830 S, 765 S cm-1• This spectrum was superimposable
with that of an authentic sample, MS: '!Y~ (relative abundance) J68
(M+, 82), 325 (87), )10 (100), 297 (60), 286 (50). FI'-NMR (d6-Acetone)
o 5.31 (1H, m, -~ 5), 6,6) (1H, d, -11?), 7,14 (1H, s, H4) and 7.26
(lH, d, -H5) ppm.
Page 150
139
5. Identification of PC-2-43-9 (Aversin}
The yellow crystals showed m.p. 200-202°0 (lit. 217°c4).
UVs AEtOH 225 (E,3.3 X 104) 250 (2.2 X 104), 284 (4.3 X 104), 310 max (9.3 x 103) and 430 (.92 x 103) nm. IR: )TKBr J440 (broad), 1635 S,
1610 M, 1590 S, 1550 W, 1472 W, 1440 W, 1418 W, 1380 M, 1350 S, 1320 S,
1290 S, 1275 M, 1240 M, 1200 W, 1170 W, 1155 M, 1110 W, 1074 W, 980 W,
940 M, 910 W cm-1• This spectrum was identical to that of the litera-
ture one. MS: ml~ (relative abundance) 368 (M+, 80), 353 (60), 339 (20)
and 325 (100). FT-NMR (CDC13) 6 2.36 (2H, m, -~ 7), 3.60 (lH, m, -~ 6),
. 4.00 and 4.04 (6H, 2S, 2-0CH3), 4.17 (2H, m, -~ 8), 6.47 (lH, d, J6Hz, .
-~ 5) 6.80 (1H, d, J3Hz, -~), 7.24 (lH, s, -H4) and 7.46 (lH, d, J3Hz,
-H5) ppm. All the spectroscopic data were in agreement with the assigned
structure. The low m.p. as compared with literature data, probably was
due to a trace amount of impurity or solvent contaminating the compound.
6. Identification of PC-2-53-5 (Versicolorin C)
The orange crystals showed m.p. >300°c (lit. m.p. >310°c2•3).
Following TLC and HPLC, it gave identical Rf value and capacity factor EtOH to those of authentic VC. UV: .f\. max 22'.3, 2.54, 265, 291, '.318 and 450 nm.
IR: )/" KBr 3390 S, 1620 S, 1605 S, 1573 M, 1465 M, 1430 M, 1400 M, 1385 S,
1515 S, 1280 M, 1255 M, 1235 M, 1190 M, 1160 S, 1088 M, 955 M, 900 M, -1 750 M cm • This spectrum is superimposable with that of an authentic
sample. MS: ml~ (relative abundance) 340 (M+, 82), 325 (60), 311 (35),
29 (100). FT-NMR (d5-acetone) 6 2.32 (2H, m, -~ 7), 3.62 (2H, m, -~ 8)
4,16 (lH, m, -~ 6), 6.5 (lH, d, J6Hz, -~ 5), 6.62 (lH, d, J2Hz, -a,), 7,13 (lH, s, -H4), 7.2J (lH, d, J2Hz, -H5) ppm,
Page 151
140
7. Identification of PC-2-40-3 (Versicolorin A}
The orange crystals showed m.p. 281° (dee.) (lit, 289°c2a,b).
Following TLC and HPLC, it gave identical Rf value and capacity factor EtOH 6 as those of authentic sample. UV: ;\ 218, 2.54, 2 4, 289, 319, and max
450 run. IR: ))" KBr 3330 S, 1625 S, 1604 S, 1390 S, 1305 S, 1290 S, -1 1220 M, 1185 M, 1164 M, 990 M, 940 M, cm • This spectrum was super-
imposable with that of an authentic sample. MS: TJJ/~ (relative abundance)
338 (M+, 100), 310 (91), 309 (85), 279 (18).
8. Trimethyl Versicolorin A
Versicolorin A (80 mg) in dry acetone (20 ml) was heated under
reflux for 5 hr. with 4 g. of anhydrous potassium caroonate and 1.2 ml
of dimethyl sulfate. The reaction mixture was cooled, evaporated under
· reduced pressure to 5 ml, and treated with 10 ml of NH40H:H20 (50:50)
mixture until no more heat was evolved to destroy the excess dimethyl-
sulfate. The aqueous solution was then extracted three times with 20 ml
portions of ethyl acetate, and the combined extracts washed with water
until neutral, dried over magnesium sulfate and evaporated to dryness.
The methylated product was separated by preparative TLC with hexane:
ethyl acetate (80:20) as the developing solvent. After recrystallization
from acetone, the major compound was identified as tri-methyl versi-
colorin A (60 mg), m.p. 2J7-2J8°C (lit. 24192a,b). UV; AEtOH 222 (E, max
2.5 x 104) 283 (3.1 x 104), 343 (5.2 x 103) and 405 (3.8 x 103) nm.
IR: ).J-KBr 2910 M, 1657 S, 1600 S, 1560 M, 1450 S, 1350 S, 1318 S,
1295 M, 1255 M, 1227 M, 1204 M, 1155 M, 1130 M, 1090 M, 1058 M, 975 S,
940 M, 750 M. MS: TJJ/~ (relative abundance) 380 (M+, 12), 365 (40),
Page 152
141
J6J (Jl), 351 (100), 337 (65), JJ6 (33), 324 (52), 323 (JO), 322 (56),
321 (Jl), 309 (27), 307 (45), J06 (50), 293 (JO), 279 (25) and 263 (25).
FT-NMR (CDC13) 6 7,50 (lH, s, -H4), 7,35 (lH, d, J2Hz, -H5) 6.80 (lH,
d, J2Hz, -~), 6,75 (lH, d, J3Hz, -~ 5), 6.49 (lH, m, -~ 8), 5.36 (lH,
m, -~ 7), 4.78 (lH, m, -~ 6), 4.04, 3.97 and 3,95 (9H, 3 s, 3-0CH3) ppm.
