1 Fluoro-alkane, -alkenes, -alkynes and –aromatics Fluoroalkanes Properties of fluoroalkanes Consider these typical bond strengths: C-C bond 88kcal/mol C-F bond 111kcal/mol C-H bond 106kcal/mol This implies that the carbon – carbon bonds are the weakest link in fluoroalkanes.
Transcript
1
Fluoro-alkane, -alkenes, -alkynes and –aromatics
Fluoroalkanes
Properties of fluoroalkanesConsider these typical bond strengths:
C-C bond 88kcal/mol
C-F bond 111kcal/mol
C-H bond 106kcal/mol
This implies that the carbon – carbon bonds are the weakest link in fluoroalkanes.
2
Features to bear in mind:
(i)Bond Strengths tend to increase as the total number of fluorines increases
I.e.
F < FF
<F
FF
C-F bond shortens (strengthens) as more F's are added
Bond strengths
3
The C-F bond shortens (gets stronger) as more fluorines are added due to an increase in back bonding (halo effect).
Some C-C bond strengthening also occurs on substitution of F for H.
For example the CF3-CH2CH3 bond is ~5kcal stronger than the CF3CH2-CH3 bond.
(i) -(CF2)n- is more stable than -(CH2)n- due to better chemical stability.
PTFE shows resistance to oxidation and microbial attack. (Generally related to BDE’s of C-H and C-F).
BDE
Fluorine makes the atoms close to it bond stronger also !!
↑ shorter ↑ stronger no significant changes
4
(iii) Electron Pair Repulsions (Intramolecular) are important to PTFE.
1,3 repulsions force the F atoms out of linearity, which is in contrast to the hydrogen containing polymer analogue.
5
Boiling PointsFluorocarbons are obviously heavier than their hydrocarbon counterparts, and thus we
might initially expect them to have correspondingly higher boiling points.
However, the boiling points of hydrocarbons and perfluorocarbons are very similar.
(B.Pts in oC)
The increase in molecular weight that increases the boiling point, therefore must be offset by the decrease in attractive intermolecular bonding forces in the fluorocarbon systems.
(I.e. the electron pairs on fluorines in one molecule have a tendency to repel the other fluorinated molecules).
PTFE has a coefficient of friction described as “wet ice sliding on wet ice”.
This low friction and oil repellency is almost certainly due to the electron pair repulsions.
Chemical InertnessThe electron clouds of the fluorines “screen” the carbon atoms of the chain, and thus
discourage the approach and attack of incoming reactants. (The carbons are the weakest link).
Thus fluorocarbons find uses as inert fluids (see later).
Carbon tetrafluoride is fantastically stable and inert.
But the hydrolysis of CF4 is predicted to be exothermic by 73kcal/mol. (i.e. very thermodynamically favorable).
7
So it is a large kinetic barrier to hydrolysis (i.e. a high activation energy) that causes the stability.
In fact under the right conditions perfluorocarbons like PTFE can generate a lot of heat (e.g. for use in pyrotechnics or car airbag inflation).
Usually this is achieved using metals → metal fluorides; an exothermic reaction).
SolubilityPerfluorocarbons have a profound ability to dissolve partially fluorinated compounds.
Also certain fluorocarbons have excellent oxygen and nitrogen solvating ability, and prompted their application as artificial blood.(“Oxygen transport fluids”)
8
Reactions of PerfluorocarbonsGenerally perfluorocarbons are very inert and unreactive.
The only reaction they are prone to undergo, usually under forcing conditions, is Defluorination
(Note that in hydrocarbon systems, reduction = increasing saturationIn fluorinated systems, reduction = increasing unsaturation)
Heating with strong reducing agents (electron donors) can afford defluorination.
These include zero valent metals and also organic electron transfer agents, like Thiolate.
CF3
F
CF3
Fe
500oCF
Birch
Reduction
9
The product with Thiolate anion was a little unexpected.
F
SPhPhS-
F
SPhPhS
PhSSPh
SPh
SPhSPh
10
Electron donation to Perfluorodecalin gives a radical anion, which expels fluoride anion to leave a tertiary radical.
Further reduction produces an anion which expels fluoride ion to generate a new C=C double bond.
This process continues until pefluoronaphthalene is generated.
As you will see later, such compounds are prone to nucleophilic attack (more precisely, Nucleophilic Aromatic Substitution), and Thiolate, being a good nucleophile, ends up substituting at every fluorinated position.
11
Fluoroalkenes
Unsaturation tends to increase reactivity of fluorocarbon systems.
E.g.
