Genetic Disruption of Calcineurin Improves Skeletal MusclePathology and Cardiac Disease in a Mouse Model ofLimb-Girdle Muscular Dystrophy*
Received for publication, October 4, 2006, and in revised form, January 12, 2007 Published, JBC Papers in Press, February 7, 2007, DOI 10.1074/jbc.M609368200
Stephanie A. Parsons‡1, Douglas P. Millay‡§, Michelle A. Sargent‡, Francisco J. Naya¶, Elizabeth M. McNally�,H. Lee Sweeney**, and Jeffery D. Molkentin‡2
From the ‡Department of Pediatrics, University of Cincinnati, Cincinnati Children’s Hospital Medical Center, Cincinnati,Ohio 45229-3039, the §Department of Molecular Genetics, University of Cincinnati, Cincinnati, Ohio 45267, the ¶Departmentof Biology, Boston University, Boston, Massachusetts 02215, the �Department of Medicine, University of Chicago, Chicago,Illinois 60637, and the **Department of Physiology, University of Pennsylvania, Philadelphia, Pennsylvania 19104
Calcineurin (Cn) is a Ca2�/calmodulin-dependent serine/threonine phosphatase that regulates differentiation-specificgene expression in diverse tissues, including the control of fiber-type switching in skeletal muscle. Recent studies have impli-cated Cn signaling in diminishing skeletal muscle pathogenesisassociated with muscle injury or disease-related muscle degen-eration. For example, use of the Cn inhibitor cyclosporine A hasbeen shown to delay muscle regeneration following toxin-in-duced injury and inhibit regeneration in the dystrophin-defi-cient mdx mouse model of Duchenne muscular dystrophy. Incontrast, transgenic expression of an activated mutant of Cn inskeletal muscle was shown to increase utrophin expression andreduce overall disease pathology inmdxmice. Here we examinethe effect of altered Cn activation in the context of the �-sarco-glycan-null (scgd�/�)mousemodel of limb-girdlemuscular dys-trophy. In contrast to results discussed in mdx mice, geneticdeletion of a loxP-targeted calcineurin B1 (CnB1) gene using askeletal muscle-specific cre allele in the scgd�/� backgroundsubstantially reduced skeletalmuscle degeneration andhistopa-thology compared with the scgd�/� genotype alone. A similarregression in scgd-dependent disease manifestation was alsoobserved in calcineurin A� (CnA�) gene-targeted mice in bothskeletal muscle and heart. Conversely, increased Cn expressionusing a muscle-specific transgene increased cardiac fibrosis,decreased cardiac ventricular shortening, and increasedmusclefiber loss in the quadriceps. Our results suggest that inhibitionof Cn may benefit select types of muscular dystrophy.
The Ca2�/calmodulin-activated serine/threonine proteinphosphatase calcineurin (Cn)3 is expressed in many different
cell types and is involved in a number of processes includinglymphocyte development and proliferation, neuronal andmus-cle development, cardiac hypertrophy, and skeletal musclefiber-type switching (1). Cn exists as a heterotrimer consistingof a 57–61-kDa catalytic A subunit (CnA) and two smaller16–19-kDa EF-hand containing Ca2�-binding proteinsreferred to as calcineurin B (CnB) and calmodulin (2). In verte-brates, three unlinked loci encode the catalytic subunit (CnA�,CnA�, andCnA�), whereas two loci encode the regulatory sub-unit (CnB1 and CnB2) (3). The CnA�, CnA�, and CnB1 genesare each expressed in a ubiquitous pattern throughout thebody, whereas CnA� and CnB2 expression are more restrictedto the brain and testis (4–7). Once activated, Cn directlydephosphorylates a family of transcription factors referred to asnuclear factor of activatedT cells (NFAT)within the cytoplasm,exposing a nuclear localization sequence and promoting trans-location into the nucleus where they participate in transcrip-tional induction events (8). There are four Cn-regulated NFATtranscription factors, NFATc1–c4, each of which is expressedin skeletal muscle (9, 10). Cn activity and the ability to activateNFATare directly antagonized by the pharmacologic inhibitorsFK506 and cyclosporine A (CsA) (8).In skeletal muscle, augmented or sustained patterns of muscle
contraction and fiber recruitment maintains or promotes theslow/oxidative fiber-typeprogramthrough increasedCa2� fluxingthat facilitates Cn activation (11–21). However, Cn-NFAT signal-ing isnotbelieved to regulatehypertrophicgrowthof skeletalmus-cle (18, 20), in contrast to itswell characterizedhypertrophic func-tion in cardiac muscle (22). Cn-NFAT signaling has also beenimplicated in regulating the differentiation, developmental matu-ration, and regeneration of skeletalmuscle (9, 13, 18, 23, 24). Con-sistent with these observations, Cn signaling can modify the pro-gression and severity of skeletal muscle pathology associated withgenetic mutations that cause Duchenne and other forms of mus-cular dystrophy (see below).Duchenne and Becker muscular dystrophies are X-linked
recessive disorders that arise due tomutations in thedystrophin
* This work was supported by a grant from the National Institutes of Health(to J. D. M.). The costs of publication of this article were defrayed in part bythe payment of page charges. This article must therefore be herebymarked “advertisement” in accordance with 18 U.S.C. Section 1734 solely toindicate this fact.
1 Supported by training grants from the National Institutes of Health Grants5T32HL007752 and 1T32AR053461-01. Current address: Wyeth Research,Collegeville, PA 19426.
2 Established Investigator of the American Heart Association. To whom corre-spondence should be addressed: 3333 Burnet Ave., Cincinnati, OH 45229.Fax: 513-636-5958; E-mail: [email protected].
3 The abbreviations used are: Cn, calcineurin; Cn*, constitutively active Cn;CnA, calcineurin A subunit; CnB, calcineurin B subunit; CsA, cyclosporine A;
gene, leading to destabilization in structural attachmentbetween the underlying contractile units with the dystrophin-glycoprotein complex (DGC) and the basal lamina, hence alter-ing the integrity of the cell membrane and promoting myofiberdegeneration (25, 26). Themdxmouse, which has a single pointmutation within an exon of the dystrophin gene, is a widelyemployed model of muscular dystrophy. Disease in the mdxmodel shows a continuum of skeletal muscle disease severitywith the diaphragm being the most affected. However, unlikehuman Duchenne patients,mdxmice live well into adulthood.Studies in mdx mice have suggested that Cn activation is pro-tective and benefits the degree of muscle pathology. For exam-ple, transgenic mice expressing a constitutively active cal-cineurin protein (Cn*) in skeletal muscle were crossed withmdxmice, showing significant reductions in the extent of myo-tube central nucleation, fiber size variability, and inflammation,as well as demonstrating an improvement in membrane integ-rity compared withmdxmice alone (27). As a potential mech-anism underlying this protective effect, Cn* transgenic miceshowed increasedNFATc1nuclear localization in skeletalmus-cle cells, and a 2-fold up-regulation in utrophin protein levelsthat helped restore the integrity of the DGC inmdxmice (27).Studies of Cn inhibition found that treatment of young mdxmice with CsA led to a severe impairment in muscle regenera-tion, as assessed by fewer centrally nucleated fibers, reducedmuscle mass, decreased force output, increased collagenreplacement, and increased inflammation relative to vehicle-treated littermates (28). In support of these findings, the mus-cle-sparing effects associated with the glucocortocoid deflaza-cort inmdxmice could be abrogated by the co-administrationof CsA (29). Thus, Cn activity appears to be protective in themdxmouse model.To further characterize the role of Cn signaling in muscular
dystrophy and to establish the relevance of Cn activity in non-dystrophin-deficient models of muscular dystrophy, weemployed �-sarcoglycan-null (scgd�/�) mice. The �-sarcogly-can gene encodes a member of the sarcoglycan complex oftransmembrane proteins that is assembled into the DGC and iscritical for maintainingmembrane stability (25, 26). The loss ofscgd causes reductions or complete loss of the othermembers ofthe sarcoglycan complex, an effect that is not uniformly seen inthe absence of dystrophin (30, 31). scgd�/� mice have cardiom-yopathy and muscular dystrophy (corresponding to a humanlimb-girdle myopathy) withmany hallmarks of progressive dis-ease including cell death, muscle regeneration, inflammation,fibrosis, and reduced survival (30, 31).Contrary to the previously observed protective influence of
Cn activation inmdxmice, here we describe an entirely oppo-site effect associatedwithCn signaling in scgd�/�mice.Geneticdisruption of Cn signaling, using two different geneticapproaches, greatly benefited both skeletal and cardiac muscledisease in scgd�/� mice, as measured by a reduction or evenabsence (depending on the degree of Cn reduction) of fibrosisin both skeletal and cardiac muscle, decreased skeletal muscledegeneration and inflammation, and improved cardiac disease.Conversely, Cn* transgenic mice showed worsened skeletalmuscle histopathology and heart disease in the scgd nullbackground.
