29
Publications

Publications

This article appeared in a journal published by Elsevier. The attachedcopy is furnished to the author for internal non-commercial researchand education use, including for instruction at the authors institution
and sharing with colleagues.
Other uses, including reproduction and distribution, or selling orlicensing copies, or posting to personal, institutional or third party
websites are prohibited.
In most cases authors are permitted to post their version of thearticle (e.g. in Word or Tex form) to their personal website orinstitutional repository. Authors requiring further information
regarding Elsevier’s archiving and manuscript policies areencouraged to visit:
http://www.elsevier.com/copyright

Author's personal copy
Characterization of 4-methyl-2-oxo-1,2-dihydroquinolin-6-yl acetate as aneffective antiplatelet agent
Nivedita Priya a, Anjali Gupta b, Karam Chand b, Prabhjot Singh a, Abha Kathuria b, Hanumantharao G. Raj a,*,Virinder S. Parmar b, Sunil K. Sharma b,*
a Department of Biochemistry, V P Chest Institute, University of Delhi, Delhi 110 007, Indiab Bioorganic Laboratory, Department of Chemistry, University of Delhi, Delhi 110 007, India
a r t i c l e i n f o
Article history:Received 27 January 2010Revised 1 April 2010Accepted 2 April 2010Available online 8 April 2010
Keywords:Quinolin-2-onesPlatelet CRTAaseAntiplatelet agentsCox-1
a b s t r a c t
We have studied earlier a membrane bound novel enzyme Acetoxy Drug: protein transacetylaseidentified as Calreticulin Transacetylase (CRTAase) that catalyzes the transfer of acetyl groups from poly-phenolic acetates (PAs) to the receptor proteins and thus modulating their biological activities. In thiscommunication, we have reported for the first time that acetoxy quinolones are endowed with antiplate-let action by virtue of causing CRTAase catalyzed activation of platelet Nitric Oxide Synthase (NOS) byway of acetylation leading to the inhibition of ADP/Arachidonic acid (AA)-dependent platelet aggregation.The correlation of specificity of platelet CRTAase to various analogues of acetoxy quinolones with intra-cellular NO and consequent effect on inhibition of platelet aggregation was considered crucial. Amongacetoxy quinolones screened, 6-AQ (4-methyl-2-oxo-1,2-dihydroquinolin-6-yl acetate/6-acetoxyquino-lin-2-one, 22) was found to be the superior substrate to platelet CRTAase and emerged as the most activeentity to produce antiplatelet action both in vitro and in vivo. 6-AQ caused the inhibition of cyclooxygen-ase-1 (Cox-1) resulting in the down regulation of thromboxane A2 (TxA2) and the inhibition of plateletaggregation. Structural modification of acetoxy quinolones positively correlated with enhancement ofintracellular NO and antiplatelet action.
� 2010 Elsevier Ltd. All rights reserved.
1. Introduction
Platelets are involved in the cellular mechanisms of primaryhomeostasis leading to the formation of blood clots. Platelets areactivated when brought into contact with agents such as collagen,thrombin, and ADP. The damage to the blood vessel walls exposesthe sub endothelium proteins, most notably collagen. Thecirculating platelets bind collagen with collagen-specific glycopro-tein Ia/IIa receptors. The adhesion is strengthened further by thelarge, multimeric circulating protein like von Willebrand factor(vWF), which forms links between the platelets glycoprotein Ib/IX/V and the collagen fibrils. This adhesion activates the platelets.ADP receptors P2Y1 and P2Y12, both belonging to theG-protein-coupled seven-transmembrane domain receptor family,
expressed on platelets, extensively bind to ADP resulting in the re-lease of dense granules containing PAF (Platelet Activating Factors)vWF, serotonin, thromboxane A2 (TxA2) which further activate theother circulating platelets.1 Numerous antiplatelet agents weredeveloped based on their ability to block the receptors responsiblefor platelet activation. Further, the agents causing the inhibition ofcyclooxygenase catalyzed TxA2 synthesis would also lead to theinhibition of platelet aggregation.2 Previous investigations carriedout in our laboratory documented for the first time, the remarkableactivation of endothelial NOS by a certain class of PAs by way ofacetylation of NOS mediated by CRTAase.3 Accordingly, PAs werefound to be effective in the inhibition of ADP induced plateletaggregation.
In the present investigation, efforts have been made to comparethe specificities of acetoxy quinolones on CRTAase mediated acti-vation of NOS and also to delineate the structure activity relation-ship (SAR) with reference to the effect of position of substitution ofacetoxy group on benzenoid ring and pyridone ring, alkyl group atC-3 position of the quinolone moiety, and substitution at N- andO- of the pyridone ring of PAs. The results clearly demonstrated6-acetoxy-4-methylquinolin-2-one (6-AQ, 22) to be the bestsubstrate to platelet CRTAase compared to the other acetoxy quin-olones resulting in inhibition of ADP induced platelet aggregation.
0968-0896/$ - see front matter � 2010 Elsevier Ltd. All rights reserved.doi:10.1016/j.bmc.2010.04.011
Abbreviations: AA, arachidonic acid; AQ, acetoxy quinolone; CDNB, 1-cloro 2,4-dinitro benzene; Cox-1, cyclooxygenase-1; Cox-2, cyclooxygenase-2; CRTAase,calreticulin transacetylase; DAMC, 7,8-diacetoxy-4-methyl coumarin; DCFH-DA,dichlorofluorescin diacetate; NOS, nitric oxide Synthase; GSH, reduced glutathione;GST, glutathione-S-transferase; NO, nitric oxide; PAs, polyphenolic acetates; PPP,platelet poor plasma; PRP, platelet rich plasma; TxA2, thromboxane A2; TxB2,thromboxane B2.
* Corresponding authors. Tel.: +91 11 27666646x191 (S.K.S.).E-mail addresses: [email protected] (H.G. Raj), [email protected]
(S.K. Sharma).
Bioorganic & Medicinal Chemistry 18 (2010) 4085–4094
Contents lists available at ScienceDirect
Bioorganic & Medicinal Chemistry
journal homepage: www.elsevier .com/locate /bmc

Author's personal copy
2. Results
In our earlier work we elucidated the role of acetoxy groups onthe benzenoid ring of chromones, coumarins, xanthones, and flav-ones in facilitating the acetylation of receptor proteins catalyzed byCRTAase. In this regard we also studied the factors, such as theproximity of the acetoxy group to the oxygen heteroatom, the roleof carbonyl group on the benzopyran nucleus, and the effect ofsubstituents on the coumarin molecule in controlling the proteinacetylation.4,5
However, the action of CRTAase on acetoxy quinolones has notbeen studied so far. Herein, we have elucidated the action of CRTA-ase on a series of acetoxy quinolones and consequent effect on theenhancement of NO levels in platelets and inhibitory effect onADP/AA induced platelet aggregation. We have compared the spec-ificities of acetoxy quinolones by varying the position of acetoxygroup on benzenoid ring, replacing 4-methyl group by acetoxygroup, and by incorporating alkyl groups of varying size at C-3position. Also, we have synthesized N- and O-substituted alkyl es-ters of acetoxy quinolin-2-ones to study the CRTAase substratespecificity. Such a study would allow us to observe the effect there-of on the rate of catalytic activity of CRTAase and the efficacy ofthese acetoxy quinolones to activate platelet NOS. The methoxyderivatives of C-3 alkyl quinolones (1–9) were synthesized viaKnorr reaction of 2-alkyl ethyl acetoacetate with anisidines.Demethylation was carried out with a mixture of hydrobromic acidand acetic acid to yield corresponding hydroxy quinolones (10–18)which were then acetylated with acetic anhydride in acetic acid toyield acetoxy derivatives 19–27 (Scheme 1). The 6- and 7-acetoxyderivatives of quinolones were further derivatized into N- (28 and29) and O-alkyl esters (30 and 31) by reaction with ethyl bromo-acetate in the presence of potassium carbonate (Scheme 2). The
formation of two isomers is due to the existence of tautomerismin the amidic bond. 4-Hydroxy quinolone (32) was prepared byreaction of aniline with malonic acid in the presence of zinc chlo-ride and phosphorus oxychloride and further its acetylation gives4-acetoxy quinolone (4-AQ, 33), as shown in Scheme 3. All thecompounds were fully characterized on the basis of their physicaland spectral data, and of total 33 quinolone derivatives, 21, that is,2, 3, 5, 6, 8, 9, 12, 14, 15, 17, 18, 21, and 23–31 are novel.
2.1. CRTAase activity
The results documented in Table 1 revealed the deferentialspecificity of platelet CRTAase to a number of acetoxy quinolonesand the position of acetoxy group played a key role in decidingspecificity of platelet CRTAase and was found to be in the order:6-AQ > 7-AQ�8-AQo 4-AQ. It is evident from the results (Table 1)that substitution of alkyl group at C-3 position resulted in drasticreduction of CRTAase activity and longer the alkyl chain greaterwas the inhibition. The substitution of ester group at N-, that is,in compounds 28 and 29 resulted in the marginal decline of CRTA-ase activity of platelets, while O-substitution in compounds 30 and31 hardly affected the activity. Moreover, substitution of 4-methylgroup with acetoxy group (compound 33) showed poor inhibitionof platelet aggregation.
NH2+
O
O
O
R
HN
O
R
O
H2SO4
HN O
R
HN O
R
HN O
R
R = H / C2H5 / C6H13 R = H / C2H5 / C6H13
1 R = H; R1 = 7-OCH3
2 R = C2H5; R1 = 7-OCH33 R = C6H13; R1 = 7-OCH34 R = H; R1 = 6-OCH3
5 R = C2H5; R1 = 6-OCH36 R = C6H13; R1 = 6-OCH37 R = H; R1 = 8-OCH3
8 R = C2H5; R1 = 8-OCH39 R = C6H13; R1 = 8-OCH3
HBr / CH3COOH Ac2O / CH3COOH
H3CO135-150 º C
100 ºC
10 R = H; R1 = 7-OH11 R = C2H5; R1 = 7-OH12 R = C6H13; R1 = 7-OH13 R = H; R1 = 6-OH14 R = C2H5; R1 = 6-OH15 R = C6H13; R1 = 6-OH16 R = H; R1 = 8-OH17 R = C2H5; R1 = 8-OH18 R = C6H13; R1 = 8-OH
19 R = H; R1 = 7-OAc20 R = C2H5; R1 = 7-OAc21 R = C6H13; R1 = 7-OAc22 R = H; R1 = 6-OAc23 R = C2H5; R1 = 6-OAc24 R = C6H13; R1 = 6-OAc25 R = H; R1 = 8-OAc26 R = C2H5; R1 = 8-OAc27 R = C6H13; R1 = 8-OAc
reflux reflux
H3CO
R1 R1 R1
1
2
3456
78 9
10
Scheme 1. Synthesis of 3-alkyl derivatives of quinolin-2-ones.
HN O BrCH2COOEt
K2CO3 / DMF
N OCH2COOEt
28 R = 7-OAc 30 R = 7-OAc29 R = 6-OAc 31 R = 6-OAc
+N OCH2COOEt
R = 7-OAcR = 6-OAc
R RR
Scheme 2. Synthesis of N- and O-substituted alkyl esters of quinolin-2-ones.
NH2HN O
OH
HN O
OAc32 33
ZnCl2, POCl3
CH2(COOH)2 (CH3CO)2O
CH3COOH
Scheme 3. Synthesis of 4-acetoxy derivative of quinolin-2-one.
4086 N. Priya et al. / Bioorg. Med. Chem. 18 (2010) 4085–4094

Author's personal copy
2.2. Enhancement of intracellular nitric oxide (NO) levels
2.2.1. Measurement of NO level by flow cytometryThe influence of acetoxy quinolones on the NO level in platelets
has been clearly brought out in Figure 1. Platelets were incubatedwith acetoxy quinolones followed by the measurement of NO lev-els by flow cytometry. It is clear from the results that 6-AQ (22)profoundly enhanced NO level in platelets as compared to 7-acet-oxy- (19) and 8-acetoxyquinolone (25) derivatives. The structural
modification of acetoxy quinolones was found to influence their ef-fect on NO production in platelets in tune with the specificity ofplatelet CRTAase to these compounds (Table 1).
2.2.2. Effect of acetoxy quinolones on platelet aggregation2.2.2.1. Platelet aggregation in vitro. The effect of acetoxy quino-lones on the ADP and AA induced platelet aggregation in vitro isshown in Figure 2. PRP was incubated with acetoxy quinolones fol-lowed by the measurement of platelet aggregation by the additionof ADP/AA. 6-AQ (22) inhibited the ADP induced platelet aggrega-tion to a greater extent as compared to 7-AQ (19) and 8-AQ (25)derivatives. The modification of carbonyl group of the quinolonemoiety (30 and 31) hardly affected the platelet aggregation whilethe presence of alkyl ester at N- (28 and 29) resulted in appreciablereduction of platelet aggregation. 6-AQ has profound effect on AAinduced platelet aggregation as compared to that induced byADP. The trend of activities followed the same manner, higher incase of 6-acetoxy as compared to 7- and 8-acetoxy derivatives.These observations have amply revealed that the structural modi-fications of acetoxy quinolones alter their effectiveness in the man-ner dependent on their specificities to CRTAase as the substrate.The 6-hydroxyquinolone (13) was devoid of antiplatelet activity.
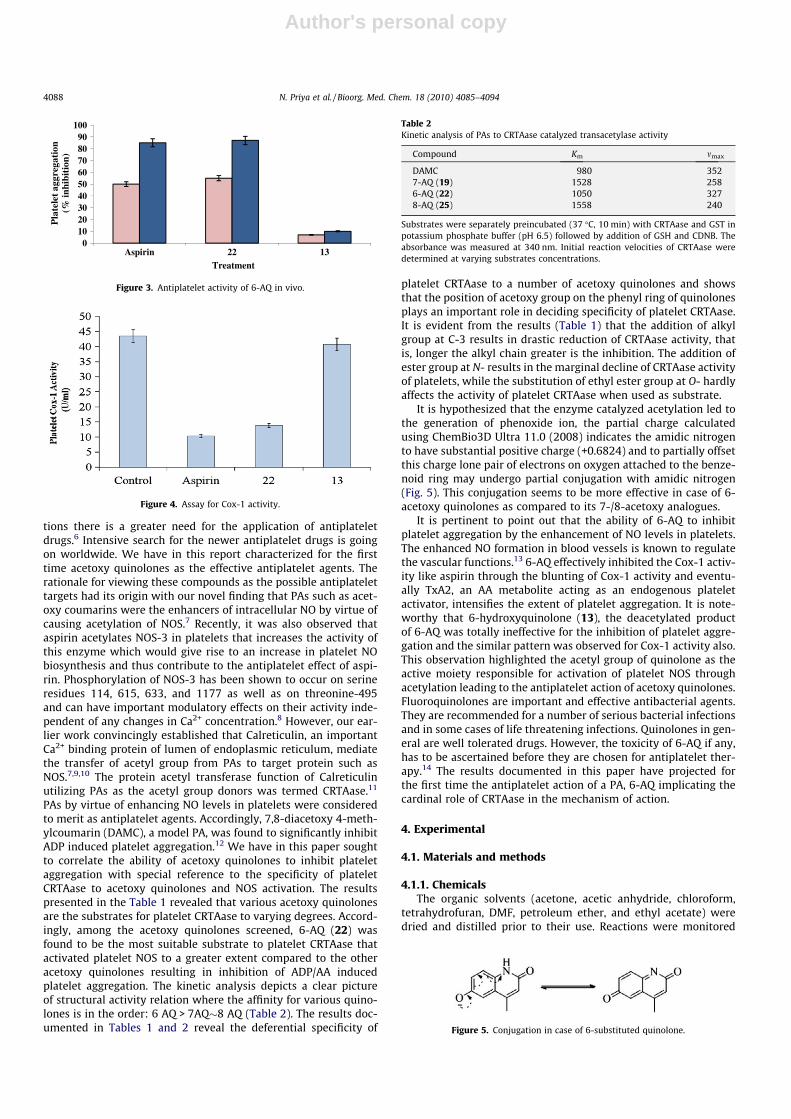
2.2.2.2. Platelet aggregation in vivo. The effect of 6-AQ (22) onthe ADP/AA induced platelet aggregation in vivo is shown in Fig-ure 3. Rats were treated with 6-AQ p.o., sacrificed after 24 h andthe platelet aggregation phenomenon was assessed by aggregom-etry. 6-AQ was also found to effectively inhibit both ADP as wellas AA induced platelet aggregation in vivo. Like aspirin, 6-AQ hasalso effectively inhibited AA induced platelet aggregation muchmore than induced by ADP. Accordingly, 6-AQ exhibited greaterantiplatelet activity in rats while the 6-hydroxy derivative (13)had no ability to inhibit platelet aggregation.
2.3. Cox-1 activity assay
The effect of 6-AQ (22) on the Cox-1 activity in vivo is shown inFigure 4. Rats were treated with 6-AQ p.o., sacrificed after 24 h andthe Cox-1 activity was measured by ELISA. 6-AQ was found toeffectively inhibit Cox-1 activity like aspirin by approximately3.5-fold. Accordingly, 6-AQ exhibited greater inhibition of Cox-1activity in rats while the 6-hydroxy derivative (13) could not.
3. Discussion
Cardiovascular diseases such as myocardial infarction, unstableangina, and deep vein thrombosis greatly contribute to the mortal-ity in the developed world. For the treatment of such heart condi-
Table 1Assay of CRTAase activity of platelets using acetoxy quinolones as the acetyl groupdonor
Acetoxy quinolones CRTAase activity (units)
19 13.25 ± 0.08520 13.0 ± 0.07321 2.0 ± 0.05228 10.0 ± 0.04430 13.2 ± 0.08122 31.0 ± 0.09523 26.0 ± 0.07124 7.0 ± 0.06713 Nil29 23.0 ± 0.07731 28.6 ± 0.26825 12 ± 0.05226 11.5 ± 0.04527 Nil33 Nil
CRTAase activity was assayed in platelet lysate as described in Section 4.1. The unitof CRTAase was expressed in terms of % inhibition of GST under the experimentalconditions. Values are mean ± SEM of five observations (p <0.01).
Figure 1. Influence of acetoxy quinolones on NO levels in washed platelets.
Figure 2. Screening of acetoxy quinolones for antiplatelet activity in vitro.
N. Priya et al. / Bioorg. Med. Chem. 18 (2010) 4085–4094 4087
Author's personal copy
tions there is a greater need for the application of antiplateletdrugs.6 Intensive search for the newer antiplatelet drugs is goingon worldwide. We have in this report characterized for the firsttime acetoxy quinolones as the effective antiplatelet agents. Therationale for viewing these compounds as the possible antiplatelettargets had its origin with our novel finding that PAs such as acet-oxy coumarins were the enhancers of intracellular NO by virtue ofcausing acetylation of NOS.7 Recently, it was also observed thataspirin acetylates NOS-3 in platelets that increases the activity ofthis enzyme which would give rise to an increase in platelet NObiosynthesis and thus contribute to the antiplatelet effect of aspi-rin. Phosphorylation of NOS-3 has been shown to occur on serineresidues 114, 615, 633, and 1177 as well as on threonine-495and can have important modulatory effects on their activity inde-pendent of any changes in Ca2+ concentration.8 However, our ear-lier work convincingly established that Calreticulin, an importantCa2+ binding protein of lumen of endoplasmic reticulum, mediatethe transfer of acetyl group from PAs to target protein such asNOS.7,9,10 The protein acetyl transferase function of Calreticulinutilizing PAs as the acetyl group donors was termed CRTAase.11
PAs by virtue of enhancing NO levels in platelets were consideredto merit as antiplatelet agents. Accordingly, 7,8-diacetoxy 4-meth-ylcoumarin (DAMC), a model PA, was found to significantly inhibitADP induced platelet aggregation.12 We have in this paper soughtto correlate the ability of acetoxy quinolones to inhibit plateletaggregation with special reference to the specificity of plateletCRTAase to acetoxy quinolones and NOS activation. The resultspresented in the Table 1 revealed that various acetoxy quinolonesare the substrates for platelet CRTAase to varying degrees. Accord-ingly, among the acetoxy quinolones screened, 6-AQ (22) wasfound to be the most suitable substrate to platelet CRTAase thatactivated platelet NOS to a greater extent compared to the otheracetoxy quinolones resulting in inhibition of ADP/AA inducedplatelet aggregation. The kinetic analysis depicts a clear pictureof structural activity relation where the affinity for various quino-lones is in the order: 6 AQ > 7AQ�8 AQ (Table 2). The results doc-umented in Tables 1 and 2 reveal the deferential specificity of
platelet CRTAase to a number of acetoxy quinolones and showsthat the position of acetoxy group on the phenyl ring of quinolonesplays an important role in deciding specificity of platelet CRTAase.It is evident from the results (Table 1) that the addition of alkylgroup at C-3 results in drastic reduction of CRTAase activity, thatis, longer the alkyl chain greater is the inhibition. The addition ofester group at N- results in the marginal decline of CRTAase activityof platelets, while the substitution of ethyl ester group at O- hardlyaffects the activity of platelet CRTAase when used as substrate.
It is hypothesized that the enzyme catalyzed acetylation led tothe generation of phenoxide ion, the partial charge calculatedusing ChemBio3D Ultra 11.0 (2008) indicates the amidic nitrogento have substantial positive charge (+0.6824) and to partially offsetthis charge lone pair of electrons on oxygen attached to the benze-noid ring may undergo partial conjugation with amidic nitrogen(Fig. 5). This conjugation seems to be more effective in case of 6-acetoxy quinolones as compared to its 7-/8-acetoxy analogues.
It is pertinent to point out that the ability of 6-AQ to inhibitplatelet aggregation by the enhancement of NO levels in platelets.The enhanced NO formation in blood vessels is known to regulatethe vascular functions.13 6-AQ effectively inhibited the Cox-1 activ-ity like aspirin through the blunting of Cox-1 activity and eventu-ally TxA2, an AA metabolite acting as an endogenous plateletactivator, intensifies the extent of platelet aggregation. It is note-worthy that 6-hydroxyquinolone (13), the deacetylated productof 6-AQ was totally ineffective for the inhibition of platelet aggre-gation and the similar pattern was observed for Cox-1 activity also.This observation highlighted the acetyl group of quinolone as theactive moiety responsible for activation of platelet NOS throughacetylation leading to the antiplatelet action of acetoxy quinolones.Fluoroquinolones are important and effective antibacterial agents.They are recommended for a number of serious bacterial infectionsand in some cases of life threatening infections. Quinolones in gen-eral are well tolerated drugs. However, the toxicity of 6-AQ if any,has to be ascertained before they are chosen for antiplatelet ther-apy.14 The results documented in this paper have projected forthe first time the antiplatelet action of a PA, 6-AQ implicating thecardinal role of CRTAase in the mechanism of action.
4. Experimental
4.1. Materials and methods
4.1.1. ChemicalsThe organic solvents (acetone, acetic anhydride, chloroform,
tetrahydrofuran, DMF, petroleum ether, and ethyl acetate) weredried and distilled prior to their use. Reactions were monitored
0102030405060708090
100
Aspirin 22 13
Pla
tele
t agg
rega
tion
(% in
hibi
tion
)
Treatment
Figure 3. Antiplatelet activity of 6-AQ in vivo.
Figure 4. Assay for Cox-1 activity.
Table 2Kinetic analysis of PAs to CRTAase catalyzed transacetylase activity
Compound Km mmax
DAMC 980 3527-AQ (19) 1528 2586-AQ (22) 1050 3278-AQ (25) 1558 240
Substrates were separately preincubated (37 �C, 10 min) with CRTAase and GST inpotassium phosphate buffer (pH 6.5) followed by addition of GSH and CDNB. Theabsorbance was measured at 340 nm. Initial reaction velocities of CRTAase weredetermined at varying substrates concentrations.
Figure 5. Conjugation in case of 6-substituted quinolone.
4088 N. Priya et al. / Bioorg. Med. Chem. 18 (2010) 4085–4094

