Glutamatergic Postsynaptic Density Protein Dysfunctions in Synaptic Plasticity and Dendritic Spines Morphology: Relevance to Schizophrenia and Other Behavioral Disorders Pathophysiology, and Implications for Novel Therapeutic Approaches Andrea de Bartolomeis & Gianmarco Latte & Carmine Tomasetti & Felice Iasevoli Received: 6 July 2013 /Accepted: 13 August 2013 # Springer Science+Business Media New York 2013 Abstract Emerging researches point to a relevant role of postsynaptic density (PSD) proteins, such as PSD-95, Homer, Shank, and DISC-1, in the pathophysiology of schizo- phrenia and autism spectrum disorders. The PSD is a thick- ness, detectable at electronic microscopy, localized at the postsynaptic membrane of glutamatergic synapses, and made by scaffolding proteins, receptors, and effector proteins; it is considered a structural and functional crossroad where multi- ple neurotransmitter systems converge, including the dopami- nergic, serotonergic, and glutamatergic ones, which are all implicated in the pathophysiology of psychosis. Decreased PSD-95 protein levels have been reported in postmortem brains of schizophrenia patients. Variants of Homer1, a key PSD protein for glutamate signaling, have been associated with schizophrenia symptoms severity and therapeutic re- sponse. Mutations in Shank gene have been recognized in autism spectrum disorder patients, as well as reported to be associated to behaviors reminiscent of schizophrenia symp- toms when expressed in genetically engineered mice. Here, we provide a critical appraisal of PSD proteins role in the pathophysiology of schizophrenia and autism spectrum disor- ders. Then, we discuss how antipsychotics may affect PSD proteins in brain regions relevant to psychosis pathophysiolo- gy, possibly by controlling synaptic plasticity and dendritic spine rearrangements through the modulation of glutamate- related targets. We finally provide a framework that may explain how PSD proteins might be useful candidates to develop new therapeutic approaches for schizophrenia and related disorders in which there is a need for new biological treatments, especially against some symptom domains, such as negative symptoms, that are poorly affected by current antipsychotics. Keywords Psychosis . Antipsychotics . Synapse . PSD-95 . Homer . Shank Introduction Schizophrenia is a complex disorder affecting nearly 1 % of the general population. A large amount of evidence suggests that schizophrenia is caused by aberrant synaptic plasticity and metaplasticity. Perturbation of regular dendritic spines architecture and function has been described in the disease [1–3]. Multiple neurotransmitter systems have been implicat- ed in schizophrenia pathophysiology, and strong evidence points out abnormalities in dopamine, glutamate, and seroto- nin neurotransmission [4, 5]. Signaling pathways activated by these neurotransmitters converge on the postsynaptic density (PSD), which is considered as a structural and functional multi-protein crossroad. PSD is an electron-dense thickening localized under postsynaptic membranes, which comprises several hundred proteins and particularly characterizes large excitatory glutamatergic synapses [6]. PSD proteins are dis- tributed in highly organized macromolecular complexes that process, integrate, and converge synaptic signals to the nucle- us [7]. Overall, PSD proteins are involved in synaptic A. de Bartolomeis (*) : G. Latte : C. Tomasetti : F. Iasevoli Laboratory of Molecular and Translational Psychiatry, Unit of Treatment Resistant Psychosis, Department of Neuroscience, Reproductive and Odontostomatologic Sciences, Section of Psychiatry, University School of Medicine “Federico II”, Via Pansini 5, 80131 Naples, Italy e-mail: [email protected]Mol Neurobiol DOI 10.1007/s12035-013-8534-3
Transcript
Glutamatergic Postsynaptic Density Protein Dysfunctionsin Synaptic Plasticity and Dendritic Spines Morphology:Relevance to Schizophrenia and Other Behavioral DisordersPathophysiology, and Implications for NovelTherapeutic Approaches
Andrea de Bartolomeis & Gianmarco Latte &
Carmine Tomasetti & Felice Iasevoli
Received: 6 July 2013 /Accepted: 13 August 2013# Springer Science+Business Media New York 2013
Abstract Emerging researches point to a relevant role ofpostsynaptic density (PSD) proteins, such as PSD-95,Homer, Shank, and DISC-1, in the pathophysiology of schizo-phrenia and autism spectrum disorders. The PSD is a thick-ness, detectable at electronic microscopy, localized at thepostsynaptic membrane of glutamatergic synapses, and madeby scaffolding proteins, receptors, and effector proteins; it isconsidered a structural and functional crossroad where multi-ple neurotransmitter systems converge, including the dopami-nergic, serotonergic, and glutamatergic ones, which are allimplicated in the pathophysiology of psychosis. DecreasedPSD-95 protein levels have been reported in postmortembrains of schizophrenia patients. Variants of Homer1, a keyPSD protein for glutamate signaling, have been associatedwith schizophrenia symptoms severity and therapeutic re-sponse. Mutations in Shank gene have been recognized inautism spectrum disorder patients, as well as reported to beassociated to behaviors reminiscent of schizophrenia symp-toms when expressed in genetically engineered mice. Here,we provide a critical appraisal of PSD proteins role in thepathophysiology of schizophrenia and autism spectrum disor-ders. Then, we discuss how antipsychotics may affect PSDproteins in brain regions relevant to psychosis pathophysiolo-gy, possibly by controlling synaptic plasticity and dendriticspine rearrangements through the modulation of glutamate-
related targets. We finally provide a framework that mayexplain how PSD proteins might be useful candidates todevelop new therapeutic approaches for schizophrenia andrelated disorders in which there is a need for new biologicaltreatments, especially against some symptom domains, suchas negative symptoms, that are poorly affected by currentantipsychotics.
Schizophrenia is a complex disorder affecting nearly 1 % ofthe general population. A large amount of evidence suggeststhat schizophrenia is caused by aberrant synaptic plasticityand metaplasticity. Perturbation of regular dendritic spinesarchitecture and function has been described in the disease[1–3]. Multiple neurotransmitter systems have been implicat-ed in schizophrenia pathophysiology, and strong evidencepoints out abnormalities in dopamine, glutamate, and seroto-nin neurotransmission [4, 5]. Signaling pathways activated bythese neurotransmitters converge on the postsynaptic density(PSD), which is considered as a structural and functionalmulti-protein crossroad. PSD is an electron-dense thickeninglocalized under postsynaptic membranes, which comprisesseveral hundred proteins and particularly characterizes largeexcitatory glutamatergic synapses [6]. PSD proteins are dis-tributed in highly organized macromolecular complexes thatprocess, integrate, and converge synaptic signals to the nucle-us [7]. Overall, PSD proteins are involved in synaptic
A. de Bartolomeis (*) :G. Latte :C. Tomasetti : F. IasevoliLaboratory of Molecular and Translational Psychiatry, Unit ofTreatment Resistant Psychosis, Department of Neuroscience,Reproductive and Odontostomatologic Sciences, Section ofPsychiatry, University School of Medicine “Federico II”,Via Pansini 5, 80131 Naples, Italye-mail: [email protected]
Mol NeurobiolDOI 10.1007/s12035-013-8534-3
plasticity and dendritic spines architecture. Rearrangements inPSD proteins multimers at synaptic spines, occurring withprecise stimulus-related spatiotemporal patterns, are currentlysupposed to underlie synaptic plasticity-related events, such aslong-term potentiation (LTP) and long-term depression (LTD)[8, 9].
According to the biological functions of PSD in humans, arecent study demonstrated that mutations in 199 human PSDgenes (the 14 % of all PSD genes) are implicated in more than200 diseases [10]. The 50 % of these diseases are primarynervous system disorders, including neurological, psychiatric,and developmental disorders [10]. Moreover, a large part ofthose mutations may principally affect cognitive and learningprocesses, as well as emotion/affective behaviors and socialinteraction. Therefore, according to this view, emerging evi-dence is accumulating that implicates PSD protein dysfunc-tions in major psychiatric disorders in which cognitive/behavioral and social processes are impaired, such as schizo-phrenia and autism spectrum disorders. Based on the N -meth-yl-D-aspartate (NMDA) receptor hypofunction hypothesis ofschizophrenia [11], several studies have found abnormalitiesin PSD proteins in brain regions where NMDA receptors arelocalized, as well as gene association studies have revealed anincreased risk for schizophrenia in patients with mutations ingenes affecting NMDA functions (for a review, see [12]).Moreover, several molecular defects in PSD componentsand in PSD-related proteins have been found in postmortembrains of schizophrenia patients, mostly in regions implicatedin the pathophysiology of the disease [13, 14]. Thus, PSDalterations may contribute to the synaptic derangements inschizophrenia [9]. The recent evidence that master organizingPSD scaffolding proteins are linked to autism spectrum disor-ders (ASD) [15] further confirms their role in synaptic plas-ticity processes potentially involved in cognition and socialinteraction.
Consistent with the putative role of PSD proteins in schizo-phrenia and autism spectrum disorders, several studies haveimplicated glutamatergic PSD components in the molecularmechanisms of action of antipsychotic drugs. Though primar-ily acting on dopamine transmission, evidence exists thatantipsychotics may also modulate glutamate-related targets,in particular the N-methyl-D-aspartate receptor (NMDAR)-interacting molecules of the PSD [16, 17]. Indeed, differentantipsychotic drugs have been shown to modulate glutamate-related molecules in PSD [16, 18–22]. Therefore, PSD pro-teins have been proposed as a target for antipsychotic action[23]. Antipsychotics may affect glutamatergic neurotransmis-sion at multiple levels, and they may regulate dendritic spineformation and synaptogenesis [24, 25], as well as the expres-sion, trafficking, and functioning of PSD-related molecules[22, 26–28].
The aim of this review is to provide a critical description ofPSD proteins role in the pathophysiology and treatment of
schizophrenia and other behavioral disorders, such as ASD.Herein, we will discuss how antipsychotics may affect PSDproteins in brain regions relevant to psychosis pathophysiolo-gy, possibly by controlling dendritic spines formation andsynaptic plasticity through the modulation of specificNMDAR-related targets. Moreover, we will provide recentinformation on how PSD molecules may be considered valu-able candidates to develop potential new therapeutic ap-proaches for schizophrenia and autism spectrum disorders.
Clinical and Preclinical Evidence of PSD ProteinsInvolvement in Schizophrenia and other BehavioralDiseases
PSD-95/SAP90 is a member of the membrane-associatedguanylate kinase family (MAGUK), which has master orga-nizing roles in the multimerization and clustering of proteincomplexes within the PSD (Fig. 1). PSD-95 protein containsin its structure three repeated PDZ (PSD-95/disc large/zonulaoccludens-1) domains, one SH3 (Src homology 3), and oneguanylate kinase (GUK) domain [5]. PDZ domains arepeptide-binding domains located at the C-terminus ofMAGUK proteins, which may enable PSD-95 to interact withseveral binding partners within the PSD, such as NMDAR and5HT receptor subunits, as well as other tyrosine kinase recep-tors and ion channels, cell adhesion molecules, and cytoplas-mic proteins [29]. SH3 and GUK are other peptide-bindingdomains, which may also form intramolecular bonds [5].
Thus, by assembling in multimers with other PSD proteins,PSD-95 enables the formation of extensive protein complexesthat organize receptors and signal transduction proteins in thePSD. Indeed, affinity-purified PSD-95 complexes have beendescribed to include 2-amino-3-(3-hydroxy-5-methyl-isoxazol-4-yl)propanoic acid receptor (AMPAR) subunits(GluR1, GluR2, GluR3, GluR4), NMDAR subunits (NR1,NR2A, NR2B), scaffolding proteins (PSD-93, Shank2,Shank3, Homer, SAPAP1, SAPAP2, SAPAP4), G proteinregulators (such as SynGAP or BRAG1), and other PSDproteins [30, 31]. Furthermore, PSD-95 may interact withdopamine D2 and serotonin 5-HT2 receptors to regulate theiractivation state [5]. PSD-95 has also been described to stabi-lize glutamate receptors in the PSD and to provide a linkbetween NMDARs and intracellular signaling molecules [9].
Given its functions, PSD-95 has been implicated in synapticplasticity processes and in the interplay among glutamatergic,dopaminergic, and serotonergic signaling pathways. Hence,aberrant PSD-95 functioning may cause abnormal glutamate
Mol Neurobiol
signaling, thereby potentially takingpart inmolecular dysfunc-tions involved in schizophrenia and behavioral disorderpathophysiology.
Fine-tuned PSD proteins interactions by PSD-95 may con-tribute to long-term changes in synaptic shape and strength. InCA1 hippocampal pyramidal neurons, activity-dependentgrowth of apical spines has been associated with the destabi-lization of PSD architecture, causing transient loss and rapidreplacement of PSD-95, together with Shank2 [32]. It has alsobeen observed that signaling through a PSD-95-mediatedpathway is required for activity-dependent synaptic enlarge-ment and for the recruitment of other architectural proteins,such as Shank [32].
PSD-95 has been supposed to participate in multiple stepsof synaptic rearrangements, based on the discovery that
different PSD-95 domains have been found implicated inthis process [32]. Morphological changes in PSD by PSD-95 may be regulated through CaMKII phosphorylation atSer73 site, which has been reported to slow down both thegrowth of apical spines and the strength of synaptic currents[32]. Therefore, PSD-95 may both trigger synaptic growthand terminate it, probably via the modulation of growth-related proteins trafficking in the PSD. Accordingly, theknockdown of PSD-95 gene expression by short hairpinRNAhas been shown to impair early and late phases of spinegrowth [32].
The overexpression of PSD-95may affect spines morphol-ogybyincreasingspinevolumeandexpandingPSDstructure,aswell as by concomitantly decreasing spines density [33]. PSD-95 overexpression may also induce the formation of multi-
Fig. 1 Complex interactions among transductional pathways in thePSD . PSD proteins elaborate and integrate multiple transductional path-ways starting at different membrane receptors (i.e., glutamate, dopamine).Scaffolding proteins (Homer, Shank, PSD-95) provide physical connec-tions among different receptors, such as ionotropic and metabotropicglutamate receptors, as well as they link these receptors to intracellularcalcium stores. Dopamine receptors activate transductional pathways thattightly intermingle with glutamatergic ones, through the action of keyPSD proteins, such as GSK3, which may participate in the elaboration ofdiverse signals (dopamine, glutamate, Wnt) and regulate neuronal sur-vival and differentiation. All these transductional pathways converge inthe end on appropriate nuclear targets via specific effectors, such asCaMK, MAPKs, or Erk, in order to fine modulate long-term activitydependent neuronal rearrangements. NMDAR , N -methyl-D-aspartate
innervated spines, i.e. dendritic spines that are connected withup to seven presynaptic terminals [33]. The formation ofmulti-innervated spines is prevented by deletion of the nitric oxidesynthase(NOS)-interactingPDZ2domainofPSD-95[33].Ithasbeen reported that NOS–PSD-95 interaction and nitric oxide(NO) signaling may promote synapse formation. Down-regulation by small interfering RNA or by pharmacologicalblockade ofNOSmayprevent the formation ofmultiple synap-ses mediated by PSD-95 overexpression [33]. Nevertheless,treatment of hippocampal slices eitherwith aNOdonor orwithcyclic guanosine monophosphate analogues has been demon-strated to induce the formation of multi-innervated spines byPSD-95 overexpression [33].
Recent studies have demonstrated that PSD-95, in cooper-ation with other PSD scaffolding proteins (i.e. Shank,Homer1, GKAP), may establish the architectural basis forglutamate receptor clustering—in particular of AMPARs—inresponse to prolonged stimuli [34]. In particular, PSD-95might have a crucial role in concentrating AMPARs in post-synaptic active zones in correspondence of presynapticglutamate-releasing sites, thereby maximizing the effects ofpostsynaptic excitatory potentials [31].
Therefore, PSD-95 enrichment within PSD may regulatemorphological and functional synaptic plasticity in multipleand complex ways. Actually, PSD-95 may trigger synapticgrowth, and its overexpressionmay block the growth of singlesynapses and favor the formation of multi-innervated spines.PSD-95 interactions may drive postsynaptic architectural re-modeling in response to stimuli. Moreover, together with theother scaffolding proteins, PSD-95 may modify glutamatereceptors postsynaptic membrane position, thus increasing ordecreasing excitatory currents even without altering the num-ber of receptors.
PSD-95 Bridges Dopamine, Glutamate, and SerotoninTransductional Pathways
PSD-95-related proteins provide a physical link between glu-tamate and dopamine systems. PSD-95 proteins may regulatedopamine D1 receptor (D1R) trafficking and function, throughthe interaction between the carboxyl-terminal tail of D1Rs andthe NH(2) terminus of PSD-95 [35]. PSD-95 may reducesurface D1R expression by promoting receptor internalization[35, 36]. Co-expression of PSD-95 and D1Rs in mammaliancells has been found to inhibit D1R-mediated cAMP accumu-lation [35]. Therefore, the overexpression of PSD-95 mayreduce D1R signaling and prevent functional hyperdo-paminergia. Accordingly, genetically engineered mice lackingfunctional PSD-95 proteins exhibit increased response to di-rect D1R agonists or to indirect dopamine agonists (e.g. am-phetamine) [35]. PSD-95 may be also implicated inpreventing the concurrent overactivation of D1Rs andNMDARs during neurotoxicity states. Indeed, PSD-95
proteins have been demonstrated to inhibit D1R-NMDARassociation and uncouple the NMDAR-dependent enhance-ment of D1R signaling [36]. Thus, PSD-95 gene expressionmay inhibit D1R function, whereas its knockdown may en-hance NMDAR-dependent D1R functioning [35, 36].Notably, the disruption of the interaction between D1Rs andPSD-95 in striatum has been described to reduce L-DOPA-induced dyskinesia in rat and macaque models [37], therebysuggesting the involvement of PSD-95/D1R complexes indopamine-related dysfunctions. Besides dopamine D1Rs,PSD-95 has been demonstrated also to physically interact withdopamine D2Rs in cell cultures [38]. However, the in vivoeffects of PSD-95/D2R interaction are still elusive at present.In the striatum, D1Rs and D2Rs are expressed in functionallydifferent neuronal populations, being D1Rs selectivelyexpressed in the medium-sized spiny neurons of the directpathway and D2Rs in the indirect pathway cells [39]. Providedthe inhibitory effects of PSD-95 binding to D1Rs on theNMDAR/D1R downstream signaling, it is possible thatPSD-95 might exert “D2-like” slowing down functions onD1R signaling in striatal neuron subtypes in which D2Rs arenot physically expressed.
PSD-95 interacts with Calcyon, a transmembrane proteinpredominantly expressed in the central nervous system [40]that may be increased in schizophrenia patients [41, 42]. PSD-95 and Calcyon have been found to form a ternary complexwith D1Rs in dendritic spines of hippocampal neurons [43].Furthermore, the PKC-dependent phosphorylation of Calcyonmay promote its association with PSD-95 and the recruitmentto plasma membrane, as well as it may enhance the internal-ization of surface D1Rs [43]. Thus, the Calcyon–PSD–95-D1R complex may represent a further mechanism linkingdopamine and glutamate transductional pathways and also apotential target for psychopharmacotherapy.