MATERIALS AND METHODS
Animal Models—scgd�/� (30), CnB1fl/f l (18, 32), myosinlight chain1f-cre (MLC-cre) (18, 33), CnA��/� (34), NFAT-luciferase (35), and muscle creatine kinase Cn*transgene (Tg)(MCK-Cn*Tg) (16) mice have been described previously. Ani-mals had free access to food and water, and all experimentationwas performed in the Cincinnati Children’s Hospital ResearchFoundation animal care facility in accordance with the guide-lines of the National Institutes of Health. Experimental proto-cols were reviewed and approved by the Institutional AnimalCare and Use Committee. Both male and female mice wereused for all analyses. For decreased Cn protein expression stud-ies, all analyses were performed onmice�8–10months of age.For increased Cn expression studies, mice 6-months of agewere examined.WesternAnalysis—Muscle protein extracts were prepared by
homogenizing tissue in cell lysis buffer containing 20 mM Tris,pH 7.4, 137mM sodium chloride, 25mM �-glycerophosphate, 2mM sodium pyrophosphate, 2 mM EDTA, 1 mM sodiumorthovanadate, 1% Triton X-100, 10% glycerol, 1 mM phenyl-methylsulfonyl fluoride, 5 �g/ml leupeptin, 5 �g/ml aprotinin,and 2mM benzamidine. Proteins (50–100�g) were resolved ona sodiumdodecyl sulfate-polyacrylamide gel, and transferred toa polyvinylidine difluoridemembrane, and immunodetected byusing an enhanced chemifluorescence kit as specified by themanufacturer (Amersham Biosciences). The following anti-bodies were used: �-tubulin monoclonal antibody (1:2500,Santa Cruz Biotechnology, Santa Cruz, CA), CnB1 rabbit poly-clonal antibody (1:500, Upstate, Waltham, MA), and cal-cineurin pan-A rabbit polyclonal antibody (1:1000, ChemiconInternational, Inc., Temecula, CA).Western blot reactivity wasquantified on a Storm860 PhosphorImager (GE Healthcare)using ImageQuant software.Determination of Hydroxyproline Content—The method
used is a modified version of that described by Woessner et al.(36). Briefly, a small piece of muscle (between 5 and 8 mg fordiaphragm and 200 and 300 mg for gastrocnemius and quadri-ceps) wasweighed and placed into a glass Pyrex tube containing500 �l of 6 N hydrochloric acid. The tubes were capped looselyand placed into a 100 °C oven for 20 min. Tubes were thencapped tightly and baked overnight to completely hydrolyze thetissue. The following day the tubes were removed, uncapped,and placed in a dessicator containing sodium hydroxide pellets.The dessicator was placed into the oven at 50 °C and connectedto a vacuum going through a sodium hydroxide trap. The tissuewas kept under vacuum until dry (24 h to 1 week) and thenresuspended in 1 ml of 5 mM hydrochloric acid. An aliquot ofsample (25�l) was combinedwith 180�l ofMilliQH2O in 12�75-mmglass tubes. To this, 100�l of chloramineT solutionwasadded (0.14 g of chloramine-T, 2 ml H2O, and 8 ml ofhydroxyproline assay buffer). Hydroxyproline assay buffer wasprepared by combining 11.4 g of sodium acetate anhydrous,7.5 g of trisodium citrate dihydrate, 40 ml of H2O (pH adjustedto 6.0), and 77 ml of isopropyl alcohol and bringing to a finalvolume of 200 ml with H2O. Following addition of chloramineT solution, 1.25 ml of Erlich’s reagent was added (6.0 g of p-di-methylaminobenzaldehyde, 18 ml of 60% perchlorate, and 78
Calcineurin Deletion Improves Muscular Dystrophy
MARCH 30, 2007 • VOLUME 282 • NUMBER 13 JOURNAL OF BIOLOGICAL CHEMISTRY 10069
ml of isopropyl alcohol). The samples were then vortexed for15 s and incubated at 55 °C for 20–25 min. Once cooled toroom temperature, sample absorbance was read at 558 nm. Astandard curve was run alongside the samples using trans-4-hydroxy-L-proline (Sigma) standards (ranging from 0 to 4 �g)to determine hydroxyproline concentration. Readings werenormalized to original tissue weight.Luciferase Assay—Excised skeletal muscle (�8 to 30mg) was
placed in 400 �l of ice-cold lysis buffer (1% Triton X-100, 100mM Tris-HCl, pH 7.8, 2 mM EDTA, 2 mM dithiothreitol, 2�g/ml aprotinin, 2�g/ml leupeptin, 1�g/ml pepstatin, and 100�g/ml phenylmethylsulfonyl fluoride). Tissue was then finelyminced on ice using scissors and sonicated. Homogenates werespun for 10 min at 14,000 � g at 4 °C. In a 96-well plate, 200 �lof ice-cold reaction buffer (4 mM ATP, pH 7.0, 15 mM magne-sium sulfate, 30 mM Tricine, pH 7.8, and 10 mM dithiothreitol)was combined with 20 �l of tissue supernatant. To measureluciferase enzymatic activity in tissue extracts, 100 �l of 1 mMluciferin (Promega, Madison, WI) was injected into each well,and light intensity wasmeasuredwith a luminometer (Microlu-mat LB 96B, Berthold, Wildbad, Germany) over 10 s andexpressed as relative light units over 10 s/�g of proteins. Lucif-erase assays were performed on skeletal muscle frommice 5 to7 weeks of age.Evans Blue Dye Uptake—Evans blue dye (10 mg/ml in PBS)
was injected intraperitoneally into 5–7-month-old mice (0.1ml/10 g body weight). The mice were sacrificed 6 h after injec-tion.Muscles were excised and embedded inO.C.T. compound(Tissue-Tek, Torrence, CA), and snap-frozen in liquid nitro-gen-cooled isopentane. Blocks were then sectioned into 8-�mthick slices, dried for 10 min, and washed briefly in PBS. Tovisualize muscle fibers, sections were then incubated with flu-orescein isothiocyanate-labeled wheat germ agglutinin (50�g/ml in 1� PBS, Sigma) in the dark for 1 h. Slides were thenwashed 3 times, 5 min each, and mounted with VectaShield(Vector Laboratories, Burlingame, CA) and coverslips.Histology—Paraffin-embedded sections (8-�m thick) were
cut at the mid-belly of the muscle. Masson trichrome stainingwas carried out at the Molecular Pathology core facility at Cin-cinnati Children’s Hospital Medical Center. Unoccupied areawas determined using NIH ImageJ software. Briefly, theunstained areawithin a quadriceps section stainedwithMassontrichrome was calculated along with the total area of the sec-tion. The ratio of the unstained area/total area was calculated,with the resulting number used as an index of “unoccupiedarea.”Echocardiography—Mice were anesthetized with 2% isoflu-
rane, and hearts were visualized using a Hewlett Packard Sonos5500 instrument and a 15-MHz transducer. Cardiac ventriculardimensions were measured on M-mode three times for thenumber of animals indicated.Analysis of Centrally Located Nuclei and Fiber Area—Dia-
phragmmuscles frommice were collected, fixed in 10% forma-lin containing PBS, and embedded in paraffin. Tissues were cutcrosswise through the mid-belly of the muscle, and embeddedcut side down.Muscle sections were cut at a thickness of 8-�m.Sections were deparaffinized in xylene and rehydrated in dilu-tions of ethanol. To visualize fibers, sections were incubated
with TRITC-labeled wheat germ agglutinin (Sigma) at 50�g/ml in 1� PBS for 1 h, washed three times for 10min each in1� PBS, with the secondwash containing 1�g/ml bis-benzam-ide for nuclei visualization, if necessary. Sections were thenmounted with VectaShield and coverslips. Fiber area analysiswas performed using ImageJ analysis software (National Insti-tutes of Health), and �250 fibers were measured per section.For central nuclei analysis, 2–3 areas at�10magnificationwereexamined and counted, averaging �650 fibers per animal.Real Time PCR—Total RNA was extracted from quadriceps
muscles using TRIzol� reagent (Invitrogen). Onemicrogram oftotal RNA from each muscle was converted into double-stranded cDNAby using the SuperScript First-strand SynthesisSystem (Invitrogen) with an oligo(dT) primer. Two microlitersof the synthesized cDNA was then used for quantitative realtime PCR. The LightCycler� FastStart DNAMasterPLUS SYBRGreen kit (RocheApplied Science)was used for amplification ofutrophin A, myocyte enhancer factor 2C (MEF2C), and 18 SrRNA using the LightCycler 2.0 System. Primers used foramplification were: MEF2C, 5�-GGCCATGGTACACCGAG-TACAACGAGC-3� and 5�-GGGGATCCCTGTGTTACCTG-CACTTGG-3�; Utrophin A, 5�-GGCAGGAAGATTGCA-CAAGT-3� and 5�-CTGCTAGCCAAGTCCCAGAG-3�; 18S,5�-AGTCCCTGCCCTTTGTACACA-3� and 5�-CGATC-CGAGGGCCTCACTA-3�. The mRNA levels of 18S weremeasured as an internal control gene. Each cDNA samplewas subjected to two individual PCR analyses using eitherthe MEF2C or utrophin A primer pair. The target geneand the internal control gene were amplified in separatetubes. The increase in fluorescence was measured in realtime during the extension step. The threshold cycle (CT) wascalculated, and the relative gene expression was calculatedusing the 2���CT method. For each sample, 18S CT values weresubtracted from Utrophin A or MEF2C CT values to derive a�CT value. The average value for wild-type (WT) muscle wasthen subtracted from the values obtained fromCn*Tg, scgd�/�,or scgd�/� Cn*Tgmuscles to derive a ��CT value. The expres-sion of these genes relative toWTwas then evaluated using thedesignation 2���CT.In Situ Ligation of Hairpin 1—Frozen diaphragm sections (7
�m thick) were incubated with a biotin-labeled blunt-end hair-pin oligo (kit component) and T4 DNA ligase (kit component)as specified in the ApopTag� Peroxidase In SituOligo LigationApoptosis Detection Kit (Chemicon International). Ligation ofthe hairpin 1 oligo was detected using streptavidin-fluoresceinisothiocyanate conjugate (Alexa, 1:400 dilution). Total nucleiand membranes were stained with TO-PRO-3 iodide (Molecu-lar Probes) and wheat germ agglutinin-TRITC (Sigma), respec-tively. Sections were analyzed using a confocal microscope,with pictures taken at�200magnification. Six pictures per ani-mal were counted for positive myonuclei. The analysis of liga-tion rates was performed in 4WT, 5 scgd�/�, 6 scgd�/� Cn*Tg,and 6 scgd�/� CnB1fl/flMLC-cre mice.Statistical Analysis—Data were expressed as themean� S.E.
