Page 1
Working document QAS/15.622
May 2015
Draft document for comment
1
2
3
GUIDANCE FOR ORGANIZATIONS PERFORMING IN VIVO 4
BIOEQUIVALENCE STUDIES. 5
PROPOSAL FOR REVISION 6
(May 2015) 7
8
DRAFT FOR COMMENT 9
10
11
12
13
14
15
16
17
18
© World Health Organization 2015 19
All rights reserved. 20
This draft is intended for a restricted audience only, i.e. the individuals and organizations having received this draft. The draft 21 may not be reviewed, abstracted, quoted, reproduced, transmitted, distributed, translated or adapted, in part or in whole, in 22 any form or by any means outside these individuals and organizations (including the organizations' concerned staff and 23 member organizations) without the permission of the World Health Organization. The draft should not be displayed on any 24 website. 25
Please send any request for permission to: 26
Dr S. Croft, Prequalification Team-Inspections, Regulation of Medicines and other Health Technologies, Department of 27 Essential Medicines and Health Products, World Health Organization, CH-1211 Geneva 27, Switzerland; 28 email: [email protected] . 29
The designations employed and the presentation of the material in this draft do not imply the expression of any opinion 30 whatsoever on the part of the World Health Organization concerning the legal status of any country, territory, city or area or 31 of its authorities, or concerning the delimitation of its frontiers or boundaries. Dotted lines on maps represent approximate 32 border lines for which there may not yet be full agreement. 33
The mention of specific companies or of certain manufacturers’ products does not imply that they are endorsed or 34 recommended by the World Health Organization in preference to others of a similar nature that are not mentioned. Errors 35 and omissions excepted, the names of proprietary products are distinguished by initial capital letters. 36 37 All reasonable precautions have been taken by the World Health Organization to verify the information contained in this 38 draft. However, the printed material is being distributed without warranty of any kind, either expressed or implied. The 39 responsibility for the interpretation and use of the material lies with the reader. In no event shall the World Health 40 Organization be liable for damages arising from its use. 41 42 This draft does not necessarily represent the decisions or the stated policy of the World Health Organization. 43
44
Should you have any comments on the attached text, please send these to Dr S. Croft, Prequalification
Team-Inspections, Regulation of Medicines and other Health Technologies ([email protected] ) with a
copy to Ms Marie Gaspard ([email protected] ) by 22 June 2015.
Medicines Quality Assurance working documents will be sent out electronically only and will
also be placed on the Medicines website for comment under “Current projects”. If you do not
already receive our draft working documents please let us have your email address (to
[email protected] ) and we will add it to our electronic mailing list.
Page 2
Working document QAS/15.622
page 2
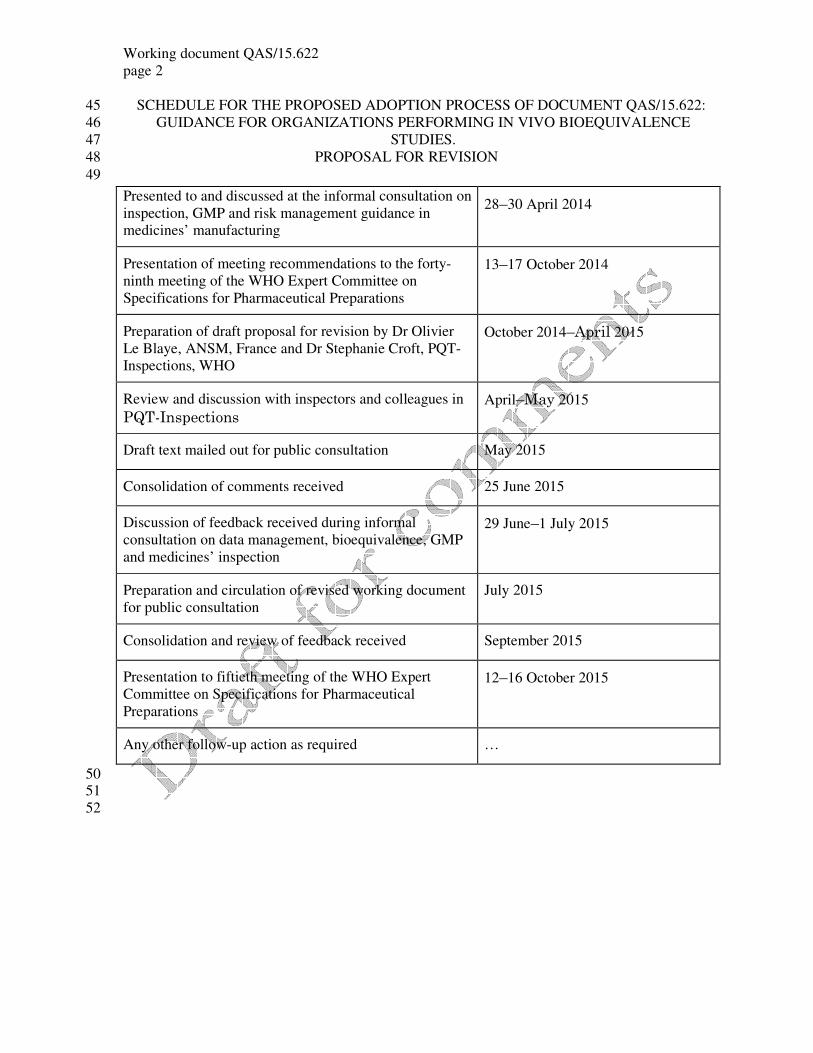
SCHEDULE FOR THE PROPOSED ADOPTION PROCESS OF DOCUMENT QAS/15.622: 45
GUIDANCE FOR ORGANIZATIONS PERFORMING IN VIVO BIOEQUIVALENCE 46
STUDIES. 47
PROPOSAL FOR REVISION 48
49
50 51
52
Presented to and discussed at the informal consultation on
inspection, GMP and risk management guidance in
medicines’ manufacturing
28–30 April 2014
Presentation of meeting recommendations to the forty-
ninth meeting of the WHO Expert Committee on
Specifications for Pharmaceutical Preparations
13–17 October 2014
Preparation of draft proposal for revision by Dr Olivier
Le Blaye, ANSM, France and Dr Stephanie Croft, PQT-
Inspections, WHO
October 2014–April 2015
Review and discussion with inspectors and colleagues in
PQT-Inspections
April–May 2015
Draft text mailed out for public consultation May 2015
Consolidation of comments received 25 June 2015
Discussion of feedback received during informal
consultation on data management, bioequivalence, GMP
and medicines’ inspection
29 June–1 July 2015
Preparation and circulation of revised working document
for public consultation
July 2015
Consolidation and review of feedback received September 2015
Presentation to fiftieth meeting of the WHO Expert
Committee on Specifications for Pharmaceutical
Preparations
12–16 October 2015
Any other follow-up action as required …
Page 3
Working document QAS/15.622
page 3
PROPOSAL FOR REVISION OF 53
GOOD TRADE AND DISTRIBUTION PRACTICES FOR 54
PHARMACEUTICAL STARTING MATERIALS1 55
56
57
BACKGROUND 58 59
During an informal consultation held in 2014 and the forty-ninth meeting of the World Health 60
Organization (WHO) Expert Committee on Specifications for Pharmaceutical Preparations 61
discussion took place regarding the possible revision of the guidance for organizations 62
performing in vivo bioequivalence studies (WHO Technical Report Series, No. 937, Annex 9, 63
2006). 64
65
It was agreed that in light of the new developments a draft for revision be prepared. 66
67
This guideline is being revised to take into consideration the revision for the multisource 68
guideline, as well as the creation of a new guideline on good data management. It will also be 69
revised to take into consideration the experience accumulated in the area of assessing and 70
inspecting bioequivalence studies since 2006. The areas with recurrent inspection findings are 71
being clarified and supplementary detail has been added in the area of bioanalysis. It also 72
includes an increased level of insistence on subject safety and data integrity. 73
74
The WHO Prequalification Project (now called Prequqlification Team (PQT)) was started in 2001 75
to assure that medicinal products supplied for procurement meet WHO norms and standards with 76
respect to quality, safety and efficacy (http://www.who.int/prequal/). Specifically it is a 77
requirement that the submitted product dossier with all its necessary contents is assessed and found 78
acceptable, and that the manufacturing sites for the finished pharmaceutical product (FPP), as well 79
as the active pharmaceutical ingredient (API), are both inspected and found to comply with WHO 80
good manufacturing practices (GMP). Since products submitted to the PQT are usually 81
multisource ("generic") products, therapeutic equivalence is generally demonstrated by performing 82
a bioequivalence study, for example in a contract research organization (also known as a clinical 83
research organization) (CRO). For prequalification of such a product it is vital that, in addition to 84
the above-mentioned requirements, the CRO used by the sponsor for bioequivalence studies is 85
compliant with respect to WHO good clinical practices (GCP) and considers relevant elements 86
from WHO good laboratory practices (GLP) and good practices for quality control laboratories to 87
ensure integrity and traceability of data. In addition, if local legal provisions exist, CROs should be 88
licensed by the respective national medicines authority. Where required by national regulations, 89
bioequivalence studies should be authorized by the national regulatory authority. Those involved in 90
the conduct and analysis of bioequivalence studies with products to be submitted for 91
prequalification therefore need to ensure that they comply with the mentioned WHO norms and 92
standards to be prepared for any inspections by WHO. 93
94
95
96
97
98
1 Good trade and distribution practices for pharmaceutical starting materials was first published in the WHO
Technical Report Series, No. 917, 2004.
