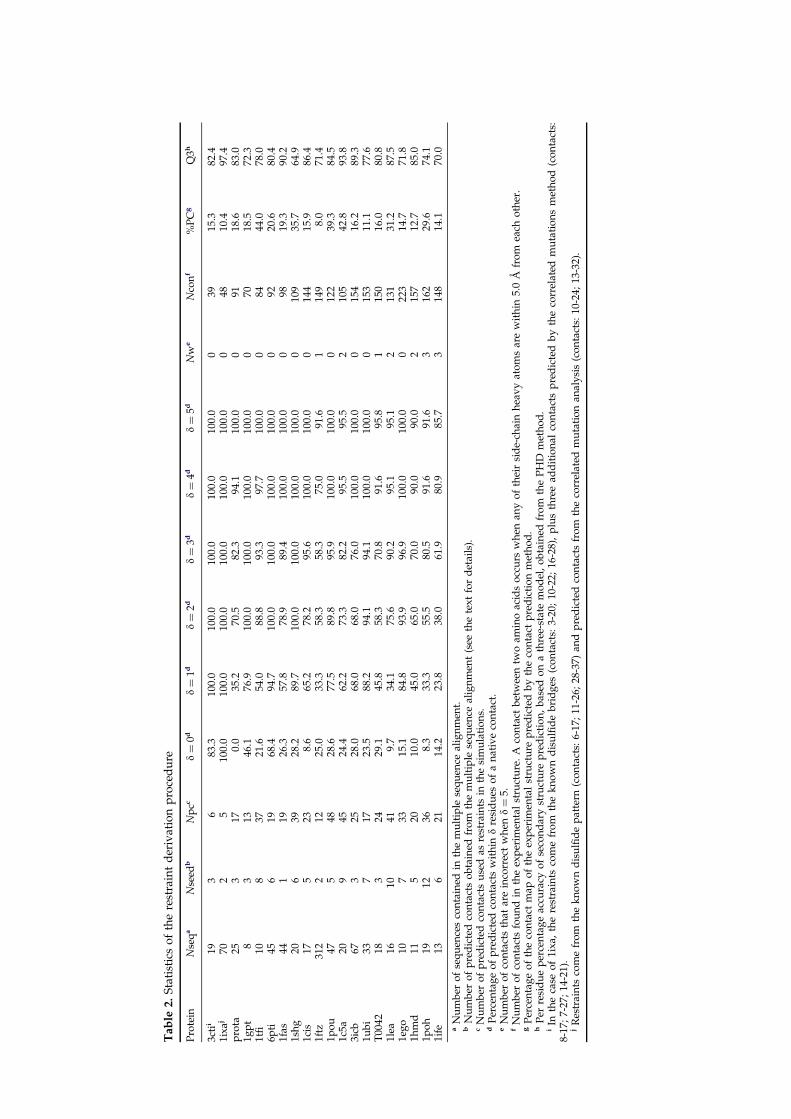
Fold Assembly of Small Proteins Using Monte Carlo Simulations Driven by Restraints Derived from Multiple Sequence Alignments Angel R. Ortiz 1 , Andrzej Kolinski 1,2 and Jeffrey Skolnick 1 * 1 Department of Molecular Biology, TPC-5, The Scripps Research Institute, 10550 North Torrey Pines Road, La Jolla CA 92037, USA 2 Department of Chemistry University of Warsaw ul. Pasteura 1, 02-093 Warsaw Poland The feasibility of predicting the global fold of small proteins by incorpor- ating predicted secondary and tertiary restraints into ab initio folding simulations has been demonstrated on a test set comprised of 20 non- homologous proteins, of which one was a blind prediction of target 42 in the recent CASP2 contest. These proteins contain from 37 to 100 residues and represent all secondary structural classes and a representative variety of global topologies. Secondary structure restraints are provided by the PHD secondary structure prediction algorithm that incorporates multiple sequence information. Predicted tertiary restraints are derived from mul- tiple sequence alignments via a two-step process. First, seed side-chain contacts are identified from correlated mutation analysis, and then a threading-based algorithm is used to expand the number of these seed contacts. A lattice-based reduced protein model and a folding algorithm designed to incorporate these predicted restraints is described. Depend- ing upon fold complexity, it is possible to assemble native-like topologies whose coordinate root-mean-square deviation from native is between 3.0 A ˚ and 6.5 A ˚ . The requisite level of accuracy in side-chain contact map prediction can be roughly 25% on average, provided that about 60% of the contact predictions are correct within 1 residue and 95% of the predictions are correct within 4 residues. Precision in tertiary contact prediction is more critical than absolute accuracy. Furthermore, only a subset of the tertiary contacts, on the order of 25% of the total, is sufficient for successful topology assembly. Overall, this study suggests that the use of restraints derived from multiple sequence alignments combined with a fold assembly algorithm holds considerable promise for the prediction of the global topology of small proteins. # 1998 Academic Press Limited Keywords: protein structure prediction; correlated mutations; side-chain contact prediction; lattice protein models; secondary structure prediction; threading *Corresponding author Introduction At present, the prediction of protein structure from amino acid sequence remains one of the major unsolved problems in molecular biology. The solution to this problem demands the develop- ment of effective conformational search algorithms and the formulation of potentials capable of recog- nizing the native state from the manifold of mis- folded structures. Reduced protein models having one or a few interaction centers per residue and statistical potentials extracted from protein struc- ture databases offer a reasonable way to address both issues (Godzik et al., 1994; Kolinski et al., 1995a,b; Kolinski & Skolnick, 1994a; Park & Levitt, 1995; Skolnick et al., 1993). Using such approaches, several successful ab initio predictions of simple topologies have been reported. For example, the conformation of short peptides such as melittin (Ripoll & Scheraga, 1990), pancreatic polypeptide inhibitor (Sun, 1993; Wallqvist & Ullner, 1994), apamin (Sun, 1993), and PthrP (Wallqvist & Ullner, 1994) have been predicted with a backbone RMSD ranging from 1.7 A ˚ to 4.5 A ˚ . Lattice folding simu- lations (Kolinski & Skolnick, 1994b) of the B domain of Protein A (Gouda et al., 1992) have Abbreviations used: RMSD, root-mean-square deviation; cRMSD, coordinate RMSD; PDB, Protein Data Bank. J. Mol. Biol. (1998) 277, 419–448 0022–2836/98/120419–30 $25.00/0/mb971595 # 1998 Academic Press Limited
Transcript
J. Mol. Biol. (1998) 277, 419±448
Fold Assembly of Small Proteins Using Monte CarloSimulations Driven by Restraints Derived fromMultiple Sequence Alignments
Angel R. Ortiz1, Andrzej Kolinski1,2 and Jeffrey Skolnick1*
1Department of MolecularBiology, TPC-5, The ScrippsResearch Institute, 10550 NorthTorrey Pines Road, La JollaCA 92037, USA2Department of ChemistryUniversity of Warsawul. Pasteura 1, 02-093 WarsawPoland
The feasibility of predicting the global fold of small proteins by incorpor-ating predicted secondary and tertiary restraints into ab initio foldingsimulations has been demonstrated on a test set comprised of 20 non-homologous proteins, of which one was a blind prediction of target 42 inthe recent CASP2 contest. These proteins contain from 37 to 100 residuesand represent all secondary structural classes and a representative varietyof global topologies. Secondary structure restraints are provided by thePHD secondary structure prediction algorithm that incorporates multiplesequence information. Predicted tertiary restraints are derived from mul-tiple sequence alignments via a two-step process. First, seed side-chaincontacts are identi®ed from correlated mutation analysis, and then athreading-based algorithm is used to expand the number of these seedcontacts. A lattice-based reduced protein model and a folding algorithmdesigned to incorporate these predicted restraints is described. Depend-ing upon fold complexity, it is possible to assemble native-like topologieswhose coordinate root-mean-square deviation from native is between3.0 AÊ and 6.5 AÊ . The requisite level of accuracy in side-chain contact mapprediction can be roughly 25% on average, provided that about 60% ofthe contact predictions are correct within �1 residue and 95% of thepredictions are correct within �4 residues. Precision in tertiary contactprediction is more critical than absolute accuracy. Furthermore, only asubset of the tertiary contacts, on the order of 25% of the total, issuf®cient for successful topology assembly. Overall, this study suggeststhat the use of restraints derived from multiple sequence alignmentscombined with a fold assembly algorithm holds considerable promise forthe prediction of the global topology of small proteins.
# 1998 Academic Press Limited
Keywords: protein structure prediction; correlated mutations; side-chaincontact prediction; lattice protein models; secondary structure prediction;threading
*Corresponding author
Introduction
At present, the prediction of protein structurefrom amino acid sequence remains one of themajor unsolved problems in molecular biology.The solution to this problem demands the develop-ment of effective conformational search algorithmsand the formulation of potentials capable of recog-nizing the native state from the manifold of mis-folded structures. Reduced protein models havingone or a few interaction centers per residue and
mean-squareD; PDB, Protein Data
971595
statistical potentials extracted from protein struc-ture databases offer a reasonable way to addressboth issues (Godzik et al., 1994; Kolinski et al.,1995a,b; Kolinski & Skolnick, 1994a; Park & Levitt,1995; Skolnick et al., 1993). Using such approaches,several successful ab initio predictions of simpletopologies have been reported. For example, theconformation of short peptides such as melittin(Ripoll & Scheraga, 1990), pancreatic polypeptideinhibitor (Sun, 1993; Wallqvist & Ullner, 1994),apamin (Sun, 1993), and PthrP (Wallqvist & Ullner,1994) have been predicted with a backbone RMSDranging from 1.7 AÊ to 4.5 AÊ . Lattice folding simu-lations (Kolinski & Skolnick, 1994b) of the Bdomain of Protein A (Gouda et al., 1992) have
# 1998 Academic Press Limited
420 Fold Prediction of Small Proteins
yielded structures with a Ca RMSD from native onthe order of 3.0 AÊ . The most accurate predictionsto date are those of the GCN4 leucine zipper,whose ®nal predicted Ca RMSD is 0.8 AÊ from thenative crystal structure (O'Shea et al., 1991; Viethet al., 1994). In general, these methods have beenmainly successful on helical proteins having simpleglobal folds. For more complex structures of natu-ral proteins, ab initio folding has failed thus far.Simpli®ed protein representations and inaccuraciesin the attendant potentials can conspire to yield aninability to identify the native topology fromalternative non-native structures, especially thosewith a substantial similarity to the native fold.Furthermore, as protein size and topological com-plexity increases, conformational samplingbecomes exponentially more problematic. Thus,alternative approaches are required that can sur-mount these dif®culties.
Introduction of secondary structure restraintsobtained from secondary structure predictionalgorithms is a natural extension of pure, restraintfree ab initio folding. This is a particularly appeal-ing idea since knowledge of the native secondarystructure elements enormously reduces the confor-mational space that must be searched. Recently,the accuracy of secondary structure prediction hasimproved from about 65% to 72%, on average(Rost & Sander, 1996b). This development lendscredence to the idea that secondary structuralelements can be identi®ed with reasonable accu-racy. Some progress is also being made in algor-ithms that can predict regions where the chainreverses global direction, viz., U-turns (Kolinskiet al., 1997). The feasibility of biasing search algor-ithms with secondary structure knowledge was®rst explored using ``exact'' secondary structure, asobserved in the experimental native conformation.In this regard, using an off-lattice model and exactknowledge of the native secondary structure,Friesner et al. (1996) have obtained very encoura-ging results, successfully folding two four-helixbundles (cytochrome b562 (B256) and myohemery-thrin (2MHR)), a large a-helical protein (myoglobin(1MBO)), and a relatively complicated a/b fold,the C-terminal domain of the L7/L12 50 S riboso-mal protein (1CTF) (Friesner & Gunn, 1996; Gunnet al., 1994). Similarly, Dandekar & Argos (1994,1996), using a genetic algorithm to search confor-mational space, obtained encouraging results for atest set of 19 small proteins, including all a andsome a/b proteins. In their studies, they have suc-ceeded in predicting a signi®cant proportion ofthese small proteins at �5 AÊ resolution. However,Dandekar & Argos have also observed that use ofpredicted secondary structure information pro-duces a substantial deterioration in the perform-ance of their prediction algorithm. As a practicalmatter, however, any successful tertiary structureassembly algorithm must be able to successfullyhandle predicted secondary structural informationwhose accuracy is at the current state-of-the-art,and tests using predicted secondary structure
should be made. Along these lines, an earlyexample of where predicted secondary structurewas incorporated as restraint information in a sub-sequent topology assembly algorithm is due toKolinski & Skolnick (1994b). The resulting pre-dicted structure of crambin had a backbone Ca
RMSD of 3.2 AÊ . A more recent example is due toSimons et al. (1997), who instead of secondarystructure predictions used an interesting techniqueto derive short-range conformational preferencesfrom multiple sequence alignments. Some a-helicalproteins could be assembled using this approach,although the potential function used by the authorsdid not allow the native-like topology to be discri-minated from alternative answers. However, it wasapparent from these and other studies that knowl-edge of secondary structure preferences alone doesnot entirely eliminate competing misfolded states.Furthermore, secondary structure bias is local innature and, therefore, does not provide a gradientin the conformational energy landscape that canfunnel the conformation towards the native state.Thus, problems with both potentials and confor-mational search protocols still remain.