Author's personal copy
by precoated TLC plates (Merck Silica Gel 60F254); the spots werevisualized either by UV light, or by spraying with 5% alcoholic FeCl3
solution. Silica gel (100–200 mesh) was used for column chroma-tography. Sodium hydride (60% dispersed in mineral oil) was sup-plied from Spectrochem. Pvt. Ltd, India. Melting points wererecorded in capillaries in sulfuric acid bath and are uncorrected.Infrared spectra were recorded on Perkin–Elmer FT-IR modelBXspectrophotometer. The 1H NMR and 13C NMR spectra were re-corded on Bruker AC-400 (400 MHz, 100 MHz) NMR spectrometerand Avance-300 spectrometer using TMS as internal standard. Thechemical shift values are on d scale and the coupling constant val-ues (J) are in Hertz. The EI/HR mass spectra were recorded on Agi-lent-6210 ES-TOF. Ethyl 2-ethyl-3-oxobutanoate, reducedglutathione (GSH), 1-chloro-2,4-dinitrobenzene (CDNB), dichloro-fluorescin diacetate (DCFH-DA), L-arginine, adenosine diphosphate(ADP), were obtained from M/S Sigma Chemical Co. St. Louis. Mo.USA. All other chemicals used were of high purity and were ob-tained from local suppliers.
4.1.2. General procedure for the synthesis of 3-alkyl-methoxy-4-methylquinolin-2-ones (1–9)
Anisidine (20 g, 164 mmol) was added drop wise to alkylatedethyl acetoacetate15 (277 mmol) and the reaction mixture wasrefluxed for 20 h. After the completion of reaction, the contentswere cooled and then poured on sodium carbonate solution. Thecompound was extracted with chloroform and the solvent wasevaporated in vacuo. 70% Sulfuric acid (40 mL) was added andthe reaction mixture was stirred at 95 �C. The progress of reactionwas monitored on TLC (5% methanol–chloroform). On completionof the reaction, the solution was cooled and poured on crushedice (500 g). The resulting precipitate was filtered and washed withwater and petroleum ether. The crude product was recrystallizedfrom ethanol to give methoxy quinolin-2-ones 1–9.16
4.1.2.1. 7-Methoxy-4-methylquinolin-2(1H)-one (1). It was ob-tained as white solid (83%); mp: 195 �C, (Literature va-lue = 196 �C);17 1H NMR (acetone-d6, 300 MHz): d 2.45 (s, 3H, C-4CH3), 3.88 (s, 3H, OCH3), 6.30 (s, 1H, H-3), 6.84 (d, 1H, J = 8.7 Hz,H-6), 6.94 (s, 1H, H-8), 7.66 (d, 1H, J = 8.9 Hz, H-5), 11.00 (br s,1H, NH); 13C NMR (DMSO-d6, 75 MHz): d 19.36 (C-4 CH3), 56.14(OCH3), 99.12, 111.12 (C-6 and C-8), 114.68 (C-3), 118.56 (C-10),127.02 (C-5), 141.26 (C-9), 148.85 (C-4), 161.80 (C-7), 162.89 (C-2); IR (KBr) mmax: 2957.41, 1658.97, 1629.05, 1549.71, 1474.57,1417.53, 1261.70, 1217.14, 1177.11, 1023.59, 856.63, 808.71,710.74 cm�1; UV (methanol) kmax: 323 and 337 nm; HRMS:C11H11O2N [M]+: 189.9665.
4.1.2.2. 3-Ethyl-7-methoxy-4-methylquinolin-2(1H)-one (2). Itwas obtained as yellow solid (65%); mp: 126 �C; 1H NMR (CDCl3,300 MHz): d 1.17 (t, 3H, J = 7.4 Hz, CH2CH3), 2.46 (s, 3H, C-4 CH3),2.81 (q, 2H, J = 7.5 Hz, CH2CH3), 3.89 (s, 3H, –OCH3), 6.78 (s, 1H,H-8), 6.81 (d, 1H, J = 9.2 Hz, H-6), 7.59 (d, 1H, J = 8.7 Hz, H-5),11.95 (br s, 1H, NH); 13C NMR (CDCl3, 75 MHz): d 13.50 (CH2CH3),14.84 (C-4 CH3), 23.74 (CH2CH3), 55.51 (OCH3), 107.09, 119.39 (C-6and C-8), 122.56 (C-10), 124.94 (C-3), 131.05 (C-5), 143.97 (C-9),147.56 (C-4), 152.31 (C-7), 160.38 (C-2); IR (KBr) mmax: 2932.78,1660.95, 1621.90, 1560.80, 1512.79, 1461.85, 1396.94, 1255.88,1223.07, 1179.61, 1141.37, 1029.98, 925.23, 842.09, 763.03 cm�1;UV (methanol) kmax: 324 and 339 nm; HRMS: C13H15O2N [M+H]+:218.3962.
4.1.2.3. 3-Hexyl-7-methoxy-4-methylquinolin-2(1H)-one (3). Itwas obtained as yellow solid (75%); mp: 140 �C; 1H NMR (CDCl3,300 MHz): d 0.91 (br s, 3H, CH2CH3), 1.26–1.55 (m, 8H,CH2(CH2)4CH3), 2.47 (s, 3H, C-4 CH3), 2.76 (t, 2H, J = 7.3 Hz,CH2CH2), 3.89 (s, 3H, OCH3), 6.81–6.86 (m, 2H, H-6 and H-8),
7.60 (d, 1H, J = 8.9 Hz, H-5), 12.29 (br s, 1H, NH); 13C NMR (CDCl3,75 MHz): d 11.80 (CH2CH3), 14.48 (C-4 CH3), 23.09, 27.27, 29.57,30.01, 32.21 ((CH2)5CH3), 55.79 (OCH3), 98.72, 111.94 (C-6 and C-8), 115.71 (C-10), 125.96 (C-3), 129.05 (C-5), 138.92 (C-9), 143.51(C-4), 160.97 (C-7), 164.76 (C-2); IR (KBr) mmax: 1660.22, 1612.15,1562.22, 1514.39, 1462.82, 1398.69, 1260.73, 1225.71, 1173.39,1028.91, 940.95, 927.99, 798.50, 607.85 cm�1; UV (methanol) kmax:325 and 339 nm; HRMS: C17H23O2N [M]+: 273.1893.
4.1.2.4. 6-Methoxy-4-methylquinolin-2(1H)-one (4). It was ob-tained as white solid (75%); mp: 260–262 �C, (Literature va-lue = 260–262 �C);18 1H NMR (DMSO-d6, 300 MHz): d 2.42 (s, 3H,C-4 CH3), 3.82 (s, 3H, OCH3), 6.39 (s, 1H, H-3), 7.13–7.18 (m, 2H,H-5 and H-7), 7.26 (dd, 1H, J = 1.8 and 5.3 Hz, H-8), 11.46 (br s,1H, NH); 13C NMR (DMSO-d6, 75 MHz): d 19.45 (C-4 CH3), 56.37(OCH3), 107.69, 117.52 (C-5 and C-7), 119.87 (C-10), 121.06 (C-8), 122.11 (C-9), 131.38 (C-3), 148.27 (C-4), 154.96 (C-6), 162.03(C-2); IR (KBr) mmax: 3433.06, 2821.76, 1653.70, 1621.98,1503.71, 1421.31, 1275.63, 1240.22, 1202.18, 1179.30, 1044.05,835.98, 628.40 cm�1; UV (methanol) kmax: 269 and 350 nm; HRMS:C11H11O2N [M]+: 189.8188.
4.1.2.5. 3-Ethyl-6-methoxy-4-methylquinolin-2(1H)-one (5). Itwas obtained as white solid (65%); mp: 204–206 �C; 1H NMR(DMSO-d6, 300 MHz): d 1.01 (t, 3H, J = 7.2 Hz, CH2CH3), 2.39 (s,3H, C-4 CH3), 2.63 (q, 2H, J = 7.2 Hz, 2H, CH2CH3), 3.79 (s, 3H,OCH3), 7.06–7.14 (m, 2H, H-5 and H-7), 7.21 (d, 1H, J = 8.7 Hz, H-8), 11.49 (br s, 1H, NH); 13C NMR (DMSO-d6, 75 MHz): d 13.21(CH2CH3), 14.52 (C-4 CH3), 19.71, (CH2CH3), 55.40 (OCH3), 106.83,116.19 (C-5 and C-7), 117.65 (C-8), 120.64 (C-10), 131.45 (C-3),132.90 (C-9), 140.99 (C-4), 154.02 (C-6), 160.85 (C-2); IR (KBr)mmax: 3434.17, 2965.75, 1642.07, 1502.12, 1462.63, 1414.19,1371.42, 1271.48, 1218.23, 1131.27, 1038.23, 927.21, 836.80,633.80 cm�1; UV (methanol) kmax: 272 and 349 nm; HRMS:C13H15O2N [M]+: 217.7774.
4.1.2.6. 3-Hexyl-6-methoxy-4-methylquinolin-2(1H)-one (6). Itwas obtained as white solid (70%); mp: 146–148 �C; 1H NMR(CDCl3, 300 MHz): d 0.91 (br s, 3H, CH2CH3), 1.26–1.57 (m, 8H,CH2(CH2)4CH3), 2.47 (s, 3H, C-4 CH3), 2.82 (t, 2H, J = 7.1 Hz,CH2CH2), 3.87 (s, 3H, OCH3), 7.08–7.11 (m, 2H, H-5 and H-7),7.35 (d, 1H, J = 8.4 Hz, H-8), 12.35 (br s, 1H, NH); 13C NMR (CDCl3,75 MHz): d 14.18 (CH2CH3), 15.21 (C-4 CH3), 22.67, 27.13, 29.08,29.56, 31.81 ((CH2)5CH3), 55.71 (OCH3), 106.57, 117.34 (C-5 andC-7), 117.73 (C-8), 121.72 (C-10), 131.51 (C-3), 132.18 (C-9),142.28 (C-4), 154.83 (C-6), 163.52 (C-2); IR (KBr) mmax: 3148.91,2928.00, 2850.90, 1656.22, 1624.10, 1560.71, 1506.61, 1462.53,1418.38, 1382.73, 1279.60, 1209.01, 1173.66, 1039.55, 855.83.804.15, 725.86, 643.24 cm�1; UV (methanol) kmax: 274 and348 nm; HRMS: C17H23O2N [M]+: 273.4577.
4.1.2.7. 8-Methoxy-4-methylquinolin-2(1H)-one (7). It was ob-tained as white solid (30%); mp: 192–194 �C, (Literature va-lue = 188–190 �C);19 1H NMR (DMSO-d6, 300 MHz): d 2.41 (s, 3H,C-4 CH3), 3.89 (s, 3H, OCH3), 6.42 (s, 1H, H-3), 7.14–7.15 (m, 2H,H-5 and H-7), 7.27–7.30 (m, 1H, H-6), 10.58 (br s, 1H, NH); 13CNMR (DMSO-d6, 75 MHz): d 18.74 (C-4 CH3), 56.07 (OCH3),110.89, 116.36 (C-5 and C-7), 120.08 (C-9), 121.36, 121.57 (C-3and C-6), 128.57 (C-10), 145.80 (C-4), 148.15 (C-8), 161.14 (C-2);IR (KBr) mmax: 3163.95, 2933.15, 1648.26, 1605.82, 1462.84,1389.90, 1265.97, 1154.88, 1049.33, 860.83, 791.04, 739.93,726.00, 636.77 cm�1; UV (methanol) kmax: 278 and 335 nm; HRMS:C11H11O2N [M]+: 189.7739.
N. Priya et al. / Bioorg. Med. Chem. 18 (2010) 4085–4094 4089

Author's personal copy
4.1.2.8. 3-Ethyl-8-methoxy-4-methylquinolin-2(1H)-one (8). Itwas obtained as white solid (20%); mp: 186–188 �C; 1H NMR(DMSO-d6, 300 MHz): d 1.03 (t, 3H, J = 7.5 Hz, CH2CH3), 2.41 (s,3H, C-4 CH3), 2.65 (q, 2H, J = 7.5 Hz, CH2CH3), 3.89 (s, 3H, OCH3),7.07–7.16 (m, 2H, H-5 and H-6), 7.32 (dd, 1H, J = 2.1 and 7.5 Hz,H-7), 10.48 (br s, 1H, NH); 13C NMR (DMSO-d6, 75 MHz): d 13.12(CH2CH3), 14.69 (C-4 CH3), 19.69 (CH2CH3), 56.02 (OCH3), 109.83,116.28 (C-5 and C-7), 120.47 (C-3), 121.33 (C-6), 126.93 (C-9),132.98 (C-10), 141.66 (C-4), 145.47 (C-8), 160.75 (C-2); IR (KBr)mmax: 3148.45, 2927.25, 1638.11, 1485.25, 1390.38, 1259.63,1214.22, 1051.94, 866.49, 766.44, 727.12, 677.02 cm�1; UV (meth-anol) kmax: 256, 281 and 333 nm; HRMS: C13H15O2N [M]+:217.1143.
4.1.2.9. 3-Hexyl-8-methoxy-4-methylquinolin-2(1H)-one (9). Itwas obtained as white solid (25%); mp: 112–114 �C; 1H NMR(CDCl3, 300 MHz): d 0.89 (br s, 3H, CH2CH3), 1.31–1.52 (m, 8H,CH2(CH2)4CH3), 2.46 (s, 3H, C-4 CH3), 2.74 (t, 2H, J = 7.2 Hz,CH2CH2), 3.96 (s, 3H, OCH3), 6.93 (d, 1H, J = 7.8 Hz, H-5), 7.12 (t,1H, J = 8.1 Hz, H-6), 7.29 (d, 1H, J = 8.4 Hz, H-7), 9.10 (br s, 1H,NH); 13C NMR (DMSO, 75 MHz): d 14.14 (CH2CH3), 15.36 (C-4CH3), 22.70, 27.22, 29.00, 29.60, 31.84 ((CH2)5CH3), 55.98 (OCH3),108.75, 116.36 (C-5 and C-7), 121.37 (C-3), 121.46 (C-6), 126.84(C-9), 132.73 (C-10), 142.63 (C-4), 145.39 (C-8), 161.64 (C-2); IR(KBr) mmax: 2924.14, 1654.17, 1647.83, 1459.82, 1271.95,1217.55, 1051.29, 862.42, 765.87, 727.19, 618.33 cm�1; UV (meth-anol) kmax: 281 and 334 nm; HRMS: C17H23O2N [M]+: 273.2189.
4.1.3. General procedure for the synthesis of 3-alkyl-hydroxy-4-methylquinolin-2-ones (10–18)
A mixture of 10 mL hydrobromic acid and acetic acid (7:3) wasadded to 1 g of 3-alkyl-methoxy-4-methyl-1H-quinolin-2-ones (1–9). The reaction mixture was refluxed for 75 h and then poured oncrushed ice. The resulting precipitate was filtered and washed withwater to yield 3-alkyl-7-hydroxyquinolin-2-ones (10–18).20
4.1.3.1. 7-Hydroxy-4-methylquinolin-2(1H)-one (10). It was ob-tained as white solid (85%); mp: >300 �C, (Literature value = 306–307 �C);21 1H NMR (DMSO-d6, 500 MHz): d 2.28 (s, 3H, C-4 CH3),6.08 (s, 1H, H-3), 6.59 (d, 1H, J = 8.8 Hz, H-6), 6.65 (d, 1H,J = 2.4 Hz, H-8), 7.45 (d, 1H, J = 8.8 Hz, H-5), 10.02 (br s, 1H, OH),11.33 (br s, 1H, NH); 13C NMR (DMSO-d6, 125 MHz): d 19.03 (C-4CH3), 100.61, 111.80 (C-6 and C-8), 113.32 (C-3), 117.50 (C-10),126.68 (C-5), 141.04 (C-9), 148.44 (C-4), 159.95 (C-7), 162.69 (C-2); IR (KBr) mmax: 3425.24, 2927.52, 2364.39, 1652.89, 1541.56(amide-II), 1474.84, 1406.35, 1255.85, 1220.96, 1074.07, 905.44,817.60, 689.94 cm�1; UV (methanol) kmax: 324 and 337 nm; HRMS:C10H9O2N [M+H]+: 176.2008.
4.1.3.2. 3-Ethyl-7-hydroxy-4-methylquinolin-2(1H)-one (11). Itwas obtained as yellow solid (70%); mp: 250–252 �C; 1H NMR(CDCl3, 300 MHz): d 1.01 (t, 3H, J = 7.3 Hz, CH2CH3), 2.35 (s, 3H,C-4 CH3), 2.59 (q, 2H, J = 7.2 Hz, CH2CH3), 6.56 (d, 1H, J = 7.6 Hz,H-6), 6.63 (s, 1H, H-8), 7.53 (d, 1H, J = 8.7 Hz, H-5), 9.87 (br s, 1H,OH), 11.34 (br s, 1H, NH); 13C NMR (CDCl3, 75 MHz): d 13.89(CH2CH3), 14.82 (C-4 CH3), 19.82 (CH2CH3), 100.21, 111.56 (C-6and C-8), 113.67 (C-10), 126.39 (C-3), 128.92 (C-5), 139.23 (C-9),142.07 (C-4), 158.85 (C-7), 162.30 (C-2); IR (KBr) mmax: 3291.86,3229.49, 2966.43, 1629.42, 1573.05, 1514.05, 1417.29, 1319.53,1256.84, 1191.89, 1066.27, 972.79, 862.40, 791.89, 690.01,657.09 cm�1; UV (methanol) kmax: 325 and 339 nm; HRMS:C12H13O2N [M+H]+: 204.3963.
4.1.3.3. 3-Hexyl-7-hydroxy-4-methylquinolin-2(1H)-one (12). Itwas obtained as yellow solid (75%); mp: 202 �C; 1H NMR (DMSO-d6, 500 MHz): d 0.85–0.87 (m, 3H, CH2CH3), 1.23–1.38 (m, 8H,
CH2(CH2)4CH3), 2.34 (s, 3H, C-4 CH3), 2.50–2.56 (m, 2H, CH2CH2),6.60–6.67 (m, 2H, H-6 and H-8), 7.51–7.54 (m, 1H, H-5), 9.86 (brs, 1H, OH), 11.35 (br s, 1H, NH); 13C NMR (CDCl3, 125 MHz): d14.82 (CH2CH3), 15.54 (C-4 CH3), 22.98, 27.02, 29.53, 29.78, 32.09((CH2)5CH3), 100.61, 111.93 (C-6 and C-8), 114.10 (C-10), 126.78(C-3), 128.13 (C-5), 139.64 (C-9), 142.67 (C-4), 159.22 (C-7),162.86 (C-2); IR (KBr) mmax: 3218.05, 2955.52, 2923.83, 1620.25,1564.06, 1510.64, 1405.93, 1325.26, 1257.31, 1191.41, 1109.40,807.53, 721.67, 692.46 cm�1; UV (methanol) kmax: 326 and340 nm; HRMS: C16H21O2N [M]+: 260.0510.
4.1.3.4. 6-Hydroxy-4-methylquinolin-2(1H)-one (13). It was ob-tained as white solid (80%); mp: 324–326 �C, (Literature va-lue = 326–330 �C);22 1H NMR (DMSO-d6, 300 MHz): d 2.34 (s, 3H,C-4 CH3), 6.36 (s, 1H, H-3), 7.00 (br s, 2H, H-5 and H-7), 7.17 (brs, 1H, H-8), 9.40 (br s, 1H, OH), 11.42 (br s, 1H, NH); 13C NMR(DMSO-d6, 75 MHz): d 19.36 (C-4 CH3), 109.41, 117.43 (C-5 andC-7), 120.39 (C-10), 121.09 (C-8), 121.80 (C-9), 132.74 (C-3),148.01 (C-4), 152.93 (C-6), 161.99 (C-2); IR (KBr) mmax: 3489.59,3187.46, 1661.73, 1634.87, 1608.22, 1513.42, 1433.01, 1294.34,1191.88, 866.12, 853.10, 810.02, 642.28 cm�1; UV (methanol) kmax:269 and 352 nm; HR MS: C10H9O2N [M]+: 175.7805.
4.1.3.5. 3-Ethyl-6-hydroxy-4-methylquinolin-2(1H)-one (14). Itwas obtained as white solid (75%); mp: 262–264 �C; 1H NMR(DMSO-d6, 300 MHz): d 0.98 (t, 3H, J = 7.2 Hz, CH2CH3), d 2.31 (s,3H, C-4 CH3), 2.59 (q, 2H, J = 7.05 Hz, CH2CH3), 6.91 (d, 1H,J = 8.4 Hz, H-7), 7.01 (d, 1H, J = 1.8 Hz, H-5), 7.10 (d, 1H,J = 8.7 Hz, H-8), 9.32 (br s, 1H, OH), 11.39 (br s, 1H, NH); 13CNMR (DMSO-d6, 75 MHz): d 13.24 (CH2CH3), 14.42 (C-4 CH3),19.68 (CH2CH3), 108.49, 116.05 (C-5 and C-7), 118.24 (C-8),120.91 (C-10), 130.33 (C-9), 132.60 (C-3), 140.63 (C-4), 151.92(C-6), 160.78 (C-2); IR (KBr) mmax: 3231.81, 2973.40, 1641.50,1620.48, 1505.72, 1480.33, 1431.71, 1392.90, 1319.33, 1279.99,1207.53, 1170.02, 939.73, 880.82, 859.94, 700.63, 642.18 cm�1;UV (methanol) kmax: 274 and 350 nm; HRMS: C12H13O2N [M]+:203.7640.
4.1.3.6. 3-Hexyl-6-hydroxy-4-methylquinolin-2(1H)-one (15). Itwas obtained as white solid (80%); mp: 221–223 �C; 1H NMR(DMSO-d6, 300 MHz): d 0.80 (br s, 3H, CH2CH3), 1.23 (br s, 8H,CH2(CH2)4CH3), 2.27 (s, 3H, C-4 CH3), 2.55 (br s, 2H, CH2CH2),6.88 (d, 1H, J = 8.1 Hz, H-7), 6.97 (s, 1H, H-5), 7.07 (d, 1H,J = 8.7 Hz, H-8), 9.24 (br s, 1H, OH), 11.34 (br s, 1H, NH); 13CNMR (DMSO-d6, 75 MHz): d 13.89 (CH2CH3), 14.70 (C-4 CH3),22.06, 26.45, 28.51, 28.89, 31.17 ((CH2)5CH3), 108.48, 116.04 (C-5and C-7), 118.21 (C-8), 120.91 (C-10), 130.32 (C-9), 131.38 (C-3),140.86 (C-4), 151.92 (C-6), 160.95 (C-2); IR (KBr) mmax: 3565.89,3143.50, 2952.85, 1647.12, 1624.99, 1504.90, 1422.73, 1317.46,1288.39, 1208.76, 1165.02, 937.98, 826.24, 645.45 cm�1; UV(methanol) kmax: 274 and 350 nm; HRMS: C16H21O2N [M]+:259.7355.
4.1.3.7. 8-Hydroxy-4-methylquinolin-2(1H)-one (16). It was ob-tained as white solid (70%); mp: 250–254 �C, (Literaturevalue = 248 �C);23 1H NMR (DMSO-d6, 300 MHz): d 2.37 (s, 3H, C-4 CH3), 6.34 (s, 1H, H-3), 6.89–6.98 (m, 2H, H-5 and H-6), 7.10(d, 1H, J = 7.8 Hz, H-7), 10.25 (br s, 2H, NH and OH); 13C NMR(DMSO-d6, 75 MHz): d 18.70 (C-4 CH3), 114.36, 114.96 (C-5 andC-7), 120.48 (C-9), 121.04, 121.60 (C-3 and C-6), 127.84 (C-10),143.67 (C-4), 148.19 (C-8), 160.91 (C-2); IR (KBr) mmax: 3401.25,1639.37, 1601.19, 1553.68, 1478.15, 1398.60, 1288.58, 1208.23,1145.95, 924.47, 861.73, 792.88, 735.68, 600.31 cm�1; UV(methanol) kmax: 254, 279 and 336 nm; HRMS: C10H9O2N [M]+:175.9968.
4090 N. Priya et al. / Bioorg. Med. Chem. 18 (2010) 4085–4094