PFIB is ten times more toxic than phosgene, whereas perfluoromethyldecalin is used as artificial blood. (A slight difference !!!)
F3C
F3CCF2
CF3
F F
PerFluoroIsoButene Perfluoromethyldecalin
12
Preparation of Fluoroalkenes
Synthesis of CF2=CF2
Industrially:
Laboratory:By controlled heating of PTFE under vacuum, you can trap TFE.
This is a unique “unzipping” of a polymer back to a monomer.
CHCl3 CHF2ClHF / SbF5
2 CHF2Cl F2C=CF2
Pt, 700oC
via
:CF2
H-Cl
carbenedimerisation
PTFE F2C=CF2
Heat, vacuum
13
At greater temperatures (700°C) and pressures, higher mass compounds are formed.
PTFE → CF2=CF2 + CF2=CFCF3 + CF2=C(CF3)2
F2C=CF2
F2C=CF2
F2C=CFCF3
:CF2
F2C=C(CF3)2
HFP
PFIB
:CF2
14
HexafluoroPropeneHFP is a very important fluoroalkene for synthetic reactions since it does not
homopolymerise (one of the few).
Commercially is produced by the controlled pyrolysis of CF2=CF2 (see previous).
In the lab it can be generated via the following synthetic scheme:
Cl
O E.C.F.
HF F3C
F2C
CF2
F
O 1) H2O
2) NaOH F3C
F2C
CF2
O- +Na
O
- F-
F3CC
CF2
heat-CO2
_
F FC CF2
F3C
F
15
Reactions of Fluorinated AlkenesFluorinated alkenes are typically electrophilic, and therefore they react readily with
nucleophiles.
E.g.
The reaction can either be nucleophilic addition or substitution.
Notice that in both cases, this reaction is Regiospecific. (CH3O- attached to the CF2 end).
It is important to be able to explain what factors influence the reactivity of the fluoroalkenes, and thus why these reactions are regioselective.
16
General Rules for Ionic Reactions of Fluoroalkenes
This highlights the different substituent effects of “fluorine” and “fluoroalkyl”.
Fδδ+
Clδ+
is more reactivethan
C Finductive withdrawaloffset by lp repulsions
C C F
_
strongly stabilizing
17
Orientation of Attack
The nucleophilic reactions are very specific for attack at the terminal =CF2 group.
If we consider the two possible intermediate anions which would be produced by attack at either end of HFP, and consider their relative stabilities, we can explain the regioselectivity of these reactions.
The RHS reaction has two favorable fluoroalkyl stabilizations and one fluorine destabilization
The LHS reaction has one favorable fluoroalkyl stabilization and two fluorine destabilizations.
Clearly the RHS reaction pathway is preferred.
18
This also explains the following regiochemistry with CTFE:
Relative Reactivity Among Fluoroalkenes
CF2=CF2 < CF2=CFCF3 < CF2=C(CF3)2
Their corresponding intermediates (and approximately their transition states) are:
Which show an increasing stability.
Hence PFIB is more reactive than HFP than TFE.
(PFIB reacts with neutral methanol).
Nuc-
FC
CFCl
F NucCF2C
Cl
F _CNucFC
Cl
F
- F-
TFE PFIBHFP
→ Increasing in Anion stability →
19
Nucleophilic Epoxidation
Sodium Hypochlorite (NaOCl) is a good nucleophilic epoxidising agent (Recall mcpba is an electrophilic epoxidising agent)
20
“Mirror Image” Chemistry
Reactions with Fluoride Ion create a Mirror Image Organic Chemistry.(Nothing to do with chirality!)
Compare:
OligomerisationAlkenes can be polymerized by acid, whereas fluoroalkenes can be oligomerised using
fluoride ion.
F- F2C C(CF3)2 (CF3)3C-
H+ H2C C(CH3)2 (CH3)3C+
Chemistry
Chemistry
21
And even for the generation of strained bicyclic systems.
Such systems can give rise to long lived anions.
E.g.
CsF
F
F_F
F
Cs+
22
The anions can be observed at low temperature by 19F NMR.
Negative sigma complexes can be formed and observed by NMR.
E.g.
CsFN
N
NF
N
N
NFF
F F_
Cs+
23
Negative Friedal Crafts ProcessesFluorinated systems can be used for a process analogous to FC alkylations, but using
anions / nucleophiles instead of cations / electrophiles.E.g.
(See fluoroaromatic section later for more detail.)