Statistical significance was determined by Student’s t test at thep � 0.05 level.
Calcineurin Deletion Improves Muscular Dystrophy
10070 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 282 • NUMBER 13 • MARCH 30, 2007
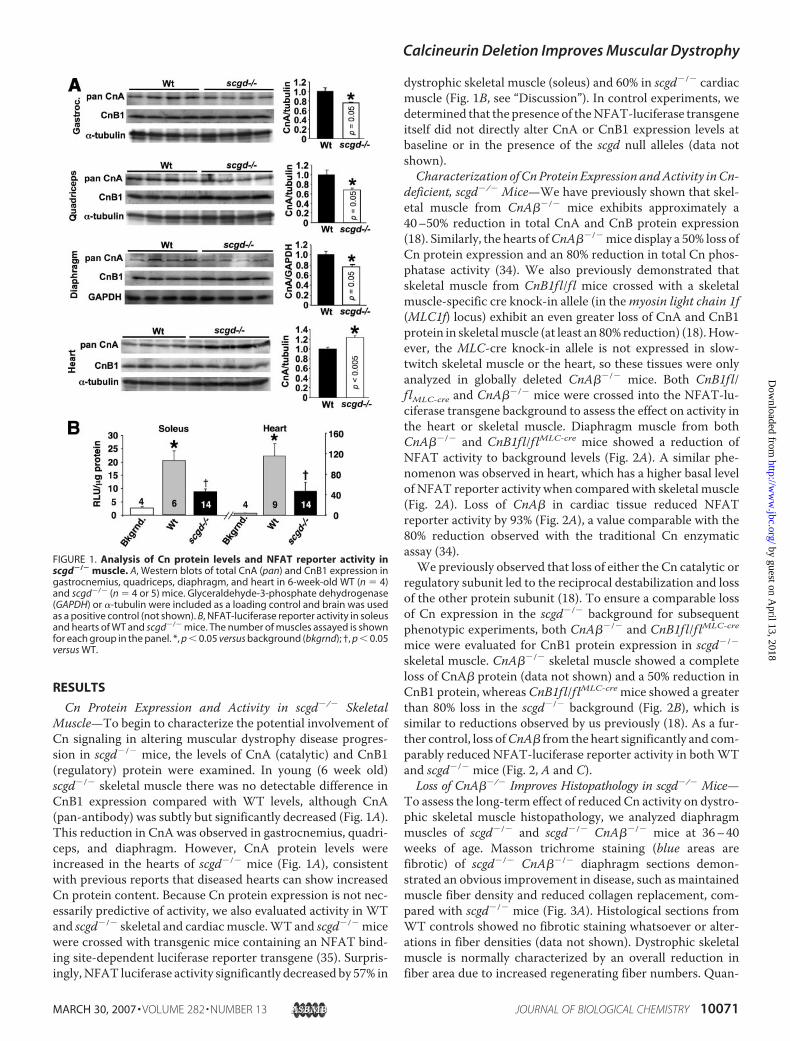
Cn Protein Expression and Activity in scgd�/� SkeletalMuscle—To begin to characterize the potential involvement ofCn signaling in altering muscular dystrophy disease progres-sion in scgd�/� mice, the levels of CnA (catalytic) and CnB1(regulatory) protein were examined. In young (6 week old)scgd�/� skeletal muscle there was no detectable difference inCnB1 expression compared with WT levels, although CnA(pan-antibody) was subtly but significantly decreased (Fig. 1A).This reduction in CnA was observed in gastrocnemius, quadri-ceps, and diaphragm. However, CnA protein levels wereincreased in the hearts of scgd�/� mice (Fig. 1A), consistentwith previous reports that diseased hearts can show increasedCn protein content. Because Cn protein expression is not nec-essarily predictive of activity, we also evaluated activity in WTand scgd�/� skeletal and cardiacmuscle.WT and scgd�/� micewere crossed with transgenic mice containing an NFAT bind-ing site-dependent luciferase reporter transgene (35). Surpris-ingly,NFAT luciferase activity significantly decreased by 57% in
dystrophic skeletal muscle (soleus) and 60% in scgd�/� cardiacmuscle (Fig. 1B, see “Discussion”). In control experiments, wedetermined that the presence of theNFAT-luciferase transgeneitself did not directly alter CnA or CnB1 expression levels atbaseline or in the presence of the scgd null alleles (data notshown).Characterization of CnProtein Expression andActivity inCn-
deficient, scgd�/� Mice—We have previously shown that skel-etal muscle from CnA��/� mice exhibits approximately a40–50% reduction in total CnA and CnB protein expression(18). Similarly, the hearts ofCnA��/�mice display a 50% loss ofCn protein expression and an 80% reduction in total Cn phos-phatase activity (34). We also previously demonstrated thatskeletal muscle from CnB1fl/f l mice crossed with a skeletalmuscle-specific cre knock-in allele (in themyosin light chain 1f(MLC1f) locus) exhibit an even greater loss of CnA and CnB1protein in skeletalmuscle (at least an 80% reduction) (18).How-ever, the MLC-cre knock-in allele is not expressed in slow-twitch skeletal muscle or the heart, so these tissues were onlyanalyzed in globally deleted CnA��/� mice. Both CnB1fl/f lMLC-cre and CnA��/� mice were crossed into the NFAT-lu-ciferase transgene background to assess the effect on activity inthe heart or skeletal muscle. Diaphragm muscle from bothCnA��/� and CnB1fl/f lMLC-cre mice showed a reduction ofNFAT activity to background levels (Fig. 2A). A similar phe-nomenon was observed in heart, which has a higher basal levelof NFAT reporter activity when compared with skeletal muscle(Fig. 2A). Loss of CnA� in cardiac tissue reduced NFATreporter activity by 93% (Fig. 2A), a value comparable with the80% reduction observed with the traditional Cn enzymaticassay (34).We previously observed that loss of either the Cn catalytic or
regulatory subunit led to the reciprocal destabilization and lossof the other protein subunit (18). To ensure a comparable lossof Cn expression in the scgd�/� background for subsequentphenotypic experiments, both CnA��/� and CnB1fl/f lMLC-cre
mice were evaluated for CnB1 protein expression in scgd�/�
skeletal muscle. CnA��/� skeletal muscle showed a completeloss of CnA� protein (data not shown) and a 50% reduction inCnB1 protein, whereas CnB1fl/f lMLC-cremice showed a greaterthan 80% loss in the scgd�/� background (Fig. 2B), which issimilar to reductions observed by us previously (18). As a fur-ther control, loss ofCnA� from the heart significantly and com-parably reduced NFAT-luciferase reporter activity in bothWTand scgd�/� mice (Fig. 2, A and C).Loss of CnA��/� Improves Histopathology in scgd�/� Mice—
To assess the long-term effect of reduced Cn activity on dystro-phic skeletal muscle histopathology, we analyzed diaphragmmuscles of scgd�/� and scgd�/� CnA��/� mice at 36–40weeks of age. Masson trichrome staining (blue areas arefibrotic) of scgd�/� CnA��/� diaphragm sections demon-strated an obvious improvement in disease, such as maintainedmuscle fiber density and reduced collagen replacement, com-pared with scgd�/� mice (Fig. 3A). Histological sections fromWT controls showed no fibrotic staining whatsoever or alter-ations in fiber densities (data not shown). Dystrophic skeletalmuscle is normally characterized by an overall reduction infiber area due to increased regenerating fiber numbers. Quan-
FIGURE 1. Analysis of Cn protein levels and NFAT reporter activity inscgd�/� muscle. A, Western blots of total CnA (pan) and CnB1 expression ingastrocnemius, quadriceps, diaphragm, and heart in 6-week-old WT (n � 4)and scgd�/� (n � 4 or 5) mice. Glyceraldehyde-3-phosphate dehydrogenase(GAPDH) or �-tubulin were included as a loading control and brain was usedas a positive control (not shown). B, NFAT-luciferase reporter activity in soleusand hearts of WT and scgd�/� mice. The number of muscles assayed is shownfor each group in the panel. *, p 0.05 versus background (bkgrnd); †, p 0.05versus WT.