Funneling can be ef®ciently obtained throughthe use of long-range (in sequence) distancerestraints. A number of workers have begun toexamine the feasibility of such an approach. Forexample, Smith-Brown et al. (1993) have attemptedto predict several protein folds by assuming exactknowledge of the secondary structure and a subsetof interresidue distance restraints encoded as abiharmonic potential. They ®nd that a considerablenumber of restraints per residue is required toassemble the fold, making the approach impracti-cal for most prediction purposes. Another interest-ing study is due to Aszodi & Taylor (1996), whoassumed correct native secondary structure and aset of simulated tertiary restraints (Aszodi et al.,1995). Here in an attempt to build the protein core,restraints were supplemented by a set of interresi-due distances based on patterns of conservedhydrophobic residues obtained from a multiplesequence alignment. Folds were then assembledusing distance geometry with a simpli®ed proteinchain model. Aszodi & Taylor (1996) were able toassemble structures below 5 AÊ RMSD when atleast N/4 restraints are used, where N is the num-ber of protein residues. However, with their force®eld, they have excessive dif®culties selecting thecorrect fold from competing alternatives. Alongsimilar lines, Mumenthaler & Braun (1995) devel-oped an interesting self-correcting distance geome-try method that tries to automatically eliminatewrongly predicted contacts derived from multiplesequence alignments. Again, the correct secondarystructure is assumed, but now totally predicted ter-tiary restraints, based on the conservation patternsof hydrophobic residues in multiple sequencealignments, are used to assemble the fold. Withthis method, encouraging results have beenobtained with the successful folding to the nativetopology in six out of eight helical proteins stu-
Fold Prediction of Small Proteins 421
died. Still, a signi®cant number of restraints arerequired in all these approaches. This poses a pro-blem because no prediction technique is availablethat can provide both the requisite number andaccuracy of secondary and tertiary restraints thatthese approaches demand for more complex folds.
One step towards addressing these problemswas made recently by Skolnick et al. (1997b). Theydeveloped a new program, called MONSSTER(MOdeling of New Structures from Secondary andTErtiary Restraints), that is able to successfully foldsmall proteins using a considerably smaller num-ber of distance restraints than previous approachesdemanded. It was found that when ``exact''restraints are available, helical proteins can befolded with roughly N/7 restraints, while all b anda/b proteins require about N/4 restraints, where Nis the number of residues in the protein chain. Ofcourse, for any particular case, the accuracydepends on restraint distribution and fold com-plexity. MONSSTER employs a lattice-basedreduced representation of the protein chain. Inaddition to secondary and tertiary restraints, thereis a potential that incorporates statistical prefer-ences for secondary structure, side-chain burialand pair interactions, together with a hydrogenbond potential. We term these non-restraint contri-butions inherent interactions. The resulting foldaccuracy is substantially degraded when theseinherent contributions to the potential are elimi-nated. Thus, these simulations indicated that thereis a complementarity between the inherent contri-butions to the potential and the supplementary butcrucial information provided by secondary and ter-tiary restraints.
The encouraging results obtained with thismodel prompted us to attempt the next logicalstep. Namely, we explored the possibility of assem-bling global protein topologies using entirely pre-dicted secondary and tertiary restraints. Predictedrestraints are noisy in nature and it is unclearwhether an algorithm that works within the limitof a very small number of correct restraints isrobust enough to successfully handle the unavoid-able presence of incorrect predictions. Extant sec-ondary structure prediction schemes provide alogical jumping off point for the incorporation ofpredicted secondary structure information. Theprotocol for predicting tertiary restraints is lessobvious. Following on the ideas of GoÈebel et al.(1994), predicted tertiary contacts are extracted onthe basis of evolutionary information contained inmultiple sequence alignments, complemented withthreading calculations (our unpublished results). Insequence alignments, since some pairs of positionsappear to exhibit a covariation in their mutationalbehavior consistent with their physical and chemi-cal properties, it has been suggested that spatiallyclose neighbors might be more likely to exhibitsuch behavior. Using statistical techniques, thiseffect has been quanti®ed by a method known ascorrelated mutation analysis (GoÈebel et al., 1994). Ithas been shown that, by applying a stringent sig-
ni®cance cut-off in the prediction of contacts bycorrelated mutations, a small number of contactscan be predicted that are a factor of 1.4 to 5.1 timesbetter than random. Previously, the number of cor-rect contacts obtained this way has been either toosmall to permit successful tertiary structure assem-bly if a high signi®cance cut-off was used to avoidfalse positives, or too noisy if the number of con-tacts selected was that demanded by existingassembly algorithms (Rost & Sander, 1996a). Here,it is shown that, for a representative set of proteins,a modi®cation of the correlated mutation analysisapproach (our unpublished results), when coupledto secondary structure prediction and followed bystructure assembly using a version of MONSSTERupdated to handle incorrect predictions, is able tobridge the gap between sequence analysis andfolding simulations. This permits the ab initio fold-ing of some complex topologies.
The outline of the remainder of the paper is asfollows. In Methods, we describe the approach fol-lowed in this work, which can be logically dividedinto two parts: secondary and tertiary restraintderivation, and fold assembly/re®nement usingMONSSTER. In the Results, we describe its appli-cation to a set of 20 representative single domainproteins. This is followed by the Discussion, whichexamines possible reasons why the currentapproach can be successful and delineates theimprovements required, in both the restraint deri-vation procedure and in the fold assembly/re®ne-ment protocol, to make this approach generallyapplicable. Finally, in the Conclusions, we sum-marize the current state-of-the-art of protein struc-ture prediction as provided by MONSSTER.
Methods
Overview
A ¯ow chart of the tertiary structure predictionprotocol is depicted in Figure 1. The procedurepresented in this work can be logically dividedinto two parts: restraint derivation and structureassembly/re®nement using the MONSSTER algor-ithm. With respect to restraint derivation, the ®rstobjective is to predict the number, location, andidentity of the dominant secondary structuralelements that will comprise the protein. These con-sist of helices and b-strands, termed here the coretopological elements of the molecule. In addition,U-turns between these secondary structureelements are predicted (Kolinski et al., 1997). Next,we try to predict the secondary structure elementsin contact. This is attempted by obtaining the mostreliable set of predicted contacts between coreelements using correlated mutation analysis. Wedenote the contacts obtained in this way as``seeds''. This protocol allows us to maximize thesignal-to-noise ratio of predicted contacts. How-ever, the number of seeds is still insuf®cient toallow successful fold assembly to occur. Hence, weexploit the fact that packing patterns between sec-
Figure 1. Flow chart of the protein fold predictionmethod.
422 Fold Prediction of Small Proteins
ondary structure elements are degenerate. Seedcontacts are ``enriched'' by ®nding the most com-patible contact map in the structural databasegiven the predicted seed and the secondary struc-ture elements involved by using a fragment clus-tering/inverse folding protocol (Godzik et al.,1992). Full details of this procedure will be givenin a forthcoming publication. The key point is thatthe resultant restraints are not particularly accurateand that approaches incorporating such restraintsmust be adjusted to account for their ambiguity.This is accomplished in an updated version ofMONSSTER designed to accommodate theinherent inaccuracies of such restraints, as isdescribed below. The general features of the force®eld and representation are the same as thosedescribed earlier (Skolnick et al., 1997b). Thus, wefocus here only on those aspects that differ fromthe previous implementation in order to adapt theprocedure to incorrect and/or ambiguousrestraints.
Restraint derivation method
Secondary structure prediction
Multiple sequence alignments for each of theproteins studied were obtained from the HSSPdatabase (Sander & Schneider, 1991). This align-ment is used as input for the PHD (Rost &Sander, 1993) secondary structure predictionmethod. For the purposes of hydrogen bondassignment, all predicted strand elements areassumed to correspond to a strand in the realsecondary structure. For helices, only thoseelements with a reliability index higher thanthree are used. Chain reversals are predicted bythe U-turn prediction algorithm LINKER devel-
oped by Kolinski et al. (1977). Because of theirreliability, elements predicted as U-turns overridePHD predictions (Kolinski et al., 1997). In prac-tice, each residue can be assigned to one of ®veconformational states: a predicted extended state,a predicted helix, a predicted U-turn, a b-(strand)state or a non-predicted state. The set of pre-dicted helices and strands comprise the putativecore elements of the protein.
Side-chain contact prediction
The prediction of residue contacts is performedin two stages. First, a correlated mutation anal-ysis (GoÈebel et al., 1994) of the multiple sequencealignment is done to identify the seed contacts.The procedure is based on de®ning an exchangematrix or other similarity measure at eachsequence position in a multiple sequence align-ment. One then calculates the correlation coef®-cient between exchange matrices at any twopositions. In the calculation of the covariancematrix, regions containing deletions and inser-tions are not considered. Here, residue compari-son is carried out using the McLachlan (1971)matrix. In this work, the same multiple sequencealignment is used for secondary structure andcorrelated mutation analysis. Only correlationsbetween elements predicted to be in core regions(and not U-turns) are considered. The rationale isthat by restricting the predictions to rigidelements of the putative core, the assumption ofcloseness in space for positions showing covari-ance in their mutational behavior might be morevalid. Correlation is measured by a Pearson-typecorrelation coef®cient:
rij � 1
N2
Xkl
�sikl ÿ hsii��sjkl ÿ hsji�sisj
�1�
Here, i and j are two different positions in a mul-tiple sequence alignment, and the indices k and lrun from 1 to the number N of sequences in thefamily. The parameter sikl (sjkl) is the comparisonscore (according to the McLachlan (1971)mutation matrix) of the amino acids of sequencesk and l at position i(j) of the alignment. Averagevalues over all aligned sequences at positions iand j are given by hsii and hsji. The parameterssi and sj correspond to the standard deviation ofthe scores in the positions i and j, respectively.A correlation coef®cient cut-off threshold of 0.5 isused for contact prediction. At most, only onecontact per each pair of core secondary structureblocks is used. Thus, this analysis delineates pre-dicted secondary structure elements in contact. Inaddition, the maximum number of seeds allowedis equal to the maximum number of expectedcontacts (ns) between the L of predicted second-ary structure elements. This value is obtainedfrom a representative database of small proteinsand is roughly given by ns � L (L ÿ 1)/4 (ourunpublished results). However, correlated
Fold Prediction of Small Proteins 423
mutation analysis only provides a few seed side-chain contacts; their number is generally insuf®-cient to assemble a protein from the unfoldedstate using MONSSTER. Thus, the number ofside-chain contacts needs to be increased.
The set of seed restraints is enriched by a com-bined structural fragment search and threadingfolding procedure (Godzik et al., 1992). All pairs ofsecondary structure elements compatible with thepredicted secondary structure types and predictedcontacts are extracted from a structural database.This structural database is the same used by Hu etal. (1997). For each tested sequence, homologues tothe full target sequence found in the database wereremoved prior to the application of the growingprocedure. To account for the inaccuracies in thecorrelated mutation analysis, a tolerance of a one-residue shift in each member of the contacting resi-due pair is allowed. Fragments are then scored bya statistical potential that considers local confor-mational propensities and the burial energy withinthe pair of fragments (Godzik et al., 1992). Pair andhigher order interactions are ignored (Godzik et al.,1992; Hu et al., 1997) to avoid the imbalancebetween intra and extra fragment interactions,which would result if such contributions wereincluded. The top ten scoring fragments are super-imposed in space by minimizing their coordinateRMSD, and then clustered on the basis of theirpair-wise RMSD. If they do not show a clear clus-tering (with an upper limit of 5.5 AÊ for the mostdivergent fragment pair), then additional side-chain contact restraints are not derived. Conver-sely, if the fragments spatially cluster, then thefragment within this cluster whose RMSD is smal-lest, with respect to all other members, is selectedand its side-chain contact map is projected onto thequery sequence. Following this procedure, thenumber of predicted contacts usually increases byabout a factor of ®ve with respect to that predictedfrom correlated mutation analysis alone. Fulldetails of this procedure will be given in a forth-coming publication (A.R.O. & J.S., unpublishedresults).
Assembly and refinement protocol
Protein model
Geometric properties. The Ca coordinates of theprotein backbone are con®ned to a set of latticepoints located on an underlying cubic lattice whoselattice spacing, a � 1.22 AÊ (Kolinski & Skolnick,1994a). Successive Ca atoms are connected by a setof 90 virtual bond vectors a.v, with {v} � {(�3, �1,�1),... (�3, �1, 0),... (�3, 0, 0),... (�2, �2, �1),...(�2, �2, 0),...}. The distance a is chosen so that themean Ca virtual bond length is 3.8 AÊ . Side-chainsare represented by a set of rotamers, each locatedat the side-chain center of mass. They are notrestricted to lattice points. With the exception ofGly, Pro and Ala, there are multiple rotamers foreach amino acid, chosen so that the center of mass
of a side-chain in real proteins will be no fartherthan 1 AÊ from some member of the rotamerlibrary. For more details, see Kolinski & Skolnick(1994a, 1997b).