Page 4
Working document QAS/15.622
page 4
Contents 99
page 100 Background 101 Introduction 3 102 1. Scope 4 103 2. Glossary 5 104 GENERAL SECTION 105 3. Organization and management 9 106 4. Computer systems 9 107
Hardware 9 108 Software 9 109 Data management 9 110
5. Quality assurance 12 111 6. Archive facilities 12 112 7. Premises 12 113 8. Personnel 13 114 CLINICAL SECTION 115 9. Clinical phase 14 116 10. Clinical laboratory 15 117 11. Ethics 16 118 12. Monitoring 16 119 13. Investigators 17 120 14. Receiving, storage and handling of investigational drug products 18 121 15. Case report forms 20 122 16. Volunteers, recruitment methods 12 123 17. Dieting 13 124 18. afety, adverse events, adverse event reporting 21 125 BIOANALYTICAL SECTION 126 19. Method development 22 127 20. Method validation 22 128 21. Sample collection, storage and handling of biological material 22 129 22. Analysis of study samples 23 130 23. Data processing and documentation 24 131 24. Good laboratory practices 24 132 PHARMACOKINETIC, STATISTICAL CALCULATIONS AND 133 REPORTING SECTION 134 25. Pharmacokinetic and statistical calculations 25 135 26. Clinical study report 25 136 REFERENCES 137 Appendix 1. Examples of the list of standard operating procedures at the contract research 138 organization 27 139
140
INTRODUCTION 141 142
Multisource pharmaceutical products need to conform to the same standards of quality, 143
efficacy and safety as required of the originator's (comparator) product. Specifically, the 144
multisource product should be therapeutically equivalent and interchangeable with the 145
comparator product. Testing the bioequivalence between a product and a suitable comparator 146
(pharmaceutically equivalent or a pharmaceutical alternative) in a pharmacokinetic study with 147
a limited number of subjects is one way of demonstrating therapeutic equivalence without 148
having to perform a clinical trial involving many patients. In such a pharmacokinetic study 149
any statement about the safety and efficacy of the test product will be a prediction based on 150
Page 5
Working document QAS/15.622
page 5 measurement of systemic concentrations, assuming that essentially similar plasma 151
concentrations of the drug will result in essentially similar concentrations at the site of action, 152
and thus an essentially similar therapeutic outcome. The bioequivalence study thus provides 153
indirect evidence of the efficacy and safety of a multisource drug product. Often this will be 154
the only evidence that the product is safe and efficacious. It is therefore crucial that the 155
bioequivalence study is performed in an appropriate manner. Several guidance documents 156
stress the importance of onsite inspections to verify compliance with standards of good 157
clinical practice.i,4
158
159
1. SCOPE 160 161
The objective of this document is to provide guidance to organizations that are involved in the 162
conduct and analysis of in vivo bioequivalence studies. This guidance has been updated relative to 163
the previous version of this document. 164
165
Bioequivalence studies should be performed in compliance with the general regulatory 166
requirements and good practices recommendations as specified in the WHO bioequivalence 167
guideline,3 GCP
4 and GLP
5 guidelines. 168
169
The text below lists general recommendations for organizations (including CROs and laboratories) 170
conducting bioequivalence studies and analysis of clinical trial samples. Recommendations for 171
facilities and equipment are listed in the respective paragraphs. Recommended documents, standard 172
operating procedures and records are listed in Appendix 1, but this is not to be considered an 173
exhaustive list – others may be necessary depending on each individual CRO’s functional and 174
compliance needs. 175
176
This document provides information on: 177
178
‒ organization and management; 179
‒ study protocols; 180
‒ clinical phase of a study; 181
‒ bioanalytical phase of a study; 182
‒ pharmacokinetic and statistical analysis; 183
‒ study report and quality assurance. 184
185
The present guideline targets organizations conducting bioequivalence studies and highlights 186
certain important aspects of the activities of such organizations. This document does not replace the 187
above-mentioned GCP or GLP or good practices for quality control laboratories guidelines, which 188
are more complete. It is therefore not a stand-alone document. For further guidance also see the 189
International Conference on Harmonisation (ICH) Tripartite Harmonised Guidelines and European 190
community (EC) regulations for GCP for trials on pharmaceutical products.1,2
191
192
193
Page 6
Working document QAS/15.622
page 6 2. GLOSSARY 194 195
The definitions given below apply to the terms used in this guidance. They may have different 196
meanings in other contexts. 197
198
adverse event 199
Any untoward medical occurrence in a clinical trial subject administered a pharmaceutical product; 200
it does not necessarily have a causal relationship with the treatment. 201
202
audit of a trial 203
A systematic examination, carried out independently of those directly involved in the trial, to 204
determine whether the conduct of a trial complies with the agreed protocol and whether the data 205
reported are consistent with the records on site, e.g. whether data reported or recorded in the case-206
report forms are consonant with those found in hospital fi les and other original records. 207
208
bioequivalence test 209
A test that determines the equivalence between the multisource product and the comparator product 210
using in vivo and/or in vitro approaches. 211
212
calibration curve samples (CCs) or calibration standards (new – definition from European 213
Medicines Agency (EMA) guideline on bioanalytical validation) 214
A matrix to which a known amount of analyte has been added or spiked. Calibration standards are 215
used to construct calibration curves. 216
217
case-report form 218
A document that is used to record data on each trial subject during the course of the trial, as defined 219
by the protocol. The data should be collected by procedures which guarantee preservation, retention 220
and retrieval of information and allow easy access for verification, audit and inspection. 221
222
comparator product (or reference product) 223
A pharmaceutical or other product (which may be a placebo) used as a reference in a clinical trial. 224
225
contract 226
A document, dated and signed by the investigator, institution and sponsor, that sets out any 227
agreements on financial matters and delegation/distribution of responsibilities. The protocol may 228
also serve as a contract when it contains such information and is signed. 229
230
contract research organization 231
A scientific organization (commercial, academic or other) to which a sponsor may transfer some of 232
its tasks and obligations. Any such transfer should be defined in writing. 233
234
ethics committee8 235
An independent body (a review board or a committee, institutional, regional or national), 236
constituted of medical professionals and non-medical members, whose responsibility is to verify 237
that the safety, integrity and human rights of the subjects participating in a particular trial are 238
protected and to consider the general ethics of the trial, thereby providing public reassurance. 239
Ethics committees should be constituted and operated so that their tasks can be executed free from 240
bias and from any influence of those who are conducting the trial. 241
242
final report 243
Page 7
Working document QAS/15.622
page 7 A comprehensive description of the trial after its completion including a description of 244
experimental methods (including statistical methods) and materials, a presentation and evaluation 245
of the results, statistical analysis and a critical, ethical, statistical and clinical appraisal. 246
247
good clinical practice 248
A standard for clinical studies which encompasses the design, conduct, monitoring, termination, 249
audit, analysis, reporting and documentation of the studies and which ensures that the studies are 250
scientifically and ethically sound and that the clinical properties of the pharmaceutical product 251
(diagnostic, therapeutic or prophylactic) under investigation are properly documented. 252
253
good laboratory practice 254
A quality system concerned with the organizational process and the conditions under which 255
nonclinical health and environmental safety studies are planned, performed, monitored, recorded, 256
archived and reported. 257
258
informed consent 259
A subject’s voluntary confirmation of willingness to participate in a particular trial, and the 260
documentation thereof. This consent should be sought only after all appropriate information has 261
been given about the trial including an explanation of its status as research, its objectives, potential 262
benefits, risks and inconveniences, alternative treatment that may be available, and of the subject’s 263
rights and responsibilities in accordance with the current revision of the Declaration of Helsinki. 264
265
inspection 266
An officially-conducted examination (i.e. review of the conduct of the trial, including quality 267
assurance, personnel involved, any delegation of authority and audit) by relevant authorities at the 268
site of investigation and/or at the site of the sponsor in order to verify adherence to good clinical 269
practices and good laboratory practices as set out in this document. 270
271
internal standard (new – definition from EMA guideline on bioanalytical validation) 272
Test compound(s) (e.g. a structurally similar analogue, or stable isotope labelled compound) added 273
to calibration standards, quality control samples and study samples at a known and constant 274
concentration to correct for experimental variability during sample preparation and analysis. 275
276
investigational labelling 277
Labelling developed specifically for products involved in a clinical trial. 278
279
investigational product (synonym: study product) 280
Any pharmaceutical product (see definition) or placebo being tested or used as a reference in a 281
clinical trial. 282
283
investigator 284
A person responsible for the trial and for the rights, health and welfare of the subjects in the trial. 285
The investigator should have qualifications and competence in accordance with local laws and 286
regulations as evidenced by an up-to-date curriculum vitae and other credentials. Decisions relating 287
to, and the provision of, medical or dental care must always be the responsibility of a clinically 288
competent person legally allowed to practise medicine or dentistry. 289
290
lower limit of quantification (new – definition from EMA guideline on bioanalytical validation) 291
Page 8
Working document QAS/15.622
page 8 The lower limit of quantification of an individual analytical procedure is the lowest amount of 292
analyte in a sample which can be quantitatively determined with pre-defined precision and 293
accuracy. 294
295
monitor 296
A person appointed by, and responsible to, the sponsor or contract research organization for the 297
monitoring and reporting of progress of the trial and for verification of data. 298
299
pharmaceutical product 300
Any substance or combination of substances which has a therapeutic, prophylactic or diagnostic 301
use, or is intended to modify physiological functions, and is presented in a dosage form suitable for 302