Calcineurin Deletion Improves Muscular Dystrophy
MARCH 30, 2007 • VOLUME 282 • NUMBER 13 JOURNAL OF BIOLOGICAL CHEMISTRY 10071
tification of fiber areas revealed the expected decrease in aver-age fiber area in scgd�/� mice relative toWT, although a trendwas observed in scgd�/� CnA��/� diaphragm such that theaverage fiber area increased (Fig. 3B). However, measurementof collagen content by assessing hydroxyproline content in abiochemical assay did show a significant reduction in fibrotictissue replacement in scgd�/� CnA��/� diaphragm comparedwith scgd�/� (Fig. 3C). Taken together, these results suggest aprotective effect of Cn inhibition on dystrophic musclepathology.Extensive and Muscle-specific Loss of Cn Improves Skeletal
Muscle Pathology in scgd�/� Mice—Examination of musclefrom scgd�/� CnB1fl/f lMLC-cre crossedmice at 36–40 weeks ofage revealed a remarkable improvement in gross musclepathology. Masson trichrome staining of diaphragm sectionsfrom scgd�/� CnB1fl/flMLC-cre mice showed no evidence offibrosis and muscle fibers appeared normal and healthy,
whereas scgd�/� diaphragms showed extensive fibrotic tissuereplacement (Fig. 4A). Histological examination of WT sec-tions showed no fibrosis (data not shown). Examination of fiberareas within the diaphragms of scgd�/� CnB1fl/flMLC-cre miceshowed a dramatic increase in average fiber area comparedwithscgd�/� mice (Fig. 4B). Quantitation of centrally nucleatedfibers in the diaphragm, which indicates the extent of ongoingregeneration, demonstrated a significant reduction in scgd�/�
CnB1fl/flMLC-cre mice compared with scgd�/� mice (Fig. 4C).Finally,measurement of hydroxyproline content showed essen-tially no increases in fibrosis in the diaphragm, quadriceps, andgastrocnemius of CnB1fl/f lMLC-cre mice, supporting the dra-matic phenotype observed byMasson trichrome staining of his-tological sections (Fig. 4D).Loss of Cn Does Not Stabilize the scgd�/� Muscle Membrane
orAlterApoptosis—Mechanistic studieswere initiated to inves-tigate why loss of Cnmight benefit muscular dystrophy diseaseprogression. The ongoing muscle degeneration/regenerationthat occurs in muscular dystrophy is associated with contrac-tile-induced damage of the sarcolemma. This damage permitspassive Ca2� influx or channel-mediated influx, which may in
FIGURE 2. NFAT reporter activity is reduced similarly in Cn-deficient andscgd�/� muscle. A, NFAT-luciferase reporter activity in diaphragm and car-diac muscle in WT, CnA��/�, and CnB1f l/f lMLC-cre mice. The number of mus-cles assayed is shown for each group in the panel. *, p 0.05 versus back-ground (bkgrnd); †, p 0.05 versus WT. B, Western blots for total CnB1 proteinin scgd�/�, scgd�/� CnA��/�, and scgd�/� CnB1fl/flMLC-cre muscle. �-Tubulinwas included as a loading control and brain extract as a positive control.Quantitation of results from this and another independent Western blot isshown below, with the number of total samples indicated in the bars. *, p 0.05 versus scgd�/�. C, cardiac NFAT-luciferase reporter activity in WT,scgd�/�, CnA��/�, and scgd�/� CnA��/� mice. The number of musclesassayed is shown for each group in the panel. *, p 0.05 versus WT; †, p 0.05versus scgd�/�.
FIGURE 3. CnA�-deficient dystrophic muscle displays improved histopa-thology and decreased fibrosis. A, representative Masson trichrome stain-ing of paraffin sections from the diaphragm of age-matched scgd�/� andscgd�/� CnA��/� mice. B, analysis of average muscle fiber area in the dia-phragm of WT, scgd�/�, and scgd�/� CnA��/� mice. The number of musclesassayed is shown for each group in the panel. *, p 0.05 versus WT. C, analysisof �g of hydroxyproline/mg of diaphragm tissue in WT, scgd�/�, CnA��/�,and scgd�/� CnA��/� mice. The number of muscles assayed is shown foreach group in the bars. *, p 0.05 versus WT; †, p 0.05 versus scgd�/�.
Calcineurin Deletion Improves Muscular Dystrophy
10072 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 282 • NUMBER 13 • MARCH 30, 2007
turn activate Ca2�-dependent signaling pathways that couldtrigger necrosis (25, 26, 37). Alternatively, increased intracellu-lar Ca2� could lead to the activation of degradative enzymesand the overload and dysfunction of Ca2� cycling and storagesystems (38). To examinewhether loss of Cn protein could alterthe characteristic increase in membrane permeability seen inmuscular dystrophy, mice were injected with Evans blue dye,which under normal conditions is a membrane-impermeantmolecule and cannot cross into skeletal muscle fibers.Whereasmuscle fromWT (not shown) and singleCnB1fl/f lMLC-cremiceshowed no Evans blue dye uptake, both scgd�/� and scgd�/�
CnB1fl/flMLC-cremice had a similar increase in susceptibility toEvans blue dye uptake (Fig. 5A). This result suggests that theamelioration of muscle disease observed in scgd�/� CnB1fl/flMLC-cremice is not associatedwith a stabilization of the plasmamembrane.Degeneration of myofibers in muscular dystrophy has been
associated with increased indexes of apoptosis, such as TUNEL
and caspase 3 cleavage. Here we performed an analysis of hair-pin 1 in situ ligation tomeasure DNA fragmentation consistentwith apoptosis in histological sections from diaphragms (6weeks old) that were also co-stained for nuclei and myofiberborders. Careful quantitation of total hairpin 1 ligation labelingshowed �0.36–0.99% of myonuclei in WT, scgd�/�, and inscgd�/� that also lacked CnB1 or that contained the Cn*TG(discussed later) (Fig. 5B). There were no significant differencesin labeling rates between the 4 groups. These results suggestthat whereas loss of CnB1 is protective to muscular dystrophyin scgd�/� mice, the mechanism appears to be independent ofapoptosis at this age.Loss of CnA� in scgd�/� Mice Is Sufficient to Reduce Fibrosis
and Improve Cardiac Disease—Mice lacking scgd also showsubstantial disease in cardiacmuscle, a feature ofmany forms ofhuman muscular dystrophy. To determine whether loss of Cnin the heart could also impart a similar beneficial effect as seenin skeletal muscle, hearts from scgd�/� and scgd�/� CnA��/�
micewere examined.Masson trichrome staining of hearts fromscgd�/� CnA��/� mice demonstrated a profound reduction infibrosis (blue staining) compared with scgd�/� hearts, whereassections fromWThearts showed no appreciable staining what-soever (Fig. 6A and data not shown). This finding was con-firmed by quantification of hydroxyproline content, showingthe dramatic reduction of collagen present in scgd�/�
CnA��/� hearts compared with scgd�/� hearts (Fig. 6B). Con-sistent with these results, the reduction in cardiac fractionalshortening characteristically observed in scgd�/� mice wasrestored to nearWT levels by deletion ofCnA� (Fig. 6C). Theseresults suggest that reduced Cn signaling is beneficial in bothdystrophic skeletal and cardiac muscle.
FIGURE 4. Skeletal muscle-specific deletion of CnB1 increases averagefiber area, decreases centrally nucleated fibers, and ameliorates fibrosisin scgd�/� mice. A, representative Masson trichrome staining of paraffin sec-tions from diaphragm of age-matched scgd�/� and scgd�/� CnB1fl/flMLC-cre
mice. B, analysis of average muscle fiber area in the diaphragm of WT,scgd�/�, and scgd�/� CnB1fl/flMLC-cre mice. The number of muscles assayed isshown for each group in the panel. *, p 0.05 versus WT; †, p 0.05 versusscgd�/�. C, quantitation of fibers containing central nuclei in the diaphragmof WT, scgd�/�, and scgd�/� CnB1fl/flMLC-cre mice. The number of musclesassayed is shown for each group in the panel. *, p 0.05 versus WT; †, p 0.05versus scgd�/�. D, analysis of �g of hydroxyproline/mg of tissue for the indi-cated muscle groups from WT, CnB1f l/f lMLC-cre, scgd�/�, and scgd�/� CnB1fl/flMLC-cre mice. The number of muscles assayed is shown for each group in thepanel. *, p 0.05 versus WT; †, p 0.05 versus scgd�/�.