9. Identification of PC-2-43-9 (6,8-Di-0-methylniduinfin) 0 ( )25 0 The orange compound showed m.p. 211-213 C, with a D - 77
(CHCl~; C ,15). UVs AEtOH 224 (~, 4.8 x 104), 251 (1.9 x 104), 288 .J max
(3.1 x 104), 314 (8.5 x 103) and 444 (8.8 x 103) nm •. IR: l)KBr 3500 s, )420 (br), 2940 M, 1680 M, 1625 S, 1600 S, 1560 I1, 1400 M, 1460 M, 1400 S1
1330 S, lJOO S, 1250 S, 1220 S, 1170 S, 1068 M, 1050 M, 1000 M, 970 M,
890 W, 850 S cm-1• 11S: I])/~ (relative abundance) 412 (M+, 64), 394 (32),
351 (18), 314 (100), 99 (42). NMR (CDc13) 6 7.46 (lH, d, J3Hz, -H5),
7.28 (lH, s, -H4), 6.86 (lH, d, J3Hz, -~), 5.30 (lH, d, J2Hz, -~ 5),
4.16 (lH, m, -8i6). 4.04 (3H, s, -OCH3), 4.oo (3H, s, -OCH3) ppm.
Acetylation of Dimethylnidurufin (Di-0-Acetyl Derivative)
Dimethylnidurufin (2 mg) was allowed to stand at room temperature
for 5 days in pyridine (0.5 ml) and Ac2o (0.2 ml). Usual work-up gave
a product showing 3 spots on TLC; purification of the crude product by
preparative TLC (hexane:ethyl acetate, 30:70) gave one major orange
product. NMR (CDClJ) 6 7.64 (lH, s, -H4), 7.42 (lH, d, J3Hz, -H5),
6.84 (lH, d, J3Hz, -~), J.98 (6H, s, 2-0CHJ)' 2.66 (3H, s, -c(o) CH3),
2.33 (JH, s, -C(O)CH3), 1.68 (3H, s, -He), 1.64 (4H, m, 2-CH2) ppm.
MS: '!!1/~ (relative abundance) 496 (M+, 10), 454 (15), 4J6 (5), 412 (70),
394 (100).
Page 153
142
10. Identification of PC-2-46-3 (3,8-dihydroxy-6-methoxy-1-
methylxanthone)
The white material showed m.p. 251-253°0 (lit. 253-255°035).
UV: AEtOH 242 (3.6 x 104), 267 (9 X 103) and 310 (2.3 x 104) nm. IR: max vKBr 3290 (s), 1660 (s), 1625 (s), 1600 (s), 1575 (s), 1505 (s), 1460
(s), 1430 (s), 1415 (s), 1295 (s), 1210 (s), 1195 (M), 1165 (s), 1145
(s), 1055 (r-1), 920 (M) 900 (M), 840 (M), 825 (s) cm -1• MS: '!!JI~ (relative abundance) Z?2 (rt, 100), 243 (55). NMR (d6-Acetone) o 6.84
(2H, s, -H4 , H5), 6.50 (lH, d, J3Hz, -H7), 6.36 (lH, d, J3Hz, -H2) and
4.01 (3H, s, -OCH3)and 2.85 (3H, s, -CH3) ppm.
11. Identification of PC-2-47-6 (Dechlorogiseofulvin)
The white crystals had m.p. 180°0 (lit. 179°36a). The com-
pound showed (a.)2~ + 376° (CHC13 0 0.96) (lit. +39036a). UV: A:~H
253 (E, 1.4x10 4), 290 (2.3x10 4) and320 (4.lxl03) nm. IR:))'KBr
2980 (s), 1715 (M), 1690 (s), 1660 (s), 1615 (s), 1585 (s), 1500 (M),
1455 (M), 1425 (M), 1350 (M), 1215 (s), 1150 (s), 1120 (s), 1055 (M),
975 (M), 965 (M), 805 (M) cm-1• MS: '!!JI~ (relative abundance) 318 (rl, 73), 287 (64), 276 (48), 181 (50), 149 (JO), 1J8 (100) and 106 (52).
NHR (OD013) o 6.21 (lH, d, JJHz, -H2), 6.03 (lH, d, JJHz, -H4), 5.52
(lH, s, -H2) 3.91 (6H, s, 2-00HJ)' 3.65 (3H, s, -OCH3•), J.25 2.20
(2H, m, -H61 ), 290, 2.~6 (lH, m, -H51 ) and 1.00 (3H, d, J7Hz, -CHJ) ppm.
12. Identification of PC-3-15-5 (Griseofulvin)
The white crystals had m.p. 215-216° (lit. 220-221°037). The
compound had UV: A~H 250 (E, 1.4 x 104), 287 (2.4 x 104) and 325
Page 154
143
(4 x 103) nm. IR: )!KBr 2940 M, 1710 S, 1660 S, 1620 S, 1590 S, 1505 M,
1470 S, 1430 M, 1402 M, 1350 S, 1)40 S, 1240 M, 1225 S, 1215 S, 1185 M,
1176 M, 1140 S, 1100 S, 1060 M, 1050 M, 995 M, 960 M, 890 M, 820 M,
800 H cm-1• MS: I]}/~ (relative abundance) 352 (M+, 51), 321 (22), 311
(10), 310 (38), 284 (14), 217 (15), 216 (17), 215 (46), 214 (43), 201
(12), 181 (21), 171 (14), 169 (24), 142 (10), 140 (25), 139 (14), 138
(100), 131 (31), 123 (16), 119 (42). NNR (d6-Acetone) & 6.56 (lH, s,
-H2), 5.64 (lH, s, -H2•), 4.14 (3H, s, -OCH3), 4104 (3H, s, -OCH3),
3.77 (3H, s, -OCHJ'), 2.90, 2.40 (3H, m, -H51 , H6 1 ) ~.92 (3H, d, J6Hz,
-CH3) ppm.
VII. Synthesis and reduction of hemiacetals of sterigmatocystin and
versicolorin A,
1. Derivatives of Versicolorin A (VA)
Synthesis of Versicolorin A-hemiacetal (Structure IV)
VA (150 mg) in acetone (150 ml) was heated under reflux for
24 hr with 1.5 ml of 10% H2so4• The reaction mixture was cooled,
evaporated under reduced pressure to 50 ml, treated with 100 ml water
and evaporated to about 100 ml to remove the bulk of the acetone. The
aqueous solution was extracted 5 times with 50 ml portions of ethyl
acetate, and the combined extracts washed with H2o until neutral, dried
over MgS04 (anhydrous), and evaporated to dryness. The crude product
(120 mg) was essentially homogeneous on TLC in system (Benzene:Ethyl
Acetate; 50:50), and on recrystallization from acetone it had m.p.