Author's personal copy
4.1.3.8. 3-Ethyl-8-hydroxy-4-methylquinolin-2(1H)-one (17). Itwas obtained as white solid (65%); mp: 212–214 �C; 1H NMR(DMSO-d6, 300 MHz): d 0.94 (t, 3H, J = 7.2 Hz, CH2CH3), 2.31 (s,3H, C-4 CH3), 2.56 (q, 2H, J = 7.2 Hz, CH2CH3), 6.83 (d, 1H,J = 7.5 Hz, H-5), 6.89–6.94 (m, 1H, H-6), 7.11 (d, 1H, J = 8.1 Hz, H-7), 10.11 (br s, 2H, NH and OH); 13C NMR (DMSO-d6, 75 MHz): d13.19 (CH2CH3), 14.68 (C-4 CH3), 19.71 (CH2CH3), 113.31, 114.90(C-5 and C-7), 120.94 (C-3), 121.47 (C-6), 126.24 (C-9), 132.63(C-10), 141.84 (C-4), 143.31 (C-8), 160.62 (C-2); IR (KBr) mmax:3178.10, 2970.45, 1924.13, 1654.16, 1606.43, 1560.17, 1606.43,1462.64, 1397.96, 1290.56, 1205.28, 1011.15, 831.96, 768.59,732.46, 680.38, 639.97 cm�1; UV (methanol) kmax: 257, 282 and334 nm; HRMS: C12H13O2N [M]+: 203.6310.
4.1.3.9. 3-Hexyl-8-hydroxy-4-methylquinolin-2(1H)-one (18). Itwas obtained as white solid (65%); mp: 174–176 �C; 1H NMR(CDCl3, 300 MHz): d 0.88–0.90 (m, 3H, CH2CH3), 1.31–1.57 (m,8H, CH2(CH2)4CH3), 2.51 (s, 3H, C-4 CH3), 2.80 (t, 2H, J = 6.9 Hz,CH2CH2), 7.09–7.26 (m, 3H, H-5, H-6 and H-7), 10.52 (br s, 2H,OH and NH); 13C NMR (CDCl3, 75 MHz): d 14.14 (CH2CH3), 15.50(C-4 CH3), 22.68, 27.14, 29.06, 29.58, 31.78 ((CH2)5CH3), 114.68,115.18 (C-5 and C-7), 122.20 (C-3), 122.98 (C-6), 125.76 (C-9),130.98 (C-10), 144.03 (C-4), 145.18 (C-8), 162.80 (C-2); IR (KBr)mmax: 3377.54, 1638.80, 1624.56, 1600.44, 1552.36, 1394.62,1278.70, 1229.22, 1200.55, 773.31, 710.18, 624.22 cm�1; UV(methanol) kmax: 255, 283 and 333 nm; HRMS: C16H21O2N [M]+:259.4653.
4.1.4. General procedure for the synthesis of 3-alkyl-4-methyl-2-oxo-1,2-dihydroquinolin-yl acetate (19–27)
A solution of 12 mL acetic anhydride and acetic acid (1:4) wasadded to 1 g of 3-alkyl-hydroxy-4-methylquinolin-2(1H)-ones10–18. The reaction mixture was refluxed for 6 h and then pouredon crushed ice.24 The resulting precipitate was filtered and washedwith water to yield acetoxy-3-alkylquinolin-2-ones 19–27.
4.1.4.1. 4-Methyl-2-oxo-1,2-dihydroquinolin-7-yl acetate(19). It was obtained as white solid (90%); mp: 258 �C, (Literature
value = 257–258 �C);21 1H NMR (DMSO-d6, 500 MHz): d 2.30 (s, 3H,C-4 CH3), 2.42 (s, 3H, OCOCH3), 6.39 (s, 1H, H-3), 6.98 (dd, 1H,J = 2.4 and J = 6.2 Hz, H-6), 7.05 (s, 1H, H-8), 7.73 (d, 1H,J = 6.1 Hz, H-5), 11.67 (br s, 1H, NH); 13C NMR (DMSO-d6,125 MHz): d 19.38 (C-4 CH3), 21.76 (OCOCH3), 108.77, 116.71 (C-6 and C-8), 118.36 (C-3), 121.21 (C-10), 126.98 (C-5), 140.43 (C-9), 148.52 (C-4), 152.59 (C-7), 162.63 (C-2), 169.82 (–OCOCH3);IR (KBr) mmax: 2927.12, 2855.27, 1751.47, 1678.55, 1561.24,1510.88, 1458.53, 1362.63, 1232.32, 116 6.29, 1025.06, 906.26,856.71, 645.56 cm�1; UV (methanol) kmax: 323 and 335 nm; HRMS:C12H11O3N [M]+: 217.2915.
4.1.4.2. 3-Ethyl-4-methyl-2-oxo-1,2-dihydroquinolin-7-yl ace-tate (20). It was obtained as yellow solid (75%); mp: 176–178 �C;1H NMR (CDCl3, 300 MHz): d 1.20 (t, 3H, J = 7.2 Hz, CH2CH3), 2.36(s, 3H, C-4 CH3), 2.50 (s, 3H, OCOCH3), 2.84 (q, 2H, J = 7.3 Hz,CH2CH3), 6.84 (d, 1H, J = 7.8 Hz, H-6), 7.14 (s, 1H, H-8), 7.70 (d,1H, J = 8.7 Hz, H-5), 12.12 (br s, 1H, NH); 13C NMR (CDCl3,75 MHz): d 13.32 (CH2CH3), 14.83 (C-4 CH3), 20.09 (OCOCH3),21.09 (CH2CH3), 108.49, 116.18 (C-6 and C-8), 118.98 (C-10),125.37 (C-3), 132.41 (C-5), 137.59 (C-9), 142.44 (C-4), 151.08 (C-7), 163.89 (C-2), 169.21 (–OCOCH3); IR (KBr) mmax: 3448.86,2931.73, 2851.67, 1770.34, 1658.08, 1562.63, 1510.84, 1370.03,1207.92, 1157.16, 1012.24, 911.90 cm�1; UV (methanol) kmax:323 and 336 nm; HRMS: C14H15O3N [M+H]+: 246.7908.
4.1.4.3. 3-Hexyl-4-methyl-2-oxo-1,2-dihydroquinolin-7-yl ace-tate (21). It was obtained as yellow solid (80%); mp: 134–136 �C;
1H NMR (DMSO-d6, 500 MHz): d 0.93 (br s, 3H, CH2CH3), 1.38–1.58 (m, 8H, CH2(CH2)4CH3), 2.35 (s, 3H, C-4 CH3), 2.49 (s, 3H,OCOCH3), 2.81 (br s, 2H, CH2CH2), 6.98 (d, 1H, J = 8.2 Hz, H-5),7.14 (s, 1H, H-8), 7.70 (d, 1H, J = 8.4 Hz, H-6), 12.25 (br s, 1H,NH); 13C NMR (CDCl3, 125 MHz): d 14.14 (CH2CH3), 15.23 (C-4CH3), 21.14 (OCOCH3), 22.65, 26.92, 28.99, 29.48, 31.73((CH2)5CH3), 108.42, 116.25 (C-6 and C-8), 119.09 (C-10), 125.53(C-3), 131.45 (C-5), 137.59 (C-9), 142.68 (C-4), 151.11 (C-7),164.01 (C-2), 169.24 (OCOCH3); IR (KBr) mmax: 3449.06, 2927.86(C-H str), 2855.18, 2367.37, 1765.40, 1656.22, 1564.92, 1511.10,1459.84, 1370.74, 1223.68, 1014.71, 924.77 cm�1; UV (methanol)kmax: 323 and 368 nm; HRMS: C18H23O3N [M]+: 301.9141.
4.1.4.4. 4-Methyl-2-oxo-1,2-dihydroquinolin-6-yl acetate25 (22). Itwas obtained as white solid (85%); mp: 270 �C; 1H NMR (DMSO-d6, 300 MHz): d 2.30 (s, 3H, C-4 CH3), 2.40 (s, 3H, OCOCH3), 6.46(s, 1H, H-3), 7.29–7.35 (m, 2H, H-5 and H-7), 7.47 (dd, 1H, J = 1.5and 7.5 Hz, H-8), 11.71 (br s, 1H, NH); 13C NMR (DMSO-d6,75 MHz): d 18.39 (C-4 CH3), 20.76 (OCOCH3), 116.27, 117.06 (C-5and C-7), 120.01 (C-10), 121.47 (C-8), 124.63 (C-3), 136.38 (C-9),144.72 (C-4), 147.43 (C-6), 161.45 (C-2), 169.53 (OCOCH3); IR(KBr) mmax: 3433.37, 2835.40, 1751.37, 1656.34, 1559.57,1502.72, 1425.27, 1373.09, 1212.40, 1166.23, 1135.37, 1012.90,905.94, 871.27, 842.91, 683.29 cm�1; UV (methanol) kmax: 264and 334 nm; HRMS: C12H11O3N [M+H]+: 218.4582.
4.1.4.5. 3-Ethyl-4-methyl-2-oxo-1,2-dihydroquinolin-6-yl ace-tate (23). It was obtained as white crystals (80%); mp: 258–260 �C; 1H NMR (DMSO-d6, 300 MHz): d 0.99 (t, 3H, J = 7.2 Hz,CH2CH3), 2.26 (s, 3H, C-4 CH3), 2.35 (s, 3H, OCOCH3), 2.62 (q, 2H,J = 7.2 Hz, CH2CH3), 7.19–7.29 (m, 2H, H-5 and H-7), 7.44 (s, 1H,H-8), 11.67 (br s, 1H, NH); 13C NMR (DMSO-d6, 75 MHz): d 13.10(CH2CH3), 14.45 (C-4 CH3), 19.68 (CH2CH3), 20.78 (OCOCH3),115.76, 116.95 (C-5 and C-7), 120.50 (C-10), 123.36 (C-8), 133.32(C-9), 134.89 (C-3), 141.59 (C-4), 144.60 (C-6), 161.15 (C-2),170.04 (OCOCH3); IR (KBr) mmax: 2934.63, 1752.29, 1644.31,1500.24, 1458.60, 1420.02, 1386.26, 1371.75, 1258.49, 1221.17,1182.79, 1046.76, 1020.62, 953.84, 897.95, 705.93, 681.39 cm�1;UV (methanol) kmax: 269 and 332 nm; HRMS: C14H15O3N [M+H]+:246.5055.
4.1.4.6. 3-Hexyl-4-methyl-2-oxo-1,2-dihydroquinolin-6-yl ace-tate (24). It was obtained as white crystals (80%); mp: 158–160 �C; 1H NMR (CDCl3, 300 MHz): d 0.90 (t, 3H, J = 6.8 Hz,CH2CH3),1.34–1.57 (m, 8H, CH2(CH2)4CH3), 2.34 (s, 3H, C-4 CH3),2.45 (s, 3H, OCOCH3), 2.80 (t, 2H, J = 7.5 Hz, CH2CH2), 7.18 (dd,1H, J = 2.4 and 8.7 Hz, H-7), 7.39–7.43 (m, 2H, H-5 and H-8)12.46 (br s, 1H, NH). 13C NMR (DMSO-d6, 75 MHz): d 14.15(CH2CH3), 15.15 (C-4 CH3), 21.11 (OCOCH3), 22.65, 27.05, 28.95,29.51, 31.76 ((CH2)5CH3), 116.53, 117.04 (C-5 and C-7), 121.63(C-10), 123.06 (C-8), 132.62 (C-3), 134.70 (C-9), 142.45 (C-4),145.38 (C-6), 163.83 (C-2), 169.90 (OCOCH3); IR (KBr) mmax:3432.83, 2926.24, 1761.26, 1656.07, 1500.98, 1369.83, 1219.35,1176.07, 1014.16, 940.47, 628.69 cm�1; UV (methanol) kmax: 269and 333 nm; HRMS: C18H23O3N [M]+: 301.6057.
4.1.4.7. 4-Methyl-2-oxo-1,2-dihydroquinolin-8-yl acetate (25). Itwas obtained as white solid (85%); mp: 242–244 �C; 1H NMR(DMSO-d6, 300 MHz): d 2.19 (s, 3H, C-4 CH3), 2.25 (s, 3H, OCOCH3),6.27 (s, 1H, H-3), 7.01 (t, 1H, J = 7.9 Hz, H-6), 7.12 (d, 1H, J = 7.7 Hz,H-5), 7.43 (d, 1H, J = 7.9 Hz, H-7), 11.27 (br s, 1H, NH); 13C NMR(DMSO-d6, 75 MHz): d 18.68 (C-4 CH3), 21.35 (OCOCH3), 121.19(C-9), 121.23, 121.48 (C-7 and C-3), 122.36, 123.84 (C-5 and C-6),131.54 (C-10), 136.74 (C-4), 148.00 (C-8), 161.63 (C-2), 169.64(OCOCH3); IR (KBr) mmax: 3431.86, 2996.30, 1763.12, 1671.14,1647.83, 1605.69, 1475.41, 1422.01, 1366.81, 1195.13, 1168.09,
N. Priya et al. / Bioorg. Med. Chem. 18 (2010) 4085–4094 4091

Author's personal copy
1138.04, 1016.39, 932.32, 869.41, 744.18, 689.19 cm�1; UV (meth-anol) kmax: 268 and 328 nm; HRMS: C12H11O3N [M+H]+: 218.1072.
4.1.4.8. 3-Ethyl-4-methyl-2-oxo-1,2-dihydroquinolin-8-yl ace-tate (26). It was obtained as white solid (80%); mp: 210–212 �C;1H NMR (DMSO-d6, 300 MHz): d 1.04 (t, 3H, J = 7.5 Hz, CH2CH3),2.37 (s, 3H, C-4 CH3), 2.44 (s, 3H, OCOCH3), 2.66 (q, 2H, J = 7.5 Hz,CH2CH3), 7.15–7.25 (m, 2H, H-5 and H-6), 7.64 (dd, 1H, J = 1.5and 7.8 Hz, H-7), 11.42 (br s, 1H, NH); 13C NMR (DMSO-d6,75 MHz): d 13.11 (CH2CH3), 14.72 (C-4 CH3), 19.67 (CH2CH3),21.35 (OCOCH3), 120.99 (C-7), 121.60 (C-3), 122.11, 122.58 (C-5and C-6), 129.99 (C-9), 133.23 (C-10), 136.46 (C-4), 141.50 (C-8),161.34 (C-2), 169.61 (OCOCH3); IR (KBr) mmax: 3162.22, 1770.93,1632.36, 1421.68, 1361.50, 1188.50, 1021.12, 955.93, 867.96,735.74, 675.53 cm�1; UV (methanol) kmax: 272 and 328 nm; HRMS:C14H15O3N [M]+: 245.4751.
4.1.4.9. 3-Hexyl-4-methyl-2-oxo-1,2-dihydroquinolin-8-yl ace-tate (27). It was obtained as white solid (80%); mp: 162–164 �C;1H NMR (CDCl3, 300 MHz): d 0.89 (t, 3H, J = 6.6 Hz, CH2CH3),1.31–1.54 (m, 8H, CH2(CH2)4CH3), 2.47 (s, 3H, C-4 CH3), 2.48 (s,3H, OCOCH3), 2.75 (t, 2H, J = 6.9 Hz, CH2CH2), 7.16–7.31 (m, 2H,H-5 and H-6), 7.56 (d, 1H, J = 8.4 Hz, H-7), 9.65–9.74 (br m, 1H,NH); 13C NMR (CDCl3, 75 MHz): d 14.11 (CH2CH3), 15.37 (C-4CH3), 21.20 (OCOCH3), 22.68, 27.20, 29.00, 29.57, 31.82((CH2)5CH3), 121.34, 121.74 (C-5 and C-7), 121.80 (C-6), 122.53(C-3), 128.95 (C-9), 132.84 (C-10), 136.37 (C-4), 142.65 (C-8),162.16 (C-2), 168.80 (OCOCH3); IR (KBr) mmax: 3428.77, 2930.79,2372.19, 1754.72, 1650.93, 1565.36, 1461.77, 1367.69, 1209.86,1017.50, 907.94, 781.27, 738.35, 698.94 cm�1; UV (methanol) kmax:273 and 327 nm; HRMS: C18H23O3N [M]+: 301.4821.
4.1.5. General procedure for the synthesis of N- and O-substitutedalkyl esters of quinolin-2-ones (28–31)
To a mixture of acetoxy-4-methylquinolin-2-ones (7.3 mmol),K2CO3 (1 g, 7.3 mmol) in anhydrous DMF (10 mL), was added ethylbromoacetate (1.2 g, 7.3 mmol) and the reaction mixture was re-fluxed for 12 h. The reaction was then cooled to room temperatureand the mixture was poured in ice cold water and extracted withethyl acetate. The crude product was purified through columnchromatography to give two isomeric products.
4.1.5.1. Ethyl 2-(7-acetoxy-4-methyl-2-oxoquinolin-1(2H)-yl)ace-tate (28). It was obtained as white crystals (60%); mp: 110 �C; 1HNMR (CDCl3, 300 MHz): d 1.25 (t, 3H, J = 7.1 Hz, CH2CH3), 2.34 (s,3H, C-4 CH3), 2.48 (s, 3H, OCOCH3), 4.23 (q, 2H, J = 7.1 Hz, CH2CH3),5.04 (s, 2H, NCH2), 6.59 (s,1H, H-3), 6.87 (s, 1H, H-8), 7.03 (d, 1H,J = 8.7 Hz, H-6), 7.73 (d, 1H, J = 8.7 Hz, H-5); 13C NMR (CDCl3,75 MHz): d 14.09 (CH2CH3), 19.23 (C-4 CH3), 21.20 (OCOCH3),43.76 (NCH2), 61.82 (CH2CH3), 107.08, 116.10 (C-6 and C-8),119.32 (C-3), 120.18 (C-10), 126.70 (C-5), 140.14 (C-9), 147.10(C-4), 152.43 (C-7), 161.73 (C-2), 168.04 (OCOCH3), 168.97(COO); IR (KBr) mmax: 2999.98, 2925.16, 1766.13, 1745.40,1658.88, 1594.89, 1437.53, 1386.80, 1371.56, 1324.86, 1243.22,1204.97, 1177.62, 1111.11, 1019.91, 958.23, 909.97, 864.10,816.25, 745.32, 714.35, 649.75, 604.10, 561.14 cm�1; UV (metha-nol) kmax: 324 nm; HRMS: C16H17O5N [M]+: 304.0316.
4.1.5.2. Ethyl 2-(7-acetoxy-4-methylquinolin-2-yloxy)acetate(30). It was obtained as white crystals (30%); mp: 98 �C; 1H NMR(CDCl3, 300 MHz): d 1.28 (br s, 3H, CH2CH3), 2.35 (s, 3H, C-4 CH3),2.62 (s, 3H, OCOCH3), 4.26 (br s, 2H, CH2CH3), 4.99 (s, 2H, OCH2CO),6.87 (s, 1H, H-3), 7.17 (d, 1H, J = 7.8 Hz, H-6), 7.51 (s, 1H, H-8), 7.88(d, 1H, J = 6.3 Hz, H-5); 13C NMR (CDCl3, 75 MHz): d 14.16(CH2CH3), 18.77 (C-4 CH3), 21.15 (OCOCH3), 61.01 (CH2CH3),62.41 (OCH2CO), 112.33, 118.89 (C-6 and C-8), 119.20 (C-3),
123.72 (C-10), 124.79 (C-5), 146.89 (C-9), 147.29 (C-4), 151.31(C-7), 161.07 (C-2), 169.24 (–OCOCH3), 169.35 (COOCH2CH3); IR(KBr) mmax: 2989.04, 2928.22, 1761.41, 1641.32, 1580.26,1516.42, 1463.00, 1426.20, 1371.03, 1210.92, 1175.98, 1136.43,1081.82, 1014.69, 943.65, 912.83, 862.57, 797.55, 692.03,599.54 cm�1; UV (methanol) kmax: 324 nm; HRMS: C16H17O5N[M]+: 303.5980.
4.1.5.3. Ethyl 2-(6-acetoxy-4-methyl-2-oxoquinolin-1(2H)-yl)ace-tate (29). It was obtained as white crystals (60%); mp: 166 �C; 1HNMR (CDCl3, 300 MHz): d 1.26 (t, 3H, J = 7.2 Hz, CH2CH3), 2.34 (s,3H, C-4 CH3), 2.45 (s, 3H, OCOCH3), 4.23 (q, 2H, J = 6.9 Hz, CH2CH3),5.08 (s, 2H, NCH2), 6.65 (s,1H, H-3), 7.10 (d, 1H, J = 9.0 Hz, H-8),7.28 (dd, 1H, J = 2.4 and 8.7 Hz, H-7), 7.44 (d, 1H, J = 2.1 Hz, H-5);13C NMR (CDCl3, 75 MHz): d 14.07 (CH2CH3), 19.07 (C-4 CH3),21.03 (OCOCH3), 43.68 (NCH2), 61.74 (CH2CH3), 114.77, 117.86(C-5 and C-7), 121.37 (C-8), 122.17 (C-10), 124.24 (C-3), 136.81(C-9), 145.36 (C-4), 146.74 (C-6), 161.35 (C-2), 168.04 (OCOCH3),169.54 (COO); IR (KBr) mmax: 3474.88, 2988.51, 1763.26, 1745.76,1664.42, 1600.02, 1570.50, 1441.61, 1424.06, 1373.46, 1313.63,1207.62, 1170.43, 1122.79, 1024.86, 953.86, 899.96, 813.98,681.48, 615.15 cm�1; UV (methanol) kmax: 276 and 336 nm; HRMS:C16H17O5N [M]+: 303.8917.
4.1.5.4. Ethyl 2-(6-acetoxy-4-methylquinolin-2-yloxy)acetate(31). It was obtained as white crystals (30%); mp: 62 �C; 1H NMR(CDCl3, 300 MHz): d 1.25–1.34 (m, 3H, CH2CH3), 2.36 (s, 3H, C-4CH3), 2.60 (s, 3H, OCOCH3), 4.21–4.30 (m, 2H, CH2CH3), 5.00 (s,2H, OCH2CO), 6.91 (s, 1H, H-3), 7.34 (dd, 1H, J = 2.7 and 9.0 Hz,H-7), 7.57 (d, 1H, J = 2.4 Hz, H-5), 7.79 (d, 1H, J = 9.0 Hz, H-8); 13CNMR (CDCl3, 75 MHz): d 14.21 (CH2CH3), 18.73 (C-4 CH3), 21.16(OCOCH3), 61.04 (CH2CH3), 62.49 (OCH2CO), 113.13, 115.17 (C-5and C-7), 124.04 (C-8), 126.00 (C-10), 129.15 (C-3), 144.02 (C-9),146.85 (C-4), 147.10 (C-6), 160.47 (C-2), 169.34 (OCOCH3),169.78 (COO); IR (KBr) mmax: 3478.18, 2990.84, 1762.23, 1750.29,1609.84, 1580.73, 1522.33, 1467.00, 1439.28, 1421.11, 1384.17,1364.61, 1341.67, 1219.46, 1168.78, 1088.15, 1036.15, 1017.43,942.95, 908.61, 887.67, 847.01, 824.95, 736.28, 620.01 cm�1; UV(methanol) kmax: 324 nm; HRMS: C16H17O5N [M]+: 304.0379.
4.1.5.5. General procedure for the synthesis of 2-oxo-1,2-dihy-droquinolin-4-yl acetate (33). Aniline (5 g, 54 mmol) was addedto a mixture of fused zinc chloride (21.5 g, 161 mmol), malonicacid (5.6 g, 54 mmol), and phosphorus oxychloride (108 mmol,16.5 g). The reaction mixture was heated at 65 �C for 36 h. It wasthen cooled and poured on ice.26 The precipitate was filtered andpurified through column chromatography using silica gel (100–200 mesh) in methanol/chloroform (1:100). A solution of aceticanhydride and acetic acid (1:4) was added to 4-hydroxyquinolin-2-one (32) and the reaction mixture was refluxed for 6 h and thenpoured on crushed ice (Scheme 3). The resulting precipitate wasfiltered and washed with water to yield 2-acetoxyquinolin-2-one(33).
4.1.5.6. 4-Hydroxyquinolin-2(1H)-one (32). It was obtained asyellow solid (70%); mp: 320 �C, (Literature value = 318–320 �C);27
1H NMR (DMSO-d6, 300 MHz): d 5.76 (s, 1H, H-3), 7.11–7.78 (m,4H, H-5, H-6, H-7 and H-8), 11.18 (br s, 1H, OH); 13C NMR(DMSO-d6, 75 MHz): d 98.18 (C-3), 114.95, 115.10 (C-6 and C-8),121.04 (C-5), 122.62 (C-10), 130.82 (C-7), 139.13 (C-9), 162.43(C-4), 163.57 (C-2); IR (KBr) mmax: 3430.12, 3094.44, 2953.09,2861.12, 2639.30, 2364.36, 1669.52, 1633.94, 1594.78, 1560.44,1506.18, 1471.81, 1420.44, 1378.93, 1332.01, 1259.95, 1235.09,1160.89, 1145.18, 1102.39, 1035.00, 909.53, 867.70, 773.96,762.96, 755.55, 671.97, 630.01 cm�1; UV (methanol) kmax: 269and 314 nm; HRMS: C9H7O2N [M]+: 161.7715.
4092 N. Priya et al. / Bioorg. Med. Chem. 18 (2010) 4085–4094

