Page 1 2015 International Good Submission Practice Workshop on Pharmaceuticals Good Submissions for Marketing Authorization of Generics in EU ‐ Expectations from the EU 17. ‐ 18. September 2015 GIS NTU Convention Center, Taipei, Taiwan Peter Bachmann
The Applicant / Marketing Authorisation Holder (MAH) has to be a legal body (company) located inside theterritory of
• the EU for a central marketing authorisation• an EEA‐Member State for a national marketingauthorisation
Page 6
… European Pharmaceutical Legislation
• Directive 2001/83/EC, as amended (human)• Directive 2001/82/EC, as amended (veterinary)to be transposed into national legislation
therefore• harmonised data requirements and assessment for a marketing authorisation are in force in the EU/EEA
but• prescription status (Rx / OTC)• reimbursement by health insuranceare within the competence of the national states
Page 7
… basic principles• no approval „light“ for known active substances, but thesame basic principles are applicable as for a medicinalproduct with a new active substance.
• each application for a marketing authorisation isassessed in line with the principles of
efficacy safety quality
and approved if the benefit‐risk‐ratio is positive
• but the amount of data within the application for a marketing authorisation may differ
Page 8
Legal Basis for MAs in the EU/EEA
Article 8 full dossier
Article 10 (1) GenericArticle 10 (3) „Hybrid“
(‚Generic‘ with additional data)Article 10 (4) Biosimilar
Article 10a well established use applicationArticle 10b combination of known constituentsArticle 10c informed consent
Page 9
Generic Medicinal Product
• an application according to Article 10 is a deviation from the normal requirements (Article 8(3))
• therefore ‐ and from a legal point of view ‐ an application for a generic marketing authorisation is not a right in its own
• it is an option if all requirements of the legislations as stated in Article 10(1), 10(3) or 10(4) are fulfilled, respectively
• each subarticle of Article 10 is selfstanding – no combinations are possible
Page 10
II.The EU
‐ Definition of a Generic ‐
Page 11
Generic Medicinal Product ‐ (1)Article 10 (1), first subparagraph
“1. …the applicant shall not be required to provide the results of preclinical tests or clinical trials ...
therefore:• the generic applicant has to provide it’s own pharmaceutical dossier to prove the quality of the (generic) medicinal product
• independent evaluation; no assessment in comparison to the pharmaceutical quality of the reference medicinal product
• Certificate of Suitability of the European Directorate for theQuality of Medicines EDQM (CEP)– only for substances with a monograph in the European
Pharmacopoeia & not for biological active substances
Page 13
Substances with a monograph in the Ph.Eur. ‐ASMF vs. CEP
Identical requirements for the documentation to besupplied, i.e.
– Demonstration that pharmacopoeial monographis able to detect and determine all impurities
Assessment: Licensing authority ASMFEDQM CEP
… but in fact assessors from the NCAs are doing theassessment in both cases
Page 14
CEP (= Certificate of Suitability)
• for an active substance with a Ph. Eur.Monography• show the suitability of the monograph• can replace CTD‐Module 3S• no commercial confidential information is provided to the applicant of the finished medicinal product
• evaluation of the submitted dossier of the active substance– at the EDQM– by assessors of the NCA
• unic CEP numbering system allows traceability
Page 15
ASMF ‐ CTD
• The "open" part of the ASMF ‐ included in section 3.2.S ofCTD Module 3
• It is the responsibility of the applicant for a MA for a MP toensure that the complete ASMF
– applicant's ("open") part and
– the active substance manufacturer's restricted("closed") part
is supplied to the authorities directly by the ASM in the CTD format, synchronised to arrive at around the same time asthe MA application.