269-Z?0°C. The orange compcund had the following properties: UV: >.EtOH max 223 ( t t 2.5 X 104), 255 (1.5 X 104), 266 (1.8 X 104), 291 (2.5 X 104),
Page 155
144
. 4 3 317 (1.1 x 10) and 4.56 (6.1 x 10) run. KBr 6 IR: 1' J440 S, 3240, 1 10 S,
1442 H, 1.395 M, 1378 S, 1350 M, 1315 S, 1282 S, 1264 M, 1230 M, 1180 H,
1050 N, 960 M, 908 N cm-1• MS: If}/~ (relative abundance) 356 6t, 18),
355 (27), J28 (60), 310 (67) 309 (73), 300 (74), 299 (100), 285 (40).
FT-NNR (d6-Acetone)s 6 7.18 (lH, d, J2Hz, -H5), 7.05 (lH, s, -H4),
6.61 (lH, d, J2Hz, -~), 6.49 (lH, d, J6Hz, -~ 5), 5.7J-5.5J (lH, m, -~8)
and 4.27-4.0l (lH, m, -~ 8) ppm.
B. Preparation of partially reduced versicolorin A-hemiacetal
(PC-3-69-11-3, structure VI-F) and versiconol (PC-J-69-11-4,
structure V)
Versicolorin A-hemiacetal (80 mg) was dissolved in 80 ml of THF,
40 ml of 0.05 N sodium phosphate buffer, pH 7.2, was added, and the
mixture cooled to o0 c. The cold solution was treated cb::opwise over
2 hr with 4 ml of buffer solution containing 20 mg of sodium borohydride.
The reaction mixture was then diluted with 30 ml of water, adjusted to
pH 6 with dilute HCl, and extracted five times with 50 ml portions of
ethyl acetate. The extract was washed, dried and evaporated to yield
a crude product which consisted largely of two new products, These
products were separated by preparative TLC with benzene:ethyl acetate
(70:JO) as the developing solvent. After recrystallization from acetone,
the more polar product was identified as versiconol* (15 mg) by compari-
son with an authentic sample, m.p. 26J-265°c (lit. 265°c12). UV: AEtOH max
224 (E, 2.2 X 104), 262 (7.9 X 103), 296 (9.4 X 103), )18 (1.1 X 104)
*The isolated compound showed identical TLC and melting point as an authentic :reference sample.
Page 156
14.5
and 450 (5.6 x lOJ) run. IR: VKBr 3420 (br), 1620 S, 1568 M, 1470 M,
1400 S, 1300 S, 1260 S, 1220 M, 1164 M, 1135 M, 1090 M, 1025 Mand
1000 M cm-1• MS: ~'E. (relative abundance), EI: 342 (M-18, 10),
340 (42), 312 (40), 311 (58), 298 (25), '297 (100). CI: 361 (M+ + 1, 11),
J44 (13), 343 (55), 342 (20), 341 (100). FT-NMR (d6-Acetone): 6 7.24
(2H, broad s, -H4 and H5), 6.64, (lH, d, 2H4 , -17), 4.16-3.84 (3H, m,
-11:i.5 and ~ 6), 3.4 (2H, 5, J6Hz, -~ 8) ppm.
The less polar product (15 mg) had m.p. >305° (dee.) after
recr.,stallization from acetone, and was named "partially reduced versi-EtOH colorin A-hemiacetal" and assigned the structure VI-F. UV: A max
223 (E, 2.4 x 104), 255 (1.2 x 104), 267 (1.4 x 104), 296 (2.2 x 104),
318 (9 x 103) and 453 (7,2 x 103) nm. IR: )}'KBr 3580 M, 3150 (br) S,
1620 S, 1590 S, 1435 M, 1393 S, 1322 S, 1285 M, 1260 S, 1200 N, 1175 M,
1030 Mand 760 M cm-1• MS: ~'E. (relative abundance). EI: J40 (M-18,
84), 312 (63), 311 (92), 297 (100). CI ;42 (23), 341 (l'lH+-18, 100).
NMR (d6-DMS0) 6 12.62 and 11.94 (2H, 2 broads, zchelated-OH), 6.98
(lH, d, J2Hz, H5). 6.88 (lH, s, -H4), 6.48 (lH, d, J2Hz, -~), 5.50 (lH, m, -11:i_5), 3,80-3.00 (3H, m, -11J.8 and 1\6) ppm.
c. Preparation of tri-methylversicolorin A-hemiacetal (PC-3-6-1)
Tri-methyl-versicolorin A (55 mg) in acetone (40 ml) was heated
under reflux for 24 hr. with o.8 ml of lCr/o sulfuric acid. The reaction
mixture was cooled, evaporated under reduced pressure to 5 ml, treated
with 20 ml of water, and extracted three times with 20 ml of portions
of methylene chloride. The combined extracts were washed until neutral,
dried over magnesium sulfate, and evaporated to dryness. The product
Page 157
146
was recrystallized from ethyl acetate to yield orange crystals ( 40 mg), o ( . o 2a,b) EtOH 2 285 ~,, d 408 m.p. 186.5 C lit. 187 C • UV: Amax 22, , ~7 an run.
IR: )]KBr )405 (br), 2965 S, 2850 M, 1705 M, 1660 S, 1650 S, 1595 S,
1560 s, 1455 s, 1420 s, 1340 s, 1320 s, lJOO s, 126o·s, 1210 11, 1160 M, -1 1120-1080 br, 2020 s, 1000 S, 965 S, 845 M, 800 Sand 750 M, cm •
MS, m/~ (relative abundance) 398 (M+, 100), 383 (JO), 382 (71), 381
(36), 380 (32), 369 (31), 367 (63), 365 (40), 351 (51), 337 (65), 328
(73), 325 (46), 323 (37), 313 (100) and Jll (50). Fl'-NMR (CDClJ): 6
7.42 (lH, s, -H4), 7.JJ (lH, d, J2Hz, -H5), 6.76 (lH~ d, J2Hz, -117), 6.48 (lH, d, J?Hz, -~ 5), 5.74 (lH, m, -1\8), 4.14 (lH, m, -1\6), 4.0J,
J.99 and J.95 (9H, J s, J-OCH3), 2.64-2.44 (2H, m, -1\7) ppm.
