Flink in Genomics Efficient and scalable processing of raw Illumina BCL data F. VERSACI L. PIREDDU G. ZANETTI – FlinkForward 2016 – 13 September 2016 Francesco Versaci (CRS4) Flink in Genomics 13 September 2016 1 / 28
Transcript
Flink in GenomicsEfficient and scalable processing of raw Illumina BCL data
F. VERSACI L. PIREDDU G. ZANETTI
– FlinkForward 2016 –
13 September 2016
Francesco Versaci (CRS4) Flink in Genomics 13 September 2016 1 / 28
Outline
1 Introduction
2 BCL to FASTQ Conversion
3 Implementation in Flink
4 Evaluation and Final Considerations
Francesco Versaci (CRS4) Flink in Genomics 13 September 2016 2 / 28
CRS4Centro di Ricerca, Sviluppo e Studi Superiori in Sardegna
CRS4
Research center in Sardinia, ItalyFocus on big data, biosciences, HPC, visual computing, energyand environment
Francesco Versaci (CRS4) Flink in Genomics 13 September 2016 3 / 28
Next-Generation SequencingCost
Genome sequencing is now much cheaper than in the pastAbout 1000 euros per whole human genome
103
104
105
106
107
108
Cos
t[U
SD]
2001 2004 2007 2010 2013 2016Year
(Data from https://www.genome.gov/sequencingcosts/)
Francesco Versaci (CRS4) Flink in Genomics 13 September 2016 4 / 28
High-throughput DNA sequencing has many applications, includingResearch into understanding human genetic diseasesMedicine, e.g., oncology, clinical pathology, . . .Human phylogenyPersonalized diagnostic applications
Huge amount of dataA single sequencer can produce 1 TB/day of data
Which need to be converted, filtered, aggregated, reconstructed,analysed, . . .
Francesco Versaci (CRS4) Flink in Genomics 13 September 2016 5 / 28
Standard pipeline
When using Illumina sequencers, the standard pipeline starts with twoprograms:bcl2fastq2 Proprietary, open-source tool by Illumina to convert raw
BCL data to FASTQ formatBWA-MEM Free (GPLv3) aligner to reconstruct the full genomic
sequence based on the short reads generated by the sequencer
ProblemParallel tools, but shared-memory (single node)To exploit more nodes data need to be distributed, there can befailures, etc.
Francesco Versaci (CRS4) Flink in Genomics 13 September 2016 6 / 28
BCL converter
In this talk we present a distributed-memory BCL converterDeveloped within the Flink frameworkWritten in ScalaEfficient (i.e., speed comparable to bcl2fastq2)ScalableCan easily be integrated into existent Hadoop/YARN workflows
Francesco Versaci (CRS4) Flink in Genomics 13 September 2016 7 / 28
Outline
1 Introduction
2 BCL to FASTQ Conversion
3 Implementation in Flink
4 Evaluation and Final Considerations
Francesco Versaci (CRS4) Flink in Genomics 13 September 2016 8 / 28
Shotgun genome sequencing
The DNA is a sequence of four bases:Adenine, Cytosine, Guanine andThymine (A, C, G and T)To reconstruct it, the genome is brokenup into short fragments (reads)The fragments are attached to a support(tile)Fluorescent molecules are iterativelyattached to bases of the DNA fragmentsbeing sequencedAt each cycle, the machine acquires(optically) a single base from all thefragments
Francesco Versaci (CRS4) Flink in Genomics 13 September 2016 9 / 28
File organization
We adopt the file structure of Illumina HiSeq 3000/4000machinesA single file refers to data obtained by specific lane, tile and cyclecombinationE.g., file L003/C80.1/s_3_1213.bcl.gz corresponds to dataread from tile t = 1213, in lane l = 3 during cycle c = 80Test dataset: 8 lanes × 112 tiles/lane × 210 cycles = 188,160gzip-compressed BCL files = about 250 GB
Francesco Versaci (CRS4) Flink in Genomics 13 September 2016 10 / 28
BCL files are arrays of bytesEach byte encodes a base (bits0-1) and a quality score (bits 2-7).filter files specify which readsshould be ignored.locs files contain somemetadata which need to beattached to each readTo get the reads from the raw BCLdata we need to perform somesort of matrix transposition
File 1
File 2
File 3
File 4
File 5
A
A
G
C
T
TCGG
CAGA
CAGA
TGAT
CGTC
Cycle 1
Cycle 2
Cycle 3
Cycle 4
Cycle 5
Loca
tion
5
Loca
tion
4
Loca
tion
3
Loca
tion
2
Loca
tion
1
A A G C T
C A A G GG G G A TG A A T C
T C C T C
Read 1Read 2etc...
