Journal of Photochemistry and Photobiology A: Chemistry xxx (2005) xxx–xxx
Fluorescence monitoring of photoinitiated polymerization reactionsSynthesis, photochemical study and behaviour as fluorescentprobes of new derivatives of 4′-dimethylaminostyryldiazines
P. Bosch∗, C. Peinado, V. Martın, F. Catalina, T. CorralesInstituto de Ciencia y Tecnologıa de Polımeros, CSIC, Juan de la Cierva 3, 28006 Madrid, Spain
Received 10 May 2005; received in revised form 6 October 2005; accepted 6 October 2005
nd micropolarity of the surroundings. They have been used as fluorescent sensors to monitor the photopolymerization reactions ofnd difunctional methacrylate monomers in bulk. The fluorescence emission band of the probes showed an increase in intensity, aypsochromic shift, as the degree of conversion increases, throughout the entire polymerization range in the different systems checke2005 Elsevier B.V. All rights reserved.
Monitoring the molecular environment of a small moleculey means of its fluorescence has been widely employed inhemistry. Such small molecules are usually designed toxamine specific properties of their immediate environmentr microenvironment by means of a shift in the maximumf their emission or changes in their emission intensity, andre referred to as fluorescent probes. Fluorescent molecularrobes have been widely studied to measure solvent polaritynd viscosity[1,2], which have been used in biochemicalpplications[3] and in materials science[4]. Recently, therere increasing literature reports describing fluorescent probes
or testing processes occurring during polymerization. Probeuorescence changes accompanying polymerization are relatedo both changes in the microviscosity and the local polarityf the medium surrounding the probe, induced by both therowth of polymer chains and the resulting polymeric solutions.ifferent types of fluorescent probes have been developed
n recent years for monitoring photoinitiated polymerization
processes: excimer forming probes[5], charge transfer (CTprobes[6] and organic salts[7]. This method offers somadvantages compared to others, such as DSC and FTIR,its high sensitivity, selectivity and it is non-invasive. Moreomonitoring of UV-curing processes may be committed onwhich is relevant from an applied point of view.
In this work, it is described the synthesis and the photopcal study of three new probes containing an heterocyclicin their structures,N,N-dimethyl-[4-(2-pyridazin-3-yl-vinyl)phenyl]-amine (DMA-2,3), N,N-dimethyl-[4-(2-pyrimidin-4-yl-vinyl)-phenyl]-amine (DMA-2,4) andN,N-dimethyl-[4-(2-pyrazin-2-yl-vinyl)-phenyl]-amine (DMA-2,5). Also, monitoing the photoinduced polymerization reactions of some mand difunctional methacrylate monomers in bulk has been utaken by means of the fluorescent probes. The structures towith the abbreviations are shown inScheme 1.
2 P. Bosch et al. / Journal of Photochemistry and Photobiology A: Chemistry xxx (2005) xxx–xxx
Scheme 1. Structures of the fluorescent probes together with the names used inthis paper. The numbers in the names refer to the position of nitrogen atoms inthe heterocyclic moiety.
grade for synthesis and spectroscopic grade for solvatochromicstudy. Monomers (cyclohexyl methacrylate (CHMA), lau-ryl methacrylate (LMA), ethylhexyl methacrylate (EHMA),hexanediol dimethacrylate (HDDMA) and diethylenglycoldimethacrylate (DEGDMA)) were purchased from Aldrich andwere purified by distillation prior to use.
Chemicals (4-dimethylaminobenzaldehyde, 3-methylpyrida-zine, 4-methylpyrimidine, 2-methylpyrazine, sodium methox-ide, n-butyl lithium and diisopropylamine), all of them ana-lytical grade, were also purchased from Aldrich and used asreceived.
Photoinitiator bis-(2,4,6-trimethylbenzoyl)-phenylphosphi-ne oxide (trademark Irg 819®) was generously gifted by CibaSC and used as received.
2.2. Synthesis
N,N-dimethyl-[4-(2-pyridazin-3-yl-vinyl)-phenyl]-amine(DMA-2,3): 10.6 mmol of 4-methylpyridazine was addeddropwise to a stirred solution of lithium diisopropylamine(LDA) (12.7 mmol) in dry THF at−40◦C under nitrogenatmosphere. After being stirred for 50 min 10.6 mmol ofN,N-dimethylaminobenzaldehyde, dissolved in 25 mL of dry THF,were dropwise added. The reaction was allowed to reach roomtemperature and maintained stirring for 20 h. After addition of1s oveM thes zin-3 asf ofc leted atedd n ofN ughcaw h ac h;a
obtained (purity higher than 95%). The overall yield was13%.
N,N-dimethyl-[4-(2-pyrimidin-4-yl-vinyl)-phenyl]-amine(DMA-2,4): was synthesized by slight modification of a reportedprocedure[8]. 0.17 mmol ofN,N-dimethylaminobenzaldehydeand 0.11 mmol of 4-methylpyrimidine were dissolved in 35 mLo Naw roomt ture( her andt Thes withhw
,
5);0);
,
eor:
( ure
5 mL of a saturated solution of NH4Cl to hydrolyze the lithiumalt and extraction with AcOEt, the organic phase was leftgSO4 overnight, filtered and evaporated. A mixture of
tarting aldehyde, 1-(4-dimethylamino-phenyl)-2-pyrida-yl-ethanol and its dehydration product (DMA-2,3) w
ound, which was dissolved in 20 mL of MeOH and 5 mLoncentrated HCl and refluxed for 7 h to allow the compehydration of the alcohol. The reaction crude was evaporissolved in water, neutralized with a saturated solutioa2CO3, extracted with ethyl acetate and purified throolumn chromatography eluting with CH2Cl2:AcOEt (1:9),fter which a mixture ofcis and trans DMA-2,3 isomersas obtained. Solid was dissolved in 40 mL of THF witatalytic amount of I2 and maintained refluxing during 2fter the isomerization reaction 0.31 g oftrans DMA-2,3 was
r
,
f dry MeOH under argon atmosphere. 0.11 mmol of MeOere added and the reaction mixture was maintained at
emperature for 30 min. A slight increase in tempera45◦C) led to heavy precipitation of yellow crystals. Teaction mixture was then cooled in an ice bath, filteredhe precipitate repeatedly washed with cold methanol.olid was purified through column chromatography elutedexane: AcOEt (1:1), after which 1.20 g of puretrans DMA-2,4ere obtained (yield: 50%).