D. Sodium borohydride reduction of tri-methylversicolorin A-
hemiacetal
Trimethylversicolorin A-hemiacetal (35 mg) was dissolved in 60 ml
of THF, 60 ml of 0.05 M sodium phosphate buffer, pH 7.2, was added, and
the mixture cooled to o0 c. 'rhe cold solution was treated dropwise over
2 hr. with 2.5 ml of buffer solution containing 8 mg of sodium boro-
hydride. The reaction mixture was then adjusted to pH 6 with dilute
HCl, and extracted three ti.mes with 100 ml portions of ethyl acetate.
The extract was washed with water until neutral, dried and evaporated
to yield a crude product which consisted largely of two new products.
These products were separated by preparative TLC with solvent system B.
The less polar product was identified as tri-methyl-isoversiconal
(5 mg), PC-J-68-1-2, m.p. 155-157°0. UV: AEtOH 224, 287, 345 and max 405 run. IR: llKBr 3450 (br), 2940 (br), 1655 S, 16)0 S, 1590 S, 1572 S, ·
Page 158
147
1550 S, 1440 M, 1412 M, 1340 S, 1J28 S, 1315 S, 1240 M, 1220 M, 1205 M,
1150 M, 1115 M, 1090 M, 1070 M, 1040 M, 1020 M, 996 Mand 875 M cm-l.
NS: m/~ (relative abundance). EI 382 (M-18, 8J), J67 (100), J65 (42,
J5J (41), JJ9 (J2), JJ5 (50) and J2J (48). CI. J8J (MH-18, 100),
J84 (35). Fr-NMR (d6-Acetone) o 7.28 (lH, s, -H4), 7.26 (lH, d, J2Hz,
-H5), 6.96 (lH, d, J2Hz, -~), 5.68 (lH, m, -~ 8), J98 (9H, J-OCHJ) ppm.
The more polar product was identified as tri-methyl-versiconol
(6 mg), PC-J-68-1-J, m.p. 2J0-2J2°C. UV: AEtOH 22J, 286, J49 and max 402 nm. IR: ))""KBr J4JO (br), J290 (br), 2960 M, 1665 S, 1648 S, 1600 S, .
1570 S, 1455 M, 14J5 M, 1J60 S, 1320 S, 1260 M, 1220 M, 1220 M, 1170 M,
114o M, 1055 Sand 995 M cm-1• MS: r.J/~ (relative abundance). EI. J84 (M-18, JO), J8J (18), J82 (42), J71 (J6), J70 (52), J69 (6J), J68 (Jl),
J67 (100), J65 (J5), 355 (55), J54 (27), 353 (86), J52 (47), J51 (45),
J41 (2J), J40 (J4), JJ9 (86), J38 (46), JJ7 (49), JJ5 (40), J25 (8J),
J24 (57), J2J (97), J21 (57), Jll (J5), JlO (J6), J09 (51), JO? (44),
296 (29), 295 (55), 290 (JO), 283 (27), 279 (JO). CI. 40J (MH~ 5),
J86 (22), 385 (MH-18, 100), J84 (24), J8J (88). Fr-NMR (d-Acetone):
o 7.37 (1H, s, -H4), 7.24 (lH, d~ J2Hz, -H5), 6.96 (lH, d, J2Hz, -H.,) and J.98 (9H, J-OCHJ) ppm.
E. Preparation of Iso-versicolorin C (Structure XVII-B)
Partially reduced versicolorin A-hemiacetal, PC-J-69-11-J, (8 mg)
was dissolved in 20 ml of THF and 0.5 ml of HCl (JO%) solution. The
THF solution was refluxed for 2 hr, cooled, diluted with 20 ml of water,
and extracted three times with 30 ml portions of ethyl acetate. The
extract was washed with water until neutral, dried and evaporated to
Page 159
148
yield an orange product. After recrystallization from ethyl acetate,
1 t showed mp >J.50° (dee.). UV, A EtOH 222 ( ~ , 2 • .5 x 104), 2.5.5 max (1.4 X 104) 26,5 (1.6 X 104), 294 (1.8 X 104), 318 (le) X 104) and
463 (6.5 x 103) nm. IRc )JKBr 3)80 (br), 2910 M, 1605 S, 1565 M,
1463 M, 1393 S, 1298 S, 1253 S, 1202 M, 1160 M, 1142 M, 1121 M, 1090 M,
1060 M, 1020 M, 844 Mand 790 M. MSc l!J!'l. (relative abundance) 340
(N+, 62), 313 (20), 312 (43), 311 (100), 298 (20), 297 (84). NMR
(d6-DMS0) o 12.40 and 11.94 (2H, 2 broad S, 2 chelated -OH), 7.02
(lH, d, JJHz, -H5), 6.91 (lH, s, -H4), 6.50 (lH, d, J3Hz, -tt.,), 5,98
(lH, distorted s, -~ 5), 4.14 (2H, m, -~ 8), 3.72 (lH, broads, -~ 6)
and 2.20 (2H, distorted s, -Hr,) ppm.
VII.2. Derivatives of Sterigmatocystin (ST)
A. Synthesis of Sterigmatocyst.in-hemiacetal (Structure VIII-A)
ST (1.0 g) in acetone (200 ml) was heated under reflux for 12 hr
with 10 ml of 10% H2so4• The reaction mixture was cooled overnight,
and the yellow-green precipitate was filtered and collected. The
filtrate was evaporated under reduced pressure to ab:>ut .50 ml to remove
the bulk of the acetone. The acetone solution was extracted 3 times
with 150 ml portions of ethyl acetate, and the combined extracts washed
with water until neutral, dried over Mgso4 (anhydrous), and evaporated
to dryness. The yellow-green solid was combined with the yellow-green
precipitate which was collected after filtering the acetone solution.