F-
F2C CFCF3 (CF3)2FC- NF
NF
C FF3CCF3
NF
C3F7
C3F7NF
C3F7
C3F7NF
C3F7
C3F7C3F7
C3F7 C3F7-
thermodynamic kinetic
24
Electrophilic Attack On Fluorinated Alkenes
Consider the following reaction:
When the reaction was performed with Deuterio-fluorosulfonic acid, there was no D incorporation.
This implies the mechanism does not involve proton (or D+) transfer from the acid.
CF3CH CH2 CF3HH3C
F3C
H
HHSO3F
(DIMER)
CF3CH CH2 CF3HH3C
F3C
H
HDSO3F
25
Here there was no change in the location of the deuterium, which again implies that the reaction is induced by the acid, but there is no H+ transferred.
.........So how does this reaction proceed?
CF3CD CH2 CF3DH3C
F3C
D
HHSO3F
26
27
Notice that the +ve charge stays well away from the powerfully electron withdrawing CF3group.
There is a Hydrogen shift (hydride, :H-) to produce a more stable allylic cation.
Certain fluorinated cations are observable by NMR.
E.g.
The NMR shifts imply (confirm) that the +ve charge lies almost totally at C1 and C3.
CF3CFF
OCH3F
pC6H4OCH3
F
F
F
+
SbF5
SO2, -60oC
28
Notice that for a positive charge:
α Fluorine is strongly stabilizing
β Fluorine is strongly destabilizing
Therefore we would (correctly) predict that the cation derived from HFP is less stable than that derived from pentafluoropropene.
CF3CF CF2F
FF
F
F
+
CF3CH CF2F
FH
F
F
+
less stable
more stable
+ve charge at C1 and C3, so an F at C2 is a β F = destabilizing
29
Free Radical Additions
For reactivity towards free radicals we observe:
CHCl=CHCl << CH2=CH2 << CCl2=CH2
This implies that substituents tend to enhance the reactivity towards free radical addition if the substituents are at the same end of the double bond.
This is a combination of steric and electronic factors.
For the radical addition of HBr to fluoroethene:
(ROOR → 2 RO• then HBr→ ROH and Br• which adds to the alkene)
H2C CFHH-Br
Peroxides BrCH2-CFH2
30
This regiochemistry indicates an intermediate radical of BrCH2CFH•
And thus it seems that an α F is more stabilizing to radicals than an α H.
Tetrafluoroethene can be easily polymerized under free radical conditions (which is unusual for a tetrasubstituted ethene).
The ΔH for polymerization for TFE is about 17kcal/mol more exothermic than for ethene. (This has to do with hybridization - see later).
F2C CF2
InInCF2-CF2
C2F4 (CF2-CF2)n
PTFE
31
Other Commercial PolymersCommercial homopolymers are
PVDF -(CH2CF2)-n
Kel-F. -(CF2CFCl)-n
HFP will not homopolymerize (for steric reasons), but can form useful commercial co-polymers with TFE, and VDF:
FEP -(CF2CF2)-x-(CF2CFCF3)-y
Viton -(CH2CF2)-x-(CF2CFCF3)-y
32
Cycloaddition Reactions
Fluoroalkenes show an unusual propensity for forming 4 membered rings via thermal 2+2 cycloaddition.
E.g.
We shall discuss the matter of why the “head to head” dimer is formed later.
Hydrocarbon ethene derivatives do not form in analogous fashion.
(Recall that a 2+2 concerted cycloaddition is thermally forbidden).
A common feature of successful 2+2 cycloadditions is a terminal =CF2 group.
F2C CFCl200oC Cl
Cl
FZn
FDechlorination
33
Mixed Cycloadditions can occur:E.g.
The observed stereochemistry of these reactions reveals the likely mechanism.
The fact that stereochemistry is lost implies a diradical pathway as the most likely mechanism. (i.e.NOT CONCERTED).
F2C CF2
H2C CH2
FF
FF
F2C CF2
C CH
D D
H
200oC CF2F2C
HHD D
andCF2F2C
DHD H
mixture of isomerspurecis
syn &anti
34
The fact that stereochemistry is lost implies a diradical pathway as the most likely mechanism.
F2C CF2
C CH
D D
H
concerted CF2F2C
HHD D
F2C CF2
DHC CHD
stepwisediradical
CF2F2C
HHD D
andCF2F2C
DHD H
Rotation
before closure
stereochemistrymaintained
stereochemistry lost
35
This diradical pathway helps explain why we observed the head to head dimer of CTFE earlier.
This implies that to a radical center, an α Chlorine is more stabilizing that an α fluorine.