N,N-dimethyl-[4-(2-pyrazin-2-yl-vinyl)-phenyl]-amineDMA-2,5): was synthesized according to the proced
P. Bosch et al. / Journal of Photochemistry and Photobiology A: Chemistry xxx (2005) xxx–xxx 3
described to obtain DMA-2,3. In this case, time of condensationreaction was 5 h and onlytrans isomer was observed afterof dehydration reaction, which was isolated through columnchromatography eluted with CH2Cl2:AcOEt (1:1). 0.74 g ofpuretrans DMA-2,4 were obtained (yield: 31%).
merU ectrw eteu its ino luor of thp e int cencqu cid(
2
thyla ampA d bU rget phot
2
1%,wt SCsp nden ons sure
lamp and twin quartz optical fiberguides. The simultaneous mea-suring of the fluorescence of the probe and the heat evolved in thepolymerization was performed as described previously, using ashortwave pass dichroic beamsplitter to separate irradiation andemission wavelengths[10], and recording a complete fluores-cence spectrum each 14 s. This dichroic beamsplitter (Lambda)shows high transmission at long wavelengths (λ > 400 nm) andhigh reflectivity at shorther wavelengths (λ < 400 nm). Thus, thebeamsplitter was placed at 45◦ respect to the plane of the cellholders in a modified head of the calorimeter. In this way, thesamples (40�L) were located in the holder of the photocalorime-ter. They were then front-face irradiated through an optical fiberwith polychromatic light (λ < 400 nm) provided by a 400 WHg-lamp (Macam-Flexicure). This light also acts as an exci-tation source for the fluorescent probes inserted as dopants intothe UV-curing formulations. The light was focused to simul-taneously irradiate the sample and the reference holders of thephotocalorimeter. Fluorescence is collected with a fiber collima-tor (Spindler and Hoyer) into an optical fiber, positioned at 90◦respect to the UV-excitacion beam and emission is recorded bya Perkin-Elmer spectrofluorimeter LS-50, operated in biolumi-nescence mode to avoid excitation light from the apparatus. Aband pass optical filter centered at 313 nm (76% of transmissionand 71% at 365 nm) was used to eliminate wavelengths short-her than 405 nm which overlap with the fluorescence emissionspectrum.
alueo cal/sf t ofp cal-c
2
atedb d byL
ν
ν
wa tso ityarta
f
f
Absorption spectra were recorded with a Perkin-ElV–vis Lambda 16 spectrophotometer. Fluorescence spere obtained on a Perkin-Elmer LS 50B spectrophotomsing 355 nm as excitation wavelength and varying the slrder to achieve better spectra for the different probes. Fescence spectra were corrected with the response curvehotomultiplier. The optical densities of all the probes wer
he range 0.02–0.05 at the absorption maximum. Fluoresuantum yields were determined by comparing[9] with thesual standard solution of quinine bisulfate in 1N sulfuric a0.546).
.4. Steady state irradiation
Samples of DMA-2,4 were irradiated under nitrogen in ecetate at 365 nm with a Kratos high pressure mercury lbsorbance of the probes during irradiation was measureV spectroscopy at the maximum wavelength of the cha
ransfer absorption band, in a Shimatzu UV-256 FS spectroometer.
.5. Polymerization proceeding and analysis
The fluorescent probe (0.002%, w/w) and photoinitiator (/w) were dissolved in the monomer to be studied. 40�L of
his formulation were placed into an aluminium pan in the Dample holder and the temperature maintained at 30◦C. Sam-les were degassed prior irradiation and then, irradiated uitrogen in situ with a MACAM-Flexicure portable irradiatiystem provided with a Sylvania 400 W Hg medium-pres
ar
-e
e
.y--
r
Incident light intensity was set constant for all runs at a vf 0.75 mcal/s for monofunctional monomers and 0.36 m
or difunctional ones. A value of 13.1 kcal/mol for the heaolymerization of the methacrylic double bond was used inulations[11].
.6. Dipole moment determination
The excited state singlet dipole moment has been estimy the solvatochromic method using the equations defineippert [12] (Eqs.(1) and(2)).
A − νF = − (µe − µg)2
4πε0hcα3 [f (D) − f (n2)] + [ν0A − ν0
F] (1)
A + νF = − (µ2e − µ2
g)
4πε0hcα3 [f (D) − f (n2)] + [ν0A + ν0
F] (2)
hereν0A andν0
F are respectively the frequencies (in cm−1) ofbsorption and emission,µg and µe are the dipole momenf ground and excited states,α the radius of Onsager cavndε0 is the permitivity in vacuum. The function [f(D) − f(n2)]epresents the oriented polarization, where the termf(D) is theotal polarization andf(n2) is the induced polarization (Eqs.(3)nd(4)).
(D) = 2(D − 1)
2D + 1(3)
(n2) = 2(n2 − 1)
2n2 + 1(4)
4 P. Bosch et al. / Journal of Photochemistry and Photobiology A: Chemistry xxx (2005) xxx–xxx
whereD andn are the dielectric constant and the refraction index,respectively.
Probes geometries have been optimized using Density Func-tional Theory (DFT) methods. It has been used standard split-valence plus polarization functions on heavy atoms 6-31G* [13]basis set and the hybrid Becke[14] three-parameterized den-sity functional using the Lee, Yang and Paar (LYP) functional(B3LYP)[15]. Because B3LYP DFT explicitly includes electroncorrelation, it allows the calculation of reliable, accurate thermo-dynamic data. Gaussian 98, Rev. A.7[16] and CS Chem3D Pro[17] programs have been employed for calculation and drawing.Different conformations have been carefully checked to obtainthe absolute minimum energy. Harmonic vibrational frequen-cies have been calculated and their positive values indicate trueminima.