After recrystallization from acetone it showed m.p. 210°-212°c. The
yellow-green compound (400 mg) had the following properties: UV:
A:~H 232 ( € , 2.8 x 104), 249 (J.4 x 104) and JZ? (7. 7 x 104) nm.
Page 160
149
IR: )!KBr y~oo s, 1650 s, 1625 s (br), 1580 (s, br), 1490 s, 1480 s, 1460 S, 1440 S, 1410 M, 1340 M, 1275 S, 1240 S, 1200 M, 1130 M, 1100 M,
-1 1075 S, 1040 M, 985 M, 920 Ivl, 900 M, 885 M, 815 S, 775 S cm • MS: + . TJ1/~ (relative abundance) 342 (M , 16), 324 (80), 313 (19), 306 (31),
296 (20), 295 (49), 278 (22), 277 (20), 267 (23), 266 (21), 265 (27),
181 (34), 169 (35), 152 (24), 151 (23), 149 (40). NHR: (d6-DMSO) o
7.51 (lH, t, J7Hz, -H5), 6.82 (lH, d, J8Hz, -H6), 6.62 (lH, d, J8Hz, -H4),
6.45 (lH, s, -8i1), 6.50-6,32 (lH, m, -I\4), 5.56-5.38 (lH, m, -H17),
4.18-4.oo (lH, m, -8i5), 3,84 (3H, s, -oCH3), 2.20 (2H, m, -8i6) ppm.
Fl'-NMR (CDCJ.3): 6 7.48 (lH, t, J7Hz, -H5), 6.81 (lH, d, J8Hz, -H6),
6.75 (1H, d, J8Hz, -H4), 6.36 (1H, s, -8i1), 5.83-5,59 (lH, m, -11i7),
6,53 (lH, d, J2Hz, -8i4), 4,32-4,07 (lH, m, -1\5), 3,99 (3H, s, -OCH3)
ppm,
C and H analysis: Calcd, for c188i4o7•o,5 H2o, C, 61,7%, H, 4.J%,:
Found, a, 61.9%, H, 4.4%.
B, Isolation of Sterigmatocystin-ethoxyacetal (Structure IX)
In the preparation of another batch of sterigmatocystin-hemiacetal,
recycled ethyl acetate was used to extract the hemiacetal from the acidic
acetone solution. After drying and evaporation of the solvent, two major
products were observed on TLC. The more polar one was identified as
sterigmatocystin-hemiacetal, and the other was a new compound. The
two compounds were separated by preparative TLC with solvent system B.
The less polar product was identified as sterigmatocystin-ethoxy acetal
(60 mg), m .p. 189-192°0. UV, A EtOH 232 ( € , 2. 7 x 104), 248 (3.3 x 104), max and 326 (1.7 x 104) nm. IR, J!KBr 2900 M (br), 1632 s, 1608 s (br),
Page 161
150
1565 S, 1478 S, 1465 S, 1440 S, 14o0 M, 1J80 M, 1J25 M, 1260 S, 1218 S,
1178 M, 1115 S, 1080 S, 1065 S, 1028 S, 946 M, 890 M (br), 810 M, 796 M,
and 760 M cm-1• MS: ry~ (relative abundance) J70 (M+, 45), J42 (22),
J41 (100), J25 (11), Jl3 (16), 297 (35), 295 (lJ), 285 (28). NMR (CDC13)
6 13.28 and lJ.20 (lH, 2S, -chelated OH), 7.46 (lH, t, J8Hz, -H5),
6.84-6.64 (2H, m, -H4 and H6), 6.50 (lH, d, J6Hz, -~ 4), 6.36 (1H,
disto~ed s, -~ 1), 5.J4 (1H, m, -~ 7), 4.18 (lH, m, -~ 5), 4.oo (JH,
s, -OcH3), 3.84-3.16 (2H, m, -OCH2-), 2.40 (2H, m, -~ 6), 1.14 and 0.89
(JH, 2t, J7Hz, -CHJ) ppm.
c. NaBH4 reduction of Sterigmatocystin-Hemiacetal
Sl'-hemiacetal (JOO mg) was dissolved in JOO ml of THF, 80 ml of
0.05 M soditun phosphate buffer, pH 7.2; was added, and the mixture
cooled to o0 c. The cold solution was treated dropwise over J hr with
50 ml of buffer solution containing 90 mg of NaBH4• The reaction mixture
was then diluted with 50 ml of water, adjusted to pH 6 with dilute HCl,
and extracted 5 times with 150 ml portions of ethyl acetate. The
extract was washed, dried, and evaporated to yield a crude product
which consisted largely of two new products. These products were
separated by preparative TLC with benzene:ethyl acetate, 50:50, as the
developing solvent. The less polar product was identified as partially-
reduced sterigmatocystin-hemiacetal (XI-C), PC-J-73-2J (80 mg), m.p.
22J-226°c, UV: A:~H 232 ( E:, 2.2 x 104), 249 (2.7 x 104) and J25
(1.5 x 104) nm. IR: )TKBr )410 (br), J260 {br), 2940 (br), 1640 s, 1610 S, 1570 S, 1495 S, 1480 S, 1480 S, 1450 S, 1410 M, 1330 M, 1295 M,
1270 M, 1230 S, 1200 M, 114o S, 1100 S, 1030 M, 1020 M, 995 M, 975 M,
Page 162
151
860 I1, 825 s, -1 cm • MS, !fY(}. (relative abundance) 344 (M+, 38), 326
(70), 314 (22), 313 (100), '297 (20), 285 (60), 283 (42), 255 (20), 253
(27), 169 (21), 149 (27), 131 (22), 119 (33). NMR (d6-DMSO) 6 13.15
(lH, s, chelated -OH), 7.54 (lH, t, J9Hz, -H5), 6.84 (lH, d, J8Hz, -H6),
6.63 (lH, d, J8Hz, -H4), 6.30 (lH, s, -~ 1), 5.64-5.42 (1H, m, -~ 7),
3.83 (3H, s, -OCH3), 3.80 (2H, m, -~ 4) ppm.