Author's personal copy
4.1.5.7. 2-Oxo-1,2-dihydroquinolin-4-yl acetate (33). It was ob-tained as yellow solid (85%); mp: 220 �C, (Literature value = 217–219 �C);28 1H NMR (DMSO-d6, 300 MHz): d 2.45 (s, 3H, –OCOCH3),6.65 (s, 1H, H-3), 7.22–7.66 (m, 4H, H-5, H-6, H-7 and H-8), 11.89(br s, 1H, NH); 13C NMR (DMSO-d6, 75 MHz): d 20.75 (–OCOCH3),98.19 (C-3), 112.28, 115.48 (C-6 and C-8), 122.03 (C-5), 122.44(C-10), 131.47 (C-7), 138.87 (C-9), 156.04 (C-4), 162.39 (C-2),172.04 (–OCOCH3); IR (KBr) mmax: 3396.31, 3009.90, 2965.78,2858.41, 1768.58, 1668.77, 1615.91, 1561.20, 1506.34, 1475.03,1434.55, 1398.53, 1370.26, 1268.81, 1189.23, 1160.47, 1143.99,1078.60, 984.19, 953.88, 887.47, 770.84, 758.32, 660.04 cm�1; UV(methanol) kmax: 264 and 326 nm; HRMS: C11H9O3N [M+H]+:204.1366.
4.2. Isolation of platelet rich plasma (PRP)
The citrated blood was used for the preparation of PRP by themethod of Vickers and Thompson.29 Venous blood (9 mL) wascollected from healthy human volunteer and mixed with 1.0 mLof 3.8% trisodium citrate and centrifuged at 180g for 10 min. Theupper two-third fraction of plasma (PRP) was transferred to an-other centrifuge tube leaving behind lower one-third layer to avoidcontamination with WBC’s and RBC’s. Platelet poor plasma (PPP)was obtained by centrifugation of the remaining sample at 2500gfor 10 min. Platelet count was determined in PRP using electroniccounter, SYSMEX Model No. FA 20 and were adjusted to250 � 106/mL with PPP.
4.3. Aggregometry
The test compounds (in appropriate concentrations) were sepa-rately preincubated with PRP in a final volume of 0.5 mL at 37 �C.Platelet aggregation was induced by the addition of ADP (15 lM)and assessed by using a platelet aggregation profiler (BIODATACORPORATION, Model No. PAP-4) and the results were expressedas the maximum percentage of light transmittance change (%max) from the baseline at the end of the recording time, usingPPP as a reference. Platelet aggregation curves were recorded for6 min and analyzed according to internationally establishedstandards.
4.4. Platelet aggregation in vivo
Male sprague dawely rats (weight 200–250 g) housed in meshcages maintained at 25 �C and illuminated at 12:12 h light dark cy-cles. The known amount of test compound was suspended inappropriate volume of saline, sonicated for 30 s and the prepara-tion was administered to the rats orally at a dose of 133 lmoles/kg. The animals were sacrificed after 24 h, and blood samples weretaken by cardiac puncture. The blood was centrifuged according toabove mentioned procedure and then analyzed for the assessmentof platelet aggregation.30
4.5. Cox-1 activity assay
Platelets lysate were prepared from the above mentioned sam-ple in 100 lL of lysis buffer (50 mmol/L tris, 150 mmol/L NaCl,10 mmol/L EGTA, 1% triton X-100, 1% sodium deoxycholate,1 mmol/L sodium vanadate, 50 mmol/L NaF, 2 mmol/L EDTA (pH8.0), 1 mmol/L phenylmethylsulfonyl fluoride, and 5 g/mL of leu-peptin/pepstatin A/aprotinin for 15 min at 4 �C and assessed forCox-1 activity.31 The assay was carried out using Cox-1 assay ELISAkit (Cayman Chemical), according to the manufacturer’s protocol.Briefly, 50 lL of each lysed samples were added to the wells, theenzymatic reaction was initiated by adding 100 lM N,N,N,N-tetra-methyl-p-phenylenediamine (TMPD) and 100 lM arachidonic acid
(saturating condition) in assay buffer. Inhibitors were added to theincubation reaction at different time intervals before the additionof TMPD and arachidonic acid. The enzyme activity was measuredcalorimetrically by monitoring the appearance of oxidized TMPD at590 nm.
4.6. Assay of platelet CRTAase
Platelet CRTAase was assayed as mentioned in the earlier pub-lications.7 The unit of CRTAase activity was expressed in terms of% inhibition of GST activity under the conditions of the assay.
4.7. Platelet measurement of NO level by flow cytometry
The method outlined by Imrich and Kobzik as described earlierwas followed for the assay of NOS in platelets by flow cytometry.32
4.8. Calculation and statistics
Calculations and statistics were performed using the graph padprism 3.02 software. The one-way analysis of variance (ANOVA)tests followed by the Turkey multiple comparisons test were used.Data were expressed as mean ± standard error. Statistical signifi-cance was calculated using the Student’s t-test. p values less than0.05 (p <0.05) were considered to be statistically significant.
Acknowledgments
The financial assistance from the Department of Scientific andIndustrial Research, University Grant Commission, Government ofIndia, and University of Delhi is gratefully acknowledged. A.G.and P.S. are the recipients of Senior Research Fellowship fromCouncil of Scientific and Industrial Research, Government of India.
Supplementary data
Supplementary data associated with this article can be found, inthe online version, at doi:10.1016/j.bmc.2010.04.011.
References and notes
1. Roberts, D. E.; Mc Nicol, A.; Bose, R. J. Biol. Chem. 2004, 297, 19421.2. Vane, J. R. Nature (New Biol.) 1971, 231, 232.3. Bansal, S.; Gaspari, M.; Raj, H. G.; Kumar, A.; Cuda, G.; Verheji, E.; Tyagi, Y. K.;
Ponan, P.; Rastogi, R. C.; Parmar, V. S. Appl. Biochem. Biotechnol. 2008, 144, 37.4. Singh, I.; Kohli, E.; Raj, H. G.; Gyanda, K.; Jain, S. K.; Tyagi, Y. K.; Gupta, G.;
Kumari, R.; Kumar, A.; Pal, G.; Prasad, A. K.; Rastogi, R. C.; Olsen, C. E.; Jain, S. C.;Parmar, V. S. Bioorg. Med. Chem. 2002, 10, 4103.
5. Raj, H. G.; Kohli, E.; Goswami, R.; Goel, S.; Rastogi, R. C.; Jain, S. C.; Wengel, J.;Olsen, C. E.; Parmar, V. S. Bioorg. Med. Chem. 2001, 9, 1085.
6. Charles, H.; Hennekens, M. D.; Mark, L.; Dyken, M. D.; Fuster, V. Circulation1997, 96, 2751.
7. Seema; Kumari, R.; Gupta, G.; Saluja, D.; Kumar, A.; Goel, S.; Tyagi, Y. K.; Gulati,R.; Vinocha, A.; Murlidhar, K.; Dwarkanath, B. K.; Rastogi, R. C.; Parmar, V. S.;Patkar, S. A.; Raj, H. G. Cell. Biochem. Biophys. 2007, 47, 53.
8. O’ Kane, P.; Xie, L.; Liu, Z.; Queen, L.; Jackson, G.; Ji, Y.; Ferro, A. Cardiovasc. Res.2009, 83, 123.
9. Kohli, E.; Gaspari, M.; Raj, H. G.; Parmar, V. S.; Sharma, S. K.; van der Greef, J.;Kumari, R.; Gupta, G.; Seema; Khurana, P.; Tyagi, Y. K.; Watterson, A. C.; Olsen,C. E. Biochim. Biophys. Acta 2004, 1698, 55.
10. Singh, P.; Ponnan, P.; Krishnan, S.; Tyagi, T. K.; Priya, N.; Bansal, S.; Scumaci, D.;Gaspari, M.; Cuda, G.; Joshi, P.; Gambhir, J. K.; Saluja, D.; Prasad, A. K.; Saso, L.;Rastogi, R. C.; Parmar, V. S.; Raj, H. G. J. Biol. 2009. doi:10.1093/jb/mvq002.
11. Raj, H. G.; Kumari, R.; Seema; Gupta, G.; Kumar, R.; Saluja, D.; Muralidhar, K.M.; Kumar, A.; Dwarkanath, B. S.; Rastogi, R. C.; Prasad, A. K.; Patkar, S. A.;Watterson, A. C.; Parmar, V. S. Pure Appl. Chem. 2006, 78, 985.
12. Khurana, P.; Kumari, R.; Vohra, P.; Kumar, A.; Seema; Gupta, G.; Raj, H. G.;Dwarkanath, B. S.; Saluja, D.; Bose, M.; Vij, A.; Chaudhary, N. K.; Adhikari, J. S.;Tyagi, Y. K.; Kohli, E. Bioorg. Med. Chem. 2006, 14, 575.
13. Loscalzo, J.; Welch, G. Prog. Cardiovasc. Dis. 1995, 38, 87.14. Stahlment, R.; Lode, H. Drugs 1999, 58, 37.15. Kathuria, A.; Gupta, A.; Priya, N.; Singh, P.; Raj, H. G.; Prasad, A. K.; Parmar, V.
S.; Sharma, S. K. Bioorg. Med. Chem. 2009, 17, 1550.
N. Priya et al. / Bioorg. Med. Chem. 18 (2010) 4085–4094 4093

Author's personal copy
16. Staskun, B. J. Org. Chem. 1964, 29, 1153.17. Fabian, W. M. F.; Niederreiter, K. S.; Uray, G.; Stadlbauer, W. J. Mol. Str. 1999,
477, 209.18. Kidwai, M.; Bansal, V. Lett. Org. Chem. 2007, 4, 519.19. Nadzan, A. M.; Rinehart, K. L., Jr. J. Am. Chem. Soc. 1977, 99, 4647.20. Pettit, G. R.; Piatak, D. M. J. Org. Chem. 1960, 25, 721.21. Traven, V. F.; Podhaluzina, N. Y.; Vasilyev, A. V.; Manaev, A. V. ARKIVOC 2000, 931.22. Holmes, R. R.; Conrady, J.; Guthrie, J.; McKay, R. J. Am. Chem. Soc. 1954, 76, 2400.23. Shoeb, H. A.; Korkor, M. I.; Tammam, G. H.; El-Amin, S. M. Can. J. Pharm. Sci.
1980, 15, 66.24. Mann, F. G.; Saunders, B. C. Practical Organic Chemistry, 4th ed.; Longman:
London, 1960. p 107.
25. Nguyen, M. T.; Pham, V. P.; Ta, V. T. Tap Chi Hoa Hoc 2005, 43, 424. Chem. Abstr.2005, 146, 274192.
26. Zhang, S.-L.; Huang, Z.-S.; Li, Y.-M.; Chan, A. S. C.; Gu, L.-Q. Tetrahedron 2008,64, 4403.
27. Park, S.-J.; Lee, J.-C.; Lee, K.-I. Bull. Korean Chem. Soc. 2007, 28, 1203.28. Selig, P.; Bach, T. Synthesis 2008, 14, 2177.29. Thompson, S. G.; Vickers, M. V. Thromb. Hameost. 1985, 53, 216.30. O’Brien, J. R. J. Clin. Pathol. 1962, 15, 446.31. Corazzi, T.; Leone, M.; Maucci, R.; Corazzi, L.; Gresele, P. J. Pharmacol. Exp. Ther.
2005, 315, 1331.32. Imrich, A.; Kobzik, L. Nitric Oxide 1997, 1, 359.
4094 N. Priya et al. / Bioorg. Med. Chem. 18 (2010) 4085–4094

Accepted Manuscript
Substrate specificity of coumarins and quinolones towards Calreticulin medi‐
ated transacetylation: Investigations on antiplatelet function
Abha Kathuria, Nivedita Priya, Karam Chand, Prabhjot Singh, Anjali Gupta,
Sarah Jalal, Shilpi Gupta, Hanumantharao G. Raj, Sunil K. Sharma
PII: S0968-0896(11)00942-4
DOI: 10.1016/j.bmc.2011.11.016
Reference: BMC 9655
To appear in: Bioorganic & Medicinal Chemistry
Received Date: 16 August 2011
Revised Date: 8 November 2011
Accepted Date: 9 November 2011
Please cite this article as: Kathuria, A., Priya, N., Chand, K., Singh, P., Gupta, A., Jalal, S., Gupta, S., Raj, H.G.,
Sharma, S.K., Substrate specificity of coumarins and quinolones towards Calreticulin mediated transacetylation:
Investigations on antiplatelet function, Bioorganic & Medicinal Chemistry (2011), doi: 10.1016/j.bmc.2011.11.016
This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers
we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and
review of the resulting proof before it is published in its final form. Please note that during the production process
errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.

Bioorganic & Medicinal Chemistry � ����� � ����� �� ������ ����� �� ���
Substrate specificity of coumarins and quinolones towards Calreticulin mediated transacetylation: Investigations on antiplatelet function
Abha Kathuria,a Nivedita Priya,b Karam Chand,a Prabhjot Singh,b Anjali Gupta,a Sarah Jalal,a
Shilpi Gupta,a Hanumantharao G. Raj,b,* and Sunil K. Sharmaa,*
aBioorganic Laboratory, Department of Chemistry, University of Delhi, Delhi 110 007, India bDepartment of Biochemistry, V P Chest Institute, University of Delhi, Delhi 110 007, India
1. Introduction
The essential role played by platelets in the pathogenesis of arterial thrombosis is well established: platelets represent a major target of therapeutic interventions aiming at decreasing the incidence and severity of cardiovascular accidents in patients. Platelets adhere to damaged endothelium by a process mediated by the interaction between membrane receptors and various ligands present in the endothelium and sub endothelium. The platelet integrin GPs Ia/IIIb and Ib/IX bind collagen and von Willebrand factor (vWF), respectively. Adhesion results in platelet activation, which in turn leads to recruitment of other platelets to the site of injury. Agonists such as thrombin, thromboxane A2, and adenosine diphosphate (ADP) are produced or released locally, causing a conformational change in membrane GPIIb/IIIa receptor, which then mediates the final obligatory steps in platelet aggregation and becoming a functional receptor for adhesive molecules such as fibronectin, vitronectin, and vWF, the last of which may mediate platelet aggregation under high shear rate conditions.1,2
Numerous antiplatelet agents have been developed based on their ability to block the receptors responsible for platelet activation. However, there are three families of agents that inhibit platelet function, with proven clinical efficacy: (i) Cyclooxygenase inhibitors, such as aspirin; (ii) ADP receptor
antagonists, such as the thienopyridine derivatives e.g. ticlopidine and clopidogrel; (iii) glycoprotein (GP) IIb/IIIa (or integrin aIIbb3) antagonists. All these drugs are used during coronary interventions and in the medical management of acute coronary syndromes, while only aspirin and the thienopyridine derivatives are used in long-term prevention of cardiovascular events.3
While minimizing ischemic recurrences, an intensified antiplatelet regimen also invariably leads to severe side-effects, e.g. gastrointestinal toxicity due to aspirin include nausea, vomiting, dyspepsia, heartburn, gastrointestinal ulceration, etc.4
Also, in recent years, the issue of resistance to antiplatelet agents, in particular aspirin and thienopyridines, has been highlighted in the medical literature.5-8 Despite the fact that the currently available drugs suffer from some drawbacks and are prone to resistance, they have a good risk-to-benefit ratio which justify the unceasing search for agents that can further improve the clinical outcome of patients with atherosclerosis through greater efficacy and/or safety. New platelet targets for potential antithrombotic drugs include several receptors and effectors that are important for platelet function. Further, the agents causing the inhibition of cyclooxygenase catalyzed TxA2 synthesis would also lead to the inhibition of platelet aggregation. The enzyme NOS generates NO, which is essential for vascular function. NOS is involved in vessel dilatation, inhibition of platelet and leukocyte adhesion,
AR TI C LE IN FO ABS TRAC T
Article history: Received Received in revised form Accepted Available online
Keywords: N-Acetylaminocoumarins Quinolin-2-ones Platelet CRTAase Antiplatelet agents Cyclooxygenase-1 (Cox-1) Thromboxane A2 (TxA2)
Calreticulin Transacetylase (CRTAase) is known to catalyze the transfer of acetyl group from polyphenolic acetates (PA) to certain receptor proteins (RP), thus modulating their activity. Herein, we studied for the first time the substrate specificity of CRTAase towards N-acetylamino derivatives of coumarins and quinolones. This study is endowed with antiplatelet action by virtue of causing CRTAase catalyzed activation of platelet Nitric Oxide Synthase (NOS) by way of acetylation leading to the inhibition of ADP/Arachidonic acid (AA)-dependent platelet aggregation. Among all the N-acetylamino/acetoxy coumarins and quinolones screened, 7-N-acetylamino-4-methylcoumarin (7-AAMC, 17) was found to be the superior substrate to platelet CRTAase and emerged as the most promising antiplatelet agent both in vitro and in vivo. Further it caused the inhibition of cyclooxygenase-1 (Cox-1) resulting in the down regulation of thromboxane A2 (TxA2), modulation of tissue factor and the inhibition of platelet aggregation. It was also found effective in the inhibition of LPS induced pro-thrombotic conditions.
2009 Elsevier Ltd. All rights reserved.

and inhibition of proliferation and migration of vascular smooth muscle cells.9 Earlier reports from our group have established the activation of platelet NOS by certain class of polyphenolic acetates (PAs) by way of acetylation of NOS mediated by CRTAase.10 Accordingly, PAs were found to be effective in the inhibition of ADP induced platelet aggregation.
In the present investigation, efforts have been made to compare the specificities of N-acetylamino and acetoxy derivatives of coumarins and quinolones on CRTAase mediated activation of NOS and also to delineate the structure activity relationship (SAR) with reference to the effect of alkyl group at C-3 position, and substitution of C-4 methyl by trifloromethyl group of the coumarin/quinolone moiety.
2. Results
In earlier reports we elucidated the role of acetoxy groups on the benzenoid ring of chromones, coumarins, xanthones, flavones, and quinolones, in facilitating the acetylation of receptor proteins catalyzed by CRTAase. Also, we studied the factors, such as the proximity of the acetoxy group to the oxygen/nitrogen heteroatom, the role of carbonyl group on the benzopyran nucleus, and the effect of substituents on these molecules in controlling the protein acetylation.11-14 However, N-acetylaminocoumarins, and quinolones have never been screened for CRTAase activity. In lieu of the above, efforts have been made to compare for the first time the specificities of heterocyclic compounds viz. N-acetylaminocoumarins/ quinolones on CRTAase mediated activation of NOS and other enzymes. We have evaluated the specificity of CRTAase towards a series of 6/7-acetoxy quinolones and its consequent effect on the enhancement of NO levels in platelets and inhibitory effect on ADP/AA induced platelet aggregation. Also, C-6/ C-8 acetyl-7-acetoxycoumarins (48-51) were included in this study to evaluate the effect of C-acetyl group on the benzenoid ring.
The results clearly demonstrated that specificity of the N-acetylamino /acetoxy derivatives for CRTAase is in the order: N-acetylaminocoumarins > acetoxyquinolones > N-acetylamino quinolones > acetyl coumarins, and among all the compounds screened 7-N-acetylamino-4-methylcoumarin (7-AAMC, 17) was found to be the best substrate to platelet CRTAase compared to the other N-acetylamino/acetoxy polyphenolic derivatives resulting in significant inhibition of ADP induced platelet aggregation.
The leading compound, 7-AAMC, when administered to the rats, was found to be effective in inhibition of Cox-1 activity and eventually inhibition of thromboxane. This compound was taken further for detailed study, i.e. in vivo LPS mediated thrombotic prone conditions and the levels of iNOS and tissue factor (TF) were measured and found to be inhibited in the case of compound treated rats.
2.1. Chemistry
2.1.1. Synthesis of 7-N-acetylamino derivatives of coumarin (17-21)
The synthesis of 7-amino-4-methyl derivatives of coumarin was carried out by following the procedure given by Atkins and Bliss.15 Herein, urethane protected m-aminophenol (3-hydroxyphenylurethane, 1) was made to react with alkylated ethyl acetoacetate (2-5) or 4,4,4-trifluoroethyl acetoacetate (6) in the presence of 70% H2SO4-C2H5OH to obtain quantitatively 3-alkyl-7-carbethoxy-4-methyl/trifluoromethylcoumarin (7-11) via Pechmann condensation. The earlier published literature procedure from our group was followed for synthesizing alkylated ethyl acetoacetate (2-alkyl ethylbutanoate) from ethyl
acetoacetate and alkyl bromide.13 The deprotection of corresponding 7-carbethoxy-4-methyl/fluorocoumarin (7-11) was carried out with a 1:1 mixture of concentrated suphuric acid and acetic acid to yield 3-alkyl-7-amino-4-methyl/trifluoromethylcoumarin (12-16) which was then acetylated using acetic anhydride and catalytic amount of N,N-dimethyl amino pyridine (DMAP) in THF to yield corresponding N-acetyl coumarin 17-21 (Scheme-1).
Scheme 1. Synthesis of 7-aminocoumarin derivatives: (i) C2H5OCOCl, Et2O, rt; (ii) H2SO4-C2H5OH (7:3), rt; (iii) H2SO4-CH3COOH (1:1), reflux; (iv) (CH3CO)2O, DMAP, THF, rt. 2.1.2. Synthesis of N-(2-oxo-1,2-dihydroquinolin-7-yl) acetamide (25-27)
Synthesis of N-(2-oxo-(4-methyl/trifloromethyl)-1,2-dihydro quinolin-7-yl)acetamide (25-27) was carried out from 1,3-diaminobenzene and substituted ethyl acetoacetate (2-3, 6) by following Conrad Limpach synthesis,16 the compound so obtained (22-24) was then subjected to acetylation (Scheme-2).
Scheme 2. Synthesis of 7-amino derivatives of quinolones: (i) 150 oC; (ii) (CH3CO)2O, CH3COOH. 2.1.3. Synthesis of acetoxy derivatives of quinolin-2(1H)-ones (38-42)
Acetoxy derivatives of quinolin-2-ones were synthesized by following the method reported earlier from our group.14 Anisidines were first reacted with substituted ethyl acetoacetate (2 and 6) via Knorr reaction to give methoxy quinolones (28–32). Subsequently demethylation, was carried out using a mixture of hydrobromic acid and acetic acid, the corresponding hydroxy quinolones (33–37) so obtained were then acetylated with acetic anhydride in acetic acid to yield acetoxy quinolones 38-42 (Scheme-3).
2.1.4. Synthesis of 6/8-acetyl-7-acetoxycoumarins (49-52)
7-Acetoxy-4-methylcoumarin (43) obtained by following the literature procedure,17 was subjected to Fries migration18 and a mixture of 8-acetyl- (44-45) and 6-acetyl- (46-47) 7-hydroxycoumarins in 9:1 ratio was obtained. The mixture was separated through column chromatography and the resulting compounds were then acetylated using acetic anhydride and catalytic amount of DMAP to yield the title compounds (Scheme-4).

Scheme 3. Synthesis of acetoxy derivatives of quinolones: (i) R2COCHR1COOC2H5, 150 oC; (ii) H2SO4, 100 oC; (iii) HBr-CH3COOH (7:3), 120 oC; (iv) (CH3CO)2O, THF, rt.
Scheme 4. Synthesis of acetoxy derivatives of acetylcoumarins: (i) CH3COCHRCOOC2H5, conc. H2SO4, rt; (ii) (CH3CO)2O, DMAP,THF, rt; (iii) AlCl3, 125-170 oC; (iv) (CH3CO)2O, DMAP, THF, rt. All the compounds were fully characterized on the basis of their physical and spectral data, and of total forty six coumarin and quinolone derivatives synthesized, twenty five, that is, 8-10, 18-20, 23, 26, 29-31, 34-36, 38-42, 45, and 47-51 are novel and reported for the first time. Though the compounds 1, 7, 11-17, 21-22, 24-25, 27-28, 32-33, 37, 44, and 46 are known in literature, however their complete spectral data is not reported. Herein, we have reported the spectral data for all these compounds in the experimental section.
2.2. Biological activity
2.2.1. CRTAase activities
The results illustrated in Figure 1 revealed the differential specificity of platelet CRTAase to a number of N-acetylamino/ acetoxy derivatives of coumarins and quinolones. The specificity of these compounds towards platelet CRTAase was found to be in the order: N-acetylaminocoumarins > acetoxy quinolones > N-acetylaminoquinolones > acetyl coumarins. It is evident from the results (Figure 1) that by incorporating an alkyl group at C-3 position of coumarins and quinolones resulted in drastic reduction of CRTAase activity, the activity decreases with increase in the size of alkyl chain. Also a decline in platelet CRTAase activity was observed on substituting the C-4 methyl by trifluoromethyl group, i.e. in compounds 21, 27, 38, and 42. Further the presence of acetyl group at C-6/C-8 position on the benzenoid ring in coumarins 48-51 resulted in poor inhibition of platelet aggregation or no inhibition at all.
Figure 1. The effect of test compounds on platelet CRTAase. CRTAase activity was assayed in platelet lysate using test compounds (50 µM). The unit of CRTAase was expressed in terms of % inhibition of GST under the experimental conditions.
2.2.2. Enhancement of intracellular nitric oxide (NO) levels
2.2.2.1. Measurement of NO level by flow cytometry
The influence of N-acetylamino or acetoxy derivatives of coumarins/quinolones on the NO level in platelets has been described in Figure 2. Platelets were incubated with N-acetylamino/acetoxy coumarins and quinolones followed by the measurement of NO levels by flow cytometry. It is evident from the results that compound 17 (7-AAMC) profoundly enhanced NO level in platelets as compared to other acetoxy derivatives. The structural modification of N-acetylaminocoumarins was found to influence their effect on NO production in platelets in tune with the specificity of platelet CRTAase to these compounds (Figure 2).
Figure 2. Effect of test compounds on platelet NOS activity. Human platelets were incubated with test componds (100 µM) along with L-arginine (100 µM) and DCFH-DA (2 µM) for 30 min at 37 °C followed by the measurement of DCF fluorescence.
2.2.2.2. Effect of acetoxy quinolones and coumarins on platelet aggregation
2.2.2.2.1. In vitro Platelet aggregation
In vitro antiplatelet activity of the test compounds is depicted in Figure 3, among all the compounds tested, compound 17 was found to be most effective in causing the inhibition of ADP and AA induced platelet aggregation. This compound at concentration 25-250 µM showed a definite trend of dose dependent inhibition of ADP/AA induced platelet aggregation. The IC50 value in vitro for ADP and AA induced platelet aggregation is 145±2.4 µM and 77±3.0 µM respectively (Table 1).