Interaction scheme
Inherent contributions. This class of terms is inde-pendent of the restraint predictions and isdesigned to capture both generic (sequence inde-pendent) and sequence-speci®c protein-like fea-tures. Many of these contributions are identical tothose described in MONSSTER (Skolnick et al.,1997b). Such terms include an amino acid pairspeci®c potential that describes the intrinsic sec-ondary structural preferences, E14, and a one-bodycentrosymmetric burial potential, E1. Here, toavoid non-physical segregation of the subunits, wehave added a packing density regularizer, Edensity
(Kolinski & Skolnick, 1997). This term is designedto ensure that the average overall density distri-bution of residues in native proteins is reproduced.This term provides a strong compressive force inthe unfolded state, but contributes negligibly incompact states. A more sensitive side-chain paircontact potential, Epair, that has been derived by amore careful analysis of the appropriate referencestate is now used (Skolnick et al., 1997a). Hydrogenbonds are Ca-based and very much in the spirit ofLevitt & Greer (1977).
Restraint contributions. Predicted secondary struc-tures and tertiary contacts are implemented intothe model in the form of restraint contributions tothe conformational energy. Furthermore, a set ofsomewhat re®ned knowledge-based restraintsdesigned to reproduce the packing of supersecond-ary structural elements is used. The implemen-tation of each type of restraint is discussed in turn.
Local secondary structure bias. Secondary struc-ture bias is incorporated into the local secondarystructure dependent terms with magnitudeEtarget,sec. As indicated above, a given residue canbe in one of ®ve conformational states assigned onthe basis of the local chain geometry. For thoseresidues having a predicted secondary structuraltype, energetic biases for the various allowed con-formational states are assigned. Turns are encodedon a generic basis, i.e. their chirality is not speci-®ed. Rather, they behave as ¯exible joints betweenregular secondary structural elements (see Skolnicket al. (1997b) for additional details).
U-turn surface bias. Regions predicted as U-turnsare assumed to lie at the protein surface. Thus, forthese residues, a penalty of 0.5 kT (with k Boltz-mann's constant and T the absolute temperature)per residue is added when they lie at or below theradius of gyration. This term of total magnitude,
424 Fold Prediction of Small Proteins
EU-turn, acts to reduce kinetic traps by segregatingthe different parts of the protein into its corre-sponding layers. Similarly, N and C-terminal resi-dues are penalized by 4 kT if they are buried (i.e.at or below the radius of gyration) to account fortheir charged ends.
Hydrogen bond mixing rules. The hydrogen bondpotential is modi®ed for those residues assigned toa predicted type of secondary structure so that theresulting hydrogen bond pattern is compatiblewith the secondary structural prediction. The mag-nitude of this term is EH-bond. More speci®cally: (1)continuous stretches of strands and extended statesor their combinations cannot form intra-elementhydrogen bonds. Strands can form hydrogenbonds only with other strands, extended states ornon-assigned states. On the other hand, extendedstates can form hydrogen bonds with all statesexcept helices. (2) For those residues assigned to behelical, hydrogen bonds beyond the ®fth neighboralong the chain are not allowed.
b-Strand cooperativity term. In trial calculations, itwas observed that predicted b-strands had con-siderable dif®culty forming b-sheets. The sameobservation has been made by other authors(Dandekar & Argos, 1996; Friesner & Gunn, 1996;Simons et al., 1997). In our case, this behaviorappears to result from a combination of the exces-sive conformational entropy of the backbone andthe highly permissive hydrogen bond scheme. Tocorrect for these effects, a cooperativity term thatstabilizes and propagates the formation of b-sheets(Eb-prop) has been included. For each predictedstrand, the hydrogen bond state of each residue inthe putative strand is scanned. If the residue ofinterest participates in two hydrogen bondsbelonging to two different b-strands, then a stabil-ization energy equal to that of the hydrogen bondcooperativity term is added. Strand residues canboth nucleate and participate in the cooperativity.In other words, blocks of secondary structure pre-dicted to be strands can be located either in thecore or at the edges of the b-sheet. Extended stateresidues can serve as cooperative hydrogen bondpartners, but cannot nucleate cooperativity; there-fore, their location in the b-sheet core is energeti-cally penalized, but not forbidden. There is nodirectionality in this cooperativity term. Thus, itcannot distinguish parallel from antiparallelarrangements of the strands, rather the ®nalarrangement is dictated by the connectivity of thechain and the predicted restraints. This hydrogenbond cooperativity term has the effect of propagat-ing the b-sheet. It also helps to bury strands pre-dicted by PHD into the core, and to locateextended states predicted by LINKER at the sur-face and at the edges of a b-sheet. Trial calculationsindicated that a small bias is adequate to success-fully build b-sheets. In the all -b and a/b proteinsstudied here, the total magnitude of this term has a
value of roughly ÿ5 kT in successfully folded struc-tures.
Tertiary restraints
Restraint function
The restraint function used in this work consistsof a simple ¯at-bottom harmonic potential. Let rij
be the actual distance between two restrained resi-dues. In practice, the restraint could operatebetween side-chain centers of mass or between theprojection of the residue pair onto the principalaxes of their respective secondary structuralelements. This situation is discussed in greaterdetail in the next section, which describes restraintsplinning. Thus, the restraint function is as follows:
Eres
� 800
� krep�rij ÿ r0ij�2
� 0
if Eres > 800
if rij > r0ij and Eres < 800
if rij < r0ij
�2�
krep � 4.0 kT/(lu)2 with lu equal to one lattice unit(1.22 AÊ ). In the case of side-chain centers ofmass, r\rm 0
ij � (hrABi � sAB) (1 � o). For a pairof residues, A and B, hrABi and sAB are the aver-age separation distance and standard deviation ofthis distance observed in a structural database.The value of o � 0.5 is used by default. In thecase of restraint splinning (see below), the value(hrABi � sAB) is substituted by the average separ-ation distance observed in a structural data basefor the packing of secondary structure elements.The values used are: 10 AÊ for a-a, 8 AÊ for a-band 6 AÊ for b-b super-secondary elements,respectively.
Restraint splinning. Most predicted seeds areshifted by at least one residue with respect to theexperimentally observed contact. Moreover, aftergrowth, the different patches of contacts can havedifferent phases. For example, suppose that onehelix is predicted to contact two other secondarystructure elements, i.e. it has two seed contacts.Because each seed is obtained and grown indepen-dently, the overall predicted contact pattern of thehelix with the two other elements could be imposs-ible. These seed contacts can lie on opposite sidesof the helix, but in fact in the folded structure theirsecondary structure partners can be on the sameside of the helix face. This effect could either pre-clude successful assembly or distort the folded con-formation such that distinction, using energycriteria from misfolded alternatives, is not possible.One way to eliminate these artifacts is to apply therestraints between the axes of the secondary struc-ture elements. This is done by smoothing the localCa chain using the method described in the Appen-dix of Skolnick et al. (1997b). This list of smoothedcoordinates is continuously updated during thesimulation.
Figure 2. Scheme of the geometric de®nitions used tode®ne the knowledge-based rules in bab supersecond-ary structures. The secondary structure elements arerepresented as blue cylinders; the strands are rep-resented as two thin cylinders and the helix as a thickcylinder. The axes of the secondary structure elementsare represented by the blue cylinders and are rep-resented by the vectors k ÿ 2, k ÿ 1 and k, respectively.The b-sheet to which the two strands belong is rep-resented as a red membrane. The vector a connects thebeginning of the last b-strand with the end of the ®rstb-strand. The vector b connects the middle of the vectora with the middle of the k ÿ 1 element; it is shown ingreen in the Figure. Note that it is shifted from its orig-inal position for display purposes. Vectors d and e,shown in magenta, are perpendicular to the planede®ned by vectors k ÿ 2 and a, and k and e, respect-ively. Figure generated with MOLMOL (Koradi et al.,1996).
Fold Prediction of Small Proteins 425
Knowledge-based restraints. Knowledge-basedinformation about the general features of proteintopology is also used (Skolnick et al., 1997b). Thisknowledge-based information acts to reduce thenumber of misfolded structures. Two types ofknowledge-based rules are considered, namely thechirality of bab units and the angle formed in bbasupersecondary structure units (Chothia &Finkelstein, 1990). The implementation used herediffers in some important aspects from thatdescribed by Skolnick et al. (1997b). First of all,because the secondary structure prediction schemecan miss an intervening element, the number ofsuccessive residues between secondary structureregions is counted. If the number of loop residuesis greater than 15 residues, it is assumed that anintervening secondary structure element has beenmissed by the secondary structure prediction algor-ithm, and the knowledge-based rules are notapplied at all. The knowledge-based rules them-selves are also implemented in a different waythan that described in our previous work. First ofall, in the bba rule, the chirality requirement iseliminated and only the angle between theelements is restricted. When predicted secondarystructure is used, this rule is not suf®ciently robustbecause strands, particularly at the edges of thefold in a/b proteins, can be missed. This results inthe inappropriate application of the chiralityrequirement of the rule. In the case of the bab rule,the vector de®nitions and restraint potential arethe same as in our previous work (Skolnick et al.,1997b). However, the de®nition of the anglesbetween elements is different from that previouslyemployed because the previous implementationmade implicit assumptions about the chain geome-try that can be violated. In particular, in the meth-od described in our previous work, the ®rst strandof the bab element was not demanded to be in theplane formed by the vector describing the orien-tation of the second strand, and the vector connect-ing the beginning of the second strand with theend of the ®rst strand. Therefore, unusual geome-tries were not penalized. These geometries did notappear in our previous work because the sparseset of restraints used in the folding simulationswas exact and always involved some contactsbetween b-sheet forming strands. However, theuse of incorrect, clustered restraints in the presentwork permits the appearance of such confor-mations. The new vector de®nitions of the babelement are shown in Figure 2. The energy penaltyfor the bab rule is then given by:
Ebab �X�Vpÿ1�k� � Vpÿ2�k��Z�k� �3�
with Z(k) � 1 if element k ÿ 2 is predicted to be b,k ÿ 1 is a and k is b, and Z(k) � 0 otherwise. Thesum is taken over the number of secondary struc-ture elements Nsec. The potentials Vp ÿ 1 and Vp ÿ 2
are given by:
Vpÿ1�k� � Z�k�Kbab�b � e�2 �4�
Vpÿ2�k� � l�k�Kbab�0:7ÿ �e � d��2 �5�where the vectors b, d and e are given in Figure 2.These vectors are obtained as follows: Let usdenote xa
b as the splinned coordinates (Skolnicket al., 1997b) of the starting (a � s) and ending(a � e) points of the secondary structure elementsb � k ÿ 2, k ÿ 1 or k, repectively (see Figure 2). Theunit vectors describing the direction of the second-ary structures of the b elements can be found as:vb � ||xe
b ÿ xsb||. We can also de®ne the vector
connecting the two strands as a � (xek ÿ 2 ÿ xs
k). Thevectors d and e can then be obtained as the follow-ing cross-products: d � (vk ÿ 2� a) and e � (vk � a).For the derivation of b, we refer to the derivationof equation (A8a) in our previous work (Skolnicket al., 1997b). The value of Z(k) � 1 if (b �e) <0, asthe connection is then left-handed, and it isZ(k) � 0 otherwise. Also, if ||e �d|| >0.7, thenl(k) � 0, otherwise l(k) � 1. This allows an angle ofup to 45� between the strands, therefore takinginto account the possible b-sheet twist (seeFigure 2). The typical value of Kbab � 20 kT.
Relative weighting of the various contributions.The total energy of a given conformation is given
426 Fold Prediction of Small Proteins
by:
E � 0:5E14 � 1:5E1 � Edensity � 2:75Epair
� Etarget;sec � EUÿturn � EHÿbond
� Ebÿprop � Eres � Eknow �6�
Conformational sampling
Sampling of conformational space occurs via astandard asymmetric Monte Carlo Metropolisscheme (Metropolis et al., 1953). Several types oflocal conformational micromodi®cations of thechain backbone and rare, small distance motions oflarger chain fragments, together with side groupequilibration cycles, are used. From 10 to 40 inde-pendent assembly simulations for each protein arecarried out, each from a fully extended initial con-formation. Each simulation starts at a reduced tem-perature of 5.0, and then the temperature is slowlylowered to 1.0. Low energy structures are thensubject to isothermal re®nement. The predictedfold is the one exhibiting the lowest averageenergy during the isothermal calculation. (In cor-rectly folded structures, this energy is roughly 5kTper residue.)