The more polar product was identified as sterigmatodiol XII,
PC-3-74-1 (120 mg). It had m.p. 208-210°C, and showed the following
properties: UV, ,._EtOH 232 ( E, 2.4 x 104), 249 (2.9 x 104) and 331 max
(1.6 x 104) nm. IR: )TKBr 3400 (br), 2940 (br), 1645 S, 1605 S, 1510 N,
1485 M, 1465 M, 1450 M, 1415 M, 1320 M, 1270 H, 1240 S, 1215 M, 1100 M,
1055 M, 1030 M, 970 S, 815 I1 cm-1• MS: !JY(}. (relative abundance) 346 (H+),
54), 316 (20), 315 (70), 297 (20), 285 (77) 283 (20), 271 (loo). FT-NI1R
(d6-Acetone): 6 7.56 (lH, t, J8Hz, -H5), 6.90 (lH, d, J7Hz, -H6), 6.66
(lH, d, J7Hz, -H4), 6.46 (lH, s, -~ 1), 4016-3.96 (3H, m, -~ 4 and ~ 5),
3.90 (3H, s, -OcH3), 3.58 (2H, t, J7Hz, -~ 7) ppm.
c and H analysis: Calcdo for Ss~807: c, 62.4%, H, 5.2%:
Founds C, 62.6%, H, 5.3%.
D. Preparation of Dihydrosterigmatocystin (Structure XIV).
Sterigmatocystin (400 mg) was hydrogenated in ethyl acetate (50 ml)
over 10% palladium catalyst on carbon (100 mg) at 25°c and atmospheric
pressure for 4 hr. The yellow-green product was recovered in the usual
manner and further purified by recrystallization from acetone. The
purified product (200 mg) had m.p. 226-227°c (lit. 227-228°c). 15 UV:
A~~H 232 ( E, 2.2 x 104), 247 (2.8 x 104) and 327 (1.4 x 104) nm. IR,
Page 163
152
VKBr 2960 M, 1638 s, 1628 s, 1615 s, 1608 s, 1575 s, 1550 s, 1490 s, 1475 S, 1468 S, 1445 S, 1408 M, 1390 M, 1270 S, 1230 S, 1195 M, llJO M,
1082 S, 1048 M cm-1• I1S: '!Y£ (relative abundance) J26 (1{, 100), JOB
(20), 297 (26). NMR (CDClJ) o lJ.04 (1H, s, chelated-OH), 7.36 (lH, t,
J8Hz, ·-H5), 6. 70 (lH, d, J7Hz, -H6), 6, 62 (1H, d, J7Hz, -H4), 6,42 (lH,
d, J5Hz, -8i.4), 6,24 (lH, s, -8i.1), 4,16 (2H, m, -8i.7), J,93 (JH, s,
-OCH3), J,6J (1H, q, J7Hz, -8i_5), 2.JO (2H,m, -~ 6) ppm,
E. Preparation of Isodihyd.ro-sterigmatocystin (Structure XIII}
Partially reduced sterigmatocystin-hemiacetal XI-C, (20 mg), was
dissolved in 50 ml of THF and 1 ml of HCl (J~fo) solution. The solution
was refluxed for 4 hr, cooled, diluted with 20 ml of water, and extracted
with three times with JO ml portions of ethyl acetate. The extract was
washed with neutral, dried and evaporated to dryness. On recrystal-
lization from ethyl acetate, the light-yellow crystals, showed m.p,
226-227°c. UV: /1.EtOH 232 ( E: , 1,9 x 104), 252 (2.4 x 104) and 325 max (1,J x 104) nm, IR: lfKBr 2880 M, 1645 S, 1610 S, 1570 S, 1492 S, 1473 S,
1458 S, 1410 H, 1J56 M, 1315 M, 1292 M, 1265 M, 1237 S, 1208 1'1, llJJ S,
112 S, 1060 H, 1040 M, 933 M, 862 M, 824 Sand 767 M cm-l, MS: "!2}~
(relative abundance) J26 (M+, 100), J08 (JJ), 297 (32), 283 (70), 265
(Jo). NHR (CDC13), o 12.96 (lH, s, chelated, -OH), 7.36 (lH, t, J7Hz,
-H5), 6.69 (lH, d, J7Hz, -H6), 6.62 (lH, d, J7Hz, -H4), 6.20 (lH, s,
-~ 1), 5.79 (1H, d, J2Hz, -~ 7), 4.19 (2H, m, -8i.4), 3,90 (JH, s, -oc~), J,88 (lH, m, -8i.5) and 2,20 (2H, m, -11J_6) ppm.
C and H analysiss Calcd. for Sa11J.4o6, c, 66,3%, H, 4,3%;
Found: C, 66.J%, H, 4.4%.
Page 164
153
F. Diazomethane methylation of partially reduced sterigmatocystin-
hemiacetal
Alcohol-free diazomethane, 47 prepared from 2.2 g of Diazald, in
anhydrous ether was added to a 50 ml ethyl acetate solution containing
15 mg partially reduced sterigmatocystin-hemiacetal. The mixed solution
was stirred for 2 hr at o0 c, and evaporated to dr,yness. The white-yellow
solid was recrystallized from ethyl acetate, and the light yellow cry-
stals obtained were identified as methylated partially reduced sterigma-
tocystin-hemiacetal (XV). The material had m.p. 202-2o4°c. UV: )..EtOH max 231 ( E., 2.1 x 104), 250 (2.7 x 104) and 329 (1.3 x 104) nm. IR:
))KBr .'.3480 S, 2950 M, 2890 M, 1660 S, 1610 S, 1570 S, 1510 M, 1470 S,
1410 M, 1325 S, 1280 M, 1240 S, 1225 S, 1190 M, 1125 S, 1090 M, 1080 M,
1065 M, 1005 M, 925 M, 825 S, 785 S, 750 M, cm-1• NS: m/~ (relative
abundance) 358 (M+, 100), )40 (14) 328 (18), 3Z7 (43), JOO (15), 299
(65), 285 (44), 273 (14), 272 (61), 255 (15), 254 (20), 242 (14) and
226 (15). NHR (CDC13/d 6-D•IS0); 6 ppm 1).02 (lH, broads, -chelated OH)~
7.51 (lH, t, J8Hz, -H5), 6.82 (lH, d, J8Hz, -H6), 6.64 (lH, d, J8Hz,
-H4), 6.50 (1H, s, -11J.1), 5.64 (1H, broad d, J4Hz, -H17), 4.80 (lH,
broads, -~ 5), 4.oo (2H, m, -~ 4), 3.80 (6H, s, 2-oc~), 2.30 (2H, m,
-~6) ppm. An impurity peak at 1.80 was also present.