• Letter of Access to the CA
Page 16
Generic Medicinal Product ‐ (1a)
Article 10 (1), first subparagraph
“1. …the applicant shall not be required to provide the results of preclinical tests or clinical trials if he can demonstrate that the medicinal product is a generic of a reference medicinal product …
Page 17
Reference Medicinal Product: Definition
Article 10 (2) (a)
“... shall mean a medicinal product authorised under Article 6, in accordance with the provisions of Article 8;”
Article 8 full dossier Article 10a well established use application Article 10b combination of known constituents Article 10c informed consent
EU / EEA
Page 18
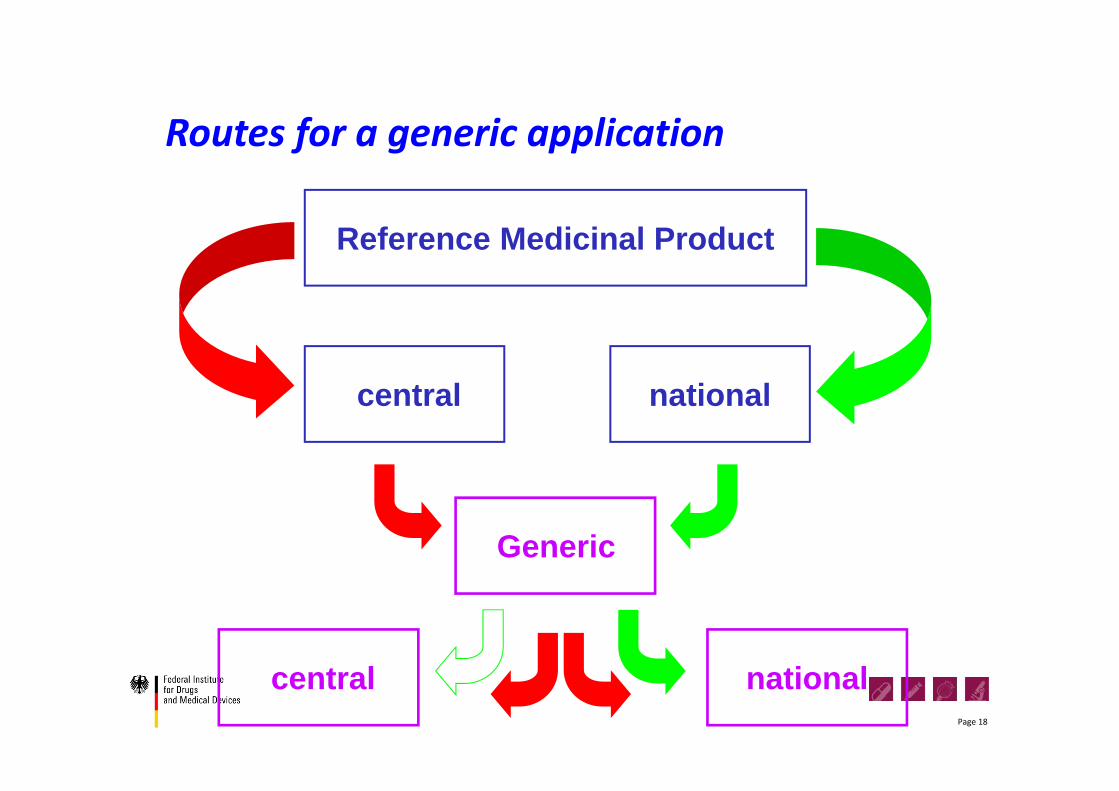
Routes for a generic application
Generic
central national
central national
Reference Medicinal Product
Page 19
Generic Medicinal Product ‐ (1b)Article 10 (1), first subparagraph
“1. …the applicant shall not be required to provide the results of preclinical tests or clinical trials if he can demonstrate that the medicinal product is a generic of a reference medicinal product which is or has been authorised under Article 6 for not less than eight years in a Member State or in the Community.”
8 years data exclusivity+ 2 years market exclusivity = 10 yearsbut we don’t care about patents
Page 20
Generic Medicinal Product ‐ (2)
Article 10 (2) b: “generic medicinal product”• same qualitative and quantitative composition in active substances
but– the different salts, esters, ethers, isomeres, mixture of isomeres, complexes or derivatives are the same active substance, unless they differ significantly in properties with regard to safety and/or efficacy.