3. Results and discussion
3.1. Synthesis
The structures of the probes are shown inScheme 1, togetherwith the abbreviations used, which refer to the position of thenitrogen atoms in the heterocyclic ring.
The synthesis of the probes consists in the well knownMichael addition of the methyl derivative of the correspond-i edb ubleb
thed weab ld nob f themp thep re ino hec ucei f thed ol iso
3.2. Absorption and emission properties
A description of the absorption and emission maxima of thenew probes in several solvents, together with the correspondingET30 andπ* solvent parameters are summarized inTable 1.For DMA-2,3 thetrans isomer was freshly obtained to assurethe photophysical behaviour corresponds not to a mixture ofcis–trans isomers.
The electronic absorption spectra present a broad band witha maximum located in the 362–400 nm regions and a shoulderaround 325 nm. The shortest wavelength band corresponds to a� → �* transition whereas the long-wavelength band, charac-terized by higher molar absorption coefficients, is attributed toan intramolecular CT transition. On varying the solvent polarity,moderate shifts in the absorption maxima are observed (up to24 nm from cyclohexane to methanol). The higher red shift of theCT absorption band in DMA-2,4 and DMA-2,5 (as comparedwith DMA-2,3) may be explained by the increasing electronaffinity of the acceptor subunit (the corresponding diazines) inthe following order:
DMA-2,3 < DMA-2,4 < DMA-2,5
and the corresponding lowering of the CT state energies.The electronic emission spectra are highly solvatochromic.
The spectra for DMA-2,4 are shown as an example (Fig. 1).reas-
i nmf s infl
ntumy uchar tokess ings iteds
rityo larityp tweent
TM l solv
λmax
41 0.0042
438 28453 .54453 .2746 .55
478 .5847 0.45482 .58
1 49 .711 52 0.541 526 .60
ng heterocycle toN,N-dimethylaminobenzaldehyde, followy dehydration of the alcohol to form the conjugated doond.
For the pyrimidine derivative, both the Michael attack andehydration reaction occur in the presence of the relativelyase sodium methoxide, and the intermediate alcohol coue isolated. This is due to the more nucleophilic nature oethylpyrimidine carbanion, produced by theortho- andpara-osition of the nitrogen atoms in the heterocyclic ring. Foryrazine and pyridazine derivatives, the nitrogen atoms artho- andmeta-positions, which lead to minor reactivity of torresponding carbanions, and then the reaction is only prodn the presence of a stronger base (LDA) and the mixture oehydration product together with the intermediate alcohbtained.
able 1aximum absorption and emission wavelengths for the probes in severa
The fluorescence maxima are strongly red shifted on incng solvent polarity showing a batochromic shift of ca. 112rom cyclohexane to methanol. Quantum yields of probeuid media are low (1.10−3 to 0.11).
It is observed as a general trend that the low emission quaields increase with solvent polarity, whilst protic solvents ss alcohols slightly quench the fluorescence emission[18]. Theed shift of their spectral position and the increase of the Shift (Table 2) and of the emission bandwidth with increasolvent polarity point out that emission comes from a CT exctate.
In order to correlate the influence of the solvent polan the absorption and emission maxima, we used the poarameters which expressed the best local interactions be
he solute and the solvent, i.e., the ET30 andπ* values[19,20]
P. Bosch et al. / Journal of Photochemistry and Photobiology A: Chemistry xxx (2005) xxx–xxx 5
Fig. 1. Fluorescence spectra of DMA-2,4 in several solvents.
where H-bonding solvents have been excluded due to the specificsolvent–solute interactions[18]. The correlations with ET30parameters are shown, as an example, inFig. 2 for all probes.As can be seen, both the absorption and emission maxima corre-late well with solvent polarity parameters, and the slopes of thelines are indicative of the sensitiveness towards solvent polar-ity. The highest solvatochromic sensitivity is observed for thestyrylpyrazine (DMA-2,5) and the scattering of some data aredue to specific solvent effects. For example, it is well known thatdioxane is not a good solvent for solvatochromic studies.
The polarity of the solvent has a larger effect on the fluores-cence spectra than on the absorption ones. Apparently, a largeeffective change in charge separation between the ground and theexcited state is achieved. A strong donor such as dimethylaminogroup can produce an intramolecular charge transfer state ICT inthese systems where the diazines may behave as acceptors. Thetwo electron pairs of the diazines nitrogen atoms are in an orbitalperpendicular to the aromatic� electron system, and therefore,it is difficult for these diazines to act as donors through the reso-nance effect. Moreover, the internal rotation of the dialkylaminogroup may contribute to the red-shifted fluorescence emission.
A three-state kinetic scheme is proposed to explain thephotophysical behaviour of these donor–acceptor substituted
Fig. 2. Plot of the maximum emission wavelengths of probes versus ET30 sol-vent parameter.
stilbenes-like compounds. It is widely accepted that, in non-substituted stilbene derivatives, a double bond twisted excitedisomer P* acts as deactivation photochemical funnel, andreduces the fluorescence quantum yield of the excited E* state.An additional third state is present if the probe has donor andacceptor subtituents. The photophysical behaviour is differentfrom that of the stilbene and the nature of this new excited stateis assigned to a ICT (intramolecular charge transfer) state, whichis the responsible of the emission. Generally, in stilbene-likemolecules, photoinduced internal rotations (leading for instanceto twisted intramolecular charge transfer) are known to play animportant role[21,22].