administration to humans. 303
304
principal investigator 305
The investigator serving as coordinator for certain kinds of clinical trials, e.g. multicentre trials. 306
307
protocol 308
A document which states the background, rationale and objectives of the trial and describes its 309
design, methodology and organization, including statistical considerations, and the conditions 310
under which it is to be performed and managed. The protocol should be dated and signed by the 311
investigator, the institution involved and the sponsor. It can also function as a contract. 312
313
quality assurance relating to clinical trials 314
Systems and quality control procedures that are established to ensure that the trial is performed and 315
the data are generated in compliance with good clinical practices and good laboratory practices. 316
These include procedures to be followed which apply to ethical and professional conduct, standard 317
operating procedures, reporting, and professional qualifications or skills of personnel. 318
319
quality control samples (new – definition from EMA guideline on bioanalytical validation) 320
A spiked sample used to monitor the performance of a bioanalytical method and to assess the 321
integrity and validity of the results of the unknown samples analysed in an individual batch. 322
323
raw data 324
All records or certified copies of original observations, clinical findings or other activities in a 325
clinical trial necessary for the reconstruction and evaluation of the trial. Such material includes 326
laboratory notes, memoranda, calculations and documents, as well as all records of data from 327
automated instruments or exact, verified copies, e.g. in the form of photocopies or microfiches. 328
Raw data can also include photographic negatives, microfilm, magnetic media (e.g. computer 329
diskettes) and optical media (CD-ROMs). 330
331
serious adverse event 332
An event that is associated with death, admission to hospital, prolongation of a hospital stay, 333
persistent or significant disability or incapacity, or is otherwise life-threatening in connection with a 334
clinical trial. 335
336
sponsor 337
An individual, a company, an institution or an organization which takes responsibility for the 338
initiation, management and/or financing of a clinical trial. When an investigator initiates and takes 339
full responsibility for a trial, the investigator then also assumes the role of the sponsor. 340
341
Page 9
Working document QAS/15.622
page 9 standard operating procedures 342
Standard, detailed, written instructions for the management of clinical trials. They provide a general 343
framework enabling the efficient implementation and performance of all the functions and activities 344
for a particular trial as described in this document. 345
346
study director 347
According to the Organisation for Economic Co-operation and Development principles of good 348
laboratory practice: the individual responsible for the overall conduct of the nonclinical health and 349
environmental safety study. In a bioequivalence trial, the individual responsible for the conduct of 350
the bioanalytical part of the study. 351
352
study product: see investigational product 353
354
test product 355
Any pharmaceutical product (see definition) or placebo being tested against the reference in a 356
clinical trial. 357
358
trial subject 359
An individual who participates in a clinical trial, either as a recipient of the pharmaceutical product 360
under investigation or as a control. The individual may be: 361
‒ a healthy person who volunteers to participate in a trial; 362
‒ a person with a condition unrelated to the use of the investigational product; 363
‒ a person (usually a patient) whose condition is relevant to the use of the investigational 364
product. 365
366
upper limit of quantification (new – definition from EMA guideline on bioanalytical validation) 367
The upper limit of quantification of an individual analytical procedure is the highest amount of 368
analyte in a sample which can be quantitatively determined with pre-defined precision and 369
accuracy. 370
371
validation 372
Action of proving and documenting, in accordance with the principles of good clinical practices 373
and good laboratory practices, that any procedure, process, equipment (including the software or 374
hardware used), material, activity or system actually and consistently leads to the expected results. 375
376
verification of data 377
The procedures carried out to ensure that the data contained in the final report match original 378
observations. These procedures may apply to raw data, data in case-report forms (in hard copy or 379
electronic form), computer printouts and statistical analysis and tables. 380
381
GENERAL SECTION 382
383
3. ORGANIZATION AND MANAGEMENT 384 385
Note: the acronym “CRO” is used throughout this document to refer not only to a contract 386
research organization (CRO), but also to any organization involved in the conduct or analysis of in 387
vivo bioequivalence studies. As defined in the International Conference on Harmonisation (ICH) 388
Tripartite Harmonised Guidelines, Guidelines for Good Clinical Practice (5), a “CRO” is a person 389
or an organization (commercial, academic or other) contracted by the sponsor to perform one or 390
more of a sponsor’s trial-related duties and functions. 391
Page 10
Working document QAS/15.622
page 10 392
3.1 Where national requirements exist as to the legal status of a CRO these have to be complied 393
with. This also applies to the research unit which is a subsidiary of the manufacturer. 394
395
3.2 The CRO should have an organization chart reflecting key positions and the names of 396
responsible persons. The organization chart should be authorized (signed and dated). 397
398
3.3 There should be job descriptions for all personnel, including a description of their 399
responsibilities. All job descriptions should be acknowledged and signed off by the staff member to 400
whom it applies (revised). 401
402
3.4 There should be a list of signatures of authorized personnel participating in each study 403
(revised). 404
405
4. COMPUTER SYSTEMS 406 407
Note: Computer systems should be qualified (hardware and software6). 408
Qualification is the planning, carrying out and recording of tests on equipment and systems, 409
which form part of the validated process, to demonstrate that it will perform as intended. 410
Since data for bioequivalence studies is often transferred electronically between 411
organizations involved in the studies, compatible software is essential (revised) 412
413
Hardware 414 415 4.1 There should be a sufficient number of computers to enable personnel to perform data entry 416
and data handling, required calculations and compiling of reports. 417
418
4.2 Computers should have sufficient capacity and memory for the intended use. 419
420
4.3 There should be access control to the trial-related information entered and stored in 421
computers. The method of access control should be specified (e.g. password protection) and a list 422
of people who have access to the database should be maintained. Secure and unique, individual-423
specific identifiers and passwords, should be used (added sentence). 424
425
Software 426 427 4.4 The software programmes selected should be suitable and validated for the intended use 428
(revised sentence). 429
430
4.5 There should be a system in place for the implementation of regular updates to key software 431
programmes (e.g. such as those used for control and data processing of chromatographic and mass 432
spectrometry systems) whenever required, following an appropriate risk assessment on the potential 433
impact that it could have on current data and on qualification/validation status (new). 434
435
4.6 Software programmes used, frequency of virus testing, storage of data and the making and 436
archiving and keeping of back-ups should be specified in writing. 437
438
4.7 The programmes used should be able to provide the required quality and management 439
information, reliably and accurately. Necessary programmes for data management include word 440
Page 11
Working document QAS/15.622
page 11 processing, data entry, databases, graphics, pharmacokinetics and statistical programmes. Self-441
designed software programmes must be suitable and validated for their intended use. 442
443
4.8 These requirements apply to all systems used in clinical bioequivalence (BE) studies. For 444
instance, the software used to obtain data such as electrocardiographs (ECGs) are considered 445
covered under the scope of this guidance (new). 446
447
Data management 448
449
4.7 Data entry includes transfer of the data from case report forms (CRF) and analytical data to 450
the computerized system for pharmacokinetic and statistical analysis and reporting. 451
452
4.8 Data entry procedures should be designed to prevent errors. The data entry process should 453
be specified in the standard operating procedure (SOP). 454
455
4.9 Double entry of the data should be performed. Data validation methodology (proof-reading, 456
double data entry, electronic logical control) should be specified in writing. 457
458
4.10 Changes made to data entered in the database should be made by authorized persons only. 459
Changes should be specified and documented. 460
461
4.11 Electronic data should be backed up at regular intervals. The reliability and 462
completeness of these back-ups should be verified – data should not be selected but 463
comprehensively backed up (new)). 464
465
4.12 All of the raw electronic data must be kept. This includes: 466
– all meta data associated to a computerized system and the equipment that is associated to it 467
(which includes the audit trails for integration, for projects and for the entire instrument); 468
– validation data and meta data in the form of their source electronic files. 469
PDF copies are not sufficient on their own, unless it can be demonstrated that these are the 470
raw data and that no alteration was possible after they were generated (new). 471
472
4.13 All electronic records obtained from high performance liquid chromatography(HPLC) 473
and mass spectrometric (MS) analysis (e.g. HPLC-MS/MS) are required to be retained, 474
maintained and backed-up. It should be ensured that back-up data are exact and complete and 475
that they are secure from alteration, inadvertent erasures or loss shall be maintained. The 476
printed paper copy of the chromatogram would not be considered a “true, exact and complete 477
copy” of the entire electronic raw data used to create that chromatogram. Printed 478
chromatograms do not generally include, for example, the sample sequence, instrument 479
method, processing method, integration settings or the full audit trail, of which all were used 480
to create the chromatogram or are associated with its validity. Therefore there should be a 481
higher emphasis on conservation of electronic data than paper data, as paper data is usually 482
not considered the true source data, except in the case of paper logbooks where the original 483
record was handwritten, for instance (new). 484
485
[Please refer to the WHO Good data management practices guidelines for more detailed guidance 486
on requirements for computerized systems – full reference to be confirmed once finalized.] (new) 487 488 5. QUALITY ASSURANCE 489 490
Page 12