PSD-95 is also essential for proper targeting and synapticmembrane stabilization of serotonin 5-HT2A and 5-HT2C re-ceptors. The interaction with PSD-95 may prevent agonist-mediated internalization of 5-HT2A receptors and enhances 5-HT2A receptor-dependent signaling [44], as well as it maypromote 5-HT2C constitutive and agonist-mediated internali-zation [45]. PSD-95 has been reported to regulate 5-HT2Areceptors surface membrane turnover, thus contributing totheir targeting to apical dendrites of pyramidal neurons.PSD-95 is also required for 5-HT2C-mediated signalingin vivo, as well as for 5-HT2A-mediated actions of hallucino-genic drugs [46]. Notably, both 5-HT2A and 5-HT2C receptorsdownstream pathways, as well as the animal behaviors medi-ated by the activation of these receptors, have been foundimpaired in mice lacking functional PSD-95 [46]. Also, treat-ment with either clozapine (the prototypical “atypical” anti-psychotic compound), M100907 (a 5-HT2A receptor antago-nist), or SR46349B (a 5-HT2A/2C antagonist), all of whichmay regularly reduce behavioral impairments induced by
Mol Neurobiol
NMDAR-blocking drugs (such as phencyclidine), is ineffec-tive in PSD-95 null mice [46]. By controlling 5-HT2A/2C
receptor trafficking and signaling, PSD-95 may contribute tothe mechanisms of action of both hallucinogenic drugs andatypical antipsychotics. Hence, PSD-95 may act as a pivotalcrossroad molecule along dopamine, glutamate, and serotoninsignaling systems and enable their interplay and reciprocaltransactivation.
PSD-95 in the Pathophysiology of Psychotic Disorders
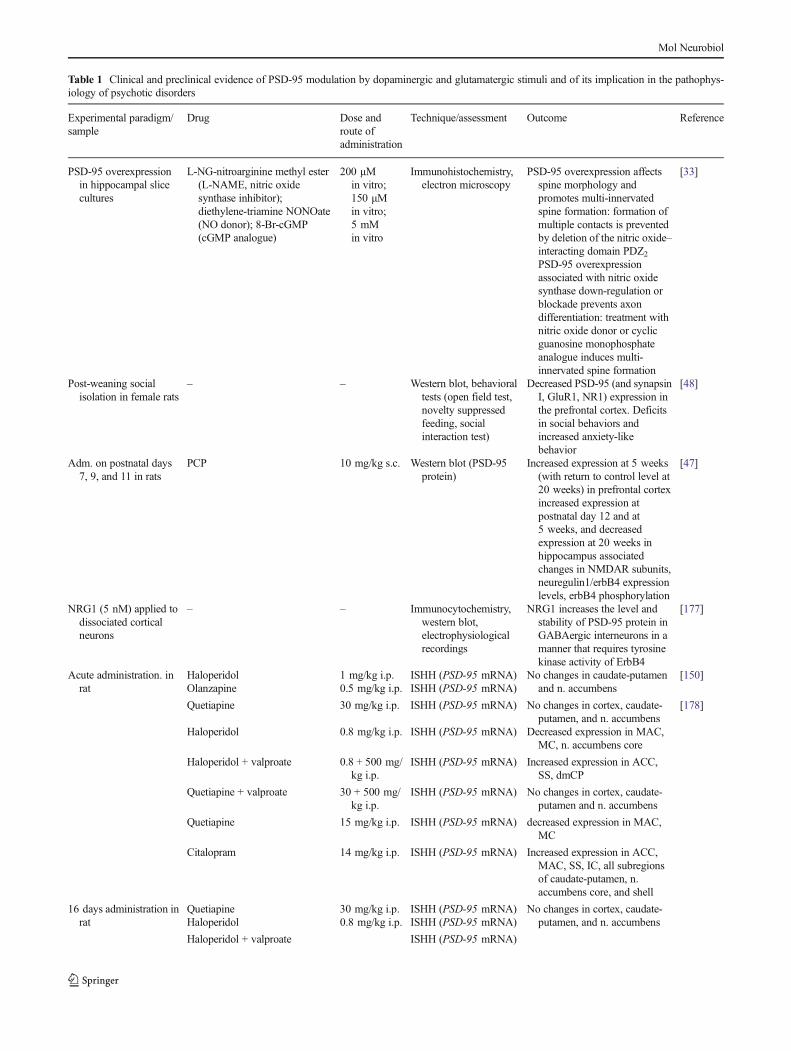
A growing number of studies have implicated PSD-95 inanimal models of psychosis, even if with the limitation ofanimal modeling in psychiatry, and in the pathophysiology ofschizophrenia, as demonstrated by postmortem human brainanalyses (Table 1). In animal studies, perinatal phencyclidine(PCP) administration in rats has been demonstrated to lead tocomplex changes in PSD-95 protein expression in both pre-frontal cortex (PFC) and hippocampus throughout the centralnervous system development. The injection of PCP in rats atpostnatal days 7, 9, and 11 may provoke alterations inNMDAR subunits, in neuregulin1/erbB4 expression levels,and in erbB4 phosphorylation that may be detected immedi-ately after the injection (postnatal day 12) and last until20 weeks after birth, thus providing new insights on abnormaldevelopmental processes putatively involved in schizophrenia[47]. Nevertheless, neuregulin1 may increase PSD-95 proteinlevels and stability; these effects being specific to GABAergicinterneurons [47]. Decreased PSD-95 protein expression hasalso been found in the PFC of isolation-reared female rats, ananimal model of neuropsychiatric disorders that include fea-tures reminiscent of anxiety- and schizophrenia-like disorders[48].
In human postmortem studies, altered PSD-95 gene andprotein expression have been reported in brain regions in-volved in schizophrenia pathophysiology. Early works dem-onstrated an increase in thalamic PSD-95 gene expression inschizophrenia patients, with a concurrent decrease inNMDAR NR1 subunits in the same region [14].Noteworthy, more recent studies by the same group havedescribed opposite regulation of PSD-95 transcripts in thethalamus of schizophrenia patients, depending on the diseaseonset age. Postmortem studies indicated that young schizo-phrenia patients have decreased PSD-95 thalamic levels andincreased NMDAR NR2B subunits [13], whereas in elderlypatients, increased PSD-95 and NR2B protein levels indorsomedial thalamus were found [49]. In the anterior cingu-late cortex, increased expression of PSD-95 transcripts butdecreased protein expression and decreased phosphorylationat Ser295 site have been reported [29, 50, 51]. DecreasedPSD-95 protein expression has also been described in thedentate molecular layer of hippocampus [52] and in a subcel-lular endoplasmic reticulum-enriched fraction from the
dorsolateral prefrontal cortex [53] of schizophrenia patients.Thus, both animal and human studies provide substantialevidence of a crucial role of PSD-95 in the normal establishingof correct glutamatergic synaptic downstream signaling dur-ing neurodevelopment. Early alterations in PSD-95 functionsmay lead to aberrant glutamatergic signaling that is putativelyat the basis of cognitive and behavioral dysfunctions in psy-chotic disorders.
Based on the putative role of PSD-95 in spine enlargementand synaptic strength, it could be hypothesized that lack offunctional PSD-95 may cause dendritic spine shrinking andabnormal synaptic plasticity, which may contribute to themolecular underpinnings of psychosis. According to thisview, it has been observed that pluripotent stem cells fromperipheral fibroblasts of schizophrenia patients may showdecreased PSD-95 protein amounts and reduced neuronalconnectivity when differentiated into neurons [19].
On the other hand, enhanced PSD-95-ErbB4 coupling hasbeen found in PFC of schizophrenia patients [54], therebysuggesting that enhanced interactions between these two mol-ecules may be involved in schizophrenia pathophysiology.ErbB4 is a member of the epidermal growth factor receptorsfamily of tyrosine kinase receptors, which are essential fornormal nervous system development [55, 56]. Despite thebiological role of PSD–95-ErbB4 interaction has not beenyet elucidated, an increased coupling of these two moleculesmay be responsible for ErbB4 signaling blockade (i.e. bypromoting ErbB4 recycling and endocytosis) or be secondaryto synaptic derangements in schizophrenia.
However, discrepant findings in postmortem studies mayderive from several technical and methodological limitations,including the potential effects of chronic antipsychotic treat-ment, differences in the type of prevailing symptoms (posi-tive, negative, or cognitive ones), total duration of the illness,patients’ kind of death, and elapsed time between death andstorage of the specimen [57, 58].
Consistently with its putative role in cognitive and behav-ioral disorders pathophysiology, PSD-95 has also been impli-cated in other neurodevelopmental diseases, such as autism.
Mice with a homozygous PSD-95 gene deletion (Dlg4−/−)show a complex phenotype reminiscent of autism, whichincludes increased repetitive behaviors, abnormal communi-cation and social behaviors, impaired motor coordination,increased stress- and anxiety-related responses [59]. Thesemutant mice also exhibit altered dendritic spine morphologyin amygdala and altered forebrain gene expression profile[59].Moreover, prenatal rat exposure to valproic acid, a modelof ASD, may lead to increased PSD-95 protein expression inmale but not female rats in hippocampus and cortex [60].These results seem in agreement with multiple studies inwhich male preponderance in risk factors for ASD is sug-gested. However, neither the hypothesis of excessive fetaltestosterone, nor of Y chromosome-linked abnormalities, nor
Mol Neurobiol
Table 1 Clinical and preclinical evidence of PSD-95 modulation by dopaminergic and glutamatergic stimuli and of its implication in the pathophys-iology of psychotic disorders
Increased expression at 5 weeks(with return to control level at20 weeks) in prefrontal cortexincreased expression atpostnatal day 12 and at5 weeks, and decreasedexpression at 20 weeks inhippocampus associatedchanges in NMDAR subunits,neuregulin1/erbB4 expressionlevels, erbB4 phosphorylation
Dihydroxyphenylglycine(DHPG, group I mGluRsagonist)
50 μMin vitro
Fluorescence in situhybridization,immunocytochemis-try,immunoprecipitation,western blot,luciferase assay
PSD-95 translation isbidirectionally regulatedthrough the control of bothmiR-125a and the fragile Xmental retardation protein(FMRP) phosphorylationstatus
[151]
Human postmortembrains ofschizophrenia patients
– – ISHH (PSD-95 mRNA) Increase in thalamic PSD-95expression, with a concurrentdecrease in NR1 subunit ofNMDA receptors
[14]
Human postmortembrains of youngschizophrenia patients
– – ISHH (PSD-95 mRNA) Decrease in thalamic PSD-95expression, with a concurrentincrease in NR2 subunit ofNMDA receptors
[13]
Human postmortembrains of elderlyschizophrenia patients
– – Western blot analysis Increase in thalamic proteinlevels of PSD-95 and of NR2subunit of NMDAreceptors
[49]
Human postmortembrains ofschizophrenia patients
– – Western blot analysis Decreased protein levels of PSD-95 and NR2 subunit of NMDAreceptors in dorsolateralprefrontal cortex andanterior cingulatecortex
[51, 53]
Human postmortembrains ofschizophrenia patients
– – Immunoautoradiography Decrease in PSD-95 proteinlevels in the dentate molecularlayer of hippocampus, notrelated to antipsychotictreatment
[52]
ACC anterior cingulate cortex; cGMP cyclic guanosine monophosphate; dlCP, vlCP, dmCP, vmCP dorsolateral, ventrolateral, dorsomedial, andventromedial caudate-putamen respectively; IC insular cortex; ISHH in situ hybridization histochemistry; MAC medial agranular cortex; MC motorcortex; NO nitric oxide; PPI prepulse inhibition; RT-PCR reverse transcription polymerase chain reaction; SS somatosensory cortex
Mol Neurobiol
of male vs. female differences in social empathizing functionsmay fully explain the prominent male ASD prevalence (for areview, see [61]). Recent results confirm the higher thresholdof female susceptibility to ASD as compared to males. Satoand colleagues [62] recently reported that Shank1 mutationsassociated with ASD phenotypes are limited to males. Thesestudies reinforce the view that alterations of PSD-95 maycause defective synaptic architecture and spine functions.
Interestingly, different genes linked to autism (such asprotocadherin 10, Pcdh10 ; fragile X mental retardation 1,Fmr1 ; myocyte enhancer factor 2, Mef2) may participate inthe degradation of PSD-95 and in synapse elimination, thussuggesting a pivotal function of PSD-95-mediated deficits insynapse elimination among the different genetic causes ofautism [63].
PSD-95 dysfunctions have also been implicated inAngelman syndrome (AS), a neurodevelopmental disorderthat includes cognitive impairment and autism.
Ubiquitin-protein ligase E3A knockout mice (a model ofAS) exhibit high Arc protein amount in response to synapticactivity, impaired LTP in the hippocampus, and deficits inlearning behaviors [64]. Impaired LTP may be due to a reduc-tion in BDNF-induced PSD-95 association with TrkB, therebyresulting in reduced PLCγ and PI3K signaling [64]. Theadministration of CN2097, a PSD-95 binding peptideinteracting with the PDZ domain, may reduce Arc/PSD-95interactions, thus restoring BDNF-induced TrkB/PSD-95complex formation and LTP induction [64].
Taken together, these findings suggest that PSD-95 mayregulate synaptic targeting and localization of several PSDproteins, which are organized as a “molecular lego” in thearchitecture of dendritic spines. Moreover, PSD-95 may rep-resent an intermediate molecule along multiple neurotransmit-ter systems, allowing their cross-talk at both structural andfunctional levels. These observations provide support forstudying PSD-95 as a candidate molecule to better dissectthe pathophysiological bases of cognitive impairment in neu-ropsychiatric disorders, such as schizophrenia or ASD.
Homer
Homer Proteins Are Multimodal Postsynaptic Adaptors thatFine Regulate PSD Architecture
Homer genes encode for a family of proteins including threeisoforms in mammals (Homer1, Homer2, and Homer3),which are predominantly localized at the PSD, where theyact as multimodal adaptors by interacting with several PSDproteins [7, 65, 66] (Fig. 1). Homer proteins are primarilyclassified into (a) constitutively expressed isoforms (i.e.Homer1b/c, Homer 2, and Homer 3), which are bimodalproteins with a N-terminal Ena/VASP (EVH) domainallowing the binding to other PSD proteins, and a C-terminal
coiled-coil domain that enables self-assembly; (b) short, non-multimerizing, activity-dependent splice variants of theHomer1 gene (Homer1a, Ania-3), which lack the C-terminaldomain and are able to interact with PSD targets but cannotself-assemble [65, 66]. These short forms are induced in animmediate-early gene-like fashion after neuronal stimulationand act as endogenous “dominant-negative” by disruptinglong Homer isoforms protein–protein interactions [65]. Onceinduced, Homer short forms cause rapid and transientrearrangements of long Homer clusters and in turn of synapticarchitecture. These transient changes have significant conse-quences for synaptic signaling, including agonist-independentactivation of group I metabotropic glutamate receptors(mGluRs) [67], changes in local Ca2+ levels [68], activation/inactivation of surface ion channels [69], and modulation ofsecond messenger signaling [9, 70].
The relative ratio of long/short Homer forms (i.e.Homer1b/c vs. Homer1a) has also been described to impaction channel functioning and synaptic signaling [66], as well assynaptic architecture, spine shape, and size. Indeed, Homer1bmay enhance Shank1B-mediated increase in spine length andwidth [71]. On the other hand, overexpression of Homer1amay reduce the number and size of dendritic spines, decreasethe density of postsynaptic proteins (such as Shank) in spines,and inhibit postsynaptic AMPAR and NMDAR currents inhippocampal neurons [72].
Homer1a is instrumental in the activity-induced reorgani-zation of both pre- and postsynaptic structures. Basically,Homer1a induction is involved in glutamate-induced biphasicchanges in the distribution of both presynaptic proteins (suchas synaptotagmin, synaptophysin, and synapsin) and postsyn-aptic proteins (such as PSD-95 and Homer1c), at least incultured hippocampal neurons [73]. Furthermore, Homer1aprotein induction may reduce group I mGluR-dependent LTDin layer VI pyramidal cells from rat visual cortex slices [74].Finally, the acute disruption of mGluR–Homer interactionshas been demonstrated to impair mGluR-dependent LTD inrat hippocampal slices [75].
Long and short Homer isoforms may cooperate to fine tunePSD-mediated synaptic plasticity. This fine tuning appearsstringently regulated in space (i.e. in PSD microdomains)and time (i.e. short Homer proteins are induced to providetime-limited rearrangements of long Homer clusters).
Homer Proteins Modulation by Dopaminergicand Glutamatergic Stimuli
Several studies have demonstrated that the expression oftranscript encoding for both long and short Homer isoformsmay be affected by psychotomimetic drugs modulating eitherdopaminergic or glutamatergic receptors. Both acute metham-phetamine and cocaine administration may induce Homer1amRNA in the neocortex of saline-pretreated rats [76]. Also,
Mol Neurobiol
increased long Homer protein amount in the nucleusaccumbens has been described after acute cocaine administra-tion [77]. Consistent with the role of Homers as scaffoldproteins at the crossroad of dopamine and glutamate system,drugs acting at NMDARs, such as the noncompetitive inhib-itor PCP, may increase Homer1a mRNA expression in ratPFC prelimbic region and in primary auditory cortex 2 h aftertreatment, as well as it may decrease Homer1a mRNA ex-pression in retrosplenial cortex and dentate gyrus 24 h aftertreatment [78]. Similarly to PCP, the NMDAR noncompetitiveantagonist ketamine may induce Homer1a mRNA in theventral striatum and in the core and the shell of the nucleusaccumbens when acutely administered at subanesthetic doses[79]. Both PCP and ketamine are NMDAR-noncompetitiveantagonists known to cause psychotic symptoms in humansand to exacerbate psychotic symptoms in schizophrenia pa-tients [80, 81]. Moreover, PCP and ketamine are believed tomimic NMDAR hypofunction, which is considered a valuableand heuristic pharmacological model of schizophrenic symp-toms [14].
Homer in the Pathophysiology of Psychotic Disorders
Given its role in activity-dependent synaptic rearrangements,Homer gene and protein expression changes in response todopaminergic and glutamatergic stimuli have been consideredas fine-tuned mechanisms to preserve synaptic homeostasis[82]. Thus, growing evidence has been provided that Homerprotein dysfunctions might be involved in the pathophysiolo-gy of neuropsychiatric disorders implicating defects in synap-tic plasticity, such as schizophrenia [83, 84] (Table 2). Thedeletion of Homer1 gene has been demonstrated to inducebehavioral and neurochemical abnormalities relevant to ani-mal models of schizophrenia, including altered performancesin sensory, motor, social, and learning/memory tests [85, 86].Homer1 knockout (KO) mice show altered food reward andreinforcement, altered antipsychotic-sensible sensorimotorgating, increased motor activation, and attenuated habituationof motor activity when exposed to a novel environment. Thesemice also have increased sensitivity to the locomotor-activating effects of MK-801 or methamphetamine, decreasedextracellular glutamate content in the nucleus accumbens, andincreased content in the PFC, as well as blunted increase inPFC extracellular glutamate after cocaine stimulation [86].