Figure 3. Antiplatelet activity of test compounds in vitro. PRP was incubated with the test compound (100 µM) for 10 min at 37 °C followed by the addition of ADP (15 µM) and platelet aggregation was monitored by aggregometry. Values are mean ± SEM of 5 observations.
Table 1. In vitro IC50 value of 7-AAMC for ADP and AA.
PRP was incubated with the test compound (25-250 µM) for 10 min at 37 °C followed by the addition of ADP (15 µM)/AA (0.5 mM) and platelet aggregation was monitored by aggregometry. Values are mean ± SEM of 5 observations. IC50 value was determined from a dose-response plot. 2.2.2.2.2. In vivo Platelet aggregation
7-AAMC was found to effectively inhibit both ADP as well as AA induced platelet aggregation in vivo like aspirin as per the dose dependent curve (Figure 4). The IC50 values of 7-AAMC in case of ADP and AA induced platelet aggregation was found to be 125.3±1.4 �mole/kg (27.2±0.30 mg/kg) and 55±1.5 �mole/kg (12±0.41 mg/kg) respectively. The optimum dose of 7-AAMC in the case of ADP as an inducer was found to be 160.2±1.7 µmole/kg (34.76±0.35 mg/kg), which had exhibited maximum inhibitory effect, as evident from the dose dependent curve. This effective concentration of 7-AAMC was used in studies related to the investigation of the mechanism of the antiplatelet activity. The inhibition of ADP induced platelet aggregation in in vivo of rats administered with ASA p.o. (125.3 �mole/kg, IC50 of 7-AAMC in case of ADP) was found to be 52% while ASA when administered at a dose of 50.6 �mole/kg (IC50 of 7-AAMC in case of AA) showed 55% of inhibition of AA induced platelet aggregation. Evidently, both 7-AAMC and ASA were found to effectively inhibit AA induced platelet aggregation to a greater extent as compared with that of ADP.
Figure 4. Dose-dependent inhibition of platelet aggregation by 7-AAMC in in vivo. Rats were administered with 7-AAMC, a single dose p.o. (10-50) mg/kg, sacrificed after 24 h, blood was drawn and ADP/AA induced platelet aggregation was measured. Values are mean ± SEM of 5 observations. For all doses of 7-AAMC (p<0.005) when compared between ADP and ASA.
2.2.3. Cox-1 activity assay
The effect of 7-AAMC on the Cox-1 activity in vivo is shown in Figure 5. Three groups of rats were treated with 7-AAMC, and ASA as against control p.o., sacrificed after 24 h and the Cox-1 activity was measured by ELISA method. 7-AAMC was found to effectively inhibit Cox-1 activity like aspirin by approximately 3.34 folds.
Figure 5. Effect of 7-AAMC on inhibition of Cox-1 activity in platelets. ASA/7-AAMC were administered to rats p.o. (160.2 µmole/kg) and sacrificed after 24 h, blood was drawn and PRP prepared. Platelet lysate was bioassayed for Cox-1 activity. Values are mean ± SEM of 5 observations. �(p < 0.001) compared to the Cox-1 activity of control.
2.2.4. Modulation of TxB2 metabolite
PRP assayed for AA induced platelet aggregation were bioassayed for TxB2 concentration by ELISA method. In control samples TxB2 concentration was 85.4 pg/mg protein and dropped to 36.2 pg/mg (0.42 folds of control) and 33.6 pg/mg (0.39 folds of control) protein in ASA and 7-AAMC treated samples respectively (Figure 6).
Figure 6. Effect of 7-AAMC on inhibition of TxB2 levels. ASA/7-AAMC were administered to rats p.o. (160.2 µmole/kg), sacrificed after 24 h and blood was drawn. PRP prepared were assessed for AA induced platelet aggregation for 10 mins. Plasma was used to analyze TxB2 level. �(p < 0.01) as compared to the control group. 2.2.5. Modulation of Tissue factor (TF) level
The administration of LPS to rats resulted in the enhancement of tissue factor level upto 6.3±0.33 folds in PBMCs (peripheral blood mononuclear cell). The Prior treatment of rats with 7-AAMC/ASA led to the remarkable decrease in LPS induced
Compound IC50 values (µM)
ADP AA
7-AAMC 145±2.4 77±3.0
ASA Nil 80±2.5
�

elevation of TF upto 1.93±0.31 and 4.95±0.25 folds respectively (Figure 7).
Figure 7. Tissue Factor Measurement in PBMCs. Rats were administered test compounds (160.2 µmole/kg) p.o. for 5 days. A group of rats treated with 7-AAMC/aspirin were administered with LPS i.p. (2.5 mg/kg b.w.) 6 h before sacrificing. Another group of rats were administered LPS alone. Lymphomonocytes were isolated from blood and TF-expression was assayed by ELISA. Control rats were treated accordingly. �p < 0.05 compared between 7-AAMC and ASA treated groups. �p < 0.001 compared between 7-AAMC and LPS alone treated groups.
2.2.6. Modification of LPS induced iNOS expression by 7-AAMC
Immunodetection of LPS-induced iNOS expressions were performed in PBMCs by western blotting. The Figure 8, clearly depicted that the samples treated with LPS alone have intense iNOS expression (lane 3) whereas there was a diminished expression of iNOS in LPS plus 7-AAMC treated samples (lane 5). The ASA treated lane showed much higher iNOS expression, compared to 7-AAMC.
Figure 8. Inhibition of iNOS expression in PBMC of rat administered p.o. with 7-AAMC. Rats were administered test compounds (160.2 µmoles/ kg) p.o. for five days. A group of rats (placebo/compound treated) were administered LPS i.p. (2.5 mg/kg b.w.) 6 h before sacrificing. Lymphomonocytes were isolated from blood and iNOS expression was assayed by Placebo administered rats were treated accordingly. Western blot was carried out using anti-iNOS antibody (1:1000, v/v). Lane 1: Control; lane 2: Prestained marker; lane 3: LPS; lane 4: ASA+LPS; lane 5: 7-AAMC+LPS.
3. Discussion
Cardiovascular diseases such as myocardial infarction, unstable angina, and deep vein thrombosis greatly contribute to the mortality in the developed world. For the treatment of such heart conditions there is a greater need for the application of anti-platelet drugs.19 Theoretically, anti-platelet agents can be developed that target each step in the platelet activation or inhibition mechanisms. Numerous new anti-platelet agents were developed based on their inhibitory effects on platelet activation. Nonetheless, the number of anti-platelet agents ready for clinical trials is still insufficient, and deleterious side effects are also ssociated with most of the existing agents. Therefore, the search for an ideal anti-platelet agent is going on worldwide. Our earlier work convincingly established that Calreticulin, an important
Ca2+ binding protein of lumen of endoplasmic reticulum, mediate the transfer of acetyl group from polyphenolic (PAs) to target protein such as NOS.20-22 The protein acetyl transferase function of Calreticulin utilizing PAs as the acetyl group donors was termed CRTAase.23 For the past many years our group and others have studied the versatility of wide variety of biologically relevant heterocyclic compounds viz. coumarins, biscoumarins, chromones, flavones, isoflavones, and xanthones towards CRTAase mediated acetylation of functional proteins leading to expression of biological and pharmacological effects.11-13, 23-27
The results suggested that the specificity of these polyphenolic acetates for CRTAase is in the order: acetoxycoumarins = acetoxychromones > acetoxyflavones = acetoxyxanthones. Further investigations on the specificity for Calreticulin transacetylase (CRTAase) with respect to the number and positions of acetoxy groups on the benzenoid ring of coumarin revealed that acetoxy groups in proximity to the oxygen heteroatom (at C-7 and C-8 positions) demonstrate a high degree of specificity to CRTAase.11 The carbonyl group on the pyran nucleus of PAs was found absolutely essential as the polyphenolic acetates (PAs) devoid of carbonyl group at C-2/C-4 position were found to support negligible activity of CRTAase, when used as the substrate.27 It has also been observed that the acetoxy phenylcoumarins (having a phenyl ring instead of a methyl group at the C-4 position, yielded reduction of CRTAase activity while acetoxy dihydrocoumarin, had no profound effect on the CRTAase activity.27 We have also studied the influence of an alkyl group at the C-3 position of the acetoxy coumarins on the CRTAase activity. The increase in size of the alkyl group at the C-3 position of coumarin nucleus resulted in the reduction of CRTAase activity and related effects.13 In our previous studies we reported that acetoxy coumarins and quinolones are endowed with antiplatelet action by virtue of causing CRTAase catalyzed activation of platelet Nitric Oxide Synthase (NOS) by way of acetylation leading to the inhibition of ADP/AA-dependent platelet aggregation.
Herein we sought to correlate for the first time the ability of amino containing heterocyclic compounds viz. N-acetylaminocoumarins/quinolones to inhibit platelet aggregation with special reference to the specificity of platelet CRTAase to these derivatives and NOS activation. The results presented in the Figure 1 revealed that the acetoxy derivatives of coumarins and quinolones are the substrates for platelet CRTAase to varying degrees. Among all the O/N-acetyl derivatives screened, 7-AAMC (17) was found to be the most suitable substrate to platelet CRTAase that activated platelet NOS to a greater extent compared to the other compounds resulting in inhibition of ADP/AA induced platelet aggregation. These results depict a structural activity relation where the affinity for various N-acetylamino/acetoxy derivatives is in the order: N-acetylaminocoumarins > acetoxy quinolones > N-acetylaminoquinolones > acetyl coumarins. The results show that the N-acetylamino functionality on the benzenoid ring of coumarins plays a crucial role in enhancing its specificity of platelet CRTAase, however it does not have any significance in case of quinolones. It is the position of acetoxy group on the benzenoid ring of quinolones that dictates their specificity for platelet CRTAase. Further C-6/C-8 acetyl derivatives of coumarins do not have much effect on the CRTAase activity. It is thus evident from the results (Figure 1) that the addition of alkyl group at C-3 position of coumarins and quinolones results in drastic reduction of CRTAase activity, and this effect increases with the size of alkyl chain. Also, the replacement of C-4 methyl group by trifloromethyl group resulted in the significant decline of CRTAase activity of platelets.
�

The ability of 7-AAMC (17) to inhibit platelet aggregation can be correlated to the enhancement of NO levels in blood platelets. The enhanced NO formation in blood vessels is known to regulate the vascular functions.26 The blunting of Cox-1 activity and eventually TxA2, an AA metabolite acting as an endogenous platelet activator, intensifies the extent of inhibition of platelet aggregation. It is noteworthy that 7-AAMC, inhibited the Cox-1 activity even better than aspirin which is so far considered to be the best antiplatelet inhibitor. While its deacetylated analog i.e. 7-amino-4-methylcoumarin (12), the synthetic precursor of 7-AAMC was totally ineffective for the inhibition of platelet aggregation and the similar pattern was observed for Cox-1 activity too. These observations highlighted the crucial role of N-acetylamino group of coumarin for activation of platelet NOS through acetylation leading to the antiplatelet action of acetylamino derivatives of coumarins. The expression of LPS induced iNOS was measured in isolated rat PBMCs. In these models, the increased iNOS production correlates with the vascular injury.28 TF expression is also induced in circulating monocytes by bacterial LPS and pro inflammatory cytokines. Binding of factor VII to the extracellular domain of TF catalyzes the generation of factor Xa and IXa, triggering thrombin generation.29 7-AAMC treated samples showed apparent decrease in LPS induced TF expression in PBMCs. However, the toxicity of 7-AAMC if any, has to be ascertained before they are chosen for antiplatelet therapy. The results documented in this manuscript have projected for the first time the antiplatelet action of a 7-AAMC implicating the cardinal role of CRTAase in the mechanism of action.
4. Conclusion
Earlier studies from our laboratory for the first time focused the attention on the versatility of polyphenolic acetates from the point of view of CRTAase mediated acetylation of functional proteins leading to the expression of biological and pharmacological effects. We have already demonstrated the ability of acetoxy derivatives of quinolones as the possible antiplatelet agents. In the present study we have taken the derivatives of coumarins, aminocoumarins and quinolones for the analysis of their antiplatelet activity. Among which 7-AAMC stood prominent and was found to overcome the thrombotic prone conditions effectively when compared to aspirin viz, inhibition of iNOS and TF. Thus, 7-AAMC could be further studied to evaluate the potentiality as an antiplatelet agent.
5. Experimental section
5.1. Chemistry
5.1.1. General
All the solvents were dried and distilled prior to their use. Reactions were monitored by precoated TLC plates (Merck silica gel 60F254); the spots were visualized either by UV light, or by spraying with 5% alcoholic FeCl3 solution. Silica gel (100-200 mesh) was used for column chromatography. Melting points were recorded in capillaries in suphuric acid bath and Buchi M-560 melting point apparatus. Infrared spectra were recorded on Perkin-Elmer FT-IR model 9 spectrophotometer. The 1H and 13C NMR spectra were recorded on Bruker AC-400 and Jeol-400 (400 MHz, 100.6 MHz) NMR spectrometer and Avance-300 (300 MHz, 75.5 MHz) spectrometer using TMS as internal standard. The chemical shift values are on � scale and the coupling constant values (J) are in Hz. The ESI MS of the known compounds and the HRMS of novel compounds were recorded on Agilent-6210 ES-TOF, JEOL JMX-SX-102A, Bruker Compass Data Analysis 4.0 (micrOTOF-Q II 10262) and Waters LCT Micromass-KC455. All the compounds synthesized were
either crystallized or purified through column chromatography using different solvents, the purity of all the compounds obtained was greater than 95%.
5.1.2. General procedure for synthesis of 3-hydroxyphenyl- urethane (1)
Ethyl chloroformate (10.0 g, 92 mmol) was added in one portion to a stirred suspension of m-aminophenol (10.0 g, 92 mmol) in 400 mL of anhydrous diethyl ether. A white precipitate (amine hydrochloride) formed immediately. The reaction mixture was stirred for 2 h at room temperature. The hydrochloride was removed by filtration. The filtrate was then evaporated to give grey colored solid. Further crystallization from petroleum ether (200 mL) gave upon cooling (0 oC) a white solid (7.0 g, 84%). Melting point = 91-92 oC (Literature value = 91-95 oC)15; UV (acetonitrile) �max: 279 and 285 nm; IR (KBr) �max: 3302.57 (NH), 3050.98, 2987.92, 1698.61 (NHCOO), 1624.18, 1558.07, 1474.68, 1251.02, 1099.55, 864.31 cm-1; 1H NMR (CDCl3, 300 MHz): � 1.30 (t, 3H, J = 7.0 Hz, H-3�), 4.22 (q, 2H, J = 7.0 Hz, H-2�), 6.56-6.64 (m, 2H, H-4 and H-6), 6.77 (s, 1H, H-2), 6.93 (brs, 1H, OH), 7.12 (t, 1H, J = 8.1 Hz, H-5), 7.37 (brs, 1H, NH); 13C NMR (CDCl3, 75.5 MHz): � 14.43 (C-3�), 61.68 (C-2�), 106.01, 110.47 and 110.76 (C-2, C-4 and C-6), 129.93 (C-5), 138.84 (C-1), 154.12 (C-3), 156.95 (C-1�). ESI MS: Calculated for C9H11NO3 [M]+. 181, found 181.
5.1.3. General procedure for the synthesis of 7-carbethoxy aminocoumarins (7-11)
A solution of 3-hydroxyphenylurethane 1 (7.0 g) and un/ substituted ethyl acetoacetate (2-6, 1.2 equivalent) suspended in 88 mL of 70% ethanolic H2SO4 was stirred at room temperature for 4-6 h. The product formation was monitored by TLC. On completion of the reaction the clear yellow solution was poured into 400 mL of ice cold water, giving a voluminous brown crystalline precipitate. The solid was filtered and then crystallized from ethanol to give 7-carbethoxy aminocoumarin (7-11).
5.1.3.1. 7-Carbethoxyamino-4-methylcoumarin (7)
The title compound (7) was prepared from ethyl acetoacetate (2) and 3-hydroxy phenylurethane (1) following the general procedure as light brown colour crystals in 83% yield. Melting point = 185-186 oC (Literature value = 186-188 oC)15; UV (acetonitrile) �max: 292 and 324 nm; IR (KBr) �max: 3289.27 (NH), 2976.16, 1730.12 (COO), 1701.80 (NHCOO), 1626.71, 1529.44, 1409.61, 1092.54, 1008.02, 862.66 cm-1; 1H NMR (DMSO-d6, 300 MHz): � 1.27 (t, 3H, J = 6.9 Hz, H-3�), 2.38 (s, 3H, C-4 CH3), 4.17 (q, 2H, J = 6.9 Hz, H-2�), 6.22 (s, 1H, H-3), 7.40 (d,1H, J = 8.7 Hz, H-6), 7.54 (s, 1H, H-8), 7.67 (d, 1H, J = 8.7 Hz, H-5), 10.12 (s,1H, NH); 13C NMR (DMSO-d6, 75.5 MHz): � 14.36 (C-3�), 17.92 (C-4 CH3), 60.68 (C-2�), 104.37, 111.78, 114.24 and 114.74 (C-3, C-6, C-8 and C-10), 125.91 (C-5), 142.82 (C-7), 153.18 and 153.32 (C-4 and C-9), 153.80 (C-1�), 160.05 (C-2); ESI MS: Calculated for C13H13NO4 [M]+. 247, found 247.
5.1.3.2. 7-Carbethoxyamino-3-ethyl-4-methylcoumarin (8)
The title compound (8) was prepared from ethyl 2-acetylbutanoate (3) and 3-hydroxyphenylurethane (1) by following the general procedure and was obtained as brown colour crystals in 42% yield. Melting point = 158-160 oC; UV (acetonitrile) �max: 295 and 324 nm; IR (KBr) �max: 3287.54 (NH), 2923.38, 2854.63, 1729.65 (COO), 1682.13 (NHCOO), 1623.45, 1539.28, 1458.34, 1235.06, 1013.77, 863.25 cm-1; 1H NMR (CDCl3, 400 MHz): � 1.12 (t, 3H, J = 7.6 Hz, -CH2CH3), 1.30 (t, 3H, J = 7.2 Hz, H-3�), 2.38 (s, 3H, C-4 CH3), 2.65 (q, 2H, J = 7.6 Hz, -CH2CH3), 4.23 (q, 2H, J = 7.0 Hz, H-2�), 7.12 (brs, 1H,

NH), 7.36 (d,1H, J = 8.4 Hz, H-6), 7.41 (d, 1H, J = 2.0 Hz, H-8), 7.49 (d, 1H, J = 8.8 Hz, H-5); 13C NMR (CDCl3, 100.6 MHz): � 13.18 (C-3�), 14.56 and 14.59 (-CH2CH3 and C-4 CH3), 20.97 (-CH2CH3), 61.73 (C-2�), 105.72, 114.43 and 116.231 (C-6, C-8 and C-10), 125.22 (C-3), 126.12 (C-5), 140.53 (C-7), 145.72 (C-4) 152.90 (C-9), 153.31 (C-1�), 162.05 (C-2); HRMS: Calculated for C15H17NO4 [M+H]+ 276.1158, found 276.1225.
5.1.3.3. 7-Carbethoxyamino-3-hexyl-4-methylcoumarin (9)
The title compound (9) was prepared from ethyl 2-acetyloctanoate (4) and 3-hydroxy phenylurethane (1) by following the general procedure and was obtained as brown colour crystals in 31% yield. Melting point = 138-140 oC; UV (acetonitrile) �max: 295 and 324 nm; IR (KBr) �max: 3257.82 (NH), 3103.62, 2924.17, 1733.34 (COO), 1682.12 (NHCOO), 1612.72, 1537.51, 1459.71, 1081.78, 876.20 cm-1; 1H NMR (Acetone-d6, 400 MHz): � 0.85 (brs, 3H, -CH2(CH2)4CH3), 1.23 (t, 3H, J = 7.6 Hz, H-3�), 1.29-1.49 (m, 8H, -CH2(CH2)4CH3), 2.39 (s, 3H, C-4 CH3), 2.58 (t, 2H, J = 7.8 Hz, -CH2(CH2)4CH3), 4.15 (q, 2H, J = 6.8 Hz, H-2�), 7.42 (dd, 1H, J = 2.4 and 8.8 Hz, H-6), 7.59 (d, 1H, J = 2.4 Hz, H-8) 7.62 (d, 1H, J = 8.8 Hz, H-5), 8.99 (brs, 1H, NH); 13C NMR (Acetone-d6, 100.6 MHz): 13.51 (C-3�), 13.98 (-CH2(CH2)4CH3 and C-4 CH3), 22.46, 27.26, 28.63, 29.21, 31.60 (-(CH2)5CH3), 60.79 (C-2�), 104.63, 114.15 and 115.52 (C-6, C-8 and C-10), 124.20 (C-3), 125.50 (C-5), 141.85 (C-7), 145.86 (C-4), 153.06 (C-9), 154.06 (C-1�), 160.96 (C-2); HRMS: Calculated for C19H25NO4 [M+H]+ 332.1856, found 332.1851.
5.1.3.4. 7-Carbethoxyamino-3-decyl-4-methylcoumarin (10)
The title compound (10) was prepared from ethyl 2-acetyldodecanoate (5) and 3-hydroxyphenylurethane (1) following the general procedure and was obtained as brown colour crystals in 28% yield. Melting point = 128-130 oC; UV (acetonitrile) �max: 295 and 324 nm; IR (KBr) �max: 3259.95 (NH), 3102.62, 2923.44, 1732.62 (COO), 1683.01 (NHCO), 1609.53, 1537.96, 1231.90, 1081.86, 875.82 cm-1; 1H NMR (DMSO-d6, 300 MHz): � 0.83 (brs, 3H, -CH2(CH2)8CH3), 1.21-1.39 (m, 19H, H-3� and -CH2(CH2)8CH3), 2.34 (s, 3H, C-4 CH3), 2.50 (brs, 2H, -CH2(CH2)4CH3), 4.14 (q, 2H, J = 6.8 Hz, H-2�), 7.36 (d, 1H, J = 8.1 Hz, H-6), 7.50 (s, 1H, H-8), 7.65 (d, 1H, J = 8.7 Hz, H-5), 10.06 (brs, 1H, NH); 13C NMR (DMSO-d6, 75.5 MHz): 13.92 (C-3�), 14.41 and 14.44 (-CH2(CH2)8CH3 and C-4 CH3), 22.09, 23.21, 26.83, 28.16, 28.70, 28.86, 28.98, 29.63, 31.28 (-(CH2)9CH3), 60.59 (C-2�), 104.10, 114.17 and 114.80 (C-6, C-8 and C-10), 123.19 (C-3), 125.72 (C-5), 141.74 (C-7), 146.43 (C-4), 152.24 (C-9), 153.32 (C-1�), 160.82 (C-2); HRMS: Calculated for C23H33NO4 [M+H]+ 388.2443, found 388.2482.
5.1.3.5. 7-Carbethoxyamino-4-trifluoromethylcoumarin (11)
The title compound (11) was prepared from 3-hydroxyphenylurethane (1) and ethyl 4,4,4-trifluoromethyl acetoacetate (6) by following the general procedure as colourless crystals in 30% yield. Melting point = 170 oC decomposed (Literature value = 172 oC)30; UV (acetonitrile) �max: 336 nm; IR (KBr) �max: 3315.20 (NH), 3082.02, 2987.55, 1744.42 (COO), 1721.96 (NHCOO), 1622.16, 1591.88, 1482.06, 1285.01, 1074.13, 867.22 cm-1; 1H NMR (CDCl3 + DMSO-d6, 300 MHz): � 1.33 (t, 3H, J = 7.2 Hz, H-3�), 4.26 (q, 2H, J = 7.1 Hz, H-2), 6.61 (s, 1H, H-3), 7.48 (dd, 1H, J = 1.5 and 9.0 Hz, H-6), 7.58 (d, 1H, J = 9.0 Hz, H-5), 7.76 (d, 1H, J = 1.5 Hz, H-8), 9.53 (s,1H, NH); 13C NMR (CDCl3 + DMSO-d6, 75.5 MHz): � 14.12 (C-3�), 60.83 (C-2�), 105.49, 107.48, 112.12 and 115.07 (C-3,C-6, C-8 and C-10), 122.99 (C-5), 125.10 (d, J = 32.4 Hz, -CF3), 141.13 - 140.70 (q, J = 275.5 Hz, C-4), 143.92 (C-7), 153.16 (C-9), 155.00 (C-1�), 158.97 (C-2); ESI MS: Calculated for C13H10F3NO4 [M]+. 301, found 301.
5.1.4. General procedure for the synthesis of 7-aminocoumarins (12-16)
7-Carbethoxy aminocoumarins 7-11 (5.0 g) were refluxed for 4 h in a mixture of concentrated H2SO4 and glacial acetic acid (1:1, 10 ml). On cooling a yellow precipitate was deposited. The mixture was poured over 100 mL of ice cold water and allowed to stand overnight. The resulting suspension was made slightly alkaline with 50% aqueous NaOH under cold conditions. The brown precipitate formed was then filtered and washed with ice cold water (3 x 50 mL). Crystallization from ethanol yielded light brown colored crystals.
5.1.4.1. 7-Amino-4-methylcoumarin (12)
The title compound (12) was obtained as brown colour crystals in 72% yield by following the general procedure. Melting point = 220-222 oC (Literature value = 220-224 oC)15; UV (acetonitrile) �max: 296 and 326 nm; IR (KBr) �max: 3412.00, 3300.14 (NH2), 3113.42, 2924.94, 1681.89 (COO), 1619.66, 1407.59, 1261.96, 1076.10, 868.00 cm-1; 1H NMR (DMSO-d6, 400 MHz): � 2.28 (s, 3H, C-4 CH3), 5.88 (s, 1H, H-3), 6.09 (brs, 2H, NH2), 6.39 (s, 1H, H-8), 6.53-6.54 (m, 1H, H-6), 7.38 (d, 1H, J = 8.8 Hz, H-5); 13C NMR (Acetone-d6, 100.6 MHz): � 18.57 (C-4 CH3), 99.06 (C-8), 108.02, 109.39 and 111.714 (C-3, C-6 and C-10), 126.75 (C-5), 153.62, 154.29 and 156.00 (C-4, C-7 and C-9), 161.28 (C-2); ESI MS: Calculated for C10H9NO2
[M+H]+ 176, found 176.
5.1.4.2. 7-Amino-3-ethyl-4-methylcoumarin (13)
The title compound (13) was obtained as brown colour crystals in 41% yield by following the general procedure. Melting point = 206-208 oC31; UV (acetonitrile) �max: 326 nm; IR (KBr) �max: 3454.05, 3356.11 (NH2), 3241.13, 3075.38, 2959.43, 1677.62 (COO), 1643.77, 1450.78, 1260.50, 1061.28, 857.59 cm-
1; 1H NMR (DMSO-d6, 400 MHz): � 1.23 (brs, 3H, -CH2CH3), 2.32 (s, 3H, C-4 CH3), 2.48 (m, 2H, -CH2CH3), 7.34-7.63 (m, 3H, H-5, H-6 and H-8); 13C NMR (Acetone-d6, 100 MHz): � 13.45 and 14.95 (-CH2CH3 and C-4 CH3), 20.78 (-CH2CH3), 99.99 (C-8), 114.69 and 115.33 (C-6 and C-10), 124.94 (C-3), 126.26 (C-5), 146.74 (C-4), 152.76 and 153.85 (C-7 and C-9), 161.19 (C-2); ESI MS: Calculated for C12H13NO2 [M]+. 203, found 203.
5.1.4.3. 7-Amino-3-hexyl-4-methylcoumarin (14)32
The title compound (14) was obtained as brown colour crystals in 26% yield by following the general procedure. Melting point = 198-200 oC; UV (acetonitrile) �max: 348 nm; IR (KBr) �max: 3447.78, 3356.61 (NH2), 3245.31, 2953.29, 2923.26, 1678.16 (COO), 1458.82, 1258.38, 1073.04, 857.62 cm-1; 1H NMR (DMSO-d6, 400 MHz): � 0.81 (brs, 3H, -CH2(CH2)4CH3), 1.23-1.34 (m, 8H, -CH2(CH2)4CH3), 2.24 (s, 3H, C-4 CH3), 2.42 (brs, 2H, -CH2(CH2)4CH3), 5.90 (brs, 2H, NH2), 6.34 (s, 1H, H-8), 6.51 (d, 1H, J = 8.4 Hz, H-6), 7.36 (d, 1H, J = 8.4 Hz, H-5); 13C NMR (DMSO-d6, 100.6 MHz): � 14.49 and 14.88 (-CH2(CH2)4CH3 and C-4 CH3), 22.62, 27.19, 28.97, 29.21, 31.69 (-(CH2)5CH3), 98.94 (C-8), 109.96 and 111.75 (C-6 and C-10), 119.34 (C-3), 126.51 (C-5), 147.80 (C-4), 152.45 and 154.32 (C-7 and C-9), 161.90 (C-2); HRMS: Calculated for C16H21NO2
[M+H]+ 260.1572, found 260.1652.
5.1.4.4. 7-Amino-3-decyl-4-methylcoumarin (15)32
The title compound (15) was obtained as brown colour crystals in 30% yield by following the general procedure. Melting point = 172-174 oC; UV (acetonitrile) �max: 339 nm; IR (KBr) �max: 3451.51, 3358.51 (NH2), 3246.34, 3091.60, 2956.00, 1676.82 (COO), 1458.40, 1263.87, 1072.48, 857.50 cm-1; 1H NMR (DMSO-d6, 300 MHz): � 0.85 (brs, 3H, -CH2(CH2)8CH3),