It is well known that the energy level of the charge transferstate is given by Eq.(5):
ECT = Eox(D) − Ered(A) + C (5)
whereEox(D) andEred(A) are the electrochemical one-electronoxidation and reduction potential of the donor and acceptorgroups, respectively andC is a constant that depends on thecharge separation degree. As the donor group is the same dialky-lamino moiety the electron withdrawing ability of each diazinemay account for the differences in the emission characteristics.
Table 2Fluorescence quantum yields and Stokes shifts of fluorescent probes in different solvents
6 P. Bosch et al. / Journal of Photochemistry and Photobiology A: Chemistry xxx (2005) xxx–xxx
Therefore, it should be expected that not only the solvent but thenature of the acceptor chromophore has strong influence on theCT fluorescence. A good correlation between the Stokes shiftin polar solvents and the reduction potential of the correspond-ing diazines[23] was found. This family of fluorescent sensorsoffers the possibility of tuning fluorescence characteristics as afunction of the electron-acceptor character of the moiety whichdepends on the relative position of the N atoms in the diazinering.
3.3. Dipole moments in the ground and excited state
The calculations of both geometrical parameters and thedipole moments in the ground state (Table 3and Scheme 2)have been performed as stated in Section2. The existence ofelectronic conjugation between the groups attached to the aro-matic benzene ring can be seen by the length of the bondsL1,L2 andL3. In the minimum energy configuration, the moleculeshave a planar structure in the ground state, as can be seen by thevalues of the dihedral angles.
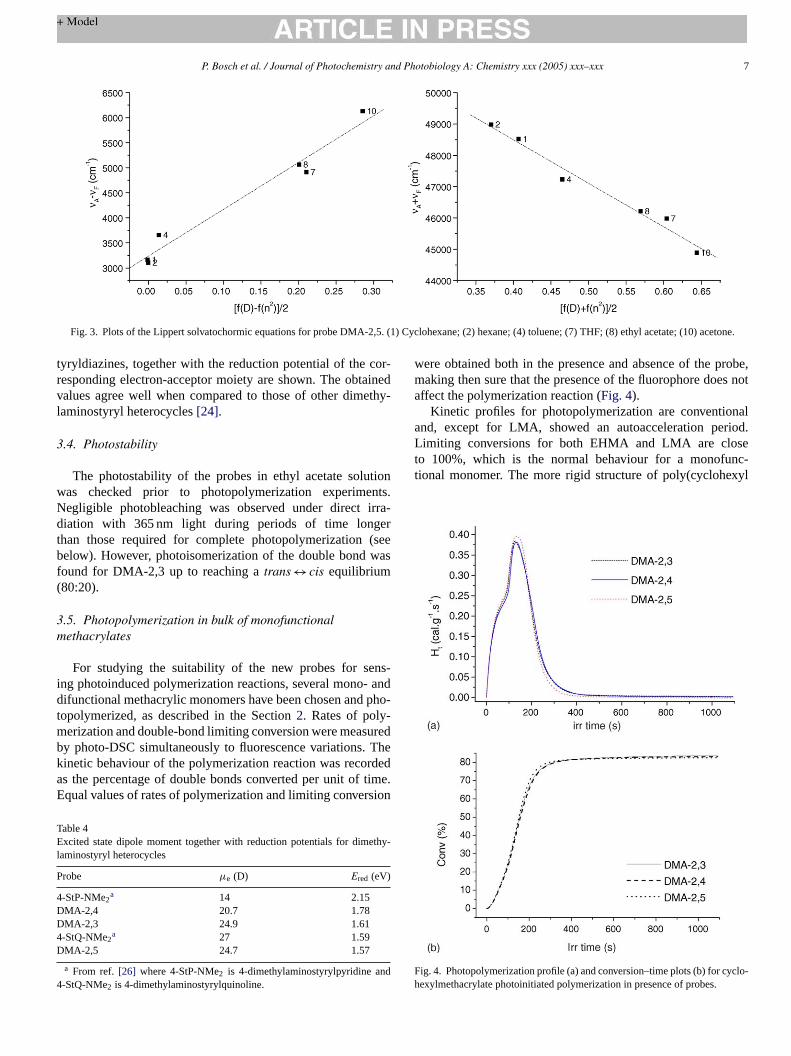
The dipole moments in the singlet excited state have beenestimated using the Lippert solvatochromic method, assumingthat the vector of ground state is co-linear to the vector of theexcited state. InFig. 3, the plots of the Lippert solvatochromicequations for DMA-2,5 are shown as an example. Ifm1 andm2are the slopes of both plots, and dividing Eq.(1) and Eq.(2), thef fg dipolem
w y:
µ
F mentso ined.I undw ty inaav nos-
Tabl
e3
Cal
cula
ted
geom
etric
alpa
ram
eter
sin
the
min
imum
ener
gyco
nfigu
ratio
n,an
dca
lcul
ated
dipo
lem
omen
tsin
the
grou
ndan
dex
cite
dst
ates
ofpr
obes
Pro
bes
Bon
dle
ngth
sa
(A)
Dih
edra
lang
lesa
(A)
µg
(D)
µe
(D)
�µ
L1,N
1C
3L
2,N
7C
8L
3,N
8C
9L
4,N
9C
10D
1,C
2N
1C
3C
4D
2,C
2N
1C
3C
5D
3,C
6N
7C
8C
9D
4,C
8N
9C
10N
2
DM
A-2
,31.
383
1.45
51.
352
1.46
16.
544
7.32
90.
420
0.05
45.
424
.919
.5D
MA
-2,4
1.38
11.
453
1.35
31.
458
4.71
95.
058
0.20
60.
012
6.6
20.7
14.1
DM
A-2
,51.
383
1.45
51.
352
1.45
86.
500
7.06
70.
499
0.05
45.