We will later prove that the stability order is:
C Cl > C F > C H
more stable
FF2C CFCl
F2C CFCl2 CF2=CFCl
Cl
Cl
36
We also see a surprising difference in butadiene / cyclobutene isomerization between fluorocarbon and hydrocarbon systems.
ΔF6 F
Δ
whereas
This behavior will be explained later.
Note that [2+2] thermal cycloaddns can and do occur, BUT NOT IN A CONCERTED PERICYCLIC MANNER.
37
For compounds with a terminal =CF2, (2+2) cycloadditions generally proceed in preference to (4+2) cycloadditions.
Another example is very revealing:
CF2CF2 CF2
CF2
CF2
CF2
(60%) (30%)
475oC
CCl2CF2
80oC
CF2
CCl2F2C CCl2 F2C CCl2
(2%) (15%) (83%)
2+2 4+2
4+2 2+2 2+2
38
Clearly the (2+2) is preferred over the (4+2) cycloaddition.
The ratio of the (2+2) cycloadducts reflects the stability of the diradical intermediates.
Firstly the dienophile generates a dichloro (not difluoro) radical.
The allylic radical with a methyl on C-3 is the more highly substituted in the important places (i.e. C-1 and C-3), and thus is more stable, and leads to the major product.
CCl2CF2
F2C CCl2
F2C CCl2
(15%)
(83%)
F2CCCl2
F2CCCl2
more stable allylic radical
39
Thermodynamic Effects of Perfluoroalkyl Groups
There is a strong thermodynamic driving force for Fluorine to be attached to an atom of high p character.
I.e. F prefers a carbon to be sp3 rather than sp2 rather than sp.
Recall the exothermicity of the polymerization of TFE to PTFE was almost 17kcal/mol higher versus the non-fluorinated ethene.
This is probably attributed to the relief of electron/electron repulsions in the starting fluoroalkene to the saturated product polymer.
C F
sp2
C F prefered over
sp3
40
A fluorine on an sp2 (C=C) both inductively withdraws and resonance donates.
These opposing features essentially cancel each other out, and F-C=C and H-C=C are about the same bond energy.
Notice the difference between F and RF as double bond substituents:
RF-C=C significantly lower the orbital energies (stronger bond).
In double bond competition reactions F-C=C is slightly more electron rich than H-C=C.(F is better π donor than σ withdrawer).
O
H
HF
F
F
F
O
HH
CF2CF2
DA is best with e- rich diene and e- poor dienophile, so we can tell which is more e- poor.
41
Recall perfluorobutadiene / cyclobutene.
In this reaction we are reducing the number of sites where F is bound to sp2 carbon
(That’s 4 → 2)
Another example of this is the fluoride ion catalyzed isomerization of hexafluorobutadiene to hexafluorobutyne.
ΔF 6 F
F6CsF
C CF3C CF3
H+
C CH3C CH3
(100oC)
ΔH = + 10kCal/mol
42
The fact that hybridization is so important to fluorocarbons is shown by the following experimentally observed bond angles for difluoromethylene groups:
This implies even sp2 C-F’s are not really sp2, but more “sp3-like”.
All the above examples highlight the fact that fluorine prefers not to be attached to unsaturated carbon.
sp3 = 109.5o
sp2 = 120o
{“sp2” C-F {
{
“sp3” C-F
43
FluoroalkynesBy definition, a fluoroalkyne has Fluorine bound to an sp hybridized carbon.
Fluorine prefers high p character atoms (e.g. sp3), thus sp is highly disfavored.
Consequently fluoroalkynes are generally very reactive. (I.e. potential for boom!)
It is tempting to attribute this to π / lone pair electron repulsions between the triple bond and fluorine atom.
Difluoroethyne is “treacherously unstable”, and whilst it has not been isolated, it has been trapped from the pyrolysis / photolysis of difluoromaleic anhydride.
sp C-F bond = short = e-s all close together
44
Monofluoroethyne polymerizes rapidly to give trifluorobenzene.
O
O
OH
FFHΔ
dangerously explosive
F F
Ftrimerization
45
Bulky groups can be used to help (kinetically) stabilize reactive compounds.