Computational details
The typical computational time of assembling a100-residue protein with MONSSTER, consisting ofa run of about 5 � 106 Monte Carlo steps, is 5.0hours on a single SGI MIPS R10000 processor run-ning at 180 MHz clock speed and using a cachesize of 32 kb. Each isothermal calculation needs anadditional 5.0 hour run.
Structural analysis
The folded structures were compared with theexperimental conformations using two sets ofmeasurements, describing the global similarity ofthe structures and the local matching of the sec-ondary structure elements. For the global simi-larity, the total coordinate root-mean-squaredeviation (cRMSD) of the two structures was calcu-lated after computing the best superposition of thepredicted structure with the experimental struc-ture, using the McLachlan algorithm. In all thecRMSD calculations, all residues of both structureswere included.
The comparison of the secondary structure pre-diction of the predicted models with that of theexperimental structure and that coming from sec-ondary structure prediction algorithms is compli-cated, and we feel that no satisfactory method canbe used at the moment. The standard method forassigning secondary structure is due to Kabsch &Sander (1983) and is implemented in the DSSP pro-gram. The method relies heavily on the hydrogenbond pattern of the structure as computed fromthe peptide plates. The Ca models of the predictedstructures lack these peptide plates. An all-atom
reconstruction of these models is possible, and hasactually been carried out (see below), but the DSSPmethod is very sensitive to small shifts in the coor-dinates in the secondary structure assignment;thus, the direct use of the DSSP assignments in thepredicted models is prone to introduce consider-able artifacts. For this reason, we adopted theRichards & Kundrot (1988) de®nition of secondarystructure, based on the pseudodihedral angles ofconsecutive Ca atoms. Still, this approach is notentirely satisfactory, as the PHD method has beendeveloped using the DSSP de®nition of secondarystructure. However, trial calculations indicated thatartifacts introduced by this approach are smallerthan when the DSSP assignment is used. Compu-tation of the Richards-Kundrot secondary structurewas carried out on the basis of reconstructed all-atom models. All-atom reconstruction of each ofthe predicted structures was carried out using theMODELLER program (Sali & Blundell, 1993) usingdefault parameter settings.
Proteins tested
The above protocol has been applied to the setof 20 small proteins listed in Table 1 sorted by sizeand named according to the entry in the Brookha-ven database (Bernstein et al., 1977). The size of theproteins ranges from the 29 amino acids of 3cti tothe 100 amino acids of 1ife. They were chosen toexamine how well the methodology performs onthe following different structural motifs: small dis-ul®de-rich proteins; all-a proteins; all-b proteinsand a/b proteins. The fold description of each ofthe proteins is presented in Table 1 according tothe SCOP database (Murzin et al., 1995). In allcases, the structures were chosen at random, withthe only constraints being that no global misassign-ment of the secondary structure prediction tookplace, i.e. no long helical segment was assigned asextended or vice versa, and that a suf®cient numberof homologous sequences, at least ten, was avail-able in the HSSP database (Sander & Schneider,1991). For the two smallest disul®de-rich proteins,3cti and 1ixa, which are substantially devoid ofsecondary structure, the same protocol wasapplied. However, here we assumed knowledge ofthe identity of the disul®de bridges, as the generalprotocol would presumably fail due to the smallpredicted content of secondary structure. Such dis-ul®de bridges are used as seeds in the tertiaryrestraint derivation protocol for contact prediction.Here, the objective is to study whether the knowl-edge of disul®de bridges, together with the proto-col we now present, can produce low resolutionmodels of small disul®de-rich proteins, which, ingeneral, are considerably less regular than largerproteins. For T0042, we also used the known disul-®de pattern, as it was available to the predictionteams in the CASP2 contest. In the case of theother 17 proteins, the native disul®de bridges werenot used as seeds in those proteins that had disul-®de bridges in their native structure. Rather, cross-
Fold Prediction of Small Proteins 427
links were chosen so as to be compatible with thepredicted contact map. To this effect, the followingalgorithm was used: ®rst, the set of all possiblepairs of disul®de bridges is computed. From thisset, the ®rst disul®de bridge is assigned as thatwith the closest contact map distance to any of thepredicted contacts. The selected pair of cysteineresidues is removed from the list of free cysteineresidues, and the list of possible disul®de bridgesis recomputed. The algorithm continues until allpairs are assigned.
All additional information in this study, such asthe derived set of predicted contacts for all pro-teins, and structural predictions, is available via theWWW on the URL http://www.scripps.edu/skolnick/ORTIZ/ortiz.html
Results
Derivation of restraints
Secondary structure restraints
The secondary structure bias for each residueused in these series of calculations has beenobtained by mixing the PHD and LINKER predic-tions, as described in Methods. The accuracy of thePHD predictions is shown in Table 2, and the ®nalstates assigned to each residue in a representativesubset of the proteins studied are shown inFigure 3. The average secondary structure predic-tion accuracy for the set of proteins used here ishigher than the expected accuracy of the method,but it is still within one standard deviation of thecurrent accuracy of PHD. Combination of the LIN-KER algorithm with the PHD method seems toimprove, on average, the quality of the predictions.This improvement comes from two sources: (1) theability of LINKER to break long PHD-predictedhelices by inserting loops in unphysically longhelices assessed on the basis of the expected pro-tein size, and (2) the ability of LINKER to predictextended states of local hydrophilic stretches,usually missed by PHD as a result of the ®tting ofPHD to the DSSP assignment of secondary struc-ture, which demands a hydrogen-bond network inthe assignment (Kabsch & Sander, 1983). Thus,PHD sometimes misses stretches of sequence withpoor b-sheet propensities, but which populateextended states. For example, the second strand in1gpt was missed by PHD, but localized as anextended state by LINKER (observe in Figure 3 thedifference between 1 and 4 states). A similar situ-ation is observed in the ®rst strand of 1t®. For1hmd, helices two and three are merged, but LIN-KER successfully corrects this overprediction(Figure 3). A similar situation was observed in 3icb(not shown). However, because of the limited accu-racy in loop positioning of the algorithm at theresidue level, the ®nal outcome of the secondarystructure assignment is usually more shifted withrespect to the experimental secondary structurethan in the original PHD prediction. Although the
overall secondary structure assignment of the pro-teins tested here can be considered to be quitegood by state-of-the-art standards, it is worthpointing out that there are still some proteins forwhich entire elements of secondary structure aremissed. Thus, in the case of 1ftz, the third helix ismissed, and an additional helix is predicted in theC terminus. Similarly, the third strand of 1shg ismissed, as is the second and third helix and thefourth strand of 1ego (Figure 3), as well as thethird strand of 1poh at the edge of the fold (notshown). There are also considerable discrepanciesbetween the predicted and observed lengths andlocations of the secondary structure elements(Figure 3).
Contact prediction
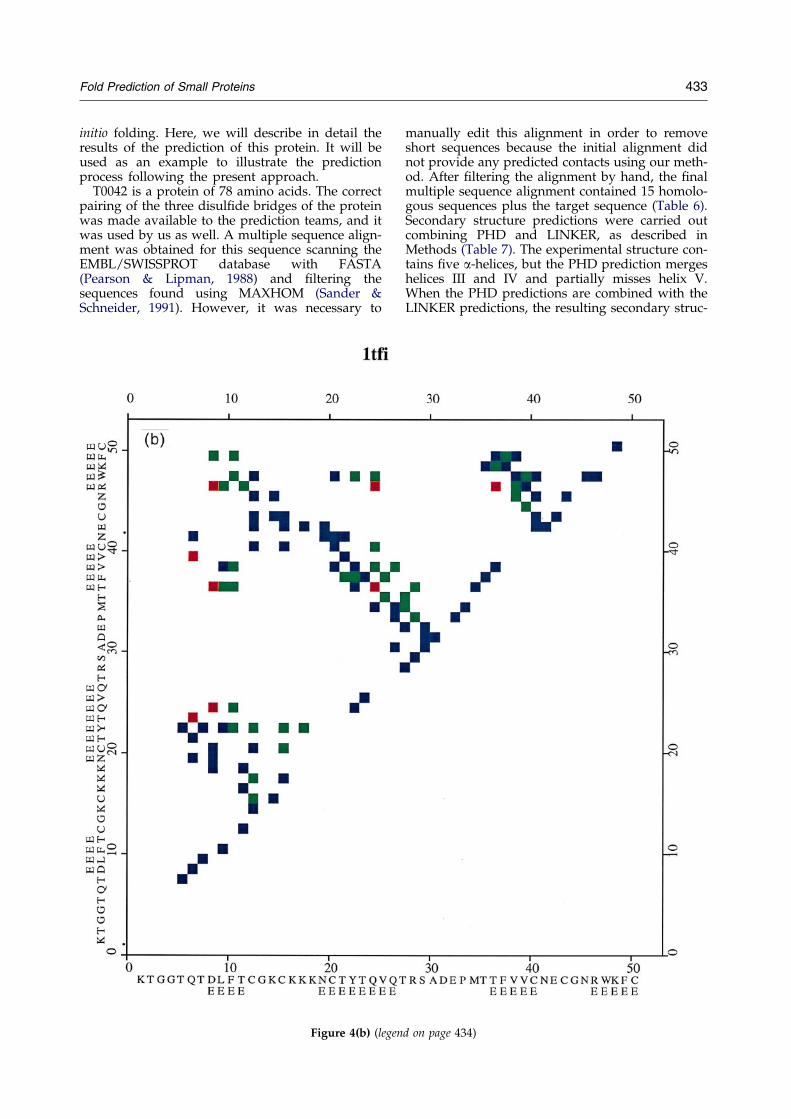
The results of side-chain contact prediction arecompiled in Tables 2 and 3. Not consideringcysteine-rich proteins where the disul®de contactpattern has been assumed to be known, the overallaccuracy of the procedure of contact prediction issimilar to that reported by other authors (GoÈebelet al., 1994; Olmea & Valencia, 1997), on the orderof 25%. Turning to the issue of precision, allowingan error of one residue in the assignment of part-ners yields an accuracy on the order of 60%; withintwo residues is 77%; within three residues is 85%;and within four residues is 95%. The average pre-diction of the contact map coverage is on the orderof 25%, which corresponds to about N/4 restraintsper N protein residues and is consistent with ourprevious ®ndings (Skolnick et al., 1997b). Thus, ingeneral, the number of contacts predicted is asmall portion of the whole protein contact mapand usually contains a signi®cant amount of noise.In some cases, wrong pairings of secondary struc-ture elements are obtained, as is the case with1poh, 1ife and 1hmd. In general, as expected, thebigger the protein, the higher the chance to assigna wrong pairing of secondary structure elements.The structures used for ``growing'' the contactspredicted by correlated mutations for each of theproteins studied are compiled in Table 3. Thestructures selected are entirely unrelated to the tar-get sequence, i.e. no remote sequence homologueswere used. With the contact derivation procedureused in this work, the ®nal predicted contacts arehighly clustered in structure; therefore, the effec-tive number of contacts that can be considered tobe independent is lower than the number of pre-dicted contacts. Some other effects are also worthnoting. Comparison of the predicted and observedcontact maps (see Figure 4) shows that, as a resultof the independent growth of each one of therestraints, frequent phase shifts for the differentrestraint subsets are observed. This problem is par-ticularly important for restraints involving a-helices. This can produce helix unwrapping andother structural distortions. As explained inMethods, we have tried to avoid this problem byintroducing the ``splinning'' procedure.
Figure 3. Secondary structure assignment for a representative subset of the proteins used in this study. The aminoacid sequence is given for each protein. The observed secondary structure in the experimental conformation accordingto the DSSP assignment (Kabsch & Sander, 1983) of three states is also shown, as is the assigned secondary structurestate in the folding simulations, according to the prediction results. 1 stands for coil assignment; 2 for helix assign-ment; 3 for a U-turn assignment; 4 for strand assignment, and 5 corresponds to no assignment of secondary structure.
430 Fold Prediction of Small Proteins
Table 3. For each of the predicted proteins, the sourcestructures used for contact map growth are shown
Protein Source structures used for contact growtha
a In the case of 3cti and 1ixa, the contact map growth step wasnot used, as a result of the insuf®cient secondary structure con-tent in these structures. Here, restraints are given by the pre-dicted contacts plus the known disul®de bridges. See alsoTable 2.
Fold Prediction of Small Proteins 431
Fold assembly and discrimination
Fold assembly is carried out starting from anextended chain; therefore, the initial restraintenergy is very large in the ®rst cycles of the algor-ithm where the ®rst motions of the chain mainlydecrease the restraint energy. Thus, compact statesare generated very quickly. Finally, the secondarystructure forms, and the adjustments of secondarystructure elements take place. In some cases,during the ®rst annealing run, the structures aretrapped in misfolded states. Then, during sub-sequent annealing runs, the correct registration ofelements takes place. This effect is particularlyobserved in a/b proteins. In the ®nal folds, typi-cally the restraint energy is close to zero as a resultof the soft implementation of restraints (Table 4),although on average the number of satis®ed pre-dicted contacts is about half the number of pre-dicted contacts. Based on restraint satisfaction, it isnot possible to discriminate among alternativeanswers (Table 4). Because the energy landscape isrugged, individual structures obtained from theassembly runs are not able to provide a reliableenergy for the particular fold they represent. As aresult, to rank order the folds, it is necessary tocarry out isothermal calculations for at least thelower part of the energy spectrum of the createdfolds (Table 4).