C and H analysis: Ca.led. for °i.9~ 807°0.5 H2o, C, 62.1; H, 5.2.
Found: C, 62.J; H, 5.1. Xanthone derivatives containing water of
crystallization were also observed by W.T. Bradner and co-workers.53
Page 165
1.
REFERENCES
a. Joseph V. Rodricks, "Mycotoxins and Other ~al Related Food Problems." Advances in Chemistry Series 19,ACS, Washington, D.C. (1976).
b. A. Ciegler, s. Kadis and S.J. Ajl, "Microbial Toxins." Vol. VI, Academic Press, New York, N.Y. (1971).
c. I.F.H. Purchase, "M cotoxins," Elsevier Scientific Publishing Company, New York, N.Y. 1974).
2. a. T. Hamasaki, Y. Hatsuda, N. Terashima and M. Renbutsu, !irf.. Biol. Chem. (Tokyo), 21,, 11 (1967).
b. L.S. Lee, J.W. Bennett, A.F. Cucullu and A.L. Ory, J. Agr. Food Chem., 24, 1167 (1976). - .
J. a. T. Hamasaki, Y. Hatsuda, N. Ferashima and M. Renbutusu, Agr. Biol. Chem. (Tokyo),~. 696 (1963).
b. J.G. Heathcote and M.F. Dutton, Tetrahedron, 25, 1497 (1969).
4. E. Bullock, D. Kirkaldy, J.C. Roberts and J.G. Underwood, J. Chem. Soc., 829 (1963),
5. G.c. Elsworthy, J,S.E. Holker, J.M. McKeown, J.B. Robinson and L.J. Mulheirn, Chem. Commun., 1069 (1970).
6. T. Hamasaki, Y. Hatsuda, M. Ishida and Y. Kiyama, Agr. Biol. Chem. (Tokyo), .ll, 444 (1971).
7, a, P.J. Aucamp and c.w. Holzapfel, J.S. Afr. Chem. Inst., 23, 40 (1970).
b, J.s.E. Holker, S,A. K~a.l, L.J. Mulhein and P.M. White,~. Commun,, 24, 911 (1966).
8. J. Grandjean, J. Jadot and J.L. Ramant, Bull. Soc. Chim. Belges, 81, 521 (1972).
9. J.C. Roberts and P. Roffey, J. Chem. Soc., 3666 (1965).
10. a. J.H. Birkinshaw and I.M.M. Hammady, Biochem. J,, .2,2, 162 (1957).
b, J.H. Birkinshaw, J.C. Roberts and P. Roffey, J. Chem. Soc., Q, 855 (1966).
154
Page 166
155
11. a, T. Hamasaki, M. Renbutusu and Y. Hatsuda, Agr. Biol. Chem. (Tokyo), ,;g_, 1513 (1967).
b. L.S. Lee, J.W. Bennett, L.A. Goldblatt and R.E. Lundin, J. Arner. Oil Chem. Soc., 48, 93 (1971).
12. Y. Hatsuda, T. Hamasaki, M. Ishid ands. Yoshikawa, Agr. Biol. ~. (Tokyo), 22,, 131 (1969).
13. a. D.F.G. Pusey and J.C. Roberts, J. Chem. Soc., 3542 (1963).
b. J.A. Donkersloot, R.I. Mateles and S.S. Yang, Biochem. Biophys. Res. Commun.,~, 1051 (1972).
14. a. J.E. Davies, D. Kirkaldy and J.C. Roberts, J. Chem. Soc., 2169 (1960).
b. E. Bullock, J.C. Roberts, and J.G. Underwood, ibid., 4179 (1962).
15. J.S.E. Holker and S.A. Kagal, Chem. Comm., 1574 (1963),
16. Y. Hatsuda, T. Hamasaki, M. Isuda, K. Matsui ands. Hara,~. Biol. Chem. (Tokyo), 2£, 521 (1972).
17. T. Hamasaki, K. l1atsui, K. Isono and Y. Hatsuda, Agr. Biol. Chem. (Tokyo), 22., 1769 (1973).
18. A.G. Brown, J. Chem. Soc., Q, 2572 (1970).
19. R.W. Richards, J. Antibiot. (Tokyo), 24 (10), 715 (1971).
20. P.J. Aucamp and c.w. Holzapfel, J.S. Afr. Chem. Inst., 22, 535 (1969). ~
21. K. Fukuyama, T. Tsuikikara, Y. JCatsube, T. Hamasaki and Y. Hatsuda, Tetrahedron Lett., J, 189 (1976).
22. K.K. Chexal, C. Fouweather, J.S.E. Holker, T.J. Simpson and K. Young, T. Chem. Soc., Perkin Trans.!, (13), 1584 (1974).
23. J. Adye and R.I. Mateles, Biochem, Biophys. Acta, 86, 418 (1964).
24. a. M. Stack and J.V. Rodricks, J. Ass. Offic. Anal. Chem., 2±., 86, (1971).
b. M. Stack and J.V. Rodricks, ibid., .2§., 1123 (1973).
25. J.R. Vercellotti, L.P. Egan and W.T. Lowry, Carbohyd. Res.,~. 261 (1972).
Page 167
26.
27.
1.56
z. Durackova, v. Betina and P. Nemec, J. Chromatogr. Sci, 116, 141 (1976)
a. L. Stoloff, s. Nesheim, L. Yin, J.V. Rodricks, M. Stack and A.D. Campbell, J. Ass. Offic. Anal. Chem., 2±, 91 (1971).
b. G. Sullivan, D.D. Maness, G.J. Yakatan and J. Scholler, J. Chromatogr., 116, 490 (1976).