However, long and short Homer1 variants are differentlyinvolved in PFC glutamate neurotransmission and in devel-opment of behaviors relevant to schizophrenia. Actually, arecent study demonstrated that the adeno-associated virus(AAV)-mediated restoration of either Homer1a or Homer1cinHomer1 KOmice may differently affect synaptic functionsand consequent behaviors [87]. Homer1c restoration in thePFC of Homer1 KO mouse reverses aberrant working mem-ory and sensorimotor function, locomotor hyperactivity in
response to a novel environment, sensitivity to cocaine, andPFC glutamate content [87]. By contrast, the AAV-mediatedrestoration ofHomer1a has been demonstrated to only reversealterations in emotional reactivity in mutant animals [87].Notably, these behavioral and neurochemical alterations havebeen found worsened by dopamine function enhancers, suchas cocaine and methamphetamine, and were prevented orattenuated by D2 receptor antagonist agents, such ashaloperidol.
Consistent with preclinical findings, several clinical studieshave implicated Homer genes in schizophrenia pathophysiol-ogy. An early study found a significant association between asingle nucleotide polymorphism (SNP) within intron 4 ofHomer1 gene and schizophrenia [88]. However, this resultwas not replicated in an extended sample by the same authors[88].
More recently, an association between Homer1 gene poly-morphisms and clinical psychopathology assessments inschizophrenia has been demonstrated. TwoHomer1 polymor-phisms (rs2290639, which is an intronic polymorphism, andrs4704560, which is a mutation in the 5′-flanking region ofHomer1 gene, and could be considered as a potential promot-er polymorphism) have been associated with scores on posi-tive and negative syndrome scale (PANSS, a rating scale forsymptoms severity assessment in schizophrenia) subscales atbaseline. Namely, the rs2290639 variant was significantlyassociated with scores on PANSS total, positive, and globalpsychopathology subscales, whereas the rs4704560 variantwas significantly associated with scores on PANSS-negativesubscale [89].
Also, a putative role for Homer2 gene in schizophreniasusceptibility has been suggested. Actually, the rs2306428polymorphic variant has been strongly associated with thedisease [90].
The role of Homer proteins in synaptic plasticity has stim-ulated further studies on their involvement in neuropsychiatricdiseases in which molecular processes underlying cognitionare considered dysfunctional, such as ASD.Homer1 gene hasbeen recognized as a novel autism-risk gene in a single nucle-otide variant analysis of blood samples from 290 unrelatednonsyndromic autism cases and 300 ethnically matched con-trols [91]. Several rare and potentially damaging variants havebeen identified in the autism population that co-segregate withthe disorder and affect functionally relevant protein regions orregulatory sequences [91]. Intriguingly, the interaction be-tween long Homer isoforms and mGluR5 is strongly dimin-ished in Fmr1 KO mice, a model of fragile X syndrome (aninherited cause of intellectual disability and autism)[92]. In this model, the genetic deletion of Homer1a mayrestore the long Homer–mGluR5 interaction and correct al-tered phenotypes, thus suggesting a potential Homer-relatedmechanism of mGluR5 dysfunction in this autism-relateddisease [93].
Mol Neurobiol
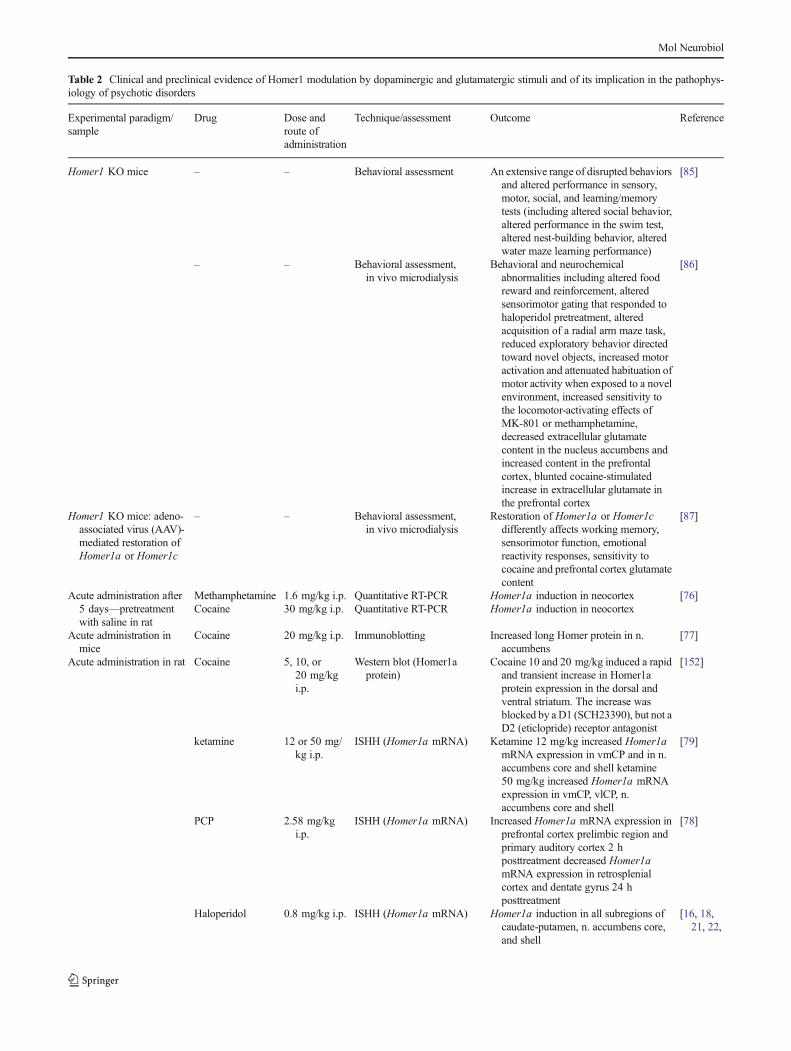
Table 2 Clinical and preclinical evidence of Homer1 modulation by dopaminergic and glutamatergic stimuli and of its implication in the pathophys-iology of psychotic disorders
Experimental paradigm/sample
Drug Dose androute ofadministration
Technique/assessment Outcome Reference
Homer1 KO mice – – Behavioral assessment An extensive range of disrupted behaviorsand altered performance in sensory,motor, social, and learning/memorytests (including altered social behavior,altered performance in the swim test,altered nest-building behavior, alteredwater maze learning performance)
[85]
– – Behavioral assessment,in vivo microdialysis
Behavioral and neurochemicalabnormalities including altered foodreward and reinforcement, alteredsensorimotor gating that responded tohaloperidol pretreatment, alteredacquisition of a radial arm maze task,reduced exploratory behavior directedtoward novel objects, increased motoractivation and attenuated habituation ofmotor activity when exposed to a novelenvironment, increased sensitivity tothe locomotor-activating effects ofMK-801 or methamphetamine,decreased extracellular glutamatecontent in the nucleus accumbens andincreased content in the prefrontalcortex, blunted cocaine-stimulatedincrease in extracellular glutamate inthe prefrontal cortex
[86]
Homer1 KO mice: adeno-associated virus (AAV)-mediated restoration ofHomer1a or Homer1c
– – Behavioral assessment,in vivo microdialysis
Restoration of Homer1a or Homer1cdifferently affects working memory,sensorimotor function, emotionalreactivity responses, sensitivity tococaine and prefrontal cortex glutamatecontent
[87]
Acute administration after5 days—pretreatmentwith saline in rat
Methamphetamine 1.6 mg/kg i.p. Quantitative RT-PCR Homer1a induction in neocortex [76]Cocaine 30 mg/kg i.p. Quantitative RT-PCR Homer1a induction in neocortex
Acute administration inmice
Cocaine 20 mg/kg i.p. Immunoblotting Increased long Homer protein in n.accumbens
[77]
Acute administration in rat Cocaine 5, 10, or20 mg/kgi.p.
Western blot (Homer1aprotein)
Cocaine 10 and 20 mg/kg induced a rapidand transient increase in Homer1aprotein expression in the dorsal andventral striatum. The increase wasblocked by a D1 (SCH23390), but not aD2 (eticlopride) receptor antagonist
[152]
ketamine 12 or 50 mg/kg i.p.
ISHH (Homer1a mRNA) Ketamine 12 mg/kg increased Homer1amRNA expression in vmCP and in n.accumbens core and shell ketamine50 mg/kg increased Homer1a mRNAexpression in vmCP, vlCP, n.accumbens core and shell
[79]
PCP 2.58 mg/kgi.p.
ISHH (Homer1a mRNA) Increased Homer1a mRNA expression inprefrontal cortex prelimbic region andprimary auditory cortex 2 hposttreatment decreased Homer1amRNA expression in retrosplenialcortex and dentate gyrus 24 hposttreatment
[78]
Haloperidol 0.8 mg/kg i.p. ISHH (Homer1a mRNA) Homer1a induction in all subregions ofcaudate-putamen, n. accumbens core,and shell
olanzapine 2.5 mg/kg i.p. ISHH (Homer1a mRNA) Homer1a induction in lateral regions ofcaudate-putamen and in n. accumbenscore
Clozapine 15 mg/kg i.p. ISHH (Homer1a mRNA) Homer1a induction in the cortex (ACC,MAC, SS, IC) and in n. accumbenscore and shell
Aripiprazole 12 or 30 mg/kg i.p.
ISHH (Homer1a mRNA) Aripiprazole 12 mg/kg: Homer1ainduction in all subregions of caudate-putamen aripiprazole 30 mg/kg:Homer1a induction in frontal andcingulate cortex and in n. accumbensshell
Quetiapine 15 or 30 mg/kg i.p.
ISHH (Homer1a mRNA) Quetiapine 15 mg/kg: no changes inHomer1a expression quetiapine30 mg/kg: Homer1a induction in thecortex (ACC, MAC, MC)
risperidone 3 mg/kg i.p. ISHH (Homer1a mRNA) Homer1a induction in lateral regions ofcaudate-putamen
ziprasidone 4 or 10 mg/kgi.p.
ISHH (Homer1a mRNA) Ziprasidone 4 mg/kg: Homer1ainduction in all caudate-putamensubregions ziprasidone 10 mg/kg:Homer1a induction in the cortex(ACC, MC, SS, IC) and in all caudate-putamen and n. accumbens subregions
No changes in Homer1a expression,increased Homer1b/c expression in thecortex (ACC, MAC, SS, IC)
267 schizophrenic patientstreated in monotherapy
– – Genotyping, weeklypsychopathology
Two Homer1 polymorphisms, rs2290639and rs4704560, associated with scores
[89]
Mol Neurobiol
In summary, Homer proteins are key proteins of the PSD,involved in postsynaptic glutamatergic signaling and in dopa-mine glutamate cross-talk. Long Homers act as scaffoldingproteins, bridging glutamate receptors with their intracellulareffectors. Short Homers disrupt these clusters in a space- andtime-controlled fashion. This balance contributes to finelyregulate multiple biological functions, such as Ca2+ dynamicsin dendritic spine microdomains, whose disruption may con-cur to dysfunctions of synaptic plasticity and aberrant behav-ioral manifestations [94].
Shank
Shank Proteins Form Functional Protein Platformsat Postsynaptic Sites
The ProSAP/Shank (named from SH3 and ankyrin domains)family of proteins is constituted by master organizing PSDscaffolding proteins that are implicated in propagating andmodulating glutamate neurotransmission [95] (Fig. 1).Shank genes derive from the same orthologous genes, withslight differences in humans and animals, thus providing andoptimal candidate for translational research on its involvementin neuropsychiatric disorders [96, 97]. Shank proteins, i.e.Shank1, Shank2, and Shank3, are coded by three differentgenes, each of them having multiple splice variants producingdifferent Shank proteins [98].
Shanks may cross-link Homer and PSD-95 in the PSD andparticipate in NMDAR, mGluR, and AMPAR downstreamsignaling [99–101]. Moreover, Shanks promote spine forma-tion, as well as maturation and enlargement of dendritic spines[101]. Through their multiple interactions, Shank proteinsenable the formation of a polymeric network complex, whichrequires assembly of Homer tetramers and Shank multimers.This structure has been proposed to serve as a functionalplatform for other PSD proteins [95].
Shank in the Pathophysiology of Psychotic Disorders
Dysfunctions of Shank proteins have been reported in severalneuropsychiatric disorders, i.e. autism, schizophrenia, andAlzheimer’s disease [102] (Table 3). Indeed, two de novomutations of Shank3 have been associated with schizophreniaand schizoaffective disorder cases [103]. These mutations arepredicted to affect Shank3 protein function. In particular, theR1117X mutation may cause Shank3 loss of function.Notably, in all the above cases, mental retardation was alsodiagnosed [103].
The Shank1 promoter variant rs3818280 has been associ-ated with impaired working memory in schizophrenia, whichdepends on patients’ genotype (CC, CT, or TT). On the digitspan task of the Wechsler Adult Intelligence Scale (WAIS-R),schizophrenia patients carrying either CT or TT genotypeshave been shown to repeat less number of sequences in bothforward and backward digit span subtests as compared topatients carrying CC genotype [104]. Similarly, in a popula-tion of at risk subjects for psychosis, subjects carrying the Tallele performed worse than those carrying CC genotype inthe forward digit span subtest [104]. Recently, differentShank1 gene deletions have been demonstrated in twounrelated ASD families from a population of European andCanadian individuals [105]. Several social-like impairmentshave been described in Shank1 (−/−) null mutant mice, includ-ing decreased levels of ultrasonic vocalizations and scent-marking behavior [106].
Mutations in the gene coding for Shank3 have been report-ed in ASD patients [67, 107–109]. Two de novo mutations(STOP and Q321R) and two inherited variations (R12C andR300C) identified in ASD patients have been reported toaffect spine development and morphology, as well as sponta-neous neuronal activity in cultured neurons [110]. Notably,mice carrying Shank3 deletions (Shank3B−/− mice) exhibitself-injurious repetitive grooming behaviors and deficits insocial interaction resembling autistic behaviors in humans
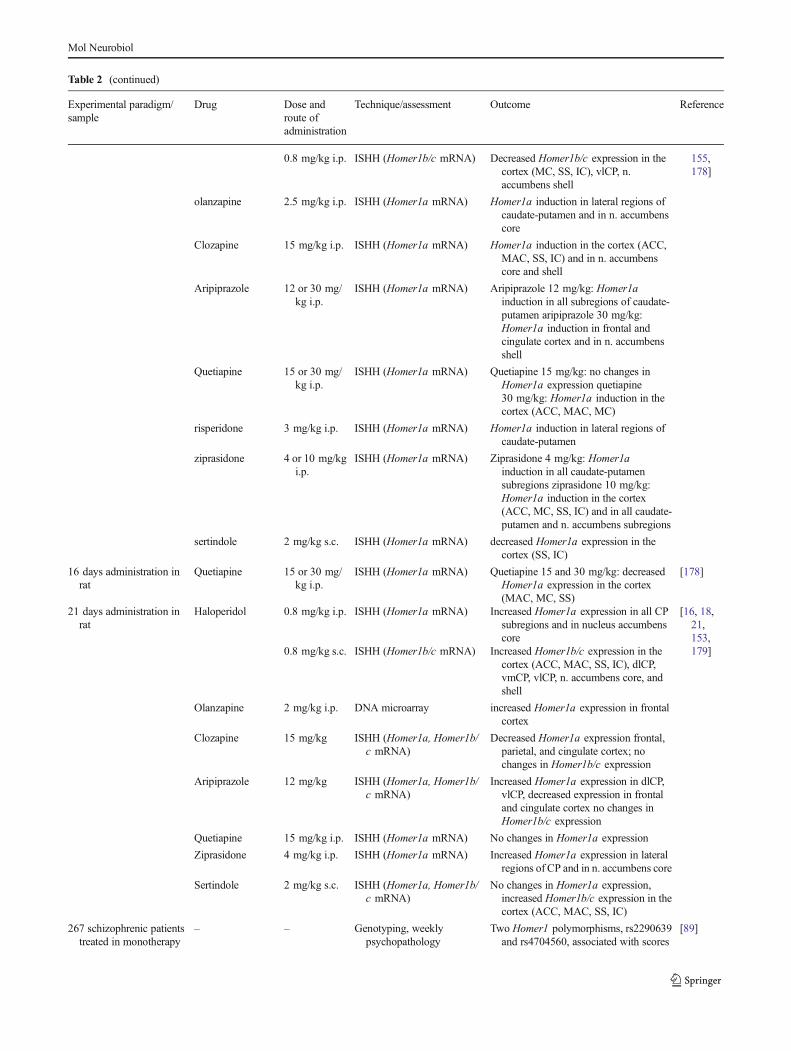
Table 2 (continued)
Experimental paradigm/sample
Drug Dose androute ofadministration
Technique/assessment Outcome Reference
with differentantipsychotics
assessment using Positiveand Negative SyndromeScale (PANSS)
on PANSS subscales at baseline 7polymorphisms associated with clinicalresponse after different 4-weekantipsychotic treatments
DNA pools of 368schizophrenia patientsand related controls
– – Genotyping One SNP (Homer 1 IVS4+18A>G)associated with schizophrenia, afinding confirmed by individual SNPgenotyping (P=0.01)
[88]
ACC anterior cingulate cortex; dlCP, vlCP, dmCP, vmCP dorsolateral, ventrolateral, dorsomedial, and ventromedial caudate-putamen, respectively; ICinsular cortex; ISHH in situ hybridization histochemistry;MAC medial agranular cortex;MC motor cortex;NO nitric oxide;PPI prepulse inhibition;RT-PCR reverse transcription polymerase chain reaction; SS somatosensory cortex
Mol Neurobiol
[111]. Shank3B−/− mice also showed reduced SAPAP3,Homer1b/c, and PSD93 protein levels and reduced GluR2,NR2A, and NR2B subunit levels in striatal PSD fractions.
These molecular alterations have been associated with overallthinner and smaller PSDs and with reduced spine density onmedium-sized striatal spiny neurons in these mice [111].