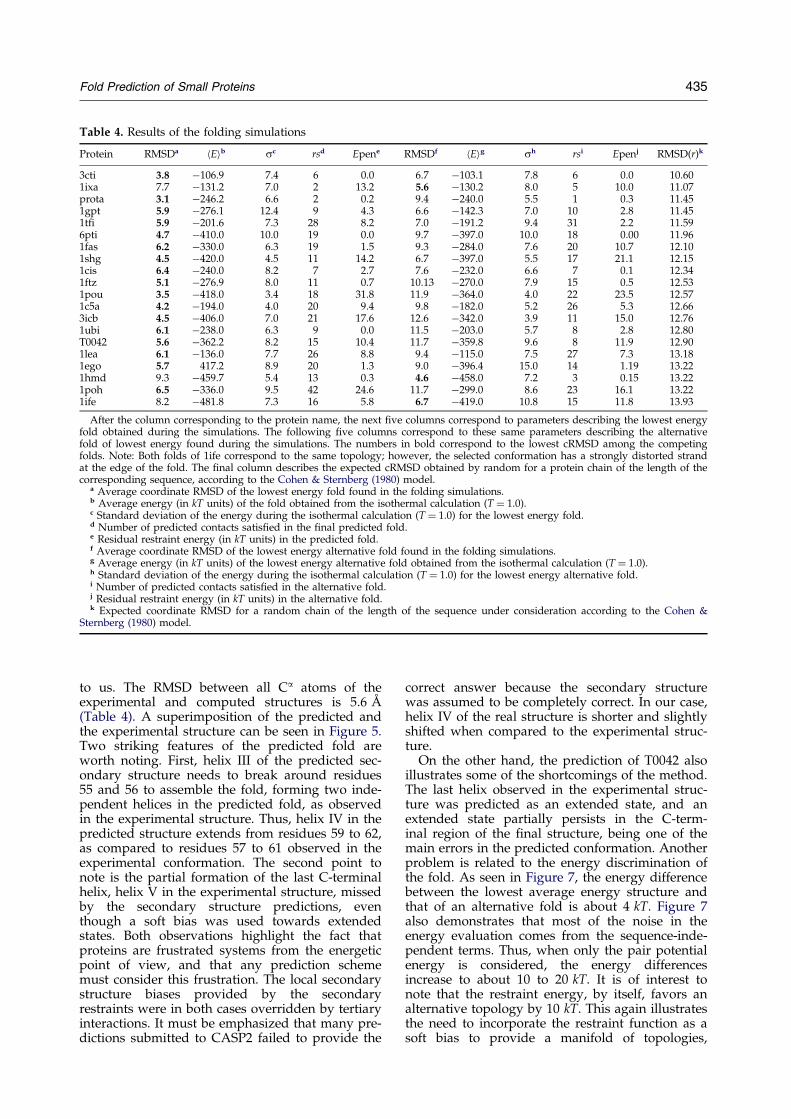
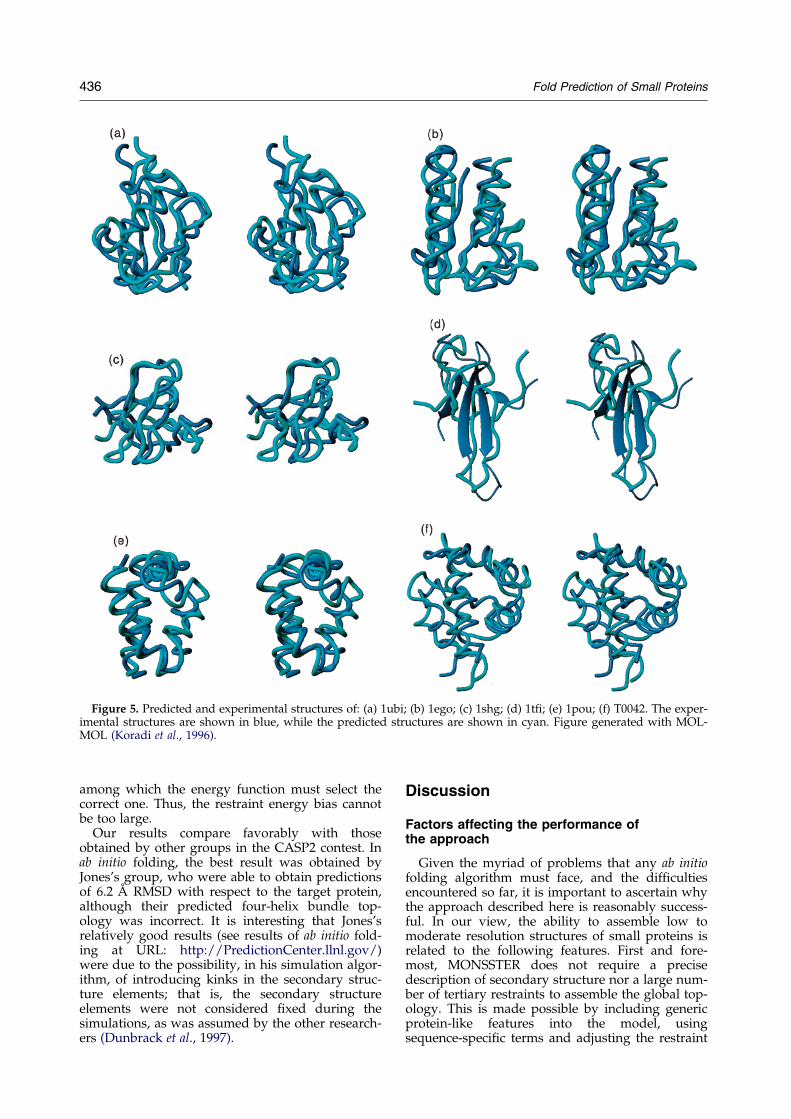
The superimposed predicted and experimentalconformations of a representative subset of theproteins tested in this work can be seen inFigure 5. Details of the resolution achieved foreach particular protein can be found in Table 4.The average cRMSD is about 5 AÊ . Thus, in com-parison with the use of ``exact'' restraints, a pricein resolution between 1.0 to 2.0 AÊ has to be
paid. When the different protein classes are con-sidered, the average cRMSD of the lowest energyset of structures ranges from about 4 AÊ for heli-cal proteins to roughly 6 AÊ for b and mixedmotif proteins. In all cases, the global topology isrecovered either as the best energy (in 17 out of20 cases) or as the next best energy alternativefold. Of the three that failed (1ixa, 1hmd and1ife), the misfolded state of 1ixa results from themisplacement of a few residues in the C-terminalregion; in the case of 1hmd, it is not possible todistinguish between the two topological mirrorimages, which are essentially isoenergetic. In thelast case, 1ife, the selected fold is actually correctin spite of the unacceptably high cRMSD. Here,a coil region shifts from the edge of the fold tothe back of the protein. These numbers could becompared with the expected cRMSD obtained byrandom for protein chains of the length of thesequences used in these folding studies (Table 4),using the expression given by Cohen &Sternberg (1980). The average value that couldhave been obtained by random is around 12 AÊ .
Turning again to the assembly process, it is inter-esting to note that the different secondary structureelements do not simply pack as rigid bodies; thatis, as shown in Figure 6, changes in secondarystructure status produced by long-range inter-actions are common in order to assemble the fold.This can be quanti®ed by the comparison of thesecondary structure predictions and the secondarystructure assignment of the predicted models(Table 5). In some cases, the secondary structureelements extend in length from the original predic-tions, as in the case of the a-helices of 6pti or the b-strands in 1gpt. In some other cases, they need toshorten to accommodate themselves into the pro-tein fold, as in the case of 1lea. And in a number ofcases, additional secondary structure elementsform. The most striking case is that of 1ubi, as anexample of a-helix formation, and 1shg forb-strands. Overall, our results suggest that ®xingthe length of secondary structure elements andtreating them as rigid bodies can have a deleter-ious effect in fold assembly. For example, as aresult of ``fraying ends'', some hydrophobic resi-dues can be exposed and some wrong contactsbetween elements can be formed, reducing or eveneliminating the energy gap with alternative folds.Some ¯exibility is required in order to correctlypack the secondary structure elements. Usuallytheir predicted length is incorrect, and there areshifts in registration with respect to the tertiaryrestraints. Therefore, if a rigid model is used tode®ne the secondary structure, the correct fold canbe missed. However, on average, the correctness ofthe secondary structure in the predicted modelsdoes not improve when compared with the orig-inal secondary structure predictions used asrestraints during the simulations (see Table 5 andFigure 6).
432 Fold Prediction of Small Proteins
The case of T0042 as a blind prediction
The second meeting on the Critical Assessmentof Techniques for Protein Structure Prediction(CASP2) was held in Asilomar, California,recently. Several protein targets were available toresearchers as blind predictions covering differentaspects of protein structure prediction: docking,homology modeling, threading and ab initio fold-ing (URL http://iris4.carb.nist.gov/casp2/). Atthe time of the meeting, the present work wasbeing carried out, and we felt that the methodwas too immature for us to participate. However,once we obtained enough experience with the
Figure 4(a) (legen
approach presented here, and in order to com-pare our results with those of other groups usingdifferent methods under similar conditions, wepursued the blind prediction of target 42 (T0042)of the meeting. It must be stressed that the pre-diction was made without knowledge of the tar-get conformation, as this structure has beenreleased only recently. (All of the numericalassessments for this and the rest of the targets, aswell as the experimental structure of some of thetargets, are available through the World WideWeb URL http://PredictionCenter.llnl.gov/.)T0042 was chosen because it was the most popu-lar target sequence for most groups doing ab
initio folding. Here, we will describe in detail theresults of the prediction of this protein. It will beused as an example to illustrate the predictionprocess following the present approach.
T0042 is a protein of 78 amino acids. The correctpairing of the three disul®de bridges of the proteinwas made available to the prediction teams, and itwas used by us as well. A multiple sequence align-ment was obtained for this sequence scanning theEMBL/SWISSPROT database with FASTA(Pearson & Lipman, 1988) and ®ltering thesequences found using MAXHOM (Sander &Schneider, 1991). However, it was necessary to
Figure 4(b) (legen
manually edit this alignment in order to removeshort sequences because the initial alignment didnot provide any predicted contacts using our meth-od. After ®ltering the alignment by hand, the ®nalmultiple sequence alignment contained 15 homolo-gous sequences plus the target sequence (Table 6).Secondary structure predictions were carried outcombining PHD and LINKER, as described inMethods (Table 7). The experimental structure con-tains ®ve a-helices, but the PHD prediction mergeshelices III and IV and partially misses helix V.When the PHD predictions are combined with theLINKER predictions, the resulting secondary struc-
d on page 434)
Figure 4. Contact maps of a representative set of proteins used in this work. The experimental contact map is shownin blue, the predicted seeds are shown in red, and the expanded contacts obtained by inverse folding are shown ingreen (see the text for details). A contact distance cut-off of 4.5 AÊ between side-chain heavy atoms is used. Contactmaps for the following proteins are shown: (a) 1ego; (b) 1t®; (c) 1pou.
434 Fold Prediction of Small Proteins
ture assignment is actually worse than the PHDprediction alone: helices III and IV remain merged,helix V is totally missed and helix II is considerablyshortened (Table 7). Using the correlated mutationanalysis, three contact seeds could be predictedfrom the multiple sequence alignment (see Table 8).One of them involves a disul®de bridge observedin this protein, which made us feel more con®dentabout the quality of the predicted seeds. Theseseeds, together with the known disul®de bridges,were used in the inverse folding calculation withthe objective of ``expanding'' the predicted con-tacts. Only two of the three seeds could ``grow''.
Thus, the ®nal number of restraints was 23(Table 9). Interestingly, one of the fragmentsselected for the enrichment process involved 2utg,which has been found by other researchers duringthe CASP2 contest to be a popular template whenglobal threading of the sequence is done. Theobtained secondary and tertiary restraints wereused as input for MONSSTER. Ten simulationswere performed. After the isothermal calculations,the lowest average energy fold was selected as thepredicted structure.