FIGURE 5. Assessment of plasma membrane integrity and apoptosis.A, confocal fluorescence of Evans blue dye uptake in the diaphragm ofCnB1f l/f lMLC-cre, scgd�/�, and scgd�/� CnB1fl/flMLC-cre mice. Histological sec-tions from three separate mice are shown (1–3). Evans blue dye uptake fluo-resces red, whereas the green fluorescence results from wheat germ aggluti-nin-fluorescein isothiocyanate staining to show membranes. B, percentage ofnuclei with hairpin 1 oligonucleotide ligation in the indicated genotypes ofmice from diaphragm histological sections (6 weeks of age). The numberof mice analyzed in each group is present under “Materials and Methods.”Approximately 1000 –3000 nuclei were counted for each animal. There wereno significant differences between the groups.
Calcineurin Deletion Improves Muscular Dystrophy
MARCH 30, 2007 • VOLUME 282 • NUMBER 13 JOURNAL OF BIOLOGICAL CHEMISTRY 10073
Characterization of Constitutively Active CnTransgenicMice—Having determined that loss of Cn expression is advantageousin dystrophic skeletal and cardiac muscle, we next wanted toassess the effects of increasedCn activity in the disease progres-sion of scgd�/� mice. Mice containing a Cn*Tg (MCK pro-moter) were used, which have high levels of expression in skel-etal muscle and slightly lower levels in the heart (16). Skeletalmuscles of these mice show increases in slowmuscle fibers andminor changes in muscle mass (16, 39). Expression of theCn*Tg protein was evident in a variety of normal skeletal mus-cles and heart (Fig. 7A). To avoid confusion, it should be notedthat the Cn* protein co-migrates with background proteins incertain muscle types, such as heart, that nonspecifically bindthe pan-CnA antibody and appear as a doublet (Fig. 7A). As acontrol, expression of the Cn*Tg was observed to be equallyrobust when expressed in the scgd�/� background (Fig. 7B).
Because expression of Cn is a well known inducer of cardiachypertrophy (22), we examined the hearts of these mice for anyincrease inmass. Even thoughmoderately expressed, theMCK-Cn* transgene was sufficient to induce a 41.3% increase in theheart-to-body weight ratio compared with WT (data notshown). When crossed with the NFAT-luciferase transgenic
mice, Cn*Tg mice showed a 100% increase in activity in theheart comparedwithWTmice (Fig. 7C). However, as presentedearlier, dystrophic heart muscle showed a significant reductionin NFAT-luciferase activity, even in the presence of the MCK-Cn* transgene (Fig. 7C) (see “Discussion”).To investigate downstreameffects of Cn-NFAT signaling, we
quantified the mRNA levels of utrophin A andMEF2C. As pre-viously observed, utrophin A mRNA levels were increased byCn activation in skeletal muscle, as was MEF2C. Utrophin A,but not MEF2C, was also up-regulated by the scgd null muta-tion, although the combination of the Cn*Tg with the scgd nullmutation produced a down-regulation relative to either allele(Fig. 7D).Expression of Cn* in scgd�/� Mice Does Not Increase Fibrosis
or Affect Fiber Area—In light of our data suggesting that loss ofCn is beneficial to scgd�/�muscle, we analyzed the histopathol-ogy of diaphragmmuscle from Cn*Tgmice. Masson trichromestaining of diaphragm sections from mice 24 weeks of age didnot show obvious differences in fibrotic tissue replacementbetween scgd�/� Cn*Tg and scgd�/� mice (Fig. 8A). Onceagain, WT histological sections were omitted because they
FIGURE 6. Loss of Cn reduces fibrosis and improves fractional shorteningin hearts of dystrophic mice. A, representative Masson trichrome staining ofparaffin sections from separate hearts of age-matched scgd�/� and scgd�/�
CnA��/� mice. Fibrosis is shown in blue. B, quantification of �g ofhydroxyproline/mg of tissue from the hearts for the indicated groups of mice.The number of hearts assayed is shown for each group in the bars. *, p 0.05versus WT; †, p 0.05 versus scgd�/�. C, echocardiographic assessment offractional shortening in the hearts of the indicated groups of mice. The num-ber of hearts measured is shown for each group in the bars. *, p 0.05 versusWT; †, p 0.05 versus scgd�/�.
FIGURE 7. Assessment of Cn* transgene expression and activity. A, West-ern blot analysis using a pan-CnA antibody to show expression of the Cn*protein in the indicated tissue of WT and MCK-Cn*Tg mice. Each lane wasgenerated from a separate muscle/animal. B, Western blot analysis of Cn*protein expression in dystrophic muscle (from scgd�/� mice) shows robustexpression of the transgene. Each lane was generated from a separate mus-cle. C, analysis of NFAT-luciferase reporter activity in hearts of mice from theindicated groups expressing the Cn*Tg. The number of hearts measured isshown for each group in the panel. *, p 0.05 versus background (bkgrnd); †,p 0.05 versus WT or Cn*Tg. D, real time PCR for utrophin A and MEF2C mRNAfrom skeletal muscle of the indicated genotypes. *, p 0.05 versus WT; †, p 0.05 versus Cn*Tg; #, p 0.05 versus scgd�/�.
Calcineurin Deletion Improves Muscular Dystrophy
10074 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 282 • NUMBER 13 • MARCH 30, 2007
showed no fibrotic staining (data not shown).However, areas ofmuscle fiber loss with suggestive fatty tissue replacement wereapparent throughout nearly every muscle section in scgd�/�
Cn*Tgmice. Although fatty tissue replacement ofmuscle is notan uncommon phenomenon in dystrophic muscle, its appear-ance was far more prevalent in the presence of the Cn*Tg (Fig.8A). Analysis of average fiber area of muscle revealed theexpected decrease in scgd�/� mice compared with WT, whichwas similarly decreased in the presence of the Cn*Tg (Fig. 8B).Quantification of hydroxyproline content confirmed the Mas-son trichrome sections, establishing that there is no significantdifference in diaphragm collagen content between scgd�/� andscgd�/� Cn*Tg mice (Fig. 8C). This conclusion is supported byanalysis ofhistopathology in thequadricepsmuscleof scgd�/� andscgd�/� Cn*Tg mice. Whereas fibrosis was similar, quadricepsshowed a noticeable increase in areas of profound fiber loss andsuggestive fatty tissue replacement in scgd�/� Cn*Tg mice com-paredwith scgd�/�micealone (Fig. 9,A andB). Sections fromWTquadricepsshowednosuchevents (datanot shown).Thus, expres-
sion of Cn* in the scgd�/� background mildly enhances histopa-thology through a specific increase in fiber loss.Increased Cardiac Fibrosis and Worsened Heart Disease in
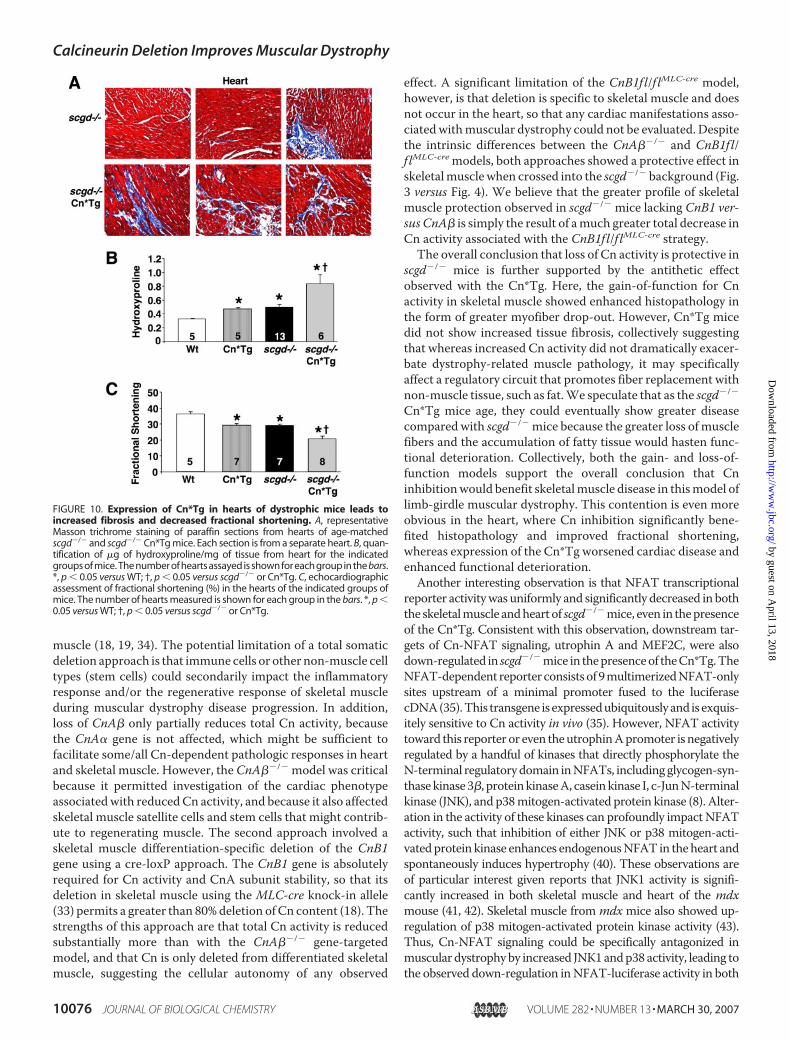
scgd�/� Cn*Tg Mice—In contrast to skeletal muscle, expres-sion of the Cn*Tg in the heart is sufficient to induce hypertro-phy and pathology by itself (22). Analysis ofMasson trichrome-stained sections showed significantly more fibrosis andmyocyte disorganization in the hearts of scgd�/� Cn*Tg micecompared with scgd�/� mice alone, whereas WT sectionsshowed no visual fibrosis (Fig. 10A, and data not shown). Quan-tification of hydroxyproline content confirmed this observa-tion, showing a 148.6% increase in collagen content in scgd�/�
Cn*Tg mice over WT hearts and an 80.4% increase overscgd�/� hearts (Fig. 10B). Furthermore, echocardiographicanalysis of cardiac fractional shortening revealed that bothscgd�/� hearts and Cn*Tg hearts have a comparable decrease,whereas hearts of scgd�/� Cn*Tg mice showed an even greaterdecrease (Fig. 10C). In conclusion, activation of Cn in cardiacmuscle significantly worsens the pathology associated withscgd�/�-dependent muscular dystrophy.