Working document QAS/15.622
page 12 5.1 The CRO should have an appropriate quality assurance (QA) system. 491
492
5.2 The QA system and the person(s) responsible for QA should operate independently from 493
those involved in all steps of the study, including: 494
‒ conducting or monitoring of the trial 495
‒ conducting bioanalysis 496
‒ performing reporting and statistical analysis. 497
As a consequence, QA personnel should not be directly involved in trial-related activities, and an 498
in-process audit by QA personnel does not replace oversight by another person when a four-eyes 499
principle has to be applied (revised). 500
501
5.3 The QA unit should be responsible for: 502
‒ verifying all activities undertaken during the study; 503
‒ ensuring that the QA systems, including SOPs of the CRO, are followed, reviewed and 504
updated; 505
‒ checking all the study data for reliability and traceability; 506
‒ planning and performing self-inspections (internal audits) at regular and defined intervals in 507
accordance with an SOP, and following up on any corrective action as required; 508
‒ ensuring that contract facilities, such as analytical laboratories, adhere to GLP. This would 509
include auditing of such facilities, and following up on any corrective action as required 510
(revised sentence); 511
‒ verifying that the trial report accurately and completely reflects the data of the study. 512
513
5.4 The CRO should allow the sponsor to monitor the studies and to perform audits of the 514
clinical and analytical study and sites. 515
516
5.5 The laboratory should have a QA unit which should be independent from the person(s) 517
responsible for analytical work and which should ensure that the analytical method in use is 518
validated and current. 519
520
5.6 Both retrospective and in-process (e.g. in bioanalysis, as the samples and standards 521
are being prepared and tested), QA verifications should be performed (new). 522
523
6. ARCHIVE FACILITIES 524 525
Note: The CRO should have sufficient and appropriately secure storage space, which should be 526
fire proof, humidity-controlled and pest-controlled, for archiving of trial-related 527
documentation and product samples (added wording) 528
529
6.1 An SOP should be in place for archiving. 530
531
6.2 Access to archive storage areas should be controlled and restricted to authorized personnel. 532
533
6.3 Records should be maintained of document access and return (new). 534
535
6.4 The length of period for which study documentation including raw data is kept in the 536
archive should be defined in the SOP and may vary depending on country requirements. 537
538
Page 13
Working document QAS/15.622
page 13 6.5 Product samples should be retained for a specific period in compliance with the applicable 539
national requirements or international recommendations as appropriate and should be defined in the 540
SOP and be specified in the contract between the sponsor and the CRO (revised). 541
542
6.6 The duration of storage of bioanalytical samples should be specified in the contract between 543
the sponsor and the CRO (new). 544
545
6.7 All data, including both paper and electronic, should be easy to retrieve and traceable (new). 546
547
7. PREMISES 548 549
7.1 Clinical trials must be carried out under conditions which ensure adequate safety for the 550
subjects. The site selected should be appropriate to the stage of development of the product and the 551
potential risk involved. 552
553
7.2 The CRO should have sufficient space to accommodate the personnel and activities required 554
to perform the studies. The trial site must have adequate facilities, including laboratories, and 555
equipment. The facilities used for the clinical phase of the study, including areas listed in paragraph 556
9.6 should be well organized in order to carry out the activities in logical order. 557
558
7.3 The entry to the facility should be restricted and controlled. There should be alarm systems 559
to detect the exit of subjects from clinical facilities or the doors should be locked (however, doors 560
should be locked only if emergency evacuation can still be ensured). Entry and exit to the facility 561
should be recorded (revised). 562
563
7.4 Sites involved in clinical activities should include a pharmacy where investigational 564
products should be stored under appropriate conditions with restricted control. Appropriate 565
entry/exit record of each visit to the pharmacy should be maintained. 566
567
7.5 Utilities such as water, air, gas and electricity should be adequate, stable and uninterrupted 568
(new). 569
570
7.6 There should be access to telephone, email and facsimile facilities to ensure proper 571
communication. The CRO should have the necessary office equipment (printer, copy-machine) to 572
perform the required activities. 573
574
7.7 Laboratory premises should be designed to suit the operations to be carried out in them. 575
Sufficient space should be given to avoid mix ups, contamination and cross-contamination. There 576
should be adequate suitable storage space for samples, standards, solvents, reagents and records. 577
578
7.8 Laboratory premises should be designed to provide adequate protection to all employees, by 579
ensuring their safety while handling or working in the presence of chemicals and biological 580
samples. This would include adequate ventilation (exhaust ventilation or other engineering controls 581
to keep the airborne concentrations of vapours below their respective threshold limit value). If this 582
is not possible then suitable respiratory equipment (e.g. when working with solvents, vapour 583
respirators that are approved/certified for their use with the specific solvents) should be provided. 584
The following protective measures should also be implemented: 585
‒ containers containing volatile organic solvents, such as those used to contain mobile phases 586
or liquid/liquid extraction solvents, should be closed with an appropriate seal; 587
Page 14
Working document QAS/15.622
page 14 ‒ volatile organic chemicals should be handled under certified fume-hoods or air extractors 588
and safety and eye showers should be available in the laboratory; 589
‒ strong acids and bases should be handled in a suitable area by staff wearing appropriate eye 590
and hand protection and should be stored in accordance with their labelled storage 591
conditions. Strong acids and strong bases should be stored separately; 592
‒ staff working in the laboratory should be familiar with and knowledgeable of the material 593
safety data sheets for the chemicals that they are handling. These should be maintained and 594
be accessible to staff at the laboratory for all chemicals in use; 595
‒ flammable solvents should be stored in appropriately ventilated storage rooms and in 596
purpose-designed cabinets (new). 597
598
7.9 Premises should have suitable systems in place to dispose of waste, to treat fumes and to 599
protect the environment in conformance to local or national regulation (new). 600
601
8. PERSONNEL 602 603
8.1 There should be a sufficient number of qualified and appropriately trained medical, 604
paramedical, technical and clerical staff to support the trial and to be able to respond effectively to 605
all reasonably foreseeable emergencies. The number of members of staff required depends on the 606
number and complexity of the trials performed by the CRO. At all stages during the trial, including 607
at night, there should be a sufficient number of appropriately qualified and trained personnel to 608
ensure that the rights, safety and well-being of the subjects are maintained, and to take care of the 609
subjects in emergency situations. 610
611
8.2 The conduct and analysis of the in vivo bioequivalence studies should involve the following 612
key persons with appropriate responsibilities: 613
614
‒ medical/scientific director 615
‒ principal investigator/investigator and co-investigators 616
‒ study director 617
‒ quality assurance manager (Note: different QA personnel may be necessary to ensure 618
adequate conduct of their functions in each respective field of expertise, such as the clinical 619
department vs the bioanalytical department of a CRO.) 620
‒ technical manager 621
‒ quality control managers (Note: this category can include technical quality control managers 622
as well as data quality control for both the clinical, bioanalytical and pharmacokinetic / 623
statistical parts.) 624
‒ clinical monitors (revised) 625
626
8.3 One person could perform more than one of the above-mentioned functions; however, the 627
person responsible for quality assurance should be independent and report to the head of the 628
organization only. 629
630
8.4 Contract workers may be employed to perform certain activities. All contract workers 631
having access to the clinical or bioanalytical areas or performing trial-related activities should be 632
provided with adequate information, training and job descriptions (revised). 633
634
8.5 Current curriculum vitae and training records should be kept for full-time and contract 635
workers. 636
637
Page 15
Working document QAS/15.622
page 15 8.6 The personnel responsible for the planning and conduct of the study should have 638
appropriate qualifications and sufficient knowledge and experience in the relevant field. 639
640
8.7 Records for training and assessment of knowledge of GCP, GLP and any other relevant area 641
or technique should be maintained (new). 642
643
8.8 There should be adequate measures in place to protect personnel from accidental 644
contamination (e.g. from accidental needle pricks) while obtaining blood samples from subjects or 645
while handling the resulting samples that are derived from blood products (e.g. plasma and its 646
extracts) or while handling or disposing of infectious waste (new). 647
648
CLINICAL SECTION (added title) 649
650
9. CLINICAL PHASE 651 652
Note: As in vivo BE trials are considered as clinical trials, specifically a Phase I study, the 653
general requirements and recommendations of GCP apply to all BE trials. Clinical trials 654
must be carried out under conditions which ensure adequate safety of the subjects. The 655
clinical phase of the study can be performed in the premises of a CRO or by contracting 656
suitable premises in a hospital. 657
658
9.1 A CRO should have rooms meeting the requirements listed in the sections below. 659
660
9.2 There should be sufficient space to accommodate the study subjects. 661
662
9.3 Where appropriate, beds should be available for the volunteers. The necessity of beds and 663
overnight stay depends on the type of trial and investigational drug and should be specified in the 664
trial protocol. Overnight stays are usually required during the night prior to dosing to ensure 665
adequately controlled conditions and that there was no outside food/medication intake within the 666
number of hours that is specified in the trial protocol (new). 667
668
9.4 Alarms should be located within arms-reach from the beds, to ensure that subjects that are 669
unable to move to a medical emergency are able to alert CRO staff (new). 670
671
9.5 Facilities for changing and storing clothes and for washing and toilet purposes should be 672
clean, well ordered, easily accessible and appropriate for the number of users. Closed toilets should 673
be alarmed and doors should be designed to ensure that they can be opened from the outside should 674