1.23-1.39 (m, 16H, -CH2(CH2)8CH3), 2.28 (s, 3H, C-4 CH3), 2.45-2.47 (m, 2H, -CH2(CH2)8CH3), 5.93 (brs, 2H, NH2), 6.39 (s, 1H, H-8), 6.56 (d, 1H, J = 8.4 Hz, H-6), 7.40 (d, 1H, J = 8.4 Hz, H-5); 13C NMR (DMSO-d6, 75.5 MHz): � 13.86 and 14.27 (-CH2(CH2)8CH3 and C-4 CH3), 22.02, 22.02, 26.58, 28.37, 28.63, 28.84, 28.92, 28.92, 31.22 (-(CH2)9CH3), 98.39 (C-8), 109.41 and 111.17 (C-6 and C-10), 118.80 (C-3), 126.89 (C-5), 147.17 (C-4), 151.85 and 153.74 (C-7 and C-9), 161.37 (C-2); HRMS: Calculated for C20H29NO2 [M+Na]+ 338.2096, found 338.2071.
5.1.4.5. 7-Amino-4-trifluoromethylcoumarin (16)
The title compound (16) was obtained as light brown colour crystals in 46% by following the general procedure. Melting point = 225-227 oC. (Literature value = 222 oC)30; UV (acetonitrile) �max: 336 nm; IR (KBr) �max: 3455.49, 3362.91 (NH2), 3092.60, 2710.67, 1710.36 (COO), 1451.57, 1222.50, 998.15, 852.77 cm-1; 1H NMR (Methanol-d4, 400 MHz): � 6.36 (s, 1H, H-3), 6.54 (d, 1H, J = 2.2 Hz, H-8), 6.64 (dd, 1H, J = 2.3 and 8.7 Hz, H-6), 7.37-7.41 (m, 1H, H-5); 13C NMR (Methanol-d4, 100.6 MHz): � 100.64 (C-8), 104.04, 108.54 and 113.51 (C-3, C-6 and C-10), 123.50 (C-5), 127.19 (d, J = 277.2 Hz, -CF3), 142.70 - 143.69 (q, J = 32.4 Hz, C-4), 155.62 and 158.35 (C-7 and C-9), 162.26 (C-2); ESI MS: Calculated for C10H6F3NO2
[M]+. 229, found 229.
5.1.5. General procedure for the synthesis of 7-N-acetylamino coumarins (17-21)
A mixture of 7-aminocoumarin (500 mg, 12-16) and DMAP (10-20 mg) was dissolved in minimum amount of THF. To this acetic anhydride was added and the resultant mixture was stirred at room temperature for 24 h. On completion of the reaction 50 mL ice cold water was added. The crude off-white solid was then filtered, washed with water and dried. The crude solid so obtained was crystallized with ethanol to give 7-N-acetylaminocoumarin derivatives.
5.1.5.1. 7-N-Acetylamino-4-methylcoumarin (17)
The title compound (17) was obtained as colourless crystals in 72% yield by following the general procedure. Melting point = 200-204 oC33; UV (acetonitrile) �max: 325 nm; IR (Nujol) �max: 3299.12 (NH), 3112.77, 2922.62, 1715.58 (COO), 1682.19 (NHCO), 1619.76, 1587.24, 1463.26, 1262.01, 1076.43, 868.84 cm-1; 1H NMR (DMSO-d6, 300 MHz): � 2.10 (s, 3H, H-2�), 2.39 (s, 3H, C-4 CH3), 6.25 (s, 1H, H-3), 7.45 (d, 1H, J = 8.4 Hz, H-6), 7.70 (d, 1H, J = 8.7 Hz, H-5), 7.75 (s, 1H, H-8), 10.38 (brs, 1H, NH); 13C NMR (DMSO-d6, 75.5 MHz): � 17.87 (C-4 CH3), 24.11 (C-2�), 105.30, 112.04, 114.72 and 114.89 (C-3, C-6, C-8 and C-10), 125.73 (C-5), 142.53 (C-7), 152.97 and 153.61 (C-4 and C-9), 159.95 (C-2), 168.99 (C-1�); ESI MS: Calculated for C12H11NO3 [M+H]+ 218, found 218.
5.1.5.2. 7-N-Acetylamino-3-ethyl-4-methylcoumarin (18)
The title compound (18) was obtained as off-white colour crystals in 67% yield by following the general procedure. Melting point = 180-182 oC; UV (acetonitrile) �max: 296 and 323 nm; IR (KBr) �max: 3289.04 (NH), 3109.24, 2974.59, 1716.33 (COO), 1689.51 (NHCO), 1618.41, 1585.07, 1427.01, 1250.93, 1089.60, 864.15 cm-1; 1H NMR (DMSO-d6, 300 MHz): � 1.08 (t, 3H, J = 7.5 Hz, -CH2CH3), 2.38 (s, 3H, H-2�), 2.45 (s, 3H, C-4 CH3), 2.58 (brs, 2H, -CH2CH3), 7.23 (d, 1H, J = 7.8 Hz, H-6), 7.34-7.41 (m, 1H, H-5), 7.53 (s, 1H, H-8), 10.05 (s, 1H, NH); 13C NMR (DMSO-d6, 75.5 MHz): � 12.87 and 14.14 (-CH2CH3 and C-4 CH3), 20.21 (-CH2CH3), 24.11 (C-2�), 105.08, 114.89 and 115.35 (C-6, C-8 and C-10), 124.70 (C-3), 125.64 (C-5), 141.48 (C-7), 146.13 (C-4), 152.04 (C-9), 160.64 (C-2), 168.93 (C-1�);
HRMS: Calculated for C14H15NO3 [M+H]+ 246.1085, found 246.1125.
5.1.5.3. 7-N-Acetylamino-3-hexyl-4-methylcoumarin (19)
The title compound (19) was obtained as light brown colour crystals in 60% yield by following the general procedure. Melting point = 160 oC; UV (acetonitrile) �max: 326 nm; IR (Nujol) �max: 3297.88 (NH), 3113.01, 2924.71, 1730.94 (COO), 1674.98 (NHCO), 1614.95, 1590.20, 1462.44, 1265.21, 1099.24, 860.80 cm-1; 1H NMR (CDCl3, 300 MHz): � 0.88 (brs, 3H, -CH2(CH2)4CH3), 1.25-1.52 (m, 8H, -CH2(CH2)4CH3), 2.25 (s, 3H, H-2�), 2.39 (s, 3H, C-4 CH3), 2.64 (t, 2H, J = 7.6 Hz, -CH2(CH2)4CH3), 7.54 (d, 1H, J = 8.7 Hz, H-6), 7.64 (s, 1H, H-8), 7.84 (d, 1H, J = 8.4 Hz, H-5), 8.35 (s, 1H, NH); 13C NMR (CDCl3, 75.5 MHz): � 13.87 and 14.42 (-CH2(CH2)4CH3 and C-4 CH3), 21.99, 24.09, 26.81, 28.09, 28.59, 31.04 (C-2�, - (CH2)5CH3), 105.09, 114.90 and 115.34 (C-6, C-8 and C-10), 123.49 (C-3), 125.64 (C-5), 141.17 (C-7), 146.38 (C-4), 152.04 (C-9), 160.80 (C-2), 168.92 (C-1�); HRMS: Calculated for C18H23NO3 [M+H]+ 302.1678, found 302.1746.
5.1.5.4. 7-N-Acetylamino-3-decyl-4-methylcoumarin (20)
The title compound (20) was obtained as light yellow colour crystals in 60% yield by following the general procedure. Melting point = 148-150 oC; UV (acetonitrile) �max: 296 and 326 nm; IR (Nujol) �max: 3321.03 (NH), 3192.27, 3107.29, 2922.50, 1732.71 (COO), 1673.95 (NHCO), 1624.77, 1614.53, 1536.35, 1256.52, 1083.00, 871.14 cm-1; 1H NMR (CDCl3, 300 MHz): � 0.87 (brs, 3H, -CH2(CH2)8CH3), 1.25-1.49 (m, 16H, -CH2(CH2)8CH3), 2.25 (s, 3H, H-2�), 2.40 (s, 3H, C-4 CH3), 2.64 (brs, 2H, -CH2(CH2)4CH3), 7.54 (d, 1H, J = 9.3 Hz, H-6), 7.64 (brs, 1H, H-5), 7.82 (s, 1H, H-8), 8.37 (s, 1H, NH); 13C NMR (CDCl3, 75.5 MHz): � 13.87 and 14.41 (-CH2(CH2)8CH3 and C-4 CH3), 20.99 (C-2�), 22.03, 24.10, 26.82, 28.10, 28.64, 28.81, 28.92, 28.92, 31.23 (-(CH2)9CH3), 105.12, 114.91 and 115.35 (C-6, C-8 and C-10), 123.50 (C-3), 125.58 (C-5), 141.48 (C-7), 146.33 (C-4), 152.06 (C-9), 160.80 (C-2), 168.91 (C-1�); HRMS: Calculated for C22H31NO3 [M+H]+ 358.2337, found 358.2377.
5.1.5.5. 7-N-Acetylamino-4-trifluoromethylcoumarin (21)
The title compound (21) was obtained as brown colour crystals in 76% yield by following the general procedure. Melting point = 178-180 oC (Literature value = 227 oC)30; UV (acetonitrile) �max: 337 nm; IR (KBr) �max: 3337.75 (NH), 3071.84, 2925.07, 1712.25 (COO), 1621.47 (NHCO), 1586.99, 1515.30, 1240.08, 1023.68, 858.46 cm-1; 1H NMR (DMSO-d6, 300 MHz): � 2.14 (s, 3H, H-2�), 6.89 (s, 1H, H-3), 7.53-7.67 (m, 2H, H-5 and H-6), 7.91 (s, 1H, H-8), 10.54 (brs, 1H, NH); 13C NMR (DMSO-d6, 75.5 MHz): � 24.19 (C-2�), 105.91, 107.98, 114.20 and 115.77 (C-3, C-6, C-8 and C-10), 123.49 (C-5), 125.35 (d, J = 276.3 Hz, -CF3), 138.98-139.82 (q, J = 31.7 Hz, C-4), 143.49 (C-7), 154.72 (C-9) 158.61 (C-2), 169.39 (C-1�); ESI MS: Calculated for C12H8F3NO3 [M+H]+ 272, found 272.
5.1.6. General procedure for the synthesis of 7-aminoquinolin-2(1H)-one (22-24)
1,3-Diaminobenzene (1.0 g, 9.3 mmol), was added to substituted ethyl acetoacetates (1.2 equivalent, 2-3, 6) and the mixture was refluxed for 20 h. It was then poured on ice (100 g) and the precipitate was filtered. The product was obtained through column chromatography using silica gel (100-200 mesh) in methanol/chloroform (1:99).
5.1.6.1. 7-Amino-4-methylquinolin-2(1H)-one (22)
The title compound (22) was obtained as yellow solid in 65% yield by following the above general procedure. Melting point:

271-272 oC (Literature value = 271 oC)34; UV (MeOH) �max: 335 and 352 nm; IR (KBr) �max: 3423.29, 3307.93 (NH2), 2920.17, 1656.23 (NHCO), 1553.03, 1474.45, 1259.63, 1069.35, 914.48, 839.04, 689.74 cm-1; 1H NMR (DMSO-d6, 300 MHz): � 2.28 (s, 3H, C-4 CH3), 5.74 (brs, 2H, NH2), 5.96 (s, 1H, H-3), 6.37 (s, 1H, H-8), 6.46 (d, 1H, J = 8.4 Hz, H-6), 7.34 (d, 1H, J = 8.7 Hz, H-5), 11.18 (brs, 1H, NH); 13C NMR (DMSO-d6, 75.5 MHz): � 18.55 (C-4 CH3), 96.85, 110.48, 110.55 and 114.67 (C-3, C-6, C-8 and C-10), 125.67 (C-5), 140.81 (C-9), 148.13 (C-4), 151.15 (C-7), 162.55 (C-2); ESI MS: Calculated for C10H10N2O [M]+ 174, found 174.
5.1.6.2. 7-Amino-3-ethyl-4-methylquinolin-2(1H)-one (23)
The title compound (23) was obtained as yellow solid in 65% yield by following the above general procedure. Melting point: 280-281 oC; UV (MeOH) �max: 336 and 349 nm; IR (KBr) �max: 3459.26, 3363.28 (NH2), 2962.89, 1624.72 (NHCO), 1555.37, 1417.38, 1333.30, 1263.33, 1058.13, 879.67, 781.94, 690.30 cm-
1; 1H NMR (DMSO-d6, 300 MHz): � 1.00 (t, 3H, J = 7.4 Hz, -CH2CH3), 2.30 (s, 3H, C-4 CH3), 2.56 (q, 2H, J = 7.4 Hz, -CH2CH3), 5.60 (brs, 2H, NH2), 6.3 (s, 1H, H-8), 6.47 (dd, 1H, J = 1.2 and 8.4 Hz, H-6), 7.38 (d, 1H, J = 8.7 Hz, H-5), 11.19 (brs, 1H, NH); 13C NMR (DMSO-d6, 75.5 MHz): � 13.73 and 14.30 (-CH2CH3 and C-4 CH3), 19.37 (-CH2CH3), 96.78, 110.53 and 110.95 (C-6, C-8 and C-10), 125.44 (C-3), 126.17 (C-5), 139.05 (C-9), 141.99 (C-4), 150.05 (C-7), 162.14 (C-2). HRMS: Calculated for C12H14N2O [M+Na]+ 225.1004, found 225.0998.
5.1.6.3. 7-Amino-4-(trifluoromethyl)quinolin-2(1H)-one (24)
The title compound (24) was obtained as yellow solid in 70% yield by following the above general procedure. Melting point: 274 oC (Literature value = 274 oC)35; UV (MeOH) �max: 352 nm; IR (KBr) �max: 3365.74, 3239.82 (NH2), 2930.56, 1647.95 (NHCO), 1554.42, 1478.31, 1282.65, 1259.44, 1175.51, 1130.41, 969.84, 868.46, 659.01 cm-1; 1H NMR (DMSO-d6, 300 MHz): � 6.06 (brs, 2H, NH2), 6.34, 6.37 (2 × s, 2H, H-3 and H-8), 6.47 (d, 1H, J = 8.4 Hz, H-6), 7.26 (d, 1H, J = 6.9 Hz, H-5), 11.73 (brs, 1H, NH); 13C NMR (DMSO-d6, 75.5 MHz): � 96.67, 103.46 (C-6 and C-8), 111.79 and 113.42 (C-3 and C-10), 121.02 (C-5), 125.08 (C-9), 136.53-137.33 (-CF3), 142.11 (C-4), 152.07 (C-7), 160.87 (C-2); HRMS: Calculated for C10H7F3N2O [M+H]+ 229.0583, found 229.0582.
5.1.7. General procedure for the synthesis of N-(2-oxo-1,2-dihydroquinolin-7-yl)acetamides (25-27)
7-Aminoquinolin-2(1H)-ones (1.0 g, 22-24) were added to a solution of acetic anhydride and acetic acid (1:4, 10 ml). The mixture was refluxed for 6 h and poured on ice. The precipitate was filtered and washed with water and ether to yield 7-N-acetylquinolin-2-ones (25-27).36
5.1.7.1. N-(4-Methyl-2-oxo-1,2-dihydroquinolin-7-yl) acetamide (25)
The title compound (25) was obtained as brown crystals in 60% yield by following the general procedure. Melting point: 300 oC (Literature value = 300 oC)34; UV (MeOH) �max: 331 and 347 nm; IR (KBr) �max: 3417.52 (NH), 3130.05, 1673.47 (NHCOCH3), 1644.18 (NHCO), 1574.00, 1453.17, 1281.91, 1074.76, 987.98, 812.16, 683.74 cm-1; 1H NMR (DMSO-d6, 300 MHz): � 2.09 (s, 3H, H-2�), 2.38 (s, 3H, C-4 CH3), 6.26 (s, 1H, H-3), 7.30 (d, 1H, J = 8.7 Hz, H-6), 7.62 (d, 1H, J = 8.7 Hz, H-5), 7.99 (s, 1H, H-8), 10.197, 11.53 (2 x brs, 2H, 2 x NHCO); 13C NMR (DMSO-d6, 75.5 MHz): � 18.44 (C-4 CH3), 24.18 (C-2�), 104.37 and 113.48 (C-6 and C-8), 115.46 and 118.89 (C-3 and C-10), 125.31 (C-5), 139.51 (C-9), 141.05 (C-4), 147.74 (C-7),
162.14 (C-2), 168.85 (C-1�); ESI MS: Calculated for C12H12N2O2 [M]+. 216, found 216.
5.1.7.2. N-(3-Ethyl-4-methyl-2-oxo-1,2-dihydroquinolin-7-yl) acetamide (26)
The title compound (26) was obtained as brown crystals in 60% yield by following the general procedure. Melting point: 305-306 oC; UV (MeOH) �max: 332 and 343 nm; IR (KBr) �max: 3448.26 (NH), 3191.35, 2964.96, 1680.38 (NHCOCH3), 1641.46 (NHCO), 1585.52, 1458.61, 1267.91, 1099.42, 956.61, 808.78, 622.31 cm-1; 1H NMR (DMSO-d6, 300 MHz): � 1.02 (t, 3H, J = 7.2 Hz, -CH2CH3), 2.08 (s, 3H, H-2�), 2.37 (s, 3H, C-4 CH3), 2.61 (q, 2H, J = 6.9 Hz, -CH2CH3), 7.29 (d, 1H, J = 8.7 Hz, H-6), 7.63 (d, 1H, J = 9.0 Hz, H-5), 7.73 (s, 1H, H-8), 10.12, 11.51 (2 x brs, 2H, 2 x NHCO); 13C NMR (DMSO-d6, 75.5 MHz): � 13.40 and 14.37 (-CH2CH3 and C-4 CH3), 19.56 (-CH2CH3), 24.16 (C-2�), 104.06, 113.36 and 115.90 (C-6, C-8 and C-10), 125.08 (C-3), 130.52 (C-5), 137.81 (C-9), 140.01 (C-4), 141.38 (C-7), 161.79 (C-2), 168.67 (C-1�); HRMS: Calculated for C14H16N2O2 [M+H]+ 245.1245, found 245.1285.
5.1.7.3. N-(2-Oxo-4-(trifluoromethyl)-1,2-dihydroquinolin-7-yl)acetamide (27)
The title compound (27) was obtained as brown crystals in 90% yield by following the general procedure. Melting point: > 300 oC35; UV (MeOH) �max: 345 nm; IR (KBr) �max: 3291.66 (NH), 3107.16, 2926.93, 1682.41 (NHCOCH3), 1658.36 (NHCO), 1612.70, 1586.81, 1468.85, 1278.43, 1250.44, 1011.56, 869.86, 654.62 cm-1; 1H NMR (DMSO-d6, 300 MHz): � 2.09 (s, 3H, H-2�), 6.79 (s, 1H, H-3), 7.35 (d, 1H, J = 9.0 Hz, H-6), 7.62 (d, 1H, J = 8.7 Hz, H-5), 7.98 (s, 1H, H-8); 13C NMR (DMSO-d6, 75.5 MHz): � 24.16 (C-2�), 104.71, 108.61 (C-6 and C-8), 114.62 (C-3), 119.16 (C-10), 120.76 (C-5), 124.84 (C-9), 136.11-136.93 (-CF3), 140.86 (C-4), 142.06 (C-7), 160.41 (C-2), 169.12 (C-1�); HRMS: Calculated for C12H9F3N2O2 [M+Na]+ 293.0508, found 293.0508.
5.1.8. Synthesis of methoxy quinolin-2(1H)-ones (28-29)
Anisidine (2 g, 16.0 mmol) was added dropwise to substituted ethyl acetoacetate (3 mL, 16.0 mmol), the mixture was refluxed for 12 h. The mixture was then cooled and poured on sodium carbonate solution. The compound was then extracted with 50 ml ethyl acetate and the solvent was evaporated. 70% Sulphuric acid (5 mL) was added to it and the solution was heated at 95 oC for 6 h. It was then poured on crushed ice (100 g) and the precipitate was filtered and washed with water and ether. The crude product so obtained was purified by column chromatography.
5.1.8.1. 6-Methoxy-4-(trifluoromethyl)quinolin-2(1H)-one (28)37
The product was obtained through column chromatography in ethyl acetate/petroleum ether (1:49). The title compound (28) was obtained as yellow solid in 70% yield by following the general procedure. Melting point: 270 oC; UV (MeOH) �max: 329 and 351 nm; IR (KBr) �max: 2925.60, 2854.60, 1617.99 (NHCO), 1560.66, 1522.43, 1496.43, 1420.29, 1385.36, 1279.82, 1255.71, 1227.75, 1193.02, 1152.09, 1102.61, 1029.68, 947.35, 917.65, 845.40, 822.41, 747.90, 725.71 and 623.49 cm-1; 1H NMR (DMSO-d6, 300 MHz): � 3.72 (s, 3H, -OCH3), 7.13 (s, 1H, H-3), 7.48-7.82 (m, 2H, H-5 and H-7), 7.95 (brs, 1H, H-8) and 12.14 (brs, 1H, NH); 13C NMR (DMSO-d6, 75.5 MHz): � 55.53 (-OCH3), 99.12, 100.01 (C-7 and C-5), 114.68 (C-3), 122.18 and 123.69 (C-8 and C-9), 130.67 (C-10), 143.96 (C-4), 144.06-145.09 (-CF3), 157.89 (C-6) and 161.55 (C-2); HRMS: Calculated for C11H8F3NO2 [M]+. 243.1819, found 243.5345.