Francesco Versaci (CRS4) Flink in Genomics 13 September 2016 11 / 28
Francesco Versaci (CRS4) Flink in Genomics 13 September 2016 12 / 28
Implementation choices
The converter is written in ScalaWe use sbt to handle compilation and dependenciesAll the code is less than 1000 linesNo fancy IDEs, just EMACS as editor
Francesco Versaci (CRS4) Flink in Genomics 13 September 2016 13 / 28
Algorithmic overview
For each lane/tile combinationBCL files corresponding to different cycles are openedconcurrentlyBases and quality scores are extracted and filteredFor each fragment a text header is added, containing variousmeta-data and an indexThen they are sorted by their indexesSince there can be read errors also in the indices, the repartitionis fuzzy: a parameter sets the numbers of allowed misinterpretedsymbolsFinally, a gzip-compressed file for each index is written to disk
Francesco Versaci (CRS4) Flink in Genomics 13 September 2016 14 / 28
DataSet vs DataStream
Our data is static and read from a storage unitThis is not a typical streaming applicationWe have tried both DataSet and DataStream structuresUsing DataStream is fasterBecause of its better overlap of I/O and computations?
Lesson Learned #1Try DataStream even if it doesn’t seem like a natural fit for yourapplication
Francesco Versaci (CRS4) Flink in Genomics 13 September 2016 15 / 28
Data granularity
BCL files are arrays of bytesIt might seem natural to process them in Flink asDataStream[Byte]But reading and writing single bytes is not efficientWe process data in bigger chunks (2048 bytes)It imposes a lower load on the streaming frameworkBetter cache locality exploitation
Lesson Learned #2Avoid fine granularity and read in larger chunks
Francesco Versaci (CRS4) Flink in Genomics 13 September 2016 16 / 28
Job granularity
The job unit (mini-job) is the processing of a lane-tilecombinationMini-jobs run for about one minute on one coreWe can choose to aggregate n mini-jobs into a Flink jobAnd assign c cores to each Flink jobE.g., aggregate n = 16 mini-jobs and run them on c = 4 coresWhat about launching one huge Flink job which handles all thework and cores?Best results with n = 2 and c = 1 (with processor SMT = 2)
Lesson Learned #3Keep Flink jobs reasonably small
Francesco Versaci (CRS4) Flink in Genomics 13 September 2016 17 / 28
Low level optimizationsUse of ByteBuffer
To extract bases and quality scores we need to perform some bitmasks and shiftsE.g., to get the quality score from byte b we can run
va l b : Byte = in . ge tva l q : Byte =
(0 x21 + ( b & 0xFC ) >>> 2 ) . toBy te
We can obtain a 8x speed-up by grouping bytes into 64-bit longsand executing the equivalent operations:
va l r : Long = in . getLongva l q : Long = 0x2121212121212121l
+ ( ( r & 0xFCFCFCFCFCFCFCFCl ) >>> 2 ) )
To interpret byte arrays as longs, we need to use the ByteBufferclass
Francesco Versaci (CRS4) Flink in Genomics 13 September 2016 18 / 28
Low level optimizationsUse of look-up tables
We need to convert bases from numeric to ASCII notationE.g., 0x0001020303020100 maps to “ACGTTGCA”We can do it efficiently by compressing the input and using it asan index in a look-up tableE.g., 0x0001020303020100 is compressed to index0b0001101111100100=0x1BE4 and searched in theprecomputed look-up tableThe table has 216 = 65536 entries
Francesco Versaci (CRS4) Flink in Genomics 13 September 2016 19 / 28
Scheduling
To schedule the Flink jobs we use Scala FuturesParallel and not blocking
/ / numTasks = number o f F l i n k jobsi m p l i c i t va l ec = s c a l a . concur ren t . Execu t ionContex t
. f romExecutor ( Execu to r s . newFixedThreadPool ( numTasks ) )va l mini Jobs : Seq [ Mini Job ] = r eade r . ge tMin i Jobs/ / f p a r = number o f mini−j obs per F l i n k jobsva l f l i n k J o b s = work . s l i d i n g ( fpa r , f p a r )