C C FCCH3
H3CCH3
t-Bu Br
H F- HBr
t-Bu
F F
t-But-Bu
F
t-Bu
F
Ft-Bu
Ft-Bu
Dewar Benzene
BenzvaleneF F
t-But-Bu
F
t-Bu
Δ
F F
t-BuF
t-Bu
t-Bu
Δ
x 3
x 3
This lead to the formation of stable Dewar benzenes and benzvalene
46
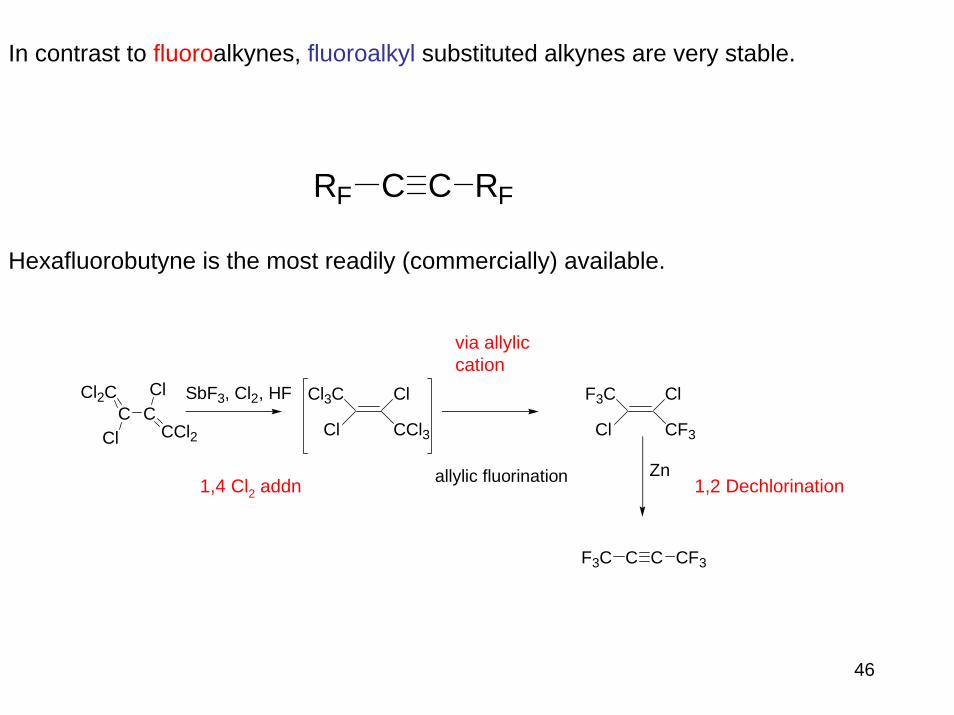
In contrast to fluoroalkynes, fluoroalkyl substituted alkynes are very stable.
Hexafluorobutyne is the most readily (commercially) available.
C C RFRF
C C CF3F3C
Cl2CC C
CCl2Cl
Cl SbF3, Cl2, HF Cl3C
Cl
Cl
CCl3
F3C
Cl
Cl
CF3
allylic fluorination Zn1,4 Cl2 addn
via allyliccation
1,2 Dechlorination
47
Reactions of Hexafluorobut-2-yne1) Trimerization
Heating the alkyne to 400oC generates hexakis(trifluoromethyl) benzene.
This benzene derivative shows some interesting chemistry, including the fact that is able to form numerous benzene isomers upon photolysis.
F3C CF3
CF3
CF3
CF3
F3CC C CF3F3C3
48
2) Nucleophilic AttackThe powerful electron withdrawing groups render the triple bond electrophilic.
This means these systems react with nucleophiles.
Reaction with Methoxide gives products of nucleophilic addition.
49
Fluoride ion reacts to give carbanions that can either be trapped, or continue to oligomerize / polymerize.
50
The polymer generated by anionic polymerization is a trifluoromethyl-polyacetylene.
It is unusual that this conjugated polymer is white – normal conjugated polyenes are colored.
UV spectra indicate that the double bonds in this polymer are not conjugated.
This is attributed to the steric requirements of the bulky trifluoromethyl groups, which force the polyene out of planarity.(so cannot be conjugated)
(Side note about the trifluoromethyl group and sterics:Steric size and steric effect are two different things:
Steric size is an intrinsic molecular property, an absolute term.
Steric effect is an extrinsic property, and depends on the nature of the transition state for a particular process.
51
Steric parameters for the trifluoromethyl group shown above, imply that the CF3group is much larger than a methyl, and in fact has a steric effect, at the least, comparable to that of a hydrocarbon iso-propyl group.)
AX
EQ
*
52
…Back to nucleophilic additions to C4F6.Addition of allyl alcohol is accompanied by the Claisen rearrangement.
The classic Claisen Rearrangement is
C CF3C CF3 H CF3
F3C O
H CF3
F3C OOH
NaOH
allyl aryl ether → ortho allyl aryl
• •
• •
53
A 6 electrocyclic process. Tautomerization of the ketone (actually a dienone) to the more stable aromatic enolic form, which we know as a phenol.