– in such cases additional information of the proof of safety and/or efficiacy of the different salt, … must be supplied by the applicant.
Page 21
Generic Medicinal Product ‐ (3)
cont.
– if the different salts, esters, ethers, isomeres, mixture of isomeres, complexes or derivatives are the same active substance differ significantly in properties with regard to safety and/or efficacy.
application according toArticle 10(3) orArticle 8
Page 22
Generic Medicinal Product ‐ (4)
Article 10 (2) b: “generic medicinal product”
same qualitative and quantitative composition in active substances
• same pharmaceutical form as the reference medicinal product ‐ but all immediate‐release oral pharmaceutical forms are the same
Page 23
… same pharmaceutical form ‐ (1)
Standard Terms (pharmaceutical forms) of the European Pharmcopoeia are applicable by law
– therefore exemption by legislation ‐ but all immediate‐release oral pharmaceutical forms are the same
– consequences for all non‐immediate‐release oral forms e.g. modified release, gastro‐resistant tablets, gastro‐resistant capsules
Page 24
Generic Medicinal Product ‐ (5)
Article 10 (2) b: “generic medicinal product”
same qualitative and quantitative composition in active substances
same pharmaceutical form as the reference medicinal product ‐ but all immediate‐release oral pharmaceutical forms are the same
• bioequivalence with the reference medicinal product or waiver of bioequivalence according to guidelines
Page 25
Bioequivalence ‐ (1)
• the way to show therapeutic equivalence between the generic and the reference medicinal product
• bioequivalence is the surrogate for efficacy• revised ‘Guideline on the Investigation of Bioequivalence’ in force since 01 August 2010– major shift from clinical to pharmaceutical relevance
(Note: Generally, same dissolution specifications)
Bioequivalence ‐ (2)
Page 27
Bioequivalence ‐ (3)
Scope of the Guideline• focuses on recommendations for bioequivalence studies for immediate release formulations with systemic action
• sets the relevant criteria under which bioavailability studies– need not be required– are waived for additional strength– are needed for specific formulations– BCS (Biopharmaceutics Classification System) based Biowaiver
Page 28
Bioequivalence ‐ (5)
cont. Scope of the Guideline
the limits• guidance on BE‐studies for modified releaseproducts, transdermal products and orally inhaled products are given in other guidelines
• scope is limited to chemical entities• the general principles outlined in this guideline are not applicable to herbal medicinal products
Page 29
… if BE can not be shown: ‘Hybrid‘
Article 10(3) of Directive 2001/83/EC“In cases where the medicinal product does not fall within the definition of a generic medicinal product as provided in paragraph 2(b) or where the bioequivalence cannot be demonstrated through bioavailability studies or in case of changes in the active substance(s), therapeutic indications, strength, pharmaceutical form or route of administration, vis‐à‐vis the reference medicinal product, the results of the appropriate pre‐clinical tests or clinical trials shall be provided.”