Table 3 Clinical and preclinical evidence of Shank implication in the pathophysiology of psychotic disorders
Experimental paradigm/sample
Drug Dose androute ofadministration
Technique/assessment Outcome Reference
Shank3 mutantsoverexpression incultured neurons
– – Immunocytochemistry, western blot,immunoprecipitation,electrophysiological recording
Two Shank3 gene de novo mutations(STOP and Q321R) and twoinherited variations (R12C andR300C) identified in patients withASD differently affect dendritic spinedevelopment, morphology andneuronal activity
[110]
Shank1 (−/−) null mutantmice
– – Behavioral assessment Several social communicationimpairments, including decreasedlevels of ultrasonic vocalizations andscent-marking behavior
[180]
Shank3B−/− mice – – Behavioral assessment, western blot,electrophysiological recording
Mice exhibited self-injurious repetitivegrooming behaviors and deficits insocial interaction resembling autisticbehaviors; altered PSD compositionin the striatum, with overall thinnerand shorter PSDs and reduced spinedensity at medium spiny neurons
[111]
Rat dissociatedhippocampal neurons
Clozapine In vitro1.0 μM
Immunocytochemistry Clozapine increased Shank1a proteindensity along primary and secondarydendrites, increased dendritic spinedensity in primary dendrites,increased the number of filopodia andmushroom dendritic spines
[24]
Haloperidol In vitro0.1 μM
Immunocytochemistry Haloperidol decreased Shank1a proteindensity along secondary dendritesand decreased the number offilopodia in secondary dendrites
285 controls and 185patients withschizophrenia orschizoaffective disorder,with unaffected parents
– – Gene screening, variation analysis,assays in zebrafish and rathippocampal neurons
Two de novo mutations (R1117X andR536W) associated withschizophrenia and schizoaffectivedisorder cases. Assays in zebrafishand rat hippocampal neuronsrevealed behavior and differentiationdefects resulting from R1117Xmutation
[103]
199 schizophreniapatients, 206 healthycontrols, 77 subjects atrisk for psychosis
– – Genotyping of coding and promotervariants in Shank1, Shank2 andShank3 genes; Wechsler AdultIntelligence Scale (WAIS-R) digitspan test
Shank1 promoter variant rs3818280was associated with impairedauditory working memory inschizophrenia, which depended onpatients’ genotype (CC, CT, or TT)
Subjects at risk for psychosis carrying aT allele performed worse than thosecarrying CC genotype in the forwarddigit span subtest
[104]
1,158 Canadian and 456European individualswith ASD
– – Genotyping Two different Shank1 gene deletions ina 4-generation family in which malecarriers haveASD and in an unrelatedASD-affected patient
[62]
ACC anterior cingulate cortex; ASD autism spectrum disorders; cGMP cyclic guanosine monophosphate; dlCP, vlCP, dmCP, vmCP dorsolateral,ventrolateral, dorsomedial, and ventromedial caudate-putamen, respectively; IC insular cortex; ISHH in situ hybridization histochemistry;MAC medialagranular cortex;MC motor cortex; NO nitric oxide; PPI prepulse inhibition; RT-PCR reverse transcription polymerase chain reaction; SS somatosen-sory cortex
Mol Neurobiol
Evidence exists that ASD-associated mutations in Shank3may impair AMPAR and NMDAR signaling and may alterneurexin–neuroligin-mediated signaling in rat hippocampalneurons [112].
These findings may suggest that Shank protein aberrationscould contribute to cognitive symptoms in schizophrenia andcould be implicated in intellectual disability. Shank proteinsappear critically involved in the regulation of dendritic spinemorphology, architecture, and function. Despite being consid-ered mere scaffolding molecules, Shank proteins are pivotal inglutamatergic signaling and their defects are prominently impli-cated in intellectual disability and autistic diseases. Impairmentof activity-dependent synaptic plasticity has been suggested inthese conditions [49], and Shank anomalies have been describedto impair glutamate-mediated neurotransmission and dendriticspine morphology [41, 112].
DISC1
DISC1 Regulates Multiple Intracellular Pathwaysin Neurogenesis and Neurodevelopment
Disrupted-in-schizophrenia 1 (DISC1 ) is a susceptibility genefor major mental disorders including schizophrenia, bipolardisorder, and major depression. DISC1 has been first charac-terized by cloning of a chromosomal translocation that segre-gated with a spectrum of major mental illnesses in a Scottishfamily [113, 114]. Besides the PSD, DISC1 has also beenfound in other subcellular localizations including centrosome,nucleus, cytoskeleton, growth cones, membranes, and mito-chondria [115].
DISC1 is involved in several signaling pathways (includ-ing NMDAR-, GABA-, GSK3β-, and Wnt-mediated signal-ing pathways) and takes part in neurogenesis and neuraldevelopment in adult brain [116]. DISC1 interacts with otherproteins, such as neuregulin and dysbindin, along the Akt/GSK signaling pathway, whose dysfunctions have been im-plicated in schizophrenia pathophysiology [13, 117, 118].Indeed, aberrant interaction among these molecules may leadto NMDAR dysfunctions during neurodevelopment [13].DISC1 has a major role during neurodevelopment, since itmay regulate neuronal progenitor proliferation [119]. It maybe hypothesized that these functions are probably exerted viamodulation of GSK3beta/beta-catenin signaling, since DISC1inhibits GSK3beta activity through a direct interaction [119](Fig. 1). GSK3 inhibitors may normalize neural progenitorproliferation and schizophrenia-related behavioral abnormali-ties caused by DISC1 loss of function [119].
Recent studies have also highlighted the role of DISC1 indopaminergic signaling via D1Rs [78], which may represent afurther potential molecular crossroad between dopaminergicand glutamatergic dysfunctions in schizophrenia. Mice carry-ing DISC1 mutations are considered valuable animal models
of schizophrenia [120]. Mutant mice carrying a putativedominant-negative form ofDISC1 (DN-DISC1 ) showedmor-phological, immunohistochemical, and behavioral abnormal-ities that may resemble schizophrenia alterations [121]. In thesame model, several dopamine-related abnormalities, such asincreased striatal D2R and DAT expression, decreased basalextracellular dopamine levels in ventral striatum, and higherdopamine increase after methamphetamine were found[122]. Moreover, in a transgenic mouse model with inducibleexpression of mutant human DISC1 restricted to forebrainregions, mild enlargement of the lateral ventricles, atten-uation of neurite outgrowth in primary cortical neurons,reduced LIS1, SNAP-25 and endogenous DISC1 proteinlevels, and gender-dependent behavioral abnormalities weredescribed [123].
DISC1 in the Pathophysiology of Psychotic Disorders
DISC1 role in schizophrenia has been originally demonstratedin a large Scottish family sample, in which a balanced trans-location of this gene originated directly disrupted transcriptsthat co-segregated with the disease [113]. Based on thesefindings, several animal models have been created reproduc-ing DISC1 Scottish mutations [124]. Mutant DISC1 expres-sion in mice has been shown to result in reduced serineracemase protein levels, thereby reducing D-serine productionand leading to behavioral abnormalities consistent with aNMDAR hypofunction [121] (Table 4). The DISC1 geneticmousemodel of schizophrenia also displays impaired synapticconnections between frontal cortical neurons [125]. Overall,the comprehensive analysis of the numerous DISC1 mutantmice revealed selective deficits in working memory and inneural circuits (i.e., prefrontal cortex, hippocampus) involvedin the pathophysiology of schizophrenia and related mentaldisorders [126, 127]. Moreover, recent studies have demon-strated that DISC1 mutants display abnormal tangential mi-gration of cortical interneurons during embryonal stages, aswell as they have selective alterations in GABAergic neuralsubpopulations in cortex and hippocampus [128]. These find-ings are strikingly consistent with the aberrant interneuronslaminar distribution patterns described in postmortem schizo-phrenia patients [129, 130].
These findings suggest that mutations of DISC1 may im-pair synaptic morphology and glutamate–dopamine signaling,with region-specific distribution. These suggestions have beenconfirmed in in vitro studies demonstrating that DISC1 mu-tations may directly affect axon and spine morphology inhippocampal and cortical neurons [131].
To confirm preclinical data onDISC1 role in schizophreniapathophysiology, several DISC1 genetic variants have beenassociated with schizophrenia in different human populations[66, 132, 133].
Mol Neurobiol
Table 4 Clinical and preclinical evidence of DISC1 modulation by dopaminergic and glutamatergic stimuli and of its implication in the pathophys-iology of psychotic disorders
Experimental paradigm/sample Drug Dose androute ofadministration
Technique/assessment Outcome Reference
DN-DISC1 transgenic mice (micecarrying a dominant-negativeform of DISC1 , DN-DISC1 ,which is expressed under theCaMKII promoter)
– – RT-PCR, in situ hybridization,in vivo magnetic resonanceimaging,immunohistochemistry,behavioral assessment
DN-DISC1 transgenic mice showlateral ventricles enlargement,decreased parvalbuminimmunoreactivity in the cortex,and several behavioralimpairments, includinglocomotor hyperactivity, alteredsensorimotor gating, alteredolfactory-associated behavior,and increased immobility in theforced swim test
[121]
– – PET scan with [11C]raclopride,autoradiography of D2Rs with[3H]-Spiperone, real-timePCR, open field and in vivomicrodialysis aftermethamphetamine,immunoblot for DAT
Several dopamine-relatedabnormalities, such as increasedstriatal D2R and DATexpression,decreased basal extracellulardopamine levels in ventralstriatum, and higher dopamineincrease after methamphetamine(1 mg/kg i.p.)
[181]
Transgenic mice model of inducibleexpression of mutant humanDISC1 restricted in forebrainareas
– – Western blot,immunoprecipitation,histopathological andimmunohistochemical assays,magnetic resonance imaging,behavioral assessment
Mutant human DISC1 transgenicmice show enlargement of lateralventricles, attenuation of neuriteoutgrowth in primary corticalneurons, reduced LIS1, SNAP-25, and endogenous DISC1protein levels, gender-dependentbehavioral abnormalities
[123]
Pyramidal neurons from medialprefrontal cortex in two mousemodels: mDISC1 mice(expressing a truncated mouseDISC1 protein throughout theentire brain) and hDISC1 mice(expressing a truncated humanDISC1 protein in forebrainregions
– – Whole-cell patch clamprecordings
In cortical pyramidal neurons fromboth models the frequency ofspontaneous EPSCs is increased.Male mice are more affected inboth models, exhibiting increasesin the ratio of excitatory toinhibitory events. Sex-specificchanges in spontaneous IPSCsare observed in the mDISC1model
[125]
Acute adm. in L100P DISC1mutant mice
Haloperidol 0.4 mg/kg i.p. Behavioral assessment Partial amelioration in PPI deficits [81]Clozapine 3 mg/kg i.p. Behavioral assessment Partial amelioration in PPI deficits
clozapine abolishes thedisruption of latent inhibition in100P/100P mice
Repeated adm. in polyI:C/DN-DISC1 transgenic mice
Haloperidol 1 mg/kg oncea day oral
Behavioral assessment Suppression of increased MK-801-induced hyperactivity
[162]
Clozapine 3 mg/kg oncea day oral
Behavioral assessment Amelioration of cognitiveimpairment suppression ofincreased MK-801-inducedhyperactivity
21 days adm. in mice Haloperidol 0.05 mg/kgi.p.
Real-time quantitative RT-PCR(DISC1 mRNA)
No changes inDISC1 expression infrontal cortex and hippocampus
[80]
Olanzapine 0.04 mg/kgi.p.
Real-time quantitative RT-PCR(DISC1 mRNA)
Increased DISC1 expression infrontal cortex and hippocampus
No changes inDISC1 expression infrontal cortex and hippocampus
– – [113]
Mol Neurobiol
Other Proteins
Several other PSD and PSD-related proteins have been foundimpaired in preclinical and clinical studies on schizophrenia.
MAGUKs
The MAGUKs are a superfamily of PSD-95-related scaffold-ing proteins comprising SAP-102, PSD-93, or SAP-97.
SAP-102 mRNA expression has been found increased inhippocampal tissue after rat isolation rearing [134], which isconsidered a valid neurodevelopmental model of schizophre-nia [135, 136]. Expression of SAP-102 has also been founddecreased in striatum of schizophrenia patients [137], whereasgene expression in thalamus has been reported differentiallyexpressed in the brain of schizophrenia patients based on theage of the subjects: decreased in young schizophrenia patients[13] and increased in older ones [14].
Table 4 (continued)
Experimental paradigm/sample Drug Dose androute ofadministration
Technique/assessment Outcome Reference
A large Scottish family carrying abalanced (1;11)(q42.1;q14.3)translocation which segregateswith schizophrenia and relatedpsychiatric disorders
DNA sequencing, PCR, northernblot analysis
The (1;11)(q42.1;q14.3)translocation disrupts two novelgenes, named DISC1 andDISC2, suggesting these genesmay be considered candidategenes for susceptibility topsychiatric illness
A set of Chinese Han individuals,including 310 schizophrenicpatients and 400 controls
– – Genotyping Three short tandem repeat loci areassociated with schizophrenia:(ATCC)n1, D1S1621, and(ATCC)n2. The short tandemrepeats occur in intronicsequences near to a critical splicejunction that gives rise to theexpression of DISC1 isoforms
[66]
Case–control study; a set of 222French Caucasian schizophrenicpatients and 151 healthyunrelated controls
The DISC1 rs3738401 missensevariant is significantly morefrequent in ultra-resistance toantipsychotic treatment than intreatment-respondingschizophrenia patients,suggesting that DISC1 variantsmay have a functional influenceon response to antipsychotictreatment
[132]
A Japanese sample of 33schizophrenia patients and 29healthy comparison subjects
– – Magnetic resonance imaging,genotyping
The DISC1 Ser704Cyspolymorphismmay be relevant tomedication effect on brainmorphology in treatedschizophrenia patients: In Serhomozygote patients, the rightmedial superior frontal gyrusvolume is correlated with thedaily dose of antipsychoticmedication
[133]
BPRS-E expanded version of the Brief Psychiatric Rating Scale; CaMKII calcium/calmodulin-dependent protein kinase II; CGI-S Clinical GlobalImpression-Severity Scale; D2R D2 dopamine receptor; DAT dopamine transporter; DIGS Diagnosis Interview for Genetic Studies; DN-DISC1dominant-negative form of DISC1 ; EPSC excitatory postsynaptic currents; GAF Global Assessment of Functioning Scale; hDISC1 human DISC1;IPSC inhibitory postsynaptic currents; LIS1 lissencephaly-1; mDISC mouse DISC1; PANSS Positive and Negative Syndrome Scale; PCR polymerasechain reaction; polyI:C/DN-DISC1 transgenic mice polyriboinosinic–polyribocytidylic acid (polyI:C)-treated DN-DISC1 transgenic mice, a mousemodel of mental disorders obtained by inducing abnormal immune response during the perinatal period in mice with overexpression of the humandominant-negative form of DISC1 ; PPI prepulse inhibition; RT-PCR reverse transcription polymerase chain reaction; SNAP-25 synaptosomal-associated protein 25
Mol Neurobiol
Increased PSD-93 transcript, but decreased protein, hasbeen reported in the anterior cingulate cortex of schizophreniapatients [50], showing pattern of expression similar to PSD-95 in this region. A decreased protein expression of SAP-97has been shown in the PFC of schizophrenia patients [138].Notably, SAP-97 transcripts have been found upregulated inrat adult neocortex after acute injection of the NMDARnoncompetitive antagonists phencyclidine and dizocilpine,but not after administration of the indirect dopamine agonistscocaine and methamphetamine [139].
Kalirin
Kalirin is a GDP/GTP exchange factor (GEF), which interactswith PSD-95 and spinophilin through its C-terminus PDZ-binding motif (Fig. 1). Kalirin is an essential protein in matureexcitatory synapses implicated in spine and synapse formation[140].
KALRN KO mice show several structural, functional, andbehavioral alterations inherent to schizophrenia pathophysiol-ogy, including (a) decreased cortical, but not hippocampal,Rac1 activity; (b) decreased cortical, but not hippocampalspines density; (c) reduced cortical AMPAR currents; (d)impaired working memory and sociability; (e) reducedprepulse inhibition response; and (f) locomotor hyperactivity,reversed by clozapine [20].
Kalirin knockdown by RNA interference in cultured neuronshas been shown to affect dendrite morphology [141]. Decreasedspine density, impaired activity-dependent spine plasticity, anddecreased complexity of dendritic trees have been reported incortical pyramidal neurons of KALRN-null mice [141].
Kalirin has been implicated in different neuropsychiatric dis-eases, above all in schizophrenia. Multiple missense rare muta-tions in KALRN gene have been proposed as a risk factor forschizophrenia [142]. Furthermore, decreased Duo (the humanortholog of the murine Kalirin-7) mRNA levels and decreasedspine density have been found in PFC of schizophrenia patients[143]. Therefore, although still scarce, the above findings suggestthat kalirin may represent a promising candidate in the researchon schizophrenia and behavioral disorders pathophysiology.
Other preclinical and clinical studies have reported alter-ations in a number of other PSD-related proteins, such asneurofilament-light [13, 14, 50], SAPAP [144], or caldendrin[145]. However, the functional role and biological relevanceof these alterations are yet to be determined.
Overall, the molecular changes of PSD and PSD-relatedproteins discussed herein suggest a defect in PSD functioningin schizophrenia. Abnormal PSD functioning may cause gluta-mate dysfunctions and aberrant interplay with other neurotrans-mitter systems relevant to schizophrenia pathophysiology, suchas the dopaminergic and the serotonergic ones [146, 147].
PSD Protein Modulation by Antipsychotic Drugs
Antipsychotic therapies impact mainly, albeit not exclusively,dopaminergic neurotransmission, since all available antipsy-chotics act as antagonists at dopamine D2 receptors, althoughwith different degrees [148]. Notably, a large part of antipsy-chotics also interact with other dopamine receptors, besidesD2Rs [149]. However, evidence is accumulating that antipsy-chotic drugs may achieve part of their effects by inducinglong-term adaptive changes at glutamatergic postsynapticsites in brain regions relevant to psychosis, such as the PFC,the striatum, and the hippocampus [46, 147, 150]. A role inantipsychotic drug action has been postulated for PSD scaf-folding proteins, which are master organizers of postsynapticCa2+ networks contributing to drug-induced neuroplasticchanges [70]. In the next paragraphs, we will discuss the mostrecent findings regarding the involvement of PSD scaffoldingproteins in the mechanisms of action of typical and atypicalantipsychotics, as well as their possible role as moleculartargets for future antipsychotic strategies.
PSD-95
Preclinical studies have shown that PSD-95 gene expressionis modulated by both acute and chronic antipsychotic treat-ments, with a region-specific pattern and with effects depen-dent on treatment duration (Table 1). A recent report hasdescribed a decrease in PSD-95 expression in selected subre-gions of the rat cortex following acute haloperidol orquetiapine administration [20]. Conversely, PSD-95 expres-sion has been found significantly increased in cortical regionsby acute co-administration of haloperidol and valproate [20].
Unlike cortical regions, in striatum, no significant changesin PSD-95 expression have been recognized in acute para-digms [16, 22]. On the other hand, a 21-days chronic treat-ment by either typical or atypical antipsychotics has beenreported to increase PSD-95 expression in this region [16,22]. Therefore, PSD-95 modulation by chronic antipsychoticsmay underlie specific synaptic plasticity changes, potentiallyrelated to prolonged treatments and affecting glutamatergicsignaling in striatum. Provided the suppressive effects of PSD-95 on D1R-mediated signaling [35, 36], the increase in PSD-95 expression may contribute to slow down D1R-mediatedoverstimulation, which in turn results from prolonged D2Rblockade by long-term antipsychotic treatments.