024
.719
.7
aN
umer
atio
nof
atom
sco
rres
pond
sto
the
posi
tions
mar
ked
inS
chem
e2.
ollowing equation is obtained (Eq.(6)), which is independent oeometrical parameters and could be used to estimate theoment in the excited state.
m1
m2= (µe − µg)2
(µ2e − µ2
g)(6)
henm2 > m1 the simplified form of this equation is given b
e = m1 + m2
m2 − m1µg (7)
rom these bases, an estimated value for the dipole mof the singlet excited states of the probes has been obta
t has to be pointed out that some interactions were foith specific solvents and not withstanding such ambiguissigning the exact dipole moment, the magnitude ofµe impliesn effective charge separation upon excitation. InTable 4, thealues of the excited dipole moment of the dimethylami
Scheme 2.
P. Bosch et al. / Journal of Photochemistry and Photobiology A: Chemistry xxx (2005) xxx–xxx 7
Fig. 3. Plots of the Lippert solvatochormic equations for probe DMA-2,5. (1) Cyclohexane; (2) hexane; (4) toluene; (7) THF; (8) ethyl acetate; (10) acetone.
tyryldiazines, together with the reduction potential of the cor-responding electron-acceptor moiety are shown. The obtainedvalues agree well when compared to those of other dimethy-laminostyryl heterocycles[24].
3.4. Photostability
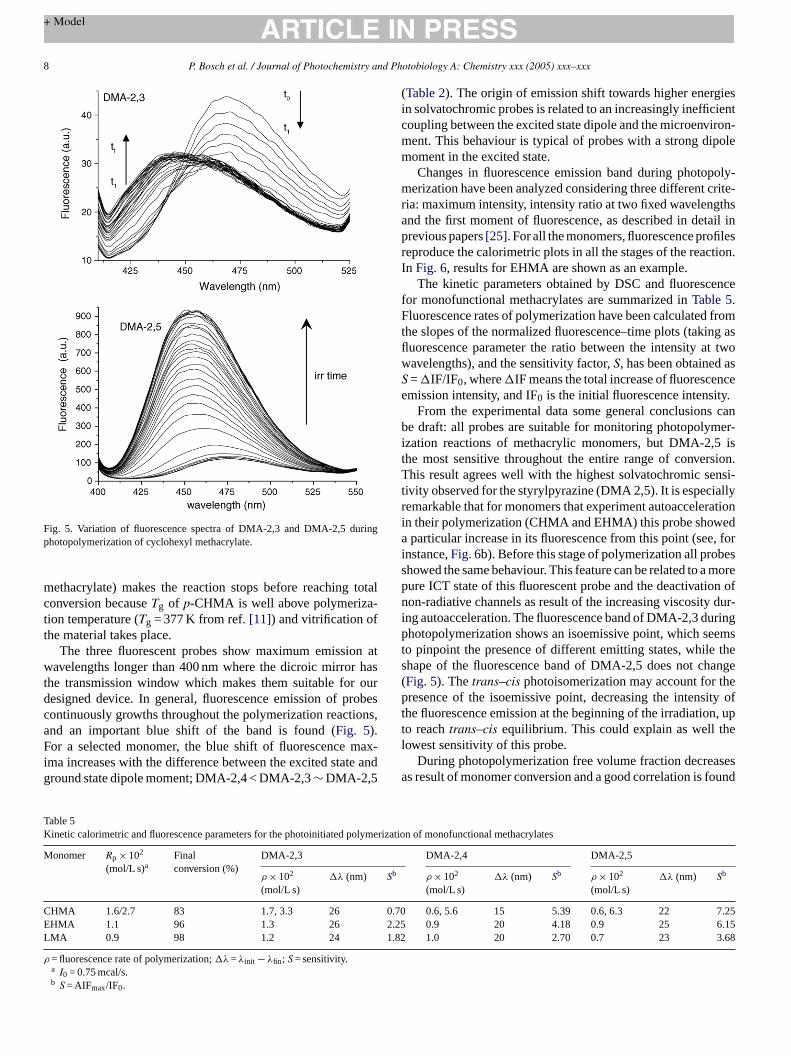
The photostability of the probes in ethyl acetate solutionwas checked prior to photopolymerization experiments.Negligible photobleaching was observed under direct irra-diation with 365 nm light during periods of time longerthan those required for complete photopolymerization (seebelow). However, photoisomerization of the double bond wasfound for DMA-2,3 up to reaching atrans ↔ cis equilibrium(80:20).
3.5. Photopolymerization in bulk of monofunctionalmethacrylates
For studying the suitability of the new probes for sens-ing photoinduced polymerization reactions, several mono- anddifunctional methacrylic monomers have been chosen and pho-topolymerized, as described in the Section2. Rates of poly-merization and double-bond limiting conversion were measuredby photo-DSC simultaneously to fluorescence variations. Thekinetic behaviour of the polymerization reaction was recordeda timeE sion
TE ethy-l
P
4DD4D
d4
were obtained both in the presence and absence of the probe,making then sure that the presence of the fluorophore does notaffect the polymerization reaction (Fig. 4).
Kinetic profiles for photopolymerization are conventionaland, except for LMA, showed an autoacceleration period.Limiting conversions for both EHMA and LMA are closeto 100%, which is the normal behaviour for a monofunc-tional monomer. The more rigid structure of poly(cyclohexyl
Fig. 4. Photopolymerization profile (a) and conversion–time plots (b) for cyclo-hexylmethacrylate photoinitiated polymerization in presence of probes.
s the percentage of double bonds converted per unit ofqual values of rates of polymerization and limiting conver
able 4xcited state dipole moment together with reduction potentials for dim
a From ref. [26] where 4-StP-NMe2 is 4-dimethylaminostyrylpyridine an-StQ-NMe2 is 4-dimethylaminostyrylquinoline.
.