The acyclic version is:
54
There is no reason for keto/enol tautomerization in the acyclic case, and the product is a rearranged ketone.)
This type of reaction is correctly labeled as a concerted pericyclic [3,3] sigmatropic rearrangement.
A sigmatropic rearrangement is defined as migration, in an uncatalyzed intramolecular process, of a σ bond, adjacent to one or more systems, to a new position in a molecule, with the π systems becoming reorganized in the process.
The order of a sigmatropic rearrangement is expressed by two numbers set in brackets: [i, j].
These numbers are determined by counting the atoms over which each end of the σ bond has moved.
55
Each of the original termini is given the number 1.
O
R
O
R
sigma bond that breaks
new sigma bond11
2
2
33 3
32
211
a [3,3] sigmatropic rearrangement
56
Cope Rearrangement ([3,3] sigmatropic )
57
58
3) Cycloaddition ReactionsThese fluoroalkyl-alkynes are also reactive dienophiles for cycloaddition reactions.
59
Fluoro-aromatic Systems
(Fluoroaromatic is used to refer to aromatic compounds with a C-F on the aromatic ring.C6H5F is a fluoroaromaticCF3-C6H5 is fluoroalkylaromatic.)
Fluoroaromatic compounds have only recently seen a large growth in their commercial applications.
Fluoroaromatic compounds now are considering key components in the pharmaceutical, agrochemical, dye and advanced materials industries.
Generally speaking there is not a great “universal” route to making fluoroaromatics.
60
There are a variety of different ways that are capable for synthesizing molecules with one or two ‘aromatic’ fluorines:
a) Elemental Fluorine / Electrophilic N-F reagents
b) Balz-Schiemann
c) Fluoride anion HALEX
To create perfluoro aromatics, the strategy of Perfluorination (saturation) followed by reduction (aromatization) is still the best.
d) High Valency Metal Fluorides (followed by reduction)
e) ECF (followed by reduction)
61
We have seen examples of most of these in the previous chapters:
a) Direct fluorination with F2 / N2, or Selectfluor:
62
c) Halex – nucleophilic aromatic substitution using fluoride.The Sanger Reagent 2,4-dinitrofluorobenzene, C6H3F(NO2)2, used to identify the end amino acid in a protein chain. It is named after Frederick Sanger (1918- ).
why this N ?
N A S
63
The two nitro groups enable nucleophilic aromatic substitution to occur very easily.
This compound can be made simply via NAS with fluoride ion.
NO2
NO2Cl
KF
NO2
NO2F
64
Without appropriate activating groups, multiple exchanges require very forcing conditions indeed.
d+e)
Cl KF
autoclaveF
NCl KF
autoclave NF
NF
ClCl+
major productwith Cl metato N remaining
hard to force itto this product
CoF3
CH3 CF3
F FHigh Temp.
Fe
CF3
F F
65
b) The Balz-Schiemann Reaction
Covered in organic II.
Perhaps still the best/simplest/easiest way to put a fluorine onto an aromatic ring.
This is still the way that most of the chemical industry does it.
NO2 NH2 N2+ Cl- N2
+BF4-
C6H6
F
N2 BF3
NaNO2
HCl
HBF4 heat
66
There are some slight variations which can offer some advantages:
i) the use of HPF6 instead of HBF4.Heating gives Ar-F + N2 + PF5
ii) Photochemical decomposition of the aryldiazonium tetrafluoroborate salt (eliminates the need to heat)
iii) The use of NaNO2 / HF (or Py-HF) to convertAr-NH2 → Ar-N2
+ F-, followed by heating.
The mechanism for the Balz-Schiemann reaction is not fully understood, and it is generally believed there are several competing processes occurring...
67
Normally we might imagine a slow RDS of aryl cation formation, followed by trapping with a nucleophile.
Ar+
Ar•
68
Fluoroaromatics and Nucleophilic Aromatic Substitution
Hexafluorobenzene undergoes NAS with nucleophiles via the addition / elimination mechanism.