It has been suggested that PSD-95 may be necessary foratypical antipsychotic action at serotonin receptors [46]. PSD-95 may participate in the clozapine molecular mechanisms ofaction in mice, since PSD-95null mice do not show theclozapine-mediated normalization of PCP-disrupted prepulseinhibition [46]. These effects are thought to involve 5-HT2A
receptors, since in PSD-95null mice, the selective antagonists
Mol Neurobiol
at 5-HT2A receptors, such as M100907 and SR46349B, failedto reverse PCP-disrupted prepulse inhibition [46]. The puta-tive role of PSD-95 in the modulation of serotonergic signal-ing has been also confirmed by the observation that theselective serotonin reuptake inhibitor antidepressants signifi-cantly increased PSD-95 expression in rat cortex and striatum[20].
Thus, PSD-95 may represent a valuable adaptor moleculethat cross-links glutamatergic, dopaminergic, and serotonergictransmission, thus providing a postsynaptic modulation ofthese systems. This particular action of PSD-95 may deservespecific attention for future antipsychotic strategies that wouldovercome the receptor level and would directly target thepostsynaptic signaling compartment.
Besides its modulation by current antipsychotic treatments,recent microRNA studies have indicated the possibility ofusing PSD-95 transcriptional regulation as a potential thera-peutic strategy for the treatment of neuropsychiatric disorders.MiR-485 has been demonstrated to negatively regulate spinesdensity, PSD-95 clustering, and surface GluR2 expression incultured hippocampal neurons, thus providing new ap-proaches to synaptic plasticity dysfunctions in severe neuro-logical diseases, such as Huntington’s or Alzheimer’s disease[120]. Notably, PSD-95 translation may be bidirectionallyregulated by mGluR-mediated signaling through the controlof phosphorylation status of both the fragile X mental retar-dation protein and the microRNA miR-125a [151].
Homer
Among Homer genes, the inducible immediate-early geneisoform Homer1a is known to respond to dopaminergic ma-nipulations [152]. A number of preclinical studies have shownthat Homer1a expression may be induced by acute antipsy-chotic administration in brain regions relevant to schizophre-nia pathophysiology and may be differently modulated bytypical and atypical antipsychotics, potentially according totheir dopaminergic receptor profile (Table 2). The high D2R-blocking antipsychotic haloperidol has been shown to induceHomer1a in all striatal subregions, with prominent impact onthe dorsal and lateral regions, and in both the core and the shellof the nucleus accumbens [18, 150, 153]. Another antipsy-chotic with high D2R affinity, (−)-sulpiride, may induceHomer1a in the ventrolateral subregion of the caudate-putamen and in the core of the accumbens [21]. Differentlyfrom typical antipsychotics, atypical antipsychotics have beendemonstrated to induce aHomer1a region-specific expressiondepending also on their affinity to receptors other than thedopaminergic ones, such as the serotonergic receptors, as wellas on the dose administered. Risperidone, which has elevatedD2R affinity but lower maximal binding effect comparedto haloperidol [106], may induce Homer1a expression inthe lateral regions of the caudate-putamen only [21].
Olanzapine-induced expression of Homer1a gene has beenreported in the core of the nucleus accumbens only [150],although high doses of the compound may also elicitHomer1a expression in the lateral regions of the caudate-putamen [21]. Ziprasidone, a high D2R affinity atypical anti-psychotic, differentially impacts Homer1a expression,according to dosage. At low doses, ziprasidone may producea striatum-specific Homer1a induction, while a wider geneinduction, spreading also to the cortex and the nucleusaccumbens, has been found at high doses, probably dependingon higher involvement of serotonergic receptors [16].Moreover, the expression of Homer1a in the striatum byziprasidone is significantly higher at high doses, which arecorrelated to the liability to extrapyramidal side effects inanimal models [154]. Also, the dopamine partial agonistaripiprazole has been demonstrated to differentially modulateHomer1a expression depending on the dose. A strong induc-tion of Homer1a expression has been observed in all caudate-putamen subregions by acute administration of lowaripiprazole doses [18]. High aripiprazole doses have no sig-nificant effects in the striatum but may induce Homer1a ex-pression in the cingulate cortex and in the inner and outerlayers of the frontal cortex [18]. These dose-dependent effectsmay be likely due to aripiprazole mixed agonist/antagonistactivity at pre- and postsynaptic D2Rs. Basically, lowaripiprazole doses are supposed to exert a prevalent antagonistactivity at postsynaptic D2Rs, thus directly inducing Homer1astriatal expression [155], whereas high doses may exert aprevalent agonist activity at presynaptic D2 autoreceptors,thereby having scarce effects on Homer1a expression [18].
Atypical antipsychotics with low D2R affinity, such as clo-zapine, or with unique D2R dissociation kinetics, such asquetiapine and sertindole, have been observed to modulateHomer1a expression with specific patterns in striatum.Indeed, clozapine has been shown to acutely induce Homer1aexpression in the nucleus accumbens only [16, 18]. Acuteclozapine may also induce Homer1a expression in the cortex(in the anterior cingulate, medial agranular, somatosensory andinsular cortices), probably due to its impact on serotonergicreceptors [16, 18]. Quetiapine, a fast dissociating D2R antago-nist, has been found to slightly induce Homer1a striatal expres-sion [153]. However, quetiapine may induce a robustHomer1aexpression in the cortex when acutely administered, whereassignificantly decreasing it in chronic paradigms [20]. This fea-ture may strengthen the hypothesis of a combined dopaminer-gic–serotonergic control of Homer1a expression in the cortex.Indeed, acute administration of the serotonergic-selective anti-psychotic sertindole has been found to reduceHomer1a expres-sion in the somatosensory and insular cortices [22].
Taken together, these lines of evidence support the view thatHomer1a could represent a molecular sensor of glutamatergicpostsynaptic involvement in the mechanism of action of antipsy-chotics. Moreover, since the above-mentioned findings suggest
Mol Neurobiol
thatHomer1a induction by antipsychoticsmay be related to theirpropensity to perturb dopamine transmission, the pattern ofHomer1a expression may be considered as a predictor of theliability of each antipsychotic to induce extrapyramidal sideeffects [153].
Homer1a has been demonstrated to preserve its expressionprofile in the striatum after chronic antipsychotic administra-tion, and it seems to be unaffected by the tolerance or desen-sitization phenomena observed for other immediate-earlygenes, such as c-fos [156, 157]. Indeed, haloperidol has beendemonstrated to modulate Homer1a expression with similarpatterns in both acute and chronic paradigms [18, 22].Nonetheless, some adaptive changes should not be ruled outafter chronic antipsychotic administration. Chronic clozapinetreatment, as opposed to acute treatment, has been reported toproduce no significant Homer1a changes in the caudate-putamen, whereas it may decrease Homer1a expression inthe cingulate cortex and in the inner layers of both frontal andparietal cortices.
In opposition to what observed after acute treatment,chronic ziprasidone may induce Homer1a expression onlyin the lateral regions of the caudate-putamen and in the coreof the nucleus accumbens [16]. Lastly, chronic aripiprazole,differently from acute administration, has been demonstratedto induce Homer1a in the lateral regions of the caudate-putamen while reducing it in the cingulate and in the innerlayers of the frontal cortex [18]. These effects might be part ofan adaptive response of the glutamatergic system to chronicantipsychotic treatment [18]. Overall, despite Homer1a mod-ulation by antipsychotics appears to be not susceptible oftolerance, acute or chronic treatment may result in differentpatterns of gene expression, probably accounting forneuroplastic adaptations triggered by prolonged treatments.
Besides the inducible isoform Homer1a , dopaminergicdrugs have also been demonstrated tomodulate the expressionof the constitutive isoforms of Homer1 gene, thus putativelyinducing direct rearrangements in synaptic architecture. Theacute administration of selective antagonists at D2Rs andD4Rs, as well as of a D2R partial agonist (i.e., terguride), hasbeen demonstrated to reduce Homer1b/c expression in thestriatum and in cortical subregions [155]. Nevertheless, theD1R selective antagonist SCH-23390 has been described toincrease Homer1b/c expression in the core of the nucleusaccumbens, while reducing it in the motor cortex [155].However, the evaluation ofHomer1b/c modulation after acuteor chronic antipsychotic administration has provided contrast-ing results. Acute haloperidol treatment, indeed, has beenobserved to reduceHomer1b/c expression in the ventrolateralcaudate-putamen, in the shell of the nucleus accumbens, andin the motor, somatosensory, and insular cortices [155],whereas no significant differences in expression have beenfound after a 16-day chronic treatment by haloperidol orquetiapine [16]. However, treatment duration may be a critical
factor to cause changes in Homer1b/c expression, which hasbeen found to increase in both striatum and cortex following a21-day haloperidol or sertindole treatment [22].
A recent work has also demonstrated that chronic treatmentwith the mood stabilizers lithium or valproate—which arewidely used as therapeutic add-on strategies to antipsychoticsin schizophrenia or in bipolar disorder—may directly impactthe expression of Homer1b/c and related synaptic genes(Shank and inositol 1,4,5 trisphosphate receptors) in corticaland subcortical regions [147]. Moreover, valproate add-on toeither haloperidol or quetiapine has been reported to elicitchanges in Homer1a expression patterns different from thoseinduced by either drugs when given alone [20]. These findingsconfirm the role of Homer1 genes in synaptic rearrangementstriggered by antipsychotic treatments.
Notably, a recent clinical study has reported that twoHomer1 polymorphisms may be associated with clinical re-sponse (expressed as improvement on PANSS subscores) inschizophrenia patients after different 4 week antipsychotictreatments [89]. Taken together, clinical and preclinical datasuggest that Homer1 genes might be crucial in the pathogen-esis and in the severity of psychotic symptoms, as well as indetermining the efficacy of antipsychotic drugs.
Shank
Several studies have demonstrated that typical and atypicalantipsychotics may differentially impact dendritic spine for-mation and synaptogenesis through the direct rearrangementof synaptic architecture, e.g. by modulating postsynaptic scaf-folding proteins, in particular Shank (Table 3). Clozapine andhaloperidol may specifically affect Shank protein density indendritic spines. In rat dissociated hippocampal neurons,in vitro clozapine administration has been found to increaseShank1a protein density along dendrites, whereas haloperidolcaused a decrease in Shank1a density [24]. The observedincrease may be explained as a possible specific increase inshaft synapses or as the formation of multiple synapses perdendritic spine. The different modulation of Shank expres-sion by clozapine and haloperidol may be consistent with adifferent impact on dendritic spine formation by the twoantipsychotics. Clozapine, indeed, but not haloperidol, hasbeen reported to increase dendritic spine density in primarydendrites and the number of filopodia in secondary den-drites and to up-regulate dendritic spines [24]. These ef-fects have been suggested to contribute, at least in part, tohigher efficacy of clozapine in treatment-resistant schizo-phrenia [158].
DISC1
As described above, DISC1 is a multifunctional scaffoldingprotein that interacts with different proteins involved in
Mol Neurobiol
neuronal migration, neurite outgrowth, cytoskeletal modula-tion, and signal transduction [159]. DISC1 participates in themaintenance of spine morphology and function by anchoringKalirin-7, a GEF for Rac1, and regulating the access ofKalirin-7 to Rac1, a step that is crucial for controlling Rac1activation in response to NMDAR stimulation [160] (Fig. 1).DISC1 may also interact with the Traf2 and Nck-interactingkinase, stabilizing the levels of postsynaptic density proteinsto regulate synaptic composition and activity [161].
Chronic administration of the atypical antipsychoticsolanzapine and risperidone at clinically relevant doses hasbeen shown to increase DISC1 mRNA expression in micefrontal cortex, whereas haloperidol and clozapine have noeffect. Olanzapine may increase DISC1 mRNA expressionalso in the hippocampus and a similar trend has been shownfor risperidone [80]. No data exist at present that may help tounderstand these differences in DISC1 gene modulation byspecific antipsychotics. Moreover, no studies have beenperformed measuring DISC1 protein modulation by differentantipsychotics. It is possible that these differences may derivefrom the specific receptor affinity profile of each antipsychot-ic. However, these results suggest that DISC1 may be impli-cated in antipsychotic mechanisms of action (Table 4), al-though additional studies are needed to dissect if its expres-sion changes may be specific for drug and for brain region.The increase in DISC1 expression by chronic atypical anti-psychotic treatment may suggest a putative impact on DISC1-mediated modulation of synaptic spines as a potential mech-anism of action of these agents.
Repeated clozapine, but not haloperidol, oral administra-tion has been reported to improve cognitive impairment in thepolyI:C-treated DN-DISC1 (polyI:C/DN-DISC1 ) transgenicmice [162], a mouse model of behavioral disorder obtained byinducing abnormal immune response during the perinatalperiod in mice through the overexpression of the humanDN-DISC1 . In this animal model, both clozapine and halo-peridol suppress the MK801-induced hyperactivity, whereasno effects on social behavior impairments have been observedwith both antipsychotics [162].
In clinical studies, DISC1 missense variants have beenassociated to schizophrenia patients who display ultra-resistance to treatments, defined as patients who continue toexperience positive psychotic symptoms despite a period ofclozapine therapy of at least 6 weeks and at least two unsuc-cessful previous trials with conventional or atypical antipsy-chotic drugs [132]. These results suggest that DISC1 variantsmay have a functional influence on the response to antipsy-chotic treatment. Indeed, a DISC1 Ser704Cys polymorphismhas been shown to affect the effects of therapies on brainmorphology in treated schizophrenia patients [133]. Patientscarrying the Cys allele variant show smaller grey mattervolume in supramarginal gyrus than Ser carriers [133]. Serhomozygotes show a direct correlation between the right
medial superior frontal gyrus volume and the daily dose (inhaloperidol equivalents) of antipsychotic medication, thusimplicating a direct role of DISC1 in the effects of antipsy-chotics on brain morphology [133].
Overall, clinical and preclinical data suggest that DISC1may represent an interesting postsynaptic molecule mediatingantipsychotic-induced cytoarchitectural plasticity.
Discussion
Understanding protein–protein interactions at PSD could helpto clarify the role of PSD proteins in normal and pathologicalsynaptic plasticity, as well as in response to pharmacologicalchallenges by psychotropic drugs. Scaffolding PSD proteinsmay modulate dendritic spines morphology and function inseveral ways. They contribute to regulate trafficking andlocalization of multiple proteins in spine microdomains, tobridge receptors to their intracellular effectors along secondmessenger pathways, and to allow cross-talk among differentneurotransmitter systems [7, 9, 146, 163]. Through this com-plex range of biological actions, PSD proteins may participateto activity-dependent long-term synaptic changes, which com-prise both enduring morphological rearrangements (e.g. spineformation, enlargement, or shrinking) and sustained changesin synaptic signaling strength (i.e., LTP or LTD [9]). PSDproteins may be considered valuable candidates in the patho-physiology of synaptic diseases, such as schizophrenia andschizophrenia-related disorders, and may represent intriguingtargets for future therapeutic approaches.
Since the PSD is primarily implicated in glutamatergicsignaling and glutamate-mediated activity-dependent synapticplasticity, abnormal PSD functioning may crucially contributeto glutamatergic dysfunctions and “synaptopathies” that havebeen hypothesized in schizophrenia [164]. PSD also repre-sents a site of integration and cross-talk between theglutamatergic and other neurotransmitter systems relevant toschizophrenia pathophysiology, such as the dopaminergic andthe serotonergic ones. Therefore, aberrant PSD functioningmay also underlie dysfunctions in dopamine–glutamate–sero-tonin cross-talk [5, 165]. It appears that loss of function or lackof some PSD proteins, such as PSD-95, Homer, or Shank,may impair glutamatergic signaling, in turn disruptingactivity-dependent synaptic plasticity as well as dendriticspine architecture and morphology. Although more studiesneed to be carried out, somemolecular mechanisms putativelyimplicating PSD in the pathophysiology of schizophrenia andbehavioral disorders, such as ASD, may be hypothesized.
The major function of PSD-95 proteins is to connectNMDARs to their intracellular effectors [30, 166]; therefore,aberrant PSD-95 may strongly affect glutamatergic transmis-sion NMDAR mediated. Dysfunctions of PSD-95 proteinsmay also increase surface D1Rs (due to disruption of the
Mol Neurobiol
PSD-95-mediated D1R internalization) [36], as well as influ-ence AMPAR signaling [166], and impair 5-HT2A/5-HT2C
receptor trafficking [43]. These abnormal neurotransmitterinteractions might lead to a multireceptor impairment, whichmay result in reduced glutamatergic signaling and increasedD1R transmission and aberrant serotonergic signaling in post-synaptic neurons. Moreover, PSD-95 has been demonstratedto recruit other PSD components to regulate dendritic spinearchitecture. The overexpression of PSD-95 not only termi-nates dendritic spine enlargement, but also promotes genera-tion of multi-innervated spines [33]. Therefore, PSD-95 dys-functions may cause synaptic shrinkage and may affect long-term changes in synaptic strength, thereby impairing synapticplasticity. On the other hand, the excessive intracellular signal-ing via the PSD-95/NOS pathway has been considered detri-mental, thus predisposing to glutamatergic hyperexcitability insome clinical conditions, e.g. stroke [167].
The different isoforms of Homer proteins are in recip-rocal and functional balance. Basically, a balanced ratiobetween long and short Homer proteins may preservepostsynaptic neuron potentials and homeostatic scaling[94, 95], as well as glutamatergic signaling [66, 168],dopamine–glutamate interplay [21], surface ion channelsopening and functioning [69], and dendritic spines archi-tecture [72, 95, 169]. Impairment of this balance, such asby genetic manipulations, have been reported to causerelevant neurochemical, structural, and behavioral alter-ations [30, 85, 87]. The loss or reduction of functionallong Homer proteins may depend on either genetic causesor overexpression of short Homer isoforms, such asHomer1a. Reduction of Homer1b/c expression or increaseof Homer1a expression may disrupt Homer1b/c-mediatedclusters that connect glutamate receptors to their intracel-lular effectors [168], thereby modifying local ionic con-centrations (due to altered Ca2+ release from internalstores and ion flow from surface ion channels) [70], andaffecting synaptic architecture, since Homer proteinsbridge PSD components to cytoskeleton [95]. Moreover,reduction of Homer1b/c expression or excessive Homer1ainduction may ultimately lead to downscaling of local poten-tials [94], thus putatively causing a reduced postsynapticneuron activation, which is at least in part functionally equiv-alent to glutamatergic hypofunction.