there be a medical emergency (new). 675
676
9.6 The study site should have the following facilities which should be separate areas where 677
appropriate: 678
‒ rooms (areas) for volunteer registration and screening; 679
‒ room (area) for volunteers (recreation area); 680
‒ room (area) for individual volunteers to obtain informed consent without compromising 681
privacy (new); 682
‒ ancillary areas for the volunteers; 683
‒ pharmacy area; 684
‒ restricted-access area for pharmaceutical operations (e.g. storage, repacking, dispensing, 685
documentation) (see also section 13); 686
Page 16
Working document QAS/15.622
page 16 ‒ rooms (areas) for dosing and administration of the drug(s) under investigation and sample 687
collection; 688
‒ room (area) for sample processing (e.g. plasma separation) and storage (freezer); 689
‒ access to controlled storage areas for study materials, medication and documentation 690
including CRFs; 691
‒ rooms (areas) in which to prepare standardized meals and a dining hall; 692
‒ availability of emergency or first-aid equipment and appropriate rescue medication for use 693
in emergencies. 694
‒ adequate facilities for the proper care of subjects who require emergency or other medical 695
care; 696
‒ archiving facilities. 697
698
9.7 Access to key documents, such as the randomization list, should be restricted to only certain 699
specific members of personnel such as the pharmacist in charge of the study. Such documents 700
should be password-secured (if electronic) or kept under lock and key (if distributed as a hard-701
copy) and their distribution should be documented (new). 702
703
9.8 Equipment used to obtain clinical measurements should be appropriately calibrated (new). 704
705
9.9 The adequate function and performance of emergency use equipment (e.g. defibrillators) 706
should be verified at appropriate intervals (new). 707
708
9.10 Requirements of good laboratory practices (5) and of section 24 of these guidelines should 709
be applied to equipment used to process and store biological samples obtained in the clinical phase 710
(e.g. centrifuges, freezers, etc.) (new). 711
712
10. CLINICAL LABORATORY 713 714
10.1 A suitable qualified clinical laboratory should be used for analysing samples. An accredited 715
laboratory should be used whenever possible (revised). 716
717
10.2 Haematological tests, urine analysis and other tests should be performed during the clinical 718
trial as specified in the study protocol. 719
720
10.3 The CRO should be supplied with information about analytical methods used in the 721
laboratory, a dated list of laboratory normal ranges and accreditation certificate of the laboratory, if 722
available. These should be available for inspection by regulatory authorities, if required (new). 723
724
10.4 A current and signed curriculum vitae of the responsible analyst should be available in the 725
laboratory information file. 726
727
10.5 Individual reports should be established by the laboratory for each subject and should be 728
included in the CRFs. Source or raw data for all tests performed should be archived by the 729
laboratory in electronic or paper formats, depending on their source and storage capacity. 730
Electronic formats are preferred (new). 731
732
10.6 Data integrity requirements apply to all tests related to the study ( full reference to be 733
confirmed once finalized.) For instance, raw data should be adequately protected from modification 734
or deletion (new). 735
736
Page 17
Working document QAS/15.622
page 17 10.7 The principles of GLP (5) and of section 24 of this guideline should be applied (new). 737
738
11. ETHICS 739 740 11.1 Independent ethics committee 741
Trials must be approved by an independent ethics committee (IEC) (or equivalent) before a study is 742
conducted, according to WHO Operational guidelines for Ethics Committees that review 743
biomedical research (8) and to the enforced legislation. This committee must be independent from 744
the sponsor, the investigator and of the CRO. The discussions, recommendations and decisions of 745
the IEC meetings should be documented in detailed minutes of the meeting. The IEC should be 746
given sufficient time for reviewing protocols, informed consent forms (ICFs) and related 747
documentation (revised). 748
749
11.2 Informed consent 750
• Information for study participants should be given in a language and on a level of 751
complexity appropriate and understandable to the subject, both orally and in writing. 752
753
• Informed consent must always be given by the subject and documented in writing before the 754
start of any trial-related activities, in accordance with GCP. If informed consent is also 755
recorded by video this recording should be retained following local legal requirements 756
(revised) 757
758
• The information must make clear that participation is voluntary and that the subject has the 759
right to withdraw from the study on his or her own initiative at any time, without having to 760
give a reason (compensation should be paid prorata temporis). If subjects who withdraw 761
from the study offer their reasons for doing so, those reasons should be included in the 762
study records. 763
764
• The subject must have access to information about insurance, and other procedures for 765
compensation or treatment should he or she be injured or disabled by participating in the 766
trial. 767
768
• The volunteers/subjects should be given opportunity to discuss their concerns with a 769
physician regarding potential side effects or reactions from the use of test product before 770
participation in the trial. They should also be given the opportunity and sufficient time to 771
discuss their concerns with their participation in the trial with individuals outside of the 772
clinical research organization, such as friends and family members, if they wish (new). 773
774
775
12. MONITORING 776 777
Note: monitoring is an essential activity to ensure the quality of the clinical trial. 778
779
12.1 The monitor should be qualified (see section 8, Personnel). The main responsibility of the 780
monitor for a bioequivalence trial is to ensure that the study is conducted in accordance with the 781
protocol, GCP, GLP and applicable ethical and regulatory requirements. This includes verification 782
of the use of correct procedures for completion of CRFs and verification of the accuracy of data 783
obtained. 784
785
Page 18
Working document QAS/15.622
page 18 12.2 The sponsor can delegate the monitoring function to the CRO. In such cases the CRO 786
should be able to arrange for the monitoring of the trial according to regulatory requirements. 787
788
12.3 The frequency of monitoring visits should be agreed to between the CRO and the sponsor. 789
However, a pre- and post-study visit as well as a monitoring visit during the conduct of the trial are 790
usually performed. The monitor should prepare a written report after each site visit and 791
communicate any issues to the CRO and to the sponsor as promptly as possible, even during 792
conduct of the study if possible, to enable prompt corrective action. Such communications and 793
corrective actions should be documented (reworded). 794
795
12.4 When the monitoring is delegated to the CRO, SOPs should be available to describe: 796
‒ the designation of monitors, who should be independent from the personnel performing the 797
trial; 798
‒ monitoring visit procedures; 799
‒ the extent of source data verification, including with regards to accountability of the 800
investigational products and adherence to the protocol. 801
The extent of the monitoring, including the number of visits to be performed, should be agreed 802
upon with the sponsor (reworded). 803
804
12.5 Separate SOPs (with checklists for the monitor) for the initiation visit, routine monitoring 805
visits and a closing visit are recommended. 806
807
12.6 Appropriate entry/exit record of each monitoring visit should be maintained (new). 808
809
13. INVESTIGATORS 810 811
13.1 The principal investigator (PI) should have the overall responsibility for the clinical conduct 812
of the study, including clinical aspects of study design, administration of the products under 813
investigation, contacts with local authorities and the ethics committee and for signing the protocol 814
and the final study report. 815
816
13.2 The investigator(s) should have appropriate qualifications, be suitably trained and 817
have experience in the conduct of bioequivalence studies (the legal status of persons 818
authorized to act as investigators differs between countries) and at least one investigator must 819
be legally allowed to practice medicine. 820
821
13.3 The medically-qualified investigator should be responsible for the integrity, health and 822
welfare of the subjects during the trial and the accurate documentation of all trial-related clinical 823
data. 824
825
13.4 The CRO is responsible for selecting investigator(s). In cases where the investigators are 826
not permanent employees of the CRO external investigators should be contracted and adequately 827
trained. 828
829
14. RECEIVING, STORAGE AND HANDLING OF INVESTIGATIONAL DRUG 830
PRODUCTS 831 832
14.1 CROs should document all the information concerning the receipt, storage, handling 833
and accountability of investigational and comparator products at any stage of the trial. CROs 834
must keep records of information about the shipment, delivery, receipt, storage (including 835
Page 19
Working document QAS/15.622
page 19 storage conditions), dispensing, administration, reconciliation, return and/or destruction of 836
any remaining pharmaceutical products. Detail of the pharmaceutical product used should 837
include dosage form and strength, lot number, expiry date and other coding that identifies the 838
specific characteristics of the product tested. Samples of the product in the original container 839
should be retained for possible confirmatory testing in the future (word replaced). 840
841
14.2 A suitable location within the CRO or a local pharmacy or hospital pharmacy should 842
assume responsibility for storage, delivery, return and record keeping of the investigational drug 843
and, when appropriate, comparator product(s). 844
845
14.3 Pharmaceutical products should be stored under appropriate storage conditions as specified 846
in the official drug information provided by the sponsor (word replaced). 847
848
14.4 All study medication should be kept in a securely locked area accessible only to authorized 849
persons. 850
851
14.5 Randomization should be performed in accordance with an SOP and records should be 852
maintained, including the randomization list and seed, if applicable. Under normal operations the 853
randomization list should be accessible only to the person who generates it, a dispensing 854
pharmacist and the statistician (both named/nominated in the protocol) and should not be circulated 855
or made available to other staff members via any medium. A system should be in place which 856
allows the PI or delegated staff to access the randomization list in case of emergencies (reworded 857
and expanded). 858
859
14.6 Labelling should be performed in accordance with the following requirements: 860
‒ the printing step should be done in a manner that reduces potential risks of mislabelling and 861
should be done in accordance with a SOP; 862
‒ each label should include the following information 863
• name of the sponsor, 864