DISCUSSION
The most dramatic result of the current study is that geneticdisruption of Cn reduced histopathology in both skeletal mus-cle and hearts of mice lacking scgd. Two genetic approacheswere used to reduce Cn activity in vivo as a means of assessingits role in scgd-deficient muscular dystrophy. The first involveda total somatic deletion of the CnA� gene, which comprises atleast 50% or more of the total Cn activity in heart and skeletal
FIGURE 8. Mild increase in diaphragm histopathology in scgd�/� Cn*Tgmice. A, representative Masson trichrome staining of paraffin sections fromseparate diaphragms of age-matched scgd�/� and scgd�/� Cn*Tg mice.B, analysis of the average fiber area in the diaphragm of WT, scgd�/�, andscgd�/� Cn*Tg mice. The number of muscles measured is shown for eachgroup in the bars. *, p 0.05 versus WT. C, quantification of �g of hydroxypro-line/mg of tissue from diaphragm for the indicated groups of mice. The num-ber of muscles assayed is shown for each group in the bars. *, p 0.05 versusWT.
FIGURE 9. Histological analysis of quadriceps muscles reveals increasedfatty tissue replacement in scgd�/� Cn*Tg mice. A, representative Massontrichrome staining of paraffin sections from quadriceps of age-matchedscgd�/� and scgd�/� Cn*Tg mice. B, histological sections from quadricepsmuscle were digitized and the area not occupied by muscle fiber was calcu-lated for the indicated genotype. *, p 0.05 versus Cn*Tg or scgd�/�. Numberof samples analyzed was 12 for WT, 6 for Cn*Tg, 10 for scgd�/�, and 6 forscgd�/� Cn*TG.
Calcineurin Deletion Improves Muscular Dystrophy
MARCH 30, 2007 • VOLUME 282 • NUMBER 13 JOURNAL OF BIOLOGICAL CHEMISTRY 10075
muscle (18, 19, 34). The potential limitation of a total somaticdeletion approach is that immune cells or other non-muscle celltypes (stem cells) could secondarily impact the inflammatoryresponse and/or the regenerative response of skeletal muscleduring muscular dystrophy disease progression. In addition,loss of CnA� only partially reduces total Cn activity, becausethe CnA� gene is not affected, which might be sufficient tofacilitate some/all Cn-dependent pathologic responses in heartand skeletal muscle. However, theCnA��/� model was criticalbecause it permitted investigation of the cardiac phenotypeassociatedwith reducedCn activity, and because it also affectedskeletal muscle satellite cells and stem cells that might contrib-ute to regenerating muscle. The second approach involved askeletal muscle differentiation-specific deletion of the CnB1gene using a cre-loxP approach. The CnB1 gene is absolutelyrequired for Cn activity and CnA subunit stability, so that itsdeletion in skeletal muscle using the MLC-cre knock-in allele(33) permits a greater than 80%deletion ofCn content (18). Thestrengths of this approach are that total Cn activity is reducedsubstantially more than with the CnA��/� gene-targetedmodel, and that Cn is only deleted from differentiated skeletalmuscle, suggesting the cellular autonomy of any observed
effect. A significant limitation of the CnB1fl/f lMLC-cre model,however, is that deletion is specific to skeletal muscle and doesnot occur in the heart, so that any cardiac manifestations asso-ciatedwithmuscular dystrophy could not be evaluated. Despitethe intrinsic differences between the CnA��/� and CnB1fl/f lMLC-cremodels, both approaches showed a protective effect inskeletalmusclewhen crossed into the scgd�/� background (Fig.3 versus Fig. 4). We believe that the greater profile of skeletalmuscle protection observed in scgd�/� mice lacking CnB1 ver-sus CnA� is simply the result of amuch greater total decrease inCn activity associated with the CnB1fl/f lMLC-cre strategy.The overall conclusion that loss of Cn activity is protective in
scgd�/� mice is further supported by the antithetic effectobserved with the Cn*Tg. Here, the gain-of-function for Cnactivity in skeletal muscle showed enhanced histopathology inthe form of greater myofiber drop-out. However, Cn*Tg micedid not show increased tissue fibrosis, collectively suggestingthat whereas increased Cn activity did not dramatically exacer-bate dystrophy-related muscle pathology, it may specificallyaffect a regulatory circuit that promotes fiber replacement withnon-muscle tissue, such as fat.We speculate that as the scgd�/�
Cn*Tg mice age, they could eventually show greater diseasecomparedwith scgd�/� mice because the greater loss ofmusclefibers and the accumulation of fatty tissue would hasten func-tional deterioration. Collectively, both the gain- and loss-of-function models support the overall conclusion that Cninhibitionwould benefit skeletalmuscle disease in thismodel oflimb-girdle muscular dystrophy. This contention is even moreobvious in the heart, where Cn inhibition significantly bene-fited histopathology and improved fractional shortening,whereas expression of the Cn*Tg worsened cardiac disease andenhanced functional deterioration.Another interesting observation is that NFAT transcriptional
reporter activitywas uniformly and significantly decreased in boththe skeletalmuscle andheartof scgd�/�mice, even in thepresenceof the Cn*Tg. Consistent with this observation, downstream tar-gets of Cn-NFAT signaling, utrophin A and MEF2C, were alsodown-regulated in scgd�/�mice in thepresenceof theCn*Tg.TheNFAT-dependent reporterconsistsof9multimerizedNFAT-onlysites upstream of a minimal promoter fused to the luciferasecDNA(35).This transgene isexpressedubiquitouslyand isexquis-itely sensitive to Cn activity in vivo (35). However, NFAT activitytoward this reporter or even theutrophinApromoter is negativelyregulated by a handful of kinases that directly phosphorylate theN-terminal regulatorydomain inNFATs, includingglycogen-syn-thasekinase3�, proteinkinaseA, caseinkinase I, c-JunN-terminalkinase (JNK), and p38mitogen-activated protein kinase (8). Alter-ation in the activity of these kinases can profoundly impactNFATactivity, such that inhibition of either JNK or p38 mitogen-acti-vatedprotein kinase enhances endogenousNFAT in theheart andspontaneously induces hypertrophy (40). These observations areof particular interest given reports that JNK1 activity is signifi-cantly increased in both skeletal muscle and heart of the mdxmouse (41, 42). Skeletal muscle frommdxmice also showed up-regulation of p38 mitogen-activated protein kinase activity (43).Thus, Cn-NFAT signaling could be specifically antagonized inmuscular dystrophyby increased JNK1andp38 activity, leading tothe observed down-regulation inNFAT-luciferase activity in both
FIGURE 10. Expression of Cn*Tg in hearts of dystrophic mice leads toincreased fibrosis and decreased fractional shortening. A, representativeMasson trichrome staining of paraffin sections from hearts of age-matchedscgd�/� and scgd�/� Cn*Tg mice. Each section is from a separate heart. B, quan-tification of �g of hydroxyproline/mg of tissue from heart for the indicatedgroups of mice. The number of hearts assayed is shown for each group in the bars.*, p 0.05 versus WT; †, p 0.05 versus scgd�/� or Cn*Tg. C, echocardiographicassessment of fractional shortening (%) in the hearts of the indicated groups ofmice. The number of hearts measured is shown for each group in the bars. *, p 0.05 versus WT; †, p 0.05 versus scgd�/� or Cn*Tg.