“Super-Generics”
Page 30
III.The EU
‐ Information and Advice forGeneric ‐
Page 31
… for central marketing authorisations – (1)
Page 32
… for central marketing authorisations – (2)
Page 33
… for national marketing authorisations – (1)
Page 34
… for national marketing authorisations – (2)
Page 35
Validation of an Application
• Administrative procedure (!) – therefore novalidation of the content of the application
Page 36
• Application not received/modules are missing • Proposed MAH not established in the EEA • Missing/incorrect fee• Application form/cover letter not signed/not signed with original signature • The application form is incorrect (eg. information missing, incorrect type of procedure, legal
basis incorrect, incorrect reference medicinal product, reference to an European reference medicinal product although there is a nationally authorised medicinal product)
• Documents in accordance with NtA, vol. 2B are missing or absence not justified (eq. Braille, Consultation with Target Patient Groups, Pharmacovigilance System, Environmental RiskAssessment, Specific Requirements for Different Application Types, Paediatric Regulation(whereapplicable))
• Annexes to the application form are missing or absence not justified (eg Declaration from the QP, TSE certificates for excipients of animal origin)
• Manufacturing licenses, GMP certificates and/or import licenses have not been updated or are missing. Proposed batch releaser is situated outside the EEA
• The comparator for the bioequivalence study does not originate from the EEA • …
Page 37
IV.The EU
‐ An Electronic World ‐
Page 38
Portals• no problem if submission to one agency only
• pure national submission: single national portal• centralised procedure: eSubmission Gateway
Page 39
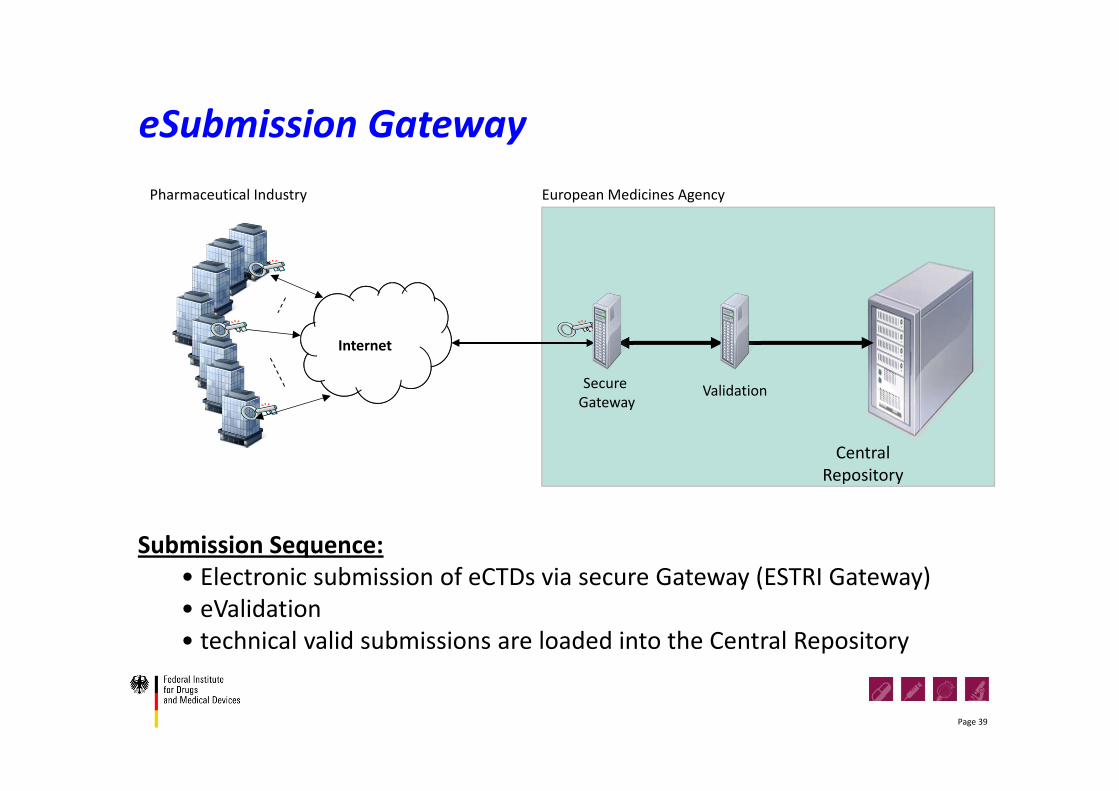
eSubmission Gateway
Submission Sequence:• Electronic submission of eCTDs via secure Gateway (ESTRI Gateway)• eValidation• technical valid submissions are loaded into the Central Repository
Pharmaceutical Industry
Internet
Secure Gateway
CentralRepository
Validation
European Medicines Agency
Page 40
National Portals – to be replaced by ‘CESP‘
Page 41
CESP – how it works
Page 42
Page 43
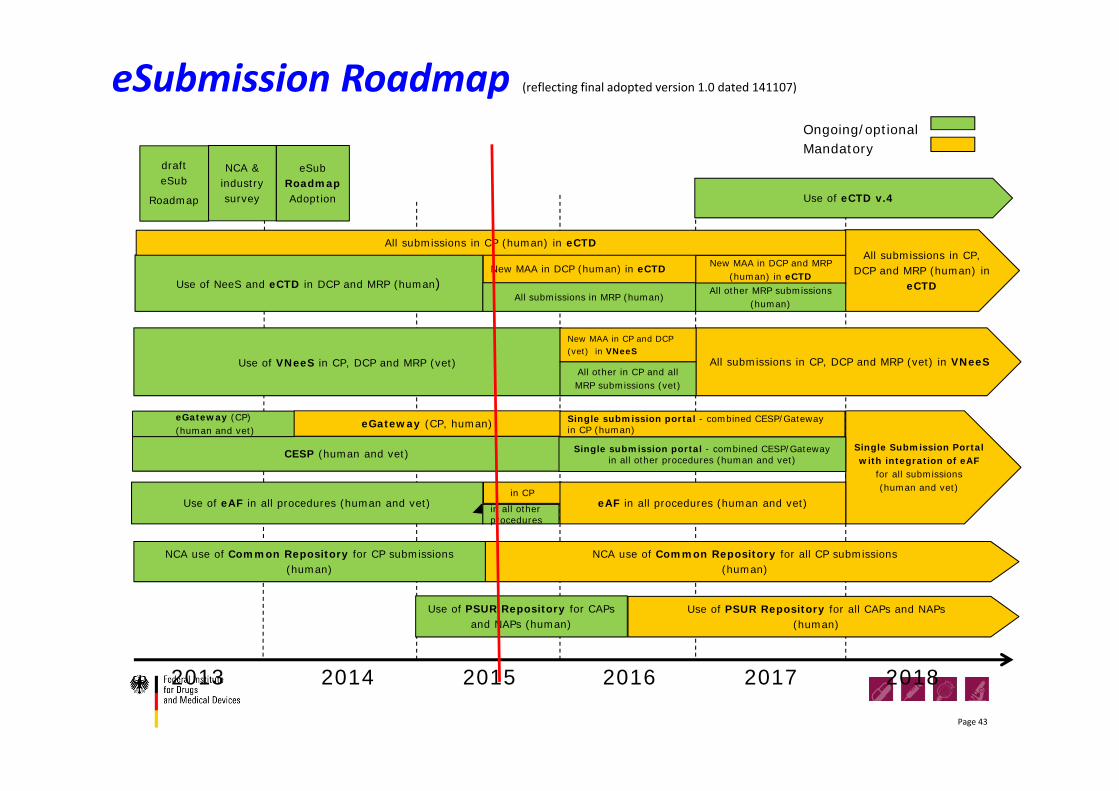
eSubmission Roadmap (reflecting final adopted version 1.0 dated 141107)
2013 2014 2015 2016 2017 2018
Use of eAF in all procedures (human and vet)
Single Submission Portal with integration of eAF
for all submissions (human and vet)
Use of VNeeS in CP, DCP and MRP (vet)
All submissions in CP, DCP and MRP (human) in
eCTD
Ongoing/optionalMandatory
Use of eCTD v.4
CESP (human and vet)
eGateway (CP)(human and vet)
eGateway (CP, human)
NCA use of Common Repository for all CP submissions(human)