Notice the ‘mirror image’ analogy of hydrocarbon systems:
69
For Example
F
F
F
F
F
F NaOCH3
CH3OH
F
OCH3
F
F
F
F
F
FF
F
CH3Li
F
FF
F
F
FCH3
F
F
FF
F
70
But what about multiple attacks, relative rates and directing effects?Electron Donating Groups (Deactivating for NAS)
Hydrogen as a Substituent:
Electron withdrawing Substituents (Activating for NAS)
F
CH3
F
F
F
F
OCH3
CH3
F
F
F
F
CH3OH
CH3ONa Rel rates
0.63 (deact)
F
H
F
F
F
F
OCH3
H
F
F
F
F
CH3OH
CH3ONa 1 (control)
F
CF3
F
F
F
F
OCH3
CF3
F
F
F
F
CH3OH
CH3ONa5,000 (act)
71
These reactions all proceed at 60oC.The relative rates for these reactions are 0.63 : 1: 5,000
Electron withdrawing substituents enhance the rate for these NAS, and electron-donating substituents lower the rate.
This is of course easy to explain by considering the relative stabilities of the intermediate carbanions formed in the endothermic RDS of these NAS processes.
But do you notice a pattern in directing effects?All three situations show a major preference for a para substitution.
This implies that it is not the substituent, BUT THE FLUORINES, that govern the position of attack and substitution.
So we should consider the basis of the orientating/directing preferences of Fluorine atoms.
It has to do with the effects of the fluorine atoms around the ring on the negatively charged σ intermediate.
72
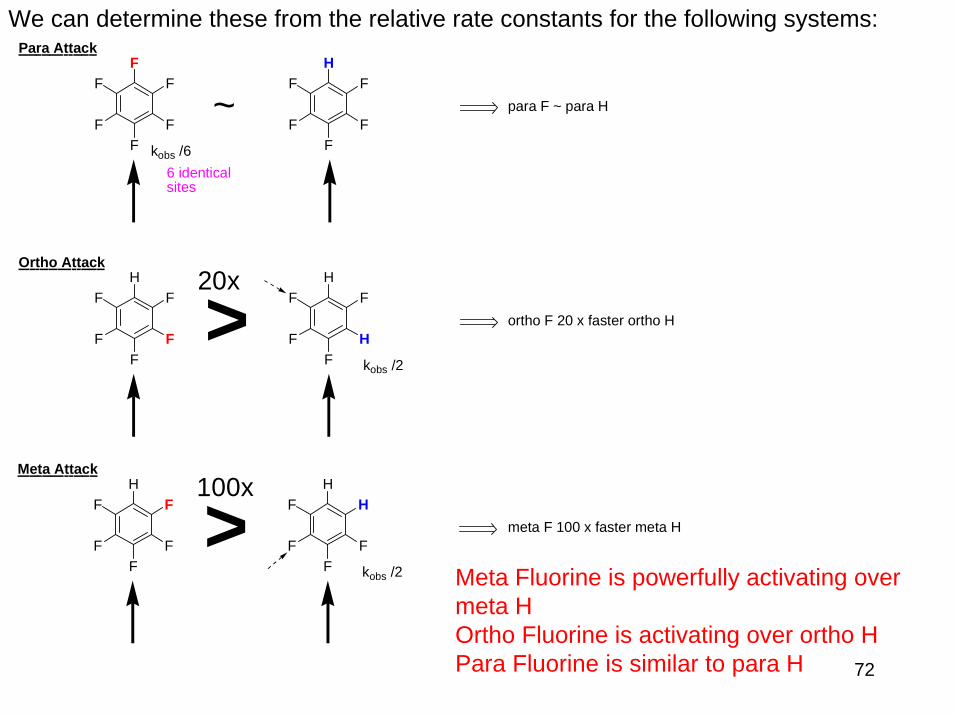
We can determine these from the relative rate constants for the following systems:
Meta Fluorine is powerfully activating over meta HOrtho Fluorine is activating over ortho HPara Fluorine is similar to para H
F
F
F
F
F
F
F
H
F
F
F
F
>
~kobs /6
para F ~ para H
Para Attack
F
H
F
F
F
F
F
H
F
F
H
F
kobs /2
ortho F 20 x faster ortho H
Ortho Attack20x
>F
H
F
F
F
F
F
H
F
H
F
F
kobs /2
meta F 100 x faster meta H
Meta Attack100x
6 identical sites
73
Remember the different effects of α and β fluorines on a carbanion center:
So when a nucleophile attacks and forms a negative sigma complex:
74
A meta F is β to the –ve charge, and thus stabilizing
A para F α to the –ve charge, and thus slightly destabilizing
This explains the 100x preference, and equivalence exhibited in relative rates.
One could assume an ortho F should also be destabilizing – however we determined it to be 20x more active!
The full picture about ortho F is a little more involved…
The initial state effects are also important.
75
The ortho Fluorine enhances the ion dipole effect – which means the approach of a nucleophile is electrostatically encouraged.