8 P. Bosch et al. / Journal of Photochemistry and Photobiology A: Chemistry xxx (2005) xxx–xxx
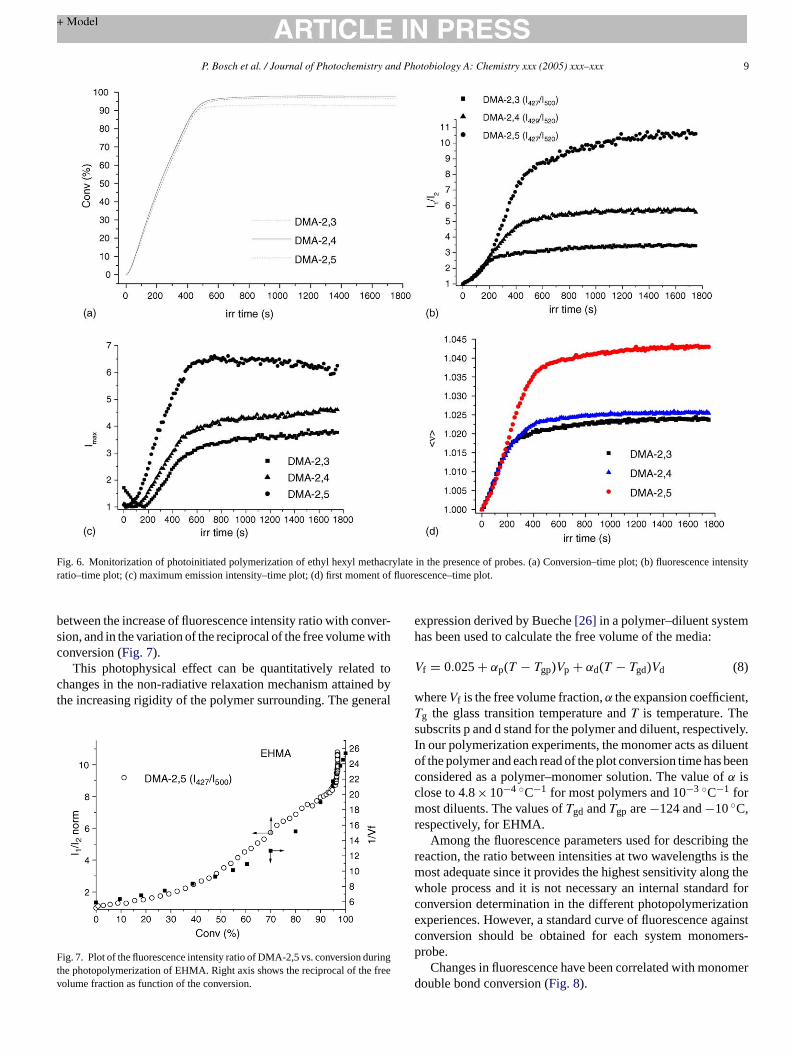
Fig. 5. Variation of fluorescence spectra of DMA-2,3 and DMA-2,5 duringphotopolymerization of cyclohexyl methacrylate.
methacrylate) makes the reaction stops before reaching totaconversion becauseTg of p-CHMA is well above polymeriza-tion temperature (Tg = 377 K from ref.[11]) and vitrification ofthe material takes place.
The three fluorescent probes show maximum emission awavelengths longer than 400 nm where the dicroic mirror hasthe transmission window which makes them suitable for ourdesigned device. In general, fluorescence emission of probecontinuously growths throughout the polymerization reactionsand an important blue shift of the band is found (Fig. 5).For a selected monomer, the blue shift of fluorescence maxima increases with the difference between the excited state anground state dipole moment; DMA-2,4 < DMA-2,3∼ DMA-2,5
(Table 2). The origin of emission shift towards higher energiesin solvatochromic probes is related to an increasingly inefficientcoupling between the excited state dipole and the microenviron-ment. This behaviour is typical of probes with a strong dipolemoment in the excited state.
Changes in fluorescence emission band during photopoly-merization have been analyzed considering three different crite-ria: maximum intensity, intensity ratio at two fixed wavelengthsand the first moment of fluorescence, as described in detail inprevious papers[25]. For all the monomers, fluorescence profilesreproduce the calorimetric plots in all the stages of the reaction.In Fig. 6, results for EHMA are shown as an example.
The kinetic parameters obtained by DSC and fluorescencefor monofunctional methacrylates are summarized inTable 5.Fluorescence rates of polymerization have been calculated fromthe slopes of the normalized fluorescence–time plots (taking asfluorescence parameter the ratio between the intensity at twowavelengths), and the sensitivity factor,S, has been obtained asS =�IF/IF0, where�IF means the total increase of fluorescenceemission intensity, and IF0 is the initial fluorescence intensity.
From the experimental data some general conclusions canbe draft: all probes are suitable for monitoring photopolymer-ization reactions of methacrylic monomers, but DMA-2,5 isthe most sensitive throughout the entire range of conversion.This result agrees well with the highest solvatochromic sensi-tivity observed for the styrylpyrazine (DMA 2,5). It is especiallyr rationi eda e, fori ess morep on ofn dur-i ringp emst thes ange( hep ity oft n, upt el
sesa ound
Table 5Kinetic calorimetric and fluorescence parameters for the photoinitiated polyme
ρ = fluorescence rate of polymerization;�λ =λinit − λfin; S = sensitivity.a I0 = 0.75 mcal/s.b S = AIFmax/IF0.
l
t
s,
-d
emarkable that for monomers that experiment autoaccelen their polymerization (CHMA and EHMA) this probe show
particular increase in its fluorescence from this point (senstance,Fig. 6b). Before this stage of polymerization all probhowed the same behaviour. This feature can be related to aure ICT state of this fluorescent probe and the deactivation-radiative channels as result of the increasing viscosity
ng autoacceleration. The fluorescence band of DMA-2,3 duhotopolymerization shows an isoemissive point, which se
o pinpoint the presence of different emitting states, whilehape of the fluorescence band of DMA-2,5 does not chFig. 5). Thetrans–cis photoisomerization may account for tresence of the isoemissive point, decreasing the intens
he fluorescence emission at the beginning of the irradiatioo reachtrans–cis equilibrium. This could explain as well thowest sensitivity of this probe.