Shank proteins have been implicated in dendritic spinesmaturation and enlargement as well as in the regulation of thesynaptic signaling strength. Transfection of the Shank3 iso-form in cerebellar granule cells has been found to induce theformation of new synapses and to increase NMDAR- andAMPAR-mediated currents [101]. It has also been reportedthat the Shank3 isoform elicits transynaptic changes in pre-synaptic protein levels and function, thereby modulating syn-aptic transmission at excitatory, glutamatergic synapses [41,112]. Expression in cultured neurons of Shank isoforms
carrying ASD-associated mutations has been found to inter-fere with excitatory signaling, in particular reducing AMPAand NMDA receptor-mediated excitatory postsynaptic cur-rents [41, 112]. On the light of these observations, it couldbe hypothesized that dysfunctions in Shank proteins maycause reduced glutamatergic signaling and may lead to severemorphological aberrations in dendritic spines.
Alterations of other PSD and PSD-related proteins havebeen less studied; however, considered their physiologicalrole it is attempting to hypothesize that their dysfunctionscould also affect glutamatergic signaling, synaptic plasticity,and dendritic spines morphology. Further studies in this fieldare warranted.
Despite the above-mentioned observations, in several stud-ies the expression of PSD genes has been found increased,rather than decreased, in schizophrenia patients [50]. Severalexplanations can be proposed for this finding. For example,methodological bias, such as the homogeneity of patients’sample selection in the case of postmortem studies (i.e.“mixed” schizophrenia diagnosis, such as schizoaffective dis-order, or not well defined pre-mortem antipsychotic treat-ment), may account for these results. Also, PSD protein geneexpression may be increased as a compensatory mechanism topreserve physiological PSD functioning. Nonetheless, it can-not be excluded that molecular mechanisms leading to schizo-phrenia may include excessive PSD protein activity (e.g.glutamatergic excitotoxicity) [170] and/or the formation ofnon-functional synapses that might impair cerebral connectiv-ity [59]. Despite the fact that PSD proteins functions havebeen mostly studied in striatal tissue of adult rats, it can behypothesized that PSD proteins may exert different molecularactions depending on the brain region, neuronal type (i.e.,striatal neurons vs. cortical neurons), and neurodevelopmentalstage. Should this be the case, it is to be expected that PSDprotein dysfunctions may be different and lead to differentconsequences based on the brain region and the age theyoccur. Recent studies have demonstrated that currently avail-able antipsychotics may modulate gene expression of somePSD proteins. Both typical and atypical antipsychotics havebeen reported to induce Homer1a expression in striatum,either after acute or chronic administration [16, 150, 153,171]. Transient Homer1a induction by antipsychotics maybe a mechanism to reduce postsynaptic hyperactivation con-sequent to stimulation of D1R by residual synaptic dopamine.Also,Homer1a inductionmay directly depend, at least in part,upon the blockade of postsynaptic D2Rs by antipsychotics.Therefore, Homer1a induction may be a sensible marker ofdopamine perturbation by antipsychotics. Consistent with thisview, Homer1a may be differently induced by antipsychoticsaccording to their dopaminergic profile. Indeed, higherHomer1a induction has been observed by antipsychoticsdisplaying a higher degree of D2R blockade [21].Furthermore, antipsychotics have also been found to acutely
Mol Neurobiol
induce other short Homer isoforms, such as Ania-3 [18, 153],or other inducible PSD-related genes, such as Arc [19, 172].
Constitutive PSD genes, such as Homer1b and PSD-95 ,have been poorly affected in acute paradigms of antipsychoticadministration [155], whereas significant changes have beenobserved after prolonged antipsychotic treatments. Expressionof both PSD-95 and Homer1b genes has been found in-creased after chronic haloperidol treatment, whereasziprasidone only mildly increased gene expression, andsertindole had no effects [16, 22]. These findings may suggestthat at least some antipsychotics may exert their chroniceffects by modulating PSD protein functions. Antipsychotic-mediated increase of these molecules may determine long-term improvements of glutamatergic signaling, as well as ofdopamine–glutamate–serotonin interplay, thus contributing torestore synaptic plasticity processes, and dendritic spinemorphology.
These suggestions could possibly open new avenues ofinvestigation for schizophrenia treatment. Current therapiesfor schizophrenia are jeopardized by high rates of poor re-sponse, in part as a result of the lack of really true innovativemechanisms of action among available antipsychotics [173].Novel therapeutic approaches are therefore mandatory toovercome, at least in part, the drawbacks of schizophreniatherapy. One possible novel therapeutic target may be repre-sented by the modulation of PSD proteins discussed herein, aswell as other PSD proteins that have been less characterizedsince now for their involvement in schizophrenia, such asGRIP, SAP102, and Preso1. Targeting PSD proteins to unveilnovel treatments for behavioral disorders should be balancedby the observation that mutations in the gene sequence ofthese proteins have been linked to pathophysiology of intel-lectual disability and autism-like phenotypes. Therefore, itwill be fundamental to consider this limitation and evaluateconsequently the strategy to undertake in designing new ther-apeutics possibly acting at the PSD.
A recent pharmaceutical strategy has pointed on the gener-ation of cell permeable peptides (CPPs), i.e., small peptidesthat may cross the blood–brain barrier and pass through neu-ron surface membrane [174]. Small inhibitory peptides areCPPs that link scaffolding proteins, such as PSD-95, by pro-tein–protein interaction. By this mechanism, small inhibitorpeptides prevent scaffolding proteins to interact with theirtargets [174]. An anti-PSD-95 small inhibitory peptide iscurrently under evaluation, also in humans, in stroke, to re-duce the PSD-95/NOS pathway stimulation consequent tocortical ischemic damage [175].
The possibility to use small inhibitory peptides also in schizo-phrenia (e.g. to block Homer1a interaction with Homer1b) hasnot been tested to date. However, it may represent an intriguingstrategy for future researches. Another putative target could bethe inhibition of PSD-95/D1R interaction selectively in thecortex, in order to increase dopamine signaling in this region,
since some symptoms of schizophrenia have been attributed tolow cortical D1R-mediated signaling [176].
Taken together, these observations indicate that PSD pro-teins are crucial molecules in synaptic architecture and func-tion, involved in neuropsychiatric disorders pathophysiology,mostly in schizophrenia and related diseases, with the possi-bility to be targeted, directly or indirectly, by novel treatmentstrategies. Finally, considering the present lack of specificpharmacological treatments for autism and the deep involve-ment of PSD proteins in autism spectrum disorders, furtherstudies exploring PSD proteins as potential targets in theseclasses of child diseases are warranted.
Acknowledgments All members of the Laboratory of Molecular andTranslational Psychiatry contributed to ideas and comments on the pres-ent work.
Conflict of Interest Andrea de Bartolomeis has received unrestrictedresearch funding from Astra Zeneca, Janssen-Cilag, and Lundbeck. Thefunding was made available to the Department of Neuroscience,University of Naples Federico II. He has received honoraria as speakerat educational activity sponsored by Astra-Zeneca Italia, Janssen-CilagItaly, Eli Lilly, and Bristol-Myers Squibb.
All other authors declare that, except for income received from ourprimary employer, no financial support or compensation has been re-ceived from any individual or corporate entity over the past 3 years forresearch or professional service, and there are no personal financialholdings that could be perceived as constituting a potential conflict ofinterest.
References
1. Balu DT, Coyle JT (2011) Neuroplasticity signaling pathwayslinked to the pathophysiology of schizophrenia. NeurosciBiobehav Rev 35(3):848–870
2. Bennett MR (2011) Schizophrenia: susceptibility genes, dendritic-spine pathology and gray matter loss. Progress in neurobiology95(3):275–300
3. Van Winkel R, Esquivel G, Kenis G, Wichers M, Collip D,Peerbooms O, Rutten B, Myin-Germeys I, Van Os J (2010)Review: genome-wide findings in schizophrenia and the role ofgene-environment interplay. CNS Neurosci Ther 16(5):e185–192
4. Lisman JE, Coyle JT, Green RW, Javitt DC, Benes FM, Heckers S,Grace AA (2008) Circuit-based framework for understanding neu-rotransmitter and risk gene interactions in schizophrenia. TrendsNeurosci 31(5):234–242
5. Zhang J, Lewis SM, Kuhlman B, Lee AL (2013) Supertertiarystructure of the MAGUK core from PSD-95. Structure 21(3):402–413
6. Kennedy MB (2000) Signal-processing machines at the postsynap-tic density. Science 290(5492):750–754
7. de Bartolomeis A, Fiore G (2004) Postsynaptic density scaffoldingproteins at excitatory synapse and disorders of synaptic plasticity:implications for human behavior pathologies. International reviewof neurobiology 59:221–254
8. Murakoshi H, Yasuda R (2012) Postsynaptic signaling during plas-ticity of dendritic spines. Trends Neurosci 35(2):135–143
9. Boeckers TM (2006) The postsynaptic density. Cell Tissue Res326(2):409–422
Mol Neurobiol
10. Bayes A, van de Lagemaat LN, Collins MO, Croning MD, WhittleIR, Choudhary JS, Grant SG (2011) Characterization of the prote-ome, diseases and evolution of the human postsynaptic density. NatNeurosci 14(1):19–21
11. SnyderMA, GaoWJ (2013)NMDAhypofunction as a convergencepoint for progression and symptoms of schizophrenia. Front CellNeurosci 7:31
12. Coyle JT, Basu A, Benneyworth M, Balu D, Konopaske G (2012)Glutamatergic synaptic dysregulation in schizophrenia: therapeuticimplications. Handb Exp Pharmacol 213:267–295
13. Clinton SM, Meador-Woodruff JH (2004) Abnormalities of theNMDA receptor and associated intracellular molecules in the thala-mus in schizophrenia and bipolar disorder. NeuropsychopharmacolOff Publ Am Coll Neuropsychopharmacol 29(7):1353–1362
14. Clinton SM, Haroutunian V, Davis KL, Meador-Woodruff JH(2003) Altered transcript expression of NMDA receptor-associated postsynaptic proteins in the thalamus of subjects withschizophrenia. Am J Psychiatry 160(6):1100–1109
15. Grabrucker AM (2013) A role for synaptic zinc in ProSAP/ShankPSD scaffold malformation in autism spectrum disorders. DevNeurobiol. doi:10.1002/dneu.22089
16. Iasevoli F, Ambesi-Impiombato A, Fiore G, Panariello F,Muscettola G, de Bartolomeis A (2011) Pattern of acute inductionof Homer1a gene is preserved after chronic treatment with first- andsecond-generation antipsychotics: effect of short-term drug discon-tinuation and comparison with Homer1a-interacting genes. JPsychopharmacol 25(7):875–887
18. Tomasetti C, Dell’Aversano C, Iasevoli F, de Bartolomeis A (2007)Homer splice variants modulation within cortico-subcortical regionsby dopamine D2 antagonists, a partial agonist, and an indirectagonist: implication for glutamatergic postsynaptic density in anti-psychotics action. Neuroscience 150(1):144–158
19. Brennand KJ, Simone A, Jou J, Gelboin-Burkhart C, Tran N,Sangar S, Li Y, Mu Y, Chen G, Yu D, McCarthy S, Sebat J, GageFH (2011) Modelling schizophrenia using human induced pluripo-tent stem cells. Nature 473(7346):221–225
20. Cahill ME, Xie Z, Day M, Photowala H, Barbolina MV, Miller CA,Weiss C, Radulovic J, Sweatt JD, Disterhoft JF, Surmeier DJ,Penzes P (2009) Kalirin regulates cortical spine morphogenesisand disease-related behavioral phenotypes. Proc Natl Acad Sci US A 106(31):13058–13063
21. Iasevoli F, Fiore G, Cicale M, Muscettola G, de Bartolomeis A(2010) Haloperidol induces higher Homer1a expression than risper-idone, olanzapine and sulpiride in striatal sub-regions. PsychiatryRes 177(1–2):255–260
22. Iasevoli F, Tomasetti C, Marmo F, Bravi D, Arnt J, deBartolomeis A (2010) Divergent acute and chronic modulationof glutamatergic postsynaptic density genes expression by the anti-psychotics haloperidol and sertindole. Psychopharmacol 212(3):329–344
23. Hashimoto R, Tankou S, Takeda M, Sawa A (2007) Postsynapticdensity: a key convergent site for schizophrenia susceptibility fac-tors and possible target for drug development. Drugs Today (Barc)43(9):645–654
24. Critchlow HM, Maycox PR, Skepper JN, Krylova O (2006)Clozapine and haloperidol differentially regulate dendritic spineformation and synaptogenesis in rat hippocampal neurons. MolCell Neurosci 32(4):356–365
25. Meshul CK, Stallbaumer RK, Taylor B, Janowsky A (1994)Haloperidol-induced morphological changes in striatum are associ-ated with glutamate synapses. Brain research 648(2):181–195
26. O’Connor JA, Muly EC, Arnold SE, Hemby SE (2007) AMPAreceptor subunit and splice variant expression in the DLPFC ofschizophrenic subjects and rhesus monkeys chronically adminis-tered antipsychotic drugs. Schizophr Res 90(1–3):28–40
27. Fumagalli F, Frasca A, Racagni G, Riva MA (2008) Dynamicregulation of glutamatergic postsynaptic activity in rat prefrontalcortex by repeated administration of antipsychotic drugs. MolPharmacol 73(5):1484–1490
28. Purkayastha S, Ford J, Kanjilal B, Diallo S, Del Rosario IJ,Neuwirth L, El Idrissi A, Ahmed Z, Wieraszko A, Azmitia EC,Banerjee P (2012) Clozapine functions through the prefrontal cortexserotonin 1A receptor to heighten neuronal activity via calmodulinkinase II-NMDA receptor interactions. J Neurochem 120(3):396–407
29. Kim E, ShengM (2004) PDZ domain proteins of synapses. Nat RevNeurosci 5(10):771–781
30. Dosemeci A, Makusky AJ, Jankowska-Stephens E, Yang X, SlottaDJ, Markey SP (2007) Composition of the synaptic PSD-95 com-plex. Mol Cell Proteom MCP 6(10):1749–1760
31. Ricciardi S, Ungaro F, Hambrock M, Rademacher N, Stefanelli G,Brambilla D, Sessa A,Magagnotti C, Bachi A, Giarda E, Verpelli C,Kilstrup-Nielsen C, Sala C, Kalscheuer VM, Broccoli V (2012)CDKL5 ensures excitatory synapse stability by reinforcing NGL-1-PSD95 interaction in the postsynaptic compartment and is im-paired in patient iPSC-derived neurons. Nat Cell Biol 14(9):911–923
32. Steiner P, HigleyMJ, XuW, Czervionke BL, Malenka RC, SabatiniBL (2008) Destabilization of the postsynaptic density by PSD-95serine 73 phosphorylation inhibits spine growth and synaptic plas-ticity. Neuron 60(5):788–802
33. Nikonenko I, Boda B, Steen S, Knott G, Welker E, Muller D(2008) PSD-95 promotes synaptogenesis and multiinnervatedspine formation through nitric oxide signaling. J Cel Biol 183(6):1115–1127
34. Macgillavry HD, Song Y, Raghavachari S, Blanpied TA (2013)Nanoscale scaffolding domains within the postsynaptic densityconcentrate synaptic AMPA receptors. Neuron 78(4):615–622
35. Zhang J, Vinuela A, Neely MH, Hallett PJ, Grant SG, Miller GM,Isacson O, Caron MG, Yao WD (2007) Inhibition of the dopamineD1 receptor signaling by PSD-95. J Biol Chem 282(21):15778–15789
36. Zhang J, Xu TX, Hallett PJ, WatanabeM, Grant SG, Isacson O, YaoWD (2009) PSD-95 uncouples dopamine-glutamate interaction inthe D1/PSD-95/NMDA receptor complex. J Neurosci Off J SocNeurosci 29(9):2948–2960
37. Porras G, Berthet A, Dehay B, Li Q, Ladepeche L, Normand E,Dovero S, Martinez A, Doudnikoff E, Martin-Negrier ML, ChuanQ, Bloch B, Choquet D, Boue-Grabot E, Groc L, Bezard E (2012)PSD-95 expression controls L-DOPA dyskinesia through dopamineD1 receptor trafficking. J Clin Investig 122(11):3977–3989
38. Sun P,Wang J, GuW, ChengW, Jin GZ, Friedman E, Zheng J, ZhenX (2009) PSD-95 regulates D1 dopamine receptor resensitization,but not receptor-mediated Gs-protein activation. Cell Res 19(5):612–624
39. Gerfen CR (1992) The neostriatal mosaic: multiple levels of com-partmental organization. Trends Neurosci 15(4):133–139
40. Xiao J, Dai R, Negyessy L, Bergson C (2006) Calcyon, a novelpartner of clathrin light chain, stimulates clathrin-mediated endocy-tosis. J Biol Chem 281(22):15182–15193