5.1.8.2. 3-Butyl-6-methoxy-4-methylquinolin-2(1H)-one (29)
The title compound (29) was obtained through column chromatography in ethyl acetate/petroleum ether (1:4). It was obtained as grey solid in 70% yield following the general procedure. Melting point: 173-176 oC; UV (acetonitrile) �max: 210, 237, 239 and 349 nm; IR (KBr) �max: 2952.38, 2928.73, 2857.02, 1654.13 (NHCO), 1624.22 (NHCO), 1505.90, 1223.80, 1460.16, 1413.99, 1036.53, 825.91, 635.93 cm-1; 1H NMR (CDCl3, 400 MHz): � 0.88 (t, 3H, J = 7.2 Hz, -CH2(CH2)2CH3), 1.36-1.45 (m, 4H, -CH2(CH2)2CH3), 2.36 (s, 3H, C-4 CH3), 2.82 (t, 2H, J = 7.3 Hz, -CH2(CH2)4CH3), 3.76 (s, 3H, -OCH3), 6.98-7.00 (m, 2H, H-5 and H-7), 7.25 (d, 1H, J = 9.5 Hz, H-8), 12.34 (brs, 1H, NH); 13C NMR (CDCl3, 100.6 MHz): � 14.06 (-CH2(CH2)2CH3), 15.16 (C-4 CH3), 22.94, 26.80, 31.29 (-(CH2)3CH3), 55.65 (-OCH3), 106.48 and 117.30 (C-5 and C-7), 117.69 and 121.66 (C-8 and C-10), 131.42 (C-3), 132.04 (C-9), 142.31 (C-4), 154.77 (C-6), 163.49 (C-2); HRMS: Calculated for C15H20NO2 [M+H]+ 246.1449, found 246.1489.
5.1.8.3. 6-Methoxy-4-methyl-3-octylquinolin-2(1H)-one (30)
The title compound (30) was obtained through column chromatography in ethyl acetate/petroleum ether (1:9). It was obtained as white solid in 65% yield following the general procedure. Melting point: 118-120 oC; UV (acetonitrile) �max: 212, 237 and 351 nm; IR (KBr) �max: 2925.61, 2849.06, 1654.00 (NHCO), 1623.85 (NHCO), 1506.12, 1417.76, 1277.61, 1208.23, 1040.08, 855.67, 642.98 cm-1; 1H NMR (CDCl3, 400 MHz): � 0.77 (t, 3H, J = 6.6 Hz, -CH2(CH2)6CH3), 1.17-1.47 (m, 12H, -CH2(CH2)6CH3), 2.35 (s, 3H, C-4 CH3), 2.70 (t, 2H, J = 7.3 Hz, -CH2(CH2)6CH3), 3.75 (s, 3H, -OCH3), 6.96-6.98 (m, 2H, H-5 and H-7), 7.24-7.26 (m, 1H, H-8), 12.43 (brs, 1H, NH); 13C NMR (CDCl3, 100.6 MHz): � 14.10 (-CH2(CH2)6CH3), 15.18 (C-4 CH3), 22.65, 27.09, 29.11, 29.31, 29.55, 29.88, 31.91 (-(CH2)7CH3), 55.65 (-OCH3), 106.50, 117.33 (C-5 and C-7), 117.67 and 121.67 (C-8 and C-10), 131.46 (C-3), 132.10 (C-9), 142.27 (C-4), 154.78 (C-6), 163.49 (C-2); HRMS: Calculated for C19H27NO2 [M+H]+ 302.2075, found 302.2115.
5.1.8.4. 3-Decyl-6-Methoxy-4-methylquinolin-2(1H)-one (31)
The title compound (31) was obtained through column chromatography in ethyl acetate/petroleum ether (1:9). It was obtained as grey solid in 65% yield following the general procedure. Melting point: 112-114 oC; UV (acetonitrile) �max: 213, 238, 274 and 350 nm; IR (KBr) �max: 2924.60, 2854.04, 1652.28 (NHCO), 1623.77 (NHCO), 1504.18, 1460.98, 1223.22, 1039.48, 932.58, 636.69 cm-1; 1H NMR (CDCl3, 400 MHz): � 0.86 (t, 3H, J = 6.6 Hz, -CH2(CH2)8CH3), 1.25-1.59 (m, 16H, -CH2(CH2)8CH3), 2.45 (s, 3H, C-4 CH3), 2.81 (t, 2H, J = 7.6 Hz, -CH2(CH2)8CH3), 3.85 (s, 3H, -OCH3), 7.06-7.09 (m, 2H, H-5 and H-7), 7.36 (d, 1H, J = 9.5 Hz, H-8), 12.60 (brs, 1H, NH); 13C NMR (CDCl3, 100.6 MHz): � 14.09 (-CH2(CH2)8CH3), 15.18 (C-4 CH3), 22.66, 27.08, 29.34, 29.60, 29.65, 29.65, 29.74, 29.87, 31.89 (-(CH2)9CH3), 55.63 (-OCH3), 106.44, 117.35 (C-5 and C-7), 117.67 (C-8), 121.65 (C-10), 131.47 (C-3), 132.09 (C-9), 142.25 (C-4), 154.75 (C-6), 163.52 (C-2); HRMS: Calculated for C21H31NO2 [M+Na]+ 352.2252, found 352.2247.
5.1.8.5. 7-Methoxy-4-(trifluoromethyl)quinolin-2(1H)-one (32)
The title compound (32) was obtained through column chromatography in ethyl acetate/petroleum ether (1:49). It was obtained by as yellow solid in 80% yield following the general procedure. Melting point: 250-252 oC, (Literature value = 252 oC)30; UV (MeOH) �max: 337 nm; IR (KBr) �max: 2977.72, 2921.33, 2855.32, 1629.65 (NHCO), 1522.80, 1483.44, 1458.58,
1400.34, 1376.35, 1326.42, 1298.56, 1271.62, 1223.69, 1186.71, 1138.76, 1122.76, 1036.98, 1013.18, 970.18, 915.63, 895.30, 839.82, 810.73, 711.90, 669.89 and 616.21 cm-1; 1H NMR (DMSO-d6, 300 MHz): � 3.84 (s, 3H, -OCH3), 6.77 (s, 1H, H-3), 6.92-6.96 (m, 2H, H-6 and H-8), 7.61 (d, 1H, J = 8.4 Hz, H-5) and 12.20 (brs, 1H, NH); 13C NMR (DMSO-d6, 75.5 MHz): � 55.54 (-OCH3), 99.79, 107.01 (C-6 and C-8), 112.00 (C-3), 118.13 (C-10), 124.37 (C-5), 125.64 (C-9), 136.23-136.64 (-CF3), 141.75 (C-4), 160.38 (C-7) and 161.63 (C-2); HRMS: Calculated for C11H8F3NO2 [M+H]+ 244.1819, found 244.3801.
5.1.9. Synthesis of hydroxy quinolin-2(1H)-one (33-37)
Methoxy quinolin-2(1H)-ones (28-32) was dissolved in a solution of hydrobromic acid/acetic acid (7:3). The mixture was refluxed for 72 h and then poured on ice. The precipitate was then filtered and washed with water and ether.
5.1.9.1. 6-Hydroxy-4-(trifluoromethyl)quinolin-2(1H)-one (33)37
The title compound (33) was obtained as yellow solid in 70% yield by following the general procedure. Melting point: 268 oC; UV (MeOH) �max: 332 and 354 nm; IR (KBr) �max: 3587.74 (OH), 3105.12, 2992.77, 2812.77, 2655.46, 1610.31 (NHCO), 1568.30, 1529.48, 1499.70, 1459.35, 1387.53, 1295.46, 1260.18, 1233.69, 1154.25, 1105.03, 950.44, 926.72, 857.70, 832.05, 732.62, 721.92 and 625.53 cm-1; 1H NMR (DMSO-d6, 300 MHz): � 7.04 (s, 1H, H-3), 7.19-7.34 (m, 2H, H-5 and H-7), 7.95 (d, 1H, J = 6.6 Hz, H-8), 10.29 (brs, 1H, OH), 11.96 (brs, 1H, NH); 13C NMR (DMSO-d6, 75.5 MHz): � 99.54 and 102.94 (C-5 and C-7), 119.95 (C-3), 122.69 and 123.45 (C-8 and C-9), 130.70 (C-10), 143.04 (C-4), 144.02-144.48 (-CF3), 156.27 (C-6), 161.11 (C-2); HRMS: Calculated for C10H6F3NO2 [M+H]+ 230.0423, found 230.0424.
5.1.9.2. 3-Butyl-6-hydroxy-4-methylquinolin-2(1H)-one (34)
The title compound (34) was obtained as white solid in 80% yield following the general procedure. Melting point: 196-198 oC; UV (acetonitrile) �max: 274, 284 and 350 nm; IR (KBr) �max: 3203.29 (OH), 2954.34, 2929.36, 1647.69 (NHCO), 1623.66 (NHCO), 1505.05, 1280.15, 1196.18, 877.85, 713.34, 642.61 cm-
1; 1H NMR (DMSO-d6, 400 MHz): � 0.77 (t, 3H, J = 6.9 Hz, -CH2(CH2)2CH3), 1.21 - 1.24 (m 4H, -CH2(CH2)2CH3), 2.21 (s, 3H, C-4 CH3), 2.40 - 2.47 (m, 2H, -CH2(CH2)2CH3), 6.88 (dd, 1H, J = 2.2 and 8.4 Hz, H-7), 6.91 (s, 1H, H-5), 7.03 (d, 1H, J = 8.8 Hz, H-8), 9.07 (brs, 1H, OH), 11.31 (brs, 1H, NH); 13C NMR (DMSO-d6, 100.6 MHz): � 13.94 (-CH2(CH2)2CH3), 14.80 (C-4 CH3), 22.45, 26.27, 30.85 (-(CH2)3CH3), 108.55, 116.14 (C-5 and C-7), 118.31 and 121.00 (C-8 and C-10), 130.38 (C-9), 131.41 (C-3), 141.01 (C-4), 152.01 (C-6), 161.04 (C-2); HRMS: Calculated for C14H17NO2 [M+H]+ 232.1293, found 232.1332.
5.1.9.3. 6-Hydroxy-4-methyl-3-octylquinolin-2(1H)-one (35)
The title compound (35) was obtained as white solid in 80% yield following the general procedure. Melting point: 205-207 oC; UV (acetonitrile) �max: 207, 235, 274 and 351 nm; IR (KBr) �max: 3265.87 (OH), 2956.23, 2926.20, 2854.98, 1647.99 (NHCO), 1623.38 (NHCO), 1503.76, 1276.79, 1205.13, 859.03, 642.02 cm-1; 1H NMR (DMSO-d6, 400 MHz): � 0.71 (t, 3H, J = 5.4 Hz, -CH2(CH2)6CH3), 1.09-1.27 (m, 12H, -CH2(CH2)6CH3), 2.20 (s, 3H, C-4 CH3), 2.47 (t, 2H, J = 6.6 Hz, -CH2(CH2)6CH3), 6.82 (dd, 1H, J = 1.4 and 8.8 Hz, H-7), 6.91 (d, 1H, J = 1.4 Hz, H-5), 7.02 (d, 1H, J = 8.8 Hz, H-8), 9.22 (brs, 1H, OH), 11.31 (brs, 1H, NH); 13C NMR (DMSO-d6, 100.6 MHz): � 13.98 (-CH2(CH2)6CH3), 14.80 (C-4 CH3), 22.16, 26.54, 28.65, 28.78, 29.01, 29.35, 31.36 (-(CH2)7CH3), 108.54, 116.14 (C-5 and C-7), 118.29 and 121.00 (C-8 and C-10), 130.38 (C-9), 131.44 (C-3),

140.98 (C-4), 152.02 (C-6), 161.06 (C-2); HRMS: Calculated for C18H25NO2 [M+H]+ 288.1919, found 288.1958.
5.1.9.4. 3-Decyl-6-Hydroxy-4-methylquinolin-2(1H)-one (36)
The title compound (36) was obtained as white solid in 80% yield, following the general procedure. Melting point: 209-211 oC; UV (acetonitrile) �max: 210, 235, 276 and 352 nm; IR (KBr) �max: 3213.80 (OH), 2959.73, 2925.88, 2852.54, 1646.53 (NHCO), 1623.06 (NHCO), 1505.04, 1279.36, 1205.61, 880.57, 642.15 cm-1; 1H NMR (DMSO-d6, 400 MHz): � 0.81 (s, 3H, J = 6.6 Hz, -CH2(CH2)8CH3), 1.19-1.37 (m, 16H, -CH2(CH2)8CH3), 2.30 (s, 3H, C-4 CH3), 2.57 (t, 2H, J = 7.3 Hz, -CH2(CH2)8CH3), 6.91 (dd, 1H, J = 2.2 and J = 8.8 Hz, H-7), 7.00 (d, 1H, J = 2.2 Hz, H-5), 7.11 (d, 1H, J = 8.8 Hz, H-8), 9.28 (brs, 1H, OH), 11.40 (brs, 1H, NH); 13C NMR (DMSO-d6, 100.6 MHz): � 13.99 (-CH2(CH2)8CH3), 14.81 (C-4 CH3), 22.17, 26.56, 28.66, 28.79, 29.09, 29.09, 29.13, 29.35, 31.70 (-(CH2)9CH3), 108.53 and 116.12 (C-5 and C-7), 118.28 and 120.98 (C-8 and C-10), 130.39 (C-9), 131.44 (C-3), 140.92 (C-4), 152.01 (C-6), 161.04 (C-2); HRMS: Calculated for C20H29NO2 [M+Na]+ 338.2096, found 338.2091.
5.1.9.5. 7-Hydroxy-4-(trifluoromethyl)quinolin-2(1H)-one (37)
The title compound (37) was obtained as yellow solid in 70% yield by following the general procedure. Melting point: > 300 oC (Literature m.p. = > 300 oC)30; UV (MeOH) �max: 338 nm; IR (KBr) �max: 3421.52 (OH), 3092.43, 2928.06, 1665.48 (NHCO), 1624.06 (NHCO), 1550.55, 1474.27, 1435.70, 1417.71, 1367.57, 1292.79, 1265.70, 1230.75, 1205.67, 1159.83, 1140.13, 1020.97, 976.95, 873.58, 842.56, 818.90, 723.79 and 661.73 cm-1; 1H NMR (DMSO-d6, 300 MHz): � 6.68 (s, 1H, H-3), 6.76-6.82 (m, 2H, H-5 and H-8), 7.53 (dd, 1H, J = 2.1 and 8.7 Hz, H-6), 10.57 (brs, 1H, OH) and 12.08 (brs, 1H, NH); 13C NMR (DMSO-d6, 75.5 MHz): � 100.70 and 106.05 (C-6 and C-8), 112.89 (C-3), 117.04 (C-10), 120.80 (C-5), 124.44 (C-9), 125.75 (C-4), 136.34-136.75 (-CF3), 141.86 (C-7) and 160.45 (C-2); HRMS: Calculated for C10H6F3NO2 [M+H]+ 230.0423, found 230.0428.
5.1.10. Synthesis of 2-Oxo-1,2-dihydro-6/7-yl acetate (38-42)
Hydroxy quinolin-2(1H)-ones (33-37) were added to a solution of acetic anhydride and acetic acid (1:4). The mixture was refluxed for 6 h and poured on ice. The precipitate was filtered and washed with water and ether.
5.1.10.1. 2-Oxo-4-(trifluoromethyl)-1,2-dihydro-6-yl acetate (38)
The title compound (38) was obtained as white crystals in 90% yield by following the general procedure. Melting point: 296 oC; UV (MeOH) �max: 284 and 317 nm; IR (KBr) �max: 3435.52, 2935.89, 1781.96 (-OCO), 1607.63 (NHCO), 1574.73, 1509.30, 1474.64, 1368.73, 1277.44, 1175.13, 1138.89, 1097.65, 1067.80, 1053.62, 1011.83, 950.61, 931.82, 895.53, 839.20, 731.04 and 669.78 cm-1; 1H NMR (DMSO-d6, 300 MHz): � 2.37 (s, 3H, -OCOCH3), 7.81 (dd, 1H, J = 2.1 and 9.0 Hz, H-7), 7.98 (d, 1H, J = 2.1 Hz, H-5), 8.01 (s, 1H, H-3), 8.29 (brs, 1H, J = 9.3 Hz, H-8); 13C NMR (DMSO-d6, 75.5 MHz): � 20.88 (-OCOCH3), 110.36, 113.19 (C-7 and C-5), 119.30 (C-3), 123.53 (C-8), 128.06 (C-9), 131.20 (C-10), 146.23-147.18 (-CF3), 150.65 (C-4), 155.69 (C-6), 168.24 (C-2) and 169.05 (-OCOCH3); HRMS: Calculated for C12H8F3NO3 [M+H]+ 272.0456, found 271.9899.
5.1.10.2. 3-Butyl-4-methyl-2-oxo-1,2-dihydroquinolin-6-yl acetate (39)
The title compound (39) was obtained as white crystals in 83% yield following the general procedure. Melting point: 180-
182 oC; UV (acetonitrile) �max: 270, 279, 334 and 336 nm; IR (KBr) �max: 2954.84, 2929.81, 2861.11, 1761.82 (-OCO), 1662.99 (NHCO), 1503.03, 1206.71, 1180.61, 921.86 cm-1; 1H NMR (CDCl3, 400 MHz): � 0.95 (t, 3H, J = 6.9 Hz, -CH2(CH2)2CH3), 1.41-1.54 (m, 4H, -CH2(CH2)2CH3), 2.31 (s, 3H, C-4 CH3), 2.43 (s, 3H, -OCOCH3), 2.79 (t, 2H, J = 7.6 Hz, -CH2(CH2)2CH3), 7.16 (dd, 1H, J = 2.2 and 8.8 Hz, H-7), 7.37-7.39 (m, 2H, H-5 and H-8), 12.39 (brs, 1H, NH). 13C NMR (DMSO-d6, 100.6 MHz): � 14.02 (-CH2(CH2)2CH3), 15.11 (C-4 CH3), 21.07 (-OCOCH3), 22.89, 26.72, 31.17 (-(CH2)3CH3), 116.47, 116.99 (C-5 and C-7), 121.57 and 123.02 (C-8 and C-10), 132.53 (C-3), 134.65 (C-9), 142.41 (C-4), 145.31 (C-6), 163.81 (C-2), 169.87 (-OCOCH3); HRMS: Calculated for C16H19NO3 [M+Na]+ 296.1263, found 296.1257.
5.1.10.3. 4-Methyl-3-octyl-2-oxo-1,2-dihydroquinolin-6-yl acetate (40)
The title compound (40) was obtained as white crystals in 80% yield following the general procedure. Melting point: 139-141 oC; UV (acetonitrile) �max: 203, 231 and 333 nm; IR (KBr) �max: 2923.00, 2852.55, 1759.66 (-OCO), 1659.74 (NHCO), 1504.25, 1212.45, 1183.04, 919.92 cm-1; 1H NMR (CDCl3, 400 MHz): � 0.84-0.86 (m, 3H, -CH2(CH2)6CH3), 1.26-1.53 (m, 12H, -CH2(CH2)6CH3), 2.31 (s, 3H, C-4 CH3), 2.42 (s, 3H, OCOCH3), 2.77 (t, 2H, J = 7.3 Hz, -CH2(CH2)2CH3), 7.15 (d, 1H, J = 8.1 Hz, H-7), 7.37-7.40 (m, 2H, H-5 and H-8), 12.49 (brs, 1H, NH). 13C NMR (DMSO-d6, 100.6 MHz): � 14.09 (-CH2(CH2)6CH3), 15.12 (C-4 CH3), 21.07 (OCOCH3), 22.63, 27.00, 28.98, 29.28, 29.50, 29.82, 31.88 (-(CH2)7CH3), 116.46, 117.01 (C-5 and C-7), 121.56 and 122.99 (C-8 and C-10), 132.56 (C-3), 134.66 (C-9), 142.39 (C-4), 145.29 (C-6), 163.82 (C-2), 169.86 (OCOCH3); HRMS: Calculated for C20H27NO3 [M+H]+ 330.2024, found 330.2064.
5.1.10.4. 3-Decyl-4-Methyl-2-oxo-1,2-dihydroquinolin-6-yl acetate (41)
The title compound (41) was obtained as white crystals in 80% yield following the general procedure. Melting point: 130-132 oC; UV (acetonitrile) �max: 269, 279, 333 and 348 nm; IR (KBr) �max: 2922.31, 2851.35, 1760.48 (-OCO), 1660.86 (NHCO), 1505.10, 1217.86, 1012.71, 927.50, 629.66 cm-1; 1H NMR (CDCl3, 400 MHz): � 0.92 (t, 3H, J = 6.9 Hz, -CH2(CH2)8CH3), 1.32-1.60 (m, 16H, -CH2(CH2)8CH3), 2.38 (s, 3H, C-4 CH3), 2.49 (s, 3H, -OCOCH3), 2.85 (t, 2H, J = 7.7 Hz, -CH2(CH2)8CH3), 7.23 (dd, 1H, J = 2.2 and 8.8 Hz, H-7), 7.44-7.48 (m, 2H, H-5 and H-8), 12.64 (brs, 1H, NH). 13C NMR (CDCl3, 100.6 MHz): � 14.09 (-CH2(CH2)8CH3), 15.11 (C-4 CH3), 21.06 (-OCOCH3), 22.65, 26.99, 28.12, 28.29, 29.32, 29.56, 29.63, 29.83, 31.87 (-(CH2)9CH3), 116.44, 117.05 (C-5 and C-7), 121.56 and 122.99 (C-8 and C-10), 132.54 (C-3), 134.67 (C-9), 142.38 (C-4), 145.28 (C-6), 163.85 (C-2), 169.86 (-OCOCH3); HRMS: Calculated for C22H31NO3 [M+Na]+ 380.2202, found 380.2196.
5.1.10.5. 2-Oxo-4-(trifluoromethyl)-1,2-dihydro-7-yl acetate (42)
The title compound (42) was obtained as white crystals in 90% yield by following the general procedure. Melting point: 190 oC; UV (MeOH) �max: 336 and 275 nm; IR (KBr) �max: 3432.60, 2925.24, 2851.40, 1766.92 (-OCO), 1683.55 (NHCO), 1617.88, 1567.98, 1521.40, 1422.24, 1373.73, 1331.68, 1289.58, 1267.15, 1207.14, 1188.00, 1166.51, 1137.41, 1020.35, 976.43, 924.25, 884.45, 856.51, 825.17, 728.52 and 653.95 cm-1; 1H NMR (DMSO-d6, 300 MHz): � 2.32 (s, 3H, -OCOCH3), 6.97 (s, 1H, H-3), 7.12 (dd, 1H, J = 1.8 and 8.7 Hz, H-6), 7.19 (d, 1H, J = 1.8 Hz, H-8) and 7.73 (d, 1H, J = 8.7 Hz, H-5); 13C NMR (DMSO-d6, 75.5 MHz): � 20.98 (-OCOCH3), 108.94, 110.91 (C-