• a statement of “for clinical trial use only”, 865
• trial reference number or study number, 866
• batch number, 867
• subject identification number (to which the product is destined to be given to), 868
• period, 869
• active ingredient and dosage, 870
• the storage conditions, 871
• expiry date (month/year) or retest date, 872
• identification of the product: Whenever possible, just prior to dosing, a check should 873
be performed of vial contents matching the information on the label; 874
‒ compliance of all labels with the randomization list, should be verified once printed, prior to 875
labelling of the containers; 876
‒ labelling should be done on the container, not on the lid, to ensure that the information is 877
not lost once the lid is removed; 878
‒ labels should be designed in such a way that two identical labels are pasted to the container 879
and that the second label can be easily cut or detached and pasted onto the CRFs at the time 880
of dosing (e.g. two labels printed side by side, with only one that is actually pasted onto the 881
container and the other one which remains attached but unpasted – it should be torn off or 882
cut with scissors at the time of dosing); 883
Page 20
Working document QAS/15.622
page 20 ‒ the empty containers should be labelled separately for the test and the reference 884
investigational products and should remain adequately segregated and placed in a secure 885
area under lock and key, to ensure absence of risk of any potential mix ups, until the 886
dispensing stage; 887
‒ label reconciliation should be performed; 888
‒ appropriate, detailed records should be maintained for each of the above steps (new). 889
890
14.7 Dispensing should be performed in accordance with the following requirements: 891
‒ the surface area on to which the product will be handled should be thoroughly cleaned prior 892
to bringing bottles of the product in the area. Any product container (full or empty), lone 893
dosage formulations, labelling materials contaminants/dirt/debris should be removed from 894
the area; 895
‒ a second person should verify that the surface area (otherwise referred to as “line”) is indeed 896
clear and clean prior to bringing and opening containers of the product: 897
‒ test and reference products should be handled using an appropriate instrument, such as a 898
spatula or spoon, as opposed to gloved hands; 899
‒ tablets should be distributed in each container in accordance with the randomization list 900
either for the comparator or for the test product. Both products should never be handled at 901
the same time. This also applies to the labelled containers; 902
‒ records should be made of this step in a manner that is similar to manufacturing batch 903
records, as described in WHO GMP guidelines, i.e. each and every step should be recorded 904
sequentially in detail; 905
‒ the surface area used to handle the product and its surroundings should be cleared and 906
cleaned immediately after and/or prior to initiating the dispensing of the next product (it is 907
important to note that this also applies to different products used in the same study); 908
‒ drug accountability and dispensing records should be maintained at all times. Each activity 909
should be documented at the time it is performed. This includes 910
• records of doses dispensed and returned or destroyed, 911
• records of cleaning and clearance of the area prior to dispensing, 912
• record of verification of adequate cleaning and clearance of the area, 913
• record of verification by a second person of each step; 914
‒ any factors that could affect the integrity of the data relating to investigational medicinal 915
products and comparators should be recorded, monitored and controlled. 916
917
[Please refer for further guidance on labelling and dispensing to the WHO good manufacturing 918
practices: supplementary guidelines for the manufacture of investigational pharmaceutical 919
products for clinical trials in humans, Annex 7, WHO Technical Report Series, No. 964, 1996.] 920
(new) 921
922
14.8 Dosing should be performed in accordance with the following requirements: 923
‒ dosing should be performed in accordance with a SOP; 924
‒ it should be performed under the supervision of the investigator or of qualified staff to 925
whom this task has been explicitly delegated in writing; 926
‒ the exact time of dosing should be documented; 927
‒ mouth check should be performed by looking under the tongue, under the lips, in the 928
corners of the mouth and between gums and cheeks, using a tongue depressor or a spatula, 929
in the case of solid oral dosage forms. For other types of dosage forms verification of 930
adequate administration should be performed by other suitable means. It should be 931
documented; 932
Page 21
Working document QAS/15.622
page 21 ‒ if more than one dosage unit is administered this should be clearly documented; 933
‒ dosing can be documented directly in the case report forms. If retranscribed in the case of 934
report forms from other documents the original documents should be retained; 935
‒ drug reconciliation, after dosing, should be verified by a second responsible person 936
(revised). 937
938
14.9 The investigator should follow the protocol requirements, randomization scheme and where 939
required, blinding. The investigator should ensure that the investigational product use is 940
documented in such a way as to ensure appropriate dosage. 941
942
15. CASE REPORT FORMS 943 944
15.1 CRFs should be used to record data on each subject during the course of the trial. 945
946
15.2 The CRO should have a procedure for designing CRFs if the sponsor requests the CRO to 947
design them. It is recommended to use a standardized format or template which should be amended 948
for each study protocol in accordance with the requirements for the particular study. The CRF 949
should be sent to the IEC for review and approval together with the study protocol, if it is not part 950
of the protocol (new sentence added). 951
952
15.3 The required data to be collected on each volunteer should be specified in the trial protocol. 953
A sample CRF should be appended to the protocol. 954
955
15.4 CRFs should be used to guarantee preservation, retention and retrieval of volunteer 956
information. CRFs should reflect the actual results obtained during the study and allow easy access 957
to verification, audit and inspection of the data. 958
959
15.5 Appropriate procedures should be established and followed to document the investigator's 960
certification of the accuracy of CRFs. Any errors or omissions should be clarified with the 961
investigator, corrected, dated and signed and explained on the CRF. 962
963
15.6 Clinical laboratory reports and all ECGs should be included with the CRFs for each subject 964
and should be submitted along with the dossier ((new) – Note: this should be proposed for inclusion 965
in the multisource guideline as well.) 966
967
968
16. VOLUNTEERS, RECRUITMENT METHODS 969 970
Note: The organization or institution performing BE studies should ideally have a pool of 971
healthy volunteers which have been medically tested and selected. Recruitment of 972
volunteers undertaken immediately before the study is not desirable since it is often 973
done in a hurry and may compromise the selection criteria, especially for safety; it 974
may also limit the time given to the subjects before they give their consent to 975
participate in the trial (revised). 976
977
16.1 Procedures for the recruitment of volunteers should be available and should include a 978
description of the potential methods that can be used by the CRO for recruitment of volunteers. A 979
database should be maintained for volunteers to avoid cross-participation and to specify a minimum 980
time interval between a volunteer’s participation in two studies. Access to the database should be 981
password controlled in order to secure confidential volunteer/subject information (revised). 982
Page 22
Working document QAS/15.622
page 22 983
16.2 Volunteer and subject identification should be ensured by reliable means. If a biometric 984
system is used for the identification of volunteers this system should be validated on a periodic 985
basis as well as after any change made to the validated system that could impact its function (new). 986
987
16.3 Informed consent of potential subjects should be obtained for any screening procedures 988
required to determine eligibility for the study, in addition to informed consent for participation in 989
the research portion of the study. 990
991
16.4 Subject selection criteria (inclusion and exclusion criteria) and recruitment procedures 992
should be described in the clinical trial protocol. 993
994
16.5 Subject screening results and trial participation should be recorded in a validated 995
database. If a regional or national database exists it should be used (the CRO should input 996
data about subject participation and should consult the database as part of screening to verify 997
that the subject complies with the exclusion period defined in the protocol for participation in 998
other studies). Access to the database should be password controlled in order to secure 999
confidential volunteer/subject information (new). 1000
1001
16.6 Ideally such a database should record and allow the users to query: 1002
– contact details; 1003
– gender; 1004
– status: eligible, disqualified, not eligible, quarantined, etc.; 1005
– date and place of last study participation, if applicable/if known; 1006
– date of last screening; 1007
– a unique code assigned to the subject which will never change; 1008
– outcome of last trial: Completed, randomised but not dosed, withdrawn for personal 1009
reason, withdrawn for medical reason, etc. 1010
These data should be backed up daily and be available for review at any time (new). 1011
1012
16.7 Medical records should be generated for each subject and should include information 1013
obtained during each screening visit and each study participation, which could be relevant for 1014
the inclusion and follow-up of the subject into subsequent trials. 1015
1016
17. DIETING 1017 1018
17.1 As meals can significantly affect absorption of drugs fasting and meals should be 1019
standardized and adequately controlled during the study days. The CRO should be able to arrange 1020
for standardized meals, snacks and drinks to study subjects as described in the clinical trial 1021
protocol. 1022
1023
17.2 Records should be maintained for timing, duration and amount of food and fluids 1024
consumed. 1025
1026
17.3 Standardized meals should be designed by a dietician with appropriate qualification, 1027
training and experience. If such services are contracted out a formal contract with terms of 1028
reference should be available (new). 1029
1030
1031
Page 23
Working document QAS/15.622
page 23 18. SAFETY, ADVERSE EVENTS, ADVERSE EVENT REPORTING 1032 1033
18.1 Appropriate study planning includes adequate evaluation of risk to the subjects. The study 1034
should be planned, organized, performed and monitored so that the safety profile will be 1035
acceptable, including to the volunteers. 1036
1037
18.2 First-aid emergency equipment and appropriate rescue medication should be available at the 1038
study site and adequate facilities of the proper care of subjects who require emergency or other 1039
medical care. 1040
1041
18.3 The investigator(s) should be responsible for medical decisions in case of adverse events 1042