All submissions in CP, DCP and MRP (vet) in VNeeS
NCA use of Common Repository for CP submissions (human)
Single submission portal - combined CESP/Gatewayin all other procedures (human and vet)
Use of NeeS and eCTD in DCP and MRP (human)
in CP
New MAA in DCP (human) in eCTD
All submissions in MRP (human)
New MAA in DCP and MRP (human) in eCTD
All other in CP and all MRP submissions (vet)
New MAA in CP and DCP (vet) in VNeeS
All other MRP submissions (human)
eAF in all procedures (human and vet)
All submissions in CP (human) in eCTD
Use of PSUR Repository for all CAPs and NAPs(human)
Use of PSUR Repository for CAPs and NAPs (human)
draft eSub
Roadmap
NCA & industry survey
eSub RoadmapAdoption
Single submission portal - combined CESP/Gatewayin CP (human)
in all other procedures
Page 44
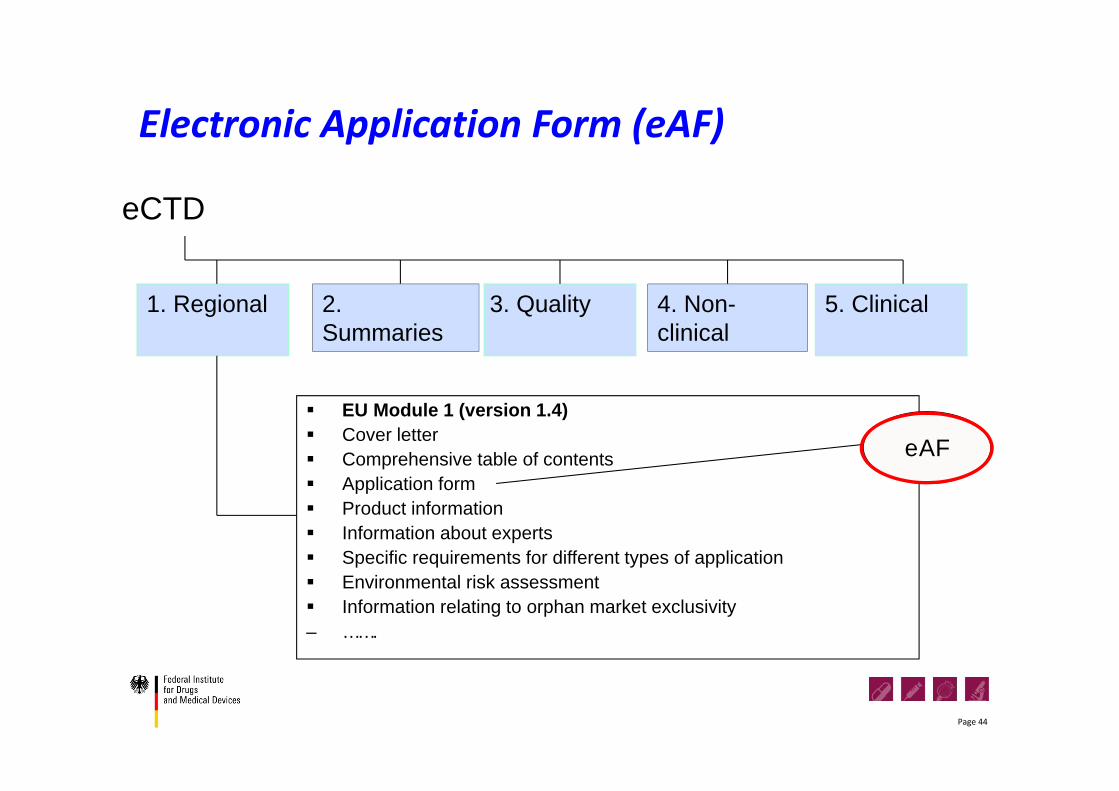
Electronic Application Form (eAF)
eCTD
3. Quality 5. Clinical1. Regional
EU Module 1 (version 1.4) Cover letter Comprehensive table of contents Application form Product information Information about experts Specific requirements for different types of application Environmental risk assessment Information relating to orphan market exclusivity– …….
eAF
4. Non-clinical
2. Summaries
Page 45
Page 46
ContactFederal Institute for Drugs and Medical DevicesExecutive Department ‘European and International Affairs’Kurt‐Georg‐Kiesinger‐Allee 3D‐53175 Bonn