Calcineurin Deletion Improves Muscular Dystrophy
10076 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 282 • NUMBER 13 • MARCH 30, 2007
heart and skeletal muscle of scgd�/� mice. If Cn activity is indeeddetrimental to muscular dystrophy disease progression, such amechanism of down-regulated NFAT activity could represent aprotective compensatory effect.It is also interesting that calcineurin activity is reduced in
skeletal muscle disease, whereas inmany forms of heart diseaseactivity and/or protein levels are significantly increased (indeedit is increased in the hearts of scgd�/� mice). However, thisdichotomous observation between heart and skeletal musclelikely reflects the highly specialized role that calcineurin playsin each tissue. For example, in the heart calcineurin directlyregulates myocyte growth, yet overexpression of the same acti-vated calcineurin transgene in skeletal muscle had no effect ongrowth or fiber size (16, 22). In contrast, calcineurin functionsas a critical regulator of fiber-type switching in skeletal muscle,whereas the heart lacks such a biologic process (16). Giventhese dramatically different functional roles, it is not surprisingthat Cn activity could be antithetically regulated during diseasein cardiac versus skeletal muscle.Our observation that Cn inhibition largely rescues skeletal
muscle and heart disease manifestations in the scgd�/� mouseis in dramatic contrast to conclusions reached in the mdxmouse. For example, muscles from mdx mice expressing thesame MCK-Cn* transgene that we used showed significantreductions in the extent of central nucleation, fiber size varia-bility, inflammation, and an improvement in membrane integ-rity (27). These results suggested that Cn activation, not inhibi-tion, is protective in dystrophic muscle. A potential reasonunderlying this difference likely relates to inherent differencesin themdx and scgd�/� mouse models themselves. Indeed, wehave previously observed that myostatin inhibition or deletionhas little effect or may be even detrimental to muscular dystro-phy in scgd�/� mice (44), whereas disease in mdx mice is par-tially rescued by myostatin inhibition/deletion (45). Both mdxand scgd�/� mice have different disease characteristics thatrelate to a different phenotypic spectrum in human musculardystrophy. For example,mdxmice are thought to have greaterregenerative capacity in their skeletal muscle compared withdystrophic humans (46, 47). Moreover, progression of diseasein human Duchenne muscular dystrophy patients leads to sig-nificantly shortened life expectancy, whereas mdxmice have anear-normal lifespan. As many as 50% of humans with Duch-ennemuscular dystrophy also develop severe and life-threaten-ing cardiomyopathy, whereas themdxmodel shows onlyminorcardiac pathology (48). Loss of �-sarcoglycan in the mousemodels a rare form of human limb-girdle muscular dystrophyreferred to as LGMD2F. However, loss of any of the proteinswithin the DGC leads to a similar disease mechanism thatinvolves destabilization of the sarcolemma and influx of Ca2�,which induces degeneration of myofibers. Thus, whereas thescgd�/� mouse employed here corresponds to a rare form ofhuman disease, its use to dissect disease progression is likelyrelevant tomany forms ofmuscular dystrophy. Indeed, scgd�/�
mice show both skeletal muscle and significant cardiac muscledisease, they show fairly widespread pathology in all skeletalmuscles examined that is reminiscent of the pathologyobserved in severe forms of human muscular dystrophy, and
they have foreshortened lifespan as seen in many human mus-cular dystrophies such as Duchenne (30).Anotherpossibility is thatdifferentmembranealterationsoccur
between themdx and scgd�/�mousemodels such thatCnactivityis differentially affected. The hypothesis has been proposed thatthe DGC and accessory structural proteins can anchor and regu-late the activation of various signaling effectors. Hence, it is possi-ble that loss of �-sarcoglycan and the other sarcoglycans with it,preferentially affects Cn signaling or an accessory signaling path-way that modulates Cn in a manner that is distinct from a loss indystrophin alone. As a final consideration, it should also be notedthat the genetic background of the mdx mouse is C57-based,whereas the scgd�/� mouse is 129Sv-based, which could alsoaccount for some variability in phenotype.ThemechanismwherebyCn inhibition protects against skel-
etal muscle and heart muscle disease in the scgd�/� back-ground is uncertain, although some evidence supports thehypothesis that cell death or limited necrosis is involved. Forexample, studies conducted in neurons, lymphocytes, andtumor cell lines have shown both pro- and anti-apoptotic rolesfor Cn activation (49). In the heart, transgenic mice expressingactivated CnA were protected from cell death following ische-mia-reperfusion injury, whereas genetic disruption of theCnA� gene enhanced myocyte death induced by ischemia-reperfusion injury (50, 51). In contrast to these studies, isopro-terenol or aldosterone stimulation promoted apoptosis in asso-ciation with Cn activation, suggesting that Cn could alsopromote cell death in cardiac myocytes (52, 53). Mechanisti-cally, Cn can localize to themitochondria in fibroblasts in asso-ciation with FK506-binding protein 38, resulting in Bcl-2 andBcl-xl redistribution and effects on cell death (54). Cn has alsobeen implicated as a direct inducer of apoptosis in primary hip-pocampal neurons by dephosphorylating the pro-apoptoticfactor Bad, resulting in its induction of mitochondrial-depend-ent cell death (55). More recently, we also observed that Cncould serve a pro-apoptotic role in cardiac myocytes and fibro-blasts through a direct regulatory effect on apoptosis signal-regulating kinase 1 (56). Whereas it has yet to be determinedwhether Cn signaling regulates cell death/necrosis in skeletalmuscle myofibers, we did observe less central nucleation andinflammation in scgd�/� mice lacking the CnB1 gene specifi-cally in skeletal muscle. However, direct assessment of apopto-sis with hairpin 1 ligation failed to reveal that skeletal musclefibers die by such a mechanism in scgd�/� mice. Instead, wehave observed a prominent increase in necrosis as the potentialmechanism underlyingmyofiber loss in scgd�/� mice.4 Thus, itis possible that Cn activation can promote myofiber death byenhancing necrosis in a model of limb-girdle muscular dystro-phy, although it may not in the mdx model if myofiber degen-eration proceeds through a different mechanism (apoptosis).Regardless of the exactmechanism, our results indicate that Cninhibition may provide some benefit in select types of muscledisease, such as limb-girdle muscular dystrophy. This interpre-tation might also suggest that CsA could be potentially of ben-efit as well (at least some aspects of the disease). However, cau-
4 D. P. Millay and J. D. Molkentin, unpublished observation.
Calcineurin Deletion Improves Muscular Dystrophy
MARCH 30, 2007 • VOLUME 282 • NUMBER 13 JOURNAL OF BIOLOGICAL CHEMISTRY 10077
tion is warranted because CsA is not a pure inhibitor ofcalcineurin, as it can also alter tissue necrosis by directly regu-lating mitochondrial permeability pore transition (57). More-over, previous clinical trials with CsA in humans with Duch-enne and Becker muscular dystrophy were equivocal, althoughmost were underpowered. Thus, whereas Cn may offer sometherapeutic value, new inhibitors may be needed or dramati-cally different time courses used in a preventative schemerather than for treating more advanced disease.
Acknowledgment—We thank Wyeth Research for support in finaliz-ing revisions to this manuscript.
REFERENCES1. Crabtree, G. R. (2001) J. Biol. Chem. 276, 2313–23162. Klee, C. B., Ren, H., andWang, X. (1998) J. Biol. Chem. 273, 13367–133703. Rusnak, F., and Mertz, P. (2000) Physiol. Rev. 80, 1483–15214. Buttini, M., Limonta, S., Luyten, M., and Boddeke, H. (1995)Histochem. J.
27, 291–2995. Jiang, H., Xiong, F., Kong, S., Ogawa, T., Kobayashi, M., and Liu, J. O.
(1997)Mol. Immunol. 34, 663–6696. Muramatsu, T., Giri, P. R., Higuchi, S., and Kincaid, R. L. (1992) Proc. Natl.
Acad. Sci. U. S. A. 89, 529–5337. Takaishi, T., Saito, N., Kuno, T., and Tanaka, C. (1991) Biochem. Biophys.
Res. Commun. 174, 393–3988. Hogan, P. G., Chen, L., Nardone, J., and Rao, A. (2003) Genes Dev. 17,
2205–22329. Abbott, K. L., Friday, B. B., Thaloor, D., Murphy, T. J., and Pavlath, G. K.