After these calculations were completed, theexperimental structure of T0042 was then available
Table 4. Results of the folding simulations
Protein RMSDa hEib sc rsd Epene RMSDf hEig sh rsi Epenj RMSD(r)k
After the column corresponding to the protein name, the next ®ve columns correspond to parameters describing the lowest energyfold obtained during the simulations. The following ®ve columns correspond to these same parameters describing the alternativefold of lowest energy found during the simulations. The numbers in bold correspond to the lowest cRMSD among the competingfolds. Note: Both folds of 1ife correspond to the same topology; however, the selected conformation has a strongly distorted strandat the edge of the fold. The ®nal column describes the expected cRMSD obtained by random for a protein chain of the length of thecorresponding sequence, according to the Cohen & Sternberg (1980) model.
a Average coordinate RMSD of the lowest energy fold found in the folding simulations.b Average energy (in kT units) of the fold obtained from the isothermal calculation (T � 1.0).c Standard deviation of the energy during the isothermal calculation (T � 1.0) for the lowest energy fold.d Number of predicted contacts satis®ed in the ®nal predicted fold.e Residual restraint energy (in kT units) in the predicted fold.f Average coordinate RMSD of the lowest energy alternative fold found in the folding simulations.g Average energy (in kT units) of the lowest energy alternative fold obtained from the isothermal calculation (T � 1.0).h Standard deviation of the energy during the isothermal calculation (T � 1.0) for the lowest energy alternative fold.i Number of predicted contacts satis®ed in the alternative fold.j Residual restraint energy (in kT units) in the alternative fold.k Expected coordinate RMSD for a random chain of the length of the sequence under consideration according to the Cohen &
Sternberg (1980) model.
Fold Prediction of Small Proteins 435
to us. The RMSD between all Ca atoms of theexperimental and computed structures is 5.6 AÊ
(Table 4). A superimposition of the predicted andthe experimental structure can be seen in Figure 5.Two striking features of the predicted fold areworth noting. First, helix III of the predicted sec-ondary structure needs to break around residues55 and 56 to assemble the fold, forming two inde-pendent helices in the predicted fold, as observedin the experimental structure. Thus, helix IV in thepredicted structure extends from residues 59 to 62,as compared to residues 57 to 61 observed in theexperimental conformation. The second point tonote is the partial formation of the last C-terminalhelix, helix V in the experimental structure, missedby the secondary structure predictions, eventhough a soft bias was used towards extendedstates. Both observations highlight the fact thatproteins are frustrated systems from the energeticpoint of view, and that any prediction schememust consider this frustration. The local secondarystructure biases provided by the secondaryrestraints were in both cases overridden by tertiaryinteractions. It must be emphasized that many pre-dictions submitted to CASP2 failed to provide the
correct answer because the secondary structurewas assumed to be completely correct. In our case,helix IV of the real structure is shorter and slightlyshifted when compared to the experimental struc-ture.
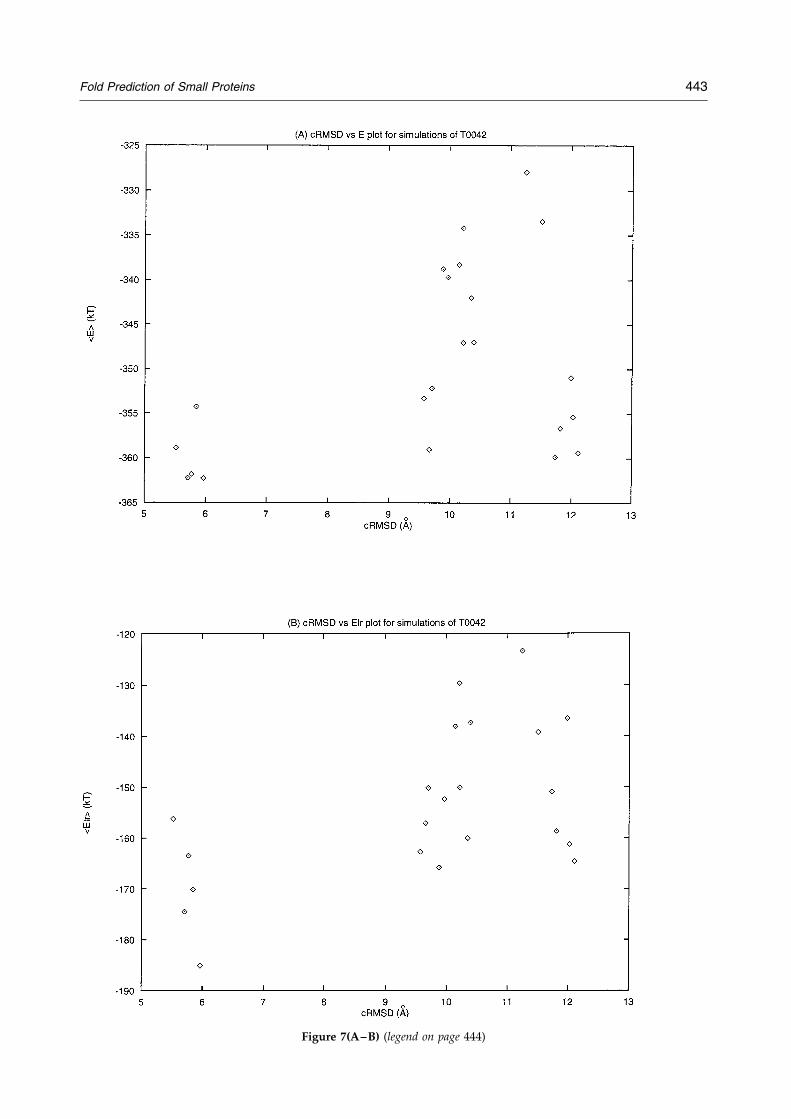
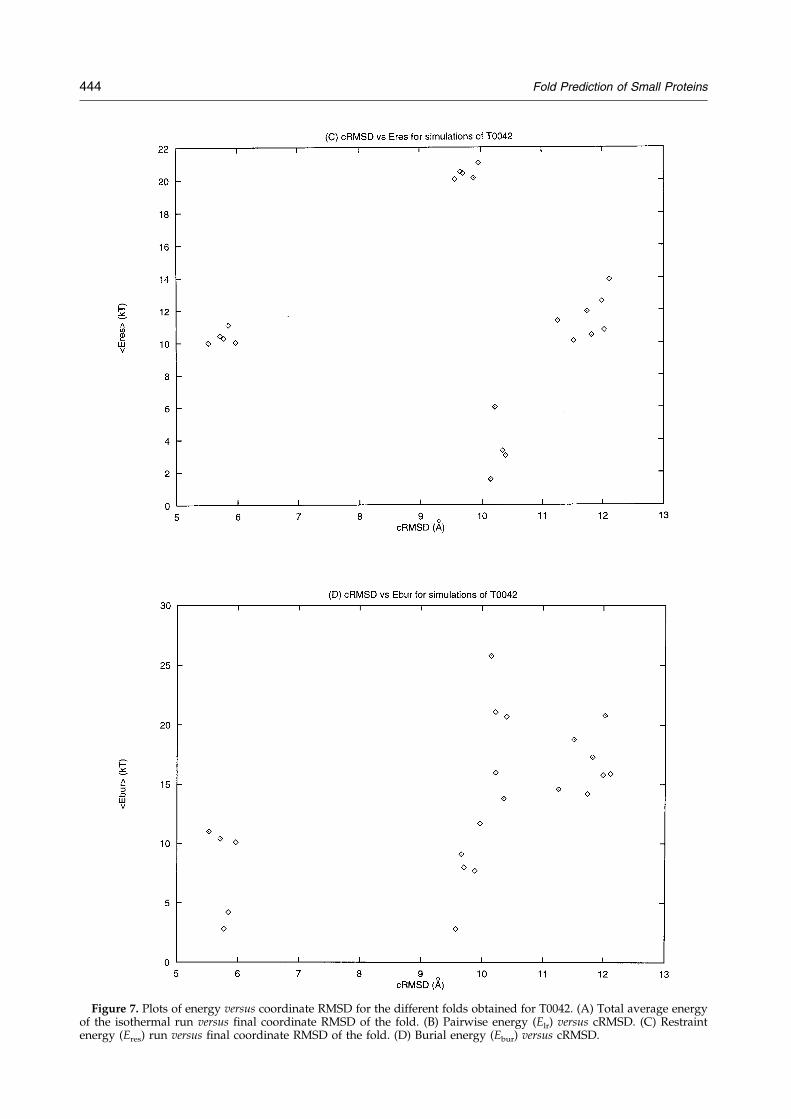
On the other hand, the prediction of T0042 alsoillustrates some of the shortcomings of the method.The last helix observed in the experimental struc-ture was predicted as an extended state, and anextended state partially persists in the C-term-inal region of the ®nal structure, being one of themain errors in the predicted conformation. Anotherproblem is related to the energy discrimination ofthe fold. As seen in Figure 7, the energy differencebetween the lowest average energy structure andthat of an alternative fold is about 4 kT. Figure 7also demonstrates that most of the noise in theenergy evaluation comes from the sequence-inde-pendent terms. Thus, when only the pair potentialenergy is considered, the energy differencesincrease to about 10 to 20 kT. It is of interest tonote that the restraint energy, by itself, favors analternative topology by 10 kT. This again illustratesthe need to incorporate the restraint function as asoft bias to provide a manifold of topologies,
Figure 5. Predicted and experimental structures of: (a) 1ubi; (b) 1ego; (c) 1shg; (d) 1t®; (e) 1pou; (f) T0042. The exper-imental structures are shown in blue, while the predicted structures are shown in cyan. Figure generated with MOL-MOL (Koradi et al., 1996).
436 Fold Prediction of Small Proteins
among which the energy function must select thecorrect one. Thus, the restraint energy bias cannotbe too large.
Our results compare favorably with thoseobtained by other groups in the CASP2 contest. Inab initio folding, the best result was obtained byJones's group, who were able to obtain predictionsof 6.2 AÊ RMSD with respect to the target protein,although their predicted four-helix bundle top-ology was incorrect. It is interesting that Jones'srelatively good results (see results of ab initio fold-ing at URL: http://PredictionCenter.llnl.gov/)were due to the possibility, in his simulation algor-ithm, of introducing kinks in the secondary struc-ture elements; that is, the secondary structureelements were not considered ®xed during thesimulations, as was assumed by the other research-ers (Dunbrack et al., 1997).
Discussion
Factors affecting the performance ofthe approach
Given the myriad of problems that any ab initiofolding algorithm must face, and the dif®cultiesencountered so far, it is important to ascertain whythe approach described here is reasonably success-ful. In our view, the ability to assemble low tomoderate resolution structures of small proteins isrelated to the following features. First and fore-most, MONSSTER does not require a precisedescription of secondary structure nor a large num-ber of tertiary restraints to assemble the global top-ology. This is made possible by including genericprotein-like features into the model, usingsequence-speci®c terms and adjusting the restraint
implementation to the expected accuracy and pre-cision of the predicted restraints. As a result, a sub-stantial number of prediction errors are tolerated.Thus, one can employ existing secondary structureprediction algorithms and can focus the tertiaryrestraint derivation process on the generation of arelatively small number of tertiary contacts ofenhanced reliability.
Turning explicitly to the latter, another import-ant aspect of this approach is the increased sig-
Figure 6 (continued on
nal-to-noise ratio in the predicted contactsobtained by restricting the analysis to residuecovariation in the predicted core secondary struc-tural elements. Such contacts are extractedthrough the use of a two-step procedure thatminimizes the appearance of wrong pairings ofsecondary structure elements and generates alocally self-consistent set of restraints. We stressthat prediction of contacts based only on thelocal threading of all possible pairs of secondary
page 438 with legend)
Figure 6. Observed and predicted secondary structures for all proteins studied in this work. Calculation of the sec-ondary structure using the three-dimensional coordinates of the protein models was done using the Richards &Kundrot (1988) method. RK refers to the secondary structure assignment of the experimental structure; PHD to thepredictions of secondary structure using the PHD method; and PRD is the secondary structure assignment based onthe coordinates of the predicted structure.
438 Fold Prediction of Small Proteins
structure elements is unreliable (Hu et al., 1997;A.R.O. & J.S., unpublished results). For example,by using only local threading, almost allb-strands would like each other and it would be,in general, impossible to discriminate the b-sheetpattern in a/b and b proteins. There is not enoughspeci®city in the potential (local secondary struc-ture plus burial) to discriminate reliably among thepreferred pairings of b-strands. These limitations oflocal threading are well known and have beenreported by us (Hu et al., 1997) and others(Hubbard & Park, 1995). It is the use of a few con-tacts derived from correlated mutations obtainedwith high reliability together with the fragmentthreading/clustering protocol of restraint growingthat yields restraints of suf®cient quality for suc-cessful structure assembly.