During photopolymerization free volume fraction decreas result of monomer conversion and a good correlation is f
P. Bosch et al. / Journal of Photochemistry and Photobiology A: Chemistry xxx (2005) xxx–xxx 9
Fig. 6. Monitorization of photoinitiated polymerization of ethyl hexyl methacrylate in the presence of probes. (a) Conversion–time plot; (b) fluorescence intensityratio–time plot; (c) maximum emission intensity–time plot; (d) first moment of fluorescence–time plot.
between the increase of fluorescence intensity ratio with conver-sion, and in the variation of the reciprocal of the free volume withconversion (Fig. 7).
This photophysical effect can be quantitatively related tochanges in the non-radiative relaxation mechanism attained bythe increasing rigidity of the polymer surrounding. The general
Fig. 7. Plot of the fluorescence intensity ratio of DMA-2,5 vs. conversion duringthe photopolymerization of EHMA. Right axis shows the reciprocal of the freevolume fraction as function of the conversion.
expression derived by Bueche[26] in a polymer–diluent systemhas been used to calculate the free volume of the media:
Vf = 0.025+ αp(T − Tgp)Vp + αd(T − Tgd)Vd (8)
whereVf is the free volume fraction,α the expansion coefficient,Tg the glass transition temperature andT is temperature. Thesubscrits p and d stand for the polymer and diluent, respectively.In our polymerization experiments, the monomer acts as diluentof the polymer and each read of the plot conversion time has beenconsidered as a polymer–monomer solution. The value ofα isclose to 4.8× 10−4 ◦C−1 for most polymers and 10−3 ◦C−1 formost diluents. The values ofTgd andTgp are−124 and−10◦C,respectively, for EHMA.
Among the fluorescence parameters used for describing thereaction, the ratio between intensities at two wavelengths is themost adequate since it provides the highest sensitivity along thewhole process and it is not necessary an internal standard forconversion determination in the different photopolymerizationexperiences. However, a standard curve of fluorescence againstconversion should be obtained for each system monomers-probe.
Changes in fluorescence have been correlated with monomerdouble bond conversion (Fig. 8).
10 P. Bosch et al. / Journal of Photochemistry and Photobiology A: Chemistry xxx (2005) xxx–xxx
Fig. 8. Fluorescence–conversion plots for the polymerization of monofunctionamethacrylates.
In these plots, the good sensitivity of the probes DMA-2,4and DMA-2,5 until final conversion can be again observed. Inaddition other significant feature can be seen: the fluorescencemission from all probes (except for the LMA/DMA-2,3 andLMA/DMA-2,4 samples) continues growing at the end of thereaction after double bond conversion has reached a constantvalue. This is interpreted in terms of an increase in rigidificationof the medium after completion of the double bond polymeriza-tion reaction, which is due to secondary crosslinking reactions.
These reactions are hydrogen atom abstraction from the poly-meric chains by the photogenerated initiator primary radicals.This has been observed before in the photocuring reaction ofacrylic adhesives[27,28]. The decrease observed in fluores-cence for the LMA/DMA-2,3 and LMA/DMA-2,4 samples isattributed to a slight photodegradation of the probes at the endof the reaction, and it is only observed at very long times for theslower LMA polymerization.
3.6. Photopolymerization in bulk of difunctionalmethacrylates
The photopolymerization reaction of HDDMA andDEGDMA has been performed in the same way than formonofunctional monomers and the same fluorescence andcalorimetric features have been found. Kinetic data are summa-rized in Table 6and fluorescence–conversion plots are shownin Fig. 9.
Rates of polymerization are higher than for monofunctionalmonomers, and complete polymerization takes place in less than200 s. Limiting conversions do not exceed 60–63%, as expectedfor crosslinking reactions. In general, the probes behave in thesame manner than for linear polymerizations, so they are alsosuitable for monitoring crosslinking reactions in where a highrigid medium is obtained. It is also observed the better sen-
Fig. 9. Fluorescence–conversion plots for the polymerization of difunctionalmethacrylates.
l
e
P. Bosch et al. / Journal of Photochemistry and Photobiology A: Chemistry xxx (2005) xxx–xxx 11
Table 6Kinetic calorimetric and fluorescence parameters for the photoinitiated polymerization of difunctional methacrylates
ρ = fluorescence rate of polymerization;�λ =λinit − λfin; S = sensitivity.a I0 = 0.36 mcal/s.b S = AIFmax/IF0.
sitivity of DMA-2,5 and an increase in fluorescence emissionof all the probes after conversion reached its limiting value. Asurprising fact is that the probes seem less sensitive towardscrosslinking polymerization reactions that for linear polymer-ization, although in the former higher rigidity is obtained. Thisfact could be seen in the sensitivity factor for these probes aswell as the small wavelength shift of the bands, which does notexceed 13 nm for the most sensitive probe. We do not have aproper explanation for this, but new experiments are currentlybeing performed, which will be published further.
A striking different behaviour is observed for DMA-2,3 probemonitoring fluorescence changes during photopolymerization ofdifunctional monomers compared to monofunctional ones. Asmentioned before fluorescence emission band shows an isoemis-sive point during the photopolymerization of monofunctionalmonomers that disappears during the photopolymerization ofdifunctional, such as DEGDMA and HDDMA. The influenceof the primary excited state on the CT emitting state mayaccount for this behaviour in the same sense that impairs thebehaviour of this probe respect to DMA-2,4 and DMA-2,5.Moreover, thetrans–cis photoisomerization depends to a largeextent on the medium, such as solvatation, temperature and,especially, viscosity. The enhacenment of viscosity has a pro-found effect slowing down isomerization. Therefore, the initialdecrease of fluorescence emission during irradiation of mono-functional monomers it is not observed for the difunctional ones,b er. Ap f thes omei
4
d thefl angi
ctiono ho-t robes shiu ac-t
hoseo elera bee
checked for following the reaction are: maximum intensity,emission ratio between intensities at two wavelengths and thefirst moment of fluorescence, from which emission ratio betweenintensities at two wavelengths has been shown to be the mostadequate throughout the entire reaction.