and for notifying the relevant health authorities, the sponsor and, when applicable, the EC, without 1043
delay in the case of serious adverse events. Appropriate timelines should be respected as governed 1044
by national regulations. 1045
1046
18.4 The CRO should have appropriate adverse event registration and reporting forms, which 1047
should be provided to the investigator; these forms can be part of the CRF. If required the 1048
respective sponsor's forms may be used. 1049
1050
BIOANALYTICAL SECTION 1051
Note: The analysis of drug concentrations may be performed in the same CRO which conducted 1052
the clinical study, or may be contracted to another laboratory or CRO. 1053
1054
19. METHOD DEVELOPMENT 1055
1056 19.1 The bioanalytical laboratory should provide detail on how an assay method was developed. 1057
The laboratory should keep a copy of any publication used to develop the bioanalytical method. 1058
The modifications and adaptations to the published method made by the laboratory should be 1059
documented. 1060
1061 19.2 Selection of the internal standard should be justifiable by sound scientific principles. In 1062
general, chemical and physical properties of the internal standard should be as close to those of the 1063
analyte as possible. Both stable isotope-labelled and non-isotope labelled internal standards are 1064
acceptable, though the use of stable isotope-labelled internal standard is recommended when mass 1065
spectrometric methods are used. The selection of a stable isotope labelled internal standard should 1066
take into consideration factors such as the isotope labelling positions in order to limit the risk of 1067
exchange reactions. 1068
1069
19.3 Method development should ensure that methods are created in a manner which will 1070
minimize any potential human error and should take into consideration the different volumes to be 1071
measured during the course of an analysis. 1072
(new section) 1073
1074
20. METHOD VALIDATION 1075 Requirements of the EMA Guideline on bioanalytical method validation (7) should be applied 1076
(new). 1077
1078 20.1 Validation requirements for the analytical method should be described in the protocol. 1079
There should be separate SOPs for analytical method validation. 1080
1081
Page 24
Working document QAS/15.622
page 24 20.2 Data to support the stability of the samples under the stated conditions and period of storage 1082
should be available preferably before the start of the study. 1083
1084
20.3 Method validation should be performed with at least one run that is comparable in length to 1085
those that are expected to be used for analysis of samples (new). 1086
1087
21. SAMPLE COLLECTION, STORAGE AND HANDLING OF BIOLOGICAL 1088
MATERIAL 1089 1090
21.1 The specification of the samples (serum, plasma or urine), sampling method, volume and 1091
number of samples should be stated in the clinical trial protocol and the information provided to the 1092
volunteer. 1093
1094
21.2 There should be documented procedures for the collection, preparation, transport and 1095
storage of samples. 1096
1097
21.3 Actual sampling times and deviations from the prespecified sampling times should be 1098
recorded. Deviations should be reported in the study report and should be taken into consideration 1099
when calculating the pharmacokinetic parameters (new sentence added). 1100
1101
21.4 Labelling of collected samples should be clear to ensure correct identification and 1102
traceability of each sample. 1103
1104
21.5 The storage conditions of samples depend on the investigational drug. However, all storage 1105
conditions (e.g. freezer temperature) should be specified in the study protocol, controlled, 1106
monitored and recorded throughout the storage period and transportation. Procedures should be in 1107
place to ensure sample integrity in case of system failures. 1108
1109
21.6 Records for the storage and retrieval of samples should be maintained. 1110
1111
21.7 It is recommended to keep duplicate or back-up samples; and store and ship them 1112
separately. 1113
1114
21.8 Local requirements for the handling and destruction of remaining biological materials 1115
should be followed. 1116
1117
22. ANALYSIS OF STUDY SAMPLES 1118
1119 Requirements of the EMA Guideline on bioanalytical method validation (7) should be applied 1120
(new). Additionally: 1121
1122 22.1 Each analytical run should include calibration curve (CC) standards, QC samples and 1123
subject samples processed simultaneously. The exact sequence of processing should be 1124
documented. All samples collected from a given subject during all trial periods should be analysed 1125
in the same run unless scientifically justified (e.g. due to the limited stability of samples, requiring 1126
the analysis of period one samples before period two is conducted) (new). 1127
1128
22.2 Equipment with an adequate capacity should be used to be able to process all samples in a 1129
run simultaneously, rather than splitting the samples into several extraction batches. However, if 1130
several extraction batches are used, each batch should include QC samples. The acceptance criteria 1131
Page 25
Working document QAS/15.622
page 25 for the analytical run should be defined in a SOP first for the full run, then if the run is acceptable, 1132
for each individual extraction batch (new). 1133
1134
22.3 The insertion of wash samples into runs after samples with a high level of concentration is 1135
recommended when there is a significant carry-over effect, but efforts should be made during 1136
method development to avoid such effects (new). 1137
1138
22.4 With regards to the use of blank plasma in the preparation of CCs and QCs: 1139
‒ the number of freeze-thaw cycles and the storage duration that a given blank plasma sample 1140
can be submitted to, should be limited as much as possible to ensure absence of degradation 1141
and/or change of its properties. Freezing blank plasma in small volumes should be 1142
considered to help limit the number of freeze-thaw cycles for any given blank plasma 1143
sample; 1144
‒ the anticoagulant that was used for the blank plasma should be documented. It should match 1145
the anticoagulant that was used in study samples, in nature and in proportion (new). 1146
1147
22.5 With regards to incurred sample reanalysis: 1148
‒ incurred sample reanalysis should be performed in line with the EMA Guideline on 1149
Bioanalytical Method Validation (2011) (7); 1150
‒ out-of-range results should be scrutinized and investigated. The impact on the validity of 1151
sample test results should be assessed (new). 1152
1153
23. DATA PROCESSING AND DOCUMENTATION (revised title) 1154
1155
23.1 Integration settings should be science-based and fully justifiable. Smoothing should be kept 1156
low enough not to mask possible interferences and changes in peak geometry (new). 1157
1158
23.2 The different iterations used to obtain a CC should be saved – if a given CC fails, it is not 1159
acceptable to exclude CCs which meet acceptance criteria or similarly, to include CC standards 1160
which do not meet criteria, just to make the calibration or the QC standards pass. The source data 1161
should contain the original, first evaluation of runs (containing all calibration samples). If several 1162
calibration samples are excluded sequentially the CC obtained at each step should be retained to 1163
document that the criteria to exclude the next sample were met. If electronic raw data are used it is 1164
acceptable to only save the final calibration if it is possible to revert to the initial calibration during 1165
an inspection. The process and criteria for acceptance and exclusion of CC standards should be 1166
described in an SOP (new). 1167
1168
23.3 If the first or last calibration sample is rejected the calibration range should be truncated, i.e. 1169
the second calibration sample becomes the lower limit of (LLOQ) in that run (or the one before last 1170
calibration sample becomes the upper limit of quantification (ULOQ). Samples with a 1171
concentration below the revised LLOQ (or above the revised ULOQ) should be reanalysed. 1172
Alternatively, the whole run may be repeated but this is not the preferred option (new). 1173
1174
23.4 Internal standard variation should be trended and used to verify result validity. Significant 1175
changes in internal standard response could signal an analytical problem which could require an 1176
investigation and/or sample reanalysis. Significant differences between the internal standard results 1177
of CC standards or QC standards vs samples could also signal problems affecting the reliability of 1178
the results (new). 1179
1180
Page 26
Working document QAS/15.622
page 26 23.5 Full audit trails should be activated at all times and on all analytical instruments in a given 1181
facility, both prior, during and after the method validation and the study of interest (new). 1182
1183
23.6 All original analytical raw data (e.g. calculations, chromatograms and their associated audit 1184
trails, etc.) should be documented in a manner that will ensure traceability with respect to the 1185
sample number, equipment used, date and time of analysis and the name(s) of the technician(s). If 1186
several audit trail files are generated all should be retained (e.g. results table audit trail, project 1187
audit trail, instrument audit trail) (revised). 1188
1189
23.7 Each data point should be traceable to a specific sample, including sample number, time of 1190
collection of the sample, time of centrifugation, if applicable, time when the sample was placed in 1191
the freezer, time of sample analysis, etc., to be able to determine whether any aberrant results might 1192
have been due to sample mishandling. 1193
1194
24. GOOD LABORATORY PRACTICES (additional title) 1195
1196 24.1 Although most GLP guidelines (5) apply formally only to non-clinical safety studies, 1197
general principles of GLP should also be followed in the analysis of biological samples from 1198
clinical trials. 1199
1200
24.2 Analysis should be performed in a laboratory with established quality assurance systems 1201
(revised). 1202
1203
24.3 Key sample storage systems or other areas requiring environmental controls should be 1204
adequately qualified, calibrated and maintained. There should be an alarm system or an adequate 1205
monitoring system to control the temperature of the critical stage areas and key sample storage 1206
systems, such as freezers. If there is an automatic alarm system it has to be tested regularly for its 1207
functionality. The daily monitoring and all the alarm checks should be documented. There should 1208
be a system in place to ensure that timely and appropriate action is taken following an alarm 1209
(revised paragraph). 1210
1211
24.4 For purposes of qualification and requalification the temperature mapping of the freezers 1212
and refrigerators should be run for between 24 and 72 hours, or more if justified. Remapping 1213
should be done after any significant modifications to the storage units (new). 1214
1215
24.5 Appropriate repairs and/or sample transfer to other equivalent storage units should be 1216
considered whenever an analysis of temperature monitoring records show unexplained variability 1217
outside normal operating limits (new). 1218
1219
24.6 Balances, other measuring devices and equipment/instruments used during the conduct of a 1220