41. Clinton SM, Ibrahim HM, Frey KA, Davis KL, Haroutunian V,Meador-Woodruff JH (2005) Dopaminergic abnormalities in selectthalamic nuclei in schizophrenia: involvement of the intracellularsignal integrating proteins calcyon and spinophilin. Am J Psychiatry162(10):1859–1871
42. Koh PO, Bergson C, Undie AS, Goldman-Rakic PS, Lidow MS(2003) Up-regulation of the D1 dopamine receptor-interacting
protein, calcyon, in patients with schizophrenia. Arch Gen Psychiatr60(3):311–319
43. Ha CM, Park D, Han JK, Jang JI, Park JY, Hwang EM, Seok H,Chang S (2012) Calcyon forms a novel ternary complex withdopamine D1 receptor through PSD-95 protein and plays a role indopamine receptor internalization. J Biol Chem 287(38):31813–31822
44. Fischer R, Fotin-MleczekM, Hufnagel H, Brock R (2005) Break onthrough to the other side-biophysics and cell biology shed light oncell-penetrating peptides. Chembiochem 6(12):2126–2142
45. Cao C, Rioult-Pedotti MS, Migani P, Yu CJ, Tiwari R, Parang K,Spaller MR, Goebel DJ, Marshall J (2013) Impairment of TrkB-PSD-95 signaling in Angelman syndrome. PLoS Biol 11(2):e1001478
46. Abbas AI, Yadav PN, Yao WD, Arbuckle MI, Grant SG, CaronMG, Roth BL (2009) PSD-95 is essential for hallucinogen andatypical antipsychotic drug actions at serotonin receptors. JNeurosci Off J Soc Neurosci 29(22):7124–7136
47. du Bois TM, Newell KA, Huang XF (2012) Perinatal phencyclidinetreatment alters neuregulin 1/erbB4 expression and activation in laterlife. Eur Neuropsychopharmacol J Eur Coll Neuropsychopharmacol22(5):356–363
48. Hermes G, Li N, Duman C, Duman R (2011) Post-weaning chronicsocial isolation produces profound behavioral dysregulation withdecreases in prefrontal cortex synaptic-associated protein expres-sion in female rats. Physiol Behav 104(2):354–359
49. Clinton SM, Haroutunian V, Meador-Woodruff JH (2006) Up-regulation of NMDA receptor subunit and post-synaptic densityprotein expression in the thalamus of elderly patients with schizo-phrenia. J Neurochem 98(4):1114–1125
50. Kristiansen LV, Beneyto M, Haroutunian V, Meador-Woodruff JH(2006) Changes in NMDA receptor subunits and interacting PSDproteins in dorsolateral prefrontal and anterior cingulate cortexindicate abnormal regional expression in schizophrenia. MolPsychiatry 11(8):737–747, 705
51. Funk AJ, Rumbaugh G, Harotunian V, McCullumsmith RE,Meador-Woodruff JH (2009) Decreased expression of NMDAreceptor-associated proteins in frontal cortex of elderly patients withschizophrenia. NeuroReport 20(11):1019–1022
52. Toro C, Deakin JF (2005) NMDA receptor subunit NRI and post-synaptic protein PSD-95 in hippocampus and orbitofrontal cortex inschizophrenia and mood disorder. Schizophr Res 80(2–3):323–330
53. Kristiansen LV, Patel SA, Haroutunian V, Meador-Woodruff JH(2010) Expression of the NR2B-NMDA receptor subunit and itsTbr-1/CINAP regulatory proteins in postmortem brain suggest al-tered receptor processing in schizophrenia. Synapse 64(7):495–502
54. Hahn CG, Wang HY, Cho DS, Talbot K, Gur RE, Berrettini WH,Bakshi K, Kamins J, Borgmann-Winter KE, Siegel SJ, Gallop RJ,Arnold SE (2006) Altered neuregulin 1-erbB4 signaling contributesto NMDA receptor hypofunction in schizophrenia. Nat Med 12(7):824–828
55. Barros CS, Calabrese B, Chamero P, Roberts AJ, Korzus E, LloydK, Stowers L, Mayford M, Halpain S, Muller U (2009) Impairedmaturation of dendritic spines without disorganization of corticalcell layers in mice lacking NRG1/ErbB signaling in the centralnervous system. Proc Natl Acad Sci U S A 106(11):4507–4512
56. Qiu C, Tarrant MK, Choi SH, Sathyamurthy A, Bose R, Banjade S,Pal A, Bornmann WG, Lemmon MA, Cole PA, Leahy DJ (2008)Mechanism of activation and inhibition of the HER4/ErbB4 kinase.Structure 16(3):460–467
57. Ferrer I, Martinez A, Boluda S, Parchi P, BarrachinaM (2008) Brainbanks: benefits, limitations and cautions concerning the use of post-mortem brain tissue for molecular studies. Cell Tissue bank 9(3):181–194
58. Pandey GN, Dwivedi Y (2010) What can post-mortem studies tellus about the pathoetiology of suicide? Futur Neurol 5(5):701–720
59. Feyder M, Karlsson RM, Mathur P, Lyman M, Bock R, MomenanR, Munasinghe J, Scattoni ML, Ihne J, Camp M, Graybeal C,Strathdee D, Begg A, Alvarez VA, Kirsch P, Rietschel M, CichonS, Walter H, Meyer-Lindenberg A, Grant SG, Holmes A (2010)Association of mouse Dlg4 (PSD-95) gene deletion and humanDLG4 gene variation with phenotypes relevant to autism spectrumdisorders andWilliams’ syndrome. Am J Psychiatry 167(12):1508–1517
60. Kim KC, Kim P, Go HS, Choi CS, Park JH, Kim HJ, Jeon SJ, DelaPena IC, Han SH, Cheong JH, Ryu JH, Shin CY (2013) Male-specific alteration in excitatory post-synaptic development and so-cial interaction in pre-natal valproic acid exposure model of autismspectrum disorder. J Neurochem 124(6):832–843
61. Baron-Cohen S, Lombardo MV, Auyeung B, Ashwin E,Chakrabarti B, Knickmeyer R (2011) Why are autism spectrumconditions more prevalent in males? PLoS Biol 9(6):e1001081
62. Sato D, Lionel AC, Leblond CS, Prasad A, Pinto D, Walker S,O’Connor I, Russell C, Drmic IE, Hamdan FF, Michaud JL, EndrisV, Roeth R, Delorme R, Huguet G, Leboyer M, RastamM, GillbergC, Lathrop M, Stavropoulos DJ, Anagnostou E, Weksberg R,Fombonne E, Zwaigenbaum L, Fernandez BA, Roberts W,Rappold GA, Marshall CR, Bourgeron T, Szatmari P, Scherer SW(2012) SHANK1 deletions in males with autism spectrum disorder.Am J Hum Genet 90(5):879–887
63. Tsai NP, Wilkerson JR, Guo W, Maksimova MA, DeMartino GN,Cowan CW, Huber KM (2012) Multiple autism-linked genes me-diate synapse elimination via proteasomal degradation of a synapticscaffold PSD-95. Cell 151(7):1581–1594
64. Abi-Dargham A (2003) Probing cortical dopamine function inschizophrenia: what can D1 receptors tell us? World Psychiatry2(3):166–171
65. Shiraishi-Yamaguchi Y, Furuichi T (2007) The Homer family pro-teins. Genome Biol 8(2):206
66. Cao F, Zhang H, Feng J, Gao C, Li S (2013) Association study ofthree microsatellite polymorphisms located in introns 1, 8, and 9 ofDISC1 with schizophrenia in the Chinese Han population. GenetTest Mol Biomarkers 17(5):407–411
67. Ango F, Prezeau L, Muller T, Tu JC, Xiao B, Worley PF, Pin JP,Bockaert J, Fagni L (2001) Agonist-independent activation of me-tabotropic glutamate receptors by the intracellular protein Homer.Nature 411(6840):962–965
68. Tu JC, Xiao B, Yuan JP, Lanahan AA, Leoffert K, Li M, Linden DJ,Worley PF (1998) Homer binds a novel proline-rich motif and linksgroup 1 metabotropic glutamate receptors with IP3 receptors.Neuron 21(4):717–726
69. Chen X, Vinade L, Leapman RD, Petersen JD, Nakagawa T,Phillips TM, Sheng M, Reese TS (2005) Mass of the postsynapticdensity and enumeration of three keymolecules. Proc Natl Acad SciU S A 102(32):11551–11556
70. ChengMC, Lu CL, Luu SU, Tsai HM, Hsu SH, Chen TT, Chen CH(2010) Genetic and functional analysis of the DLG4 gene encodingthe post-synaptic density protein 95 in schizophrenia. PloS ONE5(12):e15107
71. Sala C, Piech V, Wilson NR, Passafaro M, Liu G, Sheng M (2001)Regulation of dendritic spine morphology and synaptic function byShank and Homer. Neuron 31(1):115–130
72. Sala C, Futai K, Yamamoto K, Worley PF, Hayashi Y, Sheng M(2003) Inhibition of dendritic spine morphogenesis and synaptictransmission by activity-inducible protein Homer1a. J Neurosci OffJ Soc Neurosci 23(15):6327–6337
73. Inoue Y, Udo H, Inokuchi K, Sugiyama H (2007) Homer1a regu-lates the activity-induced remodeling of synaptic structures in cul-tured hippocampal neurons. Neuroscience 150(4):841–852
74. Ueta Y, Yamamoto R, Sugiura S, Inokuchi K, Kato N (2008) Homer1a suppresses neocortex long-term depression in a cortical layer-specific manner. J Neurophysiol 99(2):950–957
Mol Neurobiol
75. Ronesi JA, Huber KM (2008) Homer interactions are necessary formetabotropic glutamate receptor-induced long-term depression andtranslational activation. J Neurosci Off J Soc Neurosci 28(2):543–547
76. Fujiyama K, Kajii Y, Hiraoka S, Nishikawa T (2003) Differentialregulation by stimulants of neocortical expression of mrt1, arc, andhomer1amRNA in the rats treated with repeated methamphetamine.Synapse 49(3):143–149
77. Fourgeaud L,Mato S, Bouchet D, Hemar A,Worley PF,Manzoni OJ(2004) A single in vivo exposure to cocaine abolishesendocannabinoid-mediated long-term depression in the nucleusaccumbens. J Neurosci Off J Soc Neurosci 24(31):6939–6945
78. Cochran SM, Fujimura M, Morris BJ, Pratt JA (2002) Acute anddelayed effects of phencyclidine upon mRNA levels of markers ofglutamatergic and GABAergic neurotransmitter function in the ratbrain. Synapse 46(3):206–214
79. Iasevoli F, Polese D, Ambesi-Impiombato A, Muscettola G, deBartolomeis A (2007) Ketamine-related expression of glutamatergicpostsynaptic density genes: possible implications in psychosis.Neurosci Lett 416(1):1–5
80. Chiba S, Hashimoto R, Hattori S, Yohda M, Lipska B, WeinbergerDR, Kunugi H (2006) Effect of antipsychotic drugs on DISC1 anddysbindin expression in mouse frontal cortex and hippocampus. JNeural Transm 113(9):1337–1346
82. Hu JH, Park JM, Park S, Xiao B, Dehoff MH, Kim S, Hayashi T,Schwarz MK, Huganir RL, Seeburg PH, Linden DJ, Worley PF(2010) Homeostatic scaling requires group I mGluR activationmediated by Homer1a. Neuron 68(6):1128–1142
83. Szumlinski KK, Kalivas PW, Worley PF (2006) Homer proteins:implications for neuropsychiatric disorders. Curr Opin Neurobiol16(3):251–257
84. Bae JS, Kim JY, Park BL, Cheong HS, Kim JH, Shin JG, Park CS,Kim BJ, Lee CS, Kim JW, Lee M, Choi WH, Shin TM, Hwang J,Shin HD, Woo SI (2013) Lack of association between DISC1polymorphisms and risk of schizophrenia in a Korean population.Psychiatry Research 208(2):189–190
85. Jaubert PJ, Golub MS, Lo YY, Germann SL, Dehoff MH, WorleyPF, Kang SH, Schwarz MK, Seeburg PH, Berman RF (2007)Complex, multimodal behavioral profile of the Homer1 knockoutmouse. Gene Brain Behav 6(2):141–154
86. Szumlinski KK, Lominac KD, Kleschen MJ, Oleson EB, DehoffMH, Schwarz MK, Seeburg PH, Worley PF, Kalivas PW (2005)Behavioral and neurochemical phenotyping of Homer1 mutantmice: possible relevance to schizophrenia. Gene Brain behav 4(5):273–288
87. Lominac KD, Oleson EB, Pava M, Klugmann M, Schwarz MK,Seeburg PH, During MJ, Worley PF, Kalivas PW, Szumlinski KK(2005) Distinct roles for different Homer1 isoforms in behaviorsand associated prefrontal cortex function. J Neurosci Off J SocNeurosci 25(50):11586–11594
88. Norton N, Williams HJ, Williams NM, Spurlock G, Zammit S,Jones G, Jones S, Owen R, O’Donovan MC, Owen MJ (2003)Mutation screening of the Homer gene family and associationanalysis in schizophrenia. Am J Med Genet B NeuropsychiatrGenet Off Publ Int Soc Psychiatr Genet 120B(1):18–21
89. Spellmann I, Rujescu D,Musil R, Mayr A, Giegling I, Genius J, ZillP, Dehning S, Opgen-Rhein M, Cerovecki A, Hartmann AM,Schafer M, Bondy B, Muller N, Moller HJ, Riedel M (2011)Homer-1 polymorphisms are associated with psychopathology andresponse to treatment in schizophrenic patients. J Psychiatr Res45(2):234–241
90. GilksWP, Allott EH, Donohoe G, Cummings E, Gill M, Corvin AP,Morris DW (2010) Replicated genetic evidence supports a role forHOMER2 in schizophrenia. Neurosci Lett 468(3):229–233
91. Kelleher RJ 3rd, Geigenmuller U, Hovhannisyan H, Trautman E,Pinard R, Rathmell B, Carpenter R, Margulies D (2012) High-throughput sequencing of mGluR signaling pathway genes revealsenrichment of rare variants in autism. PloS ONE 7(4):e35003
92. Giuffrida R, Musumeci S, D’Antoni S, Bonaccorso CM, Giuffrida-Stella AM, Oostra BA, Catania MV (2005) A reduced number ofmetabotropic glutamate subtype 5 receptors are associated withconstitutive homer proteins in a mouse model of fragile X syn-drome. J Neurosci Off J Soc Neurosci 25(39):8908–8916
93. Ronesi JA, Collins KA, Hays SA, Tsai NP, Guo W, Birnbaum SG,Hu JH, Worley PF, Gibson JR, Huber KM (2012) Disrupted Homerscaffolds mediate abnormal mGluR5 function in a mouse model offragile X syndrome. Nat Neurosci 15(3):431–440, S431
94. de Bartolomeis A, Tomasetti C (2012) Calcium-dependent net-works in dopamine-glutamate interaction: the role of postsynapticscaffolding proteins. Mol Neurobiol 46(2):275–296
95. Hayashi MK, Tang C, Verpelli C, Narayanan R, Stearns MH, XuRM, Li H, Sala C, Hayashi Y (2009) The postsynaptic densityproteins Homer and Shank form a polymeric network structure.Cell 137(1):159–171
96. Sheng M, Kim E (2000) The Shank family of scaffold proteins. JCell Sci 113(Pt 11):1851–1856
97. Boeckers TM, Bockmann J, Kreutz MR, Gundelfinger ED (2002)ProSAP/Shank proteins—a family of higher order organizing mol-ecules of the postsynaptic density with an emerging role in humanneurological disease. J Neurochem 81(5):903–910
98. Lim S, Naisbitt S, Yoon J, Hwang JI, Suh PG, Sheng M, Kim E(1999) Characterization of the Shank family of synaptic proteins.Multiple genes, alternative splicing, and differential expression inbrain and development. J Biol Chem 274(41):29510–29518
99. Tu JC, Xiao B, Naisbitt S, Yuan JP, Petralia RS, Brakeman P, DoanA, Aakalu VK, Lanahan AA, ShengM,Worley PF (1999) Couplingof mGluR/Homer and PSD-95 complexes by the Shank family ofpostsynaptic density proteins. Neuron 23(3):583–592
100. Bertaso F, Roussignol G, Worley P, Bockaert J, Fagni L, Ango F(2010) Homer1a-dependent crosstalk between NMDA and metab-otropic glutamate receptors in mouse neurons. PloS ONE 5(3):e9755
101. Roussignol G, Ango F, Romorini S, Tu JC, Sala C, Worley PF,Bockaert J, Fagni L (2005) Shank expression is sufficient to inducefunctional dendritic spine synapses in aspiny neurons. J NeurosciOff J Soc Neurosci 25(14):3560–3570
102. Grabrucker AM, Schmeisser MJ, Schoen M, Boeckers TM (2011)Postsynaptic ProSAP/Shank scaffolds in the cross-hair ofsynaptopathies. Trends Cell Biol 21(10):594–603
103. Gauthier J, Champagne N, Lafreniere RG, Xiong L, Spiegelman D,Brustein E, Lapointe M, Peng H, Cote M, Noreau A, Hamdan FF,Addington AM, Rapoport JL, Delisi LE, Krebs MO, Joober R,Fathalli F, Mouaffak F, Haghighi AP, Neri C, Dube MP, SamuelsME, Marineau C, Stone EA, Awadalla P, Barker PA, Carbonetto S,Drapeau P, Rouleau GA (2010) De novo mutations in the geneencoding the synaptic scaffolding protein SHANK3 in patientsascertained for schizophrenia. Proc Natl Acad Sci U S A 107(17):7863–7868
104. Lennertz L, Wagner M, Wolwer W, Schuhmacher A, Frommann I,Berning J, Schulze-Rauschenbach S, Landsberg MW, SteinbrecherA, Alexander M, Franke PE, Pukrop R, Ruhrmann S, Bechdolf A,GaebelW, Klosterkotter J, Hafner H,Maier W,Mossner R (2012) Apromoter variant of SHANK1 affects auditory working memory inschizophrenia patients and in subjects clinically at risk for psycho-sis. Eur Arch Psychiatry Clin Neurosci 262(2):117–124
105. Asrar S, Jia Z (2013) Molecular mechanisms coordinating function-al and morphological plasticity at the synapse: role of GluA2/N-
Mol Neurobiol
cadherin interaction-mediated actin signaling in mGluR-dependentLTD. Cell Signal 25(2):397–402
106. Assie MB, Dominguez H, Consul-Denjean N, Newman-Tancredi A(2006) In vivo occupancy of dopamine D2 receptors by antipsy-chotic drugs and novel compounds in the mouse striatum andolfactory tubercles. Naunyn Schmiedeberg’s Arch Pharmacol373(6):441–450
107. Durand CM, Betancur C, Boeckers TM, Bockmann J, Chaste P,Fauchereau F, Nygren G, Rastam M, Gillberg IC, Anckarsater H,Sponheim E, Goubran-Botros H, Delorme R, Chabane N, Mouren-Simeoni MC, de Mas P, Bieth E, Roge B, Heron D, Burglen L,Gillberg C, Leboyer M, Bourgeron T (2007) Mutations in the geneencoding the synaptic scaffolding protein SHANK3 are associatedwith autism spectrum disorders. Nat Genet 39(1):25–27
108. Moessner R,Marshall CR, Sutcliffe JS, Skaug J, Pinto D, Vincent J,Zwaigenbaum L, Fernandez B, Roberts W, Szatmari P, Scherer SW(2007) Contribution of SHANK3 mutations to autism spectrumdisorder. Am J Hum Genet 81(6):1289–1297
109. Emamian ES, Karayiorgou M, Gogos JA (2004) Decreased phos-phorylation of NMDA receptor type 1 at serine 897 in brains ofpatients with Schizophrenia. J Neurosci Off J Soc Neurosci 24(7):1561–1564
110. Durand CM, Perroy J, Loll F, Perrais D, Fagni L, Bourgeron T,Montcouquiol M, Sans N (2012) SHANK3 mutations identified inautism lead to modification of dendritic spine morphology via anactin-dependent mechanism. Mol Psychiatry 17(1):71–84
111. Peca J, Feliciano C, Ting JT, WangW, Wells MF, Venkatraman TN,Lascola CD, Fu Z, Feng G (2011) Shank3 mutant mice displayautistic-like behaviours and striatal dysfunction. Nature 472(7344):437–442