6 and C-8), 117.40 (C-3), 121.45 (C-10), 124.27 (C-5), 125.72 (C-9), 136.00-136.42 (CF3), 140.84 (C-4), 152.56 (C-7), 160.15 (C-2) and 168.84 (-OCOCH3); HRMS: Calculated for C12H8F3NO3 [M]+. 271.1920, found 271.3652.
5.1.11. Synthesis of 8/6-acetyl-7-hydroxycoumarin (44-47)
7-Acetoxy-4-methylcoumarin17 (2 g, 9.2 mmol) and anhydrous aluminium chloride (4.5 g, 34 mmol) were taken in a round bottom flask fitted with a reflux condenser. The temperature of the reaction mixture was raised quickly to 125 oC and then slowly over a period of 2 h to 170 oC. On completion of the reaction, crushed ice was added to the reaction mixture followed by the acidification using dilute hydrochloric acid. The crude product was filtered, washed with water followed by ether and then subjected to column chromatography (petroleum ether-ethyl acetate).
5.1.11.1. 8-Acetyl-7-hydroxy-4-methylcoumarin (44)
The title compound 44 was obtained as light yellow solid in 70% yield by column chromatography using ethyl acetate - petroleum ether (1:20) as an eluent; Melting point: 162-163 oC (Literature mp = 162-163 oC)18; UV (acetonitrile) �max: 211, 308 and 350 nm; IR (Nujol) �max: 2920.95, 2853.95, 1738.43 (COCH3), 1732.03 (CO), 1610.85, 1463.28, 1371.81, 1234.08, 1087.60, 1059.53, 878.03, 658.62 cm-1; 1H NMR (CDCl3, 400 MHz): � 2.36 (s, 3H, C-4 CH3), 2.86 (s, 3H, -COCH3), 6.08 (s, 1H, H-3), 6.82 (d, 1H, J = 8.8 Hz, H-6), 7.60 (d, 1H, J = 8.8 Hz, H-5), 13.48 (brs, 1H, OH); 13C NMR (CDCl3, 100.6 MHz): � 19.08 (C-4 CH3), 33.79 (-COCH3), 109.09 (C-8), 110.89, 111.73 and 114.96 (C-3, C-6 and C-10), 131.19 (C-5), 152.95 (C-9), 154.98 (C-4), 159.19 (C-7), 166.47 (C-2), 204.21 (COCH3). ESI MS: Calculated for C12H10O4 [M]+. 218, found 218.
5.1.11.2. 8-Acetyl-3-ethyl-7-hydroxy-4-methylcoumarin (45)
The title compound 45 was obtained as light yellow solid in 70% yield by column chromatography using ethyl acetate - petroleum ether (1:20) as an eluent. Melting point: 171-172 oC; UV (acetonitrile) �max: 240, 273, 305 and 350 nm; IR (KBr) �max: 2970.32, 2935.77, 1717.83 (CO), 1614.92, 1370.28, 1215.39, 1092.50, 834.73, 647.36, 469.32 cm -1; 1H NMR (CDCl3, 400 MHz): � 1.09 (t, 3H, J = 7.3 Hz, -CH2CH3), 2.33 (s, 3H, C-4 CH3), 2.59 (q, 2H, J = 7.3 Hz, -CH2CH3), 2.86 (s, 3H, -COCH3), 6.78 (d, 1H, J = 8.7 Hz, H-6), 7.60 (d, 1H, J = 9.5 Hz, H-5), 13.34 (brs, 1H, OH); 13C NMR (CDCl3, 100.6 MHz): � 12.90 (-CH2CH3), 14.77 (C-4 CH3), 20.57 (-CH2CH3), 33.82 (-COCH3), 108.88 (C-8), 112.46 and 114.62 (C-6 and C-10), 124.14 (C-3), 131.23 (C-5), 146.03 (C-4), 153.32 (C-9), 160.07 (C-7), 165.34 (C-2), 204.24 (-COCH3); HRMS: Calculated for C14H14O4 [M+H]+ 247.0926, found 247.0965.
5.1.11.3. 6-Acetyl-7-hydroxy-4-methylcoumarin (46)
The title compound 46 was obtained as light yellow solid in 70% yield by column chromatography using ethyl acetate - petroleum ether (1:10) as an eluent. Melting point: 210-211 oC (Literature value = 211 oC)38; UV (acetonitrile) �max: 257, 297 and 342 nm; IR (KBr) �max: 3438.76 (OH), 2357.39, 1738.11 (CO), 1614.66, 1369.57, 1308.01, 1233.59, 1059.21, 876.65, 657.39, 455.93 cm-1; 1H NMR (CDCl3, 400 MHz): � 2.42 (s, 3H, C-4 CH3), 2.68 (s, 3H, -COCH3), 6.15 (s, 1H, H-3), 6.82 (s, 1H, H-8), 7.94 (s, 1H, H-5), 12.61 (brs, 1H, OH); 13C NMR (CDCl3, 100.6 MHz): � 18.57 (C-4 CH3), 26.63 (-COCH3), 105.39 (C-8), 112.86 and 112.99 (C-3 and C-10), 117.07 (C-6), 128.12 (C-5), 151.62 (C-4), 158.71 (C-9), 159.88 (C-7), 165.31 (C-2), 203.30 (-COCH3); ESI MS: Calculated for C12H10O4 [M+H]+ 219, found 219.
5.1.11.4. 6-Acetyl-3-ethyl-7-hydroxy-4-methylcoumarin (47)
The title compound 47 was obtained as light yellow solid in 70% yield by column chromatography using ethyl acetate - petroleum ether (1:10) as an eluent. Melting point: 144-146 oC; UV (acetonitrile) �max: 210, 230, 256, 305 and 341 nm; IR (KBr) �max: 3413.02 (OH), 3057.27, 2973.31, 1721.02 (CO), 1646.41, 1386.40, 1254.88, 1167.11, 1058.49, 930.50, 805.41, 599.82 cm-
1; 1H NMR (CDCl3, 400 MHz): � 1.10 (t, 3H, J = 7.3 Hz, -CH2CH3), 2.38 (s, 3H, C-4 CH3), 2.61 (q, 2H, J = 7.3 Hz, -CH2CH3), 2.67 (s, 3H, -COCH3), 6.74 (s, 1H, H-8), 7.91 (s, 1H, H-5), 12.50 (brs, 1H, OH); 13C NMR (CDCl3, 100.6 MHz): � 13.03 (-CH2CH3), 14.77 (C-4 CH3), 20.86 (-CH2CH3), 26.57 (-COCH3), 104.72 (C-8), 113.52 and 116.92 (C-6 and C-10), 125.99 (C-3), 127.88 (C-5), 144.75 (C-4), 157.27 (C-9), 160.65 (C-7), 164.16 (C-2), 203.38 (-COCH3); HRMS: Calculated for C14H14O4 [M+Na]+ 269.0790, found 269.0784.
5.1.12. Synthesis of 7-acetoxy derivative of acetyl coumarin (48-51)
A mixture of 7-hydroxycoumarin (500 mg, 44-47) and DMAP (10-20 mg) were dissolved in minimum amount of THF. To this acetic anhydride (1.0 equivalent) was added and the resultant mixture was stirred at room temperature for 24 h. On completion of the reaction 50 mL ice cold water was added. The crude off-white solid was then filtered, washed with water and dried. The crude solid so obtained was crystallized with ethanol to give 7-acetoxycoumarin derivatives.
5.1.12.1. 7-Acetoxy-8-acetyl-4-methylcoumarin (48)
The title compound 48 was obtained as light yellow solid in 98% yield; Melting point: 188-190 oC; UV (acetonitrile) �max: 212, 274 and 313 nm; IR (KBr) �max: 3082.40, 2925.76, 1769.48 (-OCO), 1740.26 (COCH3), 1698.32 (CO), 1596.00, 1381.65, 1199.64, 1054.96, 864.42, 593.44 cm-1; 1H NMR (CDCl3, 400 MHz): � 2.29 (s, 3H, -OCOCH3), 2.45 (s, 3H, C-4 CH3), 2.68 (s, 3H, -COCH3), 6.31 (s, 1H, H-3), 7.09 (d, 1H, J = 8.0 Hz, H-6), 7.68 (d, 1H, J = 8.8 Hz, H-5); 13C NMR (CDCl3, 100.6 MHz): � 18.96 (C-4 CH3), 20.86 (-OCOCH3), 32.04 (-COCH3), 111.11 and 114.71 (C-3 and C-6), 118.12 (C-10), 123.31 (C-8), 126.60 (C-5), 149.43 (C-7), 151.05 (C-4), 151.92 (C-9), 159.06 (C-2), 168.87 (-OCOCH3), 197.87 (-COCH3); HRMS: Calculated for C14H12O5 [M+H]+ 261.0718, found 261.0757.
5.1.12.2. 7-Acetoxy-8-acetyl-3-ethyl-4-methylcoumarin (49)
The title compound 49 was obtained as light yellow solid in 98% yield; Melting point: 155-157 oC; UV (Acetonitrile) �max: 276, 2849 and 314 nm; IR (KBr) �max: 3085.89, 2982.69, 1778.16 (-OCO), 1717.48 (COCH3), 1701.29 (CO), 1595.10, 1371.81, 1185.78, 1093.23, 1054.47, 891.60, 590.73 cm-1; 1H NMR (CDCl3, 400 MHz): � 1.09 (t, 3H, J = 7.3 Hz, -CH2CH3), 2.28 (s, 3H, -OCOCH3), 2.42 (s, 3H, C-4 CH3), 2.66-2.72 (m, 5H, -CH2CH3 and -COCH3), 6.06 (d, 1H, J = 8.7 Hz, H-6), 7.67 (d, 1H, J = 8.8 Hz, H-5), 13C NMR (CDCl3, 100.6 MHz): � 12.94 (-CH2CH3), 14.81 (C-4 CH3), 20.84 (-CH2CH3), 21.03 (-OCOCH3), 32.07 (-COCH3), 118.80 and 118.91 (C-6 and C-10), 122.84 (C-3), 126.51 (C-8), 127.98 (C-5), 145.06 (C-4), 148.29 (C-7), 149.54 (C-9), 159.95 (C-2), 167.97 (-OCOCH3), 198.12 (-COCH3); HRMS: Calculated for C16H16O5 [M+Na]+ 311.0895, found 311.0890.
5.1.12.3. 7-Acetoxy-6-acetyl-4-methylcoumarin (50)
The title compound 50 was obtained as light yellow solid in 98% yield; Melting point: 169-171 oC; UV (acetonitrile) �max: 207, 251 and 313 nm; IR (KBr) �max: 3091.97, 2925.08, 1756.51 (-OCO), 1685.61 (COCH3), 1631.92 (CO), 1385.77, 1268.96,

1195.18, 1138.24, 1051.53, 915.02, 572.94 cm-1; 1H NMR (CDCl3, 400 MHz): � 2.40 (s, 3H, -OCOCH3), 2.49 (s, 3H, C-4 CH3), 2.61 (s, 3H, -COCH3), 6.32 (s, 1H, H-3), 7.11 (s, 1H, H-8), 8.08 (s, 1H, H-5); 13C NMR (CDCl3, 100.6 MHz): � 18.72 (C-4 CH3), 21.21 (-OCOCH3), 29.65 (-COCH3), 112.51 (C-3), 115.38 (C-8), 117.88 (C-10), 127.33 (C-5 and C-6), 151.66, 151.76 (C-4 and C-7), 156.22 (C-9), 159.50 (C-2), 168.77 (-OCOCH3), 195.64 (-COCH3); HRMS: Calculated for C14H12O5 [M+K]+ 299.0322, found 299.0316.
5.1.12.4. 7-Acetoxy-6-acetyl-3-ethyl-4-methylcoumarin (51)
The title compound 51 was obtained as light yellow solid in 98% yield; Melting point: 128-130 oC; UV (acetonitrile) �max: 250 and 312 nm; IR (KBr) �max: 3422.06, 2973.74, 1761.00 (-OCO), 1723.04 (COCH3), 1682.15 (CO), 1618.65, 1365.45, 1198.62, 1151.04, 1057.74, 909.97, 780.07, 532.01 cm-1; 1H NMR (CDCl3, 400 MHz): � 1.16 (t, 3H, J = 7.6 Hz, -CH2CH3), 2.39 (s, 3H, -OCOCH3), 2.46 (s, 3H, C-4 CH3), 2.61 (s, 3H, -COCH3), 2.69 (q, 2H, -CH2CH3), 7.07 (s, 1H, H-8), 8.09 (s, 1H, H-5); 13C NMR (CDCl3, 100.6 MHz): � 12.95 (-CH2CH3), 14.66 (C-4 CH3), 21.12 (-CH2CH3), 21.22 (-OCOCH3), 29.65 (-COCH3), 112.04 (C-8), 118.62 (C-10), 127.05 (C-3), 127.24 (C-5), 128.70 (C-6), 144.76 (C-4), 150.72 (C-7), 154.77 (C-9), 160.46 (C-2), 168.91 (-OCOCH3), 195.86 (-COCH3); HRMS: Calculated for C16H16O5 [M+H]+ 289.1031, found 289.1071.
5.2. Biological section
5.2.1. Assay of platelet CRTAase
The assay mixture consisted of 0.25 M potassium phosphate buffer (pH 6.5), washed platelet lysate (20 µg protein), test compound (50 �M) added in 10 �L of methanol, to make up the final volume of 0.8 mL. The contents of the tube, scaled up as per the requirement were preincubated at 37 ºC for various periods. The aliquots were removed periodically into a spectrophotometer cuvette containing CDNB and GSH to make up their concentration (1 mM) in a total volume of 1 mL and the progress of the GST activity was followed at 340 nm using Cary spectrophotometer (Cary Bio100). Reactions wherein substrates replaced with vehicle alone served as controls. The unit of CRTAase was expressed in terms of % inhibition of GST under the experimental conditions.
5.2.2. Blood collection from human volunteers and preparation of platelet rich plasma (PRP)
Blood from healthy volunteers (n=45; age, 27±1.2yrs) were taken for this study after full explanation to them about the details of the experiment and taking their consent. Approval of the Ethical Committee of Vallabhbhai Patel Chest Institute, University of Delhi, Delhi, India, was obtained in the meeting held on 31-8-2007 for this study and certificate was issued on October 3rd 2007. 9 mL of venous blood was collected with 1.0 mL of 3.8% trisodium citrate from healthy human volunteers after overnight fasting and had abstained from medication including aspirin, paracetamol and alcohol. The citrated blood was used for the preparation of PRP. Platelet count was determined in PRP using electronic cell counter, SYSMEX Model FA 20 and were adjusted to 250 X 106/mL with Platelet-poor plasma (PPP).
5.2.3. Aggregometry
PRP (500 µL) was pipetted into siliconized glass cuvettes and kept at 37 °C for 2 min in the aggregometer. The test compounds dissolved in methanol (100 �M), were added in the reaction cuvette to analyse the platelet aggregation. Individual samples were then incubated at 37 °C for 10 min. After incubation period,
platelet aggregation was induced by the addition of ADP (15 �M)/AA (1mM) and assessed by using a Platelet Aggregation Profiler (BIODATA CORPORATION, Model PAP-4) and the results were expressed as the maximum percentage of light transmittance change (% max) from the baseline at the end of the recording time, using PPP as a reference. Platelet aggregation curves were recorded for 6 minutes and analysed according to internationally established standards. 7-AAMC (17), the lead compound was carried for in depth study where various concentrations (25-250 �M) were used to obtain the concentration response curves.
5.2.4. Measurement of NOS activity
PRP was incubated with test compounds (100 �M) for 10 min and the activity was triggered by the addition of ADP (15 �M) for 5 min. Platelets were paletted down and were washed twice with PBS and resuspended in the standard buffer (137 mM NaCl, 2.8 mM KCl, 1 mM MgCl2, 12 mM NaHCO3, 0.4 mM Na2HPO4, 0.35% BSA, 10 mM HEPES, 5.5 mM glucose, pH 7.4) containing 1�L of DCF-DA (dissolved in CH3OH) in a total volume of 1 mL to make the final concentration 2 �M and kept at 37 °C for 30 min and then the reaction was stopped by placing the tubes containing the reaction mixture at ice. One set of PRP aliquots were preincubated with L-NAME (100 �M)), inhibitor of NOS for 30 min before the addition of test compounds and subjected to NOS assay. Briefly NO was measured in cells labelled with the NO fluorescent dye DCFH-DA. The median fluorescence was measured using the FITC detector (525 nm).
5.2.5. Administration of test compound to the rats
Male Sprague Dawely rats (weight 200-250 g) housed in mesh cages maintained at 25 °C and illuminated at 12:12 h light/dark cycles. This study was conducted in the Department of Biochemistry, V.P. Chest Institute, University of Delhi, Delhi, India. All experimental procedures and protocols used in this investigation were reviewed and approved by the Institutional Animal Ethics Committee (IAEC). A known amount of the 7-AAMC/ASA was suspended in appropriate volume of normal saline, sonicated for 30 sec and the preparation was administered to the rats (3 to 4 per group) orally.
5.2.6. Assessment of platelet aggregation
The doses of 7-AAMC were taken at a range, (10-50) mg/kg or (46-230 µmole/kg), administered to the rats orally once and were sacrificed after 24 h, blood samples were taken by cardiac puncture followed by the measurement of ADP/AA induced platelet aggregation and IC50 was determined. The observed IC50
concentration of 7-AAMC was compared with same concentration of ASA for antiplatelet activity. The optimum concentration (concentration below saturating dose with maximum inhibitory effect) obtained from the dose response curve was used for in depth study of 7-AAMC as an antiplatelet candidate drug.
5.2.7. Assay of Cox-1 activity
Three groups of animals were taken Group 1: control; Group 2: 7-AAMC; Group 3: ASA. Control animals received orally, saline alone. The animals were separately administered 7-AAMC /ASA (160.2 µmole/kg each) p.o. and were sacrificed after 24 h. Platelets lysate were prepared as mentioned above and assessed for Cox-1 activity. The assay was carried out using Cox-1 ELISA kit (Cayman Chemical), according to the manufacturer’s protocol. Briefly, 50µL of each lysed samples were added to the wells, the enzymatic reaction was initiated by adding 100 µM N,N,N,N-tetramethyl-p-phenylenediamine (TMPD) and 100µM AA (saturating condition) in assay buffer. Inhibitors were added

to the incubation reaction at different time intervals before the addition of TMPD and AA. The enzyme activity was measured by ELISA reader, monitoring the appearance of oxidised TMPD at 590 nm.
5.2.8. Analysis of TxB2 in rat platelets
PRP (250 x 106 per mL of platelets) of rats, administered with 7-AAMC/ASA (160.2 µmole/kg) was incubated with AA for 10 min at 37 ºC. Platelets were settled down by centrifugation and plasma was taken for the estimation of TxB2 levels. TxB2 was bioassayed by ELISA method using Cayman’s Chemicals as per manufacturer’s instruction.
5.2.9. Effect of 7-AAMC (17) under thrombotic prone condition: LPS administration to rats
Rats were administered daily 7-AAMC/ASA (160.2 �mole/kg) for 5 days. The timing was chosen as per the preliminary time dependent studies carried on 7-AAMC at a concentration of 160.2 �mole/kg in order to withstand the LPS insult, and it was observed that at least 5 days were required by 7-AAMC to obtain full inhibitory activity on pro-inflammatory condition such as expression of TF/iNOS (data not shown). On the day 5, the last dose of the indicated drug was administered followed by an intra peritoneal injection of LPS, at a dose of 2.5 mg/kg and the animals were sacrificed after 6 h. PBMCs were taken for the assessment of TF and iNOS.
5.2.10. Analysis of tissue factor
The lymphocytes were prepared from rat blood by density gradient using histopack and lysed in 100 �L of lysis buffer for 5 min at 4 °C and centrifuged at 2500 x g for 10 min. The supernatant obtained was used for the assay of TF, by ELISA method using Assay Pro kit as per the manufacturers’ instructions.
5.2.11. Immunodetection of iNOS
Lysed Lymphocytes were used for western blotting method to detect the LPS induced iNOS expression. The protein concentration in the supernatant was determined by the Bradford method (Bio-Rad) and equal amount of proteins (30 µg) were denatured and separated on SDS-PAGE for western blotting. After electrophoresis, the separated protein on the gel was transblotted to a nitro cellulose membrane (Axiva) at 300 mA for 2.5 h at 4 ºC. Membrane was kept in blocking reagent (5% blotto) to block the nonspecific sites. The primary antibody was diluted (1: 1000) in Tris-buffer saline (TBS) containing 1% BSA and was incubated with the blot for 1 h at 4 ºC with moderate agitation. The membrane was extensively washed with 0.05% Tween-20 detergent in TBS, (TBST), followed by incubation with HRP conjugated secondary antibody (Sigma Chemical Co.) for same time duration at room temperature. The membrane was washed extensively with TBST/TBS and the transblotted protein bands were visualized by treating with diaminobenzidene (DAB) and hydrogen peroxide.
5.2.12. Statistical Analysis
Calculations and statistics were performed using the Graph Pad Prism 3.02 software. The one-way analysis of variance (ANOVA) tests followed by the post-hoc Turkey multiple comparisons test and Student’s tests were used. Data were expressed as mean ± standard error. P values less than 0.05 (P < 0.05) were considered to be statistically significant.
Acknowledgements
Financial support from the DU-DST Purse grant, University of Delhi and Defense Research and Development Organization (DRDO) is gratefully acknowledged. We are also thankful to the Council of Scientific and Industrial Research (CSIR), New Delhi for awarding SRF to A. Kathuria, N. Priya, K. Chand, A. Gupta and S. Gupta.
.
References and notes
1. O' Brien, J. R. Lancet 1990, 335, 711. 2. Pande, A. K.; Sethi, K. K. Heart Views 2000, 1, 258. 3. Cattaneo, M. Hematol. J. 2004, 5, S170. 4. Törüner, M. Anatol. J. Cardiol. 2007, 7, 27. 5. Cattaneo, M. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1980. 6. Patrono, C. J. Thromb. Haemost. 2003, 1, 1710. 7. Hankey, G. J.; Eikelboom, J. W. Lancet 2006, 367, 606. 8. Campbell, C. L.; Steinhubl, S. R. J. Thromb. Haemost. 2005, 3, 665. 9. Loscalzo, J.; Welch, G. Prog. Cardiovasc. Dis. 1995, 38, 87.
10. Bansal, S.; Gaspari, M.; Raj, H. G.; Kumar, A.; Cuda, G.; Verheji, E.; Tyagi, Y.K.; Ponan, P.; Rastogi, R.C.; Parmar, V.S. Appl. Biochem. Biotechnol. 2008, 144, 37.�
11. Singh, I.; Kohli, E.; Raj, H. G.; Gyanda, K.; Jain, S. K.; Tyagi, Y. K.; Gupta, G.; Kumari, R.; Kumar, A.; Pal, G.; Prasad, A. K.; Rastogi, R. C.; Olsen, C. E.; Jain, S. C.; Parmar, V. Bioorg. Med. Chem. 2002, 10, 4103.�
12. Raj, H. G.; Kohli, E.; Goswami, R.; Goel, S.; Rastogi, R. C.; Jain, S. C.; Wengel, J.; Olsen, C. E.; Parmar, V. S. Bioorg. Med. Chem. 2001, 9, 1085-1091.�13. Kathuria, A.; Gupta, A.; Priya, N.; Singh, P.; Raj, H. G.; Prasad, A. K.; Parmar, V. S.; Sharma, S.K. Bioorg. Med. Chem. 2009, 17, 1550-1556. 14. Priya, N.; Gupta, A.; Chand, K; Singh, P.; Kathuria, A.; Raj, H. G.; Parmar, V. S.; Sharma, S. K. Bioorg. Med. Chem. 2010, 18, 4085. 15. Atkins, R. L.; Bliss, D. E. J. Org. Chem. 1978, 43, 1975-1980. 16. Kaslow, C. E.; Cook, D. J. J. Am. Chem. Soc. 1945, 67, 1969. 17. Pechmann, H. V.; Duisberg, C. Chem. Ber. 1883, 16, 2119.�18. Russell, A.; Frye, J. R. Org. Synth. Coll. 1955, 3, p. 281; 1941, 21, p. 22. 19. Hennekens, C.H.; Dyken, M. L.; Fuster, V. Circulation, 1997, 96, 2751; Appl. No., 1998, 016011. 20. Seema; Kumari, R.; Gupta, G.; Saluja, D.; Kumar, A.; Goel, S.; Tyagi, Y. K.; Gulati, R.; Vinocha, A.; Murlidhar, K.; Dwarkanath, B. K.; Rastogi, R. C.; Parmar, V. S.; Patkar, S. A.; Raj, H. G. Cell. Biochem. Biophys. 2007, 47, 53.
21. Kohli, E.; Gaspari, M.; Raj, H. G.; Parmar, V. S.; Sharma, S. K.; Van der Greef, J.; Kumari, R.; Gupta, G.; Seema; Khurana, P.; Tyagi, Y. K.; Watterson, A. C.; Olsen, C. E. Biochim. Biophys. Acta 2004, 1698, 55. 22. Singh, P.; Ponnan, P.; Krishnan, S.; Tyagi, T. K.; Priya, N.; Bansal, S.; Scumaci, D.; Gaspari, M.; Cuda, G.; Joshi, P.; Gambhir, J. K.; Saluja, D.; Prasad, A. K.; Saso, L.; Rastogi, R.C.; Parmar, V. S.; Raj, H. G. J. Biochem. 2010, 147, 625.
23. Raj, H. G.; Kumari, R.; Seema; Gupta, G.; Kumar, R.; Saluja, D.; Muralidhar, K. M.; Kumar, A.; Dwarkanath, B. S.; Rastogi, R. C.; Prasad, A. K.; Patkar, S. A.; Watterson, A. C.; Parmar, V. S. Pure Appl. Chem. 2006, 78, 985. 24. Raj, H. G.; Parmar, V. S.; Jain, S. C.; Goel, S.; Singh, A.; Gupta, K.; Rohil, V.; Tyagi, Y. K.; Jha, H. N.; Olsen, C. E.; Wengel, J. Bioorg. Med. Chem. 1998, 6, 1895. 25. Raj, H. G.; Parmar, V. S.; Jain, S. C.; Goel, S.; Singh, A.;Tyagi, Y. K.; Jha, H. N.; Olsen, C. E.; Wengel, J. Bioorg. Med. Chem. 1999, 7, 369. 26. Kumar, A.; Singh, B. K.; Sharma, N. K.; Gyanda, K.; Jain, S. K., Tyagi, Y. K.; Baghel, A. S.; Pandey, M.; Sharma, S. K.; Prasad, A. K.; Jain, S. C.; Rastogi, R. C.; Raj, H. G.; Watterson, A.C.; Parmar, V.S. Eur. J. Med. Chem., 2007, 42, 447.� 27. Kumar, A.; Singh, B. K.; Tyagi, R.; Jain, S. C.;. Sharma, S. K.; Prasad, A. K.; Raj, H. G.; Rastogi, R. C.; Watterson, A. C.; Parmar, V. S. Bioorg. Med. Chem. 2005, 13, 4300. � 28. Loscalzo, J. Arterioscler. Thromb Vasc Biol. 2001, 21, 1259. 29. Toschi, V.; Gallo, R.; Lettino, M.; Fallon, J. T.; Gertz, S. D.; Fernandez- Ortiz, A.; Chesebro, J.H.; Bradimon, L.; Nemerson, Y.; Fuster, V.; Bradimon, J. J. Circulation 1997, 95, 594. 30. Bisell, E. R.; Mitchell, A. R.; Smith, R. E. J. Org. Chem. 1980, 45, 2283. 31. Dzyubenko, M. I.; Vodotyka, G. S.; Maslov, V. V.; Nikitchenko, V. M. Optikai Spektroskopiya 1975, 39, 310; Chem. Abstr. 1975, 83, 211135. 32. Kathuria, A.; Jalal, S.; Tiwari, R.; Shirazi, A. N.; Gupta, S.; Kumar, S.; Parang, K.; Sharma, S. K. Bioorg. Med. Chem. 2011 (communicated). 33. Yang, S. - P.; Han, L. -J.; Wang, D. -Q.; Ding, T. –Z. Acta Crystallographica, Sec E: Structure Reports online, 2006, E62, o5196. 34. Jayashree, B. S.; Thomas, S.; Nayak, Y. Med. Chem. Res. 2010, 19,193. 35. Uray, G.; Strohmeier, G. A.; Fabian, W. M. F. Proceedings of ECSOC-3

and proceedings of ECSOC-4, September 1-30, 1999 and 2000, 521; Chem. Abstr. 2000, 134, 366519.���36����Wallingford, V. H; Homeyer, A. H. U.S. Patent 2,358,768, 1944; Chem. Abstr. 1944, 39, 12041. 37. Zhi, L.; Tegley, C.; Pio, B.; Arjan van Oeveren, C.; Motamedi, M.; Martinborough, E.; West, S.; Higuchi, R.; Hamann, L.; Farmer, L. PCT Int. Appl. 2001, 356 pp; Chem. Abstr. 2001, 134, 207727. 38. Ramesh, P.; Das, A.T.; Mohandass, P.; Nagasathya, R. Ind. J. Chem. 2008, 47B, 1447.

GRAPHICAL ABSTRACT
Substrate specificity of coumarins and quinolones towards Calreticulin mediated transacetylation: Investigations on antiplatelet function Abha Kathuria,a Nivedita Priya,b Karam Chand,a Prabhjot Singh,b Anjali Gupta,a Sarah Jalal,a Shilpi Gupta,a Hanumantharao G. Raj,b,* and Sunil K. Sharmaa,*
aBioorganic Laboratory, Department of Chemistry, University of Delhi, Delhi 110 007, India; bDepartment of Biochemistry, V P Chest Institute, University of Delhi, Delhi 110 007, India
Leave this area blank for abstract info.

