. map ( f j => Fu tu re { run Jobs ( f j ) } )/ / conver t Seq [ Fu tu re ] to Fu tu re [ Seq ]va l f l i s t = Fu tu re . sequence ( f l i n k J o b s )s c a l a . concur ren t . Await . r e s u l t ( f l i s t , Dura t ion . I n f )
Francesco Versaci (CRS4) Flink in Genomics 13 September 2016 20 / 28
Outline
1 Introduction
2 BCL to FASTQ Conversion
3 Implementation in Flink
4 Evaluation and Final Considerations
Francesco Versaci (CRS4) Flink in Genomics 13 September 2016 21 / 28
Hardware
Experiments run on the Amazon Elastic Compute Cloud (EC2)Up to 14 instances of r3.8xlarge machinesCPUs 32 virtual cores (Xeon E5-2670 v2, 25 MB cache)RAM 250 GBDisks 2x320 GB SSDNetwork 10 Gb EthernetHDFS distributed among the n computing nodesEach datanode using its two SSD disksYARN running on the same n nodesFlink running inside Hadoop/YARN
Francesco Versaci (CRS4) Flink in Genomics 13 September 2016 22 / 28
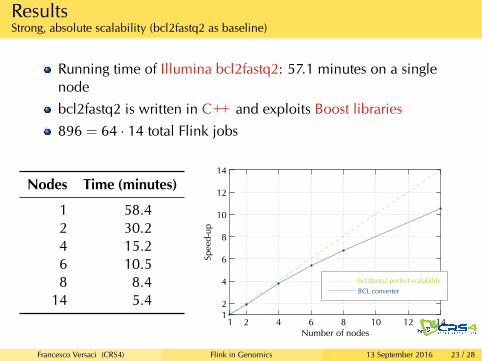
ResultsStrong, absolute scalability (bcl2fastq2 as baseline)
Running time of Illumina bcl2fastq2: 57.1 minutes on a singlenodebcl2fastq2 is written in C++ and exploits Boost libraries896 = 64 · 14 total Flink jobs
Nodes Time (minutes)
1 58.42 30.24 15.26 10.58 8.4
14 5.412
4
6
8
14
10
12
Spee
d-up
1 2 4 6 8 1410 12Number of nodes
bcl2fastq2 perfect scalabilityBCL converter
Francesco Versaci (CRS4) Flink in Genomics 13 September 2016 23 / 28
I/O vs Computations
The program is CPU-bound on the tested hardwareTotal I/O size (input+output): ≈ 500 GBI/O rate on single node: ≈ 150 MB/sI/O rate on 14 nodes: ≈ 1.6 GB/sNote: both input and output are gzip-compressed
Francesco Versaci (CRS4) Flink in Genomics 13 September 2016 24 / 28
Flink features – What we have used
Custom Flink Input/OutputFormatHadoop libraries to read/write files
import org . apache . hadoop . f s .{ F i leSys tem , FSData InputS t ream , FSDataOutputStream , Path }
import org . apache . hadoop . io . compress . z l i b .{ Zl ibCompressor , Z l i b F a c t o r y }
DataStreammap and flatMap
MapFunction and FlatMapFunction
filter
split and select
Francesco Versaci (CRS4) Flink in Genomics 13 September 2016 25 / 28
Flink features – What was not availableEfficient zip of DataStreams
ProblemGiven two data streams
va l names : DataStream [ S t r i n g ]va l ages : DataStream [ I n t ]
Join them as
va l combined : DataStream [ ( S t r i n g , I n t ) ]
Useful when reading data about the same object from differentfilesE.g., .bcl, .locs and .filter filesInverse function of
va l names = combined . map ( _ . _1 )va l ages = combined . map ( _ . _2 )
Francesco Versaci (CRS4) Flink in Genomics 13 September 2016 26 / 28
Flink features – What was not availableA smarter job scheduler
RemarkWe’re talking about Flink 1.0: it seems the new job scheduler is muchsmarter :)
It would be convenient for the job scheduler to be able toPick jobs from some (priority?) queueRuns them concurrently on the available Flink task slotsStart a new job as soon as another one finishesHandle failures and retries
Francesco Versaci (CRS4) Flink in Genomics 13 September 2016 27 / 28
Future work
Integrate our converter into Seal1 toolkit for short DNA readsmanipulation and analisysAdopt Flink also in the second stage of the pipeline, i.e., have aFlink-based aligner
Thanks for your attention!
1http://biodoop-seal.sourceforge.net/Francesco Versaci (CRS4) Flink in Genomics 13 September 2016 28 / 28
Integrate our converter into Seal1 toolkit for short DNA readsmanipulation and analisysAdopt Flink also in the second stage of the pipeline, i.e., have aFlink-based aligner
Thanks for your attention!
1http://biodoop-seal.sourceforge.net/Francesco Versaci (CRS4) Flink in Genomics 13 September 2016 28 / 28