The amount that this impacts the reactivity of the fluoroarene depends on the fluoroarene itself.
More precisely the initial state effects impact the transition state: if it is a highly reactive system the T.S. will be early, and this will be encouraged by the ortho fluorine.
A late transition state (less reactive system) is far less impacted by initial effects and so an ortho Fluorine would just show its destabilizing effect of F α to an anion.
However, almost all of these fluoroarenes are considered to be reactive systems.
F
F
F
F
F
FNuc-
δ-δ-δ+ δ+
76
Also, there has been evidence that implies that for these fluorinated sigma complexes, most of the negative charge really lies at the position para to attached nucleophile.
That is to say Lewis structure (A) is more significant / important than (B) for these systems.
This would also explain the activating effect of ortho fluorine (takes advantage of its withdrawing ability).
parameta
ortho
meta
orthoFNuc
_
parameta
ortho
meta
orthoFNuc
_
(A) (B)
77
So overall, wrt NAS:
Ortho F considered slightly activating
Meta F considered very activating
Para F considered deactivating (slightly) / comparable to H
Para attack is generally preferred.
X
Fo
m
o
m
PARA Attackall activating
X
Fp
m
ORTHO Attackmostly activating
om
X
Fm
p o
META Attackmostly activating
o
two meta, two ortho
78
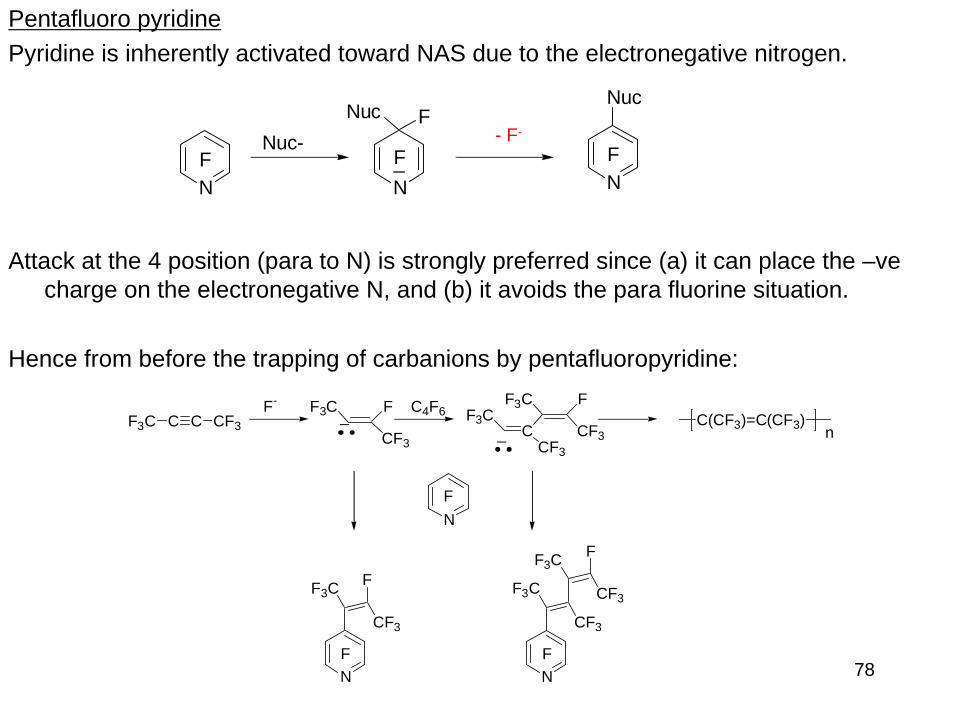
Pentafluoro pyridinePyridine is inherently activated toward NAS due to the electronegative nitrogen.
Attack at the 4 position (para to N) is strongly preferred since (a) it can place the –ve charge on the electronegative N, and (b) it avoids the para fluorine situation.
Hence from before the trapping of carbanions by pentafluoropyridine:











































































![THE EFFICACY OF TERM STRUCTURE ESTIMATION …crab.rutgers.edu/~yaari/Articles-PDF/OCR[19].pdf · the efficacy of term structure estimation techniques: a monte carl0 study mark buono,](https://static.documents.pub/doc/80x56/5b38c9cc7f8b9abd438da8be/the-efficacy-of-term-structure-estimation-crab-yaariarticles-pdfocr19pdf.jpg)
