(1998)Mol. Biol. Cell 9, 2905–291610. Hoey, T., Sun, Y. L., Williamson, K., and Xu, X. (1995) Immunity 2,
Ventura-Clapier, R. (2000) J. Biol. Chem. 275, 19653–1966012. Chin, E. R., Olson, E. N., Richardson, J. A., Yang, Q., Humphries, C., Shel-
ton, J. M., Wu, H., Zhu, W., Bassel-Duby, R., and Williams, R. S. (1998)Genes Dev. 12, 2499–2509
13. Delling, U., Tureckova, J., Lim, H. W., De Windt, L. J., Rotwein, P., andMolkentin, J. D. (2000)Mol. Cell. Biol. 20, 6600–6611
14. McCullagh, K. J., Calabria, E., Pallafacchina, G., Ciciliot, S., Serrano, A. L.,Argentini, C., Kalhovde, J. M., Lomo, T., and Schiaffino, S. (2004) Proc.Natl. Acad. Sci. U. S. A. 101, 10590–10595
15. Miyazaki, M., Hitomi, Y., Kizaki, T., Ohno, H., Haga, S., and Takemasa, T.(2004) J. Physiol. Pharmacol. 55, 751–764
16. Naya, F. J., Mercer, B., Shelton, J., Richardson, J. A., Williams, R. S., andOlson, E. N. (2000) J. Biol. Chem. 275, 4545–4548
17. Oh,M., Rybkin, I. I., Copeland, V., Czubryt,M. P., Shelton, J.M., van Rooij,E., Richardson, J. A., Hill, J. A., De Windt, L. J., Bassel-Duby, R., Olson,E. N., and Rothermel, B. A. (2005)Mol. Cell. Biol. 25, 6629–6638
18. Parsons, S. A., Millay, D. P., Wilkins, B. J., Bueno, O. F., Tsika, G. L.,Neilson, J. R., Liberatore, C.M., Yutzey, K. E., Crabtree, G. R., Tsika, R.W.,and Molkentin, J. D. (2004) J. Biol. Chem. 279, 26192–26200
19. Parsons, S. A.,Wilkins, B. J., Bueno, O. F., andMolkentin, J. D. (2003)Mol.Cell. Biol. 23, 4331–4343
20. Serrano, A. L., Murgia, M., Pallafacchina, G., Calabria, E., Coniglio, P.,Lomo, T., and Schiaffino, S. (2001) Proc. Natl. Acad. Sci. U. S. A. 98,13108–13113
21. Wu, H., Naya, F. J., McKinsey, T. A., Mercer, B., Shelton, J. M., Chin, E. R.,Simard, A. R., Michel, R. N., Bassel-Duby, R., Olson, E. N., and Williams,R. S. (2000) EMBO J. 19, 1963–1973
22. Molkentin, J. D., Lu, J. R., Antos, C. L., Markham, B., Richardson, J., Rob-bins, J., Grant, S. R., and Olson, E. N. (1998) Cell 93, 215–228
23. Kegley, K. M., Gephart, J., Warren, G. L., and Pavlath, G. K. (2001) Dev.Biol. 232, 115–126
24. Horsley, V., Friday, B. B., Matteson, S., Kegley, K. M., Gephart, J., andPavlath, G. K. (2001) J. Cell Biol. 153, 329–338
25. Durbeej, M., and Campbell, K. P. (2002) Curr. Opin. Genet. Dev. 12,349–361
26. Lapidos, K. A., Kakkar, R., and McNally, E. M. (2004) Circ. Res. 94,1023–1031
27. Chakkalakal, J. V., Harrison, M. A., Carbonetto, S., Chin, E., Michel, R. N.,and Jasmin, B. J. (2004) Hum. Mol. Genet. 13, 379–388
28. Stupka, N., Gregorevic, P., Plant, D. R., and Lynch, G. S. (2004) Acta Neu-ropathol. 107, 299–310
29. St-Pierre, S. J., Chakkalakal, J. V., Kolodziejczyk, S. M., Knudson, J. C.,Jasmin, B. J., and Megeney, L. A. (2004) FASEB J. 18, 1937–1939
30. Hack, A. A., Lam, M. Y., Cordier, L., Shoturma, D. I., Ly, C. T., Hadhazy,M. A., Hadhazy, M. R., Sweeney, H. L., and McNally, E. M. (2000) J. CellSci. 113, 2535–2544
31. Coral-Vazquez, R., Cohn, R. D., Moore, S. A., Hill, J. A., Weiss, R. M.,Davisson, R. L., Straub, V., Barresi, R., Bansal, D., Hrstka, R. F.,Williamson,R., and Campbell, K. P. (1999) Cell 98, 465–474
32. Neilson, J. R., Winslow, M. M., Hur, E. M., and Crabtree, G. R. (2004)Immunity 20, 255–266
33. Bothe, G. W., Haspel, J. A., Smith, C. L., Wiener, H. H., and Burden, S. J.(2000) Genesis 26, 165–166
34. Bueno, O. F., Wilkins, B. J., Tymitz, K. M., Glascock, B. J., Kimball, T. F.,Lorenz, J. N., andMolkentin, J. D. (2002) Proc. Natl. Acad. Sci. U. S. A. 99,4586–4591
35. Wilkins, B. J., Dai, Y. S., Bueno, O. F., Parsons, S. A., Xu, J., Plank, D. M.,Jones, F., Kimball, T. R., andMolkentin, J. D. (2004)Circ. Res. 94, 110–118
36. Woessner, J. F., Jr. (1961) Arch. Biochem. Biophys. 93, 440–44737. Allen, D. G., Whitehead, N. P., and Yeung, E. W. (2005) J. Physiol. 567,
723–73538. Berchtold, M. W., Brinkmeier, H., and Muntener, M. (2000) Physiol. Rev.
80, 1215–126539. Talmadge, R. J., Otis, J. S., Rittler, M. R., Garcia, N. D., Spencer, S. R., Lees,
S. J., and Naya, F. J. (2004) BMC Cell Biol. 5, 2840. Molkentin, J. D. (2004) Cardiovasc. Res. 63, 467–47541. Megeey, L. A., Kablar, B., Perry, R. L., Ying, C.,May, L., andRudnicki,M.A.
(1999) Proc. Natl. Acad. Sci. U. S. A. 96, 220–22542. Kolodziejczyk, S.M.,Walsh, G. S., Balazsi, K., Seale, P., Sandoz, J., Hierlihy,
A.M., Rudnicki,M.A., Chamberlain, J. S.,Miller, F. D., andMegeney, L. A.(2001) Curr. Biol. 11, 1278–1282
43. Lang, J. M., Esser, K. A., and Dupont-Versteegden, E. E. (2004) Exp. Biol.Med. (Maywood) 229, 503–511
44. Parsons, S. A.,Millay, D. P., Sargent,M.A.,McNally, E.M., andMolkentin,J. D. (2006) Am. J. Pathol. 168, 1975–1985
45. Patel, K., and Amthor, H. (2005) Neuromusc. Disord. 15, 117–12646. Tanabe, Y., Esaki, K., andNomura, T. (1986)Acta Neuropathol. 69, 91–9547. Anderson, J. E., Ovalle, W. K., and Bressler, B. H. (1987) Anat. Rec. 219,
243–25748. Finsterer, J., and Stollberger, C. (2003) Cardiology 99, 1–1949. Baines, C. P., and Molkentin, J. D. (2005) J. Mol. Cell. Cardiol. 38, 47–6250. Bueno, O. F., Lips, D. J., Kaiser, R. A., Wilkins, B. J., Dai, Y. S., Glascock,
B. J., Klevitsky, R., Hewett, T. E., Kimball, T. R., Aronow, B. J., Doevendans,P. A., and Molkentin, J. D. (2004) Circ. Res. 94, 91–99
51. DeWindt, L. J., Lim, H.W., Taigen, T.,Wencker, D., Condorelli, G., Dorn,G.W., 2nd, Kitsis, R. N., andMolkentin, J. D. (2000)Circ. Res. 86, 255–263
52. Mano, A., Tatsumi, T., Shiraishi, J., Keira, N., Nomura, T., Takeda, M.,Nishikawa, S., Yamanaka, S., Matoba, S., Kobara, M., Tanaka, H.,Shirayama, T., Takamatsu, T., Nozawa, Y., and Matsubara, H. (2004) Cir-culation 110, 317–323
53. Saito, S., Hiroi, Y., Zou, Y., Aikawa, R., Toko, H., Shibasaki, F., Yazaki, Y.,Nagai, R., and Komuro, I. (2000) J. Biol. Chem. 275, 34528–34533
54. Shirane, M., and Nakayama, K. I. (2003) Nat. Cell. Biol. 5, 28–3755. Wang, H. G., Pathan, N., Ethell, I. M., Krajewski, S., Yamaguchi, Y., Shi-
basaki, F., McKeon, F., Bobo, T., Franke, T. F., and Reed, J. C., (1999)Science 284, 339–343
56. Liu, Q.,Wilkins, B. J., Lee, Y. J., Ichijo, H., andMolkentin, J. D. (2006)Mol.Cell. Biol. 26, 3785–3797
57. Waldmeier, P. C., Zimmermann, K., Qian, T., Tintelnot-Blomley, M., andLemasters, J. J. (2003) Curr. Med. Chem. 10, 1485–1506
Calcineurin Deletion Improves Muscular Dystrophy
10078 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 282 • NUMBER 13 • MARCH 30, 2007





















![ORIGINAL ARTICLE fic Glucose-Induced Control of Insulin ... · cellular sources. Calcineurin inhibitors (FK506, cyclosporin A, and a peptide calcineurin inhibitor [CAIN]) abolished](https://static.documents.pub/doc/80x56/5e35e15c9ec8741478479276/original-article-ic-glucose-induced-control-of-insulin-cellular-sources-calcineurin.jpg)