The rate of polymerization has been measured from thefluorescence–time plots and fluorescence has been correlatedwith conversion measured simultaneously by differential scan-ning calorimetry.
All probes have been shown to be sufficiently sensitiveto follow the complete polymerization reaction of mono- anddifunctional methacrylic monomers, being DMA-2,5 the mostsensitive among them.
It is emphasized the need of a detailed solvatochromic anal-ysis to use fluorescent probes for monitoring polymerization.
References
[1] C. Reichardt, Chem. Rev. 94 (1994) 2319.[2] A. Jacobson, A. Petric, D. Hogenkamp, A. Sinur, J.R. Barrio, J. Am.
Chem. Soc. 118 (1996) 5572.[3] Fluorescence spectroscopy, imaging and probes, in: R. Kraayenhof,
A.J.W.G. Visser, H.C. Gerritsen (Eds.), Springer Series on Fluorescence,Berlin, Springer-Verlag, 2002.
[4] H. Itagaki, in: T. Tanaka (Ed.), Experimental Methods in Polymer Sci-ence, Academic Press, USA, 2000 (Chapter 3).
[5] (a) O. Peckan, Y. Yilmaz, O. Okay, Polymer 38 (1997) 1693;
;
993)
993)
995)
l. A:
cro-
999)
1996)
[ lymer
[ ook,
[
ecause the increase of viscosity in this case is much highhotopolymerization progresses, the molecular mobility otilbenoid probes becomes more restricted and the photoiszation rate diminishes[29].
. Conclusions
Three new fluorescent probes have been synthesized anuorescence emission has been characterized towards chn the polarity and viscosity of the medium.
They have been used to monitor the polymerization reaf mono- and difunctional methacrylic monomers. As p
opolymerization proceeds, the fluorescence band of the phowed an increase in intensity as well as a spectral bluentil final conversion. This behaviour allows following the re
ion by fluorescence spectroscopy.The fluorescent kinetic profiles accurately reproduce t
btained by photo-DSC, including the onset of the autoacction process. The fluorescence parameters which have
s
r-
ires
sft
-n
(b) O. Okay, D. Kaya, O. Peckan, Polymer 40 (1999) 179.[6] (a) J. Paczkowski, D.C. Neckers, Macromolecules 24 (1991) 3013
(b) J. Paczkowski, D.C. Neckers, Macromolecules 25 (1992) 548;(c) J. Paczkowski, D.C. Neckers, J. Polym. Sci. A: Chem. 31 (1841;(d) I.N. Kotchetov, D.C. Neckers, J. Imaging Sci. Technol. 37 (1156;(e) W.F. Jagger, A.A. Volkers, D.C. Neckers, Macromolecules 28 (18153;(f) R. Popielarz, S. Hu, D.C. Neckers, J. Photochem. PhotobioChem. 110 (1997) 79;(g) B. Strehmel, J.H. Malpert, A.M. Sarker, D.C. Neckers, Mamolecules 32 (1999) 7476;(h) V.F. Jagger, A.M. Sarker, D.C. Neckers, Macromolecules 32 (18791.
[7] W.F. Jager, D. Kundasheva, D.C. Neckers, Macromolecules 29 (7351.
[8] D.M. Brown, G.A.R. Kon, J. Chem. Soc. (1948) 2147.[9] D.F. Eaton, J. Photochem. Photobiol. B: Biol. 2 (1988) 523.10] C. Peinado, A. Alonso, E.F. Salvador, J. Baselga, F. Catalina, Po
fourth ed., John Wiley and Sons, 1999.12] E. Lippert, Z. Naturfosch. 10 (1995) 541.
12 P. Bosch et al. / Journal of Photochemistry and Photobiology A: Chemistry xxx (2005) xxx–xxx
[13] W.J. Hehre, L. Radom, P.V.R. Schleyer, J.A. Pople, Ab initio MolecularOrbital Theory, John Wiley and Sons, New York, 1986.
[14] A.D. Becke, J. Chem. Phys. 98 (1993) 5648.[15] C. Lee, W. Yang, R.G. Parr, Phys. Rev. B B37 (1998) 785.[16] M.J. Frisch, G.W. Trucks, H.B. Schlegel, G.E. Scuseria, M.A. Robb,
J.R. Cheeseman, V.G. Zakrzewski, J.A. Montgomery Jr., R.E. Strat-mann, J.C. Burant, S. Dapprich, J.M. Millam, A.D. Daniels, K.N. Kudin,M.C. Strain, O. Farkas, J. Tomasi, V. Barone, M. Cossi, R. Cammi, B.Mennucci, C. Pomelli, C. Adamo, S. Clifford, J. Ochterski, G.A. Peters-son, P.Y. Ayala, Q. Cui, K. Morokuma, D.K. Malick, A.D. Rabuck, K.Raghavachari, J.B. Foresman, J. Cioslowski, J.V. Ortiz, A.G. Baboul,B.B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. Gom-perts, R.L. Martin, D.J. Fox, T. Keith, M.A. Al-Laham, C.Y. Peng, A.Nanayakkara, C. Gonzalez, M. Challacombe, P.M.W. Gill, B. Johnson,W. Chen, M.W. Wong, J.L. Andres, C. Gonzalez, M. Head-Gordon,E.S. Replogle, J.A. Pople, Gaussian 98, Revision A.7, Gaussian, Inc.,Pittsburgh, PA, 1998.
[17] CS Chem3D Pro. CambridgeSoft Corporation, Cambridge, USA, 1999.[18] P. Suppan, N. Ghoneim (Eds.), Solvatochromism, The Royal Society of
Chemistry, Cambridge, UK, 1997.
[19] (a) C. Reichardt, H.F. Ebel (Eds.), Solvents and Solvent Effects inOrganic Chemistry, second ed., VCH, Basel, 1998;(a) C. Reichardt, Chem. Rev. 94 (1994) 2319.