28. j.S.E. Holker, Personal Communication.
29.- P. Y. Berger and J. Jadot, Bull. Soc. Roy. Sci. Liege., 44, 633 (1975).
JO. J,A. Schmit, R.A. Henry, R.C. Williams and J.F. Deckman, J. Chromatogr. Sci., 2, 645 (1971).
Jl. P.P. Rai, T,D. Turner and S.A. Matlin, J. Chromatogr. Sci., 110, 4ol (1975). .
32. K. Ito, A. Yamane, T. Hamasaki and Y. Hatsuda, Agr. Biol. Chem. (Tokyo), 40, 2099 (1976).
JJ. a. D,G.I. Kingston, P,N. Chen and J.R, Vercellotti, J. Chromatogr., Sci,, 118, 414 (1976).
b. K. Ito, A. Yamane, T. Hamasaki and Y. Hatsuda, Agr. Biol. ~. (Tokyo), 40, 2099 (1976).
)4. K. Hostettman and H.M. McNair, J. Chromatogr. Sci., 116, 201 (1976).
35. W.J. McMaster, A.I. Scott and s. Trippett, J. Chem. Soc., 4629 (1960).
J6. a. J. MacI'1illan, J. Chem. Soc., 1697 (1953).
b. J.A. Ballantine and R.G. Fenwick, Org, Mass. Spec., g, 1145 (1969).
37. J,F. Grove, J, MacMillan, T,P.C. Mulholland and M.A.T, Rogers, J. Chem. Soc., 3949 (1952).
38. L.A. Goldblatt, "Aflatoxin," Academic Press, New York, N.Y. (1969).
39. R.H.R. Gundu and P.A. Harlen, J. Econ. Entomol., §i, 988 (1972).
40. a. H. W. Schroeder, R.J. Cole, R.D. Grigsby and H. Hein, Jr., Appl. Microbiol., ?l., 394 (1974).
b. R.H. Cox, F. Churchill, R.J. Cole and J.W. D::>rner, ACS 28th Southeast Meeting, paper 42J, (1976).
Page 168
41.
42.
43,
44.
45.
46.
157
s.H. Ashoor and F.S. Chu, J. Agr, Food, Chem.,~. 445 (1975),
a. A,E, Pohland, M,E. Cushmac and P.J. Audrellos, J, Ass. Offic, Anal. Chem., 21,, 967 (1968).
b. c.E. Johnson and F,A, Bovey, J, Chem. PhYs., ~. 1012 (19.58),
G,A, Gray, Anal. Chem., ~. 546A (1975),
K.G.R, Pachler, P.S, Steyn, R. Vleggaar, P.L. Wessels and D,B. Scott, J. Chem. Soc. Perkin I, 1182 (1976),
R.H. Cox and R.J. Cole, J, Org. Chem., 42, 112 (1977),
G.c. Levy and G,L, Nelson, ul3c-NMR for Organic Chemists," Wiley-Interscience, New York, N,Y., (1972),
a. T.J. deBoer and H.J. Blcker, Rec, Trav. Chim. Pays-Bas,, Zl, 229 (1954).
b. Idem., Qrg, Syn., Coll., Vol. 4, 250 (1963).
48. a. R, Thomas, "Biogenesis of Antibiotic Substances," Academic Press, New York, N.Y. (1965).
b. R.I. Mateles and G.N. Wogan, Advan. Mic:robiol. Physiol., 1, 25 (1967).
c, N. Biollaz, G. B!lchi, G. Milne, J. Amer. Chem. Soc., 2,g_, 1035 (1970).
d, M,O. Noss, "Phytochemical EcologY," Academic Press, London (1972).
49. a. M.T. Lin, D.P.H. Hsieh, R.C. Yao and J.A. Donkersloot, Biochem., 12, 5167 (1973).
b. D.P,H. Hsieh, M.T. Lin and R.Y. Yao, Biochem. Biophys. Res. Commun., ~. 992 (1973).
50, D.P.H. Hsieh, M.T. Linn, R.C. Yao and R, Singh, J. Agr, Food, ~ •• 24, 1170 (1976).
51. a. J,C. Roberts, Fortschritte, Chem, Org. Naturstoffe, 21_, 119 (1973).
b. J .G. Heathcote, M.F. Cutton and J .R. Hibbert, Chem, Ind,, l, 1027 (1973).
Page 169
158
52. D.G.I. Kingston, P.N. Chen and J.R. Vercellotti, Phytochemistry, 1.2, 1037 (1976).
53. J.M. Essery, F.D. O'Herron, D.N. McGregor and W.T. Bradner, J. Med. Chem., 12,, 1339 (1976).
Page 170
The vita has been removed from the scanned document
Page 171
ME'l'ABOLITES OF ASPERGILLUS VERSICOLOR
by
Paul Ning-Chuan Chen
(ABSI'RAar)
Systematic fractionations of crude extracts from various strains
of Aspergillus versicolor were carried out by a combination of techni-
ques, including liquid-liquid partition, column chromatography and
thin-layer chromatography. As a result of these fractionations a total
of eleven metabolites were obtained in pure form. Ten of these were
identified as the known compounds, sterigmatocystin, de-0-methyl-
sterigmatocystin, 5-methoxysterigmatocystin, versicolorin A, versicolorin
C, aversin, averufin, griseofulvin, dechlorogriseofulvin and J,8-
dihydroxy-6 -methoxy-1-methylxanthonc. A new metabolite was isolated
and identified as 6,8-di-O-methylnidurufin.
Two high-performance liquid chromatography systems were developed
for the analysis of these metabolites, using microparticle silica gel
and microparticle polar bonded phase columns.
Studies were also carried out on the reduction of the hemi-acetal
derivatives of sterigmatocystin and versicolorin A. Reduction with a
limited amount of sodium borohydride yielded in each case a mixture of
two major products. The fully reduced products were identified as
diols, while the partially reduced products were found to be new
hemiacetals. The stn.icture elucidation of these compounds was carried
out with the aid of carbon magnetic resonance spectroscopy and of
chemical conversions.
Page 172
A modified pathway for the biosynthesis of versicolorin A from
averufin is proposed that incorporates nidurufin as an intermediate.