112. Arons MH, Thynne CJ, Grabrucker AM, Li D, Schoen M, CheyneJE, Boeckers TM, Montgomery JM, Garner CC (2012) Autism-associated mutations in ProSAP2/Shank3 impair synaptic transmis-sion and neurexin-neuroligin-mediated transsynaptic signaling. JNeurosci Off J Soc Neurosci 32(43):14966–14978
113. Millar JK, Wilson-Annan JC, Anderson S, Christie S, Taylor MS,Semple CA, Devon RS, St Clair DM, Muir WJ, Blackwood DH,Porteous DJ (2000) Disruption of two novel genes by a translocationco-segregating with schizophrenia. Hum Mol Genet 9(9):1415–1423
114. Porteous DJ, Millar JK, Brandon NJ, Sawa A (2011) DISC1 at 10:connecting psychiatric genetics and neuroscience. Trends Mol Med17(12):699–706
115. Soares DC, Carlyle BC, Bradshaw NJ, Porteous DJ (2011) DISC1:structure, function, and therapeutic potential for major mental ill-ness. ACS Chem Neurosci 2(11):609–632
116. Benes FM (2000) Emerging principles of altered neural circuitry inschizophrenia. Brain Res Brain Res Rev 31(2–3):251–269
117. Papaleo F, Lipska BK, Weinberger DR (2012) Mouse models ofgenetic effects on cognition: relevance to schizophrenia.Neuropharmacology 62(3):1204–1220
118. Papaleo F, Yang F, Garcia S, Chen J, Lu B, Crawley JN,WeinbergerDR (2012) Dysbindin-1 modulates prefrontal cortical activity andschizophrenia-like behaviors via dopamine/D2 pathways. MolPsychiatry 17(1):85–98
119. Mao Y, Ge X, Frank CL, Madison JM, Koehler AN, Doud MK,Tassa C, Berry EM, Soda T, Singh KK, Biechele T, Petryshen TL,Moon RT, Haggarty SJ, Tsai LH (2009) Disrupted in schizophrenia1 regulates neuronal progenitor proliferation via modulation ofGSK3beta/beta-catenin signaling. Cell 136(6):1017–1031
120. Cohen JE, Lee PR, Chen S, Li W, Fields RD (2011) MicroRNAregulation of homeostatic synaptic plasticity. Proc Natl Acad Sci US A 108(28):11650–11655
121. Hikida T, Jaaro-Peled H, Seshadri S, Oishi K, Hookway C, Kong S,Wu D, Xue R, Andrade M, Tankou S, Mori S, Gallagher M,Ishizuka K, Pletnikov M, Kida S, Sawa A (2007) Dominant-
negative DISC1 transgenic mice display schizophrenia-associatedphenotypes detected by measures translatable to humans. Proc NatlAcad Sci U S A 104(36):14501–14506
122. Boccuto L, Lauri M, Sarasua SM, Skinner CD, Buccella D,Dwivedi A, Orteschi D, Collins JS, Zollino M, Visconti P, DupontB, Tiziano D, Schroer RJ, Neri G, Stevenson RE, Gurrieri F,Schwartz CE (2013) Prevalence of SHANK3 variants in patientswith different subtypes of autism spectrum disorders. Eur J HumGenet 21(3):310–316
123. PletnikovMV, AyhanY, Nikolskaia O, XuY, OvanesovMV, HuangH, Mori S, Moran TH, Ross CA (2008) Inducible expression ofmutant human DISC1 in mice is associated with brain and behav-ioral abnormalities reminiscent of schizophrenia. Mol Psychiatry13(2):173–186, 115
124. Arguello PA, Markx S, Gogos JA, Karayiorgou M (2010)Development of animal models for schizophrenia. Dis ModelMech 3(1–2):22–26
125. Holley SM, Wang EA, Cepeda C, Jentsch JD, Ross CA, PletnikovMV, Levine MS (2013) Frontal cortical synaptic communication isabnormal in Disc1 genetic mouse models of schizophrenia.Schizophr Res 146(1–3):264–272
126. Kvajo M, McKellar H, Arguello PA, Drew LJ, Moore H,MacDermott AB, Karayiorgou M, Gogos JA (2008) A mutationin mouse Disc1 that models a schizophrenia risk allele leads tospecific alterations in neuronal architecture and cognition. Proc NatlAcad Sci U S A 105(19):7076–7081
127. Kvajo M, McKellar H, Drew LJ, Lepagnol-Bestel AM, Xiao L,Levy RJ, Blazeski R, Arguello PA, Lacefield CO, Mason CA,Simonneau M, O’Donnell JM, MacDermott AB, Karayiorgou M,Gogos JA (2011) Altered axonal targeting and short-term plasticityin the hippocampus of Disc1 mutant mice. Proc Natl Acad Sci U SA 108(49):E1349–1358
128. Lee FH, Zai CC, Cordes SP, Roder JC, Wong AH (2013) Abnormalinterneuron development in disrupted-in-schizophrenia-1 L100Pmutant mice. Mol Brain 6:20
129. BlumBP,Mann JJ (2002) The GABAergic system in schizophrenia.Int J Neuropsychopharmacol 5(2):159–179
130. Hashimoto T, Volk DW, Eggan SM, Mirnics K, Pierri JN, Sun Z,Sampson AR, Lewis DA (2003) Gene expression deficits in asubclass of GABA neurons in the prefrontal cortex of subjects withschizophrenia. J Neurosci Off J Soc Neurosci 23(15):6315–6326
131. Lepagnol-Bestel AM, Kvajo M, Karayiorgou M, Simonneau M,Gogos JA (2013) A Disc1 mutation differentially affects neuritesand spines in hippocampal and cortical neurons. Mol Cell Neurosci54:84–92
132. Mouaffak F, Kebir O, Chayet M, Tordjman S, VacheronMN, MilletB, Jaafari N, Bellon A, Olie JP, Krebs MO (2011) Association ofdisrupted in schizophrenia 1 (DISC1) missense variants with ultra-resistant schizophrenia. Pharmacogenomics J 11(4):267–273
133. Takahashi T, Suzuki M, Tsunoda M, Maeno N, Kawasaki Y, ZhouSY, Hagino H, Niu L, Tsuneki H, Kobayashi S, Sasaoka T, Seto H,Kurachi M, Ozaki N (2009) The disrupted-in-schizophrenia-1Ser704Cys polymorphism and brain morphology in schizophrenia.Psychiatry Res 172(2):128–135
134. Zhao X, Sun L, Jia H, Meng Q, Wu S, Li N, He S (2009) Isolationrearing induces social and emotional function abnormalities andalters glutamate and neurodevelopment-related gene expression inrats. Prog Neuro-psychopharmacol Biol Psychiatry 33(7):1173–1177
135. Fone KC, Porkess MV (2008) Behavioural and neurochemicaleffects of post-weaning social isolation in rodents-relevance todevelopmental neuropsychiatric disorders. Neurosci Biobehav Rev32(6):1087–1102
136. Moller M, Du Preez JL, Viljoen FP, Berk M, Emsley R, Harvey BH(2012) Social isolation rearing induces mitochondrial, immunolog-ical, neurochemical and behavioural deficits in rats, and is reversed
Mol Neurobiol
by clozapine or N-acetyl cysteine. Brain, behavior, and immunity30:156–167
137. Kristiansen LV, Meador-Woodruff JH (2005) Abnormal striatalexpression of transcripts encoding NMDA interacting PSD proteinsin schizophrenia, bipolar disorder and major depression. SchizophrRes 78(1):87–93
138. Toyooka K, Iritani S, Makifuchi T, Shirakawa O, Kitamura N,Maeda K, Nakamura R, Niizato K, Watanabe M, Kakita A,Takahashi H, Someya T, Nawa H (2002) Selective reduction of aPDZ protein, SAP-97, in the prefrontal cortex of patients withchronic schizophrenia. J Neurochem 83(4):797–806
139. Hiraoka S, Kajii Y, Kuroda Y, Umino A, Nishikawa T (2010) Thedevelopment- and phencyclidine-regulated induction ofsynapse-associated protein-97 gene in the rat neocortex. EurNeuropsychopharmacol J Eur Coll Neuropsychopharmacol 20(3):176–186
140. Mandela P, Ma XM (2012) Kalirin, a key player in synapseformation, is implicated in human diseases. Neural plasticity2012:728161
141. Xie Z, Cahill ME, Penzes P (2010) Kalirin loss results in corticalmorphological alterations. Mol Cell Neurosci 43(1):81–89
142. Kushima I, Nakamura Y, Aleksic B, IkedaM, Ito Y, Shiino T, OkochiT, Fukuo Y, Ujike H, Suzuki M, Inada T, Hashimoto R, Takeda M,Kaibuchi K, Iwata N, Ozaki N (2012) Resequencing and associationanalysis of the KALRN and EPHB1 genes and their contribution toschizophrenia susceptibility. Schizophr Bull 38(3):552–560
143. Hill JJ, Hashimoto T, Lewis DA (2006) Molecular mechanismscontributing to dendritic spine alterations in the prefrontal cortexof subjects with schizophrenia. Mol Psychiatry 11(6):557–566
144. Kajimoto Y, Shirakawa O, Lin XH, Hashimoto T, Kitamura N,Murakami N, Takumi T, Maeda K (2003) Synapse-associated pro-tein 90/postsynaptic density-95-associated protein (SAPAP) isexpressed differentially in phencyclidine-treated rats and is in-creased in the nucleus accumbens of patients with schizophrenia.Neuropsychopharmacol Off Publ Am Coll Neuropsychopharmacol28(10):1831–1839
145. Bernstein HG, Sahin J, Smalla KH, Gundelfinger ED, Bogerts B,Kreutz MR (2007) A reduced number of cortical neurons showincreased Caldendrin protein levels in chronic schizophrenia.Schizophr Res 96(1–3):246–256
146. Verpelli C, Schmeisser MJ, Sala C, Boeckers TM (2012) Scaffoldproteins at the postsynaptic density. Adv Exp Med Biol 970:29–61
147. de Bartolomeis A, Buonaguro EF, Iasevoli F (2013) Serotonin-glutamate and serotonin-dopamine reciprocal interactions as puta-tive molecular targets for novel antipsychotic treatments: fromreceptor heterodimers to postsynaptic scaffolding and effector pro-teins. Psychopharmacology 225(1):1–19
148. Seeman P (2010) Dopamine D2 receptors as treatment targets inschizophrenia. Clin Schizophr Relat Psychoses 4(1):56–73
149. Miyamoto S, Miyake N, Jarskog LF, Fleischhacker WW,Lieberman JA (2012) Pharmacological treatment of schizophrenia:a critical review of the pharmacology and clinical effects of currentand future therapeutic agents. Mol Psychiatry 17(12):1206–1227
150. de Bartolomeis A, Aloj L, Ambesi-Impiombato A, Bravi D, CaracoC, Muscettola G, Barone P (2002) Acute administration of antipsy-chotics modulates Homer striatal gene expression differentially.Brain Res Mol Brain Res 98(1–2):124–129
152. Zhang GC, Mao LM, Liu XY, Parelkar NK, Arora A, Yang L, HainsM, Fibuch EE, Wang JQ (2007) In vivo regulation of Homer1aexpression in the striatum by cocaine.Mol Pharmacol 71(4):1148–1158
153. Ambesi-Impiombato A, Panariello F, Dell’aversano C, Tomasetti C,Muscettola G, de Bartolomeis A (2007) Differential expression of
Homer 1 gene by acute and chronic administration of antipsychoticsand dopamine transporter inhibitors in the rat forebrain. Synapse61(6):429–439
154. Seeger TF, Seymour PA, Schmidt AW, Zorn SH, Schulz DW, LebelLA, McLean S, Guanowsky V, Howard HR, Lowe JA 3rd et al(1995) Ziprasidone (CP-88,059): a new antipsychotic with com-bined dopamine and serotonin receptor antagonist activity. JPharmacol Exp Ther 275(1):101–113
155. Iasevoli F, Tomasetti C, Ambesi-Impiombato A, Muscettola G, deBartolomeis A (2009) Dopamine receptor subtypes contribution toHomer1a induction: insights into antipsychotic molecular action.Prog Neuro-psychopharmacol Biol Psychiatry 33(5):813–821
156. Robinet EA, Geurts M, Maloteaux JM, Pauwels PJ (2001) Chronictreatment with certain antipsychotic drugs preserves upregulation ofregulator of G-protein signalling 2 mRNA in rat striatum as opposedto c-fos mRNA. Neurosci Lett 307(1):45–48
157. Semba J, Sakai MW, Suhara T, Akanuma N (1999) Differential effectsof acute and chronic treatment with typical and atypical neuroleptics onc-fos mRNA expression in rat forebrain regions using non-radioactivein situ hybridization. Neurochem Int 34(4):269–277
158. Leucht S, Corves C, Arbter D, Engel RR, Li C, Davis JM (2009)Second-generation versus first-generation antipsychotic drugs forschizophrenia: a meta-analysis. Lancet 373(9657):31–41
159. Johnstone M, Thomson PA, Hall J, McIntosh AM, Lawrie SM,Porteous DJ (2011) DISC1 in schizophrenia: genetic mouse modelsand human genomic imaging. Schizophr Bull 37(1):14–20
160. Hayashi-Takagi A, Takaki M, Graziane N, Seshadri S, Murdoch H,Dunlop AJ, Makino Y, Seshadri AJ, Ishizuka K, Srivastava DP, XieZ, Baraban JM, Houslay MD, Tomoda T, Brandon NJ, Kamiya A,Yan Z, Penzes P, Sawa A (2010) Disrupted-in-schizophrenia 1(DISC1) regulates spines of the glutamate synapse via Rac1. NatNeurosci 13(3):327–332
161. Wang Q, Charych EI, Pulito VL, Lee JB, Graziane NM, CrozierRA, Revilla-Sanchez R, Kelly MP, Dunlop AJ, Murdoch H, TaylorN, Xie Y, Pausch M, Hayashi-Takagi A, Ishizuka K, Seshadri S,Bates B, Kariya K, Sawa A, Weinberg RJ, Moss SJ, Houslay MD,Yan Z, Brandon NJ (2011) The psychiatric disease risk factorsDISC1 and TNIK interact to regulate synapse composition andfunction. Mol Psychiatry 16(10):1006–1023
162. Nagai T, Kitahara Y, Ibi D, Nabeshima T, Sawa A, Yamada K(2011) Effects of antipsychotics on the behavioral deficits in humandominant-negative DISC1 transgenic mice with neonatal polyI:Ctreatment. Behav Brain Res 225(1):305–310
163. Iasevoli F, Tomasetti C, de Bartolomeis A (2013) Scaffolding pro-teins of the post-synaptic density contribute to synaptic plasticity byregulating receptor localization and distribution: relevance for neu-ropsychiatric diseases. Neurochem Res 38(1):1–22
164. de Bartolomeis A, Fiore G, Iasevoli F (2005) Dopamine-glutamateinteraction and antipsychotics mechanism of action: implication fornew pharmacological strategies in psychosis. Curr Pharm Des11(27):3561–3594
165. de Bartolomeis A, Iasevoli F (2003) The Homer family and the signaltransduction system at glutamatergic postsynaptic density: potential rolein behavior and pharmacotherapy. Psychopharmacol Bull 37(3):51–83
166. de Bartolomeis A, Marmo F, Filomena Buonaguro E, Rossi R,Tomasetti C, Iasevoli F (2013) Imaging brain gene expressionprofiles by antipsychotics: region-specific action of amisulpride onpostsynaptic density transcripts compared to haloperidol. Europeanneuropsychopharmacology: the journal of the European College ofNeuropsychopharmacology, in press
167. de Bartolomeis A, SarappaC,Magara S, Iasevoli F (2012) Targetingglutamate system for novel antipsychotic approaches: relevance forresidual psychotic symptoms and treatment resistant schizophrenia.Eur J Pharmacol 682(1–3):1–11
168. de Bartolomeis A, Tomasetti C, Cicale M, Yuan PX, Manji HK(2012) Chronic treatment with lithium or valproate modulates the
Mol Neurobiol
expression of Homer1b/c and its related genes Shank and inositol1,4,5-trisphosphate receptor. Eur Neuropsychopharmacol J Eur CollNeuropsychopharmacol 22(7):527–535
169. Debono R, Topless R, Markie D, Black MA, Merriman TR (2012)Analysis of the DISC1 translocation partner (11q14.3) in geneticrisk of schizophrenia. Gen Brain Behav 11(7):859–863
170. Elias GM, Nicoll RA (2007) Synaptic trafficking of glutamate recep-tors byMAGUK scaffolding proteins. Trends Cell Biol 17(7):343–352
171. Polese D, de Serpis AA, Ambesi-Impiombato A, Muscettola G, deBartolomeis A (2002) Homer 1a gene expression modulation byantipsychotic drugs: involvement of the glutamate metabotropicsystem and effects of D-cycloserine. Neuropsychopharmacol OffPubl Am Coll Neuropsychopharmacol 27(6):906–913
172. Dracheva S, Marras SA, Elhakem SL, Kramer FR, Davis KL,Haroutunian V (2001) N-methyl-D-aspartic acid receptor expres-sion in the dorsolateral prefrontal cortex of elderly patients withschizophrenia. Am J Psychiatry 158(9):1400–1410
173. de Bartolomeis A, SarappaC,Magara S, Iasevoli F (2012) Targetingglutamate system for novel antipsychotic approaches: relevance forresidual psychotic symptoms and treatment resistant schizophrenia.Eur J Pharmacol 682(1–3):1–11
174. Zhou L, Li F, Xu HB, Luo CX, Wu HY, Zhu MM, Lu W, Ji X, ZhouQG, Zhu DY (2010) Treatment of cerebral ischemia by disruptingischemia-induced interaction of nNOSwith PSD-95. Nat Med 16(12):1439–1443
175. Minnerup J, Sutherland BA, Buchan AM, Kleinschnitz C (2012)Neuroprotection for stroke: current status and future perspectives.Int J Mol Sci 13(9):11753–11772
176. Ehrlich I, Malinow R (2004) Postsynaptic density 95 controlsAMPA receptor incorporation during long-term potentiation andexperience-driven synaptic plasticity. The Journal of neuroscience:the official journal of the Society for Neuroscience 24(4):916–927
177. Ting AK, Chen Y, Wen L, Yin DM, Shen C, Tao Y, Liu X, XiongWC, Mei L (2011) Neuregulin 1 promotes excitatory synapsedevelopment and function in GABAergic interneurons. J NeurosciOff J Soc Neurosci 31(1):15–25
178. Tomasetti C, Dell’Aversano C, Iasevoli F, Marmo F, de BartolomeisA (2011) The acute and chronic effects of combined antipsychotic-mood stabilizing treatment on the expression of cortical and striatalpostsynaptic density genes. Prog Neuro-psychopharmacol BiolPsychiatry 35(1):184–197
179. Fatemi SH, Reutiman TJ, FolsomTD, Bell C, Nos L, Fried P, PearceDA, Singh S, Siderovski DP,Willard FS, FukudaM (2006) Chronicolanzapine treatment causes differential expression of genes infrontal cortex of rats as revealed by DNA microarray technique.Neuropsychopharmacol Off Publ Am Coll Neuropsychopharmacol31(9):1888–1899
180. Wohr M, Roullet FI, Hung AY, Sheng M, Crawley JN (2011)Communication impairments in mice lacking Shank1: reducedlevels of ultrasonic vocalizations and scent marking behavior.PloS ONE 6(6):e20631
181. Jaaro-Peled H, Niwa M, Foss CA, Murai R, de Los RS, Kamiya A,Mateo Y, O’Donnell P, Cascella NG, Nabeshima T, Guilarte TR,Pomper MG, Sawa A (2013) Subcortical dopaminergic deficits in aDISC1 mutant model: a study in direct reference to human molec-ular brain imaging. Hum Mol Genet 22(8):1574–1580