trial should be periodically calibrated and verified before use (new). 1221
1222
24.7. There should be SOPs for the operation, use, calibration, checks and preventive 1223
maintenance of equipment. Records should be maintained. Equipment used during the course of the 1224
trial should be identified to be able to verify that they have been appropriately qualified and 1225
calibrated (revised). 1226
1227
24.8 Chemicals, reference substances, reagents, solvents and solutions should be labelled to 1228
indicate identity, purity concentration (if appropriate), expiry date and specific storage instructions. 1229
Information concerning source, preparation date and stability should be available (revised). 1230
Page 27
Working document QAS/15.622
page 27 1231
1232
PHARMACOKINETIC, STATISTICAL CALCULATIONS AND REPORTING 1233
SECTION 1234
1235
25. PHARMACOKINETIC AND STATISTICAL CALCULATIONS 1236 1237
25.1 The statistical model underlying any primary BE analysis should be stated in the protocol 1238
and/or a statistical analysis plan. It should be made clear which factors are fixed and which are 1239
random. It should be stated if the model is a mixed effects model, a normal linear model, etc. 1240
1241
25.2 Calculations should be made by qualified persons (see Section 8: Personnel). 1242
1243
25.3 The means of performing pharmacokinetic and statistical calculations (both software and 1244
scripts) should be specified in the study protocol and/or a statistical analysis plan and data analysis 1245
should conform to these requirements (revised). 1246
1247
25.4 Calculations should be made using validated software and scripts. Software and scripts 1248
should be validated or qualified using an SOP, ideally with datasets of varying complexity and with 1249
the alpha level(s) actually in use. Self-designed software should be demonstrated as suitable for 1250
intended use. For guidance on the use of computerized systems (5) (see Section 4: Computer 1251
Systems) (revised). 1252
1253
25.5 Data values input should be double-checked by a second qualified person as per an SOP 1254
(new). 1255
1256
25.6 The manner in which AUCinf is derived should be documented and defined in an SOP (i.e. 1257
how the points used for extrapolation are selected) (new). 1258
1259
25.7 A database of trial records should be maintained and it should preferably be locked as soon 1260
as possible after completion of the study. Once it is locked the study can be unblinded and 1261
statistical analysis performed. The dates of locking and statistical analysis should be documented, 1262
mentioned in the study report and the process should be defined in a suitable procedure (new). 1263
1264
26. CLINICAL STUDY REPORT 1265 1266
26.1 The clinical study report should reflect the complete study procedures and results in an 1267
accurate manner. 1268
1269
26.2 The clinical study report should be well written and presented. All deviations from the 1270
protocol in the performance of the study should be reported. 1271
1272
26.3 There should be no discrepancies between the results stated in the report and the actual 1273
original (raw) data. 1274
1275
26.4 The report should comply with regulatory requirements as applicable, and be presented in a 1276
standard format. The report should cover at least the items listed in the ICH guideline (8). 1277
1278
Page 28
Working document QAS/15.622
page 28 26.5 The study report should include a report on the bioanalytical part of the trial, including a 1279
description of the bioanalytical method used and the validation report of this method. 1280
1281
26.6 The procedure for approval of the clinical study report by the investigator and sponsor and 1282
for approval of the bioanalytical report by the study director should be specified (revised). 1283
1284
26.7 The report should be approved (signed and dated) by the responsible persons. 1285
1286
26.8 All monitoring and audit reports should be available before release of the final study report 1287
(revised). 1288
1289
REFERENCES (revised) 1290
1291
1. Note for Guidance on Good Clinical Practice (CPMP/ICH/135/95), ICH Topic E6, Guideline 1292
for Good Clinical Practice, Step 5, Consolidated Guideline 1.5.96. The European Agency for 1293
the Evaluation of Medicinal Products (EMEA) 2002. 1294
1295
2. Directive 2001/20/EC of the European Parliament and the Council, "approximation of the 1296
laws, regulations and administrative provisions of the Member States relating to the 1297
implementation of good clinical practice in the conduct of clinical trials on medicinal 1298
products for human use". Official Journal of the European Communities of 4 April 2001. 1299
1300
3. Multisource (generic) pharmaceutical products: guidelines on registration requirements to 1301
establish interchangeability. In: Expert Committee on Specifications for Pharmaceutical 1302
Preparations. Forty-ninth report. World Health Organization, Geneva. WHO Technical 1303
Report Series, No. 992, Annex 7, 2015, pp. 347–390. 1304
1305
4. Guidelines for good clinical practice (GCP) for trials on pharmaceutical products, WHO 1306
Technical Report Series, No. 850, 1995 (pp. 97–137). 1307
1308
5. WHO Handbook on Good Laboratory Practice/OECD Series on Principles of Good 1309
Laboratory Practice and Compliance Monitoring, Number 1: OECD Principles on Good 1310
Laboratory Practice (as revised in 1997). Organization for Economic Co-operation and 1311
Development. ENV/MC/CHEM(98)17. 26.Jan, 1998. 1312
1313
6. The Good Automated Manufacturing Practice (GAMP) Guide for Validation of Automated 1314
Systems in Pharmaceutical Manufacture (GAMP4). ISPE - International Society for 1315
Pharmaceutical Engineering, December 2001. 1316
1317
7. Guidelines on Bioanalytical Method Validation EMEA/CHMP/EWP/192217/2009 Rev.1 1318
Corr.* Committee for Medicinal Products for Human Use (CHMP), 1 February 2012. 1319
1320
8. WHO Operational guidelines for Ethics Committees that review biomedical research (7). 1321
WHO, TDR/PRD/ETHICS/2000.1. http://www.who.int/tdr/publications/ 1322
documents/ethics.pdf?ua=1). 1323
1324
1325
Page 29
Working document QAS/15.622
page 29
APPENDIX 1 (REVISED) 1326 EXAMPLES OF THE LIST OF STANDARD OPERATING PROCEDURES AT THE CONTRACT 1327
RESEARCH ORGANIZATION 1328 1329
The following are examples of the list of standard operating procedures (SOPs) that should be 1330
used at contract research organizations (CROs). This list is not exhaustive as other procedures 1331
may be necessary depending on the functional and compliance requirements at each facility. 1332
All of the documents at the CRO related to a bioequivalence/clinical trial should be controlled 1333
(version date, approved, etc.) documents. This control is easier if the documents are in the 1334
SOP format or are appended to SOPs. 1335
SOPs should be in place at least for all the critical and major operations in the bioequivalence 1336
(BE)/clinical trial. 1337
Number and name of SOP 1338
1. Conduct of BE study. 1339
2. Archiving and retrieval of documents related to BE study. 1340
3. Quality assurance of the BE study; audits of clinical and bioanalytical part of the study 1341
and the study report. 1342
4. Study files. 1343
5. Preparation and review of the protocol for the study. 1344
6. Amendment to the protocol for the study. 1345
7. Protocol deviations/violation recording and reporting. 1346
8. Sponsor/CRO quality assurance agreement in conducting the BE study. 1347
9. Study approval process by ethical committee . 1348
10. Bioavailability (BA)/BE report. 1349
11. Study report. 1350
12. Written informed consent. 1351
13. Obtaining written informed consent for screening from study volunteers. 1352
14. Allotment of identification numbers to volunteers at various stages in BE study. 1353
15. Investigator’s brochure (IB). 1354
16. Case-report form (CRF). 1355
17. Preparation of CRF, review and completion. 1356
18. Data collection and CRF completion. 1357
19. Adverse/serious adverse event monitoring, recording and reporting. 1358
20. Organization chart of the study. 1359
21. Training of the personnel. 1360
22. Responsibilities of the members of the research team. 1361
23. Monitoring of the study by the sponsor. 1362
24. Conduct of pre-study meeting. 1363
25. Study start-up. 1364
26. Subject management. 1365
27. SOP on mobilization of individuals for registration into volunteer bank. 1366
28. Eligibility criteria for registration and registration of individuals into volunteer bank. 1367
29. Handling of subject withdrawal. 1368
30. Allotment of identification numbers to volunteers at various stages in the biostudy. 1369
31. Screening of enrolled volunteers for the study. 1370
32. Collection of urine samples of subjects for detection of drugs of abuse and 1371
transportation of samples to pathology laboratory. 1372
33. Custodian duties. 1373
Page 30
Working document QAS/15.622
page 30 34. Payments to research subjects for BA/BE studies. 1374
35. Procedures for entry into and exit from clinical unit 1375
36. Handling of subject check-in and check-out. 1376
37. Housekeeping at clinical unit. 1377
38. Planning, preparation, evaluation and service of standardized meals for bio-studies. 1378
39. Distribution of meals to study subjects. 1379
40. Operation and maintenance of nurse calling system. 1380
41. Administration of oral solid dosage form of the drug to human subjects during BA/BE 1381
study. 1382
42. Cannulation of study subjects 1383
43. Collection of blood samples from study subjects. 1384
44. System for number of bio-samples. 1385
45. Recording of vital signs of subjects. 1386
46. Operation and verification of fire alarm system. 1387
47. Oxygen administration to subject from medical oxygen cylinder. 1388
48. Emergency care of subjects during BA/BE study. 1389
49. Availability of ambulance during BA/BE study. 1390
50. Centrifugation and separation of blood samples. 1391
51. Storage of plasma/serum samples. 1392
52. Segregation of bio-samples. 1393
53. Transfer of plasma/serum samples to bioanalytical laboratory. 1394
54. Procedures for washing glassware. 1395
55. Recording temperature and relative humidity of rooms. 1396
56. Instruction on operation and maintenance procedures for all the equipment in the 1397
clinical unit. 1398
57. Numbering the equipment and log books for use in the clinical unit. 1399
58. Control of access to pharmacy. 1400
59. Pharmacy area requirements. 1401
60. Authorization related to drug storage, dispensing and retrieval from storage for BE study. 1402
61. Study drug receipt, return and accountability documentation. 1403
62. Study drug receipt and return procedures. 1404
63. Storage of drugs in the pharmacy. 1405
64. Line clearance before and after dispensing. 1406
65. Documentation of line clearance and dispensing; packaging records and release of 1407
dispensed drugs. 1408
66. Retention of samples of study drugs. 1409
67. Disposal of archived study drugs. 1410
68. Disposal of biological materials. 1411
69. Procedures for bioanalytical laboratory (SOPs for the different equipment, analytical 1412
methods, reagent preparation). 1413
70. Out-of-specification (OOS) in the laboratory 1414
71. Acceptance criteria for analytical runs: acceptance of calibration curves, acceptance of 1415
the runs based on QC samples results. 1416
72. Chromatographic acceptance criteria, chromatogram integration. 1417
73. Sample re-assay. 1418
74. Pharmacokinetic data from bioanalytical data. 1419
75. Statistics in the BE study. 1420
i
***