An interesting observation made here is that it isbetter not to implement a restraint than to includegrossly wrong information. The appearance of
false positives is one of the main factors affectingthe performance of the approach. Another interest-ing outcome is that the average overall precision ofthe predicted restraints is more important than theaverage overall accuracy in low resolution foldingsimulations. This effect can be appreciated inFigure 8(A), where a correlation can be observedbetween precision at d � 2 and cRMSD for a-helicalproteins on the one hand and b-containing proteinson the other. For two of the proteins, 1ixa and 6pti,that do not lie in any of the correlation lines, thediscrepancy can be explained by the good accuracyof the contact predictions in both cases(Figure 8(B)). However, no clear correlation withthe cRMSD was found in the case of the accuracy(Figure 8(B)). Moreover, it is interesting to notethat the dependency of the quality of the predictedfold on the precision of the restraints is higher fora-helices than for b-containing proteins, something
Table 5. Accuracy of secondary structure prediction
Q3 PRD refers to the Q3 value obtained from the predicted ter-tiary structure model, while Q3 PHD refers to the Q3 valueobtained using the PHD method. Secondary structure assign-ment of the three-dimensional structures is made according tothe Richards & Kundrot (1988) de®nition.
Fold Prediction of Small Proteins 439
probably related to the dif®culties of modelingb-strands.
Comparison with previous studies
Different authors have now started to investigatethe use of multiple sequence information and/orpredicted secondary structure using different foldprediction techniques. Perhaps the ®eld that hasmost strongly pursued by this approach is thread-ing. There, as recently reported by differentauthors, the use of variability patterns in multiplesequence alignments (Defay & Cohen, 1996;Taylor, 1997) or predicted secondary structure(Rice & Eisenberg, 1997; Rost et al., 1997; Russellet al., 1996), provide considerable improvementover the single sequence approach. But, and atleast up to the best of our knowledge, the onlyreported application of predicted restraints inthreading is due to Russell et al. (1996). In theirstudy, putative contacts derived from biochemicalarguments were used as ®lters in a fold recognitiontechnique that makes use of predicted secondarystructure. However, no distance restraint infor-mation coming from multiple sequence alignmentshas been reported in threading. The reasons forthis might be that the predicted contact infor-mation has been regarded as not being reliable,and certainly it is not without post-processing, andthat introduction of distance restraints requiresincorporation of double dynamic programmingtechniques (Taylor, 1997) or Monte Carloapproaches, making the threading procedurerather computationally expensive.
On the other hand, the combined effort of theCohen and Benner groups has producedapproaches recently to fold prediction based onhierarchical building procedures that are similar inspirit to the one presented here (Gerloff et al.,1997a,b). In their case, multiple sequence align-ments are built and secondary structure is pre-dicted. Later, an analysis of compensatoryvariations found in the multiple sequence align-ment between pairs of positions is carried out. Thisallows the authors to derive relationships of close-ness in space between the secondary structureelements. In the ®nal stage of their predictionmethod, folds are searched in the database thatmeet at least a fraction of the predicted structuralfeatures. Bona ®de predictions of the C-terminaldomain of the b and g chains of ®brinogen havebeen published making use of this approach(Gerloff et al., 1997a).
Perhaps the most similar approach to fold pre-diction to that presented here has been suggestedby Aszodi & Taylor (1995). Their model studiesusing simulated restraints were very similar to ourprevious studies on the determination of the requi-site number of exact restraints necessary for suc-cessful fold assembly (Skolnick et al., 1997b).However, later studies by Aszodi & Taylor (1996)have addressed the problem of remote homologymodeling rather than fold prediction (Aszodi &Taylor, 1996), and also follow an approach similarin spirit to the one presented here. In their case,multiple sequence alignments are used to de®neconserved regions that are assumed to form part ofthe protein core. A ®tting function derived fromthe protein database is used to map restraint dis-tances from these conservation patterns. Aszodi &Taylor (1996) have shown convincingly that this isa promising technique for remote homology mod-eling.
Limitations of the current methodology
While the results described above are encoura-ging, there are problems with this approach thatmust be addressed. First and foremost, the yield ofnative topologies is only about 10 to 20%, andextraction of correct from incorrect topologiesrequires a long series of isothermal simulations.The fact that different assembly runs produce awide dispersion in the ®nal energies of the proteinmodel (even for the same overall global fold) is asignature of sampling problems. While sampling isprobably adequate for 50 to 60-residue proteins,sampling problems become acute as the size of theprotein (or more precisely, the number of topologi-cal elements) increases. Thus, the development ofbetter sampling approaches should permit us toextend the treatment to larger proteins havingmore complicated topologies. Another dif®culty isrelated to fold selection. Typically, one of two situ-ations arises. Either one must differentiate thenative topology from its topological mirror image(a fold where the chirality of the secondary struc-
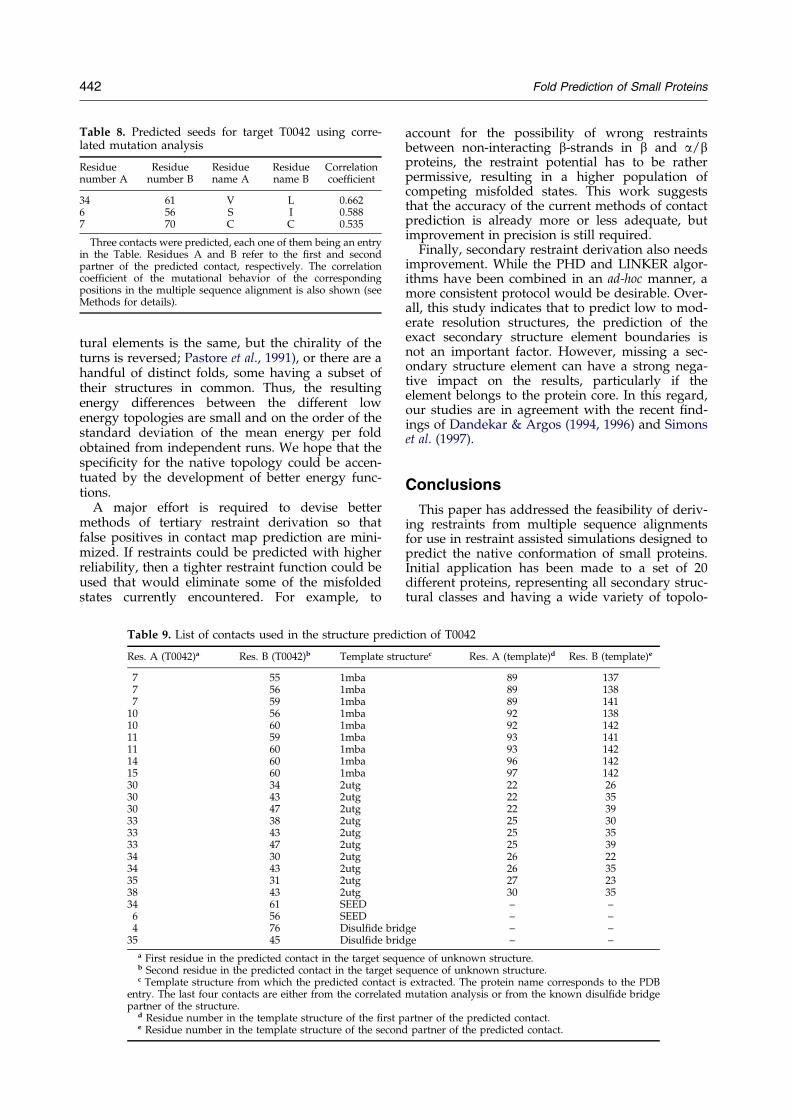
Table 8. Predicted seeds for target T0042 using corre-lated mutation analysis
Residuenumber A
Residuenumber B
Residuename A
Residuename B
Correlationcoefficient
34 61 V L 0.6626 56 S I 0.5887 70 C C 0.535
Three contacts were predicted, each one of them being an entryin the Table. Residues A and B refer to the ®rst and secondpartner of the predicted contact, respectively. The correlationcoef®cient of the mutational behavior of the correspondingpositions in the multiple sequence alignment is also shown (seeMethods for details).
442 Fold Prediction of Small Proteins
tural elements is the same, but the chirality of theturns is reversed; Pastore et al., 1991), or there are ahandful of distinct folds, some having a subset oftheir structures in common. Thus, the resultingenergy differences between the different lowenergy topologies are small and on the order of thestandard deviation of the mean energy per foldobtained from independent runs. We hope that thespeci®city for the native topology could be accen-tuated by the development of better energy func-tions.
A major effort is required to devise bettermethods of tertiary restraint derivation so thatfalse positives in contact map prediction are mini-mized. If restraints could be predicted with higherreliability, then a tighter restraint function could beused that would eliminate some of the misfoldedstates currently encountered. For example, to
Table 9. List of contacts used in the structure predic
a First residue in the predicted contact in the target sequb Second residue in the predicted contact in the target sec Template structure from which the predicted contact i
entry. The last four contacts are either from the correlatedpartner of the structure.
d Residue number in the template structure of the ®rst pe Residue number in the template structure of the second
account for the possibility of wrong restraintsbetween non-interacting b-strands in b and a/bproteins, the restraint potential has to be ratherpermissive, resulting in a higher population ofcompeting misfolded states. This work suggeststhat the accuracy of the current methods of contactprediction is already more or less adequate, butimprovement in precision is still required.
Finally, secondary restraint derivation also needsimprovement. While the PHD and LINKER algor-ithms have been combined in an ad-hoc manner, amore consistent protocol would be desirable. Over-all, this study indicates that to predict low to mod-erate resolution structures, the prediction of theexact secondary structure element boundaries isnot an important factor. However, missing a sec-ondary structure element can have a strong nega-tive impact on the results, particularly if theelement belongs to the protein core. In this regard,our studies are in agreement with the recent ®nd-ings of Dandekar & Argos (1994, 1996) and Simonset al. (1997).
Conclusions
This paper has addressed the feasibility of deriv-ing restraints from multiple sequence alignmentsfor use in restraint assisted simulations designed topredict the native conformation of small proteins.Initial application has been made to a set of 20different proteins, representing all secondary struc-tural classes and having a wide variety of topolo-
ence of unknown structure.quence of unknown structure.
s extracted. The protein name corresponds to the PDBmutation analysis or from the known disul®de bridge
artner of the predicted contact.partner of the predicted contact.
Figure 7(A±B) (legend on page 444)
Fold Prediction of Small Proteins 443
Figure 7. Plots of energy versus coordinate RMSD for the different folds obtained for T0042. (A) Total average energyof the isothermal run versus ®nal coordinate RMSD of the fold. (B) Pairwise energy (Elr) versus cRMSD. (C) Restraintenergy (Eres) run versus ®nal coordinate RMSD of the fold. (D) Burial energy (Ebur) versus cRMSD.
444 Fold Prediction of Small Proteins
Figure 8. (A) Precision versuscRMSD for the set of proteins usedin this work. The cRMSD valuesshown in bold in Table 3 are usedin this plot. The d � 2 level wasused (i.e. percentage of correctlypredicted contacts allowing for a�2 residue error in prediction withrespect to a correct contact),although similar results areobtained for d � 1 and d � 3.(B) The same plot, but usingaccuracy criteria (i.e. percentage ofcorrectly predicted contacts withoutany residue error with respect tothe correct contact, so that d � 0).
Fold Prediction of Small Proteins 445
gies. From the experience gained so far, these con-clusions can be drawn.
(1) At the level of secondary structure elements,the accuracy of existing secondary structure predic-tion algorithms appears to be acceptable for suc-cessful fold assembly, provided that some tertiaryrestraints are included in the assembly algorithm.Problems encountered with current secondarystructure prediction algorithms arise either fromentirely missing a secondary structure element ormistaking the secondary structural class (helix orb) of some elements. Depending on the position ofthe missed element in the global fold, this might ormight not prohibit fold assembly. In some cases,when the missed elements lie at the edge of thefold, the ability to assemble the native topology isnot affected. In fact, sometimes such missingelements are induced by tertiary interactions. If,
however, the element missed corresponds to a cru-cial core element (e.g. a helix between twob-strands), it can exert a strong in¯uence on thequality of the ®nal predicted structure and in theworst case, could result in a grossly incorrect pre-dicted structure.
(2) Low resolution models of small proteins canbe assembled from rather inaccurate predictions ofa subset of the total number of tertiary side-chaincontacts. These predicted side-chain contacts neednot span the entire structure, but can be stronglyclustered. On average, the required level of accu-racy in contact map prediction is on the order of25%, provided that the predictions are reasonablyprecise, i.e. 95% lie within �4 residues of correctcontacts. About 25% of the total number of side-chain contacts need to be identi®ed. This studystrongly suggests that precision is a more import-ant factor than accuracy in determining the likeli-
446 Fold Prediction of Small Proteins
hood of successful fold assembly. Althoughadditional investigation is necessary to assess itsgenerality, our results suggest that the requiredrestraints can be reliably derived from multiplesequence alignments using a combination of corre-lated mutations followed by structural ®ltration/inverse folding (additional details will be given ina forthcoming publication).
(3) The strategy presented here yields predic-tions for all fold types whose RMSD from nativeranges from 3.0 to 6.5 AÊ , depending upon foldcomplexity. Such structures are at the level of accu-racy that can be obtained when threading tech-niques are applied to match a sequence to astructure whose sequence homology lies in orbelow the twilight zone of sequence identity. In thefolding simulations, the accuracy and yield of cor-rectly assembled structures is different for thedifferent protein classes. In general, all-a proteinsare predicted with higher accuracy than a/b pro-teins, and these, in turn, have better accuracy thanall-b proteins.
(4) Because of errors in the tertiary restraints, theresulting structures exhibit shifts in registrationand distorted mutual orientations of pairs of sec-ondary structural elements. Such structural distor-tions also reduce the energy gap between theputative native conformation and alternative folds,as compared to our previous studies using ``exact''restraints. Thus, the present protocol allows for theprediction of a small number of possible nativeconformations. However, selection of the speci®cfold based on the force ®eld energy is more uncer-tain as a result of problems with the current energyfunction.
This study successfully demonstrates that, for aset of small proteins, the use of restraints derivedfrom multiple sequence alignments incorporatedinto a tertiary structure prediction algorithmallows for assembly of native-like structures. Theapproach has been shown to be capable of assem-bling low resolution tertiary structures of greatercomplexity than was previously possible. How-ever, the dif®culties encountered in the assemblyof these topologies, even when low resolutionrestraints are employed, paints a cautionary pic-ture for the likelihood of ab initio assembly of com-plex folds without restraints. Given contemporarycomputer resources, sampling algorithms andexisting force ®elds, the folding of more complextopologies is likely to be problematic. Probably,over the short term, the only way to progress is tocombine insights gained from restraint-free foldingstudies on simple folds with strategies that reducethe conformational search space.
Acknowledgments
This work is supported by grant GM-37408 from theNational Institutes of Health. A.K. also acknowledgessupport from the University of Warsaw (grant BST-34/
97) and is an International Scholar of the HowardHughes Medical Institute. A.R.O. also acknowledgessupport from the Spanish Ministry of Education, as wellas access to the computational facilities of EMBL duringthis work. A.R.O. also thanks Dr Wei-Ping Hu for shar-ing some of his source codes at the beginning of this pro-ject, as well as Drs Li Zhang, Leszek Rychlewski andAdam Godzik for useful discussions. Finally, we thankboth reviewers for their careful reading of the manu-script and useful suggestions that have signi®cantlyimproved the quality of the text.
References
Aszodi, A. & Taylor, W. R. (1996). Homology modellingby distance geometry. Folding Design, 1, 325±334.
Aszodi, A., Gradwell, M. J. & Taylor, W. R. (1995).Global fold determination from a small number ofdistance restraints. J. Mol. Biol. 251, 308±326.
Bernstein, F. C., Koetzle, T. F., Williams, G. J. B., Meyer,E. F., Jr, Brice, M. D., Rodgers, J. R., Kennard, O.,Simanouchi, T. & Tasumi, M. (1977). The ProteinData Bank: a computer-based archival ®le formacromolecular structures. J. Mol. Biol. 112, 535±542.
Chothia, C. & Finkelstein, A. (1990). The classi®cationand origins of protein folding patterns. Annu. Rev.Biochem. 59, 1007±1039.
Cohen, F. E. & Sternberg, M. J. E. (1980). On the predic-tion of protein structure: the signi®cance of root-mean-square deviation. J. Mol. Biol. 138, 321±333.
Dandekar, T. & Argos, P. (1994). Folding the main-chainof small proteins with the genetic algorithm. J. Mol.Biol. 236, 844±861.
Dandekar, T. & Argos, P. (1996). Identifying the tertiaryfold of small proteins with different topologies fromsequence and secondary structure using the geneticalgorithm and extended criteria speci®c for strandregions. J. Mol. Biol. 256, 645±660.
Defay, T. R. & Cohen, F. E. (1996). Multiple sequenceinformation for threading algorithms. J. Mol. Biol.262, 314±323.
Dunbrack, R. L., Gerloff, D. L., Bower, M., Chen, X.,Lichtarge, O. & Cohen, F. E. (1997). Meeting review:the second meeting on the critical assessment oftechniques for protein structure prediction (CASP2),Asilomar, California, December 13±16, 1996. FoldingDesign, 2, R27±R42.
Friesner, R. A. & Gunn, J. R. (1996). Computational stu-dies of protein folding. Annu. Rev. Biophys. Biomol.Struct. 25, 315±342.
Gerloff, D. L., Cohen, F. E. & Benner, S. A. (1997a).A predicted consensus structure for the C-terminusof the beta and gamma chains of ®brinogen.Proteins: Struct. Funct. Genet. 27, 279±289.
Gerloff, D. L., Cohen, F. E., Korostensky, C., Turcotte,M., Gonnet, G. H. & Benner, S. A. (1997b). A pre-dicted consensus structure for the N-terminal frag-ment of the Heat Shock Protein HSP90 family.Proteins: Struct. Funct. Genet. 27, 450±458.
Godzik, A., Skolnick, J. & Kolinski, A. (1992). A top-ology ®ngerprint approach to the inverse proteinfolding problem. J. Mol. Biol. 227, 227±238.
Godzik, A., Kolinski, A. & Skolnick, J. (1994). Latticerepresentation of globular proteins: how good arethey?. J. Comput. Chem. 14, 1194±1202.
Gouda, H., Torigoe, H., Saito, A., Sato, M., Arata, Y. &Schimada, I. (1992). Three-dimensional solutionstructure of the B-domain of staphylococcal ProteinA: comparisons of the solution and crystalstructures. Biochemistry, 40, 9665±9672.
Gunn, G. J. R., Monge, A. & Friesner, R. A. (1994).Hierarchical algorithm for computer modeling ofprotein tertiary structure: folding of myoglobin to 6.2 AÊ resolution. J. Phys. Chem. 98, 702±711.
Hu, W.-P., Godzik, A. & Skolnick, J. (1997). Sequence-structure speci®city: how does an inverse foldingapproach work?. Protein Eng. 10, 317±331.
Hubbard, T. J. & Park, J. (1995). Fold recognition and abinitio structure predictions using hidden Markovmodels and beta-strand pair potentials. Proteins:Struct. Funct. Genet. 23, 398±402.
Kabsch, W. & Sander, C. (1983). Dictionary of proteinsecondary structure: pattern recognition of hydro-gen-bonded and geometrical features. Biopolymers,22, 2577±2637.
Kolinski, A. & Skolnick, J. (1994a). Monte Carlo simu-lations of protein folding: I. Lattice model and inter-action scheme. Proteins: Struct. Funct. Genet. 18,338±352.
Kolinski, A. & Skolnick, J. (1994b). Monte Carlo simu-lations of protein folding: II. Application to proteinA, ROP, and crambin. Proteins: Struct. Funct. Genet.18, 353±366.
Kolinski, A. & Skolnick, J. (1997). Determinants of sec-ondary structure of polypeptide chains: interplaybetween short range and burial interactions. J. Chem.Phys. 107, 953±964.
Kolinski, A., Galazka, W. & Skolnick, J. (1995a).Computer design of idealized b-motifs. J. Chem.Phys. 103, 10286±10297.
Kolinski, A., Milik, M., Rycombel, J. & Skolnick, J.(1995b). A reduced model of short range inter-actions in polypeptide chains. J. Chem. Phys. 103,4312±4323.
Kolinski, A., Skolnick, J., Godzik, A. & Hu, W.-P. (1997).A method for the prediction of surface ``U''-turnsand transglobular connections in small proteins.Proteins: Struct. Funct. Genet. 27, 290±308.
Koradi, R., Billeter, M. & Wuethrich, K. (1996).MOLMOL: a program for display and analysis ofmacromolecular structures. J. Mol. Graph. 14, 51±55.
Levitt, M. & Greer, J. (1977). Automatic identi®cation ofsecondary structure in globular proteins. J. Mol.Biol. 114, 181±293.
McLachlan, A. D. (1971). Test for comparing relatedamino acid sequences. J. Mol. Biol. 61, 409±424.
Metropolis, N., Rosenbluth, A. W., Rosenbluth, M. N.,Teller, A. H. & Teller, E. (1953). Equation of statecalculations by fast computing machines. J. Chem.Phys. 51, 1087±1092.
Mumenthaler, C. & Braun, W. (1995). Predicting thehelix packing of globular proteins by self-correctingdistance geometry. Protein Sci. 4, 863±871.
Murzin, A., Brenner, S. E., Hubbard, T. & Chothia, C.(1995). SCOP: a structural classi®cation of proteindatabase for the investigation of sequences andstructures. J. Mol. Biol. 247, 536±540.
Olmea, O. & Valencia, A. (1997). Improving contact pre-dictions by the combination of correlated mutationsand other sources of sequence information. FoldingDesign, 2, S25±S32.
O'Shea, E. K., Klemm, J. D., Kim, P. S. & Alber, T.(1991). X-ray structure of the GCN4 leucine zipper,a two-stranded, parallel coiled coil. Science, 254,539±544.
Park, B. H. & Levitt, M. (1995). The complexity andaccuracy of discrete state models of proteinstructure. J. Mol. Biol. 249, 493±507.
Pastore, A., Atkinson, R. A., Saudek, V. & Williams,R. J. P. (1991). Topological mirror images in proteinstructure computation: an underestimated problem.Proteins: Struct. Funct. Genet. 10, 22±32.
Pearson, W. R. & Lipman, D. J. (1988). Improved toolsfor biological sequence comparison. Proc. Natl Acad.Sci. USA, 85, 2444±2448.
Rice, D. W. & Eisenberg, D. (1997). A 3D-1D substi-tution matrix for protein fold recognition thatincludes predicted secondary structure of thesequence. J. Mol. Biol. 267, 1026±1038.
Richards, F. M. & Kundrot, C. E. (1988). Identi®cation ofstructural motifs from protein coordinate data: sec-ondary structure and ®rst level super-secondarystructure. Proteins: Struct. Funct. Genet. 3, 71±84.
Ripoll, D. R. & Scheraga, H. A. (1990). On the multipleminima problem in the conformational analysis ofpolypeptides. IV. Application of electrostatically dri-ven Monte Carlo method to the 20±residue mem-brane bond portion of melittin. Biopolymers, 30,165±176.
Rost, B. & Sander, C. (1993). Prediction of secondarystructure at better than 70% accuracy. J. Mol. Biol.232, 584±599.
Rost, B. & Sander, C. (1996a). Bridging the proteinsequence-structure gap by structure predictions.Annu. Rev. Biophys. Biomol. Struct. 25, 113±136.
Rost, B. & Sander, C. (1996b). Progress of 1D proteinstructure prediction at last. Proteins: Struct. Funct.Genet. 23, 295±300.
Rost, B., Schneider, R. & Sander, C. (1997). Protein foldrecognition by prediction-based threading. J. Mol.Biol. 270, 471±480.
Russell, R. B., Copley, R. C. & Barton, G. J. (1996).Protein fold recognition by mapping predicted sec-ondary structures. J. Mol. Biol. 259, 349±365.
Sali, A. & Blundell, T. L. (1993). Comparative proteinmodelling by satisfaction of spatial restraints. J. Mol.Biol. 234, 779±815.
Sander, C. & Schneider, R. (1991). Database of hom-ology derived protein structures and the structuralmeaning of sequence alignment. Proteins: Struct.Funct. Genet. 9, 56±68.
Simons, K. T., Klooperberg, C., Huang, E. & Baker, D.(1997). Assembly of protein tertiary structures fromfragments with similar local sequences using simu-lated annealing and Bayesian scoring functions.J. Mol. Biol. 268, 209±225.
Skolnick, J., Kolinski, A., Brooks, C., III, Godzik, A. &Rey, A. (1993). A method for prediction of proteinstructure from sequence. Curr. Biol. 3, 414±423.
Skolnick, J., Jaroszewski, L., Kolinski, A. & Godzik, A.(1997a). Derivation and testing of pair potentials forprotein folding. When is the quasichemical approxi-mation correct?. Protein Sci. 6, 676±688.
Skolnick, J., Kolinski, A. & Ortiz, A. R. (1997b).MONSSTER: A method for folding globular pro-teins with a small number of distance restraints.J. Mol. Biol. 265, 217±241.
Smith-Brown, M. J., Kominos, D. & Levy, R. M. (1993).Global folding of proteins using a limited numberof distance restraints. Protein Eng. 6, 605±614.
448 Fold Prediction of Small Proteins
Sun, S. (1993). Reduced representation model of protein
structure prediction: statistical potential and genetic
algorithms. Protein Sci. 2, 762±785.
Taylor, W. R. (1997). Multiple sequence threading: an
analysis of alignment quality and stability. J. Mol.
Biol. 269, 902±943.
Vieth, M., Kolinski, A., Brooks, C. L., III & Skolnick, J.(1994). Prediction of the folding pathways andstructure of the GCN4 ``leucine zipper''. J. Mol. Biol.237, 361±367.
Wallqvist, A. & Ullner, M. (1994). A simpli®ed aminoacid potential for use in structure prediction ofproteins. Proteins: Struct. Funct. Genet. 18, 267±289.
Edited by F. Cohen
(Received 19 August 1997; received in revised form 5 December 1997; accepted 5 December 1997)